 Hongyi Li1,2†
Hongyi Li1,2† Yongjia Weng1,2†Shaojie Wang1,2Fang Wang1,2
Yongjia Weng1,2†Shaojie Wang1,2Fang Wang1,2 Yanqiang Wang1,2
Yanqiang Wang1,2 Pengzhou Kong1,2Ling Zhang1,2Caixia Cheng3
Pengzhou Kong1,2Ling Zhang1,2Caixia Cheng3 Heyang Cui1,2Enwei Xu4Shuqing Wei5Dinghe Guo1,2Fei Chen1,2Yanghui Bi6Yongsheng Meng7
Heyang Cui1,2Enwei Xu4Shuqing Wei5Dinghe Guo1,2Fei Chen1,2Yanghui Bi6Yongsheng Meng7 Xiaolong Cheng1,2*
Xiaolong Cheng1,2* Yongping Cui1,2*
Yongping Cui1,2*- 1Department of Pathology & Shanxi Key Laboratory of Carcinogenesis and Translational Research of Esophageal Cancer, Shanxi Medical University, Taiyuan, China
- 2Key Laboratory of Cellular Physiology, Ministry of Education, Shanxi Medical University, Taiyuan, China
- 3Department of Pathology, the First Hospital, Shanxi Medical University, Taiyuan, China
- 4Department of Pathology, Shanxi Province Cancer Hospital, Taiyuan, China
- 5Department of Thoracic Surgery (Ⅰ), Shanxi Province Cancer Hospital, Taiyuan, China
- 6The Science Research Center, Shanxi Bethone Hospital, Taiyuan, China
- 7Tumor Biobank, Shanxi Province Cancer Hospital, Taiyuan, China
Background: CDCA7 is a copy number amplified gene identified not only in esophageal squamous cell carcinoma (ESCC) but also in various cancer types. Its clinical relevance and underlying mechanisms in ESCC have remained unknown.
Methods: Tissue microarray data was used to analyze its expression in 179 ESCC samples. The effects of CDCA7 on proliferation, colony formation, and cell cycle were tested in ESCC cells. Real-time PCR and Western blot were used to detect the expression of its target genes. Correlation of CDCA7 with its target genes in ESCC and various SCC types was analyzed using GSE53625 and TCGA data. The mechanism of CDCA7 was studied by chromatin immunoprecipitation (ChIP), luciferase reporter assays, and rescue assay.
Results: The overexpression of CDCA7 promoted proliferation, colony formation, and cell cycle in ESCC cells. CDCA7 affected the expression of cyclins in different cell phases. GSE53625 and TCGA data showed CCNA2 expression was positively correlated with CDCA7. The knockdown of CCNA2 reversed the malignant phenotype induced by CDCA7 overexpression. Furthermore, CDCA7 was found to directly bind to CCNA2, thus promoting its expression.
Conclusions: Our results reveal a novel mechanism of CDCA7 that it may act as an oncogene by directly upregulating CCNA2 to facilitate tumor progression in ESCC.
Background
Esophageal cancer which accounts for 11% of diagnosed cancers was the fourth most common cancer type. In China, the dominant histologic type of esophageal cancer is esophageal squamous cell carcinoma (ESCC) which causes more than 175,000 deaths every year (1). The 5-year survival rate of ESCC which ranges from 22% to 30% still tends to be low because of the limitation of technical developments for early diagnosis and treatment (2). However, the advent and progression of next-generation sequencing (NGS) in recent years has given us some achievements on ESCC (3–7).
In our previous WGS analysis of 31 ESCC tumor tissues and matched adjacent non-tumor tissues, we identified some genes with copy number variation, including cell division cycle-associated 7 gene (CDCA7), that was amplified in 5 out of 31 ESCC patients (8). CDCA7 is located on 2q31.1. It is characterized as a c-Myc and E2F responsive gene that participates in neoplastic transformation (9, 10). It has been reported that the expression of CDCA7 is elevated in a high fraction of human lung, colon, ovary, rectum, stomach, and uterus cancer types, suggesting that CDCA7 may play a crucial role in cancer development (11–13). A recent study showed that the high expression of CDCA7 predicted poorer disease-free survival in patients with triple-negative breast cancer (TNBC) and was associated with metastatic relapse status (14). One research in lung adenocarcinoma reported that CDCA7 promoted lung adenocarcinoma proliferation through regulating the cell cycle, while its mechanism has not been completely elucidated yet (15). Meanwhile, CDCA7 as a DNA-binding protein can function as a transcription regulator to mediate the tumor-promoting effect (9).
CCNA2, which is synthesized at the beginning of S-phase (16, 17), binds and activates cyclin-dependent kinases (CDK) CDK2 and CDK1, the catalytic partners of CCNA2. The CDK2/CCNA2 complex is the machinery that drives the progression of S-phase. In the S-phase of the cell cycle, the CCNA2–CDK complex can phosphorylate key substances in the process of DNA replication, such as CDC6. This phosphorylation is crucially important for the initiation of DNA replication. It is possible that CCNA2–CDK contributes to tumorigenesis by the phosphorylation of oncoproteins and the increased expression level of CCNA2 accelerates cell proliferation once the tumor has formed (18–20). Increased expression of CCNA2 has been observed in various types of cancer such as lung, breast, liver, cervical, and others (18, 21–24). The expression level of CCNA2 is closely related with cell proliferation; thus, it is used as a proliferation marker for the molecular diagnosis of cancer (18). Meanwhile, the expression of CCNA2 appears to be of prognostic value for the prediction of survival and early relapse in many types of cancer (18, 25).
In our study, we analyzed the copy number amplification data from The Cancer Genome Atlas (TCGA) database in various types of tumors and the correlation between CDCA7 expression level and clinical variables in ESCC using the mRNA expression data from the GEO database. Furthermore, we verified that CDCA7 has as a tumor-promoting role in ESCC, and elaborated on its potential mechanisms of carcinogenesis. Our results show that CDCA7 may bind to CCNA2 to upregulate its expression. Therefore, increased CCNA2 promotes the proliferation of ESCC cells, thus promoting tumor growth. Our study provides useful clues for more effective therapeutic strategies against ESCC.
Methods
Clinical Samples
The copy number data were obtained from our study. The tumor and the matched adjacent non-tumor samples were recruited from Shanxi Cancer Hospital of Shanxi Medical University. The patients were without preoperative chemotherapy, radiotherapy, and other treatments before operation, and written consent was obtained from all of them. Hematoxylin and eosin (H&E) staining was used to diagnose these tissues, and the diagnosis was performed by at least two pathologists independently. The ESCC individuals were staged according to the American Joint Commission for Cancer (AJCC)/International Union Against Cancer (UICC) TNM staging system (eighth edition). The study was approved by the Institutional Reviewing Board (IRB) and the Research Committee of Shanxi Medical University.
Cell Lines and Cell Culture
ESCC cell lines KYSE150, KYSE180, KYSE450, and TE-1 and immortal embryonic esophageal epithelium cell lines NE3 and HET-1A used in the research were purchased from the Cell Bank of Type Culture Collection of the Chinese Academy of Sciences. The cell line 293T was from our lab. The cell lines KYSE150, KYSE180, KYSE450, and TE-1 were cultured in HyClone™ RPMI-1640 medium, and the cell lines HET-1A and 293T were cultured in HyClone™ DMEM/High Glucose medium (GE Healthcare Life Sciences, HyClone Laboratories, Logan, UT, USA). The culture was with 10% fetal bovine serum (FBS; Gibco; Thermo Fisher Scientific, Inc., Waltham, MA, USA). The cell line NE3 was cultured in a 1:1 mixture of EpiLife medium (Cascade Biologics, Inc., Portland, OR, USA) and defined keratinocyte serum-free medium (dKSFM; Gibco; Thermo Fisher Scientific, Inc., Waltham, MA, USA). All of the cell lines were cultured at 37°C, 5% CO2. The culture medium was replaced according to the cell state. Subculture was carried out when the cell fusion was about 80%–90%.
Overexpression and Knockdown of CDCA7 in ESCC Lines
SiRNAs or plasmids were transfected into the cells at the logarithmic growth phase using Lipofectamine 2000 reagent (Invitrogen, Carlsbad, CA) according to the instructions of the manufacturer. For knockdown of endogenous CDCA7, we used vectors containing the sequence 5′-GCCCTCAGAGAATTCTGTGACTGAT-3′ (CDCA7-si1) and 5′-CATCCGTGACCCTTCCGCATATAAT-3′ (CDCA7-si2). These shRNAs were cloned into the vector pHBLV-U6-Scramble-ZsGreen-Puro vector. For stable overexpression, the coding sequence (CDS) region of the CDCA7 gene was cloned into pHBLV-CMV-MCS-3FLAG-EF1-ZsGreen-T2A-PURO. The recombinant plasmids and the packaging plasmids (Hanbio Biotechnology Co., Ltd., Shanghai, China) were co-transfected into 293T cells. The lentivirus supernatant was used to infect the KYSE150, KYSE450, and KYSE180 cell lines. The negative control was the corresponding empty vectors. The KYSE150 and KYSE450 knockdown stable cell lines and the KYSE180 overexpression stable cell line were screened out for 7–14 days with 1.0, 1.0, and 0.8 µg/ml puromycin (Invitrogen; Thermo Fisher Scientific, Inc.), respectively. The efficiency of knockdown and overexpression was determined by real-time PCR and Western blot assay. We used small interference RNA (siRNA) for CCNA2 knockdown, and the siRNA sequence information is as follows: CCNA2-si1, 5′-CTATGGACATGTCAATTGT-3′; CCNA2-si2, 5′-GAGTGTTAATGAAGTACCA-3′. The CDS of CDCA7 and CCNA2 genes was cloned into the pcDNA3.1 vector with a V5 tag and 6*His tag.
MTT Assay
The MTT assay was performed using a 96-well plate with 5 × 103 transfected cells each well and cultured for 24–120 h. A 20-µl MTT solution (5 mg/ml) was added to a 200-µl culture medium each well for 4 h at 37°C. The MTT formazan crystals that remained after removing the medium were then solubilized in dimethyl sulfoxide (DMSO) for 15–20 min. The absorbance was measured by a spectrophotometer at 490 nm to show the relative number of surviving cells in each well indirectly.
Colony Formation Assay
A total of 1,000 cells/well were seeded into six-well plates and incubated at 37°C and 5% CO2 for 10–15 days. Polyformaldehyde (4%) was used to fix these cells and 1% crystal violet was used to stain these cells subsequently. The numbers of colonies containing more than 10 cells were counted.
Flow Cytometry Analysis
Cells collected were fixed with 70% alcohol and stored overnight at −20°C. Propidium iodide (PI) was used to stain the collected cells according to the instructions of the manufacturer. The stained cells were analyzed using a flow cytometer (BD Company, USA).
Immunofluorescence
KYSE150, KYSE180, and KYSE450 cells were transfected with CDCA7-V5 plasmid and empty vector, respectively. Formaldehyde (4%) was used to fix the cells for 10 min. BSA (1%) was used to incubated the cells for 1 h to block non-specific protein–protein interactions after permeabilized by 0.1% Triton X-100. The cells were incubated with the primary antibody rabbit anti-V5 (Abcam, Cambridge, UK, 2 µg/ml) overnight at 4°C. Alexa Fluor® 594 goat anti-rabbit IgG antibody (Thermo Fisher, Carlsbad, USA, 1:1,000) was used for 30 min at room temperature after washing four times in PBS. DAPI at a concentration of 0.5 µg/ml was used to stain the cell nuclei.
Chromatin Immunoprecipitation Sequencing Assay
KYSE150 cells were transfected with the V5-tagged CDCA7 plasmid for the chromatin immunoprecipitation (ChIP) assay. The assay was performed according to the instructions of the manufacturer (Millipore, Burlington, MA, USA). The DNA fragments were enriched by anti-V5 antibody (Abcam, Cambridge, UK), and the isotype IgG (Abcam, Cambridge, UK) was used as a negative control. CHIP-seq was performed by Novogene (Beijing, China). Screening and quality control of the CHIP-seq were based on standard protocol. The sequences of primers used for the amplification of CCNA2 genome regions containing a putative CDCA7 binding site are listed in Table S1.
Western Blot
The cells were lysed for 1 h with RIPA buffer containing protease and phosphatase inhibitors (Thermo Fisher Scientific) on ice. The components of RIPA buffer are as follows: 1% Triton X-100, 50 mM Tris–HCl, pH 7.6, 150 mM NaCl, 1% sodium deoxycholate, and 0.1% SDS. The lysates were centrifuged at 12,000g at 4°C for 30 min, and the total protein concentrations of supernatant were determined by the Bradford method. Fifty micrograms of protein was separated by 10% SDS-PAGE and then transferred onto polyvinylidene fluoride (PVDF) membranes (Millipore, USA). The membrane was incubated with special antibodies, including CDCA7, CCND1, CCNA2, CCNE1, and GAPDH, at 4°C overnight. The IRDye 800CW secondary antibody (Abcam, Cambridge, UK) was used to detect the blot. A relative amount of protein was normalized to GAPDH level. The antibodies used in this experiment are shown as follows: CDCA7 (Sigma, USA), CCND1 (Proteintech, Rosemont, IL, USA), CCNA2 (Proteintech, Rosemont, IL, USA), CCNE1 (Proteintech, Rosemont, IL, USA), and GAPDH (Proteintech, Rosemont, IL, USA).
RNA Extraction and Real-Time PCR
Total RNA of ESCC cells was purified using RNAiso plus (Takara, Dalian, China). Two micrograms of total RNA was used for complementary DNA (cDNA) synthesis using a PrimeScript® RT reagent kit with gDNA Eraser (Takara). TB Green® Premix Ex Taq® II kit (Takara) was used in real-time PCR according to the instruction of the manufacturer. All real-time PCR reactions were performed in triplicate with an Applied Biosystems Step One Plus (ABI, Foster City, CA, USA). The relative expression levels of the target genes were normalized to endogenous GAPDH. Quantification of the expression levels of target genes was calculated using the 2−ΔΔCt formula. The primers synthesized by Thermo Fisher are listed in Table S2.
Dual-Luciferase Reporter Assay
According to the results of ChIP-seq, we cloned the CDCA7 DNA-binding fragment with CCNA2 from the KYSE150 cell line genomic DNA. The cloned DNA fragment was constructed into the reporter plasmid of pGL3-promoter (Promega, Madison, WI, USA). Then, we divided this DNA fragment into four segments and constructed them into the pGL3-promoter vector. The different DNA fragments were cloned using the primers listed in Table S3. Six motif sequences obtained from ChIP-seq were also constructed into pGL3-promoter vector. Cells (3 × 104) were cultured in triplicate in 24-well plates for 12–24 h. Then, the pGL3 reconstruction reporter plasmids were transiently co-transfected with the pRL-TK plasmid into KYSE150 and KYSE150 knockdown cells using Lipofectamine 2000 reagent (Invitrogen, Carlsbad, CA). After transfection for 48 h, luciferase and Renilla signals were measured according to the instruction of Dual-Luciferase Reporter Assay Kit (Promega, Madison, USA).
Mouse Xenograft Assay
The effects of CDCA7 on tumorigenesis and growth in vivo were detected via mouse xenograft assay. We used 20 5- to 6-week-old female NU-Foxn1nu nude mice (Vital River Laboratory Animal Technology Co., Ltd., Beijing, China) for the mouse xenograft assay. A total of 3 × 106 KYSE150-NC cells or CDCA7-knockdown stable KYSE150 cells were used to inject into the right or left oxter of female NU-Foxn1nu nude mice, respectively. Tumor size and weight were determined with calipers and balance twice a week. The mice were executed and the tumors were removed after 28 days. The formula V = (W2 × L)/2 was used to calculate the tumor volume. V is the tumor volume, W is the tumor width, and L is the tumor length. Tumor size was presented as mean ± standard deviation (SD).
Immunohistochemistry
The isolated xenograft tumor tissues were fixed using formalin and embedded by paraffin for immunohistochemical staining. Briefly, xylene and a series of grades of alcohol were used to deparaffinize and rehydrate these sections, and the sections were then soaked with 3% H2O2 15 min. Sodium citrate buffer (pH 6.0) or Tris-EDTA buffer (pH 9.0) were used for antigen retrieval for 4 or 3 min in a pressure cooker, followed by incubation with primary antibody at 4°C overnight. The slides were incubated with second antibody at 37°C for 20 min after washing with PBS and then stained with DAB and counterstained with hematoxylin. The expression of CDCA7, Ki-67, and CCNA2 was quantitatively analyzed with Aperio Cytoplasma 2.0 software by immunohistochemistry. The antibodies used in this experiment are shown as follows: CDCA7 (Sigma, USA, 1:200 dilution), Ki-67 (Abcam, Cambridge, UK), and CCNA2 (Proteintech, Rosemont, IL, USA, 1:2,000 dilution).
Bioinformatics and Data Analysis
The mRNA expression data and the clinical information of 179 ESCC tissues and paired non-tumor tissues by microarray analysis were from a previous study by Li et al. (26) and downloaded from the GEO database (GSE53625). The copy number data were obtained from our other study.
The copy number data of CDCA7 in varied cancer types, including ESCC, ECA, lung squamous cell carcinoma (LUSC), head and neck squamous cell carcinoma (HNSC) collected from the TCGA database, were downloaded via cBioPortal for Cancer Genomics (https://www.cbioportal.org/) (27, 28). The expression data of CDCA7 and CCNA2 in different cancer types such as ESCC, LUSC, HNSC were downloaded from TCGA via Xena Browser (https://xenabrowser.net/heatmap/).
Statistical Analyses
Each of the experiment in the study was performed in triplicate, and data were presented as the mean ± SEM. Statistical Package for Social Science for Windows (SPSS, version 20.0; IBM Inc., USA) was used to analyze the experimental data. The means of two groups and more than two groups were compared using Student’s t-test and one-way ANOVA, respectively. P-value of <0.05 was considered to be statistically significant. GraphPad Prism software was used to analyze the correlations between CDCA7 and CCNA2 using non-parametric correlation (Spearman).
Results
CDCA7 Was Frequently Amplified in ESCC
In our previous study, CDCA7 was identified as one of the copy number amplification genes in ESCC (8). Here, we analyzed the copy number amplification data from TCGA through cBioPortal and found that the copy number amplification of CDCA7 existed in various kinds of tumor. Its alteration frequency was much higher in ESCC than in other tumors (Figure 1A). Furthermore, we analyzed the mRNA expression data of 179 pairs of ESCC tumors and adjacent normal tissues via microarray analysis. The mRNA expression data and the clinical information of 179 ESCC tissues and paired non-tumor tissues by microarray analysis were from a previous study by Li et al. and downloaded the from the GEO database (GSE53625) (26). We observed that CDCA7 showed statistically higher expression levels in most of the individuals compared with that of normal tissues (Figure 1B). After analyzing the copy number amplification and expression of CDCA7 in 95 ESCC patients in the TCGA database, we found that there was a correlation between the expression of CDCA7 and the copy number amplification, indicating that the CDCA7 copy number amplification may cause to increase its expression (Figure 1C).
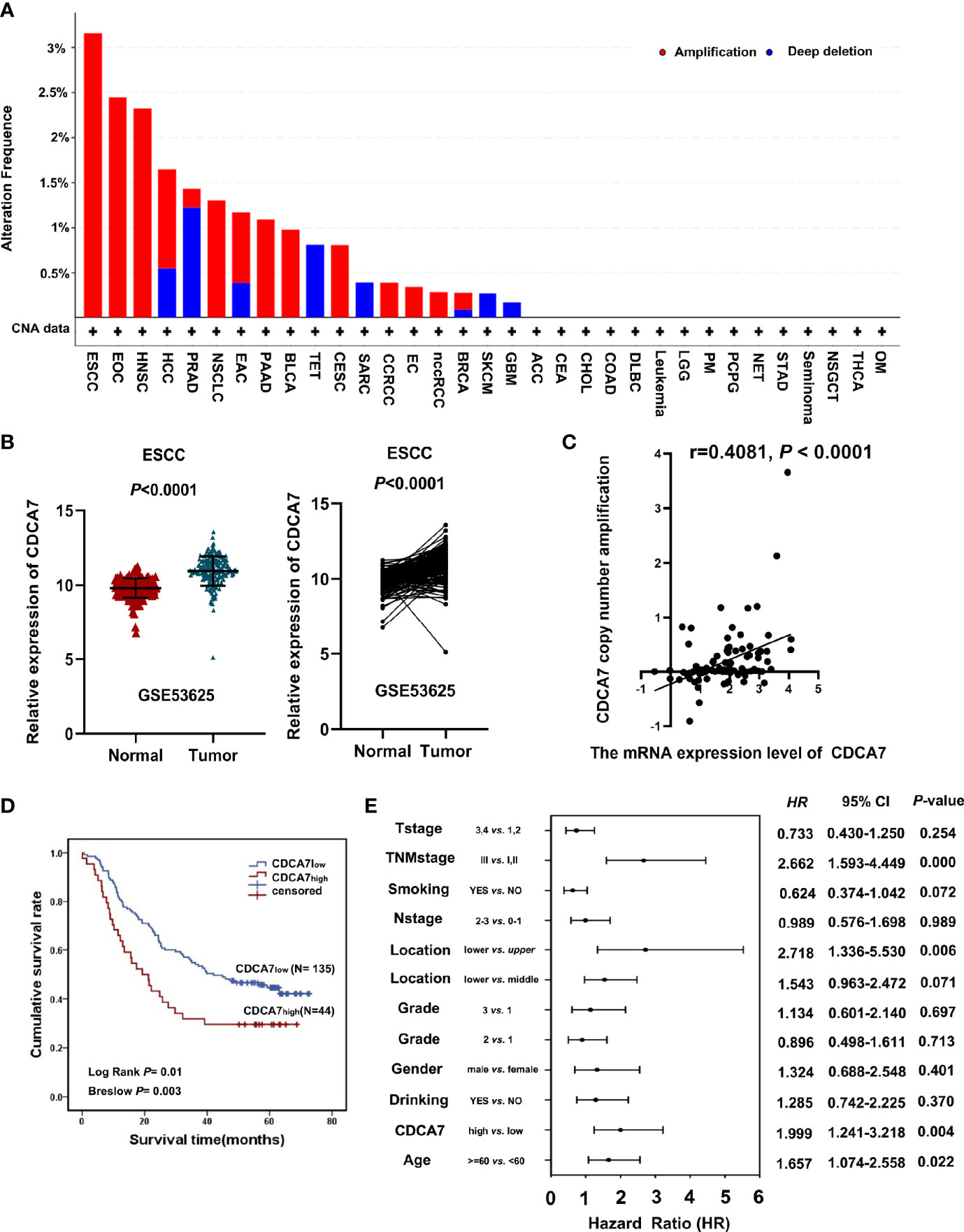
Figure 1 CDCA7 expression predicts the prognosis of esophageal squamous cell carcinoma (ESCC) patients. (A) The CDCA7 copy number amplification of various kinds of tumor in the TCGA database. (B) ESCC tumor tissues had a high CDCA7 expression compared with its non-tumor tissues using non-paired t-test and paired t-test; P < 0.001. (C) The correlation analysis between CDCA7 copy number amplification and expression (r = 0.4081, P < 0.0001). (D) The patients with CDCA7low had better survival than those with CDCA7high (log rank P = 0.01) using Kaplan–Meier survival analysis. (E) Multivariate analysis showed that TNM stage [hazard ratio (HR) = 2.662, 95% CI: 1.593–4.449, P < 0.001), location (lower vs. upper) (HR = 2.718, 95% CI: 1.336–5.530, P = 0.006), age (HR = 1.657, 95% CI: 1.074–2.558, P = 0.022), and CDCA7 expression were independent predictive factors for overall survival (HR = 1.999, 95% CI: 1.241–3.218, P = 0.004).
The cohort of 179 patients was divided into two groups according to the expression level of CDCA7. The top 25% of patients were defined as the patients with a higher level (named as CDCA7high) and the remaining 75% were defined as the patients with a lower level (named as CDCA7low) according to the expression level of CDCA7 from high to low. Then, we analyzed the correlation between the expression of CDCA7 and the clinical variables in ESCC. The results in Table 1 show that the expression of CDCA7 was related to the grade of ESCC patients (P = 0.0083). The patients with CDCA7high had a poor grade compared with the CDCA7low patients. Furthermore, the patients with CDCA7high had a worse survival than those with CDCA7low (log rank P = 0.01, Figure 1D) using Kaplan–Meier survival analysis. The multivariate analysis showed that TNM stage [hazard ratio (HR) = 2.662, 95% CI: 1.593–4.449, P < 0.001], location (lower vs. upper) (HR = 2.718, 95% CI: 1.336–5.530, P = 0.006), age (HR = 1.657, 95% CI: 1.074–2.558, P = 0.022), and CDCA7 expression (HR = 1.999, 95% CI: 1.241–3.218, P = 0.004) were independent predictive factors for overall survival (Figure 1E). Furthermore, CDCA7 was related with the survival status in patients in the male group (P = 0.001), age <60 group (P = 0.02), drinking group (P < 0.001), smoking group (P = 0.014), T1+T2 group (P = 0.019), N0+N1 group (P = 0.026), and TNM stage = III group (P = 0.025) (Figures S1, S2). Hence, we speculate the copy number amplification and high expression level of CDCA7 may promote the occurrence and development of ESCC.
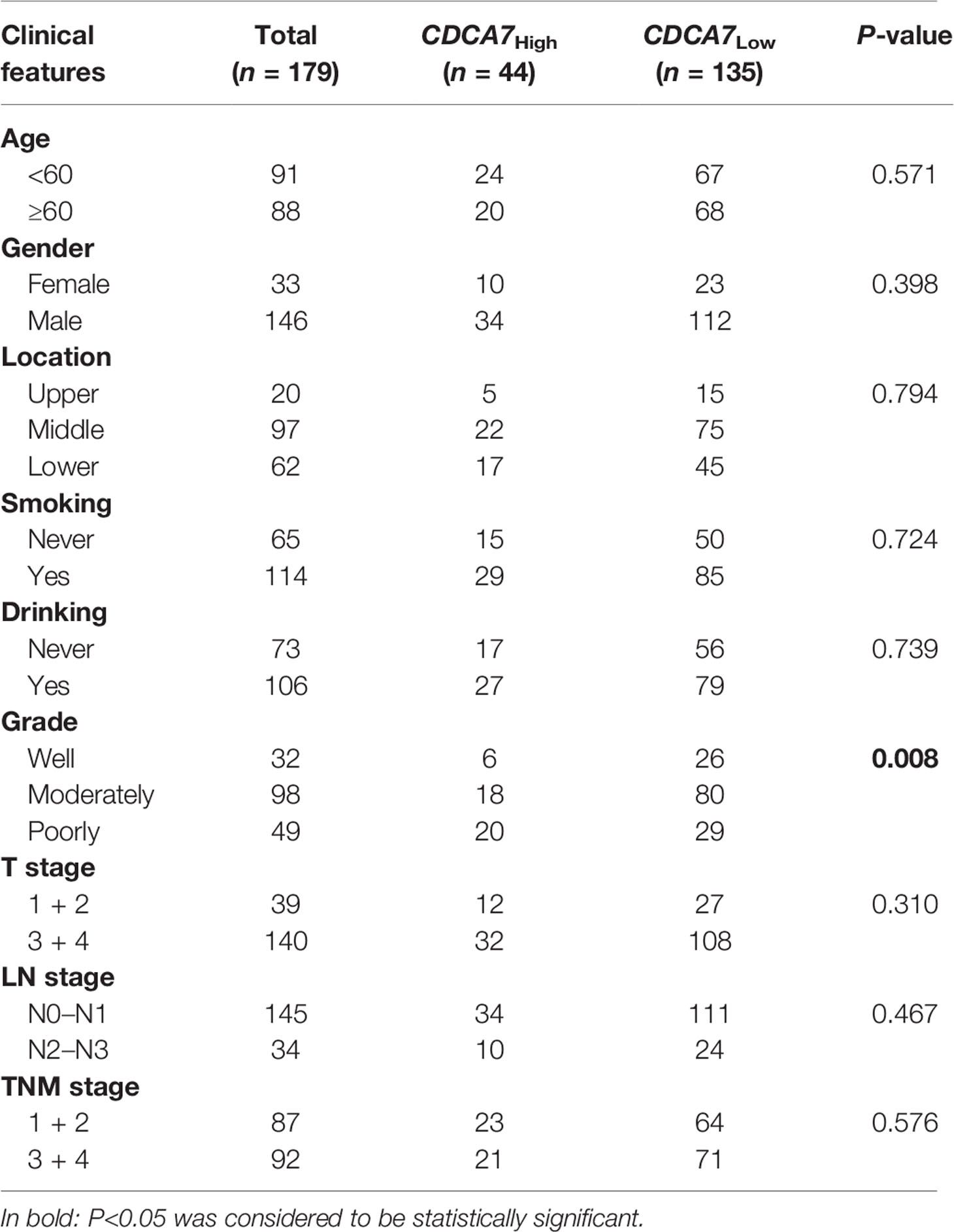
Table 1 Correlation analysis between CDCA7 copy number in ESCC and clinicopathological variables.
CDCA7 Promotes Cell Proliferation, Colony Formation, and Cell Cycle of ESCC Cells
To verify the biological roles of CDCA7 in ESCC, we first analyzed the mRNA and protein expression levels in immortal embryonic esophageal epithelium cell lines NE3 and HET-1A and ESCC cell lines including KYSE150, KYSE180, KYSE450, and TE-1 via quantitative real-time PCR (q-RTPCR) and Western blot (Figure S3). In all these cell lines, we selected KYSE150 and KESE450 as relatively high endogenous CDCA7 level cell lines for knockdown experiments. KYSE180 was selected as low endogenous CDCA7 level cell line for overexpression. The efficiency of overexpression and knockdown were verified by Western blot, respectively (Figures 2A, B). Then, we detected the changes in cell phenotypes, including proliferation, colony formation, and cell cycle. The results showed that CDCA7 silencing significantly inhibited the ability of cell proliferation and colony formation in KYSE150 and KESE450 (Figures 2C, E), while overexpression of CDCA7 increased the ability of cell proliferation and colony formation markedly (Figures 2D, F). Meanwhile, the results of flow cytometry indicated that CDCA7 overexpression decreased the proportion of G1-phase cells and increased the proportion of G2 + M-phase cells (Figure 2H). On the contrary, CDCA7 silencing significantly increased the proportion of G1-phase cells and decreased the proportion of G2 + M-phase cells (Figure 2G).
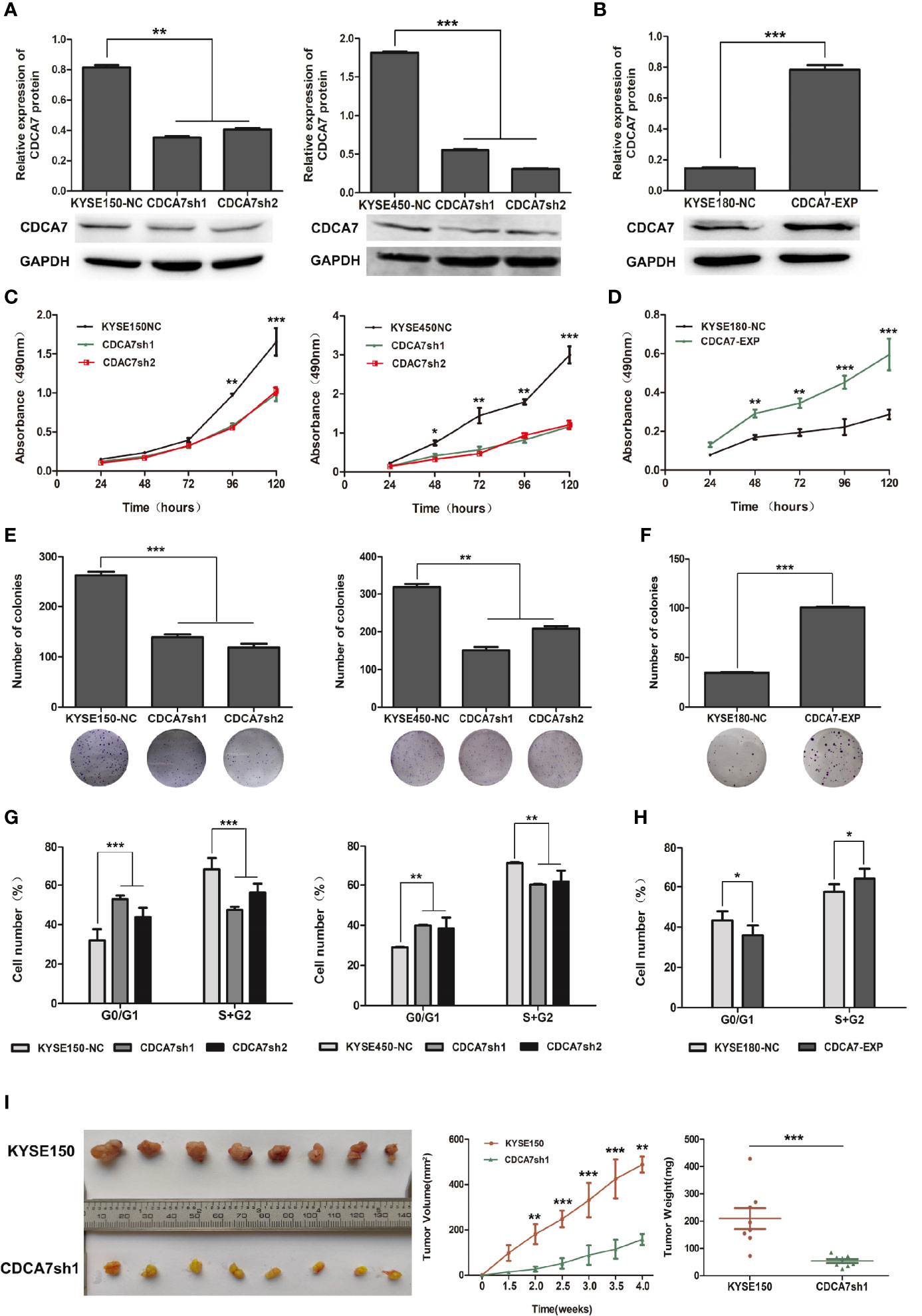
Figure 2 The effect of CDCA7 gene in ESCC cell lines. (A) The CDCA7 knockdown efficiency in KYSE150 and KYSE450 cells. (B) The CDCA7 overexpression efficiency in KYSE180 cells. (C) CDCA7 knockdown inhibited the proliferation of ESCC Cells. (D) CDCA7 overexpression promoted the proliferation of ESCC cells. (E) CDCA7 knockdown inhibited the ability of colony formation in ESCC cells. (F) CDCA7 overexpression promoted the ability of colony formation in ESCC cells. (G) CDCA7 knockdown inhibited the cell cycle of ESCC cells. (H) CDCA7 overexpression promoted the cell cycle of ESCC cells. (I) Tumor growth was inhibited significantly in the CDCA7-knockdown group compared with the control group in vivo. Left: tumor tissues in the CDCA7-knockdown group and the control group; right: tumor weight and tumor growth curve. (0.01 < P ≤ 0.05, *; 0.001 < P ≤ 0.01, **; P ≤ 0.001,***).
To confirm the oncogenic role of CDCA7 in vivo, we established a subcutaneous transplantation tumor model in female NU-Foxn1nu nude mice using stable CDCA7-knockdown KYSE150 and KYSE150 cells. Four weeks later, tumors were stripped after the mice were sacrificed. The tumor growth rate of the KYSE150 group was significantly faster than that of the CDCA7-knockdown group (Figure 2I). The results found that the mean tumor volume of the CDCA7-knockdown group and the control group was 158.74 ± 24.83 and 488.41 ± 35.84 mm3, respectively (t-test, P < 0.001, Figure 2I). The mean tumor weight of the control group and the CDCA7-knockdown group was 209.61 ± 108.84 and 54.23 ± 19.39 mg, respectively (t-test, P < 0.001, Figure 2I).
Cyclins Were Identified as CDCA7 Targets by ChIP-Sequencing
Immunofluorescence assay was performed in KYSE150, KYSE450, and KYSE180 cells to affirm whether CDCA7 expresses in the nucleus as CDCA7 was found to be a DNA-binding protein and can serve as a transcription regulator (9, 11, 14). The results showed that CDCA7 was located in both the cytoplasm and the nucleus (Figure S4).
Since CDCA7 may act as a transcription regulator, chromatin immunoprecipitation sequencing (ChIP-seq) technology was applied to screen a wide range of DNA fragments interacting with CDCA7. Genome-wide mapping of CDCA7-binding profile by ChIP-seq identified 14,930 binding events (P < 10−3), associated with 11,897 unique genes following a nearest gene annotation. As shown in Figure 3A, most (12,462/14,930) of the binding events occur at a distance about 2,000 bp from the transcriptional start site (TSS) of genes, which is generally considered to be the gene promoter region and activation region. These results suggested that CDCA7 may play a role as a transcription factor or transcription regulatory factor. Next, we performed a KEGG pathway enrichment analysis on the 11,897 unique genes which were associated with the DNA fragments obtained from ChIP-seq. The KEGG pathway enrichment analysis showed that target genes were enriched in the pathways including pathways in cancer, cell cycle pathway, PI3K–Akt signaling pathway, MAPK signaling pathway, Ras signaling pathways, and Hippo signaling pathways, which may contribute to ESCC cell proliferation and tumorigenesis (Figure 3B). Among the DNA fragments obtained from ChIP-seq, some of them were located in the promoter region of the cell cycle related genes, i.e., 3,000 bp before the transcription initiation site. These genes include CCND1, CCNE1, CCNA2, etcetera (Figure 3C). Bdg files, such as the CDCA7_V5.bdg and Control.bdg shown in Figure 3C, are the corresponding bedgraph format track files provided by the company, which are convenient to view the position distribution of reads on the genome under different resolution conditions. CDCA7_V5.bdg represents the DNA fragments that can bind to CDCA7 detected by the anti-V5 antibody, and Control.bdg is the DNA fragments of the input group. When we opened the bdg files in the UCSC database and compared CDCA7_V5.bdg with Control.bdg, the location of the peak is the binding site of CDCA7 with the three cyclins. As shown in Figure 3C, CDCA7 binds with CCND1 at the position of −3,732 to −2,502 bp, binds with CCNA2 at the position of −74 to 734 bp, and binds with CCNE1 at the positon of −53 to 925 bp from each transcription start site, respectively. At the same time, when the binding of H3K4Me1 and H3K21Ac was displayed on the genome in the UCSC database, we found that apart from CCNE1, CDCA7, and H3K4Me1, H3K27Ac shared the same binding position in CCND1 and CCNA2 genomes. These findings once again suggested that CDCA7 may play a role as a transcription factor or transcription regulator to regulate the expression of cyclins.
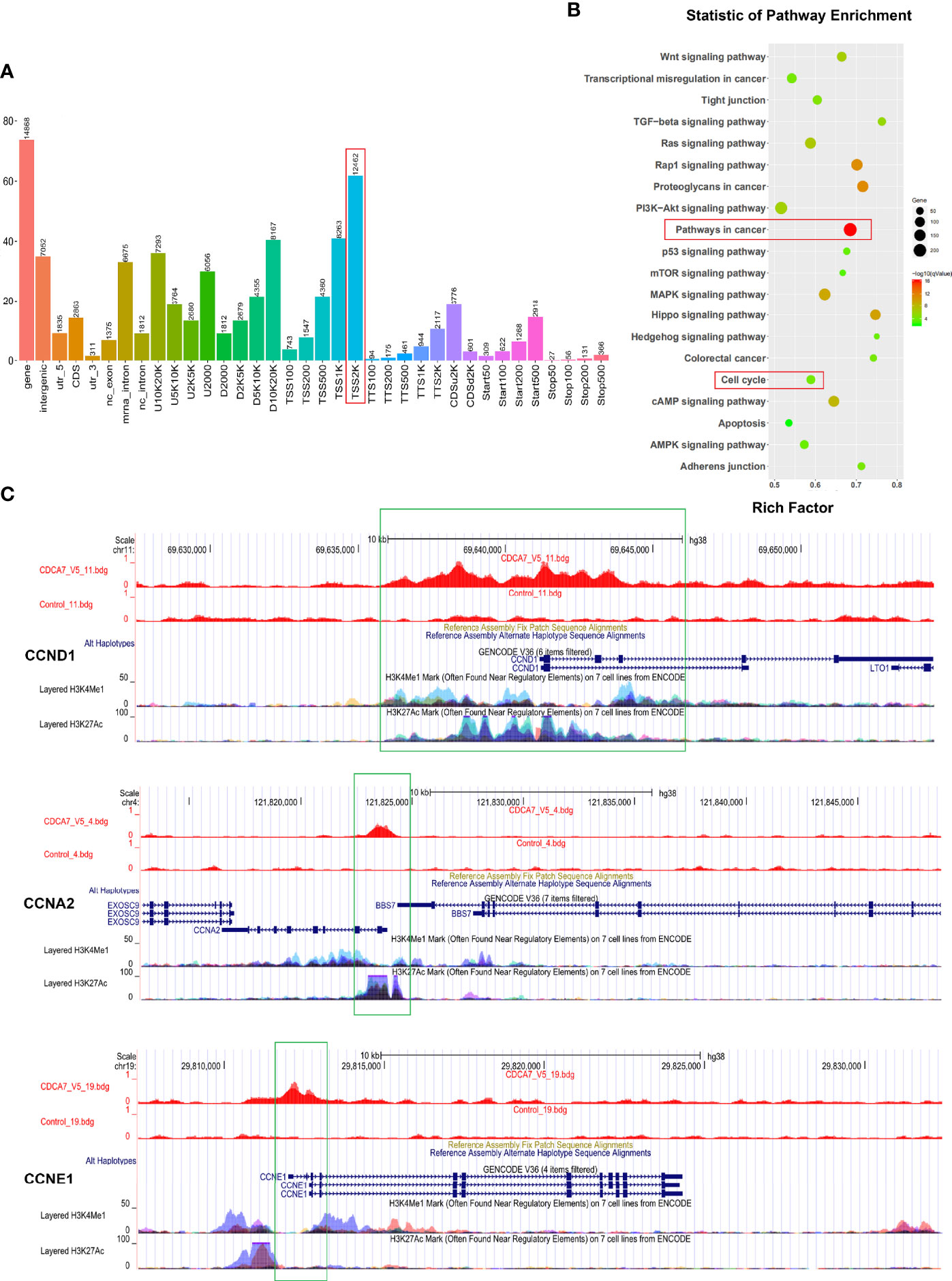
Figure 3 Cyclins may be the downstream genes regulated by CDCA7 as a transcription regulator. (A) Most of the binding sites of CDCA7 with DNA located at the position of 2,000 bp before the transcription initiation site of the genes. (B) The pathway enrichment analysis of the DNA fragments obtained from ChIP-sequencing. (C) CDCA7 binds with CCND1 at the position of −3,732 to −2,502 bp, binds with CCNA2 at the position of −74 to 734 bp, and binds with CCNE1 at the positon of −53 to 925 bp from each transcription start site, respectively.
CCNA2 May Be the Downstream Target Gene of CDCA7
Since the results of ChIP-seq showed that CDCA7 may act as a transcription regulator to regulate the expression of cyclins, we detected the mRNA and protein levels of the three cyclins in CDCA7 overexpression and knockdown stable cell lines. We found that the expression level of the CCNA2 was significantly decreased when CDCA7 was knocked down and vice versa (Figures 4A, B). Meanwhile, we verified the correlation between CDCA7 and the three cyclins using the mRNA data of 96 ESCC tissues in the TCGA database. The results (Figure 4C) showed that there was a weak positive correlation between CDCA7 and CCND1 (r = 0.2121, P = 0.038) and a strong positive correlation between CDCA7 and CCNA2 (r = 0.6527, P < 0.0001), while there was no correlation between CDCA7 and CCNE1 (r = −0.0528, P = 0.6116). Next, we analyzed the mRNA expression data of ESCC (n = 358) in GSE53625. Based on the mRNA expression data of ESCC (n = 358), CDCA7 was positively correlated with CCNA2 (r = 0.7047, P < 0.0001). Interestingly, when the mRNA expression data of other types of SCC in the TCGA database were analyzed, we found that CDCA7 was positively correlated with CCNA2 in LUSC (n = 501, r = 0.4995, P < 0.0001) and HNSC (n = 502, r = 0.5771, P < 0.0001) (Figure 4D). Immunohistochemistry was further used to detect the tumor tissue stripped from nude mice with anti-CDCA7, anti-CCNA2, and anti-Ki-67 antibodies. The results showed that the staining intensity of CCNA2 and Ki-67 in the CDCA7 knockdown group was obviously weaker than that of the KYSE150 group (Figure 4E). The H-score of CCNA2 and Ki-67 in CDCA7-knockdown group (181.344 ± 17.549 and 7.84 ± 0.200) was significantly lower than that in the control group (91.19 ± 9.07 and 58.59 ± 0.626) (t-test, P < 0.001, Figure 4E).
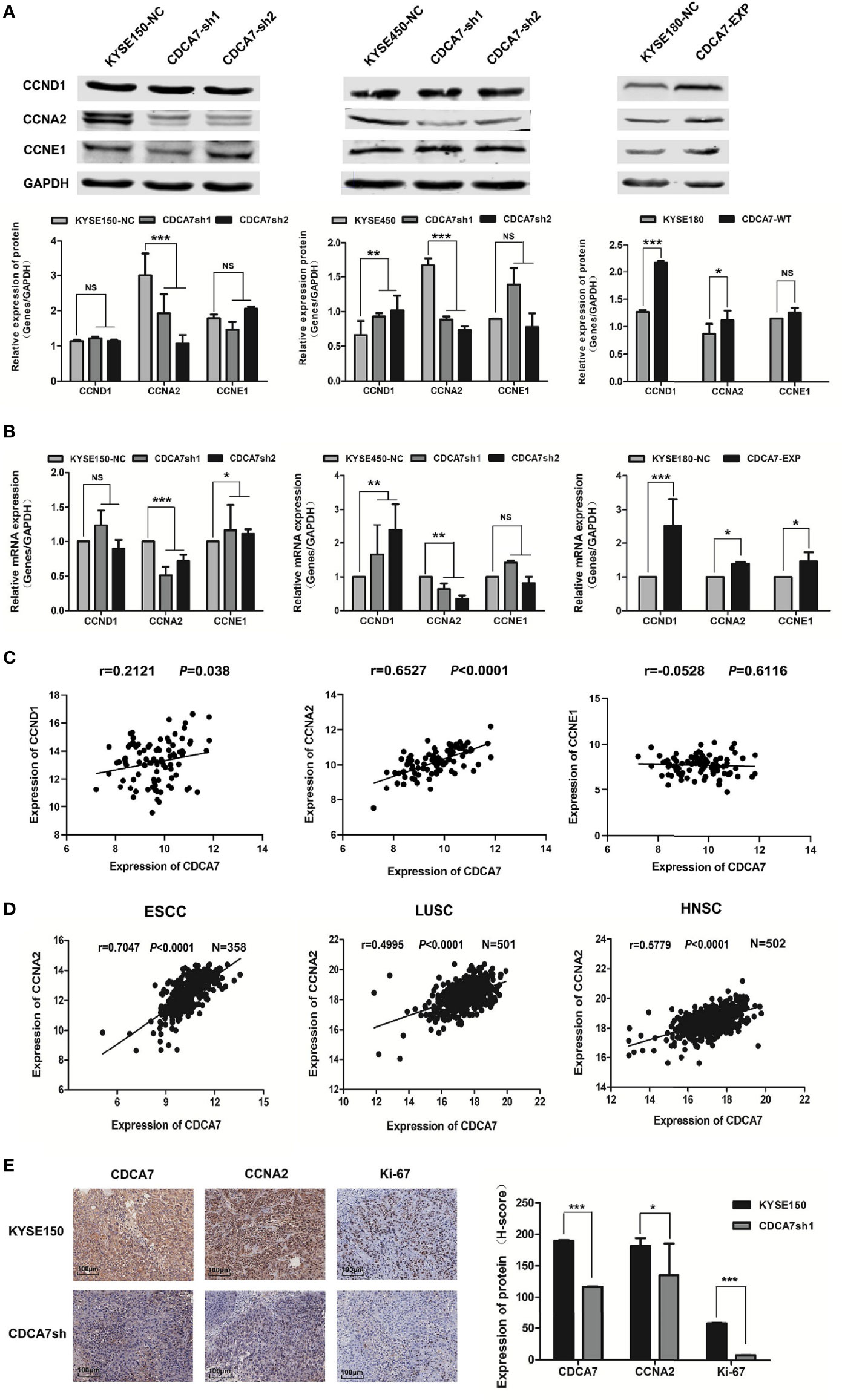
Figure 4 CCNA2 may be the downstream target gene of CDCA7. (A) The protein expression levels of CCND1, CCNA2, and CCNE1 in CDCA7 knockdown cell lines and CDCA7 overexpression cell lines. (B) The mRNA expression levels of CCND1, CCNA2, and CCNE1 in CDCA7 knockdown cell lines and CDCA7 overexpression cell lines. (C) CDCA7 was positively correlated with CCND1 expression (r = 0.2121, P = 0.038) and CCNA2 expression (r = 0.6527, P < 0.0001), while it was not correlated with CCNE1 expression (r = −0.0528, P = 0.6116). (D) The correlation of CDCA7 and CCNA2 expression in ESCC, LUSC, and HNSC; correlation coefficient (r) and P-values were shown in the figures. P < 0.05 was considered statistically significant. (E) IHC assay showed CDCA7, CCNA2, and Ki-67 expression in CDCA7 knockdown xenograft tumor tissue and the control group tissue. Scale bar = 100 μm (P > 0.05, NS; 0.01 < P ≤ 0.05, *; 0.001 < P ≤ 0.01,**; P ≤ 0.001,***).
These results revealed that the CDCA7 probably affected the cell cycle progression, occurrence, and development of cancers through regulating the expression of CCNA2.
CDCA7 Regulates CCNA2 Expression Through Binding to the Target Regions of CCNA2
The binding region of CDCA7 on CCNA2 started from position −74 to 734 bp relative to the TSS, and we constructed the −90 to 809 bp into the pGL3 promoter vector for the dual-luciferase assay subsequently. The reason why we expanded the region of the DNA fragment is the high GC content of the DNA sequence near 734 bp and there is no way to design a suitable pair of PCR primers. To explore the binding domain of CDCA7, the interval from −83 to 809 bp was divided into four segments randomly. The four segments were −90 to 130, 113–292, 275–476, and 456–809 bp. Dual-luciferase assay indicated that the 456–809-bp region of the CCNA2 was the core element regulated by CDCA7 (Figure 5A). To understand the molecular mechanism for the activity of CDCA7 in regulating gene transcription, a de novo search for DNA-enriched motifs was performed within the binding fragments and five predicted motifs were obtained. Each of the motifs corresponded to some transcription factor at different degrees (Figure 5B). To demonstrate whether CDCA7 acts its role as a transcription factor or a transcription regulator factor by binding with DNA through these motifs, we constructed the sequences of the motifs into pGL3-promoter vectors for dual-luciferase reporting experiments, the results revealed that motif-1 (5′-TAGACAAGAGTT-3′), motif-2 (5′-GTGATCAGTGCAGA-3′), motif-3 (5′-CTGGAACAGCAC-3′), motif-4 (5′-GTGTGTGTGTGT-3′), and motif-5 (5′-AGTAGTAGTA-3′) might be the functional binding sites of CDCA7 with DNA (Figure 5D). Next, we compared these motifs with the sequences from −74 to 734 bp and found five DNA fragments highly similar to the five motifs located in the 456–809-bp region (Figure 5C). We inferred that the five DNA segments may be the functional binding sites of CDCA7 with CCNA2. In order to further research whether CDCA7 directly binds to CCNA2 through these sites, ChIP-PCR was performed. We found that CDCA7 bound to the DNA fragment from 484 to 495, 641 to 654, 670 to 679, and 711 to 722 bp in the CCNA2 genomic region (Figure 5E). To identify whether CDCA7 regulates CCNA2 through these binding sites, we constructed a dual-luciferase reporter plasmid with the segments of CCNA2 from −90 and 809 bp which deleted the 484–495, 641–654, 670–679, and 711–722 bp. The results showed that CDCA7 could not activate the expression of downstream reporter gene when the four binding sites were knocked out (Figure 5F). It proved again that CDCA7 may regulate the transcription and expression of CCNA2 by binding with these four binding sites.
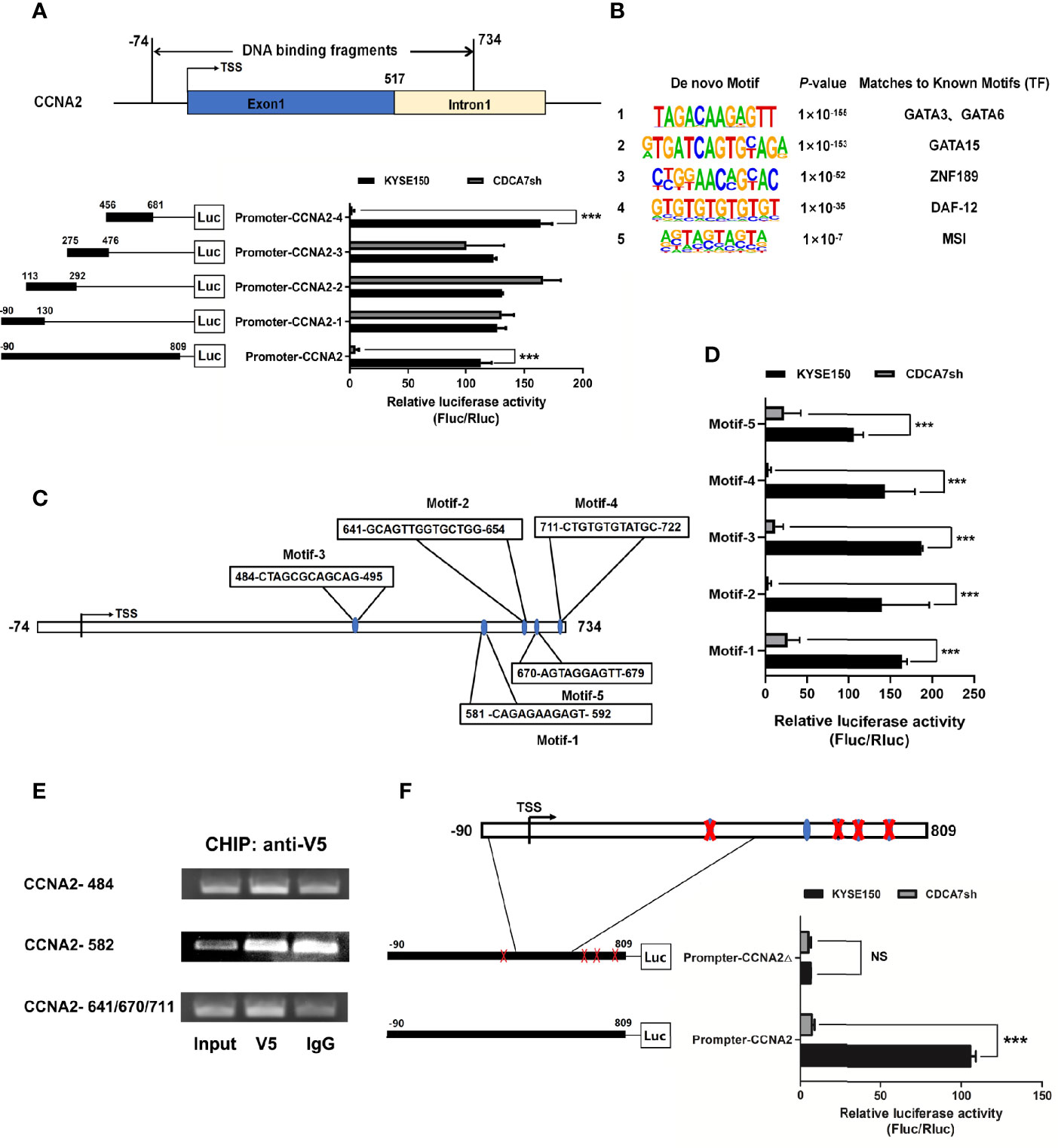
Figure 5 CDCA7 directly binds to CCNA2 and increases its transcription activity in ESCC cells. (A) CDCA7 binds with CCNA2 from −74 to 734 bp in the ChIP-seq and luciferase reporter assays showed that 456 to 809 bp of CCNA2 were the core regions regulated by CDCA7. (B) Five predicted motifs were analyzed from the DNA-binding fragments obtained from the ChIP-seq. (C) Five DNA fragments in the 456–809-bp region were highly similar to motif-1, motif-2, motif-3, motif-4, and motif-5. (D) Luciferase reporter assays showed that CDCA7 may regulate the transcription of the target genes through the five motifs. (E) ChIP-PCR showed that CDCA7 binds to the CCNA2 at the position of 484–495, 641–654, 670–679, and 711–722 bp in ESCC cells. (F) Luciferase reporter assays showed that CDCA7 could not activate the expression of downstream reporter gene when the four binding sites of 484–495, 641–654, 670–679, and 711–722 bp were knocked out. (P > 0.05, NS; P ≤ 0.001,***).
CDCA7 Promotes Cell Cycle Through Regulating CCNA2
To confirm whether CDCA7 promotes cell cycle through CCNA2, we carried out the interference and rescue experiment of CCNA2. The results showed that forced overexpression of CCNA2 in CDCA7 knockdown ESCC cells (Figure 6A) was performed, and a series of phenotype changes had been identified in ESCC cells. The results showed that CCNA2 overexpression promoted the proliferation and colony formation (Figure 6B) induced by CDCA7 knockdown in KYSE450 cells. Meanwhile, when we silenced its expression in CDCA7 overexpression ESCC cells (Figure 6C), the results of cell phenotype experiments showed that knockdown of CCNA2 can reduce cell proliferation and colony formation ability (Figure 6D) induced by CDCA7 overexpression in KYSE180 cells. These results suggested that the acceleration effect of CDCA7 on cell cycle may depend on its transcription regulation of CCNA2, and CCNA2 inhibition may partially reverse the cell proliferation progression induced by CDCA7 overexpression.
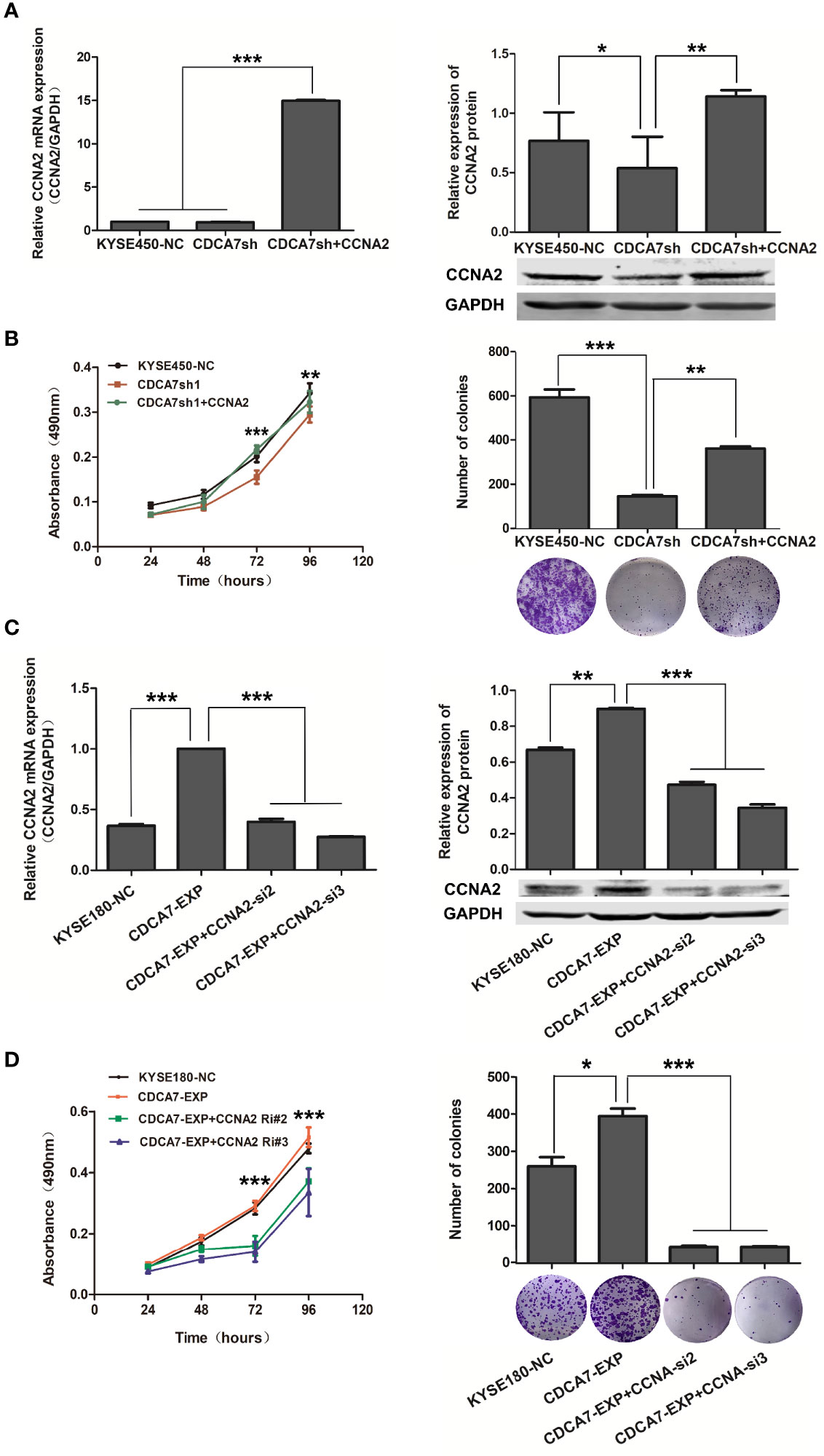
Figure 6 CDCA7 promotes S-phases via transcriptionally regulating CCNA2 expression. (A) CCNA2 overexpression in KYSE450 cell line with CDCA7 knockdown; the mRNA and protein expression levels of CCNA2 and GAPDH were detected by qRT-PCR and Western blot. GAPDH was used as a loading control. (B) CCNA2 overexpression promoted the proliferation induced by CDCA7 knockdown. (C) CCNA2 knockdown in KYSE180 cell line with CDCA7 overexpression; the mRNA and protein expression levels of CCNA2 and GAPDH were detected by Western blot. GAPDH was used as a loading control. (D) CCNA2 knockdown inhibited the proliferation induced by CDCA7 overexpression. (0.01 < P ≤ 0.05, *;0.001 < P ≤ 0.01,**,P ≤ 0.001,***).
The results indicated that CDCA7 gene might act as a tumor promoter in ESCC and its copy number amplification or increased expression may accelerate the cell cycle process and promote cell proliferation by binding to the genome of CCNA2 functional domain and increasing its expression in ESCC. When CDCA7 is knocked down or decreased, its transcription regulation effect is attenuated, and the cell cycle process and the cell proliferation of ESCC are inhibited as the expression of CCNA2 is depressed. Furthermore, the mechanism that CDCA7 acts as an oncogene possibly through regulation of cell proliferation might be applied in various types of SCC.
Discussion
Previous reports showed that overexpression of CDCA7 predicts poor prognosis and tumor progression in human breast cancer, lung adenocarcinoma and lymphoma, colorectal cancer, and pancreatic diseases (29–33). In this study, we uncovered the potential prognostic value of CDCA7, one of the copy number altered genes, for ESCC patients; revealed the tumor-promoting role of CDCA7 gene; and explored its possible mechanism in ESCC for the first time. CDCA7 was highly expressed in not only ESCC but also SCC in transcriptome sequencing data. The Kaplan–Meier survival analysis showed that patients with high expression level of CDCA7 had poor prognosis. This result reminded us that CDCA7 may be used as a candidate target to guide the individual diagnosis and a biomarker to establish a technical system for the molecular classification of ESCC.
Further functional studies reveal that CDCA7 may exert its oncogenic roles via directly binding to the position of 484–495, 641–654, 670–679, and 711–722 bp from the transcription start site of CCNA2. The data of TCGA and GSE53625 further confirmed the positive correlation between CDCA7 and CCNA2 in ESCC, indicating that the high expression level of CDCA7 may be an important driving event in the occurrence and development in ESCC.
CCNA2, which is one of the two A-type cyclins and ubiquitously expressed in cultured cells, has been reported to be upregulated in a variety of cancers (34–37). CCNA2 is considered to be the critical S-phase cyclin in mammalian cells (18, 38). CCNA2 is expressed at the beginning of the S-phase (16, 39) and existed in both the S- and G2-phases. Once synthesized, it binds with its catalytic partners, the cyclin-dependent kinases (CDK) CDK2 and CDK1, and activates its catalytic activity. The CDK2/CCNA2 complex promotes DNA replication through localizing to replication foci in the nucleus (17, 40). The complexes phosphorylate the proteins which play important roles in DNA synthesis and thus drive the S-phase progression (16, 18, 19, 41–43). In addition, a second function of CCNA2 is involved in the entry of cells into mitosis since it also is expressed at the G2-phase (44). The accumulation of CCNA2 is rate-limiting for S-phase entry, so overexpression of CCNA2 can induce cultured cell early entry into the S-phase under normal circumstances (45, 46). Indeed, inhibition of CCNA2 function by p21Cip1 during the G2-phase or injection of anti-CCNA2 antibodies into cultured fibroblasts both can block the process of cells into mitosis (41, 47).
It is known to all that disorder of the cell cycle process is one of the causes of many cancers (48–52). Cancer cells lose many of the inhibitory controls in the cell cycle because of the inactivation or mutation of suppressor genes and overexpression or amplification of oncogenes (53). The aberrant transcription of upregulation of cyclins and CDKs can result in uncontrolled cell cycle progression and mitosis. Our study showed that CCNA2 was a direct downstream target gene of CDCA7, and its expression may be activated by CDCA7 on both the transcription level and the protein level. Therefore, the copy number amplification or increase of CDCA7 may lead to a high level expression of CCNA2 to accelerate the cell cycle process. This may be a mechanism and indicate the important role of CDCA7 in ESCC. Therefore, we speculated that patients with high expression of CDCA7 could be treated with cell cycle-specific agents (CCSA) since the expression of CCNA2 and the number of cells in the proliferative phase are correspondingly increased. This study provides a theoretical and experimental foundation for the research and development of drug targets for clinical treatment of ESCC in China.
In summary, our study shows that CDCA7, a copy number amplification gene in ESCC, may act as a tumor promoter via regulating CCNA2 directly and accelerate the cell cycle process of ESCC cells. The copy number amplification may lead to tumorigenesis and progression of ESCC. Our findings provide a new insight into the molecular mechanisms involved in ESCC development. However, there are still some deficiencies in our research process. Whether the high expression level of CDCA7 is more sensitive to CCSA as we expected needs further experimental verification. Meanwhile, further in-depth research is needed to clarify the mechanism of ECSS carcinogenesis, to develop the prognostic method, and to identify feasible therapeutic targets which could be used to overcome the disease.
Data Availability Statement
Publicly available datasets were analyzed in this study. These data can be found here: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE53625, https://xenabrowser.net/, and https://www.cbioportal.org/.
Ethics Statement
The studies involving human participants were reviewed and approved by the Institutional Reviewing Board (IRB) and the Research Committee of Shanxi Medical University. The patients/participants provided their written informed consent to participate in this study. The animal study was reviewed and approved by Institutional Reviewing Board (IRB) and the Laboratory Animal Welfare Committee of Shanxi Medical University.
Author Contributions
YC and XC designed the study. HL, YJW, SJW, FW, CC, EX, SQW, DG, FC, YB, and YM acquired the data. HL, YJW, SJW, and HC analyzed the data. HL prepared the manuscript. PK, YQW, and LZ edited the manuscript. YC reviewed the manuscript. All authors contributed to the article and approved the submitted version.
Funding
The work was supported by funds from the National Natural Science Foundation of China (81602175, 81773150, 81972613), the Fund for Shanxi “1331 Project” and “1331 Project” Key Subjects Construction, the Fund for “Sanjin Scholars,” the Foundation for Youths of Shanxi Province (201901D211349, 201901D211345), and the Natural Science Foundation of Shanxi Province (201801D121306).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
This work uses data that have been provided from a previous study by Li et al. and can be downloaded from the GEO database (GSE53625).
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2021.734655/full#supplementary-material
References
1. Chen W, Zheng R, Baade P, Zhang S, Zeng H, Bray F, et al. Cancer Statistics in China, 2015. CA: Cancer J For Clin (2016) 66(2):115–32. doi: 10.3322/caac.21338
2. Allemani C, Matsuda T, Di Carlo V, Harewood R, Matz M, Nikšić M, et al. Global Surveillance of Trends in Cancer Survival 2000-14 (CONCORD-3): Analysis of Individual Records for 37 513 025 Patients Diagnosed With One of 18 Cancers From 322 Population-Based Registries in 71 Countries. Lancet (London England) (2018) 391(10125):1023–75. doi: 10.1016/S0140-6736(17)33326-3
3. Zhang L, Zhou Y, Cheng C, Cui H, Cheng L, Kong P, et al. Genomic Analyses Reveal Mutational Signatures and Frequently Altered Genes in Esophageal Squamous Cell Carcinoma. Am J Hum Genet (2015) 96(4):597–611. doi: 10.1016/j.ajhg.2015.02.017
4. Song Y, Li L, Ou Y, Gao Z, Li E, Li X, et al. Identification of Genomic Alterations in Oesophageal Squamous Cell Cancer. Nature (2014) 509(7498):91–5. doi: 10.1038/nature13176
5. Lin D, Hao J, Nagata Y, Xu L, Shang L, Meng X, et al. Genomic and Molecular Characterization of Esophageal Squamous Cell Carcinoma. Nat Genet (2014) 46(5):467–73. doi: 10.1038/ng.2935
6. Gao Y, Chen Z, Li J, Hu X, Shi X, Sun Z, et al. Genetic Landscape of Esophageal Squamous Cell Carcinoma. Nat Genet (2014) 46(10):1097–102. doi: 10.1038/ng.3076
7. Sawada G, Niida A, Uchi R, Hirata H, Shimamura T, Suzuki Y, et al. Genomic Landscape of Esophageal Squamous Cell Carcinoma in a Japanese Population. Gastroenterology (2016) 150(5):1171–82. doi: 10.1053/j.gastro.2016.01.035
8. Cheng C, Zhou Y, Li H, Xiong T, Li S, Bi Y, et al. Whole-Genome Sequencing Reveals Diverse Models of Structural Variations in Esophageal Squamous Cell Carcinoma. Am J Hum Genet (2016) 98(2):256–74. doi: 10.1016/j.ajhg.2015.12.013
9. Prescott J, Osthus R, Lee L, Lewis B, Shim H, Barrett J, et al. A Novel C-Myc-Responsive Gene, JPO1, Participates in Neoplastic Transformation. J Biol Chem (2001) 276(51):48276–84. doi: 10.1074/jbc.M107357200
10. Haggerty T, Zeller K, Osthus R, Wonsey D, Dang C. A Strategy for Identifying Transcription Factor Binding Sites Reveals Two Classes of Genomic C-Myc Target Sites. Proc Natl Acad Sci USA (2003) 100(9):5313–8. doi: 10.1073/pnas.0931346100
11. Goto Y, Hayashi R, Muramatsu T, Ogawa H, Eguchi I, Oshida Y, et al. JPO1/CDCA7, a Novel Transcription Factor E2F1-Induced Protein, Possesses Intrinsic Transcriptional Regulator Activity. Biochim Biophys Acta (2006) 1759:60–8. doi: 10.1016/j.bbaexp.2006.02.004
12. Gill R, Gabor T, Couzens A, Scheid M. The MYC-Associated Protein CDCA7 Is Phosphorylated by AKT to Regulate MYC-Dependent Apoptosis and Transformation. Mol Cell Biol (2013) 33(3):498–513. doi: 10.1128/MCB.00276-12
13. Osthus R, Karim B, Prescott J, Smith B, McDevitt M, Huso D, et al. The Myc Target Gene JPO1/CDCA7 Is Frequently Overexpressed in Human Tumors and has Limited Transforming Activity In Vivo. Cancer Res (2005) 65(13):5620–7. doi: 10.1158/0008-5472.CAN-05-0536
14. Ye L, Li F, Song Y, Yu D, Xiong Z, Li Y, et al. Overexpression of CDCA7 Predicts Poor Prognosis and Induces EZH2-Mediated Progression of Triple-Negative Breast Cancer. Int J Of Cancer (2018) 143(10):2602–13. doi: 10.1002/ijc.31766
15. Wang H, Ye L, Xing Z, Li H, Lv T, Liu H, et al. CDCA7 Promotes Lung Adenocarcinoma Proliferation via Regulating the Cell Cycle. Pathol Res Pract (2019) 215(11):152559. doi: 10.1016/j.prp.2019.152559
16. Girard F, Strausfeld U, Fernandez A, Lamb N. Cyclin A Is Required for the Onset of DNA Replication in Mammalian Fibroblasts. Cell (1991) 67(6):1169–79. doi: 10.1016/0092-8674(91)90293-8
17. Jackman M, Kubota Y, den Elzen N, Hagting A, Pines J. Cyclin A- and Cyclin E-Cdk Complexes Shuttle Between the Nucleus and the Cytoplasm. Mol Biol Cell (2002) 13(3):1030–45. doi: 10.1091/mbc.01-07-0361
18. Yam C, Fung T, Poon R. Cyclin A in Cell Cycle Control and Cancer. Cell Mol Life Sci CMLS (2002) 59(8):1317–26. doi: 10.1007/s00018-002-8510-y
19. den Elzen N, Pines J. Cyclin A Is Destroyed in Prometaphase and can Delay Chromosome Alignment and Anaphase. J Cell Biol (2001) 153(1):121–36. doi: 10.1083/jcb.153.1.121
20. Wang Y, Prives C. Increased and Altered DNA Binding of Human P53 by S and G2/M But Not G1 Cyclin-Dependent Kinases. Nature (1995) 376(6535):88–91. doi: 10.1038/376088a0
21. Ohashi R, Gao C, Miyazaki M, Hamazaki K, Tsuji T, Inoue Y, et al. Enhanced Expression of Cyclin E and Cyclin A in Human Hepatocellular Carcinomas. Anticancer Res (2001) 21:657–62.
22. Volm M, Koomägi R, Mattern J, Stammler G. Cyclin A is Associated With an Unfavourable Outcome in Patients With Non-Small-Cell Lung Carcinomas. Br J Cancer (1997) 75(12):1774–8. doi: 10.1038/bjc.1997.302
23. Bukholm I, Bukholm G, Nesland J. Over-Expression of Cyclin A is Highly Associated With Early Relapse and Reduced Survival in Patients With Primary Breast Carcinomas. Int J Cancer (2001) 93(2):283–7. doi: 10.1002/ijc.1311
24. Kanai M, Shiozawa T, Xin L, Nikaido T, Fujii S. Immunohistochemical Detection of Sex Steroid Receptors, Cyclins, and Cyclin-Dependent Kinases in the Normal and Neoplastic Squamous Epithelia of the Uterine Cervix. Cancer (1998) 82(9):1709–19. doi: 10.1002/(SICI)1097-0142(19980501)82:9<1709::AID-CNCR18>3.0.CO;2-8
25. Yasmeen A, Berdel W, Serve H, Müller-Tidow C--. And A-Type Cyclins as Markers for Cancer Diagnosis and Prognosis. Expert Rev Mol Diagn (2003) 3(5):617–33. doi: 10.1586/14737159.3.5.617
26. Li J, Chen Z, Tian L, Zhou C, He M, Gao Y, et al. LncRNA Profile Study Reveals a three-lncRNA Signature Associated With the Survival of Patients With Oesophageal Squamous Cell Carcinoma. Gut (2014) 63(11):1700–10. doi: 10.1136/gutjnl-2013-305806
27. Cerami E, Gao J, Dogrusoz U, Gross B, Sumer S, Aksoy B, et al. The Cbio Cancer Genomics Portal: An Open Platform for Exploring Multidimensional Cancer Genomics Data. Cancer Discov (2012) 2(5):401–4. doi: 10.1158/2159-8290.CD-12-0095
28. Gao J, Aksoy B, Dogrusoz U, Dresdner G, Gross B, Sumer S, et al. Integrative Analysis of Complex Cancer Genomics and Clinical Profiles Using the Cbioportal. Sci Signaling (2013) 6(269):pl1. doi: 10.1126/scisignal.2004088
29. Chhatriya B, Mukherjee M, Ray S, Saha B, Lahiri S, Halder S, et al. Transcriptome Analysis Identifies Putative Multi-Gene Signature Distinguishing Benign and Malignant Pancreatic Head Mass. J Trans Med (2020) 18(1):420. doi: 10.1186/s12967-020-02597-1
30. Li S, Huang J, Qin M, Zhang J, Liao C. High Expression of CDCA7 Predicts Tumor Progression and Poor Prognosis in Human Colorectal Cancer. Mol Med Rep (2020) 22(1):57–66. doi: 10.3892/mmr.2020.11089
31. Lewis B, Shim H, Li Q, Wu C, Lee L, Maity A, et al. Identification of Putative C-Myc-Responsive Genes: Characterization of Rcl, a Novel Growth-Related Gene. Mol Cell Biol (1997) 17(9):4967–78. doi: 10.1128/MCB.17.9.4967
32. Martín-Cortázar C, Chiodo Y, Jiménez R, Bernabé M, Cayuela M, Iglesias T, et al. CDCA7 Finely Tunes Cytoskeleton Dynamics to Promote Lymphoma Migration and Invasion. Haematologica (2020) 105(3):730–40. doi: 10.3324/haematol.2018.215459
33. Jiménez-P R, Martín-Cortázar C, Kourani O, Chiodo Y, Cordoba R, Domínguez-Franjo M, et al. CDCA7 Is a Critical Mediator of Lymphomagenesis That Selectively Regulates Anchorage-Independent Growth. Haematologica (2018) 103(10):1669–78. doi: 10.3324/haematol.2018.188961
34. Wang J, Chenivesse X, Henglein B, Bréchot C. Hepatitis B Virus Integration in a Cyclin A Gene in a Hepatocellular Carcinoma. Nature (1990) 343(6258):555–7. doi: 10.1038/343555a0
35. Pines J, Hunter T. Human Cyclin A is Adenovirus E1A-Associated Protein P60 and Behaves Differently From Cyclin B. Nature (1990) 346(6286):760–3. doi: 10.1038/346760a0
36. Ravnik S, Wolgemuth D. The Developmentally Restricted Pattern of Expression in the Male Germ Line of a Murine Cyclin A, Cyclin A2, Suggests Roles in Both Mitotic and Meiotic Cell Cycles. Dev Biol (1996) 173(1):69–78. doi: 10.1006/dbio.1996.0007
37. Wolgemuth D. Function of the A-Type Cyclins During Gametogenesis and Early Embryogenesis. Results Problems Cell Differ (2011) 53:391–413. doi: 10.1007/978-3-642-19065-0_17
38. Hochegger H, Takeda S, Hunt T. Cyclin-Dependent Kinases and Cell-Cycle Transitions: Does One Fit All? Nat Rev Mol Cell Biol (2008) 9(11):910–6. doi: 10.1038/nrm2510
39. Erlandsson F, Linnman C, Ekholm S, Bengtsson E, Zetterberg A. A Detailed Analysis of Cyclin A Accumulation at the G(1)/S Border in Normal and Transformed Cells. Exp Cell Res (2000) 259(1):86–95. doi: 10.1006/excr.2000.4889
40. Cardoso M, Leonhardt H, Nadal-Ginard B. Reversal of Terminal Differentiation and Control of DNA Replication: Cyclin A and Cdk2 Specifically Localize at Subnuclear Sites of DNA Replication. Cell (1993) 74(6):979–92. doi: 10.1016/0092-8674(93)90721-2
41. Pagano M, Pepperkok R, Verde F, Ansorge W, Draetta G. Cyclin A Is Required at Two Points in the Human Cell Cycle. EMBO J (1992) 11(3):961–71. doi: 10.1002/j.1460-2075.1992.tb05135.x
42. Zindy F, Lamas E, Chenivesse X, Sobczak J, Wang J, Fesquet D, et al. Cyclin A Is Required in S Phase in Normal Epithelial Cells. Biochem Biophys Res Commun (1992) 182(3):1144–54. doi: 10.1016/0006-291X(92)91851-G
43. Fisher D. Control of DNA Replication by Cyclin-Dependent Kinases in Development. Results Problems Cell Differ (2011) 53:201–17. doi: 10.1007/978-3-642-19065-0_10
44. Swenson K, Farrell K, Ruderman J. The Clam Embryo Protein Cyclin A Induces Entry Into M Phase and the Resumption of Meiosis in Xenopus Oocytes. Cell (1986) 47(6):861–70. doi: 10.1016/0092-8674(86)90801-9
45. Resnitzky D, Hengst L, Reed S. Cyclin A-Associated Kinase Activity Is Rate Limiting for Entrance Into S Phase and Is Negatively Regulated in G1 by p27Kip1. Mol Cell Biol (1995) 15(8):4347–52. doi: 10.1128/MCB.15.8.4347
46. Chibazakura T, Kamachi K, Ohara M, Tane S, Yoshikawa H, Roberts J. Cyclin A Promotes S-Phase Entry via Interaction With the Replication Licensing Factor Mcm7. Mol Cell Biol (2011) 31(2):248–55. doi: 10.1128/MCB.00630-10
47. Furuno N, den Elzen N, Pines J. Human Cyclin A is Required for Mitosis Until Mid Prophase. J Cell Biol (1999) 147(2):295–306. doi: 10.1083/jcb.147.2.295
49. Kaplon J, van Dam L, Peeper D. Two-Way Communication Between the Metabolic and Cell Cycle Machineries: The Molecular Basis. Cell Cycle (Georgetown Tex) (2015) 14(13):2022–32. doi: 10.1080/15384101.2015.1044172
50. Moncada S, Higgs E, Colombo S. Fulfilling the Metabolic Requirements for Cell Proliferation. Biochem J (2012) 446(1):1–7. doi: 10.1042/BJ20120427
51. Kalucka J, Missiaen R, Georgiadou M, Schoors S, Lange C, De Bock K, et al. Metabolic Control of the Cell Cycle. Cell Cycle (Georgetown Tex) (2015) 14(21):3379–88. doi: 10.1080/15384101.2015.1090068
52. Aarts M, Linardopoulos S, Turner N. Tumour Selective Targeting of Cell Cycle Kinases for Cancer Treatment. Curr Opin Pharmacol (2013) 13(4):529–35. doi: 10.1016/j.coph.2013.03.012
Keywords: CDCA7, cell cycle, CCNA2, copy number amplification, ESCC (esophageal squamous cell carcinoma)
Citation: Li H, Weng Y, Wang S, Wang F, Wang Y, Kong P, Zhang L, Cheng C, Cui H, Xu E, Wei S, Guo D, Chen F, Bi Y, Meng Y, Cheng X and Cui Y (2021) CDCA7 Facilitates Tumor Progression by Directly Regulating CCNA2 Expression in Esophageal Squamous Cell Carcinoma. Front. Oncol. 11:734655. doi: 10.3389/fonc.2021.734655
Received: 03 August 2021; Accepted: 27 September 2021;
Published: 19 October 2021.
Edited by:
Bin Qiao, Zhengzhou University, ChinaReviewed by:
Jian Zhang, Texas A&M Health Science Center, United StatesJiheng Xu, New York University, United States
Copyright © 2021 Li, Weng, Wang, Wang, Wang, Kong, Zhang, Cheng, Cui, Xu, Wei, Guo, Chen, Bi, Meng, Cheng and Cui. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yongping Cui, Y3VpeXBAc3htdS5lZHUuY24=; Xiaolong Cheng, Y2hlbmd4bEBzeG11LmVkdS5jbg==
†These authors have contributed equally to this work