 Hong-Chieh Tsai1,2
Hong-Chieh Tsai1,2 Kuo-Chen Wei1,3,4,5
Kuo-Chen Wei1,3,4,5 Pin-Yuan Chen5,6Chiung-Yin Huang3,4
Pin-Yuan Chen5,6Chiung-Yin Huang3,4 Ko-Ting Chen1,4
Ko-Ting Chen1,4 Ya-Jui Lin1,5Hsiao-Wei Cheng1,7Yi-Rou Chen1
Ya-Jui Lin1,5Hsiao-Wei Cheng1,7Yi-Rou Chen1 Hsiang-Tsui Wang7,8,9,10*
Hsiang-Tsui Wang7,8,9,10*- 1Department of Neurosurgery, Linkou Chang Gung Memorial Hospital, Taoyuan, Taiwan
- 2School of Traditional Chinese Medicine, Chang Gung University, Taoyuan, Taiwan
- 3Department of Neurosurgery, New Taipei Municipal TuCheng Hospital, Chang Gung Memorial Hospital, New Taipei City, Taiwan
- 4Neuroscience Research Center, Linkou Chang Gung Memorial Hospital, Taoyuan, Taiwan
- 5School of Medicine, Chang Gung University, Taoyuan, Taiwan
- 6Department of Neurosurgery, Keelung Chang Gung Memorial Hospital, Keelung, Taiwan
- 7Institute of Pharmacology, College of Medicine, National Yang-Ming University, Taipei, Taiwan
- 8Institute of Pharmacology, College of Medicine, National Yang Ming Chiao Tung University, Taipei, Taiwan
- 9Institute of Food Safety and Health Risk Assessment, National Yang Ming Chiao Tung University, Taipei, Taiwan
- 10Doctor Degree Program in Toxicology, Kaohsiung Medical University, Kaohsiung, Taiwan
Glioblastoma (GBM), the most lethal type of brain tumor in adults, has considerable cellular heterogeneity. The standard adjuvant chemotherapeutic agent for GBM, temozolomide (TMZ), has a modest response rate due to the development of drug resistance. Multiple studies have shown that valproic acid (VPA) can enhance GBM tumor control and prolong survival when given in conjunction with TMZ. However, the beneficial effect is variable. In this study, we analyzed the impact of VPA on GBM patient survival and its possible correlation with TMZ treatment and p53 gene mutation. In addition, the molecular mechanisms of TMZ in combination with VPA were examined using both p53 wild-type and p53 mutant human GBM cell lines. Our analysis of clinical data indicates that the survival benefit of a combined TMZ and VPA treatment in GBM patients is dependent on their p53 gene status. In cellular experiments, our results show that VPA enhanced the antineoplastic effect of TMZ by enhancing p53 activation and promoting the expression of its downstream pro-apoptotic protein, PUMA. Our study indicates that GBM patients with wild-type p53 may benefit from a combined TMZ+VPA treatment.
Introduction
Glioblastoma (GBM), the most common and most lethal type of brain tumor, accounts for 50% of malignancies in the intrinsic central nervous system and has the highest loss of potential life years compared to other cancers (1–3). Standard treatment includes surgical excision and concomitant chemoradiotherapy with temozolomide (TMZ), followed by TMZ chemotherapy. Recent studies in molecular biology have shown that, despite similarities in histological appearances, GBM harbors significant genetic, epigenetic, and gene expression heterogeneities both interpersonally and intratumorally (4–6). The heterogeneous genetic background of GBM patients results in variable sensitivities of cancer cells to TMZ treatment and, thus, differential clinical outcomes. Hypermethylation at the promoter of O6-methylguanine DNA methyltransferase (MGMT), a DNA repair gene, is associated with increased sensitivity to TMZ treatment and improved patient survival (7–11). Isocitrate dehydrogenase 1 (IDH1) mutant GBM patients have more favorable outcomes, partly due to the enhanced sensitivity to TMZ chemotherapy (12, 13). On the other hand, a dysfunctional p53 DNA response pathway is associated with TMZ resistance (10, 14, 15). Previous studies have shown that somatic alterations that deregulate p53 were found in 85%–90% of GBM tumors, including 27.9% of p53 gene mutations or deletions (16). Furthermore, p53 mutation often co-occurs with IDH1 and ATRX mutations, which are critical markers defining GBM molecular classification (17, 18). These molecular alterations have significant clinical implications in that they not only define radically different subgroups of GBM but also significantly affect tumor susceptibility to treatment and, thus, patient prognosis (19–21).
Valproic acid (VPA) is an anti-epileptic drug that is widely used to treat or prevent perioperative seizures associated with GBM (22). In addition to its anti-epileptic activity, it has been shown in some retrospective studies that VPA was capable of improving GBM patient survival when given in conjunction with TMZ (23–26). However, the results of these studies are inconsistent. Multiple mechanisms of the anticancer action of VPA have been proposed, including inhibition of histone deacetylase (HDAC), alteration in the chromatin structure, disruption of DNA repair pathways or redox regulation, and induction of autophagy (27–30). Several reports have shown that VPA induces p53-dependent radiosensitization and chemosensitization in vitro and in vivo (31–34). In the present study, we attempted to identify GBM genetic alterations that are associated with favorable outcomes of VPA treatment in GBM patients and decipher the underlying mechanism. In clinical samples, GBM patient survival with respect to VPA treatment and p53 gene status was investigated. Furthermore, the effect of TMZ alone or in combination with VPA on GBM cancer cells was examined using both p53 wild-type and p53 mutant human GBM cell lines, and its underlying molecular mechanisms were examined.
Materials and Methods
Patient Data Collection
The institutional database of Linkou Chang-Gung Memorial Hospital was used to identify patients who underwent surgical excision of GBM between January 2015 and December 2017 following guidelines approved by the IRB board (IRB# 201701979B0). Patients who underwent standard treatment for newly diagnosed GBM, which included surgical excision, TMZ combined chemoradiotherapy (CCRT), and oral TMZ chemotherapy, and patients who underwent surgical excision for recurrent GBM were included. Patients who underwent biopsy only, did not undergo surgical excision, had a diagnosis based on imaging only, did not have histopathologically proven GBM, and did not undergo standard treatment for newly diagnosed GBM were excluded. A total of 166 patients were recruited. The medical records of these patients were reviewed and followed up until December 31, 2019. Clinical details, including immunohistochemical (IHC) staining of specific markers such as MGMT, the patient’s seizure status, date of birth, date of diagnosis, date of operation, and the recorded date of disease progression or death, were recorded. Residual tumor samples were obtained from the tumor bank.
p53 and IDH1 Mutation Analysis
Genomic DNA was extracted from frozen tissues, exon 4 of IDH1 and IDH2 or exons 4–9 of p53 were PCR amplified from tumor DNA, and mutations were analyzed by sequencing analysis. The primers used in PCR and sequencing are shown in Table 1. All mutations were confirmed by sequencing both DNA strands.

Table 1 The sequencing primers of exon 4 of IDH1 and IDH2 or exon 4-9 of p53.
Cell Culture and Treatment
The GBM cell lines U87, DBTRG-05MG, U118MG, and LN229 were purchased from ATCC (Manassas, VA, USA). U87 cells were grown in minimum essential medium (MEM) supplemented with 10% fetal bovine serum (FBS) at 37°C in an atmosphere of 5% CO2. DBTRG-05MG cells were grown in RPMI-1640 supplemented with 10% FBS at 37°C in an atmosphere of 5% CO2. U118MG and LN229 cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% FBS at 37°C in an atmosphere of 5% CO2. The p53 status of these cell lines was confirmed using DNA sequencing analysis (Supplementary Figure S7). Cells at 70% confluency were washed with phosphate-buffered saline (PBS) and treated with TMZ (0–10 mM) and/or VPA (2.5 mM) in complete culture medium at 37°C in the dark.
RNA Interference
Knockdown of p53, E2F1, or HDAC2 in GBM cells by RNA interference with human p53 siRNA (Sigma, St. Louis, MO, USA), E2F1 Silencer Select siRNA (/N4390824; Life Technology Corporation, Carlsbad, CA, USA), or HDAC2 Silencer Select siRNA (AM51331; Life Technology Corporation) was carried out according to the manufacturer’s protocol using GenMute siRNA Transfection Reagent (SignaGen Laboratories, Frederick, MD, USA). The sequence of the p53 siRNA targeting p53 mRNA (NM_000546.5) was GACUCCAGUGGUAAUCUAC, and siRNA was synthesized by Sigma (St. Louis, MO, USA). p53 siRNA, E2F1 siRNA, HDAC2 siRNA, or control siRNA was transfected at a final concentration of 30 nM for 24 h, followed by TMZ or VPA treatment as described above. Western blotting and reverse transcription PCR (RT-PCR) analyses were used to verify the efficiency of transfection.
Cell Viability Assay
GBM cells were plated in 96-well culture plates and incubated in a humidified chamber at 37°C and 5% CO2 overnight before drug treatment. Cell viability was assessed 24 h after the addition of the drugs. The culture medium was then removed from each well and replaced with 150 μl of MTT (3-[4,5-dimethylthiazol-2-yl]-2,5 diphenyl tetrazolium bromide; 0.5 mg/ml) in complete medium. The cells were incubated for 2 h at 37°C. Formazan crystals formed in cells were dissolved in 50 μl of dimethyl sulfoxide (DMSO). The absorbance of the samples at 570 nm was measured using a microplate reader. The results were evaluated by measuring the optical density of the MTT solution at 570 nm.
Western Blot Analysis
The cell lysate was prepared by sonication in RIPA buffer with protease and phosphatase inhibitor cocktails. Proteins (30 µg) were separated by 8%–15% sodium dodecyl sulfate–polyacrylamide gel electrophoresis and transferred to polyvinylidene difluoride membranes, blocked in 5% milk, and incubated overnight at 4°C in primary antibodies including PARP (1:1,000; #9542, Cell Signaling, Danvers, MA, USA), E2F1 (1:1,000; #3742, Cell Signaling, Danvers, MA, USA), p53 (DO-1) (1:1,000;, cat. no. OP43, Calbiochem, San Diego, CA, USA), PUMA (1:1,000; #12450, Cell Signaling, Danvers, MA, USA), caspase 3 (1:1,000; #9662, Cell Signaling, Danvers, MA, USA), and caspase 9 (1:1,000; #9502, Cell Signaling, Danvers, MA, USA), followed by the respective anti-IgG secondary antibodies (1:3,000; Millipore, Burlington, MA, USA) for 1 h at room temperature. Membranes were developed for visualization and photography using enhanced chemiluminescence (ECL) (Millipore Corporation, Billerica, MA, USA). Optical band densities were quantified using ImageJ software, and the results were analyzed using Excel software.
Flow Cytometry Analysis of the Cell Cycle Phases
After harvesting, the cells were washed twice in ice-cold PBS and fixed in ice-cold 70% ethanol for 30 min or overnight at 4°C. The cells were washed in PBS and digested with DNase-free RNase A (50 U/ml) at 37°C for 30 min. Before flow cytometry analysis, the cells were resuspended in 500 μl propidium iodide (PI, 10 μg/ml; Sigma, St. Louis, MO, USA) for DNA staining. PI staining was used to measure the cell cycle status using a Becton-Dickinson FACScan instrument (Franklin Lakes, NJ, USA) and the Cell Quest software.
Quantitative Real-Time RT-PCR
Total RNA was isolated from the harvested cells using the TRIzol® Reagent (Thermo Fisher Scientific, Waltham, MA, USA) and underwent reverse transcription using RevertAid Reverse Transcriptase (Thermo Fisher Scientific) according to the manufacturer’s instructions. Subsequent real-time RT-PCR analysis of cDNA was performed in triplicate using SYBR green dye on the StepOnePlus™ Real-Time PCR System (Applied Biosystems, Waltham, MA, USA). The primer sequences are shown as follows (5'–3'): PUMA, CCTGGAGGGTCCTGTACAATCTC; GCAGGCACCTAATTGGGCTC; GAPDH: CCGTCTAGAAAAACCTGCC; GCCAAATTCGTTGTCATACC. To calculate the relative mRNA expression, GAPDH was used as an internal control for all quantitative RT-PCRs and compared with the control groups.
Statistical Analyses
Student’s t-tests were used to determine statistical significance, and two-tailed p-values are shown. A minimum of three independent replicate experiments were performed to justify the use of statistical tests. Survival and progression-free survival were analyzed using Kaplan–Meier survival analysis, and the log-rank test was used for comparisons between two groups. Multivariate analysis was performed using Cox regression analysis. P < 0.05 was considered statistically significant. All statistical analyses were performed using SPSS software version 20.0.
Results
TMZ Combined with VPA Is Associated With Improved Survival in GBM Patients With Wild-Type p53
To evaluate the effect of VPA on the survival of GBM patients, we retrospectively reviewed patients diagnosed with primary GBM from 2015 to 2017 who underwent curative excisional surgery. Some of these patients underwent more than one surgical excision during these 2 years. For the purpose of analysis, we only included each patient’s first operation. The demographic data are shown in Table 2. Of all 166 patients, 139 underwent surgery for newly diagnosed GBM and 27 underwent surgery for recurrent tumors. All newly diagnosed GBM patients underwent standard treatment, including surgical excision, combined chemoradiotherapy with TMZ, and TMZ chemotherapy. All recurrent GBM patients received avastin treatment after surgical excision. The median survival of all patients combined was 15.20 ± 1.17 months, with 15.67 ± 1.42 months for newly diagnosed patients and 13.10 ± 2.34 months for recurrent GBM patients.
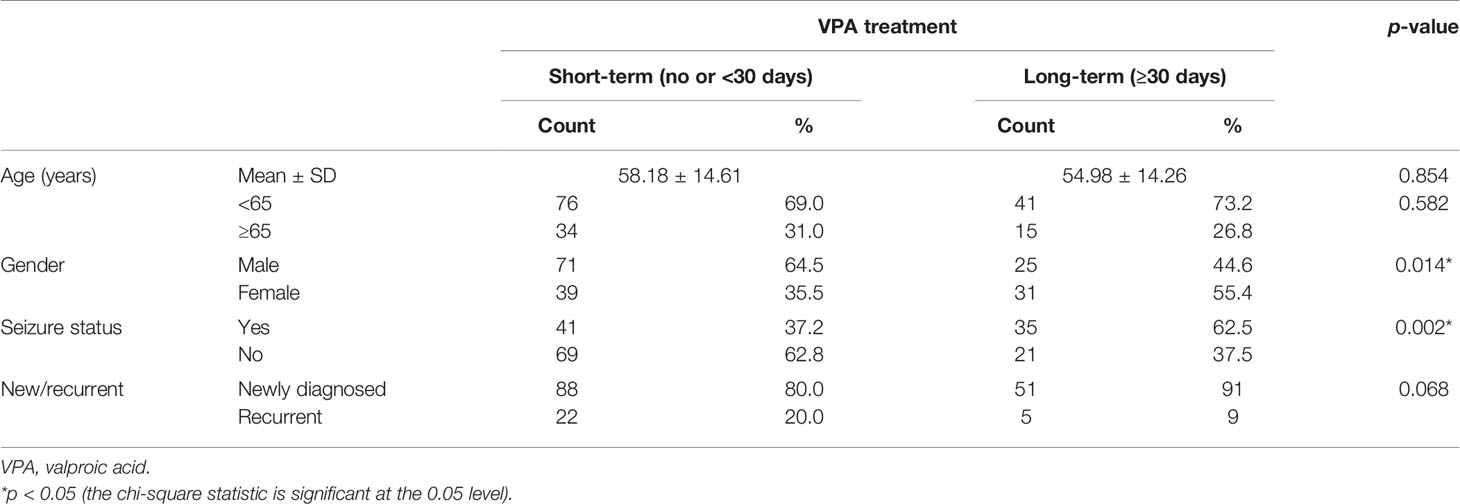
Table 2 Patient demographics and clinical characteristics of glioblastoma (GBM) patients.
Of these 166 patients, 51 out of 139 newly diagnosed GBM patients and 5 out of 27 recurrent GBM patients had VPA treatment for over 30 days. These patients were classified into the long-term VPA group. Patients who received long-term VPA treatment had significantly longer survival (Figure 1A), which is concordant with our previous report (36). Kaplan–Meier survival analyses showed that only long-term VPA treatment was associated with longer overall survival in the combined newly diagnosed and recurrent patients, while younger age (<65 years), seizure history, and long-term VPA treatment were associated with better survival in newly diagnosed GBM patients (Figure 1B and Supplementary Figures S1A–C). In the Cox regression analysis, none of these factors were independent prognostic factors (Supplementary Tables S1 and S2). We further analyzed the characteristics of patient who underwent VPA treatment and their impact on the effects of VPA treatment. There were significantly more seizure patients and more female patients in the long-term VPA group (Table 2). The effect of VPA on median survival in the different groups is shown in Supplementary Table S3. Favorable outcomes with VPA treatment were observed in patients with newly diagnosed GBM (Figure 1B) and in patients who were under 65 years old (Figure 1C), but not in those with recurrent GBM (Supplementary Figure S1D) or older patients (≥65 years old) (Supplementary Figure S1E). The effect was not affected by the patient’s gender or seizure status.
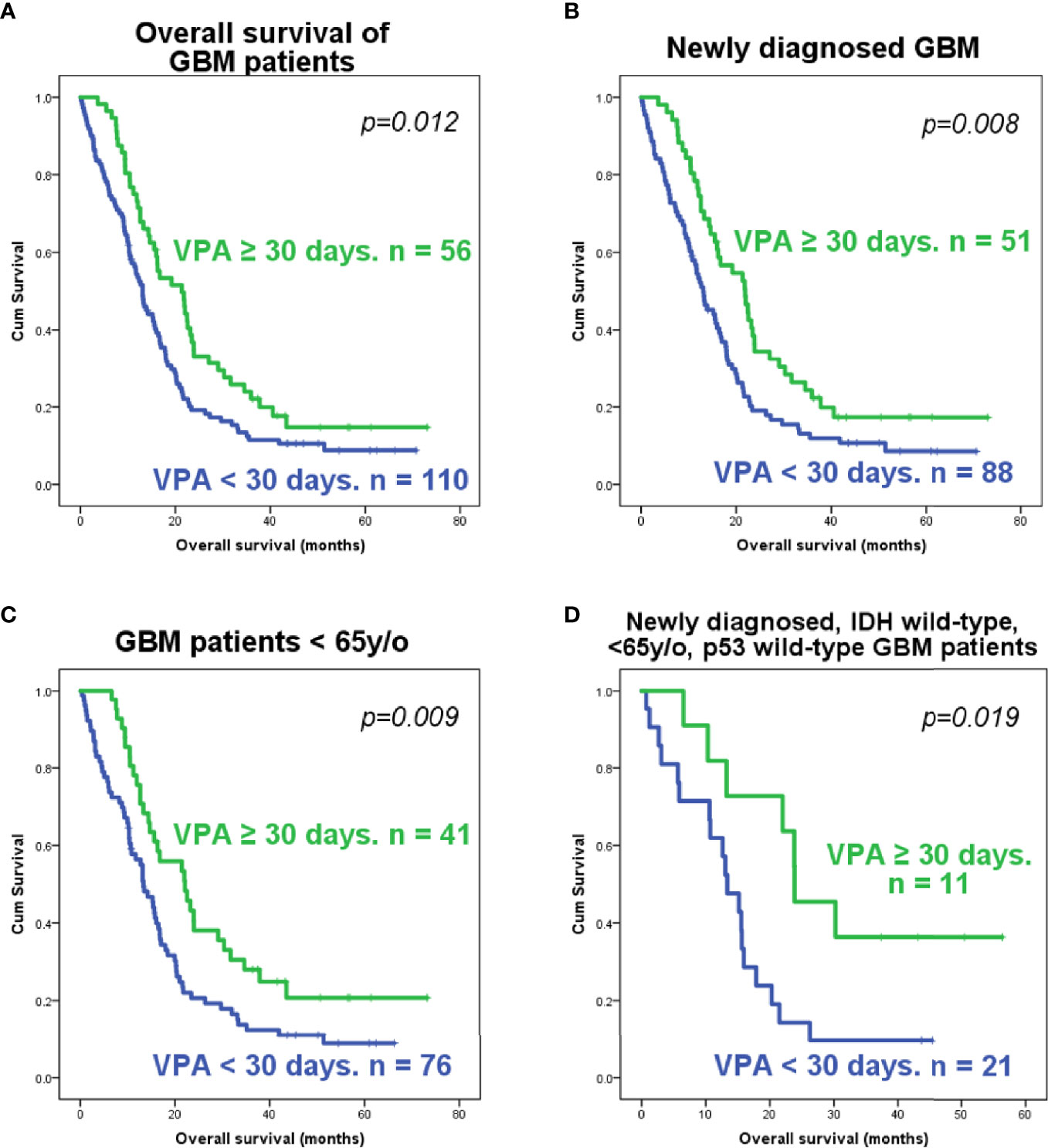
Figure 1 Kaplan–Meier analysis of the survival of glioblastoma (GBM) patients according to the valproic acid (VPA) treatment group. Survival plots of all GBM patients (A), newly diagnosed GBM patients (B), younger GBM patients (<65 years old) (C), and younger (<65 years old), newly diagnosed GBM patients with wild-type IDH1 and wild-type p53 (D). The p-value was calculated using the log-rank test in SPSS software.
To further elucidate the effects of VPA on GBM subtypes, we examined the IHC staining reports of these patients to identify the expression status of MGMT and study its correlation with the VPA treatment effect. Additionally, we tried to obtain surgical specimens from these patients and evaluated their IDH1 mutation status. MGMT promoter methylation inhibits DNA repair gene expression and is significantly associated with improved patient survival and TMZ sensitivity. Of the 166 patients recruited, IHC staining was available in 92 patients. No correlation was found between the expression of MGMT determined by IHC and patient survival (Supplementary Figure S2). Additionally, VPA treatment did not result in a statistically significant overall survival benefit in either MGMT staining-positive or MGMT staining-negative patients (Supplementary Figure S3), although there appears to be a trend toward better survival in long-term VPA-treated MGMT staining-negative newly diagnosed GBM patients (Supplementary Figure S3E).
Since several previous studies have indicated that VPA affects tumor growth via p53-dependent pathways (31–34), we also checked the p53 mutation status in these tissue samples. We were able to obtain 85 tumor tissue samples from these GBM patients. The demographic data and gene analysis results are shown in Table 3. The majority of these samples were IDH1 wild type, IDH2 wild type, and p53 wild type. Ten samples out of these 85 had IDH1R132 mutations, while none had IDH2 mutations. Of the p53 mutations identified in 11 tumors, 10 were missense mutations and one was a nonsense mutation, and all of them were identified as p53-inactivating mutations in the IARC TP53 database (R20) (37) (Supplementary Table S4 and Supplementary Figure S4). The mutational status of p53 was not significantly associated with the overall survival or progression-free survival of either newly diagnosed or recurrent GBM patients (Supplementary Figure S5). Further analysis revealed that VPA treatment is associated with improved survival in GBM patients who were under 65 years old and had newly diagnosed IDH wild-type and p53 wild-type GBM (Figure 1D), but not in patients who were under 65 years old and had newly diagnosed IDH wild-type and p53 mutant GBM, patients who were older than 65 years with newly diagnosed IDH wild-type GBM, and patients with newly diagnosed IDH mutant GBM, with recurrent GBM regardless of the p53 or IDH mutational status, and with p53 mutant or IDH mutant GBM regardless of age or recurrence status (Supplementary Figure S6).
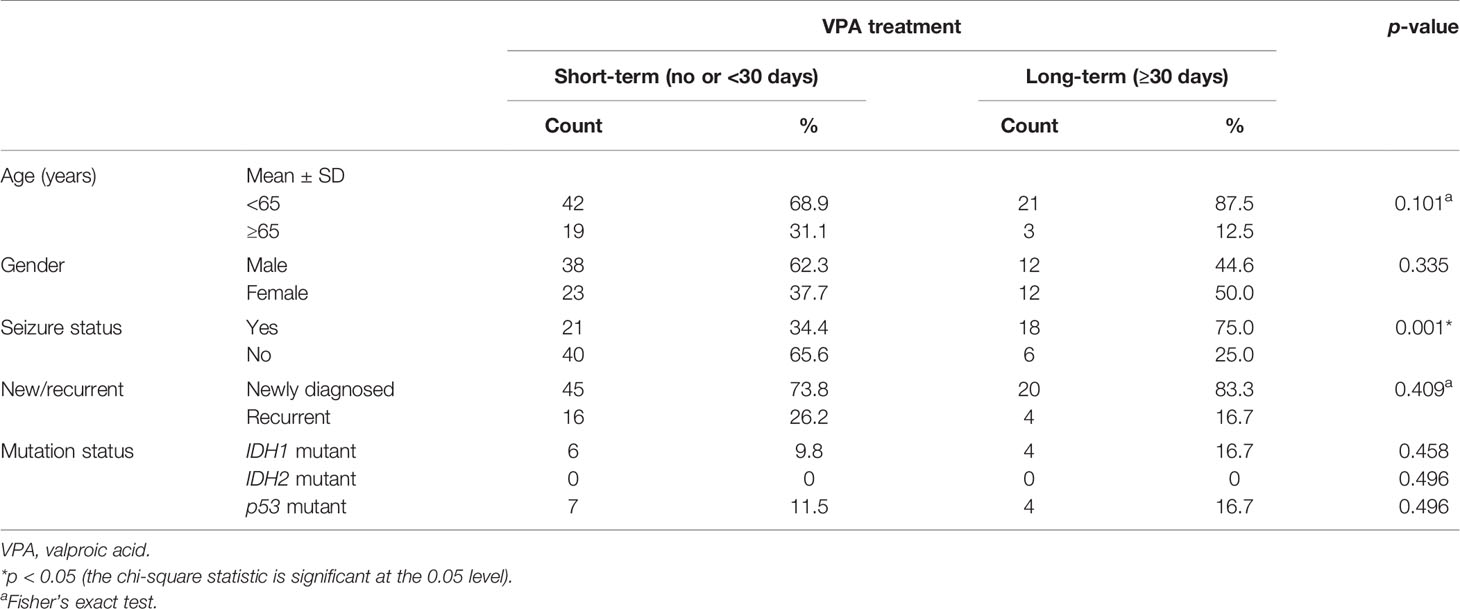
Table 3 Patient demographics and clinical characteristics of 85 glioblastoma (GBM) patients whose tumor samples underwent genetic evaluation.
In conclusion, our results indicate that the survival benefit of VPA in GBM patients may be dependent on the patient’s age, IDH mutation status, and p53 mutation status. Long-term VPA treatment may confer some degree of survival benefit in newly diagnosed and p53 wild-type GBM patients who are under 65 years old.
VPA Treatment Enhanced TMZ-Induced Cytotoxicity in GBM Cancer Cells in a p53-Dependent Manner
Since the survival benefit of TMZ combined with VPA treatment was observed in newly diagnosed GBM patients with wild-type p53 (Figure 1D), we hypothesize that VPA may exert its pro-survival effect by enhancing the anticancer activity of TMZ dependent on the p53 gene status. To determine whether VPA enhances the TMZ-mediated inhibition of GBM cancer cell proliferation and whether p53 mutation affects susceptibility to GBM, we examined the effect of TMZ alone or in combination with VPA on GBM cancer cell lines with varying p53 status. We found that co-treatment with VPA enhanced the growth inhibitory effect of TMZ in p53 wild-type GBM cells U87 and DBTRG-05MG, but did not significantly affect the growth of p53 mutant GBM cells LN229 and U118MG (Figures 2A, B and Supplementary Figure S7). This is consistent with the cell cycle analysis showing that combined treatment with TMZ and VPA increased the sub-G1 population in U87 and DBTRG-05MG cells (Figures 2C, D), indicating enhanced cellular apoptosis. The effect was not apparent in LN229 and U118MG cells (Figures 2E, F). Western blotting analysis confirmed that VPA treatment significantly increased caspase 3 and caspase 9 cleavage in U87 and DBTRG-05MG cells and increased the expression of PUMA, a pro-apoptotic protein downstream of p53 activation, as measured 24 h after TMZ treatment, but not in LN229 and U118MG cells (Figures 3A, B and Supplementary Figure S8). These results suggest that, in TMZ-induced genotoxic events, VPA increased TMZ cytotoxicity by activating p53 and enhancing the expression of PUMA. To further validate the role of p53 in VPA-induced apoptosis in combination with TMZ, we knocked down p53 in the U87 and DBTRG-05MG cell lines and evaluated the activation of the apoptosis pathway and the expression of PUMA in response to TMZ or VPA treatment. Knockdown of p53 expression in both cell lines significantly reduced the activation of the apoptotic pathway, as measured by caspase 9 and caspase 3 activation and enhanced PUMA expression (Figures 3C, D). In conclusion, our results show that the effect of VPA on GBM cancer cell proliferation is p53-dependent and that VPA enhances TMZ-induced apoptosis by promoting the expression of PUMA, a pro-apoptotic gene downstream of p53.
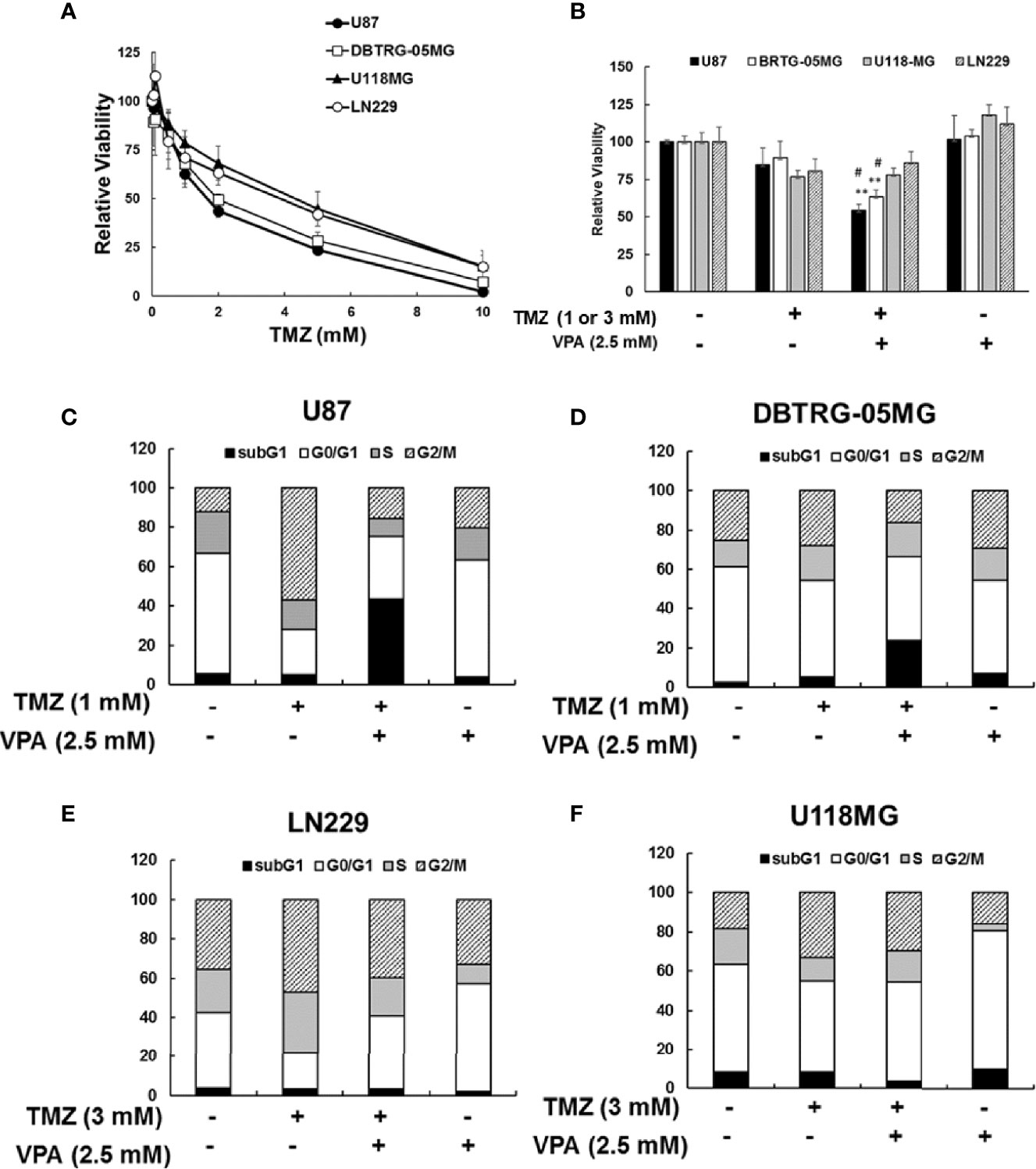
Figure 2 Cytotoxicity of temozolomide (TMZ) without or with valproic acid (VPA) in glioblastoma (GBM) cell lines. (A) Cytotoxicity of different dosages of TMZ (0–10 mM) for 24 h in p53 wild-type GBM cells (U87 and DBTRG-05MG) and p53 mutant GBM cells (U118MG and LN229). (B) Cytotoxicity of TMZ (1 or 3 mM) combined with VPA (2.5 mM) for 24 h in p53 wild-type GBM cells (U87 and DBTRG-05MG) and p53 mutant GBM cells (U118MG and LN229). Cytotoxicity was analyzed using the MTT assay, as described in Materials and Methods. Data are the mean ± SD. **p < 0.01 compared with cells without TMZ or VPA treatment; #p < 0.05 compared with cells with TMZ treatment. (C, D) Cell cycle analysis of TMZ (1 mM) combined with VPA (2.5 mM) for 24 h in p53 wild-type GBM cells (U87 and DBTRG-05MG). (E, F) Cell cycle analysis of TMZ (3 mM) combined with VPA (2.5 mM) for 24 h in p53 mutant GBM cells (U118MG and LN229). Cell cycle was analyzed using propidium iodide (PI) staining with flow cytometry, as described in Materials and Methods.
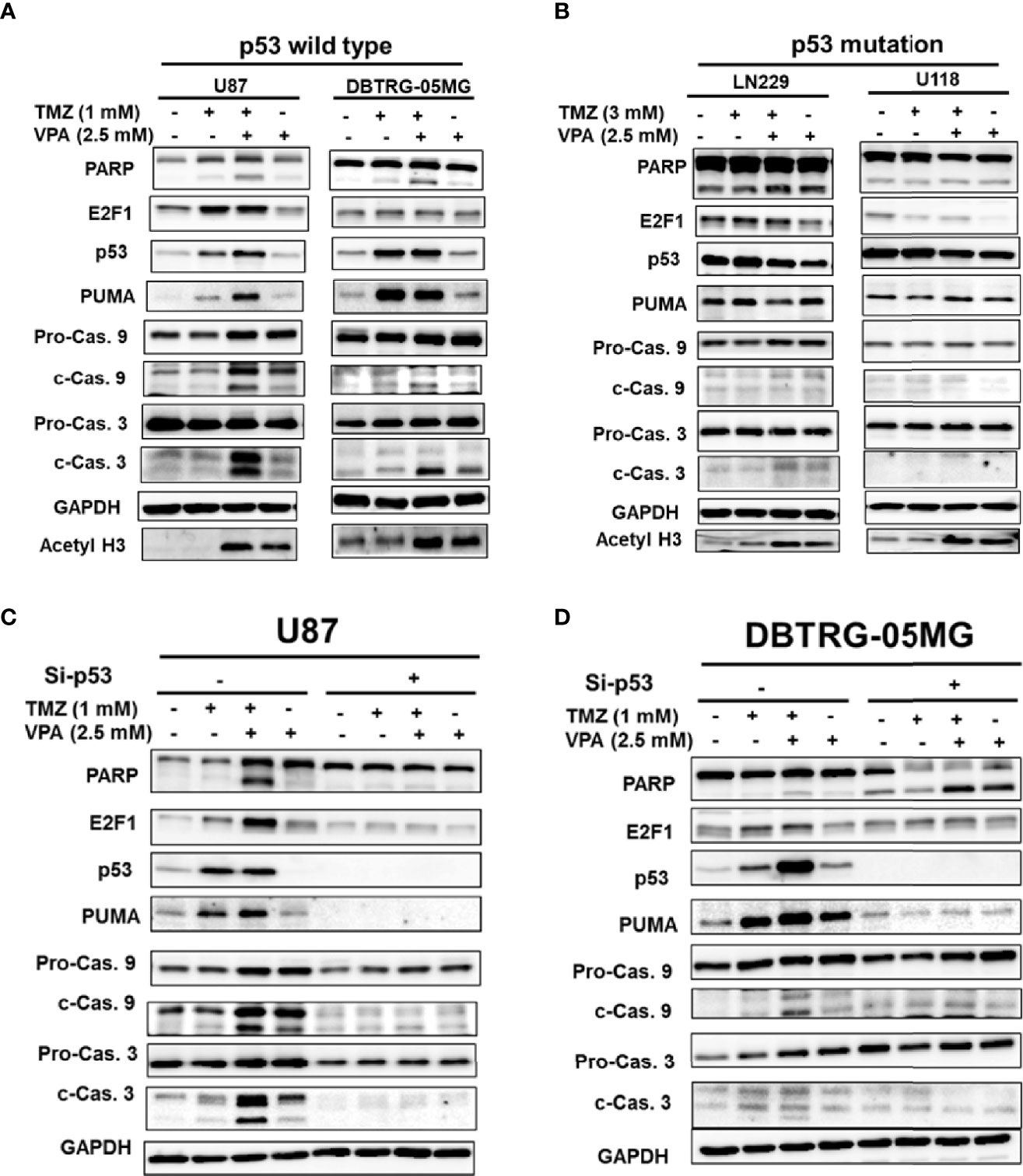
Figure 3 Temozolomide (TMZ) combined with valproic acid (VPA) enhanced cellular apoptosis through the p53–PUMA pathway in GBM cell lines. Western blot analysis of apoptosis (PARP, cleavage of caspase 9 and caspase 3), E2F1, p53, and PUMA, a downstream target of p53, in p53 wild-type GBM cells (U87 and DBTRG-05MG) (A) and p53 mutant GBM cells (U118MG and LN229) (B). p53 wild-type GBM cells (U87 and DBTRG-05MG) were treated with TMZ (1 mM), VPA (2.5 mM), or TMZ (1 mM) combined with VPA (2.5 mM) for 24 h, and p53 mutant GBM cells (U118MG and LN229) were treated with TMZ (3 mM), VPA (2.5 mM), or TMZ (3 mM) combined with VPA (2.5 mM) for 24 h. (C, D) Western blot analysis of apoptosis (PARP, cleavage of caspase 9 and caspase 3), E2F1, p53, and PUMA, a downstream target of p53, in U87 and DBTRG-05MG cells after the knockdown of p53 with siRNA (Si-p53) followed by TMZ or VPA treatment, as described above. Note that the knockdown of p53 reduced the apoptosis induced by TMZ combined with VPA in p53 wild-type cell lines.
Neither E2F1 nor HDAC2 Further Enhanced TMZ-VPA-Induced Apoptosis in p53 Wild-Type GBM Cells
We tried to further delineate the mechanisms by which VPA enhanced p53 downstream PUMA expression. Previous studies have shown that abundant crosstalk exists between the p53 and E2F1 pathways (38). In genotoxic events, there is extensive crosstalk between the MDM2–p53 and Rb–E2F1 pathways, which cooperate to initiate apoptosis. We found that the expression of E2F1 was decreased in response to TMZ or TMZ+VPA treatment after p53 knockdown (Figures 3C, D). However, E2F1 knockdown did not affect the activation of the apoptosis pathway or the expression of PUMA induced by VPA+TMZ treatment (Supplementary Figure S9). The results suggest that E2F1 activation was modulated by p53 activation, but was not necessary for TMZ+VPA to exert its pro-apoptotic function. Additionally, several studies indicated that the degradation of HDAC2 is one of the anticancer actions of VPA (28, 39) and that knockdown of HDAC2 enhances the sensitivity of GBM cancer cells to TMZ (40). Therefore, we examined the expression and the effect of HDAC2 on the apoptosis-enhancing ability of VPA+TMZ treatment. We found that HDAC2 expression did not decrease after VPA treatment for 24 h alone or in combination with TMZ (Figures 4A, B). Additionally, knockdown of HDAC2 did not abrogate but further enhanced the pro-apoptotic effect of VPA+TMZ co-treatment in either p53 wild-type GBM cell line, as inferred from the increased PARP cleavage and caspase 9 and caspase 3 activation (Figures 4A, B). A synergistic effect was not observed in either p53 mutant GBM cell line (Figures 4C, D). Interestingly, knockdown of HDAC2 in LN229 and U118MG p53 mutant cells induced cellular apoptosis without TMZ or VPA treatment (Figures 4C, D). Our results indicate that VPA may not exert its effect by inhibiting E2F1 activation or HDAC2 activity/expression. HDAC2 inhibition may induce apoptosis in p53 mutant cells via other signaling pathways.
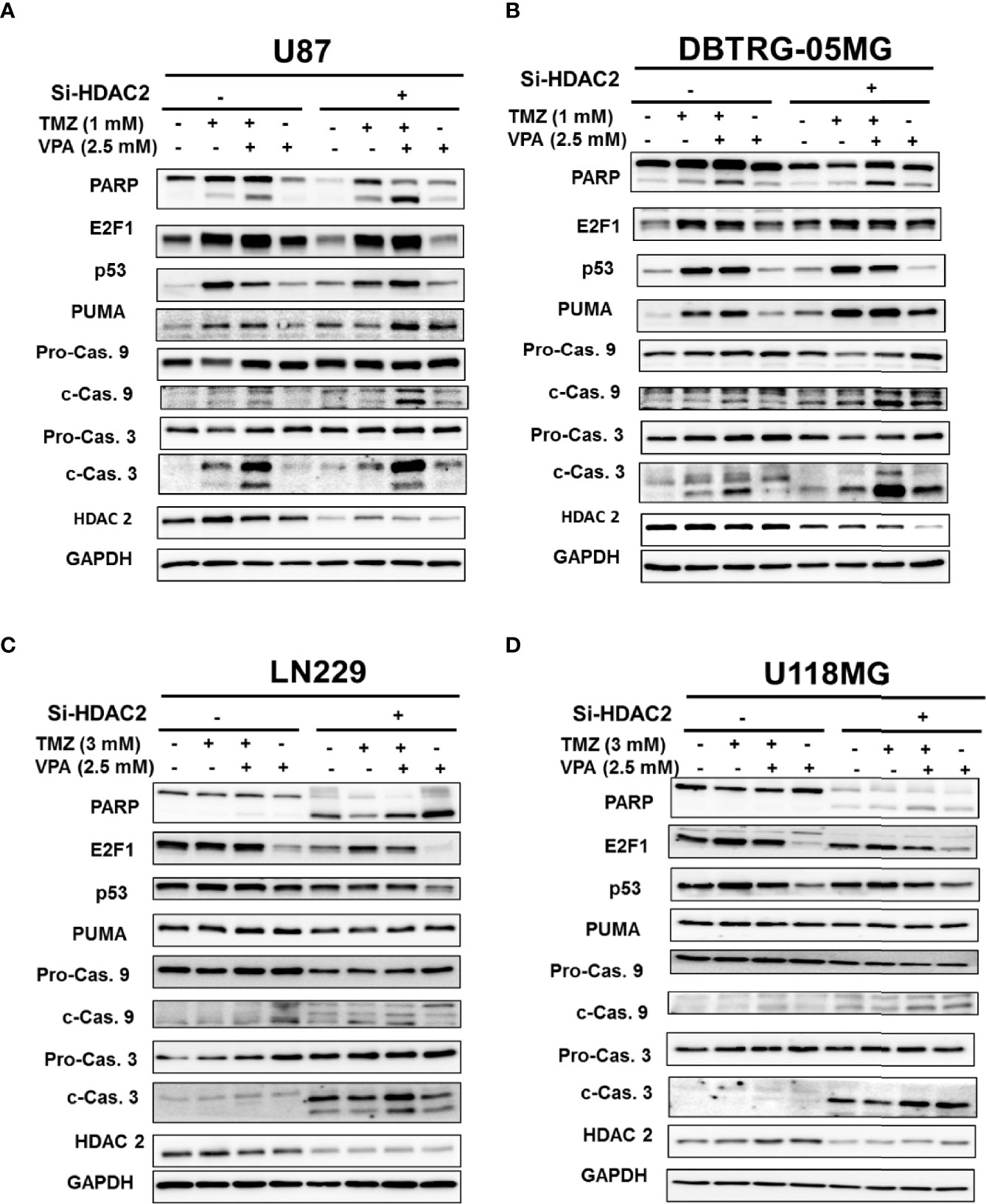
Figure 4 Knockdown of HDAC2 further enhanced the apoptosis pathway induced by temozolomide (TMZ) combined with valproic acid (VPA) in p53 wild-type cell lines. (A) Western blot analysis of apoptosis (PARP, cleavage of caspase 9 and caspase 3), E2F1, p53, and PUMA, a downstream target of p53, in p53 wild-type GBM cells, U87 (A) and DBTRG-05MG (B), and in p53 mutant GBM cells, LN229 (C) and U118MG (D), after the knockdown of HDAC2 with siRNA (Si-HDAC2) followed by TMZ or VPA treatment, as described below. p53 wild-type GBM cells (U87 and DBTRG-05MG) were treated with TMZ (1 mM), VPA (2.5 mM), or TMZ (1 mM) combined with VPA (2.5 mM) for 24 h, and p53 mutant GBM cells (U118MG and LN229) were treated with TMZ (3 mM), VPA (2.5 mM), or TMZ (3 mM) combined with VPA (2.5 mM) for 24 h. Note that the knockdown of HDAC2 induced cellular apoptosis in p53 mutant GBM cells.
Discussion
Glioblastoma (GBM) has been shown to harbor great genetic, epigenetic, and gene expression heterogeneities in both interpersonal and intratumor tissues (4–6, 41). GBM arises de novo (primary GBM) or via the dedifferentiation of lower-grade glioma (secondary GBM) (42). While distinct mutations are predominant in each subtype, alterations of the tumor suppressor p53 are the most common (43). These molecular alterations of GBM significantly affect tumor susceptibility to chemotherapy and, thus, patient prognosis (19–21). It has been shown that the expressions of p53 in newly diagnosed and recurrent GBM patients are inconsistent and can be altered upon recurrence (44, 45). Whether p53 mutation affects GBM sensitivity to chemotherapy and prognosis remains controversial (46–49). In this study, our results indicate that VPA enhances TMZ cytotoxicity by promoting apoptosis through enhancing p53 pathway activation and increasing the expression of its downstream target gene, PUMA. Wild-type p53 expression in GBM cells is necessary for VPA to exert its function. Screening of p53 mutations may help identify GBM patients who will benefit from a combined VPA and TMZ treatment.
The life expectancy of GBM patients is 12–18 months, despite advances in diagnosis and treatment (50–52). Several known prognostic factors include age, preoperative functional status, history of seizure, tumor location and size, extent of surgery, use of radiotherapy, and IDH mutation (52–56). In our study, the median survival of newly diagnosed GBM patients was 15.67 ± 1.42 months. Younger age (<65 years), seizure history, and long-term VPA treatment (>30 days) were associated with favorable outcomes (Supplementary Figures S1A, C and Figure 1A). The results are roughly concordant with other studies (52, 54). On the other hand, the median survival of recurrent GBM patients was 13.10 ± 2.34 months, which was not significantly different from that of newly diagnosed patients (p = 0.300). Since all our recurrent GBM patients underwent surgical excision, it is likely that the included patients were in better physical condition capable of undergoing surgery and had tumors that were presumably located at favorable locations suited for surgical intervention, thus improving the results. When combining newly diagnosed and recurrent GBM patients, only VPA treatment was statistically significantly associated with improved survival (Figure 1A).
VPA is used to treat seizures in our hospital. Although it has been shown that long-term use of anticonvulsants in seizure-free patients adds no clinical benefits (57, 58), some physicians in our institute were used to keeping anticonvulsants for patients who had symptoms suspicious of complex partial seizures or absence of seizures until the diagnosis was excluded. Therefore, in our dataset, there were some patients who had no definite seizure diagnosis, but who received VPA for more than 1 month (Table 2). Although long-term VPA treatment was associated with improved survival in the single variate analysis (Figure 1A), multivariate Cox regression analysis did not indicate VPA use as an independent prognostic factor (Supplementary Tables S1 and S2). Since VPA is used to treat seizures, which in itself is a favorable prognostic factor, evaluation of the VPA effect on the survival of GBM patient is confounded by the presence of seizures. Furthermore, there appeared to be more female patients in our long-term VPA group (Table 2). Gender alone does not affect treatments given to patients in our hospital. Although our seizure treatment did not differentiate between males or females, it was not shown in our study that gender significantly affected the results of GBM survival or VPA treatment (Supplementary Figure S1B), which indicates that there may be some degree of selection bias in our dataset. Further analysis indicated that VPA treatment appeared to be effective in newly diagnosed GBM patients and in younger patients (Figures 1B, C), but not in recurrent GBM or in patients over 65 years old (Supplementary Figures S1D, E). This result is also concordant with previous studies (59, 60). Interestingly, in our analysis, the benefit of long-term VPA treatment did not reach statistical significance in either the seizure or no-seizure group but in the combined group (Supplementary Table S3), indicating that the presence or absence of seizures does not significantly impact the effect of VPA. Kuo et al. reported that VPA does not improve survival in patients who are seizure-free (59). This discrepancy is likely caused by our small sample size and the patient variability and requires further investigation.
The MGMT promoter methylation status has long been recognized as a prognostic factor of GBM patient survival (61, 62). However, it was not routinely examined in our hospital because the test was not covered by our National Health Insurance program. The Clinical Pathology Department in our institute used IHC staining to detect MGMT expression, which is known to have low specificity and undetermined thresholds and may not have a strong correlation with the outcomes of GBM patients (63–65). In this study, we did not have enough resources or patient samples to perform methylation-specific PCR. Our analysis did not demonstrate a survival benefit in either MGMT staining-positive or MGMT staining-negative patients (Supplementary Figure S2). We also did not detect a pro-survival effect of VPA treatment in either MGMT staining-positive or MGMT staining-negative patients (Supplementary Figures S3A, D). In newly diagnosed MGMT-negative GBM patients, there appeared to be a trend toward improved survival in the long-term VPA treatment group, but this trend did not reach statistical significance (p = 0.052) (Supplementary Figure S3E). Interestingly, there are some reports indicating that VPA may enhance TMZ sensitivity by downregulating MGMT expression (30). Roos et al. reported that GBM cells with functionally intact p53 genes were more sensitive to TMZ treatment due to the activation of the Fas/CD95/APO-1 receptor and the subsequent apoptosis triggered by O6-methylguanine, a product of TMZ-induced DNA damage (66). Further studies evaluating the effect of MGMT promoter methylation on VPA treatment and its interaction with the p53 pathway may help better identify VPA-responsive patients. In conclusion, our data indicate a possible survival benefit of prolonged VPA treatment in younger, newly diagnosed GBM patients.
The molecular alterations of GBM have been shown to significantly affect tumor susceptibility to chemotherapy and, thus, patient prognosis (19–21). In this study, p53 mutation was detected in 11 out of the 85 patients sampled. The mutational status of p53 did not significantly affect patient survival (Supplementary Figure S5), which is concordant with previous studies (67). We also found that TMZ combined with long-term VPA treatment was effective in GBM patients with wild-type p53 (Figure 1D). Additionally, genetic alterations affecting the function of the p53 pathway, such as CDKN2A/ARF deletion or MDM2/MDM4 amplification, could be present in ~85% of all GBM patients (16) and had similar effects on the pro-survival effect of VPA treatment. Whether these genetic alterations that impair the p53 pathway activation affect VPA-induced TMZ potentiation requires further study. Since only 11 p53 mutant samples were identified (Table 3), the study may be limited by its small sample size, patient heterogeneity, and possible selection bias. However, we were able to demonstrate that VPA enhances TMZ cytotoxicity by enhancing apoptosis via the p53 pathway and the expression of the downstream target, PUMA (Figures 3A, B and Supplementary Figure S8) using p53 wild-type and p53 mutant human GBM cells.
p53 is commonly activated in response to DNA damage, genotoxicity, oncogene activation, aberrant growth signals, and hypoxia, all of which are events that can be encountered during carcinogenesis (48). p53 upregulated modulator of apoptosis (PUMA), a Bcl-2 homology 3 (BH3)-only pro-apoptotic Bcl-2 family member, was identified as a molecule that directly mediates p53-associated apoptosis (68). The PUMA protein associates with the mitochondria and induces apoptosis much earlier than the apoptosis that results from the exogenous expression of p53 when it is overexpressed in various cell lines (69). Previous studies have shown that PUMA overexpression results in massive apoptosis in GBM cells with wild-type or mutant p53, indicating that it is a therapeutic tool for GBM (70). Additionally, PUMA has been shown to increase the drug sensitivity of TMZ-resistant cells; thus, PUMA may be a suitable target for intervention to improve the therapeutic efficacy of TMZ (71). Here, we found that PUMA was further induced in p53 wild-type GBM cells, U87 cells, and DBTRG-05MG cells (Figure 3A). In our study, TMZ treatment induced p53 activation and apoptosis in p53 wild-type GBM cells, which was further enhanced by VPA treatment. For the in vitro experiments with GBM cells, we used TMZ at concentrations of 1 and 3 mM, based roughly on the IC50 at 24 h (Figure 2). This TMZ concentration induces cell death mainly via non-repaired N-alkylations, while for O6-methylguanine-induced apoptosis, much lower doses are sufficient, which are in the range of 1–50 μM (72). Under clinical conditions, the tissue concentration of TMZ is approximately 1 μg/ml (5.2 μM) and the serum concentration about 15 μg/ml (78 μM) (73, 74). In this concentration range, the main treatment effect is achieved via O6-methylguanine-induced DNA double-strand breaks and subsequent apoptosis. Whether the findings reported here are clinically relevant requires further elucidation. Additionally, there were some inconsistent reports of p53 gene activity in LN229 cells (75, 76). The majority agrees that LN229 retains at least partial p53 activity despite the mutation. In our study, we did find a missense mutation at exon 4. Treatment with TMZ failed to induce PUMA expression in LN229 cells (Figure 3B), suggesting at least a possible partial loss of p53 function. We also demonstrated that the knockdown of p53 abrogated the expression of PUMA and the cleavage of caspase 9, caspase 3, and PARP in p53 wild-type GBM cells treated with TMZ+VPA (Figures 3C, D) and reversed the pro-apoptotic effect of VPA in TMZ treatment, indicating that VPA enhanced TMZ-induced cell apoptosis via p53–PUMA pathway activation.
Several mechanisms for the anticancer effect of VPA have been proposed, including inhibition of HDAC, alteration of the chromatin structure, disruption of DNA repair pathways or redox regulation, and induction of autophagy (11, 27–30). Xie et al. reported that VPA attenuates the immunosuppressive function of myeloid-derived suppressor cells and may potentially improve the antitumor activity of CD8+ T cells (77). In malignant melanoma cells, VPA was shown to enhance IFN-β-induced caspase 8 expression, thus improving its response to TMZ treatment (78). In murine limb organogenesis, VPA was shown to induce p53 hyperacetylation through its HDAC inhibitor activity, thus enhancing p53 target gene expression (79). Among these mechanisms, VPA, as an HDAC inhibitor, has been widely recognized. It induces HDAC2 degradation and inhibits HDAC2 activity (28, 39). However, our results did not show a reduction in HDAC2 expression subsequent to VPA treatment (Figure 4). Knockdown of HDAC2 further enhanced TMZ+VPA-induced cellular apoptosis in p53 wild-type GBM cells (Figures 4A, B). Interestingly, HDAC2 silencing induced cellular apoptosis in p53 mutant GBM cells (Figures 4C, D), which is consistent with previous studies showing that silencing HDAC2 can suppress the proliferation of GBM cells (40). On the other hand, several potent HDAC inhibitors that entered clinical trials in recent years, such as vorinostat (SAHA) or trichostatin A (TSA), failed to demonstrate a significant survival benefit to patients in phase II clinical trials, either as single agents or in combination with standard TMZ treatment (80–82). This is consistent with our results that only VPA, but not SAHA or TSA, enhanced TMZ-induced apoptosis in U87 cells (Supplementary Figure S10). These results together suggest that VPA may not exert its effect by inhibiting HDAC2 activity or expression, and HDAC2 inhibition may potentiate the effect via other signaling pathways.
Conclusion
Our results indicate that the survival benefit of the combination regimen of TMZ and VPA in GBM patients is dependent on their p53 mutation status. In cellular models, our results show that VPA enhanced the antineoplastic effect of TMZ by enhancing apoptosis via activation of the p53 pathway and increasing the expression of its downstream pro-apoptotic protein, PUMA. Taken together, wild-type p53 may serve as an indicator of the effectiveness of a combined TMZ+VPA treatment in GBM.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding author.
Ethics Statement
The studies involving human participants were reviewed and approved by Institutional Review Board of Chang-Gung Medical Foundation (IRB#: 201701979B0). Written informed consent for participation was not required for this study in accordance with the national legislation and the institutional requirements.
Author Contributions
H-CT, K-CW, and H-TW designed, performed research, and analyzed data. H-CT, K-CW, P-YC, C-YH, K-TC, Y-JL, and Y-RC recruited, collected clinical specimens, and analyzed clinical data. H-CT and H-WC performed the experiments. H-CT and H-TW wrote the paper. All authors contributed to the article and approved the submitted version.
Funding
This work was funded by grants from the Ministry of Science and Technology, Taiwan [NHRI-EX110-11027PI, 109-2320-B-010-024 (H-TW) and 107-2320-B-182-044 (H-CT)], and Chang-Gung Memorial Hospital [CMRPG3K1441 (H-CT)].
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
The authors thank all members of the Brain Tumor Team, Cancer Center, Chang-Gung Memorial Hospital, Linkou, and the Neuroscience Research Center of Chang Gung Memorial Hospital, Linkou, for their invaluable help.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2021.722754/full#supplementary-material
References
1. Louis DN, Ohgaki H, Wiestler OD, Cavenee WK, Burger PC, Jouvet A, et al. The 2007 WHO Classification of Tumours of the Central Nervous System. Acta Neuropathol (2007) 114(2):97–109. doi: 10.1007/s00401-007-0243-4
2. Ostrom QT, Gittleman H, Xu J, Kromer C, Wolinsky Y, Kruchko C, et al. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2009-2013. Neuro Oncol (2016) 18(suppl_5):v1–75. doi: 10.1093/neuonc/now207
3. Rouse C, Gittleman H, Ostrom QT, Kruchko C, Barnholtz-Sloan JS. Years of Potential Life Lost for Brain and CNS Tumors Relative to Other Cancers in Adults in the United States, 2010. Neuro Oncol (2016) 18(1):70–7. doi: 10.1093/neuonc/nov249
4. Soeda A, Hara A, Kunisada T, Yoshimura S, Iwama T, Park DM. The Evidence of Glioblastoma Heterogeneity. Sci Rep (2015) 5:7979. doi: 10.1038/srep07979
5. Sottoriva A, Spiteri I, Piccirillo SG, Touloumis A, Collins VP, Marioni JC, et al. Intratumor Heterogeneity in Human Glioblastoma Reflects Cancer Evolutionary Dynamics. Proc Natl Acad Sci U S A (2013) 110(10):4009–14. doi: 10.1073/pnas.1219747110
6. Verhaak RG, Hoadley KA, Purdom E, Wang V, Qi Y, Wilkerson MD, et al. Integrated Genomic Analysis Identifies Clinically Relevant Subtypes of Glioblastoma Characterized by Abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell (2010) 17(1):98–110. doi: 10.1016/j.ccr.2009.12.020
7. Hegi ME, Diserens AC, Gorlia T, Hamou MF, de Tribolet N, Weller M, et al. MGMT Gene Silencing and Benefit From Temozolomide in Glioblastoma. N Engl J Med (2005) 352(10):997–1003. doi: 10.1056/NEJMoa043331
8. Fan CH, Liu WL, Cao H, Wen C, Chen L, Jiang G. O6-Methylguanine DNA Methyltransferase as a Promising Target for the Treatment of Temozolomide-Resistant Gliomas. Cell Death Dis (2013) 4:e876. doi: 10.1038/cddis.2013.388
9. Agnihotri S, Gajadhar AS, Ternamian C, Gorlia T, Diefes KL, Mischel PS, et al. Alkylpurine-DNA-N-Glycosylase Confers Resistance to Temozolomide in Xenograft Models of Glioblastoma Multiforme and is Associated With Poor Survival in Patients. J Clin Invest (2012) 122(1):253–66. doi: 10.1172/JCI59334
10. Bocangel DB, Finkelstein S, Schold SC, Bhakat KK, Mitra S, Kokkinakis DM. Multifaceted Resistance of Gliomas to Temozolomide. Clin Cancer Res (2002) 8(8):2725–34.
11. Chen CH, Chang YJ, Ku MS, Chung KT, Yang JT. Enhancement of Temozolomide-Induced Apoptosis by Valproic Acid in Human Glioma Cell Lines Through Redox Regulation. J Mol Med (Berl) (2011) 89(3):303–15. doi: 10.1007/s00109-010-0707-1
12. Lin L, Cai J, Tan Z, Meng X, Li R, Li Y, et al. Mutant IDH1 Enhances Temozolomide Sensitivity via Regulation of the ATM/CHK2 Pathway in Glioma. Cancer Res Treat (2021) 53(2):367–77. doi: 10.4143/crt.2020.506
13. Lu Y, Kwintkiewicz J, Liu Y, Tech K, Frady LN, Su YT, et al. Chemosensitivity of IDH1-Mutated Gliomas Due to an Impairment in PARP1-Mediated DNA Repair. Cancer Res (2017) 77(7):1709–18. doi: 10.1158/0008-5472.CAN-16-2773
14. Lee SY. Temozolomide Resistance in Glioblastoma Multiforme. Genes Dis (2016) 3(3):198–210. doi: 10.1016/j.gendis.2016.04.007
15. Forte IM, Indovina P, Iannuzzi CA, Cirillo D, Di Marzo D, Barone D, et al. Targeted Therapy Based on P53 Reactivation Reduces Both Glioblastoma Cell Growth and Resistance to Temozolomide. Int J Oncol (2019) 54(6):2189–99. doi: 10.3892/ijo.2019.4788
16. Brennan CW, Verhaak RG, McKenna A, Campos B, Noushmehr H, Salama SR, et al. The Somatic Genomic Landscape of Glioblastoma. Cell (2013) 155(2):462–77. doi: 10.1016/j.cell.2013.09.034
17. Kloosterhof NK, Bralten LB, Dubbink HJ, French PJ, van den Bent MJ. Isocitrate Dehydrogenase-1 Mutations: A Fundamentally New Understanding of Diffuse Glioma? [Review]. Lancet Oncol (2011) 12(1):83–91. doi: 10.1016/S1470-2045(10)70053-X
18. Donehower LA, Soussi T, Korkut A, Liu Y, Schultz A, Cardenas M, et al. Integrated Analysis of TP53 Gene and Pathway Alterations in The Cancer Genome Atlas. Cell Rep (2019) 28(5):1370–84.e5. doi: 10.1016/j.celrep.2019.07.001
19. Weller M, Stupp R, Reifenberger G, Brandes AA, van den Bent MJ, Wick W, et al. MGMT Promoter Methylation in Malignant Gliomas: Ready for Personalized Medicine? Nat Rev Neurol (2010) (1):39–51. doi: 10.1038/nrneurol.2009.197
20. Szopa W, Burley TA, Kramer-Marek G, Kaspera W. Diagnostic and Therapeutic Biomarkers in Glioblastoma: Current Status and Future Perspectives. BioMed Res Int (2017) 2017:8013575. doi: 10.1155/2017/8013575
21. Eckel-Passow JE, Lachance DH, Molinaro AM, Walsh KM, Decker PA, Sicotte H, et al. Glioma Groups Based on 1p/19q, IDH, and TERT Promoter Mutations in Tumors. N Engl J Med (2015) 372(26):2499–508. doi: 10.1056/NEJMoa1407279
22. Loscher W. Basic Pharmacology of Valproate: A Review After 35 Years of Clinical Use for the Treatment of Epilepsy. CNS Drugs (2002) 16(10):669–94. doi: 10.2165/00023210-200216100-00003
23. Ochiai S, Nomoto Y, Yamashita Y, Watanabe Y, Toyomasu Y, Kawamura T, et al. Roles of Valproic Acid in Improving Radiation Therapy for Glioblastoma: A Review of Literature Focusing on Clinical Evidence. Asian Pac J Cancer Prev (2016) 17(2):463–6. doi: 10.7314/APJCP.2016.17.2.463
24. Yuan Y, Xiang W, Qing M, Yanhui L, Jiewen L, Yunhe M. Survival Analysis for Valproic Acid Use in Adult Glioblastoma Multiforme: A Meta-Analysis of Individual Patient Data and a Systematic Review. Seizure (2014) 23(10):830–5. doi: 10.1016/j.seizure.2014.06.015
25. Kerkhof M, Dielemans JC, van Breemen MS, Zwinkels H, Walchenbach R, Taphoorn MJ, et al. Effect of Valproic Acid on Seizure Control and on Survival in Patients With Glioblastoma Multiforme. Neuro Oncol (2013) 15(7):961–7. doi: 10.1093/neuonc/not057
26. Watanabe S, Kuwabara Y, Suehiro S, Yamashita D, Tanaka M, Tanaka A, et al. Valproic Acid Reduces Hair Loss and Improves Survival in Patients Receiving Temozolomide-Based Radiation Therapy for High-Grade Glioma. Eur J Clin Pharmacol (2017) 73(3):357–63. doi: 10.1007/s00228-016-2167-1
27. Tseng JH, Chen CY, Chen PC, Hsiao SH, Fan CC, Liang YC, et al. Valproic Acid Inhibits Glioblastoma Multiforme Cell Growth via Paraoxonase 2 Expression. Oncotarget (2017) 8(9):14666–79. doi: 10.18632/oncotarget.14716
28. Kalal BS, Pai VR, Behera SK, Somashekarappa HM. HDAC2 Inhibitor Valproic Acid Increases Radiation Sensitivity of Drug-Resistant Melanoma Cells. Med Sci (Basel) (2019) 7(3):eng. doi: 10.3390/medsci7030051
29. Hosein AN, Lim YC, Day B, Stringer B, Rose S, Head R, et al. The Effect of Valproic Acid in Combination With Irradiation and Temozolomide on Primary Human Glioblastoma Cells. J Neurooncol (2015) 122(2):263–71. doi: 10.1007/s11060-014-1713-x
30. Ryu CH, Yoon WS, Park KY, Kim SM, Lim JY, Woo JS, et al. Valproic Acid Downregulates the Expression of MGMT and Sensitizes Temozolomide-Resistant Glioma Cells. J BioMed Biotechnol (2012) 2012:987495. doi: 10.1155/2012/987495
31. Mascaro-Cordeiro B, Oliveira ID, Tesser-Gamba F, Pavon LF, Saba-Silva N, Cavalheiro S, et al. Valproic Acid Treatment Response In Vitro is Determined by TP53 Status in Medulloblastoma. Childs Nerv Syst (2018) 34(8):1497–509. doi: 10.1007/s00381-018-3817-7
32. McCormack E, Haaland I, Venas G, Forthun RB, Huseby S, Gausdal G, et al. Synergistic Induction of P53 Mediated Apoptosis by Valproic Acid and Nutlin-3 in Acute Myeloid Leukemia. Leukemia (2012) 26(5):910–7. doi: 10.1038/leu.2011.315
33. Chen X, Wong P, Radany E, Wong JY. HDAC Inhibitor, Valproic Acid, Induces P53-Dependent Radiosensitization of Colon Cancer Cells. Cancer Biother Radiopharm (2009) 24(6):689–99. doi: 10.1089/cbr.2009.0629
34. Leitch C, Osdal T, Andresen V, Molland M, Kristiansen S, Nguyen XN, et al. Hydroxyurea Synergizes With Valproic Acid in Wild-Type P53 Acute Myeloid Leukaemia. Oncotarget (2016) 7(7):8105–18. doi: 10.18632/oncotarget.6991
35. Wang P, Dong Q, Zhang C, Kuan PF, Liu Y, Jeck WR, et al. Mutations in Isocitrate Dehydrogenase 1 and 2 Occur Frequently in Intrahepatic Cholangiocarcinomas and Share Hypermethylation Targets With Glioblastomas. Oncogene (2013) 25(25):3091–100. doi: 10.1038/onc.2012.315
36. Tsai HC, Wei KC, Tsai CN, Huang YC, Chen PY, Chen SM, et al. Effect of Valproic Acid on the Outcome of Glioblastoma Multiforme. Br J Neurosurg (2012) 26(3):347–54. doi: 10.3109/02688697.2011.638996
37. Bouaoun L, Sonkin D, Ardin M, Hollstein M, Byrnes G, Zavadil J, et al. TP53 Variations in Human Cancers: New Lessons From the IARC TP53 Database and Genomics Data. Hum Mutat (2016) 37(9):865–76. doi: 10.1002/humu.23035
38. Polager S, Ginsberg D. P53 and E2f: Partners in Life and Death. Nat Rev Cancer (2009) 9(10):738–48. doi: 10.1038/nrc2718
39. Kramer OH, Zhu P, Ostendorff HP, Golebiewski M, Tiefenbach J, Peters MA, et al. The Histone Deacetylase Inhibitor Valproic Acid Selectively Induces Proteasomal Degradation of HDAC2 [Research Support, Non-U.S. Gov’t]. EMBO J (2003) 22(13):3411–20. doi: 10.1093/emboj/cdg315
40. Zhang Z, Wang Y, Chen J, Tan Q, Xie C, Li C, et al. Silencing of Histone Deacetylase 2 Suppresses Malignancy for Proliferation, Migration, and Invasion of Glioblastoma Cells and Enhances Temozolomide Sensitivity. Cancer Chemother Pharmacol (2016) 78(6):1289–96. doi: 10.1007/s00280-016-3188-2
41. Cancer Genome Atlas Research N. Comprehensive Genomic Characterization Defines Human Glioblastoma Genes and Core Pathways. Nature (2008) 455(7216):1061–8. doi: 10.1038/nature07385
42. Louis DN, Perry A, Reifenberger G, von Deimling A, Figarella-Branger D, Cavenee WK, et al. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A Summary [Review]. Acta Neuropathol (2016) 131(6):803–20. doi: 10.1007/s00401-016-1545-1
43. Nandeesh BN, Naskar S, Shashtri AH, Arivazhagan A, Santosh V. Recurrent Glioblastomas Exhibit Higher Expression of Biomarkers With Stem-Like Properties. J Neurosci Rural Pract (2018) 9(1):86–91. doi: 10.4103/jnrp.jnrp_417_17
44. Wiewrodt D, Nagel G, Dreimuller N, Hundsberger T, Perneczky A, Kaina B. MGMT in Primary and Recurrent Human Glioblastomas After Radiation and Chemotherapy and Comparison With P53 Status and Clinical Outcome [Comparative Study Research Support, Non-U.S. Gov’t]. Int J Cancer (2008) 122(6):1391–9. doi: 10.1002/ijc.23219
45. Stark AM, Witzel P, Strege RJ, Hugo HH, Mehdorn HM. P53, Mdm2, EGFR, and Msh2 Expression in Paired Initial and Recurrent Glioblastoma Multiforme. J Neurol Neurosurg Psychiatry (2003) 74(6):779–83. doi: 10.1136/jnnp.74.6.779
46. Park CM, Park MJ, Kwak HJ, Moon SI, Yoo DH, Lee HC, et al. Induction of P53-Mediated Apoptosis and Recovery of Chemosensitivity Through P53 Transduction in Human Glioblastoma Cells by Cisplatin. Int J Oncol (2006) 28(1):119–25. doi: 10.3892/ijo.28.1.119
47. Djuzenova CS, Fiedler V, Memmel S, Katzer A, Hartmann S, Krohne G, et al. Actin Cytoskeleton Organization, Cell Surface Modification and Invasion Rate of 5 Glioblastoma Cell Lines Differing in PTEN and P53 Status. Exp Cell Res (2015) 330(2):346–57. doi: 10.1016/j.yexcr.2014.08.013
48. England B, Huang T, Karsy M. Current Understanding of the Role and Targeting of Tumor Suppressor P53 in Glioblastoma Multiforme [Review]. Tumour Biol (2013) 34(4):2063–74. doi: 10.1007/s13277-013-0871-3
49. Bieging KT, Mello SS, Attardi LD. Unravelling Mechanisms of P53-Mediated Tumour Suppression. Nat Rev Cancer (2014) 14(5):359–70. doi: 10.1038/nrc3711
50. Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, et al. Radiotherapy Plus Concomitant and Adjuvant Temozolomide for Glioblastoma. N Engl J Med (2005) 352(10):987–96. doi: 10.1056/NEJMoa043330
51. Stupp R, Hegi ME, Mason WP, van den Bent MJ, Taphoorn MJ, Janzer RC, et al. Effects of Radiotherapy With Concomitant and Adjuvant Temozolomide Versus Radiotherapy Alone on Survival in Glioblastoma in a Randomised Phase III Study: 5-Year Analysis of the EORTC-NCIC Trial. Lancet Oncol (2009) 10(5):459–66. doi: 10.1016/S1470-2045(09)70025-7
52. Wen J, Chen W, Zhu Y, Zhang P. Clinical Features Associated With the Efficacy of Chemotherapy in Patients With Glioblastoma (GBM): A Surveillance, Epidemiology, and End Results (SEER) Analysis. BMC Cancer (2021) 21(1):81. doi: 10.1186/s12885-021-07800-0
53. Lamborn KR, Chang SM, Prados MD. Prognostic Factors for Survival of Patients With Glioblastoma: Recursive Partitioning Analysis. Neuro Oncol (2004) 6(3):227–35. doi: 10.1215/S1152851703000620
54. Berendsen S, Varkila M, Kroonen J, Seute T, Snijders TJ, Kauw F, et al. Prognostic Relevance of Epilepsy at Presentation in Glioblastoma Patients. Neuro Oncol (2016) 18(5):700–6. doi: 10.1093/neuonc/nov238
55. Cohen AL, Holmen SL, Colman H. IDH1 and IDH2 Mutations in Gliomas. Curr Neurol Neurosci Rep (2013) 13(5):345. doi: 10.1007/s11910-013-0345-4
56. Cheng HB, Yue W, Xie C, Zhang RY, Hu SS, Wang Z. IDH1 Mutation is Associated With Improved Overall Survival in Patients With Glioblastoma: A Meta-Analysis. Tumour Biol: J Int Soc Oncodevel Biol Med (2013) 34(6):3555–9. doi: 10.1007/s13277-013-0934-5
57. Wali AR, Rennert RC, Wang SG, Chen CC. Prophylactic Anticonvulsants in Patients With Primary Glioblastoma. J Neurooncol (2017) 135(2):229–35. doi: 10.1007/s11060-017-2584-8
58. Wu AS, Trinh VT, Suki D, Graham S, Forman A, Weinberg JS, et al. A Prospective Randomized Trial of Perioperative Seizure Prophylaxis in Patients With Intraparenchymal Brain Tumors. J Neurosurg (2013) 118(4):873–83. doi: 10.3171/2012.12.JNS111970
59. Kuo YJ, Yang YH, Lee IY, Chen PC, Yang JT, Wang TC, et al. Effect of Valproic Acid on Overall Survival in Patients With High-Grade Gliomas Undergoing Temozolomide: A Nationwide Population-Based Cohort Study in Taiwan. Med (Baltimore) (2020) 99(28):e21147. doi: 10.1097/MD.0000000000021147
60. Lu VM, Texakalidis P, McDonald KL, Mekary RA, Smith TR. The Survival Effect of Valproic Acid in Glioblastoma and its Current Trend: A Systematic Review and Meta-Analysis. Clin Neurol Neurosurg (2018) 174:149–55. doi: 10.1016/j.clineuro.2018.09.019
61. Zhao YH, Wang ZF, Cao CJ, Weng H, Xu CS, Li K, et al. The Clinical Significance of O(6)-Methylguanine-DNA Methyltransferase Promoter Methylation Status in Adult Patients With Glioblastoma: A Meta-Analysis. Front Neurol (2018) 9:127. doi: 10.3389/fneur.2018.00127
62. Riemenschneider MJ, Hegi ME, Reifenberger G. MGMT Promoter Methylation in Malignant Gliomas. Target Oncol (2010) 5(3):161–5. doi: 10.1007/s11523-010-0153-6
63. Wang L, Li Z, Liu C, Chen L, Liu L, Hu Z, et al. Comparative Assessment of Three Methods to Analyze MGMT Methylation Status in a Series of 350 Gliomas and Gangliogliomas. Pathol Res Pract (2017) 213(12):1489–93. doi: 10.1016/j.prp.2017.10.007
64. Rodriguez FJ, Thibodeau SN, Jenkins RB, Schowalter KV, Caron BL, O’Neill BP, et al. MGMT Immunohistochemical Expression and Promoter Methylation in Human Glioblastoma. Appl Immunohistochem Mol Morphol (2008) 16(1):59–65. doi: 10.1097/PAI.0b013e31802fac2f
65. Brandner S, McAleenan A, Kelly C, Spiga F, Cheng HY, Dawson S, et al. MGMT Promoter Methylation Testing to Predict Overall Survival in People With Glioblastoma Treated With Temozolomide: A Comprehensive Meta-Analysis Based on a Cochrane Review. Neuro Oncol (2021) 23(9):1457–69. doi: 10.1093/neuonc/noab105
66. Roos WP, Batista LF, Naumann SC, Wick W, Weller M, Menck CF, et al. Apoptosis in Malignant Glioma Cells Triggered by the Temozolomide-Induced DNA Lesion O6-Methylguanine. Oncogene (2007) 26(2):186–97. doi: 10.1038/sj.onc.1209785
67. Zhang Y, Dube C, Gibert M Jr., Cruickshanks N, Wang B, Coughlan M, et al. The P53 Pathway in Glioblastoma. Cancers (Basel) (2018) 10(9):297. doi: 10.3390/cancers10090297
68. Nakano K, Vousden KH. PUMA, a Novel Proapoptotic Gene, is Induced by P53. Mol Cell (2001) 7(3):683–94. doi: 10.1016/s1097-2765(01)00214-3
69. Yu J, Zhang L. PUMA, a Potent Killer With or Without P53 [Research Support, N.I.H., Extramural Research Support, Non-U.S. Gov’t Review]. Oncogene (2008) 27 Suppl 1:S71–83. doi: 10.1038/onc.2009.45
70. Ito H, Kanzawa T, Miyoshi T, Hirohata S, Kyo S, Iwamaru A, et al. Therapeutic Efficacy of PUMA for Malignant Glioma Cells Regardless of P53 Status [Research Support, N.I.H., Extramural Research Support, Non-U.S. Gov’t Research Support, U.S. Gov’t, P.H.S.]. Hum Gene Ther (2005) 16(6):685–98. doi: 10.1089/hum.2005.16.685
71. Miao W, Liu X, Wang H, Fan Y, Lian S, Yang X, et al. P53 Upregulated Modulator of Apoptosis Sensitizes Drug-Resistant U251 Glioblastoma Stem Cells to Temozolomide Through Enhanced Apoptosis [Research Support, Non-U.S. Gov’t]. Mol Med Rep (2015) 11(6):4165–73. doi: 10.3892/mmr.2015.3255
72. He Y, Kaina B. Are There Thresholds in Glioblastoma Cell Death Responses Triggered by Temozolomide? Int J Mol Sci (2019) (7):1562. doi: 10.3390/ijms20071562
73. Ostermann S, Csajka C, Buclin T, Leyvraz S, Lejeune F, Decosterd LA, et al. Plasma and Cerebrospinal Fluid Population Pharmacokinetics of Temozolomide in Malignant Glioma Patients. Clin Cancer Res (2004) 10(11):3728–36. doi: 10.1158/1078-0432.CCR-03-0807
74. Portnow J, Badie B, Chen M, Liu A, Blanchard S, Synold TW. The Neuropharmacokinetics of Temozolomide in Patients With Resectable Brain Tumors: Potential Implications for the Current Approach to Chemoradiation. Clin Cancer Res (2009) 15(22):7092–8. doi: 10.1158/1078-0432.CCR-09-1349
75. Van Meir EG, Kikuchi T, Tada M, Li H, Diserens AC, Wojcik BE, et al. Analysis of the P53 Gene and its Expression in Human Glioblastoma Cells. Cancer Res (1994) 54(3):649–52.
76. Roth W, Fontana A, Trepel M, Reed JC, Dichgans J, Weller M. Immunochemotherapy of Malignant Glioma: Synergistic Activity of CD95 Ligand and Chemotherapeutics. Cancer Immunol Immunother (1997) 44(1):55–63. doi: 10.1007/s002620050355
77. Xie Z, Ago Y, Okada N, Tachibana M. Valproic Acid Attenuates Immunosuppressive Function of Myeloid-Derived Suppressor Cells. J Pharmacol Sci (2018) 137(4):359–65. doi: 10.1016/j.jphs.2018.06.014
78. Roos WP, Jost E, Belohlavek C, Nagel G, Fritz G, Kaina B. Intrinsic Anticancer Drug Resistance of Malignant Melanoma Cells is Abrogated by IFN-Beta and Valproic Acid. Cancer Res (2011) 71(12):4150–60. doi: 10.1158/0008-5472.CAN-10-3498
79. Paradis FH, Hales BF. Valproic Acid Induces the Hyperacetylation of P53, Expression of P53 Target Genes, and Markers of the Intrinsic Apoptotic Pathway in Midorganogenesis Murine Limbs. Birth Defects Res B Dev Reprod Toxicol (2015) 104(5):177–83. doi: 10.1002/bdrb.21149
80. Gryder BE, Sodji QH, Oyelere AK. Targeted Cancer Therapy: Giving Histone Deacetylase Inhibitors All They Need to Succeed [Research Support, N.I.H., Extramural Research Support, Non-U.S. Gov’t Review]. Future Med Chem (2012) 4(4):505–24. doi: 10.4155/fmc.12.3
81. Bailey H, Stenehjem DD, Sharma S. Panobinostat for the Treatment of Multiple Myeloma: The Evidence to Date [Review]. J Blood Med (2015) 6:269–76. doi: 10.2147/JBM.S69140
Keywords: glioblastoma, temozolomide, valproic acid, p53, PUMA, apoptosis
Citation: Tsai H-C, Wei K-C, Chen P-Y, Huang C-Y, Chen K-T, Lin Y-J, Cheng H-W, Chen Y-R and Wang H-T (2021) Valproic Acid Enhanced Temozolomide-Induced Anticancer Activity in Human Glioma Through the p53–PUMA Apoptosis Pathway. Front. Oncol. 11:722754. doi: 10.3389/fonc.2021.722754
Received: 09 June 2021; Accepted: 08 September 2021;
Published: 01 October 2021.
Edited by:
Liam Chen, University of Minnesota, United StatesReviewed by:
Bernd Kaina, Johannes Gutenberg University Mainz, GermanySalvatore Massimiliano Cardali, University of Messina, Italy
Copyright © 2021 Tsai, Wei, Chen, Huang, Chen, Lin, Cheng, Chen and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hsiang-Tsui Wang, aHR3YW5nMDFAbnljdS5lZHUudHc=