
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Oncol. , 30 September 2021
Sec. Hematologic Malignancies
Volume 11 - 2021 | https://doi.org/10.3389/fonc.2021.728613
This article is part of the Research Topic Insights in Hematologic Malignancies: 2021 View all 28 articles
 Samantha Bruno1*
Samantha Bruno1* Lorenza Bandini2Agnese Patuelli2Valentina Robustelli1,2Claudia Venturi1Manuela Mancini2
Lorenza Bandini2Agnese Patuelli2Valentina Robustelli1,2Claudia Venturi1Manuela Mancini2 Dorian Forte1Sara De Santis1Cecilia Monaldi1Alessandra Grassi2Gabriella Chiurumbolo2Stefania Paolini2Gianluca Cristiano1
Dorian Forte1Sara De Santis1Cecilia Monaldi1Alessandra Grassi2Gabriella Chiurumbolo2Stefania Paolini2Gianluca Cristiano1 Cristina Papayannidis2Chiara Sartor1Jacopo Nanni1Emanuela Ottaviani2
Cristina Papayannidis2Chiara Sartor1Jacopo Nanni1Emanuela Ottaviani2 Antonio Curti2
Antonio Curti2 Michele Cavo1,2
Michele Cavo1,2 Simona Soverini1
Simona Soverini1FMS-like tyrosine kinase 3 (FLT3) is among the most common driver genes recurrently mutated in acute myeloid leukemia (AML), accounting for approximately 30% of cases. Activating mutations of the FLT3 receptor include internal tandem duplications (ITD) that map to the auto-inhibitory juxtamembrane (JM) domain or point mutations within the tyrosine kinase domain (TKD). Several FLT3 tyrosine kinase inhibitors have been developed in the last few years, but midostaurin is currently the only one approved for the treatment of newly diagnosed patients harboring FLT3 mutations. Here we describe for the first time a novel in-frame deletion in exon 14 (JM domain) of the FLT3 gene, that we identified in a young woman with CBFb-MYH11-positive AML. We demonstrated that this novel FLT3 variant is pathogenic, since it is responsible for constitutive activation of FLT3 receptor. Finally, ex-vivo studies demonstrated that this novel mutation is sensitive to midostaurin.
Acute myeloid leukemia (AML) is a highly heterogeneous haematological malignancy characterized by a wide range of genomic alterations responsible of defective regulation of the differentiation and self-renewal programs of hematopoietic stem cells. FMS-like tyrosine kinase 3 (FLT3) is a member of class III receptor tyrosine kinases that exhibits activating mutations in approximately 30% of AML patients, making it one of the most recurrently mutated genes (1, 2). Notably, there are two types of FLT3 mutations which include the internal tandem duplications (ITD) within the auto-inhibitory juxtamembrane (JM) domain, described in about 25-30% of AML patients, and the less frequent gain-of-function mutations occurring in about 8% of AML cases as point mutations in the activation loop of the tyrosine kinase domain (TKD) (1, 2). The leukemogenic relevance and inferior overall survival of FLT3-ITD mutations have been shown, although it has been demonstrated that the prognostic impact is affected by both mutant allele burden and presence of co-existing mutations (3, 4). Indeed, high allele ratio (AR>0.5; FLT3-ITDhigh) is associated with higher disease risk, whereas low AR (<0.5; FLT3-ITDlow) is associated with intermediate risk, which becomes favourable in case of co-occurrence of the nucleophosmin 1 (NPM1) mutation (4). Concerning the FLT3-TKD mutations, their prognostic significance still remains unclear, with numerous conflicting data (3, 5–8). The clinical impact of ITD and TKD mutations of FLT3 is dependent on the subsequent constitutive activation of the tyrosine kinase, that activates its downstream signalling pathways including the signal transducer and activator of transcription 5 (STAT5), RAS/mitogen-activated protein kinase (MAPK)/extracellular-signal-regulated kinase (ERK), phosphoinositide 3-kinase (PI3K)/Serine-Threonine Kinase 1 (AKT) (9–13). These targets have been associated with increased proliferation and survival of the leukemic cell population (14). In light of the crucial pathogenetic role of FLT3 constitutive activation, small molecule inhibitors have been developed as new promising therapies to treat this subset of AML patients.
Here we describe for the first time a novel FLT3 in-frame deletion and its impact on downstream signalling pathways showing the activation effect of such mutation on FLT3 receptor. Moreover, ex-vivo study performed on mutated primary cells sought to demonstrated their sensitivity to midostaurin, a multi-targeted kinase inhibitor active against FLT3, currently approved for first-line treatment of FLT3-mutated AML (15).
A 32 years-old woman which presented with persistent fever associated with marked hyperleukocytosis [white blood cells (WBC), 319,000/mm3], anemia [hemoglobin (Hb), 5.7 g/dL] and thrombocytopenia [platelets (PLTs), 22,000/mm3] was admitted to our hospital in July 2019. Morphological analysis of bone marrow (BM) aspirate suggested a diagnosis of acute myelomonocytic leukemia with 80% of myeloid blasts. Computed tomography (TC) examination of the brain detected central nervous system (CNS) localization of blast cells. Immunophenotypic analysis of leukemic cells from the BM aspirate confirmed the expression of several markers of hematopoietic stem and progenitor cells, including CD34, CD13, CD33, CD117, MPO, CD14, CD64, and aberrant expression of CD4. Karyotype and fluorescence in situ hybridization (FISH) analyses of BM nuclei identified the inv(16)(p13;q22) chromosome rearrangement in 20/20 metaphases examined. This was confirmed by quantitative RT-PCR that detected the presence of CBFβ-MYH11 transcript type A (CBFβ-MYH11/ABL*100 = 85.37), along with over-expressed WT1 (ratio WT1/ABL*10,000 = 1409.53). Therefore, the patient was diagnosed with M4 inv(16) acute myeloid leukemia (AML) according to French-American-British (FAB) classification and was classified as favourable cytogenetic risk based on cytogenetic and molecular biology data according to ELN 2017 (4). However, in light of CNS localization at diagnosis, the patient was considered at high risk of relapse. A FLAI-5 (fludarabine-cytarabine-idarubicin) regimen was selected as induction chemotherapy, which resulted effective and well tolerated. The follow-up evaluation performed after 2 months of induction therapy showed a good response: the patient exhibited a peripheral blood count with WBC levels of 6,100/mm3, Hb levels of 10.1 g/dL and PLTs of 189,000/mm3. Accordingly, BM evaluation by morphology, immunophenotyping and molecular biology revealed complete remission (CR; CBFβ-MYH11/ABL*100 = 0.0332; WT1/ABL*10,000 = 13.24) and TC of the brain was negative for SNC localization of blast cells. Therefore, the patient continued with 1 cycle of consolidation therapy with the FLAI-5 regimen. As reported above, due to CNS localization at diagnosis, the patient underwent allogeneic stem cell transplantation (allo-SCT) receiving haploidentical peripheral blood stem cells from her brother. After transplant, she remained in complete remission until the last follow-up. Figure 1 summarizes relevant clinical course and diagnostic results.
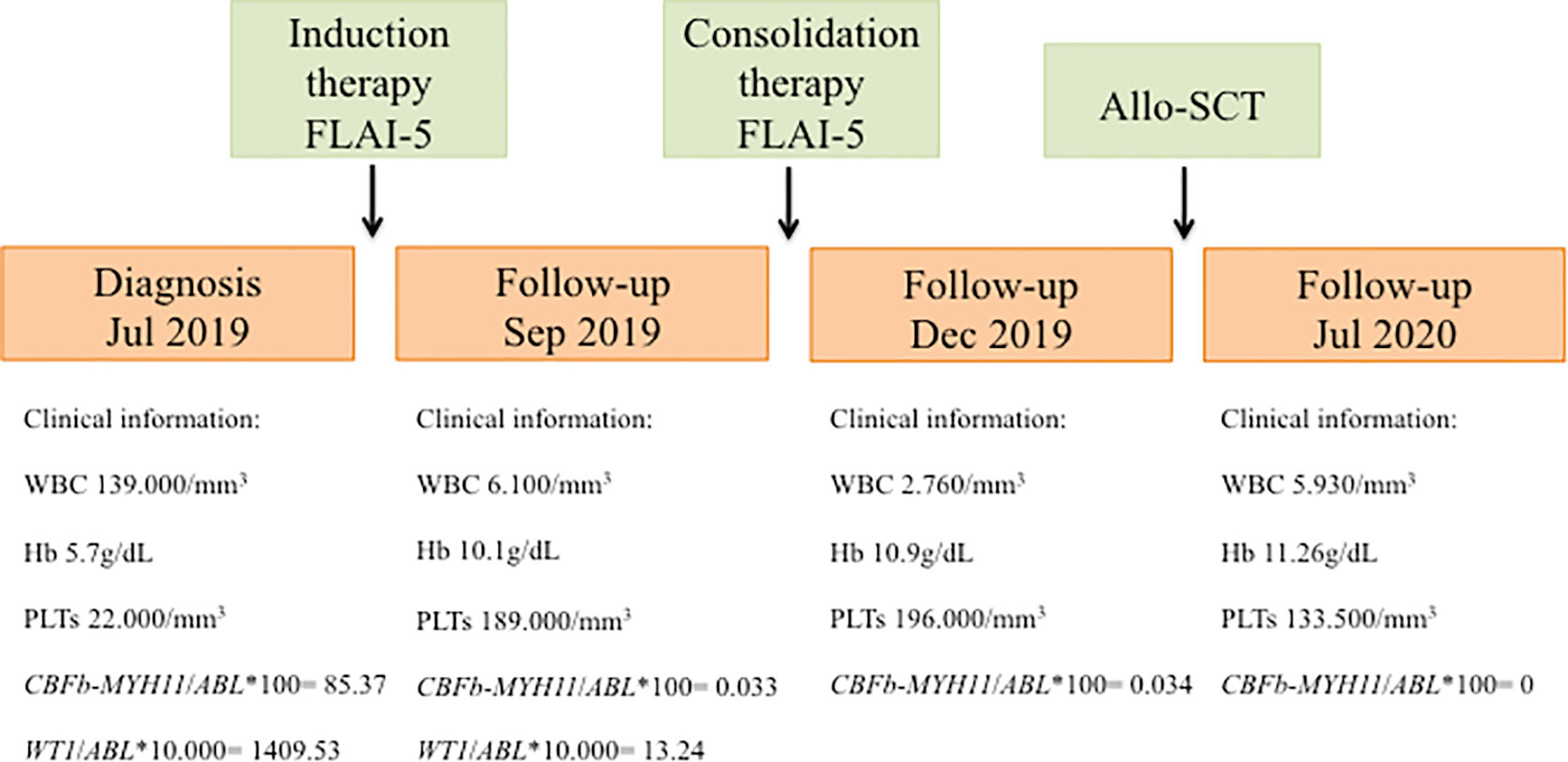
Figure 1 Schematic representation showing timeline of treatment course and clinical information. FLAI-5, fludarabine-cytarabine-idarubicin; Allo-SCT, allogeneic stem cell transplantation; WBC, white blood cells; Hb, hemoglobin; PLTs, platelets.
In light of the clinical utility of next-generation sequencing (NGS) for the assessment of the genetic landscape of AML providing diagnostic and prognostic information, the AmpliSeq Myeloid Panel (Thermofisher) was performed on primary blast cells enriched by density gradient centrifugation from the patient bone marrow sample collected at diagnosis. NGS analysis revealed a FLT3 mutation with a variant allele frequency (VAF) of 40%, represented by an in-frame deletion at exon 14 of the JM domain (c.1770_1784del15; p.Phe590_Arg595delinsLeu). Interrogation of the COSMIC database indicated that this deletion had never been reported before. The mutation was confirmed by Sanger sequencing (Figure 2) and mutant-to-wild-type allelic ratio (AR) was obtained through PCR-electrophoresis and fragment analysis (FLT3-ITD-AR=1.1). The FLT3 mutation became undetectable in the sample collected at the time of CR after induction chemotherapy.
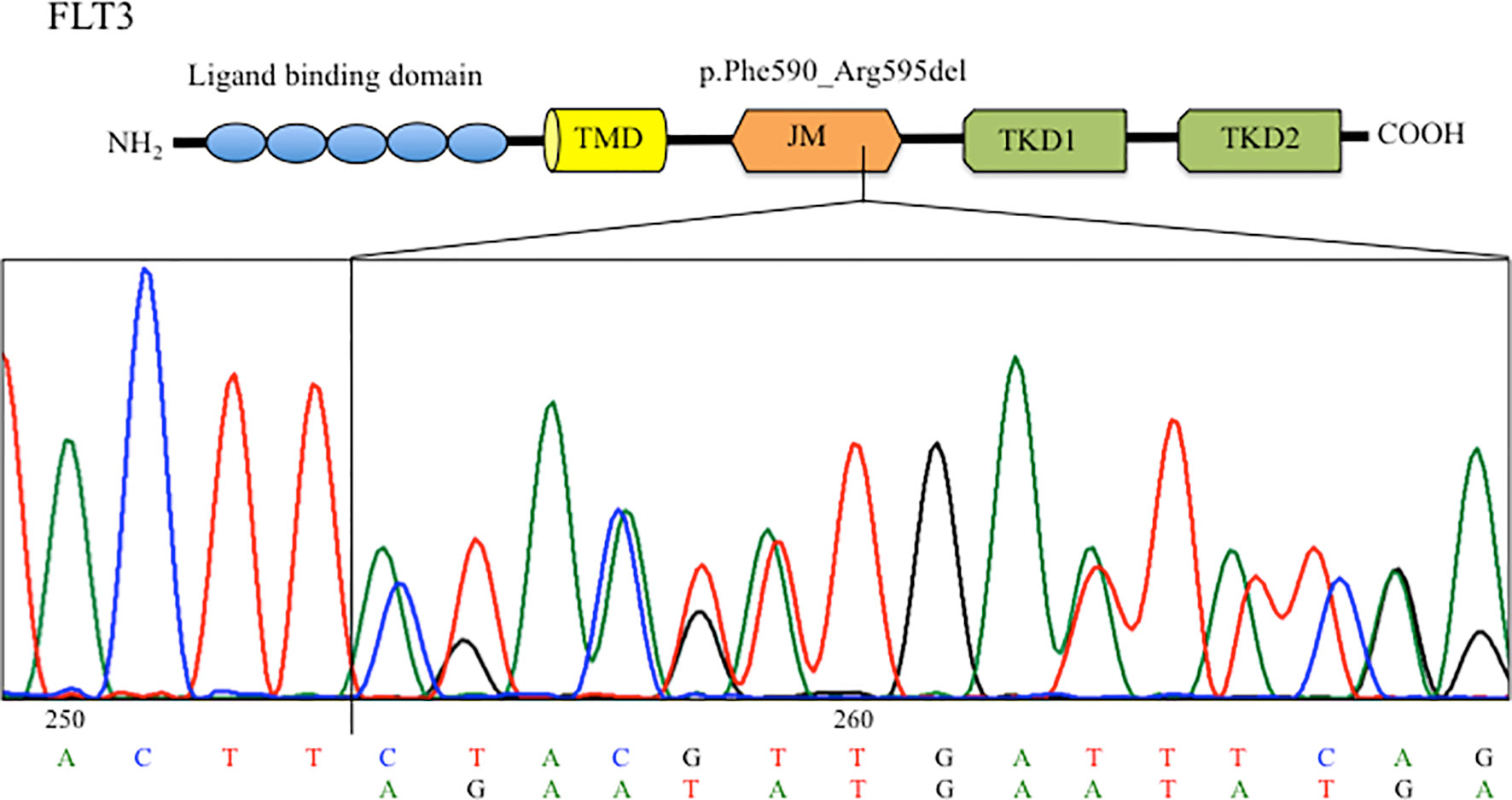
Figure 2 Sanger sequencing chromatogram showing the FLT3 deletion identified in the BM at diagnosis. TMD, transmernbrane domain; JMD, juxtamembrane domain; TKD, tyrosine kinase domain.
In order to understand whether the newly described FLT3 alteration influenced its cellular activity, we analysed by Western blot (WB) the expression and activation of some target proteins downstream of FLT3. As shown in Figure 3A, we observed that the deletion in the JM domain resulted in the constitutive activation of FLT3, with the sample at diagnosis showing increased levels of phosphorylated FLT3 and of its downstream signalling molecules. Five AML patients with wild-type (wt) FLT-3 and 5 AML patients carrying canonical FLT3-ITD mutations (with an AR ranging from 0.8 to 1.5) were used as control for protein expression levels of FLT3 downstream signalling molecules (Figure 3B). In particular, the sample at diagnosis, showed increased levels of p21, c-MYC and Cyclin D1, that were associated with the activation of STAT5 signalling pathway, together with the up-regulation of MAPK-activated proteins p-ERK and p38. In addition, the activation of FLT3 induced p27 and BIM expression, which are regulated by AKT and FOXO3A signalling, as well as the up-regulation of 14-3-3 and phosphorylated BAD (serine 112 and 136) that are responsible for the block of cell death. Results were superimposable to what we could observe in the 5 FLT3-ITD mutated samples as compared to wt FLT3 samples. As expected, the sample at remission showed a significant reduction of phosphorylated FLT3 and of the activation of all the above mentioned targets, supporting the direct link between activated signalling molecules and the observed FLT3 mutation. Lastly, to investigate the clinical relevance of the described mutation, we assessed the effects of midostaurin on the clonogenic potential of leukemic blasts cells isolated from the patient at diagnosis. Midostaurin induced a dose-dependent reduction in colony formation with an LD50 of 100nM, suggesting that the newly reported mutation is sensitive to midostaurin inhibition (Figure 4).
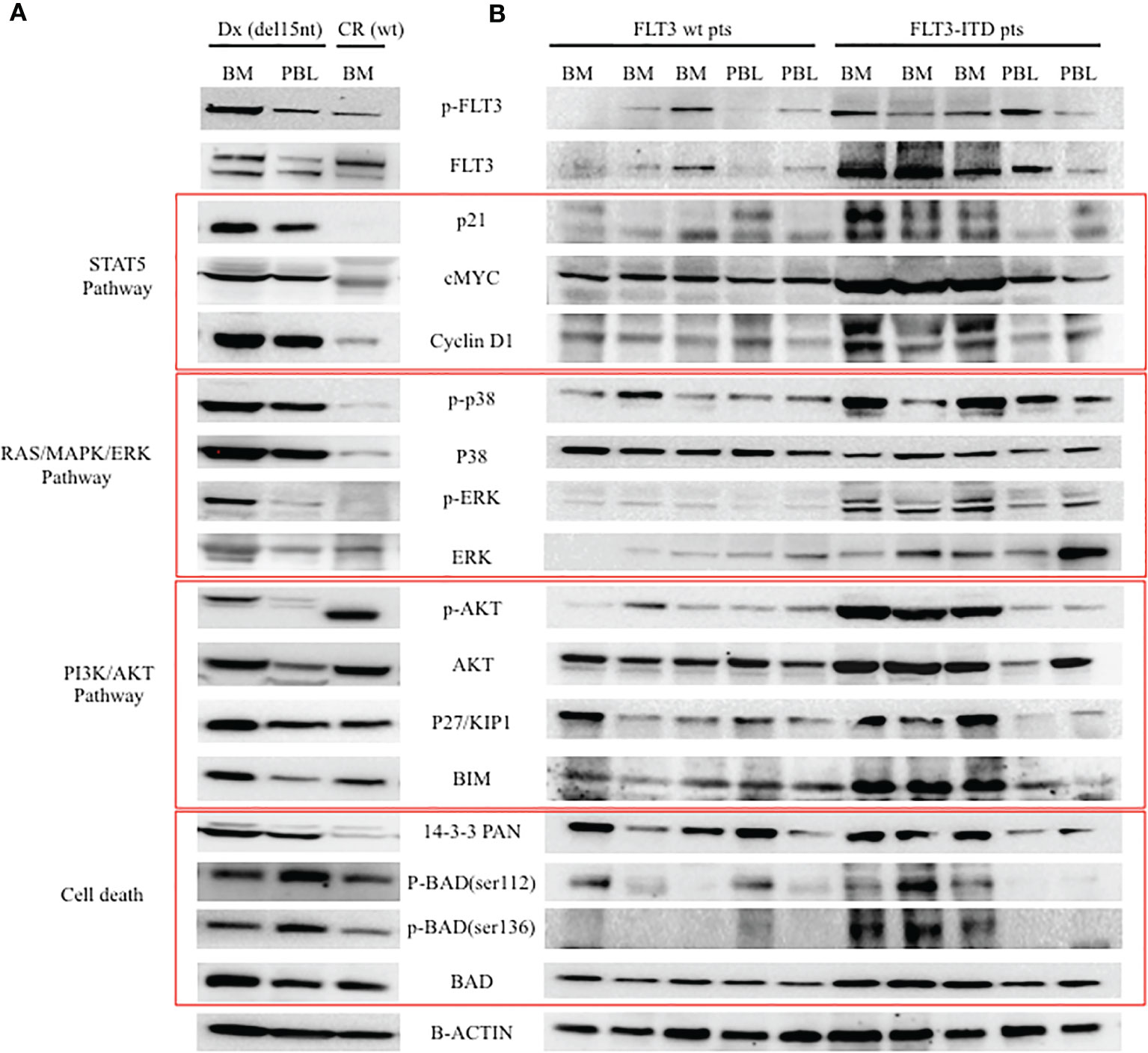
Figure 3 Western blot analysis of FLT-3 expression and of its down-stream signalling pathway. (A) Analysis of patient cells at two different time points (Dx: diagnosis with FLT3 carrying the c.1770_1784del15; CR: complete remission with wt FLT3). (B) Analysis of 5 AML patients with wt FLT3 along with 5 patients carrying FLT3-ITD, used as control of FLT3 downstream signalling. BM, bone marrow; PBL, peripheral blood.
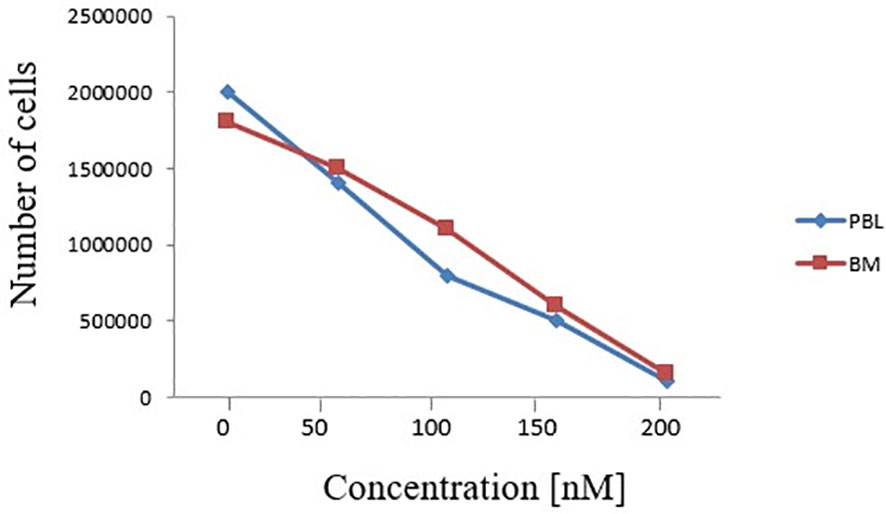
Figure 4 Effects of increasing doses of midostaurin on the clonogenic capacity of primary BM and PBL cells carrying the reported FLT3 deletion (c.1770_1784del15; p.Phe590_Arg595delinsLeu).
FLT3 is the most frequently mutated gene in AML patients, with a strong disease specificity (2), and encodes is one of the key molecules involved in the pathogenesis of AML. Indeed, FLT3 is one of the markers currently used for the molecular genetic diagnosis and risk stratification of AML (4). The clinical relevance of ITD and TKD mutations in AML pathogenesis and progression highlighted the need for specific compounds targeting FLT3 constitutive activation. Consequently, during the last years several inhibitors have been developed as target therapies to improve the outcome of FLT3-mutated patients (16, 17). In this study, we describe for the first time a novel FLT3 mutation identified in an AML patient, consisting in a deletion of 15 nucleotides in the JM domain, where the FLT3-ITDs usually localize. Interestingly, other case reports showing noncanonical FLT3 deletions have been published, and all these mutations cluster in the JM domain, a highly conserved region that mediates the activation of the FLT3 receptor (18–20). There are limited functional data for deletions in the JM domain, however existing knowledge suggest that any type of mutation, including deletions, in the JM sequence could potentially activate the FLT3 tyrosine kinase receptor, with consequent pathogenetic and clinical impact. In this study, we demonstrated by WB that the newly reported deletion indeed induces the constitutive activation of FLT3 and of its downstream effectors, including STAT5, ERK and AKT pathways. The patient showed a good response to conventional chemotherapy and underwent transplant. However, we also evaluated by ex-vivo treatment the efficacy of midostaurin, the TKI currently approved for the treatment of de novo FLT3-mutated patients, finding that midostaurin could effectively reduce the clonogenic capacity of primary blast cells. Although additional studies are needed to better characterize the functional consequences and TKI sensitivity of this deletion, our results are corroborated by recent work from Young and colleagues. They used a scanning mutagenesis approach to generated a panel of FLT3 deletions (clustering in the region comprised between K568 to V581) demonstrating that this class of mutations lead to the activation of signalling molecules downstream to FLT3 and are targetable with several TKI inhibitors (20). In conclusion, our results suggest the clinical relevance of the novel FLT3 deletion herein reported, that acts as a standard activating ITD mutation and can be successfully targeted by midostaurin. Our findings provide useful information for clinicians treating AML patients, highlighting the importance to characterize and monitor the genomic alterations of AML patients.
The original contributions presented in the study are included in the article/supplementary materials. Further inquiries can be directed to the corresponding author.
The studies involving human participants were reviewed and approved by local Ethics Committee (protocol 112/2014/U/Tess of Policlinico Sant’Orsola-Malpighi). The patients/participants provided their written informed consent to participate in this study.
SB, LB, DF, SS, EO and MM contributed to the conception, research design, drafting of the manuscript, and conceptualization methodology. LB and AP performed NGS analysis. VR, CV, SDS, and CM performed sanger sequencing and PCR-electrophoresis and fragment analysis. GaC and AG performed FISH analysis. SB and MM performed WB analysis. SB and DF performed colony-formation assay. SP, GiC, CP, CS, JN, AC, and MC provided clinical data and enrolled the patient for the study. SS and AC reviewed and edited the manuscript. EO, SS, and MC acquired the funding. MM, EO and SS provided the supervision of the study. All authors contributed to the article and approved the submitted version.
AC was employed by Novartis, Pfizer, Abbvie and acted as speaker in Advisory Board for Novartis and Abbvie. CP was employed by Astellas, Amgen and acted as speaker in Advisory Board for Abbvie, Janssen, Novartis, Pfizer and Astellas.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The authors acknowledge the supports from AIL Bologna ODV.
1. Thiede C, Steudel C, Mohr B, Schaich M, Schäkel U, Platzbecker U, et al. Analysis of FLT3-Activating Mutations in 979 Patients With Acute Myelogenous Leukemia: Association With FAB Subtypes and Identification of Subgroups With Poor Prognosis. Blood (2002) 99:4326–35. doi: 10.1182/blood.V99.12.4326
2. Papaemmanuil E, Gerstung M, Bullinger L, Gaidzik VI, Paschka P, Roberts ND, et al. Genomic Classification and Prognosis in Acute Myeloid Leukemia. N Engl J Med (2016) 374:2209–21. doi: 10.1056/NEJMoa1516192
3. Yanada M, Matsuo K, Suzuki T, Kiyoi H, Naoe T. Prognostic Significance of FLT3 Internal Tandem Duplication and Tyrosine Kinase Domain Mutations for Acute Myeloid Leukemia: A Meta-Analysis. Leukemia (2005) 19:1345–9. doi: 10.1038/sj.leu.2403838
4. Döhner H, Estey E, Grimwade D, Amadori S, Appelbaum FR, Büchner T, et al. Diagnosis and Management of AML in Adults: 2017 ELN Recommendations From an International Expert Panel. Blood (2017) 129:424–47. doi: 10.1182/blood-2016-08-733196
5. Mead AJ, Linch DC, Hills RK, Wheatley K, Burnett AK, Gale RE, et al. FLT3 Tyrosine Kinase Domain Mutations are Biologically Distinct From and Have a Significantly More Favorable Prognosis Than FLT3 Internal Tandem Duplications in Patients With Acute Myeloid Leukemia. Blood (2007) 110:1262–70. doi: 10.1182/blood-2006-04-015826
6. Whitman SP, Ruppert AS, Radmacher MD, Mrózek K, Paschka P, Langer C, et al. FLT3 D835/I836 Mutations are Associated With Poor Disease-Free Survival and a Distinct Gene-Expression Signature Among Younger Adults With De Novo Cytogenetically Normal Acute Myeloid Leukemia Lacking FLT3 Internal Tandem Duplications. Blood (2008) 111:1552–9. doi: 10.1182/blood-2007-08-107946
7. Boddu P, Kantarjian H, Borthakur G, Kadia T, Daver N, Pierce S, et al. Co-Occurrence of FLT3-TKD and NPM1 Mutations Defines a Highly Favorable Prognostic AML Group. Blood Adv (2017) 1:1546–50. doi: 10.1182/bloodadvances.2017009019
8. Perry M, Bertoli S, Rocher C, Hayette S, Ducastelle S, Barraco F, et al. FLT3-TKD Mutations Associated With NPM1 Mutations Define a Favorable-Risk Group in Patients With Acute Myeloid Leukemia. Clin Lymphoma Myeloma Leuk (2018) 18:e545–50. doi: 10.1016/j.clml.2018.06.006
9. Takahashi S. Downstream Molecular Pathways of FLT3 in the Pathogenesis of Acute Myeloid Leukemia: Biology and Therapeutic Implications. J Hematol Oncol (2011) 4:13. doi: 10.1186/1756-8722-4-13
10. Spiekermann K, Bagrintseva K, Schwab R, Schmieja K, Hiddemann W. Overexpression and Constitutive Activation of FLT3 Induces STAT5 Activation in Primary Acute Myeloid Leukemia Blast Cells. Clin Cancer Res (2003) 9:2140–50.
11. Brandts CH, Sargin B, Rode M, Biermann C, Lindtner B, Schwäble J, et al. Constitutive Activation of Akt by Flt3 Internal Tandem Duplications Is Necessary for Increased Survival, Proliferation, and Myeloid Transformation. Cancer Res (2005) 65:9643–50. doi: 10.1158/0008-5472.CAN-05-0422
12. Choudhary C, Brandts C, Schwable J, Tickenbrock L, Sargin B, Ueker A, et al. Activation Mechanisms of STAT5 by Oncogenic Flt3-ITD. Blood (2007) 110:370–4. doi: 10.1182/blood-2006-05-024018
13. Nogami A, Oshikawa G, Okada K, Fukutake S, Umezawa Y, Nagao T, et al. FLT3-ITD Confers Resistance to the PI3K/Akt Pathway Inhibitors by Protecting the mTOR/4ebp1/Mcl-1 Pathway Through STAT5 Activation in Acute Myeloid Leukemia. Oncotarget (2015) 6:9189–205. doi: 10.18632/oncotarget.3279
14. Levis M, Small D. FLT3: ITDoes Matter in Leukemia. Leukemia (2003) 17:1738–52. doi: 10.1038/sj.leu.2403099
15. Levis M. Midostaurin Approved for FLT3-Mutated AML. Blood (2017) 129:3403–6. doi: 10.1182/blood-2017-05-782292
16. Stone RM, Mandrekar SJ, Sanford BL, Laumann K, Geyer S, Bloomfield CD, et al. Midostaurin Plus Chemotherapy for Acute Myeloid Leukemia With a FLT3 Mutation. N Engl J Med (2017) 377:454–64. doi: 10.1056/NEJMoa1614359
17. Zhao J, Song Y, Liu D. Gilteritinib: A Novel FLT3 Inhibitor for Acute Myeloid Leukemia. Biomark Res (2019) 7:19. doi: 10.1186/s40364-019-0170-2
18. Chatain N, Perera RC, Rossetti G, Rossa J, Carloni P, Schemionek M, et al. Rare FLT3 Deletion Mutants may Provide Additional Treatment Options to Patients With AML: An Approach to Individualized Medicine. Leukemia (2015) 29(12):2434–8. doi: 10.1038/leu.2015.131
19. Shin S-Y, Lee S-T, Park C-H, Kim H-J, Kim S-H. Two Cases of in-Frame Deletion Mutation in Juxtamembrane Domain of the FLT3. Leuk Lymphoma (2016) 57(2):486–8. doi: 10.3109/10428194.2015.1065982
Keywords: acute myeloid leukemia, FLT3 mutation, NGS - next generation sequencing, targeted therapy, midostaurin
Citation: Bruno S, Bandini L, Patuelli A, Robustelli V, Venturi C, Mancini M, Forte D, De Santis S, Monaldi C, Grassi A, Chiurumbolo G, Paolini S, Cristiano G, Papayannidis C, Sartor C, Nanni J, Ottaviani E, Curti A, Cavo M and Soverini S (2021) Case Report: A Novel Activating FLT3 Mutation in Acute Myeloid Leukemia. Front. Oncol. 11:728613. doi: 10.3389/fonc.2021.728613
Received: 21 June 2021; Accepted: 13 September 2021;
Published: 30 September 2021.
Edited by:
Massimo Breccia, Sapienza University of Rome, ItalyReviewed by:
Tomasz Stoklosa, Medical University of Warsaw, PolandCopyright © 2021 Bruno, Bandini, Patuelli, Robustelli, Venturi, Mancini, Forte, De Santis, Monaldi, Grassi, Chiurumbolo, Paolini, Cristiano, Papayannidis, Sartor, Nanni, Ottaviani, Curti, Cavo and Soverini. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Samantha Bruno, c2FtYW50aGEuYnJ1bm8yQHVuaWJvLml0
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.