 Natalia Scaglia
Natalia Scaglia Yesica Romina Frontini-López
Yesica Romina Frontini-López Giorgia Zadra
Giorgia Zadra- 1Biochemistry Research Institute of La Plata "Professor Doctor Rodolfo R. Brenner" (INIBIOLP), National University of La Plata/National Council of Scientific and Technical Research of Argentina, La Plata, Argentina
- 2Institute of Molecular Genetics, National Research Council, Pavia, Italy
Advanced prostate cancer (PCa) represents the fifth cause of cancer death worldwide. Although survival has improved with second-generation androgen signaling and Parp inhibitors, the benefits are not long-lasting, and new therapeutic approaches are sorely needed. Lipids and their metabolism have recently reached the spotlight with accumulating evidence for their role as promoters of PCa development, progression, and metastasis. As a result, interest in targeting enzymes/transporters involved in lipid metabolism is rapidly growing. Moreover, the use of lipogenic signatures to predict prognosis and resistance to therapy has been recently explored with promising results. Despite the well-known association between obesity with PCa lethality, the underlying mechanistic role of diet/obesity-derived metabolites has only lately been unveiled. Furthermore, the role of lipids as energy source, building blocks, and signaling molecules in cancer cells has now been revisited and expanded in the context of the tumor microenvironment (TME), which is heavily influenced by the external environment and nutrient availability. Here, we describe how lipids, their enzymes, transporters, and modulators can promote PCa development and progression, and we emphasize the role of lipids in shaping TME. In a therapeutic perspective, we describe the ongoing efforts in targeting lipogenic hubs. Finally, we highlight studies supporting dietary modulation in the adjuvant setting with the purpose of achieving greater efficacy of the standard of care and of synthetic lethality. PCa progression is “a matter of fats”, and the more we understand about the role of lipids as key players in this process, the better we can develop approaches to counteract their tumor promoter activity while preserving their beneficial properties.
Introduction
Prostate cancer (PCa) is the second leading cause of cancer death in men in the US and the fifth cause worldwide (1). While primary PCa is successfully treated with surgery, about 30% of PCa cases recur. Androgen deprivation therapy (ADT) is the standard of care for androgen-sensitive metastatic PCas (mASPC). mASPC are initially responsive to ADT but will eventually develop resistance, a disease stage known as metastatic castration-resistant PCa (mCRPC) (2). Management of mCRPC was primarily based on taxanes (i.e., Docetaxel, Cabazitaxel). However, in the last decade, second-generation androgen-receptor (AR) signaling inhibitors (i.e., enzalutamide) and intra-tumor androgen synthesis inhibitors (i.e., abiraterone) have been approved for the treatment of mCRPC with improved survival benefits. Unfortunately, efficacy is not long-lasting due to the occurrence of several mechanisms of resistance, including the overexpression of AR splicing constitutive active variants (i.e., AR-V7) (3). Therapeutic strategies based on the radioactive isotope radium-223 or cell-based immunotherapy (Sipuleucel-T) are also not resolutive (2, 4). Treatments with PARP inhibitors have been recently approved by the U.S. Food and Drug Administration (FDA) for the treatment of mCRPC patients harboring tumors with defects in DNA damage response (DDR), especially in the gene BRAC2, opening a new area for precision oncology in advanced PCa (5). This is also supported by the recent approval of two genetic tests, BRACAnalysis CDx and FoundationOne CDx to identifying mCRPC who have DDR genetic alterations and thus will most likely respond to PARP inhibitors (i.e., Olaparib and Rucaparib). (https://bit.ly/2z5Lu5C; https://bwnews.pr/2ZtfCSS). Clinical trials testing immune check-point inhibitors (ICI) are ongoing in mCRPC patients with disappointing results so far (6) Thus, strategies to boost responses to ICI are currently sought. Alterations of lipid metabolism in PCa cells were first observed long time ago using radiolabeling approaches and linked to AR signaling modulation (7, 8). However, the last decades have faced a change in the perspective of lipid role in cancer development and progression. In addition of being building blocks for membrane synthesis and energy fuel, lipids have emerged as key players in mediating oncogenic signaling, endoplasmic reticulum (ER) and oxidative stresses, non-apoptotic cell death (i.e., ferroptosis), and inflammatory stimuli (9). More recently, a lot of attention has been paid on the impact of lipids on the tumor microenvironment (TME), in particular on the immune TME (10). The plasticity of lipid metabolism rewiring allows PCa cells to thrive in hostile and nutrient-deprived environments and to spread to distant tissues. As a result, new mechanisms of therapy resistance and disease progression associated with lipid metabolism rewiring have recently emerged. This has also been supported by the recent advances in analytical techniques including high-resolution mass spectrometry (MS)-based lipidomics and MS-based imaging (MSI) that allow to measure hundreds of lipid species at once, Including rare lipid species, and to provide spatial resolution. These new technologies have uncovered the complexity and heterogeneity of lipid metabolism rewiring in a way that could not have been assessed before, opening new possibilities for both biomarkers and therapeutics discovery (9). Indeed, the combination of lipid metabolism modulators with standard of care is now strongly pursued.
In this review, we describe how lipid metabolism rewiring, including alterations in both de novo lipid synthesis, uptake, transport, storage, and utilization, contributes to PCa progression and therapy resistance and we discuss how these vulnerabilities can be exploited therapeutically. We emphasize the recently uncovered role of lipid metabolism in immune TME and the potential impact of de novo lipogenesis inhibitors as immunotherapy sensitizers. This review also briefly describes the advances in measuring and imaging lipids and how these more sophisticated analytical techniques contribute to improve biomarker discovery.
This review highlights that PCa progression is “a matter of fats” and lipids are rediscovered protagonists of oncogenic signaling, stress adaptation, and tumor-TME crosstalk. The more we understand about these aspects the better we can develop strategies to counteract their tumor supportive functions while enhancing their health-promoting roles.
Lipid Metabolism Rewiring in PCa Development and Progression
Since the discovery in the mid-1990s of OA-519, an oncogenic antigen encoding for Fatty acid (FA) Synthase (FASN) highly expressed in breast cancer, research on the role of lipid metabolism in cancer has proceeded quite slowly with respect to other fields (11). However, interest for the mechanisms through which lipids promote tumorigenesis and tumor progression has been regained in the last decade, paralleled by the rapid development of high-resolution analytical techniques to interrogate the lipidome in a comprehensive and unbiased manner. While the majority of attention has been focused on the de novo FA synthesis dysregulation as hallmark of PCa development and progression, the perspective has lately changed to include many aspects of lipid metabolism spanning from FA uptake and transport, FA oxidation, lipid storage, and remodeling.
Alterations in De Novo FA Synthesis
While non-transformed prostate cells obtain the majority of lipids for membrane synthesis and energy fuel from the diet and circulation, PCa cells show an increased in de novo FA synthesis from glucose or glutamine, despite circulating lipids [reviewed in (12)]. This results in increased production of phospholipids and sphingolipids to support new membrane synthesis in proliferating PCa cells but also in a net accumulation of intra-tumor lipids mostly as triglycerides stored lipid droplets (LD) (13) (Figure 1). LD accumulation, which is associated with a more aggressive disease, provides an excellent reservoir for building blocks and energy in conditions of nutrients deprivation such as those encountered during PCa progression and metastatic spread. Moreover, LDs prevent lipotoxicity due to excessive accumulation of free FAs (see below). This increased net lipid production, known as “lipogenic phenotype”, is observed at early stages of PCa development and it is further enhanced in mCRPC. Consistently, enzymes or transcriptional factors (TFs) involved in de novo FA synthesis such as the TF sterol regulatory element-binding proteins (SREBPs), ATP citrate lyase (ACLY), Acetyl-CoA carboxylase (ACC), and FASN are overexpressed in primary PCa and especially in mCRPC. Specifically, FASN, the key lipogenic enzyme responsible for the synthesis of the 16C saturated FA palmitate from acetyl-CoA and malonyl-CoA, was found among the top ten genes overexpressed in AR-V7-driven CRPC metastases (mets) (14–16). In line with this, the interrogation of the Cancer Genome Atlas and other publicly available datasets uncovered a positive association between FASN expression and worse clinico-pathological features, including Gleason grade, tumor stage, lymph node positivity, shorter time to recurrence, cancer-free survival, and overall survival [reviewed in (17)]. As a result, great efforts are directed to exploit the lipogenic phenotype in mCRPC (see below). The work of Swinnen and coworkers has been instrumental to demonstrate the tight control of de novo FA synthesis by androgens/AR signaling, the major driver of PCa development and progression to mCRPC (7, 8). A feedforward mechanism between SREBP and AR was initially described, whereby AR promotes SREBP activation and nuclear translocation while SREBP regulates AR promoter activity and expression (18, 19). Later on, Chan and coworkers identified AR-binding site in FASN gene promoter, suggesting AR-mediated direct regulation of FASN expression (20). This evidence has been supported by immunoprecipitation sequencing (ChIP-seq) experiments that revealed AR binding sites in several lipogenic enzymes in CRPC samples, besides FASN (21). Altogether these data suggest that both indirect and direct mechanisms of AR-mediated control of the lipogenic program exist. Cai and coworkers analyzed AR cistrome and demonstrated that activation of lipid biosynthesis is a major function of AR signaling during PCa progression. Specifically, increased expression of AR-V7 turned out to be crucial for the reactivation of the lipid synthesis in CRPC, suggesting a key role of this splicing variant in regulating lipid metabolism in the CRPC setting. The authors also identified an AR-dependent lipogenic gene expression signature that predicts poor patient outcome (16). Our recent study has uncovered the existence of a reciprocal modulation between FASN and AR, in particular AR-V7, and it has proposed FASN inhibition as a non-canonical approach to indirectly antagonize AR-V7 and potentially overcome therapy resistance to enzalutamide and abiraterone (22). Overexpression of ACLY, ACC, and FASN has been consistently associated with increased PCa cell proliferation, tumor growth, migration and invasion, activation of oncogenic signaling, protection from chemotherapeutics-induced apoptosis, features that are reversed using genetic/pharmacological inhibition of enzyme activities [reviewed in (12)]. However, new roles for de novo FA synthesis in PCa progression have recently emerged. These involve post-translational modifications, DNA damage response, redox maintenance, ER and oxidative stress and resistance to ferroptosis, a lipid peroxidation-mediated non-apoptotic form of cell death [reviewed in (9)]. Palmitoylation of Wnt-1, RAS-related protein Rab-7a, alpha-tubulin, and eIF3L initiation factor are some of the post-translational modifications mediated by FASN and regulated by AR that activate oncogenic signaling in PCa (23–25). More recently, a palmitoyl-protein signature has been described in PCa derived extracellular vesicles (EVs), membrane-enclosed particles that play an important role in cancer progression as source of nutrients, signaling molecules, immune modulators, and circulating biomarkers, uncovering another potential mechanism of support to PCa progression (26).
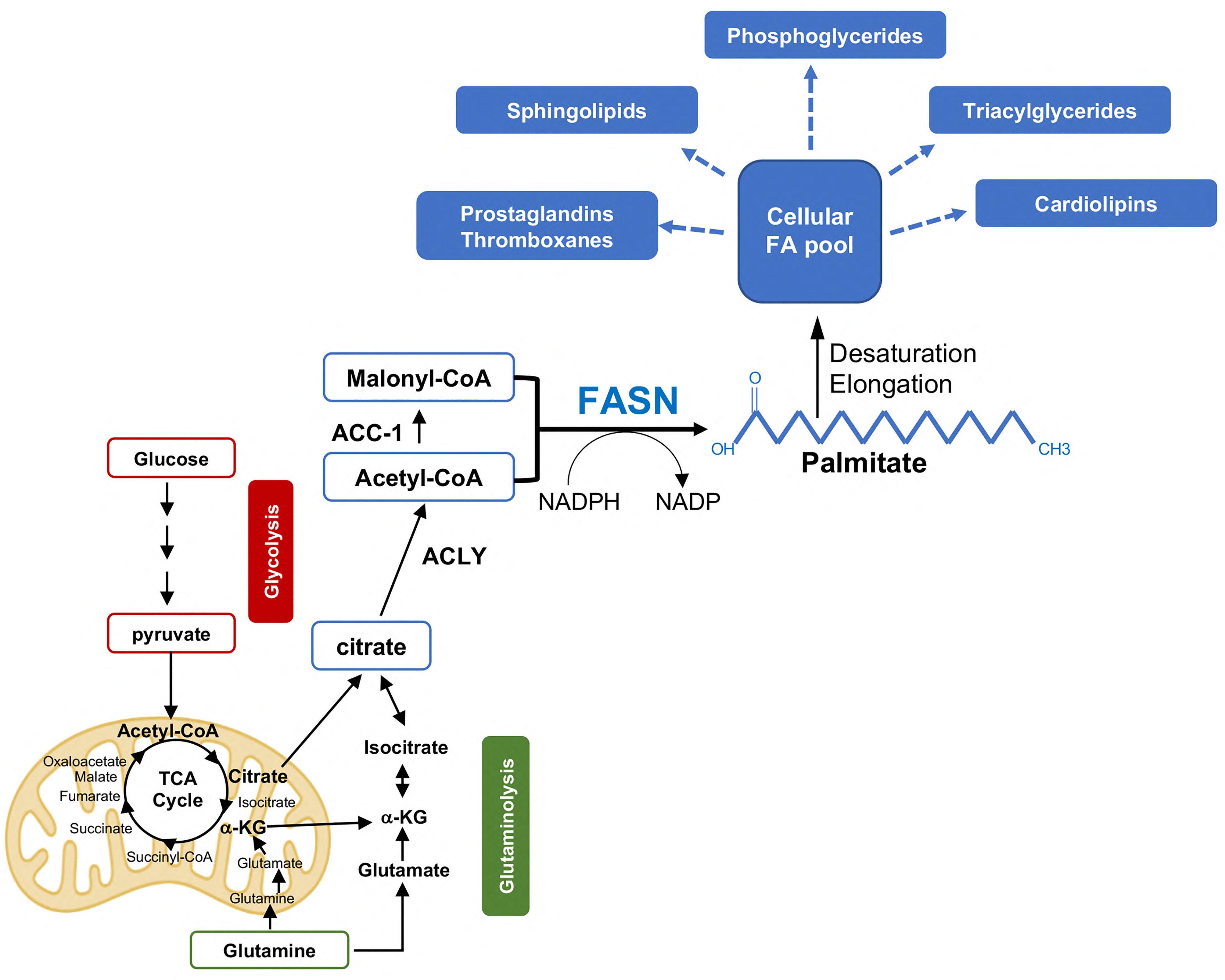
Figure 1 De novo FA synthesis in PCa progression. Acetyl-CoA derived from glucose and glutamine metabolism is exported to the cytosol in the form of citrate and reconverted to acetyl-CoA by the enzyme ATP citrate lyase (ACLY). Acetyl-CoA is then converted to Malonyl-CoA by the Acetyl coenzyme A carboxylase a (or ACC-1). Fatty acid synthase (FASN) catalyzes the condensation of Acetyl-CoA and Malonyl-CoA (in the presence of the reducing equivalent icotinamide adenine dinucleotide phosphate (NADPH)) to generate the 16-carbon FA palmitate, a saturated FA that undergoes further modifications (i.e., elongation/desaturation) to form more complex lipids. The latter serve as energy source, building blocks, and inflammatory/immune modulators to sustain PCa progression and CR. αKG, alpha-ketoglutarate; NADPH, Nicotinamide adenine dinucleotide phosphate.
In 2016, Wu and coworkers demonstrated the involvement of FASN in DNA repair and resistance to genotoxic insults. The authors found that FASN up-regulation regulates PARP-1 expression through NF-κB and SP1 modulation and increases Ku protein recruitment and DNA repair through activation of non-homologous end joining (27). Evidence for a direct interaction of FASN with MRN (MRE11-RAD50-NBS) complex has also been reported (28). By consuming NADPH, high rates of de novo FA synthesis also maintain redox balance and increased NADP/NADPH ratio, which is needed to support oxidative reactions such those in the pentose phosphate pathway for nucleotide synthesis (29). FASN expression/activity is also crucial in counteracting ER and oxidative stress in PCa by promoting saturated FA acids (SFA) synthesis and the remodeling of ER and mitochondrial membranes (22, 30–32). Furthermore, the increased production of SFAs and their acylation in phospholipids give rise to membranes characterized by a high ratio of SFA and polyunsaturated FAs (SFA/PUFA). These changes affect membrane fluidity, microdomains formation (i.e., lipid rafts), and lipid peroxidation (33, 34). SFA-enriched membranes affect the uptake of certain chemotherapeutics such as doxorubicin and promote the resistance to ionizing radiation (35, 36). Since SFAs are more resistant to lipid peroxidation, metastatic PCa cells with SFA-enriched membranes would be most likely less susceptible to oxidative stress-induced ferroptosis, an iron-dependent form of cell death induced by reactive oxygen species (ROS)-mediated lipid peroxidation (37). Targeting de novo lipogenesis and the Lands cycle has recently been shown to induce ferroptosis in KRAS-mutant lung cancer (38) and we anticipate similar results in mCRPC. These new findings have opened new possibilities for combinatorial treatments.
Alterations in FA Modelling
Once palmitate (16:0) and stearate (a 2C-elongated FA, 18:0), the most abundant SFAs, are synthesized or acquired from the diet (Figures 1, 2) they usually undergo further modifications including desaturation and elongation. Desaturation of de novo synthesized SFAs involves the introduction of a cis-double bond to the acyl chain at the delta-9 (Δ9) position by stearoyl-CoA desaturases (SCDs) to generate the monounsaturated FAs (MUFA) palmitoleate and oleate (39). As humans lack delta-12 (Δ12) and delta-15 (Δ15) desaturases, PUFAs need to be acquired from the diet. Hence, α-linoleic acid (LA, an omega-6 PUFA) and α-linolenic acid (ALA, an omega-3 FA) are essential diet-derived PUFAs, which are required for the generation of further desaturated PUFAs (i.e., arachidonic acid), eicosanoids (i.e., prostaglandins and thromboxanes), and lipoxins, all of which play crucial roles as signaling molecules and mediators of PCa progression (40). LA and ALA desaturation is primarily catalyzed by the FA desaturases FADS1-3. Two human isoforms of SCD exist, SCD1 and SCD5 (41, 42). SCD1, the most abundant SCD in human cells, is highly expressed in human PCa with respect to normal tissues (43). Consistently, PCa cells upregulate de novo FA synthesis to generate SFA and MUFA-rich phospholipids that partition into detergent-resistant lipid rafts to markedly alter signal transduction, vesicular trafficking, and cell migration (44, 45). SCD1 pharmacological inhibition with BZ36 was shown to repress proliferation of AS LNCaP and CRPC C4-2 cells in vitro and in vivo through the abrogation of phosphatidylinositol generation and consequent inhibition of AKT pathway (46). Inhibition of SCD1 was also shown to activate 5' AMP-activated protein kinase (AMPK) and glycogen synthase kinase-3 (GSK3β), resulting in decreased β-catenin transcriptional activity (46).
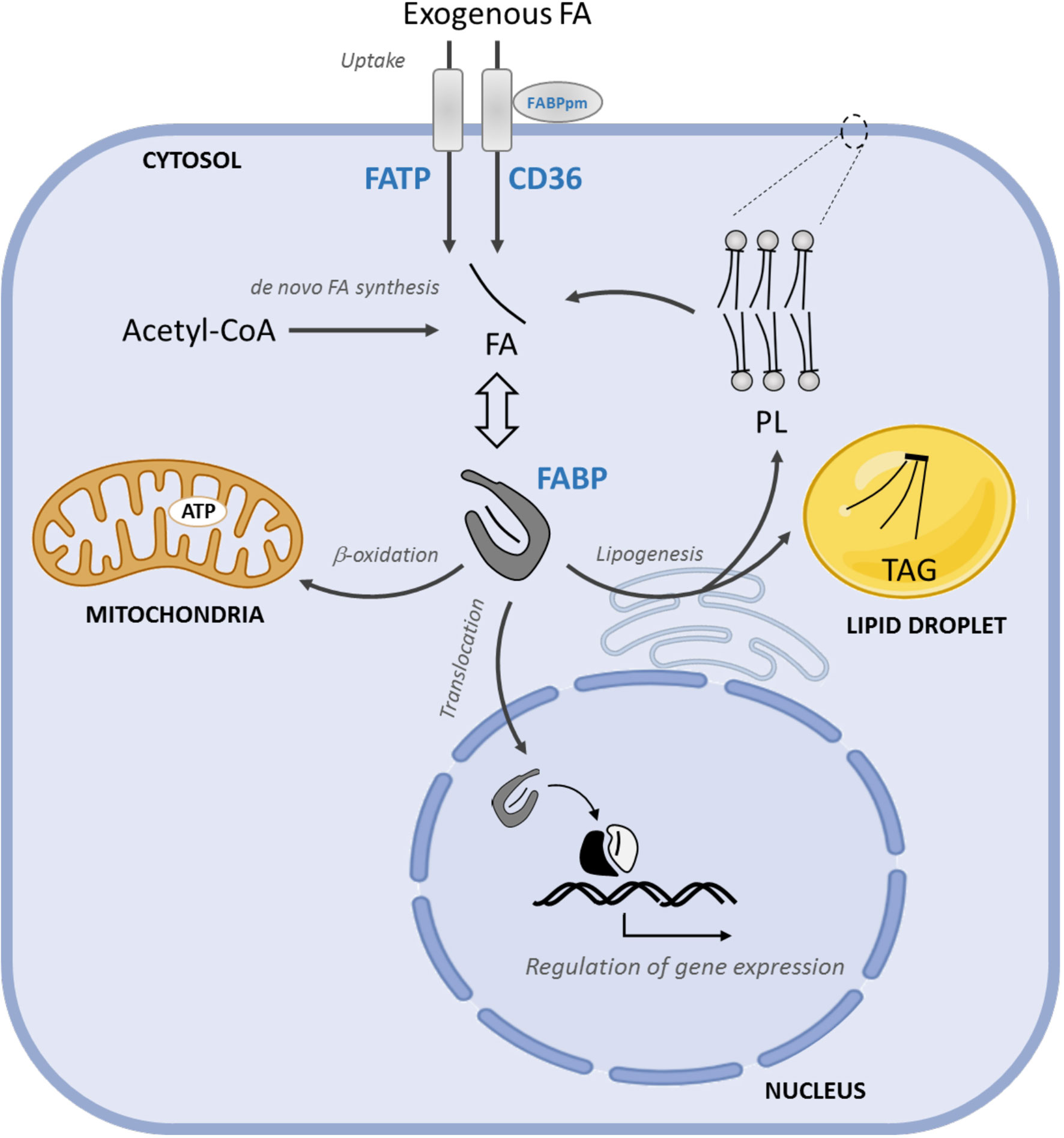
Figure 2 FA uptake, intracellular transport, and FAO in PCa progression. The uptake of exogenous FA is mediated by membrane FA transporters, CD36, FATPs and FABPpm. FABPs solubilize cytosolic FAs and coordinate their intracellular transport towards FABPs solubilize cytosolic FAs towards storage (mainly TAG) or structural (principally PL). Moreover, FABPs coordinate their intracellular transport lipids to the mitochondria for energy supply and to the nucleus where FA regulate gene expression storage (mainly TAG) or structural (principally PL) lipids, to the mitochondria for energy supply and to the nucleus, where FA regulate gene expression. FATP, FA transport proteins; FABPs, FA binding proteins; TAG, triacylglycerides; PL, phospholipids.
SCD1 silencing also results in changes in the composition of cardiolipins, the major constituents of mitochondria membranes. As a result, alterations of mitochondria membrane favor the release of cytochrome c and the induction of apoptosis (43). Thus, overexpression of SCD-1 may represent a protective mechanism to apoptosis that PCa cells adopt, especially during stress. As an oxygen and NADPH-consuming process, desaturation occurrence is particularly challenging during cancer progression where hypoxic conditions are frequently observed. To overcome this, cancer cells tend to accumulate MUFAs in LDs, hydrolyze LDs, and assemble MUFA into PLs under hypoxic conditions. While the increase in MUFA incorporation in cellular membranes enhances their fluidity, it also reduces their PUFA/MUFA ratio, providing a robust protection from ferroptosis (47, 48). Thus, SCD-1 inhibitors are currently tested in the preclinical setting to induce ferroptosis (9, 48, 49).
Besides desaturation, FAs undergo elongation, a process that is catalyzed by a class of enzymes called elongases (ELOngation of Very Long fatty acids; ELOVLs), comprising seven members (ELOVL 1-7). ELOVLs add two carbon units to the carboxyl end of FA chains. While their precise functions are still not fully clarified, ELOVL-1, -3, and -6 predominantly elongate SFAs and MUFAs, ELOVL-2 and -4 elongate PUFAs, ELOVL-5 elongates MUFAs and PUFAs, and ELOVL-7 elongates SFAs and PUFAs (50). ELOVL-7 was the first elongase identified as overexpressed in human PCa tissues with respect to adjacent non-tumoral tissues (51). ELOVL-7 is induced by androgens and when overexpressed in LNCaP xenograft promotes tumor growth in mice fed high-fat diet (HFD). In vitro FA elongation assay and FA composition analysis showed that ELOVL-7 is preferentially involved in FA elongation of very-long-chain SFAs included in phospholipids and neutral lipids (i.e., cholesterol ester) and when silenced it reduces androgens synthesis and CRPC tumor growth (51). Both ELOV-7 and ELOVL-5 are among the lipogenic genes overexpressed following AR reactivation and thus considered critical for the progression to CRPC (16). Consistently, Centenera and coworkers showed a significant increase in AR-regulated elongation of fatty acyl chain phospholipids, mediated by ELOVL-5, in both PCa cells and patient-derived explants. ELOVL-5 silencing markedly altered mitochondrial morphology and function, leading to enhanced ROS generation and suppression of PCa cell proliferation, 3D growth, in vivo tumor growth, and metastasis formation. These features were rescued by the supplementation of cis-vaccenic MUFA, a direct product of ELOVL-5 elongation. These data suggest that lipid elongation is a metastasis-promoter metabolic pathway, which is targetable via ELOVL-5 (52). Aside from membrane lipid elongation, ELOVL-5 has more recently been involved in the generation of eicosanoids, inflammatory lipids with potent pro-tumorigenic signaling effects (9).
Alterations in FA Uptake
The cellular uptake of free FAs, either derived from the hydrolysis of triacylglycerols (TAGs) in very low-density lipoproteins (VLDL), chylomicron or adipocytes, require their transport across the plasma membrane. The mechanisms and the identity of the proteins involved in this process are still not fully understood (53, 54). This is use, in part, to the use of bulky fluorescent, non-metabolizable FA analogs or indirect measurement of FA uptake. So far, the best characterized mediators of FA uptake are the scavenger receptor CD36, membrane-associated FA binding protein (FABPpm), and transmembrane FA transport proteins (FATPs) (Figure 2).
CD36 (also known as FAT, SCARB3, SR-B2, GP4 and others) is a ubiquitously expressed plasma membrane glycoprotein that binds diverse ligands, including FA, thrombospondin, oxidized low-density lipoproteins (LDL) and anionic phospholipids. CD36 is involved in FA uptake, clearance of apoptotic cells, and angiogenesis and it has been implicated in several diseases, including cancer (55–58). In skeletal muscles, CD36 has also been found in the outer mitochondrial membrane, where it might be involved in FA oxidation (FAO) under muscle contraction (i.e., exercise), although this aspect is still controversial (59, 60).
CD36 drives tumor progression in glioblastoma, melanoma, oral, and other carcinomas and it is required for stem cell self-renewal, tumor initiation, and metastatic potential in preclinical models (61, 62). CD36 is overexpressed more commonly in mets than in primary tumors and associated with poor prognosis (62–65). Furthermore, CD36 mRNA levels positively correlate with epithelial-mesenchymal transition (EMT) in several cancers, including PCa (63). In human PCa, CD36 protein was detected in both epithelial and stromal cells and equally expressed in tumor and adjacent normal regions, preventing its use as diagnostic biomarker (65). The discrepancy between the mRNA and protein findings may be ascribed to post-transcriptional mechanisms (66). Once in the cytosol, the fate of a FA largely depends on the cell metabolic status and ongoing signaling activation, resulting in FA incorporation in structural (mainly membrane PL) or storage lipids (in the form of TAG), in FA employment as second messenger or inflammatory molecule, and in FA use as fuel. The assessment of CD36 protein or mRNA levels has been used as FAO proxy in some studies. This a priori association is, however, often misleading, as in the case of PCa where enhanced FA uptake in human PCa and patient-derived xenografts (PDX) results in increased incorporation of FAs into complex lipids without FAO alteration. Consistently, CD36 ablation in PTEN knockout (KO) PCa mouse model failed to alter FAO (65). As expected, FA uptake impairment increases de novo FA synthesis as a compensatory mechanism, prompting the concomitant use of FA uptake and synthesis inhibitors. Accordingly, the combination of CD36 and FASN inhibitors significantly reduced PCa proliferation in vivo and in patient-derived PCa explants and it increased sensitivity to ionizing radiation, suggesting a potential synergistic effect in the clinical setting (65–68)
FABPpm is located in the outer plasma membrane leaflet and in mitochondria, displaying different function in each compartment (54). Despite the name, FABPpm is not related to the cytosolic FA binding proteins (see below). FABPpm expression is regulated by androgens in AS PCa cells, while its expression and function in CRPC cells is still largely unknown (69).
FATP1-6, also known as solute carrier family 27 (SLC27A1-6), are differentially expressed in a wide variety of tissues with different subcellular localizations. Their role as FA transporters and their function are still not fully clarified (54, 70). FATP1 is involved in FA metabolism and cancer progression (71–73). FATPs expression is highly heterogeneous in PCa tissues and cell lines and it varies across databases and detection methodologies (64, 74). The expression of FATP-6 is increased in enzalutamide-resistant LNCaP cells compared to the parental cells but no association with prognosis was observed in the clinical setting (75). Thus, further investigation on the role of FATP1-6 in PCa progression is required.
Besides those described above, other mechanisms for scavenging lipids from the extracellular milieu (76) may be involved. Recently, VLDL endocytosis has been described in breast cancer as a new mechanism to acquire exogenous FA, which may potentially occur also in advanced PCa (74, 77). The plasticity of cancer cells to obtain FA sources should be carefully taken into account during the planning and the design of therapeutic strategies. Dual targeting of FA uptake and synthesis holds promise for translation into the clinical setting.
Alterations in FA Transport
Once in the cytosol, free FAs bind to FA binding proteins (FABPs), which increase their solubility in the intracellular aqueous milieu. FABPs are small (~15kDa) proteins that bind medium and long chain FAs as well as other lipophilic molecules, including eicosanoids, bile salts, lysophospholipids, and retinoic acid [reviewed in (78, 79)]. So far, ten different FABPs have been described in humans (FABP1-9, and the less characterized FABP12) showing tissue specificity and both redundant and distinctive functions (80). Acting as lipid chaperones, FABPs coordinate intracellular transport and lipid metabolism, and serve as sensors to signal FA supply to the nucleus (Figure 2).
FABP5 is the most characterized and highly expressed FABP in human PCas and cell lines, especially in CRPC cells (81–88). Different mechanisms account for FABP5 upregulation, including a positive feedback loop mediated by include proliferator-activated receptors (PPAR) PPARβ/δ (89), CpG island hypomethylation (85), and gene amplification (90). The latter is highly frequent in advanced CRPC (88). In human PCas, a positive correlation between FABP5 expression and androgen signaling responsive genes was observed. While FABP5 mRNA did not correlate with clinico-pathological features, FABP5 protein levels were significantly associated with high Gleason score and reduced patient survival (82, 84). Furthermore, FABP5 mRNA, protein, and serum levels were all increased in lymph node mets, suggesting FABP5 as a potential prognostic biomarker (91).
Consistent with a role in PCa progression, genetic or pharmacological inhibition of FABP5 decreased cell proliferation, colony-formation, invasive potential of PC3 and the more aggressive PC3-M cells. In vivo, tumor growth, mets formation, vascular endothelial growth factor (VEGF) expression, and microvessel density were also significantly reduced (81, 82, 92).
One of the main functions of FABPs is to escort both exogenous and de novo synthesized FAs towards nuclear receptors, such as the PPARs and modulate the expression of genes involved in cell survival, growth, migration, and invasion [reviewed in (93)]. A direct interaction between FABP5 and PPARβ/δ or PPARγ has been demonstrated using in vitro and cell-based assays (94, 95) and it accounts for some of the pro-tumoral effects of FABP5 in PCa (89, 89, 95–98). In 2016, Forootan and coworkers showed that FABP5 promotes VEGF expression and angiogenesis through FABP5-mediated FA transport to PPARγ. In CRPC, this mechanism overcomes the canonical AR-mediated regulation of VEGF/angiogenesis, suggesting FABP5/FA/PPARγ pathway as a potential therapeutic target (97). PPAR-independent FABP5-mediated regulation of gene expression has also been described in PCa (98, 99). Other FABP5-mediated oncogenic mechanisms include the activation of SREBP-1c and the hypoxia-inducible factor 1-alpha (HIF-1α), although their roles in PCa has not been explored yet (100, 101).
FABP5 is also secreted by adipocytes, and it may potentialy contribute to the tumor supportive role of periprostatic fat (102). Finally, FABP5 has been found in urinary extracellular vesicles, where it may serve as a prognostic PCa biomarker (103).
Both pro-tumorigenic and anti-tumorigenic roles have been ascribed to FABP4 according to tumor type and TME. While FABP4 acts as a tumor suppressor when ectopically expressed in DU145 PCa cells, recent studies suggest FABP4 involvement in adipose-PCa crosstalk. According to this model, FABP4 promotes FA release from adipocyte TAG to fuel mets formation while cancer cells induce changes in adipocyte metabolism to promote FA release (104, 105). Consistently, Herroon and collaborators showed that adipocyte-derived conditioned media increases FABP4 and CD36 expression in PCa and breast cancer cells, their proliferation, invasion, and LD accumulation. This was also associated with a significant increase in several cytokines, VEGF, and HIF-1α. Conversely, inhibition of FABP4 impaired adipocyte-derived conditioned media-induced invasion (106). Furthermore, HFD was shown to induce FABP4 expression in PC3 bone tumors but not in subcutaneous ones, indicating that bone marrow-derived adipocytes may promote specific metabolic alterations in PCa bone mets (106). Oncomine data and immunohistochemistry (IHC) confirmed increased expression of FABP4 mRNA and protein in bone mets, especially in areas enriched for infiltrating adipocytes (64, 106). Mechanistically, FABP4 expression is dependent on PPARγ, which in turn is activated by FA/FABP4, suggesting the existence of a feedforward mechanism that sustains high FABP4 levels in PCa cells.
Similar to FABP5, FABP4 is also secreted by adipocytes and it plays a role as adipokine. Circulating FABP4 levels correlates with obesity and some features of the metabolic syndrome in both mice and humans (107–109) and may impact PCa progression. Indeed, serum FABP4 levels were associated with high Gleason grade (110, 111). Several evidence suggest a link between FABP4 intracellular levels and PCa aggressiveness. Ectopic expression of FABP4 promotes DU145 PCa cell invasion in vitro, while in vivo FABP4 knockdown (KD) reduces tumor growth and lung mets formation (112). Haung and coworkers also uncovered FABP4-mediated tumor/TME crosstalk that sustains PCa invasive potential. According to this, not only PCa-secreted FABP4 increases PCa invasiveness by upregulating matrix metalloproteinases (MMP 2 and 9) but it also induces stromal cells to secrete interleukin-8 and -6, further promoting PCa invasiveness (110). Conversely, FABP4 inhibition was shown to decrease HFD-induced mets, adipocyte infiltration, reactive fibroblasts and serum IL-8. Altogether, these data support a critical role for FABP4 in shaping the TME and promoting PCa progression (110). Since both FABP 4 and 5 are also expressed in macrophages and endothelial cells, they may contribute to tumor-TME crosstalk through other mechanisms.
Although less characterized, other FABPs are involved in PCa onset and progression. FABP4, FABP5, FABP8, FABP9, and FABP12 loci were found in a commonly amplified region within the chromosome 8 (8q21.13), frequently observed in human PCas mets. In line with this, increased mRNA levels of FABP 4, 8, 9, and 12 were associated with increased Gleason score and PCa recurrence (90). In 2020, Liu and coworkers also demonstrated that FABP12 promotes EMT and PCa cell motility, at least in part, through a PPARγ-dependent pathway (113), while FABP9 suppression inhibits PC3 cell invasive potential in PPARγ-independent manner (86).
Altogether these data strongly support a role for FABPs in PCa progression and their potential use as therapeutic targets (9).
Alterations in FA oxidation
While the majority of reports describe AR-mediated regulation of de novo FA synthesis in PCa progression, evidence is accumulating that both FA synthesis and FAO are regulated by AR signaling and contribute to castration resistance (CR) in a fine-tuned manner. For FAO to occur, FAs need to be converted to fatty acyl-CoAs by long chain Acyl-CoA Synthetases (ACSLs) and to cross the outer mitochondrial membrane. The latter is mediated by Carnitine palmitoyltransferase 1 (CPT-1), specifically the isoform CPT-1A. CPT-1A allows FA-CoAs across the mitochondrial membrane through the conversion to FA-carnitine, a rate-limiting step for FAO [reviewed in (12, 114)]. FAO is transcriptionally regulated by the PPAR family which mainly activate the expression of CPT-1 and other FAO enzymes in response to glucose deficiency, and post-translationally via the allosteric inhibition of CPT-1 by malonyl-CoA. The latter is mediated by the activation of AMPK, which phosphorylates and prevent ACC-2 (or ACCβ, the isoform expressed in the mitochondria) to synthesize malonyl-CoA [reviewed in (12, 114)]. Once in the mitochondria, FAs are oxidized to acetyl-CoA, which is used for energy production, generation of reducing equivalents to maintain redox homeostasis, or as substrate for new anabolic processes. During hypoxia or in response to drug treatment, cancer cells appear to favor FAO to rapidly generate ATP and NADH and promote survival. Indeed, targeting FAO with etomoxir was shown to reduce hypoxic areas in combination with radiation in metastatic PCa sphere (115).
The group led by Schlaepfer has been instrumental in uncovering the role of FAO in PCa progression to CRPC. In 2017, the authors showed that CPT1A isoform is abundant in high-grade PCa compared to benign tissues, and they demonstrated a synergistic effect in combining CPT-1A inhibitors with anti-androgen therapy. Mechanistically, the authors uncovered that CPT1A inhibition decreases AKT and inositol polyphosphate-5-phosphatase K (INPP5K) activation, resulting in increased AR activity and sensitivity to enzalutamide. Combination of FAO inhibitors (etomoxir, ranolazine, and perhexiline) with enzalutamide displayed a synergistic inhibitory effect, suggesting that co-targeting FAO and AR may have anti-cancer efficacy in mCRPC clinical setting (116). In 2019, the same group provided evidence for a new link between FAO and CR. The authors demonstrated that androgen withdrawal (which mimics the standard of care therapy for metastatic PCa) increases CPT-1A expression and FAO activity, which supports CRPC growth and antiandrogen resistance by supplying acetyl groups for histone acetylation (117). In a follow-up study, the authors showed that CPT-1A overexpression promotes antioxidant defenses, which foster PCa progression (118). Finally, last year, the group put forward the involvement of FAO in immunomodulation. Using the TRAMPC1 PCa model, the authors demonstrated that FAO inhibition with ranolazine decreases Tim3 content in CD8+ tumor-infiltrating T cells, increases macrophages, and decreases blood myeloid immunosuppressive monocytes, suggesting that targeting FAO stimulates anti-cancer immunity (118).
Besides CPT-1A, other FAO enzymes are involved in PCa progression. Combining proteomics and metabolomics, Biomme and coworkers identified the mitochondrial 2,4-dienoyl-CoA reductase (DECR1), an auxiliary FAO enzyme, as critical for CRPC. DECR1 participates in redox homeostasis by controlling the ratio between saturated and unsaturated phospholipids. As a result, DECR1 KO induced ER stress and sensitized CRPC cells to ferroptosis. Furthermore, DECR1 deletion impaired lipid metabolism and reduced CRPC tumor growth in vivo (119). Similar results were obtained by Nassar and coworkers using different models. The authors confirmed DECR1 KD-mediated cellular accumulation of PUFAs, enhanced mitochondrial oxidative stress, and lipid peroxidation. Specifically, DECR1 KD selectively inhibited PUFA oxidation, resulting in the suppression of proliferation, migration of PCa cells (including those resistant to enzalutamide), and metastasis formation in mouse xenograft models (120). These new findings implicate PUFA oxidation via DECR1 as an unexplored facet of FAO to promote PCa progression.
Yajun and coworkers also uncovered the involvement of the FAO regulator nuclear envelope protein Sun2 in PCa progression. The authors found a reduction of Sun2 expression in PCa tissues compared with adjacent normal tissues, which correlated with higher Gleason grade, postoperative T stage, lymph node invasion, and shorter PCa-free and overall survival. Sun2 silencing increased FAO activity, feature that was reversed by the use of etomoxir, suggesting a new role for Sun2 in promoting PCa progression through FAO modulation (121). Finally, Itkonen and coworkers identified enoyl-CoA-isomerase 2 (ECl2), a novel AR target involved in FAO. ECl2 was found overexpressed in PCa samples and associated with poor outcome, suggesting its possible involvement in PCa progression (122).
Alterations in Lipid Storage
Under excess of nutrients, de novo or acquired FAs are incorporated in TAGs and accumulate as LDs, organelles composed by deposits of TAGs and cholesterol esters, and surrounded by a monolayer of PLs. LDs represent a reservoir and source of lipids for cancer cells, particularly under stress conditions such as hypoxia (123). Increased abundance of LDs is a feature of many aggressive cancers, including PCa [reviewed in (9)]. The terminal step in TAG biosynthesis is catalyzed by acyl-CoA:diacylglycerol acyltransferase (DGAT) enzymes, which transfer an acyl chain from fatty acyl CoA to diacyl glycerol (DAG). DGAT1 is overexpressed in PCa compared to normal epithelium and a recent study demonstrated that inhibition of DGAT1 reduces cell proliferation and migration in vitro and tumor growth in vivo by regulating intracellular lipids and non-centrosomal microtubule-organizing center (MTOC) protein GM130 (124). Similar results were also independently obtained by Mitra and coworkers (125). Using label-free Raman spectroscopy, Yue and coworkers demonstrated an aberrant accumulation of esterified cholesterol in LDs in high-grade PCa and mets due to the loss of the tumor suppressor PTEN, the activation of PI3K/AKT pathway, and the consequent activation of TF SREBP and LDL receptor (LDL-R). LD accumulation required the occurrence of cholesterol esterification. As a result, pharmacological and genetic inhibition of cholesterol esterification using cholesterol acyltransferase (ACAT) significantly suppressed cancer proliferation, migration, invasion, and tumor growth in vivo (126). This finding suggests ACAT as a potential target in PTEN mutated/deleted CRPC, which account for around 70% of CRPCs.
TAGs in LDs are sequentially hydrolyzed by three different lipases, the adipose triglyceride lipase (ATGL), the HS lipase (HSL), and the monoacylglycerol lipase (MAGL) [reviewed in (9)]. In 2011, Nomura and coworkers showed that MAGL is increased in androgen-independent human PCa cell lines, and that pharmacological or genetic inhibition of MAGL impairs PCa aggressiveness. Furthermore, MAGL was found as part of an EMT and stem-like gene signature, suggesting MAGL as a potential therapeutic target in advanced PCa (127). These data highlight LDs are critical players in supporting PCa progression, especially under stress.
Alterations in Phospholipid Synthesis and Membrane Remodeling
FAs are essential building blocks for PLs. Early studies showed that a substantial fraction of the FAs acquired by PCa end up in PLs, which together with cholesterol and sphingolipids are the major constituents of membranes. In 2003, Swinnen and coworkers demonstrated that FASN plays a major role in the synthesis of PLs partitioning into detergent-resistant membrane microdomains, the latter being involved in key cellular processes including signal transduction, intracellular trafficking, cell polarization, and cell migration (45). PLs can be synthesized de novo but they can also be dynamically remodeled. For de novo PL synthesis, FAs are first incorporated in phosphatidic acid (PA) followed by phosphatidylcholine (PC), and phosphatidylethanolamines (PE) synthesis through the Kennedy pathway, although PE can also be generated from phosphatidylserines (PS) by headgroup exchange. PS is synthesized in the ER by headgroup exchange from PC and PE. Phosphatidylinositol (PI) is indirectly synthesized from PA, while cardiolipins (CL) are synthesized locally [reviewed in (9)]. PLs remodeling is catalyzed by phospholipases which can release acyl chains at different positions depending on the subclass of enzymes (PLA, PLC, PLD), while PL reacylation is catalyzed by a class of acyltransferases such as lysophosphatidylcholine acyl transferases (LPCAT). Our group demonstrated that de novo PC synthesis is required for cell cycle completion, upon cell division (128). Many enzymes involved in PL synthesis and remodeling are highly dysregulated in PCa. Lipin-1, a phosphatidic acid phosphatase (PAP) that regulates the rate-limiting step in PL synthesis is overexpressed in high-grade PCa and in PCa cells resistant to chemotherapy (i.e., Docetaxel). cBioPortal data also showed that patients with Lipin-1 amplification are characterized by decreased survival. Lipin-1 KD decreased both PCa cell proliferation and migration through RhoA activation, increased PA levels, and induced autophagy through the inhibition of PI3K/AKT/mTORC1 pathway., Lipin-1 depletion with propranolol sensitized cancer cells to rapamycin, suggesting new combination therapies (129, 130). Choline kinase alpha (ChoKa), the first enzyme of the Kennedy pathway, is also overexpressed in several cancer including PCa (131). Priolo and coworkers showed that the oncogene MYC increases ChoKa expression, as well as lipid synthesis (132). In line with this, positron emission tomography (PET) with PL-precursors 11C-choline or 18F-fluoro-choline has shown promising results in the detection of PCa recurrence and mets (133). Asim and coworkers demonstrated that ChoKa expression is regulated by androgens and it is positively associated with tumor stage. The authors also uncovered a role for ChoKa as a chaperone that binds to AR ligand-binding domain (LBD), enhancing AR stability. Consistently, ChoKa inhibition decreased AR protein levels and AR transcriptional program, and inhibited the growth of PCa cell lines, human PCa explants, and tumor xenografts (134), suggesting ChoKa as a marker of tumor progression and a potential therapeutic target. PLs remodeling is catalyzed by phospholipases, including PLA2, which is also involved in the generation of signaling FAs such as arachidonic acid (AA, see below) and lysophospholipids (LysoPLs). Phospholipase A2 Group IIA (PLA2G2A), especially the secretory form, is overexpressed in almost all human PCa specimens and correlate with high tumor grade. Blocking sPLA2-IIa function compromises CRPC cell growth, highlighting sPLA2 as a potential therapeutic target for CRPC. Serum sPLA2-IIa levels were increased in PCa patients and associated with high Gleason score and advanced disease stage, suggesting that serum sPLA2-IIa may serve as a PCa prognostic biomarker. A recent report also associated the expression of PLA2G2A with ferroptosis resistance through PUFA depletion in PCa membranes (135, 136). LysoPLs can stimulate PCa cell migration through several mechanisms, including the activation of the cationic channel T transient receptor potential vanilloid 2 (TRPV2), and the activation of lysophosphatidic acid (LPA)/LPA-R/mitogen-activated protein kinase (MAPK) pathway (137, 138). LysoPLs are also substrates for MAGL, whose expression is dysregulated in aggressive PCa (see above). LysoPLs can be reacylated by enzymes such as ysophosphatidylcholine acyl transferases (LPCATs). Grupp and coworkers demonstrated that the expression of lysophosphatidylcholine acyltransferase 1 (LPCAT1), a key enzyme in Lands’ cycle remodeling pathway, correlates with PCa progression and resistance to chemotherapy (i.e., Paclitaxel) and might be used as prognostic biomarker of clinical outcomes and biochemical recurrence (139). LPCAT1 mediates CRPC growth via nuclear re-localization and Histone H4 palmitoylation in an androgen-dependent fashion, increasing mRNA synthesis rates. Silencing of LPCAT1 reduced the proliferation and CRPC cell invasive potential, suggesting this enzyme as a potential therapeutic candidate in CRPC (140).
Alterations in Cholesterol Metabolism
Cholesterol is a major constituent of cell membranes, LDs, and a precursor of androgens synthesis. It is evident that alterations in cholesterol synthesis and metabolism are associated with PCa pathogenesis and progression (141, 142). PCa cells can acquire cholesterol from exogenous sources, including circulating lipoproteins (i.e., VLDL, and LDL) and exosomes, from intra-cellular storage (i.e., LDs), and from de novo cholesterol synthesis. All these processes are significantly altered in PCa, especially in aggressive PCa and CRPC. As mentioned above, Yue and coworkers demonstrated an aberrant accumulation of esterified cholesterol in LDs of high-grade PCa and mets due to PTEN loss-mediated activation of the PI3K/AKT pathway, and consequent increase of SREBP and LDL-R (126). However, low levels of LDL-R and high squalene monooxygenase (SQLE) expression were recently detected in high Gleason grade-human PCas and associated with lethal disease. According to these new results, PCas that progress to lethal disease rely on de novo cholesterol synthesis (via SQLE), rather than transcellular uptake (via LDL-R) or cholesterol esterification (via Sterol O-Acyltransferase 1, SOAT1) (142). The association of SQLE overexpression with lethal disease was validated in a second study from the same group looking at three different prospective cohorts (143). Absolute SQLE expression was associated with lethal cancer independently of Gleason grade and stage and with increased histologic markers of angiogenesis. SQLE expression at PCa diagnosis was found to be prognostic for lethal PCa both after prostatectomy and in a watchful waiting setting (143). Conversely, vitamin D-regulated catabolic enzyme sterol-27-hydroxylase (CYP27A1), which converts cholesterol to 27-hydroxycholesterol was detected at low levels in tumors characterized by high Gleason grade and high expression of cholesterol synthesis enzymes, including SQLE. Low expression of CYP27A1 was also associated with higher risk of lethal cancer, independent of SQLE (144). Altogether, these data support the notion that intra-tumor cholesterol accumulation (via increased synthesis or reduced catabolism) is a feature of lethal PCa. As expected, the key enzymes for cholesterol synthesis, 3-hydroxy-3-methyl-glutaryl-coenzyme A reductase (HMGCR, the first-rate limiting enzyme) and 3-hydroxy-3-methyl-glutaryl-coenzyme A synthetase (HMGCS) are regulated by androgens and upregulated in PCa, especially in CRPC and contribute to CR [reviewed in (12)]. HMGCS and HMGCR were found overexpressed in stromal cells when co-cultured with PCa cells to support PCa progression, suggesting that HMGCS and HMGCR in both PCa epithelium and stroma, might serve as theraputic targets (145).
The Role of Lipids as Signaling Mediators in PCa
Besides their function as building blocks and energy suppliers, lipids can function as intra- and extracellular messengers and mediators of malignant behavior. Several classes of lipids are involved in signaling, including sphingolipids and eicosanoids. Tumor-promoting functions have been described for several sphingolipids, including sphingosine, spingosine-1-phosphate (S1P), ceramide, and ceramide-1-phosphate (C1P) (146). By using isotopic FA labeling strategy coupled with metabolomic profiling platforms to comprehensively map palmitic acid incorporation into complex lipids in cancer cells, Louie and coworkers elucidated that cancer cells, including PCa cells, and tumors robustly incorporate and remodel exogenous palmitate into structural and oncogenic glycerophospholipids, but mostly in sphingolipids and ether lipids. FA incorporation into oxidative pathways was reduced in aggressive PCa cells, and instead shunted into pathways for generating signaling lipids such as ceramide and sphingomyelin, suggesting a role for sphingolipids in PCa progression (147). In line with this, Increased levels of S1P were found in more aggressive PC. Pharmacological inhibition (with ABC294640) of sphingosine kinase 2 (SphK2), one of the two Sphk isoforms that catalyzes the synthesis of S1P from sphingosine, effectively reduced CRPC cell proliferation and xenograft tumor growth by targeting AR and the oncogene MYC (148). Classically, ceramide induces senescence and growth inhibition in cancer. However, recent studies suggested that ceramide effects are context dependent and rely on downstream effectors, which can both promote or inhibit tumor growth (149). Along the line, increased expression of acid ceramidase (AC) was observed in PCa. AC significantly altered the expression of ceramide species without affecting the total levels. In AC-overexpressing DU145 cells, low levels of C14-C20 ceramides (long chain ceramides) and elevated levels of C24, C24:1 ceramides (very long chain ceramides) were indeed detected. This was associated with increased proliferation, migration and augmented tumorigenicity in vivo, which were reversed by pharmacological or genetic AC inhibition (150, 151). Although AC-mediated oncogenic mechanisms are still unknown, it is likely that AC-induced very long chain ceramide species promote cell growth while long chain ceramides induce cell apoptosis [reviewed in (151)]. Consistently, LC/MS-based lipidomics in plasma from patients with primary PCa, mHSPC, and mCRPC, showed that elevated circulating ceramide levels are associated with poor outcomes across tumor stages progression from localized PCa, mHSPC, to mCRPC. Patients with elevated ceramide levels were more likely to have metastatic relapse, therapeutic failure (ADT/docetaxel), and shorter overall survival. The authors also validated a previously published prognostic 3-lipid signature with potential clinical traslation (152). Both ceramide and C1P are activators of PLA2, an enzyme that releases AA for subsequent conversion to prostaglandins, molecules involved in inflammation, immunity, and tumor growth modulation (see below). Increased levels of prostaglandins, like PGE2, are associated with enhanced PCa proliferation and invasion, which can be reversed by the use of cyclooxygenases (COX) inhibitors, suggesting the involvement of PGE2 in PCa progression. Contrasting results have been however obtained, highlighting the need for more validation studies (153, 154). Phosphoinositides represent another class of critical signaling molecules and central mediators of the PI3K/Akt/mTORC1 signaling axis. Activation of PI3K results in the rapid conversion of PI(4,5)P2 into PI(3,4,5)P3, leading to AKT activation. PI(4,5)P2 it-self can also play a major role in recruiting cytosolic proteins, facilitating processes like fusion, membrane budding, and the formation of signaling platforms [reviewed in (9)]. Finally, glycerolipid-derived mediators, such as DAG, LysoPA and LysoPC are involved in cancer progression. DAG, generated from the hydrolysis of PI(4,5)P2, functions as a second messenger that triggers the oncogenic activation of protein kinase C (PKC). Sustained levels of DAG and activated PKC signaling were reported as a mechanism of resistance to FASN inhibitors, suggesting the assessment of DAG as predictive biomarker of FASN activity and the therapeutic combination of FASN and PKC inhibitors (155).
Oncogenic and Environmental Regulation of Lipid Rewiring in PCa
Lipid metabolism rewiring is very dynamic. Cancer cells, including PCa cells, adapt their metabolism in response to changes in nutrients supply, hormonal status, growth factors stimuli as well as epi/genetic alterations in oncogenes (i.e., MYC, PI3K/AKT) or tumor suppressor genes (i.e., PTEN, p53, RB), commonly found in mCRPC, as comprehensively described in our recent review (31). Here, we focus on the impact of systemic metabolism and environmental factors, in particular diet, to PCa metabolism rewiring and disease progression.
Both obesity and sustained consumption of fat-enriched diets alter nutrient gradient in the TME, which may favor cancer cells/TME metabolic symbiosis, inflammation, cancer progression, and chemoresistance (10). SFA-enriched diet is sufficient to promote mCRPC in the nonmetastatic PTEN KO mouse model via an aberrant lipogenic program orchestrated by SREBP (156). In Hi-MYC mouse model, HFD-induced obesity (enriched for SFA) amplifies a c-MYC-mediated oncogenic transcriptional signature, which is associated with lethality in patients (156–158). Using the same Hi-MYC mouse model, Blando and coworkers also showed that HFD-induced obesity enhances, whereas 30% caloric restriction reduces growth factor (AKT/mTORC1 and STAT3) and inflammatory (NFκB and cytokines) signaling and PCa progression (159). Consistently, reduced dietary fat intake was shown to delay PCa progression to CRPC and to prolong survival in xenograft models, suggesting low-fat diet as a promising adjuvant intervention during ADT (160). Besides SFAs, the ratio between omega-3 (n-3)/omega-6 (n-6) PUFAs also affect PCa progression. Omega-3 but not omega-6 PUFAs slowed down the growth of CRPC in PTEN KO mouse model in part by accelerating proteasome-dependent degradation of AR protein (161). In line with this, an isocaloric 20% kcal fat diet consisting of n-6 and n-3 FAs in a ratio of 1:1 (n-3 diet) reduced tumor growth rates, tumor volumes, and serum PSA levels in LAPC-4 xenografts with respect to n-6 FAs-based diet (n-6 diet). n-3 diet-tumors were characterized by low proliferation, increased apoptosis, and reduced levels of COX-2, PGE-2, and VEGF. Furthermore, LAPC-4 cells proliferation in medium containing n-3 diet serum was reduced by 22% with respect to n-6 diet (162). Several clinical trials are ongoing to evaluate the effect of n-3 PUFA in patients with advanced PCas, as well as in active surveillance and PCa prevention (NCT00458549, NCT03753334, NCT03753334, NCT00253643, NCT02176902, NCT02333435). Results from these studies will be valuable to understand whether nutrition intervention should be implemented in the management of PCa patients prior to or along with ADT/AR signaling inhibitors.
Obesity is also associated with increased fat storage in the adipose tissue. Interestingly, several tumors grow in anatomic proximity to adipose cells. This is the case of PCa, which grows adjacent to the peri-prostatic adipose tissue (PPAT) and develops mets in fatty bone marrow [reviewed in (163)]. Adipocytes can act as driving force to promote PCa cells migration to PPAT. Laurent et al. demonstrated that PPAT-derived adipocytes secrete the chemokine CCL7, which diffuses to the peripheral zone of the prostate, stimulating the migration of CCR3 expressing PCa cells. The latter is reversed by CCR3/CCL7 axis inhibition. In human PCas, CCR3 receptor expression is associated with higher occurrence of aggressive disease with extended local dissemination and biochemical recurrence (164). CCR3 is also potentially involved in the homing of PCa cells to the bone. Using in vitro migration assays, the same authors demonstrated that soluble factors released by human primary bone-marrow-derived adipocytes drive the directed migration of PCa cells in a CCR3-dependent manner. Furthermore, Oncomine microarray database uncovered increased levels of CCR3 mRNA in bone mets with respect to primary tumors, while IHC experiments demonstrated overexpression of CCR3 in bone versus visceral mets (165). Altogether, this evidence suggests the potential benefit of CCR3 antagonists in the treatment of advanced PCa. In a recent review, Nassar et al. not only describe the role of PPAT as a source of FAs and mitogens but also uncover the existence of a crosstalk between PCa and PPAT that sustains PCa pathogenesis and progression (166). In line with this, MRI-based PPAT measurements have provided new useful information in the prediction of PCa progression. Peri-prostatic fat area (PPFA) and PPFA to prostate area ratio (PPFA/PA) was reported as independent predictor of PCa, lymph node mets, Gleason score, tumor stage, and proliferation index (i.e., Ki-67) (167, 168). Thus, PPFA measurements along with transrectal ultrasound-guided biopsy may improve PCa detection and risk stratification. PPAT volume has been recently also associated with reduced progression-free survival in men with PCa on active surveillance and with poor response to ADT in patients with advanced PCa (169). These results highlight the crucial role of PPAT in PCa progression and the clinical value of MRI-based measurements of PPAT to predict prognosis and therapy response.
The Role of Lipids in Mediating Tumor-TME Crosstalk
While the role of FAs in promoting inflammation and mediating inflammatory signaling has been largely characterized, more recent data suggest a key role for FAs in immune metabolism [reviewed in (10, 170)]. Both FA synthesis and oxidation are important regulators of immune responses. FA synthesis plays a role in antigen presentation and T cell activation, whereas FAO is a key feature of CD8 memory T cells (170). The source of lipids used for FAO in memory T cells is cell type specific. Central memory CD8 T cells cannot effectively take up lipids and rely on lipolysis for FA supply, whereas tissue resident memory CD8 T cells require uptake of exogenous lipids for their survival and proliferation (171–173). In contrast to CD8 T cells, naïve and memory CD4 T cells require FA uptake and synthesis for full activation and proliferation (174). Regulatory T cells (Treg) and M2-like macrophages rely on lipid-dependent catabolism. Treg cells predominantly use FAO-fueled oxidative phosphorylation (OXPHOS) to generate energy and FAO inhibition with etomoxir suppresses Foxp3 expression in Treg cells without affecting T effector cells (Teff) cells. Thus, Treg cells display a survival advantage in low-glucose and lipid-rich environments over Teff cells and are well adapted to reside in fat tissue and lipid-rich TME, which is consistent with their increased frequency in the TME (175, 176). FAO is also required for the maturation and function of IL-4-induced anti-inflammatory M2 macrophages, which uptake FAs through CD36 and FATP1 to maintain their phenotype (177–180). FAO alterations and LD accumulation are also linked with dendritic cells dysfunction, highlighting the importance of lipids in antigen presentation (181–183).
Michelet and coworkers showed that HFD-induced obesity induces PPAR-driven lipid accumulation in Natural Killer (NK)cells, causing a complete ‘paralysis’ of their cellular metabolism and trafficking, resulting in blunted antitumor responses (184). Similarly, the integration of single-cell RNA sequencing, multiplexed immunofluorescence IHC, and mass-spectrometry approaches in vivo, uncovered that HFD-induced obesity impairs CD8+ T cell function in TME due to a distinct metabolic adaptation to obesity by the tumor and T cells. While tumor cells increase fat uptake, tumor infiltrating CD8+ T cells do not, leading to altered FA partitioning in HFD tumors, which impaired CD8+T cell infiltration and function. Analysis of human cancers revealed similar transcriptional changes in CD8+ T cell markers, suggesting the potential of lipid metabolism interventions to improve cancer immunotherapy (185). In contrast, obese cancer patients seem to respond to ICI, a phenomenon known as “Obesity paradox” (186). Thus, further studies are needed to clearly understand the impact of obesity and obesogenic HFD on immune therapy efficacy in patients.
Cancer cells not only suppress tumor immune surveillance, but they can also hijack the immune system to support their growth. For instance, ovarian cancer cells promote the efflux of cholesterol from macrophages which in turn drives a pro-tumoral M2 phenotype (187). Moreover, it has been reported that cancer cells can also promote tumor-associated myeloid-derived suppressor cells (MDSCs) to produce PGE2, an oxylipin with immune suppressive functions. This seems to occur through a cancer-dependent increase of Fatty acid transport protein 2 (FATP2) expression, which allow AA transport in MDSCs for PGE2 synthesis (188).
While the role of lipids in PCa immune TME has not been carefully investigated, early preliminary data showed increased expression of immune checkpoint PD-1, PD-L1, and PD-L2 in tumor tissues from PTEN KO mice fed HFD, suggesting an opportunity for ICI (189). Considering that the response to ICI has been so far disappointing, understanding whether obesity may boost response to ICI and “paradoxically” favor the use of immune therapy is crucial to identify a subset of mCRPC patients, who may potentially respond to immune therapies.
Exploiting Lipid Metabolism Rewiring for Therapeutic Intervention
In light of the aforementioned changes in lipid metabolism during PCa progression, huge efforts have been directed on tackling enzymes and transporters involved in all the aspects of lipid metabolism (from FA uptake transport, de novo FA/cholesterol synthesis, sphingolipid and phospholipids synthesis, to lipid storage and lipolysis). Recently published reviews from our group and others have provided an exhaustive description of the small molecules/compounds targeting lipid metabolism tested so far in oncology (9, 31). Here, we emphasize those compounds that have already been approved for clinical use or are currently tested in clinical trials.
Inhibitors of De Novo FA Synthesis
The majority of therapeutic efforts have been focused on FASN, resulting in the development of several FASN inhibitors (i.e., Orlistat, C75, cerulenin, C93, Fasnall) with good results in the preclinical setting. Unfortunately, off-target effects, poor solubility and pharmacokinetics, and untoward side effects, including important weight loss, prevented their clinical translation [reviewed in (31)]. The development of TVB-2640, an orally available inhibitor of FASN β-ketoacyl-reductase domain has changed the perspective. A phase I clinical trial in cancer patients has been completed, showing the safety and efficacy of TVB-2640 in solid malignancies (NCT02223247). Combined with paclitaxel, TVB-2640 provided positive results in heavily pretreated breast cancer patients, while the non-orally available analog TVB-3166 was effective in mCRPC preclinical models (190–192). Phase II trials are now investigating TVB-2640 in several solid tumor types including HER-2 positive advanced breast cancer in combination with trastuzumab. (NCT03032484, NCT03179904, NCT02980029, NCT03808558). Our group also characterized a new oral-available small molecule irreversible FASN inhibitor (IPI-9119) with potential clinical translation. In the preclinical setting, we demonstrated that selective FASN inhibition antagonizes the growth of mCRPC, in part by inducing ER stress-mediated downregulation of AR-FL and AR-V7 protein levels and their transcriptional activity. As a result, IPI-9119 improved the response to enzalutamide in mCRPC cell lines and organoid models. Our data support FASN repression as a non-canonical approach to inhibit AR-V7, thus overcoming current resistance to standard of care for mCRPC. Multiplex immunofluorescence analysis combined with digital pathology of mCRPC tumor microarrays confirmed FASN/AR-V7 co-expression in about 80% of mCRCP patients resistant to enzalutamide and abiraterone, highlighting this patient subset as the ideal candidate for the treatment with FASN inhibitors (22). Carefully designed clinical trials are still needed to adequately define the timing, combinations, and the suitable population to test.
Inhibitors of FA Oxidation
Targeting FAO in mCRPC has recently gained a lot of attention. Iglesias-Gato and coworkers have recently identified a subgroup of bone mets characterized by elevated expression of FAO enzymes, and thus potentially responsive to FAO inhibitors. These findings also underline the urgent need for adequate patient stratification when metabolic therapies are considered as therapeutic approaches (193). Combinations of FAO inhibitors (etomoxir, ranolazine, and perhexiline) and enzalutamide have been tested in mCRPC cell and xenograft models with positive results. Unfortunately, etomoxir use in the clinical setting has been terminated due to toxic side effects, mostly hepatotoxicity. In contrast, ranolazine and perhexiline are already approved for the treatment of heart diseases in Europe, US, and Australia (194), opening a potential safe avenue for the combinations of FAO and AR signaling inhibitors in mCRPC.
Inhibitors of Cholesterol Synthesis
Statins are commonly used to lower cholesterol levels and reduce cardiovascular risk. Statins use in the prevention of cancer risk has been evaluated with conflicting results. Their potential use in combination with the standard of care in the treatment of PCa has recently gained attention (31).
A clinical trial designed to test whether atorvastatin (an HMGCR inhibitor) delays the development of CR during ADT in metastatic or recurrent PCas is currently ongoing (NCT04026230). In the preclinical setting, HMGCR inhibition with simvastatin enhances the efficacy of enzalutamide and decreases AR/AR-Vs protein levels via inhibition of mTOR pathway (195).
A recent meta-analysis evaluated the effects of statins use on treatment outcomes (i.e., overall survival and cancer‐specific survival) among patients with advanced PCa treated with ADT or AR signaling inhibitors. Statin use was associated with lower risk of all‐cause mortality and cancer‐specific mortality in advanced PCa patients treated with ADT, whereas inconsistent results were obtained with AR signaling inhibitors (196). Thus, future studies are still required to establish the efficacy of statins in combination with AR signaling inhibitors in mCRPC patients.
Application of Lipidomics and Mass-Spectrometry Imaging in PCa Research
Despite the crucial role of lipid metabolism in PCa progression and resistance to endocrine therapies, lipidomics studies have only recently reached the spotlight most likely due to the methodological challenge of analyzing simultaneously diverse lipid classes and molecular species and technical issues associated with these analytical techniques. An outstanding review has recently highlighted the current advances of lipidomics and mass-spectrometry imaging in cancer research and their critical role in precision medicine (9). Here, we briefly summarize studies using these technologies for the identification of new predictive/prognostic biomarkers in PCa.
Lin et al. performed LC/MS-based lipidomics in plasma samples from a discovery cohort of CRPC patients and identified forty-six lipids, predominantly sphingolipids, associated with poor prognosis. The authors derived a prognostic three-lipid signature (ceramide d18:1/24:1, sphingomyelin d18:2/16:0, phosphatidylcholine 16:0/16:0) as independent prognostic factor (197). More recently, the same group detected elevated circulating ceramide species in association with poorer clinical outcomes across the PCa progression and validated the three-lipid prognostic signature in an independent cohort (152). These studies not only identified an easily detectable prognostic biomarker but also highlighted the crucial role of sphingolipids in PCa progression. Similarly, Butler et al. profiled PCa cell lines, xenografts, and patient-derived explants under treatment with androgen and AR signaling inhibitors. Significant changes in lipid elongation for multiple phospholipid classes in response to androgen treatment were identified and reversed by enzalutamide, suggesting the utility of lipidomics to predict response to endocrine therapies (198). Lipidomics and transcriptomics integration in PCa and adjacent normal tissues also identified a strong accumulation of cholesteryl esters (CEs) most likely due to increased expression of scavenger receptor class B type I (SR-BI). CE accumulation was associated with disease progression and mets formation. In a discovery set, CE robustly differentiated PCa from normal tissue. In a validation set, CEs not only potently distinguished PCa from normal tissue, but it also discriminated PCa from benign prostatic hyperplasia (BPH) superior to PSA, suggesting CE, particularly, cholesteryl oleate, as a biomarker for PCa detection (199). Furthermore, targeted lipidomics in EVs derived from prostate and PCa cell lines uncovered differences in the molecular lipid species associated with PCa progression. These differences highlight the importance of characterizing the EV lipidome, which may lead to improved prognostic biomarkers (200). Despite the high resolution, sensitivity, and specificity of LC/MS-based lipidomics, these technologies fail to provide spatial information and to integrate the information of biomarker expression with tissue pathology and compartment distribution. The development of MSI has overcome this limitation. MSI thus represents an important step forward for the evaluation of metabolic reprogramming occurring in the TME. Matrix-assisted laser desorption ionization (MALDI)-MSI, where the sample is mixed with a UV-absorbing crystalline matrix material and ionized by the laser beam, is the most commonly used MSI method (201). Our group applied MALDI-MSI to investigate changes in lipid metabolism associated with gleason score. We detected increased levels of 31 lipids, including several phosphatidylcholines, PA, phosphatidylserines, phosphatidylinositols, and cardiolipins, in Gleason score 4 + 3 compared with Gleason score 3 + 4, suggesting these analytes as potential biomarkers of PCa aggression worth further validation. Interestingly, we identified lipid changes in both regions of high tumor cell density, and in regions of tissue that appeared histologically “benign”, implying the occurrence of precancerous lipid changes with prognostic significance (202). Using a similar approach, Andersen et al. identified increased levels of metabolites crucial for lipid metabolism in PCa, including metabolites involved in the carnitine shuttle as well as building blocks for de novo lipogenesis (203). The feasibility of spatial and rapid detection of metabolites associated with PCa onset and progression showcases MALDI-MSI as a promising and innovative diagnostic/prognosis tool in the clinical setting.
Discussion
Lipid metabolism rewiring is highly dynamic throughout the course of PCa progression. Intracellular lipid changes due to either environmental cues or de novo FA synthesis/FAO increase PCa cells fitness and their capability to adapt to oxidative stress, hypoxia, ER stress, to maintain redox balance, and to counteract ferroptosis and genotoxic insults. Recent evidence also supports the role of lipids as key players in shaping TME metabolism, in particular immune metabolism. This is especially exacerbated by obesity or consumption of HFD diet, conditions in which cancer cells hijack lipids (with the support of tumor-surrounding adipose cells or cancer-associated fibroblasts) for their own benefit, impairing anti-tumor immunity. The rapid advance of lipidomics and MALDI-MSI has allowed to gain, a previously unforeseen, awareness of the dynamicity and adaptability of lipid rewiring during PCa progression, taking into account the influence of systemic metabolism and tumor-TME crosstalk. In the imminent future we anticipate the integration of MALDI-MSI, spatial transcriptomics, and digital pathology will further advance our current understanding of the biology of lipids in PCa progression and will offer opportunities for the identification of new druggable targets. Unfortunately, we still have a long road ahead to validate lipids as biomarkers and to translate the lipid-metabolism targeting drugs available so far in the clinical setting. The journey has started long time ago, but we are now fully equipped with the adequate models (i.e., patient-derived organoids, explants, xenografts, co-culture systems, immune-competent mouse models, etc.), technologies, and bioinformatics support to rapidly move forward.
PCa is “a matter of fats”. The big challenge is to carefully identify and target those lipid and pathways that are tumor-friends while preserving those that protect our health and longevity.
Author Contributions
All authors contributed to the article and approved the submitted version.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
The authors thank Prof. Betina Corsico (Instituto de Investigaciones Bioquímicas de La Plata, Argentina) for the insights and helpful discussions on FABPs. NS is a researcher of the National Scientific and Technical Research Council (CONICET) Argentina. This work was supported by Agencia Nacional de Promoción Científica y Tecnológica (ANPCyT, grant number PICT-2017-2548 to NS). YF-L is a fellow from ANPCyT.
References
1. Siegel RL, Miller KD, Fuchs HE, Jemal A. Cancer Statistics, 2021. CA Cancer J Clin (2021) 71(1):7–33. doi: 10.3322/caac.21654
2. Teo MY, Rathkopf DE, Kantoff P. Treatment of Advanced Prostate Cancer. Annu Rev Med (2019) 70:479–99. doi: 10.1146/annurev-med-051517-011947
3. Tilki D, Schaeffer EM, Evans CP. Understanding Mechanisms of Resistance in Metastatic Castration-Resistant Prostate Cancer: The Role of the Androgen Receptor. Eur Urol Focus (2016) 2(5):499–505. doi: 10.1016/j.euf.2016.11.013
4. Smith M, Parker C, Saad F, Miller K, Tombal B, Ng QS, et al. Addition of Radium-223 to Abiraterone Acetate and Prednisone or Prednisolone in Patients With Castration-Resistant Prostate Cancer and Bone Metastases (ERA 223): A Randomised, Double-Blind, Placebo-Controlled, Phase 3 Trial. Lancet Oncol (2019) 20(3):408–19. doi: 10.1016/S1470-2045(18)30860-X
5. Ku SY, Gleave ME, Beltran H. Towards Precision Oncology in Advanced Prostate Cancer. Nat Rev Urol (2019) 16(11):645–54. doi: 10.1038/s41585-019-0237-8
6. Kim TJ, Koo KC. Current Status and Future Perspectives of Checkpoint Inhibitor Immunotherapy for Prostate Cancer: A Comprehensive Review Int J Mol Sci (2020) 21(15):5484. doi: 10.3390/ijms21155484
7. Swinnen JV, Van Veldhoven PP, Esquenet M, Heyns W, Verhoeven G. Androgens Markedly Stimulate the Accumulation of Neutral Lipids in the Human Prostatic Adenocarcinoma Cell Line LNCaP. Endocrinology (1996) 137(10):4468–74. doi: 10.1210/endo.137.10.8828509
8. Swinnen JV, Ulrix W, Heyns W, Verhoeven G. Coordinate Regulation of Lipogenic Gene Expression by Androgens: Evidence for a Cascade Mechanism Involving Sterol Regulatory Element Binding Proteins. Proc Natl Acad Sci USA (1997) 94(24):12975–80. doi: 10.1073/pnas.94.24.12975
9. Butler LM, Perone Y, Dehairs J, Lupien LE, de Laat V, Talebi A, et al. Lipids and Cancer: Emerging Roles in Pathogenesis, Diagnosis and Therapeutic Intervention. Adv Drug Delivery Rev (2020) 159:245–93. doi: 10.1016/j.addr.2020.07.013
10. Peck B, Schulze A. Lipid Metabolism at the Nexus of Diet and Tumor Microenvironment. Trends Cancer (2019) 5(11):693–703. doi: 10.1016/j.trecan.2019.09.007
11. Jensen V, Ladekarl M, Holm-Nielsen P, Melsen F, Soerensen FB. The Prognostic Value of Oncogenic Antigen 519 (OA-519) Expression and Proliferative Activity Detected by Antibody MIB-1 in Node-Negative Breast Cancer. J Pathol (1995) 176(4):343–52. doi: 10.1002/path.1711760405
12. Zadra G, Photopoulos C, Loda M. The Fat Side of Prostate Cancer. Biochim Biophys Acta (2013) 1831(10):1518–32. doi: 10.1016/j.bbalip.2013.03.010
13. Roman M, Wrobel TP, Panek A, Paluszkiewicz C, Kwiatek WM. Lipid Droplets in Prostate Cancer Cells and Effect of Irradiation Studied by Raman Microspectroscopy. Biochim Biophys Acta Mol Cell Biol Lipids (2020) 1865(9):158753. doi: 10.1016/j.bbalip.2013.03.010
14. Ettinger SL, Sobel R, Whitmore TG, Akbari M, Bradley DR, Gleave ME, et al. Dysregulation of Sterol Response Element-Binding Proteins and Downstream Effectors in Prostate Cancer During Progression to Androgen Independence. Cancer Res (2004) 64(6):2212–21. doi: 10.1158/0008-5472.can-2148-2
15. Sharp A, Coleman I, Yuan W, Sprenger C, Dolling D, Rodrigues DN, et al. Androgen Receptor Splice Variant-7 Expression Emerges With Castration Resistance in Prostate Cancer. J Clin Invest (2019) 129(1):192–208. doi: 10.1172/JCI122819
16. Han W, Gao S, Barrett D, Ahmed M, Han D, Macoska JA, et al. Reactivation of Androgen Receptor-Regulated Lipid Biosynthesis Drives the Progression of Castration-Resistant Prostate Cancer. Oncogene (2018) 37(6):710–21. doi: 10.1038/onc.2017.385
17. Mah CY, Nassar ZD, Swinnen JV, Butler LM. Lipogenic Effects of Androgen Signaling in Normal and Malignant Prostate. Asian J Urol (2020) 7(3):258–70. doi: 10.1016/j.ajur.2019.12.003
18. Heemers H, Maes B, Foufelle F, Heyns W, Verhoeven G, Swinnen JV. Androgens Stimulate Lipogenic Gene Expression in Prostate Cancer Cells by Activation of the Sterol Regulatory Element-Binding Protein Cleavage Activating Protein/Sterol Regulatory Element-Binding Protein Pathway. Mol Endocrinol (2001) 15(10):1817–28. doi: 10.1210/mend.15.10.0703
19. Huang WC, Li X, Liu J, Lin J, Chung LW. Activation of Androgen Receptor, Lipogenesis, and Oxidative Stress Converged by SREBP-1 Is Responsible for Regulating Growth and Progression of Prostate Cancer Cells. Mol Cancer Res (2012) 10(1):133–42. doi: 10.1158/1541-7786.MCR-11-0206
20. Chan SC, Selth LA, Li Y, Nyquist MD, Miao L, Bradner JE, et al. Targeting Chromatin Binding Regulation of Constitutively Active AR Variants to Overcome Prostate Cancer Resistance to Endocrine-Based Therapies. Nucleic Acids Res (2015) 43(12):5880–97. doi: 10.1093/nar/gkv262
21. Sharma NL, Massie CE, Ramos-Montoya A, Zecchini V, Scott HE, Lamb AD, et al. The Androgen Receptor Induces a Distinct Transcriptional Program in Castration-Resistant Prostate Cancer in Man. Cancer Cell (2013) 23(1):35–47. doi: 10.1016/j.ccr.2012.11.010
22. Zadra G, Ribeiro CF, Chetta P, Ho Y, Cacciatore S, Gao X, et al. Inhibition of De Novo Lipogenesis Targets Androgen Receptor Signaling in Castration-Resistant Prostate Cancer. Proc Natl Acad Sci USA (2019) 116(2):631–40. doi: 10.1073/pnas.1808834116
23. Fiorentino M, Zadra G, Palescandolo E, Fedele G, Bailey D, Fiore C, et al. Overexpression of Fatty Acid Synthase is Associated With Palmitoylation of Wnt1 and Cytoplasmic Stabilization of Beta-Catenin in Prostate Cancer. Lab Invest (2008) 88(12):1340–8. doi: 10.1038/labinvest.2008.97
24. Li W, Zhang J, Zou L, Cui J, Su F, Jin J, et al. Palmitoylome Profiling Indicates That Androgens Regulate the Palmitoylation of Alphatubulin in Prostate Cancerderived LNCaP Cells and Supernatants. Oncol Rep (2019) 42(6):2788–96. doi: 10.3892/or.2019.7333
25. Cui L, Liu M, Lai S, Hou H, Diao T, Zhang D, et al. Androgen Upregulates the Palmitoylation of Eif3l in Human Prostate LNCaP Cells. Onco Targets Ther (2019) 12:4451–9. doi: 10.2147/OTT.S193480
26. Mariscal J, Vagner T, Kim M, Zhou B, Chin A, Zandian M, et al. Comprehensive Palmitoyl-Proteomic Analysis Identifies Distinct Protein Signatures for Large and Small Cancer-Derived Extracellular Vesicles. J Extracell Vesicles (2020) 9(1):1764192. doi: 10.1080/20013078.2020.1764192
27. Wu X, Dong Z, Wang CJ, Barlow LJ, Fako V, Serrano MA, et al. FASN Regulates Cellular Response to Genotoxic Treatments by Increasing PARP-1 Expression and DNA Repair Activity via NF-kappaB and SP1. Proc Natl Acad Sci USA (2016) 113(45):E6965–E73. doi: 10.1073/pnas.1609934113
28. Trujillo KM, Yuan SS, Lee EY, Sung P. Nuclease Activities in a Complex of Human Recombination and DNA Repair Factors Rad50, Mre11, and P95. J Biol Chem (1998) 273(34):21447–50. doi: 10.1074/jbc.273.34.21447
29. Hosios AM, Vander Heiden MG. The Redox Requirements of Proliferating Mammalian Cells. J Biol Chem (2018) 293(20):7490–8. doi: 10.1074/jbc.TM117.000239
30. Little JL, Wheeler FB, Fels DR, Koumenis C, Kridel SJ. Inhibition of Fatty Acid Synthase Induces Endoplasmic Reticulum Stress in Tumor Cells. Cancer Res (2007) 67(3):1262–9. doi: 10.1158/0008-5472.CAN-06-1794
31. Chetta P, Zadra G. Metabolic Reprogramming as an Emerging Mechanism of Resistance to Endocrine Therapies in Prostate Cancer. Cancer Drug Resist (2021) 4:143–62. doi: 10.20517/cdr.2020.54
32. Migita T, Ruiz S, Fornari A, Fiorentino M, Priolo C, Zadra G, et al. Fatty Acid Synthase: A Metabolic Enzyme and Candidate Oncogene in Prostate Cancer. J Natl Cancer Inst (2009) 101(7):519–32. doi: 10.1093/jnci/djp030
33. Di Vizio D, Adam RM, Kim J, Kim R, Sotgia F, Williams T, et al. Caveolin-1 Interacts With a Lipid Raft-Associated Population of Fatty Acid Synthase. Cell Cycle (2008) 7(14):2257–67. doi: 10.4161/cc.7.14.6475
34. Rysman E, Brusselmans K, Scheys K, Timmermans L, Derua R, Munck S, et al. De Novo Lipogenesis Protects Cancer Cells From Free Radicals and Chemotherapeutics by Promoting Membrane Lipid Saturation. Cancer Res (2010) 70(20):8117–26. doi: 10.1158/0008-5472.CAN-09-3871
35. Rae C, Haberkorn U, Babich JW, Mairs RJ. Inhibition of Fatty Acid Synthase Sensitizes Prostate Cancer Cells to Radiotherapy. Radiat Res (2015) 184(5):482–93. doi: 10.1667/RR14173.1
36. Chuang HY, Lee YP, Lin WC, Lin YH, Hwang JJ. Fatty Acid Inhibition Sensitizes Androgen-Dependent and -Independent Prostate Cancer to Radiotherapy via FASN/NF-KappaB Pathway. Sci Rep (2019) 9(1):13284. doi: 10.1038/s41598-019-49486-2
37. Lee JY, Kim WK, Bae KH, Lee SC, Lee EW. Lipid Metabolism and Ferroptosis. Biol (Basel) (2021) 10(3):184. doi: 10.3390/biology10030184
38. Bertolacci C, Andreani C, Vias Do Vale G, Berto S, Melegari M, Crouch AC, et al. Targeting De Novo Lipogenesis and the Lands Cycle Induces Ferroptosis in KRAS-Mutant Lung Cancer. bioRxiv (2021) 03.18.434804. doi: 10.1101/2021.03.18.434804
39. Enoch HG, Catala A, Strittmatter P. Mechanism of Rat Liver Microsomal Stearyl-CoA Desaturase. Studies of the Substrate Specificity, Enzyme-Substrate Interactions, and the Function of Lipid. J Biol Chem (1976) 251(16):5095–103. doi: 10.1016/S0021-9258(17)33223-4
40. Panagiotopoulos AA, Kalyvianaki K, Castanas E, Kampa M. Eicosanoids in Prostate Cancer. Cancer Metastasis Rev (2018) 37(2-3):237–43. doi: 10.1007/s10555-018-9750-0
41. Igal RA. Stearoyl CoA Desaturase-1: New Insights Into a Central Regulator of Cancer Metabolism. Biochim Biophys Acta (2016) 1861(12 Pt A):1865–80. doi: 10.1016/j.bbalip.2016.09.009
42. Igal RA, Sinner DI. Stearoyl-CoA Desaturase 5 (SCD5), a Delta-9 Fatty Acyl Desaturase in Search of a Function. Biochim Biophys Acta Mol Cell Biol Lipids (2021) 1866(1):158840. doi: 10.1016/j.bbalip.2020.158840
43. Peck B, Schug ZT, Zhang Q, Dankworth B, Jones DT, Smethurst E, et al. Inhibition of Fatty Acid Desaturation Is Detrimental to Cancer Cell Survival in Metabolically Compromised Environments. Cancer Metab (2016) 4:6. doi: 10.1186/s40170-016-0146-8
44. Staubach S, Hanisch FG. Lipid Rafts: Signaling and Sorting Platforms of Cells and Their Roles in Cancer. Expert Rev Proteomics (2011) 8(2):263–77. doi: 10.1586/epr.11.2
45. Swinnen JV, Van Veldhoven PP, Timmermans L, De Schrijver E, Brusselmans K, Vanderhoydonc F, et al. Fatty Acid Synthase Drives the Synthesis of Phospholipids Partitioning Into Detergent-Resistant Membrane Microdomains. Biochem Biophys Res Commun (2003) 302(4):898–903. doi: 10.1016/s0006-291x(03)00265-1
46. Fritz V, Benfodda Z, Rodier G, Henriquet C, Iborra F, Avances C, et al. Abrogation of De Novo Lipogenesis by Stearoyl-CoA Desaturase 1 Inhibition Interferes With Oncogenic Signaling and Blocks Prostate Cancer Progression in Mice. Mol Cancer Ther (2010) 9(6):1740–54. doi: 10.1158/1535-7163.MCT-09-1064
47. Magtanong L, Ko PJ, To M, Cao JY, Forcina GC, Tarangelo A, et al. Exogenous Monounsaturated Fatty Acids Promote a Ferroptosis-Resistant Cell State. Cell Chem Biol (2019) 26(3):420–32 e9. doi: 10.1016/j.chembiol.2018.11.016
48. Luis G, Godfroid A, Nishiumi S, Cimino J, Blacher S, Maquoi E, et al. Tumor Resistance to Ferroptosis Driven by Stearoyl-CoA Desaturase-1 (SCD1) in Cancer Cells and Fatty Acid Biding Protein-4 (FABP4) in Tumor Microenvironment Promote Tumor Recurrence. Redox Biol (2021) 43:102006. doi: 10.1016/j.redox.2021.102006
49. Tesfay L, Paul BT, Konstorum A, Deng Z, Cox AO, Lee J, et al. Stearoyl-CoA Desaturase 1 Protects Ovarian Cancer Cells From Ferroptotic Cell Death. Cancer Res (2019) 79(20):5355–66. doi: 10.1158/0008-5472.CAN-19-0369
50. Jump DB. Mammalian Fatty Acid Elongases. Methods Mol Biol (2009) 579:375–89. doi: 10.1007/978-1-60761-322-0_19
51. Tamura K, Makino A, Hullin-Matsuda F, Kobayashi T, Furihata M, Chung S, et al. Novel Lipogenic Enzyme ELOVL7 Is Involved in Prostate Cancer Growth Through Saturated Long-Chain Fatty Acid Metabolism. Cancer Res (2009) 69(20):8133–40. doi: 10.1158/0008-5472.CAN-09-0775
52. Centenera MM, Scott JS, Machiels J, Nassar ZD, Miller DC, Zinonos I, et al. ELOVL5 Is a Critical and Targetable Fatty Acid Elongase in Prostate Cancer. Cancer Res (2021) 81(7):1704–18. doi: 10.1158/0008-5472.CAN-20-2511
53. Kampf JP, Kleinfeld AM. Is Membrane Transport of FFA Mediated by Lipid, Protein, or Both? An Unknown Protein Mediates Free Fatty Acid Transport Across the Adipocyte Plasma Membrane. Physiol (Bethesda) (2007) 22:7–14. doi: 10.1152/physiol.00011.2006
54. Glatz JF, Luiken JJ, Bonen A. Membrane Fatty Acid Transporters as Regulators of Lipid Metabolism: Implications for Metabolic Disease. Physiol Rev (2010) 90(1):367–417. doi: 10.1152/physrev.00003.2009
55. Febbraio M, Abumrad NA, Hajjar DP, Sharma K, Cheng W, Pearce SF, et al. A Null Mutation in Murine CD36 Reveals an Important Role in Fatty Acid and Lipoprotein Metabolism. J Biol Chem (1999) 274(27):19055–62. doi: 10.1074/jbc.274.27.19055
56. Silverstein RL, Febbraio M. CD36, a Scavenger Receptor Involved in Immunity, Metabolism, Angiogenesis, and Behavior. Sci Signal (2009) 2(72):re3. doi: 10.1126/scisignal.272re3
57. Glatz JF, Luiken JJ. From Fat to FAT (CD36/SR-B2): Understanding the Regulation of Cellular Fatty Acid Uptake. Biochimie (2017) 136:21–6. doi: 10.1016/j.biochi.2016.12.007
58. Wang J, Li Y. CD36 Tango in Cancer: Signaling Pathways and Functions. Theranostics (2019) 9(17):4893–908. doi: 10.7150/thno.36037
59. Schenk S, Horowitz JF. Coimmunoprecipitation of FAT/CD36 and CPT I in Skeletal Muscle Increases Proportionally With Fat Oxidation After Endurance Exercise Training. Am J Physiol Endocrinol Metab (2006) 291(2):E254–60. doi: 10.1152/ajpendo.00051.2006
60. Smith BK, Jain SS, Rimbaud S, Dam A, Quadrilatero J, Ventura-Clapier R, et al. FAT/CD36 Is Located on the Outer Mitochondrial Membrane, Upstream of Long-Chain Acyl-CoA Synthetase, and Regulates Palmitate Oxidation. Biochem J (2011) 437(1):125–34. doi: 10.1042/BJ20101861
61. Hale JS, Otvos B, Sinyuk M, Alvarado AG, Hitomi M, Stoltz K, et al. Cancer Stem Cell-Specific Scavenger Receptor CD36 Drives Glioblastoma Progression. Stem Cells (2014) 32(7):1746–58. doi: 10.1002/stem.1716
62. Pascual G, Avgustinova A, Mejetta S, Martin M, Castellanos A, Attolini CS, et al. Targeting Metastasis-Initiating Cells Through the Fatty Acid Receptor CD36. Nature (2017) 541(7635):41–5. doi: 10.1038/nature20791
63. Nath A, Chan C. Genetic Alterations in Fatty Acid Transport and Metabolism Genes are Associated With Metastatic Progression and Poor Prognosis of Human Cancers. Sci Rep (2016) 6:18669. doi: 10.1038/srep18669
64. Chkourko Gusky H, Diedrich J, MacDougald OA, Podgorski I. Omentum and Bone Marrow: How Adipocyte-Rich Organs Create Tumour Microenvironments Conducive for Metastatic Progression. Obes Rev (2016) 17(11):1015–29. doi: 10.1111/obr.12450
65. Watt MJ, Clark AK, Selth LA, Haynes VR, Lister N, Rebello R, et al. Suppressing Fatty Acid Uptake has Therapeutic Effects in Preclinical Models of Prostate Cancer. Sci Transl Med (2019) 11(478):eaau5758. doi: 10.1126/scitranslmed.aau5758
66. Niculite CM, Enciu AM, Hinescu ME. CD 36: Focus on Epigenetic and Post-Transcriptional Regulation. Front Genet (2019) 10:680. doi: 10.3389/fgene.2019.00680
67. Kuemmerle NB, Rysman E, Lombardo PS, Flanagan AJ, Lipe BC, Wells WA, et al. Lipoprotein Lipase Links Dietary Fat to Solid Tumor Cell Proliferation. Mol Cancer Ther (2011) 10(3):427–36. doi: 10.1158/1535-7163.MCT-10-0802
68. Rae C, Fragkoulis GI, Chalmers AJ. Cytotoxicity and Radiosensitizing Activity of the Fatty Acid Synthase Inhibitor C75 Is Enhanced by Blocking Fatty Acid Uptake in Prostate Cancer Cells. Adv Radiat Oncol (2020) 5(5):994–1005. doi: 10.1016/j.adro.2020.06.022
69. Pinthus JH, Lu JP, Bidaisee LA, Lin H, Bryskine I, Gupta RS, et al. Androgen-Dependent Regulation of Medium and Long Chain Fatty Acids Uptake in Prostate Cancer. Prostate (2007) 67(12):1330–8. doi: 10.1002/pros.20609
70. Anderson CM, Stahl A. SLC27 Fatty Acid Transport Proteins. Mol Aspects Med (2013) 34(2-3):516–28. doi: 10.1016/j.mam.2012.07.010
71. Blask DE, Sauer LA, Dauchy RT, Holowachuk EW, Ruhoff MS, Kopff HS. Melatonin Inhibition of Cancer Growth In Vivo Involves Suppression of Tumor Fatty Acid Metabolism via Melatonin Receptor-Mediated Signal Transduction Events. Cancer Res (1999) 59(18):4693–701.
72. Zhang M, Di Martino JS, Bowman RL, Campbell NR, Baksh SC, Simon-Vermot T, et al. Adipocyte-Derived Lipids Mediate Melanoma Progression via FATP Proteins. Cancer Discov (2018) 8(8):1006–25. doi: 10.1158/2159-8290.CD-17-1371
73. Mendes C, Lopes-Coelho F, Ramos C, Martins F, Santos I, Rodrigues A, et al. Unraveling FATP1, Regulated by ER-Beta, as a Targeted Breast Cancer Innovative Therapy. Sci Rep (2019) 9(1):14107. doi: 10.1038/s41598-019-50531-3
74. Tousignant KD, Rockstroh A, Taherian Fard A, Lehman ML, Wang C, McPherson SJ, et al. Lipid Uptake Is an Androgen-Enhanced Lipid Supply Pathway Associated With Prostate Cancer Disease Progression and Bone Metastasis. Mol Cancer Res (2019) 17(5):1166–79. doi: 10.1158/1541-7786.MCR-18-1147
75. Verma S, Shankar E, Chan ER, Gupta S. Metabolic Reprogramming and Predominance of Solute Carrier Genes During Acquired Enzalutamide Resistance in Prostate Cancer. Cells (2020) 9(12):2535. doi: 10.3390/cells9122535
76. Kamphorst JJ, Cross JR, Fan J, de Stanchina E, Mathew R, White EP, et al. Hypoxic and Ras-Transformed Cells Support Growth by Scavenging Unsaturated Fatty Acids From Lysophospholipids. Proc Natl Acad Sci USA (2013) 110(22):8882–7. doi: 10.1073/pnas.1307237110
77. Lupien LE, Bloch K, Dehairs J, Traphagen NA, Feng WW, Davis WL, et al. Endocytosis of Very Low-Density Lipoproteins: An Unexpected Mechanism for Lipid Acquisition by Breast Cancer Cells. J Lipid Res (2020) 61(2):205–18. doi: 10.1194/jlr.RA119000327
78. Storch J, Corsico B. The Emerging Functions and Mechanisms of Mammalian Fatty Acid-Binding Proteins. Annu Rev Nutr (2008) 28:73–95. doi: 10.1146/annurev.nutr.27.061406.093710
79. Furuhashi M, Hotamisligil GS. Fatty Acid-Binding Proteins: Role in Metabolic Diseases and Potential as Drug Targets. Nat Rev Drug Discovery (2008) 7(6):489–503. doi: 10.1038/nrd2589
80. Liu RZ, Li X, Godbout R. A Novel Fatty Acid-Binding Protein (FABP) Gene Resulting From Tandem Gene Duplication in Mammals: Transcription in Rat Retina and Testis. Genomics (2008) 92(6):436–45. doi: 10.1016/j.ygeno.2008.08.003
81. Adamson J, Morgan EA, Beesley C, Mei Y, Foster CS, Fujii H, et al. High-Level Expression of Cutaneous Fatty Acid-Binding Protein in Prostatic Carcinomas and Its Effect on Tumorigenicity. Oncogene (2003) 22(18):2739–49. doi: 10.1038/sj.onc.1206341
82. Morgan EA, Forootan SS, Adamson J, Foster CS, Fujii H, Igarashi M, et al. Expression of Cutaneous Fatty Acid-Binding Protein (C-FABP) in Prostate Cancer: Potential Prognostic Marker and Target for Tumourigenicity-Suppression. Int J Oncol (2008) 32(4):767–7.
83. Tolle A, Suhail S, Jung M, Jung K, Stephan C. Fatty Acid Binding Proteins (FABPs) in Prostate, Bladder and Kidney Cancer Cell Lines and the Use of IL-FABP as Survival Predictor in Patients With Renal Cell Carcinoma. BMC Cancer (2011) 11:302. doi: 10.1186/1471-2407-11-302
84. Forootan FS, Forootan SS, Malki MI, Chen D, Li G, Lin K, et al. The Expression of C-FABP and PPARgamma and Their Prognostic Significance in Prostate Cancer. Int J Oncol (2014) 44(1):265–75. doi: 10.3892/ijo.2013.2166
85. Kawaguchi K, Kinameri A, Suzuki S, Senga S, Ke Y, Fujii H. The Cancer-Promoting Gene Fatty Acid-Binding Protein 5 (FABP5) is Epigenetically Regulated During Human Prostate Carcinogenesis. Biochem J (2016) 473(4):449–61. doi: 10.1042/BJ20150926
86. Al Fayi MS, Gou X, Forootan SS, Al-Jameel W, Bao Z, Rudland PR, et al. The Increased Expression of Fatty Acid-Binding Protein 9 in Prostate Cancer and its Prognostic Significance. Oncotarget (2016) 7(50):82783–97. doi: 10.18632/oncotarget.12635
87. Myers JS, von Lersner AK, Sang QX. Proteomic Upregulation of Fatty Acid Synthase and Fatty Acid Binding Protein 5 and Identification of Cancer- and Race-Specific Pathway Associations in Human Prostate Cancer Tissues. J Cancer (2016) 7(11):1452–64. doi: 10.7150/jca.15860
88. Nitschke K, Erben P, Waldbillig F, Abdelhadi A, Weis CA, Gottschalt M, et al. Clinical Relevance of Gene Expression in Localized and Metastatic Prostate Cancer Exemplified by FABP5. World J Urol (2020) 38(3):637–45. doi: 10.1007/s00345-019-02651-8
89. Morgan E, Kannan-Thulasiraman P, Noy N. Involvement of Fatty Acid Binding Protein 5 and PPARbeta/delta in Prostate Cancer Cell Growth. PPAR Res (2010) 2010:234629. doi: 10.1155/2010/234629
90. Liu RZ, Godbout R. An Amplified Fatty Acid-Binding Protein Gene Cluster in Prostate Cancer: Emerging Roles in Lipid Metabolism and Metastasis. Cancers (Basel) (2020) 12(12):3823. doi: 10.3390/cancers12123823
91. Pang J, Liu WP, Liu XP, Li LY, Fang YQ, Sun QP, et al. Profiling Protein Markers Associated With Lymph Node Metastasis in Prostate Cancer by DIGE-Based Proteomics Analysis. J Proteome Res (2010) 9(1):216–26. doi: 10.1021/pr900953s
92. Al-Jameel W, Gou X, Forootan SS, Al Fayi MS, Rudland PS, Forootan FS, et al. Inhibitor SBFI26 Suppresses the Malignant Progression of Castration-Resistant PC3-M Cells by Competitively Binding to Oncogenic FABP5. Oncotarget (2017) 8(19):31041–56. doi: 10.18632/oncotarget.16055
93. O’Sullivan SE, Kaczocha M. FABP5 as a Novel Molecular Target in Prostate Cancer. Drug Discovery Today (2020) S1359–6446(20):30375–5. doi: 10.1016/j.drudis.2020.09.018
94. Tan NS, Shaw NS, Vinckenbosch N, Liu P, Yasmin R, Desvergne B, et al. Selective Cooperation Between Fatty Acid Binding Proteins and Peroxisome Proliferator-Activated Receptors in Regulating Transcription. Mol Cell Biol (2002) 22(14):5114–27. doi: 10.1128/MCB.22.14.5114-5127.2002
95. Carbonetti G, Wilpshaar T, Kroonen J, Studholme K, Converso C, d’Oelsnitz S, et al. FABP5 Coordinates Lipid Signaling That Promotes Prostate Cancer Metastasis. Sci Rep (2019) 9(1):18944. doi: 10.1038/s41598-019-55418-x
96. Bao Z, Malki MI, Forootan SS, Adamson J, Forootan FS, Chen D, et al. A Novel Cutaneous Fatty Acid-Binding Protein-Related Signaling Pathway Leading to Malignant Progression in Prostate Cancer Cells. Genes Cancer (2013) 4(7-8):297–314. doi: 10.1177/1947601913499155
97. Forootan FS, Forootan SS, Gou X, Yang J, Liu B, Chen D, et al. Fatty Acid Activated PPARgamma Promotes Tumorigenicity of Prostate Cancer Cells by Up Regulating VEGF via PPAR Responsive Elements of the Promoter. Oncotarget (2016) 7(8):9322–39. doi: 10.18632/oncotarget.6975
98. Senga S, Kobayashi N, Kawaguchi K, Ando A, Fujii H. Fatty Acid-Binding Protein 5 (FABP5) Promotes Lipolysis of Lipid Droplets, De Novo Fatty Acid (FA) Synthesis and Activation of Nuclear Factor-Kappa B (NF-Kappab) Signaling in Cancer Cells. Biochim Biophys Acta Mol Cell Biol Lipids (2018) 1863(9):1057–67. doi: 10.1016/j.bbalip.2018.06.010
99. Senga S, Kawaguchi K, Kobayashi N, Ando A, Fujii H. A Novel Fatty Acid-Binding Protein 5-Estrogen-Related Receptor Alpha Signaling Pathway Promotes Cell Growth and Energy Metabolism in Prostate Cancer Cells. Oncotarget (2018) 9(60):31753–70. doi: 10.18632/oncotarget.25878
100. Li P, Yu M, Zhou C, Qi H, Wen X, Hou X, et al. FABP5 is a Critical Regulator of Methionine- and Estrogen-Induced SREBP-1c Gene Expression in Bovine Mammary Epithelial Cells. J Cell Physiol (2018) 234(1):537–49. doi: 10.1002/jcp.26762
101. Seo J, Jeong DW, Park JW, Lee KW, Fukuda J, Chun YS. Fatty-Acid-Induced FABP5/HIF-1 Reprograms Lipid Metabolism and Enhances the Proliferation of Liver Cancer Cells. Commun Biol (2020) 3(1):638. doi: 10.1038/s42003-020-01367-5
102. Hwang HH, Moon PG, Lee JE, Kim JG, Lee W, Ryu SH, et al. Identification of the Target Proteins of Rosiglitazone in 3T3-L1 Adipocytes Through Proteomic Analysis of Cytosolic and Secreted Proteins. Mol Cells (2011) 31(3):239–46. doi: 10.1007/s10059-011-0026-6
103. Fujita K, Kume H, Matsuzaki K, Kawashima A, Ujike T, Nagahara A, et al. Proteomic Analysis of Urinary Extracellular Vesicles From High Gleason Score Prostate Cancer. Sci Rep (2017) 7:42961. doi: 10.1038/srep42961
104. Coe NR, Simpson MA, Bernlohr DA. Targeted Disruption of the Adipocyte Lipid-Binding Protein (Ap2 Protein) Gene Impairs Fat Cell Lipolysis and Increases Cellular Fatty Acid Levels. J Lipid Res (1999) 40(5):967–72. doi: 10.1016/S0022-2275(20)32133-7
105. Baar RA, Dingfelder CS, Smith LA, Bernlohr DA, Wu C, Lange AJ, et al. Investigation of In Vivo Fatty Acid Metabolism in AFABP/aP2(-/-) Mice. Am J Physiol Endocrinol Metab (2005) 288(1):E187–93. doi: 10.1152/ajpendo.00256.2004
106. Herroon MK, Rajagurubandara E, Hardaway AL, Powell K, Turchick A, Feldmann D, et al. Bone Marrow Adipocytes Promote Tumor Growth in Bone via FABP4-Dependent Mechanisms. Oncotarget (2013) 4(11):2108–23. doi: 10.18632/oncotarget.1482
107. Xu A, Wang Y, Xu JY, Stejskal D, Tam S, Zhang J, et al. Adipocyte Fatty Acid-Binding Protein is a Plasma Biomarker Closely Associated With Obesity and Metabolic Syndrome. Clin Chem (2006) 52(3):405–13. doi: 10.1373/clinchem.2005.062463
108. Cao H, Sekiya M, Ertunc ME, Burak MF, Mayers JR, White A, et al. Adipocyte Lipid Chaperone AP2 Is a Secreted Adipokine Regulating Hepatic Glucose Production. Cell Metab (2013) 17(5):768–78. doi: 10.1016/j.cmet.2013.04.012
109. Schlottmann I, Ehrhart-Bornstein M, Wabitsch M, Bornstein SR, Lamounier-Zepter V. Calcium-Dependent Release of Adipocyte Fatty Acid Binding Protein From Human Adipocytes. Int J Obes (Lond) (2014) 38(9):1221–7. doi: 10.1038/ijo.2013.241
110. Huang M, Narita S, Inoue T, Koizumi A, Saito M, Tsuruta H, et al. Fatty Acid Binding Protein 4 Enhances Prostate Cancer Progression by Upregulating Matrix Metalloproteinases and Stromal Cell Cytokine Production. Oncotarget (2017) 8(67):111780–94. doi: 10.18632/oncotarget.22908
111. Harraz AM, Atia N, Ismail A, Shady A, Farg H, Gabr H, et al. Evaluation of Serum Fatty Acid Binding Protein-4 (FABP-4) as a Novel Biomarker to Predict Biopsy Outcomes in Prostate Biopsy Naive Patients. Int Urol Nephrol (2020) 52(8):1483–90. doi: 10.1007/s11255-020-02426-w
112. Uehara H, Takahashi T, Oha M, Ogawa H, Izumi K. Exogenous Fatty Acid Binding Protein 4 Promotes Human Prostate Cancer Cell Progression. Int J Cancer (2014) 135(11):2558–68. doi: 10.1002/ijc.28903
113. Liu RZ, Choi WS, Jain S, Dinakaran D, Xu X, Han WH, et al. The FABP12/PPARgamma Pathway Promotes Metastatic Transformation by Inducing Epithelial-to-Mesenchymal Transition and Lipid-Derived Energy Production in Prostate Cancer Cells. Mol Oncol (2020) 14(12):3100–20. doi: 10.1002/1878-0261.12818
114. Schlaepfer IR, Joshi M. CPT1A-Mediated Fat Oxidation, Mechanisms, and Therapeutic Potential. Endocrinology (2020) 161(2):bqz046. doi: 10.1210/endocr/bqz046
115. Dheeraj A, Agarwal C, Schlaepfer IR, Raben D, Singh R, Agarwal R, et al. A Novel Approach to Target Hypoxic Cancer Cells via Combining Beta-Oxidation Inhibitor Etomoxir With Radiation. Hypoxia (Auckl) (2018) 6:23–33. doi: 10.2147/HP.S163115
116. Flaig TW, Salzmann-Sullivan M, Su LJ, Zhang Z, Joshi M, Gijon MA, et al. Lipid Catabolism Inhibition Sensitizes Prostate Cancer Cells to Antiandrogen Blockade. Oncotarget (2017) 8(34):56051–65. doi: 10.18632/oncotarget.17359
117. Joshi M, Stoykova GE, Salzmann-Sullivan M, Dzieciatkowska M, Liebman LN, Deep G, et al. CPT1A Supports Castration-Resistant Prostate Cancer in Androgen-Deprived Conditions. Cells (2019) 8(10):bqz046. doi: 10.3390/cells8101115
118. Joshi M, Kim J, D’Alessandro A, Monk E, Bruce K, Elajaili H, et al. CPT1A Over-Expression Increases Reactive Oxygen Species in the Mitochondria and Promotes Antioxidant Defenses in Prostate Cancer. Cancers (Basel) (2020) 12(11):3431. doi: 10.3390/cancers12113431
119. Blomme A, Ford CA, Mui E, Patel R, Ntala C, Jamieson LE, et al. 2,4-Dienoyl-CoA Reductase Regulates Lipid Homeostasis in Treatment-Resistant Prostate Cancer. Nat Commun (2020) 11(1):2508. doi: 10.1038/s41467-020-16126-7
120. Nassar ZD, Mah CY, Dehairs J, Burvenich IJ, Irani S, Centenera MM, et al. Human DECR1 is an Androgen-Repressed Survival Factor That Regulates PUFA Oxidation to Protect Prostate Tumor Cells From Ferroptosis. Elife (2020) 9:e54166. doi: 10.7554/eLife.54166
121. Yajun C, Chen Y, Xiaosa L, Xiao W, Jia C, Zhong W, et al. Loss of Sun2 Promotes the Progression of Prostate Cancer by Regulating Fatty Acid Oxidation. Oncotarget (2017) 8(52):89620–30. doi: 10.18632/oncotarget.19210
122. Itkonen HM, Brown M, Urbanucci A, Tredwell G, Ho Lau C, Barfeld S, et al. Lipid Degradation Promotes Prostate Cancer Cell Survival. Oncotarget (2017) 8(24):38264–75. doi: 10.18632/oncotarget.16123
123. Cruz ALS, Barreto EA, Fazolini NPB, Viola JPB, Bozza PT. Lipid Droplets: Platforms With Multiple Functions in Cancer Hallmarks. Cell Death Dis (2020) 11(2):105. doi: 10.1038/s41419-020-2297-3
124. Nardi F, Franco OE, Fitchev P, Morales A, Vickman RE, Hayward SW, et al. DGAT1 Inhibitor Suppresses Prostate Tumor Growth and Migration by Regulating Intracellular Lipids and Non-Centrosomal MTOC Protein Gm130. Sci Rep (2019) 9(1):3035. doi: 10.1038/s41598-019-39537-z
125. Mitra R, Le TT, Gorjala P, Goodman OB Jr. Positive Regulation of Prostate Cancer Cell Growth by Lipid Droplet Forming and Processing Enzymes DGAT1 and ABHD5. BMC Cancer (2017) 17(1):631. doi: 10.1186/s12885-017-3589-6
126. Yue S, Li J, Lee SY, Lee HJ, Shao T, Song B, et al. Cholesteryl Ester Accumulation Induced by PTEN Loss and PI3K/AKT Activation Underlies Human Prostate Cancer Aggressiveness. Cell Metab (2014) 19(3):393–406. doi: 10.1016/j.cmet.2014.01.019
127. Nomura DK, Lombardi DP, Chang JW, Niessen S, Ward AM, Long JZ, et al. Monoacylglycerol Lipase Exerts Dual Control Over Endocannabinoid and Fatty Acid Pathways to Support Prostate Cancer. Chem Biol (2011) 18(7):846–56. doi: 10.1016/j.chembiol.2011.05.009
128. Scaglia N, Tyekucheva S, Zadra G, Photopoulos C, Loda M. De Novo Fatty Acid Synthesis at the Mitotic Exit is Required to Complete Cellular Division. Cell Cycle (2014) 13(5):859–68. doi: 10.4161/cc.27767
129. Brohee L, Demine S, Willems J, Arnould T, Colige AC, Deroanne CF. Lipin-1 Regulates Cancer Cell Phenotype and is a Potential Target to Potentiate Rapamycin Treatment. Oncotarget (2015) 6(13):11264–80. doi: 10.18632/oncotarget.3595
130. Ingram LM, Finnerty MC, Mansoura M, Chou CW, Cummings BS. Identification of Lipidomic Profiles Associated With Drug-Resistant Prostate Cancer Cells. Lipids Health Dis (2021) 20(1):15. doi: 10.1186/s12944-021-01437-5
131. Challapalli A, Trousil S, Hazell S, Kozlowski K, Gudi M, Aboagye EO, et al. Exploiting Altered Patterns of Choline Kinase-Alpha Expression on Human Prostate Tissue to Prognosticate Prostate Cancer. J Clin Pathol (2015) 68(9):703–9. doi: 10.1136/jclinpath-2015-202859
132. Priolo C, Pyne S, Rose J, Regan ER, Zadra G, Photopoulos C, et al. AKT1 and MYC Induce Distinctive Metabolic Fingerprints in Human Prostate Cancer. Cancer Res (2014) 74(24):7198–204. doi: 10.1158/0008-5472.CAN-14-1490
133. Von Eyben FE, Kairemo K. Acquisition With (11)C-Choline and (18)F-Fluorocholine PET/CT for Patients With Biochemical Recurrence of Prostate Cancer: A Systematic Review and Meta-Analysis. Ann Nucl Med (2016) 30(6):385–92. doi: 10.1007/s12149-016-1078-7
134. Asim M, Massie CE, Orafidiya F, Pertega-Gomes N, Warren AY, Esmaeili M, et al. Choline Kinase Alpha as an Androgen Receptor Chaperone and Prostate Cancer Therapeutic Target. J Natl Cancer Inst (2016) 108(5):djv371. doi: 10.1093/jnci/djv371
135. Dong Z, Liu Y, Scott KF, Levin L, Gaitonde K, Bracken RB, et al. Secretory Phospholipase A2-IIa is Involved in Prostate Cancer Progression and may Potentially Serve as a Biomarker for Prostate Cancer. Carcinogenesis (2010) 31(11):1948–55. doi: 10.1093/carcin/bgq188
136. Tousignant KD, Rockstroh A, Poad BLJ, Talebi A, Young RSE, Taherian Fard A, et al. Therapy-Induced Lipid Uptake and Remodeling Underpin Ferroptosis Hypersensitivity in Prostate Cancer. Cancer Metab (2020) 8:11. doi: 10.1186/s40170-020-00217-6
137. Monet M, Gkika D, Lehen’kyi V, Pourtier A, Vanden Abeele F, Bidaux G, et al. Lysophospholipids Stimulate Prostate Cancer Cell Migration via TRPV2 Channel Activation. Biochim Biophys Acta (2009) 1793(3):528–39. doi: 10.1016/j.bbamcr.2009.01.003
138. Hao F, Tan M, Xu X, Han J, Miller DD, Tigyi G, et al. Lysophosphatidic Acid Induces Prostate Cancer PC3 Cell Migration via Activation of LPA(1), P42 and P38alpha. Biochim Biophys Acta (2007) 1771(7):883–92. doi: 10.1016/j.bbalip.2007.04.010
139. Grupp K, Sanader S, Sirma H, Simon R, Koop C, Prien K, et al. High Lysophosphatidylcholine Acyltransferase 1 Expression Independently Predicts High Risk for Biochemical Recurrence in Prostate Cancers. Mol Oncol (2013) 7(6):1001–11. doi: 10.1016/j.molonc.2013.07.009
140. Han C, Yu G, Mao Y, Song S, Li L, Zhou L, et al. LPCAT1 Enhances Castration Resistant Prostate Cancer Progression via Increased mRNA Synthesis and PAF Production. PloS One (2020) 15(11):e0240801. doi: 10.1371/journal.pone.0240801
141. Krycer JR, Brown AJ. Cholesterol Accumulation in Prostate Cancer: A Classic Observation From a Modern Perspective. Biochim Biophys Acta (2013) 1835(2):219–29. doi: 10.1016/j.bbcan.2013.01.002
142. Stopsack KH, Gerke TA, Andren O, Andersson SO, Giovannucci EL, Mucci LA, et al. Cholesterol Uptake and Regulation in High-Grade and Lethal Prostate Cancers. Carcinogenesis (2017) 38(8):806–11. doi: 10.1093/carcin/bgx058
143. Stopsack KH, Gerke TA, Sinnott JA, Penney KL, Tyekucheva S, Sesso HD, et al. Cholesterol Metabolism and Prostate Cancer Lethality. Cancer Res (2016) 76(16):4785–90. doi: 10.1158/0008-5472.CAN-16-0903
144. Khan NA, Stopsack KH, Allott EH, Gerke T, Giovannucci EL, Mucci LA, et al. Intratumoral Sterol-27-Hydroxylase (CYP27A1) Expression in Relation to Cholesterol Synthesis and Vitamin D Signaling and Its Association With Lethal Prostate Cancer. Cancer Epidemiol Biomarkers Prev (2019) 28(6):1052–8. doi: 10.1158/1055-9965.EPI-18-1083
145. Ashida S, Kawada C, Inoue K. Stromal Regulation of Prostate Cancer Cell Growth by Mevalonate Pathway Enzymes HMGCS1 and HMGCR. Oncol Lett (2017) 14(6):6533–42. doi: 10.3892/ol.2017.7025
146. Ogretmen B. Sphingolipid Metabolism in Cancer Signalling and Therapy. Nat Rev Cancer (2018) 18(1):33–50. doi: 10.1038/nrc.2017.96
147. Louie SM, Roberts LS, Mulvihill MM, Luo K, Nomura DK. Cancer Cells Incorporate and Remodel Exogenous Palmitate Into Structural and Oncogenic Signaling Lipids. Biochim Biophys Acta (2013) 1831(10):1566–72. doi: 10.1016/j.bbalip.2013.07.008
148. Venant H, Rahmaniyan M, Jones EE, Lu P, Lilly MB, Garrett-Mayer E, et al. The Sphingosine Kinase 2 Inhibitor ABC294640 Reduces the Growth of Prostate Cancer Cells and Results in Accumulation of Dihydroceramides In Vitro and In Vivo. Mol Cancer Ther (2015) 14(12):2744–52. doi: 10.1158/1535-7163.MCT-15-0279
149. Saddoughi SA, Ogretmen B. Chapter Two - Diverse Functions of Ceramide in Cancer Cell Death and Proliferation. In: Norris JS, editor. Advances in Cancer Research, vol. 117. Cambridge, Massachusetts, United States: Academic Press (2013).
150. Saad AF, Meacham WD, Bai A, Anelli V, Elojeimy S, Mahdy AE, et al. The Functional Effects of Acid Ceramidase Overexpression in Prostate Cancer Progression and Resistance to Chemotherapy. Cancer Biol Ther (2007) 6(9):1455–60. doi: 10.4161/cbt.6.9.4623
151. Liu X, Cheng JC, Turner LS, Elojeimy S, Beckham TH, Bielawska A, et al. Acid Ceramidase Upregulation in Prostate Cancer: Role in Tumor Development and Implications for Therapy. Expert Opin Ther Targets (2009) 13(12):1449–58. doi: 10.1517/14728220903357512
152. Lin HM, Huynh K, Kohli M, Tan W, Azad AA, Yeung N, et al. Aberrations in Circulating Ceramide Levels are Associated With Poor Clinical Outcomes Across Localised and Metastatic Prostate Cancer. Prostate Cancer Prostatic Dis (2021) 202. doi: 10.1038/s41391-021-00338-z
153. Xu S, Zhou W, Ge J, Zhang Z. Prostaglandin E2 Receptor EP4 is Involved in the Cell Growth and Invasion of Prostate Cancer via the cAMPPKA/PI3KAkt Signaling Pathway. Mol Med Rep (2018) 17(3):4702–12. doi: 10.3892/mmr.2018.8415
154. Youlin K, Weiyang H, Simin L, Xin G. Prostaglandin E2 Inhibits Prostate Cancer Progression by Countervailing Tumor Microenvironment-Induced Impairment of Dendritic Cell Migration Through LXRalpha/CCR7 Pathway. J Immunol Res (2018) 2018:5808962. doi: 10.1155/2018/5808962
155. Benjamin DI, Li DS, Lowe W, Heuer T, Kemble G, Nomura DK. Diacylglycerol Metabolism and Signaling Is a Driving Force Underlying FASN Inhibitor Sensitivity in Cancer Cells. ACS Chem Biol (2015) 10(7):1616–23. doi: 10.1021/acschembio.5b00240
156. Chen M, Zhang J, Sampieri K, Clohessy JG, Mendez L, Gonzalez-Billalabeitia E, et al. An Aberrant SREBP-Dependent Lipogenic Program Promotes Metastatic Prostate Cancer. Nat Genet (2018) 50(2):206–18. doi: 10.1038/s41588-017-0027-2
157. Ellwood-Yen K, Graeber TG, Wongvipat J, Iruela-Arispe ML, Zhang J, Matusik R, et al. Myc-Driven Murine Prostate Cancer Shares Molecular Features With Human Prostate Tumors. Cancer Cell (2003) 4(3):223–38. doi: 10.1016/S1535-6108(03)00197-1
158. Labbe DP, Zadra G, Yang M, Reyes JM, Lin CY, Cacciatore S, et al. High-Fat Diet Fuels Prostate Cancer Progression by Rewiring the Metabolome and Amplifying the MYC Program. Nat Commun (2019) 10(1):4358. doi: 10.1038/s41467-019-12298-z
159. Blando J, Moore T, Hursting S, Jiang G, Saha A, Beltran L, et al. Dietary Energy Balance Modulates Prostate Cancer Progression in Hi-Myc Mice. Cancer Prev Res (Phila) (2011) 4(12):2002–14. doi: 10.1158/1940-6207.CAPR-11-0182
160. Ngo TH, Barnard RJ, Cohen P, Freedland S, Tran C, deGregorio F, et al. Effect of Isocaloric Low-Fat Diet on Human LAPC-4 Prostate Cancer Xenografts in Severe Combined Immunodeficient Mice and the Insulin-Like Growth Factor Axis. Clin Cancer Res (2003) 9(7):2734–43.
161. Wang S, Wu J, Suburu J, Gu Z, Cai J, Axanova LS, et al. Effect of Dietary Polyunsaturated Fatty Acids on Castration-Resistant Pten-Null Prostate Cancer. Carcinogenesis (2012) 33(2):404–12. doi: 10.1093/carcin/bgr290
162. Kobayashi N, Barnard RJ, Henning SM, Elashoff D, Reddy ST, Cohen P, et al. Effect of Altering Dietary Omega-6/Omega-3 Fatty Acid Ratios on Prostate Cancer Membrane Composition, Cyclooxygenase-2, and Prostaglandin E2. Clin Cancer Res (2006) 12(15):4662–70. doi: 10.1158/1078-0432.CCR-06-0459
163. Estève D, Roumiguié M, Manceau C, Milhas D, Muller C. Periprostatic Adipose Tissue: A Heavy Player in Prostate Cancer Progression. Curr Opin Endocrine Metab Res (2020) 10:29–35. doi: 10.1016/j.coemr.2020.02.007
164. Laurent V, Toulet A, Attane C, Milhas D, Dauvillier S, Zaidi F, et al. Periprostatic Adipose Tissue Favors Prostate Cancer Cell Invasion in an Obesity-Dependent Manner: Role of Oxidative Stress. Mol Cancer Res (2019) 17(3):821–35. doi: 10.1158/1541-7786.MCR-18-0748
165. Guerard A, Laurent V, Fromont G, Esteve D, Gilhodes J, Bonnelye E, et al. The Chemokine Receptor CCR3 Is Potentially Involved in the Homing of Prostate Cancer Cells to Bone: Implication of Bone-Marrow Adipocytes. Int J Mol Sci (2021) 22(4):1994. doi: 10.3390/ijms22041994
166. Nassar ZD, Aref AT, Miladinovic D, Mah CY, Raj GV, Hoy AJ, et al. Peri-Prostatic Adipose Tissue: The Metabolic Microenvironment of Prostate Cancer. BJU Int (2018) 121(Suppl 3):9–21. doi: 10.1111/bju.14173
167. Zhai T, Hu L, Ma W, Chen X, Luo M, Jin L, et al. Peri-Prostatic Adipose Tissue Measurements Using MRI Predict Prostate Cancer Aggressiveness in Men Undergoing Radical Prostatectomy. J Endocrinol Invest (2021) 44(2):287–96. doi: 10.1007/s40618-020-01294-6
168. Zhai TS, Jin L, Hu LT, Kadier A, Zhou Z, Liu X, et al. Impact of Peri-Prostatic Fat Measurements Using MRI on the Prediction of Prostate Cancer With Transrectal Ultrasound-Guided Biopsy. Urol Oncol (2020) 38(2):37 e1– e9. doi: 10.1016/j.urolonc.2019.10.008
169. Salji M, Hendry J, Patel A, Ahmad I, Nixon C, Leung HY. Peri-Prostatic Fat Volume Measurement as a Predictive Tool for Castration Resistance in Advanced Prostate Cancer. Eur Urol Focus (2018) 4(6):858–66. doi: 10.1016/j.euf.2017.01.019
170. Dyck L, Lynch L. Cancer, Obesity and Immunometabolism - Connecting the Dots. Cancer Lett (2018) 417:11–20. doi: 10.1016/j.canlet.2017.12.019
171. Pearce EL, Walsh MC, Cejas PJ, Harms GM, Shen H, Wang LS, et al. Enhancing CD8 T-Cell Memory by Modulating Fatty Acid Metabolism. Nature (2009) 460(7251):103–7. doi: 10.1038/nature08097 10.1038/nature08097.
172. O’Sullivan D, van der Windt GJ, Huang SC, Curtis JD, Chang CH, Buck MD, et al. Memory CD8(+) T Cells Use Cell-Intrinsic Lipolysis to Support the Metabolic Programming Necessary for Development. Immunity (2014) 41(1):75–88. doi: 10.1016/j.immuni.2014.06.005
173. Pan Y, Tian T, Park CO, Lofftus SY, Mei S, Liu X, et al. Survival of Tissue-Resident Memory T Cells Requires Exogenous Lipid Uptake and Metabolism. Nature (2017) 543(7644):252–6. doi: 10.1038/nature21379
174. Angela M, Endo Y, Asou HK, Yamamoto T, Tumes DJ, Tokuyama H, et al. Fatty Acid Metabolic Reprogramming via mTOR-Mediated Inductions of PPARgamma Directs Early Activation of T Cells. Nat Commun (2016) 7:13683. doi: 10.1038/ncomms13683
175. Michalek RD, Gerriets VA, Jacobs SR, Macintyre AN, MacIver NJ, Mason EF, et al. Cutting Edge: Distinct Glycolytic and Lipid Oxidative Metabolic Programs are Essential for Effector and Regulatory CD4+ T Cell Subsets. J Immunol (2011) 186(6):3299–303. doi: 10.4049/jimmunol.1003613
176. Kohlgruber AC, LaMarche NM, Lynch L. Adipose Tissue at the Nexus of Systemic and Cellular Immunometabolism. Semin Immunol (2016) 28(5):431–40. doi: 10.1016/j.smim.2016.09.005
177. Vats D, Mukundan L, Odegaard JI, Zhang L, Smith KL, Morel CR, et al. Oxidative Metabolism and PGC-1beta Attenuate Macrophage-Mediated Inflammation. Cell Metab (2006) 4(1):13–24. doi: 10.1016/j.cmet.2006.05.011
178. Huang SC, Everts B, Ivanova Y, O’Sullivan D, Nascimento M, Smith AM, et al. Cell-Intrinsic Lysosomal Lipolysis Is Essential for Alternative Activation of Macrophages. Nat Immunol (2014) 15(9):846–55. doi: 10.1038/ni.2956
179. Wang HW, Joyce JA. Alternative Activation of Tumor-Associated Macrophages by IL-4: Priming for Protumoral Functions. Cell Cycle (2010) 9(24):4824–35. doi: 10.4161/cc.9.24.14322
180. Johnson AR, Qin Y, Cozzo AJ, Freemerman AJ, Huang MJ, Zhao L, et al. Metabolic Reprogramming Through Fatty Acid Transport Protein 1 (FATP1) Regulates Macrophage Inflammatory Potential and Adipose Inflammation. Mol Metab (2016) 5(7):506–26. doi: 10.1016/j.molmet.2016.04.005
181. Herber DL, Cao W, Nefedova Y, Novitskiy SV, Nagaraj S, Tyurin VA, et al. Lipid Accumulation and Dendritic Cell Dysfunction in Cancer. Nat Med (2010) 16(8):880–6. doi: 10.1038/nm.2172
182. Ramakrishnan R, Tyurin VA, Veglia F, Condamine T, Amoscato A, Mohammadyani D, et al. Oxidized Lipids Block Antigen Cross-Presentation by Dendritic Cells in Cancer. J Immunol (2014) 192(6):2920–31. doi: 10.4049/jimmunol.1302801
183. Macia L, Delacre M, Abboud G, Ouk TS, Delanoye A, Verwaerde C, et al. Impairment of Dendritic Cell Functionality and Steady-State Number in Obese Mice. J Immunol (2006) 177(9):5997–6006. doi: 10.4049/jimmunol.177.9.5997
184. Michelet X, Dyck L, Hogan A, Loftus RM, Duquette D, Wei K, et al. Metabolic Reprogramming of Natural Killer Cells in Obesity Limits Antitumor Responses. Nat Immunol (2018) 19(12):1330–40. doi: 10.1038/s41590-018-0251-7
185. Ringel AE, Drijvers JM, Baker GJ, Catozzi A, Garcia-Canaveras JC, Gassaway BM, et al. Obesity Shapes Metabolism in the Tumor Microenvironment to Suppress Anti-Tumor Immunity. Cell (2020) 183(7):1848–66 e26. doi: 10.1016/j.cell.2020.11.009
186. Wang Z, Aguilar EG, Luna JI, Dunai C, Khuat LT, Le CT, et al. Paradoxical Effects of Obesity on T Cell Function During Tumor Progression and PD-1 Checkpoint Blockade. Nat Med (2019) 25(1):141–51. doi: 10.1038/s41591-018-0221-5
187. Goossens P, Rodriguez-Vita J, Etzerodt A, Masse M, Rastoin O, Gouirand V, et al. Membrane Cholesterol Efflux Drives Tumor-Associated Macrophage Reprogramming and Tumor Progression. Cell Metab (2019) 29(6):1376–89 e4. doi: 10.1016/j.cmet.2019.02.016
188. Veglia F, Tyurin VA, Blasi M, De Leo A, Kossenkov AV, Donthireddy L, et al. Fatty Acid Transport Protein 2 Reprograms Neutrophils in Cancer. Nature (2019) 569(7754):73–8. doi: 10.1038/s41586-019-1118-2
189. Yang S, Zhang Q, Liu S, Wang AR, You Z. PD-1, PD-L1 and PD-L2 Expression in Mouse Prostate Cancer. Am J Clin Exp Urol (2016) 4(1):1–8.
190. Falchook G, Infante J, Arkenau H-T, Patel MR, Dean E, Borazanci E, et al. First-In-Human Study of the Safety, Pharmacokinetics, and Pharmacodynamics of First-In-Class Fatty Acid Synthase Inhibitor Tvb-2640 Alone and With A Taxane In Advanced Tumors. EClinical Medicine (2021) 34:100797. doi: 10.1016/j.eclinm.2021.100797
191. Brenner AJ, Falchook G, Patel M, Infante JR, Arkenau H-T, Dean EM, et al. Abstract P6-11-09: Heavily Pre-Treated Breast Cancer Patients Show Promising Responses in the First in Human Study of the First-in-Class Fatty Acid Synthase (FASN) Inhibitor, TVB-2640 in Combination With Paclitaxel [abstract]. In: Proceedings of the 2016 San Antonio Breast Cancer Symposium; 2016 Dec 6-10; San Antonio, TX: Philadelphia (PA), AACR. Cancer Res (2017) 77(4 Suppl):Abstract nr P6-11-09. doi: 10.1158/1538-7445.SABCS16-P6-11-09
192. Ventura R, Mordec K, Waszczuk J, Wang Z, Lai J, Fridlib M, et al. Inhibition of De Novo Palmitate Synthesis by Fatty Acid Synthase Induces Apoptosis in Tumor Cells by Remodeling Cell Membranes, Inhibiting Signaling Pathways, and Reprogramming Gene Expression. EBioMedicine (2015) 2(8):808–24. doi: 10.1016/j.ebiom.2015.06.020
193. Iglesias-Gato D, Thysell E, Tyanova S, Crnalic S, Santos A, Lima TS, et al. The Proteome of Prostate Cancer Bone Metastasis Reveals Heterogeneity With Prognostic Implications. Clin Cancer Res (2018) 24(21):5433–44. doi: 10.1158/1078-0432.CCR-18-1229
194. Carracedo A, Cantley LC, Pandolfi PP. Cancer Metabolism: Fatty Acid Oxidation in the Limelight. Nat Rev Cancer (2013) 13(4):227–32. doi: 10.1038/nrc3483
195. Kong Y, Cheng L, Mao F, Zhang Z, Zhang Y, Farah E, et al. Inhibition of Cholesterol Biosynthesis Overcomes Enzalutamide Resistance in Castration-Resistant Prostate Cancer (CRPC). J Biol Chem (2018) 293(37):14328–41. doi: 10.1074/jbc.RA118.004442
196. Yang H, Pang L, Hu X, Wang W, Xu B, Zhang X, et al. The Effect of Statins on Advanced Prostate Cancer Patients With Androgen Deprivation Therapy or Abiraterone/Enzalutamide: A Systematic Review and Meta-Analysis. J Clin Pharm Ther (2020) 45(3):488–95. doi: 10.1111/jcpt.13092200
197. Lin HM, Mahon KL, Weir JM, Mundra PA, Spielman C, Briscoe K, et al. A Distinct Plasma Lipid Signature Associated With Poor Prognosis in Castration-Resistant Prostate Cancer. Int J Cancer (2017) 141(10):2112–20. doi: 10.1002/ijc.30903
198. Butler LM, Mah CY, Machiels J, Vincent AD, Irani S, Mutuku S, et al. Lipidomic Profiling of Clinical Prostate Cancer Reveals Targetable Alterations in Membrane Lipid Composition. bioRxiv. doi: 10.1101/2020.10.27.356634
199. Li J, Ren S, Piao HL, Wang F, Yin P, Xu C, et al. Integration of Lipidomics and Transcriptomics Unravels Aberrant Lipid Metabolism and Defines Cholesteryl Oleate as Potential Biomarker of Prostate Cancer. Sci Rep (2016) 6:20984. doi: 10.1038/srep20984
200. Brzozowski JS, Jankowski H, Bond DR, McCague SB, Munro BR, Predebon MJ, et al. Lipidomic Profiling of Extracellular Vesicles Derived From Prostate and Prostate Cancer Cell Lines. Lipids Health Dis (2018) 17(1):211. doi: 10.1186/s12944-018-0854-x
201. Kurreck A, Vandergrift LA, Fuss TL, Habbel P, Agar NYR, Cheng LL. Prostate Cancer Diagnosis and Characterization With Mass Spectrometry Imaging. Prostate Cancer Prostatic Dis (2018) 21(3):297–305. doi: 10.1038/s41391-017-0011-z
202. Randall EC, Zadra G, Chetta P, Lopez BGC, Syamala S, Basu SS, et al. Molecular Characterization of Prostate Cancer With Associated Gleason Score Using Mass Spectrometry Imaging. Mol Cancer Res (2019) 17(5):1155–65. doi: 10.1158/1541-7786.MCR-18-1057
Keywords: lipid metabolism, fatty acids, prostate cancer, castration resistance, obesity, microenvironment, lipidomics
Citation: Scaglia N, Frontini-López YR and Zadra G (2021) Prostate Cancer Progression: as a Matter of Fats. Front. Oncol. 11:719865. doi: 10.3389/fonc.2021.719865
Received: 03 June 2021; Accepted: 12 July 2021;
Published: 27 July 2021.
Edited by:
Marianna Kruithof-de Julio, University of Bern, SwitzerlandReviewed by:
Andrea Lunardi, University of Trento, ItalyMaolake Aerken, University at Buffalo, United States
Copyright © 2021 Scaglia, Frontini-López and Zadra. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Giorgia Zadra, Z2lvcmdpYS56YWRyYUBpZ20uY25yLml0