 Sihao Zheng
Sihao Zheng Qibin Song
Qibin Song Pingfeng Zhang
Pingfeng Zhang- Cancer Center, Renmin Hospital of Wuhan University, Wuhan, China
Cancer immunotherapy has accomplished significant progresses on treatment of various cancers in the past decade; however, recent studies revealed more and more heterogeneity in tumor microenvironment which cause unneglectable therapy resistance. A central phenomenon in tumor malignancy is metabolic dysfunctionality; it reprograms metabolic homeostasis in tumor and stromal cells thus affecting metabolic modifications on specific proteins. These posttranslational modifications include glycosylation and palmitoylation, which usually alter the protein localization, stability, and function. Many of these proteins participate in acute or chronic inflammation and play critical roles in tumorigenesis and progression. Therefore, targeting these metabolic modifications in immune checkpoints and inflammation provides an attractive therapeutic strategy for certain cancers. In this review, we summarize the recent progresses on metabolic modifications in this field, focus on the mechanisms on how glycosylation and palmitoylation regulate innate immune and inflammation, and we further discuss designing new immunotherapy targeting metabolic modifications. We aim to improve immunotherapy or targeted-therapy response and achieve more accurate individual therapy.
Introduction
In addition to conventional cancer therapies such as surgery, radiotherapy, chemotherapy, and targeted therapy, immunotherapy is playing a more and more important role in cancer treatment. Several types of modern cancer immunotherapies have been developed in the past decades, including immune checkpoint therapy (ICT), adoptive cell therapy, and cancer vaccines, especially since the first immune checkpoint inhibitor ipilimumab was developed and approved by the Food and Drug Administration (FDA) in 2011 (1, 2). The cancer cells reprogram their metabolic pathways to adapt to the tumor microenvironment and suppress the immune system which is supposed to recognize the cancer cells (through cancer antigens) and attack them. Cancer immunotherapy harnesses the patients’ own immune system to target cancer cells, thus it has high specificity and efficacy and has already achieved significant outcomes in clinical practice on certain cancer types (3, 4). Immune checkpoints are molecules on certain immune cells that need to be activated (or inactivated) to license an immune response. Since 2011, several immune checkpoint inhibitors have been approved by the US FDA for second- and even first-line treatment against various cancer types, such as lung cancer, renal cell carcinoma, bladder cancer, gastric cancer, and many others; these include the CTLA-4–blocking antibody ipilimumab (5), three antibodies against Programmed Cell Death Receptor-1 (PD-1, CD279) [pembrolizumab (6), nivolumab (7), and cemiplimab (8)], and three antibodies targeting Programmed Cell Death Ligand-1 (PD-L1, CD274) [atezolizumab (9), avelumab (10), and durvalumab (11)]. An increasing number of clinical trials have proven that using antibodies targeting immune checkpoints has longer follow-up than other agents and leads to improved survival with durable clinical response that can last more than a decade for patients with advanced carcinoma (1, 12–15).
Despite significant advances in clinical practice, ICT still faces outstanding challenges that need to be addressed urgently. First of all, ICT is not effective against all cancer types, furthermore, even within a “responsive” cancer type, not every patient is responsive, thus it is difficult to determine which cohort of patients should be suitable for ICT. Taking pharmaceutical treatment of PD-1 as an example, clinical data showed that only about 20% PD-L1–positive NSCLC (which accounts for 85% of all lung cancers) patients respond objectively to ICT (16, 17). The reason of the phenomenon called de novo resistance mainly encompasses immune evasion by other checkpoint pathway and incapable of T-cell infiltration to the tumor microenvironment, which is linked to innate immunity especially inflammation. Surprisingly, several clinical trials reveal that the survival benefits for NSCLC patients from anti-PD1/PD-L1 immunotherapy is independent of PD-L1 expression (18–23), thus the true predictive value of PD-L1 is limited (24). This may attribute to the failure of accurate measurement and scoring of PD-L1 protein expression with antibodies. Secondly, a substantial proportion of patients who initially respond to ICT ultimately relapse with lethal, drug-resistant disease months or years later, which is referred to as acquired resistance. Recent studies suggested that main determinants of acquired resistance are consist of function of interferon signaling pathways (25, 26), expression of antigen-presenting molecules (27), and immune-evasive oncogenic signaling pathways (28). In addition, ICT also elicits inflammatory side effects, which are often termed immune-related adverse events and primarily harm organs including lung, gastrointestinal tract, endocrine glands, skin, and liver (29).
Emerging data indicated that traditional single drug treatment is not sufficient for improving the progression-free survival of patients with tumor, and the efficacy of therapy in combination with anti-CTLA4, anti-PD1, or anti-PDL1 antibodies, as well as with other immunotherapies, chemotherapies, radiation therapies, and targeted therapies is better than that of treatment with single drug. Indeed, combinations of chemotherapy and/or targeted therapy with ICT have been approved as first-line treatments for several conditions, including lung cancer and advanced renal cell carcinoma (30, 31). However, as lung cancer is the major thoracic cancer which is the leading cause of cancer-related death in the world and only have an overall 5-year survival rate of about 20% (32), it leaves a huge gap for new treatments to fill thus reducing the health burden. With its high incidence and mortality, it is of great fundamental and clinical significance to further explore the regulatory mechanism inside the tumor microenvironment (TME) in order to find new biomarkers that can accurately predict the response to ICT.
It has been well established that TME has been significantly changed by altered tumor energy metabolism, leading to complexity for cancer treatments. The reprogramed glucose metabolism in cancer cells have been elegantly summarized in many reviews to explore the potential direct therapeutic opportunities targeting the metabolism pathways (33–35). Indirectly, the altered metabolism will change many metabolite concentration, and these metabolites have been identified to serve as signaling molecules; recent studies have shown that they are substrates for several newly identified modifications such as succinylation (36), hydroxybutyrylation (37), lactylation (38, 39), crotonylation (40), and many others (41). While these metabolic modifications are not in the scope of this review, the focus of our discussion is on glycosylation and palmitoylation, which use saccharide and saturated fatty acid as substrates for protein modifications. Glucose from extracellular sources is uptaken by cells and degraded to provide energy mainly by the anaerobic and aerobic pathways of glycolysis [also known as tricarboxylic acid cycle (TCA)]. A branch pathway called hexosamine biosynthetic pathway (HBP) uses fructose-6-P (F6P) from glycosylation to produce a nucleotide sugar UDP-N-acetylglucosamine (UDP-GlcNAc), which is the key substrate used for the glycolysis of proteins (34, 42). In cancer cells, this HBP pathway is usually upregulated with increased glycolysis, leading to a higher level of protein glycosylation and also increases heterogenity (43). Intracellular palmitic acid either comes from endogenous fatty acids (FA) synthesized by fatty acid synthase (FASN; only happening in normal liver and adipose tissue), or from exogenous fatty acids supplied by CD36-mediated uptake. It is well documented that cancer cells also have a high demands for lipids via increased FA synthesis (44). Palmitic acid (PA) not only provides energy to cells by β-oxidation but also serves as a substance involved in palmitoylation (45). However, protein palmitoylation level is not only determined by PA concentration but also by the specific palmitoyl S-acyltransferases ZDHHC enzyme which catalyzes the reaction, thus creating a complicated situation in cancer cells (46).
Like many other classic posttranslational modification (PTM) such as methylation, acetylation, phosphorylation, ubiquitination, etc., glycosylation and palmitoylation modulate protein characteristics such as structure, activity, subcellular localization, stability, and protein-protein interactions. Glycosylation involves in many fundamental molecular and cell biological processes such as cell-cell adhesion, cell-matrix interaction, cell metabolism and signaling, immune surveillance, tumor angiogenesis, etc. (43) Since palmitoylation adds a 16-carbon fatty acyl group onto cysteine side chain of protein, other than affecting the innate properties of target proteins which have critical roles in inflammation and cancer development, it most commonly increases membrane binding of target proteins and also manipulates the membrane properties. Palmitoylation plays an important role in many immunological processes such as inflammation signaling, phagocytosis, endothelial and epithelial integrity, etc (47). An increasing body of evidence showed that glycosylation and palmitoylation play important roles in tumor immunology especially PD-1/PD-L1 pathway and inflammation. In this review, we summarize the recent progresses on how these two metabolic modifications regulate immune checkpoint and inflammatory effect, aimed at discovering new cancer targets in TME for more accurate individual cancer treatments.
The Metabolic Modifications in Tumor Microenvironment
The tumor microenvironment (TME) is a complicated ecology consisting of many cells that co-evolve with cancer cells to influence the development and progression of cancer and noncellular components. In addition to malignant cells, there are many cell types including nutritional supportive cells such as stromal cells, endothelial cells, adipocytes, fibroblasts, tumor vasculature, and immune cells such as lymphocytes, dendritic cells, cancer-associated fibroblasts, macrophage, platelets, and others. Malignant cells have accumulated enough mutations to acquire the tumor hallmarks including growth, immune suppression, immortality, tumor-promoting inflammation, invasion and metastasis, angiogenesis, genome instabilities, dysregulated metabolism, etc. (48). There are various complex cell-cell communications inside TME, thus many cells reprogrammed their metabolic pathways to adapt to the acidic, hypoxic environment to provide energetic support to cancer cells, to help cancer cells evade immune surveillance, to help malignant cells to spread and invade, and so on. The extracellular matrix (ECM) in TME which is composed of active tissue components such as glycoproteins, collagens, and enzymes can influence cell adhesion, proliferation, communication, and migration (49). The composition and structure of the TME vary among cancer types and among patients, contributing to tumor heterogeneity which affects the response to cancer treatments. In the following, we focus on the most recent progresses on how the metabolic changes of lipids and carbohydrates affect immunosuppression and inflammation in TME.
Cell metabolism has been well connected to cancer a century ago since Otto Warburg discovered the aerobic glycolysis in cancer, known as the Warburg effect (50). Compared with normal cells, cancer cells develop a great capacity to grow, proliferate, and survive under stress conditions, which need a lot of energy and materials for membrane formation, biomass accumulation, immune suppression, etc. They modify several pathways in the metabolism of carbohydrates, lipids, proteins, and nucleotides, thus each cell types have a specific set of enzymes and metabolites to achieve a favorable microenvironment. Over the past 20 years, there is growing evidence that metabolism is closely linked to the development of tumor and tumor microenvironment. For example, it has been well established that cancer cells use hyperactivated PI3K-AKT pathway (activated by hormone insulin signal) to drive glucose uptake by upregulating glucose transporters GLUT1; this favors glycolysis pathway and stimulates a large amount of pyruvate and lactate production (51, 52), leading to acetyl-coenzyme A formation which could be used for ATP synthesis or for de novo lipogenesis (53, 54). On the other hand, the increased glutamine and glucose uptake by cancer cells and immune cells (55, 56) not only contributes to increased glycolysis but also increases flux into the metabolic branch HBP pathways, and the end‐product of HBP is uridine diphosphate (UDP)‐GlcNAc, which is a critical metabolite used for O-GlcNAcylation as well as for N‐glycosylation (57). Actually, there is direct evidence demonstrating that (UDP)‐GlcNAc increased 12 times in breast cancer (58).
As cancer cells are rapidly proliferating cells, they need to form a large amount of new membrane, thus they have increased endothelial lipid de novo synthesis, which is referred as reprogramming of fatty acid metabolism (55). Actually, studies have shown that FASN (catalyzes the synthesis of palmitate) was elevated in many human cancers (59, 60). Acetyl-coenzyme A is also the substrate for the de novo fatty acid synthesis; FASN converts dietary carbohydrates to long-chain saturated fatty acids through acetyl-CoA (61), resulting in the accumulation of palmitate acid in tumor cells. In addition, cancer cells increase the expression of transmembrane proteins responsible for the uptake of exogenous FA, including CD36 (also known as FAT), the fatty acid transporter family (FATP), or the soluble carrier protein family 27 (SLC27) (62). Fatty acids are major components of cell membrane; saturated and monounsaturated fatty acids are biomarkers in several types of cancer, thus they open interesting perspectives for biomarker discovery and nutritional strategies to control cancer. Palmitic acid is the most abundant lipid acids in cells (20%~30% of total fatty acids) with a concentration ranging from 0.3 to 4.1 mM (63); it is strictly regulated, and abnormal concentration increase will trigger higher level of protein lipidation such as palmitoylation. The growing understanding in glycobiology and lipid metabolism has been summarized in many other review papers (43, 64–66). As many PTM are sensitive to the concentration of their metabolite substrate, they are tightly linked to and regulated by cell metabolism; this is beyond the scope of this review but well reviewed elsewhere (67). Next, we only discuss how glycosylation and palmitoylation affect inflammation and cancer immunotherapy.
Glycosylation and Palmitoylation on Tumor Immune Checkpoint
Immunotherapies using checkpoint inhibitors tend to shrink tumors in patients with different cancers and are linked to durable responses and low toxicity levels. Therefore, they are regarded as promising interventions that can help in managing cancer. The PD-1/PD-L1 axis and cytotoxic T-lymphocyte-associated protein 4 (CTLA4), the two most well-characterized immune checkpoint signaling pathway to date, utilize quite different mechanisms to hinder T-cell–dependent immunity. Previous research has established that CTLA4 primarily prevents T-cell activation. Activation of T cells requires two signals, interaction between T-cell receptor (TCR) and the antigen-MHC complexes on the antigen-presenting cell (APC) surface and the engagement of costimulatory molecule CD28 with B7 molecules on APC (4). Upon activation, T cells proliferate and differentiate, and they express immune checkpoints such as CTLA-4 and PD-1. CTLA4 outcompetes CD28 for binding to B7 molecules and abrogate T-cell responses (68, 69). Different from CTLA4, the PD-1 association with PD-L1 plays an inhibitory function mediated by the tyrosine phosphatase SHP-2, which modulates signaling molecules downstream of the TCR binding with antigen-MHC I complexes, thus suppressing T-cell cytotoxic effect against target cells and leads to T-cell exhaustion and death (70, 71). It becomes evident that overexcessive immune response is repressed by these regulatory mechanism aiming at protecting organism from self-immune disease.
Tumor ICT, also known as tumor immune checkpoint blockade, becomes a new treatment for cancer since the first antibody targeting CTLA-4 was discovered by Nobel laureate James P. Allison (72, 73). Different from radiotherapy, chemotherapy, or targeted therapy that directly attack certain tumor cells, ICT leverages the cytotoxic potential of the immune system against cancer cells. Through blocking inhibitory regulation pathway by harnessing antibody, ICT unleash T-cell antitumor response. Unfortunately, the protective inhibitory mechanism is hijacked by various types of tumor cells to attenuate T-cell response and escape antitumor immune surveillance. Further research on the mechanism of de novo and acquired resistance to ICT and effective judgment on which cohort of patients are suitable for the use of ICT will lay a foundation for combined pharmacotherapy to treat cancer. In multiple mouse models, pharmacological inhibition of oncogenic signaling pathways, such as Wnt signaling pathway, CDK4-CDK6-dependent cell cycle, and MAPK signaling pathway can result in reversal resistance to anti-PD1 therapy. Moreover, combination therapy with anti-CTLA4 plus anti-PD1 monoclonal antibodies leads to the enhancement both effector CD4+ and CD8+ T-cell responses (72). Taken together, fundamental research in this field not only holds promise in broadening the impact and efficacy of immune checkpoint blockade but also attenuates immune-related adverse events (74). Among many ways to tackle the problems in the field, we focus on how two metabolic modifications, glycosylation and palmitoylation, impact protein trafficking, folding, and stability, ultimately altering its biochemical and biophysical properties to influence immunosuppression and inflammation. The modifications we discussed are summarized in Table 1.

Table 1 Glycosylation and palmitoylation associated with adaptive immunity.
Glycosylation on PD-L1
Protein glycosylation is characterized as a universal PTM that shows some glycans are covalently attached to a polypeptide backbone via glycosidic bond by glycosyl transferase. According to which residue is added, glycosylation can be divided into O-glycosylation and N-glycosylation (82). O-glycosylation takes place in the Golgi apparatus. It is mostly initiated by linking a single N-acetylgalactosamine (GalNAc) to Ser or Thr residue, and usually can be extended by fucosylation and sialylation resulting in producing various “cores” and different terminal structures (83). In contrast, N-glycosylation occurs on N-acetylglucosamine (GlcNAc) linked to the consensus NXT motif (Asn-X-Ser/Thr); it is initiated in the endoplasmic reticulum and finished in the Golgi apparatus. It shares a common pentasaccharide core region that can be further modified by GlcNAc, Gal, and sialic acid as terminal structures (84). Glycans dictate proteolysis patterns and directly mediate ligand-receptor interactions, oncogenic signaling transduction, immune recognition, migration, and both cell-cell and cell-matrix adhesions.
PD-L1 is a single-pass type-I membrane protein generally expressed on the cellular membrane in tumor cells and also on the host immune cells. By interacting with its receptor PD1 through its extracellular domain, PD-L1 blocks effective T-cell attacks. It has been identified by mass spectrometry and sequence analysis that PD-L1 is principally modified by N-glycan at four glycosylation sites (N35, N192, N200, N219) in the extracellular domain (85); these modifications are predominantly correlated with its stability and interaction with PD1, representing the functional form of PD-L1. At the earliest in 2016, the Hung group discovered that PD-L1 N-glycosylation enhances its stability by abolishing the interaction with GSK3β, which can phosphorylate PD-L1 at T180 and S184 sites thus mediating 26S proteasome degradation of PD-L1 by β-TrCP (85). Consistently, mutations of PD-L1 at three N-glycosylation sites (N192, N200, N219) attenuate its association with GSK3β and decrease its protein level. They further identified that PD-L1 N-glycosylation is catalyzed by β-1,3-N-acetylglucosaminyltransferase3 (B3GNT3), which is initiated by EGFR signaling downstream the transcription factor TCF4, and boosts the interaction with PD-1 in triple-negative breast cancer (TNBC) (75). Accordingly, knockout of B3GNT3 in TNBC diminishes PD-L1 N-glycosylation and prevents tumor growth. The glycosylation stabilizes PD-L1 expression not only via EGFR signaling but also by epithelial–mesenchymal transition (EMT) in TNBC. EMT transcriptionally upregulates N-glycosyltransferase STT3 by Wnt/β-catenin pathway, thus leading to elevated PD-L1 N-glycosylation in cancer stem-like cells (86). In this pathway, PD-L1 is phosphorylated at Y112 by IL-6-activated JAK1, then recruits STT3A for the subsequent PD-L1 glycosylation (76) (Figure 1). However, the correlation between B3GNT3-dependent glycosylation and STT3-mediated glycosylation remains an open question.
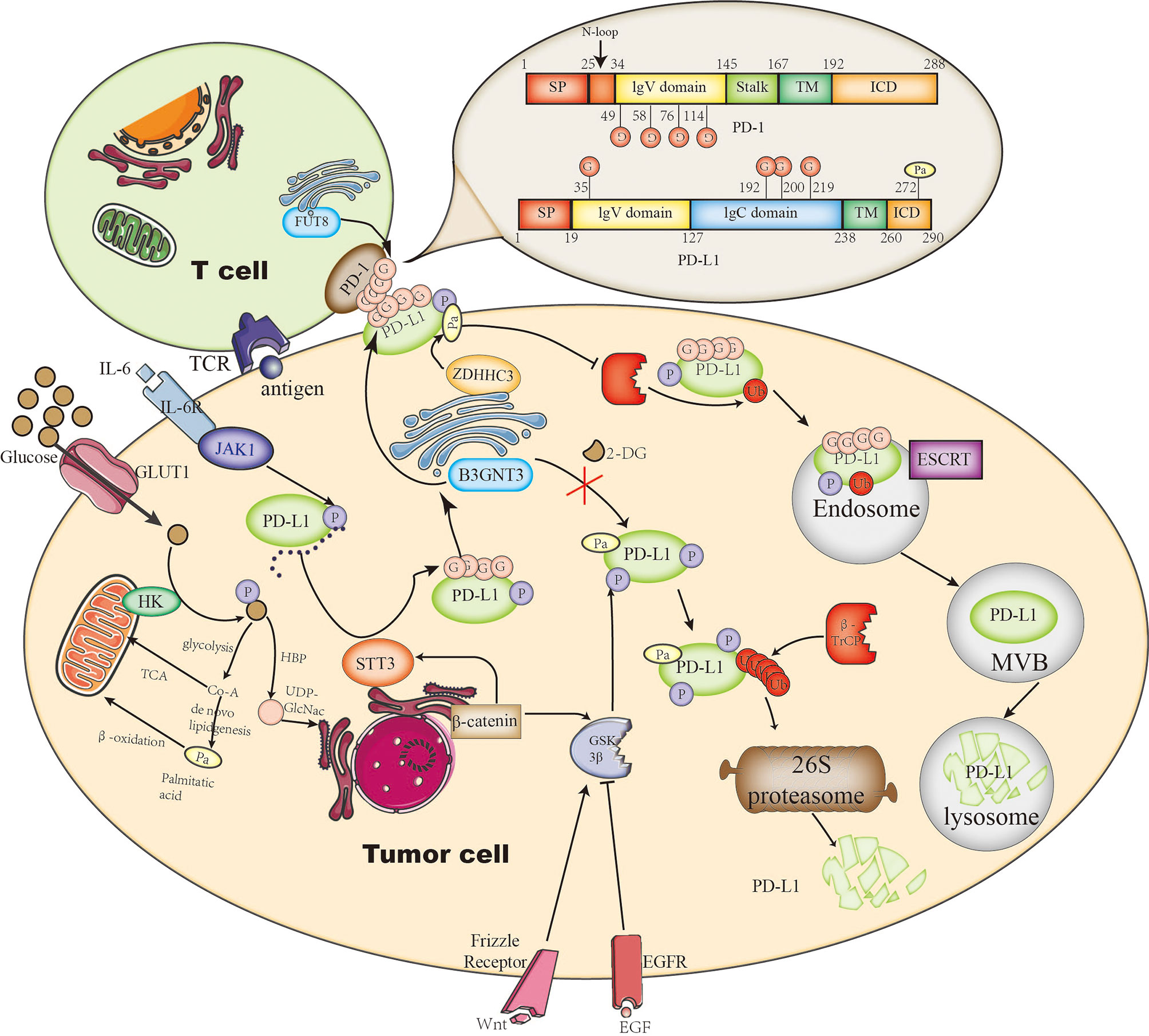
Figure 1 The roles of glycosylation and palmitoylation of PD-1/PD-L1. In T cells, PD-1 is glycosylated by FUT8, leading to its stable expression on the cell surface and causing a stronger interaction with glycosylated PD-L1. On the other hand, PD-L1 is glycosylated or degraded through different pathways. It can be glycosylated by STT3 in ER or by B3GNT3 in the Golgi apparatus. Glycosylation helps to maintain its stability by preventing GSK3β-dependent phosphorylation and downstream β-Trcp-mediated 26S proteasome degradation. In addition, PD-L1 palmitoylation catalyzed by ZDHHC3 stabilizes PD-L1 through blocking PD-L1 mono-ubiquitination, otherwise ubiquitinated PD-L1 will be sorted by endosomal sorting complexes required for transport (ESCRT) to the multivesicular body (MVB) for degradation. Glycosylation is affected by the branch glucose metabolism pathway HBP, which directly provides the glycosylation substrate (UDP)-GlcNAc. Palmitoylation is affected by the lipid synthesis which leads to palmitate acid storage.
PD-L1 has been found glycosylated in many cancer types including breast cancer, melanoma, lung cancer, colon cancer, etc. (85), suggesting a universal feature for all cancers. However, the glycosylation pathway could be cancer-type dependent (through particular glycosyltransferases). For example, in some subtypes of B-cell lymphoma, glycosyltransferase 1 domain-containing 1 (GLT1D1) is significantly upregulated and enhances PD-L1 stability through N-glycosylation; downregulation of GLT1D1 caused a decrease in glycosylated PD-L1 protein, leading to an increase in cytotoxic T-cell infiltration in the tumor microenvironment (77). It suggests that GLT1D1 probably is a promising biomarker for lymphoma; further studies on cancer-type–dependent glycosylation pathways are needed. Interestingly, drug-conjugated STM108, an antibody that is specially developed, recognizes glycosylated PD-L1 at N192 site and leads to PD-L1 internalization and degradation, inducing a potent antitumor effect as well as a bystander-killing effect on adjacent cancer cells lacking PD-L1 expression without any detectable toxicity (75). This suggests that targeting glycosylated PD-L1 is a potential strategy of immunotherapy, and the glycosylation pathways could also serve as targets or biomarkers for early diagnosis.
One of the biggest problems of ICT is how to accurately classify patients suitable for immunotherapy to achieve higher response rate. As PD-L1 is heavily glycosylated and glycosylation is heterogenous, it complicates the situation, because glycosylation not only stimulates interaction with PD-1 but also prevents binding to PD-L1 antibody (75). This has been proposed to illustrate why PD-L1–negative patients still have a favorable clinical response to ICT (87). By removing N-glycans from PD-L1, the Hung group discovered that deglycosylation significantly improves anti-PD-L1 antibody binding affinity, thus resulting in a more accurate PD-L1 quantification and prediction of clinical outcome in breast cancer (88). Deglycosylation can be achieved by using a glucose analog 2-deoxyglucose (2-DG) to block PD-L1 glycosylation and trap them inside the endoplasmic reticulum. 2-DG has been used to reverse PD-L1 expression on the cell surface of TNBC mediated by PARP inhibitor (upregulates PD-L1), and it has showed a potent antitumor activity (89). In addition, it enhances 4-1BB–mediated antitumor immunity via PD-L1 deglycosylation (4‐1BB engages with 4‐1BB ligand (4‐1BBL) or agonist antibody to stimulate CD8+ T-cell activity or NK cell growth) (90). Furthermore, it has been shown that the combination of 2-DG and EGFR tyrosine kinase inhibitor (TKI) gefitinib inhibits glycosylation of PD-L1 in TNBC. These studies suggest that 2-DG blocking PD-L1 glycosylation is a universal pathway and combination therapy could also be useful for lung cancer. Very interestingly, a more recent study confirmed this idea in lung cancer specimens (91). Next, we may use deglycosylation to provide a more reliable way to quantify PD-L1 level and guide anti-PD-1/PD-L1 immunotherapy.
Glycosylation on PD-1
PD-1 is also a single-pass type-I membrane protein, unlike PD-L1, it is usually expressed on the cellular membrane of T lymphocyte. Upon T-cell activation, TCR-dependent signaling initiates the transcription of PD-1, and IFN-γ–mediated signaling causes durable PD-1 expression (92). Completely opposite to PD-L1, GSK3β facilitates PD-1 expression by inhibiting T-bet induction, which suppresses transcription of PD-1 along with B-lymphocyte–induced maturation protein 1 (Blimp-1) after initial transcription. Similar to PD-L1, four N-glycosylation sites on the extracellular domain of PD-1 (N49, N58, N74, and N116) have also been identified by mass spectrometry analysis; they are also associated with stable cell-surface expression of PD-1 (78). Okada et al. has identified Fut8 as the core fucosyltransferase to fucosylate PD-1 and positively regulates cell-surface PD-1 expression, thus inhibition of Fut8 reduces PD-1 cell-surface expression and promotes T-cell antitumor activity in melanoma (78). Similarly, Zhang et al. discovered that core fucosylation was significantly upregulated in lung adenocarcinoma and demonstrated that de-core fucosylation of PD-1 via Fut8 knockout enhances CD8+ cytotoxic T-lymphocyte (CTL) activation and cytotoxicity in lung adenocarcinoma (93). Furthermore, the glycosylation of PD-1 at N58 is critical for PD-1 cell-surface expression and stability and is essential to mediate its interaction with PD-L1 (79). Based on the glycosylation research on PD-1, an adenine base editor (ABE) induced mutation at PD-1 N74 site, downregulating PD-1 expression in CAR-T cells and enhancing CAR-T cell cytotoxic functions in vitro and in vivo (94) (Figure 1).
Very recently, a new monoclonal antibody STM418 targeting N58-glycosylated PD-1 was developed; it shows higher binding affinity to PD-1 than previous FDA-approved antibodies and induces much stronger T-cell antitumor immunity (79). This is not a unique instance, a co-crystal structure of PD-1 glycosylated at N58 with monoclonal antibody MW11-h317 Fab provides the structure basis for molecular interaction for PD-1 N-glycosylation recognization (95). Another newly developed PD-1 antibody, known as camrelizumab and currently undergoing phase II/III trials, also selectively binds to N58-glycosylated PD-1 to inhibit PD-1/PD-L1 pathway (96). These studies suggest that targeting glycosylated PD-1 may improve immunotherapy response.
Although we have known the N-glycosylation enzymes and sites on PD-1/PD-L1, the exact composition of glycome on PD-1/PD-L1 is elusive thus far; further details revealing glycome may disclose more potential molecular targets. Though biochemical experiments and animal models have demonstrated that target PD-1/PD-L1 N-glycosylation not only improves the accuracy of disease diagnosis but also the effectiveness of treatment, more clinical trials are needed to verify its efficacy and safety. Additionally, unlike N-glycosylation, less of the O-glycosylation enzymes and sites have been discovered on PD-1/PD-L1. GCNT3 is involved in O-glycosylation and impacts the clinical outcome of colon and ovarian cancers (97); however, it is unclear whether it could also affect PD-1/PD-L1 O-glycosylation in modulating immune checkpoint. The rapid progress in this field in both basic and clinical research will not only advance our ability to identify patients who are most likely to benefit from certain immunotherapy but also help to predict response in the clinic and even lead to new treatments.
Palmitoylation on PD-L1
Palmitoylation is a lipidation process that covalently attaches a palmitate acid to residues on a protein via three different ways. (1) S-palmitoylation, linking to cysteine residues by thioester linkages, (2) O-palmitoylation, linking to serine/threonine residues by oxyester linkages, and (3) N-palmitoylation, linking to primary amino groups by amide linkages. The S-palmitoylation is the only reversible reaction which is catalyzed by endomembrane-bound palmitoyl acyltransferases (PATs); mammals comprise 23 distinct enzymes which contain the zinc finger DHHC-type domain, while very few protein palmitoylation is self-catalyzed (98–101). Contrary to a variety of PATs, only six depalmitoylation enzymes, APT1, APT2, PPT1, ABHD17A, ABHD17B, and ABHD17C, are reported to catalyze protein S-deacylation thus far (102). Emerging evidence has illustrated that S-palmitoylation regulates numerous biological processes through affecting protein migration, membrane localization, stability, and interactions (99, 103).
In 2019, two groups reported simultaneously that S-palmitoylation maintains PD-L1 stability and inhibits T-cell cytotoxic response (80, 81). Both of them identified C272 as the palmitoylation site on PD-L1, substituting C272 with alanine abolishes PD-L1 palmitoylation in tumor cells. Interestingly, the Hung group use breast tumor cells and find DHHC9 as the palmitoyl acyltransferases (80), while the Xu group identified DHHC3 as the PAT for PD-L1 in colorectal cancer (CRC) (81). The latter further proposed that the S-palmitoylation regulates PD-L1 stability by suppressing the mono-ubiquitination of PD-L1, thereby blocking the lysosomal degradation of PD-L1 via preventing its trafficking to the MVB by ESCRT, causing increased cell surface expression of PD-L1 and thus suppressing T-cell cytotoxicity (Figure 1). Interestingly, based on the idea that DHHC recognize their substrate by amino acid sequence, a competitive polypeptide is designed towards more inhibition specificity for PD-L1 than the commonly used universal palmitoylation inhibitor 2-BP, indeed the expression of PD-L1 was reduced by this inhibitor in tumor cells (81). This idea could be useful in developing research tools or clinical drugs for other palmitoylation targets; further research on improving specificity, stability, and efficacy of this kind of peptide inhibitors to target the palmitoylation in other malignant cancers such as lung cancer in encouraging.
Innate Immunity, Inflammation, and Immunotherapy
The innate immunity consists of many types of cells and soluble molecules in tissues and blood that constantly prevent microbes from invading and eliminating other offending agents. The cellular sensors for pathogen- and damage-associated molecular patterns in innate immune cells are called pattern recognition receptors (PRRs). Five main classes of PRRs have been described: membrane-bound TLRs and CLRs, cytoplasmic NLRs and RLRs, and several DNA sensors such as cGAS (104, 105). Once activated, PRRs initiate multiple innate immune signaling pathways, leading to the production of type I interferons (IFN-I) and proinflammatory cytokines, which is referred to as inflammation (106). Considering that the human body does not develop a new set of PRRs for identifying cancer cells, it is usually accepted that antitumor immunity shares the same PRRs with the innate immunity machine.
Indeed, inflammation is a sophisticated process closely associated with a variety of tumors, which tends to form an inflammatory environment surrounding it. There is considerable evidence indicating that chronic inflammation plays a critical role for tumorigenesis, for example, colorectal cancer can develop on the ground of inflammatory bowel disease (IBD) (107), chronic hepatitis B greatly increases the risk of hepatic carcinoma (108), and even lung cancer caused by smoking is attributed to irritant-induced chronic inflammation, not genetic mutations (109). The inflammatory cells recruited to the tumor microenvironment releases a majority of cytokines, which support the survival and proliferation of tumor cells (110–112). Nonetheless, it is controversial how inflammation influences the outcome of ICT, because some articles reported inflammation promotes the activation of T cell but others were opposite (113–115). Next, we will discuss the molecular mechanisms of how palmitoylation regulates inflammation.
Palmitoylation Regulates Innate Immunity and Inflammation
TLR4, a class of pattern recognition receptor in sentinel cells of innate immunity such as macrophage, dendritic cells, and neutrophils, belongs to the Toll-like receptor family. TLR4 recognizes LPS of gram-negative bacteria and is involved in inflammasome activation through activating downstream NF-kB signaling to release inflammatory factors TNF-α, IL-1β, and IL-18 (116). Earlier in 2016, Wei et al. have shown that FASN is indispensable for diet-induced inflammation, suggesting fatty acid metabolism plays a possible role in modulating inflammatory response (117). A series of follow-up research has proven that the saturated fatty acid palmitate is not a TLR4 agonist but participates in palmitoylation of C113 on MYD88, a downstream of TLR4, and this palmitoylation promotes MYD88 association with IRAF4 to activate the downstream NF-κB signaling pathway, thus inducing altered cellular metabolism and inflammation in neutrophils (118, 119) (Figure 2). Pharmacological inhibition of MYD88 palmitoylation in neutrophils suppressed TLR-induced inflammation and enhanced the chemotactic activity of neutrophils, improving the survival of mice with sepsis.
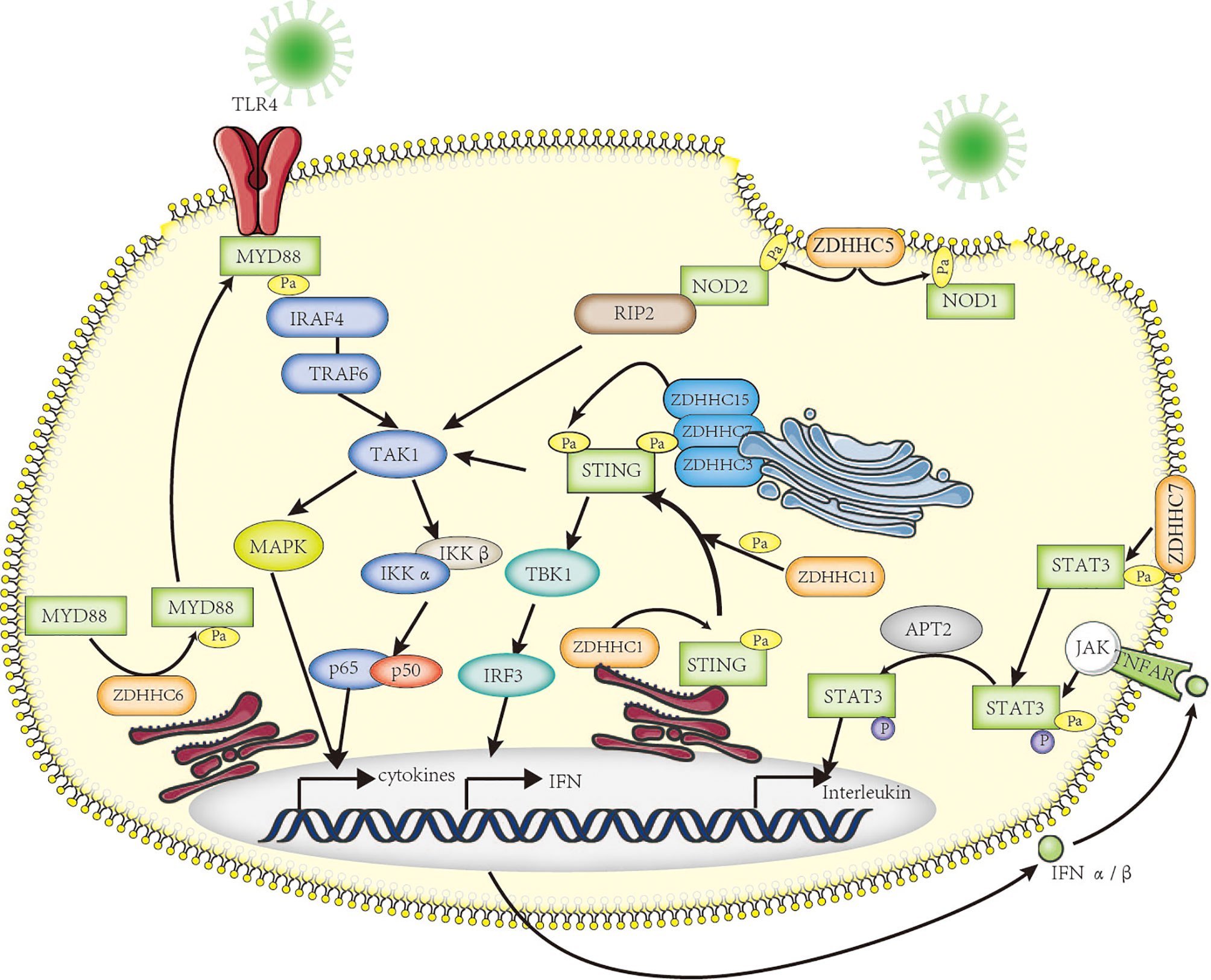
Figure 2 The roles of palmitoylation in inflammation. Palmitoylation affects inflammatory response in many ways. MYD88 is palmitoylated by ZDHHC6, facilitating downstream NF-κB inflammation pathway activation by mediating association with IRAF-4. NOD1/2 is palmitoylated by ZDHHC5, which is required for membrane recruitment and downstream signaling. Five palmitoyl-transferases (localized at different cell compartments including ER and Golgi apparatus) positively regulate STING-dependent inflammatory response. The STAT3 palmitoylation–depalmitoylation cycle, catalyzed by DHHC7 and APT2, controls phosphorylation of STAT3 by JAK2 and contributes to STAT3 nuclear translocation, which is essential for IL-17–induced inflammation.
NOD1 and NOD2, another class of intracellular pattern-recognition receptors in macrophage, belong to the NOD-like receptor family that recognizes peptidoglycans associated with microorganisms and is involved in host defense through activating downstream NF-kB and p38 MAPK signaling to release chemokine CXCL-1 and inflammatory factor IL-6 (120). It has been known that dysregulation of NOD1/2 function leads to severe immunologic and inflammatory diseases such as Crohn’s disease (CD) and Blau syndrome (121). The Neculai group provided substantial evidence showing that NOD1/2 are S-palmitoylated by ZDHHC5, and this S-palmitoylation is required for membrane localization and to induce NF-kB signaling in response to peptidoglycans (122) (Figure 2).
STING is a central adaptor in innate immune responses to DNA viruses, which are recognized by cytosolic DNA sensor cGAS leading to secretion of type I IFNs through the activation of TANK-binding kinase 1 (TBK1) and, subsequently, of the transcription factor IFN regulatory factor 3 (IRF3) and nuclear factor kappa B (NF-κB) (123, 124). It has been reported that STING can be regulated by ER-associated ZDHHC1 (125) and ZDHHC11 in innate immune responses against DNA viruses (126). However, Mukai et al. demonstrated that STING is palmitoylated in the Golgi apparatus at C89/91 by ZDHHC3, ZDHHC7, and ZDHHC15; this modification is essential for STING activation and the subsequent activation of IFN-β or NF-κb (127). More interestingly, covalently bound small-molecule inhibitors targeting STING palmitoylation was developed recently; they further confirmed that palmitoylation of STING is essential for its assembly into multimeric complexes at the Golgi apparatus and the activation of downstream STING-triggered inflammatory signaling (128) (Figure 2).
One more advanced progress on palmitoylation studies is the STAT3 palmitoylation–depalmitoylation cycle. The palmitoylation on Cys108 of STAT3 catalyzed by DHHC7 promotes membrane recruitment and phosphorylation by JAK2, then Acyl protein thioesterase 2 (APT2) depalmitoylates phosphorylated STAT3, this leads to nuclear translocation of p-STAT3, which eventually facilitates STAT3-mediated IL-17 transcription and differentiation of TH17 cells (129). Dysfunctional differentiation of TH17 cells has an important pathogenic role in IBD, including ulcerative colitis and Crohn’s disease; therefore, both DHHC7 and APT2 could be new therapeutic targets for IBD treatment.
All these palmitoylation and depalmitoylation examples (Table 2) underscore the direct evidence that palmitoylation regulates innate immunity and inflammation. Given that TME has altered metabolism pathways for lipid synthesis and causes a higher palmitate concentration, it is reasonable to speculate that at least in some circumstance a higher level of palmitoylation on cancer-related protein targets occurs, thus positively regulating tumor survival and immune suppression. Tumor cells secrete many immunosuppressive factors to directly alter T-cell effector functions and inhibit innate immune cells, preventing them from sustaining efficient antitumor immune responses. Additionally, inflammatory immune cells in TME such as tumor-associated macrophage generate a tolerant environment that suppresses T-cell response, thereby leading to immune escape. Understanding the palmitoylation-mediated regulatory mechanism of inflammation activation and tumor evasion will lead to further understand how cancer immunotherapy can be manipulated. Thus, targeting palmitoylation to regulate inflammation could provide new ways for cancer treatments.
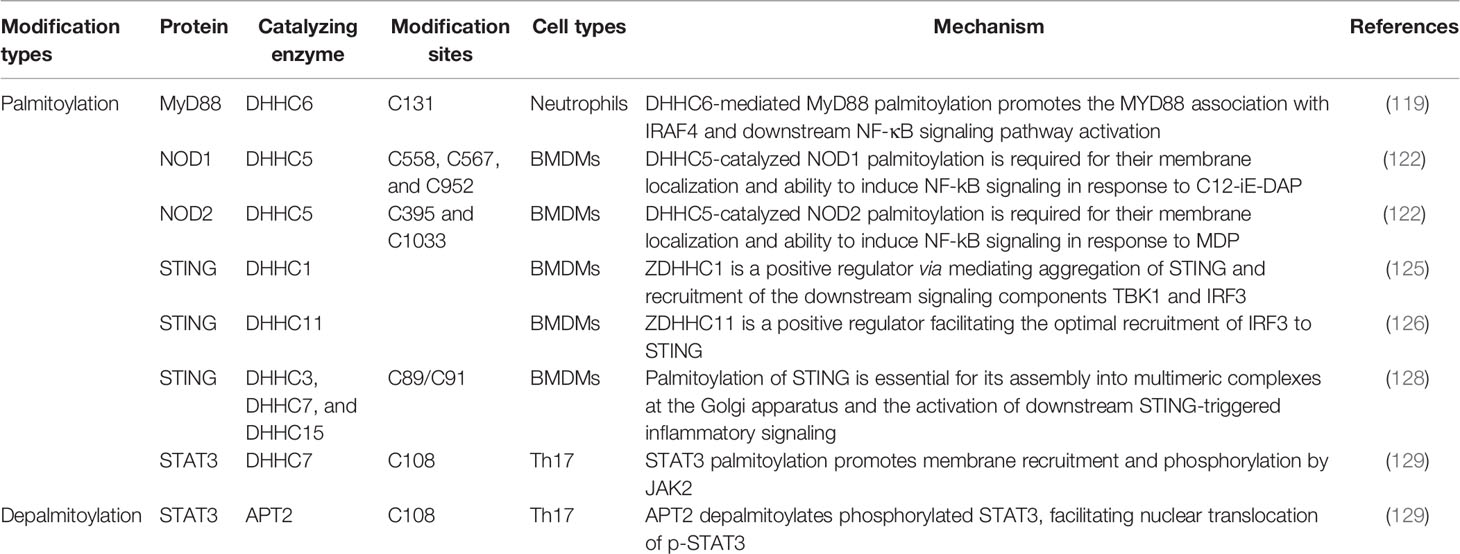
Table 2 Palmitoylation and depalmitoylation related to inflammation.
New Therapeutic Opportunities
ICT revolutionized the oncology field and opened a new door with the hope of curing cancer. It has achieved great success in some cancer types such as melanoma; however, in some other cancer types such as lung cancers, ICT is still facing great challenges because of their unpredictable responses and the risk of relapse. The regulatory mechanisms of PTMs in tumor cells and tumor microenvironment have been a hot research topic for several decades and achieved great progress in the last decade. The basic research in this field disclosed more secrets of tumor heterogeneity in TME, revealing many regulatory mechanisms on tumorigenesis and immune suppression and suggesting a number of new ways for cancer treatments, especially for those cancers with a low response rate or low survival rate, such as lung cancer. It is promising to target many components in glycosylation/deglycosylation and palmitoylation/depalmitoylation pathways; this includes the upstream material acquiring or biosynthesis pathways, the direct glycotransferases, palmitoylases/depalmitoylases, and many downstream effector proteins such as PD-1/PD-L1, NOD1/2, STAT3, etc. Due to the urgent need to achieve a more predictable immunotherapy outcome and benefit from more advanced research methods on glycosylation and palmitoylation, we expect more groundbreaking findings in this area, which will improve the outcomes of current immunotherapies via stratifying cancer patients or combinational therapies and provide more new therapeutic opportunities. In this regard, in the era of “big data,” a more accurate precision cancer immunotherapy is just around the corner.
Author Contributions
SZ and PZ wrote the review. QS discussed the review. All authors proofread the review. All authors contributed to the article and approved the submitted version.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
SZ was supported by grant from the Natural Science Foundation of China (NSFC) (82002939).
References
1. Robert C. A Decade of Immune-Checkpoint Inhibitors in Cancer Therapy. Nat Commun (2020) 11:3801. doi: 10.1038/s41467-020-17670-y
2. Shi Y, Tomczak K, Li J, Ochieng JK, Lee Y, Haymaker C, et al. Next-Generation Immunotherapies to Improve Anticancer Immunity. Front Pharmacol (2020) 11:566401. doi: 10.3389/fphar.2020.566401
3. Iwai Y, Hamanishi J, Chamoto K, Honjo T. Cancer Immunotherapies Targeting the PD-1 Signaling Pathway. J BioMed Sci (2017) 24:26. doi: 10.1186/s12929-017-0329-9
4. Sharma P, Allison JP. The Future of Immune Checkpoint Therapy. Science (2015) 348:56–61. doi: 10.1126/science.aaa8172
5. Lipson EJ, Drake CG. Ipilimumab: An Anti-CTLA-4 Antibody for Metastatic Melanoma. Clin Cancer Res (2011) 17(22):6958–62. doi: 10.1158/1078-0432.CCR-11-1595
6. Garon EB, Rizvi NA, Hui R, Leighl N, Balmanoukian AS, Eder JP, et al. Pembrolizumab for the Treatment of Non-Small-Cell Lung Cancer. N Engl J Med (2015) 372(21):2018–28. doi: 10.1056/NEJMoa1501824
7. Robert C, Long GV, Brady B, Dutriaux C, Maio M, Mortier L, et al. Nivolumab in Previously Untreated Melanoma Without BRAF Mutation. N Engl J Med (2015) 72(4):320–30. doi: 10.1056/NEJMoa1412082
8. Ahmed SR, Petersen E, Patel R, Migden MR. Cemiplimab-rwlc as First and Only Treatment for Advanced Cutaneous Squamous Cell Carcinoma. Expert Rev Clin Pharmacol (2019) 12(10):947–51. doi: 10.1080/17512433.2019.1665026
9. Horn L, Mansfield AS, Szczesna A, Havel L, Krzakowski M, Hochmair MJ, et al. First-Line Atezolizumab Plus Chemotherapy in Extensive-Stage Small-Cell Lung Cancer. N Engl J Med (2018) 379(23):2220–9. doi: 10.1056/NEJMoa1809064
10. D'Angelo SP, Russell J, Lebbe C, Chmielowski B, Gambichler T, Grob JJ, et al. Efficacy and Safety of First-Line Avelumab Treatment in Patients With Stage IV Metastatic Merkel Cell Carcinoma: A Preplanned Interim Analysis of a Clinical Trial. JAMA Oncol (2018) 4(9):e180077. doi: 10.1001/jamaoncol.2018.0077
11. Antonia SJ, Villegas A, Daniel D, Vicente D, Murakami S, Hui R, et al. Durvalumab after Chemoradiotherapy in Stage III Non-Small-Cell Lung Cancer. N Engl J Med (2017) 377(20):1919–29. doi: 10.1056/NEJMoa1709937
12. Eroglu Z, Kim DW, Wang X, Camacho LH, Chmielowski B, Seja E, et al. Long Term Survival With Cytotoxic T Lymphocyte-Associated Antigen 4 Blockade Using Tremelimumab. Eur J Cancer (2015) 51:2689–97. doi: 10.1016/j.ejca.2015.08.012
13. Xin Yu J, Hubbard-Lucey VM, Tang J. Immuno-Oncology Drug Development Goes Global. Nat Rev Drug Discov (2019) 18:899–900. doi: 10.1038/d41573-019-00167-9
14. Hamid O, Robert C, Daud A, Hodi FS, Hwu WJ, Kefford R, et al. Five-Year Survival Outcomes for Patients With Advanced Melanoma Treated With Pembrolizumab in KEYNOTE-001. Ann Oncol (2019) 30:582–8. doi: 10.1093/annonc/mdz011
15. Robert C, Ribas A, Hamid O, Daud A, Wolchok JD, Joshua AM, et al. Durable Complete Response After Discontinuation of Pembrolizumab in Patients With Metastatic Melanoma. J Clin Oncol (2018) 36:1668–74. doi: 10.1200/JCO.2017.75.6270
16. Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, Havel JJ, et al. Cancer Immunology. Mutational Landscape Determines Sensitivity to PD-1 Blockade in Non-Small Cell Lung Cancer. Science (2015) 348:124–8. doi: 10.1126/science.aaa1348
17. Herbst RS, Soria JC, Kowanetz M, Fine GD, Hamid O, Gordon MS, et al. Predictive Correlates of Response to the Anti-PD-L1 Antibody MPDL3280A in Cancer Patients. Nature (2014) 515:563–7. doi: 10.1038/nature14011
18. Paz-Ares L, Luft A, Vicente D, Tafreshi A, Gumus M, Mazieres J, et al. Pembrolizumab Plus Chemotherapy for Squamous Non-Small-Cell Lung Cancer. N Engl J Med (2018) 379:2040–51. doi: 10.1056/NEJMoa1810865
19. Gandhi L, Rodriguez-Abreu D, Gadgeel S, Esteban E, Felip E, De Angelis F, et al. Pembrolizumab Plus Chemotherapy in Metastatic Non-Small-Cell Lung Cancer. N Engl J Med (2018) 378:2078–92. doi: 10.1056/NEJMoa1801005
20. Hellmann MD, Paz-Ares L, Bernabe Caro R, Zurawski B, Kim SW, Carcereny Costa E, et al. Nivolumab Plus Ipilimumab in Advanced Non-Small-Cell Lung Cancer. N Engl J Med (2019) 381:2020–31. doi: 10.1056/NEJMoa1910231
21. Wu YL, Lu S, Cheng Y, Zhou C, Wang J, Mok T, et al. Nivolumab Versus Docetaxel in a Predominantly Chinese Patient Population With Previously Treated Advanced NSCLC: CheckMate 078 Randomized Phase III Clinical Trial. J Thorac Oncol (2019) 14:867–75. doi: 10.1016/j.jtho.2019.01.006
22. Brahmer J, Reckamp KL, Baas P, Crino L, Eberhardt WE, Poddubskaya E, et al. Nivolumab Versus Docetaxel in Advanced Squamous-Cell Non-Small-Cell Lung Cancer. N Engl J Med (2015) 373:123–35. doi: 10.1056/NEJMoa1504627
23. Borghaei H, Gettinger S, Vokes EE, Chow LQM, Burgio MA, de Castro Carpeno J, et al. Five-Year Outcomes From the Randomized, Phase III Trials CheckMate 017 and 057: Nivolumab Versus Docetaxel in Previously Treated Non-Small-Cell Lung Cancer. J Clin Oncol (2021) 39:723–33. doi: 10.1200/JCO.20.01605
24. Chiang AC, Herbst RS. Frontline Immunotherapy for NSCLC - the Tale of the Tail. Nat Rev Clin Oncol (2020) 17:73–4. doi: 10.1038/s41571-019-0317-y
25. Gao J, Shi LZ, Zhao H, Chen J, Xiong L, He Q, et al. Loss of IFN-Gamma Pathway Genes in Tumor Cells as a Mechanism of Resistance to Anti-CTLA-4 Therapy. Cell (2016) 167:397–404.e399. doi: 10.1016/j.cell.2016.08.069
26. Sucker A, Zhao F, Pieper N, Heeke C, Maltaner R, Stadtler N, et al. Acquired IFNgamma Resistance Impairs Anti-Tumor Immunity and Gives Rise to T-Cell-Resistant Melanoma Lesions. Nat Commun (2017) 8:15440. doi: 10.1038/ncomms15440
27. Sade-Feldman M, Jiao YJ, Chen JH, Rooney MS, Barzily-Rokni M, Eliane JP, et al. Resistance to Checkpoint Blockade Therapy Through Inactivation of Antigen Presentation. Nat Commun (2017) 8:1136. doi: 10.1038/s41467-017-01062-w
28. Spranger S, Gajewski TF. Impact of Oncogenic Pathways on Evasion of Antitumour Immune Responses. Nat Rev Cancer (2018) 18:139–47. doi: 10.1038/nrc.2017.117
29. Postow MA, Sidlow R, Hellmann MD. Immune-Related Adverse Events Associated With Immune Checkpoint Blockade. N Engl J Med (2018) 378:158–68. doi: 10.1056/NEJMra1703481
30. Demaria O, Cornen S, Daeron M, Morel Y, Medzhitov R, Vivier E. Harnessing Innate Immunity in Cancer Therapy. Nature (2019) 574:45–56. doi: 10.1038/s41586-019-1593-5
31. Herbst RS, Giaccone G, de Marinis F, Reinmuth N, Vergnenegre A, Barrios CH, et al. Atezolizumab for First-Line Treatment of PD-L1-Selected Patients With NSCLC. N Engl J Med (2020) 383:1328–39. doi: 10.1056/NEJMoa1917346
32. Siegel RL, Miller KD, Jemal A. Cancer Statistics, 2020. CA Cancer J Clin (2020) 70:7–30. doi: 10.3322/caac.21590
33. Ward PS, Thompson CB. Metabolic Reprogramming: A Cancer Hallmark Even Warburg did Not Anticipate. Cancer Cell (2012) 21:297–308. doi: 10.1016/j.ccr.2012.02.014
34. Hay N. Reprogramming Glucose Metabolism in Cancer: Can it be Exploited for Cancer Therapy? Nat Rev Cancer (2016) 16:635–49. doi: 10.1038/nrc.2016.77
35. Ganapathy-Kanniappan S. Taming Tumor Glycolysis and Potential Implications for Immunotherapy. Front Oncol (2017) 7:36. doi: 10.3389/fonc.2017.00036
36. Yang Y, Gibson GE. Succinylation Links Metabolism to Protein Functions. Neurochem Res (2019) 44:2346–59. doi: 10.1007/s11064-019-02780-x
37. Xie Z, Zhang D, Chung D, Tang Z, Huang H, Dai L, et al. Metabolic Regulation of Gene Expression by Histone Lysine Beta-Hydroxybutyrylation. Mol Cell (2016) 62:194–206. doi: 10.1016/j.molcel.2016.03.036
38. Zhang D, Tang Z, Huang H, Zhou G, Cui C, Weng Y, et al. Metabolic Regulation of Gene Expression by Histone Lactylation. Nature (2019) 574:575–80. doi: 10.1038/s41586-019-1678-1
39. Izzo LT, Wellen KE. Histone Lactylation Links Metabolism and Gene Regulation. Nature (2019) 574:492–3. doi: 10.1038/d41586-019-03122-1
40. Bos J, Muir TW. A Chemical Probe for Protein Crotonylation. J Am Chem Soc (2018) 140:4757–60. doi: 10.1021/jacs.7b13141
41. Trefely S, Lovell CD, Snyder NW, Wellen KE. Compartmentalised Acyl-CoA Metabolism and Roles in Chromatin Regulation. Mol Metab (2020) 38:100941. doi: 10.1016/j.molmet.2020.01.005
42. Chiaradonna F, Ricciardiello F, Palorini R. The Nutrient-Sensing Hexosamine Biosynthetic Pathway as the Hub of Cancer Metabolic Rewiring. Cells (2018) 7. doi: 10.3390/cells7060053
43. Pinho SS, Reis CA. Glycosylation in Cancer: Mechanisms and Clinical Implications. Nat Rev Cancer (2015) 15:540–55. doi: 10.1038/nrc3982
44. Snaebjornsson MT, Janaki-Raman S, Schulze A. Greasing the Wheels of the Cancer Machine: The Role of Lipid Metabolism in Cancer. Cell Metab (2020) 31:62–76. doi: 10.1016/j.cmet.2019.11.010
45. Carta G, Murru E, Banni S, Manca C. Palmitic Acid: Physiological Role, Metabolism and Nutritional Implications. Front Physiol (2017) 8:902. doi: 10.3389/fphys.2017.00902
46. Ko PJ, Dixon SJ. Protein Palmitoylation and Cancer. EMBO Rep (2018) 19. doi: 10.15252/embr.201846666
47. Lin H. Protein Cysteine Palmitoylation in Immunity and Inflammation. FEBS J (2021). doi: 10.1111/febs.15728
48. Hanahan D, Weinberg RA. Hallmarks of Cancer: The Next Generation. Cell (2011) 144:646–74. doi: 10.1016/j.cell.2011.02.013
49. Yue B. Biology of the Extracellular Matrix: An Overview. J Glaucoma (2014) 23:S20–23. doi: 10.1097/IJG.0000000000000108
50. Vaupel P, Multhoff G. Revisiting the Warburg Effect: Historical Dogma Versus Current Understanding. J Physiol (2021) 599:1745–57. doi: 10.1113/JP278810
51. Waldhart AN, Dykstra H, Peck AS, Boguslawski EA, Madaj ZB, Wen J, et al. Phosphorylation of TXNIP by AKT Mediates Acute Influx of Glucose in Response to Insulin. Cell Rep (2017) 19:2005–13. doi: 10.1016/j.celrep.2017.05.041
52. Elstrom RL, Bauer DE, Buzzai M, Karnauskas R, Harris MH, Plas DR, et al. Akt Stimulates Aerobic Glycolysis in Cancer Cells. Cancer Res (2004) 64:3892–9. doi: 10.1158/0008-5472.CAN-03-2904
53. Asgari Y, Zabihinpour Z, Salehzadeh-Yazdi A, Schreiber F, Masoudi-Nejad A. Alterations in Cancer Cell Metabolism: The Warburg Effect and Metabolic Adaptation. Genomics (2015) 105:275–81. doi: 10.1016/j.ygeno.2015.03.001
54. Long J, Zhang CJ, Zhu N, Du K, Yin YF, Tan X, et al. Lipid Metabolism and Carcinogenesis, Cancer Development. Am J Cancer Res (2018) 8:778–91.
55. Koundouros N, Poulogiannis G. Reprogramming of Fatty Acid Metabolism in Cancer. Br J Cancer (2020) 122:4–22. doi: 10.1038/s41416-019-0650-z
56. Reinfeld BI, Madden MZ, Wolf MM, Chytil A, Bader JE, Patterson AR, et al. Cell-Programmed Nutrient Partitioning in the Tumour Microenvironment. Nature (2021). doi: 10.1038/s41586-021-03442-1
57. Wells L, Vosseller K, Hart GW. Glycosylation of Nucleocytoplasmic Proteins: Signal Transduction and O-GlcNAc. Science (2001) 291:2376–8. doi: 10.1126/science.1058714
58. Oikari S, Kettunen T, Tiainen S, Hayrinen J, Masarwah A, Sudah M, et al. UDP-Sugar Accumulation Drives Hyaluronan Synthesis in Breast Cancer. Matrix Biol (2018) 67:63–74. doi: 10.1016/j.matbio.2017.12.015
59. Kuhajda FP, Jenner K, Wood FD, Hennigar RA, Jacobs LB, Dick JD, et al. Fatty Acid Synthesis: A Potential Selective Target for Antineoplastic Therapy. Proc Natl Acad Sci USA (1994) 91:6379–83. doi: 10.1073/pnas.91.14.6379
60. Zhou W, Simpson PJ, McFadden JM, Townsend CA, Medghalchi SM, Vadlamudi A, et al. Fatty Acid Synthase Inhibition Triggers Apoptosis During S Phase in Human Cancer Cells. Cancer Res (2003) 63:7330–7.
61. Wu X, Qin L, Fako V, Zhang JT. Molecular Mechanisms of Fatty Acid Synthase (FASN)-Mediated Resistance to Anti-Cancer Treatments. Adv Biol Regul (2014) 54:214–21. doi: 10.1016/j.jbior.2013.09.004
62. Su X, Abumrad NA. Cellular Fatty Acid Uptake: A Pathway Under Construction. Trends Endocrinol Metab (2009) 20:72–7. doi: 10.1016/j.tem.2008.11.001
63. Abdelmagid SA, Clarke SE, Nielsen DE, Badawi A, El-Sohemy A, Mutch DM, et al. Comprehensive Profiling of Plasma Fatty Acid Concentrations in Young Healthy Canadian Adults. PLoS One (2015) 10:e0116195. doi: 10.1371/journal.pone.0116195
64. Mereiter S, Balmana M, Campos D, Gomes J, Reis CA. Glycosylation in the Era of Cancer-Targeted Therapy: Where Are We Heading? Cancer Cell (2019) 36:6–16. doi: 10.1016/j.ccell.2019.06.006
65. Ghazarian H, Idoni B, Oppenheimer SB. A Glycobiology Review: Carbohydrates, Lectins and Implications in Cancer Therapeutics. Acta Histochem (2011) 113:236–47. doi: 10.1016/j.acthis.2010.02.004
66. Walther TC, Farese RV Jr. Lipid Droplets and Cellular Lipid Metabolism. Annu Rev Biochem (2012) 81:687–714. doi: 10.1146/annurev-biochem-061009-102430
67. Tarazona OA, Pourquie O. Exploring the Influence of Cell Metabolism on Cell Fate Through Protein Post-Translational Modifications. Dev Cell (2020) 54:282–92. doi: 10.1016/j.devcel.2020.06.035
68. Leach DR, Krummel MF, Allison JP. Enhancement of Antitumor Immunity by CTLA-4 Blockade. Science (1996) 271:1734–6. doi: 10.1126/science.271.5256.1734
69. Chambers CA, Kuhns MS, Egen JG, Allison JP. CTLA-4-Mediated Inhibition in Regulation of T Cell Responses: Mechanisms and Manipulation in Tumor Immunotherapy. Annu Rev Immunol (2001) 19:565–94. doi: 10.1146/annurev.immunol.19.1.565
70. Freeman GJ, Long AJ, Iwai Y, Bourque K, Chernova T, Nishimura H, et al. Engagement of the PD-1 Immunoinhibitory Receptor by a Novel B7 Family Member Leads to Negative Regulation of Lymphocyte Activation. J Exp Med (2000) 192:1027–34. doi: 10.1084/jem.192.7.1027
71. Dong H, Strome SE, Salomao DR, Tamura H, Hirano F, Flies DB, et al. Tumor-Associated B7-H1 Promotes T-Cell Apoptosis: A Potential Mechanism of Immune Evasion. Nat Med (2002) 8:793–800. doi: 10.1038/nm730
72. Sharma P, Allison JP. Dissecting the Mechanisms of Immune Checkpoint Therapy. Nat Rev Immunol (2020) 20:75–6. doi: 10.1038/s41577-020-0275-8
73. Ledford H, Else H, Warren M. Cancer Immunologists Scoop Medicine Nobel Prize. Nature (2018) 562:20–1. doi: 10.1038/d41586-018-06751-0
74. Kalbasi A, Ribas A. Tumour-Intrinsic Resistance to Immune Checkpoint Blockade. Nat Rev Immunol (2020) 20:25–39. doi: 10.1038/s41577-019-0218-4
75. Li CW, Lim SO, Chung EM, Kim YS, Park AH, Yao J, et al. Eradication of Triple-Negative Breast Cancer Cells by Targeting Glycosylated PD-L1. Cancer Cell (2018) 33:187–201.e110. doi: 10.1016/j.ccell.2018.01.009
76. Chan LC, Li CW, Xia W, Hsu JM, Lee HH, Cha JH, et al. IL-6/JAK1 Pathway Drives PD-L1 Y112 Phosphorylation to Promote Cancer Immune Evasion. J Clin Invest (2019) 129:3324–38. doi: 10.1172/JCI126022
77. Liu X, Zhang Y, Han Y, Lu W, Yang J, Tian J, et al. Overexpression of GLT1D1 Induces Immunosuppression Through Glycosylation of PD-L1 and Predicts Poor Prognosis in B-Cell Lymphoma. Mol Oncol (2020) 14:1028–44. doi: 10.1002/1878-0261.12664
78. Okada M, Chikuma S, Kondo T, Hibino S, Machiyama H, Yokosuka T, et al. Blockage of Core Fucosylation Reduces Cell-Surface Expression of PD-1 and Promotes Anti-Tumor Immune Responses of T Cells. Cell Rep (2017) 20:1017–28. doi: 10.1016/j.celrep.2017.07.027
79. Sun L, Li CW, Chung EM, Yang R, Kim YS, Park AH, et al. Targeting Glycosylated PD-1 Induces Potent Antitumor Immunity. Cancer Res (2020) 80:2298–310. doi: 10.1158/0008-5472.CAN-19-3133
80. Yang Y, Hsu JM, Sun L, Chan LC, Li CW, Hsu JL, et al. Palmitoylation Stabilizes PD-L1 to Promote Breast Tumor Growth. Cell Res (2019) 29:83–6. doi: 10.1038/s41422-018-0124-5
81. Yao H, Lan J, Li C, Shi H, Brosseau JP, Wang H, et al. Inhibiting PD-L1 Palmitoylation Enhances T-Cell Immune Responses Against Tumours. Nat BioMed Eng (2019) 3:306–17. doi: 10.1038/s41551-019-0375-6
82. Ohtsubo K, Marth JD. Glycosylation in Cellular Mechanisms of Health and Disease. Cell (2006) 126:855–67. doi: 10.1016/j.cell.2006.08.019
83. Christiansen MN, Chik J, Lee L, Anugraham M, Abrahams JL, Packer NH, et al. Cell Surface Protein Glycosylation in Cancer. Proteomics (2014) 14:525–46. doi: 10.1002/pmic.201300387
84. Munkley J, Elliott DJ. Hallmarks of Glycosylation in Cancer. Oncotarget (2016) 7:35478–89. doi: 10.18632/oncotarget.8155
85. Li CW, Lim SO, Xia W, Lee HH, Chan LC, Kuo CW, et al. Glycosylation and Stabilization of Programmed Death Ligand-1 Suppresses T-Cell Activity. Nat Commun (2016) 7:12632. doi: 10.1038/ncomms12632
86. Hsu JM, Xia W, Hsu YH, Chan LC, Yu WH, Cha JH, et al. STT3-Dependent PD-L1 Accumulation on Cancer Stem Cells Promotes Immune Evasion. Nat Commun (2018) 9:1908. doi: 10.1038/s41467-018-04313-6
87. Wang YN, Lee HH, Hsu JL, Yu D, Hung MC. The Impact of PD-L1 N-Linked Glycosylation on Cancer Therapy and Clinical Diagnosis. J BioMed Sci (2020) 27:77. doi: 10.1186/s12929-020-00670-x
88. Lee HH, Wang YN, Xia W, Chen CH, Rau KM, Ye L, et al. Removal of N-Linked Glycosylation Enhances PD-L1 Detection and Predicts Anti-PD-1/PD-L1 Therapeutic Efficacy. Cancer Cell (2019) 36:168–78.e164. doi: 10.1016/j.ccell.2019.06.008
89. Shao B, Li CW, Lim SO, Sun L, Lai YJ, Hou J, et al. Deglycosylation of PD-L1 by 2-Deoxyglucose Reverses PARP Inhibitor-Induced Immunosuppression in Triple-Negative Breast Cancer. Am J Cancer Res (2018) 8:1837–46.
90. Kim B, Sun R, Oh W, Kim AMJ, Schwarz JR, Lim SO. Saccharide Analog, 2-Deoxy-D-Glucose Enhances 4-1BB-Mediated Antitumor Immunity via PD-L1 Deglycosylation. Mol Carcinog (2020) 59:691–700. doi: 10.1002/mc.23170
91. Mei J, Xu J, Yang X, Gu D, Zhou W, Wang H, et al. A Comparability Study of Natural and Deglycosylated PD-L1 Levels in Lung Cancer: Evidence From Immunohistochemical Analysis. Mol Cancer (2021) 20:11. doi: 10.1186/s12943-020-01304-4
92. Terawaki S, Chikuma S, Shibayama S, Hayashi T, Yoshida T, Okazaki T, et al. IFN-Alpha Directly Promotes Programmed Cell Death-1 Transcription and Limits the Duration of T Cell-Mediated Immunity. J Immunol (2011) 186:2772–9. doi: 10.4049/jimmunol.1003208
93. Zhang N, Li M, Xu X, Zhang Y, Liu Y, Zhao M, et al. Loss of Core Fucosylation Enhances the Anticancer Activity of Cytotoxic T Lymphocytes by Increasing PD-1 Degradation. Eur J Immunol (2020) 50:1820–33. doi: 10.1002/eji.202048543
94. Shi X, Zhang D, Li F, Zhang Z, Wang S, Xuan Y, et al. Targeting Glycosylation of PD-1 to Enhance CAR-T Cell Cytotoxicity. J Hematol Oncol (2019) 12:127. doi: 10.1186/s13045-019-0831-5
95. Wang M, Wang J, Wang R, Jiao S, Wang S, Zhang J, et al. Identification of a Monoclonal Antibody That Targets PD-1 in a Manner Requiring PD-1 Asn58 Glycosylation. Commun Biol (2019) 2:392. doi: 10.1038/s42003-019-0642-9
96. Liu K, Tan S, Jin W, Guan J, Wang Q, Sun H, et al. N-Glycosylation of PD-1 Promotes Binding of Camrelizumab. EMBO Rep (2020) e51444. doi: 10.15252/embr.202051444
97. Fernandez LP, Sanchez-Martinez R, Vargas T, Herranz J, Martin-Hernandez R, Mendiola M, et al. The Role of Glycosyltransferase Enzyme GCNT3 in Colon and Ovarian Cancer Prognosis and Chemoresistance. Sci Rep (2018) 8:8485. doi: 10.1038/s41598-018-26468-4
98. Rana MS, Lee CJ, Banerjee A. The Molecular Mechanism of DHHC Protein Acyltransferases. Biochem Soc Trans (2019) 47:157–67. doi: 10.1042/BST20180429
99. Tabaczar S, Czogalla A, Podkalicka J, Biernatowska A, Sikorski AF. Protein Palmitoylation: Palmitoyltransferases and Their Specificity. Exp Biol Med (Maywood) (2017) 242:1150–7. doi: 10.1177/1535370217707732
100. Gottlieb CD, Linder ME. Structure and Function of DHHC Protein S-Acyltransferases. Biochem Soc Trans (2017) 45:923–8. doi: 10.1042/BST20160304
101. Hentschel A, Zahedi RP, Ahrends R. Protein Lipid Modifications–More Than Just a Greasy Ballast. Proteomics (2016) 16:759–82. doi: 10.1002/pmic.201500353
102. Won SJ, Cheung See Kit M, Martin BR. Protein Depalmitoylases. Crit Rev Biochem Mol Biol (2018) 53:83–98. doi: 10.1080/10409238.2017.1409191
103. Ernst AM, Toomre D, Bogan JS. Acylation – A New Means to Control Traffic Through the Golgi. Front Cell Dev Biol (2019) 7:109. doi: 10.3389/fcell.2019.00109
104. Takeuchi O, Akira S. Pattern Recognition Receptors and Inflammation. Cell (2010) 140:805–20. doi: 10.1016/j.cell.2010.01.022
105. Brubaker SW, Bonham KS, Zanoni I, Kagan JC. Innate Immune Pattern Recognition: A Cell Biological Perspective. Annu Rev Immunol (2015) 33:257–90. doi: 10.1146/annurev-immunol-032414-112240
106. Gong T, Liu L, Jiang W, Zhou R. DAMP-Sensing Receptors in Sterile Inflammation and Inflammatory Diseases. Nat Rev Immunol (2020) 20:95–112. doi: 10.1038/s41577-019-0215-7
107. Triantafillidis JK, Nasioulas G, Kosmidis PA. Colorectal Cancer and Inflammatory Bowel Disease: Epidemiology, Risk Factors, Mechanisms of Carcinogenesis and Prevention Strategies. Anticancer Res (2009) 29:2727–37.
108. Wong VW, Yu J, Cheng AS, Wong GL, Chan HY, Chu ES, et al. High Serum Interleukin-6 Level Predicts Future Hepatocellular Carcinoma Development in Patients With Chronic Hepatitis B. Int J Cancer (2009) 124:2766–70. doi: 10.1002/ijc.24281
109. Punturieri A, Szabo E, Croxton TL, Shapiro SD, Dubinett SM. Lung Cancer and Chronic Obstructive Pulmonary Disease: Needs and Opportunities for Integrated Research. J Natl Cancer Inst (2009) 101:554–9. doi: 10.1093/jnci/djp023
110. Sparmann A, Bar-Sagi D. Ras-Induced Interleukin-8 Expression Plays a Critical Role in Tumor Growth and Angiogenesis. Cancer Cell (2004) 6:447–58. doi: 10.1016/j.ccr.2004.09.028
111. Coussens LM, Werb Z. Inflammation and Cancer. Nature (2002) 420:860–7. doi: 10.1038/nature01322
112. Greten FR, Grivennikov SI. Inflammation and Cancer: Triggers, Mechanisms, and Consequences. Immunity (2019) 51:27–41. doi: 10.1016/j.immuni.2019.06.025
113. Ammirante M, Luo JL, Grivennikov S, Nedospasov S, Karin M. B-Cell-Derived Lymphotoxin Promotes Castration-Resistant Prostate Cancer. Nature (2010) 464:302–5. doi: 10.1038/nature08782
114. Vakkila J, Lotze MT. Inflammation and Necrosis Promote Tumour Growth. Nat Rev Immunol (2004) 4:641–8. doi: 10.1038/nri1415
115. Zitvogel L, Apetoh L, Ghiringhelli F, Kroemer G. Immunological Aspects of Cancer Chemotherapy. Nat Rev Immunol (2008) 8:59–73. doi: 10.1038/nri2216
116. Hayward JA, Mathur A, Ngo C, Man SM. Cytosolic Recognition of Microbes and Pathogens: Inflammasomes in Action. Microbiol Mol Biol Rev (2018) 82. doi: 10.1128/MMBR.00015-18
117. Wei X, Song H, Yin L, Rizzo MG, Sidhu R, Covey DF, et al. Fatty Acid Synthesis Configures the Plasma Membrane for Inflammation in Diabetes. Nature (2016) 539:294–8. doi: 10.1038/nature20117
118. Lancaster GI, Langley KG, Berglund NA, Kammoun HL, Reibe S, Estevez E, et al. Evidence That TLR4 Is Not a Receptor for Saturated Fatty Acids But Mediates Lipid-Induced Inflammation by Reprogramming Macrophage Metabolism. Cell Metab (2018) 27:1096–110.e1095. doi: 10.1016/j.cmet.2018.03.014
119. Kim YC, Lee SE, Kim SK, Jang HD, Hwang I, Jin S, et al. Toll-Like Receptor Mediated Inflammation Requires FASN-Dependent MYD88 Palmitoylation. Nat Chem Biol (2019) 15:907–16. doi: 10.1038/s41589-019-0344-0
120. Philpott DJ, Sorbara MT, Robertson SJ, Croitoru K, Girardin SE. NOD Proteins: Regulators of Inflammation in Health and Disease. Nat Rev Immunol (2014) 14:9–23. doi: 10.1038/nri3565
121. Hugot JP, Chamaillard M, Zouali H, Lesage S, Cezard JP, Belaiche J, et al. Association of NOD2 Leucine-Rich Repeat Variants With Susceptibility to Crohn’s Disease. Nature (2001) 411:599–603. doi: 10.1038/35079107
122. Lu Y, Zheng Y, Coyaud E, Zhang C, Selvabaskaran A, Yu Y, et al. Palmitoylation of NOD1 and NOD2 Is Required for Bacterial Sensing. Science (2019) 366:460–7. doi: 10.1126/science.aau6391
123. Zhong B, Yang Y, Li S, Wang YY, Li Y, Diao F, et al. The Adaptor Protein MITA Links Virus-Sensing Receptors to IRF3 Transcription Factor Activation. Immunity (2008) 29:538–50. doi: 10.1016/j.immuni.2008.09.003
124. Zhong B, Zhang L, Lei C, Li Y, Mao AP, Yang Y, et al. The Ubiquitin Ligase RNF5 Regulates Antiviral Responses by Mediating Degradation of the Adaptor Protein MITA. Immunity (2009) 30:397–407. doi: 10.1016/j.immuni.2009.01.008
125. Zhou Q, Lin H, Wang S, Wang S, Ran Y, Liu Y, et al. The ER-Associated Protein ZDHHC1 Is a Positive Regulator of DNA Virus-Triggered, MITA/STING-Dependent Innate Immune Signaling. Cell Host Microbe (2014) 16:450–61. doi: 10.1016/j.chom.2014.09.006
126. Liu Y, Zhou Q, Zhong L, Lin H, Hu MM, Zhou Y, et al. ZDHHC11 Modulates Innate Immune Response to DNA Virus by Mediating MITA-IRF3 Association. Cell Mol Immunol (2018) 15:907–16. doi: 10.1038/cmi.2017.146
127. Mukai K, Konno H, Akiba T, Uemura T, Waguri S, Kobayashi T, et al. Activation of STING Requires Palmitoylation at the Golgi. Nat Commun (2016) 7:11932. doi: 10.1038/ncomms11932
128. Haag SM, Gulen MF, Reymond L, Gibelin A, Abrami L, Decout A, et al. Targeting STING With Covalent Small-Molecule Inhibitors. Nature (2018) 559:269–73. doi: 10.1038/s41586-018-0287-8
Keywords: metabolic modifications, glycosylation, palmitoylation, inflammation, cancer immunotherapy, PD-1/PD-L1
Citation: Zheng S, Song Q and Zhang P (2021) Metabolic Modifications, Inflammation, and Cancer Immunotherapy. Front. Oncol. 11:703681. doi: 10.3389/fonc.2021.703681
Received: 05 May 2021; Accepted: 20 August 2021;
Published: 24 September 2021.
Edited by:
Vincenzo Desiderio, Second University of Naples, ItalyReviewed by:
Virginia Tirino, Università della Campania Luigi Vanvitelli, ItalyLuigi Mele, University of Naples Federico II, Italy
Copyright © 2021 Zheng, Song and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Pingfeng Zhang, cGluZ2ZlbmcuemhhbmdAd2h1LmVkdS5jbg==