 Osmel Companioni
Osmel Companioni Cristina Mir
Cristina Mir Yoelsis Garcia-Mayea
Yoelsis Garcia-Mayea Matilde E. LLeonart
Matilde E. LLeonart- 1Biomedical Research in Cancer Stem Cells Group, Vall d’Hebron Research Institute (VHIR), Universitat Autònoma de Barcelona, Barcelona, Spain
- 2Spanish Biomedical Research Network Center in Oncology, CIBERONC, Madrid, Spain
Sphingolipids are an extensive class of lipids with different functions in the cell, ranging from proliferation to cell death. Sphingolipids are modified in multiple cancers and are responsible for tumor proliferation, progression, and metastasis. Several inhibitors or activators of sphingolipid signaling, such as fenretinide, safingol, ABC294640, ceramide nanoliposomes (CNLs), SKI-II, α-galactosylceramide, fingolimod, and sonepcizumab, have been described. The objective of this review was to analyze the results from preclinical and clinical trials of these drugs for the treatment of cancer. Sphingolipid-targeting drugs have been tested alone or in combination with chemotherapy, exhibiting antitumor activity alone and in synergism with chemotherapy in vitro and in vivo. As a consequence of treatments, the most frequent mechanism of cell death is apoptosis, followed by autophagy. Aslthough all these drugs have produced good results in preclinical studies of multiple cancers, the outcomes of clinical trials have not been similar. The most effective drugs are fenretinide and α-galactosylceramide (α-GalCer). In contrast, minor adverse effects restricted to a few subjects and hepatic toxicity have been observed in clinical trials of ABC294640 and safingol, respectively. In the case of CNLs, SKI-II, fingolimod and sonepcizumab there are some limitations and absence of enough clinical studies to demonstrate a benefit. The effectiveness or lack of a major therapeutic effect of sphingolipid modulation by some drugs as a cancer therapy and other aspects related to their mechanism of action are discussed in this review.
Introduction
Sphingolipids are key structural components of cellular membranes containing a backbone of sphingosine (aliphatic amino alcohol) as the base of their structures. They are synthesized, metabolized and trafficked among several cell organelles. Sphingolipids are remarkably diverse and have crucial roles in maintaining barrier function and fluidity, as well as regulating the cell cycle, cell motility, differentiation, adhesion, and apoptosis (1).
Sphingolipids include ceramides, sphingomyelins, cerebrosides, sulfatides, globosides and gangliosides (Figure 1). De novo sphingolipid synthesis begins with the formation of 3-keto-dihydrosphingosine by serine palmitoyltransferase (SPT). Next, 3-keto-dihydrosphingosine is reduced to form dihydrosphingosine, which is acylated by a ceramide synthase (CerS) to form dihydroceramide. CerS enzymes have different affinities for acyl-CoA substrates, resulting in the generation of dihydroceramides with differing chain lengths (C14-C26). Dihydroceramides are then desaturated to form ceramides (2, 3). De novo generated ceramide is the central hub of the sphingolipid pathway and subsequently has several fates (Figure 2). It is phosphorylated by ceramide kinase (CK) to form ceramide-1-phosphate or it can be glycosylated by glucosylceramide synthase to form glycosphingolipids (cerebrosides, globosides, gangliosides). In addition, ceramide can be converted to sulfatides by the action of galactosylceramide synthase followed by cerebroside sulfotransferase (CST). Additionally, ceramide is also converted to sphingomyelin by the addition of a phosphorylcholine headgroup by sphingomyelin synthase (SMS). Finally, ceramide may be degraded by ceramidase (CDase) to form sphingosine. Sphingosine may be phosphorylated by sphingosine kinase 1/2 (SPHK1/SPHK2) to form sphingosine-1-phosphate (S1P), which has a prosurvival role and is critical for immunomodulation (1, 4, 5) (Figure 2).
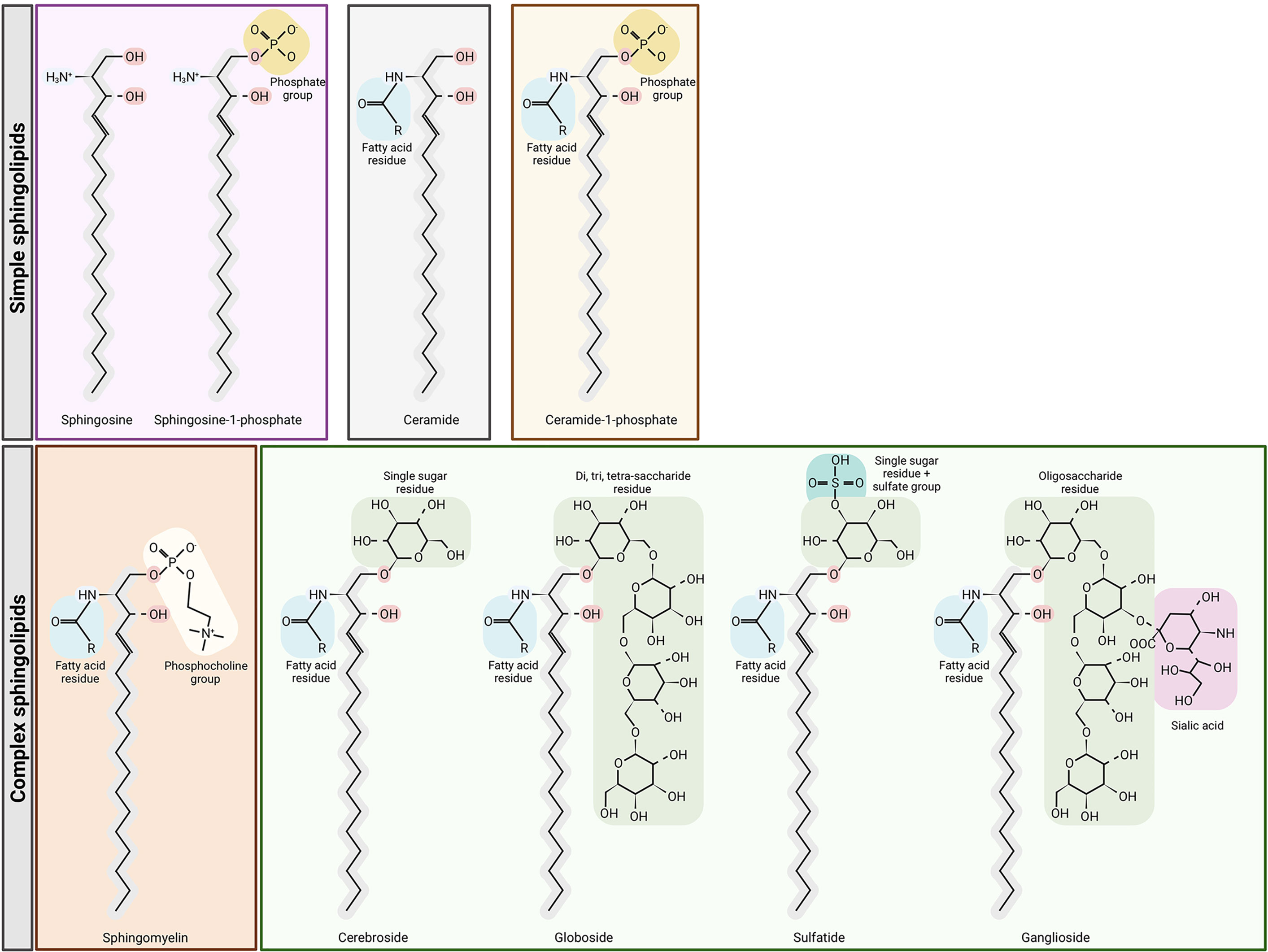
Figure 1 Structures of sphingolipids. Sphingosine is the base for simple sphingolipids. Ceramide contains a fatty acid residue. The addition of a phosphate group to sphingosine or ceramide yields sphingosine-1-phosphate and ceramide-1-phosphate, respectively. Complex sphingolipids are synthesized through ceramide modifications. The addition of a phosphocholine group to ceramide yields sphingomyelin, but the addition of glucose or galactose to ceramide yields glycosphingolipids and sulfatides. Figure created with BioRender.com.
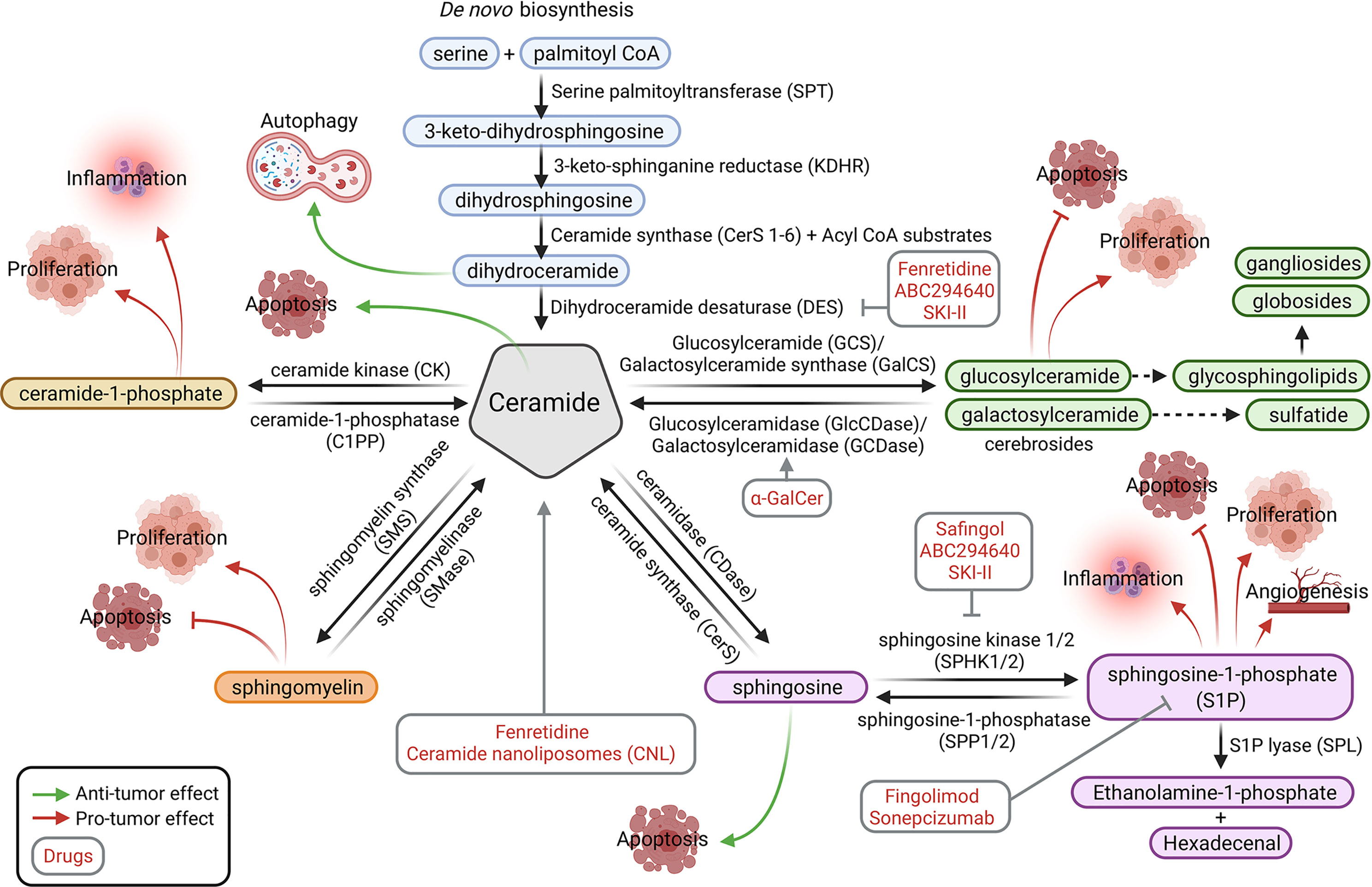
Figure 2 Sphingolipid metabolic pathways. The sphingolipid modulators (in red) and affected tumor processes (processes by which molecules promote tumor growth are indicated with red arrows, whereas those that inhibit tumor growth are indicated with green arrows) are shown. Ceramide is the central molecule that is synthesized through the de novo pathway or other catabolic pathways through conversion from ceramide-1-phosphate, sphingomyelin, sphingosine, glycosphingoplidis, or sulfatides (glucosyl or galactosyl-ceramide). Figure created with BioRender.com.
SPHK1/2 are overexpressed in numerous cancer cell types, but catabolic pathways allow the reversion of S1P to ceramide by sphingosine-1-phosphatase (SPP1/2) and ceramide synthase. The complex glycosphingolipids are hydrolyzed to glucosylceramide and galactosylceramide. These lipids are then hydrolyzed by beta-glucosidases and beta-galactosidases (GCDase) to regenerate ceramide. Similarly, sphingomyelin may be degraded by sphingomyelinase (SMase) and ceramide-1-phosphate by ceramide-1-phosphatase (C1PP) to form ceramide (4) (Figure 2).
In addition to their roles in the organization of the plasma membrane, sphingolipids also play roles as key molecules in signaling processes [for reviews, see (1, 4)]. A classic example is the increase in ceramide and sphingosine levels caused by chemotherapy, radiation, and/or oxidative stress and the subsequent induction of apoptosis by these molecules. In contrast, sphingosine-1-phosphate displays antiapoptotic and prosurvival properties. Because some of these enzymes regulate the abundance of sphingolipids, their aberrant expression or activity exerts a negative effect on cancer (5). Thus, numerous studies have been performed targeting the enzymes that catabolize ceramide, generate S1P, or regulate sphingolipid levels. Generally, different strategies have been used to exploit the potential antitumor effects of sphingolipids. Among them, we highlight the following biological processes: autophagic cell death, apoptosis induction, including mitochondrial activation (mitophagy), proliferation inhibition, and cell cycle arrest, and effects on angiogenesis and migration (Figure 3).
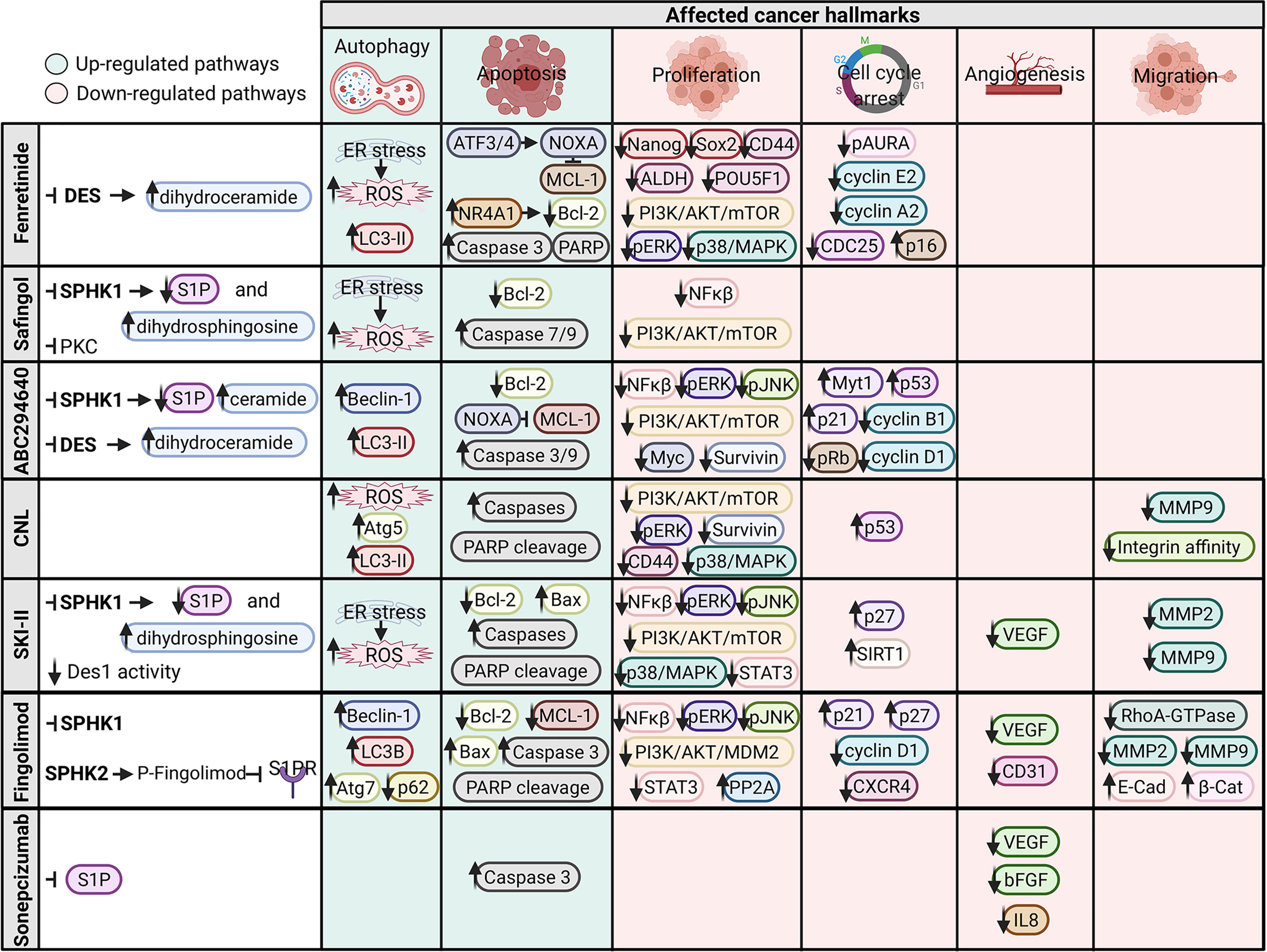
Figure 3 Signaling pathways and cancer hallmarks affected by sphingolipid modulators. Upregulated pathways are indicated in green, and downregulated pathways are indicated in red. Figure created with BioRender.com.
Chemotherapy and Sphingolipid-Related Drugs
In general, chemotherapy is an effective treatment for cancer due to its ability to kill highly proliferative cells. Chemotherapeutic agents induce stress in cancer cells at the cytoplasmic level (i.e., reactive oxygen species [ROS] production and nuclear DNA damage), and in response, ceramide levels are increased followed by the induction of apoptotic cell death. For example, daunorubicin, etoposide, and gemcitabine have all been described as inducers of de novo ceramide generation (6). Chemotherapy resistance has also been linked to altered sphingolipid metabolism, favoring the production of lipid species that ultimately lead to cell survival (7). In this sense, many inhibitors or modulators of sphingolipid metabolism have been developed to kill tumors and reverse chemotherapy resistance (8). These drugs have been employed in preclinical studies using cancer cell lines and orthotopic mouse models, as well as in clinical trials (Table 1). In the next sections, this review highlights the drugs most frequently used to target sphingolipid signaling, indicates their mechanisms of action and discusses their successes and limitations in preclinical and clinical trials of cancer treatment. The main results from published preclinical and clinical trials are summarized in Table 1.

Table 1 Summary of preclinical and clinical studies of inhibitors/modulators of sphingolipids and cancer.
Fenretinide
Fenretinide (N-(4-hydroxyphenyl)retinamide; 4-HPR) reduces the de novo synthesis of ceramide by targeting dihydroceramide desaturase (DES) while inducing an increase in dihydroceramide levels. This enzyme is responsible for the desaturation of dihydroceramide, the final step in the de novo synthesis of ceramide lipid species from dihydroceramide precursors. Dihydroceramides induce autophagy and inhibit cell growth by inducing cell cycle arrest in cancer cells (215, 216). In addition to DESs, other enzymes are fenretinide targets (i.e., CerS5).
Fenretinide treatment induces cell death through the following mechanisms: apoptosis (increased cleavage of caspases and PARP proteins; induction of NR4A1 expression, which interacts with Bcl-2, exposing aBH3 domain and a pro-apoptotic function; and induction of ATF3 expression, ATF4 expression, and NOXA transcription) (9, 11–14, 16, 18, 43); autophagy (increased LC3-II levels) (9); endoplasmic reticulum stress and accumulation of reactive oxygen species (ROS) (11, 14, 16–18, 43); repression of mammalian target of rapamycin (mTOR) signaling and a subsequent reduction in Erk1/2 activity (9); ceramide production (9, 17); antitumor activity against CSCs (reduced CD44, ALDH, Nanog, Sox2, and POU5F1 expression) (10, 16, 18); induction of cell cycle arrest (decreased p-AURA, CDC25, cyclin E2, and cyclin A2 levels and increased p16 levels) (9, 11, 12, 18); and p38-MAPK signaling (19) (Figure 3). Several articles have shown that fenretinide preferentially targets CSCs when sphere formation and stemness markers are analyzed (9–11, 14–16).
Preclinical studies have indicated the antitumor activity of fenretinide in vitro and in vivo in several tumor types in the absence of toxicity in mice. However, clinical trials have reported some mild side effects of fenretinide, such as musculoskeletal complaints (55), diarrhea, reversible night blindness, allergic reaction (21), and dermatological disorders (40). Furthermore, fenretinide lacks antitumor activity in most studies (n=13) but has been shown to stabilize the disease or exert protective effects on some cancers (n=6), mostly breast cancer. Fenretinide preferentially accumulates in fatty tissues, such as the breast, which may contribute to its effectiveness against breast cancer (42). Fenretinide has shown a lack of activity against other cancers. For example, fenretinide does not reduce the time to recurrence of renal carcinoma, consistent with low intratumor drug concentrations (33). Additionally, fenretinide does not substantially modulate the levels of several biomarkers in prostate cancer, including transforming growth factor alpha (TGF-α), insulin-like growth factor 1 (IGF-I), insulin-like growth factor binding protein 3 (IGFBP-3), sex hormone binding globulin (SHBG), and prostate-specific antigen (PSA), which are indicative of insufficient biological activity (36, 37). The remarkable hydrophobicity of this drug may be one of the factors responsible for its lack of effectiveness in clinical trials. Better formulations, such as encapsulation into nanocarriers for oral administration, have been reported to be a feasible option to increase its activity (13, 217).
However, fenretinide induces a positive hormonal (47) and metabolic profile in premenopausal women (50) and exerts a beneficial effect on total serum cholesterol and HDL levels (53). These beneficial effects have been observed in some cancers, such as breast cancer, but not in others, thereby indicating a possible specificity of fenretinide for this tumor type. Interestingly, there are some correlations between oncogenic alterations and the efficacy of this drug. For example, the sensitivity of Ewing’s sarcoma cells to fenretinide-induced cell death is decreased following downregulation of the oncogenic fusion protein EWS-Fli1 and p38(MAPK) activity (218). Also, fenretinide caused induction of oncogene c-Fos expression, whereas such an effect was not observed in resistant cells to fenretinide-induced apoptosis (219).
Also, the combination of fenretidine and ABT-263 (Bcl-2 inhibitor) induces the apoptosis of a large number of HNSCC cells, regardless of the human papillomavirus (HPV) or p53 status. The primary targets of apoptosis induced by these drugs are MCL1 (a Bcl-2 family apoptosis regulator), and Bcl-2 like 1 (Bcl-XL) (220). Remarkably, the nanomicellar combination of lenalidomide–fenretinide suppresses tumor growth in a MYCN-amplified neuroblastoma tumor mediated by increased expression of GD2, a disialoganglioside expressed on tumors of neuroectodermal origin (221). Moreover, treatment with a combination of fenretinide, tocilizumab, and reparixin significantly suppresses IL-6 release, IL-8 release, stem cell gene expression, and invasion in CSC populations (222), which may be due to increased ceramide levels and decreased IL6 and CXCR1/2 levels.
Safingol
Safingol [(2S, 3S)]-2-aminoctadecane-1,3-diol] is an inhibitor of SPHK1, PKCβ-I, PKCδ, PKCϵ, PI3K, and glucose uptake (223). Safingol also affects the balance of ceramide/dihydroceramide levels. The inhibitory effects on signaling, particularly on PKCϵ and PI3k, concomitant with the presence of ROS (67) synergize to induce apoptosis (decreased Bcl-2 levels and increased caspase cleavage) (59, 60, 62–65, 68) and/or autophagy (63, 67) (Figure 3). According to preclinical studies, the combination of safingol with conventional chemotherapy agents, such as doxorubicin (67), irinotecan (66), and mitomycin C (65), potentiates their effects, inducing apoptotic cell death and ROS production in different cell lines. Additionally, the administration of safingol in combination with bortezomib inhibits lung tumor growth and metastasis (through the modulation of NF-κB signaling) in orthotopic syngeneic mouse models (69). Unfortunately, hepatic toxicity, renal toxicity, changes in liver histology, and decreases in body weight have been observed in mice treated with safingol (56, 57). Two out of two clinical trials have indicated stable disease or minor responses to safingol in a subgroup of patients (73, 74). However, hepatic toxicity has been observed in a clinical trial of safingol (73), resulting in few additional clinical trials of this drug. In resistant cancer types, such as gastroesophageal cancer, treatment with the combination of safingol with other chemotherapeutic agents, such as cisplatin, has been proposed to potentially overcome cytotoxic drug resistance. This conclusion is based on the following observations: i) cisplatin resistance correlates with increased SPHK1 expression and with decreased sphingosine-1-phosphate lyase 1 (SGPL1) expression; and ii) the survival of patients treated with chemotherapy prior to surgery but not patients treated with surgery alone (70).
ABC294640
ABC294640 (opaganib, Yeliva, 3-(4-chlorophenyl)-N-(pyridin-4-ylmethyl)adamantane-1-carboxamide) is a selective inhibitor of both SPHK2 and DES1 that decreases the synthesis of the pro-proliferative and antiapoptotic lipid S1P, which may eventually lead to the induction of apoptosis and inhibition of cell proliferation in cancer cells overexpressing SPHK2 (75–77, 82). In vitro studies have indicated that ABC294640 reduces the proliferation and viability of several cancer cell lines and mouse xenografts without any toxic side effects. The decrease in proliferation is mediated by inhibition of SPHK2 activity (82, 85, 97), S1P depletion (76, 79, 84, 85, 95, 97, 99), accumulation of ceramide (79, 84, 85, 89, 99), induction of apoptosis (increased caspase cleavage, decreased Bcl-2 levels, and decreased NOXA transcription) (76, 78, 79, 84–91, 94–96, 98, 99, 102, 103), induction of autophagy (increased LC3-II and beclin-1 levels) (77, 83, 98), estrogen/androgen receptor signaling (decrease in progesterone or androgen receptor levels) (75, 81, 82), cell cycle arrest (increased Myt1, p-cdc2, p53, and p21 levels and decreased pRb, cyclin B1, and cyclin D1 levels) (81, 85–87, 100, 101), and modulation of cell survival pathways (decrease or inhibition of NF-κB, pERK1/2, pJNK, pAKT, c-Myc, and survivin expression, as well as p21-activated kinase 1 (PAK1)/p-Lin-11/Isl-1/Mec-3 kinase 1 (LIMK1)/Cofilin1 signaling) (77, 78, 81, 82, 84, 87, 90–92, 100–103) (Figure 3). The combination of ABC294640 with other drugs, such as regorafenib, sorafenib, PDMP, and ABT-199, induces synergistic potentiation of the treatment effect, reducing chemoresistance in various cancer types (98, 99, 103, 104). For example, SPHK2/SPP1 arbitrates regorafenib resistance by activating signal transducer and activator of transcription 3 (STAT3) and nuclear factor kappa light chain enhancer of activated B cells (NF-κB). SPHK2 targeting by ABC294640 significantly reduces resistance to regorafenib in an in vivo model of hepatocellular carcinoma (HCC) (104).
Overall, only one clinical trial for ABC294640 has been reported, and some reversible toxicities (nausea, vomiting, diarrhea, fatigue and nervous system disorders) were documented. These side effects are likely due to off-target effects. The efficacy evaluation indicated stable disease in a subgroup of patients (40%), partial response (7%) and progressive disease (53%) (93).
Ceramide Nanoliposomes
Ceramide nanoliposomes (CNLs) are lipid-based nanoparticle formulations composed of ceramide encapsulated within nanoliposomes, inducing apoptosis in the target cells due to lysosomal membrane permeabilization that leads to the leakage of hydrolytic enzymes into the cytoplasm or by conferring PI3K and PKCζ tumor-suppressive activities (107, 224). Interestingly, CNLs have also been reported to target the Warburg effect in chronic lymphocytic leukemia in vitro and in vivo (106). Ceramide alone is insoluble and has a short half-life; therefore, nanoliposomes increase its solubility and half-life. Upon administration, CNLs accumulate in the tumor environment due to enhanced permeation and retention caused by the ‘leakiness’ of the tumor vasculature (225). No targeting effect on a tumor marker or tropism of CNL for a particular tissue has been observed. However, one method for increasing the specificity of ceramide derivatives for mitochondria (to induce apoptosis by inducing cytochrome c release) is the introduction of a positive charge on the fatty acid residue by adding a pyridine structure. Pyridine-ceramides localized more readily to the mitochondria, altering their structures and functions and inducing pancreatic cancer cell death (226).
Preclinical assays with cell lines and xenografts show that CNLs potentiate the effect of chemotherapy (114–116, 120); reduce tumor proliferation mediated by apoptosis (increased cleavage of PARP and caspases) (110–112, 114, 116–119, 121, 123), autophagy (increased LC3-II and Atg5 levels) (117, 122), necrosis (106), necroptosis (109), anoikis (108), mitophagy (mitochondrial membrane permeabilization) (116, 119, 121–123), and cell cycle arrest (increased p53 expression) (116, 119); increase ROS levels (110); inhibit lysosomal function (116, 122); inhibit integrin affinity (105, 107); and target CD44 receptor (108), survivin (111), PI3K (107, 114), MAPK (105, 114), mammalian target of rapamycin (mTOR) (112, 121), Akt and Erk1/2 (110, 115) signaling (Figure 3). For example, Shaw et al. indicated that the combination of C6-CNLs with chloroquine (an inhibitor of lysosomal function and therefore an autophagy inhibitor) significantly increases apoptosis in response to ceramide by avoiding the repair of mitochondrial damage (122).
To our knowledge, two clinical trials have tested the efficacy of CNLs in cancer. In the first trial, only one patient with cutaneous breast cancer manifested a partial response, yielding a response rate of 4% and a median progression-free survival of 2 months. Topical ceramides were also well tolerated, with no grade 3 or 4 toxicities reported (113). Another clinical trial (phase I) with C6-CNLs concluded that the combination of ceramide and vinblastine is safe and has the potential to treat the heterogeneous nature of acute myelogenous leukemia (AML) through the induction of apoptotic pathways (118); therefore, phase II studies may be conducted.
SKI-II
SKI-II (SKi, SphK-I2, 4-[[4-(4-chlorophenyl)-1,3-thiazol-2-yl]amino]phenol) is a highly selective inhibitor of both SPHK1 and SPHK2 (227). In vitro studies have shown that SKI-II decreases cancer cell proliferation by inducing apoptosis (increased PARP cleavage, increased caspase cleavage, decreased Bcl-2 expression, and increased Bax levels) (124, 126, 129, 130, 135–137, 142, 144–146), autophagy (137), necrosis (136), endoplasmic reticulum stress, oxidative stress, and cell cycle arrest [increased levels of p27 and sirtuin-1 (SIRT1)] (124, 130, 145). In addition, SKI-II has been shown to decrease sphingomyelin and S1P levels (130, 136), inhibit chemotaxis (131), increase ceramide levels (126, 130, 137, 138, 142), and/or increase the activation of other crucial signaling pathways, including transcription factor NF-κB (124, 126, 144, 146), the Janus kinase 1-signal transducer and activator of transcription 1 axis (JAK-STAT) (130, 134, 145), mitogen-activated protein kinase 1 (MAPK) (125, 141, 146), Akt (125, 142–144, 228), Erk1/2 (141, 143–145), c-Jun NH2-terminal kinase 1 (JNK1) (191), tripartite motif containing 14 (TRIM14), metalloproteinases (MMP2 and MMP9), vascular endothelial growth factor (VEGF) (134), estrogens (128), Wnt family member 5A (Wnt5A) concomitant with β-catenin (132), epidermal growth factor receptor (EGFR), insulin-like growth factor binding protein 3 (IGFBP-3) (139), focal adhesion kinase (FAK), and insulin-like growth factor 1 receptor (IGF-1R) (146) (Figure 3). Sensitization of cell lines to SKI-II along with chemotherapy has also been observed (139, 141, 143). Unfortunately, clinical trials of this drug have not been conducted.
α-Galactosylceramide (α-GalCer)
The last decade has revolutionized cancer therapy with the development of immunotherapy, producing good outcomes in patients with a fatal diagnosis. α-GalCer (KRN-7000, α-galactosylceramide-pulsed antigen presenting cells) is a glycosphingolipid and synthetic iNKT (invariant Natural Killer T) cell ligand. Dendritic cells are pulsed with α-GalCer and administered to patients for achieving effective presentation and activation to iNKT cells (172). In other approaches, dendritic cells are mixed with iNKT cells or peptides derived from cancer antigens (154). Dendritic cells (DC) capture antigens and present them to several types of T-cells for their activation. Invariant natural killer T (iNKT/type I NKT) cells are a subset of T cells endowed with innate and adaptive effector functions. They are characterized by the expression of invariant T cell receptor chain Vα24-Jα18, which recognizes lipid antigens presented by CD1d (229). They exhibit powerful cytotoxic activity mediated by perforin/granzyme B. In addition to their direct antitumor effect, iNKT cells also regulate the damaging activities of NK cells, CD8+ T cells, B cells and innate cells by release of a wide variety of pro-inflammatory cytokines (153, 154, 172).
Preclinical and clinical trials using α-GalCer have shown that this therapy is safe, exhibits durable activation, and increases the number of iNKT, NK, tumor-specific, CD4+, CD8+ T, and B cells (148, 149, 151, 153–156, 160, 161, 169, 172, 173, 175). This activation is associated with increased serum levels of cytokines that stimulate the growth and function of T cells [IL-12 (150, 175) and IL-2 receptors (175)] and other factors that enhance natural killer cell activity (i.e., interferon gamma [IFN-γ] (150, 155, 156, 158, 161, 163, 172), CD16 (175), and tumor necrosis factor α [TNF-α]) and immune cell maturation (GMCSF) (164). In eleven out of twelve completed phase I-II clinical trials, tumor regression, stable disease, partial response or increased median survival time were observed in a subgroup of patients (153, 157, 159, 160, 162–169). These promising clinical findings are associated with the activation of natural killer cells, cytotoxic CD8+ T cells and CD4+ T cells, which are the most relevant immune responses to cancer (230).
Attempts to improve efficacy of iNKT treatments have focused on transduced with CARs (chimeric antigen receptors) (NCT03294954; NCT03774654), chemical modifications to the α-GalCer to stabilize interactions with CD1d, optimizing presentation through encapsulation in particulate vectors, making structural changes that help binding to CD1d, injecting agonists covalently attached to recombinant CD1d. Also, facilitate formation of resident memory CD8+ T cells could find a role in this therapy.
Fingolimod
Fingolimod (FTY720, Gilenya, 2-amino-2-[2-(4-octylphenyl)ethyl]propane-1,3-diol) is a functional antagonist of the sphingosine-1-phosphate receptor (S1PR) and structural analog of sphingosine (1). Fingolimod causes the internalization of S1PR, which sequesters T lymphocytes in lymph nodes (absent in the periphery) (231), preventing them from contributing to inflammatory and autoimmune reactions. The most universal mechanism for its potential anticancer function is limiting the conversion of sphingosine to S1P (7). Fingolimod is effective at reducing inflammatory relapses in patients with multiple sclerosis (232). Fingolimod also shifts macrophages to an anti-inflammatory M2 phenotype and modulates their proliferation, morphology, and cytokine release (233). Preclinical studies of fingolimod have indicated that this drug is safe, potentiates the effect of chemotherapy (192, 195, 196, 200, 201, 234), and suppresses tumor growth by inducing apoptosis (increased cleavage of PARP and caspases, decreased Bcl-2 and Mcl-1 levels, and increased Bax levels) (176, 177, 179, 181, 182, 187, 189, 194–199, 203, 204, 206), autophagy (increased LC3-II, beclin-1, and Atg7 levels, and decreased p62 expression) (197, 203, 208, 209), necrosis (183, 210), cell cycle arrest (increased levels of cell cycle inhibitory proteins [p27 and p21]); and decreased expression of cyclin D1 and C-X-C motif chemokine receptor 4 [CXCR4]) (186, 187, 197, 204, 209). Fingolimod also increases ceramide levels (181, 204), the proteasomal degradation of SPHK1 (182), inactivation of RhoA-GTPase (178), histone deacetylase (HDAC) activity (185), multidrug resistance protein 1 (ABCB1) levels (195), protein phosphatase 2A (PP2A) reactivation (196, 205, 206), and modulation of signaling pathways (VEGF (176, 186, 199), MMP2, MMP9, CD31, E-cadherin, β-catenin (176), estrogens (187), JNK (191), NF-κB (206), STAT3 (201, 206), AMP-activated protein kinase (AMPK) (208), mTOR (208), Erk1/2 (182, 186, 189, 191, 196, 197, 206), and PI3K/Akt (179, 180, 194, 196, 197, 206) (Figure 3).
However, no clinical trials have assessed the effectiveness of fingolimod in cancer, potentially due to the impairment of cytotoxic CD8+ T and CD4+ T cell trafficking and activation, which precludes tumor infiltration to kill cancer cells. Fingolimod blocks the immunosurveillance of B cells by suppressing the migration of tumor-specific Th1 cells from lymph nodes to the incipient tumor site, thereby preventing Th1-mediated activation of tumoricidal macrophages (235). Furthermore, it impairs the ability of cytotoxic CD8+ T cells to kill their target cells and reduces IFNγ and Granzyme B levels in splenic CD8+ T cells (236, 237). Thus, an effective action of this drug in clinical trials is not anticipated, as T cells are the main cells involved in the immune response to tumors.
Sonepcizumab
Sonepcizumab (LT1009) is a humanized monoclonal antibody against S1P. Sonepcizumab slows tumor progression in murine models with orthotopic tumors by blocking the function of proangiogenic growth factors (decreased VEGF, bFGF, and IL-8 levels) and inducing apoptosis (increased caspase cleavage). Additionally, sonepcizumab inhibits tumor vascularization in vitro and in vivo, and it neutralizes S1P-induced stimulation of proliferation in multiple cell lines (213) (Figure 3). A phase II study of sonepcizumab was terminated because it failed to meet its primary progression-free survival endpoint in patients with metastatic renal cell carcinoma who received three prior therapies. However, researchers were encouraged by the overall survival (21.7 months) and safety profile of sonepcizumab, and they advised “further investigation in combination with VEGF-directed agents or checkpoint inhibitors”. Ten percent of patients achieved a partial response, with a median duration of response of 5.9 months. No grade 3/4 treatment-related adverse events were observed in >5% of patients (214).
An increase in systemic S1P concentrations was detected following sonepcizumab treatment, suggesting that S1P signaling was still active, which might explain the limited efficacy of the drug in the clinic. Thus, future studies are needed to improve the neutralization of S1P signaling. In addition, studies testing the efficacy of this drug in combination with SPHK1/2 inhibitors or S1PR2 antagonists are warranted (1).
Conclusions
Sphingolipid-targeting drugs have been tested against several hematological malignancies and solid tumors, alone or in combination with chemotherapy, and have produced some encouraging results (42, 47, 48, 50, 52, 54). Treatments targeting sphingolipid exhibit antitumor activity in vitro and in vivo, inducing apoptosis or occasionally autophagy, as well as several other mechanisms of cell death. Among these agents, the most effective and promising treatments in clinical trials are fenretinide and α-galactosylceramide. Some plausible explanations for the partial success of these safe drugs in clinical trials have been proposed. Fenretinide accumulation in breast tissue along with the induction of apoptosis or autophagy (in caspase-defective breast cancer cells) by dihydroceramide may be responsible for its success. Researchers presumed that its accumulation in breast tissue (and not in other tissues) might be related to hormone-associated pathways that are active in these cancer types. Regarding α-galactosylceramide, the induction of an antitumor immune response mediated by iNKT, NK, T cells and B cells is the functional mechanism. Among several anticancer therapies, immune checkpoint inhibitors occupy a relevant place because of the activation of the antitumor function of T cells (238), which indirectly indicates an important role for the adaptive immune system in the efficacy of anticancer treatments. However, despite different proposals (mutations that prevent T cells from entering the tumor, inhibition of T cell activation pathways, etc.), researchers have not yet clearly determined why immunotherapy is not efficient against some types of tumors.
Current research gaps in the other drugs are associated with side effects, modest findings or the absence of clinical trials. For example, safingol and ABC294640 induced side effects on humans in clinical trials, which may be the main reason for the limited number of clinical trials. Safingol is an inhibitor of several enzymes (SPHK1, PKCβ-I, PKCδ, PKCϵ, and PI3K) and glucose uptake (223), which are needed for the proper function of normal tissues. Targeted therapy against protein kinases relies on the upregulation/activation of these molecules in particular tumors. For example, imatinib is a specific inhibitor of the constitutively active Bcr-Abl tyrosine kinase and is used to treat leukemia with the Philadelphia chromosome (Bcr-Abl) (239). Therefore, we understand that off-target effects of sanfingol due to the inhibition of several enzymes and glucose uptake are likely responsible for the hepatic toxicity observed in mouse and human studies. Potential developments in this field to alleviate this limitation might include some chemical modifications designed to increase the specificity for SPHK1 or targeting an upregulated sphingolipid in a specific tumor. Nevertheless, their use is expected to vary depending on the type of cancer, which in turn is determined by the levels of aberrant sphingolipids expressed in each type of tissue, among other factors. In addition, glucose uptake is a universal and vital step for obtaining ATP through glycolysis and oxidative phosphorylation.
CNLs are already being investigated in clinical trials, but the expected results were very modest, potentially because of a lack of CNL tropism for a specific tumor tissue type (i.e., breast). No clinical trials for SKI-II and fingolimod have been reported. For the latter, an effective action in cancer clinical trials is not expected, as this immunosuppressive drug impairs the tissue infiltration and activation of cytotoxic CD8+ T and CD4+ cells, which are the most relevant cells involved in the immune response to tumors. Clinical studies confirm this fact, as spontaneous regression of T cell lymphoma has been observed in patients with multiple sclerosis after discontinuing fingolimod (240).
With respect to sonepcizumab, an increase in systemic S1P concentrations was observed in a clinical trial (214), although it is a monoclonal antibody against S1P. Treatment with this drug resulted in a reduction in the absolute serum lymphocyte levels, which was expected based on the known effect of S1P blockade on peripheral lymphocyte trafficking (214). Moreover, upregulation of the S1PR1-STAT3 pathway enables myeloid cells to intravasate and mediate tumor proliferation and metastasis (241). In addition, S1PR1 signaling in T cells drives Treg accumulation in tumors, limits CD8+ T cell recruitment and activation, and promotes tumor growth (242, 243). Therefore, sonepcizumab does not provide effective S1P blockade in clinical trials, and the potential tumor infiltration of Tregs and myeloid cells and reduction of lymphocyte numbers fosters tumor growth.
The exhaustive characterization at several levels, including immunity, pharmacodynamics, pharmacokinetics, dosing and metabolomics, is required in preclinical studies before entering clinical trials. The most relevant factor associated with side effects is the presence of off-target effects, which might be improved by chemical modification of these drugs or new synthesis to increase specificity. For this task, the use of molecular docking based on three-dimensional protein structures would be able to develop new and more specific drugs. In addition, the lack of tissue-specific targeting and hydrophobicity of the drugs precludes an effective action. The use of aberrant sphingolipids in specific tumors as targets and nanocarriers or chemical modifications are solutions to these issues.
Aberrant sphingolipid signaling is a consequence (not the cause) of carcinogenesis due to mutations in crucial oncogenes and tumor suppressor genes. Hence, effective treatment with sphingolipid modulating drugs should be based on multiple therapeutic combinations, including immunotherapy (activates antitumor immune CD4+ and CD8+ T cells) and conventional chemotherapy Interestingly, conventional chemotherapy (i.e., tamoxifen) is active against SPHK1 and GCS; thus, the use of tamoxifen might be beneficial in patients who have acquired resistance to these enzymes. One opportunity is based on the fact that many chemotherapeutic agents modulate ceramide levels; therefore, the rational use of these agents with sphingolipid inhibitors could increase lethal levels of ceramide that are more effective at killing the tumor. Overall, an increased understanding of the mechanisms by which sphingolipids control cancer cell signaling together with in-depth studies using animal models will fill these gaps and improve future anticancer therapy based on these compounds.
Author Contributions
OC write the manuscript. CM designed the figures accordingly to the literature. YG-M revised the manuscript. ML revised and re-write the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by grants from the Instituto de Salud Carlos III (ISCIII; PI20/00556 and CP03/00101 [ML]) and CIBERONC (ML). This work was also co-financed by the European Regional Fund (ERDF) and AECC (Spanish Association of Cancer Research) (Founding Ref. GC16173720CARR [ML]). YG-M and CM were supported by the VHIR and iP-FIS (ISCIII) fellowships, respectively.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We thank T. Moline and R. Somoza from the VHIR. We thank J.A. Leal for his reading assistance and comments.
References
1. Ogretmen B. Sphingolipid Metabolism in Cancer Signalling and Therapy. Nat Rev Cancer (2018) 18:33–50. doi: 10.1038/nrc.2017.96
2. Pewzner-Jung Y, Ben-Dor S, Futerman AH. When Do Lasses (Longevity Assurance Genes) Become CerS (Ceramide Synthases)? J Biol Chem (2006) 281:25001–5. doi: 10.1074/jbc.R600010200
3. Causeret C, Geeraert L, van der Hoeven G, Mannaerts GP, Van Veldhoven PP. Further Characterization of Rat Dihydroceramide Desaturase: Tissue Distribution, Subcellular Localization, and Substrate Specificity. Lipids (2000) 35:1117–25. doi: 10.1007/s11745-000-0627-6
4. Hannun YA, Obeid LM. Principles of Bioactive Lipid Signalling: Lessons From Sphingolipids. Nat Rev Mol Cell Biol (2008) 9:139–50. doi: 10.1038/nrm2329
5. Ogretmen B, Hannun YA. Biologically Active Sphingolipids in Cancer Pathogenesis and Treatment. Nat Rev Cancer (2004) 4:604–16. doi: 10.1038/nrc1411
6. Perry DK, Carton J, Shah AK, Meredith F, Uhlinger DJ, Hannun YA. Serine Palmitoyltransferase Regulates De NovoCeramide Generation During Etoposide-Induced Apoptosis. J Biol Chem (2000) 275:9078–84. doi: 10.1074/jbc.275.12.9078
7. Shaw J, Costa-Pinheiro P, Patterson L, Drews K, Spiegel S, Kester M. Novel Sphingolipid-Based Cancer Therapeutics in the Personalized Medicine Era. Adv Cancer Res (2018) 140:327–66. doi: 10.1016/bs.acr.2018.04.016
8. Beckham TH, Cheng JC, Marrison ST, Norris JS, Liu X. Interdiction of Sphingolipid Metabolism to Improve Standard Cancer Therapies. Adv Cancer Res 1–36. doi: 10.1016/B978-0-12-394274-6.00001-7
9. Orienti I, Francescangeli F, De Angelis ML, Fecchi K, Bongiorno-Borbone L, Signore M, et al. A New Bioavailable Fenretinide Formulation With Antiproliferative, Antimetabolic, and Cytotoxic Effects on Solid Tumors. Cell Death Dis (2019) 10:529. doi: 10.1038/s41419-019-1775-y
10. Orienti I, Salvati V, Sette G, Zucchetti M, Bongiorno-Borbone L, Peschiaroli A, et al. A Novel Oral Micellar Fenretinide Formulation With Enhanced Bioavailability and Antitumour Activity Against Multiple Tumours From Cancer Stem Cells. J Exp Clin Cancer Res (2019) 38:373. doi: 10.1186/s13046-019-1383-9
11. Liu L, Liu J, Wang H, Zhao H, Du Y. Fenretinide Targeting of Human Colon Cancer Sphere Cells Through Cell Cycle Regulation and Stress−Responsive Activities. Oncol Lett (2018) 16:5339–48. doi: 10.3892/ol.2018.9296
12. Xiong J, Kuang X, Lu T, Liu X, Cheng B, Wang W, et al. Fenretinide-Induced Apoptosis of Acute Myeloid Leukemia Cells via NR4A1 Translocation Into Mitochondria and Bcl-2 Transformation. J Cancer (2019) 10:6767–78. doi: 10.7150/jca.32167
13. Wang Y, Ding Y, Wang C, Gao M, Xu Y, Ma X, et al. Fenretinide-Polyethylene Glycol (PEG) Conjugate With Improved Solubility Enhanced Cytotoxicity to Cancer Cell and Potent In Vivo Efficacy. Pharm Dev Technol (2020) 25:962–70. doi: 10.1080/10837450.2020.1765377
14. Du Y, Xia Y, Pan X, Chen Z, Wang A, Wang K, et al. Fenretinide Targets Chronic Myeloid Leukemia Stem/Progenitor Cells by Regulation of Redox Signaling. Antioxid Redox Signal (2014) 20:1866–80. doi: 10.1089/ars.2012.4935
15. Wang H, Zhang Y, Du Y. Ovarian and Breast Cancer Spheres Are Similar in Transcriptomic Features and Sensitive to Fenretinide. BioMed Res Int (2013) 2013:1–11. doi: 10.1155/2013/510905
16. Zhang H, Mi J-Q, Fang H, Wang Z, Wang C, Wu L, et al. Preferential Eradication of Acute Myelogenous Leukemia Stem Cells by Fenretinide. Proc Natl Acad Sci (2013) 110:5606–11. doi: 10.1073/pnas.1302352110
17. Morad SAF, Davis TS, Kester M, Loughran TP, Cabot MC. Dynamics of Ceramide Generation and Metabolism in Response to Fenretinide – Diversity Within and Among Leukemia. Leuk Res (2015) 39:1071–8. doi: 10.1016/j.leukres.2015.06.009
18. Bassani B, Bartolini D, Pagani A, Principi E, Zollo M, Noonan DM, et al. Fenretinide (4-HPR) Targets Caspase-9, ERK 1/2 and the Wnt3a/β-Catenin Pathway in Medulloblastoma Cells and Medulloblastoma Cell Spheroids. PloS One (2016) 11:e0154111. doi: 10.1371/journal.pone.0154111
19. Zhang L, Huang D, Shao D, Liu H, Zhou Q, Gui S, et al. Fenretinide Inhibits the Proliferation and Migration of Human Liver Cancer HepG2 Cells by Downregulating the Activation of Myosin Light Chain Kinase Through the P38−MAPK Signaling Pathway. Oncol Rep (2018) 40:518–26. doi: 10.3892/or.2018.6436
20. Villablanca JG, Krailo MD, Ames MM, Reid JM, Reaman GH, Reynolds CP. Phase I Trial of Oral Fenretinide in Children With High-Risk Solid Tumors: A Report From the Children’s Oncology Group (CCG 09709). J Clin Oncol (2006) 24:3423–30. doi: 10.1200/JCO.2005.03.9271
21. Kummar S, Gutierrez ME, Maurer BJ, Reynolds CP, Kang M, Singh H, et al. Phase I Trial of Fenretinide Lym-X-Sorb Oral Powder in Adults With Solid Tumors and Lymphomas. Anticancer Res (2011) 31:961–6.
22. Rotmensz N, De Palo G, Formelli F, Costa A, Marubini E, Campa T, et al. Long-Term Tolerability of Fenretinide (4-HPR) in Breast Cancer Patients. Eur J Cancer Clin Oncol (1991) 27:1127–31. doi: 10.1016/0277-5379(91)90309-2
23. Mohrbacher AM, Yang AS, Groshen S, Kummar S, Gutierrez ME, Kang MH, et al. Phase I Study of Fenretinide Delivered Intravenously in Patients With Relapsed or Refractory Hematologic Malignancies: A California Cancer Consortium Trial. Clin Cancer Res (2017) 23:4550–5. doi: 10.1158/1078-0432.CCR-17-0234
24. Maurer BJ, Kang MH, Villablanca JG, Janeba J, Groshen S, Matthay KK, et al. Phase I Trial of Fenretinide Delivered Orally in a Novel Organized Lipid Complex in Patients With Relapsed/Refractory Neuroblastoma: A Report From the New Approaches to Neuroblastoma Therapy (NANT) Consortium. Pediatr Blood Cancer (2013) 60:1801–8. doi: 10.1002/pbc.24643
25. Decensi A, Bruno S, Torrisi R, Parodi S, Polizzi A. Pilot Study of High Dose Fenretinide and Vitamin A Supplementation in Bladder Cancer. Eur J Cancer (1994) 30:1909–10. doi: 10.1016/0959-8049(94)00336-4
26. Colombo N, Formelli F, Cantu MG, Parma G, Gasco M, Argusti A, et al. A Phase I-II Preoperative Biomarker Trial of Fenretinide in Ascitic Ovarian Cancer. Cancer Epidemiol Biomarkers Prev (2006) 15:1914–9. doi: 10.1158/1055-9965.EPI-06-0183
27. Decensi A, Bonanni B, Baglietto L, Guerrieri-Gonzaga A, Ramazzotto F, Johansson H, et al. A Two-By-Two Factorial Trial Comparing Oral With Transdermal Estrogen Therapy and Fenretinide With Placebo on Breast Cancer Biomarkers. Clin Cancer Res (2004) 10:4389–97. doi: 10.1158/1078-0432.CCR-04-0087
28. Decensi A, Formelli F, Torrisi R, Costa A. Breast Cancer Chemoprevention: Studies With 4-HPR Alone and in Combination With Tamoxifen Using Circulating Growth Factors as Potential Surrogate Endpoints. J Cell Biochem (1993) 53:226–33. doi: 10.1002/jcb.240531142
29. De Palo G, Veronesi U, Camerini T, Formelli F, Mascotti G, Boni C, et al. Can Fenretinide Protect Women Against Ovarian Cancer? J Natl Cancer Inst (1995) 87:146–7. doi: 10.1093/jnci/87.2.146
30. Formelli F, Camerini T, Cavadini E, Appierto V, Villani MG, Costa A, et al. Fenretinide Breast Cancer Prevention Trial: Drug and Retinol Plasma Levels in Relation to Age and Disease Outcome. Cancer Epidemiol Biomarkers Prev (2003) 12:34–41.
31. Puntoni M, Petrera M, Campora S, Garrone E, Defferrari C, Torrisi R, et al. Prognostic Significance of VEGF After Twenty-Year Follow-Up in a Randomized Trial of Fenretinide in Non–Muscle-Invasive Bladder Cancer. Cancer Prev Res (2016) 9:437–44. doi: 10.1158/1940-6207.CAPR-15-0345
32. Moore MM, Stockler M, Lim R, Mok TSK, Millward M, Boyer MJ. A Phase II Study of Fenretinide in Patients With Hormone Refractory Prostate Cancer: A Trial of the Cancer Therapeutics Research Group. Cancer Chemother Pharmacol (2010) 66:845–50. doi: 10.1007/s00280-009-1228-x
33. Vaishampayan U, Heilbrun LK, Parchment RE, Jain V, Zwiebel J, Boinpally RR, et al. Phase II Trial of Fenretinide in Advanced Renal Carcinoma. Invest New Drugs (2005) 23:179–85. doi: 10.1007/s10637-005-5864-7
34. Puduvalli VK, Yung WKA, Hess KR, Kuhn JG, Groves MD, Levin VA, et al. Phase II Study of Fenretinide (NSC 374551) in Adults With Recurrent Malignant Gliomas: A North American Brain Tumor Consortium Study. J Clin Oncol (2004) 22:4282–9. doi: 10.1200/JCO.2004.09.096
35. Serrano D, Baglietto L, Johansson H, Mariette F, Torrisi R, Onetto M, et al. Effect of the Synthetic Retinoid Fenretinide on Circulating Free Prostate-Specific Antigen, Insulin-Like Growth Factor-I, and Insulin-Like Growth Factor Binding Protein-3 Levels in Men With Superficial Bladder Cancer. Clin Cancer Res (2005) 11:2083–8. doi: 10.1158/1078-0432.CCR-04-1549
36. Cheung E, Pinski J, Dorff T, Groshen S, Quinn DI, Reynolds CP, et al. Oral Fenretinide in Biochemically Recurrent Prostate Cancer: A California Cancer Consortium Phase II Trial. Clin Genitourin Cancer (2009) 7:43–50. doi: 10.3816/CGC.2009.n.008
37. Weiss HL, Urban DA, Grizzle WE, Cronin KA, Freedman LS, Kelloff GJ, et al. Bayesian Monitoring of a Phase 2 Chemoprevention Trial in High-Risk Cohorts for Prostate Cancer. Urology (2001) 57:220–3. doi: 10.1016/S0090-4295(00)00979-1
38. Modiano MR, Dalton WS, Lippman SM, Joffe L, Booth AR, Meyskens FL. Phase II Study of Fenretinide (N-[4-Hydroxyphenyl]retinamide) in Advanced Breast Cancer and Melanoma. Invest New Drugs (1990) 8:317–9. doi: 10.1007/BF00171846
39. Schneider BJ, Worden FP, Gadgeel SM, Parchment RE, Hodges CM, Zwiebel J, et al. Phase II Trial of Fenretinide (NSC 374551) in Patients With Recurrent Small Cell Lung Cancer. Invest New Drugs (2009) 27:571–8. doi: 10.1007/s10637-009-9228-6
40. Camerini T, Mariani L, De Palo G, Marubini E, Di Mauro MG, Decensi A, et al. Safety of the Synthetic Retinoid Fenretinide: Long-Term Results From a Controlled Clinical Trial for the Prevention of Contralateral Breast Cancer. J Clin Oncol (2001) 19:1664–70. doi: 10.1200/JCO.2001.19.6.1664
41. Sabichi AL, Lerner SP, Atkinson EN, Grossman HB, Caraway NP, Dinney CP, et al. Phase III Prevention Trial of Fenretinide in Patients With Resected Non Muscle-Invasive Bladder Cancer. Clin Cancer Res (2008) 14:224–9. doi: 10.1158/1078-0432.CCR-07-0733
42. Veronesi U, Mariani L, Decensi A, Formelli F, Camerini T, Miceli R, et al. Fifteen-Year Results of a Randomized Phase III Trial of Fenretinide to Prevent Second Breast Cancer. Ann Oncol (2006) 17:1065–71. doi: 10.1093/annonc/mdl047
43. Nguyen TH, Koneru B, Wei S-J, Chen WH, Makena MR, Urias E, et al. Fenretinide via NOXA Induction, Enhanced Activity of the BCL-2 Inhibitor Venetoclax in High BCL-2–Expressing Neuroblastoma Preclinical Models. Mol Cancer Ther (2019) 18:2270–82. doi: 10.1158/1535-7163.MCT-19-0385
44. Wang Y, Ding Y, Xu Y, Wang C, Ding Y, Gao M, et al. Mixed Micelles of TPGS and Soluplus ® for Co-Delivery of Paclitaxel and Fenretinide: In Vitro and In Vivo Anticancer Study. Pharm Dev Technol (2020) 25:865–73. doi: 10.1080/10837450.2020.1753770
45. Orienti I, Nguyen F, Guan P, Kolla V, Calonghi N, Farruggia G, et al. A Novel Nanomicellar Combination of Fenretinide and Lenalidomide Shows Marked Antitumor Activity in a Neuroblastoma Xenograft Model. Drug Des Devel Ther (2019) 13:4305–19. doi: 10.2147/DDDT.S221909
46. Khathayer F, Taylor MA, Ray SK. Synergism of 4HPR and SAHA Increases Anti-Tumor Actions in Glioblastoma Cells. Apoptosis (2020) 25:217–32. doi: 10.1007/s10495-020-01590-9
47. Johansson H, Bonanni B, Gandini S, Guerrieri-Gonzaga A, Cazzaniga M, Serrano D, et al. Circulating Hormones and Breast Cancer Risk in Premenopausal Women: A Randomized Trial of Low-Dose Tamoxifen and Fenretinide. Breast Cancer Res Treat (2013) 142:569–78. doi: 10.1007/s10549-013-2768-7
48. Cobleigh MA, Dowlatshahi K, Deutsch TA, Mehta RG, Moon RC, Minn F, et al. Phase I/II Trial of Tamoxifen With or Without Fenretinide, an Analog of Vitamin A, in Women With Metastatic Breast Cancer. J Clin Oncol (1993) 11:474–7. doi: 10.1200/JCO.1993.11.3.474
49. Macis D, Gandini S, Guerrieri-Gonzaga A, Johansson H, Magni P, Ruscica M, et al. Prognostic Effect of Circulating Adiponectin in a Randomized 2 × 2 Trial of Low-Dose Tamoxifen and Fenretinide in Premenopausal Women at Risk for Breast Cancer. J Clin Oncol (2012) 30:151–7. doi: 10.1200/JCO.2011.35.2237
50. Johansson H, Gandini S, Guerrieri-Gonzaga A, Iodice S, Ruscica M, Bonanni B, et al. Effect of Fenretinide and Low-Dose Tamoxifen on Insulin Sensitivity in Premenopausal Women at High Risk for Breast Cancer. Cancer Res (2008) 68:9512–8. doi: 10.1158/0008-5472.CAN-08-0553
51. Serrano D, Gandini S, Guerrieri-Gonzaga A, Feroce I, Johansson H, Macis D, et al. Quality of Life in a Randomized Breast Cancer Prevention Trial of Low-Dose Tamoxifen and Fenretinide in Premenopausal Women. Cancer Prev Res (2018) 11:811–8. doi: 10.1158/1940-6207.CAPR-18-0073
52. Decensi A, Robertson C, Guerrieri-Gonzaga A, Serrano D, Cazzaniga M, Mora S, et al. Randomized Double-Blind 2 × 2 Trial of Low-Dose Tamoxifen and Fenretinide for Breast Cancer Prevention in High-Risk Premenopausal Women. J Clin Oncol (2009) 27:3749–56. doi: 10.1200/JCO.2008.19.3797
53. Zujewski J, Pai L, Wakefield L, Giusti R, Dorr FA, Flanders C, et al. Tamoxifen and Fenretinide in Women With Metastatic Breast Cancer. Breast Cancer Res Treat (1999) 57:277–83. doi: 10.1023/A:1006216409688
54. Veronesi U, De Palo G, Marubini E, Costa A, Formelli F, Mariani L, et al. Randomized Trial of Fenretinide to Prevent Second Breast Malignancy in Women With Early Breast Cancer. J Natl Cancer Inst (1999) 91:1847–56. doi: 10.1093/jnci/91.21.1847
55. Rao RD, Cobleigh MA, Gray R, Graham ML, Norton L, Martino S, et al. Phase III Double-Blind, Placebo-Controlled, Prospective Randomized Trial of Adjuvant Tamoxifen vs. Tamoxifen and Fenretinide in Postmenopausal Women With Positive Receptors (EB193): An Intergroup Trial Coordinated by the Eastern Cooperative Oncology Group. Med Oncol (2011) 28:39–47. doi: 10.1007/s12032-010-9682-1
56. Carfagna MA, Young KM, Susick RL Jr. Sex Differences in Rat Hepatic Cytolethality of the Protein Kinase C Inhibitor Safingol: Role of Biotransformation. Toxicol Appl Pharmacol (1996) 137:173–81. doi: 10.1006/taap.1996.0070
57. Morales PR, Dillehay DL, Moody SJ, Pallas DC, Pruett S, Allgood JC, et al. Safingol Toxicology After Oral Administration to TRAMP Mice: Demonstration of Safingol Uptake and Metabolism by N-Acylation and N-Methylation. Drug Chem Toxicol (2007) 30:197–216. doi: 10.1080/01480540701375018
58. Ling LU, Tan KB, Lin H, Chiu GNC. The Role of Reactive Oxygen Species and Autophagy in Safingol-Induced Cell Death. Cell Death Dis (2011) 2:e129–12. doi: 10.1038/cddis.2011.12
59. Tsukamoto S, Huang Y, Kumazoe M, Lesnick C, Yamada S, Ueda N, et al. Sphingosine Kinase-1 Protects Multiple Myeloma From Apoptosis Driven by Cancer-Specific Inhibition of RTKs. Mol Cancer Ther (2015) 14:2303–12. doi: 10.1158/1535-7163.MCT-15-0185
60. Hamada M, Wakabayashi K, Masui A, Iwai S, Imai T, Yura Y. Involvement of Hydrogen Peroxide in Safingol-Induced Endonuclease G-Mediated Apoptosis of Squamous Cell Carcinoma Cells. Int J Mol Sci (2014) 15:2660–71. doi: 10.3390/ijms15022660
61. Tan K-B, Ling L-U, Bunte RM, Chng W-J, Chiu GNC. In Vivo Efficacy of a Novel Liposomal Formulation of Safingol in the Treatment of Acute Myeloid Leukemia. J Control Release (2012) 160:290–8. doi: 10.1016/j.jconrel.2011.11.002
62. Noda T, Iwai S, Hamada M, Fujita Y, Yura Y. Induction of Apoptosis of Detached Oral Squamous Cell Carcinoma Cells by Safingol. Possible Role of Bim, Focal Adhesion Kinase and Endonuclease G. Apoptosis (2009) 14:287–97. doi: 10.1007/s10495-009-0319-9
63. Coward J, Ambrosini G, Musi E, Truman J-P, Haimovitz-Friedman A, Allegood JC, et al. Safingol (L- Threo -Sphinganine) Induces Autophagy in Solid Tumor Cells Through Inhibition of PKC and the PI3-Kinase Pathway. Autophagy (2009) 5:184–93. doi: 10.4161/auto.5.2.7361
64. Amin HM, Ergin M, Denning MF, Quevedo ME, Alkan S. Characterization of Apoptosis Induced by Protein Kinase C Inhibitors and its Modulation by the Caspase Pathway in Acute Promyelocytic Leukaemia. Br J Haematol (2000) 110:552–62. doi: 10.1046/j.1365-2141.2000.02207.x
65. Schwartz GK, Haimovitz-Friedman A, Dhupar SK, Ehleiter D, Maslak P, Lai L, et al. Potentiation of Apoptosis by Treatment With the Protein Kinase C-Specific Inhibitor Safingol in Mitomycin C-Treated Gastric Cancer Cells. J Natl Cancer Inst (1995) 87:1394–9. doi: 10.1093/jnci/87.18.1394
66. Chiu. The Role of Protein Kinase C in the Synergistic Interaction of Safingol and Irinotecan in Colon Cancer Cells. Int J Oncol (2009) 35:1463–71. doi: 10.3892/ijo_00000465
67. Ling LU, Tan KB, Chiu GNC. Role of Reactive Oxygen Species in the Synergistic Cytotoxicity of Safingol-Based Combination Regimens With Conventional Chemotherapeutics. Oncol Lett (2011) 2:905–10. doi: 10.3892/ol.2011.330
68. Tsukamoto S, Kumazoe M, Huang Y, Lesnick C, Kay NE, Shanafelt TD, et al. SphK1 Inhibitor Potentiates the Anti-Cancer Effect of EGCG on Leukaemia Cells. Br J Haematol (2017) 178:155–8. doi: 10.1111/bjh.14119
69. Acharya S, Yao J, Li P, Zhang C, Lowery FJ, Zhang Q, et al. Sphingosine Kinase 1 Signaling Promotes Metastasis of Triple-Negative Breast Cancer. Cancer Res (2019) 79:4211–26. doi: 10.1158/0008-5472.CAN-18-3803
70. Matula K, Collie-Duguid E, Murray G, Parikh K, Grabsch H, Tan P, et al. Regulation of Cellular Sphingosine-1-Phosphate by Sphingosine Kinase 1 and Sphingosine-1-Phopshate Lyase Determines Chemotherapy Resistance in Gastroesophageal Cancer. BMC Cancer (2015) 15:762. doi: 10.1186/s12885-015-1718-7
71. Hoffmann TK, Leenen K, Hafner D, Balz V, Gerharz CD, Grund A, et al. Antitumor Activity of Protein Kinase C Inhibitors and Cisplatin in Human Head and Neck Squamous Cell Carcinoma Lines. Anticancer Drugs (2002) 13:93–100. doi: 10.1097/00001813-200201000-00011
72. Tan K-B, Ling L-U, Bunte RM, Chng W-J, Chiu GN. Liposomal Codelivery of a Synergistic Combination of Bioactive Lipids in the Treatment of Acute Myeloid Leukemia. Nanomedicine (2014) 9:1665–79. doi: 10.2217/nnm.13.123
73. Dickson MA, Carvajal RD, Merrill AH, Gonen M, Cane LM, Schwartz GK. A Phase I Clinical Trial of Safingol in Combination With Cisplatin in Advanced Solid Tumors. Clin Cancer Res (2011) 17:2484–92. doi: 10.1158/1078-0432.CCR-10-2323
74. Schwartz GK, Ward D, Saltz L, Casper ES, Spiess T, Mullen E, et al. A Pilot Clinical/Pharmacological Study of the Protein Kinase C-Specific Inhibitor Safingol Alone and in Combination With Doxorubicin. Clin Cancer Res (1997) 3:537–43.
75. Antoon JW, White MD, Meacham WD, Slaughter EM, Muir SE, Elliott S, et al. Antiestrogenic Effects of the Novel Sphingosine Kinase-2 Inhibitor ABC294640. Endocrinology (2010) 151:5124–35. doi: 10.1210/en.2010-0420
76. French KJ, Zhuang Y, Maines LW, Gao P, Wang W, Beljanski V, et al. Pharmacology and Antitumor Activity of ABC294640, a Selective Inhibitor of Sphingosine Kinase-2. J Pharmacol Exp Ther (2010) 333:129–39. doi: 10.1124/jpet.109.163444
77. Chumanevich AA, Poudyal D, Cui X, Davis T, Wood PA, Smith CD, et al. Suppression of Colitis-Driven Colon Cancer in Mice by a Novel Small Molecule Inhibitor of Sphingosine Kinase. Carcinogenesis (2010) 31:1787–93. doi: 10.1093/carcin/bgq158
78. Antoon JW, White MD, Slaughter EM, Driver JL, Khalili HS, Elliott S, et al. Targeting NFĸB Mediated Breast Cancer Chemoresistance Through Selective Inhibition of Sphingosine Kinase-2. Cancer Biol Ther (2011) 11:678–89. doi: 10.4161/cbt.11.7.14903
79. Qin Z, Dai L, Trillo-Tinoco J, Senkal C, Wang W, Reske T, et al. Targeting Sphingosine Kinase Induces Apoptosis and Tumor Regression for KSHV-Associated Primary Effusion Lymphoma. Mol Cancer Ther (2014) 13:154–64. doi: 10.1158/1535-7163.MCT-13-0466
80. Gestaut MM, Antoon JW, Burow ME, Beckman BS. Inhibition of Sphingosine Kinase-2 Ablates Androgen Resistant Prostate Cancer Proliferation and Survival. Pharmacol Rep (2014) 66:174–8. doi: 10.1016/j.pharep.2013.08.014
81. Schrecengost RS, Keller SN, Schiewer MJ, Knudsen KE, Smith CD. Downregulation of Critical Oncogenes by the Selective SK2 Inhibitor ABC294640 Hinders Prostate Cancer Progression. Mol Cancer Res (2015) 13:1591–601. doi: 10.1158/1541-7786.MCR-14-0626
82. Venant H, Rahmaniyan M, Jones EE, Lu P, Lilly MB, Garrett-Mayer E, et al. The Sphingosine Kinase 2 Inhibitor ABC294640 Reduces the Growth of Prostate Cancer Cells and Results in Accumulation of Dihydroceramides In Vitro and In Vivo. Mol Cancer Ther (2015) 14:2744–52. doi: 10.1158/1535-7163.MCT-15-0279
83. Dai L, Bai A, Smith CD, Rodriguez PC, Yu F, Qin Z. ABC294640, A Novel Sphingosine Kinase 2 Inhibitor, Induces Oncogenic Virus–Infected Cell Autophagic Death and Represses Tumor Growth. Mol Cancer Ther (2017) 16:2724–34. doi: 10.1158/1535-7163.MCT-17-0485
84. Zhou J, Chen J, Yu H. Targeting Sphingosine Kinase 2 by ABC294640 Inhibits Human Skin Squamous Cell Carcinoma Cell Growth. Biochem Biophys Res Commun (2018) 497:535–42. doi: 10.1016/j.bbrc.2018.02.075
85. Xu L, Jin L, Yang B, Wang L, Xia Z, Zhang Q, et al. The Sphingosine Kinase 2 Inhibitor ABC294640 Inhibits Cervical Carcinoma Cell Growth. Oncotarget (2018) 9:2384–94. doi: 10.18632/oncotarget.23415
86. Dai L, Smith CD, Foroozesh M, Miele L, Qin Z. The Sphingosine Kinase 2 Inhibitor ABC294640 Displays Anti-Non-Small Cell Lung Cancer Activities In Vitro and In Vivo. Int J Cancer (2018) 142:2153–62. doi: 10.1002/ijc.31234
87. Song K, Dai L, Long X, Cui X, Liu Y, Di W. Sphingosine Kinase 2 Inhibitor ABC294640 Displays Anti-Epithelial Ovarian Cancer Activities In Vitro and In Vivo. Onco Targets Ther (2019) 12:4437–49. doi: 10.2147/OTT.S208519
88. Ding X, Zhang Y, Huang T, Xu G, Peng C, Chen G, et al. Targeting Sphingosine Kinase 2 Suppresses Cell Growth and Synergizes With BCL2/BCL-XL Inhibitors Through NOXA-Mediated MCL1 Degradation in Cholangiocarcinoma. Am J Cancer Res (2019) 9:546–61.
89. Dai L, Chen J, Lin Z, Wang Z, Mu S, Qin Z. Targeting Sphingosine Kinase by ABC294640 Against Diffuse Intrinsic Pontine Glioma (DIPG). J Cancer (2020) 11:4683–90. doi: 10.7150/jca.46269
90. Venkata JK, An N, Stuart R, Costa LJ, Cai H, Coker W, et al. Inhibition of Sphingosine Kinase 2 Downregulates the Expression of C-Myc and Mcl-1 and Induces Apoptosis in Multiple Myeloma. Blood (2014) 124:1915–25. doi: 10.1182/blood-2014-03-559385
91. LeBlanc FR, Pearson JM, Tan S, Cheon H, Xing JC, Dunton W, et al. Sphingosine Kinase-2 Is Overexpressed in Large Granular Lymphocyte Leukaemia and Promotes Survival Through Mcl-1. Br J Haematol (2020) 190:405–17. doi: 10.1111/bjh.16530
92. Shi W, Ma D, Cao Y, Hu L, Liu S, Yan D, et al. SphK2/S1P Promotes Metastasis of Triple-Negative Breast Cancer Through the PAK1/LIMK1/Cofilin1 Signaling Pathway. Front Mol Biosci (2021) 8:598218. doi: 10.3389/fmolb.2021.598218
93. Britten CD, Garrett-Mayer E, Chin SH, Shirai K, Ogretmen B, Bentz TA, et al. A Phase I Study of ABC294640, a First-in-Class Sphingosine Kinase-2 Inhibitor, in Patients With Advanced Solid Tumors. Clin Cancer Res (2017) 23:4642–50. doi: 10.1158/1078-0432.CCR-16-2363
94. Antoon JW, White MD, Driver JL, Burow ME, Beckman BS. Sphingosine Kinase Isoforms as a Therapeutic Target in Endocrine Therapy Resistant Luminal and Basal-A Breast Cancer. Exp Biol Med (2012) 237:832–44. doi: 10.1258/ebm.2012.012028
95. White MD, Chan L, Antoon JW, Beckman BS. Targeting Ovarian Cancer and Chemoresistance Through Selective Inhibition of Sphingosine Kinase-2 With ABC294640. Anticancer Res (2013) 33:3573–80.
96. Yang J, Yang C, Zhang S, Mei Z, Shi M, Sun S, et al. ABC294640, a Sphingosine Kinase 2 Inhibitor, Enhances the Antitumor Effects of TRAIL in non-Small Cell Lung Cancer. Cancer Biol Ther (2015) 16:1194–204. doi: 10.1080/15384047.2015.1056944
97. Xun C, Chen M-B, Qi L, Tie-Ning Z, Peng X, Ning L, et al. Targeting Sphingosine Kinase 2 (SphK2) by ABC294640 Inhibits Colorectal Cancer Cell Growth In Vitro and In Vivo. J Exp Clin Cancer Res (2015) 34:94. doi: 10.1186/s13046-015-0205-y
98. Ding X, Chaiteerakij R, Moser CD, Shaleh H, Boakye J, Chen G, et al. Antitumor Effect of the Novel Sphingosine Kinase 2 Inhibitor ABC294640 Is Enhanced by Inhibition of Autophagy and by Sorafenib in Human Cholangiocarcinoma Cells. Oncotarget (2016) 7:20080–92. doi: 10.18632/oncotarget.7914
99. Guan S, Liu YY, Yan T, Zhou J. Inhibition of Ceramide Glucosylation Sensitizes Lung Cancer Cells to ABC294640, a First-in-Class Small Molecule SphK2 Inhibitor. Biochem Biophys Res Commun (2016) 476:230–6. doi: 10.1016/j.bbrc.2016.05.102
100. McNaughton M, Pitman M, Pitson SM, Pyne NJ, Pyne S. Proteasomal Degradation of Sphingosine Kinase 1 and Inhibition of Dihydroceramide Desaturase by the Sphingosine Kinase Inhibitors, SKi or ABC294640, Induces Growth Arrest in Androgen-Independent LNCaP-AI Prostate Cancer Cells. Oncotarget (2016) 7:16663–75. doi: 10.18632/oncotarget.7693
101. Lewis CS, Voelkel-Johnson C, Smith CD. Suppression of C-Myc and RRM2 Expression in Pancreatic Cancer Cells by the Sphingosine Kinase-2 Inhibitor ABC294640. Oncotarget (2016) 7:60181–92. doi: 10.18632/oncotarget.11112
102. Leili H, Nasser S, Nadereh R, Siavoush D, Pouran K. Sphingosine Kinase-2 Inhibitor ABC294640 Enhances Doxorubicin-Induced Apoptosis of NSCLC Cells via Altering Survivin Expression. Drug Res (Stuttg) (2018) 68:45–53. doi: 10.1055/s-0043-117181
103. Sundaramoorthy P, Gasparetto C, Kang Y. The Combination of a Sphingosine Kinase 2 Inhibitor (ABC294640) and a Bcl-2 Inhibitor (ABT-199) Displays Synergistic Anti-Myeloma Effects in Myeloma Cells Without a T(11;14) Translocation. Cancer Med (2018) 7:3257–68. doi: 10.1002/cam4.1543
104. Shi W, Zhang S, Ma D, Yan D, Zhang G, Cao Y, et al. Targeting SphK2 Reverses Acquired Resistance of Regorafenib in Hepatocellular Carcinoma. Front Oncol (2020) 10:694. doi: 10.3389/fonc.2020.00694
105. Heakal Y, Kester M. Nanoliposomal Short-Chain Ceramide Inhibits Agonist-Dependent Translocation of Neurotensin Receptor 1 to Structured Membrane Microdomains in Breast Cancer Cells. Mol Cancer Res (2009) 7:724–34. doi: 10.1158/1541-7786.MCR-08-0322
106. Ryland LK, Doshi UA, Shanmugavelandy SS, Fox TE, Aliaga C, Broeg K, et al. C6-Ceramide Nanoliposomes Target the Warburg Effect in Chronic Lymphocytic Leukemia. PloS One (2013) 8:e84648. doi: 10.1371/journal.pone.0084648
107. Zhang P, Fu C, Hu Y, Dong C, Song Y, Song E. C6-Ceramide Nanoliposome Suppresses Tumor Metastasis by Eliciting PI3K and Pkcζ Tumor-Suppressive Activities and Regulating Integrin Affinity Modulation. Sci Rep (2015) 5:9275. doi: 10.1038/srep09275
108. Haakenson JK, Khokhlatchev AV, Choi YJ, Linton SS, Zhang P, Zaki PM, et al. Lysosomal Degradation of CD44 Mediates Ceramide Nanoliposome-Induced Anoikis and Diminished Extravasation in Metastatic Carcinoma Cells. J Biol Chem (2015) 290:8632–43. doi: 10.1074/jbc.M114.609677
109. Zhang X, Kitatani K, Toyoshima M, Ishibashi M, Usui T, Minato J, et al. Ceramide Nanoliposomes as a MLKL-Dependent, Necroptosis-Inducing, Chemotherapeutic Reagent in Ovarian Cancer. Mol Cancer Ther (2018) 17:50–9. doi: 10.1158/1535-7163.MCT-17-0173
110. Li G, Liu D, Kimchi ET, Kaifi JT, Qi X, Manjunath Y, et al. Nanoliposome C6-Ceramide Increases the Anti-Tumor Immune Response and Slows Growth of Liver Tumors in Mice. Gastroenterology (2018) 154:1024–1036.e9. doi: 10.1053/j.gastro.2017.10.050
111. Liu X, Ryland L, Yang J, Liao A, Aliaga C, Watts R, et al. Targeting of Survivin by Nanoliposomal Ceramide Induces Complete Remission in a Rat Model of NK-LGL Leukemia. Blood (2010) 116:4192–201. doi: 10.1182/blood-2010-02-271080
112. Jiang F, Jin K, Huang S, Bao Q, Shao Z, Hu X, et al. Liposomal C6 Ceramide Activates Protein Phosphatase 1 to Inhibit Melanoma Cells. PloS One (2016) 11:e0159849. doi: 10.1371/journal.pone.0159849
113. Jatoi A, Suman VJ, Schaefer P, Block M, Loprinzi C, Roche P, et al. A Phase II Study of Topical Ceramides for Cutaneous Breast Cancer. Breast Cancer Res Treat (2003) 80:99–104. doi: 10.1023/A:1024409123726
114. Tran MA, Smith CD, Kester M, Robertson GP. Combining Nanoliposomal Ceramide With Sorafenib Synergistically Inhibits Melanoma and Breast Cancer Cell Survival to Decrease Tumor Development. Clin Cancer Res (2008) 14:3571–81. doi: 10.1158/1078-0432.CCR-07-4881
115. Jiang Y, DiVittore NA, Kaiser JM, Shanmugavelandy SS, Fritz JL, Heakal Y, et al. Combinatorial Therapies Improve the Therapeutic Efficacy of Nanoliposomal Ceramide for Pancreatic Cancer. Cancer Biol Ther (2011) 12:574–85. doi: 10.4161/cbt.12.7.15971
116. Morad SAF, Levin JC, Shanmugavelandy SS, Kester M, Fabrias G, Bedia C, et al. Ceramide–Antiestrogen Nanoliposomal Combinations—Novel Impact of Hormonal Therapy in Hormone-Insensitive Breast Cancer. Mol Cancer Ther (2012) 11:2352–61. doi: 10.1158/1535-7163.MCT-12-0594
117. Adiseshaiah PP, Clogston JD, McLeland CB, Rodriguez J, Potter TM, Neun BW, et al. Synergistic Combination Therapy With Nanoliposomal C6-Ceramide and Vinblastine is Associated With Autophagy Dysfunction in Hepatocarcinoma and Colorectal Cancer Models. Cancer Lett (2013) 337:254–65. doi: 10.1016/j.canlet.2013.04.034
118. Barth BM, Wang W, Toran PT, Fox TE, Annageldiyev C, Ondrasik RM, et al. Sphingolipid Metabolism Determines the Therapeutic Efficacy of Nanoliposomal Ceramide in Acute Myeloid Leukemia. Blood Adv (2019) 3:2598–603. doi: 10.1182/bloodadvances.2018021295
119. Morad SAF, Madigan JP, Levin JC, Abdelmageed N, Karimi R, Rosenberg DW, et al. Tamoxifen Magnifies Therapeutic Impact of Ceramide in Human Colorectal Cancer Cells Independent of P53. Biochem Pharmacol (2013) 85:1057–65. doi: 10.1016/j.bcp.2013.01.015
120. Su X, Song H, Niu F, Yang K, Kou G, Wang X, et al. Co-Delivery of Doxorubicin and PEGylated C16-Ceramide by Nanoliposomes for Enhanced Therapy Against Multidrug Resistance. Nanomedicine (2015) 10:2033–50. doi: 10.2217/nnm.15.50
121. Morad SAF, MacDougall MR, Abdelmageed N, Kao L-P, Feith DJ, Tan S-F, et al. Pivotal Role of Mitophagy in Response of Acute Myelogenous Leukemia to a Ceramide-Tamoxifen-Containing Drug Regimen. Exp Cell Res (2019) 381:256–64. doi: 10.1016/j.yexcr.2019.05.021
122. Shaw JJP, Boyer TL, Venner E, Beck PJ, Slamowitz T, Caste T, et al. Inhibition of Lysosomal Function Mitigates Protective Mitophagy and Augments Ceramide Nanoliposome–Induced Cell Death in Head and Neck Squamous Cell Carcinoma. Mol Cancer Ther (2020) 19:2621–33. doi: 10.1158/1535-7163.MCT-20-0182
123. Watters RJ, Fox TE, Tan S-F, Shanmugavelandy S, Choby JE, Broeg K, et al. Targeting Glucosylceramide Synthase Synergizes With C 6 -Ceramide Nanoliposomes to Induce Apoptosis in Natural Killer Cell Leukemia. Leuk Lymphoma (2013) 54:1288–96. doi: 10.3109/10428194.2012.752485
124. Li P-H, Wu J-X, Zheng J-N, Pei D-S. A Sphingosine Kinase-1 Inhibitor, SKI-II, Induces Growth Inhibition and Apoptosis in Human Gastric Cancer Cells. Asian Pac J Cancer Prev (2015) 15:10381–5. doi: 10.7314/APJCP.2014.15.23.10381
125. French KJ, Upson JJ, Keller SN, Zhuang Y, Yun JK, Smith CD. Antitumor Activity of Sphingosine Kinase Inhibitors. J Pharmacol Exp Ther (2006) 318:596–603. doi: 10.1124/jpet.106.101345
126. Antoon JW. Dual Inhibition of Sphingosine Kinase Isoforms Ablates TNF-Induced Drug Resistance. Oncol Rep (2012) 27:1779–86. doi: 10.3892/or.2012.1743
127. Sinha UK, Schorn VJ, Hochstim C, Chinn SB, Zhu S, Masood R. Increased Radiation Sensitivity of Head and Neck Squamous Cell Carcinoma With Sphingosine Kinase 1 Inhibition. Head Neck (2011) 33:178–88. doi: 10.1002/hed.21418
128. Antoon JW, Meacham WD, Bratton MR, Slaughter EM, Rhodes LV, Ashe HB, et al. Pharmacological Inhibition of Sphingosine Kinase Isoforms Alters Estrogen Receptor Signaling in Human Breast Cancer. J Mol Endocrinol (2011) 46:205–16. doi: 10.1530/JME-10-0116
129. Leroux ME, Auzenne E, Evans R, Hail N, Spohn W, Ghosh SC, et al. Sphingolipids and the Sphingosine Kinase Inhibitor, SKI II, Induce BCL-2-Independent Apoptosis in Human Prostatic Adenocarcinoma Cells. Prostate (2007) 67:1699–717. doi: 10.1002/pros.20645
130. LeBlanc FR, Liu X, Hengst J, Fox T, Calvert V, Petricoin EF, et al. Sphingosine Kinase Inhibitors Decrease Viability and Induce Cell Death in Natural Killer-Large Granular Lymphocyte Leukemia. Cancer Biol Ther (2015) 16:1830–40. doi: 10.1080/15384047.2015.1078949
131. Lepannetier S, Zanou N, Yerna X, Emeriau N, Dufour I, Masquelier J, et al. Sphingosine-1-Phosphate-Activated TRPC1 Channel Controls Chemotaxis of Glioblastoma Cells. Cell Calcium (2016) 60:373–83. doi: 10.1016/j.ceca.2016.09.002
132. Liu H, Zhang C-X, Ma Y, He H-W, Wang J-P, Shao R-G. SphK1 Inhibitor SKI II Inhibits the Proliferation of Human Hepatoma HepG2 Cells via the Wnt5A/β-Catenin Signaling Pathway. Life Sci (2016) 151:23–9. doi: 10.1016/j.lfs.2016.02.098
133. Jakobi K, Beyer S, Koch A, Thomas D, Schwalm S, Zeuzem S, et al. Sorafenib Treatment and Modulation of the Sphingolipid Pathway Affect Proliferation and Viability of Hepatocellular Carcinoma In Vitro. Int J Mol Sci (2020) 21:2409. doi: 10.3390/ijms21072409
134. Jin Z, Li H, Hong X, Ying G, Lu X, Zhuang L, et al. TRIM14 Promotes Colorectal Cancer Cell Migration and Invasion Through the SPHK1/STAT3 Pathway. Cancer Cell Int (2018) 18:202. doi: 10.1186/s12935-018-0701-1
135. Yang L, Weng W, Sun Z-X, Fu X-J, Ma J, Zhuang W-F. SphK1 Inhibitor II (SKI-II) Inhibits Acute Myelogenous Leukemia Cell Growth In Vitro and In Vivo. Biochem Biophys Res Commun (2015) 460:903–8. doi: 10.1016/j.bbrc.2015.03.114
136. Bhat VK, Bernhart E, Plastira I, Fan K, Ghaffari-Tabrizi-Wizsy N, Wadsack C, et al. Pharmacological Inhibition of Serine Palmitoyl Transferase and Sphingosine Kinase-1/-2 Inhibits Merkel Cell Carcinoma Cell Proliferation. J Invest Dermatol (2019) 139:807–17. doi: 10.1016/j.jid.2018.10.024
137. Noack J, Choi J, Richter K, Kopp-Schneider A, Régnier-Vigouroux A. A Sphingosine Kinase Inhibitor Combined With Temozolomide Induces Glioblastoma Cell Death Through Accumulation of Dihydrosphingosine and Dihydroceramide, Endoplasmic Reticulum Stress and Autophagy. Cell Death Dis (2014) 5:e1425–5. doi: 10.1038/cddis.2014.384
138. Casson L, Howell L, Mathews LA, Ferrer M, Southall N, Guha R, et al. Inhibition of Ceramide Metabolism Sensitizes Human Leukemia Cells to Inhibition of BCL2-Like Proteins. PloS One (2013) 8:e54525. doi: 10.1371/journal.pone.0054525
139. Martin JL, de Silva HC, Lin MZ, Scott CD, Baxter RC. Inhibition of Insulin-Like Growth Factor–Binding Protein-3 Signaling Through Sphingosine Kinase-1 Sensitizes Triple-Negative Breast Cancer Cells to EGF Receptor Blockade. Mol Cancer Ther (2014) 13:316–28. doi: 10.1158/1535-7163.MCT-13-0367
140. Ochnik AM, Baxter RC. Insulin-Like Growth Factor Receptor and Sphingosine Kinase are Prognostic and Therapeutic Targets in Breast Cancer. BMC Cancer (2017) 17:820. doi: 10.1186/s12885-017-3809-0
141. Liu Y, Zhu Z, Cai H, Liu Q, Zhou H, Zhu Z. SKI-II Reverses the Chemoresistance of SGC7901/DDP Gastric Cancer Cells. Oncol Lett (2014) 8:367–73. doi: 10.3892/ol.2014.2083
142. Yang Y, Ji C, Cheng L, He L, Lu C, Wang R, et al. Sphingosine Kinase-1 Inhibition Sensitizes Curcumin-Induced Growth Inhibition and Apoptosis in Ovarian Cancer Cells. Cancer Sci (2012) 103:1538–45. doi: 10.1111/j.1349-7006.2012.02335.x
143. Wu X, Wu Q, Zhou X, Huang J. SphK1 Functions Downstream of IGF-1 to Modulate IGF-1-Induced EMT, Migration and Paclitaxel Resistance of A549 Cells: A Preliminary In Vitro Study. J Cancer (2019) 10:4264–9. doi: 10.7150/jca.32646
144. Zhang C, He H, Zhang H, Yu D, Zhao W, Chen Y, et al. The Blockage of Ras/ERK Pathway Augments the Sensitivity of SphK1 Inhibitor SKI II in Human Hepatoma HepG2 Cells. Biochem Biophys Res Commun (2013) 434:35–41. doi: 10.1016/j.bbrc.2013.03.070
145. Li Y, Gao Y, Liang B, Nie W, Zhao L, Wang L. Combined Effects on Leukemia Cell Growth by Targeting Sphingosine Kinase 1 and Sirtuin 1 Signaling. Exp Ther Med (2020) 20:1–1. doi: 10.3892/etm.2020.9392
146. Grbčić P, Tomljanović I, Klobučar M, Kraljević Pavelić S, Lučin K, Sedić M. Dual Sphingosine Kinase Inhibitor SKI-II Enhances Sensitivity to 5-Fluorouracil in Hepatocellular Carcinoma Cells via Suppression of Osteopontin and FAK/IGF-1R Signalling. Biochem Biophys Res Commun (2017) 487:782–8. doi: 10.1016/j.bbrc.2017.04.100
147. Gebremeskel S, Nelson A, Walker B, Oliphant T, Lobert L, Mahoney D, et al. Natural Killer T Cell Immunotherapy Combined With Oncolytic Vesicular Stomatitis Virus or Reovirus Treatments Differentially Increases Survival in Mouse Models of Ovarian and Breast Cancer Metastasis. J Immunother Cancer (2021) 9:e002096. doi: 10.1136/jitc-2020-002096
148. Liu Y, Wang Z, Yu F, Li M, Zhu H, Wang K, et al. The Adjuvant of α-Galactosylceramide Presented by Gold Nanoparticles Enhances Antitumor Immune Responses of MUC1 Antigen-Based Tumor Vaccines. Int J Nanomedicine (2021) 16:403–20. doi: 10.2147/IJN.S273883
149. Wagner AK, Gehrmann U, Hiltbrunner S, Carannante V, Luu TT, Näslund TI, et al. Soluble and Exosome-Bound α-Galactosylceramide Mediate Preferential Proliferation of Educated NK Cells With Increased Anti-Tumor Capacity. Cancers (Basel) (2021) 13:298. doi: 10.3390/cancers13020298
150. Grabowska J, Stolk DA, Nijen Twilhaar MK, Ambrosini M, Storm G, van der Vliet HJ, et al. Liposomal Nanovaccine Containing α-Galactosylceramide and Ganglioside GM3 Stimulates Robust CD8+ T Cell Responses via CD169+ Macrophages and Cdc1. Vaccines (2021) 9:56. doi: 10.3390/vaccines9010056
151. Meng M, Chen S, Gao X, Liu H, Zhao H, Zhang J, et al. Activation of Invariant Natural Killer T Cell Subsets in C57BL/6J Mice by Different Injection Modes of α-Galactosylceramide. Iran J Allergy Asthma Immunol (2020) 19:35–44. doi: 10.18502/ijaai.v19i1.2416
152. Wang Y, Bhave MS, Yagita H, Cardell SL. Natural Killer T-Cell Agonist α-Galactosylceramide and PD-1 Blockade Synergize to Reduce Tumor Development in a Preclinical Model of Colon Cancer. Front Immunol (2020) 11:581301. doi: 10.3389/fimmu.2020.581301
153. Ishibashi F, Sakairi Y, Iwata T, Moriya Y, Mizobuchi T, Hoshino H, et al. A Phase I Study of Loco-Regional Immunotherapy by Transbronchial Injection of α-Galactosylceramide-Pulsed Antigen Presenting Cells in Patients With Lung Cancer. Clin Immunol (2020) 215:108457. doi: 10.1016/j.clim.2020.108457
154. Gasser O, Sharples KJ, Barrow C, Williams GM, Bauer E, Wood CE, et al. A Phase I Vaccination Study With Dendritic Cells Loaded With NY-ESO-1 and α-Galactosylceramide: Induction of Polyfunctional T Cells in High-Risk Melanoma Patients. Cancer Immunol Immunother (2018) 67:285–98. doi: 10.1007/s00262-017-2085-9
155. Nagato K, Motohashi S, Ishibashi F, Okita K, Yamasaki K, Moriya Y, et al. Accumulation of Activated Invariant Natural Killer T Cells in the Tumor Microenvironment After α-Galactosylceramide-Pulsed Antigen Presenting Cells. J Clin Immunol (2012) 32:1071–81. doi: 10.1007/s10875-012-9697-9
156. Kurosaki M, Horiguchi S, Yamasaki K, Uchida Y, Motohashi S, Nakayama T, et al. Migration and Immunological Reaction After the Administration of αgalcer-Pulsed Antigen-Presenting Cells Into the Submucosa of Patients With Head and Neck Cancer. Cancer Immunol Immunother (2011) 60:207–15. doi: 10.1007/s00262-010-0932-z
157. Kunii N, Horiguchi S, Motohashi S, Yamamoto H, Ueno N, Yamamoto S, et al. Combination Therapy of In Vitro -Expanded Natural Killer T Cells and α-Galactosylceramide-Pulsed Antigen-Presenting Cells in Patients With Recurrent Head and Neck Carcinoma. Cancer Sci (2009) 100:1092–8. doi: 10.1111/j.1349-7006.2009.01135.x
158. Motohashi S, Ishikawa A, Ishikawa E, Otsuji M, Iizasa T, Hanaoka H, et al. A Phase I Study of In Vitro Expanded Natural Killer T Cells in Patients With Advanced and Recurrent Non–Small Cell Lung Cancer. Clin Cancer Res (2006) 12:6079–86. doi: 10.1158/1078-0432.CCR-06-0114
159. Okamoto Y, Fujikawa A, Kurosaki M, Yamasaki K, Sakurai D, Horiguchi S, et al. Nasal Submucosal Administration of Antigen-Presenting Cells Induces Effective Immunological Responses in Cancer Immunotherapy. Adv Otorhinolaryngol (2011) 72:149–52. doi: 10.1159/000324775
160. Uchida T, Horiguchi S, Tanaka Y, Yamamoto H, Kunii N, Motohashi S, et al. Phase I Study of α-Galactosylceramide-Pulsed Antigen Presenting Cells Administration to the Nasal Submucosa in Unresectable or Recurrent Head and Neck Cancer. Cancer Immunol Immunother (2008) 57:337–45. doi: 10.1007/s00262-007-0373-5
161. Chang DH, Osman K, Connolly J, Kukreja A, Krasovsky J, Pack M, et al. Sustained Expansion of NKT Cells and Antigen-Specific T Cells After Injection of α-Galactosyl-Ceramide Loaded Mature Dendritic Cells in Cancer Patients. J Exp Med (2005) 201:1503–17. doi: 10.1084/jem.20042592
162. Ishikawa A, Motohashi S, Ishikawa E, Fuchida H, Higashino K, Otsuji M, et al. A Phase I Study of Alpha-Galactosylceramide (KRN7000)-Pulsed Dendritic Cells in Patients With Advanced and Recurrent non-Small Cell Lung Cancer. Clin Cancer Res (2005) 11:1910–7. doi: 10.1158/1078-0432.CCR-04-1453
163. Nicol AJ, Tazbirkova A, Nieda M. Comparison of Clinical and Immunological Effects of Intravenous and Intradermal Administration of α-GalactosylCeramide (KRN7000)-Pulsed Dendritic Cells. Clin Cancer Res (2011) 17:5140–51. doi: 10.1158/1078-0432.CCR-10-3105
164. Giaccone G, Punt CJA, Ando Y, Ruijter R, Nishi N, Peters M, et al. A Phase I Study of the Natural Killer T-Cell Ligand Alpha-Galactosylceramide (KRN7000) in Patients With Solid Tumors. Clin Cancer Res (2002) 8:3702–9.
165. Exley MA, Friedlander P, Alatrakchi N, Vriend L, Yue S, Sasada T, et al. Adoptive Transfer of Invariant NKT Cells as Immunotherapy for Advanced Melanoma: A Phase I Clinical Trial. Clin Cancer Res (2017) 23:3510–9. doi: 10.1158/1078-0432.CCR-16-0600
166. Nieda M, Okai M, Tazbirkova A, Lin H, Yamaura A, Ide K, et al. Therapeutic Activation of Vα24+Vβ11+ NKT Cells in Human Subjects Results in Highly Coordinated Secondary Activation of Acquired and Innate Immunity. Blood (2004) 103:383–9. doi: 10.1182/blood-2003-04-1155
167. Okai M, Nieda M, Tazbirkova A, Horley D, Kikuchi A, Durrant S, et al. Human Peripheral Blood Vα24 + Vβ11 + NKT Cells Expand Following Administration of α-Galactosylceramide-Pulsed Dendritic Cells. Vox Sang (2002) 83:250–3. doi: 10.1046/j.1423-0410.2002.00217.x
168. Motohashi S, Nagato K, Kunii N, Yamamoto H, Yamasaki K, Okita K, et al. A Phase I-II Study of α-Galactosylceramide-Pulsed IL-2/GM-CSF-Cultured Peripheral Blood Mononuclear Cells in Patients With Advanced and Recurrent Non-Small Cell Lung Cancer. J Immunol (2009) 182:2492–501. doi: 10.4049/jimmunol.0800126
169. Yamasaki K, Horiguchi S, Kurosaki M, Kunii N, Nagato K, Hanaoka H, et al. Induction of NKT Cell-Specific Immune Responses in Cancer Tissues After NKT Cell-Targeted Adoptive Immunotherapy. Clin Immunol (2011) 138:255–65. doi: 10.1016/j.clim.2010.11.014
170. Toyoda T, Kamata T, Tanaka K, Ihara F, Takami M, Suzuki H, et al. Phase II Study of α-Galactosylceramide-Pulsed Antigen-Presenting Cells in Patients With Advanced or Recurrent non-Small Cell Lung Cancer. J Immunother Cancer (2020) 8:e000316. doi: 10.1136/jitc-2019-000316
171. Saka H, Kitagawa C, Ichinose Y, Takenoyama M, Ibata H, Kato T, et al. A Randomized Phase II Study to Assess the Effect of Adjuvant Immunotherapy Using α-GalCer-Pulsed Dendritic Cells in the Patients With Completely Resected Stage II–IIIA non-Small Cell Lung Cancer: Study Protocol for a Randomized Controlled Trial. Trials (2017) 18:429. doi: 10.1186/s13063-017-2103-4
172. Stolk DA, de Haas A, Vree J, Duinkerken S, Lübbers J, van de Ven R, et al. Lipo-Based Vaccines as an Approach to Target Dendritic Cells for Induction of T- and iNKT Cell Responses. Front Immunol (2020) 11:990. doi: 10.3389/fimmu.2020.00990
173. Grasso C, Field CS, Tang C-W, Ferguson PM, J Compton B, Anderson RJ, et al. Vaccines Adjuvanted With an NKT Cell Agonist Induce Effective T-Cell Responses in Models of CNS Lymphoma. Immunotherapy (2020) 12:395–406. doi: 10.2217/imt-2019-0134
174. Fu S, He K, Tian C, Sun H, Zhu C, Bai S, et al. Impaired Lipid Biosynthesis Hinders Anti-Tumor Efficacy of Intratumoral iNKT Cells. Nat Commun (2020) 11:438. doi: 10.1038/s41467-020-14332-x
175. Richter J, Neparidze N, Zhang L, Nair S, Monesmith T, Sundaram R, et al. Clinical Regressions and Broad Immune Activation Following Combination Therapy Targeting Human NKT Cells in Myeloma. Blood (2013) 121:423–30. doi: 10.1182/blood-2012-06-435503
176. Chua C-W, Lee DT-W, Ling M-T, Zhou C, Man K, Ho J, et al. FTY720, a Fungus Metabolite, Inhibits In Vivo Growth of Androgen-Independent Prostate Cancer. Int J Cancer (2005) 117:1039–48. doi: 10.1002/ijc.21243
177. Azuma H, Horie S, Muto S, Otsuki Y, Matsumoto K, Morimoto J, et al. Selective Cancer Cell Apoptosis Induced by FTY720; Evidence for a Bcl-Dependent Pathway and Impairment in ERK Activity. Anticancer Res (2003) 23:3183–93.
178. Zhou C, Ling M-T, Kin-Wah Lee T, Man K, Wang X, Wong Y-C. FTY720, a Fungus Metabolite, Inhibits Invasion Ability of Androgen-Independent Prostate Cancer Cells Through Inactivation of RhoA-GTPase. Cancer Lett (2006) 233:36–47. doi: 10.1016/j.canlet.2005.02.039
179. Shen Y, Cai M, Xia W, Liu J, Zhang Q, Xie H, et al. FTY720, a Synthetic Compound From Isaria Sinclairii, Inhibits Proliferation and Induces Apoptosis in Pancreatic Cancer Cells. Cancer Lett (2007) 254:288–97. doi: 10.1016/j.canlet.2007.03.013
180. Chua C-W, Chiu Y-T, Yuen H-F, Chan K-W, Man K, Wang X, et al. Suppression of Androgen-Independent Prostate Cancer Cell Aggressiveness by FTY720: Validating Runx2 as a Potential Antimetastatic Drug Screening Platform. Clin Cancer Res (2009) 15:4322–35. doi: 10.1158/1078-0432.CCR-08-3157
181. Pchejetski D, Bohler T, Brizuela L, Sauer L, Doumerc N, Golzio M, et al. FTY720 (Fingolimod) Sensitizes Prostate Cancer Cells to Radiotherapy by Inhibition of Sphingosine Kinase-1. Cancer Res (2010) 70:8651–61. doi: 10.1158/0008-5472.CAN-10-1388
182. Tonelli F, Lim KG, Loveridge C, Long J, Pitson SM, Tigyi G, et al. FTY720 and (S)-FTY720 Vinylphosphonate Inhibit Sphingosine Kinase 1 and Promote its Proteasomal Degradation in Human Pulmonary Artery Smooth Muscle, Breast Cancer and Androgen-Independent Prostate Cancer Cells. Cell Signal (2010) 22:1536–42. doi: 10.1016/j.cellsig.2010.05.022
183. Zhang N, Qi Y, Wadham C, Wang L, Warren A, Di W, et al. FTY720 Induces Necrotic Cell Death and Autophagy in Ovarian Cancer Cells: A Protective Role of Autophagy. Autophagy (2010) 6:1157–67. doi: 10.4161/auto.6.8.13614
184. McGowan EM, Alling N, Jackson EA, Yagoub D, Haass NK, Allen JD, et al. Evaluation of Cell Cycle Arrest in Estrogen Responsive MCF-7 Breast Cancer Cells: Pitfalls of the MTS Assay. PloS One (2011) 6:e20623. doi: 10.1371/journal.pone.0020623
185. Hait NC, Avni D, Yamada A, Nagahashi M, Aoyagi T, Aoki H, et al. The Phosphorylated Prodrug FTY720 Is a Histone Deacetylase Inhibitor That Reactivates Erα Expression and Enhances Hormonal Therapy for Breast Cancer. Oncogenesis (2015) 4:e156–6. doi: 10.1038/oncsis.2015.16
186. Kalhori V, Magnusson M, Asghar MY, Pulli I, Törnquist K. FTY720 (Fingolimod) Attenuates Basal and Sphingosine-1-Phosphate-Evoked Thyroid Cancer Cell Invasion. Endocr Relat Cancer (2016) 23:457–68. doi: 10.1530/ERC-16-0050
187. Allam RM, Al-Abd AM, Khedr A, Sharaf OA, Nofal SM, Khalifa AE, et al. Fingolimod Interrupts the Cross Talk Between Estrogen Metabolism and Sphingolipid Metabolism Within Prostate Cancer Cells. Toxicol Lett (2018) 291:77–85. doi: 10.1016/j.toxlet.2018.04.008
188. Li J, Wang S-W, Zhang D-S, Sun Y, Zhu C-Y, Fei Q, et al. FTY720-Induced Enhancement of Autophagy Protects Cells From FTY720 Cytotoxicity in Colorectal Cancer. Oncol Rep (2016) 35:2833–42. doi: 10.3892/or.2016.4668
189. Ubai T, Azuma H, Kotake Y, Inamoto T, Takahara K, Ito Y, et al. FTY720 Induced Bcl-Associated and Fas-Independent Apoptosis in Human Renal Cancer Cells In Vitro and Significantly Reduced In Vivo Tumor Growth in Mouse Xenograft. Anticancer Res (2007) 27:75–88.
190. Shen Y, Wang X, Xia W, Wang C, Cai M, Xie H, et al. Antiproliferative and Overadditive Effects of Rapamycin and FTY720 in Pancreatic Cancer Cells In Vitro. Transplant Proc (2008) 40:1727–33. doi: 10.1016/j.transproceed.2008.03.150
191. Nagaoka Y, Otsuki K, Fujita T, Uesato S. Effects of Phosphorylation of Immunomodulatory Agent FTY720 (Fingolimod) on Antiproliferative Activity Against Breast and Colon Cancer Cells. Biol Pharm Bull (2008) 31:1177–81. doi: 10.1248/bpb.31.1177
192. Mousseau Y, Mollard S, Faucher-Durand K, Richard L, Nizou A, Cook-Moreau J, et al. Fingolimod Potentiates the Effects of Sunitinib Malate in a Rat Breast Cancer Model. Breast Cancer Res Treat (2012) 134:31–40. doi: 10.1007/s10549-011-1903-6
193. Zhang N, Dai L, Qi Y, Di W, Xia P. Combination of FTY720 With Cisplatin Exhibits Antagonistic Effects in Ovarian Cancer Cells: Role of Autophagy. Int J Oncol (2013) 42:2053–9. doi: 10.3892/ijo.2013.1906
194. Rosa R, Marciano R, Malapelle U, Formisano L, Nappi L, D’Amato C, et al. Sphingosine Kinase 1 Overexpression Contributes to Cetuximab Resistance in Human Colorectal Cancer Models. Clin Cancer Res (2013) 19:138–47. doi: 10.1158/1078-0432.CCR-12-1050
195. Xing Y, Wang ZH, Ma DH, Han Y. FTY720 Enhances Chemosensitivity of Colon Cancer Cells to Doxorubicin and Etoposide via the Modulation of P-Glycoprotein and Multidrug Resistance Protein 1. J Dig Dis (2014) 15:246–59. doi: 10.1111/1751-2980.12131
196. Cristóbal I, Manso R, Rincón R, Caramés C, Senin C, Borrero A, et al. PP2A Inhibition Is a Common Event in Colorectal Cancer and Its Restoration Using FTY720 Shows Promising Therapeutic Potential. Mol Cancer Ther (2014) 13:938–47. doi: 10.1158/1535-7163.MCT-13-0150
197. Marvaso G, Barone A, Amodio N, Raimondi L, Agosti V, Altomare E, et al. Sphingosine Analog Fingolimod (FTY720) Increases Radiation Sensitivity of Human Breast Cancer Cells In Vitro. Cancer Biol Ther (2014) 15:797–805. doi: 10.4161/cbt.28556
198. Woo SM, Seo BR, Min K, Kwon TK. FTY720 Enhances TRAIL-Mediated Apoptosis by Up-Regulating DR5 and Down-Regulating Mcl-1 in Cancer Cells. Oncotarget (2015) 6:11614–26. doi: 10.18632/oncotarget.3426
199. Kim H-S, Yoon G, Ryu J-Y, Cho Y-J, Choi J-J, Lee Y-Y, et al. Sphingosine Kinase 1 Is a Reliable Prognostic Factor and a Novel Therapeutic Target for Uterine Cervical Cancer. Oncotarget (2015) 6:26746–56. doi: 10.18632/oncotarget.4818
200. Wang Q, Alshaker H, Böhler T, Srivats S, Chao Y, Cooper C, et al. Core Shell Lipid-Polymer Hybrid Nanoparticles With Combined Docetaxel and Molecular Targeted Therapy for the Treatment of Metastatic Prostate Cancer. Sci Rep (2017) 7:5901. doi: 10.1038/s41598-017-06142-x
201. Katsuta E, Yan L, Nagahashi M, Raza A, Sturgill JL, Lyon DE, et al. Doxorubicin Effect is Enhanced by Sphingosine-1-Phosphate Signaling Antagonist in Breast Cancer. J Surg Res (2017) 219:202–13. doi: 10.1016/j.jss.2017.05.101
202. Grenald SA, Doyle TM, Zhang H, Slosky LM, Chen Z, Largent-Milnes TM, et al. Targeting the S1P/S1PR1 Axis Mitigates Cancer-Induced Bone Pain and Neuroinflammation. Pain (2017) 158:1733–42. doi: 10.1097/j.pain.0000000000000965
203. Li Y, Hu T, Chen T, Yang T, Ren H, Chen M. Combination Treatment of FTY720 and Cisplatin Exhibits Enhanced Antitumour Effects on Cisplatin-Resistant Non-Small Lung Cancer Cells. Oncol Rep (2017) 39:565–72. doi: 10.3892/or.2017.6111
204. Kreitzburg KM, Fehling SC, Landen CN, Gamblin TL, Vance RB, Arend RC, et al. FTY720 Enhances the Anti-Tumor Activity of Carboplatin and Tamoxifen in a Patient-Derived Xenograft Model of Ovarian Cancer. Cancer Lett (2018) 436:75–86. doi: 10.1016/j.canlet.2018.08.015
205. Velmurugan BK, Lee C-H, Chiang S-L, Hua C-H, Chen M-C, Lin S-H, et al. PP2A Deactivation Is a Common Event in Oral Cancer and Reactivation by FTY720 Shows Promising Therapeutic Potential. J Cell Physiol (2018) 233:1300–11. doi: 10.1002/jcp.26001
206. Lankadasari MB, Aparna JS, Mohammed S, James S, Aoki K, Binu VS, et al. Targeting S1PR1/STAT3 Loop Abrogates Desmoplasia and Chemosensitizes Pancreatic Cancer to Gemcitabine. Theranostics (2018) 8:3824–40. doi: 10.7150/thno.25308
207. Nagahashi M, Yamada A, Katsuta E, Aoyagi T, Huang W-C, Terracina KP, et al. Targeting the SphK1/S1P/S1PR1 Axis That Links Obesity, Chronic Inflammation, and Breast Cancer Metastasis. Cancer Res (2018) 78:1713–25. doi: 10.1158/0008-5472.CAN-17-1423
208. Booth L, Roberts JL, Spiegel S, Poklepovic A, Dent P. Fingolimod Augments Pemetrexed Killing of Non-Small Cell Lung Cancer and Overcomes Resistance to ERBB Inhibition. Cancer Biol Ther (2019) 20:597–607. doi: 10.1080/15384047.2018.1538616
209. Ota K, Okuma T, Lorenzo A, Yokota A, Hino H, Kazama H, et al. Fingolimod Sensitizes EGFR Wild−Type Non−Small Cell Lung Cancer Cells to Lapatinib or Sorafenib and Induces Cell Cycle Arrest. Oncol Rep (2019) 42:231–42. doi: 10.3892/or.2019.7140
210. Niemelä E, Desai D, Niemi R, Doroszko M, Özliseli E, Kemppainen K, et al. Nanoparticles Carrying Fingolimod and Methotrexate Enables Targeted Induction of Apoptosis and Immobilization of Invasive Thyroid Cancer. Eur J Pharm Biopharm (2020) 148:1–9. doi: 10.1016/j.ejpb.2019.12.015
211. Zou X, Jiang Z, Li L, Huang Z. Selenium Nanoparticles Coated With pH Responsive Silk Fibroin Complex for Fingolimod Release and Enhanced Targeting in Thyroid Cancer. Artif Cells Nanomedicine Biotechnol (2021) 49:83–95. doi: 10.1080/21691401.2021.1871620
212. Rupp T, Pelouin O, Genest L, Legrand C, Froget G, Castagné V. Therapeutic Potential of Fingolimod in Triple Negative Breast Cancer Preclinical Models. Transl Oncol (2021) 14:100926. doi: 10.1016/j.tranon.2020.100926
213. Visentin B, Vekich JA, Sibbald BJ, Cavalli AL, Moreno KM, Matteo RG, et al. Validation of an Anti-Sphingosine-1-Phosphate Antibody as a Potential Therapeutic in Reducing Growth, Invasion, and Angiogenesis in Multiple Tumor Lineages. Cancer Cell (2006) 9:225–38. doi: 10.1016/j.ccr.2006.02.023
214. Pal SK, Drabkin HA, Reeves JA, Hainsworth JD, Hazel SE, Paggiarino DA, et al. A Phase 2 Study of the Sphingosine-1-Phosphate Antibody Sonepcizumab in Patients With Metastatic Renal Cell Carcinoma. Cancer (2017) 123:576–82. doi: 10.1002/cncr.30393
215. Rahmaniyan M, Curley RW, Obeid LM, Hannun YA, Kraveka JM. Identification of Dihydroceramide Desaturase as a Direct in Vitro Target for Fenretinide. J Biol Chem (2011) 286:24754–64. doi: 10.1074/jbc.M111.250779
216. Mody N, Mcilroy GD. The Mechanisms of Fenretinide-Mediated Anti-Cancer Activity and Prevention of Obesity and Type-2 Diabetes. Biochem Pharmacol (2014) 91:277–86. doi: 10.1016/j.bcp.2014.07.012
217. Nieto K, Mallery SR, Schwendeman SP. Microencapsulation of Amorphous Solid Dispersions of Fenretinide Enhances Drug Solubility and Release From PLGA In Vitro and In Vivo. Int J Pharm (2020) 586:119475. doi: 10.1016/j.ijpharm.2020.119475
218. Myatt SS, Burchill SA. The Sensitivity of the Ewing’s Sarcoma Family of Tumours to Fenretinide-Induced Cell Death Is Increased by EWS-Fli1-Dependent Modulation of P38mapk Activity. Oncogene (2008) 27:985–96. doi: 10.1038/sj.onc.1210705
219. Appierto V, Villani MG, Cavadini E, Lotan R, Vinson C, Formelli F. Involvement of C-Fos in Fenretinide-Induced Apoptosis in Human Ovarian Carcinoma Cells. Cell Death Differ (2004) 11:270–9. doi: 10.1038/sj.cdd.4401349
220. Britt EL, Raman S, Leek K, Sheehy CH, Kim SW, Harada H. Combination of Fenretinide and ABT-263 Induces Apoptosis Through NOXA for Head and Neck Squamous Cell Carcinoma Treatment. PloS One (2019) 14:e0219398. doi: 10.1371/journal.pone.0219398
221. Orienti I, Farruggia G, Nguyen F, Guan P, Calonghi N, Kolla V, et al. Nanomicellar Lenalidomide–Fenretinide Combination Suppresses Tumor Growth in an MYCN Amplified Neuroblastoma Tumor. Int J Nanomedicine (2020) 15:6873–86. doi: 10.2147/IJN.S262032
222. Mallery SR, Wang D, Santiago B, Pei P, Bissonnette C, Jayawardena JA, et al. Fenretinide, Tocilizumab, and Reparixin Provide Multifaceted Disruption of Oral Squamous Cell Carcinoma Stem Cell Properties: Implications for Tertiary Chemoprevention. Mol Cancer Ther (2019) 18:2308–20. doi: 10.1158/1535-7163.MCT-19-0361
223. Buehrer BM, Bell RM. Inhibition of Sphingosine Kinase In Vitro and in Platelets. Implications for Signal Transduction Pathways. J Biol Chem (1992) 267:3154–9. doi: 10.1016/S0021-9258(19)50708-6
224. Koshkaryev A, Piroyan A, Torchilin VP. Increased Apoptosis in Cancer Cells In Vitro and In Vivo by Ceramides in Transferrin-Modified Liposomes. Cancer Biol Ther (2012) 13:50–60. doi: 10.4161/cbt.13.1.18871
225. Barth BM, Sharma R, Altinoğlu EI, Morgan TT, Shanmugavelandy SS, Kaiser JM, et al. Bioconjugation of Calcium Phosphosilicate Composite Nanoparticles for Selective Targeting of Human Breast and Pancreatic Cancers In Vivo. ACS Nano (2010) 4:1279–87. doi: 10.1021/nn901297q
226. Beckham TH, Lu P, Jones EE, Marrison T, Lewis CS, Cheng JC, et al. LCL124, a Cationic Analog of Ceramide, Selectively Induces Pancreatic Cancer Cell Death by Accumulating in Mitochondria. J Pharmacol Exp Ther (2013) 344:167–78. doi: 10.1124/jpet.112.199216
227. Truman J-P, García-Barros M, Obeid LM, Hannun YA. Evolving Concepts in Cancer Therapy Through Targeting Sphingolipid Metabolism. Biochim Biophys Acta - Mol Cell Biol Lipids (2014) 1841:1174–88. doi: 10.1016/j.bbalip.2013.12.013
228. Bhat VK, Bernhart E, Plastira I, Fan K, Ghaffari-Tabrizi-Wizsy N, Wadsack C, et al. Pharmacological Inhibition of Serine Palmitoyl Transferase and Sphingosine Kinase-1/-2 Inhibits Merkel Cell Carcinoma Cell Proliferation. J Invest Dermatol (2019) 139:807–17. doi: 10.1016/j.jid.2018.10.024
229. Bellone M, Ceccon M, Grioni M, Jachetti E, Calcinotto A, Napolitano A, et al. iNKT Cells Control Mouse Spontaneous Carcinoma Independently of Tumor-Specific Cytotoxic T Cells. PloS One (2010) 5:e8646. doi: 10.1371/journal.pone.0008646
230. Lan H-R, Du W-L, Liu Y, Mao C-S, Jin K-T, Yang X. Role of Immune Regulatory Cells in Breast Cancer: Foe or Friend? Int Immunopharmacol (2021) 96:107627. doi: 10.1016/j.intimp.2021.107627
231. Chi H. Sphingosine-1-Phosphate and Immune Regulation: Trafficking and Beyond. Trends Pharmacol Sci (2011) 32:16–24. doi: 10.1016/j.tips.2010.11.002
232. Sanford M. Fingolimod: A Review of Its Use in Relapsing-Remitting Multiple Sclerosis. Drugs (2014) 74:1411–33. doi: 10.1007/s40265-014-0264-y
233. Schilling T, Miralles F, Eder C. TRPM7 Regulates Proliferation and Polarisation of Macrophages. J Cell Sci (2014) 127:4561–6. doi: 10.1242/jcs.151068
234. Zheng T, Meng X, Wang J, Chen X, Yin D, Liang Y, et al. PTEN- and P53-Mediated Apoptosis and Cell Cycle Arrest by FTY720 in Gastric Cancer Cells and Nude Mice. J Cell Biochem (2010) 111:218–28. doi: 10.1002/jcb.22691
235. Lorvik KB, Bogen B, Corthay A. Fingolimod Blocks Immunosurveillance of Myeloma and B-Cell Lymphoma Resulting in Cancer Development in Mice. Blood (2012) 119:2176–7. doi: 10.1182/blood-2011-10-388892
236. Ntranos A, Hall O, Robinson DP, Grishkan IV, Schott JT, Tosi DM, et al. FTY720 Impairs CD8 T-Cell Function Independently of the Sphingosine-1-Phosphate Pathway. J Neuroimmunol (2014) 270:13–21. doi: 10.1016/j.jneuroim.2014.03.007
237. Ottenlinger F, Schwiebs A, Pfarr K, Wagner A, Grüner S, Mayer CA, et al. Fingolimod Targeting Protein Phosphatase 2A Differently Affects IL-33 Induced IL-2 and IFN-γ Production in CD8 + Lymphocytes. Eur J Immunol (2016) 46:941–51. doi: 10.1002/eji.201545805
238. Sanghera C, Sanghera R. Immunotherapy – Strategies for Expanding Its Role in the Treatment of All Major Tumor Sites. Cureus (2019) 11:e5938. doi: 10.7759/cureus.5938
239. Attwood MM, Fabbro D, Sokolov AV, Knapp S, Schiöth HB. Trends in Kinase Drug Discovery: Targets, Indications and Inhibitor Design. Nat Rev Drug Discov (2021). doi: 10.1038/s41573-021-00252-y
240. Froehlich A, Schmidt S, Landsberg J, Bieber T, Wenzel J. Spontaneous Regression of Tumor-Stage Cutaneous T-Cell Lymphoma in a Multiple Sclerosis Patient After Discontinuing Fingolimod. Mult Scler J (2018) 24:1785–7. doi: 10.1177/1352458518774444
241. Deng J, Liu Y, Lee H, Herrmann A, Zhang W, Zhang C, et al. S1PR1-STAT3 Signaling Is Crucial for Myeloid Cell Colonization at Future Metastatic Sites. Cancer Cell (2012) 21:642–54. doi: 10.1016/j.ccr.2012.03.039
242. Rathinasamy A, Domschke C, Ge Y, Böhm H-H, Dettling S, Jansen D, et al. Tumor Specific Regulatory T Cells in the Bone Marrow of Breast Cancer Patients Selectively Upregulate the Emigration Receptor S1P1. Cancer Immunol Immunother (2017) 66:593–603. doi: 10.1007/s00262-017-1964-4
Keywords: cancer, sphingolipids, preclinical, clinical studies, therapy
Citation: Companioni O, Mir C, Garcia-Mayea Y and LLeonart ME (2021) Targeting Sphingolipids for Cancer Therapy. Front. Oncol. 11:745092. doi: 10.3389/fonc.2021.745092
Received: 21 July 2021; Accepted: 30 September 2021;
Published: 19 October 2021.
Edited by:
Ciro Isidoro, University of Eastern Piedmont, ItalyReviewed by:
François Trottein, Centre National de la Recherche Scientifique (CNRS), FranceMegan Young, Penn State College of Medicine, United States
Riccardo Ghidoni, University of Milan, Italy
Copyright © 2021 Companioni, Mir, Garcia-Mayea and LLeonart. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Matilde E. LLeonart, bWF0aWxkZS5sbGVvbmFydEB2aGlyLm9yZw==