 Yangyang Wang1
Yangyang Wang1 Jihan Wang
Jihan Wang
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Oncol., 27 October 2021
Sec. Gastrointestinal Cancers: Gastric and Esophageal Cancers
Volume 11 - 2021 | https://doi.org/10.3389/fonc.2021.754788
This article is part of the Research TopicThe Microbiome in Hepatobiliary and Intestinal DiseaseView all 18 articles
Altered human microbiome characteristic has been linked with esophageal carcinoma (ESCA), analysis of microbial profiling directly derived from ESCA tumor tissue is beneficial for studying the microbial functions in tumorigenesis and development of ESCA. In this study, we identified the intratumor microbiome signature and investigated the correlation between microbes and clinical characteristics of patients with ESCA, on the basis of data and information obtained from The Cancer Microbiome Atlas (TCMA) and The Cancer Genome Atlas (TCGA) databases. A total of 82 samples were analyzed for microbial composition at various taxonomic levels, including 40 tumor samples of esophageal squamous cell carcinoma (ESCC), 20 tumor samples of esophageal adenocarcinoma (EAD), and 22 adjacent normal samples. The results showed that the relative abundance of several microbes changed in tumors compared to their paired normal tissues, such as Firmicutes increased significantly while Proteobacteria decreased in tumor samples. We also identified a microbial signature composed of ten microbes that may help in the classification of ESCC and EAD, the two subtypes of ESCA. Correlation analysis demonstrated that compositions of microbes Fusobacteria/Fusobacteriia/Fusobacteriales, Lactobacillales/Lactobacillaceae/Lactobacillus, Clostridia/Clostridiales, Proteobacteria, and Negativicutes were correlated with the clinical characteristics of ESCA patients. In summary, this study supports the feasibility of detecting intratumor microbial composition derived from tumor sequencing data, and it provides novel insights into the roles of microbiota in tumors. Ultimately, as the second genome of human body, microbiome signature analysis may help to add more information to the blueprint of human biology.
Esophageal carcinoma (ESCA) is a common type of cancer and one of the leading causes of mortality associated with the gastrointestinal tract. There are two main histological subtypes of ESCA, esophageal squamous cell carcinoma (ESCC) and esophageal adenocarcinoma (EAD). The two subtypes differ significantly in incidence, geographic distribution, and etiology. ESCC accounts for almost 90% of the ESCA incidence each year, and the geographic distribution of ESCC varies greatly, with the highest incidence rates occurring in Asia, especially China. Approximately half of all ESCC cases worldwide is reported in China, and these high rates are mainly due to China’s large population (1). In the West, EAD represents the main histological subtype and its incidence has increased rapidly over the past 30 years (2). Although the prognosis of EAD has slightly improved over the last few decades, it is still worse than that of most other cancer types. Moreover, since most patients are diagnosed at late stages, the motility of esophageal carcinoma remains high; in most countries, approximately only 15%-25% of patients survive 5 years.
The etiology of ESCA is multifactorial and includes cigarette smoking, alcohol consumption, and low fruit/vegetable intake for ESCC and gastroesophageal reflux disease (GERD), Barrett’s esophagus (BE), obesity, low fruit/vegetable intake, and cigarette smoking for EAD. The current understanding of these risk factors cannot fully explain the etiology of ESCC and EAD. Microbiota have recently emerged as novel tumorigenesis regulators and biomarkers in disease and multiple types of cancer, including ESCA (3–6). Microbial dysbiosis contributes to cancer susceptibility through complex mechanisms, including inducing inflammation and immune disfunction and interfering with anticancer drug pharmacodynamics. Dysbiosis of the gut microbiota (GM) has been studied in ESCA patients (3, 7). In addition, investigation of the esophageal microbiota is a relatively new approach in the field of ESCA (8). Several studies have indicated alterations in the esophageal microbiota in esophagitis, BE, EAD, and ESCC (7, 9, 10). There is evidence that the microbial composition of the esophagus is diverse, with gram-positive bacteria dominating in healthy conditions, while gram-negative bacteria predominating in disease status including GERD and BE (8). Exploring esophageal microbiota changes will help us better understand the tumor pathophysiology and provide potential diagnostic and/or therapeutic approaches for ESCA.
Recently, The Cancer Microbiome Atlas (TCMA) revealed a pan-cancer analysis identifying tissue-resident microbiota (11). The sample types that were analyzed for microbial prevalence were derived from The Cancer Genome Atlas (TCGA) program, and over 20,000 primary cancer and matched normal samples spanning 33 cancer types were molecularly characterized. Until now, the TCMA has been a resource for exploring the tissue-resident microbiota prevalence in several cancer types, including tissues of the oropharynx, esophagus, stomach, and colorectum. Previously, we explored the microbiota signature in four major types of gastrointestinal cancer, and the results demonstrated that the microbial profile is highly site-specific and notably differed between upper and lower gastrointestinal tumors (12). Several other studies have also investigated the intratumor microbiota derived from TCGA sequences of different cancer types (13, 14). In the study of a TCGA breast cancer cohort, the results indicated an increased Proteobacteria presence in tumor tissues, while the composition of Actinobacteria was elevated in the adjacent normal tissues (15). Rodriguez et al. detected the global microbial composition in tumor and adjacent normal tissues across 9 TCGA cancer cohorts (16). Microbiome analysis from tumor tissues as well as human blood samples will also reveal a new class of microbial-based cancer diagnostics (17). Overall, exploring the intratumor microbial signature will help improve our knowledge of the host-microbiota interaction, which is important to understand the linkage of dysbiosis with chronic inflammation and processes that influence tumorigenesis.
Understanding the relationship between clinical phenotype information and multiomics data such as the genome or microbiome is critical for human biological and medical research. To the best of our knowledge, no studies have been conducted to investigate the comprehensive microbial signature or its relationship with the clinicopathological characterization of ESCA. Here, we profiled the microbiome of patients with ESCA from the TCMA, and also analyzed the clinical phenotype and survival data of the corresponding samples from the TCGA. The global microbial composition at the phylum, class, order, family, and genus levels of tumor and noncancerous adjacent normal tissues was calculated to analyze the differential microbes. We further evaluated the correlation between the microbes and the clinical variables of the tumors. Specifically, we identified the microbial signatures related to cancer subtype, tumor stage, and survival status. We believe that the intratumor microbial study will provide a better understanding of dysbiosis and establish a new foundation for studying host-microbiota interactions and the role of microbiota in the tumorigenesis of esophageal carcinoma.
In this study, the microbiota profiles of samples from 82 cases (including 40 ESCC tumors, 20 EAD tumors, and 22 noncancerous adjacent tissues used as normal samples) at the phylum, class, order, family, and genus levels were obtained from the TCMA database (https://tcma.pratt.duke.edu/); the corresponding clinical phenotype information and survival data for the 82 patients were obtained from the TCGA program (for phenotype information, https://gdc-hub.s3.us-east-1.amazonaws.com/download/TCGA-ESCA.GDC_phenotype.tsv.gz; for survival data, https://gdc-hub.s3.us-east-1.amazonaws.com/download/TCGA-ESCA.survival.tsv). Figure 1A displays an overview of the study design, and the clinical information about the patient samples is summarized in Table 1.
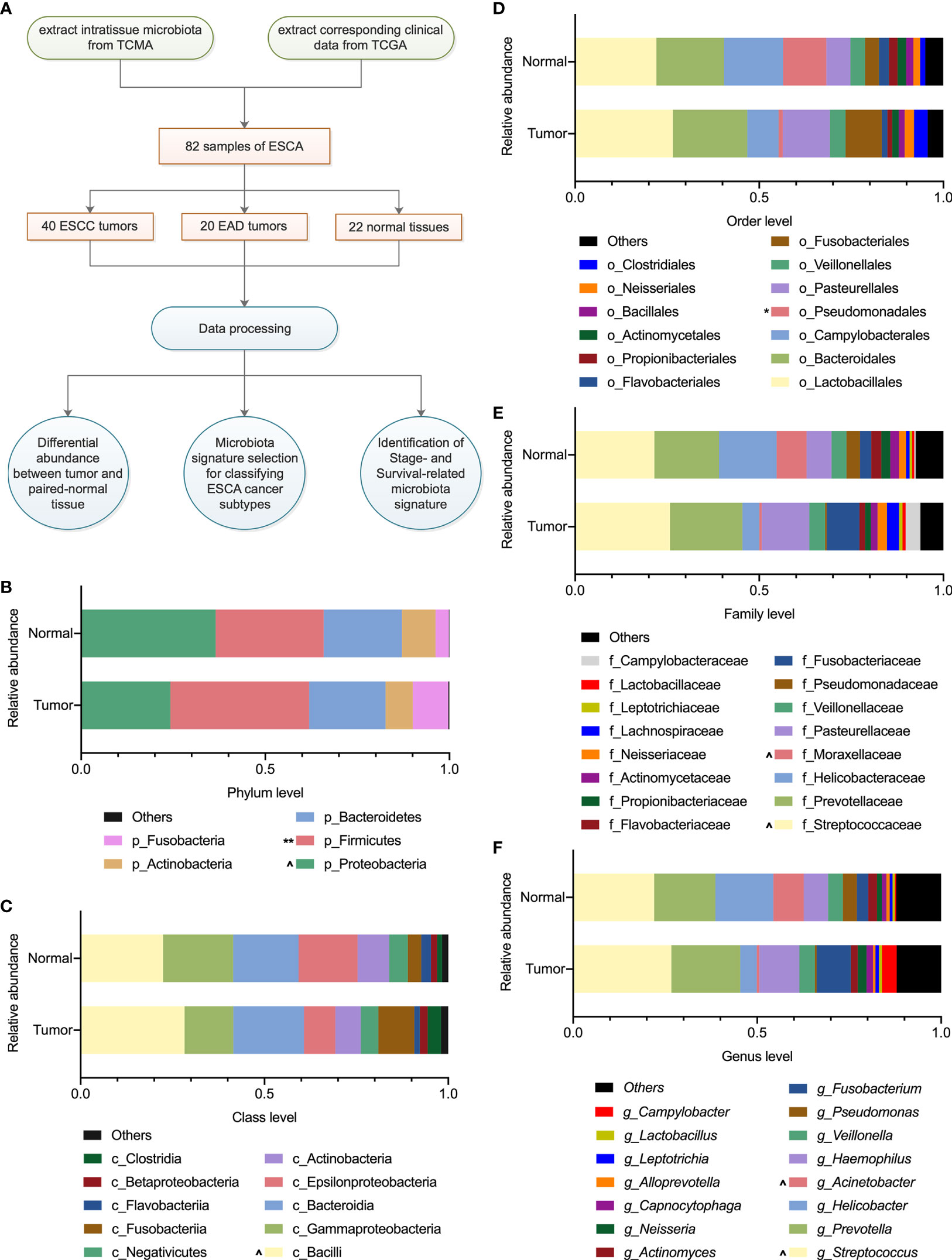
Figure 1 Overview of the study design and microbiota profiling. (A) Schematic overview of the study design. TCMA, the cancer microbiome atlas; TCGA, the cancer genome atlas; ESCA, esophageal carcinoma; ESCC, esophageal squamous cell carcinoma; EAD, esophageal adenocarcinoma. (B–F) The composition of microbiota with an average abundance > 1% in tumor and paired normal tissues at the phylum, class, order, family, and genus levels, respectively. The relative microbial abundance in tumors compared with normal samples, ^0.1 < P < 0.01, *P < 0.05, **P < 0.01 in paired-samples t-test.

Table 1 Clinical characteristics of cases in this study (information derived from the TCGA database).
The relative abundance of the microbiota composition at the phylum, class, order, family, and genus levels of each sample was calculated, and the microbial composition with an average relative abundance > 1% was selected for further analysis. In this study, there were 22 pairs of strictly matched tumor-adjacent normal samples of ESCA. The paired-samples t-test was used to analyze the differential microbial composition in tumors and their normal tissues, with a false discovery rate (FDR)-adjusted P-value < 0.05 considered significant. In addition, linear discriminant analysis effect size (LEfSe) analysis was performed by using OECloud tools at https://cloud.oebiotech.cn. Specifically, the nonparametric factorial Kruskal-Wallis (KW) sum-rank test and Wilcoxon rank-sum test were used to identify taxa biomarkers for tumor and normal samples, and linear discriminant analysis (LDA) was further performed to evaluate the microbial effects for each group. The microbes with LDA values > 2 and P < 0.1 were considered significantly enriched in that group.
There were 40 ESCC and 20 EAD tumor samples in the esophageal carcinoma group. We investigated whether these two cancer subtypes could be classified based on the tumor microbiota profile. The SHapley Additive exPlanations (SHAP) (18, 19) theoretic approach was performed for microbial feature selection to identify the more important microbial profile, which may predict the classification of the two different cancer subtypes. The global microbiota (as the variables to distinguish the two cancer subtypes) importance scores were evaluated and visualized by SHAP, and we then selected the top ten most important microbial features for further analysis. Principal component analysis (PCA) and partial least squares discrimination analysis (PLS-DA) were performed by using the packages “FactoMineR” and “mixOmics” in R version 4.0.2, respectively.
The microbiota profile of ESCA at the phylum, class, order, family, and genus levels derived from the TCMA database and the corresponding clinical information of all the samples obtained from the TCGA database were integrated for correlation analysis. Specifically, the Pearson cor.test () function in R version 4.0.2 was performed to analyze the correlation between the relative abundance of specific microbiota and tumor stage. Kaplan‐Meier survival analysis was performed to assess the survival-related microbiota. The “survival” and “survminer” packages in R version 4.0.2 were used for survival analysis and curve visualization based on the microbial composition.
There were 82 samples of esophageal carcinoma (Table 1) in the TCMA database. A total of 11, 22, 38, 67, and 221 taxa were obtained for each sample at the phylum, class, order, family, and genus levels, respectively. Table S1 summarizes the global microbial profiling at each taxonomic level. We then explored the differential microbial compositions between tumor and normal tissues. Overall, there were 5, 10, 13, 16, and 15 microbial taxa with an average relative abundance > 1% at the phylum, class, order, family, and genus levels, respectively (Figures 1B–F). At the phylum level, the intratissue microbiota was dominated by Proteobacteria (36.5% for normal tissue, 24.2% for tumor tissue) and Firmicutes (29.3% for normal tissue, 37.7% for tumor tissue), followed by Bacteroidetes (21.2% for normal tissue, 20.7% for tumor tissue), Actinobacteria (9.2% for normal tissue, 7.4% for tumor tissue), and Fusobacteria (3.6% for normal tissue, 9.7% for tumor tissue). The microbial composition of Firmicutes increased significantly (P < 0.01), while that of Proteobacteria decreased (0.1 < P < 0.05) in tumor samples compared with their paired normal tissues (Figure 1B). As Figures 1C–F show, the difference in microbial composition profiling in tumors was not obviously significant compared with that in normal tissues, except that the relative abundance of Pseudomonadales was less abundant in tumors than in normal samples at the order level (P < 0.05, Figure 1D).
LEfSe analysis helps to identify specific enriched microbial biomarkers for different groups. As Figure 2 shows, the compositional abundances of Fusobacteria/Fusobacteriia/Fusobacteriales were higher in tumor tissues, while compositional abundances of Proteobacteria, Moraxellaceae, Acinetobacter, and Flavobacteriia/Flavobacteriales/Flavobacteriaceae were enriched in normal tissues. Specifically, the compositional positive ratio of order Fusobacteriales was 63.6% (14 of 22) for tumor and 68.2% (15 of 22) for normal tissue, while the average abundance of Fusobacteriales for the positive samples was higher (0.1 < P < 0.05) in tumor (14 positive samples with an average abundance of 15.4%) than that in normal tissues (15 positive samples with an average abundance of 5.6%). In contrast, a higher compositional positive ratio of Flavobacteriaceae (5 of 22 for tumor vs. 12 of 22 for normal, P < 0.05) and Moraxellaceae (2 of 22 for tumor vs. 6 of 22 for normal, P < 0.01) at the family level as well as Acinetobacter (2 of 22 for tumor vs. 6 of 22 for normal, P < 0.01) at the genus level was detected more in normal tissues than in tumors.

Figure 2 LEfSe analysis identifying tumor- and normal-enriched microbiota. (A) The LDA score of specific microbial taxa in the tumor group and normal group. Threshold LDA score is 2. (B) Cladogram showing tumor- and normal-enriched microbial taxa. LEfSe, linear discriminant analysis effect size; LDA, linear discriminant analysis.
As the ESCA tumor samples were histologically subdivided into ESCC and EAD subtypes, we further investigated whether the intratumor microbial signature was somehow subtype-correlated in esophageal carcinoma. The SHAP (Shapley additive explanations) approach was applied to select the most valuable features in predicting the different groups, as it provided reference information about feature ranking and feature selection. Previously, a total of 59 microbial taxa were identified with an average relative composition > 1% at different taxonomic levels. To prioritize microbes of the 59 taxa, we relied on feature importance (contribution) obtained from SHAP to evaluate the whole importance value of each individual microbe in predicting cancer subtypes. A signature containing the top 10 microbial features was identified from the 59 microbial taxa as predictable factors, including Actinobacteria, Fusobacteria, Bacilli, Epsilonproteobacteria, Negativicutes, Bacillales, Pasteurellales, Fusobacteriaceae, Lactobacillaceae, and Streptococcaceae (Figure 3A). PCA (Figure S1A) and PLS-DA (Figure S1B) of all 59 microbial profiles were performed before feature ranking and selection. A relatively improved separate model was observed when performing PCA (Figure 3B) and PLS-DA (Figure 3C) based on the ten microbial features obtained from SHAP after feature ranking and selection.
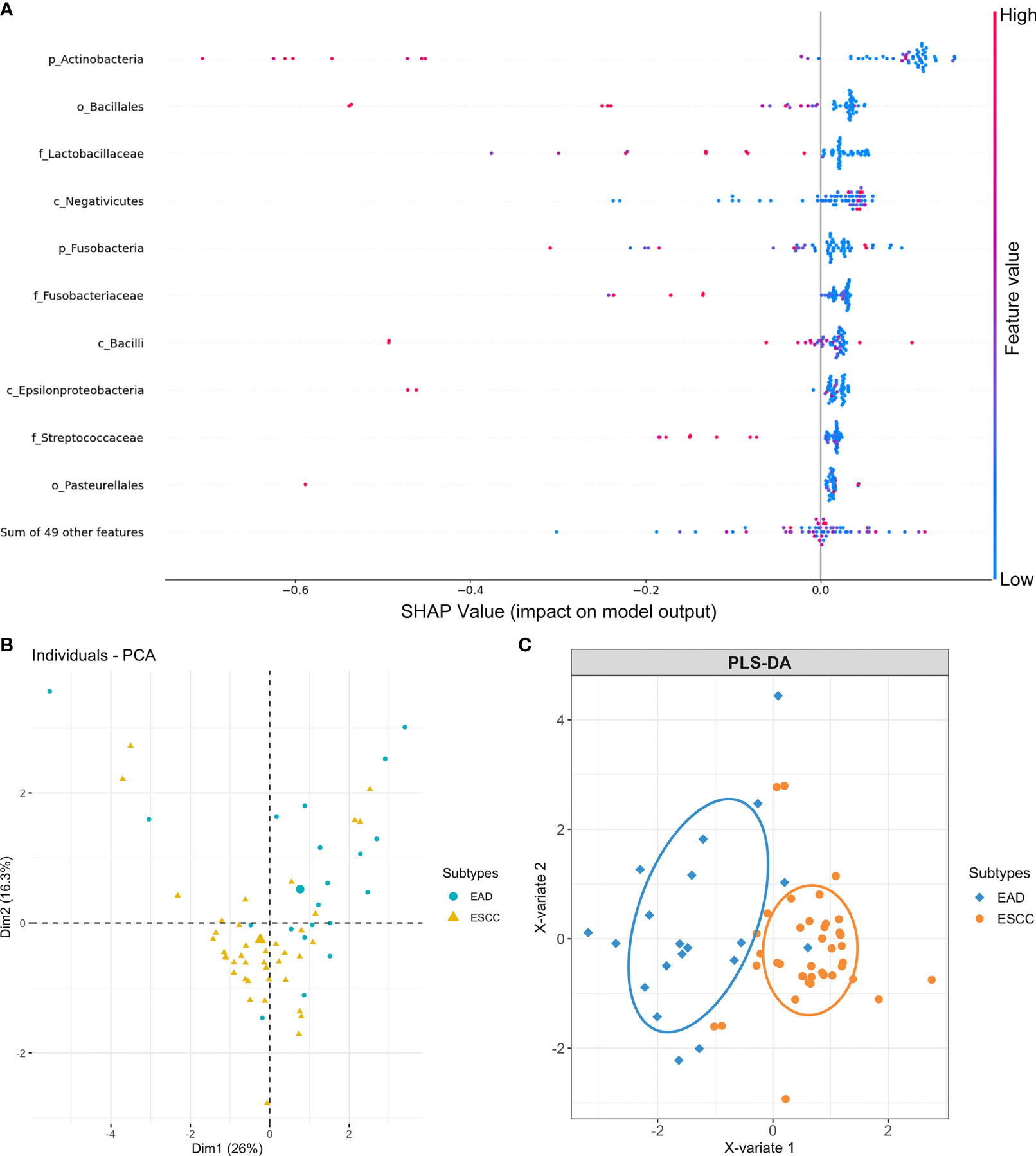
Figure 3 Microbial feature selection in predicting different cancer subtypes of esophageal carcinoma. (A) Summary plots of the importance values of the top 10 predictable microbes. (B) PCA plots and (C) PLS-DA plots based on the top 10 microbial features display the EAD and ESCC subtypes of esophageal carcinoma.
The TCGA database contains comprehensive clinical characterization of multiple types of cancer. Table 1 summarizes the clinicopathological information of the 82 cases analysed in this study. We next investigated whether there were specific microbes associated with the clinicopathological variables of ESCA patients. The results showed that the composition of Fusobacteria/Fusobacteriales was positively correlated (P < 0.01), while the relative abundance of Lactobacillales was negatively correlated (0.05 < P < 0.1) with the tumor stage status of ESCA (Figure 4). The survival analysis indicated that the enrichment of several microbes could reflect the overall survival probability of patients (Figure 5). Detailed information about the survival status of tumor patients and the microbial composition were summarized in Table S2. High abundances of Proteobacteria, Negativicutes, and Lactobacillaceae/Lactobacillus were associated with better prognosis (P < 0.05), while a high composition of Clostridia/Clostridiales and Fusobacteriia/Fusobacteriales reflected poorer prognosis (P < 0.05). The eight microbes were then applied in a multivariate Cox regression analysis. Four microbes were identified as independent prognostic factors of ESCA patients (P < 0.05), as shown in Table 2.
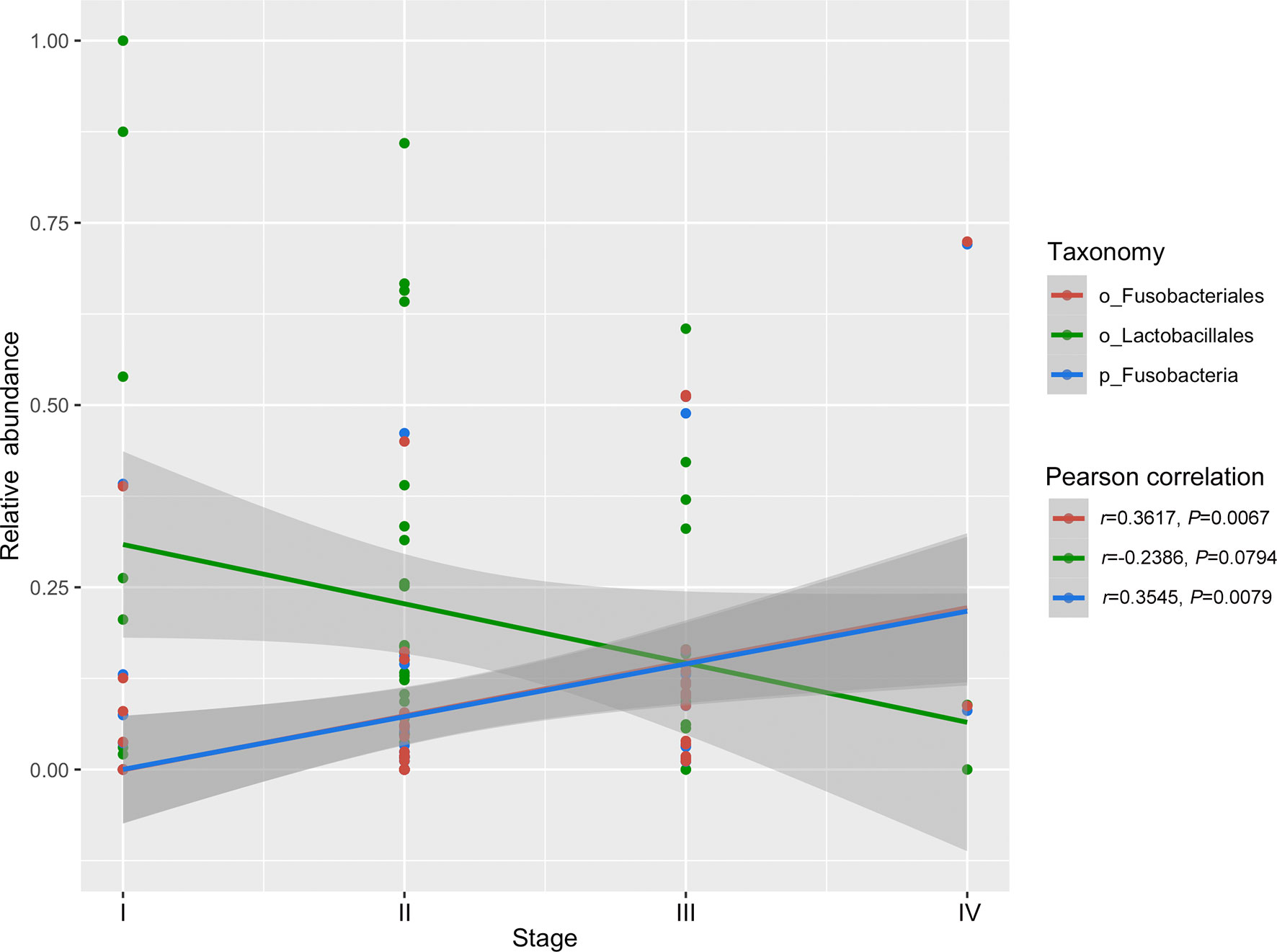
Figure 4 Correlation of specific microbes with the tumor stage status of ESCA. The analysis was performed using Pearson correlation in R.
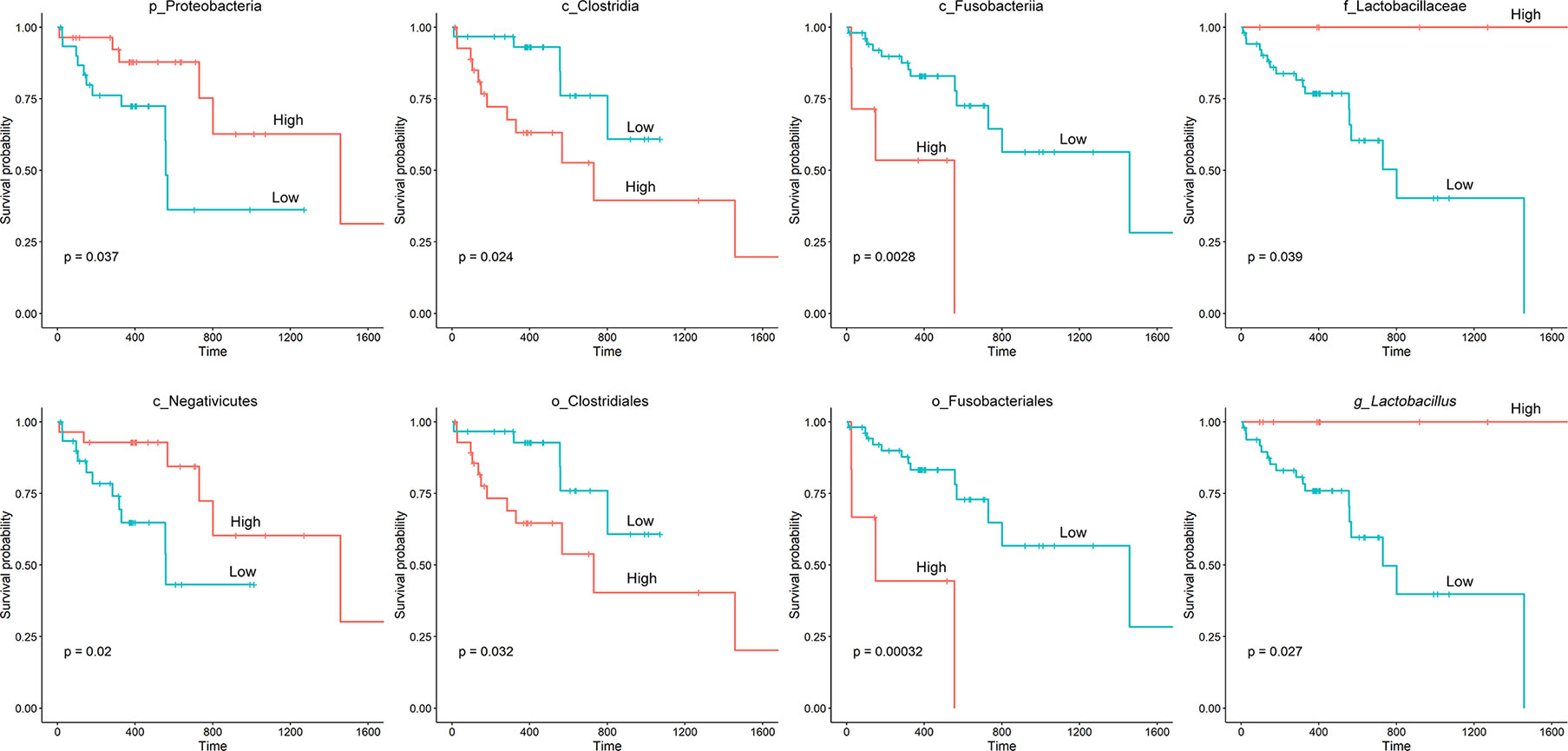
Figure 5 Kaplan‐Meier survival curves based on microbial composition. The “survival” and “survminer” packages in R were used for survival analysis. The process included determining the optimal cut-off point of variables, categorizing variables into “high” and “low” subgroups based on microbial abundance, fitting survival curves and visualization.
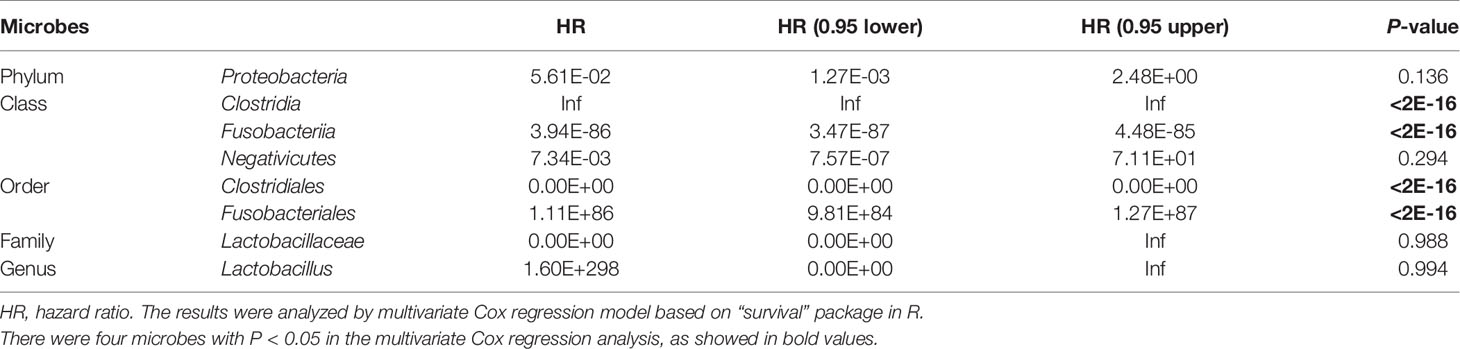
Table 2 Multivariate Cox regression analysis of the independent significance of eight microbes as prognostic factors.
It is essential to assess clinical-pathological prognostic factors such as lymph nodes, esophageal wall size and infiltration, and metastasis in relation to the microbiota under investigation. The information about lymph nodes in the current study was relatively incomplete (Table S3). As a result, we did not conduct the lymph node microbiota correlation analysis, which will require more attention in future research.
Studies on the bacterial or viral composition of human tumors using sequencing data from databases such as the TCGA have recently emerged (14, 20). Investigation of the intratumor microbiota will provide valuable information for better understanding the occurrence and progression of tumors. In addition to recent studies about microbiota changes in esophageal disease, our research performed a more comprehensive investigation of microbial characteristics in ESCA.
After microbial detection and calculation at different taxonomic levels, we found differential microbial abundance in tumor and normal samples of ESCA. Consistent with other studies, the relative abundance of Firmicutes and Fusobacteria increased, while that of Proteobacteria decreased in esophageal tumors compared with normal tissues (21–23). In another study, the enriched composition of Firmicutes and the unenriched composition of Proteobacteria were reported to be associated with BE (24). The LEfSe analysis in this study also indicated Fusobacteria/Fusobacteriia/Fusobacteriales as tumor-enriched microbes, suggesting that it might be a potential biomarker for the tumorigenesis and development of ESCA. In general, the alteration of microbial abundance at the class, order, family, and genus levels in tumors compared with adjacent normal tissue was not significant.
The human genome has been referred to as the blueprint of human biology (25). It is well established that cancer genome signature analysis helps to predict different cancer systems and their subtypes and contributes to precision medicine (26–28). The microbiome, as the second genome of the human body, plays crucial roles in health and disease (25, 29). A study reported that the intracellular microbiome of human tumors is tumor type-specific across multiple types of tumors, and intratumor bacteria or their predicted functions correlate with tumor types, subtypes, patient smoking status, and the response to immunotherapy (30). The microbiome could also be a potential biomarker/rule for subgrouping different cancer subtypes and used as a factor for exploring the complicated microenvironment components associated with tumorigenesis (31). In our research, we further identified a signature containing 10 microbial features that was somehow predictive of ESCC and EAD, the two subtypes of ESCA, by applying the SHAP approach. The human oral cavity harbours the second most abundant microbiota after the gut microbiota in the gastrointestinal tract (32). Microorganisms that exist in the oral cavity and its contiguous extensions (stopping at the distal esophagus) are all considered the oral microbiome and are altered within different oral structures and tissues (33). Here, we provide evidence that the microbial signature could be cancer subtype-related.
We then examined the relationship between specific microbes and the clinical index of ESCA patients by integrating the TCMA microbiome profile with clinical data from the TCGA. There were few links between tumor stage and microbial abundance, except that the composition of tumor-enriched Fusobacteria/Fusobacteriales was found to be positively correlated with tumor stage. Fusobacteria contribute to the formation of a proinflammatory microenvironment that promotes the colorectal neoplasia progression by recruiting tumor-infiltrating immune cells (34). In addition to its role in in colorectal cancer, Fusobacteria has been reported to be enriched in various cancer types, including oral, stomach, and breast cancer (35, 36). Studies have demonstrated that breast cancer colonized by Fusobacterium nucleatum accelerates tumor growth and metastasis (36). Furthermore, the high relative abundance of Fusobacteriia/Fusobacteriales correlated with a poorer prognosis in ESCA patients in our study, indicating that targeting Fusobacteria may be beneficial for the treatment of not only colorectal cancer but also other types of cancers. Thus, exploring the intratumor microbiome signature will facilitate the discovery of novel microbial biomarkers for cancer research.
In this study, we conducted a comprehensive analysis of the intratumor microbiome in ESCA samples. Taken together, there are differences in the abundance of several microbial taxa between the tumor and adjacent normal tissues, and the potential functions of these microbes in ESCA merit further study. We also identified the intratumor microbiota signatures that were correlated with the subtype, tumor stage, and survival status of ESCA. We expect that our research will facilitate a better understanding of the intratumor microbiome of ESCA and identify potential biomarkers for the disease, as well as provide a novel perspective on the role of the microbiome in tumors, since studies of genome variation and disease risk will necessitate the integration of human and microbial genomic data.
Our study has limitations; the number of tumor and paired normal tissue samples in the subgroups was relatively small and did not allow us to make any generalizable conclusions. Large-scale and mechanistic studies are needed to further confirm the results of this study.
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding author.
YW and JW designed, performed the study, and co-wrote the manuscript. HG and XG contributed to data and statistical analysis, and reviewed the manuscript. All authors contributed to the article and approved the submitted version.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2021.754788/full#supplementary-material
Supplementary Figure 1 | PCA and PLS-DA plots based on the 59 microbial taxa displaying EAD and ESCC subtypes of esophageal carcinoma. (A) PCA plots. (B) PLS-DA plots.
1. Abnet CC, Arnold M, Wei WQ. Epidemiology of Esophageal Squamous Cell Carcinoma. Gastroenterology (2018) 154(2):360–73. doi: 10.1053/j.gastro.2017.08.023
2. Coleman HG, Xie SH, Lagergren J. The Epidemiology of Esophageal Adenocarcinoma. Gastroenterology (2018) 154(2):390–405. doi: 10.1053/j.gastro.2017.07.046
3. Meng C, Bai C, Brown TD, Hood LE, Tian Q. Human Gut Microbiota and Gastrointestinal Cancer. Genomics Proteomics Bioinf (2018) 16(1):33–49. doi: 10.1016/j.gpb.2017.06.002
4. Helmink BA, Khan MAW, Hermann A, Gopalakrishnan V, Wargo JA. The Microbiome, Cancer, and Cancer Therapy. Nat Med (2019) 25(3):377–88. doi: 10.1038/s41591-019-0377-7
5. Picardo SL, Coburn B, Hansen AR. The Microbiome and Cancer for Clinicians. Crit Rev Oncol Hematol (2019) 141:1–12. doi: 10.1016/j.critrevonc.2019.06.004
6. Liu H, Pan LL, Lv S, Yang Q, Zhang H, Chen W, et al. Alterations of Gut Microbiota and Blood Lipidome in Gestational Diabetes Mellitus With Hyperlipidemia. Front Physiol (2019) 10:1015. doi: 10.3389/fphys.2019.01015
7. Baba Y, Iwatsuki M, Yoshida N, Watanabe M, Baba H. Review of the Gut Microbiome and Esophageal Cancer: Pathogenesis and Potential Clinical Implications. Ann Gastroenterol Surg (2017) 1(2):99–104. doi: 10.1002/ags3.12014
8. Corning B, Copland AP, Frye JW. The Esophageal Microbiome in Health and Disease. Curr Gastroenterol Rep (2018) 20(8):39. doi: 10.1007/s11894-018-0642-9
9. Lv J, Guo L, Liu JJ, Zhao HP, Zhang J, Wang JH. Alteration of the Esophageal Microbiota in Barrett's Esophagus and Esophageal Adenocarcinoma. World J Gastroenterol (2019) 25(18):2149–61. doi: 10.3748/wjg.v25.i18.2149
10. Deshpande NP, Riordan SM, Castano-Rodriguez N, Wilkins MR, Kaakoush NO. Signatures Within the Esophageal Microbiome Are Associated With Host Genetics, Age, and Disease. Microbiome (2018) 6(1):227. doi: 10.1186/s40168-018-0611-4
11. Dohlman AB, Arguijo Mendoza D, Ding S, Gao M, Dressman H, Iliev ID, et al. The Cancer Microbiome Atlas: A Pan-Cancer Comparative Analysis to Distinguish Tissue-Resident Microbiota From Contaminants. Cell Host Microbe (2021) 29(2):281–98.e285. doi: 10.1016/j.chom.2020.12.001
12. Wang J, Wang Y, Li Z, Gao X, Huang D. Global Analysis of Microbiota Signatures in Four Major Types of Gastrointestinal Cancer. Front Oncol (2021) 11:685641. doi: 10.3389/fonc.2021.685641
13. Chakladar J, Kuo SZ, Castaneda G, Li WT, Gnanasekar A, Yu MA, et al. The Pancreatic Microbiome Is Associated With Carcinogenesis and Worse Prognosis in Males and Smokers. Cancers (Basel) (2020) 12(9):2672. doi: 10.3390/cancers12092672
14. Uriarte-Navarrete I, Hernandez-Lemus E, de Anda-Jauregui G. Gene-Microbiome Co-Expression Networks in Colon Cancer. Front Genet (2021) 12:617505. doi: 10.3389/fgene.2021.617505
15. Thompson KJ, Ingle JN, Tang X, Chia N, Jeraldo PR, Walther-Antonio MR, et al. A Comprehensive Analysis of Breast Cancer Microbiota and Host Gene Expression. PloS One (2017) 12(11):e0188873. doi: 10.1371/journal.pone.0188873
16. Rodriguez RM, Hernandez BY, Menor M, Deng Y, Khadka VS. The Landscape of Bacterial Presence in Tumor and Adjacent Normal Tissue Across 9 Major Cancer Types Using TCGA Exome Sequencing. Comput Struct Biotechnol J (2020) 18:631–41. doi: 10.1016/j.csbj.2020.03.003
17. Poore GD, Kopylova E, Zhu Q, Carpenter C, Fraraccio S, Wandro S, et al. Microbiome Analyses of Blood and Tissues Suggest Cancer Diagnostic Approach. Nature (2020) 579(7800):567–74. doi: 10.1038/s41586-020-2095-1
18. Ogami C, Tsuji Y, Seki H, Kawano H, To H, Matsumoto Y, et al. An Artificial Neural Network-Pharmacokinetic Model and its Interpretation Using Shapley Additive Explanations. CPT Pharmacometrics Syst Pharmacol (2021) 10(7):760–8. doi: 10.1002/psp4.12643
19. Tseng PY, Chen YT, Wang CH, Chiu KM, Peng YS, Hsu SP, et al. Prediction of the Development of Acute Kidney Injury Following Cardiac Surgery by Machine Learning. Crit Care (2020) 24(1):478. doi: 10.1186/s13054-020-03179-9
20. Cantalupo PG, Katz JP, Pipas JM. Viral Sequences in Human Cancer. Virology (2018) 513:208–16. doi: 10.1016/j.virol.2017.10.017
21. Zhao Q, Yang T, Yan Y, Zhang Y, Li Z, Wang Y, et al. Alterations of Oral Microbiota in Chinese Patients With Esophageal Cancer. Front Cell Infect Microbiol (2020) 10:541144. doi: 10.3389/fcimb.2020.541144
22. Yamamura K, Baba Y, Nakagawa S, Mima K, Miyake K, Nakamura K, et al. Human Microbiome Fusobacterium Nucleatum in Esophageal Cancer Tissue Is Associated With Prognosis. Clin Cancer Res (2016) 22(22):5574–81. doi: 10.1158/1078-0432.CCR-16-1786
23. Liu Y, Baba Y, Ishimoto T, Iwatsuki M, Hiyoshi Y, Miyamoto Y, et al. Progress in Characterizing the Linkage Between Fusobacterium Nucleatum and Gastrointestinal Cancer. J Gastroenterol (2019) 54(1):33–41. doi: 10.1007/s00535-018-1512-9
24. May M, Abrams JA. Emerging Insights Into the Esophageal Microbiome. Curr Treat Options Gastroenterol (2018) 16(1):72–85. doi: 10.1007/s11938-018-0171-5
25. Grice EA, Segre JA. The Human Microbiome: Our Second Genome. Annu Rev Genomics Hum Genet (2012) 13:151–70. doi: 10.1146/annurev-genom-090711-163814
26. Linehan WM, Ricketts CJ. The Cancer Genome Atlas of Renal Cell Carcinoma: Findings and Clinical Implications. Nat Rev Urol (2019) 16(9):539–52. doi: 10.1038/s41585-019-0211-5
27. Cancer Genome Atlas N. Comprehensive Molecular Portraits of Human Breast Tumours. Nature (2012) 490(7418):61–70.
28. Dienstmann R, Vermeulen L, Guinney J, Kopetz S, Tejpar S, Tabernero J. Consensus Molecular Subtypes and the Evolution of Precision Medicine in Colorectal Cancer. Nat Rev Cancer (2017) 17(2):79–92. doi: 10.1038/nrc.2016.126
29. Parida S, Sharma D. The Microbiome-Estrogen Connection and Breast Cancer Risk. Cells (2019) 8(12). doi: 10.3390/cells8121642
30. Nejman D, Livyatan I, Fuks G, Gavert N, Zwang Y, Geller LT, et al. The Human Tumor Microbiome Is Composed of Tumor Type-Specific Intracellular Bacteria. Science (2020) 368(6494):973–80. doi: 10.1126/science.aay9189
31. Chen L, Li Z, Zeng T, Zhang YH, Liu D, Li H, et al. Identifying Robust Microbiota Signatures and Interpretable Rules to Distinguish Cancer Subtypes. Front Mol Biosci (2020) 7:604794. doi: 10.3389/fmolb.2020.604794
32. Verma D, Garg PK, Dubey AK. Insights Into the Human Oral Microbiome. Arch Microbiol (2018) 200(4):525–40. doi: 10.1007/s00203-018-1505-3
33. Dewhirst FE, Chen T, Izard J, Paster BJ, Tanner ACR, Yu WH, et al. The Human Oral Microbiome. J Bacteriol (2010) 192(19):5002–17. doi: 10.1128/JB.00542-10
34. Kostic AD, Chun E, Robertson L, Glickman JN, Gallini CA, Michaud M, et al. Fusobacterium Nucleatum Potentiates Intestinal Tumorigenesis and Modulates the Tumor-Immune Microenvironment. Cell Host Microbe (2013) 14(2):207–15. doi: 10.1016/j.chom.2013.07.007
35. Dong Z, Zhang C, Zhao Q, Huangfu H, Xue X, Wen S, et al. Alterations of Bacterial Communities of Vocal Cord Mucous Membrane Increases the Risk for Glottic Laryngeal Squamous Cell Carcinoma. J Cancer (2021) 12(13):4049–63. doi: 10.7150/jca.54221
Keywords: esophageal carcinoma, intratumor microbiota, The Cancer Microbiome Atlas, The Cancer Genome Atlas, microbial biomarker
Citation: Wang Y, Guo H, Gao X and Wang J (2021) The Intratumor Microbiota Signatures Associate With Subtype, Tumor Stage, and Survival Status of Esophageal Carcinoma. Front. Oncol. 11:754788. doi: 10.3389/fonc.2021.754788
Received: 24 August 2021; Accepted: 11 October 2021;
Published: 27 October 2021.
Edited by:
Phillipp Hartmann, University of California, San Diego, United StatesReviewed by:
Rafael Medrano, Oncology Hospital National Medical Center Siglo XXI IMSS, MexicoCopyright © 2021 Wang, Guo, Gao and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jihan Wang, amloYW53YW5nQG53cHUuZWR1LmNu
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.