 Kellsye P. Fabian
Kellsye P. Fabian Benjamin Wolfson†
Benjamin Wolfson† James W. Hodge
James W. Hodge
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oncol. , 23 August 2021
Sec. Cancer Immunity and Immunotherapy
Volume 11 - 2021 | https://doi.org/10.3389/fonc.2021.728018
This article is part of the Research Topic Oxidative Stress in Breast Cancer View all articles
Cancer treatment has rapidly entered the age of immunotherapy, and it is becoming clear that the effective therapy of established tumors necessitates rational multi-combination immunotherapy strategies. But even in the advent of immunotherapy, the clinical role of standard-of-care chemotherapy regimens still remains significant and may be complementary to emerging immunotherapeutic approaches. Depending on dose, schedule, and agent, chemotherapy can induce immunogenic cell death, resulting in the release of tumor antigens to stimulate an immune response, or immunogenic modulation, sensitizing surviving tumor cells to immune cell killing. While these have been previously defined as distinct processes, in this review we examine the published mechanisms supporting both immunogenic cell death and immunogenic modulation and propose they be reclassified as similar effects termed “immunogenic cell stress.”
Treatment-induced immunogenic cell stress is an important result of cytotoxic chemotherapy and future research should consider immunogenic cell stress as a whole rather than just immunogenic cell death or immunogenic modulation. Cancer treatment strategies should be designed specifically to take advantage of these effects in combination immunotherapy, and novel chemotherapy regimens should be designed and investigated to potentially induce all aspects of immunogenic cell stress.
For the past 100 years cancer treatment has undergone rapid evolutions, from surgery to radiotherapy and chemotherapy, to hormonal and molecular targeted therapies and in recent decades immunotherapy. While Paul Ehrlich first hypothesized that the immune system targets nascent tumors for destruction in 1909, it wasn’t until the 1950s that the concept of tumor neo-antigens and the immune surveillance hypothesis was developed (1). The maturation of immunotherapeutic strategies has shed new light on the role of the immune system in effective cancer therapy and reinforced the belief that harnessing the patient’s immune system is a necessary factor in effective cancer treatment. It has also led to the reassessment of the impact of pre-existing cancer treatments, especially radiotherapy and chemotherapy strategies. While different chemotherapeutics have wide-ranging effects on the immune system itself (2), the impact of chemotherapy treatment on tumor immunogenicity and the interplay between tumor and immune response are also significant and may be important for the development of future clinical strategies.
The 20th century’s evolution of cancer treatment began with the discovery of X rays in 1895, which quickly resulted in their use to treat breast cancer in 1896. The first half of the 20th century saw the rapid discovery of new radiation sources and their application to cancer treatment (3). In combination with surgery, radiotherapy became the standard of care for cancer therapy until the 1960s (4). In parallel with the development of radiotherapy strategies, research that occurred during World Wars I and II resulted in the development of the first chemotherapeutic drugs. Throughout the latter half of the last century novel chemotherapeutic agents were created; to date there are five conventional types of chemotherapy: alkylating agents, antimetabolites, cytotoxic antibiotics, mitotic inhibitors, and topoisomerase targeting agents (5) (Table 1).
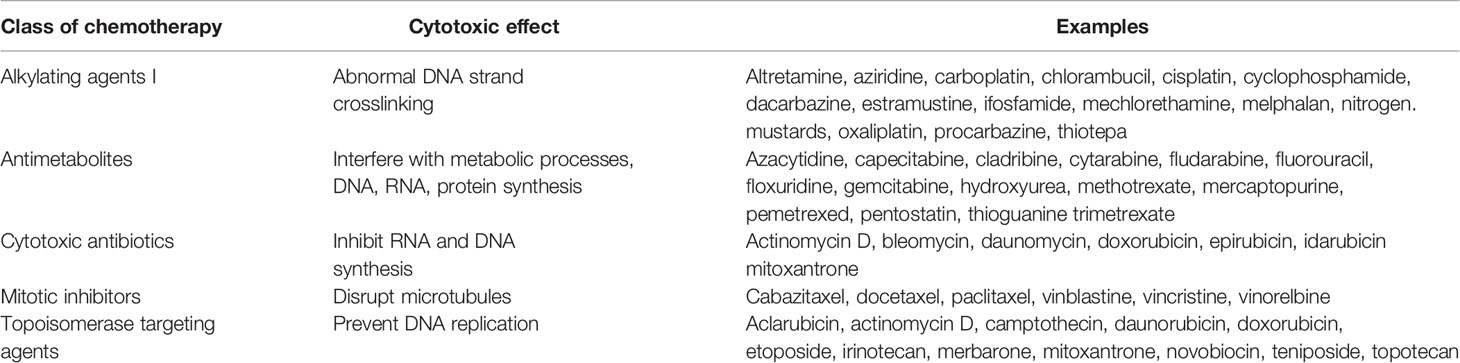
Table 1 Classes of conventional chemotherapy agents.
Despite their diverse targets and functions, each class of conventional chemotherapy causes cell death through essentially similar mechanisms: they induce DNA damage, disrupt DNA synthesis or repair, or target the basic functions of cell division. However, the diversity of targets means the classes synergize effectively. In the early 1960s the first combination chemotherapy trials were performed, combining nitrogen mustard, vincristine, methotrexate and prednisone (MOMP) or nitrogen mustard, vincristine, prednisone and procarbazine (MOPP). In advanced Hodgkin’s lymphoma, MOPP resulted in complete remission in 80% of patients with no relapse in 60% of patients, and the era of combination chemotherapy was born (4, 6).
By targeting cell division, conventional chemotherapy induces results in the death of rapidly proliferating cells, one of the primary hallmarks of cancerous cells (7). However, by targeting all rapid proliferating cells, chemotherapy also results in patient toxicities and morbidities, including immunosuppression. From the beginnings of chemotherapy research, agents cytotoxic to tumors have also been shown to be immunosuppressive, including nitrogen mustard, which was shown to induce lymphopenia, granulocytopenia and thrombocytopenia early in its development (8). Elimination or depletion of immune cells has remained a frequently observed side effect of many other chemotherapies developed since then. Interestingly, depending on chemotherapy dose and schedule, researchers have shown that chemotherapy-mediated immune cell depletion can be beneficial for the development of a vaccine-stimulated tumor antigen-specific immune response (9–11). Some chemotherapies have also been shown to have direct immunostimulant effects, increasing the maturation and cytotoxic potential of certain immune cell populations (12). In addition to its effects on the immune system itself, chemotherapy has recently been shown to impact the interaction between tumors and the immune system, primarily through cell stress-related processes called immunogenic modulation and immunogenic cell death (ICD).
The DNA damage, inhibition of DNA synthesis and repair or disruption of cell division caused by chemotherapies damage cells by taking advantage of their cell stress mechanisms (Figure 1). If this results in cell death it often induces immunogenic cell death, wherein damage-associated molecular patterns (DAMPs) or “eat me” signals are upregulated to promote immune cell phagocytosis and tumor antigen processing. Common DAMPs include membrane translocation of calreticulin (CRT), release of the pro-inflammatory protein high mobility group box 1 (HMGB1), heat shock protein (HSP) translocation, secretion of adenosine triphosphate (ATP) and type 1 interferons (IFN) (13). Immunogenic cell death and the resulting antigen processing promote a tumor-specific immune system similar to a vaccine. If chemotherapy-induced cell stress does not result in death, it can promote the expression of pro-apoptotic and immune cell engaging molecules such as Fas, tumor necrosis factor (TNF)-related apoptosis-inducing ligand (TRAIL) receptors and increased antigen presentation that sensitize surviving tumor cells to immune cell killing through the process of immunogenic modulation (14). This enables an effective immune response against tumor cells that are resistant to cytotoxic chemotherapy. While immunogenic cell death and immunogenic modulation have previously been believed to be separate processes (14), it is becoming increasingly clear that they are parts of the same spectrum of immunogenic cell stress.
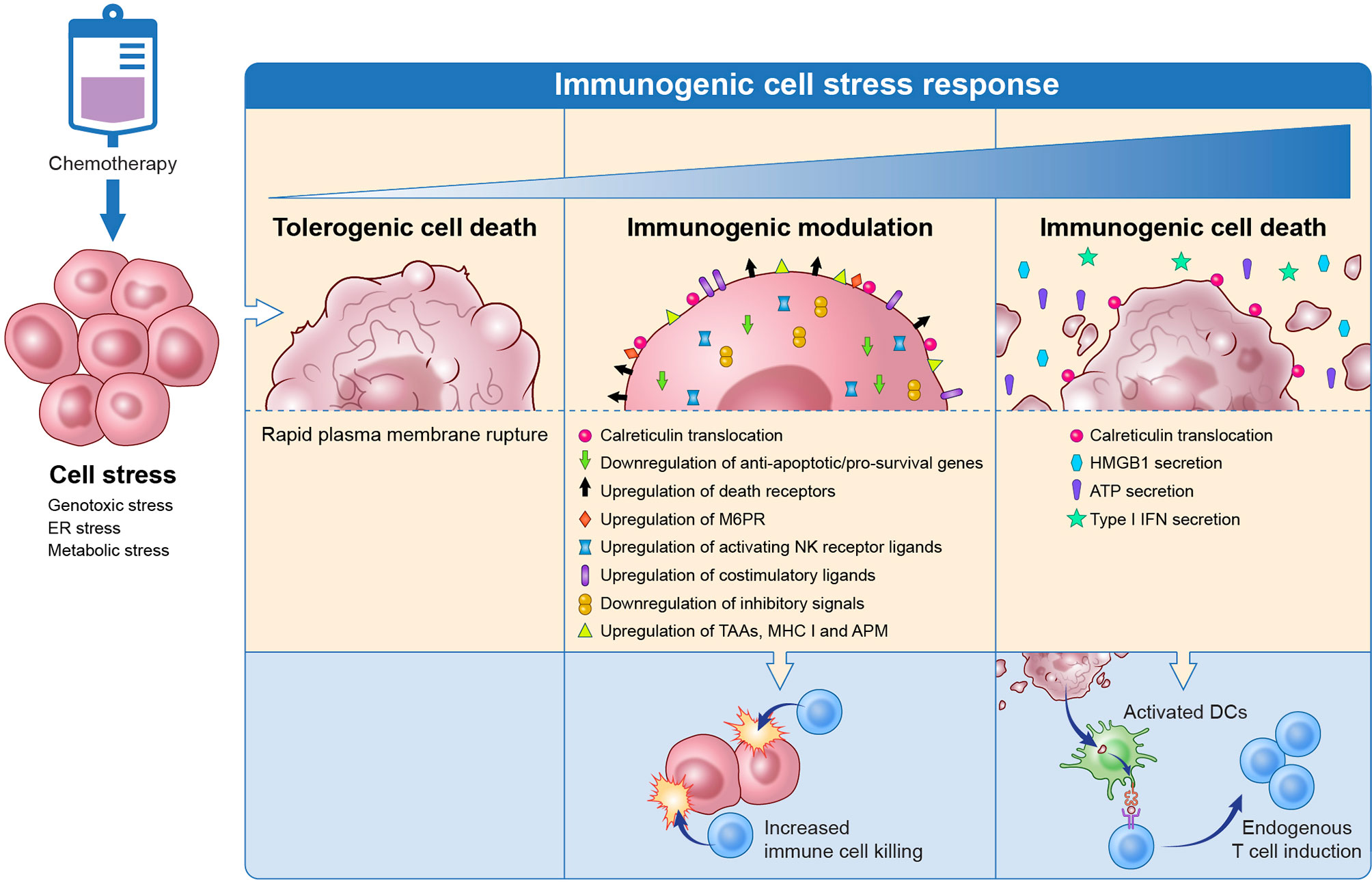
Figure 1 Chemotherapy induces immunogenic cell stress resulting in a spectrum of downstream events from tolerogenic cell death to immune modulation to immunogenic cell death. Tolerogenic cell death induced by chemotherapy is a non-inflammatory cell death that involves the rapid rupture of the plasma membrane and does not trigger immune cells. On the other end of the spectrum, chemotherapy can cause immunogenic cell death that results in the release of DAMPs that promote immune cell phagocytosis, tumor antigen processing, and antigen presentation to T cells. Tumor cells that were not eradicated with chemotherapy undergo immunogenic modulation wherein the tumor surface phenotype is altered to promote T cell targeting of the tumor cell. DAMP, damage-associated molecular patterns; TAA, tumor-associated antigen; MHC, major histocompatibility complex; APM, antigen processing machinery; M6PR, mannose-6-phosphate receptor; NK, natural killer; HMGB1, high mobility group box 1; ATP, adenosine triphosphate; IFN, interferon. Adapted from Ref (14).
For effective immunotherapy it is necessary to target four different modes of the tumor-immune system interaction. Immunotherapy strategies must induce a tumor antigen-specific T-cell population, expand the number of antigen-specific T cells and promote their migration to the tumor microenvironment, enable a prolonged immune response, and ensure an evolving immune response to prevent tumor escape (15, 16). Depending on the mechanisms engaged, immunogenic cell stress can induce antigen-specific T cells through immunogenic cell death or enable increased immune cell killing of tumor cells through immunogenic modulation. However, as not all chemotherapeutic strategies induce immunogenic cell stress, it is essential for clinicians to rationally design combinations of chemotherapy and immunotherapy for maximum synergy between these two treatment modes. While significant work has investigated these strategies in vitro and in vivo, few clinical trials currently exist that are explicitly interrogating the immunogenic effects of chemotherapy and how they could support novel immunotherapeutic strategies.
In this review we will discuss the current bodies of work investigating chemotherapy-induced immunogenic modulation and immunogenic cell death and the important similarities and differences in these processes. We posit that, rather than separate processes, immunogenic modulation and immunogenic cell death should be considered as the results of a spectrum of immunogenic cell stress that can be utilized in combination with immunotherapy to develop novel and effective clinical strategies. Finally, we will examine the state of clinical trials investigating the role of immunogenic cell stress and look to the future of combination immunotherapy utilizing standard-of-care agents that both induce immunogenic cell stress and immuno-oncology agents.
Cell stress is the fundamental process governing the induction of chemotherapy-mediated ICD and immunogenic modulation (17, 18). Cell stress is largely governed by the endoplasmic reticulum (ER), an organelle involved in crucial cellular functions such as calcium (Ca2+) homeostasis, lipid biosynthesis, and the proper folding and assembly of both secretory and transmembrane proteins (19). Misfolded proteins impede cellular functions, so the quality control performed by the endoplasmic reticulum has evolved to correct misfolded proteins or, if terminally misfolded, destroy them or the cell itself (20). This occurs through the unfolded protein response (UPR), which halts protein translation and increases expression of chaperone proteins to promote proper folding, which if unsuccessful induces cellular apoptosis.
The UPR is facilitated by the ER transmembrane proteins inositol-requiring enzyme 1α (IRE1α), protein kinase R-like ER kinase (PERK), and activating transcription factor 6α (ATF6α) (21). In unstressed cells, these proteins are inactive and are bound to the ER chaperone protein BiP (22–24); however, under conditions of ER stress, BiP dissociates from these UPR sensors to bind misfolded proteins and promote correct folding. These UPR sensors are then free to oligomerize and drive the UPR, halting further protein translation and upregulating stress response gene expression.
While the UPR is an effective quality control process in healthy cells, during tumor development adverse conditions such as accelerated cell division, hypoxia, metabolic stress, and acidosis disrupt the protein folding capacity of the ER (25). This constant sublethal UPR activation allows the tumor cells to resolve misfolded proteins, thereby promoting cell survival and tumor progression in harsh microenvironmental conditions (26). However, the reliance of tumors on the UPR also makes it a tempting target for targeted molecular therapy (27), as well as a potential source for biomarkers of cancer in undiagnosed patients (28, 29).
While low-level activation of the UPR is essential for tumor survival, intense ER stress, like that caused by chemotherapy, results in the accumulation of the misfolded proteins and ultimately triggers cell death. However, chemotherapeutic agents do not target the ER directly, and do not directly induce the UPR (Tables 1 and 2). Immunogenic cell stress inducers such as anthracyclines (doxorubicin and mitoxantrone), oxaliplatin, and cyclophosphamide primarily target the DNA or DNA replication machinery proteins while agents such as cisplatin and taxanes target DNA and microtubules. This frequently results in inhibition of transcription, translation, and cell replication, inducing ER stress through secondary or ‘collateral’ effects (18, 30). In this way chemotherapeutic targeting of an organelle or cell structure of interest also impacts the ER and causes stress via mechanisms that have not yet been fully elucidated.
The ER stress stimulated by chemotherapeutic agents results in the activation of all the UPR pathways; however, only the PERK signaling pathway appears to be involved in immunogenic cell stress. Initiation of PERK results in the phosphorylation of the eukaryotic translation initiation factor 2a (eIF2a), which reduces protein translation and influx of newly synthesized proteins into the ER (21, 31, 32). This is followed by the activation of caspase-8, which cleaves the ER-resident membrane protein BAP31, and the activation of pro-apoptotic proteins Bax and Bak (33), which are key players in mitochondrial dysfunction (34). Finally, CRT (a common DAMP as previously mentioned) is translocated from the ER lumen to the plasma surface via the Golgi apparatus that is dependent on vesicle-associated membrane protein 1 (VAMP1) and synaptosomal-associated protein 25 (SNAP25) (33). CRT translocation enhances killing of the stressed cell by increasing immune cell recognition, increasing the immunogenicity of the stressed cell (35). Genetic inhibition of ERN1 (IRE1α) and ATF6 (ATF6α) did not affect CRT exposure while disruption of the PERK pathway blocked the CRT translocation induced by anthracyclines and oxaliplatin, demonstrating that PERK is mandatory for ICD (33).
The importance of ER stress in chemotherapy-induced immunogenic cell stress has been demonstrated in elegant experiments that blocked ER stress mechanisms and examined the resulting immunogenicity. Treatment with antioxidants inhibits the immunogenicity of dying cells, demonstrating the impact and importance of reactive oxygen-species (ROS)- mediated stress in the immunological activity of some anti-cancer agents (33, 36). Likewise, silencing or pharmacological blocking of molecular components of the ER stress response diminishes the immunogenicity of cell death triggered by ICD-inducing therapies (33, 37), and the intensity and quality of danger signaling during ICD has been shown to be affected by the strength and kinetics of the ER stress induced by anticancer treatments (17, 18).
Immunogenic cell stress is a spectrum of cellular mechanisms ranging from those that sensitize a living cell to immune-mediated killing to those that induce immunogenic cell death. The characteristics of a chemotherapeutic compound that make it capable of inducing immunogenic modulation versus immunogenic cell death remain unknown, and it has been demonstrated that compounds of substantial structural and functional similarity such as oxaliplatin [capable of inducing ICD (38)] and cisplatin [incapable of inducing ICD (39)] have differential immunogenic cell stress effects (40, 41). It is likely that this represents a difference in the levels of cell stress induction; however, further work is necessary to interrogate the continuum of immunogenic cell stress.
According to the Nomenclature Committee on Cell Death, ICD is “a form of regulated cell death that is sufficient to activate an adaptive immune response in immunocompetent hosts” (42). A variety of cytotoxic chemotherapies have been demonstrated to potentiate the spatiotemporally defined process of ICD, including idarubicin, epirubicin, doxorubicin, mitoxantrone, oxaliplatin, bortezomib, and cyclophosphamide (41).
Chemotherapy-driven ICD is characterized by the secretion or surface exposure of DAMPs by dying tumor cells (13, 43). Cognate receptor binding of DAMPs on antigen-presenting cells (APCs) promotes the recruitment, activation, and maturation of APCs, resulting in the uptake of tumor-associated antigens (TAAs). This is followed by the migration of APCs to the draining lymph nodes where they present the antigens to the cytotoxic T lymphocytes (CTLs), hence stimulating an anticancer response. The best-studied DAMPs pivotal for ICD are CRT, ATP, HMBG1, and type-I IFNs (13, 18, 42).
CRT, a Ca2+ binding chaperone protein mainly located in the lumen of the ER, is involved in the regulation of Ca2+ signaling and homeostasis, as well as major histocompatibility complex (MHC) class I assembly (44). Cells undergoing ICD translocate CRT and its cofactor, ERp57, to the plasma membrane (ecto-CRT) (33). This occurs during the pre-apoptotic stage, prior to caspase-3 cleavage, phosphatidylserine (PS) externalization, and plasma membrane permeabilization (33, 37). Both ecto-CRT and PS provide a potent “eat me” signal to macrophages and dendritic cells (DCs); however, only ecto-CRT triggers ICD while PS mediates the clearance of apoptotic cells and debris without activating an immune response (37, 45–48). Ecto-CRT binds CD91 on the APCs, promoting phagocytosis, tumor antigen presentation and subsequent activation of anti-tumor CTLs. In addition, ligation of ecto-CRT to CD91 on APCs promotes proinflammatory cytokine production and Th17 priming (49). Depletion of CRT using short interfering RNA (siRNA) results in the reduction of oxiplatin-induced immune response, whereas surface adsorption of recombinant CRT restores the immunogenicity of CRT-depleted cells undergoing ICD, implicating the key role of ecto-CRT in determining cell death as immunogenic (37, 38).
Extracellular ATP is an important molecule involved in numerous autocrine and paracrine cell signaling pathways (50). Similar to CRT translocation, the release of ATP into the extracellular space during chemotherapy-induced ICD typically occurs in the pre-apoptotic stage (51, 52). Pre-mortem autophagy appears to be required (but not sufficient) for the optimal release of ATP as chemotherapy failed to promote ATP secretion and anticancer immune response in autophagy-deficient tumors (52). Chemotherapy activates caspase-dependent secretion of ATP via lysosomal exocytosis, plasma membrane blebbing, and pannexin 1 channels (53). Once secreted, extracellular ATP serves as a “find me” signal to APCs via ATP binding to the P2Y2 receptor, which directs APC chemotaxis (54, 55). Furthermore, ATP signals through the P2RX7 receptor on the surface of DCs, activates the NLRP3 (NOD-, LRR- and pyrin domain-containing protein 3) inflammasome, and promotes IL-1b production (56). IL-1b stimulates DC activation and antigen presentation, both of which are necessary for the priming of tumor-specific CTLs. In preclinical models, chemotherapy is relatively inefficient against tumors deficient for P2RX7, caspase-1, IL-1b, or IL-1R (56). Furthermore, overexpression of CD39, an ectonucleotidase that hydrolyzes ATP, abrogated the chemotherapy-induced immunogenicity of dying tumor cells (57). In contrast, CD39 blockade improved the anti-tumor activity of immunogenic chemotherapy (58). In the clinical setting, it was observed that breast cancer patients with loss-of-function allele of P2RX7 had unfavorable disease outcomes relative to individuals with the normal allele (56). Altogether, these findings indicate that ATP is a crucial component of ICD-elicited immunogenicity.
HMGB1 is a non-histone chromatin-binding protein that is associated with DNA organization and transcription regulation (59). DCs are able to actively secrete their own nuclear HMGB1, where it acts as a signaling molecule for maturation, migration, and polarization of naïve T cells (60, 61). In contrast, HMGB1 is passively released in the extracellular space during the late apoptotic/necrotic stage of ICD, when both the nuclear and plasma membranes have been permeabilized (62). The precise stress response that promotes HMGB1 release is yet to be elucidated (13). Extracellular HMGB1 can bind to different pathogen recognition receptors (PRR) on myeloid cells such as the receptor for advanced glycation end products (RAGE) and Toll-like receptor 4 (TLR4) (63–65). HMGB1-mediated signaling via TLR4 signaling through the adaptor protein MYD88 is required and sufficient for ICD based on numerous genetic and pharmacological experiments (64, 66). The HMGB1-TLR4 interaction and downstream signaling inhibit the fusion between phagosome and lysosome, which facilitate processing and cross-presentation of tumor antigens by DCs. Blocking or deletion of critical components of the pathway abrogates chemotherapy-driven ICD (64, 66). Furthermore, breast cancer, colon cancer, and head and neck squamous cell carcinoma patients carrying TLR4 loss-of-function polymorphisms had poorer clinical outcomes with chemotherapy (38, 64, 67).
Type I IFNs have classically been strongly linked with antiviral immune responses; recent studies, however, have revealed that they also play a key role in therapy-driven anti-cancer immunity (68). The ICD activity of anthracyclines appears to involve the generation of nucleic acid stress in the form of double-stranded RNA molecules that trigger the endosomal PRR TLR3 (69). TLR3 activation, in turn, stimulates the rapid production of type I IFNs in the malignant cells. Autocrine and paracrine signaling of type I IFN promote the production of CXCL10, which acts as a chemoattractant for T cells. Type I IFN is also important in cancer immunosurveillance and in the cross-priming capacity of APCs (68, 70). Defects in type I IFN signaling impair chemotherapy-driven ICD. Tumors that are genetically deficient for TLR3 or type I IFN receptor are less susceptible to doxorubicin (69). In contrast, expression of MX1 (a gene signature downstream of type I IFN signaling) is a predictive biomarker for complete response to neoadjuvant anthracycline-based chemotherapy in breast carcinoma. Furthermore, MX1 expression also predicted metastasis-free survival with neoadjuvant chemotherapy in breast cancer patients with poor prognosis (69).
As discussed here and in the previous sections, DAMP emission by cells undergoing ICD is associated with the activation of the intracellular stress response. However, the exact mechanisms of DAMP release and signaling remain to be fully elucidated. Several other DAMPs have also been linked to ICD but have not been extensively described. Anthracycline treatment induces the release of annexin A1 (ANXA1), which mediates the interaction of APCs to the malignant cells undergoing ICD through its cognate receptor formyl peptide receptor 1 (FPR1) (71). Similar to CRT, heat shock proteins (HSP)70 and HSP90 are exposed by chemotherapy and serve as “eat me” signal to DCs (72, 73). Furthermore, mitochondrial transcription factor A (TFAM) is a structural homolog of HMGB1, and during ICD it promotes APC maturation and recruitment via the RAGE receptor (74, 75). Future research is likely to identify more immunogenic DAMPs, which may serve as novel biomarkers of immunogenic cell death.
In addition to inducing cell death and the release of DAMPs, chemotherapy can also increase the susceptibility of cancer cells to immune effectors through a process called immunogenic modulation (15, 76). Immunogenic modulation includes alterations in the biology of tumor cells, such as enhanced antigen presentation, changes in surface marker expression, and upregulation of pro-apoptotic molecules (14). This results in sensitizing tumor cells to killing by both CTLs and members of the innate immune response, promoting the destruction of malignant cells that were not eradicated by treatment. This has been demonstrated to result in increased benefit in the clinic. Patients who received chemotherapy and had disease recurrence had higher clinical benefit when treated with immunotherapy compared to individuals who were not previously treated with chemotherapy, demonstrating chemotherapy’s potential to enhance the efficacy of cytotoxic immune effectors (15, 76).
As with immunogenic cell death, immunogenic modulation is not a direct result of chemotherapy, but is instead the result of activation of cell stress pathways. While ICD is the result of chemotherapy-mediated cell stress-inducing cell death, cell stress occurs on a spectrum and does not always result in death of the cell. Immunogenic modulation is therefore the result of an active, but not cytotoxic, cell stress response to chemotherapy. One of the primary functions of cellular immunity is to destroy damaged, infected and malignant cells. Cell stress can prompt both innate and adaptive immune responses through a variety of mechanisms, including upregulation of MHC class I and NKG2D ligands MICA and MICB (77), as well as inducing pro-apoptotic signaling through Fas (78) and TRAIL receptors (79, 80).
Chemotherapy-induced cell stress promotes tumor antigenicity by upregulating the expression and presentation of tumor neoantigens or tumor-associated antigens on the tumor cells. Chemotherapy treatment has been shown to increase the expression of carcinoembryonic antigen (CEA) in colon and breast carcinoma cells (5-fluorouracil and docetaxel) and cancer-testis antigens in renal cell carcinoma and ovarian cancer cells (5′-aza-2′deoxycytidine) (14, 81–83). The observed increase in TAA expression in these studies was also associated with upregulated MHC class I expression and enhanced sensitivity of the chemotherapy-treated neoplastic cells to TAA-specific CTLs. Other chemotherapeutic agents such as cyclophosphamide, oxaliplatin, and gemcitabine have also been found to increase the expression of MHC class I (84). Docetaxel, paclitaxel, and doxorubicin were shown to promote the expression of the components of the MHC class I antigen processing machinery, including calnexin, LMP2, LMP7, TAP1, TAP2 and tapasin in cancer cells (14, 85). Furthermore, chemotherapy treatment can also induce epitope spreading by revealing weaker tumor antigenic epitopes and thus eliciting CTL responses against both dominant and subdominant TAA epitopes (86).
Chemotherapy can also alter the surface phenotype of malignant cells to increase tumor susceptibility to CTL attack. Paclitaxel, cisplatin, and doxorubicin treatment rendered tumor cells more sensitive to CTL killing by upregulating mannose-6-phosphate receptors (M6PR) on the tumor cell surface, which augments cell membrane permeability to granzyme B (87). Consequently, antigen-specific CTLs were also able to induce a strong anti-tumor response against neighboring tumor cells that did not express the tumor antigens. Chemotherapy also has the capability of inducing the expression of costimulatory molecules such as CD80 and inhibiting the expression of checkpoint molecules PD-L1 and PD-L2 on the tumor cell surface, resulting in enhanced recognition and killing by CTLs (88–90).
Innate immune cell activity against tumor cells can also be promoted with chemotherapy. In addition to providing danger signal during ICD, ecto-CRT has also been associated with improved IL-15 trans-presentation to natural killer (NK) cells (91). Hence, chemotherapy-induced CRT translocation to the plasma membrane may also sensitize the tumor cells to NK cell cytolysis. Chemotherapeutic agents have also been shown to sensitize tumor cells to NK cell cytolysis through the induction of ligands on the tumor surface such as MICA/B, ULPBs and B7-H6 that bind activating NK receptors (92).
In addition to promoting increased interaction between tumor cells and cytotoxic immune cells, chemotherapy can also upset the pro-survival signaling in the tumor cells. Different chemotherapies have been shown to stimulate the expression of death receptors on the surface of tumor cells (93), including FAS (also known as CD95), and TRAIL-R1 and TRAIL-R2 (also known as DR4 and DR5) on a large panel of cancer cells. When these death receptors interact with their cognate ligands, apoptosis is triggered. Hence the upregulation of death receptors may render cancer cells more susceptible targets for NK and T cells that express and secrete death ligands, thereby resulting in improved immune clearance (93). Conversely, anti-apoptotic and/or pro-survival genes, such as those belonging to the Bcl-2 gene family, have been shown to be downregulated by chemotherapy (94, 95).
Taken together, these observations indicate an immunogenic role for chemotherapy that, while distinct from ICD, is a result of cellular stress pathways. This is particularly advantageous since not all cancer cells can be eradicated with chemotherapy. Immunogenic modulation to sensitize malignant cells to adaptive and innate immune attack is an additional layer of chemotherapy-mediated immunogenic cell stress that can be exploited in the clinic for novel chemotherapy-immunotherapy combination therapy.
While chemotherapy-mediated immunogenic cell stress has been demonstrated consistently in the laboratory, few studies have investigated its effects in the clinic. Clinical progress has been hampered by the ongoing studies for the most effective chemotherapy dose and schedule to induce immunogenic cell stress, lack of systemic biomarkers and difficulty obtaining appropriate patient samples (96). However, as immunotherapy strategies are maturing, more trials are investigating the direct immunogenic effects of standard-of-care chemotherapy as well as novel chemotherapy treatment strategies to determine the best methods for these proven agents to effectively synergize with immuno-oncology agents. For example, preliminary data from the CheckRad-8 study (NCT03426657) demonstrated that induction treatment with a single cycle of cisplatin and docetaxel combined with durvalumab and tremelimumab resulted in pathologic complete response in the rebiopsy (48%) and increased intratumoral CD8+ cells (45%), indicating the feasibility and antitumor activity of this chemo-immunotherapy combination (97). Two recent review papers have identified 161 ongoing or recently completed clinical trials utilizing at least one chemotherapeutic agent that has been previously demonstrated to induce ICD (41, 98). Furthermore, we recently published an additional review discussing clinical trials that include immunogenic modulation and immunogenic cell death inducing agents in combination immunotherapy strategies on a cancer vaccine backbone (15).
One of the most important considerations when utilizing immunogenic cell stress in clinical strategies is the optimal chemotherapy dose and schedule to induce immunogenic cell stress and to combine it with immuno-oncology agents. Most chemotherapeutic agents are used at or near their maximum tolerated dose, which although effective at inducing tumor cell death often results in significant host toxicity, including immune suppression (99). To combat this, alternative chemotherapy dosing schedules have been investigated. The most common alternative is metronomic chemotherapy, a strategy wherein frequent low doses of chemotherapy are delivered to the patient. Metronomic chemotherapy is better tolerated, especially by older or infirm patients, and while it still has been shown to deplete immune cells in certain contexts (100), it also induces immunogenic cell death (85, 101). Some have also proposed the use of medium-dose intermittent chemotherapy, wherein chemotherapy is given at a dose high enough to be cytotoxic in a majority of tumor cells, but not so high that it induces significant immunosuppression (102, 103). These different doses and schedules have been reviewed in detail previously (102, 104, 105), and therefore will not be here. Medium-dose intermittent chemotherapies have yet to enter the clinic, and while there are approximately 130 clinical trials examining metronomic chemotherapy (clinicaltrials.gov), we have identified nine trials utilizing metronomic chemotherapy in immunotherapy combination strategies (Table 2). To date no data have been published.
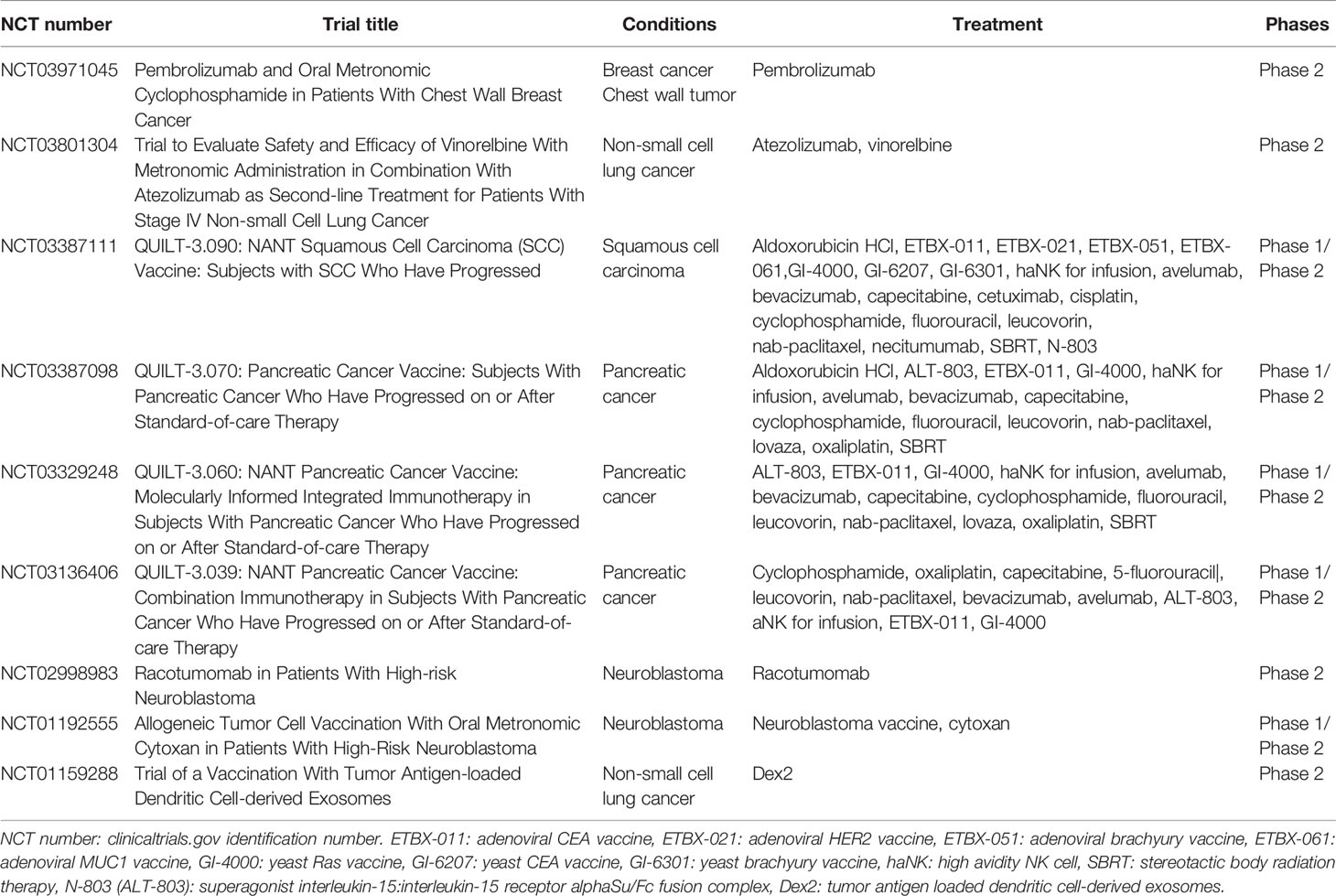
Table 2 Clinical trials combining metronomic chemotherapy with immuno-oncology agents.
Several studies have been published examining defined immunogenic cell stress biomarkers in the clinic. In lung cancer patients, investigators found that serum levels of CRT, an aforementioned DAMP indicative of cell stress, were significantly increased compared to healthy controls, and further overexpressed in lung cancer patients who had received chemotherapy compared to those who had not (chemotherapy type not specified) (106). However, no comparison between serum calreticulin and patient response was made. In ovarian carcinoma and non-small cell lung cancer (NSCLC), chemotherapy-independent CRT exposure was associated with increased immune cell infiltrates in the tumor and superior overall survival (107, 108), potentially lending clinical significance to the importance of CRT translocation in immunogenic cell stress. In acute myeloid leukemia (AML), researchers found that translocation of calreticulin and upregulation of HSP70 and HSP90 were not increased by treatment with the immunogenic cell death-inducing agents anthracycline, idarubicin or daunorubicin; however, they did find an increase in calreticulin exposure in AML blasts compared to healthy controls. Moreover, patients with high calreticulin exposure had improved disease outcome, regardless of chemotherapy, compared to calreticulin low patients. Interestingly, the investigators also reported a decrease in serum levels of the immunogenic cell stress marker HMGB1 following chemotherapy treatment, which they hypothesized might have been due to decreased numbers of abnormal cells releasing HMGB1 (109). Serum HMGB1 has also been shown to decrease following treatment in breast cancer, and was demonstrated to correlate with treatment efficacy (p=0.053) (110). However, a separate study reported that complete loss of HMGB1 is linked to poor response in breast cancer. Immunohistochemistry examination of breast cancer tumors from patients treated with adjuvant anthracycline showed that tumors with no nuclear HMGB1 staining in the majority of their cells were significantly associated with a negative impact on overall and progression-free survival (111).
These findings make it clear that further investigation into effective biomarkers and the dynamics of immunogenic cell stress in the clinic is required to validate published pre-clinical findings. We have identified five clinical trials (Table 3) focused on observation and biomarker detection. The first, a completed study of patients with non-small cell lung cancer (NCT02921854) is looking for exosomal or molecular markers of ICD in the serum of patients following high-dose radiotherapy or concurrent cisplatin and radiotherapy. However, no data are available at this time. An additional completed trial (NCT01513408) is investigating biomarkers in non-small cell lung cancer; no results have been reported yet.
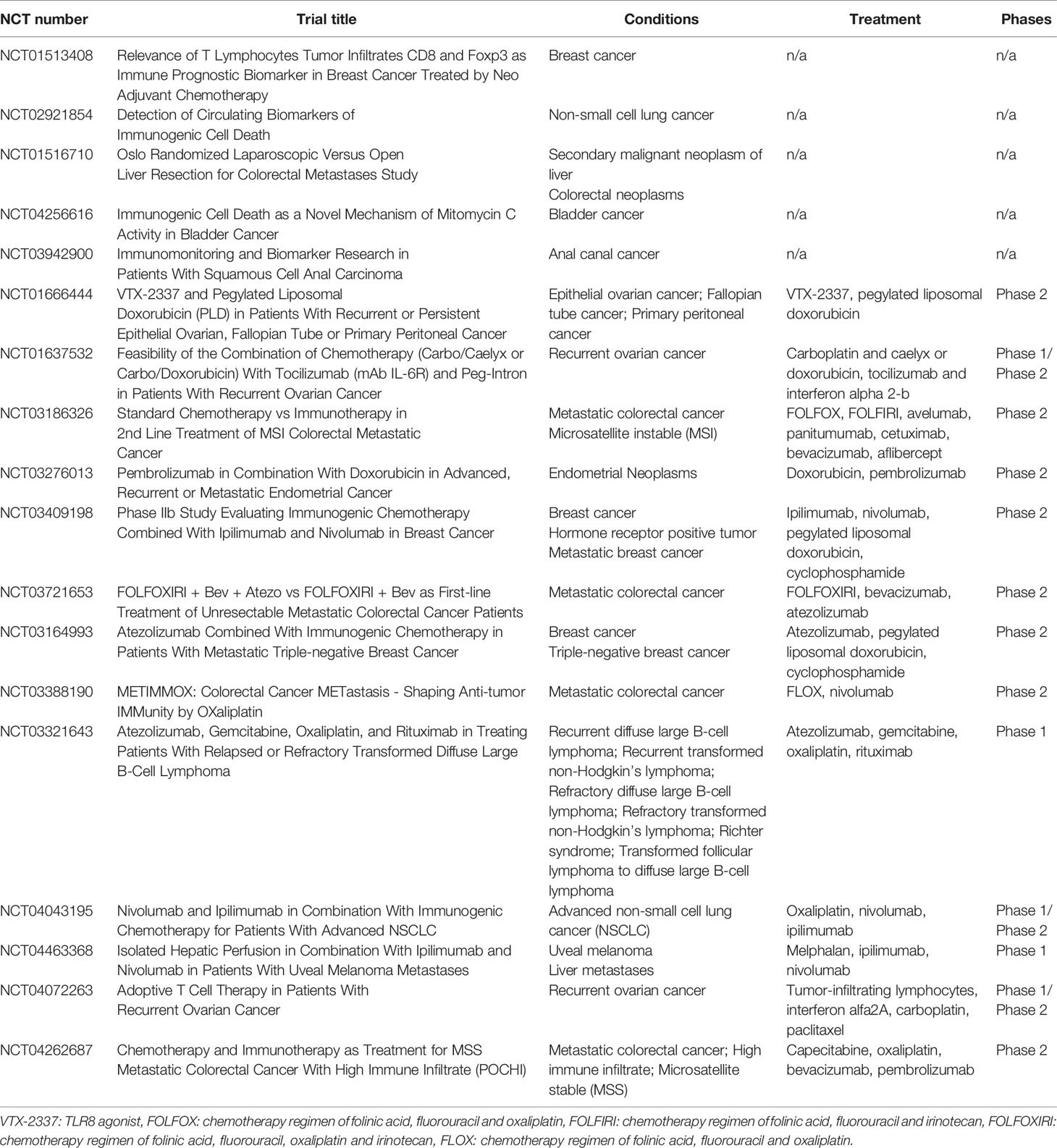
Table 3 Clinical trials investigating immunogenic cell death.
One active observational trial investigated patients with liver metastases from colorectal carcinoma (NCT01516710). While the main purpose of the study was to compare laparoscopic versus liver resection techniques for identifying colorectal metastases, secondary outcomes included investigating the metastases for markers of ICD. Using deep sequencing, the authors found that patients who had received neoadjuvant chemotherapy (which included known ICD inducer oxaliplatin in 11/15 patients) had a gene signature including genes related to toll-like receptor signaling, IFN response and leukocyte infiltration (112). In a further study investigating this patient population, it was shown that while there was no association between neoadjuvant chemotherapy and intratumoral T-cell density within colorectal liver metastases, there was a significant increase in intratumoral T-cell density in patients who received neoadjuvant chemotherapy fewer than 9.5 weeks before liver metastases resection compared to both patients with a longer interval and those who did not receive chemotherapy. This result is highly interesting and could be an important data point when designing clinical strategies meant to take advantage of chemotherapy-induced immunogenic cell death (113).
Although it is undeniable that chemotherapeutic agents induce ICD and immunogenic modulation, it remains unclear how these processes contribute to the clinical efficacy of chemotherapies. Clues to the importance of immunogenic cell stress in attaining chemotherapy-mediated therapeutic benefit can be gleaned from a randomized phase III trial comparing upfront oxaliplatin and 5-fluorouracil combination to sequential chemotherapy with single agent 5-fluorouracil until failure, followed by oxaliplatin and 5-fluorouracil combination in colorectal cancer (NCT00126256). In this study, oxaliplatin treatment increased the progression-free survival and overall survival of patients with normal TLR4 allele but did not improve clinical outcomes in patients with loss-of-function TLR4 allele. Without chemotherapy treatment, no differences in disease-free survival were observed among patients with normal or variant TLR4 allele. The results show that TLR4, a receptor for HMGB1, may be a prognostic factor but only in the context of immunogenic chemotherapy. The data indicate that immunogenic cell stress, specifically ICD, may indeed be a contributor to the clinical efficacy of oxaliplatin (38).
We identified an additional 13 clinical trials (Table 3) explicitly examining chemotherapy-induced immunogenic cell death, including two that are completed, four that are active and seven that are currently recruiting or yet to begin. Only one of the completed trials has published the complete results of the trial. This phase II trial (NCT01666444) investigated the combination of the toll-like receptor 8 (TLR8) agonist motolimod with ICD-inducing chemotherapeutic pegylated liposomal doxorubicin in women with recurrent or persistent ovarian cancer. Motolimod has previously been shown to activate NK cells, promote antibody-dependent cellular cytotoxicity, increase IFNγ production and drive the maturation of dendritic cells. Therefore, investigators hypothesized that motolimod treatment would synergize with the induction of ICD in patients. The investigators found that while motolimod combined with doxorubicin was well tolerated, there was no significant improvement in overall survival or progression-free survival compared to placebo. They observed that despite the lack of efficacy, motolimod did increase plasma expression of inflammatory cytokines and chemokines, including IL-1β, IL-6 and TNF-⍺. Interestingly, patients who experienced injection site reaction were determined to have a longer overall survival (19.8 months) than those who did not (13.3 months). It is possible this is demonstrative of overall patient immune response, and may indicate the need for patient selection for treatment with motolimod in this context (114).
A second phase I/II trial in ovarian cancer investigated the combination of the ICD inducing chemotherapeutics carboplatin or doxorubicin in combination with tocilizumab, an anti-IL-6R antibody, and pegylated IFN-⍺ (NCT01637532). IL-6 promotes the polarization of macrophages into immunosuppressive M-2 like macrophages and has been demonstrated to recruit T-regulator (Treg) cells, both of which work to inhibit an anti-tumor immune environment. Furthermore, IFN-⍺ promotes DC maturation. The investigators hypothesized that in combination with ICD, anti-IL-6 and IFN-⍺ would promote an anti-tumor immune reaction. While the complete data have yet to be published, this trial reported no dose-limiting toxicities (DLTs), and found that IL-6 was effectively blocked when 8mg/kg tocilizumab was delivered (115). The survival results from the phase II portion of the trial have yet to be released.
The efficacy of inducing immunogenic cell stress and immunogenic cell death in the clinic remains to be verified but immunogenic cell stress inducing agents will remain key parts of cancer treatment. It is likely they will play a role in the development of rationally designed immunotherapy strategies in the future.
In many indications chemotherapeutic agents remain the standard-of-care therapy despite their high potential for toxicity. While the discovery of immuno-oncology agents has resulted in a revolution in cancer treatment, clinicians have also realized that monotherapy treatment is insufficient in many patients. This has led to the development of combination immunotherapy treatment strategies and, importantly, to the rational combination of immuno-oncology agents with standard-of-care chemotherapy agents. While these often synergize well, with further research into the mechanisms and optimum dose/schedule by which chemotherapy agents induce immunogenic cell stress, they may be applied in highly effective targeted combinations with existing and future immuno-oncology agents.
Significant work has demonstrated that many chemotherapy agents can promote either immunogenic modulation or immunogenic cell death. It has become increasingly clear that these are the result of the same mechanistic pathways underneath the umbrella of immunogenic cell stress. Through cell stress mechanisms, chemotherapy sensitizes tumor cells to immune cell killing and increases the likelihood of tumor antigens released by dead tumor cells stimulating the immune system, resulting in increased numbers and infiltration of tumor-specific T cells and other immune cells necessary for immune-mediated tumor resolution.
While the cell stress response is a necessary mechanism for cellular health and quality control in response to intrinsic errors, there is a wide spectrum of cell stress responses depending on the level and duration of the stress. In the case of cells treated with cytotoxic chemotherapy, this means that the specifics of the cell stress response are dependent on the utilized dose, number of treatments, and other factors (116). However, the full dynamics of the cell stress response in relation to chemotherapy remain unknown, and further investigation is necessary to better enable clinicians to strategically utilize chemotherapy to induce immunogenic cell stress.
Furthermore, it remains unknown why some chemotherapeutic drugs induce ICD, some immunogenic modulation, some both and some neither. The exact mechanism of cell stress induced by a chemotherapy regimen is likely the driving factor for whether it will result in immunogenic cell stress or not, and as more is discovered about cell stress mechanisms it is possible these questions will find definitive answers.
In this review we have focused on the roles of chemotherapy in inducing immunogenic cell stress, but it should not be ignored that many other agents have been shown to have similar effects that also synergize with immuno-oncology agents, and depending on cancer indication or patient status these agents should also be considered for combination immunotherapy. In addition to chemotherapy, radiation is the best characterized inducer of immunogenic cell death (117), although recent findings have also demonstrated it is possible to induce local immunogenic cell death through photodynamic therapy (118). While high dose, stereotactic ablative body radiotherapy appears to be the most effective at inducing ICD, further research into the optimal dose, schedule and potential combinations with radiosensitizers and immuno-oncology agents is necessary (119–121). Multiple modalities of sublethal radiation including radiotherapy, external beam radiation, radiolabeled antibodies and brachytherapy have also been demonstrated to be effective inducers of immunogenic modulation (122, 123). Immunogenic modulation has also been demonstrated after treatment with endocrine deprivation agents (124, 125) and small molecule inhibitors (126–128). These findings make it clear that many cancer therapies, applied sublethally, can induce immunogenic cell stress that sensitizes tumor cells to immune killing. It should also be noted that there is potential to utilize immunogenic cell stress inducing therapies strictly as immuno-oncology agents in combination with standard-of-care therapies, for instance to abrogate the immunosuppressive effects of high-dose chemotherapy.
As cancer therapy strategies include immuno-oncology agents to greater degrees, it is becoming more crucial to identify the potential immunogenic effects of well characterized standard-of-care therapies. While much happened during the development of chemotherapy, immunotherapy is rapidly entering the age of combination therapy, which will include previously defined standard-of-care chemotherapy. By employing chemotherapy not just as an anti-cancer agent but also as immunogenic cell stress-inducing agent, clinicians will add another immuno-oncology tool to their toolbox, resulting in improved clinical success and patient recovery.
All authors contributed to the writing of this article and approved the submitted version.
This research was supported by the Intramural Research Program of the Center for Cancer Research, National Cancer Institute (NCI), National Institutes of Health.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The authors thank Debra Weingarten for her assistance in the preparation of this manuscript.
1. Dunn GP, Old LJ, Schreiber RD. The Three Es of Cancer Immunoediting. Annu Rev Immunol (2004) 22:329–60. doi: 10.1146/annurev.immunol.22.012703.104803
2. Zitvogel L, Apetoh L, Ghiringhelli F, Kroemer G. Immunological Aspects of Cancer Chemotherapy. Nat Rev Immunol (2008) 8(1):59–73. doi: 10.1038/nri2216
3. Lederman M. The Early History of Radiotherapy: 1895–1939. Int J Radiat Oncol Biol Phys (1981) 7(5):639–48. doi: 10.1016/0360-3016(81)90379-5
4. DeVita VT, Chu E. A History of Cancer Chemotherapy. Cancer Res (2008) 68(21):8643. doi: 10.1158/0008-5472.CAN-07-6611
5. Shields M. Chapter 14 - Chemotherapeutics. In: Badal Mccreath S, Delgoda R, editors. Pharmacognosy: Fundamentals, Applications and Strategies. Boston: Academic Press (2016). p. 295–313.
6. Devita VT Jr., Serpick AA, Carbone PP. Combination Chemotherapy in the Treatment of Advanced Hodgkin’s Disease. Ann Intern Med (1970) 73(6):881–95. doi: 10.7326/0003-4819-73-6-881
7. Hanahan D, Weinberg RA. The Hallmarks of Cancer. Cell (2000) 100(1):57–70. doi: 10.1016/s0092-8674(00)81683-9
8. Gilman A, Philips FS. The Biological Actions and Therapeutic Applications of the B-Chloroethyl Amines and Sulfides. Science (1946) 103(2675):409. doi: 10.1126/science.103.2675.409
9. Montero E, Valdes M, Avellanet J, Lopez A, Perez R, Lage A. Chemotherapy Induced Transient B-Cell Depletion Boosts Antibody-Forming Cells Expansion Driven by an Epidermal Growth Factor-Based Cancer Vaccine. Vaccine (2009) 27(16):2230–9. doi: 10.1016/j.vaccine.2009.02.018
10. Bracci L, Schiavoni G, Sistigu A, Belardelli F. Immune-Based Mechanisms of Cytotoxic Chemotherapy: Implications for the Design of Novel and Rationale-Based Combined Treatments Against Cancer. Cell Death Differ (2014) 21(1):15–25. doi: 10.1038/cdd.2013.67
11. Emens LA, Asquith JM, Leatherman JM, Kobrin BJ, Petrik S, Laiko M, et al. Timed Sequential Treatment With Cyclophosphamide, Doxorubicin, and an Allogeneic Granulocyte-Macrophage Colony-Stimulating Factor-Secreting Breast Tumor Vaccine: A Chemotherapy Dose-Ranging Factorial Study of Safety and Immune Activation. J Clin Oncol (2009) 27(35):5911–8. doi: 10.1200/jco.2009.23.3494
12. Shurin MR, Naiditch H, Gutkin DW, Umansky V, Shurin GV. Chemoimmunomodulation: Immune Regulation by the Antineoplastic Chemotherapeutic Agents. Curr Med Chem (2012) 19(12):1792–803. doi: 10.2174/092986712800099785
13. Galluzzi L, Vitale I, Warren S, Adjemian S, Agostinis P, Martinez AB, et al. Consensus Guidelines for the Definition, Detection and Interpretation of Immunogenic Cell Death. J Immunother Cancer (2020) 8(1):e000337. doi: 10.1136/jitc-2019-000337
14. Hodge JW, Garnett CT, Farsaci B, Palena C, Tsang K-Y, Ferrone S, et al. Chemotherapy-Induced Immunogenic Modulation of Tumor Cells Enhances Killing by Cytotoxic T Lymphocytes and Is Distinct From Immunogenic Cell Death. Int J Cancer (2013) 133(3):624–36. doi: 10.1002/ijc.28070
15. Wolfson B, Hodge JW. Next Generation Therapeutic Strateg-Es: Evolving Cancer Immunotherapy Through Agents That Engage, Expand and Enable the Anti-Tumor Immune Response. ImmunoMedicine (2021) 00):e1020. doi: 10.1002/imed.1020
16. Fabian KP, Padget MR, Fujii R, Schlom J, Hodge JW. Differential Combination Immunotherapy Requirements for Inflamed (Warm) Tumors Versus T Cell Excluded (Cool) Tumors: Engage, Expand, Enable, and Evolve. J Immunother Cancer (2021) 9(2):e001691. doi: 10.1136/jitc-2020-001691
17. Rufo N, Garg AD, Agostinis P. The Unfolded Protein Response in Immunogenic Cell Death and Cancer Immunotherapy. Trends Cancer (2017) 3(9):643–58. doi: 10.1016/j.trecan.2017.07.002
18. Krysko DV, Garg AD, Kaczmarek A, Krysko O, Agostinis P, Vandenabeele P. Immunogenic Cell Death and Damps in Cancer Therapy. Nat Rev Cancer (2012) 12(12):860–75. doi: 10.1038/nrc3380
19. Almanza A, Carlesso A, Chintha C, Creedican S, Doultsinos D, Leuzzi B, et al. Endoplasmic Reticulum Stress Signalling – From Basic Mechanisms to Clinical Applications. FEBS J (2019) 286(2):241–78. doi: 10.1111/febs.14608
20. Meusser B, Hirsch C, Jarosch E, Sommer T. ERAD: The Long Road to Destruction. Nat Cell Biol (2005) 7(8):766–72. doi: 10.1038/ncb0805-766
21. Ron D, Walter P. Signal Integration in the Endoplasmic Reticulum Unfolded Protein Response. Nat Rev Mol Cell Biol (2007) 8(7):519–29. doi: 10.1038/nrm2199
22. Bertolotti A, Zhang Y, Hendershot LM, Harding HP, Ron D. Dynamic Interaction of Bip and ER Stress Transducers in the Unfolded-Protein Response. Nat Cell Biol (2000) 2(6):326–32. doi: 10.1038/35014014
23. Shen J, Chen X, Hendershot L, Prywes R. ER Stress Regulation of ATF6 Localization by Dissociation of Bip/GRP78 Binding and Unmasking of Golgi Localization Signals. Dev Cell (2002) 3(1):99–111. doi: 10.1016/S1534-5807(02)00203-4
24. Wang M, Wey S, Zhang Y, Ye R, Lee AS. Role of the Unfolded Protein Response Regulator GRP78/Bip in Development, Cancer, and Neurological Disorders. Antioxid Redox Signal (2009) 11(9):2307–16. doi: 10.1089/ars.2009.2485
25. Chen X, Cubillos-Ruiz JR. Endoplasmic Reticulum Stress Signals in the Tumour and its Microenvironment. Nat Rev Cancer (2021) 21(2):71–88. doi: 10.1038/s41568-020-00312-2
26. Wang M, Kaufman RJ. The Impact of the Endoplasmic Reticulum Protein-Folding Environment on Cancer Development. Nat Rev Cancer (2014) 14(9):581–97. doi: 10.1038/nrc3800
27. Ojha R, Amaravadi RK. Targeting the Unfolded Protein Response in Cancer. Pharmacol Res (2017) 120:258–66. doi: 10.1016/j.phrs.2017.04.003
28. Samanta S, Tamura S, Dubeau L, Mhawech-Fauceglia P, Miyagi Y, Kato H, et al. Clinicopathological Significance of Endoplasmic Reticulum Stress Proteins in Ovarian Carcinoma. Sci Rep (2020) 10(1):2160. doi: 10.1038/s41598-020-59116-x
29. Huang J, Pan H, Wang J, Wang T, Huo X, Ma Y, et al. Unfolded Protein Response in Colorectal Cancer. Cell Biosci (2021) 11(1):26. doi: 10.1186/s13578-021-00538-z
30. Humeau J, Sauvat A, Cerrato G, Xie W, Loos F, Iannantuoni F, et al. Inhibition of Transcription by Dactinomycin Reveals a New Characteristic of Immunogenic Cell Stress. EMBO Mol Med (2020) 12(5):e11622. doi: 10.15252/emmm.201911622
31. Harding HP, Zhang Y, Ron D. Protein Translation and Folding Are Coupled by an Endoplasmic-Reticulum-Resident Kinase. Nature (1999) 397(6716):271–4. doi: 10.1038/16729
32. Marciniak SJ, Garcia-Bonilla L, Hu J, Harding HP, Ron D. Activation-Dependent Substrate Recruitment by the Eukaryotic Translation Initiation Factor 2 Kinase PERK. J Cell Biol (2006) 172(2):201–9. doi: 10.1083/jcb.200508099
33. Panaretakis T, Kepp O, Brockmeier U, Tesniere A, Bjorklund A-C, Chapman DC, et al. Mechanisms of Pre-Apoptotic Calreticulin Exposure in Immunogenic Cell Death. EMBO J (2009) 28(5):578–90. doi: 10.1038/emboj.2009.1
34. Wei MC, Zong WX, Cheng EH, Lindsten T, Panoutsakopoulou V, Ross AJ, et al. Proapoptotic BAX and BAK: A Requisite Gateway to Mitochondrial Dysfunction and Death. Science (2001) 292(5517):727–30. doi: 10.1126/science.1059108
35. Malamas AS, Gameiro SR, Knudson KM, Hodge JW. Sublethal Exposure to Alpha Radiation (223Ra Dichloride) Enhances Various Carcinomas’ Sensitivity to Lysis by Antigen-Specific Cytotoxic T Lymphocytes Through Calreticulin-Mediated Immunogenic Modulation. Oncotarget (2016) 7(52):86937–47. doi: 10.18632/oncotarget.13520
36. Menger L, Vacchelli E, Adjemian S, Martins I, Ma Y, Shen S, et al. Cardiac Glycosides Exert Anticancer Effects by Inducing Immunogenic Cell Death. Sci Transl Med (2012) 4(143):143ra99. doi: 10.1126/scitranslmed.3003807
37. Obeid M, Tesniere A, Ghiringhelli F, Fimia GM, Apetoh L, Perfettini J-L, et al. Calreticulin Exposure Dictates the Immunogenicity of Cancer Cell Death. Nat Med (2007) 13(1):54–61. doi: 10.1038/nm1523
38. Tesniere A, Schlemmer F, Boige V, Kepp O, Martins I, Ghiringhelli F, et al. Immunogenic Death of Colon Cancer Cells Treated With Oxaliplatin. Oncogene (2010) 29(4):482–91. doi: 10.1038/onc.2009.356
39. Martins I, Kepp O, Schlemmer F, Adjemian S, Tailler M, Shen S, et al. Restoration of the Immunogenicity of Cisplatin-Induced Cancer Cell Death by Endoplasmic Reticulum Stress. Oncogene (2011) 30(10):1147–58. doi: 10.1038/onc.2010.500
40. Wang Y-J, Fletcher R, Yu J, Zhang L. Immunogenic Effects of Chemotherapy-Induced Tumor Cell Death. Genes Dis (2018) 5(3):194–203. doi: 10.1016/j.gendis.2018.05.003
41. Vanmeerbeek I, Sprooten J, De Ruysscher D, Tejpar S, Vandenberghe P, Fucikova J, et al. Trial Watch: Chemotherapy-Induced Immunogenic Cell Death in Immuno-Oncology. Oncoimmunology (2020) 9(1):1703449. doi: 10.1080/2162402x.2019.1703449
42. Galluzzi L, Vitale I, Aaronson SA, Abrams JM, Adam D, Agostinis P, et al. Molecular Mechanisms of Cell Death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ (2018) 25(3):486–541. doi: 10.1038/s41418-017-0012-4
43. Legrand AJ, Konstantinou M, Goode EF, Meier P. The Diversification of Cell Death and Immunity: Memento Mori. Mol Cell (2019) 76(2):232–42. doi: 10.1016/j.molcel.2019.09.006
44. Gelebart P, Opas M, Michalak M. Calreticulin, a Ca2+-Binding Chaperone of the Endoplasmic Reticulum. Int J Biochem Cell Biol (2005) 37(2):260–6. doi: 10.1016/j.biocel.2004.02.030
45. Li MO, Sarkisian MR, Mehal WZ, Rakic P, Flavell RA. Phosphatidylserine Receptor Is Required for Clearance of Apoptotic Cells. Science (2003) 302(5650):1560. doi: 10.1126/science.1087621
46. Gardai SJ, McPhillips KA, Frasch SC, Janssen WJ, Starefeldt A, Murphy-Ullrich JE, et al. Cell-Surface Calreticulin Initiates Clearance of Viable or Apoptotic Cells Through Trans-Activation of LRP on the Phagocyte. Cell (2005) 123(2):321–34. doi: 10.1016/j.cell.2005.08.032
47. Panaretakis T, Joza N, Modjtahedi N, Tesniere A, Vitale I, Durchschlag M, et al. The Co-Translocation of Erp57 and Calreticulin Determines the Immunogenicity of Cell Death. Cell Death Differ (2008) 15(9):1499–509. doi: 10.1038/cdd.2008.67
48. Obeid M. ERP57 Membrane Translocation Dictates the Immunogenicity of Tumor Cell Death by Controlling the Membrane Translocation of Calreticulin. J Immunol (2008) 181(4):2533. doi: 10.4049/jimmunol.181.4.2533
49. Pawaria S, Binder RJ. CD91-Dependent Programming of T-Helper Cell Responses Following Heat Shock Protein Immunization. Nat Commun (2011) 2(1):521. doi: 10.1038/ncomms1524
50. Novak I. ATP as a Signaling Molecule: The Exocrine Focus. Physiology (2003) 18(1):12–7. doi: 10.1152/nips.01409.2002
51. Martins I, Tesniere A, Kepp O, Michaud M, Schlemmer F, Senovilla L, et al. Chemotherapy Induces ATP Release From Tumor Cells. Cell Cycle (2009) 8(22):3723–8. doi: 10.4161/cc.8.22.10026
52. Michaud M, Martins I, Sukkurwala AQ, Adjemian S, Ma Y, Pellegatti P, et al. Autophagy-Dependent Anticancer Immune Responses Induced by Chemotherapeutic Agents in Mice. Science (2011) 334(6062):1573. doi: 10.1126/science.1208347
53. Martins I, Wang Y, Michaud M, Ma Y, Sukkurwala AQ, Shen S, et al. Molecular Mechanisms of ATP Secretion During Immunogenic Cell Death. Cell Death Differ (2014) 21(1):79–91. doi: 10.1038/cdd.2013.75
54. Elliott MR, Chekeni FB, Trampont PC, Lazarowski ER, Kadl A, Walk SF, et al. Nucleotides Released by Apoptotic Cells Act as a Find-Me Signal to Promote Phagocytic Clearance. Nature (2009) 461(7261):282–6. doi: 10.1038/nature08296
55. Ma Y, Adjemian S, Mattarollo Stephen R, Yamazaki T, Aymeric L, Yang H, et al. Anticancer Chemotherapy-Induced Intratumoral Recruitment and Differentiation of Antigen-Presenting Cells. Immunity (2013) 38(4):729–41. doi: 10.1016/j.immuni.2013.03.003
56. Ghiringhelli F, Apetoh L, Tesniere A, Aymeric L, Ma Y, Ortiz C, et al. Activation of the NLRP3 Inflammasome in Dendritic Cells Induces IL-1beta-Dependent Adaptive Immunity Against Tumors. Nat Med (2009) 15(10):1170–8. doi: 10.1038/nm.2028
57. Michaud M, Sukkurwala AQ, Martins I, Shen S, Zitvogel L, Kroemer G. Subversion of the Chemotherapy-Induced Anticancer Immune Response by the Ecto-Atpase CD39. Oncoimmunology (2012) 1(3):393–5. doi: 10.4161/onci.19070
58. Perrot I, Michaud H-A, Giraudon-Paoli M, Augier S, Docquier A, Gros L, et al. Blocking Antibodies Targeting the CD39/CD73 Immunosuppressive Pathway Unleash Immune Responses in Combination Cancer Therapies. Cell Rep (2019) 27(8):2411–25.e9. doi: 10.1016/j.celrep.2019.04.091
59. Klune JR, Dhupar R, Cardinal J, Billiar TR, Tsung A. HMGB1: Endogenous Danger Signaling. Mol Med (2008) 14(7):476–84. doi: 10.2119/2008-00034.Klune
60. Dumitriu IE, Baruah P, Valentinis B, Voll RE, Herrmann M, Nawroth PP, et al. Release of High Mobility Group Box 1 by Dendritic Cells Controls T Cell Activation via the Receptor for Advanced Glycation End Products. J Immunol (2005) 174(12):7506. doi: 10.4049/jimmunol.174.12.7506
61. Dumitriu IE, Bianchi ME, Bacci M, Manfredi AA, Rovere-Querini P. The Secretion of HMGB1 Is Required for the Migration of Maturing Dendritic Cells. J Leukoc Biol (2007) 81(1):84–91. doi: 10.1189/jlb.0306171
62. Scaffidi P, Misteli T, Bianchi ME. Release of Chromatin Protein HMGB1 by Necrotic Cells Triggers Inflammation. Nature (2002) 418(6894):191–5. doi: 10.1038/nature00858
63. Messmer D, Yang H, Telusma G, Knoll F, Li J, Messmer B, et al. High Mobility Group Box Protein 1: An Endogenous Signal for Dendritic Cell Maturation and Th1 Polarization. J Immunol (2004) 173(1):307. doi: 10.4049/jimmunol.173.1.307
64. Apetoh L, Ghiringhelli F, Tesniere A, Criollo A, Ortiz C, Lidereau R, et al. The Interaction Between HMGB1 and TLR4 Dictates the Outcome of Anticancer Chemotherapy and Radiotherapy. Immunol Rev (2007) 220(1):47–59. doi: 10.1111/j.1600-065X.2007.00573.x
65. Apetoh L, Ghiringhelli F, Tesniere A, Obeid M, Ortiz C, Criollo A, et al. Toll-Like Receptor 4-Dependent Contribution of the Immune System to Anticancer Chemotherapy and Radiotherapy. Nat Med (2007) 13(9):1050–9. doi: 10.1038/nm1622
66. Nayagom B, Amara I, Habiballah M, Amrouche F, Beaune P, de Waziers I. Immunogenic Cell Death in a Combined Synergic Gene- and Immune-Therapy Against Cancer. Oncoimmunology (2019) 8(12):e1667743–e. doi: 10.1080/2162402X.2019.1667743
67. Bergmann C, Bachmann HS, Bankfalvi A, Lotfi R, Pütter C, Wild CA, et al. Toll-Like Receptor 4 Single-Nucleotide Polymorphisms Asp299Gly and Thr399Ile in Head and Neck Squamous Cell Carcinomas. J Transl Med (2011) 9:139. doi: 10.1186/1479-5876-9-139
68. Zitvogel L, Galluzzi L, Kepp O, Smyth MJ, Kroemer G. Type I Interferons in Anticancer Immunity. Nat Rev Immunol (2015) 15(7):405–14. doi: 10.1038/nri3845
69. Sistigu A, Yamazaki T, Vacchelli E, Chaba K, Enot DP, Adam J, et al. Cancer Cell–Autonomous Contribution of Type I Interferon Signaling to the Efficacy of Chemotherapy. Nat Med (2014) 20(11):1301–9. doi: 10.1038/nm.3708
70. Bek S, Stritzke F, Wintges A, Nedelko T, Böhmer DFR, Fischer JC, et al. Targeting Intrinsic RIG-I Signaling Turns Melanoma Cells Into Type I Interferon-Releasing Cellular Antitumor Vaccines. Oncoimmunology (2019) 8(4):e1570779–e. doi: 10.1080/2162402X.2019.1570779
71. Vacchelli E, Ma Y, Baracco EE, Sistigu A, Enot DP, Pietrocola F, et al. Chemotherapy-Induced Antitumor Immunity Requires Formyl Peptide Receptor 1. Science (2015) 350(6263):972–8. doi: 10.1126/science.aad0779
72. Spisek R, Charalambous A, Mazumder A, Vesole DH, Jagannath S, Dhodapkar MV. Bortezomib Enhances Dendritic Cell (DC)–Mediated Induction of Immunity to Human Myeloma via Exposure of Cell Surface Heat Shock Protein 90 on Dying Tumor Cells: Therapeutic Implications. Blood (2007) 109(11):4839–45. doi: 10.1182/blood-2006-10-054221
73. Fucikova J, Kralikova P, Fialova A, Brtnicky T, Rob L, Bartunkova J, et al. Human Tumor Cells Killed by Anthracyclines Induce a Tumor-Specific Immune Response. Cancer Res (2011) 71(14):4821–33. doi: 10.1158/0008-5472.Can-11-0950
74. Krysko DV, Agostinis P, Krysko O, Garg AD, Bachert C, Lambrecht BN, et al. Emerging Role of Damage-Associated Molecular Patterns Derived From Mitochondria in Inflammation. Trends Immunol (2011) 32(4):157–64. doi: 10.1016/j.it.2011.01.005
75. Yang M, Li C, Zhu S, Cao L, Kroemer G, Zeh H, et al. TFAM is a Novel Mediator of Immunogenic Cancer Cell Death. Oncoimmunology (2018) 7(6):e1431086. doi: 10.1080/2162402x.2018.1431086
76. Hodge JW, Kwilas A, Ardiani A, Gameiro SR. Attacking Malignant Cells That Survive Therapy. OncoImmunology (2013) 2(12):e26937. doi: 10.4161/onci.26937
77. Groh V, Bahram S, Bauer S, Herman A, Beauchamp M, Spies T. Cell Stress-Regulated Human Major Histocompatibility Complex Class I Gene Expressed in Gastrointestinal Epithelium. Proc Natl Acad Sci USA (1996) 93(22):12445–50. doi: 10.1073/pnas.93.22.12445
78. Denning TL, Takaishi H, Crowe SE, Boldogh I, Jevnikar A, Ernst PB. Oxidative Stress Induces the Expression of Fas and Fas Ligand and Apoptosis in Murine Intestinal Epithelial Cells. Free Radic Biol Med (2002) 33(12):1641–50. doi: 10.1016/s0891-5849(02)01141-3
79. Dufour F, Rattier T, Constantinescu AA, Zischler L, Morlé A, Ben Mabrouk H, et al. TRAIL Receptor Gene Editing Unveils TRAIL-R1 as a Master Player of Apoptosis Induced by TRAIL and ER Stress. Oncotarget (2017) 8(6):9974–85. doi: 10.18632/oncotarget.14285
80. Sullivan GP, O’Connor H, Henry CM, Davidovich P, Clancy DM, Albert ML, et al. TRAIL Receptors Serve as Stress-Associated Molecular Patterns to Promote ER-Stress-Induced Inflammation. Dev Cell (2020) 52(6):714–30.e5. doi: 10.1016/j.devcel.2020.01.031
81. Correale P, Aquino A, Giuliani A, Pellegrini M, Micheli L, Cusi MG, et al. Treatment of Colon and Breast Carcinoma Cells With 5-Fluorouracil Enhances Expression of Carcinoembryonic Antigen and Susceptibility to HLA-a(*)02.01 Restricted, CEA-Peptide-Specific Cytotoxic T Cells In Vitro. Int J Cancer (2003) 104(4):437–45. doi: 10.1002/ijc.10969
82. Adair SJ, Hogan KT. Treatment of Ovarian Cancer Cell Lines With 5-Aza-2′-Deoxycytidine Upregulates the Expression of Cancer-Testis Antigens and Class I Major Histocompatibility Complex-Encoded Molecules. Cancer Immunol Immunother (2009) 58(4):589–601. doi: 10.1007/s00262-008-0582-6
83. Coral S, Sigalotti L, Altomonte M, Engelsberg A, Colizzi F, Cattarossi I, et al. 5-Aza-2′-Deoxycytidine-Induced Expression of Functional Cancer Testis Antigens in Human Renal Cell Carcinoma: Immunotherapeutic Implications. Clin Cancer Res (2002) 8(8):2690–5.
84. Liu WM, Fowler DW, Smith P, Dalgleish AG. Pre-Treatment With Chemotherapy Can Enhance the Antigenicity and Immunogenicity of Tumours by Promoting Adaptive Immune Responses. Br J Cancer (2010) 102(1):115–23. doi: 10.1038/sj.bjc.6605465
85. Kaneno R, Shurin GV, Kaneno FM, Naiditch H, Luo J, Shurin MR. Chemotherapeutic Agents in Low Noncytotoxic Concentrations Increase Immunogenicity of Human Colon Cancer Cells. Cell Oncol (Dordr) (2011) 34(2):97–106. doi: 10.1007/s13402-010-0005-5
86. Jackaman C, Majewski D, Fox SA, Nowak AK, Nelson DJ. Chemotherapy Broadens the Range of Tumor Antigens Seen by Cytotoxic CD8+ T Cells In Vivo. Cancer Immunol Immunother (2012) 61(12):2343–56. doi: 10.1007/s00262-012-1307-4
87. Ramakrishnan R, Assudani D, Nagaraj S, Hunter T, Cho HI, Antonia S, et al. Chemotherapy Enhances Tumor Cell Susceptibility to CTL-Mediated Killing During Cancer Immunotherapy in Mice. J Clin Invest (2010) 120(4):1111–24. doi: 10.1172/jci40269
88. Sojka DK, Donepudi M, Bluestone JA, Mokyr MB. Melphalan and Other Anticancer Modalities Up-Regulate B7-1 Gene Expression in Tumor Cells. J Immunol (2000) 164(12):6230. doi: 10.4049/jimmunol.164.12.6230
89. Ghebeh H, Lehe C, Barhoush E, Al-Romaih K, Tulbah A, Al-Alwan M, et al. Doxorubicin Downregulates Cell Surface B7-H1 Expression and Upregulates its Nuclear Expression in Breast Cancer Cells: Role of B7-H1 as an Anti-Apoptotic Molecule. Breast Cancer Res (2010) 12(4):R48. doi: 10.1186/bcr2605
90. Lesterhuis WJ, Punt CJ, Hato SV, Eleveld-Trancikova D, Jansen BJ, Nierkens S, et al. Platinum-Based Drugs Disrupt STAT6-Mediated Suppression of Immune Responses Against Cancer in Humans and Mice. J Clin Invest (2011) 121(8):3100–8. doi: 10.1172/jci43656
91. Truxova I, Kasikova L, Salek C, Hensler M, Lysak D, Holicek P, et al. Calreticulin Exposure on Malignant Blasts Correlates With Improved Natural Killer Cell-Mediated Cytotoxicity in Acute Myeloid Leukemia Patients. Haematologica (2020) 105(7):1868–78. doi: 10.3324/haematol.2019.223933
92. Zingoni A, Fionda C, Borrelli C, Cippitelli M, Santoni A, Soriani A. Natural Killer Cell Response to Chemotherapy-Stressed Cancer Cells: Role in Tumor Immunosurveillance. Front Immunol (2017) 8:1194. doi: 10.3389/fimmu.2017.01194
93. Elrod HA, Sun S-Y. Modulation of Death Receptors by Cancer Therapeutic Agents. Cancer Biol Ther (2008) 7(2):163–73. doi: 10.4161/cbt.7.2.5335
94. Gameiro SR, Caballero JA, Higgins JP, Apelian D, Hodge JW. Exploitation of Differential Homeostatic Proliferation of T-Cell Subsets Following Chemotherapy to Enhance the Efficacy of Vaccine-Mediated Antitumor Responses. Cancer Immunol Immunother (2011) 60(9):1227–42. doi: 10.1007/s00262-011-1020-8
95. Hodge JW, Ardiani A, Farsaci B, Kwilas AR, Gameiro SR. The Tipping Point for Combination Therapy: Cancer Vaccines With Radiation, Chemotherapy, or Targeted Small Molecule Inhibitors. Semin Oncol (2012) 39(3):323–39. doi: 10.1053/j.seminoncol.2012.02.006
96. Rapoport BL, Anderson R. Realizing the Clinical Potential of Immunogenic Cell Death in Cancer Chemotherapy and Radiotherapy. Int J Mol Sci (2019) 20(4): doi: 10.3390/ijms20040959
97. Hecht M, Gostian AO, Eckstein M, Rutzner S, von der Grün J, Illmer T, et al. Safety and Efficacy of Single Cycle Induction Treatment With Cisplatin/Docetaxel/Durvalumab/Tremelimumab in Locally Advanced HNSCC: First Results of Checkrad-CD8. J ImmunoTherapy Cancer (2020) 8(2):e001378. doi: 10.1136/jitc-2020-001378
98. Garg AD, More S, Rufo N, Mece O, Sassano ML, Agostinis P, et al. Trial Watch: Immunogenic Cell Death Induction by Anticancer Chemotherapeutics. Oncoimmunology (2017) 6(12):e1386829. doi: 10.1080/2162402x.2017.1386829
99. Rasmussen L, Arvin A. Chemotherapy-Induced Immunosuppression. Environ Health Perspect (1982) 43:21–5. doi: 10.1289/ehp.824321
100. Chen CA, Ho CM, Chang MC, Sun WZ, Chen YL, Chiang YC, et al. Metronomic Chemotherapy Enhances Antitumor Effects of Cancer Vaccine by Depleting Regulatory T Lymphocytes and Inhibiting Tumor Angiogenesis. Mol Ther (2010) 18(6):1233–43. doi: 10.1038/mt.2010.34
101. Simsek C, Esin E, Yalcin S. Metronomic Chemotherapy: A Systematic Review of the Literature and Clinical Experience. J Oncol (2019) 2019:5483791. doi: 10.1155/2019/5483791
102. Wu J, Waxman DJ. Immunogenic Chemotherapy: Dose and Schedule Dependence and Combination With Immunotherapy. Cancer Lett (2018) 419:210–21. doi: 10.1016/j.canlet.2018.01.050
103. Du B, Waxman DJ. Medium Dose Intermittent Cyclophosphamide Induces Immunogenic Cell Death and Cancer Cell Autonomous Type I Interferon Production in Glioma Models. Cancer Lett (2020) 470:170–80. doi: 10.1016/j.canlet.2019.11.025
104. Chen YL, Chang MC, Cheng WF. Metronomic Chemotherapy and Immunotherapy in Cancer Treatment. Cancer Lett (2017) 400:282–92. doi: 10.1016/j.canlet.2017.01.040
105. Emens LA, Middleton G. The Interplay of Immunotherapy and Chemotherapy: Harnessing Potential Synergies. Cancer Immunol Res (2015) 3(5):436. doi: 10.1158/2326-6066.CIR-15-0064
106. Liu R, Gong J, Chen J, Li Q, Song C, Zhang J, et al. Calreticulin as a Potential Diagnostic Biomarker for Lung Cancer. Cancer Immunol Immunother (2012) 61(6):855–64. doi: 10.1007/s00262-011-1146-8
107. Fucikova J, Becht E, Iribarren K, Goc J, Remark R, Damotte D, et al. Calreticulin Expression in Human Non–Small Cell Lung Cancers Correlates With Increased Accumulation of Antitumor Immune Cells and Favorable Prognosis. Cancer Res (2016) 76(7):1746. doi: 10.1158/0008-5472.CAN-15-1142
108. Kasikova L, Hensler M, Truxova I, Skapa P, Laco J, Belicova L, et al. Calreticulin Exposure Correlates With Robust Adaptive Antitumor Immunity and Favorable Prognosis in Ovarian Carcinoma Patients. J Immunother Cancer (2019) 7(1):312. doi: 10.1186/s40425-019-0781-z
109. Fucikova J, Truxova I, Hensler M, Becht E, Kasikova L, Moserova I, et al. Calreticulin Exposure by Malignant Blasts Correlates With Robust Anticancer Immunity and Improved Clinical Outcome in AML Patients. Blood (2016) 128(26):3113–24. doi: 10.1182/blood-2016-08-731737
110. Stoetzer OJ, Fersching DMI, Salat C, Steinkohl O, Gabka CJ, Hamann U, et al. Circulating Immunogenic Cell Death Biomarkers HMGB1 and RAGE in Breast Cancer Patients During Neoadjuvant Chemotherapy. Tumor Biol (2013) 34(1):81–90. doi: 10.1007/s13277-012-0513-1
111. Ladoire S, Enot D, Andre F, Zitvogel L, Kroemer G. Immunogenic Cell Death-Related Biomarkers: Impact on the Survival of Breast Cancer Patients After Adjuvant Chemotherapy. Oncoimmunology (2015) 5(2):e1082706–e. doi: 10.1080/2162402X.2015.1082706
112. Østrup O, Dagenborg VJ, Rødland EA, Skarpeteig V, Silwal-Pandit L, Grzyb K, et al. Molecular Signatures Reflecting Microenvironmental Metabolism and Chemotherapy-Induced Immunogenic Cell Death in Colorectal Liver Metastases. Oncotarget (2017) 8(44):76290–304. doi: 10.18632/oncotarget.19350
113. Dagenborg VJ, Marshall SE, Yaqub S, Grzyb K, Boye K, Lund-Iversen M, et al. Neoadjuvant Chemotherapy Is Associated With a Transient Increase of Intratumoral T-Cell Density in Microsatellite Stable Colorectal Liver Metastases. Cancer Biol Ther (2020) 21(5):432–40. doi: 10.1080/15384047.2020.1721252
114. Monk BJ, Brady MF, Aghajanian C, Lankes HA, Rizack T, Leach J, et al. A Phase 2, Randomized, Double-Blind, Placebo- Controlled Study of Chemo-Immunotherapy Combination Using Motolimod With Pegylated Liposomal Doxorubicin in Recurrent or Persistent Ovarian Cancer: A Gynecologic Oncology Group Partners Study. Ann Oncol (2017) 28(5):996–1004. doi: 10.1093/annonc/mdx049
115. Dijkgraaf EM, Santegoets SJ, Reyners AK, Goedemans R, Wouters MC, Kenter GG, et al. A Phase I Trial Combining Carboplatin/Doxorubicin With Tocilizumab, an Anti-IL-6R Monoclonal Antibody, and Interferon-α2b in Patients With Recurrent Epithelial Ovarian Cancer. Ann Oncol (2015) 26(10):2141–9. doi: 10.1093/annonc/mdv309
116. Davies KJ. The Broad Spectrum of Responses to Oxidants in Proliferating Cells: A New Paradigm for Oxidative Stress. IUBMB Life (1999) 48(1):41–7. doi: 10.1080/713803463
117. Demaria S, Golden EB, Formenti SC. Role of Local Radiation Therapy in Cancer Immunotherapy. JAMA Oncol (2015) 1(9):1325–32. doi: 10.1001/jamaoncol.2015.2756
118. Li W, Yang J, Luo L, Jiang M, Qin B, Yin H, et al. Targeting Photodynamic and Photothermal Therapy to the Endoplasmic Reticulum Enhances Immunogenic Cancer Cell Death. Nat Commun (2019) 10(1):3349. doi: 10.1038/s41467-019-11269-8
119. Golden EB, Frances D, Pellicciotta I, Demaria S, Helen Barcellos-Hoff M, Formenti SC. Radiation Fosters Dose-Dependent and Chemotherapy-Induced Immunogenic Cell Death. Oncoimmunology (2014) 3:e28518–e. doi: 10.4161/onci.28518
120. Sia J, Szmyd R, Hau E, Gee HE. Molecular Mechanisms of Radiation-Induced Cancer Cell Death: A Primer. Front Cell Dev Biol (2020) 8:41. doi: 10.3389/fcell.2020.00041
121. Demaria S, Guha C, Schoenfeld J, Morris Z, Monjazeb A, Sikora A, et al. Radiation Dose and Fraction in Immunotherapy: One-Size Regimen Does Not Fit All Settings, So How Does One Choose? J Immunother Cancer (2021) 9(4):e002038. doi: 10.1136/jitc-2020-002038
122. Kwilas A, Donahue R, Bernstein M, Hodge J. In the Field: Exploiting the Untapped Potential of Immunogenic Modulation by Radiation in Combination With Immunotherapy for the Treatment of Cancer. Front Oncol (2012) 2:104. doi: 10.3389/fonc.2012.00104
123. Gameiro SR, Jammeh ML, Wattenberg MM, Tsang KY, Ferrone S, Hodge JW. Radiation-Induced Immunogenic Modulation of Tumor Enhances Antigen Processing and Calreticulin Exposure, Resulting in Enhanced T-Cell Killing. Oncotarget (2014) 5(2):403–16. doi: 10.18632/oncotarget.1719
124. Ardiani A, Gameiro SR, Kwilas AR, Donahue RN, Hodge JW. Androgen Deprivation Therapy Sensitizes Prostate Cancer Cells to T-Cell Killing Through Androgen Receptor Dependent Modulation of the Apoptotic Pathway. Oncotarget (2014) 5(19):9335–48. doi: 10.18632/oncotarget.2429
125. Kwilas AR, Ardiani A, Gameiro SR, Richards J, Hall AB, Hodge JW. Androgen Deprivation Therapy Sensitizes Triple Negative Breast Cancer Cells to Immune-Mediated Lysis Through Androgen Receptor Independent Modulation of Osteoprotegerin. Oncotarget (2016) 7(17):23498–511. doi: 10.18632/oncotarget.8274
126. Grenga I, Kwilas AR, Donahue RN, Farsaci B, Hodge JW. Inhibition of the Angiopoietin/Tie2 Axis Induces Immunogenic Modulation, Which Sensitizes Human Tumor Cells to Immune Attack. J Immunother Cancer (2015) 3:52. doi: 10.1186/s40425-015-0096-7
127. Donahue RN, Duncan BB, Fry TJ, Jones B, Bachovchin WW, Kiritsy CP, et al. A Pan Inhibitor of DASH Family Enzymes Induces Immunogenic Modulation and Sensitizes Murine and Human Carcinoma Cells to Antigen-Specific Cytotoxic T Lymphocyte Killing: Implications for Combination Therapy With Cancer Vaccines. Vaccine (2014) 32(26):3223–31. doi: 10.1016/j.vaccine.2014.04.008
128. Fenerty KE, Padget M, Wolfson B, Gameiro SR, Su Z, Lee JH, et al. Immunotherapy Utilizing the Combination of Natural Killer- and Antibody Dependent Cellular Cytotoxicity (ADCC)-Mediating Agents With Poly (ADP-Ribose) Polymerase (PARP) Inhibition. J Immunother Cancer (2018) 6(1):133. doi: 10.1186/s40425-018-0445-4
Keywords: chemotherapy, immunogenic cell death, immunogenic modulation, cancer, cell stress, immunotherapy
Citation: Fabian KP, Wolfson B and Hodge JW (2021) From Immunogenic Cell Death to Immunogenic Modulation: Select Chemotherapy Regimens Induce a Spectrum of Immune-Enhancing Activities in the Tumor Microenvironment. Front. Oncol. 11:728018. doi: 10.3389/fonc.2021.728018
Received: 21 June 2021; Accepted: 29 July 2021;
Published: 23 August 2021.
Edited by:
Dmitri V. Krysko, Ghent University, BelgiumReviewed by:
Oliver Kepp, Institut National de la Santé et de la Recherche Médicale (INSERM), FranceCopyright © 2021 Fabian, Wolfson and Hodge. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: James W. Hodge, amgyNDFkQG5paC5nb3Y=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.