
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Oncol. , 18 August 2021
Sec. Pharmacology of Anti-Cancer Drugs
Volume 11 - 2021 | https://doi.org/10.3389/fonc.2021.712824
This article is part of the Research Topic Role of Phytochemicals and Structural Analogs in Cancer Chemoprevention and Therapeutics View all 14 articles
 Shailima Rampogu1,2†
Shailima Rampogu1,2† Seong Min Kim3†Baji Shaik4†Gihwan Lee1Ju Hyun Kim4
Seong Min Kim3†Baji Shaik4†Gihwan Lee1Ju Hyun Kim4 Gon Sup Kim3Keun Woo Lee1*
Gon Sup Kim3Keun Woo Lee1* Myeong Ok Kim2*
Myeong Ok Kim2*Background: Breast cancer is one of the major causes of mortalities noticed in women globally. DDX3 has emerged as a potent target for several cancers, including breast cancer to which currently there are no reported or approved drugs.
Methods: To find effective cancer therapeutics, three compounds were computationally designed tweaking the structure of natural compound butein. These compounds were synthesized and evaluated for their anticancer property in MCF-7 and MDA-MB-231 cell lines targeting DDX3. The in silico molecular docking studies have shown that the compounds have occupied the binding site of the human DDX3 target. Furthermore, to investigate the cell viability effect of 3a, 3b, and 3c on MCF-7 and MDA-MB-231 cell lines, the cell lines were treated with different concentrations of compounds for 24 and 48 h and measured using MTT assay.
Results: The cell viability results showed that the have induced dose dependent suppression of DDX3 expression. Additionally, 3b and 3c have reduced the expression of DDX3 in MCF-7 and MDA-MD-231 cell lines. 3b or 3c treated cell lines increased apoptotic protein expression. Both the compounds have induced the apoptotic cell death by elevated levels of cleaved PARP and cleaved caspase 3 and repression of the anti-apoptosis protein BCL-xL. Additionally, they have demonstrated the G2/M phase cell cycle arrest in both the cell lines. Additionally, 3c decreased PI3K and AKT levels.
Conclusions: Our results shed light on the anticancer ability of the designed compounds. These compounds can be employed as chemical spaces to design new prospective drug candidates. Additionally, our computational method can be adapted to design new chemical scaffolds as plausible inhibitors.
RNA helicases are a group of proteins possessing a unique motif, called the DEAD/H (Asp-Glu-Ala-Asp/His) (1). One of the members of the RNA helicase family, DDX3, demonstrates a wide range of roles in the process of cellular biogenesis that includes cell survival, cell-cycle regulation, cellular differentiation, and apoptosis (2). The genome of human encodes for two genes, namely DDX3X and DDX3Y. DDX3Y is present on Y-chromosome and demonstrates its role in male fertility. The gene DDX3X is present on the X-chromosome bands, p11.3– > p11.23 (2) and acts as a oncogene or tumor suppressor (1). DDX3 modulates the expression of genes at various levels. It takes part in transcriptional regulation of gene promoters, engages in splicing, performs the nuclear export of RNA, demonstrates a role in translational regulation (3). The structure of DDX3 is made up of 12 conserved motifs and two recA like domains. The X-ray structure of DDX3 was cocrystallized with the AMP located at the nucleotide-binding pocket (2). The residues from the Q motif, Arg202, and Gln207, hold the adenine group and the phosphate group is held by the P-loop residues, namely Gly227, Ser228, Gly229, Lys230, and Thr231 (2). This binding site has been exploited widely by the researchers to design and discover new chemical compounds (4–6).
DDX3 is a well-studied protein and is a known validated target to design and develop antiviral and anticancer drugs (7). DDX3 communicates with several viral and human proteins and their complexes via the RNA and demonstrates a double role in viral replication. Intriguingly, it works as a cofactor for viral replication and acts as a mediator of the innate immunity system (7). The association of DDX3 in cancer has been emerging in the recent times (8) and has demonstrated a significant role in progression of malignancies besides playing a key role in tumorigenesis to metastasis (8–17). Although the role of DDX3 is noticed in several cancers (1), in the current study, we focused on breast cancer targeting two cell lines, MCF-7 and MDA-MB-231, respectively.
DDX3 demonstrates an oncogenic role in breast cancer and upon its elevation may promote cell growth and proliferation (1, 10) and corresponds to distant metastasis (18), whose knockdown represses the tumor volume in vivo (19), thus demonstrating an oncogenic role in breast cancer (1). Several inhibitors were employed to target DDX3 in breast cancer. The combination of DDX3 and PARP inhibitors have brought about synthetic lethality noticed in BRCA1-proficient breast cancer (20). Another compound RK-33 has induced radiosensitization in breast cancer via the mitochondrial translation inhibition (21).
In the current investigation, we have used the butein derivatives to potentially target DDX3 in MCF-7 and MDA-MB-231 cell lines. Butein, a flavonoid, is known to have immense therapeutic potential (22), besides being an anticancer agent (23). In lung cancer cells, butein promotes apoptosis and represses the expression of cyclooxygenase-2 (24). It is reported that butein triggers the suppression of breast cancer growth by causing the reduction of the reactive oxygen species (ROS) production (25) and hindering the AKT phosphorylation. Furthermore, it was mechanistically noticed that butein elicits apoptosis by a series of mechanisms, thereby demonstrating the anti-proliferative activity (26) and can further act as aromatase inhibitor with the IC50 value of 3.7 μM (27). It also subsides the proliferative ability of the breast cancer cells by the formation of reactive oxygen species and regulation of ERK and p38 functions (26). Estrogen has been linked with the initiation and the progression of the disease. It was evidenced that butein could efficiently inhibit the aromatase when tested on MCF-7 cells with a minimum inhibitory concentration showing less than 5 µM (27). The purified phenolic-rich EtOAC fraction (PPEF) extracts of medicinal plant Rhus verniciflua Stokes (RVS) that contained butein, demonstrated apoptosis in breast cancer cell lines (28). Fibroblasts, the cockroaches of the human body (29), are found at all the stages of cancer, further playing a major role in promoting the growth of breast cancer. The phytochemical butein has inhibited the clonogenic growth of UACC-812, which was co-cultured with fibroblasts. Mechanistically, it can be speculated that the chalcone butein compound might intrude between the fibroblast and the breast cancer cells at as low as 2.5 µg/ml (30). It is evidenced that butein downregulates the CXC chemo receptor-4 (CXCR4) by transcriptional regulation demonstrated by the downregulation of mRNA expression, suppression of NF-κB activation, and suppression of chromatin immunoprecipitation activity, thereby hindering the process of metastasis (31). It was reported that butein repressed the formation of osteoclasts prompted by MDA-MB-231 cell lines (32).
Motivated by the aforementioned reports, in the current investigation, we have synthesized butein analogs and have tested them against MCF-7 and MDA-MB-231 cell lines, respectively. The primary objective of the study is to evaluate the anticancer potential of the new compounds and to assess their ability as DDX3 inhibitors.
The butein structure was downloaded from the PubChem database (33) in the 2D format and was subsequently exported on to the Discovery Studio v18 (hereinafter referred to as DS). The compound was minimized by employing the Full Minimization protocol accessible with DS.
Using the Discovery Studio, the B ring of butein was modified to obtain new structures as described in Figure 1, and three compounds were designed and synthesized.
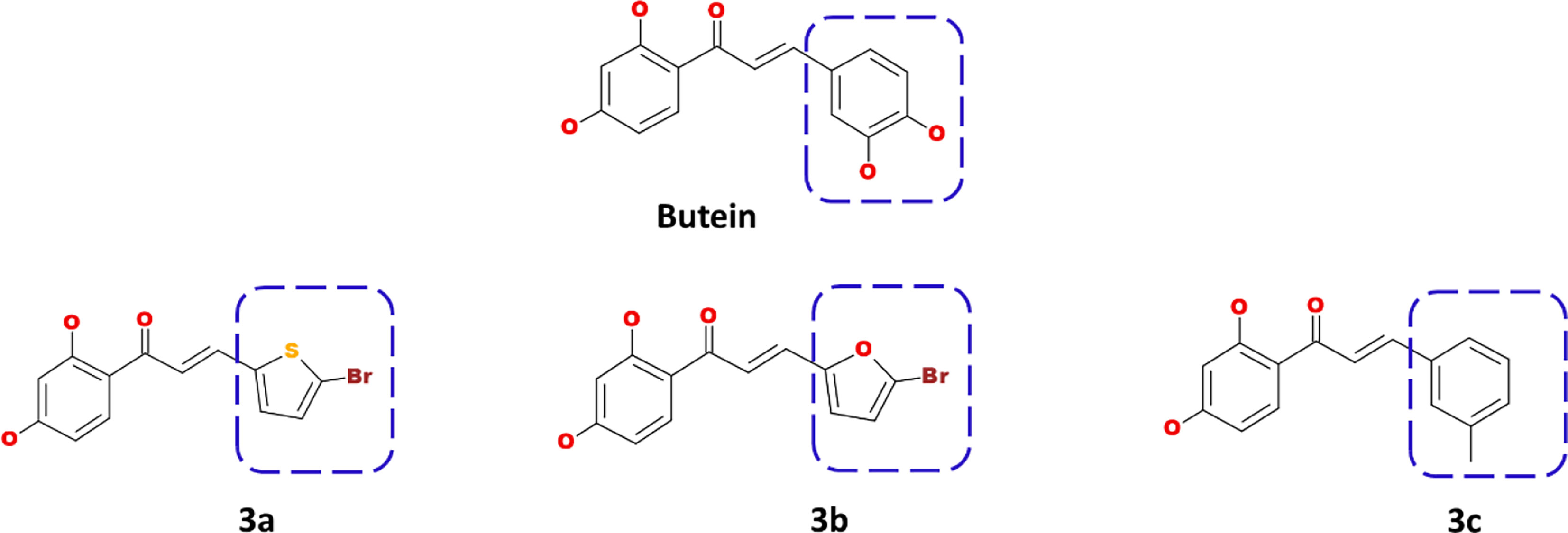
Figure 1 Computational designing of butein compounds modifying the ring B as indicated in dotted box.
To elucidate on their binding affinity toward the target DDX3, the molecular docking was performed using the CDOCKER program (34–37) available on DS. The CDOCKER utilized a CHARMm-based molecular dynamics (MD) strategy to dock ligands into a receptor-binding site. Correspondingly, Random Ligand Conformations are generated using high-temperature MD. These conformations are then translated into the binding site. The candidate poses are subsequently created using random rigid-body rotations followed by simulated annealing. A final minimization is then used to refine the ligand poses (34, 38).
The protein for the current study is the human DEAD-box RNA helicase DDX3X bearing the PDB code 2I4I cocrystallized with the AMP (39). The protein was initially prepared by initiating the Clean Protein tool available with the DS. Correspondingly, the heteroatoms and the water molecules were dislodged and were minimized using the Minimization tool available under the Minimize and Refine Protein protocol. The active site was marked for all the atoms and the residues around the cocrystallized AMP (39).
The synthesized butein-like compounds were docked into the active site of the protein permitting the generation of 50 conformers for each compound along with the butein compound. The best pose was selected from the largest cluster demonstrating a higher dock score, read according to the -CDOCKER interaction energy and the key residue interactions.
Unless otherwise noted, all reactions were carried out under an atmosphere of argon in oven-dried flasks. All reagents and chemicals were purchased from Sigma Aldrich Co., TCI, or Alfa Aesar Co. Solvents, such as tetrahydrofuran (THF) and methylene chloride, were used after distillation following standard purification procedures. 1H NMR spectra were recorded on a Bruker DRX-300 and chemical shifts (δ) for 1H NMR spectra are given in ppm relative to TMS. Column chromatography was performed on Merck silica gel (40–63 mesh).
To a solution of 2′,4′-dihydroxyacetophenone (0.76 g, 5.0 mmol) in dry CH2Cl2 (5 mL) triethylamine (2.4 mL, 17.5 mmol) and 4-dimethylamino pyridine (0.06g, 0.5 mmol) were added, and then tertbutyldimethylsilyl chloride (2.11g, 14.0 mmol) dissolved in CH2Cl2 (9 mL) was added dropwise. After the completion of addition, the reaction mixture was allowed to stirred overnight at room temperature. The reaction mixture was quenched by the addition of water. The organic fractions were collected, and the aqueous phase was extracted with CH2Cl2. The combined organic fractions were washed with water and brine, dried over anhydrous MgSO4, and concentrated under reduced pressure. The residue was purified by short column chromatography on silica gel using n-hexanes as the eluent to afford compound 2 (1.56 g, 82% yield) as a colorless oil; 1H NMR (300 MHz, Chloroform-d) δ 7.62 (d, J = 8.6 Hz, 1H), 6.47 (dd, J = 8.6, 2.3 Hz, 1H), 6.32 (d, J = 2.3 Hz, 1H), 2.57 (s, 3H), 1.00 (s, 9H), 0.97 (s, 9H), 0.28 (s, 6H), 0.21 (s, 6H).
To a solution of compound 2 (1.0 mmol) in dry THF (3 ml) under argon was added lithium diisopropylamide (LDA, 1.0 M, 1.1 ml, 1.1 mmol) dropwise at −78°C. The reaction mixture was allowed to warm to −20°C over 30 min, and then cooled to −78°C. The solution of substituted aldehyde (1.0 mmol) in dry THF (3 ml) was added dropwise at −78°C, and the resulting reaction mixture was allowed to warm to room temperature stirred overnight and then quenched by addition of sat. aqueous NH4Cl. The organic fraction was collected, and the aqueous layer was extracted with diethyl ether. The combined organic fractions were washed with brine, dried over anhydrous MgSO4, and evaporated under reduced pressure to afford the monohydroxy protected chalcones. The monohydroxy protected chalcones were purified by using CH2Cl2/MeOH as eluent.
The monohydroxy protected chalcone derivatives (0.2 mmol) were dissolved in THF (5 ml), and tetrabutylammonium fluoride (TBAF, 1.0 M, 0.4 mmol) was added. The resulting reaction mixture was stirred at room temperature for 30 min. and quenched by addition of water. The organic fraction was collected, and the aqueous layer was extracted with ethyl acetate three times. The combined organic fractions were washed with brine, dried over anhydrous MgSO4, and evaporated under reduced pressure. The residues were purified column chromatography on silica gel using CH2Cl2/MeOH as eluent to afford compounds 3a-3c in 70% to 85% yields.
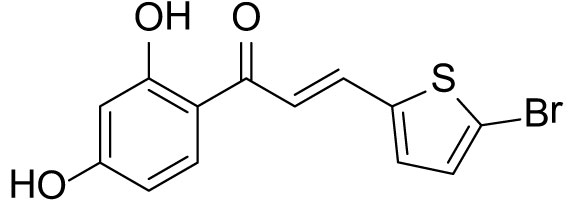
The compound (3a) was synthesized by following the general procedure using monohydroxy-protected chalcone (0.10 g, 0.23 mmol) dissolved in THF (8 mL) and tetrabutylammonium fluoride (TBAF, 1.0 M, 0.46 mL, 0.46 mmol). The 3a was purified using silica gel column CH2Cl2/MeOH 10: 1, v/v. Rf. Value = 0.5. Yellow solid (63 mg, 85%); 1H NMR (300 MHz, Chloroform-d) δ 13.28 (s, 1H), 7.87 (dd, J = 15.1, 0.6 Hz, 1H), 7.75 (d, J = 8.5 Hz, 1H), 7.23 (d, J = 15.1 Hz, 1H), 7.12 (d, J = 3.9 Hz, 1H), 7.07 (d, J = 3.9 Hz, 1H), 6.46–6.40 (m, 2H), 5.53 (s, 1H).
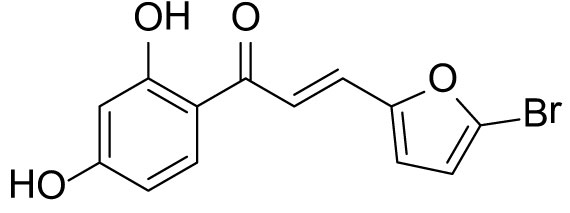
The compound (3b) was synthesized by following general procedure using monohydroxy-protected chalcone (0.10 g, 0.24 mmol) dissolved in THF (8 mL) and tetrabutylammonium fluoride (TBAF, 1.0 M, 0.48 mL, 0.48 mmol). The 3b was purified using silica gel column CH2Cl2/MeOH 10: 1, v/v. Rf. Value =0.5. Yellow solid (52 mg, 70%); 1H NMR (300 MHz, Chloroform-d) δ 13.34 (s, 1H), 7.84 (d, J = 8.5 Hz, 1H), 7.59–7.38 (m, 2H), 6.67 (d, J = 3.5 Hz, 1H), 6.48–6.40 (m, 3H), 5.30 (s, 1H).
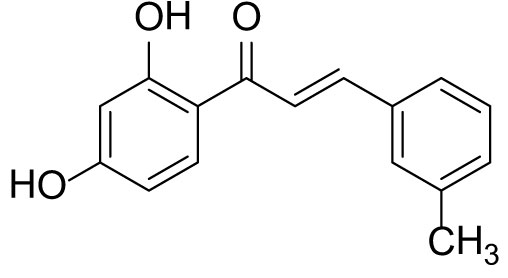
The compound (3c) was synthesized by following general procedure using monohydroxy protected chalcone (0.03 g, 0.08 mmol), was dissolved in THF (2 mL) and tetrabutylammonium fluoride (TBAF, 1.0 M, 0.16 mL, 0.16 mmol). The 3c was purified using silica gel column CH2Cl2/MeOH 10: 1, v/v. Rf. Value = 0.5. Yellow solid (16 mg, 77%) (40); 1H NMR (300 MHz, Chloroform-d) δ 13.36 (s, 1H), 7.90–7.83 (m, 2H), 7.56 (d, J = 15.5 Hz, 1H), 7.46 (d, J = 6.9 Hz, 2H), 7.23–7.35 (m, 2H), 6.47–6.41 (m, 2H), 5.47 (s, 1H), 2.41 (s, 3H); CAS. No. 1385668-91-6.
Human MCF-7 and MDA-MB-231 cell lines were purchased from the Korean Cell Line Bank (KCLB, Seoul, Korea). The MCF-7 and MDA-MB-231 cell lines were maintained in RPMI-1640 medium (Gibco, Life Technologies, Carlsbad, CA, USA) containing 10% (v/v) fetal bovine serum (FBS, Gibco) and 1% penicillin–streptomycin (Gibco) at 37°C in a humidified atmosphere of 5% CO2 (41).
The cell viability assay was conducted as described before (42) the cells were seeded in 48 well, plated at a density of 5 × 104 cells in 500 µL medium per well for 16 h. The cultured cells were treated with various concentration for 24, 48, and 72 h. After incubation, the cells were added with 55 µl of 5 mg/ml 3-(4, 5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide (MTT; Duchefa Biochemie, Haarlem, The Netherlands) solution for 2 h, and the medium was removed, which was followed by lysis with DMSO. The absorbance at 570 nm was measured with PowerWave HT microplate spectrophotometer (BioTek, Winooski, VT, USA).
The Western blot analysis was performed as described before (43). After 48 h treatment of each compounds, the cells were lysed with RIPA buffer (50 mM Tris-HCl pH 7.5, 0.1% SDS, 1% Triton X-100, 150 mM NaCl, 0.5% Sodium deoxycholate, and 2 mM EDTA) containing protease/phosphatase inhibitor cocktail (Thermo Fisher Scientific, Waltham, MA, USA) at 4°C for 1 h. The supernatants were collected and quantified using BCA protein assay kit (Thermo Fisher Scientific) according to the manufacturer’s instructions. To identify the molecular weight, we used Regular Range Protein marker (PM2510; SMOBIO Technology. Inc., Taiwan) and precisionplus protein dual color standard marker (Bio-Rad catalog no 1610374). Protein of 10–20 µg concentration was loaded on 8–12% sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE), and then transferred to polyvinylidene difluoride membrane (PVDF, ATTO, Tokyo, Japan). The membranes were blocked with TBS-T buffer (Tris-buffered saline containing 0.1% Tween 20) containing 5% (w/v) skim milk power for 1 h at 25°C, and then incubated with primary antibodies for 16 h at 4°C. After incubation with the secondary antibody for 3 h at 25°C, the membranes was detected using Clarity™ Western ECL Blotting Substrates (Bio-Rad, Hercules, CA, USA).
The data were expressed as mean ± standard error of mean (SEM) and analyzed by GraphPad Prism Version 4.0b (GraphPad Software Inc., La Jolla, CA, USA), for statistical significance using one-way analysis of variance (ANOVA). p < 0.05 was considered as statistically significant. All experiments were performed in triplicates.
The prepared butein compound was used as the starting structure (parent structure) to computationally design new compounds. Subsequently, modifying the ring B has yielded three compounds as shown in Figure 1.
The synthesized compounds were assessed for the binding affinity studies using the CDOCKER program. These compounds were docked into the ATP binding pocket of the DDX3 to understand their potentiality along with butein for comparative study. The results have shown that the modified compounds have demonstrated a better to comparable dock score than the parent compound butein as shown in Table 1.

Table 1 Molecular dock score of butein and its analogues with target DDX3.
Further, the compounds have shown interactions with the key residues positioning at the binding pocket as elucidated in Figure 2. The compound butein has formed hydrogen bond with the residue Arg202, as displayed in Figure 3A. Additionally, the key residue Tyr200 has formed the π-π stacked interaction. The 3a has prompted hydrogen bond interactions with the residues, Thr201 and Gln207, as shown in Figure 3B, along with the π-π stacked interaction by Tyr200. The compound 3b has interacted via hydrogen bonds with the residues, Thr231, Ala232, Gln281, and Glu285, respectively, as represented in Figure 3C. The key residue Tyr200 has participated by π-alkyl interaction holding the compound at the binding pocket. The compound 3c has formed hydrogen bond interactions with the residues, Thr231, Ala232, and Gln281 residues. The residue Thr231 has formed two hydrogen bonds with the ligand as illustrated in Figure 3D. The key residue Tyr200 has demonstrated the π-π stacked interaction. Several other residues have clamped the ligands at the binding pocket as shown in Supplementary 1A–C respectively.
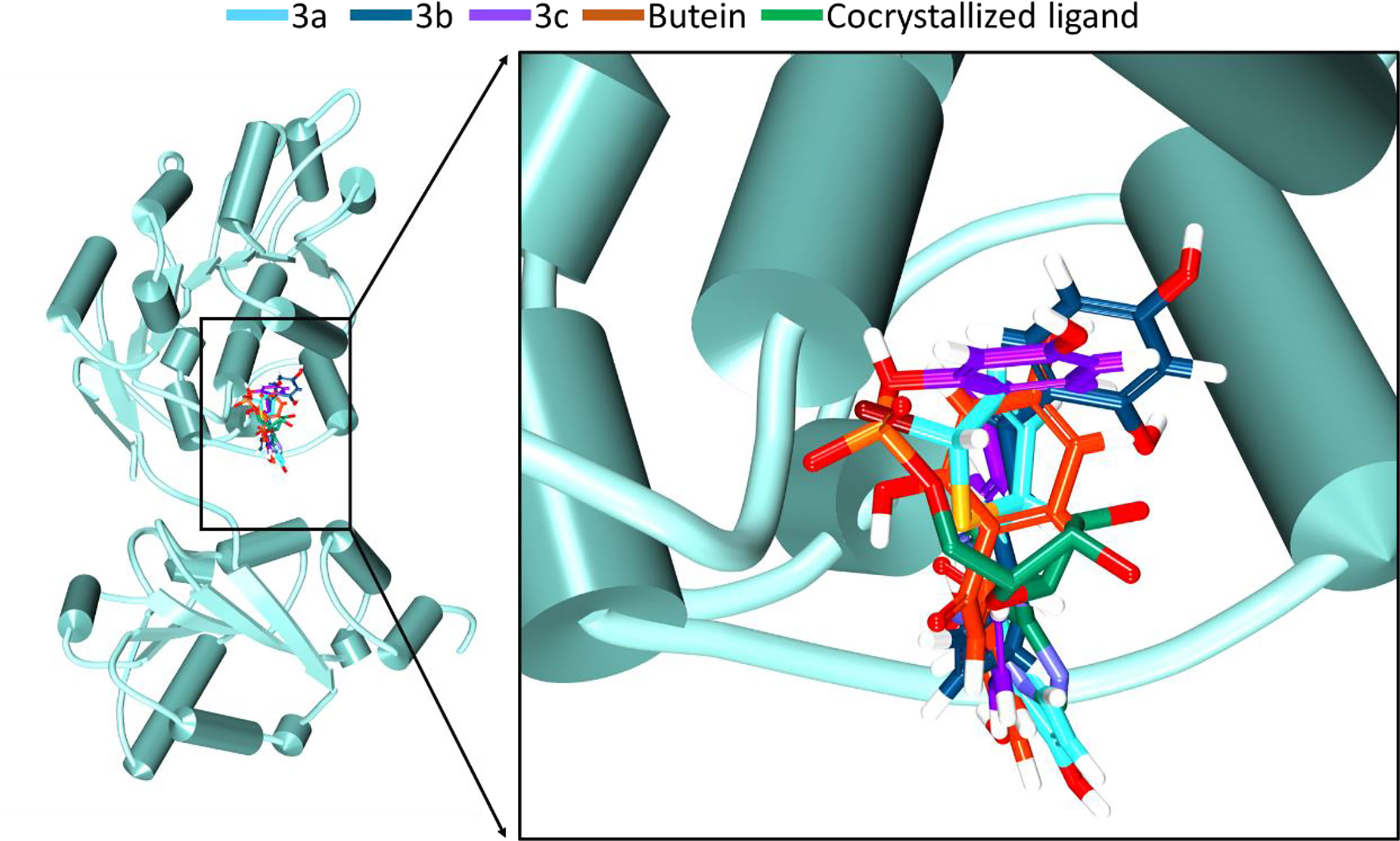
Figure 2 Accommodation of the synthesized compounds at the ATP binding pocket of DDX3 in comparison with butein and the cocrystallized ligand. The new compounds have displayed a similar binding mode as that of the cocrystallized ligand.
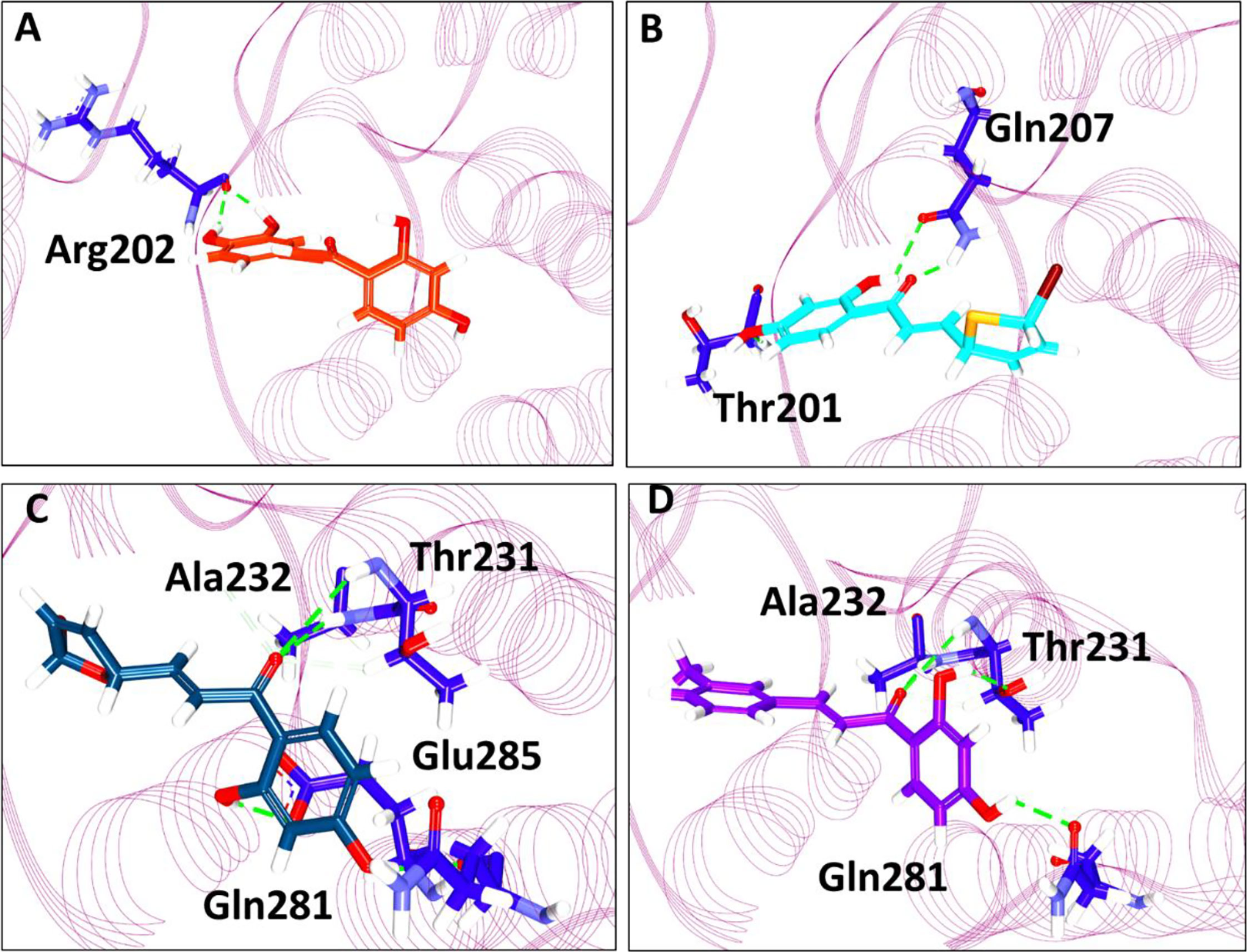
Figure 3 Intermolecular interaction between the protein and the small molecules. (A) Interaction of compound butein. (B) Hydrogen bond interaction between DDX3 and 3a. (C) Molecular interactions between 3b and DDX3. (D) Hydrogen bond interaction of DDX3 and 3c.
The syntheses of chalcones are illustrated in Scheme 1. All the chalcones were synthesized by aldol condensation reaction using an appropriate aldehyde and ketone as previously described in the literature method with modification. The dihydroxy ketone was protected by using TBSCl. Dihydroxy protected compound by reacting with aldehyde in the presence of LDA base resulted in the condensation product, which on desilylation by TBAF, the desired chalcones were obtained. The 1H NMR spectrum data of the three compounds is provided as, Supplementary Figures 2–4 for 3a, 3b, and 3c, respectively.
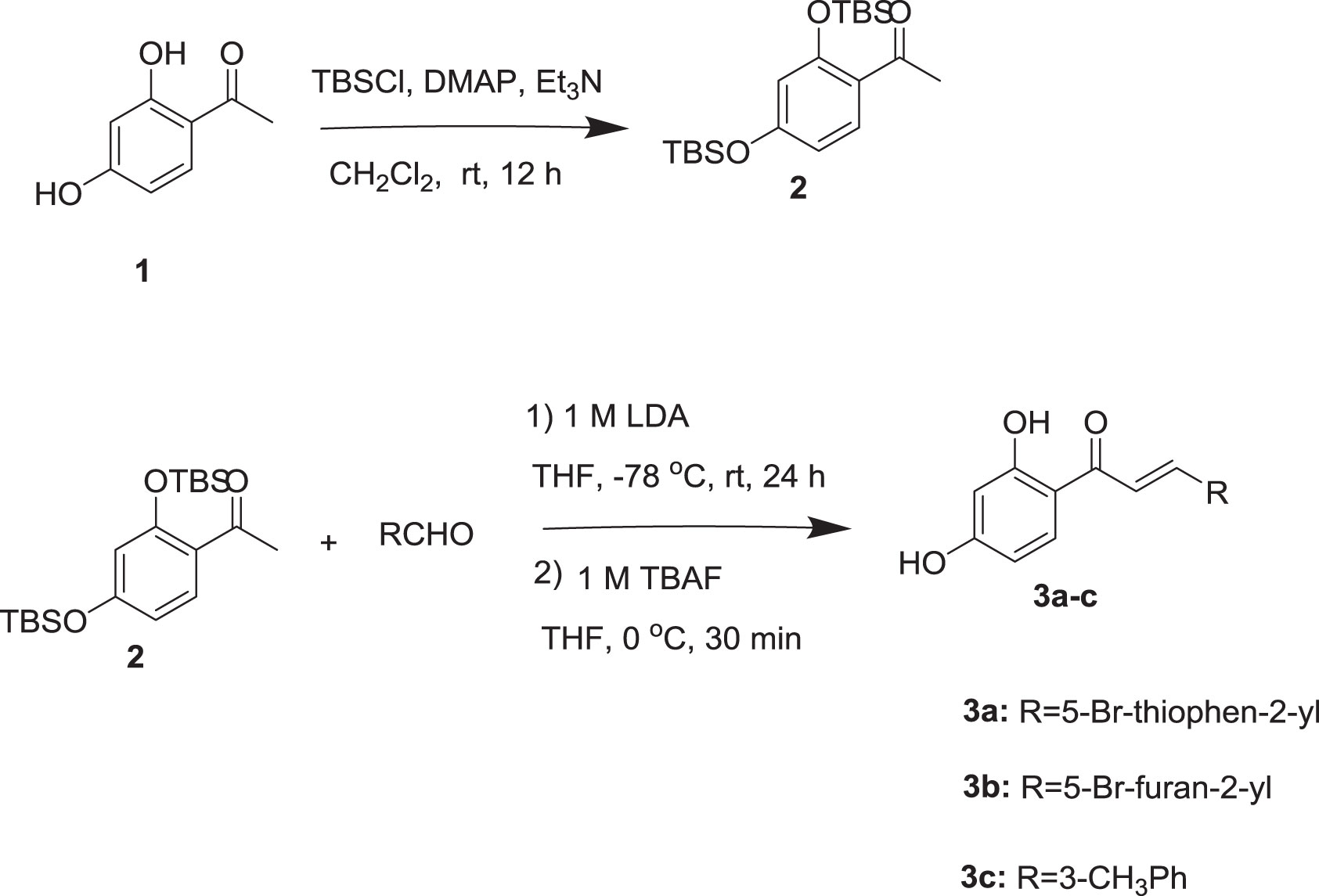
Scheme 1 Synthesis of chalcones.
To evaluate the anti-proliferative effect of 3b and 3c, MTT assays were carried out on the MDA-MB-231 and MCF-7 representing the human breast cancer cell lines. 3b and 3c were treated with various concentration for 24, 48, and 72 h in the MDA-MB-231 and MCF-7, respectively. The cell viability on the MDA-MB-231 and MCF-7 cells was markedly decreased by 3b and 3c treatment in a dose- and time-dependent manner as shown in Figures 4A, B. The IC50 values of 3b were recorded as 58.23 μM and 37.74 μM at 48 h in MCF-7 and MDA-MB-231 cell line, respectively. Also, IC50 values of 3c were shown as 22.72 μM and 20.51 μM at 48 h in MCF-7 and MDA-MB-231 cell line, respectively. These results showed that 3b and 3c have anti-proliferative effect in both human cell lines. Therefore, we used 20 and 30 μM of 3a, 25 and 50 μM of 3b, and 10 and 20 μM of 3c in our subsequent experiments.
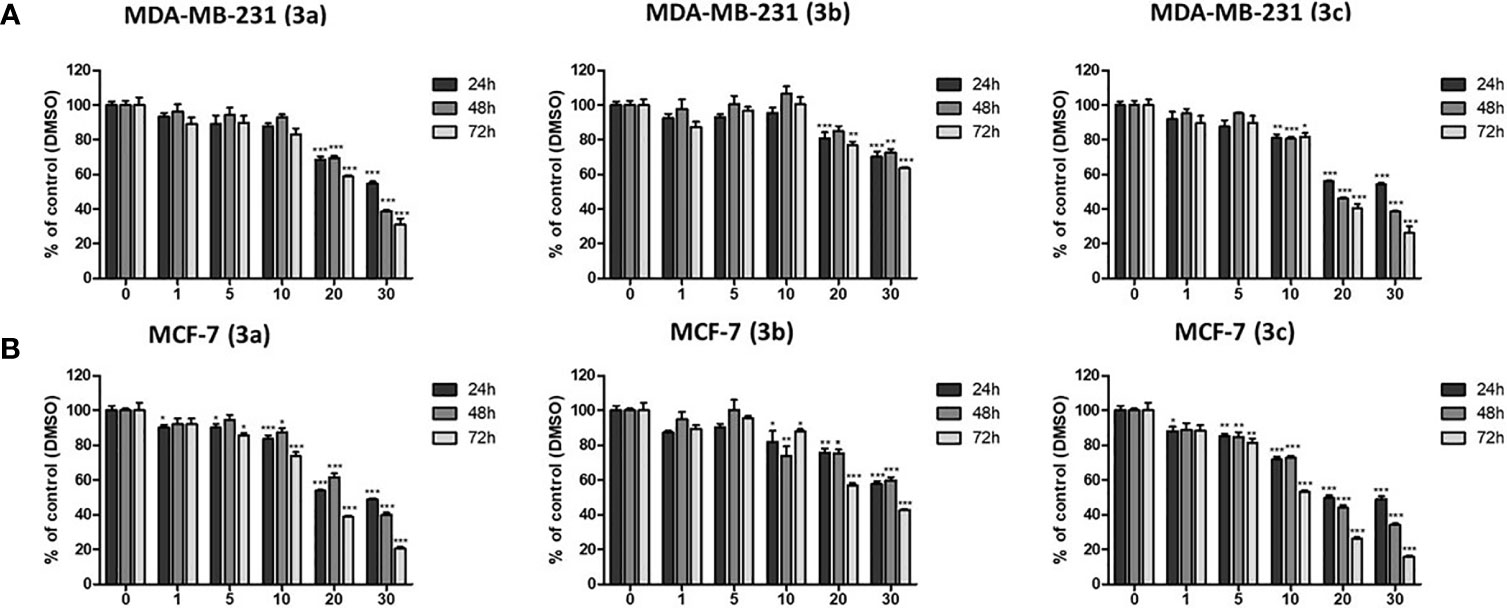
Figure 4 The cytotoxic effects on 3a, 3b, and 3c in MDA-MB-231, and MCF-7 cell lines. (A) The MDA-MB-231 and (B) MCF-7 cells were treated with each compounds as various concentrations (0–30 μM) for 24, 48, and 72 h. Then, the cell viability was measured by MTT assay. *p < 0.05 vs. untreated group; **p < 0.01 vs. untreated group; ***p < 0.001 vs. untreated group.
Based on cell viability results, we also identified the DDX3 protein expression. The two cell lines, which include MCF-7 and MDA-MB-231 cells were treated with indicated concentration of 3a, 3b, or 3c, respectively. The DDX3 protein expression was decreased dose-dependently in 3b or 3c treated MCF-7 and MDA-MB-231 cell lines as shown in Figures 5A, B, and Supplementary Figure 5. This data indicated that 3b and 3c could suppress DDX3 protein expression in MCF-7 and MDA-MB-231 cell lines. Since 3a did not show any significant reduction of DDX3 expression, as shown in Supplementary Figure 7, we have not proceeded with the compound further.
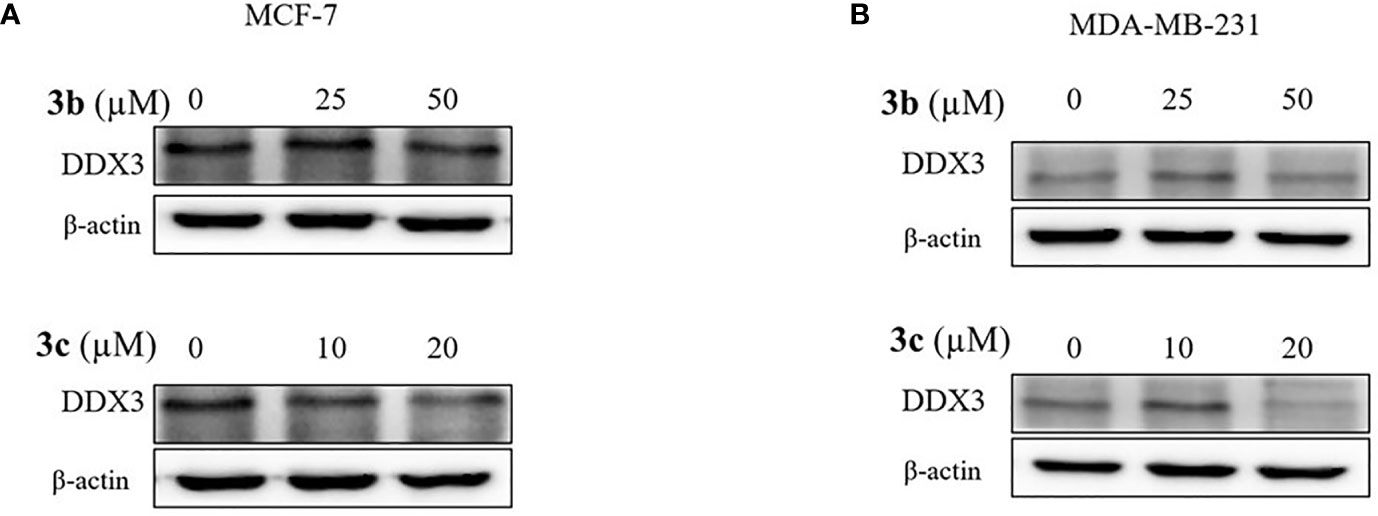
Figure 5 DDX3 protein expression on 3b and 3c in MCF-7 and MDA-MB-231 cell lines. The 3b and 3c were treated with indicated concentration (A) MCF-7 (B) MDA-MB-231 cell lines for 48 h. Control group (0 μM) was treated with same amount of DMSO. Western blot analysis were conducted to identify the DDX3 protein expression. The β-actin protein was used as loading control.
To determine the cell death and cell cycle arrest effect on 3b and 3c in human breast cancer cell lines, we identified the apoptosis and cell cycle related protein expression in 3b and 3c treated MCF-7 and MDA-MB-231 cell lines. The apoptosis marker proteins, such as cleaved PARP and cleaved caspase 3 were increased, and anti-apoptosis protein, BCL-xL, was decreased in compound use to treat both cell lines, as shown Figures 6A, B, and Supplementary Figure 6. In addition, G2/M cell cycle arrest-related protein including CDK1 and cyclinB1, were inhibited in compound treated both cell lines. Those data suggest that 3b and 3c can induce apoptotic cell death and G2/M phase cell cycle arrest in MCF-7 and MDA-MB-231 cell lines, indicating anti-cancer effect, as shown in Figures 6A, B.
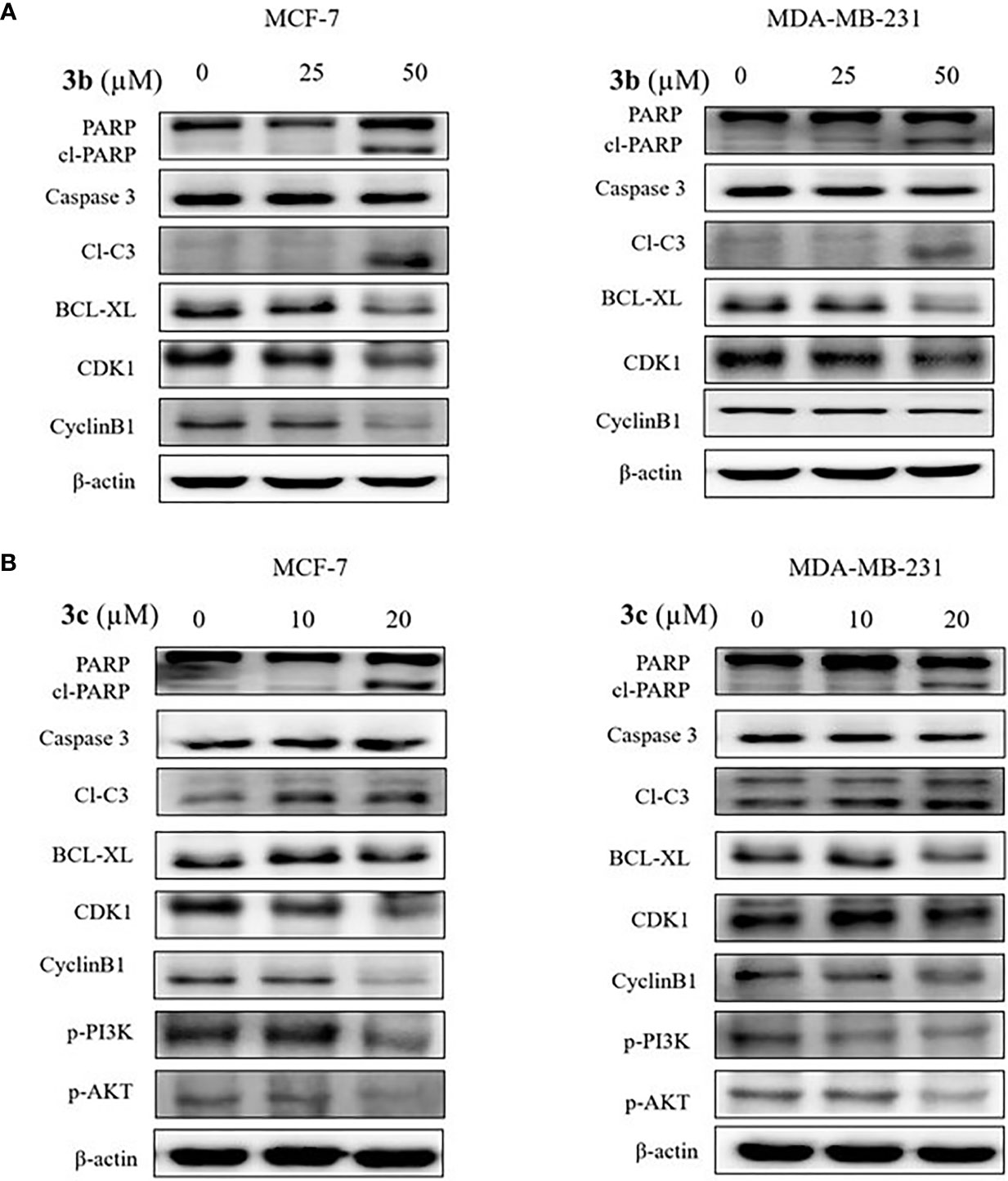
Figure 6 The apoptotic cell death effects of 3b and 3c in MCF-7 and MDA-MB-231 cell lines. The indicated concentration of 3b was treated in the MCF-7 and MDA-MB-231 cell lines for 48 h. (A) The indicated concentration of 3c was treated in the MCF-7 and MDA-MB-231 cell lines for 48 h. (B) Control group (0 μM) was treated with same amount of DMSO. The apoptotic related protesins (PARP, cleaved PARP, cleaved caspase 3, BCL-XL), cell cycle related proteins (CDK1 and CyclineB1), p-PI3K and p-AKT expression levels were analyzed using Western blot. The β-actin protein was used as loading control.
In addition, PI3K/AKT signaling pathway is an important intercellular signal involving apoptotic cell death and cell cycle arrest. Therefore, we examined the phosphorylated PI3K and AKT protein expression in the 3b and 3c treated human breast cancer cells. 3b was not affective to PI3K/AKT protein change. The results showed that inhibition of PI3K and AKT activation was induced in the 3c treated both cell lines, as shown in Figure 6B.
With an objective to find effective therapeutics to breast cancer, the current investigation has proceeded by tweaking the natural compound butein. Correspondingly, we have obtained three compounds. These three compounds, namely 3a, 3b, and 3c, were docked into the active site of the protein target 2I4I to gain insights into the atomistic interactions. Alluringly, the three compounds have rendered an interaction with the key residue Tyr200, via π-π stalk interaction with 3a and 3c, whereas 3b has prompted a π-alkyl interaction as shown in Supplementary Figure 1. Interaction with this residue is regarded to be potential as reported in previous reports (2, 4, 44). Additionally, it was reported that binding with Tyr200 phenyl ring and the imidazole-diazepine ring amplifies the binding (19). It was further noted that the compounds were accommodated at the binding pocket prompted by various residues.
To elucidate on their anti-proliferative activity in cancer cells, we proceeded with the in vitro MTT assay. This assay has shown a remarkable decline in the cell viability when compared with the untreated cells. The 3a has shown an IC50 value of 27.44 and 26.06 μM in MCF-7 and MDA-MD-231 cell lines for 48 h, whereas the 3b and 3c have demonstrated 58.23 μM and 37.74 μM and 22.72 μM and 20.51 μM, respectively, in MCF-7 and MDA-MD-231 cell lines recorded at 48 hrs. Although, 3a showed relatively stronger IC50 value, it has not repressed the DDX3 expression. This gives rise to the notion that 3a might act on other target other than DDX3, while the 3b and 3c have decreased the expression of DDX3 in both the cell lines. This finding sheds light on the identification of new inhibitors against DDX3.
Fundamentally, the flavonoids promote the anticancer mechanism by instigating the apoptosis and cell cycle arrest (42). The apoptosis was studies by the expression of cleaved PARP and cleaved caspase 3 (42). Caspases broadly belong to the protease family that manifests specificity to aspartic acids. These caspases are pivotal players of apoptosis (42). Among the caspases, Caspase-8, -9, and -10 are called the instigators of apoptosis as they are capable of activating other caspases while the caspase-3, -6, and -7 are grouped as executioners because of their ability to cleave important substrates thereby killing the cell (45). One such substrate is the poly (ADP-Ribose) polymerase (PARP) that is instrumental in maintaining DNA stability and repair (45, 46). The PARP being cleaved into two fragments serves as an indication of functional caspase activation (45). Our identified compounds have triggered the elevation of the apoptosis marker protein, such as cleaved PARP and cleaved caspase 3.
The antiapoptosis protein (apoptosis inhibitor) is an eminent member of BCL-2 family of apoptosis regulators (47). This protein is elevated in certain cancers, and its role in particular to breast cancer corresponds to increased metastatic potential than the primary tumor growth (48, 49) and is a target against invasive cancer cells (50). Our finding has shown that the level of BCL-xL was downregulated by both the compounds in two cell lines. These results signify the ability of the compounds to induce apoptosis cell death.
We further extended our study to assess the effect of 3b and 3c on cell cycle. Cell cycle has four phases that transvers from quiescence (G0 phase) to proliferation (G1, S, G2, and M phases). The process of cell cycle occurs by the activation of cyclin-dependent kinase (CDK) and its corresponding cofactor the cyclins (51). Among the innumerable factors that are involved in the process, the activation of CDK-cyclin heterodimeric complexes are considered as the paramount steps. Furthermore, the activation of the kinases (CDKs) is closely governed by the binding to cyclins (51). Correspondingly, the cell cycle proteins, cyclin B1 and CDK1, are interconnected with the G2/M phase. The primary factor cyclin B1 switches on the mitosis could compose compound with CDK1 to adjust the G2/M phase (51). Our findings have demonstrated that 3b and 3c have arrested the G2/M phase and suppressed the proteins CDK1 and cyclin B1 (51). The corresponding downregulation of CDK1 and cyclin B1 may reduce the formation of CDK1-cyclin B1 complex, thereby leading to the arrest of G2/M phase. These findings put forth that 3b and 3c have arrested the G2/M phase by repressing the expression of proteins CDK1 and cyclin B1.
The phosphatidylinositol 3-kinase (PI3K)/protein kinase B (AKT) signaling pathway is believed to be associated with the modulation of several cellular physiological processes (52) and has an important role in the regulation of cell proliferation, the cell cycle and apoptosis (52). It is a well-known fact that inhibition of PI3K/AKT signal pathway favors the process of apoptosis in cancer cell (53, 54). Likewise, 3c inhibited the expression level of PI3K and its downstream target AKT that is remarkably associated with cellular apoptosis. In a noteworthy observation, it was only 3c that could inhibit the PI3K/AKT signal pathway. The proposed mechanism of inhibition is represented in Figure 7, and all the full blots are provided as Supplementary Files.
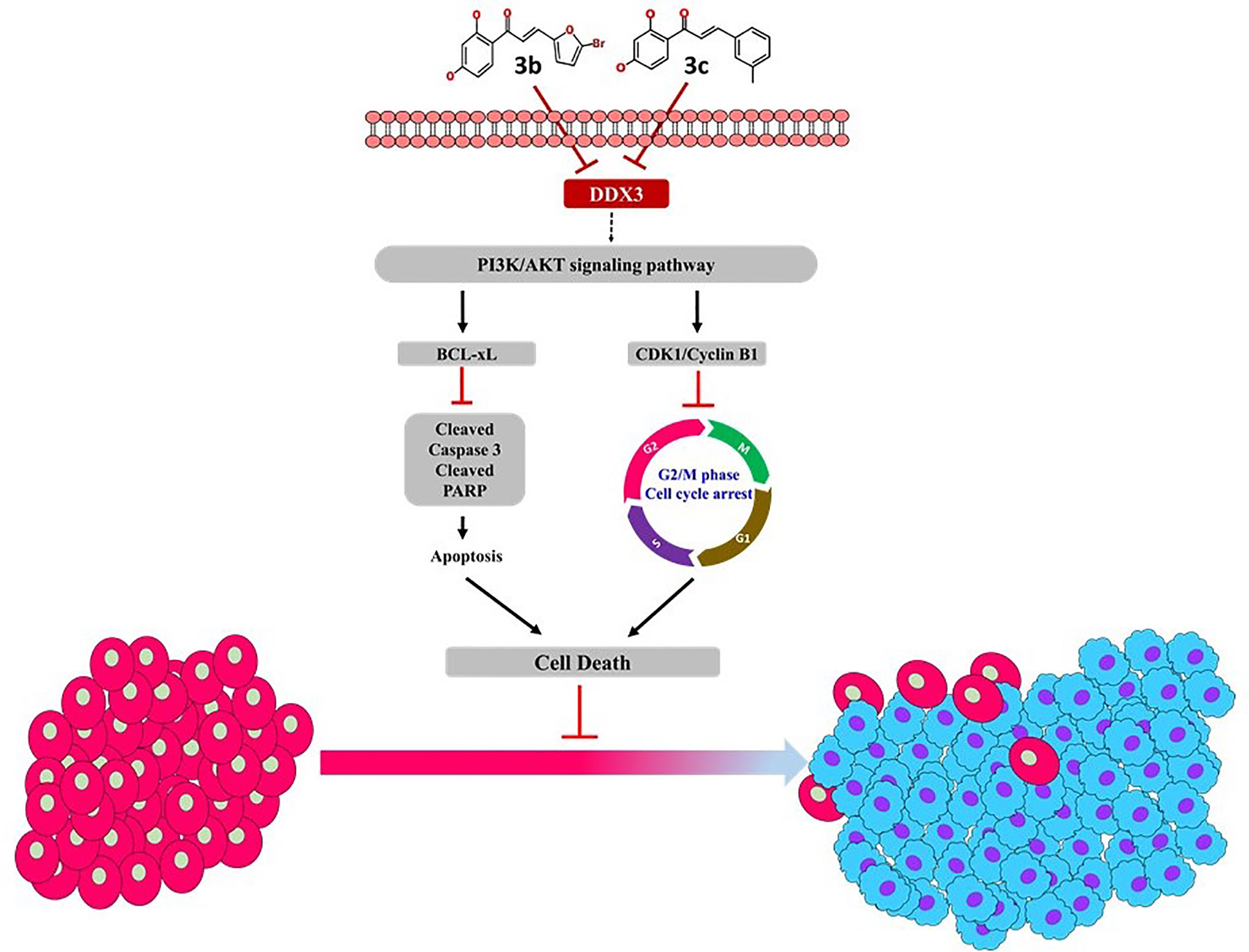
Figure 7 Proposed mechanism of 3b and 3c.
In conclusion, the present manuscript predominantly focuses on finding new therapeutics targeting DDX3. DDX3 is widely noted target that is over expressed in several cancers. In here, the phytocompound butein has been modified at ring B position to obtain new derivatives. These new compounds have repressed DDX3 expression in two cell lines, illuminating their potential to target DDX3. Additionally, these compounds have arrested the cell cycle proteins and have shown apoptosis, remarkably extending their usability in cancer treatments. Taken together, we propose two butein derivatives as novel DDX3 inhibitors that can additionally serve as scaffolds for designing new compounds.
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding authors.
SR, GL, MK, and KL conceived the idea. SR and GL performed the computational designing and molecular docking. BS and JK have synthesized the compounds. SK and GK have performed the MTT assay and the western blot analysis. SR, BS, and SK wrote the manuscript. All authors contributed to the article and approved the submitted version.
This research was supported by the Bio & Medical Technology Development Program of the National Research Foundation (NRF) and funded by the Korean government (MSIT) (No. NRF-2018M3A9A7057263). This research was supported by the Neurological Disorder Research Program of the National Research Foundation (NRF) funded by the Korean Government (MSIT) (2020M3E5D9080660).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2021.712824/full#supplementary-material
DEAD/H, Asp-Glu-Ala-Asp/His; BRCA1, breast cancer 1; CXCR4, CXC chemo receptor-4; CDK1, cyclin-dependent kinase 1; KCLB, Korean cell line bank; IC50, half-maximal inhibitory concentration; MD, molecular dynamics; PPEF, purified phenolic-rich EtOAC fraction; PARP, poly (ADP-ribose) polymerase; SEM, standard error of mean; TNFα, tumor necrosis factor alpha; THF, tetrahydrofuran; TBAF, tetrabutylammonium fluoride.
1. He Y, Zhang D, Yang Y, Wang X, Zhao X, Zhang P, et al. A Double-Edged Function of DDX3, as an Oncogene or Tumor Suppressor, in Cancer Progression (Review). Oncol Rep (2018) 39:883–92. doi: 10.3892/or.2018.6203
2. Bol GM, Xie M, Raman V. DDX3, A Potential Target for Cancer Treatment. Mol Cancer (2015) 14:1–16. doi: 10.1186/s12943-015-0461-7
3. Ariumi Y. Multiple Functions of DDX3 RNA Helicase in Gene Regulation, Tumorigenesis, and Viral Infection. Front Genet (2014) 5:423. doi: 10.3389/fgene.2014.00423
4. Botlagunta M, Kollapalli B, Kakarla L, Gajarla SP, Gade SP, Dadi CL, et al. In Vitro Anti-Cancer Activity of Doxorubicin Against Human RNA Helicase, DDX3. Bioinformation (2016) 12:347–53. doi: 10.6026/97320630012347
5. Bol GM, Vesuna F, Xie M, Zeng J, Aziz K, Gandhi N, et al. Targeting DDX 3 With a Small Molecule Inhibitor for Lung Cancer Therapy. EMBO Mol Med (2015) 7:648–69. doi: 10.15252/emmm.201404368
6. Rampogu S, Kim SM, Son M, Baek A, Park C, Lee G, et al. A Computational Approach With Biological Evaluation: Combinatorial Treatment of Curcumin and Exemestane Synergistically Regulates Ddx3 Expression in Cancer Cell Lines. Biomolecules (2020) 10(6):857. doi: 10.3390/biom10060857
7. Kukhanova MK, Karpenko IL, Ivanov AV. DEAD-Box RNA Helicase DDX3: Functional Properties and Development of DDX3 Inhibitors as Antiviral and Anticancer Drugs. Molecules (2020) 25(4):1015. doi: 10.3390/molecules25041015
8. Mo J, Liang H, Su C, Li P, Chen J, Zhang B. DDX3X: Structure, Physiologic Functions and Cancer. Mol Cancer (2021) 20:38. doi: 10.1186/s12943-021-01325-7
9. Chang PC, Chi CW, Chau GY, Li FY, Tsai YH, Wu JC, et al. DDX3, a DEAD Box RNA Helicase, Is Deregulated in Hepatitis Virus-Associated Hepatocellular Carcinoma and Is Involved in Cell Growth Control. Oncogene (2006) 25:1991–2003. doi: 10.1038/sj.onc.1209239
10. Botlagunta M, Vesuna F, Mironchik Y, Raman A, Lisok A, Winnard P, et al. Oncogenic Role of DDX3 in Breast Cancer Biogenesis. Oncogene (2008) 27:3912–22. doi: 10.1038/onc.2008.33
11. Wu DW, Lin PL, Cheng YW, Huang CC, Wang L, Lee H. DDX3 Enhances Oncogenic KRAS-Induced Tumor Invasion in Colorectal Cancer Via the β-Catenin/ZEB1 Axis. Oncotarget (2016) 7:22687–99. doi: 10.18632/oncotarget.8143
12. Wu DW, Lee MC, Wang J, Chen CY, Cheng YW, Lee H. DDX3 Loss by p53 Inactivation Promotes Tumor Malignancy Via the MDM2/Slug/E-cadherin Pathway and Poor Patient Outcome in Non-Small-Cell Lung Cancer. Oncogene (2014) 33:1515–26. doi: 10.1038/onc.2013.107
13. Liang S, Yang Z, Li D, Miao X, Yang L, Zou Q, et al. The Clinical and Pathological Significance of Nectin-2 and DDX3 Expression in Pancreatic Ductal Adenocarcinomas. Dis Markers (2015) 2015:379568. doi: 10.1155/2015/379568
14. Miao X, Yang Z-L, Xiong L, Zou Q, Yuan Y, Li J, et al. Nectin-2 and DDX3 Are Biomarkers for Metastasis and Poor Prognosis of Squamous Cell/Adenosquamous Carcinomas and Adenocarcinoma of Gallbladder. Int J Clin Exp Pathol (2013) 6:179–90. doi: 10.3109/07357907.2012.756113
15. Chen HH, Yu HI, Yang MH, Tarn WY. Ddx3 Activates Cbc-eIF3–Mediated Translation of uORF-Containing Oncogenic mRNAs to Promote Metastasis in HNSCC. Cancer Res (2018) 78(16):4512–23. doi: 10.1158/0008-5472.CAN-18-0282
16. Phung B, Cieśla M, Sanna A, Guzzi N, Beneventi G, Cao Thi Ngoc P, et al. The X-Linked Ddx3x RNA Helicase Dictates Translation Reprogramming and Metastasis in Melanoma. Cell Rep (2019) 27:3573–86. doi: 10.1016/j.celrep.2019.05.069
17. Brandimarte L, Pierini V, Di Giacomo D, Borga C, Nozza F, Gorello P, et al. New MLLT10 Gene Recombinations in Pediatric T-Acute Lymphoblastic Leukemia. Blood (2013) 121:5064–7. doi: 10.1182/blood-2013-02-487256
18. Heerma van Voss MR, Schrijver WAME, ter Hoeve ND, Hoefnagel LD, Manson QF, van der Wall E, et al. The Prognostic Effect of DDX3 Upregulation in Distant Breast Cancer Metastases. Clin Exp Metastasis (2017) 34:85–92. doi: 10.1007/s10585-016-9832-8
19. Xie M, Vesuna F, Botlagunta M, Bol GM, Irving A, Bergman Y, et al. NZ51, a Ring-Expanded Nucleoside Analog, Inhibits Motility and Viability of Breast Cancer Cells by Targeting the RNA Helicase DDX3. Oncotarget (2015) 6:29901–13. doi: 10.18632/oncotarget.4898
20. Heerma van Voss MR, Brilliant JD, Vesuna F, Bol GM, van der Wall E, van Diest PJ, et al. Combination Treatment Using DDX3 and PARP Inhibitors Induces Synthetic Lethality in BRCA1-Proficient Breast Cancer. Med Oncol (2017) 34(3):33. doi: 10.1007/s12032-017-0889-2
21. Heerma van Voss MR, Vesuna F, Bol GM, Afzal J, Tantravedi S, Bergman Y, et al. Targeting Mitochondrial Translation by Inhibiting DDX3: A Novel Radiosensitization Strategy for Cancer Treatment. Oncogene (2017) 34:63. doi: 10.1038/onc.2017.308
22. Padmavathi G, Roy NK, Bordoloi D, Arfuso F, Mishra S, Sethi G, et al. Butein in Health and Disease: A Comprehensive Review. Phytomedicine (2017) 25:118–27. doi: 10.1016/j.phymed.2016.12.002
23. Bordoloi D, Monisha J, Roy NK, Padmavathi G, Banik K, Harsha C, et al. An Investigation on the Therapeutic Potential of Butein, A Tretrahydroxychalcone Against Human Oral Squamous Cell Carcinoma. Asian Pac J Cancer Prev (2019) 20:3437–46. doi: 10.31557/APJCP.2019.20.11.3437
24. Li Y, Ma C, Qian M, Wen Z, Jing H, Qian D. Butein Induces Cell Apoptosis and Inhibition of Cyclooxygenase-2 Expression in A549 Lung Cancer Cells. Mol Med Rep (2014) 9:763–7. doi: 10.3892/mmr.2013.1850
25. Cho SG, Woo SM, Ko SG. Butein Suppresses Breast Cancer Growth by Reducing a Production of Intracellular Reactive Oxygen Species. J Exp Clin Cancer Res (2014) 33(1):51. doi: 10.1186/1756-9966-33-51
26. Yang L-H, Ho Y-J, Lin J-F, Yeh C-W, Kao S-H, Hsu L-S. Butein Inhibits the Proliferation of Breast Cancer Cells Through Generation of Reactive Oxygen Species and Modulation of ERK and p38 Activities. Mol Med Rep (2012) 6:1126–32. doi: 10.3892/mmr.2012.1023
27. Wang Y, Chan FL, Chen S, Leung LK. The Plant Polyphenol Butein Inhibits Testosterone-Induced Proliferation in Breast Cancer Cells Expressing Aromatase. Life Sci (2005) 77:39–51. doi: 10.1016/j.lfs.2004.12.014
28. Jin Hye K, Chan Hwa J, Bo-Hyoung J, Ho Yeon G, Jong-Hyeong P, You-Kyung C, et al. Selective Cytotoxic Effects on Human Cancer Cell Lines of Phenolic-Rich Ethyl-Acetate Fraction From Rhus Verniciflua Stokes. Am J Chin Med (2009) 37:609–20. doi: 10.1142/S0192415X09007090
29. Kalluri R. The Biology and Function of Fibroblasts in Cancer. Nat Rev Cancer (2016) 16:582–98. doi: 10.1038/nrc.2016.73
30. Samoszuk M, Tan J, Chorn G. The Chalcone Butein From Rhus Verniciflua Stokes Inhibits Clonogenic Growth of Human Breast Cancer Cells Co-Cultured With Fibroblasts. BMC Complement Altern Med (2005) 5:5. doi: 10.1186/1472-6882-5-5
31. Chua AWL, Hay HS, Rajendran P, Shanmugam MK, Li F, Bist P, et al. Butein Downregulates Chemokine Receptor CXCR4 Expression and Function Through Suppression of NF-κb Activation in Breast and Pancreatic Tumor Cells. Biochem Pharmacol (2010) 80:1553–62. doi: 10.1016/j.bcp.2010.07.045
32. Sung B, Cho SG, Liu M, Aggarwal BB. Butein, a Tetrahydroxychalcone, Suppresses Cancer-Induced Osteoclastogenesis Through Inhibition of Receptor Activator of Nuclear Factor-Kappab Ligand Signaling. Int J Cancer (2011) 129:2062–72. doi: 10.1002/ijc.25868
33. Kim S, Chen J, Cheng T, Gindulyte A, He J, He S, et al. PubChem 2019 Update: Improved Access to Chemical Data. Nucleic Acids Res (2019) 47:D1102–9. doi: 10.1093/nar/gky1033
34. Wu G, Robertson DH, Brooks CL, Vieth M. Detailed Analysis of Grid-Based Molecular Docking: A Case Study of CDOCKER - A Charmm-Based MD Docking Algorithm. J Comput Chem (2003) 24:1549–62. doi: 10.1002/jcc.10306
35. Rampogu S, Gajula RG, Lee G, Kim MO, Lee KW. Unravelling the Therapeutic Potential of Marine Drugs as SARS-CoV-2 Inhibitors: An Insight From Essential Dynamics and Free Energy Landscape. Comput Biol Med (2021) 135:104525. doi: 10.1016/j.compbiomed.2021.104525
36. Rampogu S, Lee KW. Old Drugs for New Purpose—Fast Pace Therapeutic Identification for SARS-CoV-2 Infections by Pharmacophore Guided Drug Repositioning Approach. Bull Korean Chem Soc (2021) 212–26. doi: 10.1002/bkcs.12171
37. Rampogu S, Lee KW. Pharmacophore Modelling-Based Drug Repurposing Approaches for SARS-CoV-2 Therapeutics. Front Chem (2021) 9:636362. doi: 10.3389/fchem.2021.636362
38. Rampogu S, Lee G, Kulkarni AM, Kim D, Yoon S, Kim MO, et al. Computational Approaches to Discover Novel Natural Compounds for SARS-CoV-2 Therapeutics. ChemistryOpen (2021) 10:593–9. doi: 10.1002/open.202000332
39. Högbom M, Collins R, van den Berg S, Jenvert RM, Karlberg T, Kotenyova T, et al. Crystal Structure of Conserved Domains 1 and 2 of the Human DEAD-Box Helicase DDX3X in Complex With the Mononucleotide Amp. J Mol Biol (2007) 372:150–9. doi: 10.1016/j.jmb.2007.06.050
40. Ameta KL, Rathore NS, Kumar B. Synthesis of Some Novel Chalcones and Their Facile One-Pot Conversion to 2-Aminobenzene-1, 3-Dicarbonitriles Using Malononitrile. Analele Univ Bucuresti Chim (2011) 20(1):15–24.
41. Lee JH, Lee HJ, Sim DY, Jung JH, Kim KR, Kim SH. Apoptotic Effect of Lambertianic Acid Through AMPK/FOXM1 Signaling in MDA-MB231 Breast Cancer Cells. Phyther Res (2018) 32:1755–63. doi: 10.1002/ptr.6105
42. Kim SM, Ha SE, Lee HJ, Rampogu S, Vetrivel P, Kim HH, et al. Sinensetin Induces Autophagic Cell Death Through p53-Related Ampk/Mtor Signaling in Hepatocellular Carcinoma Hepg2 Cells. Nutrients (2020) 12(8):2462. doi: 10.3390/nu12082462
43. Kim SM, Vetrivel P, Ha SE, Kim HH, Kim JA, Kim GS. Apigetrin Induces Extrinsic Apoptosis, Autophagy and G2/M Phase Cell Cycle Arrest Through PI3K/AKT/mTOR Pathway in AGS Human Gastric Cancer Cell. J Nutr Biochem (2020) 83:108427. doi: 10.1016/j.jnutbio.2020.108427
44. Lin TC. DDX3X Multifunctionally Modulates Tumor Progression and Serves as a Prognostic Indicator to Predict Cancer Outcomes. Int J Mol Sci (2020) 21(1):281. doi: 10.3390/ijms21010281
45. Puig B, Tortosa A, Ferrer I. Cleaved Caspase-3, Caspase-7 and Poly (ADP-Ribose) Polymerase Are Complementarily But Differentially Expressed in Human Medulloblastomas. Neurosci Lett (2001) 306:85–8. doi: 10.1016/S0304-3940(01)01873-0
46. Parsons JL, Dianova II, Allinson SL, Dianov GL. Poly(ADP-Ribose) Polymerase-1 Protects Excessive DNA Strand Breaks From Deterioration During Repair in Human Cell Extracts. FEBS J (2005) 272:2012–21. doi: 10.1111/j.1742-4658.2005.04628.x
47. Youle RJ, Strasser A. The BCL-2 Protein Family: Opposing Activities That Mediate Cell Death. Nat Rev Mol Cell Biol (2008) 9:47–59. doi: 10.1038/nrm2308
48. Martin SS, Ridgeway AG, Pinkas J, Lu Y, Reginato MJ, Koh EY, et al. A Cytoskeleton-Based Functional Genetic Screen Identifies Bcl-xL as an Enhancer of Metastasis, But Not Primary Tumor Growth. Oncogene (2004) 23:4641–5. doi: 10.1038/sj.onc.1207595
49. Fernández Y, España L, Mañas S, Fabra A, Sierra A. Bcl-x(L) Promotes Metastasis of Breast Cancer Cells by Induction of Cytokines Resistance. Cell Death Differ (2000) 7:350–9. doi: 10.1038/sj.cdd.4400662
50. Keitel U, Scheel A, Thomale J, Halpape R, Kaulfuß S, Scheel C, et al. Bcl-Xl Mediates Therapeutic Resistance of a Mesenchymal Breast Cancer Cell Subpopulation. Oncotarget (2014) 5:11778–91. doi: 10.18632/oncotarget.2634
51. Shi-Yong G, Jun L, Xiao-Ying Q, Nan Zhu Y-BJ. Downregulation of Cdk1 and CyclinB1 Expression Contributes to Oridonin-Induced Cell Cycle Arrest at G2/M Phase and Growth Inhibition in SGC-7901 Gastric Cancer Cells. Asian Pac J Cancer Prev (2014) 15:6437–41. doi: 10.7314/APJCP.2014.15.15.6437
52. Shi X, Wang J, Lei Y, Cong C, Tan D, Zhou X. Research Progress on the PI3K/AKT Signaling Pathway in Gynecological Cancer (Review). Mol Med Rep (2019) 19:4529–35. doi: 10.3892/mmr.2019.10121
53. Hussain AR, Al-Rasheed M, Manogaran PS, Al-Hussein KA, Platanias LC, Al Kuraya K, et al. Curcumin Induces Apoptosis Via Inhibition of PI3′-kinase/AKT Pathway in Acute T Cell Leukemias. Apoptosis (2006) 11:245–54. doi: 10.1007/s10495-006-3392-3
Keywords: anticancer agents, butein, DDX3, cell cycle, apoptosis
Citation: Rampogu S, Kim SM, Shaik B, Lee G, Kim JH, Kim GS, Lee KW and Kim MO (2021) Novel Butein Derivatives Repress DDX3 Expression by Inhibiting PI3K/AKT Signaling Pathway in MCF-7 and MDA-MB-231 Cell Lines. Front. Oncol. 11:712824. doi: 10.3389/fonc.2021.712824
Received: 21 May 2021; Accepted: 10 June 2021;
Published: 18 August 2021.
Edited by:
Raghuram Kandimalla, James Graham Brown Cancer Center, United StatesReviewed by:
Rajalakshmi Manikkam, Holy Cross College, IndiaCopyright © 2021 Rampogu, Kim, Shaik, Lee, Kim, Kim, Lee and Kim. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Myeong Ok Kim, bW9raW1AZ251LmFjLmty; Keun Woo Lee, a3dsZWVAZ251LmFjLmty
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.