 Sagun Parakh
Sagun Parakh Joseph Nicolazzo
Joseph Nicolazzo Andrew M Scott
Andrew M Scott Hui Kong Gan
Hui Kong Gan- 1Department of Medical Oncology, Austin Hospital, Heidelberg, VIC, Australia
- 2Tumour Targeting Laboratory, Olivia Newton-John Cancer Research Institute, Heidelberg, VIC, Australia
- 3School of Cancer Medicine, La Trobe University, Heidelberg, VIC, Australia
- 4Drug Delivery, Disposition and Dynamics, Monash Institute of Pharmaceutical Sciences, Monash University, Parkville, VIC, Australia
- 5Department of Medicine, University of Melbourne, Heidelberg, VIC, Australia
- 6Department of Molecular Imaging and Therapy, Austin Health, Heidelberg, VIC, Australia
Glioblastoma (GBM) is an aggressive and fatal malignancy that despite decades of trials has limited therapeutic options. Antibody drug conjugates (ADCs) are composed of a monoclonal antibody which specifically recognizes a cellular surface antigen linked to a cytotoxic payload. ADCs have demonstrated superior efficacy and/or reduced toxicity in a range of haematological and solid tumors resulting in nine ADCs receiving regulatory approval. ADCs have also been explored in patients with brain tumours but with limited success to date. While earlier generations ADCs in glioma patients have had limited success and high toxicity, newer and improved ADCs characterised by low immunogenicity and more effective payloads have shown promise in a range of tumour types. These newer ADCs have also been tested in glioma patients, however, with mixed results. Factors affecting the effectiveness of ADCs to target the CNS include the blood brain barrier which acts as a physical and biochemical barrier, the pro-cancerogenic and immunosuppressive tumor microenvironment and tumour characteristics like tumour volume and antigen expression. In this paper we review the data regarding the ongoing the development of ADCs in glioma patients as well as potential strategies to overcome these barriers to maximise their therapeutic potential.
Introduction
Glioblastoma (GBM) is an aggressive fatal disease characterised by complex molecular heterogeneity and aggressive infiltrative growth. Despite s decades of trials testing novel agents, the median survival remains unchanged at 14 - 17 months only (1–4). Multiple strategies have been explored with limited success to improve the efficacy of chemotherapy in GBM, including novel formulations, direct administration into the central nervous system (CNS) and targeted vascular disruption; unfortunately, these have often resulted in higher toxicity rates without significantly improving patient outcomes (5–7).
Antibody drug conjugates (ADCs) are a new but proven class of highly potent therapeutics, composed of a monoclonal antibody which specifically recognizes a cellular surface antigen linked to a cytotoxic payload (8). This results in a number of advantages: reduced toxicity due to more targeted delivery of cytotoxic therapy directly into the tumours; enhanced cell kill from the ability of use more toxic drugs that cannot be safely administered systemically; and the additive/synergistic benefit of combined tumour kill from the antibody and the payload respectively (9, 10). The ultimate efficacy of ADCs though relies on the complex interplay between three vital components: antibody, linker and payload. Early failures in the development of ADCs were due in part to challenges associated with these components, however recent advances have resulted in notable successes, resulting in nine ADCs receiving regulatory approval by the Food and Drug Administration in the USA and four ADCs by the European Medicines Agency (8, 11).
ADCs have also been explored for patients with brain tumours but with limited success to date. In particular, the apparent failure of two recent high-profile ADCs has resulted in a lessening of interest to this approach in glioma patients currently (12, 13). In this article, we will review the development of ADCs in glioma patients and summarise the data supporting their on-going development. We will discuss potential strategies to maximise their therapeutic potential by increasing their penetration through the blood-brain barrier (BBB), selection of more biologically relevant targets in the brain and its microenvironment, novel methods of drug targeting, newer payloads and better patient selection.
Early ADCs in Glioma Therapy
The first generation of ADCs tested in glioma patients comprised mainly immunotoxins and radioimmunotherapy (Table 1). Immunotoxins are antibodies conjugated to naturally occurring bacterial toxins, such as Pseudomonas aeruginosa exotoxin A and diphtheria toxin. Radioimmunoconjugates utilise isotopes such as iodine-125 or iodine-131 as payloads. These commonly targeted the EGFR axis (either the receptor itself or its mutants and ligands) due the relatively high prevalence of these targets in gliomas and their likely role as an oncogenic pathway in glioma. Targeting the EGFRvIII mutation was particularly attractive. This is comprised of an in-frame deletion of exons 2-7 that results in a truncated by constitutively active receptor (24). Furthermore, the EGFRvIII mutation is relatively frequent (in 20-40% of GBM tumours) but shows a tumour restricted expression pattern compared to wildtype EGFR (24). However, other targets of these early ADCs included IL-13Rα2 receptor, IL4 and transferrin. Unfortunately, these early ADCs were found to be ineffective due to a number of problems including high immunogenicity, unstable linkers, inefficient deliver due via early convection delivery systems, biomarker limitations to address tumour heterogeneity and toxicity (25–27).
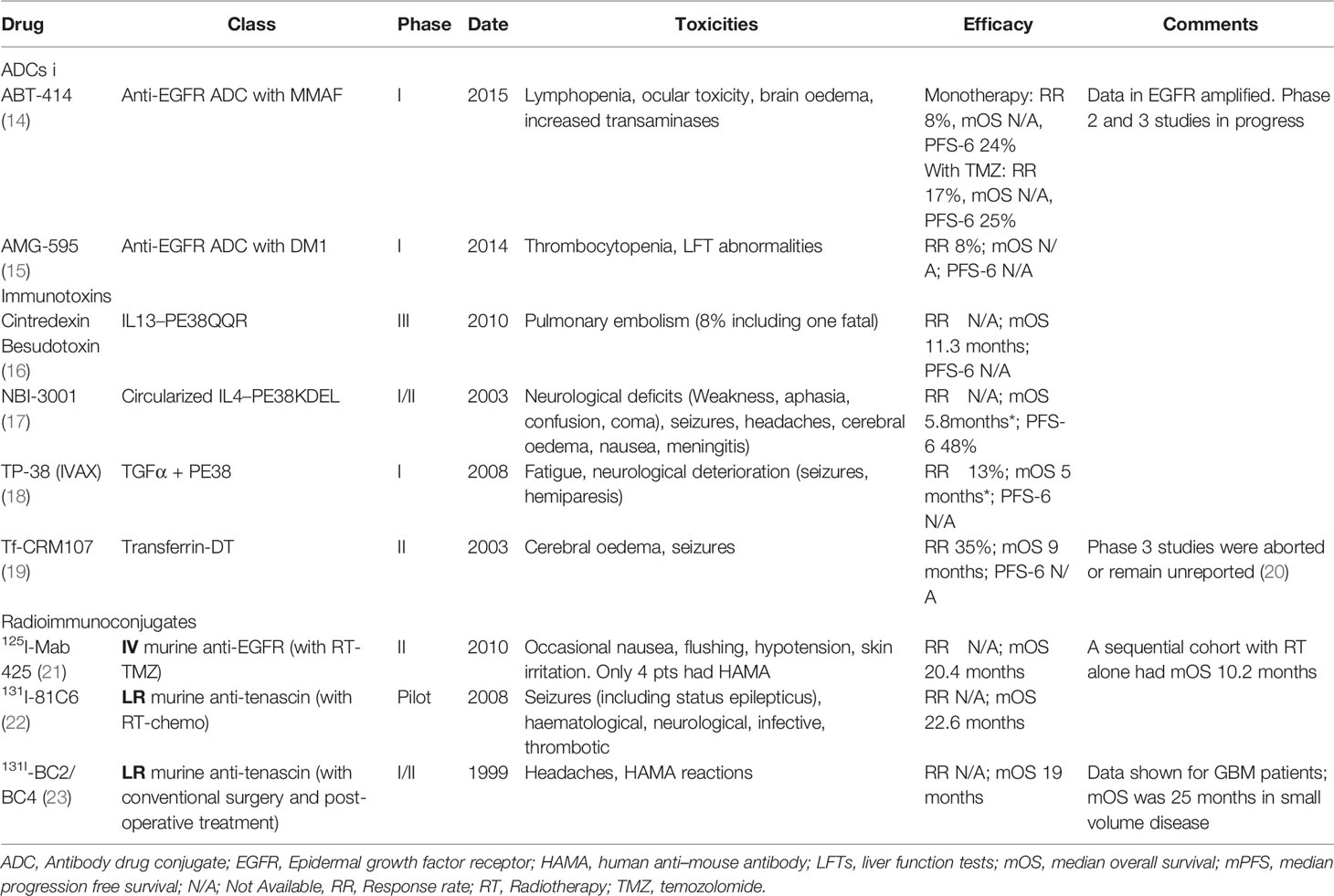
Table 1 Selected ADCs, immunotoxins and radioimmunoconjugates in high grade gliomas.
Newer ADCs in Non-Glioma Therapy
Subsequently, improved ADCs were generated which were characterised by low immunogenicity (usually with chimeric, humanised or fully human antibodies) and more effective payloads (Table 2). The success of these newer generation ADCs has been shown in haematological as well as in triple negative breast cancer (TNBC). These include brentuximab vedotin, an anti-CD30, antibody conjugated with monomethyl auristatin E (MMAE), an auristatin payload which disrupts microtubules. This has been shown to improve patient outcomes as consolidation after autologous stem cell transplant in patients with Hodgkin’s lymphoma (37), and the subsequently in combination therapy with chemotherapy in newly diagnosed patients (38). It has also been shown to be effective in patients with CD30-positive T-cell lymphoma (39, 40) and anaplastic large cell lymphoma (41). Trastuzumab emtansine, which carries also carries a microtubule targeting payload, DM1, has shown efficacy in patients with HER2-positive breast cancer and is the first ADC to be approved in solid tumors (42, 43). Other examples of successful ADCs utilising DNA-damaging payloads include inotuzumab ozogamicin with a caliceamicin payload in CD-22 positive ALL (44) and gemtuzumab ozogamicin with a caliceamicin payload in AML (45). Another highly promising class of payloads are is those targeting topoisomerase, such as Sacituzumab govitecan (SG) against Trop 2 and bearing the SN38 payload. SG has shown significant activity in TNBC with improvements in PFS and OS compared to chemotherapy alone (46). In addition, SG has demonstrated activity in intracranial xenograft models and demonstrated activity in patients with recurrent GBM in a single centre pilot study (47).
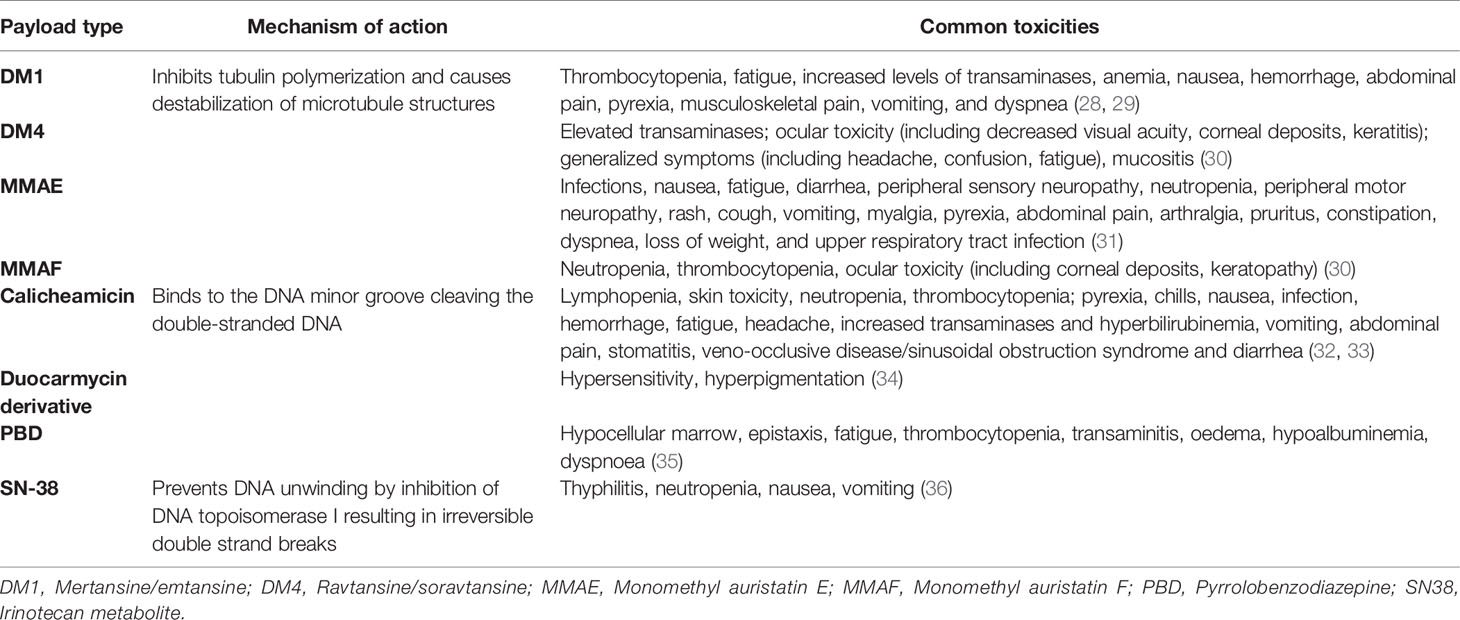
Table 2 Common toxicities associated with antibody drug conjugates.
Newer ADCs in Glioma Therapy
In addition to their use in extra-cranial malignancies as described above, these newer ADCs have also been tested in glioma patients with mixed results. As before, targeting EGFR remained highly attractive due to the high frequency of abnormalities in this pathway in high grade gliomas. Furthermore, several highly specific and novel antibodies against EGFR and EGFRvIII had been developed which promised more selective targeting. The monoclonal antibody 806 (mAb806) is a murine anti-EGFR antibody that selectively targets a cryptic epitope of the EGFR which is only exposed under certain conditions, including where wild-type EGFR is highly over-expressed, where there are autocrine loops and/or harbor there are specific mutations which expose the epitope e.g. the EGFRvIII deletion variant. As these conditions are essential tumour restricted, mAb806 and derivative constructs are also tumour restricted with no normal tissue binding. This in this way, there avoid the toxicity typically associated with other systemic EGFR drugs inhibitors (48, 49). ABT-806, the humanized form of mAb806, has shown to be well tolerated and devoid of conventional anti-EGFR toxicities like rash and diarrhoea. Furthermore, biodistribution studies of 111In-ABT-806 showed no normal tissue uptake highlighting the tumor-specific nature of mAb806 (49–52). Depatuxizumab mafodotin (Depatux-M) is an EGFR targeting ADC comprising of mAb806 linked to the anti-microtubule toxin monomethyl auristatin F (MMAF). Depatux-M has shown promising in-vivo activity in tumor models overexpressing wild type EGFR, EGFR amplification, or EGFRvIII mutation (53). Depatuxizumab mafodotin was also found to improve anti-tumour efficacy when combined with radiotherapy and temozolomide in preclinical models (53). The combination was also subsequently confirmed to be safe when tested in a Phase 1 study with newly diagnosed GBM with patients (54), and hence proceed to Phase 3 testing in the INTELLANCE I trial. Unfortunately, the addition of Depatux-M to standard chemo-irradiation with TMZ in newly diagnosed EGFR amplified glioblastoma patients was eventually discontinued for futility (12).
In contrast to the negative results in newly diagnosed patients, anti-EGFR ADCs targeting glioma with EGFR over-expression or EGFRvIII showed clear signals of efficacy in patients with relapsed glioma after chemo-radiation. Depatux-M was evaluated in the randomised phase II INTELLANCE 2 study in patients with EGFR amplified recurrent GBM (55, 56). In this study, the combination of Depatux-M with temozolomide (TMZ) demonstrated a strong trend towards substantial benefit in overall survival compared to the chemotherapy arm (HR 0.71, p=0.062) (57). The benefit of Depatux-M was highest in patients relapsing more than 16 weeks after the start of the last TMZ cycle. No evidence of efficacy in the monotherapy arm was observed in the subgroup with the MGMT promoter unmethylated tumors. These results are given added weight by the results of a Phase I/II study with AMG 595, an ADC comprising a fully human, anti-EGFRvIII monoclonal antibody linked via a non-cleavable linker to the maytansinoid DM1. AMG 595 has shown promising preclinical activity in assays including orthotopic murine models (58). In a phase I/II study of AMG 595 in patients with recurrent glioma expressing EGFRvIII (NCT01475006), the most common adverse events were thrombocytopenia (50%) and fatigue (25%); grade ≥ 3 treatment-related AEs occurred in 17 patients (53%). However, it is important to note that two patients had partial responses; 15 (47%) had stable disease, including one patient who was on treatment for 15 months (59). Unfortunately, development of this drug has also been discontinued.
Future Directions for the Development of ADCs in Glioma
The disappointing results of INTELLANCE 1 has rightly given pause and reconsideration to the role of ADCs in patients with gliomas. It has prompted reconsideration of reason why ADCs may not be suitable for use in patients with gliomas, including the relatively high toxicity when targeting the EGFR family with certain payloads, and the concern that these drugs are unable to penetrate the blood brain barrier to reach glioma tumour cells. One key concern is whether the results of INTELLANCE 1 should be allowed to overshadow the results of INTELLANCE 2 and the AMG-595 study. Much data suggest that recurrent gliomas are different disease from newly diagnosed GBM with changes in its genetic and molecular phenotype (60–66). While the further development of Depatux-M has been terminated by the company, the results of the INTELLANCE 2 study are intriguing about the possible use of this class of ADCs based on the mAb806 antibody particularly when compared to other drugs tested in GBM, such as immunotherapy, which have been universally disappointing in their lack of efficacy (Table 3). Formal testing in a phase III would be reasonable but understandably, improved ADCs with a better toxicity profile would be selected if possible. Also, better patient selection is clearly required to identify the subset of patients who clearly have exceptional sensitivity of these ADCs as has been seen with in trials with Depatux-M to date (unpublished data). In a preclinical study, disruption of BBB through the over-expression of vascular endothelial growth factor or avoiding the BBB entirely by direct intra-tumoral injection resulted in improved efficacy of Deptux-M (77). In addition, suppression of EGFR or expression of an EGFR variant lacking the binding epitope and upregulation of compensatory signaling pathways associated with altered EGFR expression and known to function in parallel or downstream from EGFR were identified as potential mechanisms of resistance to Depatux-M.
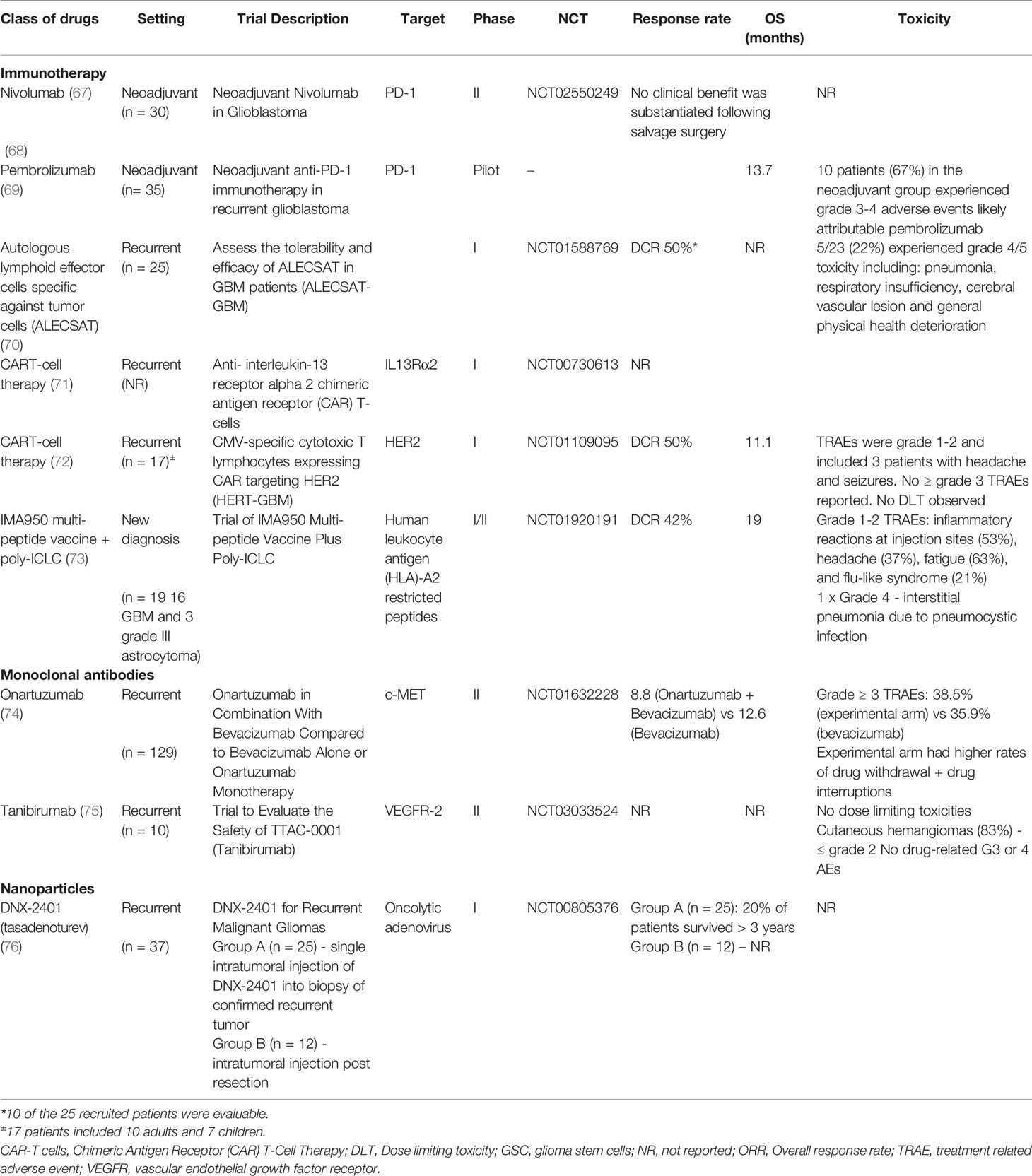
Table 3 Selected trials of therapeutic agents tested in glioma.
Improved Drug Delivery Through BBB
One of the main reasons for the ineffectiveness of therapeutic agents intended to target the CNS following peripheral administration is the restrictive nature of the BBB. The BBB is formed by endothelial cells, connected by tight junctions, which continuously interact with surrounding cells like astrocytes, pericytes, and perivascular macrophages, forming the so-called neurovascular unit (78). Primary brain tumors, in particular glioblastoma, cause disruption in the integrity of the BBB as evidenced by the accumulation of gadolinium-based magnetic resonance imaging (MRI) contrast agents within tumor regions. However this disruption is heterogeneous and there is also a clinically significant portion of the tumor with an intact BBB which affects the distribution and efficacy of drugs exposed to this region of the tumor (79). Disrupting the BBB more completely would clearly be useful for treating gliomas with ADCs, amongst other drugs. There has also been recent interest in chemical-induced BBB disruption which has led to increased CNS exposure of ADCs. For example, NEO110, which is a high purity version of the natural monoterpene perillyl alcohol, has been shown to increase the brain delivery of T-DM1 in a mouse model harbouring intracranial HER2+ breast cancer, leading to a significantly greater survival (80). The clinical translation of BBB-disruptors such as NEO110 requires evaluation before such an approach may be considered appropriate for enhancing ADC penetration into the human brain.
In addition to being a physical barrier, the BBB also acts as a biochemical barrier through the function of efflux transporters, such as P-glycoprotein (P-gp) (81) and breast cancer resistance protein (BCRP) (82). While these efflux transporters protect the brain from potentially harmful xenobiotics, they recognise many therapeutics, including a large number of anti-cancer drugs, therefore, limiting their access to the CNS (83). In addition, elimination of the cytotoxic payloads from the cellular cytoplasm by the ATP-binding cassette (ABC) transporters contribute to lower efficacy and resistance to ADCs (26). Increased MDR1 expression has shown to contribute to resistance to auristatin and maytansinoids based ADC analogues, leading to poorer patient outcomes (84, 85). Strategies to overcome drug efflux from cells include using agents that are poor efflux substrates such as hydrophilic compounds or switching from a non-cleavable linker to a protease cleavable and using newer design drugs such as bispecific and biparatopic antibodies can increase cellular internalization (26). Another mechanism that affects the therapeutic effectiveness of ADCs involves defects in the internalization pathway and reduced cell surface trafficking (86). Following internalisation, degradation of ADCs in lysosomes may be impaired by reduced lysosomal proteolytic or acidification function and/or loss of lysosomal transporter expression, resulting in failure of cleavage of cytotoxic payload from ADCs (87). Loss of lysosomal transporter expression, e.g., SLC46A3 has also been reported as a mechanism of innate and acquired resistance to PBD and DM1 bearing ADCs (88). Other potential mechanisms of escape include selection pressure and downregulation of antigens, loss of antigen expression or mutations in antigen as well as presence of ligands for antigens and resistance to ADC and acquired or innate insensitivity to the payload.
With increasing insight into the biology of the BBB and the discovery of novel transporters for trafficking of endogenous compounds, there has been significant interest in attaching natural ligands of these transporter systems to chemotherapeutics to increase their CNS access. One such ligand is angiopep-2, a 19 amino acid peptide targeting the low-density lipoprotein receptor-related protein 1 (LRP1). This has been conjugated to paclitaxel, amongst other anticancer agents. This construct of angiopep-2 and paclitaxel (GRN1005) was shown to be safe and somewhat effective in patients with advanced solid tumours (89), and a subsequent Phase I study showed that GRN1005 had similar toxicity to paclitaxel and some activity in recurrent glioma (90). These techniques were then applied to antibodies, albeit with a modified version of angiopep-2 that also is considered to exploit LRP1 to traverse the BBB i.e. melanotransferrin. Administration of melanotransferrin-trastuzumab conjugate (BT2111) reduced the number of HER2+ breast cancer metastases in the brain (by 68%) with tumours being 46% smaller in BT2111 treated mice relative to control mice (91). To the authors knowledge, there have been no studies where either Angiopep-2 or melanotransferrin have been conjugated to ADCs for the purposes of increasing CNS access, however, based on the results with trastuzumab, it is expected that utilising these shuttle protein approaches should result in increased ADC brain uptake and efficacy.
Lastly, drug penetration into tumours is not just impacted by the BBB. Physico-chemical properties such as tumour volume could be modulated to increase ADC penetration. Larger tumours have increased interstitial pressures, more impaired circulation/lymphatics and increased necrotic areas that limit the ability of ADCs to penetrate the tumours (92–94). Data from the M12-356 Phase 1 study of Depatux-M provides evidence of this problem in glioma patients (95). Preclinical imaging and biodistribution studies showed specific and significantly higher tumor uptake of zirconium-89 labelled Depatux-M (89Zr-Depatux-M) in mice with smaller tumor volume versus those with larger volumes. Concordantly, mice with smaller tumor volumes at treatment commencement had significantly better growth inhibition and significantly longer overall survival compared to mice with large tumors at treatment commencement. These findings were supported by an analysis of tumor volumes on outcomes in the M12-356 study; patients with large tumors had significantly worse response rates and overall survival. These findings strongly support strategies that would reduce tumor size and/or interstitial pressure to increase efficacy of ADCs in brain tumors (96–98). The tumour microenvironment (TME) in GBM is complex; it is characteristically immunosuppressive and made up of numerous cell types surrounded by a distinctive extra-cellular matrix (99). Dynamic changes in the cellular and metabolic composition within the TME can result in treatment resistant and tumor recurrence (100). Given the role of TME in tumor growth and blood vessel formation, strategies being investigated include targeting antigens of the TME instead of tumor specific antigens as well as overcoming the inherent immunosuppressive effects and making tumors more immune competent (99, 101, 102). Antigens of the TME are likely more accessible and targeting them allows ADCs to accumulate within tumors and release their payload based on TME-specific factors (103, 104). For example, CD25, CD205, B7-H3 are targets found in the TME for which specific ADCs are in clinical development in a number of non-CNS tumour types (105).
Approaches to Reduce ADC Size/Polarity
The antibody component of an ADC is vital for binding to the desired antigen with high affinity and specificity, maintaining a long half-life and releasing the toxic payload into tumor cells; however its large size presents a physical barrier to efficient extravasation across blood vessel walls and diffusion through tumors (106). This has led to the development of smaller formats as carriers of toxic payloads and include: antibody fragments, peptides, natural ligands, and small molecules (107). Several drug conjugates using smaller targeting domains are being evaluated in clinical trials (NCT02936323, NCT03221400, NCT03486730).
Aptamers are short single-stranded nucleic acids (RNA or ssDNA) that represent a novel imaging and drug-delivery strategy based on their sensitivity and specificity, ease of modification and low immunogenicity (108). Aptamers can be physically conjugated either by intercalating or covalent linking to novel therapeutics such as noncoding RNA or cytotoxic payloads like doxorubicin (109). Recent studies demonstrating the ability of aptamers to cross the BBB and deliver payloads to tumors (110) has resulted in considered interest in the potential role of aptamers in the management of gliomas. To date, several studies have evaluated the role of aptamer-based therapies as well as aptamer-based conjugates (non-coding RNA, nanoparticles, chemotherapeutics) in a number preclinical glioma models (111). The majority of studies targeted EGFR/EGFRvIII while others looked at other GBM-associated proteins including Tenascin-C, EphB2/3 receptors, nucleolin, vascular endothelial growth factor receptor (VEGF), and integrin α5β1 (108, 111). While the results in early preclinical studies are encouraging, further optimization of aptamers with regards to their sensitivity to body-fluid nucleases, CpG toxicity, and stability remain to be optimized.
Nanoparticles are promising carriers for drug delivery to the brain due to their unique characteristics which include their small size and specific and homogenous tumor targeting. Various classes of nanoparticles, including metallic, polymeric and lipid nanoparticles can be readily modified to effectively carry drugs across the BBB. By attaching toxic payloads to nanoparticles, tumour-specific targets expressed in GBM cells and responsible for tumorigenesis can be targeted; examples of these include antigens (i.e., A2B5), differentiation clusters (i.e., CD15, CD33, CD44, or CD133), receptors of cytokines (i.e., interleukin13 receptor), and several proteins (i.e., EGFR, Integrin-a6, α5β3, ανβ3 or L1CAM). An alternative to the classical antibody-drug conjugate is the antibody-mediated delivery of a drug containing nanoparticles. In a recent example, panitumumab/Vectibix was attached to a 400nm nanoparticle (minicell) derived from Salmonella typhimurium in the attempt to deliver an effective dose of doxorubicin to 14 patients with recurrent GBM (112). This study showed that EGFR targeting antibody-coated nanoparticles containing chemotherapeutic drugs could be delivered in recurrent GBM patients.
ADCs Against Novel Targets
Selecting an appropriate target antigen is a critical step for the success of an ADC. Ideally, the appropriate target antigen should tumor-specific and homogenous in expression. The ideal target is one which is strongly and homogenous expressed on tumor cells, absent on normal tissue and efficient internalisation when bound (113–115). The potency of ADCs is dependent on the ability of the antibody-antigen complex to internalize, release the payload within the target cells and exert the cytotoxic effect. This dependency on antigen expression levels and finite internalization limits the therapeutic potential of ADCs as well as contributes to off-target toxicities (27).Some strategies to overcome the requirement of internalisation include the targeting of non-internalising receptors and extracellular matrix targets. Furthermore, the identification of tumour microenvironment targets also raises the possibility of therapeutic approaches with ADCs which do not target tumor cells alone (8, 26, 115). Novel approaches to developing non-internalization ADCs include diabody-based ADCs against non-internalizing targets and anti-tumor angiogenesis ADC which have shown promise in preclinical studies (116, 117).
The cell surface Notch ligand delta-like 3 (DLL3) inhibits Notch pathway activation and has shown to be expressed on the cell surface of several tumor types including gliomas where DLL3 expression inversely correlated with outcome (118, 119). In brain tumors, DLL3 has been shown to be most intensely and homogeneously overexpressed in IDH-mutant gliomas compared to other glioma subtypes (120). Interestingly, in gliomas DLL3 overexpression is not a consequence of DLL3 mutations or gene amplification (120). DLL3 is not expressed in adult normal tissues (119), making it an attractive therapeutic target. Rovalpituzumab teserine (Rova-T; SC16LD6.5) is an ADC consisting of a monoclonal antibody targeting DLL3, a cathepsin-cleavable linker, and a PBD payload (119). The first-in-human clinical trial of Rova-T in small cell lung cancer (SCLC) demonstrated encouraging activity albeit significant toxicity related to the payload (121). The phase 3 study (TAHOE) in recurrent SCLC however was halted early due futility (122). An active phase 3 trial of Rova-T in the maintenance setting (MERU) is ongoing (NCT03033511). In a phase 1/2 study (NCT02709889) patients with relapsed DLL3-positive (>1% by IHC) advanced solid tumors received Rova-T at 0.2, 0.3, or 0.4 mg/kg every 6-weeks for dose escalation in disease specific cohorts (123). The study enrolled 200 patients including 23 patients with GBM. The recommended phase 2 dose was 0.3 mg/kg q6wk for 2 cycles in all cohorts. The most common adverse events were fatigue, nausea, thrombocytopenia, pleural effusion and peripheral oedema. The objective response rate (ORR) was 11% including one complete response in the GBM cohort.
Increasingly, it is also becoming apparent that targeting the tumour microenvironment may be feasible in glioma and may have advantages over targeting tumours directly. Leucine-rich repeat containing 15 (LRRC15) is a type I membrane protein with low expression in normal tissue but is highly expressed on cancer associated fibroblasts within the tumor stroma as well as directly on cancer cells including GBM (124). ABBV-085 is an ADC composed of an anti-LRRC15 humanized monoclonal antibody conjugated to MMAE via a protease cleavable valine–citrulline linker. ABBV-085 has demonstrated significant antitumor activity in multiple LRRC15 cancer-positive models, including GBM. ABBV-085 also showed enhanced activity in combination with other therapies including cytotoxic chemotherapy, radiation, immunotherapy and targeted therapies (124).
Another promising target in the tumour microenvironment is the Eph family. Eph receptor tyrosine kinases and their cell-associated ephrin ligands have been implicated in the growth and progression of a large range of cancers and are increasingly recognized as important therapeutic anti-cancer targets (125). EphA2 and EphA3 are commonly expressed in GBM, including in regions of tumor neovasculature, tumor-associated immune cells, and tumor-infiltrating cells (126), and associated with poorer outcomes in GBM patients (127). MEDI-547 is an ADC comprising an EphA2 targeted monoclonal antibody (1C1) conjugated via a non-cleavable linker to the auristatin derivative maleimidocaproyl-monomethyl auristatin phenylalanine (mcMMAF). MEDI-547 displayed encouraging antitumor activity in preclinical models (128) however clinical development was halted due to due to treatment-related adverse events during the early phase studies (129). Despite these results, given the overexpression of EphA2 in many tumor types targeting EphA2 for toxin delivery remains a promising therapeutic strategy. MM-310, an anti-EphA2 immuno-liposome containing docetaxel prodrug has shown superior tumor penetration and anti-tumor activity in a range of xenograft models compared to free docetaxel and significantly with lower toxicity (130). An ADC directed against EphA3, another member of this family, is also being pursued. An ADC based on the IIIA4 mAb, and utilizing the microtubule inhibitor maytansine (IIIA4-USAN), has highly effective in killing preclinical GBM models compared to the naked antibody (131). Similarly, anti-EphA3 bound nanoparticles loaded with the DNA alkylation agent temozolomide showed specific tumor targeting and potent anti-tumor effects in a rat glioma model (132). A phase 1 study of the Ifabotuzumab, the humaneered version of IIIA4, has shown the drug is safe, is able to successfully target the tumour microenvironment in all patients tested but without normal tissue binding (133); an ADC based on Ifabotuzumab is therefore an attractive prospect.
Novel ADCs With Improved Payloads
Coupled with the above strategies are strategies to utilise newer payloads with increased therapeutic ratios. A number of known issues can adverse impact ADCs. Linkers are an essential interface between antibody and drug payload and are critical in stability, site of conjugation and final drug/antibody ratio (DAR): parameters that impact on toxicity, efficacy and pharmacokinetic properties of ADCs (8, 30). Current linkers may release payloads in the circulation which can lead to off-target toxicity. The development of novel linkers and advances in linker technology is beyond the scope of this review but has been discussed elsewhere in detail (134, 135). In addition, the IgG1 isotype of some of these ADCs can engage the Fc-gamma receptors (FcγR), which can trigger a target-independent, FcγR–dependent internalization in FcγR-positive cells resulting in toxic effects on these untargeted healthy cells (136). In addition, each class of payload often has characteristics toxicities (Table 2). A number of newer payloads are being tested. Pyrrolobenzodiazepines (PBDs) are DNA−crosslinking agents that exert their biological activity by binding in the minor groove of DNA with enhanced potent anti-cancer activity compared to auristatins or maytansinoids (137, 138). PBD dimers exhibit significant cell permeability, potentially enabling bystander killing of neighboring tumor cells (139). A number of PBD-conjugated ADCs are being developed and in clinical trials for both solid and hematological tumors (140). For example, ABBV-321 (serclutamab talirine) is a highly selective next-generation EGFR-targeting ADC which incorporates a PBD dimer toxin conjugated to the EGFR-targeting ABT-806 affinity-matured AM1 antibody (10). It is expected that the highly selective nature of ABBV-321 would differentiate it from previously developed antibody PBD conjugates that lack a therapeutic window. In a number of xenograft cell line and patient-derived xenograft tumour models including in GBM, ABBV-321 exhibited potent anti-tumor activity (10). ABBV-321 is currently under clinical investigation in patients with advanced solid tumors (141).
Deruxtecan (DXd) is a potent topoisomerase I inhibitor with a short half-life and ability to elicit a bystander killing effect on neighboring tumour cells indicating low concern in terms of systemic toxicity and importantly may assist in overcoming intratumoral heterogeneity of cancer cells (142, 143). In addition to its by stander effect, DXd is cell membrane permeable and therefore may enter nearby cells, even those without strong HER2 expression, making it effective in low HER2-expressing cancer cells and overcome tumour heterogeneity. Bioconjugation of DXd to a humanized monoclonal antibody specifically targeting HER2 via a cleavable tetrapeptide-based linker, made it possible to obtain the conjugate Trastuzumab Deruxtecan (DS-8201a, T-DXd) with a homogeneous DAR of 7.7. Trastuzumab deruxtecan has shown impressive response rates in early phase studies in tumours with high and low HER2 expression as well as HER2 mutant cancers (144, 145). The most common side effects were gastrointestinal and haematological, however potentially fatal adverse event of interstitial lung disease (ILD) was reported in 13.6% of patients in the phase II trial, including four patients who died due to lung injury (146). The mechanism of lung injury is not well understood and predictive biomarkers for response as well as identifying patients at risk of developing toxicity lung injury are required. Another ADC carrying the DXd payload is patritumab deruxtecan which uses the same linker-payload system as trastuzumab deruxtecan and is conjugated to the anti-HER3 monoclonal antibody patritumab. In a phase I study, patritumab deruxtecan demonstrated impressive responses in patients who were heavily pretreated with a median number of four regimens, including EGFR targeting tyrosine kinase inhibitors (147). Notably, its efficacy is observed regardless of the resistance mechanisms for EGFR-TKIs, including C797S secondary EGFR mutation, MET amplification, HER2 mutation, BRAF fusion, and PIK3CA mutation (147).
Lastly, there is on-going interest in utilising new radioisotopes in antibody payload delivery. Both the EphA2 mAb IF7 coupled to Lutetium-177 and then anti-EphA3 antibody IIIA4 linked to an α-particle-emitting Bismuth-213 payload showed therapeutic effect in EphA2 and EphA3 expressing leukemia models (148, 149). In GBM models, treatment with IIIA4 labelled with the β-particle-emitting Lutetium-177 showed dose-dependent tumor cell killing and tumor growth inhibition in vivo, compared to unlabelled antibody (150).
Combinatorial Treatment Approaches to Address Heterogeneity and Resistance
Cancer cells are constantly under strong selection and evolutionary pressures, resulting in the emergence of subclones and heterogeneity in gene expression and antigen expression (151). This in turn can affect ADC efficacy which is correlated with the level of target antigen expression (114). In a phase II study, patients with early-stage HER2-positive breast cancer received six cycles of trastuzumab emtansine (T-DM1), in combination with pertuzumab in the neoadjuvant setting (152). In this study complete pathological response (pCR) was higher in those with HER2 scores of 3+ versus 2+ and no pCR was seen among the patients classified as having HER2 heterogeneity, indicating that heterogeneity of target antigen expression is an important factor in patient selection. GBM is characterised by significant heterogeneity even at the single cell level (153). Studies have reported substantial inter- and intra-tumoral variation in the levels of EGFR expression and mutation. Furthermore, EGFR mutations are frequently lost or gained between the initial tumor and recurrence while molecular alterations, such as EGFR amplification, remain persistent unchanged (60). Recently studies have also shown that EGFRvIII can be eliminated from extrachromosomal DNA of tumor cells as a resistance mechanism when tumor cells are treated with EGFR TKIs. However, upon drug removal, EGFRvIII reappears (154). Data from clinical studies suggest that substantial inter - and intra-tumoral variation in the levels of EGFR and EGFRvIII expression and mutation contribute to therapeutic resistance (155, 156).
Combinatorial approaches is one approach to address tumour heterogeneity and resistance in gliomas, and as already demonstrated improved responses and survival rates in other tumor types (157). Dual targeting of both wildtype EGFR and EGFRvIII has been shown to have more effective anti-tumor activity in intracranial murine glioma models than single targeting of either variant alone (158). The anti-EGFR TKI AG1478 has shown to increase mAb 806-reactive dimers on the surface of cells overexpressing EGFR, and the combination has of mAb806 and AG1478 resulted in enhanced anti-tumour activity in xenograft models (159). Furthermore, Orellana et al. demonstrated that stabilizing the inactive kinase conformation with lapatinib correlated convincingly with increased binding of ABT-806 (160), further supporting the approach of targeting both EGFR and its variants.
Combinatorial strategies with ADCs may also effectively address the issue of tumour heterogeneity. Combining ABBV-321 and Depatux-M resulted in greater tumor growth inhibition in an EGFR-overexpressing GBM PDX model compared to either monotherapy treatment (161). It is also possible to incorporate two payloads into an ADC using multi-loading linkers, with improvements in conjugation efficiency and ADC homogeneity. Levengood et al. developed a dual‐auristatin ADCs containing both monomethyl auristatin E (MMAE) and monomethyl auristatin F (MMAF) and showed superior therapeutic benefit in preclinical anaplastic large cell lymphoma models refractory to ADCs comprised of the individual auristatin components (162). Anami et al. developed an ADC composed of anti-HER2 antibody conjugated to the MMAF payload via branched linkers and compared with the ADC composed of linear linkers. Their results demonstrated compared to linear linkers, branched linkers were highly stable in the human plasma, having high cell specificity and antigen-binding efficiency, and more significant in vitro cell killing potency (163).
Immunogenic cell death of tumor cells induced by cytotoxic compounds used as payloads in ADCs can be potent stimulators of effector T-cell recruitment to tumors and can directly result in dendritic cell activation and maturation (164). Indeed, infiltration of T cells has been observed in tumor biopsy specimens from patients after treatment with ado-trastuzumab emtansine (T-DM1) (165). Based on the induction of antibody dependent cellular cytotoxicity (ADCC) by anti-HER2 therapies, preclinical studies addressed the potential synergistic effect of ado-trastuzumab emtansine (T-DM1) and ICIs. Preclinical studies have shown the combination of HER2-targeting ADCs with ICI resulted in curative responses despite primary resistance to immunotherapy model (166–168). Exploration of tumor specimens from patients enrolled on phase 1 study (M12-356, NCT01800695) (169) has revealed a significant association between T-cell activity and response to ABT-414 treatment (170). This raises the possibility that combination of ADCs with currently approved immunotherapy are very likely to be effective.
Improved Patient Selection
In order to maximize the therapeutic potential of ADCs and limit exposure of ADCs to patients unlikely to benefit, more sophisticated biomarkers to select patients are needed. The current strategy of identifying patients is based on tumor target expression, which can be challenging due to multiple factors including: tumor heterogeneity, assay sensitivity, and accuracy, potential changes in target expression after multiple therapies, and difficulties in determining threshold levels for target expression that correlate with efficacy (171). In most trials, patients are preselected based target expression on archival tumor tissue. The most commonly employed methodology to determine target expression is by immunohistochemistry (IHC) which does possess limitations including lacks standardisation, reproducibility and, most importantly, correlation with clinical outcome. For example, while antigen expression levels have shown to correlate with response, ADCs have also shown clinical activity in patients with low levels of target antigen expression cancers due to imperfect assays (144, 145). Furthermore, emerging data suggest that selection based on payload sensitivity may add additional value above simply target expression. In the phase 1 study (M12-356, NCT01800695) of Depatux-M in patients with newly diagnosed GBM and recurrent GBM with EGFR amplification, responses in patients with recurrent GBM correlated with EGFR amplification however not all patients with EGFR amplification responded (169). Detailed examination of tumors [RNASeq, WES, immunohistochemistry (IHC)] from patients enrolled in the M12-356 trial revealed that mutations for tubulin genes were differentially expressed in responders vs non-responders, with TTLL2, TTLL4, TUBB2A, TUBB2B, TUBG1 and TUBGCP2 mutations (1-3 per tumor) overexpressed in non-responders (172). Preliminary synthetic lethality siRNA experiments have shown that mutations of these genes render sensitive GBM lines resistant to ABT-414 treatment. Furthermore, preliminary analysis of pre-treatment tumor samples with RNASeq has also revealed that responding patients had a higher number of tumor infiltrating lymphocytes (TIL) compared to non-responders, and CD3E expression. This was confirmed by immunohistochemistry analysis of CD3+ cells, with higher CD3+ cells in responding tumors. Preliminary T cell subset IHC analysis has also shown that CD8 cells are more frequently observed in responders, and that CD4 T cells are more abundant in non-responders. These data are highly suggestive of the immune microenvironment of GBM tumors playing a role in the responsiveness of GBM to ABT-414 treatment.
Lastly, molecular imaging by single photon computed tomography (SPECT) or positron emission tomography (PET), or immuno-PET, has been successfully employed to identify target expression, drug distribution and in-vivo target delivery and interlesional target heterogeneity (26). The humanized EphA2 mAb 1C1, libelled with 64Cu, was used for positron emission tomography (PET) imaging of eight tumor models with different EphA2 expression levels, showing good correlation between tumor uptake and EphA2 expression (125). Imaging of the conformational form of EGFR expressed only on tumors has also been demonstrated in GBM patients with 111In-ch806 and ABT-806 (173, 174). The ability to image the target of ADCs has the potential to improve selection of patients and increase therapeutic outcomes in patients with gliomas.
Conclusion
ADCs have shown to be effective and safe therapies, expanding the armamentarium in several haematological and solid tumors and in many cases transforming treatment paradigms. To date, their potential in glioma patients has not been established. Early ADCs were clearly ineffective due to limitations of early ADCs in general which affected their efficacy in all tumours. More recent ADCs like ABT-414 and AMG-595 clearly show the potential of these drugs in glioma, especially in recurrent gliomas. Clearly, more sophisticated strategies in their use will be needed if we are to make ADCs therapeutically useful for most glioma patients and to improve their therapeutic ratio. A number of such strategies are already available. Improvements in the ADC technology, building on the results of results to date, is clearly needed. These should focus on improvements in selecting targets and payloads. The use of adjunctive strategies is also appealing, seeking to improve drug access to tumours across the BBB and to better select patients. Lastly, combinatorial strategies are likely if we are to substantially improve outcomes in these patients and ADCs could have a major contribution to make in such strategies.
Author Contributions
All authors contributed to conception and design of the review. All authors contributed to manuscript revision, read, and approved the submitted version.
Funding
We would like to acknowledge our funding sources including: the Cancer Council Victoria (grant No 1164229), the Victorian Cancer Agency (CRE in Brain Cancer and ECF Fellowship for SP), the Operational Infrastructure Support from the Victorian Government. The support of AMS (National Health and Medical Research Council Investigator Fellowship No 1177837) is also acknowledged.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, et al. Radiotherapy Plus Concomitant and Adjuvant Temozolomide for Glioblastoma. N Engl J Med (2005) 352(10):987–96. doi: 10.1056/NEJMoa043330
2. Stupp R, Hegi ME, Mason WP, van den Bent MJ, Taphoorn MJB, Janzer RC, et al. Effects of Radiotherapy With Concomitant and Adjuvant Temozolomide Versus Radiotherapy Alone on Survival in Glioblastoma in a Randomised Phase III Study: 5-Year Analysis of the EORTC-NCIC Trial. Lancet Oncol (2009) 10(5):459–66. doi: 10.1016/S1470-2045(09)70025-7
3. Chinot OL, Wick W, Mason W, Henriksson R, Saran F, Nishikawa R, et al. Bevacizumab Plus Radiotherapy-Temozolomide for Newly Diagnosed Glioblastoma. N Engl J Med (2014) 370(8):709–22. doi: 10.1056/NEJMoa1308345
4. Gilbert MR, Dignam JJ, Armstrong TS, Wefel JS, Blumenthal DT, Vogelbaum MA, et al. A Randomized Trial of Bevacizumab for Newly Diagnosed Glioblastoma. N Engl J Med (2014) 370(8):699–708. doi: 10.1056/NEJMoa1308573
5. Muldoon LL, Pagel MA, Netto JP, Neuwelt EA. Intra-Arterial Administration Improves Temozolomide Delivery and Efficacy in a Model of Intracerebral Metastasis, But Has Unexpected Brain Toxicity. J Neurooncol (2016) 126(3):447–54. doi: 10.1007/s11060-015-2000-1
6. Chowdhary SA, Ryken T, Newton HB. Survival Outcomes and Safety of Carmustine Wafers in the Treatment of High-Grade Gliomas: A Meta-Analysis. J Neurooncol (2015) 122(2):367–82. doi: 10.1007/s11060-015-1724-2
7. Brastianos PK, Batchelor TT. VEGF Inhibitors in Brain Tumors. Clin Adv Hematol Oncol (2009) 7(753-760):768.
8. Parakh S, Parslow AC, Gan HK, Scott AM. Antibody-Mediated Delivery of Therapeutics for Cancer Therapy. Expert Opin Drug Deliv (2016) 13(3):401–19. doi: 10.1517/17425247.2016.1124854
9. Sievers EL, Senter PD. Antibody-Drug Conjugates in Cancer Therapy. Annu Rev Med (2013) 64:15–29. doi: 10.1146/annurev-med-050311-201823
10. Anderson MG, Falls HD, Mitten MJ, Oleksijew A, Vaidya KS, Boghaert ER, et al. Targeting Multiple EGFR-Expressing Tumors With a Highly Potent Tumor-Selective Antibody–Drug Conjugate. Mol Cancer Ther (2020) 19(10):2117–25. doi: 10.1158/1535-7163.MCT-20-0149
11. Hafeez U, Parakh S, Gan HK, Scott AM. Antibody–Drug Conjugates for Cancer Therapy. Molecules (2020) 25(20):4764. doi: 10.3390/molecules25204764
12. Lassman AB, Pugh SL, Wang TJC, Aldape K, Gan HK, Preusser M, et al. (2019). Depatuxizumab Mafodotin (ABT-414) in EGFR-Amplified Newly Diagnosed GBM: A Randomized, Double-Blind, Phase III, International Clinical Trial, in: Annual Meeting of the Society for Neuro-Oncology, . AZ, USA: Phoenix.
13. Rosenthal M, Curry R, Reardon DA, Rasmussen E, Upreti VV, Damore MA, et al. Safety, Tolerability, and Pharmacokinetics of Anti-EGFRvIII Antibody-Drug Conjugate AMG 595 in Patients With Recurrent Malignant Glioma Expressing EGFRvIII. Cancer Chemother Pharmacol (2019) 84(2):327–36. doi: 10.1007/s00280-019-03879-2
14. Lassman A, Gan H, Fichtel L, Merrell R, Van Den Bent M, Kumthekar P, et al. A Phase 1 Study Evaluating ABT-414 With Temozolomide (TMZ) or Concurrent Radiotherapy (RT) and TMZ in Glioblastoma (GBM)(S43. 006). Neurology (2015) 84(14 Supplement):S43. 006.
15. Rosenthal M, Curry R, Reardon DA, Rasmussen E, Upreti VV, Damore MA, et al. Safety, Tolerability, and Pharmacokinetics of Anti-EGFRvIII Antibody-Drug Conjugate AMG 595 in Patients With Recurrent Malignant Glioma Expressing EGFRvIII. Mol Cancer Ther (2015) 14(7):1614–24. doi: 10.1007/s00280-019-03879-2
16. Kunwar S, Chang S, Westphal M, Vogelbaum M, Sampson J, Barnett G, et al. Phase III Randomized Trial of CED of IL13-PE38QQR vs Gliadel Wafers for Recurrent Glioblastoma. Neuro-Oncol (2010) 12(8):871–81. doi: 10.1093/neuonc/nop054
17. Weber F, Asher A, Bucholz R, Berger M, Prados M, Chang S, et al. Safety, Tolerability, and Tumor Response of IL4-Pseudomonas Exotoxin (NBI-3001) in Patients With Recurrent Malignant Glioma. J Neurooncol (2003) 64(1-2):125–37. doi: 10.1007/BF02700027
18. Sampson JH, Akabani G, Archer GE, Berger MS, Coleman RE, Friedman AH, et al. Intracerebral Infusion of an EGFR-Targeted Toxin in Recurrent Malignant Brain Tumors. Neuro-Oncol (2008) 10(3):320–9. doi: 10.1215/15228517-2008-012
19. Weaver M, Laske DW. Transferrin Receptor Ligand-Targeted Toxin Conjugate (Tf-CRM107) for Therapy of Malignant Gliomas. J Neurooncol (2003) 65(1):3–13. doi: 10.1023/A:1026246500788
20. Tortorella S, Karagiannis TC. Transferrin Receptor-Mediated Endocytosis: A Useful Target for Cancer Therapy. J Membr Biol (2014) 247(4):291–307. doi: 10.1007/s00232-014-9637-0
21. Li L, Quang TS, Gracely EJ, Kim JH, Emrich JG, Yaeger TE, et al. A Phase II Study of Anti-Epidermal Growth Factor Receptor Radioimmunotherapy in the Treatment of Glioblastoma Multiforme. J Neurosurg (2010) 113(2):192–8. doi: 10.3171/2010.2.JNS091211
22. Reardon DA, Zalutsky MR, Akabani G, Coleman RE, Friedman AH, Herndon JE 2nd, et al. A Pilot Study: 131I-Antitenascin Monoclonal Antibody 81c6 to Deliver a 44-Gy Resection Cavity Boost. Neuro-Oncol (2008) 10(2):182–9. doi: 10.1215/15228517-2007-053
23. Riva P, Franceschi G, Frattarelli M, Riva N, Guiducci G, Cremonini AM, et al. 131I Radioconjugated Antibodies for the Locoregional Radioimmunotherapy of High-Grade Malignant Glioma–Phase I and II Study. Acta Oncol (1999) 38(3):351–9. doi: 10.1080/028418699431438
24. Gan HK, Cvrljevic AN, Johns TG. The Epidermal Growth Factor Receptor Variant III (EGFRvIII): Where Wild Things Are Altered. FEBS J (2013) 280(21):5350–70. doi: 10.1111/febs.12393
25. Gan HK, van den Bent M, Lassman AB, Reardon DA, Scott AM. Antibody-Drug Conjugates in Glioblastoma Therapy: The Right Drugs to the Right Cells. Nat Rev Clin Oncol (2017) 14(11):695–707. doi: 10.1038/nrclinonc.2017.95
26. Hafeez U, Parakh S, Gan HK, Scott AM. Antibody–Drug Conjugates for Cancer Therapy. Molecules (2020) 25(20):4764. doi: 10.3390/molecules25204764
27. Tang H, Liu Y, Yu Z, Sun M, Lin L, Liu W, et al. The Analysis of Key Factors Related to Adcs Structural Design. Front Pharmacol (2019) 10:373. doi: 10.3389/fphar.2019.00373
28. Krop IE, Beeram M, Modi S, Jones SF, Holden SN, Yu W, et al. Phase I Study of Trastuzumab-DM1, an HER2 Antibody-Drug Conjugate, Given Every 3 Weeks to Patients With HER2-Positive Metastatic Breast Cancer. J Clin Oncol (2010) 28(16):2698–704. doi: 10.1200/JCO.2009.26.2071
29. Shen K, Ma X, Zhu C, Wu X, Jia H. Safety and Efficacy of Trastuzumab Emtansine in Advanced Human Epidermal Growth Factor Receptor 2–Positive Breast Cancer: A Meta-Analysis. Sci (2016) 6(1):1–8. doi: 10.1038/srep23262
30. Donaghy H. Effects of Antibody, Drug and Linker on the Preclinical and Clinical Toxicities of Antibody-Drug Conjugates. MAbs (2016) 8(4):659–71. Taylor & Francis. doi: 10.1080/19420862.2016.1156829
31. Oak E, Bartlett NL. A Safety Evaluation of Brentuximab Vedotin for the Treatment of Hodgkin Lymphoma. Expert Opin Drug Saf (2016) 15(6):875–82. doi: 10.1080/14740338.2016.1179277
32. Sievers EL, Larson RA, Stadtmauer EA, Estey E, Löwenberg B, Dombret H, et al. Efficacy and Safety of Gemtuzumab Ozogamicin in Patients With CD33-Positive Acute Myeloid Leukemia in First Relapse. J Clin Oncol (2001) 19(13):3244–54. doi: 10.1200/JCO.2001.19.13.3244
33. Kantarjian HM, DeAngelo DJ, Stelljes M, Martinelli G, Liedtke M, Stock W, et al. Inotuzumab Ozogamicin Versus Standard Therapy for Acute Lymphoblastic Leukemia. N Engl J Med (2016) 375(8):740–53. doi: 10.1056/NEJMoa1509277
34. Owonikoko TK, Hussain A, Stadler WM, Smith DC, Sznol M, Molina AM, et al. A Phase 1 Multicenter Open-Label Dose-Escalation Study of BMS-936561 (MDX-1203) in Clear Cell Renal Cell Carcinoma (ccRCC) and B-Cell Non Hodgkin Lymphoma (B-NHL). J Clin Oncol (2014) 32(15_suppl):2558. doi: 10.1200/jco.2014.32.15_suppl.2558
35. Mantaj J, Jackson PJ, Rahman KM, Thurston DE. From Anthramycin to Pyrrolobenzodiazepine (PBD)-Containing Antibody–Drug Conjugates (ADCs). Angew Chem Int Edition (2017) 56(2):462–88. doi: 10.1002/anie.201510610
36. Dotan E, Starodub A, Berlin J, Lieu CH, Guarino MJ, Marshall J, et al. A New Anti-CEA-SN-38 Antibody-Drug Conjugate (ADC), IMMU-130, Is Active in Controlling Metastatic Colorectal Cancer (mCRC) in Patients (Pts) Refractory or Relapsing After Irinotecan-Containing Chemotherapies: Initial Results of a Phase I/II Study. J Clin Oncol (2015) 35(29):3338–46. doi: 10.1200/jco.2015.33.15_suppl.2505
37. Moskowitz CH, Nademanee A, Masszi T, Agura E, Holowiecki J, Abidi MH, et al. Brentuximab Vedotin as Consolidation Therapy After Autologous Stem-Cell Transplantation in Patients With Hodgkin’s Lymphoma at Risk of Relapse or Progression (AETHERA): A Randomised, Double-Blind, Placebo-Controlled, Phase 3 Trial. Lancet (2015) 385(9980):1853–62. doi: 10.1016/S0140-6736(15)60165-9
38. Connors JM, Jurczak W, Straus DJ, Ansell SM, Kim WS, Gallamini A, et al. Brentuximab Vedotin With Chemotherapy for Stage III or IV Hodgkin’s Lymphoma. New Engl J Med (2018) 378(4):331–44. doi: 10.1056/NEJMoa1708984
39. Horwitz S, O’Connor OA, Pro B, Illidge T, Fanale M, Advani R, et al. Brentuximab Vedotin With Chemotherapy for CD30-Positive Peripheral T-Cell Lymphoma (ECHELON-2): A Global, Double-Blind, Randomised, Phase 3 Trial. Lancet (London England) (2019) 393(10168):229–40. doi: 10.1016/S0140-6736(18)32984-2
40. Prince HM, Kim YH, Horwitz SM, Dummer R, Scarisbrick J, Quaglino P, et al. Brentuximab Vedotin or Physician’s Choice in CD30-Positive Cutaneous T-Cell Lymphoma (ALCANZA): An International, Open-Label, Randomised, Phase 3, Multicentre Trial. Lancet (2017) 390(10094):555–66. doi: 10.1016/S0140-6736(17)31266-7
41. Pro B, Advani R, Brice P, Bartlett NL, Rosenblatt JD, Illidge T, et al. Five-Year Results of Brentuximab Vedotin in Patients With Relapsed or Refractory Systemic Anaplastic Large Cell Lymphoma. Blood (2017) 130(25):2709–17. doi: 10.1182/blood-2017-05-780049
42. Krop IE, Kim S-B, Martin AG, Lorusso PM, Ferrero J-M, Badovinac-Crnjevic T, et al. Trastuzumab Emtansine Versus Treatment of Physician’s Choice in Patients With Previously Treated HER2-Positive Metastatic Breast Cancer (TH3RESA): Final Overall Survival Results From a Randomised Open-Label Phase 3 Trial. Lancet Oncol (2017) 18(6):743–54. doi: 10.1016/S1470-2045(17)30313-3
43. Verma S, Miles D, Gianni L, Krop IE, Welslau M, Baselga J, et al. Trastuzumab Emtansine for HER2-Positive Advanced Breast Cancer. N Engl J Med (2012) 367(19):1783–91. doi: 10.1056/NEJMoa1209124
44. Kantarjian HM, DeAngelo DJ, Stelljes M, Martinelli G, Liedtke M, Stock W, et al. Inotuzumab Ozogamicin Versus Standard Therapy for Acute Lymphoblastic Leukemia. New Engl J Med (2016) 375(8):740–53. doi: 10.1056/NEJMoa1509277
45. Castaigne S, Pautas C, Terré C, Raffoux E, Bordessoule D, Bastie J-N, et al. Effect of Gemtuzumab Ozogamicin on Survival of Adult Patients With De-Novo Acute Myeloid Leukaemia (ALFA-0701): A Randomised, Open-Label, Phase 3 Study. Lancet (2012) 379(9825):1508–16. doi: 10.1016/S0140-6736(12)60485-1
46. Bardia A, Hurvitz SA, Tolaney SM, Loirat D, Punie K, Oliveira M, et al. Sacituzumab Govitecan in Metastatic Triple-Negative Breast Cancer. N Engl J Med (2021) 384(16):1529–41. doi: 10.1056/NEJMoa2028485
47. Brenner A, Floyd J, Pandey R, Chiou J, Surapreneni P, Kaklamani V, et al. Delivery and Activity of Sn-38 by Sacituzumab Govitecan in CNS Tumors. Ann Oncol (2020) 31(suppl_4):S396–408. doi: 10.1016/j.annonc.2020.08.482
48. Johns TG, Adams TE, Cochran JR, Hall NE, Hoyne PA, Olsen MJ, et al. Identification of the Epitope for the Epidermal Growth Factor Receptor-Specific Monoclonal Antibody 806 Reveals That It Preferentially Recognizes an Untethered Form of the Receptor. J Biol Chem (2004) 279(29):30375–84. doi: 10.1074/jbc.M401218200
49. Gan HK, Burgess AW, Clayton AH, Scott AM. Targeting of a Conformationally Exposed, Tumor-Specific Epitope of EGFR as a Strategy for Cancer Therapy. Cancer Res (2012) 72(12):2924–30. doi: 10.1158/0008-5472.CAN-11-3898
50. Cleary JM, Reardon DA, Azad N, Gandhi L, Shapiro GI, Chaves J, et al. A Phase 1 Study of ABT-806 in Subjects With Advanced Solid Tumors. Invest New Drugs (2015) 33(3):671–8. doi: 10.1007/s10637-015-0234-6
51. Cleary JM, Yee LK-C, Azad N, Carducci M, Cosgrove D, Limaye S, et al. A Phase 1 Study of ABT-806, a Humanized Recombinant Anti-EGFR Monoclonal Antibody, in Patients With Advanced Solid Tumors. Cancer Res (2012) 72(8 Suppl):2506. doi: 10.1158/1538-7445.AM2012-2506
52. Luwor RB, Johns TG, Murone C, Huang HS, Cavenee WK, Ritter G, et al. Monoclonal Antibody 806 Inhibits the Growth of Tumor Xenografts Expressing Either the De2–7 or Amplified Epidermal Growth Factor Receptor (EGFR) But Not Wild-Type EGFR. Cancer Res (2001) 61(14):5355–61.
53. Phillips AC, Boghaert ER, Vaidya KS, Mitten MJ, Norvell S, Falls HD, et al. ABT-414, an Antibody-Drug Conjugate Targeting a Tumor-Selective EGFR Epitope. Mol Cancer Ther (2016) 15(4):661–9. doi: 10.1158/1535-7163.MCT-15-0901
54. Reardon DA, Lassman AB, van den Bent M, Kumthekar P, Merrell R, Scott AM, et al. Efficacy and Safety Results of ABT-414 in Combination With Radiation and Temozolomide in Newly Diagnosed Glioblastoma. Neuro-oncol (2016) 19:965–75. doi: 10.1093/neuonc/now257
55. Van Den Bent MJ, French P, Eoli M, Sepúlveda JM, Walenkamp AME, Frenel J-S, et al. Updated Results of the INTELLANCE 2/EORTC Trial 1410 Randomized Phase II Study on Depatux–M Alone, Depatux-M in Combination With Temozolomide (TMZ) and Either TMZ or Lomustine (LOM) in Recurrent EGFR Amplified Glioblastoma (NCT02343406). J Clin Oncol (2018) 36(15_suppl):2023. doi: 10.1200/JCO.2018.36.15_suppl.2023
56. van den Bent M, Eoli M, Sepulveda JM, Smits M, Walenkamp A, Frenel J-S, et al. Ltbk-04 First Results of the Randomized Phase Ii Study on Depatux–M Alone, Depatux-M in Combination With Temozolomide and Either Temozolomide or Lomustine in Recurrent Egfr Amplified Glioblastoma: First Report From Intellance 2/Eortc Trial 1410. Neuro-Oncol (2017) 19(suppl_6):vi316–vi. doi: 10.1093/neuonc/nox213
57. Van Den Bent M, Eoli M, Sepulveda JM, Smits M, Walenkamp A, Frenel J-S, et al. INTELLANCE 2/EORTC 1410 Randomized Phase II Study of Depatux-M Alone and With Temozolomide vs Temozolomide or Lomustine in Recurrent EGFR Amplified Glioblastoma. Neuro-oncol (2020) 22(5):684–93. doi: 10.1093/neuonc/noz222
58. Hamblett KJ, Kozlosky CJ, Siu S, Chang WS, Liu H, Foltz IN, et al. AMG 595, an Anti-EGFRvIII Antibody Drug Conjugate, Induces Potent Anti-Tumor Activity Against EGFRvIII Expressing Glioblastoma. Mol Cancer Ther (2015) 14(7):1614–24. doi: 10.1158/1535-7163
59. Rosenthal M, Curry R, Reardon DA, Rasmussen E, Upreti VV, Damore MA, et al. Safety, Tolerability, and Pharmacokinetics of Anti-EGFRvIII Antibody–Drug Conjugate AMG 595 in Patients With Recurrent Malignant Glioma Expressing EGFRvIII. Cancer Chemother Pharmacol (2019) 84(2):327–36. doi: 10.1007/s00280-019-03879-2
60. Barthel FP, Johnson KC, Varn FS, Moskalik AD, Tanner G, Kocakavuk E, et al. Longitudinal Molecular Trajectories of Diffuse Glioma in Adults. Nature (2019) 576(7785):112–20. doi: 10.1038/s41586-019-1775-1
61. Eskilsson E, Verhaak RGW. Longitudinal Genomic Characterization of Brain Tumors for Identification of Therapeutic Vulnerabilities. Neuro-Oncol (2016) 18(8):1037–9. doi: 10.1093/neuonc/now064
62. Sottoriva A, Spiteri I, Piccirillo SGM, Touloumis A, Collins VP, Marioni JC, et al. Intratumor Heterogeneity in Human Glioblastoma Reflects Cancer Evolutionary Dynamics. Proc Natl Acad Sci (2013) 110(10):4009–14. doi: 10.1073/pnas.1219747110
63. Kim J, Lee IH, Cho HJ, Park CK, Jung YS, Kim Y, et al. Spatiotemporal Evolution of the Primary Glioblastoma Genome. Cancer Cell (2015) 28(3):318–28. doi: 10.1016/j.ccell.2015.07.013
64. Johnson BE, Mazor T, Hong C, Barnes M, Aihara K, McLean CY, et al. Mutational Analysis Reveals the Origin and Therapy-Driven Evolution of Recurrent Glioma. Science (2014) 343(6167):189–93. doi: 10.1126/science.1239947
65. Wang J, Cazzato E, Ladewig E, Frattini V, Rosenbloom DI, Zairis S, et al. Clonal Evolution of Glioblastoma Under Therapy. Net Genet (2016) 48(7):768–76. doi: 10.1038/ng.3590
66. Weller M, Butowski N, Tran DD, Recht LD, Lim M, Hirte H, et al. Rindopepimut With Temozolomide for Patients With Newly Diagnosed, EGFRvIII-Expressing Glioblastoma (ACT IV): A Randomised, Double-Blind, International Phase 3 Trial. Lancet Oncol (2017) 18(10):1373–85. doi: 10.1093/neuonc/now212.068
67. Schalper KA, Rodriguez-Ruiz ME, Diez-Valle R, López-Janeiro A, Porciuncula A, Idoate MA, et al. Neoadjuvant Nivolumab Modifies the Tumor Immune Microenvironment in Resectable Glioblastoma. Nat Med (2019) 25(3):470–6. doi: 10.1038/s41591-018-0339-5
68. Reardon DA, Kim T-M, Frenel J-S, Santoro A, Lopez J, Subramaniam DS, et al. ATIM-35. Results of the Phase IB KEYNOTE-028 Multi-Cohort Trial of Pembrolizumab Monotherapy in Patients With Recurrent PD-L1-Positive Glioblastoma Multiforme (GBM). Neuro-Oncol (2016) 18(suppl_6):vi25–6. doi: 10.1093/neuonc/now212.100
69. Cloughesy TF, Mochizuki AY, Orpilla JR, Hugo W, Lee AH, Davidson TB, et al. Neoadjuvant Anti-PD-1 Immunotherapy Promotes a Survival Benefit With Intratumoral and Systemic Immune Responses in Recurrent Glioblastoma. Nat Med (2019) 25(3):477–86. doi: 10.1038/s41591-018-0337-7
70. Kirkin AF, Dzhandzhugazyan KN, Guldberg P, Fang JJ, Andersen RS, Dahl C, et al. Adoptive Cancer Immunotherapy Using DNA-Demethylated T Helper Cells as Antigen-Presenting Cells. Nat Commun (2018) 9(1):1–12. doi: 10.1038/s41467-018-03217-9
71. Brown CE, Alizadeh D, Starr R, Weng L, Wagner JR, Naranjo A, et al. Regression of Glioblastoma After Chimeric Antigen Receptor T-Cell Therapy. N Engl J Med (2016) 375(26):2561–9. doi: 10.1056/NEJMoa1610497
72. Ahmed N, Brawley V, Hegde M, Bielamowicz K, Kalra M, Landi D, et al. Her2-Specific Chimeric Antigen Receptor–Modified Virus-Specific T Cells for Progressive Glioblastoma: A Phase 1 Dose-Escalation Trial. JAMA Oncol (2017) 3(8):1094–101. doi: 10.1001/jamaoncol.2017.0184
73. Migliorini D, Dutoit V, Allard M, Grandjean Hallez N, Marinari E, Widmer V, et al. Phase I/II Trial Testing Safety and Immunogenicity of the Multipeptide IMA950/poly-ICLC Vaccine in Newly Diagnosed Adult Malignant Astrocytoma Patients. Neuro-Oncol (2019) 21(7):923–33. doi: 10.1093/neuonc/noz040
74. Cloughesy T, Finocchiaro G, Belda-Iniesta C, Recht L, Brandes AA, Pineda E, et al. Randomized, Double-Blind, Placebo-Controlled, Multicenter Phase II Study of Onartuzumab Plus Bevacizumab Versus Placebo Plus Bevacizumab in Patients With Recurrent Glioblastoma: Efficacy, Safety, and Hepatocyte Growth Factor and O (6)-Methylguanine-DNA Methyltransferase Biomarker Analyses. J Clin Oncol (2017) 35(3):343–51. doi: 10.1200/JCO.2015.64.7685
75. Cher L, Nowak A, Iatropoulos G, Lee WS, Lee SY, Shim SR, et al. ACTR-75. A Multicenter, 3-Arm, Open-Label, Phase IIa Clinical Trial to Evaluate Safety and Efficacy of Tanibirumab (VEGFR2 mAB), in Patients With Recurrent GBM Assessed With K-Trans and Initial Area Under the Gadolinium Concentration-Time Curve (IAUGC). Neuro Oncol (2017) 19(Suppl 6):17. doi: 10.1093/neuonc/nox168.062
76. Lang FF, Conrad C, Gomez-Manzano C, Yung WA, Sawaya R, Weinberg JS, et al. Phase I Study of DNX-2401 (Delta-24-RGD) Oncolytic Adenovirus: Replication and Immunotherapeutic Effects in Recurrent Malignant Glioma. J Clin Oncol (2018) 36(14):1419. doi: 10.1200/JCO.2017.75.8219
77. Marin B-M, Porath KA, Jain S, Kim M, Conage-Pough JE, Oh J-H, et al. Heterogeneous Delivery Across the Blood-Brain Barrier Limits the Efficacy of an EGFR-Targeting Antibody Drug Conjugate in Glioblastoma. Neuro-Oncol (2021) 29:noab133. doi: 10.1093/neuonc/noab133
78. Abbott NJ, Patabendige AA, Dolman DE, Yusof SR, Begley DJ. Structure and Function of the Blood–Brain Barrier. Neurobiol Dis (2010) 37(1):13–25. doi: 10.1016/j.nbd.2009.07.030
79. Sarkaria JN, Hu LS, Parney IF, Pafundi DH, Brinkmann DH, Laack NN, et al. Is the Blood–Brain Barrier Really Disrupted in All Glioblastomas? A Critical Assessment of Existing Clinical Data. Neuro-Oncol (2018) 20(2):184–91. doi: 10.1093/neuonc/nox175
80. Wang W, He H, Marin-Ramos NI, Zeng S, Swenson SD, Cho H-Y, et al. Enhanced Brain Delivery and Therapeutic Activity of Trastuzumab After Blood-Brain Barrier Opening by NEO100 in Mouse Models of Brain-Metastatic Breast Cancer. Neuro Oncol (2021) 23(10):1656–67. doi: 10.1093/neuonc/noab204
81. Schinkel AH. P-Glycoprotein, a Gatekeeper in the Blood-Brain Barrier. Adv Drug Deliv Rev (1999) 36(2-3):179–94. doi: 10.1016/S0169-409X(98)00085-4
82. Nicolazzo JA, Katneni K. Drug Transport Across the Blood-Brain Barrier and the Impact of Breast Cancer Resistance Protein (ABCG2). Curr Topics Med Chem (2009) 9(2):130–47. doi: 10.2174/156802609787521580
83. Kim M, Kizilbash SH, Laramy JK, Gampa G, Parrish KE, Sarkaria JN, et al. Barriers Tor Effective Drug Treatment for Brain Metastases: A Mulltifactorial Problem in the Delivery of Precision Medicine. Pharm Res (2018) 35(9):177. doi: 10.1007/s11095-018-2455-9
84. Loganzo F, Tan X, Sung M, Jin G, Myers JS, Melamud E, et al. Tumor Cells Chronically Treated With a Trastuzumab–Maytansinoid Antibody–Drug Conjugate Develop Varied Resistance Mechanisms But Respond to Alternate Treatments. Mol Cancer Ther (2015) 14(4):952–63. doi: 10.1158/1535-7163.MCT-14-0862
85. Chen R, Hou J, Newman E, Kim Y, Donohue C, Liu X, et al. CD30 Downregulation, MMAE Resistance, and MDR1 Upregulation Are All Associated With Resistance to Brentuximab Vedotin. Mol Cancer Ther (2015) 14(6):1376–84. doi: 10.1158/1535-7163.MCT-15-0036
86. Sung M, Golas J, Wang F, King L, Myers J, Rosfjord E, et al. Caveolae-Mediated Endocytosis as a Novel Mechanism of Resistance to Trastuzumab Emtansine (T-Dm1). Mol Cancer Ther (2018) 17(1):243–53. doi: 10.1158/1535-7163.MCT-17-0403
87. Ríos-Luci C, García-Alonso S, Díaz-Rodríguez E, Nadal-Serrano M, Arribas J, Ocaña A, et al. Resistance to the Antibody-Drug Conjugate T-DM1 Is Based in a Reduction in Lysosomal Proteolytic Activity. Cancer Res (2017) 77(17):4639. doi: 10.1158/0008-5472.CAN-16-3127
88. Kinneer K, Meekin J, Tiberghien AC, Tai Y-T, Phipps S, Kiefer CM, et al. SLC46A3 as a Potential Predictive Biomarker for Antibody-Drug Conjugates Bearing Noncleavable Linked Maytansinoid and Pyrrolobenzodiazepine Warheads. Clin Cancer Res Off J Am Assoc Cancer Res (2018) 24(24):6570. doi: 10.1158/1078-0432.CCR-18-1300
89. Kurzrock R, Gabrail N, Chandhasin C, Moulder S, Smith C, Brenner A, et al. Safety, Pharmacokinetics, and Activity of GRN1005, a Novel Conjugate of Angiopep-2, a Peptide Facilitating Brain Penetration, and Paclitaxel, in Patients With Advanced Solid Tumors. Mol Cancer Ther (2012) 11(2):308–16. doi: 10.1158/1535-7163.MCT-11-0566
90. Drappatz J, Brenner A, Wong ET, Eichler A, Schiff D, Groves MD, et al. Phase I Study of GRN1005 in Recurrent Malignant Glioma. Clin Cancer Res (2013) 19(6):1567–76. doi: 10.1158/1078-0432.CCR-12-2481
91. Nounou MI, Adkins CE, Rubinchik S, Terrell-Hall TB, Afroz M, Vitalis T, et al. Anti-Cancer Antibody Trastuzumab-Melanotransferrin Conjugate (BT2111) for the Treatment of Metastatic HER2+ Breast Cancer Tumors in the Brain: An In-Vivo Study. Pharm Res (2016) 33(12):2930–42. doi: 10.1007/s11095-016-2015-0
92. Leu AJ, Berk DA, Lymboussaki A, Alitalo K, Jain RK. Absence of Functional Lymphatics Within a Murine Sarcoma: A Molecular and Functional Evaluation. Cancer Res (2000) 60(16):4324–7.
93. Munson JM, Shieh AC. Interstitial Fluid Flow in Cancer: Implications for Disease Progression and Treatment. Cancer Manag Res (2014) 6:317. doi: 10.2147/CMAR.S65444
94. Harder BG, Blomquist MR, Wang J, Kim AJ, Woodworth GF, Winkles JA, et al. Developments in Blood-Brain Barrier Penetrance and Drug Repurposing for Improved Treatment of Glioblastoma. Front Oncol (2018) 8:462. doi: 10.3389/fonc.2018.00462
95. Gan H, Seow A, Lau E, Sze-Ting L, Ameratunga M, Perchyonok Y, et al. ACTR-55. Tumour Volume as a Predictor of Response to Ant-EGFR ADC ABT-414. Neuro-Oncol (2018) 20(suppl_6):vi24. doi: 10.1093/neuonc/noy148.087
96. Trédan O, Galmarini CM, Patel K, Tannock IF. Drug Resistance and the Solid Tumor Microenvironment. J Natl Cancer Inst (2007) 99(19):1441–54. doi: 10.1093/jnci/djm135
97. Böckelmann LC, Schumacher U. Targeting Tumor Interstitial Fluid Pressure: Will It Yield Novel Successful Therapies for Solid Tumors? Expert Opin Ther Targets (2019) 23(12):1005–14. doi: 10.1080/14728222.2019.1702974
98. Heldin C-H, Rubin K, Pietras K, Östman A. High Interstitial Fluid Pressure—an Obstacle in Cancer Therapy. Nat Rev Cancer (2004) 4(10):806–13. doi: 10.1038/nrc1456
99. Fanelli GN, Grassini D, Ortenzi V, Pasqualetti F, Montemurro N, Perrini P, et al. Decipher the Glioblastoma Microenvironment: The First Milestone for New Groundbreaking Therapeutic Strategies. Genes (2021) 12(3):445. doi: 10.3390/genes12030445
100. Abels ER, Maas SL, Tai E, Ting DT, Broekman ML, Breakefield XO, et al. GlioM&M: Web-Based Tool for Studying Circulating and Infiltrating Monocytes and Macrophages in Glioma. Sci Rep (2020) 10(1):1–11. doi: 10.1038/s41598-020-66728-w
101. Butler M, Prasad S, Srivastava SK. Targeting Glioblastoma Tumor Microenvironment. In: Tumor Microenvironments in Organs. Switzerland: Springer (2020). p. 1–9.
102. Ali S, Borin TF, Piranlioglu R, Ara R, Lebedyeva I, Angara K, et al. Changes in the Tumor Microenvironment and Outcome for TME-Targeting Therapy in Glioblastoma: A Pilot Study. PloS One (2021) 16(2):e0246646. doi: 10.1371/journal.pone.0246646
103. Lucas AT, Price LS, Schorzman AN, Storrie M, Piscitelli JA, Razo J, et al. Factors Affecting the Pharmacology of Antibody–Drug Conjugates. Antibodies (2018) 7(1):10. doi: 10.3390/antib7010010
104. Mathur R, Weiner GJ. Picking the Optimal Target for Antibody-Drug Conjugates. Am Soc Clin Oncol Educ Book (2013) 33(1):e103–7. doi: 10.14694/EdBook_AM.2013.33.e103
105. Boni V, Sharma MR, Patnaik A. The Resurgence of Antibody Drug Conjugates in Cancer Therapeutics: Novel Targets and Payloads. Am Soc Clin Oncol Educ Book (2020) 40:e58–74. doi: 10.1200/EDBK_281107
106. Sarkaria JN, Hu LS, Parney IF, Pafundi DH, Brinkmann DH, Laack NN, et al. Is the Blood–Brain Barrier Really Disrupted in All Glioblastomas? A Critical Assessment of Existing Clinical Data. Neuro-Oncol (2017) 20(2):184–91. doi: 10.1093/neuonc/nox175
107. Coats S, Williams M, Kebble B, Dixit R, Tseng L, Yao N-S, et al. Antibody–drug Conjugates: Future Directions in Clinical and Translational Strategies to Improve the Therapeutic Index. Clin Cancer Res (2019) 25(18):5441–8. doi: 10.1158/1078-0432.CCR-19-0272
108. Amero P, Khatua S, Rodriguez-Aguayo C, Lopez-Berestein G. Aptamers: Novel Therapeutics and Potential Role in Neuro-Oncology. Cancers (2020) 12(10):2889. doi: 10.3390/cancers12102889
109. Nimjee SM, White RR, Becker RC, Sullenger BA. Aptamers as Therapeutics. Annu Rev Pharmacol Toxicol (2017) 57:61–79. doi: 10.1146/annurev-pharmtox-010716-104558
110. Monaco I, Camorani S, Colecchia D, Locatelli E, Calandro P, Oudin A, et al. Aptamer Functionalization of Nanosystems for Glioblastoma Targeting Through the Blood–Brain Barrier. J Med Chem (2017) 60(10):4510–6. doi: 10.1021/acs.jmedchem.7b00527
111. Nuzzo S, Brancato V, Affinito A, Salvatore M, Cavaliere C, Condorelli G. The Role of RNA and DNA Aptamers in Glioblastoma Diagnosis and Therapy: A Systematic Review of the Literature. Cancers (2020) 12(8):2173. doi: 10.3390/cancers12082173
112. Whittle JR, Lickliter JD, Gan HK, Scott AM, Simes J, Solomon BJ, et al. First in Human Nanotechnology Doxorubicin Delivery System to Target Epidermal Growth Factor Receptors in Recurrent Glioblastoma. J Clin Neurosci Off J Neurosurg Soc Australas (2015) 22(12):1889–94. doi: 10.1016/j.jocn.2015.06.005
113. Lambert JM, Morris CQ. Antibody–drug Conjugates (ADCs) for Personalized Treatment of Solid Tumors: A Review. Adv Ther (2017) 34(5):1015–35. doi: 10.1007/s12325-017-0519-6
114. Perez EA, Hurvitz SA, Amler LC, Mundt KE, Ng V, Guardino E, et al. Relationship Between HER2 Expression and Efficacy With First-Line Trastuzumab Emtansine Compared With Trastuzumab Plus Docetaxel in TDM4450g: A Randomized Phase II Study of Patients With Previously Untreated HER2-Positive Metastatic Breast Cancer. Breast Cancer Res (2014) 16(3):1–10. doi: 10.1186/bcr3661
115. Scott AM, Wolchok JD, Old LJ. Antibody Therapy of Cancer. Nat Rev Cancer (2012) 12(4):278–87. doi: 10.1038/nrc3236
116. Rossin R, Versteegen RM, Wu J, Khasanov A, Wessels HJ, Steenbergen EJ, et al. Chemically Triggered Drug Release From an Antibody-Drug Conjugate Leads to Potent Antitumour Activity in Mice. Nat Commun (2018) 9(1):1–11. doi: 10.1038/s41467-018-03880-y
117. Seaman S, Zhu Z, Saha S, Zhang XM, Yang MY, Hilton MB, et al. Eradication of Tumors Through Simultaneous Ablation of CD276/B7-H3-Positive Tumor Cells and Tumor Vasculature. Cancer Cell (2017) 31(4):501–15.e8. doi: 10.1016/j.ccell.2017.03.005
118. Peng SL, Saunders L, Bheddah S, Williams S, Aggarwal RR, Shea JE, et al. Metastatic Melanoma, Glioblastoma and High-Grade Extrapulmonary Neuroendocrine Carcinomas (NECs) as Novel Indications for Rovalpituzumab Tesirine: A Delta-Like Protein 3 (DLL3)-Targeted Antibody-Drug Conjugate (ADC). J Clin Oncol (2016) 34(15_suppl):11611. doi: 10.1200/JCO.2016.34.15_suppl.11611
119. Saunders LR, Bankovich AJ, Anderson WC, Aujay MA, Bheddah S, Black K, et al. A DLL3-Targeted Antibody-Drug Conjugate Eradicates High-Grade Pulmonary Neuroendocrine Tumor-Initiating Cells In Vivo. Sci Trans Med (2015) 7(302):302ra136–302ra136. doi: 10.1126/scitranslmed.aac9459
120. Spino M, Kurz SC, Chiriboga L, Serrano J, Zeck B, Sen N, et al. Cell Surface Notch Ligand DLL3 Is a Therapeutic Target in Isocitrate Dehydrogenase–Mutant Glioma. Clin Cancer Res (2019) 25(4):1261–71. doi: 10.1158/1078-0432.CCR-18-2312
121. Rudin CM, Pietanza MC, Bauer TM, Ready N, Morgensztern D, Glisson BS, et al. Rovalpituzumab Tesirine, a DLL3-Targeted Antibody-Drug Conjugate, in Recurrent Small-Cell Lung Cancer: A First-in-Human, First-in-Class, Open-Label, Phase 1 Study. Lancet Oncol (2017) 18(1):42–51. doi: 10.1016/S1470-2045(16)30565-4
122. Blackhall F, Jao K, Greillier L, Cho BC, Penkov K, Reguart N, et al. Efficacy and Safety of Rovalpituzumab Tesirine Compared With Topotecan as Second-Line Therapy in DLL3-High SCLC: Results From the Phase 3 TAHOE Study. J Thorac Oncol (2021) 16(9):1547–58. doi: 10.1016/j.jtho.2021.02.009
123. Mansfield AS, Hong DS, Hann CL, Farago AF, Beltran H, Waqar SN, et al. A Phase I/II Study of Rovalpituzumab Tesirine in Delta-Like 3-Expressing, Advanced Solid Tumors. J Clin Oncol (2020) 38(15_suppl):3552. doi: 10.1038/s41698-021-00214-y
124. Purcell JW, Tanlimco SG, Hickson J, Fox M, Sho M, Durkin L, et al. LRRC15 Is a Novel Mesenchymal Protein and Stromal Target for Antibody–Drug Conjugates. Cancer Res (2018) 78(14):4059–72. doi: 10.1158/0008-5472.CAN-18-0327
125. Janes PW, Vail ME, Gan HK, Scott AM. Antibody Targeting of Eph Receptors in Cancer. Pharmaceuticals (2020) 13(5):88. doi: 10.3390/ph13050088
126. Wykosky J, Gibo DM, Stanton C, Debinski W. EphA2 as a Novel Molecular Marker and Target in Glioblastoma Multiforme. Mol Cancer Res (2005) 3(10):541–51. doi: 10.1158/1541-7786.MCR-05-0056
127. Liu F, Park PJ, Lai W, Maher E, Chakravarti A, Durso L, et al. A Genome-Wide Screen Reveals Functional Gene Clusters in the Cancer Genome and Identifies EphA2 as a Mitogen in Glioblastoma. Cancer Res (2006) 66(22):10815–23. doi: 10.1158/0008-5472.CAN-06-1408
128. Jackson D, Gooya J, Mao S, Kinneer K, Xu L, Camara M, et al. A Human Antibody–Drug Conjugate Targeting EphA2 Inhibits Tumor Growth In Vivo. Cancer Res (2008) 68(22):9367–74. doi: 10.1158/0008-5472.CAN-08-1933
129. Annunziata CM, Kohn EC, LoRusso P, Houston ND, Coleman RL, Buzoianu M, et al. Phase 1, Open-Label Study of MEDI-547 in Patients With Relapsed or Refractory Solid Tumors. Invest New Drugs (2013) 31(1):77–84. doi: 10.1007/s10637-012-9801-2
130. Kamoun WS, Kirpotin DB, Huang ZR, Tipparaju SK, Noble CO, Hayes ME, et al. Antitumour Activity and Tolerability of an EphA2-Targeted Nanotherapeutic in Multiple Mouse Models. Nat Biomed Eng (2019) 3(4):264–80. doi: 10.1038/s41551-019-0385-4
131. Offenhäuser C, Al-Ejeh F, Puttick S, Ensbey KS, Bruce ZC, Jamieson PR, et al. EphA3 Pay-Loaded Antibody Therapeutics for the Treatment of Glioblastoma. Cancers (2018) 10(12):519. doi: 10.3390/cancers10120519
132. Chu L, Wang A, Ni L, Yan X, Song Y, Zhao M, et al. Nose-To-Brain Delivery of Temozolomide-Loaded PLGA Nanoparticles Functionalized With Anti-EPHA3 for Glioblastoma Targeting. Drug Deliv (2018) 25(1):1634–41. doi: 10.1080/10717544.2018.1494226
133. Gan H, Cher L, Inglis P, Lwin Z, Lau E, Wichmann C, et al. Abstract CT063: Preliminary Findings of a Phase I Safety and Bioimaging Trial of KB004 (Ifabotuzumab) in Patients With Glioblastoma. Cancer Res (2019) 79(13 Supplement):CT063–CT. doi: 10.1158/1538-7445.AM2019-CT063
134. Su Z, Xiao D, Xie F, Liu L, Wang Y, Fan S, et al. Antibody‒drug Conjugates: Recent Advances in Linker Chemistry. Acta Pharm Sin B (2021) 12:687926. doi: 10.1016/j.apsb.2021.03.042
135. Su D, Zhang D. Linker Design Impacts Antibody-Drug Conjugate Pharmacokinetics and Efficacy via Modulating the Stability and Payload Release Efficiency. Front Pharmacol (2021) 12:1558. doi: 10.3389/fphar.2021.687926
136. Joubert N, Beck A, Dumontet C, Denevault-Sabourin C. Antibody–Drug Conjugates: The Last Decade. Pharmaceuticals (2020) 13(9):245. doi: 10.3390/ph13090245
137. Jeffrey SC, Burke PJ, Lyon RP, Meyer DW, Sussman D, Anderson M, et al. A Potent Anti-CD70 Antibody-Drug Conjugate Combining a Dimeric Pyrrolobenzodiazepine Drug With Site-Specific Conjugation Technology. Bioconjug Chem (2013) 24(7):1256–63. doi: 10.1021/bc400217g
138. Hartley JA, Flynn MJ, Bingham JP, Corbett S, Reinert H, Tiberghien A, et al. Pre-Clinical Pharmacology and Mechanism of Action of SG3199, the Pyrrolobenzodiazepine (PBD) Dimer Warhead Component of Antibody-Drug Conjugate (ADC) Payload Tesirine. Sci (2018) 8(1):1–10. doi: 10.1038/s41598-018-28533-4
139. Li F, Emmerton KK, Jonas M, Zhang X, Miyamoto JB, Setter JR, et al. Intracellular Released Payload Influences Potency and Bystander-Killing Effects of Antibody-Drug Conjugates in Preclinical Models. Cancer Res (2016) 76(9):2710–9. doi: 10.1158/0008-5472.CAN-15-1795
140. Saber H, Simpson N, Ricks TK, Leighton JK. An FDA Oncology Analysis of Toxicities Associated With PBD-Containing Antibody-Drug Conjugates. Regul Toxicol Pharmacol (2019) 107:104429–36. doi: 10.1016/j.yrtph.2019.104429
141. Carneiro BA, Bestvina CM, Shacham-Shmueli E, Gan HK, Beck JT, Robinson R, et al. Phase I Study of the Antibody-Drug Conjugate ABBV-321 in Patients With Non-Small Cell Lung Cancer and Squamous Head and Neck Cancer With Overexpression of the Epidermal Growth Factor Receptor. J Clin Oncol (2020) 38(15_suppl):TPS3649. doi: 10.1200/JCO.2020.38.15_suppl.TPS3649
142. Ogitani Y, Hagihara K, Oitate M, Naito H, Agatsuma T. Bystander Killing Effect of DS-8201a, A Novel Anti-Human Epidermal Growth Factor Receptor 2 Antibody–Drug Conjugate, in Tumors With Human Epidermal Growth Factor Receptor 2 Heterogeneity. Cancer Sci (2016) 107(7):1039–46. doi: 10.1111/cas.12966
143. Ogitani Y, Aida T, Hagihara K, Yamaguchi J, Ishii C, Harada N, et al. DS-8201a, a Novel HER2-Targeting ADC With a Novel DNA Topoisomerase I Inhibitor, Demonstrates a Promising Antitumor Efficacy With Differentiation From T-Dm1. Clin Cancer Res (2016) 22(20):5097–108. doi: 10.1158/1078-0432.CCR-15-2822
144. Linehan AS, Fitzpatrick OM, Morris PG. Profile of Trastuzumab Deruxtecan in the Management of Patients With HER2-Positive Unresectable or Metastatic Breast Cancer: An Evidence-Based Review. Breast Cancer: Targets Ther (2021) 13:151. doi: 10.2147/BCTT.S245024
145. Tsurutani J, Iwata H, Krop I, Jänne PA, Doi T, Takahashi S, et al. Targeting HER2 With Trastuzumab Deruxtecan: A Dose-Expansion, Phase I Study in Multiple Advanced Solid Tumors. Cancer Discov (2020) 10(5):688–701. doi: 10.1158/2159-8290.CD-19-1014
146. Modi S, Saura C, Yamashita T, Park YH, Kim S-B, Tamura K, et al. Trastuzumab Deruxtecan in Previously Treated HER2-Positive Breast Cancer. N Engl J Med (2020) 382(7):610–21. doi: 10.1056/NEJMoa1914510
147. Yu H, Baik C, Gold K, Hayashi H, Johnson M, Koczywas M, et al. LBA62 Efficacy and Safety of Patritumab Deruxtecan (U3-1402), A Novel HER3 Directed Antibody Drug Conjugate, in Patients (Pts) With EGFR-Mutated (EGFRm) NSCLC. Ann Oncol (2020) 31:S1189–90. doi: 10.1016/j.annonc.2020.08.2295
148. Charmsaz S, Beckett K, Smith FM, Bruedigam C, Moore AS, Al-Ejeh F, et al. EphA2 Is a Therapy Target in EphA2-Positive Leukemias But Is Not Essential for Normal Hematopoiesis or Leukemia. PloS One (2015) 10(6):e0130692. doi: 10.1371/journal.pone.0130692
149. Charmsaz S, Al-Ejeh F, Yeadon T, Miller K, Smith FM, Stringer BW, et al. EphA3 as a Target for Antibody Immunotherapy in Acute Lymphoblastic Leukemia. Leukemia (2017) 31(8):1779–87. doi: 10.1038/leu.2016.371
150. Day BW, Stringer BW, Al-Ejeh F, Ting MJ, Wilson J, Ensbey KS, et al. EphA3 Maintains Tumorigenicity and Is a Therapeutic Target in Glioblastoma Multiforme. Cancer Cell (2013) 23(2):238–48. doi: 10.1016/j.ccr.2013.01.007
151. McGranahan N, Swanton C. Biological and Therapeutic Impact of Intratumor Heterogeneity in Cancer Evolution. Cancer Cell (2015) 27(1):15–26. doi: 10.1016/j.ccell.2014.12.001
152. Metzger Filho O, Viale G, Trippa L, Li T, Yardley DA, Mayer IA, et al. HER2 Heterogeneity as a Predictor of Response to Neoadjuvant T-DM1 Plus Pertuzumab: Results From a Prospective Clinical Trial. J Clin Oncol (2019) 37(15_suppl):502. doi: 10.1200/JCO.2019.37.15_suppl.502
153. Yu K, Hu Y, Wu F, Guo Q, Qian Z, Hu W, et al. Surveying Brain Tumor Heterogeneity by Single-Cell RNA-Sequencing of Multi-Sector Biopsies. Natl Sci Rev (2020) 7(8):1306–18. doi: 10.1093/nsr/nwaa099
154. Nathanson DA, Gini B, Mottahedeh J, Visnyei K, Koga T, Gomez G, et al. Targeted Therapy Resistance Mediated by Dynamic Regulation of Extrachromosomal Mutant EGFR DNA. Science (2014) 343(6166):72–6. doi: 10.1126/science.1241328
155. Francis JM, Zhang C-Z, Maire CL, Jung J, Manzo VE, Adalsteinsson VA, et al. EGFR Variant Heterogeneity in Glioblastoma Resolved Through Single-Nucleus Sequencing. Cancer Discov (2014) 4(8):956–71. doi: 10.1158/2159-8290.CD-13-0879
156. Felsberg J, Hentschel B, Kaulich K, Gramatzki D, Zacher A, Malzkorn B, et al. Epidermal Growth Factor Receptor Variant III (EGFRvIII) Positivity in EGFR-Amplified Glioblastomas: Prognostic Role and Comparison Between Primary and Recurrent Tumors. Clin Cancer Res (2017) 23(22):6846–55. doi: 10.1158/1078-0432.CCR-17-0890
157. Gadgeel S, Rodríguez-Abreu D, Speranza G, Esteban E, Felip E, Dómine M, et al. Updated Analysis From KEYNOTE-189: Pembrolizumab or Placebo Plus Pemetrexed and Platinum for Previously Untreated Metastatic Nonsquamous Non-Small-Cell Lung Cancer. J Clin Oncol (2020) 38(14):1505–17. doi: 10.1200/JCO.19.03136
158. Chandramohan V, Bao X, Yu X, Parker S, McDowall C, Yu Y-R, et al. Improved Efficacy Against Malignant Brain Tumors With EGFRwt/EGFRvIII Targeting Immunotoxin and Checkpoint Inhibitor Combinations. J Immunother Cancer (2019) 7(1):1–14. doi: 10.1186/s40425-019-0614-0
159. Gan HK, Walker F, Burgess AW, Rigopoulos A, Scott AM, Johns TG. The Epidermal Growth Factor Receptor (EGFR) Tyrosine Kinase Inhibitor AG1478 Increases the Formation of Inactive Untethered EGFR Dimers: Implications for Combination Therapy With Monoclonal Antibody 806. J Biol Chem (2007) 282(5):2840–50. doi: 10.1074/jbc.M605136200
160. Orellana L, Thorne AH, Lema R, Gustavsson J, Parisian AD, Cordeiro TN, et al. Oncogenic Mutations at the EGFR Ectodomain Structurally Converge to Remove a Steric Hindrance on a Kinase-Coupled Cryptic Epitope. Proc Natl Acad Sci (2019) 116(20):10009–18. doi: 10.1073/pnas.1821442116
161. Anderson MG, Falls HD, Mitten MJ, Oleksijew A, Vaidya KS, Boghaert ER, et al. Targeting Multiple EGFR-Expressing Tumors With a Highly Potent Tumor-Selective Antibody-Drug Conjugate. Mol Cancer Ther (2020) 19(10):2117–25. doi: 10.1158/1535-7163.MCT-20-0149
162. Levengood MR, Zhang X, Hunter JH, Emmerton KK, Miyamoto JB, Lewis TS, et al. Orthogonal Cysteine Protection Enables Homogeneous Multi-Drug Antibody–Drug Conjugates. Angew Chem Int Edition (2017) 56(3):733–7. doi: 10.1002/anie.201608292
163. Anami Y, Xiong W, Gui X, Deng M, Zhang CC, Zhang N, et al. Enzymatic Conjugation Using Branched Linkers for Constructing Homogeneous Antibody–Drug Conjugates With High Potency. Org Biomol Chem (2017) 15(26):5635–42. doi: 10.1039/C7OB01027C
164. Gerber H-P, Sapra P, Loganzo F, May C. Combining Antibody–Drug Conjugates and Immune-Mediated Cancer Therapy: What to Expect? Biochem Pharmacol (2016) 102:1–6. doi: 10.1016/j.bcp.2015.12.008
165. Griguolo G, Pascual T, Dieci MV, Guarneri V, Prat A. Interaction of Host Immunity With HER2-Targeted Treatment and Tumor Heterogeneity in HER2-Positive Breast Cancer. J Immunother Cancer (2019) 7(1):1–14. doi: 10.1186/s40425-019-0548-6
166. Khattar M, Traore T, Horton K, Gallery M, Brauer P, Riceberg J, et al. Synergy of an Anti-HER2 ADC TAK-522 (XMT-1522) in Combination With Anti-PD1 Monoclonal Antibody (mAb) in a Syngeneic Breast Cancer Model Expressing Human HER2. Cancer Res (2018) 78(13 Suppl):LB-294. doi: 10.1158/1538-7445.AM2018-LB-294
167. Müller P, Kreuzaler M, Khan T, Thommen DS, Martin K, Glatz K, et al. Trastuzumab Emtansine (T-DM1) Renders HER2+ Breast Cancer Highly Susceptible to CTLA-4/PD-1 Blockade. Sci Trans Med (2015) 7(315):315ra188–315ra188. doi: 10.1126/scitranslmed.aac4925
168. Rios-Doria J, Harper J, Rothstein R, Wetzel L, Chesebrough J, Marrero A, et al. Antibody–Drug Conjugates Bearing Pyrrolobenzodiazepine or Tubulysin Payloads Are Immunomodulatory and Synergize With Multiple Immunotherapies. Cancer Res (2017) 77(10):2686–98. doi: 10.1158/0008-5472.CAN-16-2854
169. Reardon DA, Lassman AB, van den Bent M, Kumthekar P, Merrell R, Scott AM, et al. Efficacy and Safety Results of ABT-414 in Combination With Radiation and Temozolomide in Newly Diagnosed Glioblastoma. Neuro-Oncol (2017) 19(7):965–75. doi: 10.1093/neuonc/now257
170. Lassman AB, Gan HK, Roberts-Rapp L, Ansell P, Merrell R, Kumthekar P, et al. (2017). Identifying the Correct Patient Population for Depatuxizumab Mafodotin (ABT-414): Biomarker Assays for Epidermal Growth Factor Receptor (EGFR) in Patients With Glioblastoma. In: World Federation of Neuro-Oncology. Zurich, 4-7 May, 2017.
171. Ileana Dumbrava E, Meric-Bernstam F, Yap TA. Challenges With Biomarkers in Cancer Drug Discovery and Development. Expert Opin Drug Discov (2018) 13(8):685–90. doi: 10.1080/17460441.2018.1479740
172. Lassman A, Roberts-Rapp L, He L, Lu X, van den Bent M, Papadopoulos K, et al. PATH-29. Molecular Determinants Associated With Response and Resistance to Depatuxizumab Mafodotin (ABT-414) In Patients With Recurrent GLIOBLASTOMA. Neuro-Oncol (2017) 19(suppl_6):vi176–7. doi: 10.1093/neuonc/nox168.719
173. Scott AM, Lee F-T, Tebbutt N, Herbertson R, Gill SS, Liu Z, et al. A Phase I Clinical Trial With Monoclonal Antibody Ch806 Targeting Transitional State and Mutant Epidermal Growth Factor Receptors. Proc Natl Acad Sci (2007) 104(10):4071–6. doi: 10.1073/pnas.0708144104
Keywords: antibody drug conjugates (ADC), glioma, glioblastoma, blood brain barrier, tumour microenvironment, biomarkers, molecular imaging
Citation: Parakh S, Nicolazzo J, Scott AM and Gan HK (2021) Antibody Drug Conjugates in Glioblastoma – Is There a Future for Them? Front. Oncol. 11:718590. doi: 10.3389/fonc.2021.718590
Received: 01 June 2021; Accepted: 15 November 2021;
Published: 03 December 2021.
Edited by:
David Nathanson, UCLA David Geffen School of Medicine, United StatesReviewed by:
Terry Calvin Burns, Mayo Clinic, United StatesGerardo Caruso, University Hospital of Policlinico G. Martino, Italy
Copyright © 2021 Parakh, Nicolazzo, Scott and Gan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hui Kong Gan, aHVpLmdhbkBvbmpjcmkub3JnLmF1