 Jiangnan He
Jiangnan He Michelle K.Y. Siu
Michelle K.Y. Siu Hextan Y. S. Ngan
Hextan Y. S. Ngan- Departments of Obstetrics and Gynaecology, Li Ka Shing (LKS) Faculty of Medicine, The University of Hong Kong, Pok Fu Lam, Hong Kong, SAR China
Cholesterol is an essential substance in mammalian cells, and cholesterol metabolism plays crucial roles in multiple biological functions. Dysregulated cholesterol metabolism is a metabolic hallmark in several cancers, beyond the Warburg effect. Reprogrammed cholesterol metabolism has been reported to enhance tumorigenesis, metastasis and chemoresistance in multiple cancer types, including ovarian cancer. Ovarian cancer is one of the most aggressive malignancies worldwide. Alterations in metabolic pathways are characteristic features of ovarian cancer; however, the specific role of cholesterol metabolism remains to be established. In this report, we provide an overview of the key proteins involved in cholesterol metabolism in ovarian cancer, including the rate-limiting enzymes in cholesterol biosynthesis, and the proteins involved in cholesterol uptake, storage and trafficking. Also, we review the roles of cholesterol and its derivatives in ovarian cancer and the tumor microenvironment, and discuss promising related therapeutic targets for ovarian cancer.
1 Introduction
Ovarian cancer is one of the most aggressive malignancies worldwide (1). Due to the lack of obvious symptoms of early-stage ovarian cancer, newly diagnosed patients often present in advanced stages of disease, leading to the designation “silent killer” (2). Epithelial ovarian cancer can be classified into type I and type II ovarian tumors mainly on the basis of their cellular morphology and genetic alterations (3). Type I tumors consist of low grade serous, endometrioid, clear cell, and mucinous carcinomas, which are genetically characterized by BRAF, Kras, PTEN, or PI3KCA mutations primarily affecting PI3K/AKT/mTOR signaling (4–7). However, type II tumors mainly include high grade serous and undifferentiated carcinomas, typically with TP53 mutation and BRCA1/2 mutation (3, 8).
Metabolism in ovarian cancer shows heterogeneity, because the viability of ovarian cancer cells is maintained in a manner dependent not solely on metabolism but on the outside environment. Accumulating evidence indicates not only the active expression of aerobic glycolysis or oxidative phosphorylation (OXPHOS) in ovarian cancer but also aberrant lipid metabolism, which is strongly associated with ovarian cancer progression (9–12).
Patients with late-stage disease commonly display tumor metastases with an accumulation of ascites. The tumor microenvironment (TME) in ovarian cancer is composed of non-malignant cells, mainly including cancer-associated fibroblasts (CAF), cancer-associated adipocytes (CAA), immune-related cells, malignant cells, and secreted cytokines or other soluble molecules in ascites, which facilitate immunosuppression through crosstalk interactions among one another (13). Given that the major site of metastasis is the omentum, the TME in ovarian cancer is different from that in other cancers and is characterized as an adipocyte- and lipid-rich milieu, which has been shown to contribute to tumorigenesis, tumor immune escape, chemoresistance, and cancer recurrence (13–15). Other typical features of the tumor microenvironment include an insufficient supply of glucose and oxygen, which are non-beneficial for survival of tumor cells. To overcome this limitation, tumor cells and tumor-associated cells act in concert to develop reprogrammed adaptive metabolism (16). Ovarian tumor cells in this lipid-rich environment also tend to predominantly utilize lipid-dominant and alternative metabolic pathways (17). In addition, studies using co-culture of adipocytes and ovarian tumor cells have indicated that adipocytes promote tumor growth and metastasis of ovarian tumors, on the basis of the stimulation of adipocytes by the altered lipid metabolism in ovarian cancer, thus resulting in upregulation of lipid uptake from adipocytes and lipolysis in ovarian cancer cells (14).
Fatty acids and cholesterol are two main types of lipids. Multiple fatty acids and enzymes involved in fatty acid metabolism, such as fatty acid-binding protein 4 (FABP4), CD36 and stearoyl-CoA desaturase 1 (SCD1), significantly enhance ovarian cancer proliferation, survival, drug resistance and metastasis, and even contribute to stemness maintenance (14, 18–21). Recently, considerable evidence supporting the importance of reprogrammed cholesterol metabolism in ovarian cancer has been reported. Highly expressed proteins and enzymes involved in cholesterol metabolism promote ovarian cancer progression; cholesterol and its derivatives also contribute to proliferation and chemoresistance in ovarian cancer and have roles in the immunosuppressive tumor microenvironment (22–25). Here, we have systematically summarized the most recent findings on cholesterol and its derivatives in ovarian cancer, with the aim of comprehensively understanding their specific functions to facilitate the identification of novel markers and therapeutic targets.
2 Overview of Cholesterol Metabolism
Cholesterol is a fundamental metabolite of mammalian cells to maintain structural integrity and fluidity of the plasma membrane, and regulates cells or cell-to-cell interactions by mediating alterations in signaling involved in cell proliferation, immunity, and inflammation (26). Several routes of cholesterol metabolism within cells have been determined (Figure 1), including (i) de novo cholesterol synthesis, (ii) exogenous cholesterol uptake, (iii) cholesterol storage, (iv) cholesterol conversion, and (v) cholesterol trafficking (27).
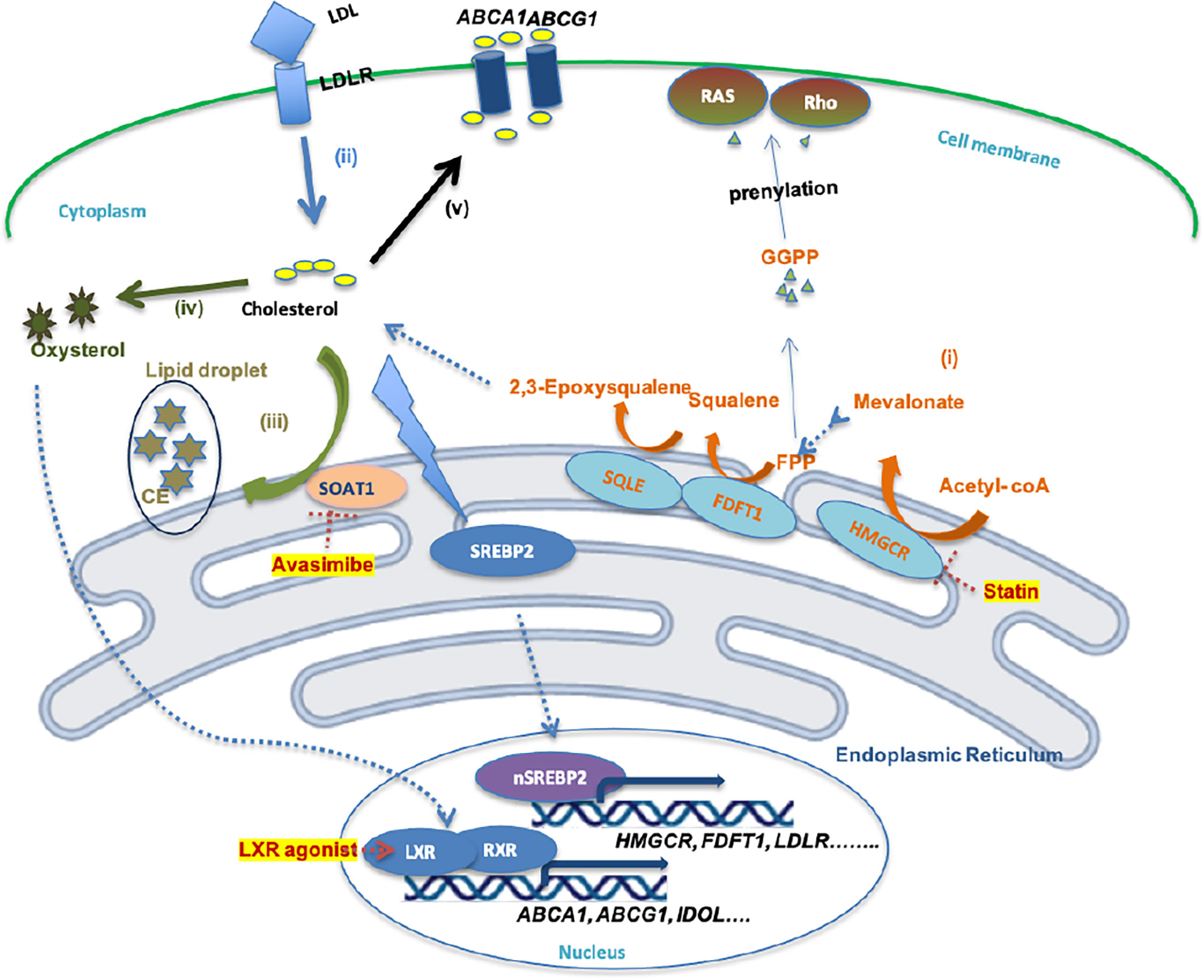
Figure 1 Schematic illustration of cholesterol metabolism homeostasis and potential drugs. (i)Cholesterol bio synthesis. (ii) Cholesterol uptake. (iii) Cholesterol storage. (iv) Cholesterol conversion. (v) Cholesterol efflux. (i) De novo cholesterol synthesis involves nearly 30 enzymatic reactions, in which HMGR and SQLE are two key rate-limiting enzymes. FPP and GGPP, intermediates in this process, contribute to the prenylation of RAS and Rho proteins, which is necessary for RAS and Rho signaling activation. (ii) Cholesterol uptake is mediated by LDL-LDLR binding, which is followed by endocytosis of LDL by cells. However, high cholesterol accumulation leads to intracellular lipo-toxicity. High intracellular cholesterol levels suppress SREBP2 transcription factor activity, thereby restricting the expression of enzymes involved in cholesterol synthesis or cholesterol uptake. (iii) Excess cholesterol is converted into cholesterol ester by SOAT1 enzyme, then stored in lipid droplets. (iv) Excess cholesterol is converted to oxysterol through multiple enzymatic or non-enzymatic process. (v) Oxysterol activates LXR-RXR signaling and results in expression of ABCA1, ABCG1, and IDOL, which promote the cholesterol efflux pathway.
(i) De novo cholesterol synthesis is initiated from acetyl-CoA via a complex enzymatic process. Within these reactions, 3-hydroxy-3-methylglutaryl-CoA (HMG-CoA) reductase (HMGCR), farnesyl-diphosphate farnesyltransferase 1 (FDFT1) and squalene epoxidase (SQLE) are key rate-limiting enzymes that convert HMG-CoA to mevalonate and squalene to 2,3-epoxysqualene (27). HMGCR, FDFT1 and SQLE are transcriptionally regulated by sterol regulatory element-binding protein 2 (SREBP2) (28). (ii) Mammalian cells take up exogenous cholesterol via low-density lipoprotein (LDL)-LDL receptor (LDLR) interactions, which internalizes cholesterol via endocytosis (12). However, free intracellular cholesterol levels require stringent control within the cytoplasm, because high levels lead to lipo-toxicity (26). An increased free cholesterol concentration >5% activates binding of SREBP cleavage-activating protein (SCAP) and Insig-1 on the endoplasmic reticulum (ER) membrane, leading to the retention of the SCAP-SREBP complex in the ER and preventing cholesterol/fatty acid synthesis and transportation, and thus lipid toxicity (29). (iii) Sterol O-acyltransferase (SOAT) is allosterically activated by elevated intracellular free cholesterol levels, promoting the conversion of cholesterols to cholesterol esters (CE), which is stored in lipid droplets (LD) (30). (iv) Oxysterol from excess cholesterol as a ligand directly activates the liver X receptor (LXR) transcription factor to regulate the (v) cholesterol efflux pathway by mediating the expression of the ATP-binding cassette (ABC) transporters, such as ABCA1 and ABCG1 (31). Excess cholesterol is exported outside the cell by ABC transporters at the cell surface, among which ABCA1 and ABCG1 are ubiquitously expressed in human cells (32). The cholesterol exported by ABCA1 is loaded onto lipid-free apolipoprotein A-I, thus producing nascent high-density lipoprotein (HDL), which in turn is converted into mature HDL by lecithin:cholesterol acyltransferase (LCAT) in the plasma (33). However, cholesterol exported by ABCG1 can directly become mature HDL (33), which can be ingested by liver cells or steroidogenic cells via binding to the HDL receptor, Scavenger receptor type B1 (SR-B1), thus resulting in selective CE uptake for subsequent synthesis of bile salts or steroid hormones (33, 34).
3 Proteins Involved in Cholesterol Metabolism in Ovarian Cancer
Several abnormally expressed proteins mediate cholesterol metabolism alterations to promote tumor cell viability, proliferation, migration, and invasion in ovarian cancer (Table 1). Therefore, the development of strategies targeting such proteins could lay the foundation for novel therapeutic treatment options.
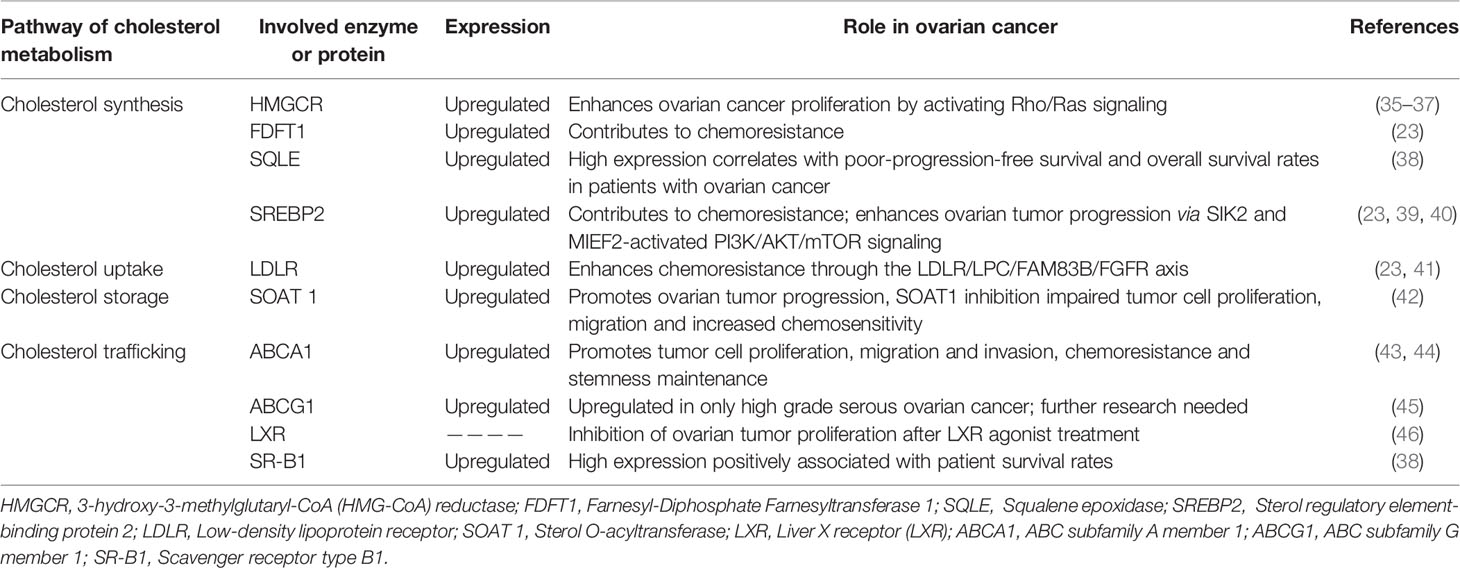
Table 1 The roles of enzymes and proteins involved in cholesterol metabolism in ovarian cancer.
3.1 Cholesterol Biosynthesis
3.1.1 HMG-CoA Reductase (HMGCR)
HMGCR, a glycoprotein located in the ER, is one of rate-limiting enzymes in the mevalonate pathway that catalyzes the generation of mevalonate from HMG-CoA with the consumption of two NAPDH molecules (26, 47). In addition to cholesterol generation to meet nutritional and membrane structure demands, intermediates of the mevalonate pathway are essential for the regulation of well-characterized oncogene-mediated signaling molecules, such as farnesyl pyrophosphate (FPP) and geranylgeranyl pyrophosphate (GGPP), which are essential for prenylation of the small GTPase proteins, Ras and Rho (48). Prenylation of Ras or Rho is critical for their membrane localization and activity (49). Oncogenic roles of HMGCR have been reported in various tumor types, including gastric, liver, and breast cancers (50–53).
The Keto and Wolf groups have reported higher HMGCR expression in cell lines and primary cultures from ovarian cancer than in normal ovarian epithelial cell lines and ovarian tissues (35, 54). Immunohistochemical expression was observed in the majority of ovarian cancer tissues (55). Mechanistically, gain-of-function TP53 variants displayed ectopic expression in SKOV-3, while the lack of endogenous p53 expression or native TP53 mutations in OVCAR-3 resulted in elevated HMGCR mRNA levels and protein expression (36, 54). HMGCR inhibition with specific statin-like drugs has been found to inhibit monolayer and ovarian tumor spheroid cellular proliferation and tumor growth in xenograft mouse models, enhance autophagy, induce cellular arrest in G0/G1, promote extrinsic and mitochondrial (intrinsic) apoptosis with increased activity of caspase-3, 8, 9 and elevated Poly (ADP-ribose) polymerase (PARP) cleavage, and increase the sensitivity of ovarian cancer cells to carboplatin (35, 37, 56, 57). However, the addition of GGPP or mevalonate instead of cholesterol rescued the anti-proliferative effect mediated by statin and activated Ras/Rho signaling (35, 37).
3.1.2 Farnesyl-Diphosphate Farnesyltransferase 1 (FDFT1)
FDFT1, also known as squalene synthase, is located in the ER and acts downstream of HMGCR to synthesize squalene from FPP (58). The role of FDFT1 in cancer development is ambiguous at present. Reports to date suggest oncogenic effects of FDFT1, such as promoting proliferation, increasing anti-apoptotic protein levels, and preventing ferroptosis by increasing squalene levels in some cancer types, and conversely, plays an anti-oncogenic role in other cancers. For instance, overexpression of FDFT1 has been shown to induce the suppression of glycolysis through the blockage of AKT/mTOR/HIF-1α signaling in colorectal cancer (59).
FDFT1 is highly expressed in ovarian cancer. Zheng et al. showed a nearly 7-fold upregulation of FDFT1 in A2780 cisplatin-resistant ovarian cancer cells relative to sensitive cells (23). Interestingly, siRNA mediated FDFT1 inhibition in A2780 cells slightly augmented tumor cell proliferation, while its overexpression impaired migration and invasion of SKOV3 and 3AO cells (60). These findings may be attributed to squalene accumulation inducing cytotoxicity within cells (61).
3.1.3 Squalene Epoxidase (SQLE)
SQLE, also known as squalene monooxygenase, is located in the ER and is a rate-limiting enzyme in the mevalonate pathway that mediates the conversion of squalene into 2,3-epoxysqualene via usage of NADPH and a molecular oxygen (62). SQLE overexpression has been observed in multiple cancers, including breast cancer, liver and lung cancer, and is correlated with their aggressive behaviors and poorer prognosis (63–67). High expression and gain of the SQLE locus have been reported in ovarian cancer (63). Furthermore, the Kaplan-Meier analysis of ovarian cancer has shown that high SQLE expression is correlated with relatively poor progression-free survival and overall survival rates in patients with ovarian cancer (38). Therefore, further research on the potential involvement of SQLE in the pathogenesis of ovarian cancer should be considered.
3.1.4 Sterol Regulatory Element-Binding Protein 2 (SREBP2)
SREBP2 is a key transcription factor of enzymes involved in cholesterol synthesis and transport, including HMGCR, FDFT1, SQLE and LDLR (28). Under conditions of cholesterol sufficiency, SREBP2 is located in the ER in an inactive state (28). Upon depletion of cholesterol, SREBP2 translocates to the Golgi apparatus and is cleaved by site 1 protease (S1P) and S2P to an active state. The active protein subsequently enters the nucleus to bind other regulatory factors at the promotor regions of target genes (68).
SREBP2 can enhance chemotherapeutic drug resistance in ovarian cancer cells via the upregulation of cholesterol synthesis (23). Zheng et al. (23) reported that the levels of SREBP2 and SREBP-targeted genes, such as HMGCR, FDFT1 and LDLR, in A2780 cells were proportionally correlated with cisplatin doses. Karashcuk and co-workers additionally reported that SREBP2 mediates ovarian cancer recurrence and escape from cell cycle arrest after paclitaxel treatment (69). The inhibition of SREBP2 with CRISPR technology in OVCAR-8 cell lines led to slower recovery rates of cell growth following paclitaxel treatment, compared to control cells (70). Thus, targeting of SREBP2 may improve drug sensitivity and lower the recurrence of ovarian cancer.
In addition to being regulated by the free intracellular cholesterol level, SREBP2 expression is also regulated by the PI3K/AKT/mTOR signaling pathway (71). In the cholesterol synthesis pathway, SREBP2 is upregulated by salt-inducible kinase 2 (SIK2), an AMPK-related kinase, and mitochondrial elongation factor 2 (MIEF2)-activated PI3K/AKT or ROS/AKT/mTOR signaling, thus, leading to the promotion of ovarian tumor growth (39, 40).
3.2 Cholesterol Uptake
3.2.1 Low-Density Lipoprotein Receptor (LDLR)
Binding of LDLR, a transmembrane glycoprotein located on the cell plasma membrane, to LDL facilitates cholesterol uptake via endocytosis (72). Based on TCGA data, high LDLR expression is significantly associated with poor overall survival rates of ovarian tumor patients (22). IHC findings have revealed a strong intensity of LDLR in endometrioid and clear cell types of ovarian cancer (41). Zheng et al. showed that LDLR was upregulated in an ovarian cancer cell line resistant to cisplatin (23). Silencing of LDLR improved the sensitivity of ovarian tumor cells to cisplatin treatment by mediating the LPC (lysophosophatidylcholine)/FAM83B (family with sequence similarity 83 member B)/FGFR (fibroblast growth factor receptor) axis (41). Therefore, LDLR may be recognized as a marker of cisplatin treatment response to ovarian tumors, and in particular, the endometrioid and clear-cell types.
3.3 Cholesterol Storage
3.3.1 Sterol O-Acyltransferase (SOAT)
Sterol O-acyltransferase (SOAT), also designated as acyl-coenzyme A cholesterol acyltransferase (ACAT), converts cholesterol and acyl-CoA to cholesterol esters (CE) in the ER, which are then stored in lipid droplets (33). SOAT exists as two isoforms, including SOAT1 and SOAT2. SOAT1 is generally detectable in all tissues, while SOAT2 is limited to the liver or intestinal tissue (73). SOAT1, but not SOAT2, is expressed highly in liver cancer, brain cancer, prostate cancer, and pancreatic cancer tissues and associated with their low overall survival rates (74–77). These findings suggest an oncogenic role for SOAT1 and support its utility as a potential therapeutic target.
Ayyagari et al. (42) reported higher SOAT1 expression levels than SOAT2 expression levels, and elevated CE levels in ovarian cancer cell lines compared to normal cell lines. Mechanistically, SOAT1 inhibition by shRNA or Avasimibe suppressed proliferation, migration and invasion of SKOV3, OC-314, and IGROV-1 cell lines by promoting mitochondrial apoptosis; the cells showed decreased mitochondrial potential, high activity of caspase 3/7, and increased ROS and p53 expression, regardless of mutation status. Furthermore, knockdown of SOAT1 improved the cisplatin sensitivity of ovarian cancer cells (42).
Of note, SOAT1 deficiency in CD8+ T cells augments their tumor-killing ability via increasing the cholesterol content on the plasma membrane and subsequently promoting T-cell receptor (TCR) clustering and immunological synapse formation in CD8+ T cells (78). SOAT1 depletion in mesothelin-directed chimeric antigen receptor T cells (CART) can strengthen their anti-tumor response against pancreatic carcinoma in vitro or in vivo (79). Therefore, SOAT1 inhibition may mediate dual anti-tumor effects in cancer treatment in terms of tumor inhibition and immunity enhancement, and is likely to have value in combination with immunotherapy.
3.4 Cholesterol Trafficking
3.4.1 ABC Subfamily A Member 1 (ABCA1)
ATP-binding cassette (ABC) transporters in the cell membrane mainly consisting of ABC subfamily A member 1 (ABCA1) and ABC subfamily G members 1, 5 and 8 (ABCG1, ABCG5, ABCG8) contribute to cholesterol efflux (80). Unlike ABCG5 and ABCG8 that are restricted to hepatocytes and enterocytes, ABCA1 and ABCG1 are ubiquitously expressed throughout the body (81). Hedditch et al. reported that high ABCA1 expression in ovarian cancer tissue was significantly correlated with poor survival outcomes of patients. In terms of functional analyses, depletion of ABCA in A2780, 27/87 and SKOV3 ovarian cancer cell lines via siRNA attenuated colony formation, migration, and invasion (43). Moreover, ABCA1 promoted ovarian cancer drug resistance and tumorigenesis. Silencing of ABCA1 in MCP2 platinum-resistant cells led to improved cisplatin sensitivity (82). In addition, ABCA1 was upregulated in EPCAM+CD45+ tumor cells derived from ascites of patients with ovarian cancer with aggressive features (44). Chou et al. showed that hypermethylation of ABCA1 was correlated with a poorer prognosis of ovarian cancer patients (83). Specifically, in vitro treatment of MCP3 and HeyC2 cell lines with shABCA1, or an in vivo HeyC2 cell-based xenograft mouse model mimicking hypermethylation enhanced tumor cell growth (83). Thus, the roles of ABCA1 in ovarian cancer require further investigation.
3.4.2 ATP Binding Cassette Subfamily G Member 1 (ABCG1)
ABCG1, located at the cell membrane surface, mediates cholesterol export from cells by mature HDL. High expression of ABCG1 has been observed in pancreatic cancer, breast cancer, lung cancer, and colon cancer (84–87). ABCG1 promotes cell proliferation, migration, and invasion in lung cancer cells, and is associated with expression of anti-apoptotic proteins (B-cell lymphoma 2 (BCL2) or Myeloid-cell leukemia 1 (MCL1), stemness markers (CD133 and ALDH), and proliferative markers (such as c-Myc) (87). ABCG1 inhibition by knockdown suppresses tumor growth in a colon tumor mouse model by blocking extracellular vesicle (EV) lipid efflux, thereby leading to the accumulation of EVs, which mediate cellular toxicity (86). In addition, ABCG1 is associated with tumor immunity. ABCG1 contributes to the macrophage phenotype shift from M1 to M2 (88). Macrophages with ABCG1 deficiency have higher cytotoxicity with NF-κB activation (88). In addition, depletion of ABCG1 causes hyperproliferation of CD4+ T cells in the peripheral blood in mice (89). These findings illustrate that ABCG1 may be a promising anti-tumor target. However, the high expression of ABCG1 has been observed in only high grade serous ovarian carcinoma (HGSC) (45). Its detailed mechanisms in ovarian cancer should be further explored.
3.4.3 Liver X Receptor (LXR)
LXR, which belongs to the nuclear receptor family, plays an important role in maintaining intracellular cholesterol homeostasis (31). LXR is activated by LXR agonists and subsequently forms a heterodimer with retinoid X receptor (RXR). This LXR-RER heterodimer combined with co-activator binds LXR-responsive-elements (LXREs) in the nucleus and mediates the expression of cholesterol metabolism-related genes, such as ABCA1, ABCG1, and inducible degrader of LDLR (IDOL) (90). LXR activation mediates anti-tumor effects in multiple cancers (91). LXR activation induces expression of inducible degrader of LDLR (IDOL), which decreases the LDLR expression induced by EGFR/SREBP-1 signaling in glioblastoma tumor cells (92). Likewise, LXR activation by its agonists significantly suppresses ovarian tumor cell proliferation (46).
3.4.4 Scavenger Receptor Type B1 (SR-B1)
SR-B1 recognizes HDL and then selectively takes up CEs into cells without the apolipoprotein moiety. SR-BI is commonly expressed in the liver cells and steroidogenic cells. SR-BI is highly expressed in multiple cancer cell lines including ovarian cancer cells lines (93). High SR-BI expression has been observed in lung cancer and breast cancer, and it is associated with malignancy and poor prognosis (94, 95). SR-BI is recognized as a biomarker of melanoma progression in patients and has been associated with STAT5 expression in clinical samples (96). However, SR-B1 expression in patients with ovarian cancer patients is positively correlated with survival rate (38). To provide further clarification, its detailed mechanisms require further exploration.
4 Roles of Cholesterol and Cholesterol Derivatives in Ovarian Cancer and Tumor Microenvironment
4.1 Cholesterol
Previous studies have shown high cholesterol levels in ascites fluid in ovarian tumors (97). Helzlsouer et al. initially reported that the cholesterol concentration in blood was proportionally correlated with the risk of ovarian cancer (98). In addition, LDL, a main blood carrier of cholesterol, and large amounts of cholesterol are associated with aggressiveness and poor survival outcomes of ovarian cancer (99). In the murine ID8 model of ovarian cancer, mice subjected to a high cholesterol diet exhibited increased tumor growth compared to that observed in the control groups (24). Dysregulated cholesterol homeostasis has been reported to enhance platinum resistance in ovarian cancer (22). Besides, high cholesterol levels in aggressive ascites were shown to contribute to cisplatin resistance in ovarian tumor cells by activating an LXR α/β nuclear receptor, with sequential upregulation of multidrug resistance protein 1 (MDR1) (100). High cholesterol loading in mitochondria perturbs mitochondrial function, inhibiting mitochondrial membrane permeabilization and the release of cytochrome c, a pro-apoptotic signal, thus contributing to chemotherapy resistance in liver cancer cells (101). The effects of dysregulated cholesterol homeostasis in mitochondria on drug resistance in ovarian cancer requires further investigation.
Cholesterol also influences energetic metabolism, thus contributing to tumor progression. In breast cancer cells, exogenous cholesterol alters metabolic pathways and consequently enhances cell proliferation in an estrogen-related receptor alpha-dependent manner, thus increasing oxidative phosphorylation and the tricarboxylic acid cycle (TCA) cycle (102). Aerobic glycolysis has been found to be augmented by exogenous cholesterol in only triple-negative breast cancer cell lines (102). Furthermore, high mitochondrial cholesterol loading increases hexokinase translocation to the mitochondria and may contribute to aerobic glycolysis in cancer cells (103). However, the relationship between cholesterol and energetic metabolism is less clear in ovarian cancer and requires further study.
Other than its effects on tumor cells, cholesterol may contribute to the immunosuppressive TME. Evidence has shown that cholesterol influences tumor-associated macrophages (TAM) in the microenvironment. Peritoneal TAMs in the ovarian cancer mouse model were reported to show increased cholesterol efflux activated by high molecular weight hyaluronic acid secretion from ID8 ovarian tumor cells, in turn, augmenting IL-4/PI3K/Akt/STAT6 signaling. However, attenuation of the IFN-γ-induced gene signature in TAM contributes to immunosuppression and the energetic needs of tumors (25). Specific knockout of ABCA1 or ABCG1 with the inhibition of cholesterol efflux in TAM effectively reversed the pro-tumorigenic effect of TAM in ovarian cancer, which could be applied to develop a novel therapeutic strategy. In addition, a large amount of cholesterol secreted from tumor cells impairs the cytotoxicity of CD8+ effector T cells and induces exhausted CD8+ T cells. HMGCR knockdown or statin treatment in B16 melanoma cells significantly decreases the frequency of exhausted CD8+ T cells at tumor sites (104). Mechanistically, high cholesterol augments endoplasmic reticulum (ER) stress in CD8+ T cells and consequently results in XBP-1 activation, which elevates the expression of immune checkpoint proteins, such as T cell immunoglobulin and mucin domain-containing protein 3 (TIM-3), Programmed cell death protein 1 (PD-1), Lymphocyte activation gene 3 protein (LAG-3), and 2B4 (CD244), in CD8+ T cells (104).
4.2 Oxysterol
Oxysterol, the hydroxylation product of cholesterol, participates in numerous cellular processes, such as cell signaling, membrane fluidity, and the activation of membrane proteins, similar to cholesterol (105). The 27-Hydroxycholesterol (27HC), a type of oxysterol, is catalyzed from cholesterol by Cytochrome P450 Family 27 Subfamily A Member 1 (CYP27A1) (106). High CYP27A1 expression is associated with poor prognosis at the early stages of disease and poorer progression-free survival but serves as a positive predictor in late-stage ovarian cancer (24). Functionally, exogenous 27HC treatment could abolish the proliferative capacity of ovarian cancer cell lines via LXR activation-induced cholesterol efflux in tumor cells. Intriguingly, however, CYP27A1 or exogenous 27HC treatment in the ovarian cancer mouse model has also been shown to augment peritoneal tumor spread and carboplatin resistance, consistent with Kaplan–Meier analyses of CYP27A1 in ovarian cancer patients (24). These data suggest that CYP27A1 and its product 27HC promote ovarian cancer progression by influencing the tumor microenvironment, rather than intrinsic effects on the tumor itself.
Reprogrammed macrophage patterns have been observed in ovarian tumors in the presence of exogenous 27HC, including increased concentrations of monocytic myeloid-derived suppressor cells (MDSC) and decreases in antigen-presenting macrophages (24). Genetic depletion of CYP27A1 could reverse the immunosuppressive effect of 27HC. In addition, a combination of the CYP27A1 inhibitor, GW273297X, and anti-PDL1 antibodies induced a significant decrease in ovarian tumor growth in mouse models compared to either treatment alone (24). In addition, the oxysterol secreted by tumor cells has been shown to impair the antigen presentation of dendritic cells (DC) by LXR-α signaling activation, thus mediating downregulation of the expression of C-C chemokine receptor type 7 (CCR7), a lymphoid homing marker of DC, on the DC cell surface (107). LXR-α activation in DCs compromises DC migration to lymph nodes, thus decreasing T cell priming (107). Treatment with Sulfotransferase 2B1b (SULT2B1b), an LXR ligand-inactive enzyme, relieves the CCR7 inhibition in DCs, and restores DC function and the anti-tumor response (107). LXR signaling activation suppresses the proliferation and expansion of T cells (108). However, Tavazoie et al. have shown that LXR activation suppressed the immunosuppressive effect of myeloid-derived suppressor cells (MDSC) by inducing Apolipoprotein E (ApoE) expression and consequently augmenting the T cell killing ability (109).
Another type of oxysterol, 25-hydroxycholesterol (25HC), is synthesized by cholesterol 25-hydroxylase (CH25H) (110). The specific role of 25-hydroxycholesterol (25HC) in ovarian cancer remains to be established. The 25HC is reported to stimulate the proliferation of BG-1 ovarian cancer cells in an estrogen receptor-α-activation-dependent manner (110). However, 25HC combined with statin reduced the viability of OVCAR-8 and SKOV3 cell lines via the suppression of SREBP2. SREBP2 suppression was greater following the combined treatment compared to that observed with statin treatment alone (111). Therefore, future studies should focus on the precise mechanisms of action of 25HC in ovarian cancer.
5 Potential Therapeutic Drugs in Ovarian Cancer
5.1 Statins
Statins are specific inhibitors of HMGCR that block the mevalonate pathway (112). Statins were originally used to lower the cholesterol level in blood and were found to be well-tolerated. Freed-Pastor et al. demonstrated that upregulation of the mevalonate pathway was mostly mediated by TP53 mutations, which is a dominant genetic mutation profile in ovarian cancer. Several reports have confirmed that lipophilic statins (Table 2), such as simvastatin and lovastatin, but not hydrophilic statins, significantly suppressed cell viability and proliferation, stemness, invasion and migration, and enhance mitochondrial apoptosis and chemotherapeutic sensitivity of ovarian cancer cell lines and primary ovarian cancer samples derived from patients or mouse models, without causing damage to normal cells (35, 37, 54, 56, 57). Statins exert pluripotent effects against cancer. Göbel and co-workers showed that lipophilic statins attenuated the expression of IL-6, IL-8, Vascular endothelial growth factor (VEGF), and Transforming growth factor beta (TGF-β), which contributed to ovarian tumor progression (115). Meanwhile, statin treatment of ovarian cancer cell lines activated c-Jun N-terminal kinase (JNK) signaling and induced the pro-apoptotic protein, Bim, reduced c-Myc phosphorylation, and blocked Ras/Rho signaling (37, 57, 113).

Table 2 Potential drugs for targeting cholesterol metabolism in ovarian cancer.
In addition to promoting resistance against tumor growth, statins are reported to enhance antigen presentation in dendritic cells (DC) and T cell cytotoxic functions in a B16 melanoma mouse model by attenuating Rab5 protein prenylation by GGPP or FPP, which are involved in the endosomal trafficking process; thus, reducing antigen internalization and degradation at the cell surface (116). Notably, the combination of statin with anti-PD1 antibodies exerted a stronger synergistic anti-tumor effect comparison with the alone treatment (116). Therefore, statin-like drugs present potent therapeutic options for ovarian cancer.
Several clinical trials (NCT04457089 and NCT00585052) on ovarian cancer have been conducted. Statins, and in particular, lipophilic statins are clearly associated with a lower risk of ovarian cancer occurrence (117).
5.2 Avasimibe
Avasimibe, an inhibitor of SOAT and a cholesterol-lowering drug, can suppress CE generation. Accumulating preclinical studies have revealed the inhibitory effects of Avasimibe on tumor growth in various cancer types, including hepatocellular carcinoma, glioblastoma, pancreatic adenocarcinoma, and prostate cancer through the regulation of intracellular cholesterol metabolism (74–77). Ayyagari et al. (42) demonstrated the anti-tumor growth effect of Avasimibe in ovarian cancer cell lines.
In addition to its tumor suppression activity, Avasimibe is also an immunomodulatory agent. Avasimibe can augment the tumor-killing function of CD8+ T cells via the enhancement of T cell receptor (TCR) signaling and immunological synapse formation of CD8+ T cells by enhancing the cholesterol level of the plasma membrane (78). Consequently, many combined therapeutic strategies have been developed for Avasimibe and other immunotherapies, such as anti-PD1 antibodies, cancer stem cell-dendritic cell (CSC-DC) vaccines, and Kras peptide vaccines (78, 118, 119).
In earlier atherosclerosis clinical trials, Avasimibe exhibited a well-tolerated safety profile (120), and thus, should be strongly considered for ovarian cancer clinical trials.
5.3 LXR Agonist
Liver X receptor, a nuclear receptor, senses alterations in cholesterol metabolism (33). Natural ligands of this receptor include different types of oxysterol, such as 22-hydroxycholesterol (22HC), 20-hydroxycholesterol (20HC), and 27HC (121). The 27HC is known to exert anti-proliferative effects in ovarian tumors. GW3965 and T0901317 are synthetic LRX ligands (121) and Curtarello et al. showed that GW3965 could significantly promote apoptosis of SKOV3 and IGROV1 cell lines by reducing LD accumulation under hypoxic conditions to a similar degree as the anti-VEGF antibody, bevacizumab (114). Combination treatment with GW3965 and bevacizumab further promoted treatment efficiency. Additionally, T0901317 exerted a significant inhibitory effect on ovarian cancer cells in a dose- and time-dependent manner via interactions with farnesoid-X receptor (FXR), rather than via LXR activation (46). However, limited research has focused on the involvement of LXR, and further experiments are required to determine the role of LXR agonists in ovarian cancer.
6 Conclusions
Numerous studies have implicated essential enzymes or proteins involved in cholesterol metabolism in ovarian cancer, thus, supporting the theory that aberrant cholesterol metabolism contributes to disease progression. Cholesterol and oxysterol, its derivative, are not only intrinsic tumor-promoting factors but also extrinsic tumor-promoting factors via reprogramming the tumor microenvironment. Elevated levels of cholesterol and oxysterol also contribute to the immunosuppressive environment. High cholesterol level in TME contributes to the generation of exhausted CD8+T cells. Oxysterol can reprogramme the TAM pattern and influence the antigen presentation ability of DC in the tumor site. Thus, drugs targeting cholesterol metabolism may present potential treatments and even overcome the immunotherapy resistance, as a combined therapy with immune checkpoint blockades. Further studies are needed to clarify the specific roles and associated mechanisms of action of these proteins in the pathogenesis of ovarian cancer to facilitate the development of therapeutic clinical agents.
Author Contributions
Writing the original draft preparation, JH. Writing—review and editing, MS and KC. Supervision, MS, HN, and KC. All authors contributed to the article and approved the submitted version.
Funding
The work was funded by the University of Hong Kong(202011159187).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Torre LA, Trabert B, DeSantis CE, Miller KD, Samimi G, Runowicz CD, et al. Ovarian Cancer Statistics, 2018. CA Cancer J Clin (2018) 68(4):284–96. doi: 10.3322/caac.21456
2. Lengyel E. Ovarian Cancer Development and Metastasis. Am J Pathol (2010) 177(3):1053–64. doi: 10.2353/ajpath.2010.100105
3. Shih Ie M, Kurman RJ. Ovarian Tumorigenesis: A Proposed Model Based on Morphological and Molecular Genetic Analysis. Am J Pathol (2004) 164(5):1511–8. doi: 10.1016/s0002-9440(10)63708-x
4. Kaldawy A, Segev Y, Lavie O, Auslender R, Sopik V, Narod SA. Low-Grade Serous Ovarian Cancer: A Review. Gynecologic Oncol (2016) 143(2):433–8. doi: 10.1016/j.ygyno.2016.08.320
5. Pierson WE, Peters PN, Chang MT, Chen LM, Quigley DA, Ashworth A, et al. An Integrated Molecular Profile of Endometrioid Ovarian Cancer. Gynecologic Oncol (2020) 157(1):55–61. doi: 10.1016/j.ygyno.2020.02.011
6. Gemignani ML, Schlaerth AC, Bogomolniy F, Barakat RR, Lin O, Soslow R, et al. Role of KRAS and BRAF Gene Mutations in Mucinous Ovarian Carcinoma. Gynecologic Oncol (2003) 90(2):378–81. doi: 10.1016/S0090-8258(03)00264-6
7. Wiegand KC, Shah SP, Al-Agha OM, Zhao Y, Tse K, Zeng T, et al. ARID1A Mutations in Endometriosis-Associated Ovarian Carcinomas. New Engl J Med (2010) 363(16):1532–43. doi: 10.1056/NEJMoa1008433
8. Kurman RJ, Shih Ie M. The Dualistic Model of Ovarian Carcinogenesis: Revisited, Revised, and Expanded. Am J Pathol (2016) 186(4):733–47. doi: 10.1016/j.ajpath.2015.11.011
9. Siu MKY, Jiang YX, Wang JJ, Leung THY, Han CY, Tsang BK, et al. Hexokinase 2 Regulates Ovarian Cancer Cell Migration, Invasion and Stemness via FAK/ERK1/2/MMP9/NANOG/SOX9 Signaling Cascades. Cancers (2019) 11(6):813. doi: 10.3390/cancers11060813
10. Siu MKY, Jiang Y-x, Wang J-j, Leung THY, Ngu SF, Cheung ANY, et al. PDK1 Promotes Ovarian Cancer Metastasis by Modulating Tumor-Mesothelial Adhesion, Invasion, and Angiogenesis via α5β1 Integrin and JNK/IL-8 Signaling. Oncogenesis (2020) 9(2):24. doi: 10.1038/s41389-020-0209-0
11. Shen L, Zhou L, Xia M, Lin N, Ma J, Dong D, et al. Pgc1α Regulates Mitochondrial Oxidative Phosphorylation Involved in Cisplatin Resistance in Ovarian Cancer Cells via Nucleo-Mitochondrial Transcriptional Feedback. Exp Cell Res (2021) 398(1):112369. doi: 10.1016/j.yexcr.2020.112369
12. Chen RR, Yung MMH, Xuan Y, Zhan S, Leung LL, Liang RR, et al. Targeting of Lipid Metabolism With a Metabolic Inhibitor Cocktail Eradicates Peritoneal Metastases in Ovarian Cancer Cells. Commun Biol (2019) 2:281. doi: 10.1038/s42003-019-0508-1
13. Worzfeld T, Pogge von Strandmann E, Huber M, Adhikary T, Wagner U, Reinartz S, et al. The Unique Molecular and Cellular Microenvironment of Ovarian Cancer. Front Oncol (2017) 7:24. doi: 10.3389/fonc.2017.00024
14. Nieman KM, Kenny HA, Penicka CV, Ladanyi A, Buell-Gutbrod R, Zillhardt MR, et al. Adipocytes Promote Ovarian Cancer Metastasis and Provide Energy for Rapid Tumor Growth. Nat Med (2011) 17(11):1498–503. doi: 10.1038/nm.2492
15. Khan SM, Funk HM, Thiolloy S, Lotan TL, Hickson J, Prins GS, et al. In Vitro Metastatic Colonization of Human Ovarian Cancer Cells to the Omentum. Clin Exp Metastasis (2010) 27(3):185–96. doi: 10.1007/s10585-010-9317-0
16. Riscal R, Skuli N, Simon MC. Even Cancer Cells Watch Their Cholesterol! Mol Cell (2019) 76(2):220–31. doi: 10.1016/j.molcel.2019.09.008
17. Ahmed N, Escalona R, Leung D, Chan E, Kannourakis G. Tumour Microenvironment and Metabolic Plasticity in Cancer and Cancer Stem Cells: Perspectives on Metabolic and Immune Regulatory Signatures in Chemoresistant Ovarian Cancer Stem Cells. Semin Cancer Biol (2018) 53:265–81. doi: 10.1016/j.semcancer.2018.10.002
18. Ji Z, Shen Y, Feng X, Kong Y, Shao Y, Meng J, et al. Deregulation of Lipid Metabolism: The Critical Factors in Ovarian Cancer. Front Oncol (2020) 10:593017. doi: 10.3389/fonc.2020.593017
19. Ladanyi A, Mukherjee A, Kenny HA, Johnson A, Mitra AK, Sundaresan S, et al. Adipocyte-Induced CD36 Expression Drives Ovarian Cancer Progression and Metastasis. Oncogene (2018) 37(17):2285–301. doi: 10.1038/s41388-017-0093-z
20. Tesfay L, Paul BT, Konstorum A, Deng Z, Cox AO, Lee J, et al. Stearoyl-CoA Desaturase 1 Protects Ovarian Cancer Cells From Ferroptotic Cell Death. Cancer Res (2019) 79(20):5355–66. doi: 10.1158/0008-5472.CAN-19-0369
21. Li J, Condello S, Thomes-Pepin J, Ma X, Xia Y, Hurley TD, et al. Lipid Desaturation Is a Metabolic Marker and Therapeutic Target of Ovarian Cancer Stem Cells. Cell Stem Cell (2017) 20(3):303–14.e5. doi: 10.1016/j.stem.2016.11.004
22. Criscuolo D, Avolio R, Calice G, Laezza C, Paladino S, Navarra G, et al. Cholesterol Homeostasis Modulates Platinum Sensitivity in Human Ovarian Cancer. Cells (2020) 9(4):828. doi: 10.3390/cells9040828
23. Zheng L, Li L, Lu Y, Jiang F, Yang XA. SREBP2 Contributes to Cisplatin Resistance in Ovarian Cancer Cells. Exp Biol Med (Maywood NJ) (2018) 243(7):655–62. doi: 10.1177/1535370218760283
24. He S, Ma L, Baek AE, Vardanyan A, Vembar V, Chen JJ, et al. Host CYP27A1 Expression Is Essential for Ovarian Cancer Progression. Endocrine-Related Cancer (2019) 26(7):659–75. doi: 10.1530/ERC-18-0572
25. Goossens P, Rodriguez-Vita J, Etzerodt A, Masse M, Rastoin O, Gouirand V, et al. Membrane Cholesterol Efflux Drives Tumor-Associated Macrophage Reprogramming and Tumor Progression. Cell Metab (2019) 29(6):1376–89.e4. doi: 10.1016/j.cmet.2019.02.016
26. Luo J, Yang H, Song BL. Mechanisms and Regulation of Cholesterol Homeostasis. Nat Rev Mol Cell Biol (2020) 21(4):225–45. doi: 10.1038/s41580-019-0190-7
27. Xu H, Zhou S, Tang Q, Xia H, Bi F. Cholesterol Metabolism: New Functions and Therapeutic Approaches in Cancer. Biochim Biophys Acta Rev Cancer (2020) 1874(1):188394. doi: 10.1016/j.bbcan.2020.188394
28. Eberlé D, Hegarty B, Bossard P, Ferré P, Foufelle F. SREBP Transcription Factors: Master Regulators of Lipid Homeostasis. Biochimie (2004) 86(11):839–48. doi: 10.1016/j.biochi.2004.09.018
29. Radhakrishnan A, Goldstein JL, McDonald JG, Brown MS. Switch-Like Control of SREBP-2 Transport Triggered by Small Changes in ER Cholesterol: A Delicate Balance. Cell Metab (2008) 8(6):512–21. doi: 10.1016/j.cmet.2008.10.008
30. Long T, Sun Y, Hassan A, Qi X, Li X. Structure of Nevanimibe-Bound Tetrameric Human ACAT1. Nature (2020) 581(7808):339–43. doi: 10.1038/s41586-020-2295-8
31. Wang B, Tontonoz P. Liver X Receptors in Lipid Signalling and Membrane Homeostasis. Nat Rev Endocrinol (2018) 14(8):452–63. doi: 10.1038/s41574-018-0037-x
32. Phillips MC. Molecular Mechanisms of Cellular Cholesterol Efflux. J Biol Chem (2014) 289(35):24020–9. doi: 10.1074/jbc.R114.583658
33. Chang TY, Chang CC, Ohgami N, Yamauchi Y. Cholesterol Sensing, Trafficking, and Esterification. Annu Rev Cell Dev Biol (2006) 22:129–57. doi: 10.1146/annurev.cellbio.22.010305.104656
34. Krieger M. Charting the Fate of the “Good Cholesterol”: Identification and Characterization of the High-Density Lipoprotein Receptor SR-Bi. Annu Rev Biochem (1999) 68:523–58. doi: 10.1146/annurev.biochem.68.1.523
35. Kato S, Smalley S, Sadarangani A, Chen-Lin K, Oliva B, Brañes J, et al. Lipophilic But Not Hydrophilic Statins Selectively Induce Cell Death in Gynaecological Cancers Expressing High Levels of HMGCoA Reductase. J Cell Mol Med (2010) 14(5):1180–93. doi: 10.1111/j.1582-4934.2009.00771.x
36. Abdullah MI, Abed MN, Richardson A. The Interplay Between TP53 and Mevalonate Pathway in Ovarian Cancer. Ann Oncol (2017) 28:v6. doi: 10.1093/annonc/mdx361.020
37. Kobayashi Y, Kashima H, Wu RC, Jung JG, Kuan JC, Gu J, et al. Mevalonate Pathway Antagonist Suppresses Formation of Serous Tubal Intraepithelial Carcinoma and Ovarian Carcinoma in Mouse Models. Clin Cancer Res (2015) 21(20):4652–62. doi: 10.1158/1078-0432.CCR-14-3368
38. Gyorffy B, Lánczky A, Szállási Z. Implementing an Online Tool for Genome-Wide Validation of Survival-Associated Biomarkers in Ovarian-Cancer Using Microarray Data From 1287 Patients. Endocrine-Related Cancer (2012) 19(2):197–208. doi: 10.1530/ERC-11-0329
39. Zhao S, Cheng L, Shi Y, Li J, Yun Q, Yang H. MIEF2 Reprograms Lipid Metabolism to Drive Progression of Ovarian Cancer Through ROS/AKT/mTOR Signaling Pathway. Cell Death Dis (2021) 12(1):18. doi: 10.1038/s41419-020-03336-6
40. Zhao J, Zhang X, Gao T, Wang S, Hou Y, Yuan P, et al. SIK2 Enhances Synthesis of Fatty Acid and Cholesterol in Ovarian Cancer Cells and Tumor Growth Through PI3K/Akt Signaling Pathway. Cell Death Dis (2020) 11(1):25. doi: 10.1038/s41419-019-2221-x
41. Chang WC, Wang HC, Cheng WC, Yang JC, Chung WM, Ho YP, et al. LDLR-Mediated Lipidome-Transcriptome Reprogramming in Cisplatin Insensitivity. Endocrine-Related Cancer (2020) 27(2):81–95. doi: 10.1530/ERC-19-0095
42. Ayyagari VN, Wang X, Diaz-Sylvester PL, Groesch K, Brard L. Assessment of Acyl-CoA Cholesterol Acyltransferase (ACAT-1) Role in Ovarian Cancer Progression-An In Vitro Study. PloS One (2020) 15(1):e0228024. doi: 10.1371/journal.pone.0228024
43. Hedditch EL, Gao B, Russell AJ, Lu Y, Emmanuel C, Beesley J, et al. ABCA Transporter Gene Expression and Poor Outcome in Epithelial Ovarian Cancer. J Natl Cancer Institute (2014) 106(7):dju149. doi: 10.1093/jnci/dju149
44. Akhter MZ, Sharawat SK, Kumar V, Kochat V, Equbal Z, Ramakrishnan M, et al. Aggressive Serous Epithelial Ovarian Cancer Is Potentially Propagated by EpCAM(+)CD45(+) Phenotype. Oncogene (2018) 37(16):2089–103. doi: 10.1038/s41388-017-0106-y
45. Elsnerova K, Mohelnikova-Duchonova B, Cerovska E, Ehrlichova M, Gut I, Rob L, et al. Gene Expression of Membrane Transporters: Importance for Prognosis and Progression of Ovarian Carcinoma. Oncol Rep (2016) 35(4):2159–70. doi: 10.3892/or.2016.4599
46. Rough JJ, Monroy MA, Yerrum S, Daly JM. Anti-Proliferative Effect of LXR Agonist T0901317 in Ovarian Carcinoma Cells. J Ovarian Res (2010) 3:13. doi: 10.1186/1757-2215-3-13
47. Liscum L, Finer-Moore J, Stroud RM, Luskey KL, Brown MS, Goldstein JL. Domain Structure of 3-Hydroxy-3-Methylglutaryl Coenzyme A Reductase, a Glycoprotein of the Endoplasmic Reticulum. J Biol Chem (1985) 260(1):522–30. doi: 10.1016/S0021-9258(18)89764-2
48. Gruenbacher G, Thurnher M. Mevalonate Metabolism in Cancer. Cancer Lett (2015) 356(2 Pt A):192–6. doi: 10.1016/j.canlet.2014.01.013
49. Wang M, Casey PJ. Protein Prenylation: Unique Fats Make Their Mark on Biology. Nat Rev Mol Cell Biol (2016) 17(2):110–22. doi: 10.1038/nrm.2015.11
50. Chushi L, Wei W, Kangkang X, Yongzeng F, Ning X, Xiaolei C. HMGCR is Up-Regulated in Gastric Cancer and Promotes the Growth and Migration of the Cancer Cells. Gene (2016) 587(1):42–7. doi: 10.1016/j.gene.2016.04.029
51. Dong L, Xue L, Zhang C, Li H, Cai Z, Guo R. HSP90 Interacts With HMGCR and Promotes the Progression of Hepatocellular Carcinoma. Mol Med Rep (2019) 19(1):524–32. doi: 10.3892/mmr.2018.9667
52. Clendening JW, Pandyra A, Boutros PC, El Ghamrasni S, Khosravi F, Trentin GA, et al. Dysregulation of the Mevalonate Pathway Promotes Transformation. Proc Natl Acad Sci USA (2010) 107(34):15051–6. doi: 10.1073/pnas.0910258107
53. Göbel A, Breining D, Rauner M, Hofbauer LC, Rachner TD. Induction of 3-Hydroxy-3-Methylglutaryl-CoA Reductase Mediates Statin Resistance in Breast Cancer Cells. Cell Death Dis (2019) 10(2):91. doi: 10.1038/s41419-019-1322-x
54. de Wolf E, Abdullah MI, Jones SM, Menezes K, Moss DM, Drijfhout FP, et al. Dietary Geranylgeraniol can Limit the Activity of Pitavastatin as a Potential Treatment for Drug-Resistant Ovarian Cancer. Sci Rep (2017) 7(1):5410. doi: 10.1038/s41598-017-05595-4
55. Brennan DJ, Brändstedt J, Rexhepaj E, Foley M, Pontén F, Uhlén M, et al. Tumour-Specific HMG-CoAR Is an Independent Predictor of Recurrence Free Survival in Epithelial Ovarian Cancer. BMC Cancer (2010) 10:125. doi: 10.1186/1471-2407-10-125
56. Kato S, Liberona MF, Cerda-Infante J, Sánchez M, Henríquez J, Bizama C, et al. Simvastatin Interferes With Cancer ‘Stem-Cell’ Plasticity Reducing Metastasis in Ovarian Cancer. Endocrine-Related Cancer (2018) 25(10):821–36. doi: 10.1530/ERC-18-0132
57. Jones HM, Fang Z, Sun W, Clark LH, Stine JE, Tran AQ, et al. Atorvastatin Exhibits Anti-Tumorigenic and Anti-Metastatic Effects in Ovarian Cancer In Vitro. Am J Cancer Res (2017) 7(12):2478–90.
58. Tansey TR, Shechter I. Squalene Synthase: Structure and Regulation. Prog Nucleic Acid Res Mol Biol (2001) 65:157–95. doi: 10.1016/S0079-6603(00)65005-5
59. Ha NT, Lee CH. Roles of Farnesyl-Diphosphate Farnesyltransferase 1 in Tumour and Tumour Microenvironments. Cells (2020) 9(11):2352. doi: 10.3390/cells9112352
60. Lu J, Zhou Y, Zheng X, Chen L, Tuo X, Chen H, et al. 20(S)-Rg3 Upregulates FDFT1 via Reducing miR-4425 to Inhibit Ovarian Cancer Progression. Arch Biochem Biophysics (2020) 693:108569. doi: 10.1016/j.abb.2020.108569
61. Mahoney CE, Pirman D, Chubukov V, Sleger T, Hayes S, Fan ZP, et al. A Chemical Biology Screen Identifies a Vulnerability of Neuroendocrine Cancer Cells to SQLE Inhibition. Nat Commun (2019) 10(1):96. doi: 10.1038/s41467-018-07959-4
62. Gill S, Stevenson J, Kristiana I, Brown AJ. Cholesterol-Dependent Degradation of Squalene Monooxygenase, a Control Point in Cholesterol Synthesis Beyond HMG-CoA Reductase. Cell Metab (2011) 13(3):260–73. doi: 10.1016/j.cmet.2011.01.015
63. Brown DN, Caffa I, Cirmena G, Piras D, Garuti A, Gallo M, et al. Squalene Epoxidase is a Bona Fide Oncogene by Amplification With Clinical Relevance in Breast Cancer. Sci Rep (2016) 6:19435. doi: 10.1038/srep19435
64. Helms MW, Kemming D, Pospisil H, Vogt U, Buerger H, Korsching E, et al. Squalene Epoxidase, Located on Chromosome 8q24.1, is Upregulated in 8q+ Breast Cancer and Indicates Poor Clinical Outcome in Stage I and II Disease. Br J Cancer (2008) 99(5):774–80. doi: 10.1038/sj.bjc.6604556
65. Liu D, Wong CC, Fu L, Chen H, Zhao L, Li C, et al. Squalene Epoxidase Drives NAFLD-Induced Hepatocellular Carcinoma and Is a Pharmaceutical Target. Sci Trans Med (2018) 10(437):eaap9840. doi: 10.1126/scitranslmed.aap9840
66. Sui Z, Zhou J, Cheng Z, Lu P. Squalene Epoxidase (SQLE) Promotes the Growth and Migration of the Hepatocellular Carcinoma Cells. Tumour Biol: J Int Soc Oncodevelopmental Biol Med (2015) 36(8):6173–9. doi: 10.1007/s13277-015-3301-x
67. Ge H, Zhao Y, Shi X, Tan Z, Chi X, He M, et al. Squalene Epoxidase Promotes the Proliferation and Metastasis of Lung Squamous Cell Carcinoma Cells Though Extracellular Signal-Regulated Kinase Signaling. Thorac Cancer (2019) 10(3):428–36. doi: 10.1111/1759-7714.12944
68. Brown MS, Radhakrishnan A, Goldstein JL. Retrospective on Cholesterol Homeostasis: The Central Role of Scap. Annu Rev Biochem (2018) 87:783–807. doi: 10.1146/annurev-biochem-062917-011852
69. Karashchuk G, Brodsky AS. Abstract A58: Transcription Factor SREBP2 Mediates Ovarian Cancer Drug Resistance and Recurrence. Clinical Cancer Res (2018) 24(15 Supplement):A58–A. doi: 10.1158/1557-3265.OVCA17-A58
70. Karashchuk G, Karashchuk N, Caksa S, Smith TS, Brodsky AS. Abstract 4409: Cholesterol Pathway Determines Ovarian Cancer Drug Resistance Through Transcription Factor SREBP2. Cancer Res (2017) 77(13 Supplement):4409–. doi: 10.1158/1538-7445.AM2017-4409
71. Düvel K, Yecies JL, Menon S, Raman P, Lipovsky AI, Souza AL, et al. Activation of a Metabolic Gene Regulatory Network Downstream of mTOR Complex 1. Mol Cell (2010) 39(2):171–83. doi: 10.1016/j.molcel.2010.06.022
72. Jeon H, Blacklow SC. Structure and Physiologic Function of the Low-Density Lipoprotein Receptor. Annu Rev Biochem (2005) 74:535–62. doi: 10.1146/annurev.biochem.74.082803.133354
73. Chang TY, Li BL, Chang CC, Urano Y. Acyl-Coenzyme A:cholesterol Acyltransferases. Am J Physiol Endocrinol Metab (2009) 297(1):E1–9. doi: 10.1152/ajpendo.90926.2008
74. Geng F, Cheng X, Wu X, Yoo JY, Cheng C, Guo JY, et al. Inhibition of SOAT1 Suppresses Glioblastoma Growth via Blocking SREBP-1-Mediated Lipogenesis. Clin Cancer Res (2016) 22(21):5337–48. doi: 10.1158/1078-0432.CCR-15-2973
75. Jiang Y, Sun A, Zhao Y, Ying W, Sun H, Yang X, et al. Proteomics Identifies New Therapeutic Targets of Early-Stage Hepatocellular Carcinoma. Nature (2019) 567(7747):257–61. doi: 10.1038/s41586-019-0987-8
76. Oni TE, Biffi G, Baker LA, Hao Y, Tonelli C, Somerville TDD, et al. SOAT1 Promotes Mevalonate Pathway Dependency in Pancreatic Cancer. J Exp Med (2020) 217(9):e20192389. doi: 10.1084/jem.20192389
77. Lee HJ, Li J, Vickman RE, Li J, Liu R, Durkes AC, et al. Cholesterol Esterification Inhibition Suppresses Prostate Cancer Metastasis by Impairing the Wnt/β-Catenin Pathway. Mol Cancer Res (2018) 16(6):974–85. doi: 10.1158/1541-7786.MCR-17-0665
78. Yang W, Bai Y, Xiong Y, Zhang J, Chen S, Zheng X, et al. Potentiating the Antitumour Response of CD8(+) T Cells by Modulating Cholesterol Metabolism. Nature (2016) 531(7596):651–5. doi: 10.1038/nature17412
79. Zhao L, Liu Y, Zhao F, Jin Y, Feng J, Geng R, et al. Inhibition of Cholesterol Esterification Enzyme Enhances the Potency of Human Chimeric Antigen Receptor T Cells Against Pancreatic Carcinoma. Mol Ther Oncolytics (2020) 16:262–71. doi: 10.1016/j.omto.2020.01.008
80. Rosenson RS, Brewer HB Jr., Davidson WS, Fayad ZA, Fuster V, Goldstein J, et al. Cholesterol Efflux and Atheroprotection: Advancing the Concept of Reverse Cholesterol Transport. Circulation (2012) 125(15):1905–19. doi: 10.1161/CIRCULATIONAHA.111.066589
81. Graf GA, Yu L, Li WP, Gerard R, Tuma PL, Cohen JC, et al. ABCG5 and ABCG8 are Obligate Heterodimers for Protein Trafficking and Biliary Cholesterol Excretion. J Biol Chem (2003) 278(48):48275–82. doi: 10.1074/jbc.M310223200
82. Chou J-L, Chen L-Y, Su H-Y, Lin S-J, Huang Y-W, Deatherage DE, et al. Abstract 83: Hypomethylation of TGF-Beta Target Gene, ABCA1 in Ovarian Cancer and Cancer Initialing Cell and Is Associated With Poor Prognosis in Cancer Patients. Cancer Res (2011) 71(8 Supplement):83–. doi: 10.1158/1538-7445.AM2011-83
83. Chou JL, Huang RL, Shay J, Chen LY, Lin SJ, Yan PS, et al. Hypermethylation of the TGF-β Target, ABCA1 Is Associated With Poor Prognosis in Ovarian Cancer Patients. Clin Epigenet (2015) 7(1):1. doi: 10.1186/s13148-014-0036-2
84. Mohelnikova-Duchonova B, Brynychova V, Oliverius M, Honsova E, Kala Z, Muckova K, et al. Differences in Transcript Levels of ABC Transporters Between Pancreatic Adenocarcinoma and Nonneoplastic Tissues. Pancreas (2013) 42(4):707–16. doi: 10.1097/MPA.0b013e318279b861
85. Hlaváč V, Brynychová V, Václavíková R, Ehrlichová M, Vrána D, Pecha V, et al. The Expression Profile of ATP-Binding Cassette Transporter Genes in Breast Carcinoma. Pharmacogenomics (2013) 14(5):515–29. doi: 10.2217/pgs.13.26
86. Namba Y, Sogawa C, Okusha Y, Kawai H, Itagaki M, Ono K, et al. Depletion of Lipid Efflux Pump ABCG1 Triggers the Intracellular Accumulation of Extracellular Vesicles and Reduces Aggregation and Tumorigenesis of Metastatic Cancer Cells. Front Oncol (2018) 8:376. doi: 10.3389/fonc.2018.00376
87. Tian C, Huang D, Yu Y, Zhang J, Fang Q, Xie C. ABCG1 as a Potential Oncogene in Lung Cancer. Exp Ther Med (2017) 13(6):3189–94. doi: 10.3892/etm.2017.4393
88. Sag D, Cekic C, Wu R, Linden J, Hedrick CC. The Cholesterol Transporter ABCG1 Links Cholesterol Homeostasis and Tumour Immunity. Nat Commun (2015) 6(1):6354. doi: 10.1038/ncomms7354
89. Armstrong AJ, Gebre AK, Parks JS, Hedrick CC. ATP-Binding Cassette Transporter G1 Negatively Regulates Thymocyte and Peripheral Lymphocyte Proliferation. J Immunol (Baltimore Md: 1950) (2010) 184(1):173–83. doi: 10.4049/jimmunol.0902372
90. Willy PJ, Umesono K, Ong ES, Evans RM, Heyman RA, Mangelsdorf DJ. LXR, a Nuclear Receptor That Defines a Distinct Retinoid Response Pathway. Genes Dev (1995) 9(9):1033–45. doi: 10.1101/gad.9.9.1033
91. Bilotta MT, Petillo S, Santoni A, Cippitelli M. Liver X Receptors: Regulators of Cholesterol Metabolism, Inflammation, Autoimmunity, and Cancer. Front Immunol (2020) 11:584303. doi: 10.3389/fimmu.2020.584303
92. Guo D, Reinitz F, Youssef M, Hong C, Nathanson D, Akhavan D, et al. An LXR Agonist Promotes Glioblastoma Cell Death Through Inhibition of an EGFR/AKT/SREBP-1/LDLR-Dependent Pathway. Cancer Discov (2011) 1(5):442–56. doi: 10.1158/2159-8290.CD-11-0102
93. Shahzad MM, Mangala LS, Han HD, Lu C, Bottsford-Miller J, Nishimura M, et al. Targeted Delivery of Small Interfering RNA Using Reconstituted High-Density Lipoprotein Nanoparticles. Neoplasia (New York NY) (2011) 13(4):309–19. doi: 10.1593/neo.101372
94. Yuan B, Wu C, Wang X, Wang D, Liu H, Guo L, et al. High Scavenger Receptor Class B Type I Expression is Related to Tumor Aggressiveness and Poor Prognosis in Breast Cancer. Tumour Biol: J Int Soc Oncodevelopmental Biol Med (2016) 37(3):3581–8. doi: 10.1007/s13277-015-4141-4
95. Feng H, Wang M, Wu C, Yu J, Wang D, Ma J, et al. High Scavenger Receptor Class B Type I Expression Is Related to Tumor Aggressiveness and Poor Prognosis in Lung Adenocarcinoma: A STROBE Compliant Article. Medicine (2018) 97(13):e0203. doi: 10.1097/MD.0000000000010203
96. Kinslechner K, Schörghofer D, Schütz B, Vallianou M, Wingelhofer B, Mikulits W, et al. Malignant Phenotypes in Metastatic Melanoma Are Governed by SR-BI and Its Association With Glycosylation and STAT5 Activation. Mol Cancer Res (2018) 16(1):135–46. doi: 10.1158/1541-7786.MCR-17-0292
97. Penet MF, Krishnamachary B, Wildes FB, Mironchik Y, Hung CF, Wu TC, et al. Ascites Volumes and the Ovarian Cancer Microenvironment. Front Oncol (2018) 8:595. doi: 10.3389/fonc.2018.00595
98. Helzlsouer KJ, Alberg AJ, Norkus EP, Morris JS, Hoffman SC, Comstock GW. Prospective Study of Serum Micronutrients and Ovarian Cancer. J Natl Cancer Institute (1996) 88(1):32–7. doi: 10.1093/jnci/88.1.32
99. Li AJ, Elmore RG, Chen IY, Karlan BY. Serum Low-Density Lipoprotein Levels Correlate With Survival in Advanced Stage Epithelial Ovarian Cancers. Gynecologic Oncol (2010) 116(1):78–81. doi: 10.1016/j.ygyno.2009.09.027
100. Kim S, Lee M, Dhanasekaran DN, Song YS. Activation of Lxrα/β by Cholesterol in Malignant Ascites Promotes Chemoresistance in Ovarian Cancer. BMC Cancer (2018) 18(1):1232. doi: 10.1186/s12885-018-5152-5
101. Montero J, Morales A, Llacuna L, Lluis JM, Terrones O, Basañez G, et al. Mitochondrial Cholesterol Contributes to Chemotherapy Resistance in Hepatocellular Carcinoma. Cancer Res (2008) 68(13):5246–56. doi: 10.1158/0008-5472.CAN-07-6161
102. Ghanbari F, Fortier A-M, Park M, Philip A. Cholesterol-Induced Metabolic Reprogramming in Breast Cancer Cells Is Mediated via the Errα Pathway. Cancers (Basel) (2021) 13(11):2605. doi: 10.3390/cancers13112605
103. Campbell AM, Chan SHP. Mitochondrial Membrane Cholesterol, the Voltage Dependent Anion Channel (VDAC), and the Warburg Effect. J Bioenergetics Biomembranes (2008) 40(3):193–7. doi: 10.1007/s10863-008-9138-x
104. Ma X, Bi E, Lu Y, Su P, Huang C, Liu L, et al. Cholesterol Induces CD8(+) T Cell Exhaustion in the Tumor Microenvironment. Cell Metab (2019) 30(1):143–56.e5. doi: 10.1016/j.cmet.2019.04.002
105. Kloudova A, Guengerich FP, Soucek P. The Role of Oxysterols in Human Cancer. Trends Endocrinol Metabol: TEM (2017) 28(7):485–96. doi: 10.1016/j.tem.2017.03.002
106. van Reyk DM, Brown AJ, Hult’en LM, Dean RT, Jessup W. Oxysterols in Biological Systems: Sources, Metabolism and Pathophysiological Relevance. Redox Report: Commun Free Radical Res (2006) 11(6):255–62. doi: 10.1179/135100006X155003
107. Villablanca EJ, Raccosta L, Zhou D, Fontana R, Maggioni D, Negro A, et al. Tumor-Mediated Liver X Receptor-Alpha Activation Inhibits CC Chemokine Receptor-7 Expression on Dendritic Cells and Dampens Antitumor Responses. Nat Med (2010) 16(1):98–105. doi: 10.1038/nm.2074
108. Bensinger SJ, Bradley MN, Joseph SB, Zelcer N, Janssen EM, Hausner MA, et al. LXR Signaling Couples Sterol Metabolism to Proliferation in the Acquired Immune Response. Cell (2008) 134(1):97–111. doi: 10.1016/j.cell.2008.04.052
109. Tavazoie MF, Pollack I, Tanqueco R, Ostendorf BN, Reis BS, Gonsalves FC, et al. LXR/ApoE Activation Restricts Innate Immune Suppression in Cancer. Cell (2018) 172(4):825–40.e18. doi: 10.1016/j.cell.2017.12.026
110. Lappano R, Recchia AG, De Francesco EM, Angelone T, Cerra MC, Picard D, et al. The Cholesterol Metabolite 25-Hydroxycholesterol Activates Estrogen Receptor α-Mediated Signaling in Cancer Cells and in Cardiomyocytes. PloS One (2011) 6(1):e16631. doi: 10.1371/journal.pone.0016631
111. Casella C, Miller DH, Lynch K, Brodsky AS. Oxysterols Synergize With Statins by Inhibiting SREBP-2 in Ovarian Cancer Cells. Gynecologic Oncol (2014) 135(2):333–41. doi: 10.1016/j.ygyno.2014.08.015
112. Istvan ES, Deisenhofer J. Structural Mechanism for Statin Inhibition of HMG-CoA Reductase. Sci (New York NY) (2001) 292(5519):1160–4. doi: 10.1126/science.1059344
113. Liu H, Liang SL, Kumar S, Weyman CM, Liu W, Zhou A. Statins Induce Apoptosis in Ovarian Cancer Cells Through Activation of JNK and Enhancement of Bim Expression. Cancer Chemother Pharmacol (2009) 63(6):997–1005. doi: 10.1007/s00280-008-0830-7
114. Curtarello M, Tognon M, Venturoli C, Silic-Benussi M, Grassi A, Verza M, et al. Rewiring of Lipid Metabolism and Storage in Ovarian Cancer Cells After Anti-VEGF Therapy. Cells (2019) 8(12):1601. doi: 10.3390/cells8121601
115. Göbel A, Zinna VM, Dell’Endice S, Jaschke N, Kuhlmann JD, Wimberger P, et al. Anti-Tumor Effects of Mevalonate Pathway Inhibition in Ovarian Cancer. BMC Cancer (2020) 20(1):703. doi: 10.1186/s12885-020-07164-x
116. Xia Y, Xie Y, Yu Z, Xiao H, Jiang G, Zhou X, et al. The Mevalonate Pathway Is a Druggable Target for Vaccine Adjuvant Discovery. Cell (2018) 175(4):1059–73.e21. doi: 10.1016/j.cell.2018.08.070
117. Akinwunmi B, Vitonis AF, Titus L, Terry KL, Cramer DW. Statin Therapy and Association With Ovarian Cancer Risk in the New England Case Control (NEC) Study. Int J Cancer (2019) 144(5):991–1000. doi: 10.1002/ijc.31758
118. Chen X, Song Q, Xia L, Xu X. Synergy of Dendritic Cell Vaccines and Avasimibe in Treatment of Head and Neck Cancer in Mice. Med Sci Monitor: Int Med J Exp Clin Res (2017) 23:4471–6. doi: 10.12659/MSM.905814
119. Pan J, Zhang Q, Palen K, Wang L, Qiao L, Johnson B, et al. Potentiation of Kras Peptide Cancer Vaccine by Avasimibe, a Cholesterol Modulator. EBioMedicine (2019) 49:72–81. doi: 10.1016/j.ebiom.2019.10.044
120. Pal P, Gandhi H, Giridhar R, Yadav MR. ACAT Inhibitors: The Search for Novel Cholesterol Lowering Agents. Mini Rev Med Chem (2013) 13(8):1195–219. doi: 10.2174/1389557511313080007
Keywords: cholesterol metabolism, tumor microenvironment, carcinogenesis, therapeutic targets, ovarian cancer
Citation: He J, Siu MKY, Ngan HYS and Chan KKL (2021) Aberrant Cholesterol Metabolism in Ovarian Cancer: Identification of Novel Therapeutic Targets. Front. Oncol. 11:738177. doi: 10.3389/fonc.2021.738177
Received: 08 July 2021; Accepted: 15 October 2021;
Published: 08 November 2021.
Edited by:
Tuuli Käämbre, National Institute of Chemical Physics and Biophysics, EstoniaReviewed by:
Sofia Avnet, University of Bologna, ItalyCinzia Domenicotti, Università di Genova, Italy
Copyright © 2021 He, Siu, Ngan and Chan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Karen K. L. Chan, a2tsY2hhbkBoa3UuaGs=
†These authors have contributed equally to this work