 Wanru Zhang1†
Wanru Zhang1† Yaping An1†Xiali Qin1†Xuemei Wu1Xinyu Wang1
Yaping An1†Xiali Qin1†Xuemei Wu1Xinyu Wang1 Huiqin Hou1Xueli Song1
Huiqin Hou1Xueli Song1 Tianyu Liu1
Tianyu Liu1 Bangmao Wang1
Bangmao Wang1 Xuan Huang2*
Xuan Huang2* Hailong Cao1*
Hailong Cao1*- 1Department of Gastroenterology and Hepatology, General Hospital, Tianjin Medical University, Tianjin Institute of Digestive Diseases, Tianjin Key Laboratory of Digestive Diseases, Tianjin, China
- 2Department of Gastroenterology, The First Affiliated Hospital of Zhejiang Chinese Medical University, Hangzhou, China
Accumulating evidence from studies in humans and animal models has elucidated that gut microbiota, acting as a complex ecosystem, contributes critically to colorectal cancer (CRC). The potential mechanisms often reported emphasize the vital role of carcinogenic activities of specific pathogens, but in fact, a series of metabolites produced from exogenous dietary substrates or endogenous host compounds occupy a decisive position similarly. Detrimental gut microbiota-derived metabolites such as trimethylamine-N-oxide, secondary bile acids, hydrogen sulfide and N-nitroso compounds could reconstruct the ecological composition and metabolic activity of intestinal microorganisms and formulate a microenvironment that opens susceptibility to carcinogenic stimuli. They are implicated in the occurrence, progression and metastasis of CRC through different mechanisms, including inducing inflammation and DNA damage, activating tumorigenic signaling pathways and regulating tumor immunity. In this review, we mainly summarized the intimate relationship between detrimental gut microbiota-derived metabolites and CRC, and updated the current knowledge about detrimental metabolites in CRC pathogenesis. Then, multiple interventions targeting these metabolites for CRC management were critically reviewed, including diet modulation, probiotics/prebiotics, fecal microbiota transplantation, as well as more precise measures such as engineered bacteria, phage therapy and chemopreventive drugs. A better understanding of the interplay between detrimental microbial metabolites and CRC would hold great promise against CRC.
Introduction
Colorectal cancer (CRC) is a leading cause of cancer-related deaths worldwide and the morbidity and mortality of CRC are still rising (1). The mechanism of the initiation and progression of CRC has not been fully elucidated yet, but it is generally believed to be the result of the extensive and complex interaction between genetic and environmental factors. Epidemiological data have demonstrated that adverse environmental exposures, including overweight and obesity, high-fat diet (HFD), smoking and high consumption of alcohol, perform predominant roles in carcinogenesis (2, 3). Significantly, these controllable factors could have a strong impact on the structure and function of gut microbiota.
The microbiota that inhabits the human gut is a key factor in maintaining the stability of the intestinal microecosystem by participating in the immune regulation, substance metabolism, nutrient absorption in the human body, directly or indirectly (4). Usually, the gut microbiome engages in mutualistic relationships with the host, whereas it may mediate the occurrence and development of some diseases, which depends on both environment and the susceptibility of the host. Due to next generation sequencing technologies, such as 16S rRNA, 18S rRNA, internal transcribed spacer (ITS) sequencing, shotgun metagenomic sequencing, metatranscriptomic sequencing and virome sequencing, identifying the characterization of host-microbiota interactions has tremendous progress and the intestinal microbiota is indicated to be closely related to CRC (5, 6). The proposed pathogens which are candidates for CRC mainly include Fusobacterium nucleatum, Escherichia coli, Bacteroides fragilis, Streptococcus bovis, Enterococcus faecalis and Peptostreptococcus anaerobius (7). Also, the influence of the vaster microbial community, particularly its metabolome, has been connected to CRC (Table 1). The gut microbiota, the “new organ” of the human organism, has an enormous metabolic potential (8), and its metabolic capacity greatly exceeds that of human cells (9). About 50% of the metabolites found in feces and urine are derived from, or modified by the gut microbiota (10). Metabolomics, which is the qualitative and quantitative assessment of the metabolites (small molecules<1.5 kDa) in cells, tissues, organs, or biological fluids, has discovered thousands of microbiota-derived metabolites and further expanded our knowledge on the effects of specific metabolites in carcinogenic or cancer-promoting activity (11, 12). Common gut microbiota-derived metabolites mainly include amino acids and their by-products, lipids and lipid-like metabolites, bile acids (BAs) derivatives, and other metabolites produced by the degradation of carnitine and choline (11, 13, 14). These metabolites are altered in CRC patients and some of them have been confirmed to have both local and systemic effects on promoting the risk, initiation and progression of CRC (Table 2).
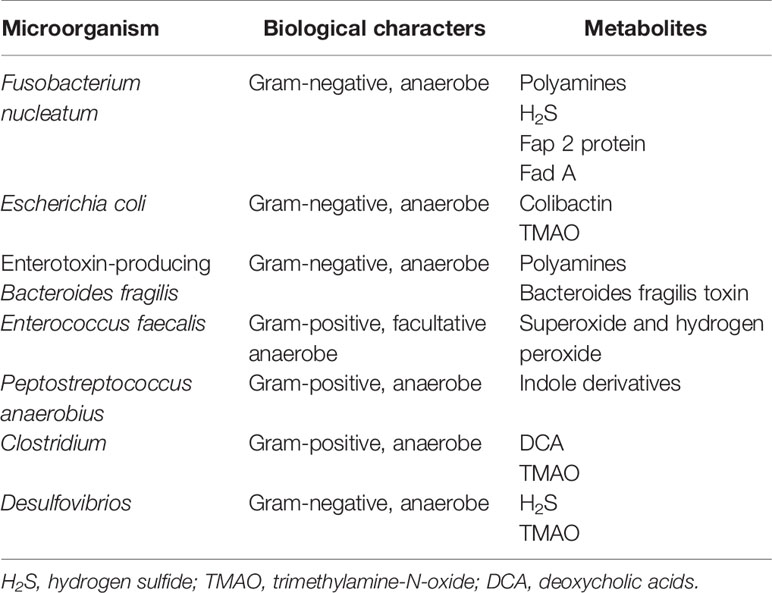
Table 1 Summary of individual microbes and their corresponding metabolites probably involved in colorectal cancer.
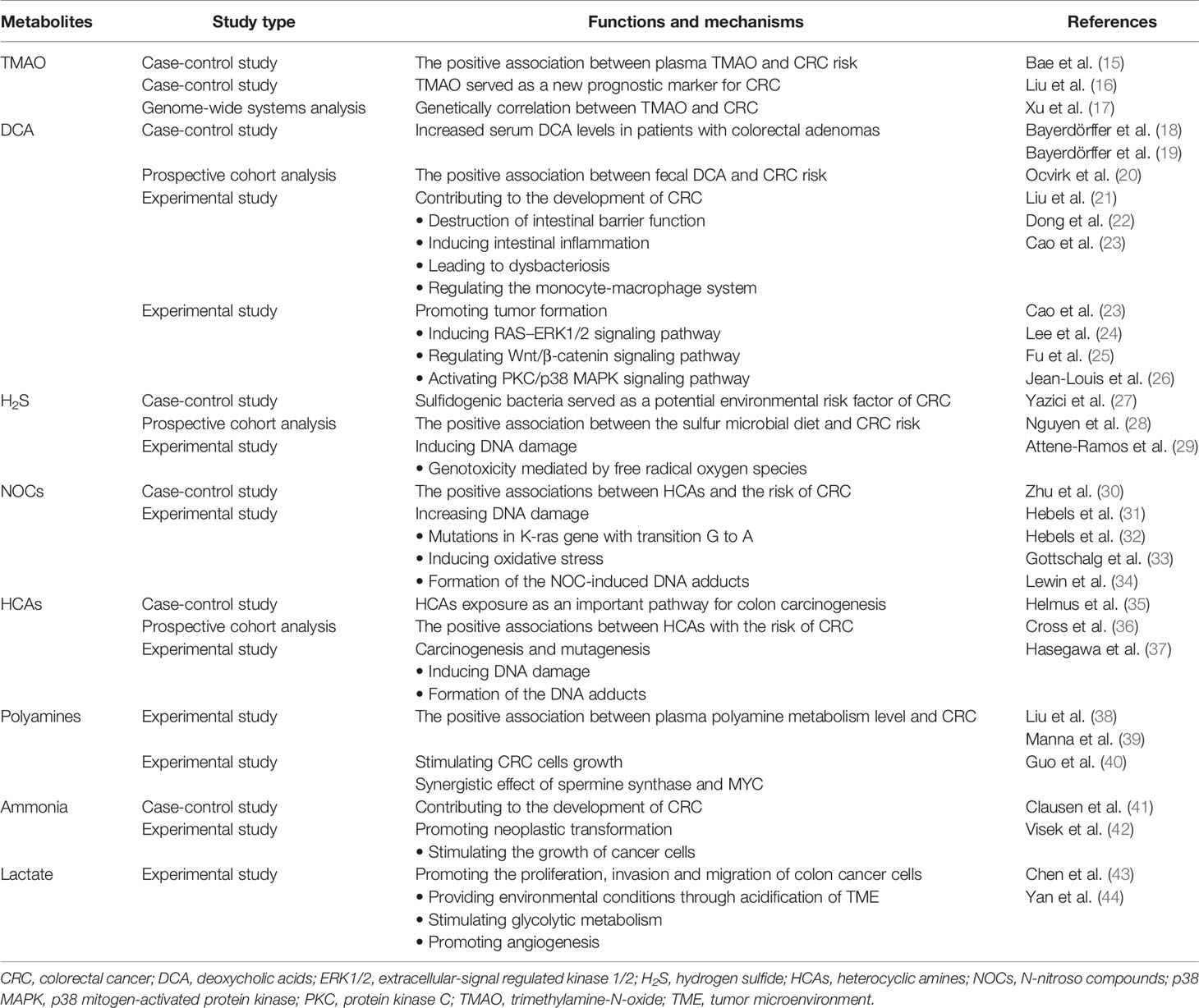
Table 2 Summary of studies related to detrimental microbiota-derived metabolites involved in CRC.
The current review highlighted the recent advances on the mechanisms by which detrimental metabolites from gut microbiota modulated the development and progression of CRC. Meanwhile, multiple potential therapeutic approaches for CRC were summarized. Targeting small molecule metabolites may contribute to providing promising therapeutic strategies for CRC.
Interaction of Gut Microbiota, Metabolites, and CRC
The potential genetic changes of CRC have been well confirmed up to now. At least three major molecular pathways can lead to CRC (chromosomal instability pathway, microsatellite instability and CpG island methylation pathway). However, in addition to the existence of several essential mutations, the occurrence and development of CRC also depend on the close interaction of mutagenized cells with the tumor microenvironment (TME) (45). During the development of CRC, tumor-promoting inflammation induced by gut microbiota usually involves the first two, characterized by the exaggerated production of cytokines by resident innate immune cells and the establishment of an immunosuppressive TME. Aberrant inflammation is considered as one of the hallmarks of cancer. During the development of CRC, tumor-promoting inflammation induced by gut microbiota usually involves the first two, characterized by the exaggerated production of cytokines by resident innate immune cells and the establishment of an immunosuppressive TME (45–47). These proinflammatory mediators interact with epithelia to compromise barrier function and further amplify the response by recruiting and activating additional immune cells. The breakdown of the intestinal barrier triggers the invasion of pathogenic microorganisms and their metabolites from the intestinal lumen, and leads to contact between intestinal epithelial cells (IECs) and components of the microbiota that might have pro-tumorigenic characteristics. Ensuring sustained immune response may trigger a cascade of inflammatory changes and promote carcinogenesis (47–49). The causal and complicit roles of microbes in cancer through their effect on the host’s immune system is defined as the immuno-oncology-microbiome (IOM) axis (50).
Pattern recognition receptors (PRRs) are essential components of the host immune system, which can recognize the conserved molecular patterns specific to microorganisms called pathogen-associated molecular patterns (PAMPs) such as lipopolysaccharide (LPS) to trigger diverse innate immune responses (51). The sub-families of PRRs mainly include several types of recognition receptors: the toll-like receptors (TLRs), the NOD-like receptors (NLR), C-type lectin receptors (CLRs), and RIG-I-like receptors (RLRs) (51). Intestinal mucosal epithelial cells and immune cells recognize intestinal microorganisms and their products mainly through TLRs, which are regulated by the intestinal microbiota and by circadian rhythmicity (52–55). In spite of different receptors, the vast majority of TLRs’ signaling is initiated by the myeloid differentiation primary-response protein-88 (MyD88) and leads to activation of downstream signaling pathways and recruitment of transcription factors such as nuclear factor-Kappa B (NF-κB), mitogen-associated protein kinase (MAPK), and interferon (IFN) regulatory factors and subsequent generation of cytokines and chemokines. The expression of TLR2 in colon cancer is significantly upregulated and the TLR2 agonists significantly enhance the proliferation, migration, and invasion of CRC cells (56). TLR4 can produce trophic factors and vascular growth factors through the TLR4/MyD88/NF-κB signaling pathway (57) and promote tumor proliferation through TLR4/Cyclooxygenase 2 (COX2)/prostaglandin E2 (PGE2). TLR9 could induce downstream signals to recruit inflammatory factors, such as IL(interleukin)-8, TGF-β, PGE2, and other immunosuppressive molecules, leading to the continuous state of inflammation, the escape of tumor immunity, and the unlimited proliferation of tumor cells (58).
Immune elimination and immune escape are hallmarks of cancer. The TME is usually at an immunosuppressive state. A range of microbial derivatives and metabolites can mediate host immune disorders via affecting the differentiation, proliferation, maturation and effector function of innate and adaptive immune cells, thereby influencing tumor surveillance (47). Polyamine synthesis is essential to induce cytotoxic activity and T-cell proliferation. Supplementation with spermidine or L-arginine promotes homeostatic differentiation of Treg cells with a beneficial role in the context of gut inflammation (59). However, polyamines possibly have opposing roles depending on their concentrations, since elevated polyamine levels in cancer have been shown to diminish the antitumor immune responses by resulting in various defects in immune cell function, including inhibition of lymphocyte proliferation (60–62). Moreover, TME rich in fatty acids can inhibit the function of effector T cells and M1-polarization of macrophages, and facilitate the differentiation of T regulatory cells (Tregs) and M2-like macrophages (63). Notably, only a relatively narrow segment of the studies has elucidated the reciprocal interaction between metabolites and tumor immunity, and their influences on immunosuppression remain poorly characterized.
Detrimental Microbiota-Derived Metabolites in Intestinal Carcinogenesis
One of the primary modes of interaction between the gut microbiota and the CRC is through metabolites. Specific classes of detrimental microbiota-derived metabolites such as Trimethylamine-N-oxide (TMAO), BAs, hydrogen sulfide (H2S), N-nitroso compounds (NOCs) form a complex metabolic network related to the etiology and severity of CRC (Figure 1). Once these metabolites pass the mucosal barrier, they can act directly on IECs or influence immune responses in the intestinal stroma, trigger the release of pro-inflammatory signals, such as tumor necrosis factor (TNF) and IL-17, or lead to immunosuppression in the TME, which can further promote tumorigenesis. Furthermore, microbial metabolites can induce tumorigenesis by inducing DNA damage and activating the intracellular tumorigenic signaling pathways.
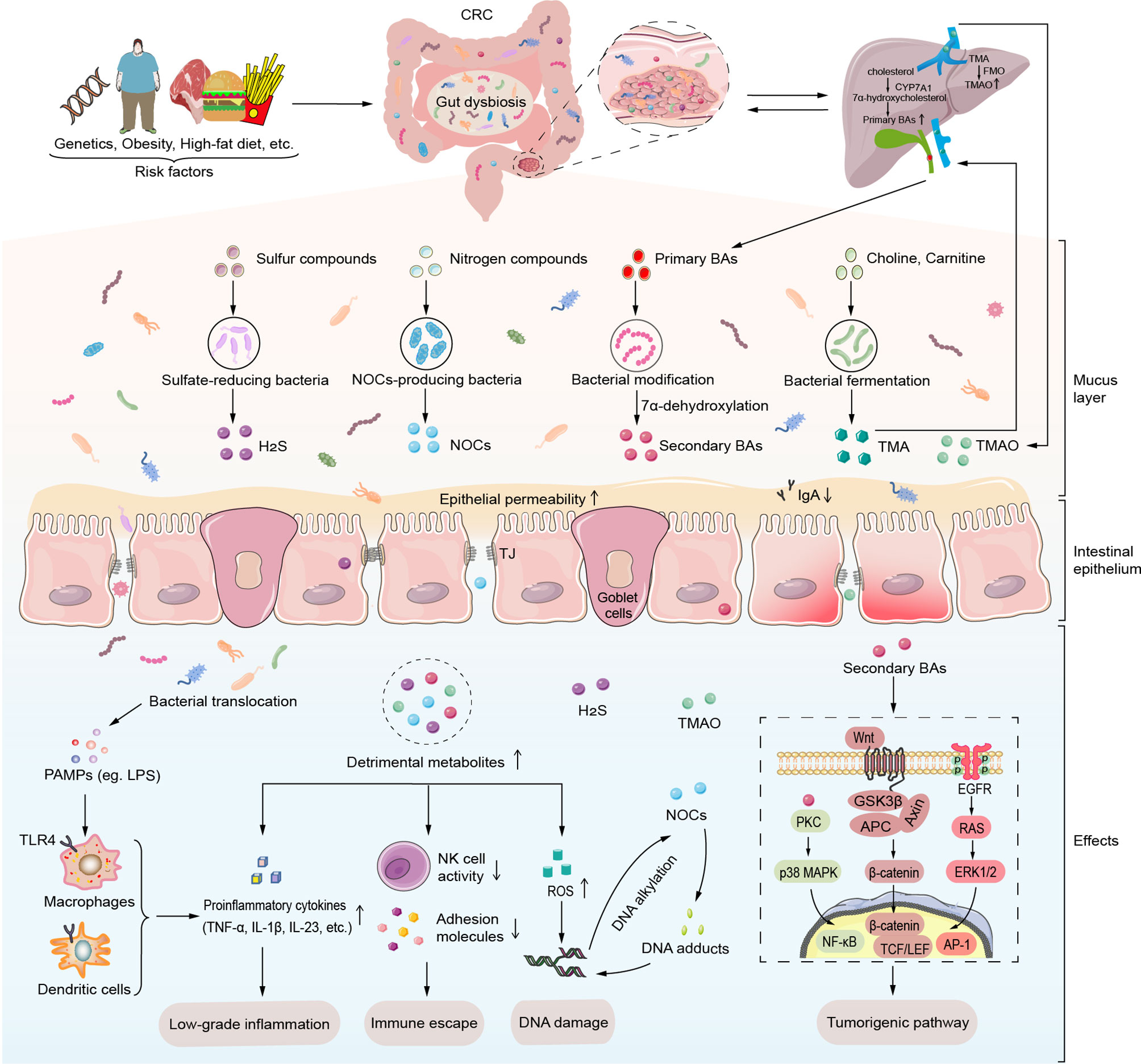
Figure 1 Detrimental gut microbial metabolites induce intestinal carcinogenesis. Various risk factors such as host genetics, obesity, and high-fat diet induce gut microbiota dysbiosis. Alteration of gut microbiota and its metabolites lead to mucus layer thickness reduction, high intestinal permeability and ensuing translocation of commensal microbiota and its metabolites. Maleficent bacteria overgrowth produces amounts of PAMPs like LPS to recognize the TLR4 of macrophages and dendritic cells, which then release certain proinflammatory cytokines (TNF-α, IL-1β, IL-23, etc.). Moreover, detrimental gut microbiota-derived metabolites such as secondary BAs, TMAO, H2S, and NOCs induce carcinogenesis through low-grade inflammation, immune escape, DNA damage, and activation of tumorigenic signals. Particularly, secondary BAs contribute to the progression of CRC via specific intracellular transduction pathways such as PKC-p38 MAPK signaling pathway, EGFR-ERK1/2 signaling pathway and Wnt/β-catenin signaling pathway. In addition to inducing ROS production, NOCs can involve in DNA damage by DNA alkylation and DNA adducts. AP-1, activator protein-1; APC, adenomatous polyposis coli; BAs, bile acids; CRC, colorectal cancer; CYP7A1, cholesterol 7 α hydroxylase; EGFR, epidermal growth factor receptor; ERK1/2, extracellular signal-regulated kinase 1/2; FMO, flavin monooxygenase; GSK3β, glycogen synthase kinase 3β; H2S, hydrogen sulfide; IL-1β, interleukin-1β; IL-23, interleukin-23; LEF, Lymphatic enhancement factor; LPS, lipopolysaccharide; NF-κB, factor-Kappa B; NOCs, N-nitroso compounds; p38 MAPK, p38 mitogen-activated protein kinase; PAMPs, pathogen-associated molecular patterns; PKC, protein kinase C; ROS, reactive oxygen species; TCF, T cell factor; TJ, tight junctions; TLR, Toll-like receptor; TMA, trimethylamine; TMAO, trimethylamine-N-oxide; TNF-α, tumor necrosis factor-α.
TMAO and CRC
With the deepening of research on microbial metabolites, TMAO has attracted more attention for its influence on many aspects of health in general. The generation of TMAO is dependent on the gut microbiota, which metabolizes phosphatidylcholine, choline, and carnitine from foods (red meat, egg yolk, milk as typical foods of HFD) to produce trimethylamine (TMA). TMA is carried through the portal circulation to the liver, where it is oxidized by the host hepatic flavin monooxygenase (FMO) to produce TMAO which is then released into circulation and eventually eliminated in the kidney (64–66). Consequently, TMAO is dependent upon dietary consumption of precursors, gut microbial enzymatic activity and host genetics (67), and may be a vital mediator among diet, gut microbiota metabolism and the risk of CRC.
TMAO might be a potential early diagnosis prognostic biomarker for CRC. One study that enrolled 835 matched case-control female pairs from the Women’s Health Initiative Observational Study as research objects showed that TMAO was positively connected with CRC risk in age-adjusted analyses (15). Similarly, Liu et al. found the serum TMAO level of CRC patients was significantly higher than that of healthy controls and was related to tumor progression and distant metastasis (16). In addition, an unbiased data-driven network-based systems approach demonstrated that TMAO was genetically associated with CRC. They found that TMAO might share dozens of genetic pathways including the immune system, cell cycle and Wnt signaling with CRC (17). Although the potential relationship between TMAO and CRC has been investigated, direct evidence for promoting CRC is currently lacking. Inflammation, oxidative stress and DNA damage could be potential molecular mechanisms between TMAO and carcinogenesis. Recently, a dose-response meta-analysis summarized the results of studies on the relationship between circulating TMAO concentrations and inflammation risk, and then revealed a non-linear positive association between increased TMAO concentrations and C-reactive protein (68). Yue et al. demonstrated that TMAO could trigger the activation of the nod-like receptor family pyrin domain containing 3 (NLRP3) inflammasome and inhibit the autophagy gene ATG16L1-induced autophagy to promote the progression of inflammatory bowel disease (IBD) (69). These results implied that inflammation might be a possible factor connecting TMAO and CRC. Oxidative stress could conduce to CRC development by leading tumor cells to be insensitive to antiproliferative signals, apoptosis and anchorage-independent cell growth, and altering the invasion and migration of tumor cells through epigenetic and metabolic mechanisms (70). It has been discovered that increased levels of TMAO in the circulation are related to oxidative stress via inducing the production of superoxide, a reactive oxygen species (ROS) (71). Moreover, TMAO involves in the formation of NOCs which cause DNA damage and epigenetic changes, suggesting a potential role of DNA damage in TMAO-contributed carcinogenesis (72). However, the effort of TMAO on the pathogenesis of CRC can be more accurately elucidated through long-term studies of multi-center populations and large samples that include dietary investigation and comprehensive analysis of intestinal target microbial detection as well as combined with animal experiments.
Secondary BAs and CRC
Primary BAs are synthesized from cholesterol in hepatocytes by cholesterol 7 α hydroxylase (CYP7A1) and released into the intestinal tract with the bile. About 95% of BAs in the intestine are reabsorbed and transported back to the liver via the portal. The rest escapes enterohepatic circulation and then enters the colon, where they are transformed by the intestinal bacteria through 7α-dehydroxylation into secondary BAs (73). Bacteria capable of producing secondary bile acids belong to the B. fragilis, Bacteroides vulgatus, Clostridium perfringens, Eubacterium, Lactobacillus and Bifidobacterium (74). The perturbations of the intestinal microbiota composition can strongly impact BA metabolism. It has been reported that interplay between BAs and gut microbiota could mediate the malignant transformation of colorectal adenomas (74), and the elevated levels of secondary BAs, especially deoxycholic acid (DCA) play a critical role in this process. In two small case-control studies from the 1990s, the serum concentration of DCA in colorectal adenoma patients was showed significantly higher compared with healthy individuals (18, 19). Consistent with this, a prospective cohort analysis investigated the association between gut microbial co-metabolism and the risk of CRC in Alaska Native and rural African people. Data manifested that fecal concentrations of the DCA were more than 2-fold higher in Alaska Native than that in rural African participants (20). Several experimental findings supported these clinical data. Our group has unveiled partial mechanisms of DCA promoting the pathogenesis of CRC using a mouse model of gastrointestinal tumorigenesis. Data showed that DCA brought about an increase in the number and volume of intestinal adenomas in Apcmin/+ mice, resulting in impaired intestinal barrier function and intestinal inflammation, and subsequently promoted intestinal carcinogenesis via activating tumor-related signaling pathways. (21–23).
In addition, some related tumorigenic signaling pathways by which DCA promotes the development of CRC have been identified and studied intensively (73). First of all, DCA triggered tyrosine phosphorylation and activated the epidermal growth factor receptor (EGFR) signaling pathway of tyrosine kinases in a ligand-dependent manner, which then led to the activation of RAS-extracellular-signal regulated kinase 1/2 (ERK1/2) signaling. ERK1/2 could induce the activation of activator protein 1 (AP-1) to mediate cell proliferation and differentiation (24). Moreover, DCA-induced Wnt signaling is able to induce an inflammatory response and stimulate the proliferation of CRC cells, which also plays a crucial role in the progression of CRC (23, 25). DCA can affect the development of cancer by causing β-catenin to be released and enter the cytoplasm where it translocates to the nucleus and stimulates transcription factors such as the T cell factor/lymphoid enhancer factor (TCF/LEF) family (75). Besides, perturbation of the plasma membrane by DCA also activates protein kinase C (PKC). PKC subsequently induces the activation of p38 mitogen-activated protein kinase (p38 MAPK) and NF-κB, which play indispensable roles in the regulation of immune and inflammatory responses. Activated NF-κB translocates into the nucleus, where it contributes to the expression of IL-1β (26, 76).
BAs receptors also play critical roles in CRC. Farnesoid X receptor (FXR) and Takeda G protein-coupled receptor 5 (TGR5) are the two most critical receptors of BAs, which mediate the regulation of BAs on downstream signaling pathways and cell functions (74). The regulation of the microbiota-FXR axis is one of the main mechanisms that gut microbiota affects and participates in the metabolism of BAs. Colonization of GF mice with a human microbiota from feces can reduce total BA levels and induce expression of FXR and its downstream target genes in the intestine (77). Currently, it is generally accepted that the deletion and decrease of FXR expression in the colon and rectum are associated with CRC development and metastasis (78, 79). FU et al. observed that down-regulation of FXR promoted the progression of CRC, whereas selective activation of intestinal FXR could restrict intestinal cancer stem cell proliferation and profoundly increased survival in APCmin/+ mouse models of adenoma (25). Moreover, a study has already found that the TGR5 mRNA expression level in intestinal tumor tissues was significantly higher than that in normal tissues (80). Nevertheless, the role of TGR5 in intestinal carcinogenesis remains to be explored.
H2S and CRC
H2S produced by fermenting different S-containing substrates notably cysteine through both the gut microbiota and endogenous enzymes is emerging as a key regulator of gut health, including CRC and IBD (81, 82). Gut luminal H2S production appears to be dependent on the action of sulfate-reducing bacteria (SRB). Therefore, the upregulation of H2S and sulfidogenic bacteria may be potential environmental risk factors contributing to CRC development (82).
Previous studies have shown that African Americans contain substantially higher abundances of SRB and Bilophila wadsworthia in the colon, which may be the reason why the incidence of CRC in African Americans is higher than in non-Hispanic whites (27). In particular, the abundance of sulfur-metabolizing bacteria correlated positively with fat and protein intake. Long-term persistence to a dietary pattern related to sulfidogenic bacteria in stool can cause an elevated risk of distant CRC (28). These data support the conception that H2S participates in colorectal carcinogenesis. The important carcinogenic mechanism may be the cytotoxicity and genotoxicity of H2S. H2S has been reported to induce genomic DNA damage that could be radical-mediated, which is directly related to the induction of cumulative mutations of CRC (29). In addition, H2S is cytotoxic to intestinal epithelium cells and causes mucosal damage (83, 84). In addition, H2S is cytotoxic to intestinal epithelium cells and causes mucosal damage (29, 83). An increase in SRB and H2S concentration could reduce disulfide bonds in the mucus network, thereby allowing luminal bacteria and their products to penetrate and contact with the host epithelial lining and immune cells to induce apoptosis in epithelial cells and inflammatory activation in immune cells (85, 86). What’s intriguing is that there indeed exists conflicting data on the protective and pathological effects of H2S in the gastrointestinal tract. It has been argued that H2S can control the inflammation and facilitate correction of microbiota biofilm dysbiosis and mucus layer reconstitution (87, 88). Also, H2S could inhibit proliferation and promote protective autophagy in colon epithelial cells via the AMPK pathway (89). These controversies may relate to the bell-shaped dose-response curve of H2S. Overall, further studies are needed to determine how H2S produced by the gut microbiota contributes to CRC pathogenesis.
NOCs and CRC
NOCs, including N-nitrosamines and N-nitrosamides, are among the most potent experimental procarcinogens mainly derived from the fermentation of proteins of red and processed meat by facultative and anaerobic colonic bacteria (90, 91). Types of dietary intake and bacterial colonization largely affect the formation of NOCs in the intestine. The high levels of red meat consumption would increase the total amount of NOCs and consequently account for the epidemiologic association between red meat consumption and CRC (92). In addition, advances from clinical studies further substantiated the hypothesis that NOCs intake might be associated with a higher CRC incidence in humans (30).
N-nitrosamines are pro-carcinogens and have no direct mutagenic effect on the cells of organs and tissues. However, cytochrome P450 family 2 subfamily E member 1 (CYP2E1)-mediated hydroxylation of N-nitrosamines can result in the formation of carcinogenic surfactant diazomethane and ultimately lead to the generation of DNA-reactive methyl carbocation (93). N-nitrosamides are direct carcinogens that can interact with the cellular macromolecule. The mechanism of NOCs inducing carcinogenesis is known to relate to DNA damage. On the one hand, NOCs are very likely to react with the nucleophilic center of DNA bases, causing DNA alkylation and inducing the K-ras gene and TP53 gene in epithelial cells to undergo G→A transitions (31, 94). Similarly, oxidative damage is another important form of DNA damage caused by nitrosamine exposure (32). On the other hand, the generation of extremely reactive alkylating agents like diazoacetate contributes to the formation of the NOC-induced DNA adducts which are the main executor of DNA-damaging and carcinogenic properties (33, 34). The imbalance between DNA damage and DNA repair determines the initiation and progression of CRC to some extent. By way of example, if not or incorrectly repaired by O(6)-Methylguanine-DNA-methyltransferase (MGMT), the NOC-specific DNA adduct O6-carboxymethyl-2′-deoxy-guanosine (O6-CMdG) would cause dramatic biological consequences, such as accumulating on the DNA of intestinal cells, promoting the shedding of colon cells, and leading to the early stage of CRC (95, 96). Therefore, how to reduce or eliminate the content of NOCs may provide a good prospect for the prevention of CRC.
Other Metabolites and CRC
A diverse reservoir of metabolites from gut microbiota includes the well-known substances mentioned above, the more underestimated examples such as heterocyclic amines (HCAs), polyamine, ammonia and lactate, which also participate in the initiation and promotion of CRC.
Another important metabolite is HCAs, which can be significantly produced by gut microbiota during fermentation of red meat and processed meat. Evidence is available that individual differences in the gut microbiota have a strong impact on the genotoxic and carcinogenic properties of HCAs. Bacteroides strains tend to help convert HCAs into DNA-reactive carcinogens, whereas probiotic lactobacilli binding HCAs can reduce their mutagenic effect (97, 98). Numerous studies have implicated HCAs as an oncogenic driver in CRC, due to the association between elevated HCAs levels and high risk of CRC (35, 36). HCAs have been reported to induce frameshift mutations, microsatellite instability, strand breaks and oxidative base damage (99). Particularly, HCA-induced DNA adducts which are considered as the main executor of DNA-damaging and carcinogenic properties can generate mutations in the colon (37).
Most tumors have a greatly increased need for polyamines to meet their huge metabolic demands. Intracellular polyamine levels are maintained through tightly regulated pathways of biosynthesis, decomposition, absorption, and output. Polyamines and their metabolites in urine and plasma are often regarded as possible biomarkers of occurrence and progression in CRC (12). A piece of strong evidence is that the levels of putrescine and agmatine were described to increase with the progression of CRC (38, 39). Additionally, Guo et al. found the synergistic effect of spermine synthase and oncogene MYC increased polyamine metabolites, suggesting dysregulation of polyamine metabolism has been linked to the development of CRC (40).
According to reports, ammonia is one of the potential carcinogenic products of protein fermentation. Amino acid deamination by the gut bacteria and urea hydrolysis by bacterial urease are the main sources of ammonia in the intestine (100, 101). Continuous exposure of colonocytes to free ammonia may contribute to the development of CRC (41). The concentration of ammonia in the human intestinal cavity progressively increases from the right colon to the left colon, with the highest levels in the region of the colon where cell proliferation and the incidence of polyps and cancer are the highest (102). Animal experiments showed that after long-term ammonium chloride perfusion into the colon of rats, the distal colon mucosa displayed histological damages with the epithelial tissue disordered and the pathologic intestinal epithelial shedding (103). Besides, excessive ammonia enhances cell proliferation in the colon mucosa, which makes neoplastic transformation more efficient (42).
In tumors, both hypoxia and oncogene expression can stimulate glycolytic metabolism. As a result, the production of lactate seems to be a common energy metabolism pathway in cancer cells (104). Lactate can stimulate angiogenesis and transfer nutrients such as oxygen and glucose to cancer cells, and consequently inducing colon cell proliferation, invasion and migration (43, 44). Furthermore, the presence of lactate in the TME leads to extreme acidic conditions, inhibits the cytotoxic and effector functions of T cells and advances an immune escape of CRC, and even hampers the efficacy of several chemotherapeutic agents resulting in a poor prognosis (105, 106).
Clinical Transformation of the Gut Metabolome in CRC
Metabolomics has been increasingly used to identify biomarkers in disease with great potential for clinical translation. With relevant technological advances and new analytical and bioinformatics tools development, metabolomics can already provide sensitive and highly reproducible platforms allowing the quantification of the known compound or comprehensive analysis of all the measurable analytes in a sample (107). A large amount of metabolomics data has been accumulated to be used to investigate the interaction between the host and microorganisms from the perspective of metabolism. It was found that several gut microbiome-associated serum metabolites (GMSM) have changed significantly through integrated analysis of the serum metabolites and fecal metagenomics of patients with CRC and adenoma, which can efficiently discriminate patients with CRC and adenoma from normal individuals (108). Dysregulation of these metabolites indicates the possibility of common diagnostic biomarkers for CRC. However, there is still a need for better non-invasive biomarkers for CRC, especially for the early stages of the disease, including the adenoma step and the initial CRC stages. The content of several classes of bioactive lipids, including polyunsaturated fatty acids, secondary BAs and sphingolipids (109, 110) increased in adenoma patients. Most of these metabolites show directionally consistent changes in patients with CRC, indicating that these changes may represent early events of carcinogenesis (111). Rao J et al. pointed out that three metabolites (hydroquinone, leucenol and sphingomyelin) are positively and significantly correlated with CEA and/or CA 19-9, which may be potential biomarkers for advanced CRC (112). Furthermore, dynamic shifts of microbial metabolism have been confirmed in patients with different stages of colorectal neoplasia. Yachida et al. showed that branched-chain amino acids and phenylalanine were significantly increased in patients with intramucosal carcinomas, and BAs were significantly elevated both in patients with multiple polypoid adenomas or intramucosal carcinomas (113). These data provided evidence for the functional importance of the gut metabolome in CRC and implied a potential role of the gut microbiota-derived metabolites in the early diagnosis and prognosis of CRC. The term “pathogenic function (pathofunction)” has been proposed to evaluate specific features of host bacterial communities based on the detrimental metabolites produced by the gut microbiota. The pathofunction of the gut microbiota may contribute to the development of precision treatment directing gut microbiota to increase host health. However, universal biomarkers for CRC detection have not been identified due to the high variability of the microbiota and their metabolites between individuals. A standardized sample preparation and metabolomics analysis methods warrant further exploration.
Manipulation of Metabolites in CRC Treatment
In view of the importance of the mechanistic link between detrimental gut microbiota-derived metabolites and CRC, targeted metabolomics has been a remarkable research topic in recent years. There have been several beneficial metabolites [i.e., short-chain fatty acids (SCFAs)] profiling studies for preventing and treating CRC, however, fewer studies have been involved in CRC therapeutic explorations by targeting detrimental microbial-derived metabolites. In our review, we put effort to mention those studies which have linked regulating excess detrimental microbial-derived metabolites in the diagnosis, prevention and treatment of CRC. Pay more attention to how to use these studies for clinical application has great significance for the diagnosis, prognosis, therapeutics, and prevention of CRC.
Diet Modulation
Diet is an important environmental determinant of metabolism. Diet-mediated changes in whole-body metabolism and systemic nutrient availability can affect the overall performance of TME (2, 114). Metabolites derived from gut microbes are the key executors of diet affecting the host physiology, either inhibiting or accelerating tumor growth. Red and processed meat might increase CRC risk by increasing the formation of detrimental microbial metabolites such as TMAO, NOCs and HCAs. On the contrary, dietary fiber (DF) enriched in fruits, vegetables and whole grains is associated with a lower risk of CRC (115). A comprehensive meta-analysis of prospective studies and clinical trials provided convincing evidence that fiber intake was related to the risk of CRC. Dose-response relationships suggested greater effects on CRC risk reduction for higher intakes of DF (116). Importantly, DF can protect against CRC by altering not only microbial composition but also the concentration and availability of metabolites. The link between DF and BAs may be identified as a potential mechanism to explain the protective role of DF in CRC. In a comparative study in African American individuals, switching from low-fiber, high-fat to high-fiber, low-fat diet resulted in significant improvement in the microbiota and metabolome associated with CRC risk. Further exploration revealed that it was mainly due to an increase of saccharolytic fermentation and butyrogenesis and inhibition of secondary BAs synthesis (117). Consistent with this, a recent study investigated concentrations of fecal and serum BAs in vegans and omnivores, showing a vegan diet low in fat and high in fiber was positively correlated with fecal BA concentrations (118). Additionally, Li et al. have suggested that supplying the DF diet to mice can reduce TMA and TMAO outputs via inhibiting intestinal microbial TMA lyase activity (119). Thus, DF intervention for individuals with various clinical statuses is expected to become an emerging treatment against CRC.
Probiotics/Prebiotics
Probiotics are well-known biologically active candidates for the treatment of several diseases and unhealthy conditions by positively regulating the gut microbiota (120). Lactobacilli, Clostridium butyricum and Lactobacillus rhamnosus GG (LGG) are some of the most studied and well-characterized among probiotics. On the one hand, probiotics are commensal live bacteria that reduce intestinal inflammation and improve intestinal barrier function to achieve the prevention and treatment of CRC. On the other hand, probiotics can remove carcinogenic metabolites and toxins from the gastrointestinal tract, compete with spoilage bacteria and pathogenic bacteria and maintain the balance of gut microbiota (121, 122). Detrimental metabolites are mainly derived from the gut microbiota, such as Bacteroides, Clostridium (clusters XIVa and XI), Eubacterium, Enterobacter, Campylobacter jejuni and SRB. Supplementing probiotics can reduce the colonization of these bacteria, which may reduce the level of detrimental metabolites in the intestine or body, and thus inhibit intestinal tumor development. Probiotics can regulate intestinal bacteria related to proteolysis, reduce harmful protein fermentation, thus reducing the toxicity of metabolites (123). It was found that supplementing with Lactobacillus or Bifidobacterium or both can significantly reduce serum cholesterol. The main mechanism is the use of bile salt hydrolyzing enzymes to decouple the bile, and then precipitation of cholesterol along with the decoupled bile (124, 125). Moreover, Our previous studies have found that Clostridium butyricum antagonized against HFD-induced intestinal carcinogenesis and decreased the production of secondary BAs via down-regulating the abundance of Clostridium (126).
Fructooligosaccharides and galactooligosaccharides and inulin are frequently reported prebiotics. Prebiotics are non-digestible selectively fermented DF that can be used as substrates by gut microbiota to promote the metabolism of lipids, proteins and minerals, and produce metabolites that are potentially protective of gut functionality (127). In addition, prebiotics promote favorable bacteria of the indigenous intestinal flora of humans, or also improve the survival of probiotics that have been ingested at the same time, thereby indirectly reducing potentially pathogenic bacteria and/or detrimental metabolites in the intestine. Interestingly, the introduction of prebiotics improves the viability of probiotics. Compared with used alone, the combination of prebiotics and probiotics has been reported to reduce the expression of genes involved in carcinogenic pathways and drug resistance, and decrease the levels of the metabolite lactate, thus disturbing the growth of cancer cells and benefiting for CRC treatment (128). However, clinical trials evaluating the efficacy of probiotics for CRC are still limited and there are no specific criteria for selecting probiotics or prebiotics, thus systematic evaluation of different strains is required.
Fecal Microbiota Transplantation
Fecal microbial transplantation (FMT) is an emerging treatment scheme for transplanting gut microbiota from healthy donors to patients through various channels, so as to restore the intestinal microbial diversity of patients and achieve the therapeutic purpose (129). The significant differences in gut microbiota composition between cancer patients and healthy individuals show the therapeutic potentials of microbiota modulation. It is reasonable to support the hypothesis that FMT is prospective in cancer management as well as cancer-treatment associated complications, which is being actively explored (130, 131). The natural gut microbiome could promote host fitness, eliminate excessive inflammation and protect against mutagen/inflammation-induced colorectal tumorigenesis (132). FMT affects metabolism by resolving dysbiosis, or directly transferring primary and secondary BAs, which may underlie the observed effects of FMT in CRC. At this stage, the uncertainty and safety of the efficacy of FMT still should not be ignored. In-depth experiments and clinical trials are essential for its development and improvement.
Engineered Bacteria
Therapeutic approaches that use live tumor-targeting bacteria are ushering in a new era of cancer therapy. Bacteria can be engineered to act as therapeutic delivery payloads following simple genetic rules or complex synthetic bioengineering principles to realize the functions of delivering drug molecule or immunoregulatory factors to tumors, destroying tumor matrix, and silencing tumor genes (133). Compared with non-precision approaches such as the use of probiotics, engineered bacteria can be more accurately mitigate the effects of detrimental microbial-derived metabolites that are significant in the pathogenesis of CRC. A recent study developed engineered Escherichia colistrain that could convert systemic ammonia into L-arginine in a mouse model, thus exhibiting the potential for microbiota-based therapeutics to more precisely regulate metabolism (134). Engineered bacteria can be applied either as a monotherapy or a complement to other anticancer therapies to obtain improved antitumor activities. Some of them have passed clinical tests and shown greatly encouraging results. In fact, the success of therapy depends on the functional stability, clinical potency and safety of the engineered bacteria (135). Meanwhile, we must identify appropriate gut-adapted strains and consider performance metrics when deploying such bacteria in vivo (136).
Phage Therapy
Phage therapy may be a promising therapeutic tool for modulating the gut microbiota and gut metabolome as well as stimulating systemic anticancer immune response (137). Bacteriophages are viruses ubiquitous in the human intestinal tract that infect and replicate within bacteria and usually have species-level specificity. By directly knockdowning their bacterial targets or further inducing cascading effects on non-susceptible species through inter-bacterial interactions, the therapeutic effect of bacteriophages can be utilized to modulate the gut microbiota, which in turn has a potential impact on gut metabolome such as significant changes in bile salts (138, 139). The link between phage and microbial metabolites provides an interesting therapeutic avenue. Manipulation of microbiota by lytic phage can be used to selectively reduce detrimental microbial metabolites (140). For instance, phage predation of E. faecalis reduced tyramine, which can induce ileal contractions (141).
Chemopreventive Drugs
The value of chemoprevention strategies for preventing early stages or recurrence of CRC or new polyp formation has been widely noted in recent decades. Several studies to date have supported the chemopreventive potency of some promising agents, particularly those targeting metabolic pathways. Inhibition of the formation of detrimental metabolites appears, therefore, to be a rational target in chemoprevention.
Ursodeoxycholic acid (UDCA) is one of the BAs that has different effects on CRC compared with DCA. A population-based study indicated that the use of UDCA was associated with a lower risk of CRC (142). UDCA treatment affected the microbial community composition in men, as manifested by increased abundance of Faecalibacterium prausnitzii and decreased Ruminococcus gnavus, which partially explained the UDCA action to inhibit adenoma development (143). Additionally, the hydrophobicity of BAs may be an important determinant of their carcinogenic properties (144). One possible biologic mechanism by which UDCA acts therapeutically may be to increase the hydrophilicity of the biliary pool, dilute the concentration of toxic secondary BAs in the biliary pool such as DCA and lithocholic acid (LCA) and then prevent colonic neoplastic transformation (145). However, only low-dose UDCA is recommended for the prevention of colorectal neoplasia. The hypothesis that UDCA may increase the risk of developing neoplasia was first proposed by Eaton et al., who showed that high-dose UDCA can increase the incidence of colorectal neoplasia in patients with ulcerative colitis and primary sclerosing cholangitis (146). Obviously, more studies are needed for UDCA applications.
CRC and diabetes mellitus share various clinical risk factors leading to an interest in anti-diabetic agents as potential chemopreventive drugs for CRC. Metformin, as the first-line oral medicine for type 2 diabetes, has been shown to significantly reduce colorectal adenoma and cancer incidence and improving survival outcomes (147, 148). In the context of CRC, the protective effects of metformin are likely multi-factorial. One of the antitumor effects of metformin is an indirect effect resulting from systemic metabolic changes, including decreases in plasma glucose. Metformin had strong effects on the gut microbiome, and this was reversed when the drug was removed (149). Exposure to metformin modulated gut microbiota in patients with type 2 diabetes and increased the concentration of BA glycoursodeoxycholic acid and tauroursodeoxycholic acid, which were known antagonists of the FXR. These changes inhibited intestinal FXR signaling and improved metabolic dysfunction, hinting the alterations in microbiome composition and the subsequent impact on secondary BAs production may be an important factor in the prevention of CRC by metformin (150). The other is a direct effect on tumor cells. The liver kinase B1 (LKB1)-dependent activation of AMPK and reduction of mammalian target of rapamycin (mTOR) activity may be significant contributors to the inhibitory effects of metformin on cancer cell growth and proliferation (151).
The polyamine metabolic pathway is a potential target for cancer chemoprevention. A polyamine-blocking therapy (PBT) by targeting their synthesis and transport can exert antitumor effects. It not only altered the levels of polyamines in many tumors, but also relieves the immunosuppression in the TME, characterized by an increase in granzyme B+, IFN-γ+ CD8+ T-cells, and decreased immunosuppressive cell levels (60, 152). Ornithine decarboxylase (ODC) and S-adenosylmethionine decarboxylase (AdoMetDC), as the rate-limiting enzyme of polyamine biosynthesis, are the key factors in the regulation of polyamine levels. Difluoromethylornithine (DFMO) is an irreversible inhibitor of ODC and recognized as a chemopreventive and chemotherapeutic agent to decreases cancer hallmarks including enhanced cell proliferation and apoptosis resistance (153). To date, several phase II and Phase II studies on DFMO in the treatment of CRC have been reported. DFMO may prevent CRC by reversing Ca2+ channel remodeling, activating a synergistic re-expression of aberrantly silenced tumor-suppressor genes and modulating DNA hypomethylation (154, 155). To inhibit polyamine synthesis, many AdoMetDC inhibitors have been discovered from the first-generation inhibitor methylglyoxal bis (MGBG) to the third-generation inhibitor AbeAdo and tested in clinical trials treating cancers (156). However, none of them has been finally approved for clinical use due to low efficiency or strong side effects. In addition, given that the polyamine transport system is upregulated in cancers, a range of polyamine analogs and polyamine-like structures have been synthesized. They can interrupt polyamine biosynthesis and compete for uptake, and thus reduce the normal polyamine content required for cell growth (157).
Conclusion and Perspectives
A convergence of basic research, epidemiological and clinical studies is illuminating the contribution of detrimental gut microbiota-derived metabolites to the initiation and progression of CRC. The microbial metabolites described above, specifically TMAO, BAs, H2S and NOCs, work as critical signaling molecules that mediate crosstalk between the microbes and host, and play pivotal roles in colorectal carcinogenesis. They represent potential biomarkers for the early diagnosis and prognosis of CRC.
Although it is known that the detrimental gut microbiota metabolites contribute to intestinal malignant lesions in numerous ways, most of them have not yet been functionally characterized. In particular, the in-depth molecular mechanisms involved in the interaction between metabolites and CRC, as well as direct synergy between metabolites remain to be elucidated. Of note, there are still controversial findings that parts of the detrimental metabolites exhibit both the properties of anti-carcinogenic and pro-tumorigenic, which may depend on many factors, such as their luminal concentrations, the duration of the colonic stasis, interactions with other metabolites and tumor developmental stage. However, based on large-scale epidemiological studies and clinical outcome trials, we still believe that targeted regulation of detrimental metabolites to eliminate or reduce their concentration is expected to be effective strategies for the prevention and treatment of CRC. Furthermore, more studies are necessary to ensure the functional stability, clinical potency and safety of management measures.
Author Contributions
WZ, YA, and XQ were the major contributors to the writing and revision of the manuscript. XMW, XYW, and HH performed the literature search and data analysis. XS and TL critically revised the manuscript. BW supervised the entire project. XH and HC were involved in the study design and the critical review and revision of the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the grants from the National Natural Science Foundation of China (82070545 and 81970477) and the Key Project of Science and Technology Pillar Program of Tianjin (20YFZCSY00020).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abbreviations
BAs, bile acids; CRC, colorectal cancer; DCA, deoxycholic acid; DF, dietary fiber; DFMO, difluoromethylornithine; FMT, fecal microbiota transplantation; FXR, farnesoid X receptor; H2S, hydrogen sulfide; HCAs, heterocyclic amines; HFD, high-fat diet; IL, interleukin; LPS, lipopolysaccharide; MAPK, mitogen-associated protein kinase; NF-κB, nuclear factor-Kappa B; NOCs, N-nitroso compounds; PAMPs, pathogen-associated molecular patterns; PRRs, pattern recognition receptors; ROS, reactive oxygen species; SRB, sulfate-reducing bacteria; TGR5, Takeda G protein-coupled receptor 5; TLRs, Toll-like receptors; TMA, trimethylamine; TMAO, trimethylamine-N-oxide; TME, tumor microenvironment; UDCA, ursodeoxycholic acid.
References
1. Siegel RL, Miller KD, Jemal A. Cancer Statistics. CA Cancer J Clin (2020) 70:7–30. doi: 10.3322/caac.21590
2. Song M, Chan AT, Sun J. Influence of the Gut Microbiome, Diet, and Environment on Risk of Colorectal Cancer. Gastroenterology (2020) 158:322–40. doi: 10.1053/j.gastro.2019.06.048
3. Kerr J, Anderson C, Lippman SM. Physical Activity, Sedentary Behaviour, Diet, and Cancer: An Update and Emerging New Evidence. Lancet Oncol (2017) 18(8):e457–457.e471. doi: 10.1016/S1470-2045(17)30411-4
4. Wieczorska K, Stolarek M, Stec R. The Role of the Gut Microbiome in Colorectal Cancer: Where Are We? Where Are We Going. Clin Colorectal Cancer (2020) 19(1):5–12. doi: 10.1016/j.clcc.2019.07.006
5. Gao B, Chi L, Zhu Y, Shi X, Tu P, Li B, et al. An Introduction to Next Generation Sequencing Bioinformatic Analysis in Gut Microbiome Studies. Biomolecules (2021) 11(4):530. doi: 10.3390/biom11040530
6. Heo G, Lee Y, Im E. Interplay Between the Gut Microbiota and Inflammatory Mediators in the Development of Colorectal Cancer. Cancers (2021) 13(4):734. doi: 10.3390/cancers13040734
7. Cheng Y, Ling Z, Li L. The Intestinal Microbiota and Colorectal Cancer. Front Immunol (2020) 11:615056. doi: 10.3389/fimmu.2020.615056
8. Tremaroli V, Bäckhed F. Functional Interactions Between the Gut Microbiota and Host Metabolism. Nature (2012) 489(7415):242–9. doi: 10.1038/nature11552
9. Cani PD, Van Hul M, Lefort C, Depommier C, Rastelli M, Everard A. Microbial Regulation of Organismal Energy Homeostasis. Nat Metab (2012) 1(1):34–46. doi: 10.1038/s42255-018-0017-4
10. Zheng X, Xie G, Zhao A, Zhao L, Yao C, Chiu NH, et al. The Footprints of Gut Microbial-Mammalian Co-Metabolism. J Proteome Res (2011) 10(12):5512–22. doi: 10.1021/pr2007945
11. Clos-Garcia M, Garcia K, Alonso C, Iruarrizaga-Lejarreta M, D'Amato M, Crespo A, et al. Integrative Analysis of Fecal Metagenomics and Metabolomics in Colorectal Cancer. Cancers (2020) 12(5):1142. doi: 10.3390/cancers12051142
12. Yang Y, Misra BB, Liang L, Bi D, Weng W, Wu W, et al. Integrated Microbiome and Metabolome Analysis Reveals a Novel Interplay Between Commensal Bacteria and Metabolites in Colorectal Cancer. Theranostics (2019) 9(14):4101–14. doi: 10.7150/thno.35186
13. Mardinoglu A, Shoaie S, Bergentall M, Ghaffari P, Zhang C, Larsson E, et al. The Gut Microbiota Modulates Host Amino Acid and Glutathione Metabolism in Mice. Mol Syst Biol (2015) 11:834. doi: 10.15252/msb.20156487
14. Hashim N, Ab-Rahim S, Suddin LS, Saman M, Mazlan M. Global Serum Metabolomics Profiling of Colorectal Cancer. Mol Clin Oncol (2019) 11(1):3–14. doi: 10.3892/mco.2019.1853
15. Bae S, Ulrich CM, Neuhouser ML, Malysheva O, Bailey LB, Xiao L, et al. Plasma Choline Metabolites and Colorectal Cancer Risk in the Women's Health Initiative Observational Study. Cancer Res (2014) 74:7442–52. doi: 10.1158/0008-5472.CAN-14-1835
16. Liu X, Liu H, Yuan C, Zhang Y, Wang W, Hu S, et al. Preoperative Serum TMAO Level Is a New Prognostic Marker for Colorectal Cancer. Biomark Med (2017) 11:443–7. doi: 10.2217/bmm-2016-0262
17. Xu R, Wang Q, Li L. A Genome-Wide Systems Analysis Reveals Strong Link Between Colorectal Cancer and Trimethylamine N-Oxide (TMAO), a Gut Microbial Metabolite of Dietary Meat and Fat. BMC Genomics (2015) 16 Suppl 7:S4. doi: 10.1186/1471-2164-16-S7-S4
18. Bayerdörffer E, Mannes GA, Richter WO, Ochsenkühn T, Wiebecke B, Köpcke W, et al. Increased Serum Deoxycholic Acid Levels in Men With Colorectal Adenomas. Gastroenterology (1993) 104:145–51. doi: 10.1016/0016-5085(93)90846-5
19. Bayerdörffer E, Mannes GA, Ochsenkühn T, Dirschedl P, Wiebecke B, Paumgartner G. Unconjugated Secondary Bile Acids in the Serum of Patients With Colorectal Adenomas. Gut (1995) 36:268–73. doi: 10.1136/gut.36.2.268
20. Ocvirk S, Wilson AS, Posma JM, Li JV, Koller KR, Day GM, et al. A Prospective Cohort Analysis of Gut Microbial Co-Metabolism in Alaska Native and Rural African People at High and Low Risk of Colorectal Cancer. Am J Clin Nutr (2020) 111:406–19. doi: 10.1093/ajcn/nqz301
21. Liu L, Dong W, Wang S, Zhang Y, Liu T, Xie R, et al. Deoxycholic Acid Disrupts the Intestinal Mucosal Barrier and Promotes Intestinal Tumorigenesis. Food Funct (2018) 9:5588–97. doi: 10.1039/c8fo01143e
22. Dong W, Liu L, Dou Y, Xu M, Liu T, Wang S, et al. Deoxycholic Acid Activates Epidermal Growth Factor Receptor and Promotes Intestinal Carcinogenesis by ADAM17-Dependent Ligand Release. J Cell Mol Med (2018) 22:4263–73. doi: 10.1111/jcmm.13709
23. Cao H, Xu M, Dong W, Deng B, Wang S, Zhang Y, et al. Secondary Bile Acid-Induced Dysbiosis Promotes Intestinal Carcinogenesis. Int J Cancer (2017) 140:2545–56. doi: 10.1002/ijc.30643
24. Lee HY, Crawley S, Hokari R, Kwon S, Kim YS. Bile Acid Regulates MUC2 Transcription in Colon Cancer Cells via Positive EGFR/PKC/Ras/ERK/CREB, PI3K/Akt/IkappaB/NF-kappaB and P38/MSK1/CREB Pathways and Negative JNK/c-Jun/AP-1 Pathway. Int J Oncol (2010) 36:941–53. doi: 10.3892/ijo_00000573
25. Fu T, Coulter S, Yoshihara E, Oh TG, Fang S, Cayabyab F, et al. FXR Regulates Intestinal Cancer Stem Cell Proliferation. Cell (2019) 176:1098–112.e18. doi: 10.1016/j.cell.2019.01.036
26. Jean-Louis S, Akare S, Ali MA, Mash EA Jr, Meuillet E, Martinez JD. Deoxycholic Acid Induces Intracellular Signaling Through Membrane Perturbations. J Biol Chem (2006) 281:14948–60. doi: 10.1074/jbc.M506710200
27. Yazici C, Wolf PG, Kim H, Cross TL, Vermillion K, Carroll T, et al. Race-Dependent Association of Sulfidogenic Bacteria With Colorectal Cancer. Gut (2017) 66:1983–94. doi: 10.1136/gutjnl-2016-313321
28. Nguyen LH, Ma W, Wang DD, Cao Y, Mallick H, Gerbaba TK, et al. Association Between Sulfur-Metabolizing Bacterial Communities in Stool and Risk of Distal Colorectal Cancer in Men. Gastroenterology (2020) 158:1313–25. doi: 10.1053/j.gastro.2019.12.029
29. Attene-Ramos MS, Wagner ED, Gaskins HR, Plewa MJ. Hydrogen Sulfide Induces Direct Radical-Associated DNA Damage. Mol Cancer Res (2007) 5:455–9. doi: 10.1158/1541-7786.MCR-06-0439
30. Zhu Y, Wang PP, Zhao J, Green R, Sun Z, Roebothan B, et al. Dietary N-Nitroso Compounds and Risk of Colorectal Cancer: A Case-Control Study in Newfoundland and Labrador and Ontario, Canada. Br J Nutr (2014) 111:1109–17. doi: 10.1017/S0007114513003462
31. Hebels DG, Briedé JJ, Khampang R, Kleinjans JC, de Kok TM. Radical Mechanisms in Nitrosamine- and Nitrosamide-Induced Whole-Genome Gene Expression Modulations in Caco-2 Cells. Toxicol Sci (2010) 116:194–205. doi: 10.1093/toxsci/kfq121
32. Hebels DG, Jennen DG, Kleinjans JC, de Kok TM. Molecular Signatures of N-Nitroso Compounds in Caco-2 Cells: Implications for Colon Carcinogenesis. Toxicol Sci (2009) 108:290–300. doi: 10.1093/toxsci/kfp035
33. Gottschalg E, Scott GB, Burns PA, Shuker DE. Potassium Diazoacetate-Induced P53 Mutations In Vitro in Relation to Formation of O6-Carboxymethyl- and O6-Methyl-2'-Deoxyguanosine DNA Adducts: Relevance for Gastrointestinal Cancer. Carcinogenesis (2007) 28:356–62. doi: 10.1093/carcin/bgl150
34. Lewin MH, Bailey N, Bandaletova T, Bowman R, Cross AJ, Pollock J, et al. Red Meat Enhances the Colonic Formation of the DNA Adduct O6-Carboxymethyl Guanine: Implications for Colorectal Cancer Risk. Cancer Res (2006) 66:1859–65. doi: 10.1158/0008-5472.CAN-05-2237
35. Helmus DS, Thompson CL, Zelenskiy S, Tucker TC, Li L. Red Meat-Derived Heterocyclic Amines Increase Risk of Colon Cancer: A Population-Based Case-Control Study. Nutr Cancer (2013) 65:1141–50. doi: 10.1080/01635581.2013.834945
36. Cross AJ, Ferrucci LM, Risch A, Graubard BI, Ward MH, Park Y, et al. A Large Prospective Study of Meat Consumption and Colorectal Cancer Risk: An Investigation of Potential Mechanisms Underlying This Association. Cancer Res (2010) 70:2406–14. doi: 10.1158/0008-5472.CAN-09-3929
37. Hasegawa R, Sano M, Tamano S, Imaida K, Shirai T, Nagao M, et al. Dose-Dependence of 2-Amino-1-Methyl-6-Phenylimidazo[4,5-B]-Pyridine (PhIP) Carcinogenicity in Rats. Carcinogenesis (1993) 14:2553–7. doi: 10.1093/carcin/14.12.2553
38. Liu R, Lin X, Li Z, Li Q, Bi K. Quantitative Metabolomics for Investigating the Value of Polyamines in the Early Diagnosis and Therapy of Colorectal Cancer. Oncotarget (2017) 9:4583–92. doi: 10.18632/oncotarget.22885
39. Manna SK, Tanaka N, Krausz KW, Haznadar M, Xue X, Matsubara T, et al. Biomarkers of Coordinate Metabolic Reprogramming in Colorectal Tumors in Mice and Humans. Gastroenterology (2014) 146:1313–24. doi: 10.1053/j.gastro.2014.01.017
40. Guo Y, Ye Q, Deng P, Cao Y, He D, Zhou Z, et al. Spermine Synthase and MYC Cooperate to Maintain Colorectal Cancer Cell Survival by Repressing Bim Expression. Nat Commun (2020) 11:3243. doi: 10.1038/s41467-020-17067-x
41. Clausen MR, Mortensen PB. Fecal Ammonia in Patients With Adenomatous Polyps and Cancer of the Colon. Nutr Cancer (1992) 18:175–80. doi: 10.1080/01635589209514217
42. Visek WJ. Diet and Cell Growth Modulation by Ammonia. Am J Clin Nutr (1978) 31:S216–216S220. doi: 10.1093/ajcn/31.10.S216
43. Chen C, Wei M, Wang C, Sun D, Liu P, Zhong X, et al. Long Noncoding RNA KCNQ1OT1 Promotes Colorectal Carcinogenesis by Enhancing Aerobic Glycolysis via Hexokinase-2. Aging (Albany NY) (2020) 12:11685–97. doi: 10.18632/aging.103334
44. Yan XL, Zhang XB, Ao R, Guan L. Effects of shRNA-Mediated Silencing of PKM2 Gene on Aerobic Glycolysis, Cell Migration, Cell Invasion, and Apoptosis in Colorectal Cancer Cells. J Cell Biochem (2017) 118:4792–803. doi: 10.1002/jcb.26148
45. Schmitt M, Greten FR. The Inflammatory Pathogenesis of Colorectal Cancer. Nat Rev Immunol (2021) 21(10):653–67. doi: 10.1038/s41577-021-00534-x
46. Zhou CB, Zhou YL, Fang JY. Gut Microbiota in Cancer Immune Response and Immunotherapy. Trends Cancer (2021) 7(7):647–60. doi: 10.1016/j.trecan.2021.01.010
47. Wang G, Huang S, Wang Y, Cai S, Yu H, Liu H, et al. Bridging Intestinal Immunity and Gut Microbiota by Metabolites. Cell Mol Life Sci (2019) 76(20):3917–37. doi: 10.1007/s00018-019-03190-6
48. Shalapour S, Karin M. Cruel to Be Kind: Epithelial, Microbial, and Immune Cell Interactions in Gastrointestinal Cancers. Annu Rev Immunol (2020) 38:649–71. doi: 10.1146/annurev-immunol-082019-081656
49. Ngwa VM, Edwards DN, Philip M, Chen J. Microenvironmental Metabolism Regulates Antitumor Immunity. Cancer Res (2019) 79(16):4003–8. doi: 10.1158/0008-5472.CAN-19-0617
50. Sepich-Poore GD, Zitvogel L, Straussman R, Hasty J, Wargo JA, Knight R. The Microbiome and Human Cancer. Science (2021) 371(6536):eabc4552. doi: 10.1126/science.abc4552
51. Amarante-Mendes GP, Adjemian S, Branco LM, Zanetti LC, Weinlich R, Bortoluci KR. Pattern Recognition Receptors and the Host Cell Death Molecular Machinery. Front Immunol (2018) 9:2379. doi: 10.3389/fimmu.2018.02379
52. Brandão I, Hörmann N, Jäckel S, Reinhardt C. TLR5 Expression in the Small Intestine Depends on the Adaptors MyD88 and TRIF, But is Independent of the Enteric Microbiota. Gut Microbes (2015) 6:202–6. doi: 10.1080/19490976.2015.1034417
53. Burgueño JF, Abreu MT. Epithelial Toll-Like Receptors and Their Role in Gut Homeostasis and Disease. Nat Rev Gastroenterol Hepatol (2020) 17:263–78. doi: 10.1038/s41575-019-0261-4
54. Hörmann N, Brandão I, Jäckel S, Ens N, Lillich M, Walter U, et al. Gut Microbial Colonization Orchestrates TLR2 Expression, Signaling and Epithelial Proliferation in the Small Intestinal Mucosa. PloS One (2014) 9:e113080. doi: 10.1371/journal.pone.0113080
55. Mukherji A, Kobiita A, Ye T, Chambon P. Homeostasis in Intestinal Epithelium is Orchestrated by the Circadian Clock and Microbiota Cues Transduced by TLRs. Cell (2013) 153:812–27. doi: 10.1016/j.cell.2013.04.020
56. Meng S, Li Y, Zang X, Jiang Z, Ning H, Li J. Effect of TLR2 on the Proliferation of Inflammation-Related Colorectal Cancer and Sporadic Colorectal Cancer. Cancer Cell Int (2020) 20:95. doi: 10.1186/s12935-020-01184-0
57. Yesudhas D, Gosu V, Anwar MA, Choi S. Multiple Roles of Toll-Like Receptor 4 in Colorectal Cancer. Front Immunol (2014) 5:334. doi: 10.3389/fimmu.2014.00334
58. Alvarez-Arellano L, Cortés-Reynosa P, Sánchez-Zauco N, Salazar E, Torres J, Maldonado-Bernal C. TLR9 and NF-κB are Partially Involved in Activation of Human Neutrophils by Helicobacter Pylori and its Purified DNA. PloS One (2014) 9:e101342. doi: 10.1371/journal.pone.0101342
59. Carriche GM, Almeida L, Stüve P, Velasquez L, Dhillon-LaBrooy A, Roy U, et al. Regulating T-Cell Differentiation Through the Polyamine Spermidine. J Allergy Clin Immunol (2021) 147:335–48.e11. doi: 10.1016/j.jaci.2020.04.037
60. Alexander ET, Minton A, Peters MC, Phanstiel O4, Gilmour SK. A Novel Polyamine Blockade Therapy Activates an Anti-Tumor Immune Response. Oncotarget (2017) 8(48):84140–52. doi: 10.18632/oncotarget.20493
61. Janakiram NB, Mohammed A, Bryant T, Zhang Y, Brewer M, Duff A, et al. Potentiating NK Cell Activity by Combination of Rosuvastatin and Difluoromethylornithine for Effective Chemopreventive Efficacy Against Colon Cancer. Sci Rep (2016) 6:37046. doi: 10.1038/srep37046
62. Tsujinaka S, Soda K, Kano Y, Konishi F. Spermine Accelerates Hypoxia-Initiated Cancer Cell Migration. Int J Oncol (2011) 38(2):305–12. doi: 10.3892/ijo.2010.849
63. Vats D, Mukundan L, Odegaard JI, Zhang L, Smith KL, Morel CR, et al. Oxidative Metabolism and PGC-1beta Attenuate Macrophage-Mediated Inflammation. Cell Metab (2006) 4(1):13–24. doi: 10.1016/j.cmet.2006.05.011
64. Janeiro MH, Ramírez MJ, Milagro FI, Martínez JA, Solas M. Implication of Trimethylamine N-Oxide (TMAO) in Disease: Potential Biomarker or New Therapeutic Target. Nutrients (2018) 10(10):1398. doi: 10.3390/nu10101398
65. Velasquez MT, Ramezani A, Manal A, Raj DS. Trimethylamine N-Oxide: The Good, the Bad and the Unknown. Toxins (2016) 8(11):326. doi: 10.3390/toxins8110326
66. Wang Z, Klipfell E, Bennett BJ, Koeth R, Levison BS, Dugar B, et al. Gut Flora Metabolism of Phosphatidylcholine Promotes Cardiovascular Disease. Nature (2011) 472(7341):57–63. doi: 10.1038/nature09922
67. Li J, Li Y, Ivey KL, Wang DD, Wilkinson JE, Franke A, et al. Interplay Between Diet and Gut Microbiome, and Circulating Concentrations of Trimethylamine N-Oxide: Findings From a Longitudinal Cohort of US Men. Gut (2011) gutjnl–2020-322473. doi: 10.1136/gutjnl-2020-322473
68. Farhangi MA, Vajdi M. Novel Findings of the Association Between Gut Microbiota-Derived Metabolite Trimethylamine N-Oxide and Inflammation: Results From a Systematic Review and Dose-Response Meta-Analysis. Crit Rev Food Sci Nutr (2020) 60(16):2801–23. doi: 10.1080/10408398.2020.1770199
69. Yue C, Yang X, Li J, Chen X, Zhao X, Chen Y, et al. Trimethylamine N-Oxide Prime NLRP3 Inflammasome via Inhibiting ATG16L1-Induced Autophagy in Colonic Epithelial Cells. Biochem Biophys Res Commun (2015) 490:541–51. doi: 10.1016/j.bbrc.2017.06.075
70. Zińczuk J, Maciejczyk M, Zaręba K, Pryczynicz A, Dymicka-Piekarska V, Kamińska J, et al. Pro-Oxidant Enzymes, Redox Balance and Oxidative Damage to Proteins, Lipids and DNA in Colorectal Cancer Tissue. Is Oxidative Stress Dependent on Tumour Budding and Inflammatory Infiltration. Cancers (Basel) (2020) 12:1636. doi: 10.3390/cancers12061636
71. Li T, Chen Y, Gua C, Li X. Elevated Circulating Trimethylamine N-Oxide Levels Contribute to Endothelial Dysfunction in Aged Rats Through Vascular Inflammation and Oxidative Stress. Front Physiol (2017) 8:350. doi: 10.3389/fphys.2017.00350
72. Oellgaard J, Winther SA, Hansen TS, Rossing P, von Scholten BJ. Trimethylamine N-Oxide (TMAO) as a New Potential Therapeutic Target for Insulin Resistance and Cancer. Curr Pharm Des (2017) 23:3699–712. doi: 10.2174/1381612823666170622095324
73. Jia W, Xie G, Jia W. Bile Acid-Microbiota Crosstalk in Gastrointestinal Inflammation and Carcinogenesis. Nat Rev Gastroenterol Hepatol (2018) 15:111–28. doi: 10.1038/nrgastro.2017.119
74. Liu T, Song X, Khan S, Li Y, Guo Z, Li C, et al. The Gut Microbiota at the Intersection of Bile Acids and Intestinal Carcinogenesis: An Old Story, Yet Mesmerizing. Int J Cancer (2020) 146:1780–90. doi: 10.1002/ijc.32563
75. Javed Z, Khan K, Sadia H, Raza S, Salehi B, Sharifi-Rad J, et al. LncRNA & Wnt Signaling in Colorectal Cancer. Cancer Cell Int (2020) 20:326. doi: 10.1186/s12935-020-01412-7
76. Anwer MS. Intracellular Signaling by Bile Acids. J Biosci (Rajshari) (2012) 20:1–23. doi: 10.3329/jbs.v20i0.17647
77. Wahlström A, Kovatcheva-Datchary P, Ståhlman M, Khan MT, Bäckhed F, Marschall HU. Induction of Farnesoid X Receptor Signaling in Germ-Free Mice Colonized With a Human Microbiota. J Lipid Res (2017) 58:412–9. doi: 10.1194/jlr.M072819
78. Yu J, Li S, Guo J, Xu Z, Zheng J, Sun X. Farnesoid X Receptor Antagonizes Wnt/β-Catenin Signaling in Colorectal Tumorigenesis. Cell Death Dis (2020) 11:640. doi: 10.1038/s41419-020-02819-w
79. Torres J, Bao X, Iuga AC, Chen A, Harpaz N, Ullman T, et al. Farnesoid X Receptor Expression Is Decreased in Colonic Mucosa of Patients With Primary Sclerosing Cholangitis and Colitis-Associated Neoplasia. Inflamm Bowel Dis (2013) 19:275–82. doi: 10.1097/MIB.0b013e318286ff2e
80. Liu L, Yang M, Dong W, Liu T, Song X, Gu Y, et al. Gut Dysbiosis and Abnormal Bile Acid Metabolism in Colitis-Associated Cancer. Gastroenterol Res Pract (2021) 2021:6645970. doi: 10.1155/2021/6645970
81. Kushkevych I, Cejnar J, Treml J, Dordević D, Kollar P, Vítězová M. Recent Advances in Metabolic Pathways of Sulfate Reduction in Intestinal Bacteria. Cells (2020) 9:698. doi: 10.3390/cells9030698
82. Blachier F, Beaumont M, Kim E. Cysteine-Derived Hydrogen Sulfide and Gut Health: A Matter of Endogenous or Bacterial Origin. Curr Opin Clin Nutr Metab Care (2019) 22:68–75. doi: 10.1097/MCO.0000000000000526
83. Roediger WE, Duncan A, Kapaniris O, Millard S. Reducing Sulfur Compounds of the Colon Impair Colonocyte Nutrition: Implications for Ulcerative Colitis. Gastroenterology (1993) 104:802–9. doi: 10.1016/0016-5085(93)91016-b
84. Roediger WE, Moore J, Babidge W. Colonic Sulfide in Pathogenesis and Treatment of Ulcerative Colitis. Dig Dis Sci (1997) 42:1571–9. doi: 10.1023/a:1018851723920
85. Figliuolo VR, Coutinho-Silva R, Coutinho C. Contribution of Sulfate-Reducing Bacteria to Homeostasis Disruption During Intestinal Inflammation. Life Sci (2018) 215:145–51. doi: 10.1016/j.lfs.2018.11.009
86. Ijssennagger N, van der Meer R, van Mil S. Sulfide as a Mucus Barrier-Breaker in Inflammatory Bowel Disease. Trends Mol Med (2016) 22:190–9. doi: 10.1016/j.molmed.2016.01.002
87. Qin M, Long F, Wu W, Yang D, Huang M, Xiao C, et al. Hydrogen Sulfide Protects Against DSS-Induced Colitis by Inhibiting NLRP3 Inflammasome. Free Radic Biol Med (2019) 137:99–109. doi: 10.1016/j.freeradbiomed.2019.04.025
88. Motta JP, Flannigan KL, Agbor TA, Beatty JK, Blackler RW, Workentine ML, et al. Hydrogen Sulfide Protects From Colitis and Restores Intestinal Microbiota Biofilm and Mucus Production. Inflamm Bowel Dis (2015) 21:1006–17. doi: 10.1097/MIB.0000000000000345
89. Wu YC, Wang XJ, Yu L, Chan FK, Cheng AS, Yu J, et al. Hydrogen Sulfide Lowers Proliferation and Induces Protective Autophagy in Colon Epithelial Cells. PloS One (2012) 7:e37572. doi: 10.1371/journal.pone.0037572
90. Crowe W, Elliott CT, Green BD. A Review of the In Vivo Evidence Investigating the Role of Nitrite Exposure From Processed Meat Consumption in the Development of Colorectal Cancer. Nutrients (2019) 11:2673. doi: 10.3390/nu11112673
91. Hughes R, Cross AJ, Pollock JR, Bingham S. Dose-Dependent Effect of Dietary Meat on Endogenous Colonic N-Nitrosation. Carcinogenesis (2001) 22:199–202. doi: 10.1093/carcin/22.1.199
92. Bingham SA, Hughes R, Cross AJ. Effect of White Versus Red Meat on Endogenous N-Nitrosation in the Human Colon and Further Evidence of a Dose Response. J Nutr (2002) 132:3522S–5S. doi: 10.1093/jn/132.11.3522S
93. Yang CS, Yoo JS, Ishizaki H, Hong JY. Cytochrome P450IIE1: Roles in Nitrosamine Metabolism and Mechanisms of Regulation. Drug Metab Rev (1990) 22:147–59. doi: 10.3109/03602539009041082
94. Gilsing AM, Fransen F, de Kok TM, Goldbohm AR, Schouten LJ, de Bruïne AP, et al. Dietary Heme Iron and the Risk of Colorectal Cancer With Specific Mutations in KRAS and APC. Carcinogenesis (2013) 34:2757–66. doi: 10.1093/carcin/bgt290
95. Cascella M, Bimonte S, Barbieri A, Del Vecchio V, Caliendo D, Schiavone V, et al. Dissecting the Mechanisms and Molecules Underlying the Potential Carcinogenicity of Red and Processed Meat in Colorectal Cancer (CRC): An Overview on the Current State of Knowledge. Infect Agent Cancer (2018) 13:3. doi: 10.1186/s13027-018-0174-9
96. Fahrer J, Kaina B. O6-Methylguanine-DNA Methyltransferase in the Defense Against N-Nitroso Compounds and Colorectal Cancer. Carcinogenesis (2013) 34:2435–42. doi: 10.1093/carcin/bgt275
97. Mai V, McCrary QM, Sinha R, Glei M. Associations Between Dietary Habits and Body Mass Index With Gut Microbiota Composition and Fecal Water Genotoxicity: An Observational Study in African American and Caucasian American Volunteers. Nutr J (2013) 8:49. doi: 10.1186/1475-2891-8-49
98. Knasmüller S, Steinkellner H, Hirschl AM, Rabot S, Nobis EC, Kassie F. Impact of Bacteria in Dairy Products and of the Intestinal Microflora on the Genotoxic and Carcinogenic Effects of Heterocyclic Aromatic Amines. Mutat Res (2001) 480-481:129–38. doi: 10.1186/1475-2891-8-49
99. Murata M, Kobayashi M, Kawanishi S. Mechanism of Oxidative DNA Damage Induced by a Heterocyclic Amine, 2-Amino-3,8-Dimethylimidazo[4,5f]Quinoxaline. Jpn J Cancer Res (2010) 90:268–75. doi: 10.1111/j.1349-7006.1999.tb00743.x
100. Kim E, Coelho D, Blachier F. Review of the Association Between Meat Consumption and Risk of Colorectal Cancer. Nutr Res (2013) 33:983–94. doi: 10.1016/j.nutres.2013.07.018
101. Blachier F, Mariotti F, Huneau JF, Tomé D. Effects of Amino Acid-Derived Luminal Metabolites on the Colonic Epithelium and Physiopathological Consequences. Amino Acids (2007) 33:547–62. doi: 10.1007/s00726-006-0477-9
102. Macfarlane GT, Gibson GR, Cummings JH. Comparison of Fermentation Reactions in Different Regions of the Human Colon. J Appl Bacteriol (1992) 72:57–64. doi: 10.1111/j.1365-2672.1992.tb04882.x
103. Lin HC, Visek WJ. Colon Mucosal Cell Damage by Ammonia in Rats. J Nutr (1991) 121:887–93. doi: 10.1093/jn/121.6.887
104. Baltazar F, Afonso J, Costa M, Granja S. Lactate Beyond a Waste Metabolite: Metabolic Affairs and Signaling in Malignancy. Front Oncol (2020) 10:231. doi: 10.3389/fonc.2020.00231
105. Cheng C, Xie Z, Li Y, Wang J, Qin C, Zhang Y. PTBP1 Knockdown Overcomes the Resistance to Vincristine and Oxaliplatin in Drug-Resistant Colon Cancer Cells Through Regulation of Glycolysis. BioMed Pharmacother (2018) 108:194–200. doi: 10.1016/j.biopha.2018.09.031
106. Haas R, Smith J, Rocher-Ros V, Nadkarni S, Montero-Melendez T, D'Acquisto F, et al. Lactate Regulates Metabolic and Pro-Inflammatory Circuits in Control of T Cell Migration and Effector Functions. PloS Biol (2015) 13:e1002202. doi: 10.1371/journal.pbio.1002202
107. Amberg A, Riefke B, Schlotterbeck G, Ross A, Senn H, Dieterle F, et al. NMR and MS Methods for Metabolomics. Methods Mol Biol (2017) 1641:229–58. doi: 10.1007/978-1-4939-7172-5_13
108. Chen F, Dai X, Zhou CC, Li KX, Zhang YJ, Lou XY, et al. Integrated Analysis of the Faecal Metagenome and Serum Metabolome Reveals the Role of Gut Microbiome-Associated Metabolites in the Detection of Colorectal Cancer and Adenoma. Gut (2021) gutjnl–2020-323476. doi: 10.1136/gutjnl-2020-323476
109. Johnson EL, Heaver SL, Waters JL, Kim BI, Bretin A, Goodman AL, et al. Sphingolipids Produced by Gut Bacteria Enter Host Metabolic Pathways Impacting Ceramide Levels. Nat Commun (2020) 11:2471. doi: 10.1038/s41467-020-16274-w
110. Wieland Brown LC, Penaranda C, Kashyap PC, Williams BB, Clardy J, Kronenberg M, et al. Production of α-Galactosylceramide by a Prominent Member of the Human Gut Microbiota. PloS Biol (2013) 11:e1001610. doi: 10.1371/journal.pbio.1001610
111. Kim M, Vogtmann E, Ahlquist DA, Devens ME, Kisiel JB, Taylor WR, et al. Fecal Metabolomic Signatures in Colorectal Adenoma Patients Are Associated With Gut Microbiota and Early Events of Colorectal Cancer Pathogenesis. mBio (2020) 11(1):e03186–19. doi: 10.1128/mBio.03186-19
112. Rao J, Wan X, Tou F, He Q, Xiong A, Chen X, et al. Molecular Characterization of Advanced Colorectal Cancer Using Serum Proteomics and Metabolomics. Front Mol Biosci (2021) 8:687229. doi: 10.3389/fmolb.2021.687229
113. Yachida S, Mizutani S, Shiroma H, Shiba S, Nakajima T, Sakamoto T, et al. Metagenomic and Metabolomic Analyses Reveal Distinct Stage-Specific Phenotypes of the Gut Microbiota in Colorectal Cancer. Nat Med (2019) 25:968–76. doi: 10.1038/s41591-019-0458-7
114. Lien EC, Vander Heiden MG. A Framework for Examining How Diet Impacts Tumour Metabolism. Nat Rev Cancer (2019) 19(11):651–61. doi: 10.1038/s41568-019-0198-5
115. Hullings AG, Sinha R, Liao LM, Freedman ND, Graubard BI, Loftfield E. Whole Grain and Dietary Fiber Intake and Risk of Colorectal Cancer in the NIH-AARP Diet and Health Study Cohort. Am J Clin Nutr (2020) 112(3):603–12. doi: 10.1093/ajcn/nqaa161
116. Reynolds A, Mann J, Cummings J, Winter N, Mete E, Te Morenga L. Carbohydrate Quality and Human Health: A Series of Systematic Reviews and Meta-Analyses. Lancet (2019) 393(10170):434–45. doi: 10.1016/S0140-6736(18)31809-9
117. O'Keefe SJ, Li JV, Lahti L, Ou J, Carbonero F, Mohammed K, et al. Fat, Fibre and Cancer Risk in African Americans and Rural Africans. Nat Commun (2015) 6:6342. doi: 10.1038/ncomms7342
118. Trefflich I, Marschall HU, Giuseppe RD, Ståhlman M, Michalsen A, Lampen A, et al. Associations Between Dietary Patterns and Bile Acids-Results From a Cross-Sectional Study in Vegans and Omnivores. Nutrients (2019) 12(1):47. doi: 10.3390/nu12010047
119. Li Q, Chen H, Zhang M, Wu T, Liu R, Zhang Z. Potential Correlation Between Dietary Fiber-Suppressed Microbial Conversion of Choline to Trimethylamine and Formation of Methylglyoxal. J Agric Food Chem (2019) 67(48):13247–57. doi: 10.1021/acs.jafc.9b04860
120. Eslami M, Yousefi B, Kokhaei P, Hemati M, Nejad ZR, Arabkari V, et al. Importance of Probiotics in the Prevention and Treatment of Colorectal Cancer. J Cell Physiol (2019) 234(10):17127–43. doi: 10.1002/jcp.28473
121. Piewngam P, Zheng Y, Nguyen TH, Dickey SW, Joo HS, Villaruz AE, et al. Pathogen Elimination by Probiotic Bacillus via Signalling Interference. Nature (2018) 62(7728):532–7. doi: 10.1038/s41586-018-0616-y
122. Pagnini C, Saeed R, Bamias G, Arseneau KO, Pizarro TT, Cominelli F. Probiotics Promote Gut Health Through Stimulation of Epithelial Innate Immunity. Proc Natl Acad Sci U S A (2010) 107(1):454–9. doi: 10.1073/pnas.0910307107
123. Wang J, Ji H. Influence of Probiotics on Dietary Protein Digestion and Utilization in the Gastrointestinal Tract. Curr Protein Pept Sci (2019) 20(2):125–31. doi: 10.2174/1389203719666180517100339
124. Khare A, Gaur S. Cholesterol-Lowering Effects of Lactobacillus Species. Curr Microbiol (2020) 77:638–44. doi: 10.1007/s00284-020-01903-w
125. Hou G, Peng W, Wei L, Li R, Yuan Y, Huang X, et al. Lactobacillus Delbrueckii Interfere With Bile Acid Enterohepatic Circulation to Regulate Cholesterol Metabolism of Growing-Finishing Pigs via Its Bile Salt Hydrolase Activity. Front Nutr (2020) 7:617676. doi: 10.3389/fnut.2020.617676
126. Chen D, Jin D, Huang S, Wu J, Xu M, Liu T, et al. Clostridium Butyricum, a Butyrate-Producing Probiotic, Inhibits Intestinal Tumor Development Through Modulating Wnt Signaling and Gut Microbiota. Cancer Lett (2020) 469:456–67. doi: 10.1016/j.canlet.2019.11.019
127. Sanders ME, Merenstein DJ, Reid G, Gibson GR, Rastall RA. Probiotics and Prebiotics in Intestinal Health and Disease: From Biology to the Clinic. Nat Rev Gastroenterol Hepatol (2019) 16:605–16. doi: 10.1038/s41575-019-0173-3
128. Greenhalgh K, Ramiro-Garcia J, Heinken A, Ullmann P, Bintener T, Pacheco MP, et al. Integrated In Vitro and In Silico Modeling Delineates the Molecular Effects of a Synbiotic Regimen on Colorectal-Cancer-Derived Cells. Cell Rep (2019) 27:1621–32.e9. doi: 10.1016/j.celrep.2019.04.001
129. Ooijevaar RE, Terveer EM, Verspaget HW, Kuijper EJ, Keller JJ. Clinical Application and Potential of Fecal Microbiota Transplantation. Annu Rev Med (2019) 70:335–51. doi: 10.1146/annurev-med-111717-122956
130. Perillo F, Amoroso C, Strati F, Giuffrè MR, Díaz-Basabe A, Lattanzi G, et al. Gut Microbiota Manipulation as a Tool for Colorectal Cancer Management: Recent Advances in Its Use for Therapeutic Purposes. Int J Mol Sci (2020) 21(15):5389. doi: 10.3390/ijms21155389
131. Chen D, Wu J, Jin D, Wang B, Cao H. Fecal Microbiota Transplantation in Cancer Management: Current Status and Perspectives. Int J Cancer (2019) 145:2021–31. doi: 10.1002/ijc.32003
132. Rosshart SP, Vassallo BG, Angeletti D, Hutchinson DS, Morgan AP, Takeda K, et al. Wild Mouse Gut Microbiota Promotes Host Fitness and Improves Disease Resistance. Cell (2017) 171:1015–28.e13. doi: 10.1016/j.cell.2017.09.016
133. Zhou S, Gravekamp C, Bermudes D, Liu K. Tumour-Targeting Bacteria Engineered to Fight Cancer. Nat Rev Cancer (2018) 18:727–43. doi: 10.1038/s41568-018-0070-z
134. Kurtz CB, Millet YA, Puurunen MK, Perreault M, Charbonneau MR, Isabella VM, et al. An Engineered E. Coli Nissle Improves Hyperammonemia and Survival in Mice and Shows Dose-Dependent Exposure in Healthy Humans. Sci Transl Med (2019) 11:eaau7975. doi: 10.1126/scitranslmed.aau7975
135. Riglar DT, Silver PA. Engineering Bacteria for Diagnostic and Therapeutic Applications. Nat Rev Microbiol (2018) 16:214–25. doi: 10.1038/nrmicro.2017.172
136. Landry BP, Tabor JJ. Engineering Diagnostic and Therapeutic Gut Bacteria. Microbiol Spectr (2017) 5(5). doi: 10.1128/microbiolspec.BAD-0020-2017
137. Sabino J, Hirten RP, Colombel JF. Review Article: Bacteriophages in Gastroenterology-From Biology to Clinical Applications. Aliment Pharmacol Ther (2020) 51:53–63. doi: 10.1111/apt.15557
138. Rasmussen TS, Koefoed AK, Jakobsen RR, Deng L, Castro-Mejía JL, Brunse A, et al. Bacteriophage-Mediated Manipulation of the Gut Microbiome - Promises and Presents Limitations. FEMS Microbiol Rev (2020) 44:507–21. doi: 10.1093/femsre/fuaa020
139. Hsu BB, Gibson TE, Yeliseyev V, Liu Q, Lyon L, Bry L, et al. Dynamic Modulation of the Gut Microbiota and Metabolome by Bacteriophages in a Mouse Model. Cell Host Microbe (2019) 25:803–14.e5. doi: 10.1016/j.chom.2019.05.001
140. Paule A, Frezza D, Edeas M. Microbiota and Phage Therapy: Future Challenges in Medicine. Med Sci (Basel) (2018) 6:86. doi: 10.3390/medsci6040086
141. Marcobal A, De las Rivas B, Landete JM, Tabera L, Muñoz R. Tyramine and Phenylethylamine Biosynthesis by Food Bacteria. Crit Rev Food Sci Nutr (2012) 52:448–67. doi: 10.1080/10408398.2010.500545
142. Huang WK, Hsu HC, Liu JR, Yang TS, Chen JS, Chang JW, et al. The Association of Ursodeoxycholic Acid Use With Colorectal Cancer Risk: A Nationwide Cohort Study. Medicine (Baltimore) (2016) 95:e2980. doi: 10.1097/MD.0000000000002980
143. Pearson T, Caporaso JG, Yellowhair M, Bokulich NA, Padi M, Roe DJ, et al. Effects of Ursodeoxycholic Acid on the Gut Microbiome and Colorectal Adenoma Development. Cancer Med (2019) 8:617–28. doi: 10.1002/cam4.1965
144. Powell AA, LaRue JM, Batta AK, Martinez JD. Bile Acid Hydrophobicity is Correlated With Induction of Apoptosis and/or Growth Arrest in HCT116 Cells. Biochem J (2001) 356:481–6. doi: 10.1042/0264-6021:3560481
145. Martinez JD, Stratagoules ED, LaRue JM, Powell AA, Gause PR, Craven MT, et al. Different Bile Acids Exhibit Distinct Biological Effects: The Tumor Promoter Deoxycholic Acid Induces Apoptosis and the Chemopreventive Agent Ursodeoxycholic Acid Inhibits Cell Proliferation. Nutr Cancer (1998) 31:111–8. doi: 10.1080/01635589809514689
146. Eaton JE, Silveira MG, Pardi DS, Sinakos E, Kowdley KV, Luketic VA, et al. High-Dose Ursodeoxycholic Acid is Associated With the Development of Colorectal Neoplasia in Patients With Ulcerative Colitis and Primary Sclerosing Cholangitis. Am J Gastroenterol (2011) 106:1638–45. doi: 10.1038/ajg.2011.156
147. Huang WK, Chang SH, Hsu HC, Chou WC, Yang TS, Chen JS, et al. Postdiagnostic Metformin Use and Survival of Patients With Colorectal Cancer: A Nationwide Cohort Study. Int J Cancer (2020) 147:1904–16. doi: 10.1002/ijc.32989
148. Ng CW, Jiang AA, Toh E, Ng CH, Ong ZH, Peng S, et al. Metformin and Colorectal Cancer: A Systematic Review, Meta-Analysis and Meta-Regression. Int J Colorectal Dis (2020) 35:1501–12. doi: 10.1007/s00384-020-03676-x
149. Bryrup T, Thomsen CW, Kern T, Allin KH, Brandslund I, Jørgensen NR, et al. Metformin-Induced Changes of the Gut Microbiota in Healthy Young Men: Results of a non-Blinded, One-Armed Intervention Study. Diabetologia (2019) 62:1024–35. doi: 10.1007/s00125-019-4848-7
150. Shikata K, Ninomiya T, Kiyohara Y. Diabetes Mellitus and Cancer Risk: Review of the Epidemiological Evidence. Cancer Sci (2013) 104:9–14. doi: 10.1111/cas.12043
151. Li M, Li X, Zhang H, Lu Y. Molecular Mechanisms of Metformin for Diabetes and Cancer Treatment. Front Physiol (2018) 9:1039. doi: 10.3389/fphys.2018.01039
152. Hanus M, Parada-Venegas D, Landskron G, Wielandt AM, Hurtado C, Alvarez K, et al. Immune System, Microbiota, and Microbial Metabolites: The Unresolved Triad in Colorectal Cancer Microenvironment. Front Immunol (2021) 12:612826. doi: 10.3389/fimmu.2021.612826
153. Alexiou GA, Lianos GD, Ragos V, Galani V, Kyritsis AP. Difluoromethylornithine in Cancer: New Advances. Future Oncol (2017) 13:809–19. doi: 10.2217/fon-2016-0266
154. Gutiérrez LG, Hernández-Morales M, Núñez L, Villalobos C. Inhibition of Polyamine Biosynthesis Reverses Ca2+ Channel Remodeling in Colon Cancer Cells. Cancers (Basel) (2019) 11(1):83. doi: 10.3390/cancers11010083
155. Wu Y, Steinbergs N, Murray-Stewart T, Marton LJ, Casero RA. Oligoamine Analogues in Combination With 2-Difluoromethylornithine Synergistically Induce Re-Expression of Aberrantly Silenced Tumour-Suppressor Genes. Biochem J (2012) 442:693–701. doi: 10.1042/BJ20111271
156. Zhang Y, Zheng Q, Zhou Y, Liu S. Repurposing Clinical Drugs as AdoMetDC Inhibitors Using the SCAR Strategy. Front Pharmacol (2020) 11:248. doi: 10.3389/fphar.2020.00248
Keywords: colorectal cancer, gut microbiota, metabolites, tumor immunity, intracellular signal transduction
Citation: Zhang W, An Y, Qin X, Wu X, Wang X, Hou H, Song X, Liu T, Wang B, Huang X and Cao H (2021) Gut Microbiota-Derived Metabolites in Colorectal Cancer: The Bad and the Challenges. Front. Oncol. 11:739648. doi: 10.3389/fonc.2021.739648
Received: 11 July 2021; Accepted: 29 September 2021;
Published: 18 October 2021.
Edited by:
Divya P. Kumar, JSS Academy of Higher Education and Research, IndiaReviewed by:
Christoph Reinhardt, Johannes Gutenberg University Mainz, GermanySara De Dosso, Oncology Institute of Southern Switzerland (IOSI), Switzerland
Sharareh Moghim, Isfahan University of Medical Sciences, Iran
Copyright © 2021 Zhang, An, Qin, Wu, Wang, Hou, Song, Liu, Wang, Huang and Cao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hailong Cao, Y2FvaGFpbG9uZ0B0bXUuZWR1LmNu; Xuan Huang, aHVhbmd4dWFuQHpjbXUuZWR1LmNu
†These authors have contributed equally to this work