 Zhiwei Chen1†
Zhiwei Chen1† Yinyin Xie1†
Yinyin Xie1† Dan Liu2
Dan Liu2 Ping Liu1Fei Li3,4
Ping Liu1Fei Li3,4 Zhanglin Zhang5
Zhanglin Zhang5 Mengmeng Zhang1
Mengmeng Zhang1 Xiaolin Wang1
Xiaolin Wang1 Yuanliang Zhang1
Yuanliang Zhang1 Xiaojian Sun1
Xiaojian Sun1 Qiuhua Huang1*
Qiuhua Huang1*- 1Shanghai Institute of Hematology, State Key Laboratory of Medical Genomics, National Research Center for Translational Medicine at Shanghai, Ruijin Hospital Affiliated to Shanghai Jiao Tong University School of Medicine, Shanghai, China
- 2Key Laboratory of Systems Biomedicine, Ministry of Education, Shanghai Center for Systems Biomedicine, Shanghai Jiao Tong University, Shanghai, China
- 3Department of Hematology, The First Affiliated Hospital of Nanchang University, Nanchang, China
- 4Institute of Hematology, Jiangxi Academy of Clinical Medical Sciences, Nanchang, China
- 5Department of Transfusion, The First Affiliated Hospital of Nanchang University, Nanchang, China
Background: Leukemic stem cell (LSC) is thought to be responsible for chronic myelogenous leukemia (CML) initiation and relapse. However, the inherent regulation of LSCs remains largely obscure. Herein, we integratedly analyzed miRNA and gene expression alterations in bone marrow (BM) Lin-Sca1+c-Kit+ cells (LSKs) of a tet-off inducible CML mouse model, Scl/tTA-BCR/ABL (BA).
Methods: Scl/tTA and TRE-BA transgenic mice were crossed in the presence of doxycycline to get double transgenic mice. Both miRNA and mRNA expression profiles were generated from BM LSKs at 0 and 3 weeks after doxycycline withdrawal. The target genes of differentially expressed miRNAs were predicted, followed by the miRNA-mRNA network construction. In vitro and in vivo experiments were further performed to elucidate their regulation and function in CML progression.
Results: As a result of the integrated analysis and experimental validation, an anti-apoptotic pathway emerged from the fog. miR-142a was identified to be downregulated by enhanced ERK-phosphorylation in BA-harboring cells, thereby relieving its repression on Ciapin1, an apoptosis inhibitor. Moreover, miR-142a overexpression could partially rescue the abnormal anti-apoptotic phenotype and attenuate CML progression.
Conclusion: Taken together, this study explored the miRNA-mRNA regulatory networks in murine CML LSKs and demonstrated that ERK-miR-142a-Ciapin1 axis played an essential role in CML pathogenesis.
Introduction
Chronic myelogenous leukemia (CML), a malignant clonal disorder of hematopoietic stem cells (HSCs), is driven by BCR/ABL1 (BA) fusion gene created by translocation t (9; 22)(q34; q11) (1). With the persistent survival, proliferation, and myeloid differentiation advantages, BA-harboring HSCs could generate an overwhelming proportion of myeloid cells in both peripheral blood (PB) and bone marrow (BM) (2). CML is mainly treated by tyrosine kinase inhibitors. However, later-stage CML patients usually suffer from resistance to these inhibitors (3, 4). Hence, the molecular pathogenesis and therapeutic targets of CML still need to be studied comprehensively.
MicroRNAs (miRNA, miR-) are short single-strand, non-coding RNAs with a length of 18-24bp, which was reported for the first time in 1993 (5). miRNAs participate in post-transcriptional regulation by inducing mRNA degradation or repressing mRNA translation. In recent years, several miRNAs were clarified as regulators of hematopoiesis or leukemogenesis. Among them, miR-17-92 is reported to be involved in the regulation of Pu.1-dependent macrophage differentiation (6). miR-146a homozygous knockout mice developed myeloid sarcoma and lymphoma, revealing the tumor suppressor role of miR-146a (7, 8). miR-125b is highly expressed in HSCs, promoting HSC expansion by providing an anti-apoptotic signal (9). miR-142 acts as an oncogene in human T-cell acute lymphoblastic leukemia by accelerating proliferation and inducing glucocorticoid resistance (10). Enforced expression of miR-142-3p could promote myeloid differentiation of AML HSPCs (11).
It has also been reported that miRNAs can be used as biomarkers for CML diagnosis and prognosis. Highly expressed miR-486-5p was detected in CML progenitors. Inhibiting miR-486-5p could reduce the growth of CML progenitors and enhance apoptosis following treatment with imatinib (12). L Venturini et al. demonstrated that BA–c-MYC–miR-17-92 pathway could mediate enhanced miRNA expression in CML CD34+ cells at the chronic phase, resulting in the acceleration of disease progression (13). miR-223 expression was shown to be negatively correlated with BA in CML patients, affecting myeloid cell proliferation and transcriptional factor by targeting MEF2C and PTBP2 (14). Downregulation of miR-199b relates to imatinib resistance in CML patients with the deletion of 9q34.1 (15). miR-451 was demonstrated to be inversely associated with BA expression and acted as a predictor of imatinib therapy response in CML (16). miR-142-3p was observed to be upregulated in imatinib-treated K562 (17).
To systematically study the roles of miRNAs in the pathogenesis of CML, herein, we parallelly analyzed BA-induced miRNA and gene expression alterations during CML development using a hematopoietic stem/progenitor cell-specific inducible transgenic murine model, Scl/tTA-BA (18). Based on the target gene prediction and integrated analysis of dysregulated miRNA and mRNA, we built several miRNA-mRNA interaction networks, aiming to elucidate their regulation and function in CML progression.
Materials and Methods
Mice Keeping
Transgenic mice used in this study were maintained in the B6 background. Scl/tTA-BA mice were fed with doxycycline (Clontech, USA) in the drinking water (20mg/L) for routine keeping. The animal protocol was reviewed and approved by the Animal Care Committee of Shanghai Jiao Tong University School of Medicine (No. B-2017-010).
Cell Sorting
BM cells were flushed from tibias and femurs of both legs of each donor into cold DMEM containing 2% FBS and 100ng/mL penicillin/streptomycin. Lineage-positive cells were depleted using the Lineage Cell Depletion Kit (Miltenyi Biotec, Germany). Lineage-negative cells were then stained with Biotin Mouse Lineage Panel (BD Pharmingen, USA), Streptavidin APC-Cy7 (BD Pharmingen, USA), Sca1-PE (BD Pharmingen, USA), and c-Kit-APC (Biolegend, USA) antibody mixture. LSKs were sorted by BD FACSAria (BD Biosciences, USA).
miRNA microarray and RNA-seq
Total RNA was isolated from BM LSKs using the miRNeasy Mini RNA Isolation Kit (Qiagen, Germany) according to the manual description. RNA integrity number of the total RNA was checked by an Agilent Bioanalyzer 2100 (Agilent Technologies, USA).
For miRNA microarray, miRNAs in total RNA were labeled by miRNA Complete Labeling and Hyb Kit (Agilent Technologies, USA) according to the manufacturer’s instruction. Hybridization was performed using Agilent Mouse miRNA (8*60K) V21.0 microarray. Slides were scanned by Agilent Microarray Scanner (Agilent Technologies, USA) and Feature Extraction software 10.7 (Agilent Technologies, USA) with default settings. Raw data were normalized by the Quantile algorithm that was included in the AgiMicroRna “R” package (19).
For RNA-seq, SMARTer Ultra Low Input RNA for Illumina Kit (Clontech, USA) was used to prepare cDNAs for library construction. Paired-end 100-bp sequencing was performed on the Illumina HiSeq 2500 system (Illumina, USA). The obtained sequence reads (Fastq files) were checked by FastQC software. Clean reads with a minimum length of 25nt were obtained by trimming the raw reads and deleting the low-quality reads, and then the clean reads were mapped to the mouse (GRCm38) genome sequence with 2-bp mismatch using the TopHat software2.0.9 (20). The gene expression levels were estimated by the Fragments Per Kilobase of exon model per Million mapped reads. EdgeR was used for differentially expressed gene analysis (21).
Bioinformatic Analysis
The gene ontology terms enriched in differentially expressed genes were determined using DAVID bioinformatics resources (22). Gene set enrichment analysis (GSEA) was conducted on RNA-seq data (Broadinstitute, Cambridge, USA) (23). Gene set files were generated according to fingerprint genes of specific hematopoietic cells that were reported by Stuart M. Chambers et al. (24). The miRNA target genes were predicted by the miRwalk2.0 database using a newly developed algorithm named ‘miRWalk’ as well as with already established programs for putative miRNA binding sites (25).
Stable Cell Line Generation
32Dcl3 (32D) cells were infected separately by pMSCV-BA-IRES-mCherry and pMSCV-IRES-mCherry retrovirus supernatants in the culture medium supplemented with 8μg/mL polybrene (Sigma-Aldrich, USA). mCherry+ cells were isolated by flow cytometry after 48 hours and sorted again after one week to generate the stable cell line expressing BA-IRES-mCherry (32D-BA) or mCherry alone (32D-mCherry). 32D-BA cells were infected separately by pMSCV-Ciapin1-IRES-puro, pMSCV-IRES-puro, pLKO-sh-Ciapin1, and pLKO-scramble virus supernatants, after 48 hours, 1μg/ml puromycin (Beyotime, China) was added in culture medium for 3 days to select the stable cell line expressing Ciapin1 (32D-BA-Ciapin1), puro alone (32D-BA-puro), Ciapin1 knockdown (BA-sh-Ciapin1), and scramble alone (BA-sh-scramble). 32D and 32D-mCherry cells were maintained in RPMI 1640 medium (Gibco, China) supplemented with 10% fetal bovine serum (Gibco, Australia) and 10% IL-3 containing WEHI-3B conditional medium. 32D-BA, 32D-BA-Ciapin1, 32D-BA-puro, BA-sh-Ciapin1, and BA-sh-scramble cells were maintained in RPMI 1640 supplemented with 10% fetal bovine serum.
Quantitative Reverse Transcription PCR (qRT-PCR)
Total RNA was isolated with TRIzol reagent and further purified with the miRNeasy Micro Kit (Qiagen, Germany) according to the manufacturer’s instruction. For miRNA qRT-PCR, Mir-X™ miRNA First-Strand Synthesis Kit (Clontech, USA) was used to generate cDNAs. The entire sequence of mature miRNA (21–23nt) was used as a miRNA-specific 5’primer. The mRQ 3’Primer was supplied in the Kit. The Delta-delta Ct method was used to determine the level of each miRNA relative to the level of sno202 snRNA.
For qRT-PCR, cDNAs were synthesized using the PrimeScript™ RT Reagent Kit with gDNA Eraser Kit (Takara, Japan). The gene expression level was determined by SYBR Green II qPCR. Rn18s was used as an internal control. The following primer-pairs were used for the target gene amplification: Rn18s forward primer 5’ GCAATTATTCC CCATGAACG 3’ and reverse primer 5’ GGCCTCACTAAAC-CATCCAA 3’; Bcl2 forward primer 5’ ATGCCTTTGTGGAA CTATATGGC 3’ and reverse primer 5’ GGTATGCACCCAGA GTGATGC 3’; Birc2 forward primer 5’ TGTGGCCTGATGTTG GATAAC 3’ and reverse primer 5’ GGTGACGAATGTG-CAAATCTACT 3’; Ciapin1 forward primer 5’ GGAGTTTG-GGATCTCCCCTG 3’ and reverse primer 5’ ACCCGACA GAATGACATCGAA 3’; Xiap forward primer 5’ CGAGCTGG GTTTCTTTATACCG 3’ and reverse primer 5’ GCAATTTG GGGA-TATTCTCCTGT 3’.
Cell Viability and Apoptosis Assay
miRNA mimics and the negative control (NC) oligonucleotides were synthesized by Shanghai GenePharma Co., Ltd (Shanghai, China). The viability of 32D-BA cells was assayed using the CCK-8 Kit (Dojindo, Japan). Cells were plated in triplicate at a density of 1×104/well in a 96-well plate at 24h hours after transfection with miRNA mimics. CCK-8 reagent (10μL) was added to each well at 24, 48, and 72h, respectively. The optical density at 450nm was measured after one hour of incubation. Apoptosis ability of murine BM cells was assessed using annexin V/PI (BD Pharmingen, USA) staining.
Luciferase Reporter Assay
3´untranslated region (3´UTR) of the target gene was inserted into the pmirGlo vector, and an empty vector was used as the control. Murine pre-miR-142a sequence, ~100nt of 5’- and 3’-flanking region were amplified from genomic locus by PCR and cloned into pMSCV-IRES-mCherry vector. Transient transfection was performed in 293T cells with 0.8μg pMSCV- miR142a-IRES-mCherry plasmid and 0.2 μg pmirGlo-control or pmirGlo-3’UTR reporter construct. The fluorescent activity was measured consecutively using Dual-Luciferase assays (Promega, UK) 48 hours after transfection, according to the instructions of the manufacturer.
Western Blot Analysis
Cells were counted and lysed in 1×SDS running buffer, heated at 100°C for 10 minutes and centrifuged to remove debris. Protein samples were separated on polyacrylamide gels, transferred onto a PVDF membrane and then blotted with the following primary antibodies: anti-β-actin antibody (Proteintech, China), anti-Bcl-2 antibody (Cell Signaling Technology, USA), anti-Birc2 antibody (Cell Signaling Technology, USA), anti-Ciapin1 antibody (Proteintech, China), anti-Erk1/2 antibody (Cell Signaling Technology, USA), anti-Phospho-Erk1/2 (Cell Signaling Technology, USA), anti-Pu.1 antibody (Cell Signaling Technology, USA), and anti-Xiap antibody (Cell Signaling Technology, USA). HRP-labeled goat anti-mouse IgG (Proteintech, China) or goat anti-rabbit IgG (Proteintech, China) was used as a secondary antibody.
Viral Vectors Construction and Infection
293T cells were transfected with the pCDH-EF1-miR142a-T2A-copGFP plasmid accompanied by pMD2.G and psPAX2, or pMSCV-BA-IRES-mCherry plasmid accompanied by EcoPack using Lipofectamine 2000 (Invitrogen, USA) as per the manufacturer’s protocol. Viral supernatant was collected 48 hours after transfection and concentrated by centrifugation.
BM Transplantation
Balb/c donor mice were treated with 5-fluorouracil 5 days before harvest of BM from tibias and femurs. BM cells were infected by the pCDH-EF1-miR142a-T2A-copGFP and pMSCV-BA-IRES-mCherry virus twice. 1.1×106 total infected cells were resuspended in 200μl PBS and injected into the tail vein of each lethally irradiated female Balb/c recipient (6-8 weeks old). PB cells were analyzed for the expression level of mCherry, Mac1, and Gr1, as well as the leukocyte number two weeks later.
Statistical Analysis
Graphpad Prism software was used to create graphs and perform statistical analyses. The significance of differences in results, including apoptosis rates, mRNA levels, WBC counts, and percentages of mCherry+ cells in PB after BMT was calculated by the student’s unpaired t-test.
Nomenclature
32Dcl23 cell line: RRID: CVCL_6G54
Doxycycline: Clontech Cat# 631311
Lineage Cell Depletion Kit, mouse: Miltenyi Biotec Cat# 130-090-858
Biotin Mouse Lineage Panel: BD Pharmingen™ Cat# 559971
Streptavidin APC-Cy™7: BD Pharmingen™ Cat# 554063
Sca-1 PE: BD Biosciences Cat# 561076, RRID: AB_2034020
CD117 (c-kit)-APC: BioLegend Cat# 135108
miRNeasy Mini Kit: QIAGEN Cat# 217004
Mir-X™ miRNA First-Strand Synthesis Kit: Clontech# 638313
PrimeScript™ RT Reagent Kit with gDNA Eraser Kit: Takara# RR047A
Cell Counting Kit-8: Dojindo# CK04-05
Dual-Luciferase assays: Promega# E1910
β-Actin antibody (C4): Proteintech Cat# 60008-1-Ig, RRID: AB_2289225
Bcl-2 (124) antibody: Cell Signaling Technology Cat# 15071, RRID : AB_2744528
Birc-2 antibody: Cell Signaling Technology Cat# 4952, RRID : AB_2063660
CIAPIN1 antibody: Proteintech Cat# 12638-1-AP, RRID : AB_2079665
Pu.1 antibody: Cell Signaling Technology Cat #2258, RRID : AB_2186909
Xiap antibody: Cell Signaling Technology Cat# 2042, RRID : AB_2214870
Results
Induced Expression of BA Altered miRNA and Gene Profiles of Murine BM LSKs
To get insight into the inherent regulation of CML LSCs, a tet-off inducible transgenic murine CML model, Scl-tTA-BA (18), was chosen for integrated analysis of the miRNA-mRNA regulatory network in BM LSKs. All mice developed a CML-like disease after doxycycline withdrawal (Figure S1). As CML is a stem cell-derived disease, the number and proportion of BM HSPC were analyzed three weeks after doxycycline withdrawal. The proportion and the absolute number of BM LSKs significantly increased in BA-harboring mice compared with that in the control group (Figure 1A). To choose the appropriate point of time for subsequent analysis, the percentage of PB granulocytes was followed up after doxycycline withdrawal, showing that the granulocyte percentage rose from 8% at week-0 to 50% at week-3 (Figure 1B). Furthermore, parallel detection of GFP+ proportion in BM LSKs of Scl/tTA-GFP mice was performed as a reference for the evaluation of BA protein level (26). At week-0, there were no GFP+ cells in LSKs, while almost half of the LSKs were GFP+ at week-3 (Figure 1C). Therefore, we sorted BM LSKs at week-0 and week-3 after doxycycline withdrawal and extracted RNA for subsequent miRNA microarray and RNA-seq experiments (Figure 1D).
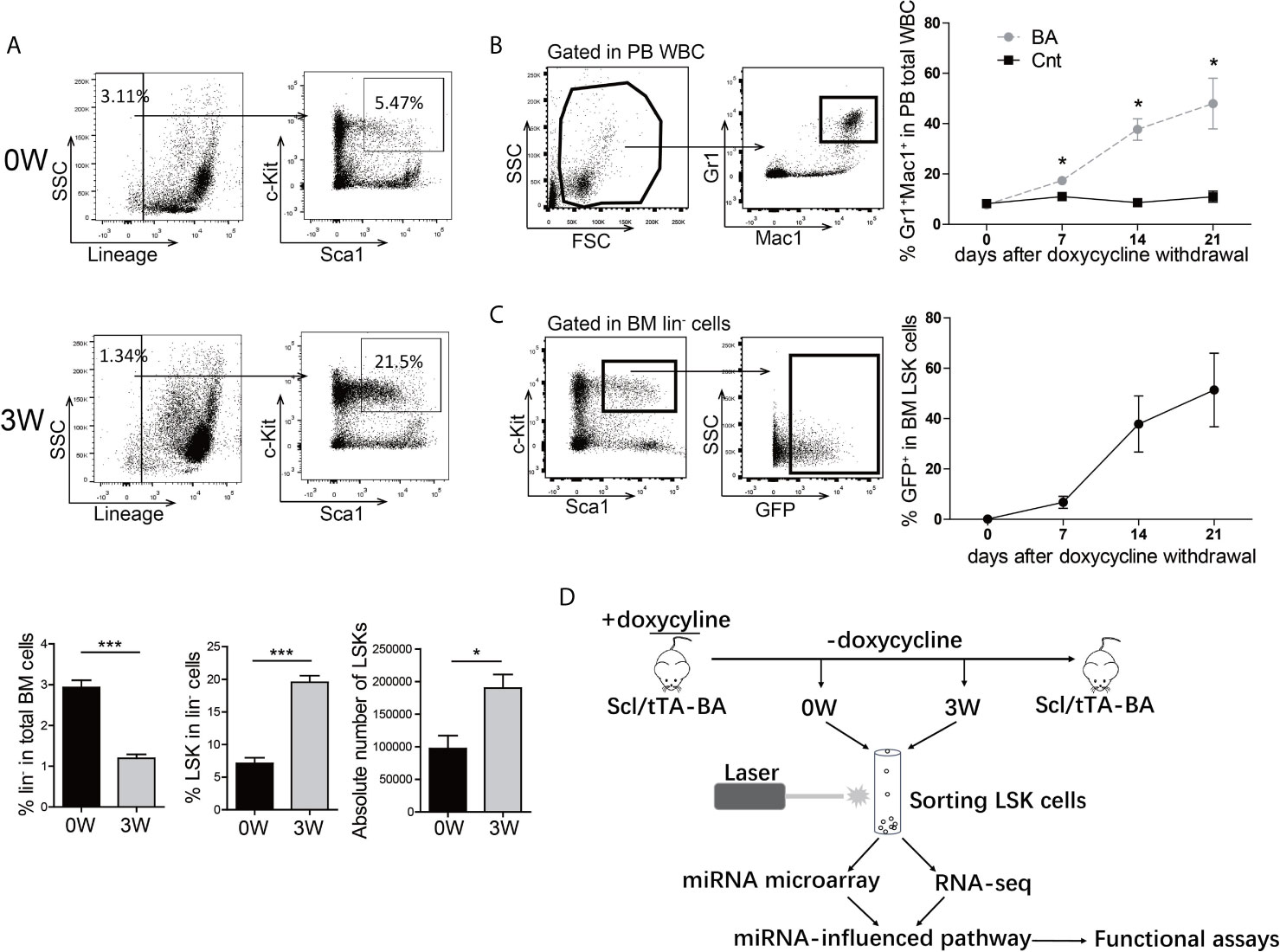
Figure 1 BCR/ABL1 (BA) interferes with the proportion of LSKs in murine bone marrow (BM). (A) Proportion and absolute number analysis of BM Lin-Sca-1+c-Kit+ (LSK) cells in Scl/tTA-BA mice before and after doxycycline withdrawal. (B) Follow-up analysis of granulocyte proportion in peripheral blood (PB) of Scl/tTA-BA mice after doxycycline withdrawal. (C) Follow-up analysis of GFP+ cell proportion in LSKs of Scl/tTA-GFP mice. (D) Schematic design of the experiment. Results are mean ± standard error of the mean (SEM), *p < 0.05, ***p < 0.001 (A, B) was assessed by unpaired t-test.
Compared with mice fed on doxycycline (week-0), there are 52 miRNAs upregulated with fold change≥1.5 and 53 miRNAs downregulated with fold change ≤ 0.67 at week-3 (Figures S2 and 2A). The dysregulated miRNAs are listed in Supplementary Table S1 and Figure 2B. Some of them, such as miRNA-26a-5p, miR-30c-5p, miR-142a-5p, miR-142a-3p, and miR-126a-3p, miR-130a-3p, and miR-34a-5p, have been known to play important roles in solid cancer or leukemia development (27–34). To further validate the array data, qRT-PCR was used to detect the expression level of BA and several selected downregulated miRNAs in LSK samples. As expected, BA expression level was significantly upregulated at week-3 after doxycycline withdrawal (Figure 2C). For miRNAs, consistent with array data, miR-126a-3p, miR-130a-3p, miR-142a-5p, miR-142a-3p, miR-29c-3p, and miR-30c-5p decreased significantly following BA expression. Another three miRNAs also had a decreasing tendency, though showed no statistical difference compared with that in the un-withdrawal sample (Figure 2D). Besides, a murine IL-3-dependent myeloid cell line, 32D, was also used to validate array data. Stable cell lines 32D-BA and 32D-mCherry were constructed (Figure 2E). Enforced expression of BA resulted in significant downregulation of most selected miRNAs (Figure 2F). Overall, our results demonstrated that induced expression of BA altered miRNA profiles of murine BM LSKs.
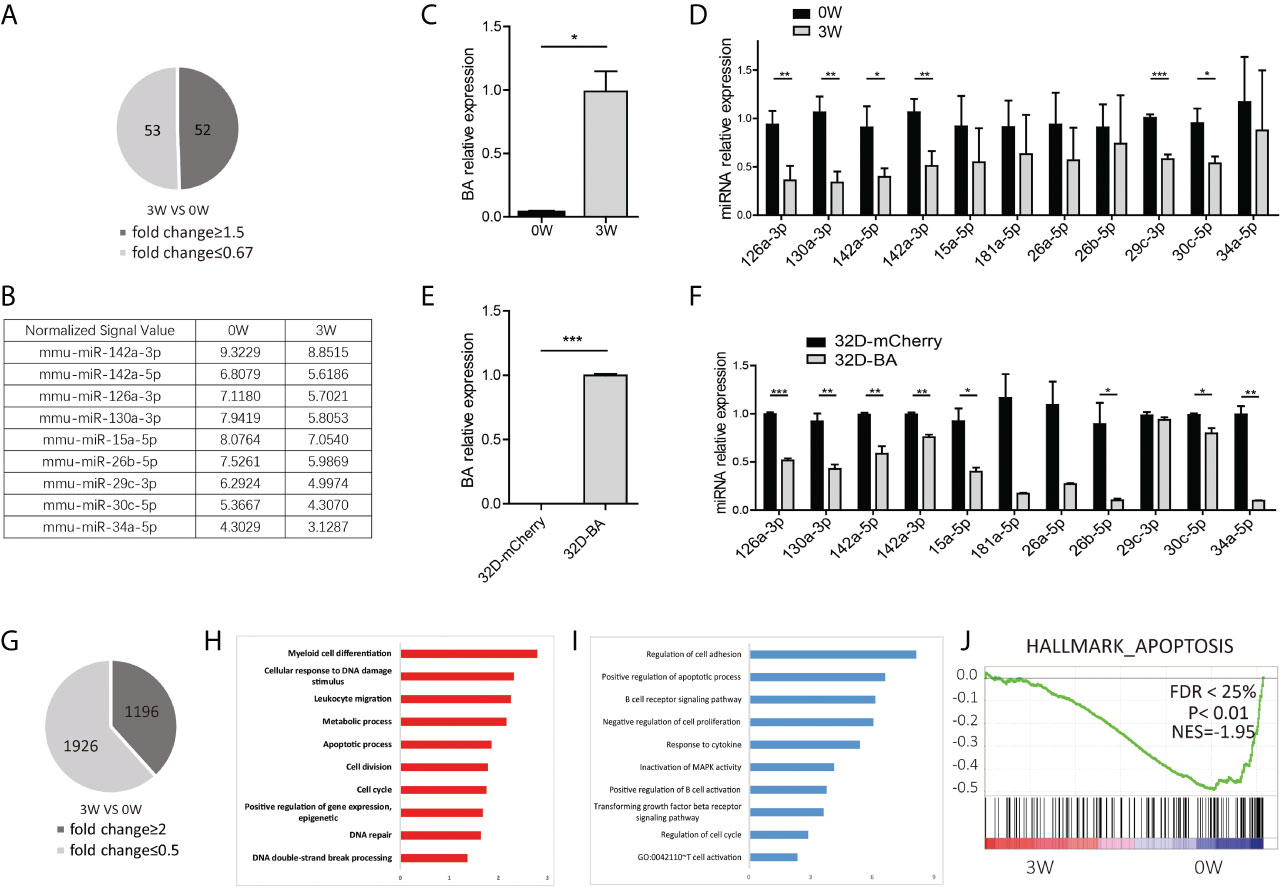
Figure 2 miRNA and mRNA alteration of BM LSKs in Scl/tTA-BA mice. (A) Number of dysregulated miRNAs detected by miRNA array (fold-change ≥1.5 or ≤0.67). (B) Normalized signal values of representative differentially expressed miRNAs. (C) Expression detection of BA in BM LSKs of Scl/tTA-BA mice. (D) qRT-PCR confirmation of expression level of selected miRNAs in BM LSKs from Scl/tTA-BA mice. (E) Expression detection of BA in 32D-BA cell line. (F) qRT-PCR confirmation of expression level of selected miRNAs in 32D-BA cell line. (G) The number of dysregulated genes detected by RNA-seq (fold-change≥2 or ≤0.5). (H, I) GO analysis of differentially expressed genes between BA-harboring and normal BM LSKs. (J) GSEA showing that the apoptosis pathway was remarkably downregulated in BM LSKs at week-3 after doxycycline withdrawal. Results are mean ± SEM, *p < 0.05, **p < 0.01, ***p < 0.001 (C–F) was assessed by unpaired t-test.
Meanwhile, global gene expression analysis was performed on BM LSKs of Scl-tTA-BA mice (Figure S3). LSKs were also collected at week-0 or week-3 after doxycycline withdrawal. After data preprocessing, we identified 3122 differentially expressed genes that contain 1196 upregulated and 1926 downregulated genes (Figure 2G). The top 100 dysregulated mRNAs are listed in Supplementary Table S2. GO analysis indicated that upregulated genes are involved in a broad range of biological process functions, including myeloid cell differentiation, cellular response to DNA damage stimulus, leukocyte migration, metabolic process, apoptosis, and cell cycle etc. (Figure 2H). Downregulated genes were mainly enriched in the regulation of cell adhesion, positive regulation of the apoptotic process, and negative regulation of cell proliferation etc. (Figure 2I). In line with this, GSEA on a customized gene set file, a gene expression fingerprint of specific hematopoietic cells from Chambers et al. (24), also showed significant depletion of the apoptosis signature in LSKs harbored BA fusion gene (Figure 2J).
Integrative Analysis of miRNA−mRNA Network Revealed an Enhanced Cell Survival Probability of CML LSKs
Next, basing on the data above, we performed an integrated analysis of dysregulated miRNAs and genes. First, target genes of miRNAs were predicted using miRwalk2.0 database. Second, we overlapped the predicted target genes with dysregulated genes detected by RNA-seq and proposed a regulatory network of miRNA-mRNA (Figure 3A). 431 interactions were built between 14 downregulated miRNAs and 299 upregulated genes (Figure 3B). The edges that connect nodes showed the interaction of miRNAs and mRNAs, and the nodes represent miRNAs or genes, red indicating upregulation, and green indicating downregulation. We also built 318 interactions between 8 upregulated miRNAs and 271 downregulated mRNAs (Figure 3C). Among the dysregulated ones shown in Figures 3B, C, several miRNAs and their target genes were known to be involved in CML progression (35–38).
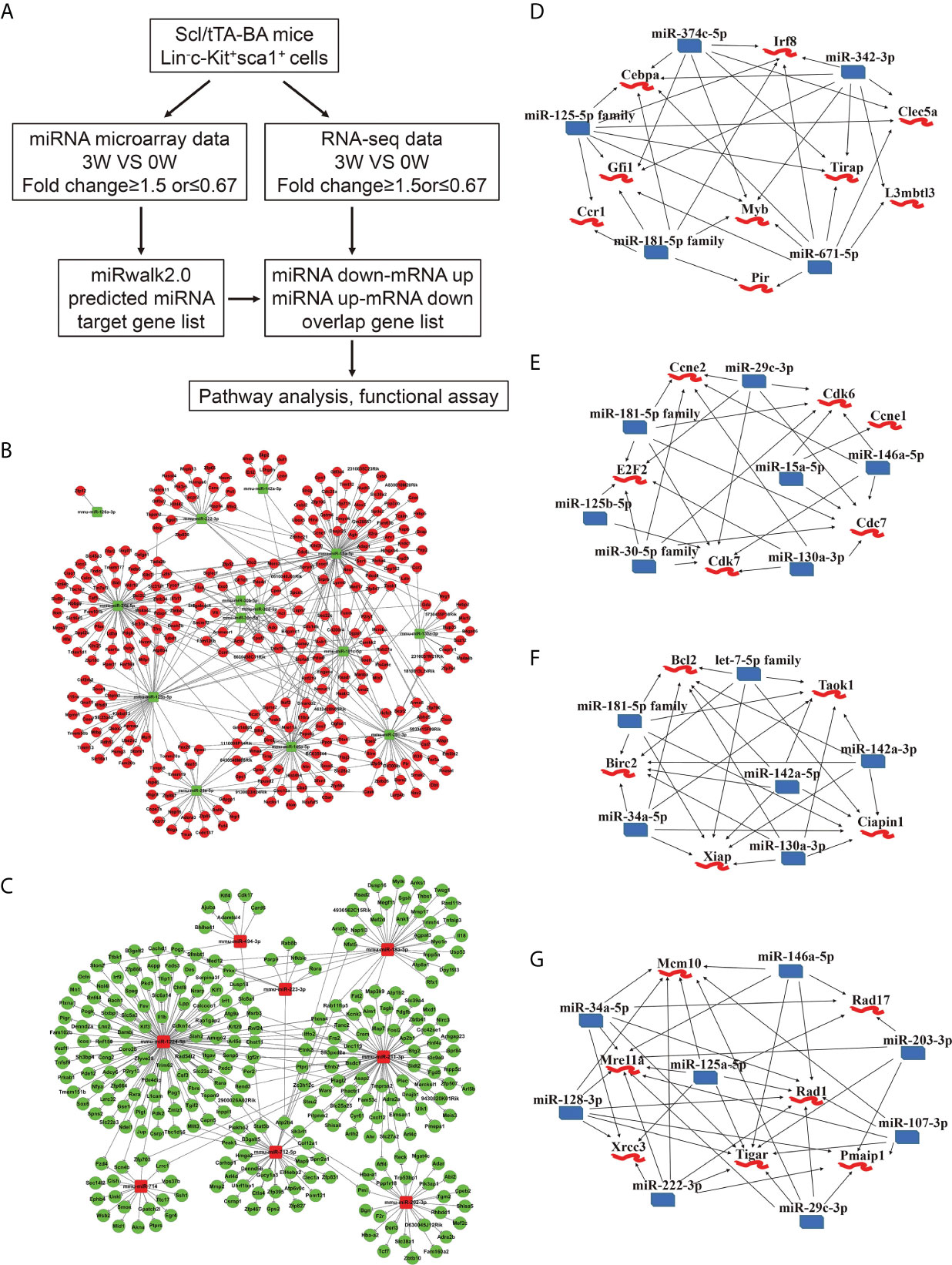
Figure 3 Integrated analysis of miRNA-mRNA regulatory networks in murine CML LSKs. (A) Integrated analysis flowchart of the screened miRNAs and predicted target genes. (B, C) The correlation networks showing 431 interactions built between 14 downregulated miRNAs and 299 possible target genes (B), and 318 interactions between 8 upregulated miRNAs and 271 possible target genes (C). (D–G) Visualization of the regulatory networks enriched for four pathways including myeloid differentiation (D), cell cycle (E), anti-apoptotic pathway (F), and DNA damage (G).
As reported, CML progression can be promoted by multiple pathways including RAS, PI3K/AKT, ERK, MYC, JAK/STAT, myeloid differentiation, apoptosis, cell cycle, and genomic instability (39). In the light of the information uncovered by integrated analysis, we especially focused our attention on four pathways, including anti-apoptotic, cell cycle, myeloid differentiation, and DNA damage pathway. A regulatory network emerged, gathering miR-374c-5p, miR-342-3p, miR-671-5p, miR-181-5p family, and miR-125-5p family. All these miRNAs are downregulated following BA expression. Cebpa, Gfi1, Myb, Irf8, and Tirap etc., whose upregulation has been reported to regulate myeloid differentiation positively, are involved in this network (Figure 3D). Furthermore, the interactions between downregulated miR-181-5p family, miR-29c-3p, miR-15a-5p, miR-146a-5p, miR-130a-3p, miR-125b-5p, and miR-30-5p family and upregulated genes Ccne1, Ccne2, Cdk6, Cdk7, Cdc7, and E2F2 were identified. The predicted target genes in this network are all involved in the cell cycle (Figure 3E). miR-130a-3p, miR-142a-5p, miR-142a-3p, miR-34a-5p, miR-181-5p family, let-7-5p family, and highly expressed genes, including Bcl2, Birc2, Ciapin1, Xiap, and Taok1, were weaved into a network related to the anti-apoptotic process, which implicated that BA expression could promote cell survival (Figure 3F). Last but not least, we displayed a network of several downregulated miRNAs (miR-146a-5p, miR-203-3p, miR-107-3p, miR-29c-3p, miR-222-3p, miR-125a-5p, miR-128-3p, and miR-34a-5p) and their predicted target genes (Mcm10, Rad17, Rad1, Pmaip1, Tigar, Mre11a, and Xrcc3), which is involved in the cellular response to DNA damage stimulus (Figure 3G).
To evaluate the biologic function of these miRNAs in BA-expressing cells, we transfected some of the mimics into 32D-BA cells and performed a CCK-8 assay for cell viability analysis. The viability of 32D-BA cells could be suppressed by miR-130a-3p, miR-142a-5p, miR-30c-5p, and miR-34a-5p (Figure 4A). The transfection efficiency of these mimics was determined by qRT-PCR analysis (Figure 4B). These data indicated that the downregulation of these miRNAs might promote cell survival. Apoptosis analysis was then performed on BM cells collected from Scl-tTA-BA mice. A significant decrease in the proportion of apoptotic cells in LSKs was detected in both early and late apoptosis at week-3 after doxycycline withdrawal compared with that at week-0 (Figure 4C). Besides, we observed that the early apoptosis was suppressed in granulocytes following BA expression, while the late apoptosis was not disturbed (Figure 4D).
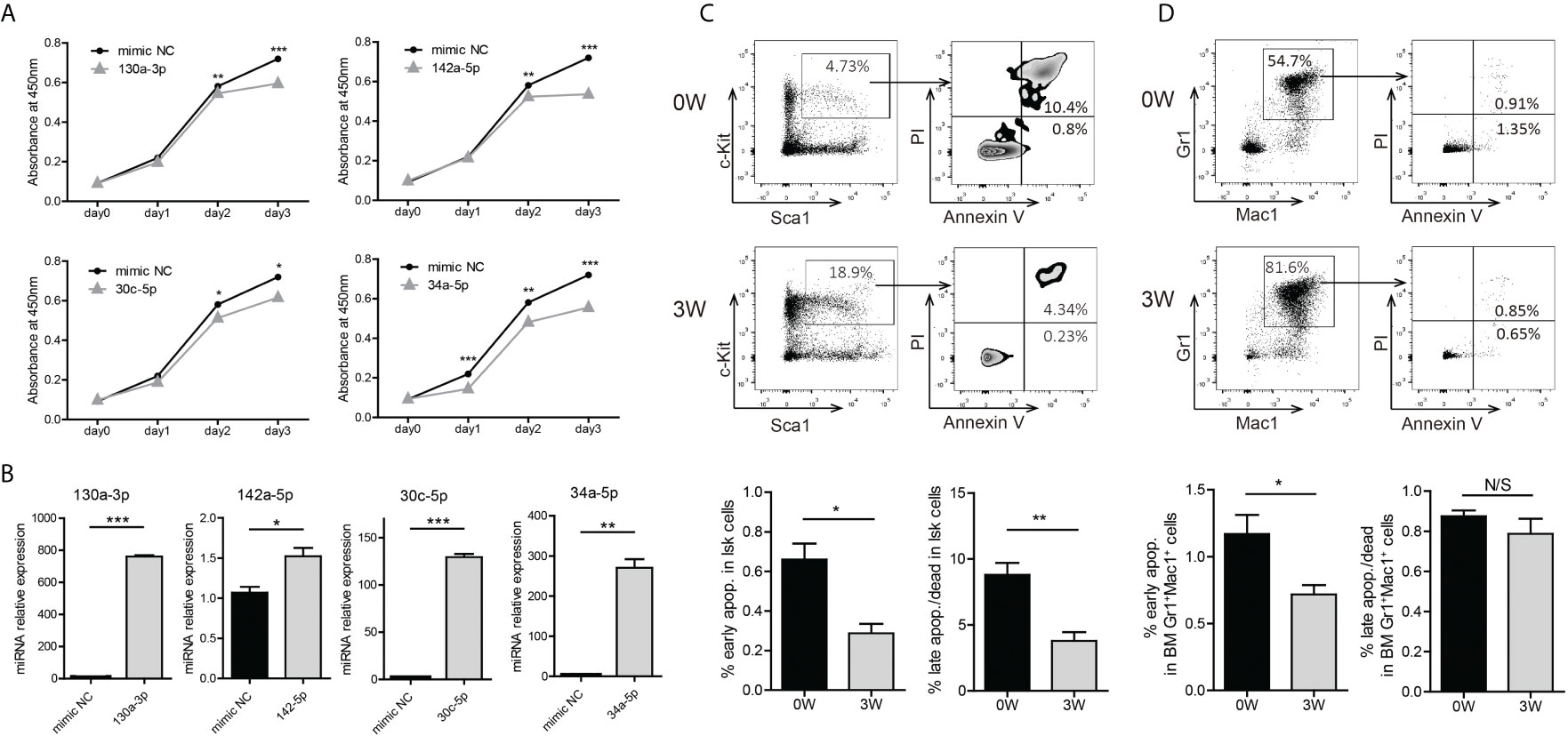
Figure 4 Induction of BA expression enhanced cell survival probability of murine BM LSKs. (A) Cell viability analysis of 32D-BA cells transfected with selected miRNAs by CCK8 assays. (B) Validation of transfection efficiency of miRNAs by qRT-PCR experiments. (C, D) Apoptosis analysis of BM LSKs (C) and granulocytes (D) via Annexin V/PI staining (n=3). Results are mean ± SEM, *p < 0.05, **p < 0.01, ***p < 0.001 and N/S (not significant) (A–D) was assessed by unpaired t-test.
Downregulation of miR-142a Promoted the Survival of BA-Harboring Cells by Relieving Its Repression on Ciapin1
As both miR-142a-5p and miR-142a-3p were involved in a network related to the anti-apoptotic process and detected to be downregulated in CML LSKs and 32D-BA cells (Figures 2D, F), we then conducted enforced overexpression of miR-142a (OE142a) in 32D-BA cells by infection with pCDH-EF1 lentivirus encoding miR-142a-T2A-GFP. Our results showed that miR-142a overexpression could gradually reduce the survival ability of 32D-BA cells (Figure 5A) and significantly promoted cell apoptosis (Figure 5B). The overexpression levels of miR-142a-5p and miR-142a-3p were confirmed by qRT-PCR (Figure 5C). These results suggested that miR-142a overexpression induced cell apoptosis in BA-harboring cells.
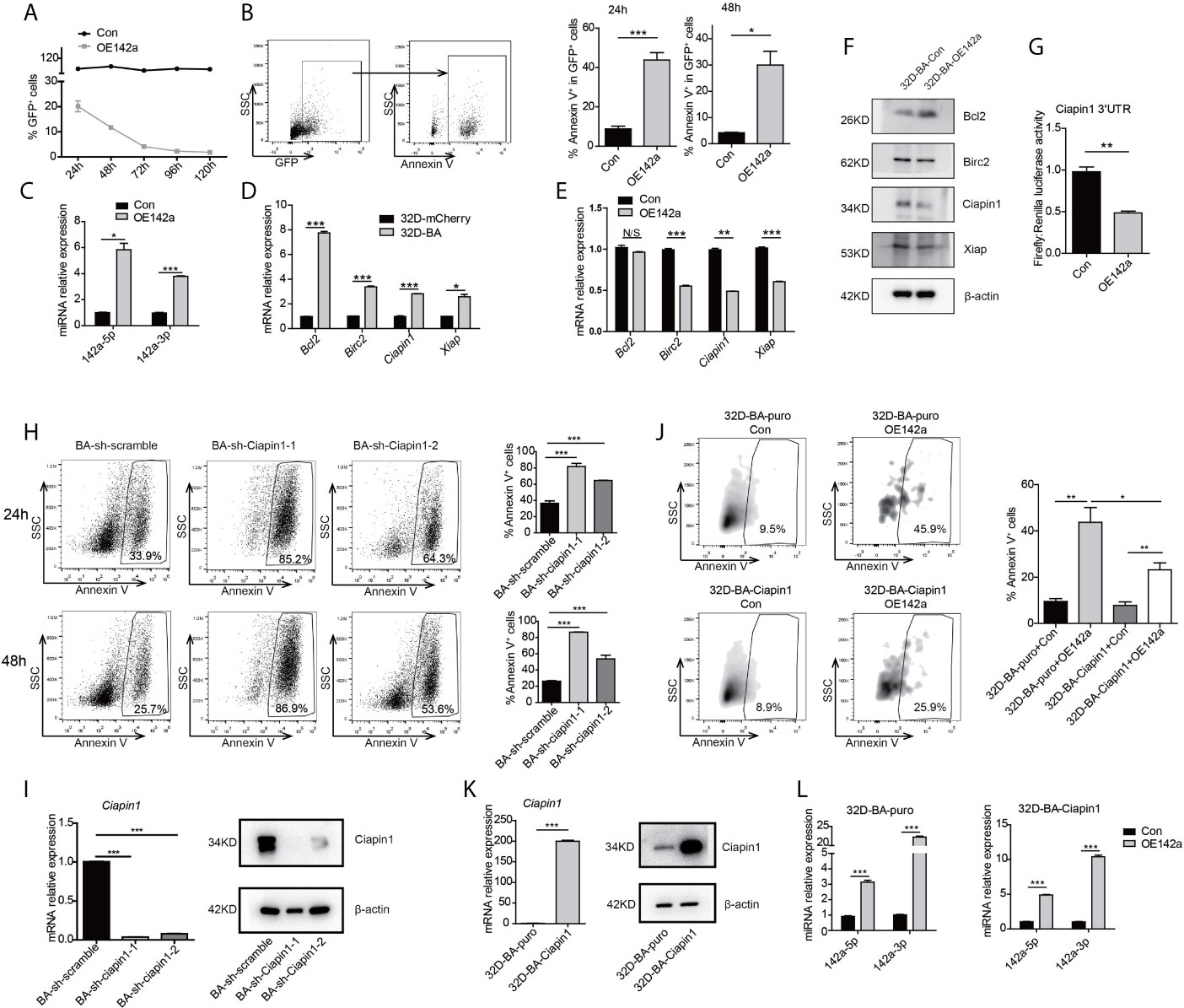
Figure 5 Overexpression of miR-142a impaired cell viability partially through targeting Ciapin1. (A) Follow-up analysis of GFP+ cell proportion in 32D-BA cells infected with pCDH-EF1-miR142a-T2A-copGFP virus. (B) Apoptosis analysis of 32D-BA cells at 24h and 48h after lentivirus infection. (C) Validation of miR-142a-5p and miR-142a-3p expression by qRT-PCR. (D, E) qRT-PCR detection of anti-apoptotic gene Bcl2, Birc2, Ciapin1, and Xiap in 32D-BA cells (D) and miR-142a-overexpressed (OE142a) 32D-BA cells (E). (F) Western blot detection of anti-apoptotic proteins Bcl2, Birc2, Ciapin1, and Xiap in 32D-BA cells infected with lentivirus encoding miR-142a. (G) Identification of miR-142a target gene by luciferase reporter assay. (H) Apoptosis analysis of 32D-BA cells at 24h and 48h after Ciapin1 knockdown. (I) Determination of Ciapin1 expression by qRT-PCR and western blot analysis. (J) Comparative analysis of apoptosis between 32D-BA-puro+OE142a and 32D-BA-Ciapin1+OE142a cells via Annexin V staining. (K) Validation of Ciapin1 expression by qRT-PCR and Western blot assay. (L) Validation of miR-142a-5p and miR-142a-3p expression in 32D-BA-puro and 32D-BA-Ciapin1 cells by qRT-PCR. Results are mean ± SEM, *p < 0.05, **p < 0.01, ***p < 0.001 and N/S (not significant) (B–E, G–L) was assessed by unpaired t-test.
In line with our prediction of anti-correlated miRNA-target gene pairs, the mRNA level of anti-apoptotic genes Bcl2, Birc2, Ciapin1, and Xiap were observed to be upregulated in 32D-BA cells but downregulated by miR-142a overexpression (Figures 5D, E). Of note, the protein level of Ciapin1 was also found to be downregulated by miR-142a overexpression (Figure 5F). Furthermore, luciferase reporter assays revealed that Ciapin1 might be the direct target of miR-142a (Figure 5G). To verify the involvement of Ciapin1 in the miR-142a-mediated cellular effect, we treated 32D-BA cells with shRNA-Ciapin1 and detected the cell apoptosis ratio. As expected, a significant increase in apoptosis was observed (Figures 5H, I). Next, we co-overexpressed Ciapin1 and miR-142a in 32D-BA cells, finding that Ciapin1 could partially rescue the impaired cell survival ability induced by miR-142a (Figure 5J). The overexpression levels of Ciapin1 and miR-142a were shown in Figures 5K, L. Taken together, these findings suggest that the downregulation of miR-142a contributes to the enhanced anti-apoptotic ability of BA-harboring cells by relieving its repression on Ciapin1.
Phosphorylated ERK Inhibits the Expression of miR-142a in BA-Harboring Cells
Previous studies have shown that Pu.1 was critical for miR-142 gene expression in murine dendritic cells, and PU.1 expression was severely impaired in CML patients (40, 41). It’s of interest whether Pu.1 was critical for miR-142 gene expression in BA-harboring cells. To verify this point, we detected the expression level of Pu.1 in 32D-BA cells, showing only a slight decrease of Pu.1 mRNA level as compared with that in 32D-mCherry cells (Figure S4A). Subsequently, 32D-BA cells were transducted with flag-Pu.1-expressing retroviral supernatants (Figure S4B). Pu.1 overexpression induced a slight increase in miR-142a-5p expression but no significant difference in miR-142a-3p expression (Figure S4C). These data indicated that Pu.1 might not be the critical reason for miR-142a downregulation in BA-harboring cells.
Another study has reported the bimodal regulation of miR-142a by the ERK pathway in mice embryonic stem cells (42). Does ERK play a pivotal role in the downregulation of miR-142a in BA-harboring cells? To address this point, we checked the expression level of ERK in 32D-BA cells. Highly phosphorylated ERK was showed in 32D-BA cells when comparing with 32D-mCherry cells (Figure 6A). Next, we treated 32D-BA cells with an ERK inhibitor (SCH772984) for 24 hours, a significant decrease of ERK phosphorylation was observed as compared with DMSO treated group (Figure 6B). Meanwhile, both miR-142a-5p and miR-142a-3p were upregulated with the treatment of different dosages (1μM, 2μM, and 5μM) (Figure 6C). Moreover, the mRNA level of Ciapin1, as well as the protein level, was repressed in ERK inhibitor-treated 32D-BA cells (Figure 6D), along with the significant increase of apoptosis rate (Figure 6E). Lastly, we treated stable 32D-BA-Ciapin1 cells with 5μM SCH772984 to examine whether Ciapin1 overexpression can rescue the impaired cell survival ability induced by ERK inhibitor in 32D-BA cells. Indeed, a significant decrease in apoptosis was observed at 72h after treatment (Figure 6F). These results supported that miR-142a was partially downregulated by BA-triggered ERK phosphorylation and then promoted leukemia cell survival through relieving its repression on Ciapin1.
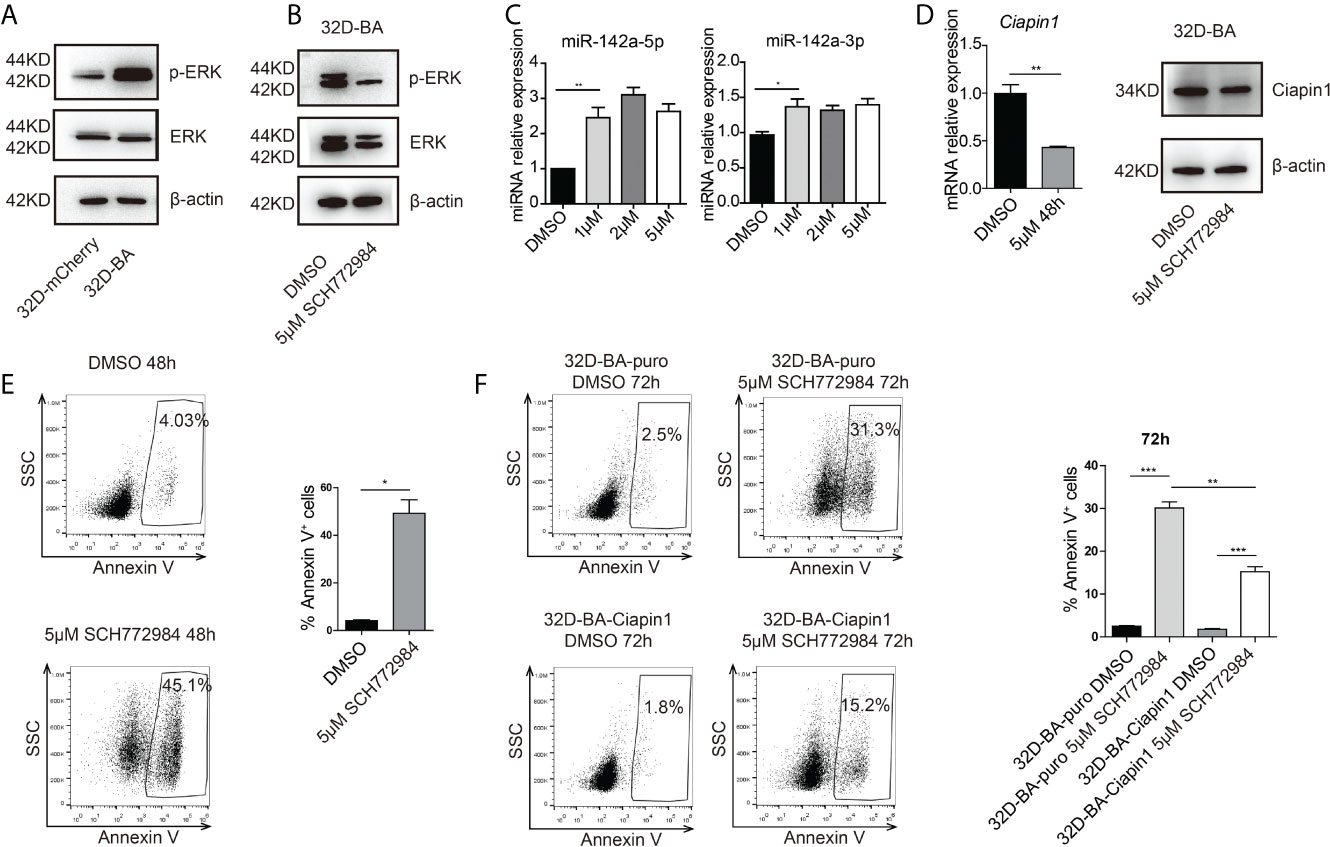
Figure 6 Phosphorylated (P) ERK inhibited the expression of miR-142a in BA-harboring cells. (A) Western blot detection of total and p-ERK protein level in 32D-BA and 32D-mCherry cells. (B) The ERK inhibitor SCH772984 inhibited the p-ERK level in 32D-BA cell line. (C) qRT-PCR detection of miR-142a-5p and miR-142a-3p level in 32D-BA cells with ERK inhibitor treatment. (D) qRT-PCR and Western blot analysis of Ciapin1 expression in 32D-BA cells with ERK inhibitor treatment. (E) Apoptosis analysis of 32D-BA cells with ERK inhibitor treatment. (F) Apoptosis analysis of 32D-BA-Ciapin1 cells with ERK inhibitor treatment. Results are mean ± SEM, *p < 0.05, **p < 0.01, ***p < 0.001 (C–F) was assessed by unpaired t-test.
miR-142a May Act As a Suppressor in CML Progression
Next, we assessed the effect of miR-142a overexpression on CML progression in vivo. BM cells from 5-fluorouracil treated healthy Balb/c mice were collected and co-infected with the pCDH-EF1-miR142a-T2A-copGFP and pMSCV-BA-IRES-mCherry viral supernatant, then transplanted into lethally irradiated wild-type Balb/c recipients (Figure 7A). The pCDH-EF1-T2A-copGFP and pMSCV-IRES-mCherry virus were used as empty controls. We then closely monitored the WBC count and the percentage of mCherry+ (BA positive) cells in PB. As our results showed, miR-142a overexpression reduced the WBC count (Figure 7B) as well as the ratio of BA positive cells in recipients (Figure 7C). Though Mice in both BA+OE142a group and BA+control group showed a CML-like phenotype (Figure 7D), overexpression of miR-142a significantly prolonged the survival of mice (Figure 7E). These results suggest that miR-142a may act as a suppressor in CML progression.
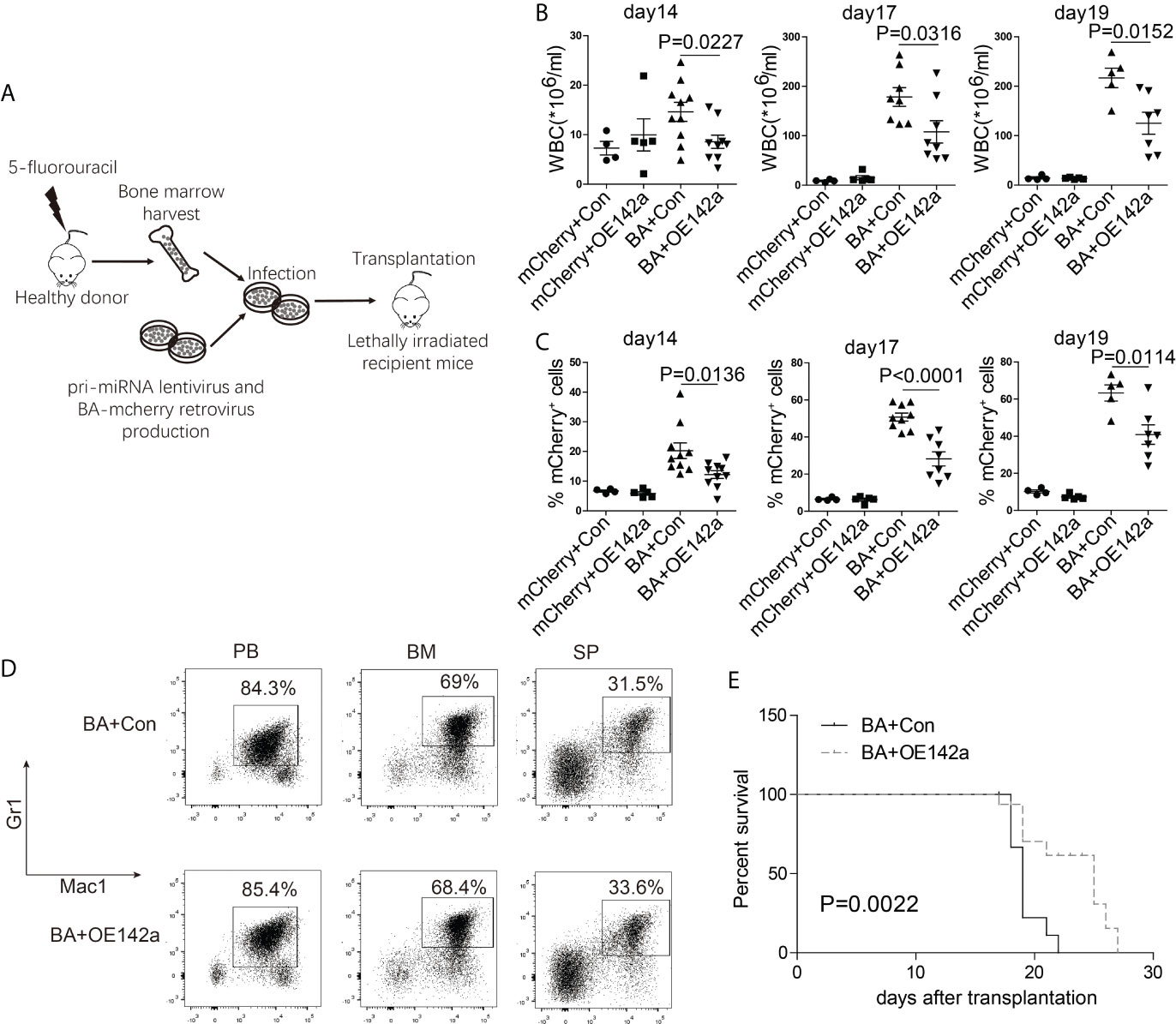
Figure 7 miR-142a may act as a suppressor in CML progression. (A) Schematic of miR-142a treatment on BA-induced CML mice. (B) Follow-up analysis of the PB white blood cell (WBC) count after transplantation. (C) Follow-up analysis of the proportion of mCherry+ cells in PB. (D) Immunophenotype analysis of mice in BA and BA+miR-142a group. (E) Kaplan-Meier survival curve of mice inoculated with miR-142a overexpression or control lentivirus. Results are mean ± SEM, p value was assessed by unpaired t-test.
Discussion
In this study, we parallelly analyzed BA-induced miRNA and gene alterations to systematically study the roles of miRNAs in the pathogenesis of CML by using a hematopoietic stem/progenitor cell-specific tet-off inducible transgenic murine model, Scl/tTA-BA, in which doxycycline withdrawal can induce BA expression in HSPCs, leading to a CML-like phenotype. This model provides an incomparable advantage for studying CML pathogenesis.
On the basis of the target gene prediction and integrated analysis of dysregulated miRNA and mRNA, we built several miRNA-mRNA interaction networks, including the apoptosis process, cell cycle, myeloid differentiation, and DNA damage. Interestingly, a group of differentially displayed genes in CML LSCs (CD34+/CD38-) and CML progenitors (CD34+/CD38+) generated from CML patients also appeared to be involved in these four processes (43). As shown in the result, miR-130a-3p, miR-142a-5p, miR-142a-3p, miR-34a-5p, miR-181-5p family, and let-7-5p family were weaved into the apoptosis network. Subsequent experiments further confirmed that both early and late apoptosis of BM LSKs in BA-induced mice were decreased compared with the control mice (Figure 4C). In line with this, miR-181a has been reported to induce apoptosis in CML (44), and miR-34a was found to be associated with apoptosis regulation in solid tumours (45, 46).
According to the information uncovered by integrated analysis of our miRNA and mRNA-seq data, miR-142a was negatively correlated with an apoptosis inhibitor, Ciapin1, which much interested us. miR-142 is specifically expressed in hematopoietic cells and dynamically regulated during hematopoiesis commitment (47); the expressions of miR-142-3p and miR-142-5p have been known to be positively correlated with the degree of cell differentiation (48, 49). Usually, miR-142 dysregulation is accompanied by hematopoiesis development disorder and leukemia genesis. It is thought to be an oncogene for its capability to accelerate proliferation and induce glucocorticoid resistance in human T-cell acute lymphoblastic leukemia (10). Enforced expression of miR-142-3p will promote myeloid differentiation of HSPCs from AML patients (11). The upregulation of miR-142-3p is positively correlated with drug sensitivity in AML (50). However, the expression level of miR-142 in CML is inconclusive. Letizia Venturini’s data showed that in several cases, miR-142-3p was downregulated, while miR-142-5p was upregulated in CD34+ cells (11). miR-142-3p was also observed to be upregulated in imatinib-treated K562 (17). However, in other data (GSE28825), both miR-142-5p and miR-142-3p showed a downward trend after imatinib treatment on K562 (51). Our data showed that both miR-142a-5p and miR-142a-3p were downregulated after doxycycline withdrawal in BM LSKs of BA-induced CML mice, which may partially take responsibility for the pro-survival effect on BA-harboring cells. Indeed, we observed that enforced overexpression of miR-142a could induce apoptosis of 32D-BA cells and prolong the survival of BA-induced CML mice. Our results suggested that miR-142a might act as a suppressor in CML progression.
miR-142 downregulation has been found in multiple diseases with diverse mechanisms. In mouse dendritic cells, Pu.1, Runx1, and C/EBPβ constitutively occupy miR-142 promoter region (40). The expression level of PU.1 in chronic phase CML patients was downregulated, and it could be rescued by treatment with interferon-alpha or imatinib (41). On the basis of this, we were interested in whether Pu.1 was critical for miR-142a expression in BA-expressing cells. However, our result showed that enforced overexpression of Pu.1 in 32D-BA cells couldn’t rescue the abnormal miR-142a expression induced by BA.
Another study reported that inhibiting the ERK pathway can upregulate miR-142a expression in mice embryonic stem cells (42). Treatment with MAPK inhibitor compounds could block the downregulation of both miR-142-5p and miR-142-3p in cardiac myocytes (52). Consistent with this, we identified that both miR-142a-5p and miR-142a-3p were upregulated in 32D-BA cells following the treatment with ERK inhibitor (Figure 6C). Along with this, cell apoptosis was also increased. These data indicated that miR-142a expression was partially inhibited by highly phosphorylated and activated ERK in BA-harboring cells.
Epigenetic modifications are also involved in the regulation of miR-142 expression. In human nasopharyngeal carcinoma, EZH2 recruits DNA methyltransferase DNMT1 and induces hypermethylation in the promoter region of miR-142, thereby inhibiting its expression (53). In Systemic lupus erythematosus CD4+ T cells, the miR-142 expression is downregulated by BCL-6 through modulating histone methylation and acetylation on the promoter of miR-142 (54). But whether these regulation mechanisms exist in BA-harboring cells is unclear, which may require further investigation.
Overall, this study is the first to systematically identify the miRNA-mRNA regulation network of LSKs in an inducible CML model. Our results showed that the ERK-miR-142a-Ciapin1 axis partially prevented BA-harboring cells from apoptosis. However, whether the integrated regulatory axis exists in CML patient samples, especially in the CD34+ subpopulation, remains unknown. It will be an important point for us in the next step.
Conclusion
Collectively, this study explored the miRNA-mRNA regulatory networks of BM LSKs in an inducible CML murine model and suggested that the ERK-miR-142a-Ciapin1 axis might play an essential role in enhancing the anti-apoptotic ability of CML cells.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding author.
Ethics Statement
The animal study was reviewed and approved by The Animal Care Committee of Shanghai Jiao Tong University School of Medicine (No. B-2017-010).
Author Contributions
ZC and YX equally contributed to the design of the study, the performance of the experiments. DL, PL, FL, ZZ, MZ, and XW performed the experiments. ZC, YX, YZ, XS, and QH analyzed the data. ZC and QH wrote the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the National Natural Science Foundation of China (81900149), Shanghai Municipal Education Commission-Gaofeng Clinical Medicine Grant Support (20152506), and Samuel Waxman Cancer Research Foundation.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We thank all colleagues in our lab for their help and support. We thank Dr. D. Tenen (Harvard University) for the generous gift of the Scl/Tta, TRE-BCR/ABL, and TRE-H2B/GFP transgenic mice.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2021.718731/full#supplementary-material
References
1. Sawyers CL. Chronic Myeloid Leukemia. N Engl J Med (1999) 340(17):1330–40. doi: 10.1056/NEJM199904293401706
2. Holyoake TL, Vetrie D. The Chronic Myeloid Leukemia Stem Cell: Stemming the Tide of Persistence. Blood (2017) 129(12):1595–606. doi: 10.1182/blood-2016-09-696013
3. Sawyers CL, Hochhaus A, Feldman E, Goldman JM, Miller CB, Ottmann OG, et al. Imatinib Induces Hematologic and Cytogenetic Responses in Patients With Chronic Myelogenous Leukemia in Myeloid Blast Crisis: Results of a Phase II Study. Blood (2002) 99(10):3530–9. doi: 10.1182/blood.V99.10.3530
4. Hochhaus A, Kreil S, Corbin AS, La Rosee P, Muller MC, Lahaye T, et al. Molecular and Chromosomal Mechanisms of Resistance to Imatinib (STI571) Therapy. Leukemia (2002) 16(11):2190–6. doi: 10.1038/sj.leu.2402741
5. Lee RC, Feinbaum RL, Ambros V. The C. Elegans Heterochronic Gene Lin-4 Encodes Small RNAs With Antisense Complementarity to Lin-14. Cell (1993) 75(5):843–54. doi: 10.1016/0092-8674(93)90529-Y
6. Pospisil V, Vargova K, Kokavec J, Rybarova J, Savvulidi F, Jonasova A, et al. Epigenetic Silencing of the Oncogenic miR-17-92 Cluster During PU.1-directed Macrophage Differentiation. EMBO J (2011) 30(21):4450–64. doi: 10.1038/emboj.2011.317
7. Zhao JL, Rao DS, Boldin MP, Taganov KD, O’Connell RM, Baltimore D. NF-Kappab Dysregulation in microRNA-146a-deficient Mice Drives the Development of Myeloid Malignancies. Proc Natl Acad Sci USA (2011) 108(22):9184–9. doi: 10.1073/pnas.1105398108
8. Starczynowski DT, Kuchenbauer F, Wegrzyn J, Rouhi A, Petriv O, Hansen CL, et al. MicroRNA-146a Disrupts Hematopoietic Differentiation and Survival. Exp Hematol (2011) 39(2):167–78.e164. doi: 10.1016/j.exphem.2010.09.011
9. Ooi AG, Sahoo D, Adorno M, Wang Y, Weissman IL, Park CY. MicroRNA-125b Expands Hematopoietic Stem Cells and Enriches for the Lymphoid-Balanced and Lymphoid-Biased Subsets. Proc Natl Acad Sci USA (2010) 107(50):21505–10. doi: 10.1073/pnas.1016218107
10. Lv M, Zhang X, Jia H, Li D, Zhang B, Zhang H, et al. An Oncogenic Role of miR-142-3p in Human T-cell Acute Lymphoblastic Leukemia (T-ALL) by Targeting Glucocorticoid Receptor-Alpha and cAMP/PKA Pathways. Leukemia (2012) 26(4):769–77. doi: 10.1038/leu.2011.273
11. Wang XS, Gong JN, Yu J, Wang F, Zhang XH, Yin XL, et al. MicroRNA-29a and microRNA-142-3p are Regulators of Myeloid Differentiation and Acute Myeloid Leukemia. Blood (2012) 119(21):4992–5004. doi: 10.1182/blood-2011-10-385716
12. Wang LS, Li L, Li L, Chu S, Shiang KD, Li M, et al. MicroRNA-486 Regulates Normal Erythropoiesis and Enhances Growth and Modulates Drug Response in CML Progenitors. Blood (2015) 125(8):1302–13. doi: 10.1182/blood-2014-06-581926
13. Venturini L, Battmer K, Castoldi M, Schultheis B, Hochhaus A, Muckenthaler MU, et al. Expression of the miR-17-92 Polycistron in Chronic Myeloid Leukemia (CML) CD34+ Cells. Blood (2007) 109(10):4399–405. doi: 10.1182/blood-2006-09-045104
14. Agatheeswaran S, Singh S, Biswas S, Biswas G, Chandra Pattnayak N, Chakraborty S. Bcr-ABL Mediated Repression of miR-223 Results in the Activation of MEF2C and PTBP2 in Chronic Myeloid Leukemia. Leukemia (2013) 27(7):1578–80. doi: 10.1038/leu.2012.339
15. Joshi D, Chandrakala S, Korgaonkar S, Ghosh K, Vundinti BR. Down-Regulation of miR-199b Associated With Imatinib Drug Resistance in 9q34.1 Deleted BCR/ABL Positive CML Patients. Gene (2014) 542(2):109–12. doi: 10.1016/j.gene.2014.03.049
16. Scholl V, Hassan R, Zalcberg IR. Mirna-451: A Putative Predictor Marker of Imatinib Therapy Response in Chronic Myeloid Leukemia. Leuk Res (2012) 36(1):119–21. doi: 10.1016/j.leukres.2011.08.023
17. Espadinha AS, Prouzet-Mauleon V, Claverol S, Lagarde V, Bonneu M, Mahon FX, et al. A Tyrosine kinase-STAT5-miR21-PDCD4 Regulatory Axis in Chronic and Acute Myeloid Leukemia Cells. Oncotarget (2017) 8(44):76174–88. doi: 10.18632/oncotarget.19192
18. Koschmieder S, Gottgens B, Zhang P, Iwasaki-Arai J, Akashi K, Kutok JL, et al. Inducible Chronic Phase of Myeloid Leukemia With Expansion of Hematopoietic Stem Cells in a Transgenic Model of BCR-ABL Leukemogenesis. Blood (2005) 105(1):324–34. doi: 10.1182/blood-2003-12-4369
19. Lopez-Romero P. Pre-Processing and Differential Expression Analysis of Agilent microRNA Arrays Using the AgiMicroRna Bioconductor Library. BMC Genomics (2011) 12:64. doi: 10.1186/1471-2164-12-64
20. Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL. TopHat2: Accurate Alignment of Transcriptomes in the Presence of Insertions, Deletions and Gene Fusions. Genome Biol (2013) 14(4):R36. doi: 10.1186/gb-2013-14-4-r36
21. Robinson MD, McCarthy DJ, Smyth GK. edgeR: A Bioconductor Package for Differential Expression Analysis of Digital Gene Expression Data. Bioinformatics (2010) 26(1):139–40. doi: 10.1093/bioinformatics/btp616
22. Huang da W, Sherman BT, Lempicki RA. Systematic and Integrative Analysis of Large Gene Lists Using DAVID Bioinformatics Resources. Nat Protoc (2009) 4(1):44–57. doi: 10.1038/nprot.2008.211
23. Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. Gene Set Enrichment Analysis: A Knowledge-Based Approach for Interpreting Genome-Wide Expression Profiles. Proc Natl Acad Sci USA (2005) 102(43):15545–50. doi: 10.1073/pnas.0506580102
24. Chambers SM, Boles NC, Lin KY, Tierney MP, Bowman TV, Bradfute SB, et al. Hematopoietic Fingerprints: An Expression Database of Stem Cells and Their Progeny. Cell Stem Cell (2007) 1(5):578–91. doi: 10.1016/j.stem.2007.10.003
25. Dweep H, Gretz N. miRWalk2.0: A Comprehensive Atlas of microRNA-target Interactions. Nat Methods (2015) 12(8):697. doi: 10.1038/nmeth.3485
26. Wilson A, Laurenti E, Oser G, van der Wath RC, Blanco-Bose W, Jaworski M, et al. Hematopoietic Stem Cells Reversibly Switch From Dormancy to Self-Renewal During Homeostasis and Repair. Cell (2008) 135(6):1118–29. doi: 10.1016/j.cell.2008.10.048
27. Miyamoto K, Seki N, Matsushita R, Yonemori M, Yoshino H, Nakagawa M, et al. Tumour-Suppressive miRNA-26a-5p and miR-26b-5p Inhibit Cell Aggressiveness by Regulating PLOD2 in Bladder Cancer. Br J Cancer (2016) 115(3):354–63. doi: 10.1038/bjc.2016.179
28. Han X, Zhen S, Ye Z, Lu J, Wang L, Li P, et al. A Feedback Loop Between miR-30a/c-5p and DNMT1 Mediates Cisplatin Resistance in Ovarian Cancer Cells. Cell Physiol Biochem (2017) 41(3):973–86. doi: 10.1159/000460618
29. Jia L, Xi Q, Wang H, Zhang Z, Liu H, Cheng Y, et al. miR-142-5p Regulates Tumor Cell PD-L1 Expression and Enhances Anti-Tumor Immunity. Biochem Biophys Res Commun (2017) 488(2):425–31. doi: 10.1016/j.bbrc.2017.05.074
30. Wang Z, Liu Z, Fang X, Yang H. Mir-142-5p Suppresses Tumorigenesis by Targeting PIK3CA in Non-Small Cell Lung Cancer. Cell Physiol Biochem (2017) 43(6):2505–15. doi: 10.1159/000484459
31. Shen WW, Zeng Z, Zhu WX, Fu GH. MiR-142-3p Functions as a Tumor Suppressor by Targeting CD133, ABCG2, and Lgr5 in Colon Cancer Cells. J Mol Med (Berl) (2013) 91(8):989–1000. doi: 10.1007/s00109-013-1037-x
32. Du C, Lv Z, Cao L, Ding C, Gyabaah OA, Xie H, et al. MiR-126-3p Suppresses Tumor Metastasis and Angiogenesis of Hepatocellular Carcinoma by Targeting LRP6 and PIK3R2. J Transl Med (2014) 12:259. doi: 10.1186/s12967-014-0259-1
33. Hu B, Zhang H, Wang Z, Zhang F, Wei H, Li L. Lncrna CCAT1/miR-130a-3p Axis Increases Cisplatin Resistance in Non-Small-Cell Lung Cancer Cell Line by Targeting SOX4. Cancer Biol Ther (2017) 18(12):974–83. doi: 10.1080/15384047.2017.1385679
34. Gao J, Li N, Dong Y, Li S, Xu L, Li X, et al. miR-34a-5p Suppresses Colorectal Cancer Metastasis and Predicts Recurrence in Patients With Stage II/III Colorectal Cancer. Oncogene (2015) 34(31):4142–52. doi: 10.1038/onc.2014.348
35. Yu Y, Yang L, Zhao M, Zhu S, Kang R, Vernon P, et al. Targeting microRNA-30a-mediated Autophagy Enhances Imatinib Activity Against Human Chronic Myeloid Leukemia Cells. Leukemia (2012) 26(8):1752–60. doi: 10.1038/leu.2012.65
36. Mosakhani N, Mustjoki S, Knuutila S. Down-Regulation of miR-181c in Imatinib-Resistant Chronic Myeloid Leukemia. Mol Cytogenet (2013) 6(1):27. doi: 10.1186/1755-8166-6-27
37. Li Q, Wu Y, Zhang Y, Sun H, Lu Z, Du K, et al. miR-125b Regulates Cell Progression in Chronic Myeloid Leukemia Via Targeting BAK1. Am J Transl Res (2016) 8(2):447–59.
38. Zhu X, Zhao H, Lin Z, Zhang G. Functional Studies of miR-130a on the Inhibitory Pathways of Apoptosis in Patients With Chronic Myeloid Leukemia. Cancer Gene Ther (2015) 22(12):573–80. doi: 10.1038/cgt.2015.50
39. Steelman LS, Pohnert SC, Shelton JG, Franklin RA, Bertrand FE, McCubrey JA. Jak/Stat, Raf/MEK/ERK, PI3K/Akt and BCR-ABL in Cell Cycle Progression and Leukemogenesis. Leukemia (2004) 18(2):189–218. doi: 10.1038/sj.leu.2403241
40. Sun Y, Sun J, Tomomi T, Nieves E, Mathewson N, Tamaki H, et al. Pu.1-dependent Transcriptional Regulation of miR-142 Contributes to its Hematopoietic Cell-Specific Expression and Modulation of IL-6. J Immunol (2013) 190(8):4005–13. doi: 10.4049/jimmunol.1202911
41. Albajar M, Gutierrez P, Richard C, Rosa-Garrido M, Gomez-Casares MT, Steegmann JL, et al. PU.1 Expression is Restored Upon Treatment of Chronic Myeloid Leukemia Patients. Cancer Lett (2008) 270(2):328–36. doi: 10.1016/j.canlet.2008.05.024
42. Sladitschek HL, Neveu PA. The Bimodally Expressed microRNA miR-142 Gates Exit From Pluripotency. Mol Syst Biol (2015) 11(12):850. doi: 10.15252/msb.20156525
43. Herrmann H, Sadovnik I, Cerny-Reiterer S, Rulicke T, Stefanzl G, Willmann M, et al. Dipeptidylpeptidase IV (CD26) Defines Leukemic Stem Cells (LSC) in Chronic Myeloid Leukemia. Blood (2014) 123(25):3951–62. doi: 10.1182/blood-2013-10-536078
44. Fei J, Li Y, Zhu X, Luo X. miR-181a Post-Transcriptionally Downregulates Oncogenic RalA and Contributes to Growth Inhibition and Apoptosis in Chronic Myelogenous Leukemia (CML). PloS One (2012) 7(3):e32834. doi: 10.1371/journal.pone.0032834
45. Welch C, Chen Y, Stallings RL. MicroRNA-34a Functions as a Potential Tumor Suppressor by Inducing Apoptosis in Neuroblastoma Cells. Oncogene (2007) 26(34):5017–22. doi: 10.1038/sj.onc.1210293
46. Liu C, Kelnar K, Liu B, Chen X, Calhoun-Davis T, Li H, et al. The microRNA miR-34a Inhibits Prostate Cancer Stem Cells and Metastasis by Directly Repressing CD44. Nat Med (2011) 17(2):211–5. doi: 10.1038/nm.2284
47. Chen CZ, Li L, Lodish HF, Bartel DP. MicroRNAs Modulate Hematopoietic Lineage Differentiation. Science (2004) 303(5654):83–6. doi: 10.1126/science.1091903
48. O’Connell RM, Chaudhuri AA, Rao DS, Gibson WS, Balazs AB, Baltimore D. MicroRNAs Enriched in Hematopoietic Stem Cells Differentially Regulate Long-Term Hematopoietic Output. Proc Natl Acad Sci USA (2010) 107(32):14235–40. doi: 10.1073/pnas.1009798107
49. Bissels U, Wild S, Tomiuk S, Hafner M, Scheel H, Mihailovic A, et al. Combined Characterization of microRNA and mRNA Profiles Delineates Early Differentiation Pathways of CD133+ and CD34+ Hematopoietic Stem and Progenitor Cells. Stem Cells (2011) 29(5):847–57. doi: 10.1002/stem.627
50. Zhang Y, Liu Y, Xu X. Upregulation of Mir-142-3p Improves Drug Sensitivity of Acute Myelogenous Leukemia Through Reducing P-Glycoprotein and Repressing Autophagy by Targeting Hmgb1. Transl Oncol (2017) 10(3):410–8. doi: 10.1016/j.tranon.2017.03.003
51. Rokah OH, Granot G, Ovcharenko A, Modai S, Pasmanik-Chor M, Toren A, et al. Downregulation of miR-31, miR-155, and miR-564 in Chronic Myeloid Leukemia Cells. PloS One (2012) 7(4):e35501. doi: 10.1371/journal.pone.0035501
52. Sharma S, Liu J, Wei J, Yuan H, Zhang T, Bishopric NH. Repression of miR-142 by p300 and MAPK is Required for Survival Signalling Via gp130 During Adaptive Hypertrophy. EMBO Mol Med (2012) 4(7):617–32. doi: 10.1002/emmm.201200234
53. Li Y, He Q, Wen X, Hong X, Yang X, Tang X, et al. Ezh2-DNMT1-mediated Epigenetic Silencing of miR-142-3p Promotes Metastasis Through Targeting ZEB2 in Nasopharyngeal Carcinoma. Cell Death Differ (2019) 26(6):1089–106. doi: 10.1038/s41418-018-0208-2
Keywords: chronic myelogenous leukemia, miRNA-mRNA network, apoptosis, miR-142a, Ciapin1
Citation: Chen Z, Xie Y, Liu D, Liu P, Li F, Zhang Z, Zhang M, Wang X, Zhang Y, Sun X and Huang Q (2021) Downregulation of miR-142a Contributes to the Enhanced Anti-Apoptotic Ability of Murine Chronic Myelogenous Leukemia Cells. Front. Oncol. 11:718731. doi: 10.3389/fonc.2021.718731
Received: 01 June 2021; Accepted: 05 July 2021;
Published: 27 July 2021.
Edited by:
Cyrus Khandanpour, University Hospital Münster, GermanyReviewed by:
Qunye Zhang, Shandong University, ChinaYang Liang, Sun Yat-sen University Cancer Center (SYSUCC), China
Copyright © 2021 Chen, Xie, Liu, Liu, Li, Zhang, Zhang, Wang, Zhang, Sun and Huang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Qiuhua Huang, SFFIMTA2MzJAcmpoLmNvbS5jbg==
†These authors have contributed equally to this work and share first authorship