 Kai-Yen Peng1
Kai-Yen Peng1 Shih-Sheng Jiang2Yu-Wei Lee1Fang-Yu Tsai2Chia-Chi Chang1,3Li-Tzong Chen2,4,5
Shih-Sheng Jiang2Yu-Wei Lee1Fang-Yu Tsai2Chia-Chi Chang1,3Li-Tzong Chen2,4,5 B. Linju Yen1*
B. Linju Yen1*- 1Regenerative Medicine Research Group, Institute of Cellular & System Medicine, National Health Research Institutes (NHRI), Zhunan, Taiwan
- 2National Institute of Cancer Research, NHRI, Zhunan, Taiwan
- 3Graduate Institute of Life Sciences, National Defense Medical Center, Taipei, Taiwan
- 4Department of Oncology, National Cheng Kung University Hospital, College of Medicine, National Cheng Kung University, Tainan, Taiwan
- 5Division of Hematology/Oncology, Department of Internal Medicine, Kaohsiung Medical University Hospital, Kaohsiung, Taiwan
Over 90% of colorectal cancer (CRC) patients have mutations in the Wnt/β-catenin pathway, making the development of biomarkers difficult based on this critical oncogenic pathway. Recent studies demonstrate that CRC tumor niche-stromal cells can activate β-catenin in cancer-initiating cells (CICs), leading to disease progression. We therefore sought to elucidate the molecular interactions between stromal and CRC cells for the development of prognostically relevant biomarkers. Assessment of CIC induction and β-catenin activation in CRC cells with two human fibroblast cell-conditioned medium (CM) was performed with subsequent mass spectrometry (MS) analysis to identify the potential paracrine factors. In vitro assessment with the identified factor and in vivo validation using two mouse models of disease dissemination and metastasis was performed. Prediction of additional molecular players with Ingenuity pathway analysis was performed, with subsequent in vitro and translational validation using human CRC tissue microarray and multiple transcriptome databases for analysis. We found that fibroblast-CM significantly enhanced multiple CIC properties including sphere formation, β-catenin activation, and drug resistance in CRC cells. MS identified galectin-1 (Gal-1) to be the secreted factor and Gal-1 alone was sufficient to induce multiple CIC properties in vitro and disease progression in both mouse models. IPA predicted SOX9 to be involved in the Gal-1/β-catenin interactions, which was validated in vitro, with Gal-1 and/or SOX9—particularly Gal-1high/SOX9high samples—significantly correlating with multiple aspects of clinical disease progression. Stromal-secreted Gal-1 promotes CIC-features and disease dissemination in CRC through SOX9 and β-catenin, with Gal-1 and SOX9 having a strong clinical prognostic value.
Introduction
Colorectal cancer (CRC) is one of most common cancers worldwide, with its incidence and mortality rising in individuals age 50 and younger (1). While overall decreases in the CRC risk factors of alcohol intake and smoking along with an increased screening has helped reduce its incidence for several decades, alarmingly, in recent years, incidence rates have been on the increase in younger adults 50 years and under (2, 3). Survival rates of CRC strongly correlate with stage, with the 5-year survival for stage I or localized disease (excluding carcinoma-in-situ) close to 90%, but decreasing to less than 20% for stage IV or metastatic disease (4, 5). Thus, understanding of the specific molecular factors involved in CRC metastases is important for the control of this globally prevalent cancer.
The Wnt/β-catenin pathway is central to CRC, with the initial step in carcinogenesis determined to be mutations in the adenomatous polyposis coli gene, which then results in the activation of β-catenin, a transcription factor critical in the maintenance of the normal intestinal stem cell compartment as well as the cells-of-origin or carcinoma-initiating cells (CICs) for CRC (6–8). Over 90% of patients have mutations in the Wnt/β-catenin pathway, making components of this pathway ironically not useful as prognostic markers (9); rather, the functional triggering of the pathway appears to be a more robust evidence for disease progression (10). Paracrine factors secreted by non-cancerous cells within the tumor microenvironment including stromal cells play important roles in the tumorigenesis of CRC. Fibroblasts, which are key components of the stroma, can promote tumorigenic and metastatic capacity in CRC CICs through the upregulation of β-catenin activity (11–13). Despite such recent key findings, there has not been much effort in using the tumor niche to search for prognostic biomarkers. We therefore became interested in elucidating the molecular interactions between tumor niche stromal cells and CRC metastasis, and to develop prognostically relevant biomarkers based on these interactions.
In this study, we determined the interactions of fibroblast-secreted factors on CRC progression, and found galectin-1 (Gal-1) to be highly secreted by two lines of human fibroblasts as determined by mass spectrometry (MS) analyses of the fibroblast-conditioned medium (CM). Gal-1 is a glycoprotein encoded by the LGALS1 gene and known to exert immunomodulatory effects including mediating tumor-immune escape (14). We found that the secreted Gal-1 has prominent direct tumor-promoting effects in CRC including enhancing CIC features and β-catenin activity in vitro, as well as in vivo tumor dissemination and disease progression. Moreover, as predicted by the Ingenuity Pathway Analysis (IPA), we validated the involvement of SOX9 (15)—a newly identified marker for aggressive CRC based on a recent large-scale integrative analysis—in Gal-1/β-catenin interactions. Analyses using human CRC transcriptomic databases and immunohistological staining of tissue array corroborated the strong clinical relevance of Gal-1 and SOX9—particularly Gal-1high/SOX9high samples—as significantly and prognostically correlated with disease presence and progression.
Materials and Methods
Cell Culture
The human CRC cell lines KM12C was obtained from the Korean Cell Line Bank (catalog no.: 80015) (16, 17). The cells were cultured as recommended in a complete medium consisting of Dulbecco’s Modified Eagle Medium: Nutrient Mixture F-12 (DMEM/F-12) supplemented with 10% fetal bovine serum (FBS), 2 mM L-glutamine, and 100 U/ml penicillin-streptomycin (all from Invitrogen-Thermo Fisher Scientific, Waltham, MA, USA). The human fibroblast cell lines MRC-5, derived from fetal lung tissue, and WS1, derived from fetal midscapular skin, were obtained from the Bioresource Collection and Research Center (BCRC, Hsinchu, Taiwan) and were cultured as recommended in the Minimum Essential Medium (MEM) with 10% FBS, 2 mM L-glutamine, and 100 U/ml penicillin-streptomycin (all from Invitrogen). Conditioned medium (CM) was collected from the cell culture after 48 hours of culturing. All cell lines were authenticated using a short-tandem repeat profiling.
Invasion Assay
Cells were treated with mitomycin C (10 ug/ml) for 2 hours to inhibit proliferation, and then 1x105 cells were seeded on Matrigel-coated chambers containing 75% Matrigel (Sigma-Aldrich, MO, USA; plates with 8.0-μm pores, BD Bioscience, Franklin Lakes, NJ, USA). After CM or treatment with recombinant protein for 48 hours, detection by light microscopy (Leica Microsystems, Wetzlar, Germany) for quantification of invading cells in polycarbonate membranes was performed. Each chamber was sampled at nine different sites. Images were quantified for the number of invading cells using the Image J software (National Institutes of Health (NIH), USA).
Quantitative Real-Time Polymerase Chain Reaction
qPCR was performed as previously reported (18). Briefly, RNA was extracted from cells with TRIzol (Invitrogen), and converted to cDNA with the ImProm-ll Reverse Transcriptase system (Promega, Madison, WI, USA) according to the protocol of the manufacturer. qPCR was carried out with Fast SYBR® Green Master Mix containing Thermo-Start DNA polymerase on the ABI 7500 Real-Time PCR System (both from Applied Biosystems-Thermo Fisher Scientific). Relative gene expression levels were analyzed as indicated by the manufacturer. The specific primers used are shown in Supplementary Table S1.
Western Blot
Western blot was performed as previously reported (18). Cells were collected from a 10-cm2 dish (8 x 105 cells/dish) and detected for whole cell or nuclear proteins, which was isolated with the nuclear extraction kit (Millipore-Merck, Darmstadt, Germany) according to the recommendations of the manufacturer. Extracted proteins were separated by electrophoresis on a 12.5% SDS-polyacrylamide gel and transferred to a nitrocellulose membrane. The membranes were blotted with antibodies against β-catenin (1:1,000; Cat. No.610153; BD Bioscience), Gal-1 (1:1,000; Cat. No.437400; Invitrogen), Slug (1:1,000; Cat. No. ABE993; Millipore), E-cadherin (1:1,000; Cat. No. MAB3199; Millipore), SOX9 (1:1,000; Cat. No. ab3697; Abcam), GAPDH (1:8,000; Cat. No.14-9523-80; eBioscience, CA, USA), α-Tubulin (1:8,000; Cat. No.14-4502-82; eBioscience), or Histone H1 (1:1,000; Cat. No. ab125027; Abcam). Detection was performed using horseradish peroxidase (HRP)-conjugated secondary antibodies and chemiluminescent HRP substrate (Millipore).
Small-Interfering RNA Knockdown Experiments
Gene knockdown experiments were performed as previously reported (18). Lipofectamine RNAiMAX Reagent (Invitrogen) was used to transfect siRNA specific for galectin-1 (LGALS1) or a non-target control (Invitrogen) into cells according to the recommendations of the manufacturer. After 48 hours, RT-PCR and Western blot were used to confirm the expression levels of LGALS1 in transfected cells. For short hairpin RNA (shRNA) knockdown experiments, MISSION TRC shRNA Lentiviral Particles (Sigma-Aldrich) containing LGALS1 or SOX9 shRNA or shLuc were used to infect the cells, which were seeded on 24-wells plates (1 x 105 cells/well) for 48 hours. The infected cells were treated with puromycin (2 ug/ml; Invitrogen) for two weeks to select the stably infected cells.
Sphere Formation
For sphere formation, cells were seeded in 6-cm2 dishes coated with 0.4% agarose gel (6 x 105 cells/well), and grown in serum-free DMEM/F-12 containing 20 ng/ml human recombinant epidermal growth factor (EGF) and 10 ng/ml basic fibroblast growth factor (bFGF; both from Peprotech, Rocky Hill, NJ, USA) for 72 hours (10). For each condition, 15 sites were randomly sampled with light microscopy (Leica Microsystems) and then quantified for the number of spheres (>30 μm) using the Image J software.
Mass Spectrometric Analysis
MS analysis on the secretome of fibroblasts was performed by Proteomics Core Lab of Chang Gung University (Taoyuan, Taiwan) as previously reported (19). Briefly, CM (24 ml) was harvested from the fibroblasts cultured in a T175 flask for 48 hours. CM was concentrated by centrifugation in Amicon Ultra-15 tubes (molecular weight cutoff 10 KDa, Millipore) five times at 4,000 g for 30 minutes each time. Proteins were separated by 2D gel electrophoresis and subjected to silver staining. Protein bands were extracted and analyzed for peptide mass by MS with MS/MS used to analyze CM protein profiles.
Enzyme Linked Immunosorbent Assay
ELISA was performed as previously reported (20). Briefly, mouse monoclonal anti-Gal-1 antibody (1:500; Cat. No.437400; Invitrogen) was coated in 96-well plates at 4°C overnight. CM was added into the wells for 2 hours at room temperature. After PBST (PBS with 0.1% Tween 20) wash, wells were incubated in biotinylated rabbit anti-Gal-1 antibody (1:2,000; Cat No.500-P210; Peprotech) for 1 hour. Subsequently, HRP-conjugated streptavidin (1:200; R&D systems, MN, USA) and TMB substrate (Invitrogen) were used to detect biotinylated signaling. Finally, the reaction was stopped by 2N H2SO4 and absorbance was measured by optical density at 450 nm. Recombinant human Gal-1 (Peprotech) was used as a positive control. The detection range of the standard curve was from 0 to 20,000 pg/ml.
Luciferase Reporter Assay
The β-catenin-mediated transcriptional activation reporter plasmids of TOPFlash and TOPFlash mutant (contains mutated TCF/LEF binding sites) were obtained from Addgene (Cambridge, MA, USA). Reporter plasmids were transfected with renilla reporter plasmids (for internal control) into cells by using Lipofectamine 2000 reagent (Invitrogen). After 48 hours of transfection, Dual-Luciferase Reporter System (Promega) was used to measure the luciferase activity.
Immunofluorescence Staining
IF was performed as previously reported (21). Briefly, cells were fixed with 4% paraformaldehyde and permeabilized with 0.1% Triton-X 100 (Sigma-Aldrich) for 20 minutes. After blocking, cells were incubated with primary antibodies against E-cadherin (1:100; Cat. No. MAB3199; Millipore) and β-catenin (1:100; Cat. No.610153; BD Biosciences) at room temperature for 2 hours, and then incubated for 1 hour with FITC-conjugated secondary antibodies (1:200; Santa Cruz Biotechnology, Santa Cruz, CA, USA). Nuclei were stained with Hoechst 33342 (Sigma-Aldrich) and cells were visualized by fluorescence microscopy (Olympus, Tokyo, Japan).
Drug Resistance Assay
Cells were seeded in a 24-well plate (4 x 104 cells/plate) and pretreated with CM (1:1 mixed with culture medium) or human Gal-1 recombinant protein (rhGal1) for 24 hours. Cisplatin (25 µM; Sigma-Aldrich) was then added to the plates with assessment for cell viability after 48 hours using the 3-(4,5-Dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide (MTT) assay (Sigma-Aldrich).
Ingenuity Pathway Analysis (IPA)
IPA (QIAGEN, Redwood, CA, USA) was used to infer the potential pathways in CRC disease progression involving LGALS1, CTNNB1, and Twist1. The pathway explorer of IPA was used to analyze the direct and indirect interactions of these three genes by utilizing Ingenuity Pathways Knowledge Base.
In Vivo Tumor Dissemination and Metastases Experiments
Animal experimentation was performed in accordance with protocols approved by the Institutional Animal Care and Use Committee (approval number: 1080102). All animals were obtained from the National Laboratory Animal Center of Taiwan (Taipei, Taiwan). A rapid metastatic tumor dissemination study was performed (22). WS1 fibroblasts and KM12C CRC cells were labeled respectively with 5 μM of the fluorescent cellular dyes 1,1’-Dioctadecyl-3,3,3’,3’-Tetramethylindocarbocyanine Perchlorate (DiI) and 3,3’-Dioctadecyloxacarbocyanine Perchlorate (DiO) (both from Invitrogen) for 5 minutes. WS1 (3 x 105 cells) were co-cultured with KM12C (3 x 105 cells) in a 1:1 ratio for 24 hours and injected into the tail vein of C57BL/6 mice (6–8 weeks old). Mice were then sacrificed 24 hours after injection. The lungs were extracted for sectioning (0.5 mm thickness) with the detection of labeled cells with fluorescence microscopy (Olympus). The fluorescence intensity of images was measured by the Image J software. For in vivo metastatic experiments, KM12C (3 x 105 cells) co-cultured with WS1 silenced for short-hairpin RNA (shRNA) of non-target sequences (shC-WS1; 3 x 105 cells) or with WS1silenced with shRNA specific for Gal-1 (shGal-WS1; 3 x 105 cells) for one day, then injected into the tail veins of NOD-SCID mice (6–8 weeks old) and followed for up to 6 weeks with weekly measurement of the body weight. Mice were then sacrificed with lung and spleen tissues collected for histological examination.
Immunohistochemistry of Mouse and Human Tissue
Tissue samples from mice were fixed with 10% formaldehyde and embedded with the optimal cutting temperature (OCT) compound prior to frozen sectioning (Sakura Finetek, Tokyo, Japan). Tissue sections were stained with anti-human histone H1 antibody (1:100; Cat. No. ab125027; Abcam) followed by peroxidase detection (Pierce-Thermo Fisher Scientific) to detect human cells in murine lung and spleen sections. Human CRC tissue arrays were obtained from SUPER BIO CHIPS (Seoul, Korea). The tissue slides were dewaxed with xylene, rehydrated in ethanol, and subsequently stained with antibodies against human Gal-1 (1:100; Cat. No. 437400; Invitrogen) and SOX9 (1:100; Cat. No. AB5535; Millipore).
Public Microarray Gene Expression Dataset Analyses
CRC transcriptome datasets including GSE33113, GSE17536, and GSE9348 were downloaded from Gene Expression Omnibus (GEO) databases of the National Center for Biotechnology Information (https://www.ncbi.nlm.nih.gov/gds/). The Cancer Genome Atlas (TCGA) database was obtained from the NIH (https://cancergenome.nih.gov). Information on the public gene expression datasets used in this study are listed in Supplementary Table S2. GSE33113 and GSE17536 were used to analyze the expression of CTNNB1 and LGALS1 (23, 24). For comparing the gene expression levels between normal colon tissues and CRC tissue, Oncomine (http://www.oncomine.org) was utilized to analyze for the expression levels of LGALS1 and SOX9 in the Kaiser Colon database (25). To analyze the expression levels of LGALS1 and SOX9 with respect to early stages of CRC compared to normal colon tissue, GSE9348 and TCGA were used (26, 27), while GSE17536 and TCGA were used to analyze for the stage-specific expression of LGALS1, SOX9, and CTNNB1 (24, 27).
Statistical Analyses
All experiments were performed at least in triplicate, with data were represented as mean ± SEM. Statistical analyses were performed using the Student’s t test for comparisons of two variables and ANOVA for comparisons of more than two variables. For CRC patient transcriptome databases GSE33113, GSE7536, GSE9348, and TCGA, Student’s t test was used for the analysis of differences in the specific gene expression levels at each stage of CRC. A value of p < 0.05 was defined as statistically significant.
Results
Fibroblast-Secreted Factors Significantly Promote Multiple Cancer-Initiating Cell Features in Colorectal Cells
To assess whether fibroblast-derived paracrine factors are involved in CRC progression, the CRC cell line KM12C was cultured in the CM of two human fibroblast cell lines, MRC-5, and WS1, and assessed for a number of CIC properties including invasive capacity, epithelial-mesenchymal transition (EMT), β-catenin translocation, sphere formation, and drug resistance; these functional assays have been shown to be more relevant to disease progression than CIC markers such as CD133 (28). When cultured in MRC-5- and WS1-CM, the invasive capacity of KM12C was significantly increased (Figure 1A) and expression of Twist1, a critical transcription factor involved in EMT, was increased significantly up to 2-fold (Figure 1B). Moreover, we found that after culturing in either MRC-5- or WS1-CM in particular, β-catenin protein levels in KM12C were increased (Figure 1C) with the occurrence of nuclear translocation (Figure 1D), which has been reported to enhance CRC tumorigenesis and CIC formation (12). In addition, the sphere formation capacity as well as drug resistance were significantly increased in KM12C after culturing in either MRC-5- or WS1-CM (Figures 1E, F). We found that KM12C, which was pretreated with MRC-5- or WS1-CM demonstrated a significantly increased resistance to cisplatin-induced cell death, particularly after WS1-CM pretreatment. MRC-5- and WS1-CM also increased CD29 and CD44 expressions, two well-studied CIC markers, as assessed by flow cytometric analysis (29, 30), in KM12C (Supplementary Figure S1). These findings therefore demonstrate that fibroblast-derived paracrine factors significantly promote multiple CIC features in CRC cells.
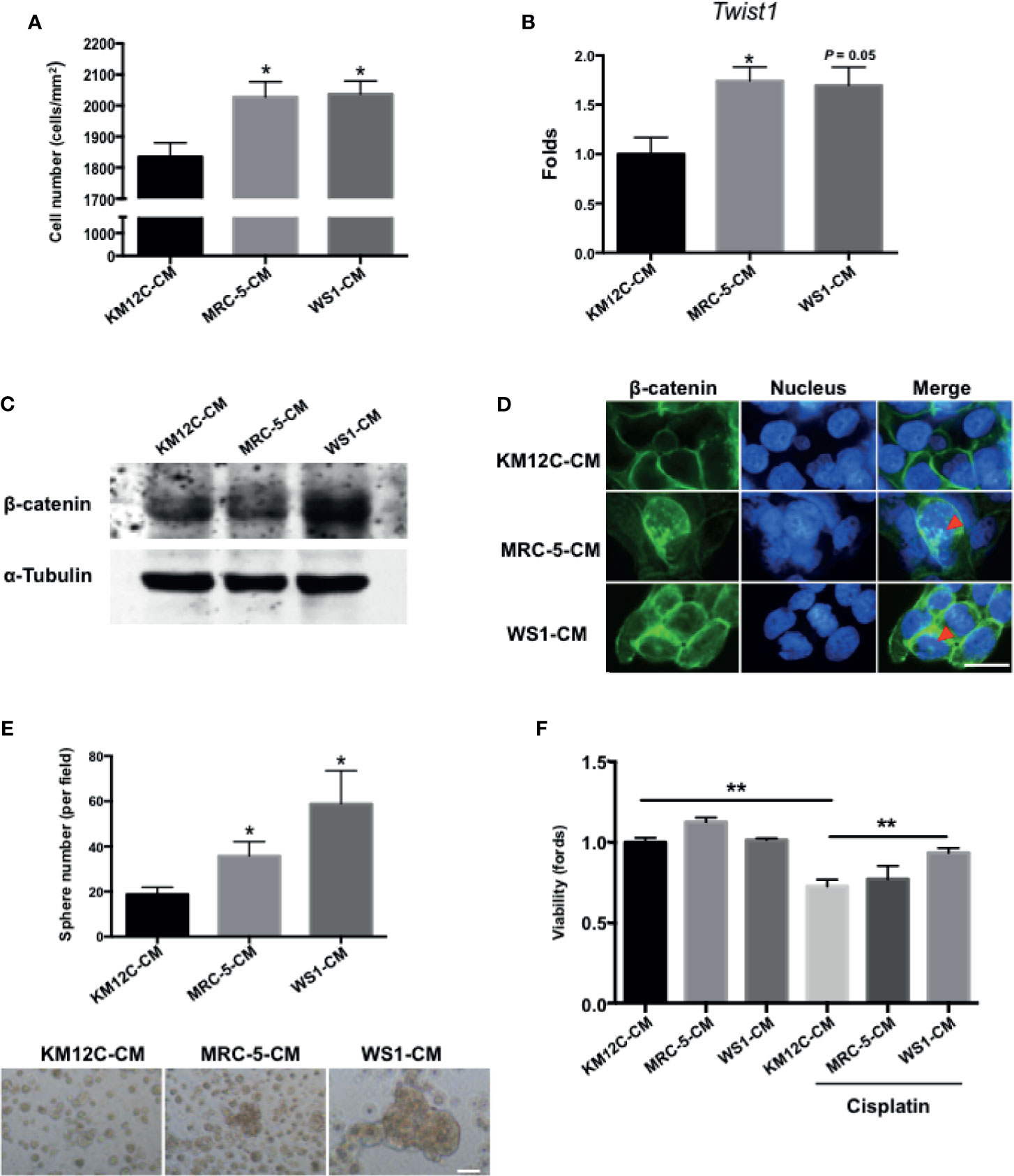
Figure 1 Fibroblast-derived paracrine factors significantly promote multiple cancer-initiating cell (CIC) features in colorectal cancer (CRC) cells. (A) Invasion capacity of the CRC cell line KM12C (KM12C) after culturing in conditioned media (CM) of two human fibroblast cell lines MRC-5 and WS1 for 48 hours; control, KM12C-CM. (B) Quantitative real-time PCR (qPCR) analysis for the gene expression levels of Twist1 in KM12C after culturing in MRC-5- or WS1-CM; control, KM12C-CM. (C) Western blot for the β-catenin levels in KM12C after culturing in MRC-5- or WS1-CM; control, KM12C-CM. (D) Immunofluorescent (IF) staining for β-catenin subcellular localization (green fluorescence) in KM12C after culturing in MRC-5- or WS1-CM; control, KM12C-CM; arrows indicate nuclear β-catenin. Hoechst 33342 was used to detect cell nuclei (blue fluorescence); scale bar, 10 μm. (E) Sphere formation capacity of KM12C after culturing in MRC-5- or WS1-CM for 72 hours; control, KM12C-CM. Quantitative results (top panel) and representative images (bottom panel) are shown; scale bar, 30 μm. (F) Drug resistance capacity of KM12C to cisplatin (25 µM) after pretreatment with either MRC-5- or WS1-CM (control, KM12C-CM) for 24 hours. Cell viability was assessed 48 hours after drug treatment. All results are shown as mean ± SEM of three independent experiments. *p < 0.05 and ** compared to control.
Fibroblast-Secreted Gal-1 Significantly Promotes Multiple Cancer-Initiating Cell Features in Colorectal Cancer Cells
To identify the specific fibroblast-derived paracrine factor(s) responsible for enhancing multiple CIC features, MS/MS was used to analyze the secretome of MRC-5- and WS1-CM, and Gal-1 was identified as the most highly secreted protein by both fibroblast populations (Supplementary Figure S2), which we confirmed with Western blot as well as ELISA (Figure 2A). While Gal-1 (LGALS-1) is well known to be involved in cancer immune evasion through modulating specific subpopulations of immune cells, there have been no reports of this protein directly targeting the cancer cell itself to promote CIC features. We therefore assessed whether Gal-1 could be the paracrine factor in fibroblast-CM directly responsible for promoting multiple CIC features in CRC cells. We found that the addition of recombinant human Gal-1 protein (rhGal-1) significantly enhanced the invasive capacity of KM12C (Figure 2B). Moreover, the addition of rhGal-1 promoted the EMT of KM12C in a dose-dependent fashion as evidenced by significant increases in the gene expression of Twist1 with a decreased expression of E-cadherin (Figure 2C). This was also seen at the protein level with an increased expression of Slug, another transcription factor involved in EMT, along with a concomitant decreased expression of E-cadherin (Figure 2D); IF staining also demonstrated a loss of E-cadherin expression at the cell junctions with the addition of rhGal-1 (Figure 2E). In addition, the sphere formation capacity (Figure 2F) as well as drug resistance (Figure 2G) were both significantly enhanced by rhGal-1 in a dose-dependent fashion. Addition of rhGal-1 also increased the expression of CD29 and CD44 in KM12C as well as HCT-116, another well-studied CRC line (Supplementary Figure S3). To further verify the role of fibroblast-secreted Gal-1 in promoting CIC features, we generated Gal-1-knockdown WS-1 fibroblasts (siGal-WS1) using gene-specific siRNA (siRNA-I; Figure 2H, left panel). We found that KM12C cultured in siGal-WS1-CM demonstrated a significant decreased capacity for invasion, compared to KM12C cultured in control non-target siRNA knockdown WS1-CM (siC; Figure 2H, right panel). Correspondingly, KM12C cultured in siGal-WS1-CM compared to siC-WS1-CM also showed a significantly decreased capacity in terms of sphere formation (Figure 2I) as well as drug resistance (Figure 2J). These results collectively demonstrate that fibroblast-secreted Gal-1 is involved in promoting multiple CIC features of CRC cells.
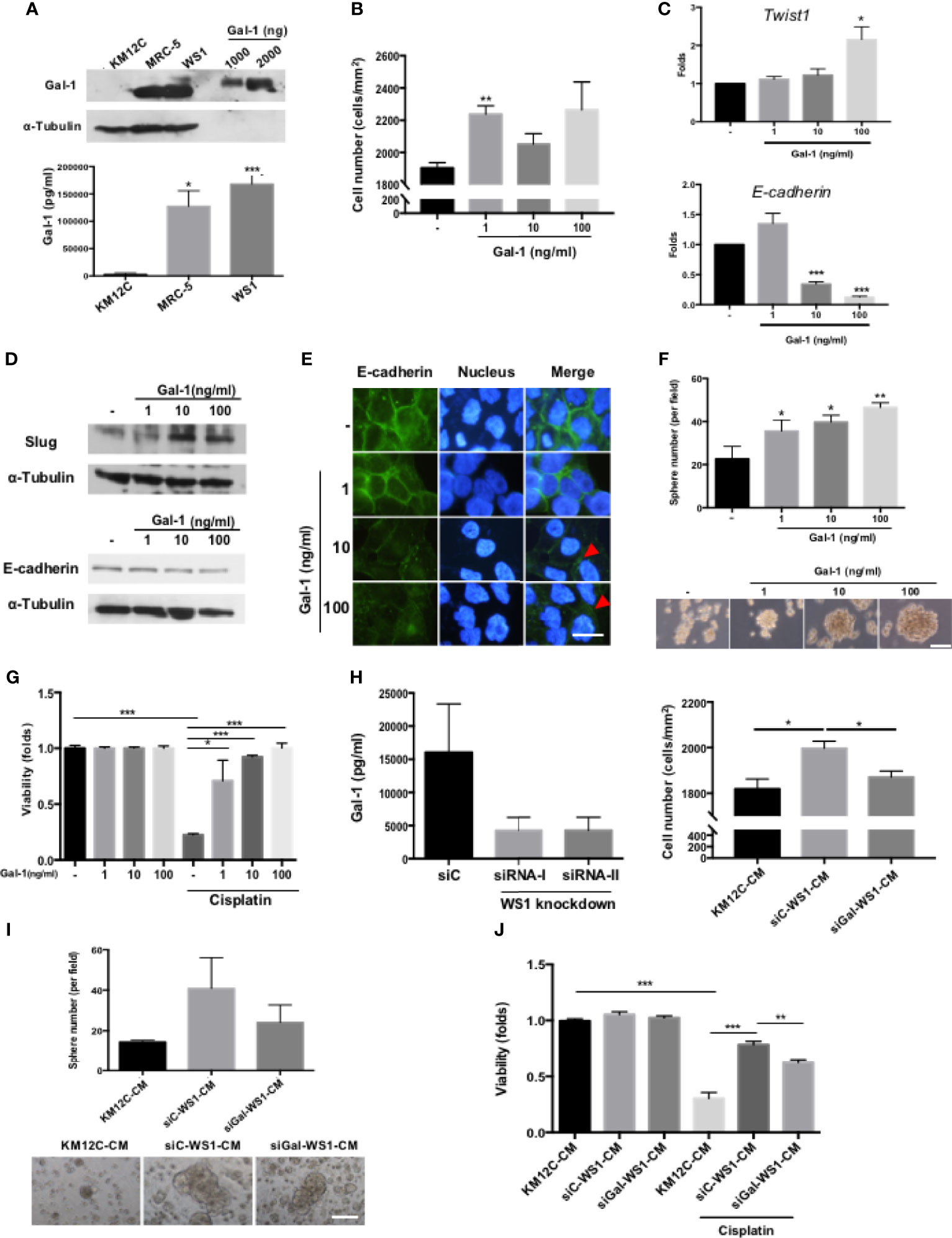
Figure 2 Fibroblast-secreted galectin-1 (Gal-1) significantly promotes multiple CIC features in CRC cells. (A) Expression of endogenous Gal-1 in MRC-5 and WS1 fibroblasts as detected through Western blot (top panel; recombinant human Gal-1 protein (rhGal-1) used as positive control) and ELISA (bottom panel). (B) Invasion capacity of KM12C with the addition of increasing doses of rhGal-1. (C) qPCR analysis for the gene expression of Twist1 and E-cadherin and (D) Western blot for protein expression of Slug and E-cadherin in KM12C with the addition of increasing doses of rhGal-1. (E) IF staining of E-cadherin (green fluorescence) in KM12C with the addition of increasing doses of rhGal-1 for 48 hours. Nuclei were stained with Hoechst 33342 (blue fluorescence); scale bar, 10 μm. (F) Sphere formation capacity of KM12C with the addition of increasing doses of rhGal-1; quantitative results (top panel) and representative images (bottom panel) are shown; scale bar, 30 μm. (G) Drug resistance capacity of KM12C to cisplatin (25 µM) after pretreatment with increasing dosages of rhGal-1 for 24 hours. Cell viability was assessed 48 hours after drug treatment. (H) Left panel: Validation of Gal-1 knockdown using small interfering RNA (siRNA) specific for Gal-1 (siRNA-I & siRNA-II) in WS1 fibroblasts. Non-target siRNA was used as a negative control. After 48 hours, CM from siRNA-I, siRNA-II, and siC were removed and analyzed by ELISA. Right panel: Invasion capacity of KM12C after culturing in siGal-WS1-CM compared to siC-WS1-CM for 48 hours. (I) Sphere formation capacity of KM12C cultured in KM12C-CM, siC-WS1-CM, or siGal-WS1-CM for 72 hours; quantitative results (top panel) and representative images (bottom panel) are shown; scale bar, 30 μm. (J) Drug resistance capacity of KM12C to cisplatin (25 µM) after pretreatment with KM12C-CM, siGal-WS1-CM, or siC-WS1-CM for 24 hours. Cell viability was assessed 48 hours after drug treatment; control, KM12C without cisplatin treatment. All results are shown as the mean ± SEM of three independent experiments. *p < 0.05; **p < 0.01, and ***p < 0.005 compared to the control.
Fibroblast-Secreted Gal-1 Significantly Increases Metastases and Tumor Dissemination of Colorectal Cancer Cells In Vivo
To assess whether the CIC features induced by fibroblast-secreted Gal-1 are involved in CRC disease progression and metastasis, we used two mouse models of metastases to validate our in vitro findings: a longer-term metastatic disease model using immunocompromised mice and a rapid lung tumor dissemination model using wild type mice. To assess whether fibroblast-secreted Gal-1 promoted metastatic disease, we generated stable clones of WS1 silenced for Gal-1 expression using short hairpin RNA (shRNA) specific for LGALS1 (shGal-WS1). We then injected KM12C co-cultured with either shGal-WS1 (KM+shGal-WS1) or with non-target shRNA-silenced WS1 (KM+shC-WS1) into the tail vein of immunocompromised mice for the evaluation of metastatic growths in the lung. As negative and positive controls, we injected mice with WS1 only (shC-WS1; without KM12C) or KM12C only (KM), respectively. After 40 days of follow-up, mice injected with KM+shC-WS1 demonstrated significant decreases in body weight and were nearly moribund when compared to either mice injected with KM+shGal-WS1 or even KM12C only; WS1 only-injected mice, on the other hand, were healthy and demonstrated an increased weight gain over time (Figure 3A). Lung and spleen tissue sections from these mice demonstrated a significantly higher number of human cells in the mice injected with KM+shC-WS1 compared to those injected with KM+shGal-WS1 (Figures 3B, C; top panel: representative sections, and bottom panel: quantitative results). To ascertain that tumor dissemination was affected by fibroblast-secreted Gal-1, we co-cultured KM12C with either siGal-WS1 (KM+siGal-WS1) or with siC-WS-1 (KM+siC-WS1), and injected cells into the tail vein of C57BL/6 mice which were sacrificed after 24 hours to assess for tumor dissemination within the lungs. Tumor seeding was more significant in the mice injected with KM+siC-WS1 compared with KM+siGal-WS1 (Figure 3D). To assess for clinical relevance, we analyzed the human CRC transcriptome databases which contain recurrence information (GSE33113 and GSE17536; Supplementary Table S2) and found that high expression levels of LGALS1, but not β-catenin (CTNNB1), correlate significantly with a high risk of metastasis and/or recurrence within 3 years (Figure 3E). Thus, these results demonstrate that fibroblast-secreted Gal-1 significantly promotes metastatic disease progression and tumor dissemination in mouse models, as well as correlate to human CRC disease recurrence.
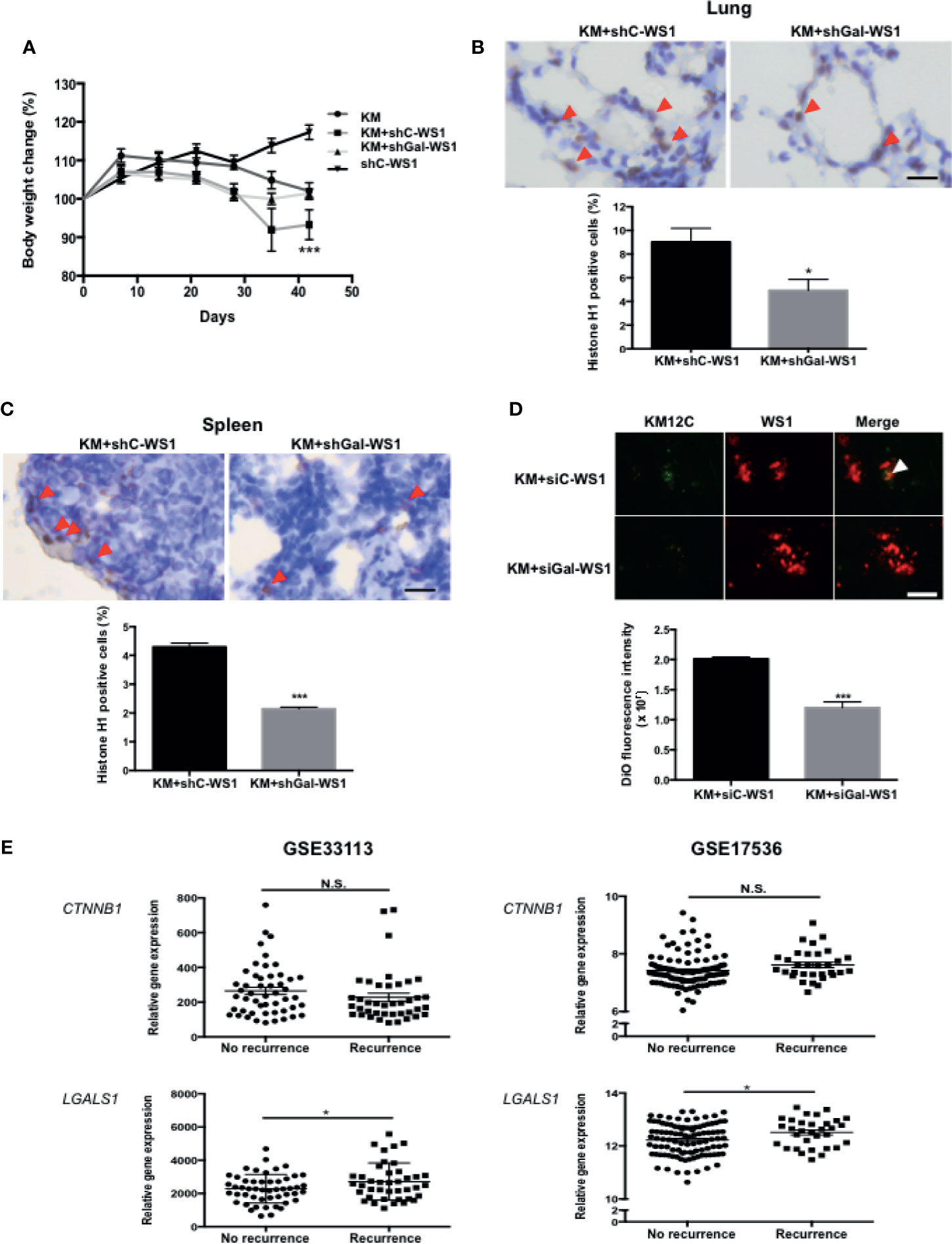
Figure 3 Fibroblast-secreted Gal-1 significantly increases metastasis and tumor dissemination of CRC cells in vivo. (A) Body weight of NOD-SCID mice 40 days after tail vein injection of KM12C only (3 x 105 cells); KM12C (3 x 105 cells) admixed with WS1 silenced for with short-hairpin RNA (shRNA) of non-target sequences (shC-WS1; 3 x 105 cells); KM12C (3 x 105 cells) admixed with WS1 silenced with shRNA specific for Gal-1 (shGal-WS1; 3 x 105 cells); or shC-WS1 only (3 x 105 cells). Each condition consisted of three mice, with their body weight measured every 7 days. Immunohistochemistry (IHC) staining for human histone H1 (brown nuclei) in (B) mouse lung and (C) spleen tissue sections; representative sections (top panel) and quantitative results (bottom panel) are shown, with arrows indicating human Histone H1(+) cells; scale bar, 20 μm. (D) Visualization of fluorescently labeled co-cultured cells KM12C (3 x 105 cells; green fluorescence, labeled with DiO), and siC- or siGal-WS1 (3 x 105 cells; red fluorescence, labeled with with DiI) in lung sections 24 hours after injection into the tail vein of C57BL/6 mice. Top panel, representative images; bottom panel; quantitative results. Arrows indicate KM12C; scale bar, 100 μm. All results are shown as the mean ± SEM of three independent experiments. *p < 0.05, and ***p < 0.005 compared to the control. (E) Analyses of Gal-1 (LGALS1) and β-catenin (CTNNB1) expression with regard to disease recurrence in the public dataset GSE33113 and GSE17536 of gene expression omnibus (GEO); *p < 0.05; N.S., not significant.
Gal-1 Promotes β-catenin Expression, Nuclear Accumulation, and Activity in Colorectal Cancer Cells
Wnt/β-catenin signaling is the central pathway involved in CRC pathogenesis, with the activation of the pathway being a feature of CICs and correlating with a more aggressive disease outcome. We therefore assess whether secreted Gal-1 can activate this pathway in CRC cells. We found that treatment of KM12C with exogenous rhGal-1 induced a cytoplasmic to nuclear translocation of β-catenin, as evidenced by IF staining (Figure 4A). This was corroborated by the Western blot data, in which both total as well as nuclear β-catenin levels were increased with increasing doses of rhGal-1 (Figure 4B). To further ascertain for the activation of β-catenin activity, we performed luciferase reporter assay by transducing either the wild type or mutant TOPFlash reporter into KM12C and then treating with rhGal-1. We found that all doses of rhGal-1 significantly induce reporter activity over baseline in the wild type but not the mutant promoter (Figure 4C). β-catenin has also been found to promote EMT (31), and we found that the addition of rhGal-1 strongly induced the expression of the EMT transcription factor Twist1 in KM12C, which could be suppressed with XAV-939 (XAV), an inhibitor of the β-catenin pathway (Figure 4D). XAV also decreased Gal-1-induced CD29 expression in HCT-116 (Supplementary Figure S4). These results therefore demonstrate that secreted Gal-1 could activate β-catenin activity in CRC cells.
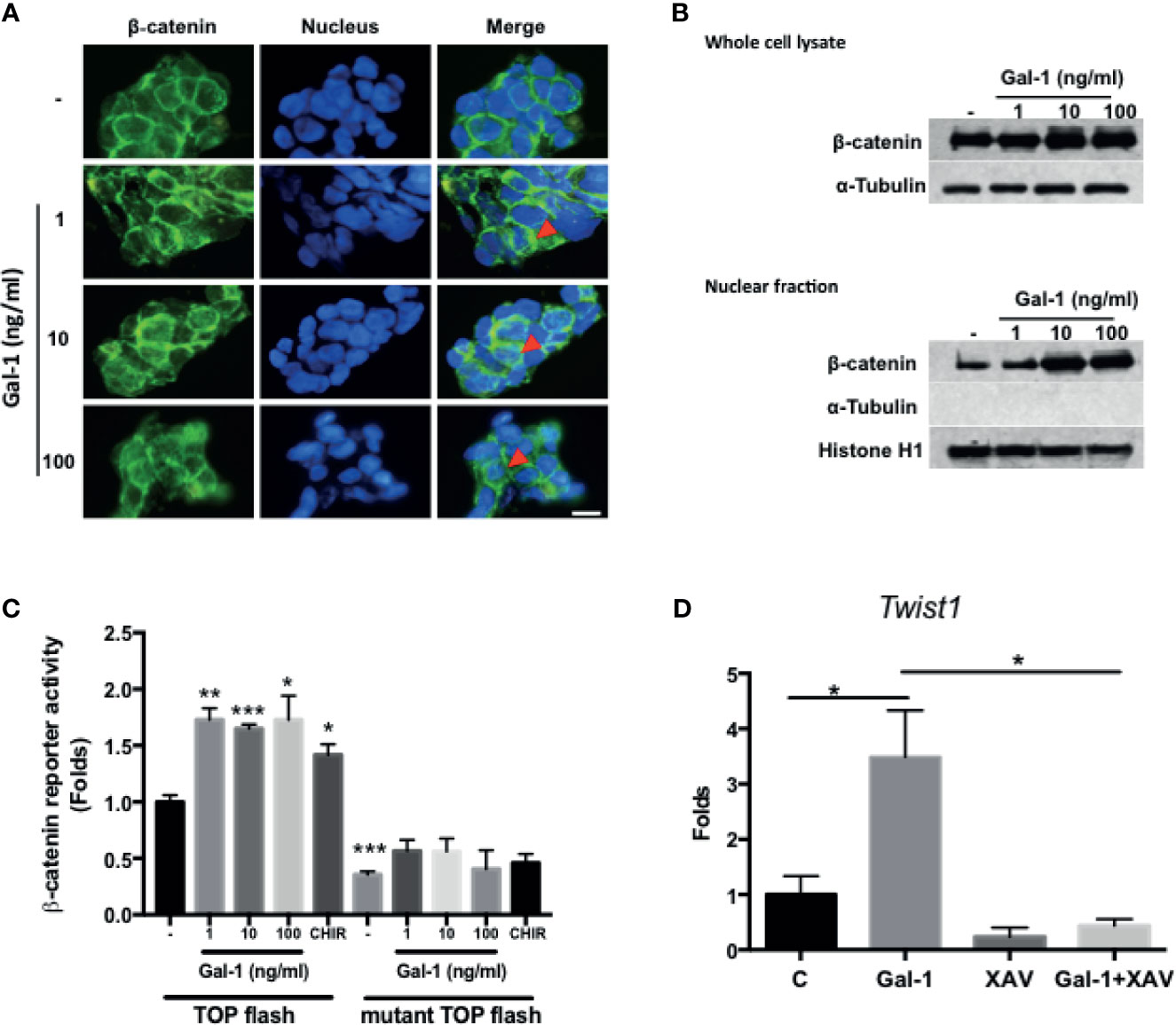
Figure 4 Gal-1 promotes β-catenin expression, nuclear translocation, and activity in CRC cells. (A) IF staining for β-catenin (green fluorescence) in KM12C with the addition of increasing doses of rhGal-1 for 48 hours. Nuclei were stained with Hoechst 33342 (blue fluorescence). Arrows show nuclear β-catenin; scale bar, 10 μm. (B) Western blot for β-catenin levels in whole cell lysate (top panel) and nuclear fraction (bottom panel) of KM12C with the addition of increasing doses of rhGal-1 for 48 hours; for nuclear fraction, histone H1 is used as the positive control and α-Tubulin as the negative control. (C) Luciferase reporter assay for β-catenin activity in KM12C with the addition of increasing doses of rhGal-1. TOPFlash plasmids (β-catenin promoter reporter construct containing TCF/LEF binding sites; please see Materials and Methods) and TOPFlash mutant plasmids (β-catenin promoter reporter construct containing mutated TCF/LEF binding sites; please see Materials and Methods) were transduced into KM12C, with the luciferase activity measured 48 hours later; addition of the Wnt/β-catenin agonist CHIR-99021 (CHIR; 0.3 µM) was used as a positive control. (D) qPCR analysis for the gene expression of Twist1 in KM12C after treatment with rhGal-1 (100 ng/ml) and without or with the Wnt/β-catenin antagonist XAV-939 (XAV; 10 µM) for 24 hours. All results are shown as the mean ± SEM of three independent experiments. *p < 0.05; **p < 0.01, and ***p < 0.005 compared to the control.
SOX9 Is a Critical Mediator Involved in Gal-1-Induced Upregulation of β-catenin Activity and Cancer-Initiating Cell Features
Given the inherent complexity of the Wnt/β-Catenin signaling pathway, we were interested in further elucidating the details in the role of Gal-1 within this critical CRC pathway. To identify/search for other mediators involved in LGALS1/CTNNB1 interactions, we performed IPA with the addition of TWIST1—a downstream target of β-catenin and one of the most important EMT transcription factors—to the analyses. Using this model, SOX9 was predicted as a key candidate gene within this interaction (Figure 5A). The role of this transcription factor in CRC remains unclear despite the very recent large-scale genomic data identifying this gene to be a significant and novel somatic recurrently mutated gene in this cancer (32). We found that the co-culture of both types of fibroblast-CM increased the protein expression of SOX9 in KM12C (Figure 5B). Moreover, the addition of rhGal-1 to KM12C not only enhanced the overall SOX9 protein expression levels, but also increased the nuclear levels of the transcription factor (Figure 5C). To investigate the role of SOX9 in CIC formation, we generated SOX9-silenced KM12C (shSOX9-KM) and confirmed the efficiency of knockdown by Western blot, which identified the shSOX9-II clone as having a more efficient knockdown. Compared to non-target knockdown KM12C (shC-KM), shSOX9-KM had a significantly decreased capacity for sphere formation; moreover, while the addition of rhGal-1 significantly improved the shSOX9-KM sphere formation capacity, this was still significantly less than the capacity of rhGal-1-treated shC-KM (Figure 5D). SOX9 also contributes to Gal-1-mediated drug resistance, since we found that shSOX9-KM was significantly more sensitive to cisplatin compared to shC-KM even with rhGal-1 pretreatment (Figure 5E; a decrease of 1.00- to 0.12-fold for cell viability in shSOX9-KM compared with 1.00- to 0.60-fold in shC-KM). To assess whether β-catenin was involved in Gal-1/SOX9 interaction, we analyzed for changes in the expression of Twist1 as a downstream gene of β-catenin in shSOX9- and shC-KM cells with the addition of rhGal-1 and with or without β-Catenin antagonism (Figure 5F). We found that the levels of Twist1 are significantly increased in shC-KM after the addition of rhGal-1, which could then be significantly suppressed to below baseline levels when the β-catenin antagonist XAV was applied; simultaneous addition of rhGal-1 and XAV in shC-KM restored Twist1 expression to baseline levels. In shSOX9-KM, however, the baseline expression of Twist1 was lower than the baseline levels in shC-KM; moreover, neither the addition of rhGal-1 nor XAV to shSOX9-KM increased Twist1 levels. In terms of invasive capacity, migration capacity was decreased in shSOX9-KM compared to shC-KM (Figure 5G), but treatment with rhGal-1 increased the migration capacity significantly in shC-KM and non-significantly in shSOX9-KM. Importantly, rhGal-1-induced migration was abrogated after the treatment with XAV in shSOX9-KM but not shC-KM. Together, these results demonstrate that SOX9 is an important mediator involved in Gal-1-induced upregulation of β-catenin signaling activity as well as the augmentation of CIC features in CRC.
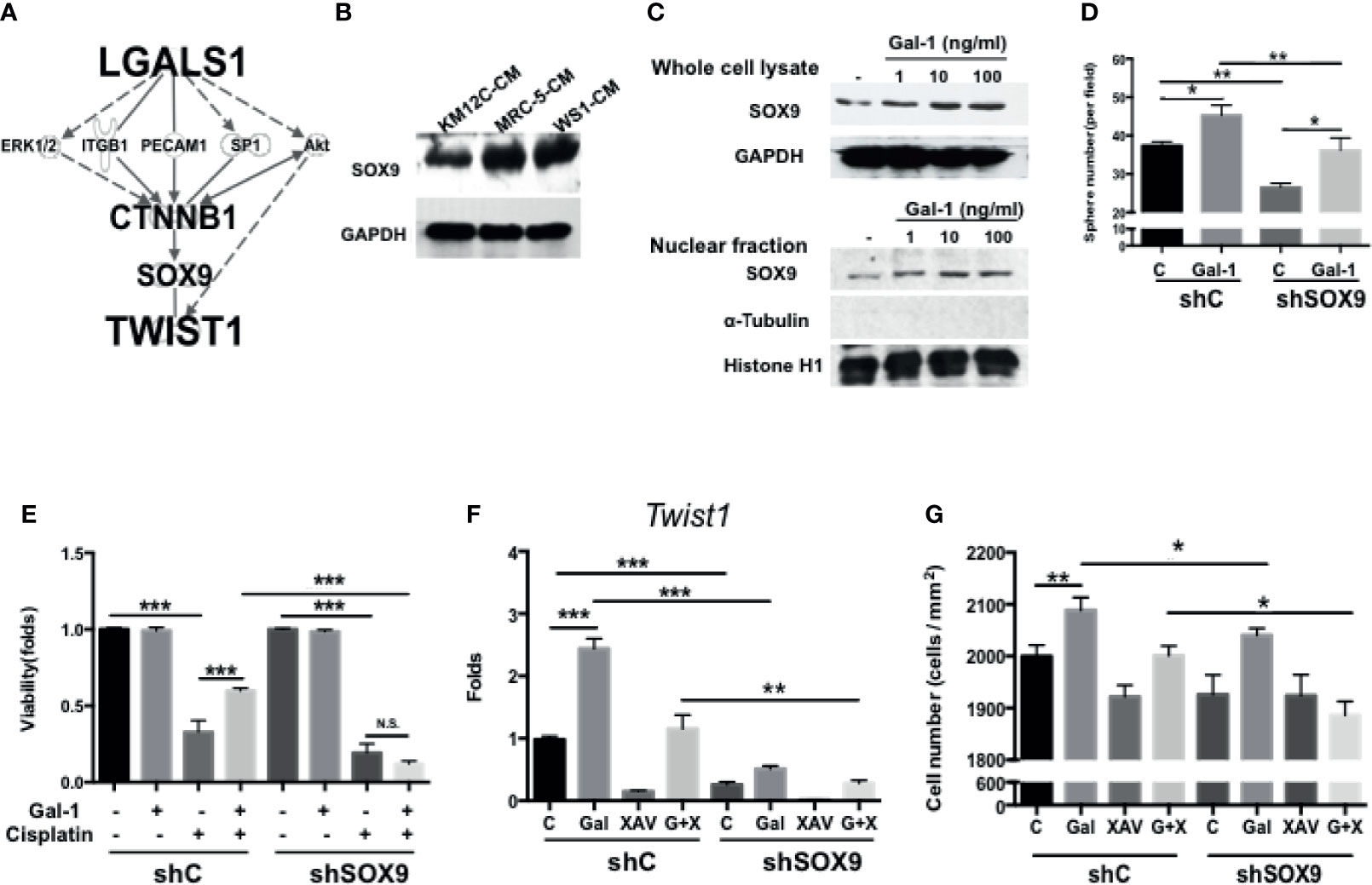
Figure 5 SOX9 is a critical mediator involved in Gal-1-induced upregulation of β-catenin activity and CIC features. (A) Ingenuity Pathway Analysis (IPA) for the prediction of candidate mediators within the LGALS1/CTNNB1/Twist1 axis. IPA database revealed the several major pathways which might be involved in tumor development and metastasis. According to the IPA results and literature review, SOX9 was selected and confirmed whether it is the downstream gene of Gal-1 by Western blot. (B) Western blot for the analysis of SOX9 protein levels in KM12C after culturing in MRC-5- or WS1-CM; KM12C-CM was used as the control. Internal control: GAPDH. (C) Western blot for SOX9 levels in whole cell lysate (top panel) and nuclear fraction (bottom panel) of KM12C with addition of increasing doses of rhGal-1 for 48 hours; for nuclear protein blot, histone H1 is used as the positive control and α-Tubulin as the negative control. (D) Sphere formation capacity of shC-KM and shSOX9-II-KMC12 (shSOX9-KM) after treating with rhGal-1 (100 ng/ml) for 72 hours. (E) Drug resistance capacity of shC- and shSOX9-KM to cisplatin (25 µM) after pretreatment with rhGal-1 (100 ng/ml) for 24 hours. Cell viability was assessed 48 hours after drug treatment. (F) qPCR analysis for the gene expression of Twist1 in shC- and shSOX9-KM after treatment with Gal-1 (100 ng/ml) and XAV (10 µM). (G) Invasion capacity of shC- and shSOX9-KM with addition of rhGal-1 (100 ng/ml) and XAV (10 µM). All results are shown as the mean ± SEM of three independent experiments. *p < 0.05; **p < 0.01, and ***p < 0.005 compared to the control. N.S., not significant.
High Expression of Gal-1 and SOX9 Correlate With Clinical Colorectal Cancer (CRC) Outcome
Our results indicate that both Gal-1 and SOX9 promote CIC features, which involve the β-catenin pathway in CRC cells. To assess whether Gal-1 and/or SOX9 expression is clinically relevant for CRC, we analyzed for the expression of either one or both of these genes to various measured clinical parameters in publicly available CRC databases. We first searched the ONCOMINE database of published microarray data with matched normal and cancer specimens, and found, in the Kaiser Colon database, that a higher expression of both LGALS1 and SOX9 are seen in CRC samples than in normal colon samples (Figure 6A). To further study whether the expression patterns of either or both genes are correlated with more detailed clinical outcomes, we analyzed the gene expression profiles of two datasets from GSE9348 and The Cancer Genome Atlas (TCGA) (Supplementary Table S2) which includes early-stage CRC samples and adjacent normal tissue. In both databases, both LGALS1 and SOX9 were significantly expressed at higher levels in early-stage CRC tissue compared to adjacent normal tissue, especially SOX9 (Figure 6B). Moreover, in databases with stage-specific gene expression information, such as GSE17536 and TCGA (Supplementary Table S2), analyses revealed that the percentage of CRC samples with a high expression of LGALS1/SOX9 (LGALS1high/SOX9high) correlated with an increasing CRC stage (Figure 6C): with increasing stage, LGALS1high/SOX9high CRC samples increased from 8.3% to 35.9% and from 8.9% to 20.0% in the GSE17536 and TCGA databases, respectively. On the other hand, neither CTNNB1/LGALS1 highly expressed (CTNNB1high/LGALS1high) nor CTNNB1high/SOX9high highly expressed CRC samples correlate with the CRC stage (Supplementary Figure S5). To verify the protein expression, we performed IHC of Gal-1 and SOX9 in a human CRC tissue microarray, which included 40 primary lesions, 10 metastatic lesions, and 9 normal colon samples (Figure 6D). The tissue array staining revealed that both Gal-1 and SOX9 protein expression were more prominent in CRC samples compared to normal tissue. Furthermore, distinct patterns of Gal-1 vs. SOX9 expression within CRC samples could be seen: Gal-1 expression appeared to increase with an increasing disease progression, while SOX9 expression appear to more strongly correlate with the presence of any cancerous lesion. Most critically, survival analyses based on expression levels of SOX9 and Gal-1 demonstrate that CRC patients with SOX9high/Gal-1high expression have a significantly shorter survival compared with patients with SOX9low/Gal-1low expression (Figure 6E and Supplementary Figure S5). Collectively, these analyses of clinical data/samples not only demonstrate that Gal-1 and/or SOX9 overexpression strongly correlate with disease presence, but also with the stage of CRC. Moreover, the presence of both Gal-1 and SOX9 together are strong predictors of a worse outcome in terms of disease survival. Along with our in vitro and mouse in vivo data, these results therefore demonstrate that stromal cell-derived Gal-1 directly target CRC cells to promote CIC features and disease progression through SOX9 and β-catenin (Figure 7).
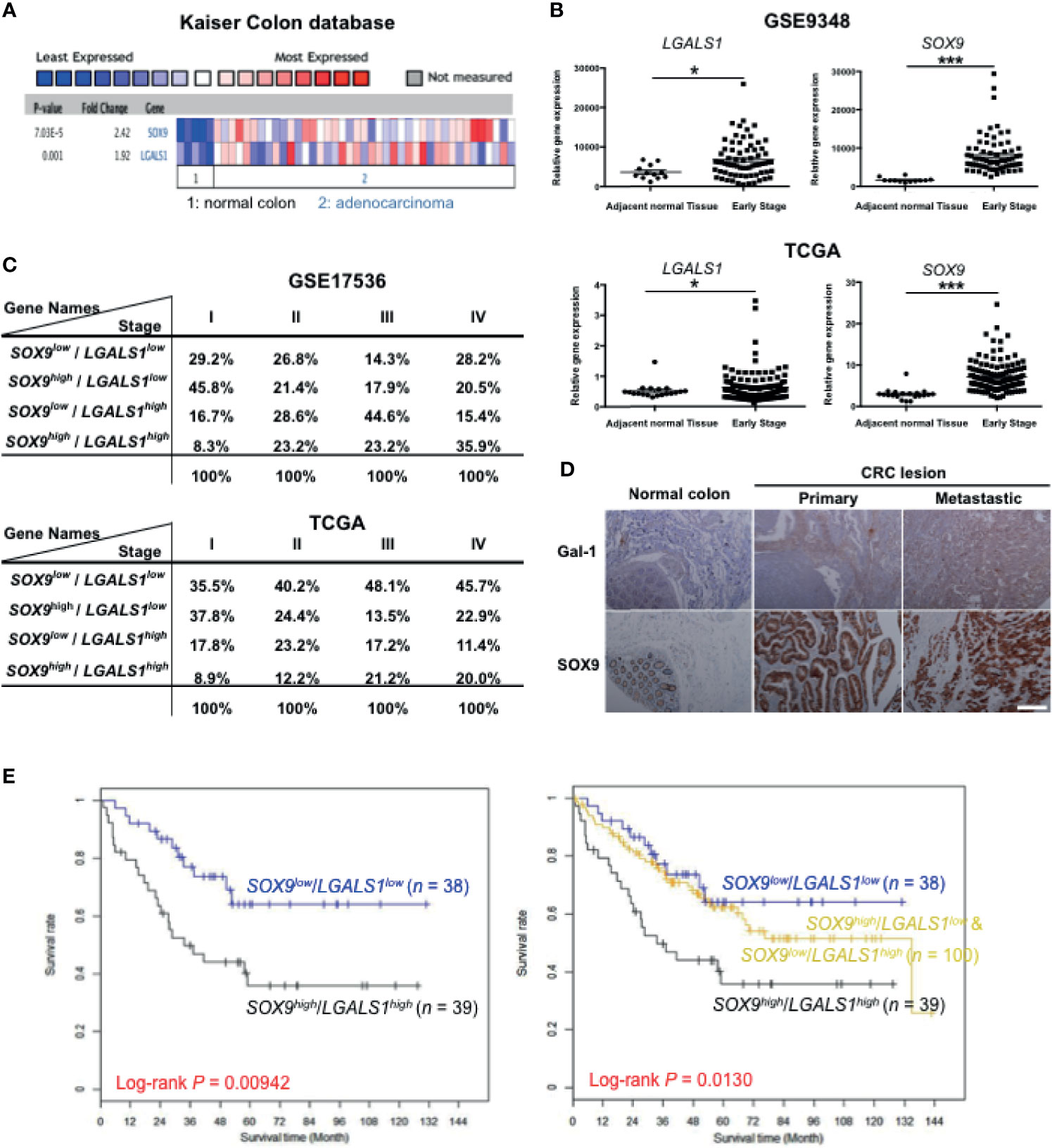
Figure 6 High expression of Gal-1 and SOX9 correlate with clinical CRC outcome. (A) ONCOMINE assessment of the expression levels of LGALS1 and SOX9 in the Kaiser Colon database. (B) Analysis of LGALS1 or SOX9 expression levels in tumor tissue compared to adjacent normal tissue using GSE9348 (top panel) and The Cancer Genome Atlas (TCGA) databases (bottom panel). *p < 0.05 and ***p < 0.005 for early-stage lesions compared to adjacent normal tissue. (C) Analysis of LGALS1 and SOX9 expression levels to the CRC stage using the GSE17536 and TCGA datasets. (D) Immunohistological staining of Gal-1 and SOX9 on CRC tumor samples, which included 40 primary lesions (primary), 10 metastatic lesions (metastatic), and 9 normal colon samples; scale bar, 100 μm. (E) Kaplan-Meier survival curves of four groups of CRC patients as stratified by median expression levels of SOX9 and Gal-1 in tumor tissue: SOX9low/Gal-1low, n = 38; SOX9low/Gal-1high & SOX9high/Gal-1low, n = 100; and SOX9high/Gal-1high, n = 39. Survival analyses was performed for two groups: SOX9high/Gal-1high versus SOX9low/Gal-1low (left-side graph); or for three groups: SOX9high/Gal-1high versus SOX9high/Gal-1low + SOX9low/Gal-1high versus SOX9low/Gal-1low (right-side graph).
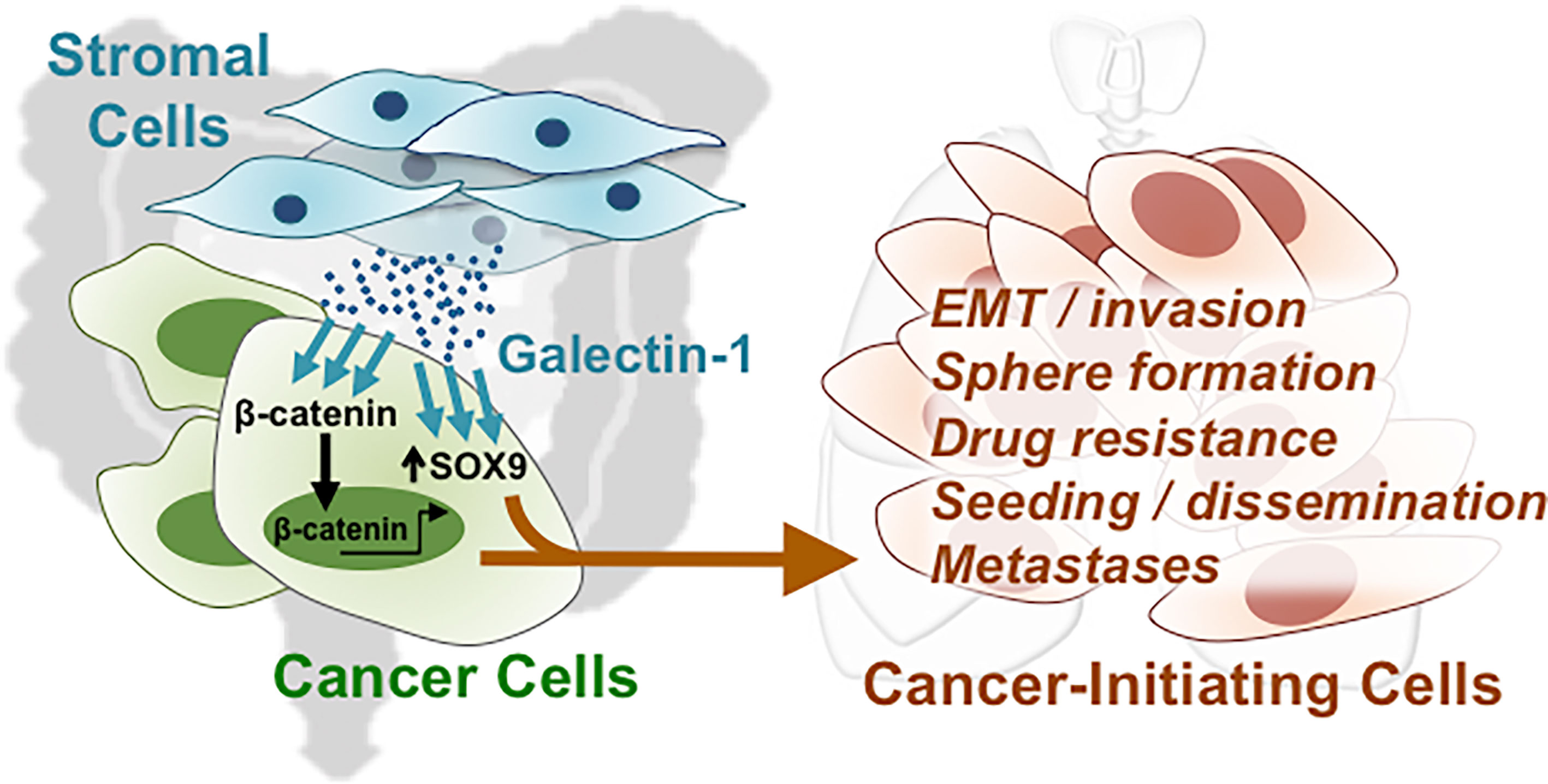
Figure 7 Direct targeting of stromal-secreted Gal-1 on CRC cells promote CIC features and disease progression through SOX9 and β-catenin.
Discussion
CRC is one of the most common diseases worldwide, and alarmingly in developed nations such as the USA, the incidence and mortality of CRC has begun to increase in younger individuals after decades of decrease for the population at large. While early-stage lesions are amenable to screening and treatment, late and metastatic stage disease still have a dismal 5-year survival of less than 15% (2–5). Thus, there is a critical need for the discovery of biomarkers for early diagnosis as well as relapse. The microenvironment or niche of the solid tumor is increasingly seen to be a crucial partner in mediating disease progression (12, 13, 33–35); we therefore embarked on studying the molecular interactions between tumor niche-stromal fibroblast cells and CRC cells for the discovery of prognostically relevant biomarkers. Our data demonstrate that fibroblast-secreted Gal-1 promotes multiple CIC features in CRC cells including activating β-catenin in vitro, promoting metastasis and tumor dissemination in vivo, as well as significantly correlating with clinical recurrence and disease progression. These findings strongly suggest that fibroblast-secreted Gal-1 could be involved in promoting the presence of disseminated tumor cells (DTCs), which represent cancer cells that have undergone EMT and can disseminate to distant organ to seed metastatic growths (36, 37). Indeed, we found that fibroblast-secreted Gal-1 enhanced EMT-related gene expression in vitro in CRC cells, and increased the number of injected human CRC cells in the lungs in both short-term and long-term in vivo mouse models (Figures 2C, D and 3C, D); moreover, high expression of Gal-1 in CRC patients correlated significantly with metastasis and recurrence (Figure 3E). Importantly, using transcriptome data and pathway prediction, we found that SOX9, a novel CRC driver (32), not only was mechanistically involved in Gal-1/β-catenin interactions but also is a highly relevant biomarker, especially when evaluated in conjunction with Gal-1. These findings collectively demonstrate that tumor niche-derived paracrine factors are not only important in the maintenance of CRC CICs, but can also be prognostically relevant in evaluating clinical disease progression. Our study also outlines the translational utility of niche/non-cancer cell type-based in vitro molecular findings to serve as biomarkers, especially given that most patient genomic and transcriptomic databases are derived from bulk tumor specimens that include non-cancer elements of the stroma and immune system.
Gal-1 is a member of the β-galactoside-binding protein family and known to modulate cancer-associated immunosuppression and angiogenesis (14, 20, 38). While the immunomodulatory mechanisms of Gal-1 have been clearly elucidated, its direct role on carcinogenesis has not been studied in much detail even for CRC in which a positive correlation with a worsening disease status has been reported (39, 40). Given the central role of the Wnt/β-catenin pathway in CRC, we postulated that tumor niche-derived paracrine factors might promote disease progression through interactions with this pathway. Our findings on the capacity of Gal-1 to activate β-catenin and induce CRC CIC features not only provide a mechanistic evidence for Gal-1 having direct interactions with CRC cells, but also support previously published clinical correlative findings of Gal-1 to be mainly expressed within the CRC stroma and not the cancer cell itself (38, 39), which we found as well (Figure 2A) (16). Critically, our in vivo findings strongly support Gal-1 as having an important role in CRC dissemination, which was bore out in analyses of human CRC databases revealing a high Gal-1 expression to be predictive of recurrence. Since Gal-1 is a secreted molecule and released in the circulation, our findings implicate that this marker could be potentially useful as a biomarker in CRC; however, further studies with patient samples are necessary to validate this possibility. Our report therefore provides further molecular understanding on previously reported disease-promoting correlations of Gal-1 in CRC.
We found SOX9, a transcription factor belonging to the Sry-related HMG-box family, to be involved in Gal-1/β-catenin interactions in CRC. An important regulator for numerous developmental processes including in the gastrointestinal epithelium (41, 42), SOX9 has been categorized as belonging in the broader Wnt/β-catenin pathway (32). While SOX9 is known to be transcriptionally repressed by β-catenin in cartilage development (43, 44) and found to contribute to a number of cancer types including CRC (15, 45, 46),, only recently through a comprehensive molecular characterization of CRC has mutations in this transcription factor been implicated in any type of human cancers (32). Previous reports on the role of SOX9 in CRC have yielded mixed results, such as a tumor suppressor (47, 48) and having oncogenic functions (15, 49). These conflicting results have been speculated to be due to a complex relationship of SOX9 dosage on function (50). Intriguingly, these studies rarely take into consideration the fact that SOX9 is a key transcription factor in developmental/non-neoplastic EMT processes, as well as in neoplastic disease (51, 52). We found SOX9 to be involved in a Gal-1/β-catenin-mediated enhancement of a number of in vitro CIC features including EMT-related gene expression (Figure 5); moreover, analyses of multiple human CRC transcriptome databases as well as tissue microarray immunohistological staining demonstrated a significant correlation of high SOX9 expression to tumor presence, which is highly suggestive of its utility as a CRC biomarker. Critically, the simultaneous use of both Gal-1 and SOX9 is strongly correlated with a worse survival in CRC patients (Figure 6E). Our findings therefore not only contribute to a molecular understanding on the roles of Gal-1 and SOX9 in the central CRC pathway of Wnt/β-catenin, but also reveal these molecules as useful prognostic markers in transcriptomic databases.
In summary, we found that fibroblast-secreted Gal-1 significantly enhanced multiple in vitro CRC CIC properties including enhancing EMT and activating β-catenin, as well as promoting in vivo metastatic disease and tumor dissemination, and clinical recurrence. Bioinformatics pathway analyses predicted SOX9, a recently discovered aggressive CRC marker, as being involved in Gal-1/β-catenin interactions, which was validated in vitro. Moreover, Gal-1 or SOX9 but not β-catenin are prognostically correlated with disease presence and progression. Critically, a high expression of both Gal-1 and SOX9 is correlated with a significantly worse disease survival. Our findings highlight the critical role of the tumor niche-stromal component of CRC in disease progression and for discovery of prognostic markers and drug targets.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Ethics Statement
The studies involving human participants were reviewed and approved by the National Health Research Institutes (human cell line use) and GEO and TCGA (public bioinformatics data). The animal study was reviewed and approved by the Institutional Animal Care and Use Committee, NHRI.
Author Contributions
Conception and design: K-YP and BY. Development of methodology: K-YP, S-SJ, and BY. Acquisition of data (provided animals, acquired and managed patients, provided facilities, etc.): Y-WL and F-YT. Analysis and interpretation of data (e.g., statistical analysis, biostatistics, computational analysis): K-YP, S-SJ, F-YT, C-CC, and BY. Writing, review, and/or revision of the manuscript: K-YP, S-SJ, C-CC, L-TC, and BY. All authors contributed to the article and approved the submitted version.
Funding
This study was supported in part by grants from the NHRI (10A1-CSPP06), the Ministry of Health & Welfare (MOHW110-TDU-B-212-144026), and the Taiwan Ministry of Science & Technology (MoST108-2314-B-400-035-MY3 and MoST110-2740-B-400-002).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
The authors would like to thank the NHRI Microarray Core Facility for the technical support.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2021.716055/full#supplementary-material
Supplementary Figure 1 | Flow cytometric analysis for surface marker expression of CD29 (top panel) and CD44 (bottom panel) in KM12C treated with fibroblast-CM (Conditioned Medium).
Supplementary Figure 2 | SDS-PAGE analysis of conditioned media from MRC-5 and WS1.
Supplementary Figure 3 | Flow cytometry analysis for surface marker expression of CD29 and CD44 in KM12C (left panel) and HCT-116 (right panel) after Gal-1(100ng/ml) treatment.
Supplementary Figure 4 | Flow cytometric analysis for surface marker expression of CD29 in HCT-116 after Gal-1(100ng/ml) and XAV(10uM) treatment.
Supplementary Figure 5 | Analyses of the data sets GSE17536 and TCGA for stage-specific expression of CTNNB1/LGALS1 or SOX9/CTNNB1.
Supplementary Table 1 | qPCR primer sequence information.
Supplementary Table 2 | Information on the public microarray gene expression profiles used within this study.
Abbreviations
CRC, Colorectal cancer; CICs, Cancer-initiating cells; CM, Conditioned medium; MS, Mass spectrometry; Gal-1, Galectin-1; IPA, Ingenuity pathway analysis; DMEM/F-12, Dulbecco’s Modified Eagle Medium: Nutrient Mixture F-12; FBS, Fetal bovine serum; MEM, Minimum essential medium; qPCR, Quantitative real-time polymerase chain reaction; siRNA, small interference RNA; shRNA, short hairpin RNA; EGF, Epidermal growth factor; bFGF, basic fibroblast growth factor; HRP, Horseradish peroxidase; ELISA, Enzyme linked immunosorbent assay; IF, Immunofluorescence staining; MTT, 3-(4,5-Dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide; DiI, 1,1’-Dioctadecyl-3,3,3’,3’-Tetramethylindocarbocyanine Perchlorate; DiO, 3,3’-Dioctadecyloxacarbocyanine Perchlorate; IHC, Immunohistochemistry; OCT, Optimal cutting temperature; GEO, Gene expression omnibus; TCGA, The Cancer Genome Atlas; EMT, Epithelial-mesenchymal transition; rhGal-1, Recombinant human Gal-1 protein; siC-WS1, Non-target siRNA control in WS1; siGal-WS1, siRNA specific for Gal-1-knockdown in WS1; shC-WS1, Non-target shRNA control in WS1; shGal-WS1, shRNA specific for Gal-1-knockdown in WS1; KM, KM12C; XAV: XAV939; shC-KM, Non-target shRNA control in KM12C; shSOX9-KM, shRNA specific for SOX9- knockdown in KM12C; DTCs, Disseminated tumor cells.
References
1. Siegel R, Desantis C, Jemal A. Colorectal Cancer Statistics, 2014. CA Cancer J Clin (2014) 64(2):104–17. doi: 10.3322/caac.21220
2. Shin A, Kim KZ, Jung KW, Park S, Won YJ, Kim J, et al. Increasing Trend of Colorectal Cancer Incidence in Korea, 1999-2009. Cancer Res Treat (2012) 44(4):219–26. doi: 10.4143/crt.2012.44.4.219
3. Siegel RL, Fedewa SA, Anderson WF, Miller KD, Ma J, Rosenberg PS, et al. Colorectal Cancer Incidence Patterns in the United States, 1974-2013. J Natl Cancer Inst (2017) 109(8):1–6. doi: 10.1093/jnci/djw322
4. Bockelman C, Engelmann BE, Kaprio T, Hansen TF, Glimelius B. Risk of Recurrence in Patients With Colon Cancer Stage II and III: A Systematic Review and Meta-Analysis of Recent Literature. Acta Oncol (2015) 54(1):5–16. doi: 10.3109/0284186X.2014.975839
5. Chen PC, Lee JC, Wang JD. Estimation of Life-Year Loss and Lifetime Costs for Different Stages of Colon Adenocarcinoma in Taiwan. PloS One (2015) 10(7):e0133755. doi: 10.1371/journal.pone.0133755
6. Markowitz SD, Bertagnolli MM. Molecular Origins of Cancer: Molecular Basis of Colorectal Cancer. N Engl J Med (2009) 361(25):2449–60. doi: 10.1056/NEJMra0804588
7. Fodde R, Smits R, Clevers HAPC. Signal Transduction and Genetic Instability in Colorectal Cancer. Nat Rev Cancer (2001) 1(1):55–67. doi: 10.1038/35094067
8. Barker N, Ridgway RA, van Es JH, van de Wetering M, Begthel H, van den Born M, et al. Crypt Stem Cells as the Cells-of-Origin of Intestinal Cancer. Nature (2009) 457(7229):608–11. doi: 10.1038/nature07602
9. Walther A, Johnstone E, Swanton C, Midgley R, Tomlinson I, Kerr D. Genetic Prognostic and Predictive Markers in Colorectal Cancer. Nat Rev Cancer (2009) 9(7):489–99. doi: 10.1038/nrc2645
10. Ricci-Vitiani L, Lombardi DG, Pilozzi E, Biffoni M, Todaro M, Peschle C, et al. Identification and Expansion of Human Colon-Cancer-Initiating Cells. Nature (2007) 445(7123):111–5. doi: 10.1038/nature05384
11. Le NH, Franken P, Fodde R. Tumour-Stroma Interactions in Colorectal Cancer: Converging on Beta-Catenin Activation and Cancer Stemness. Br J Cancer (2008) 98(12):1886–93. doi: 10.1038/sj.bjc.6604401
12. Vermeulen L, De Sousa EMF, van der Heijden M, Cameron K, de Jong JH, Borovski T, et al. Wnt Activity Defines Colon Cancer Stem Cells and is Regulated by the Microenvironment. Nat Cell Biol (2010) 12(5):468–76. doi: 10.1038/ncb2048
13. Calon A, Espinet E, Palomo-Ponce S, Tauriello DV, Iglesias M, Cespedes MV, et al. Dependency of Colorectal Cancer on a TGF-Beta-Driven Program in Stromal Cells for Metastasis Initiation. Cancer Cell (2012) 22(5):571–84. doi: 10.1016/j.ccr.2012.08.013
14. Dalotto-Moreno T, Croci DO, Cerliani JP, Martinez-Allo VC, Dergan-Dylon S, Mendez-Huergo SP, et al. Targeting Galectin-1 Overcomes Breast Cancer-Associated Immunosuppression and Prevents Metastatic Disease. Cancer Res (2013) 73(3):1107–17. doi: 10.1158/0008-5472.CAN-12-2418
15. Matheu A, Collado M, Wise C, Manterola L, Cekaite L, Tye AJ, et al. Oncogenicity of the Developmental Transcription Factor Sox9. Cancer Res (2012) 72(5):1301–15. doi: 10.1158/0008-5472.CAN-11-3660
16. Morikawa K, Walker SM, Nakajima M, Pathak S, Jessup JM, Fidler IJ. Influence of Organ Environment on the Growth, Selection, and Metastasis of Human Colon Carcinoma Cells in Nude Mice. Cancer Res (1988) 48(23):6863–71.
17. Medico E, Russo M, Picco G, Cancelliere C, Valtorta E, Corti G, et al. The Molecular Landscape of Colorectal Cancer Cell Lines Unveils Clinically Actionable Kinase Targets. Nat Commun (2015) 6:7002. doi: 10.1038/ncomms8002
18. Wang CH, Wang TM, Young TH, Lai YK, Yen ML. The Critical Role of ECM Proteins Within the Human MSC Niche in Endothelial Differentiation. Biomaterials (2013) 34(17):4223–34. doi: 10.1016/j.biomaterials.2013.02.062
19. Weng LP, Wu CC, Hsu BL, Chi LM, Liang Y, Tseng CP, et al. Secretome-Based Identification of Mac-2 Binding Protein as a Potential Oral Cancer Marker Involved in Cell Growth and Motility. J Proteome Res (2008) 7(9):3765–75. doi: 10.1021/pr800042n
20. Thijssen VL, Barkan B, Shoji H, Aries IM, Mathieu V, Deltour L, et al. Tumor Cells Secrete Galectin-1 to Enhance Endothelial Cell Activity. Cancer Res (2010) 70(15):6216–24. doi: 10.1158/0008-5472.CAN-09-4150
21. Peng KY, Lee YW, Hsu PJ, Wang HH, Wang Y, Liou JY, et al. Human Pluripotent Stem Cell (PSC)-Derived Mesenchymal Stem Cells (MSCs) Show Potent Neurogenic Capacity Which Is Enhanced With Cytoskeletal Rearrangement. Oncotarget (2016) 7(28):43949–59. doi: 10.18632/oncotarget.9947
22. Chaturvedi A, Hoffman LM, Jensen CC, Lin YC, Grossmann AH, Randall RL, et al. Molecular Dissection of the Mechanism by Which EWS/FLI Expression Compromises Actin Cytoskeletal Integrity and Cell Adhesion in Ewing Sarcoma. Mol Biol Cell (2014) 25(18):2695–709. doi: 10.1091/mbc.E14-01-0007
23. de Sousa EMF, Colak S, Buikhuisen J, Koster J, Cameron K, de Jong JH, et al. Methylation of Cancer-Stem-Cell-Associated Wnt Target Genes Predicts Poor Prognosis in Colorectal Cancer Patients. Cell Stem Cell (2011) 9(5):476–85. doi: 10.1016/j.stem.2011.10.008
24. Smith JJ, Deane NG, Wu F, Merchant NB, Zhang B, Jiang A, et al. Experimentally Derived Metastasis Gene Expression Profile Predicts Recurrence and Death in Patients With Colon Cancer. Gastroenterol (2010) 138(3):958–68. doi: 10.1053/j.gastro.2009.11.005
25. Kaiser S, Park YK, Franklin JL, Halberg RB, Yu M, Jessen WJ, et al. Transcriptional Recapitulation and Subversion of Embryonic Colon Development by Mouse Colon Tumor Models and Human Colon Cancer. Genome Biol (2007) 8(7):R131. doi: 10.1186/gb-2007-8-7-r131
26. Hong Y, Downey T, Eu KW, Koh PK. Cheah PY. A ‘Metastasis-Prone’ Signature for Early-Stage Mismatch-Repair Proficient Sporadic Colorectal Cancer Patients and Its Implications for Possible Therapeutics. Clin Exp Metastasis (2010) 27(2):83–90. doi: 10.1007/s10585-010-9305-4
27. Cancer Genome Atlas Research N. Comprehensive Genomic Characterization Defines Human Glioblastoma Genes and Core Pathways. Nature (2008) 455(7216):1061–8. doi: 10.1038/nature07385
28. Shmelkov SV, Butler JM, Hooper AT, Hormigo A, Kushner J, Milde T, et al. CD133 Expression is Not Restricted to Stem Cells, and Both CD133+ and CD133- Metastatic Colon Cancer Cells Initiate Tumors. J Clin Invest (2008) 118(6):2111–20. doi: 10.1172/JCI34401
29. Brakebusch C, Fassler R. Beta 1 Integrin Function In Vivo: Adhesion, Migration and More. Cancer Metastasis Rev (2005) 24(3):403–11. doi: 10.1007/s10555-005-5132-5
30. Chanmee T, Ontong P, Kimata K, Itano N. Key Roles of Hyaluronan and Its CD44 Receptor in the Stemness and Survival of Cancer Stem Cells. Front Oncol (2015) 5:180. doi: 10.3389/fonc.2015.00180
31. Song Y, Li ZX, Liu X, Wang R, Li LW, Zhang Q. The Wnt/beta-Catenin and PI3K/Akt Signaling Pathways Promote EMT in Gastric Cancer by Epigenetic Regulation via H3 Lysine 27 Acetylation. Tumour Biol (2017) 39(7):1010428317712617. doi: 10.1177/1010428317712617
32. Cancer Genome Atlas N. Comprehensive Molecular Characterization of Human Colon and Rectal Cancer. Nature (2012) 487(7407):330–7. doi: 10.1038/nature11252
33. Peddareddigari VG, Wang D, Dubois RN. The Tumor Microenvironment in Colorectal Carcinogenesis. Cancer Microenviron (2010) 3(1):149–66. doi: 10.1007/s12307-010-0038-3
34. Guleng B, Tateishi K, Ohta M, Kanai F, Jazag A, Ijichi H, et al. Blockade of the Stromal Cell-Derived Factor-1/CXCR4 Axis Attenuates In Vivo Tumor Growth by Inhibiting Angiogenesis in a Vascular Endothelial Growth Factor-Independent Manner. Cancer Res (2005) 65(13):5864–71. doi: 10.1158/0008-5472.CAN-04-3833
35. Morris JS, Kopetz S. Tumor Microenvironment in Gene Signatures: Critical Biology or Confounding Noise? Clin Cancer Res (2016) 22(16):3989–91. doi: 10.1158/1078-0432.CCR-16-1044
36. Kang Y, Pantel K. Tumor Cell Dissemination: Emerging Biological Insights From Animal Models and Cancer Patients. Cancer Cell (2013) 23(5):573–81. doi: 10.1016/j.ccr.2013.04.017
37. Rhim AD, Mirek ET, Aiello NM, Maitra A, Bailey JM, McAllister F, et al. EMT and Dissemination Precede Pancreatic Tumor Formation. Cell (2012) 148(1-2):349–61. doi: 10.1016/j.cell.2011.11.025
38. Liu FT, Rabinovich GA. Galectins as Modulators of Tumour Progression. Nat Rev Cancer (2005) 5(1):29–41. doi: 10.1038/nrc1527
39. Sanjuan X, Fernandez PL, Castells A, Castronovo V, van den Brule F, Liu FT, et al. Differential Expression of Galectin 3 and Galectin 1 in Colorectal Cancer Progression. Gastroenterol (1997) 113(6):1906–15. doi: 10.1016/S0016-5085(97)70010-6
40. Sheng TH, Rong LX, Li ZY, Bo J, Lei S. Tissue and Serum Galectin-1 Expression in Patients With Colorectal Carcinoma. Hepatogastroenterol (2012) 59(114):389–94. doi: 10.5754/hge09232
41. Blache P, van de Wetering M, Duluc I, Domon C, Berta P, Freund JN, et al. SOX9 Is an Intestine Crypt Transcription Factor, Is Regulated by the Wnt Pathway, and Represses the CDX2 and MUC2 Genes. J Cell Biol (2004) 166(1):37–47. doi: 10.1083/jcb.200311021
42. Mori-Akiyama Y, van den Born M, van Es JH, Hamilton SR, Adams HP, Zhang J, et al. SOX9 Is Required for the Differentiation of Paneth Cells in the Intestinal Epithelium. Gastroenterol (2007) 133(2):539–46. doi: 10.1053/j.gastro.2007.05.020
43. Akiyama H, Kim JE, Nakashima K, Balmes G, Iwai N, Deng JM, et al. Osteo-Chondroprogenitor Cells Are Derived From Sox9 Expressing Precursors. Proc Natl Acad Sci USA (2005) 102(41):14665–70. doi: 10.1073/pnas.0504750102
44. Topol L, Chen W, Song H, Day TF, Yang Y. Sox9 Inhibits Wnt Signaling by Promoting Beta-Catenin Phosphorylation in the Nucleus. J Biol Chem (2009) 284(5):3323–33. doi: 10.1074/jbc.M808048200
45. Jiang SS, Fang WT, Hou YH, Huang SF, Yen BL, Chang JL, et al. Upregulation of SOX9 in Lung Adenocarcinoma and Its Involvement in the Regulation of Cell Growth and Tumorigenicity. Clin Cancer Res (2010) 16(17):4363–73. doi: 10.1158/1078-0432.CCR-10-0138
46. Larsimont JC, Youssef KK, Sanchez-Danes A, Sukumaran V, Defrance M, Delatte B, et al. Sox9 Controls Self-Renewal of Oncogene Targeted Cells and Links Tumor Initiation and Invasion. Cell Stem Cell (2015) 17(1):60–73. doi: 10.1016/j.stem.2015.05.008
47. Prevostel C, Rammah-Bouazza C, Trauchessec H, Canterel-Thouennon L, Busson M, Ychou M, et al. SOX9 Is an Atypical Intestinal Tumor Suppressor Controlling the Oncogenic Wnt/ss-Catenin Signaling. Oncotarget (2016) 7(50):82228–43. doi: 10.18632/oncotarget.10573
48. Shi Z, Chiang CI, Mistretta TA, Major A, Mori-Akiyama Y. SOX9 Directly Regulates IGFBP-4 in the Intestinal Epithelium. Am J Physiol Gastrointest Liver Physiol (2013) 305(1):G74–83. doi: 10.1152/ajpgi.00086.2013
49. Shi Z, Chiang CI, Labhart P, Zhao Y, Yang J, Mistretta TA, et al. Context-Specific Role of SOX9 in NF-Y Mediated Gene Regulation in Colorectal Cancer Cells. Nucleic Acids Res (2015) 43(13):6257–69. doi: 10.1093/nar/gkv568
50. Prevostel C, Blache P. The Dose-Dependent Effect of SOX9 and Its Incidence in Colorectal Cancer. Eur J Cancer (2017) 86:150–57. doi: 10.1016/j.ejca.2017.08.037
51. Sakai D, Suzuki T, Osumi N, Wakamatsu Y. Cooperative Action of Sox9, Snail2 and PKA Signaling in Early Neural Crest Development. Development (2006) 133(7):1323–33. doi: 10.1242/dev.02297
Keywords: cancer-initiating cells (CICs), tumor stroma, galectin-1, SOX9, biomarkers
Citation: Peng K-Y, Jiang S-S, Lee Y-W, Tsai F-Y, Chang C-C, Chen L-T and Yen BL (2021) Stromal Galectin-1 Promotes Colorectal Cancer Cancer-Initiating Cell Features and Disease Dissemination Through SOX9 and β-Catenin: Development of Niche-Based Biomarkers. Front. Oncol. 11:716055. doi: 10.3389/fonc.2021.716055
Received: 28 May 2021; Accepted: 12 August 2021;
Published: 10 September 2021.
Edited by:
Nathaniel Weygant, Fujian University of Traditional Chinese Medicine, ChinaReviewed by:
Tae Il Kim, Yonsei University College of Medicine, South KoreaShing Yau Tam, Hong Kong Polytechnic University, Hong Kong, SAR China
Zhiyun Cao, Fujian University of Traditional Chinese Medicine, China
Copyright © 2021 Peng, Jiang, Lee, Tsai, Chang, Chen and Yen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: B. Linju Yen, Ymx5ZW5AbmhyaS5vcmcudHc=