
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Oncol. , 26 August 2021
Sec. Cancer Molecular Targets and Therapeutics
Volume 11 - 2021 | https://doi.org/10.3389/fonc.2021.703087
 Xiaorui Li1†
Xiaorui Li1† Yaru Feng1†Fengqin Shang1Zhuoying Yu1Tieshan Wang2
Yaru Feng1†Fengqin Shang1Zhuoying Yu1Tieshan Wang2 Jing Zhang1Zhiru Song1
Jing Zhang1Zhiru Song1 Ping Wang3Bingjie Shi1*
Ping Wang3Bingjie Shi1* Jianxun Wang1,4*
Jianxun Wang1,4*Multiple myeloma (MM) is a tumor type characterized by the unregulated proliferation of clonal plasma cells in the bone marrow. Immunotherapy based on chimeric antigen receptor T cell (CAR-T) therapy has achieved exciting success in the treatment of hematological malignant tumors. CD38 is highly and evenly expressed in MM and is an attractive target for MM treatment. Here, we successfully constructed two novel second-generation CAR-T cells targeting CD38 by retroviral vector transduction. CD38 CAR-T cells could be activated effectively after stimulation with CD38-positive tumor cells and secrete cytokines such as IFN-γ and TNF-α to promote tumor cell apoptosis in in vitro experiments. Real-time fluorescence monitoring experiments, luciferase detection experiments and flow cytometry experiments revealed the efficient and specific killing abilities of CD38 CAR-T cells against CD38-positive tumor cells. The proliferation ability of CD38 CAR-T cells in vitro was higher than that of untransduced T cells. Further antitumor experiments in vivo showed that CD38 CAR-T cells could be quickly activated to secrete IFN-γ and eliminate tumors. Thus, novel CD38-targeted second-generation CAR-T cells have efficient and specific antitumor activity and may become a novel therapy for the clinical treatment of MM.
Multiple myeloma (MM) is a hematologic malignancy characterized by the uncontrolled expansion of plasma cells in bone marrow (1). The clinical manifestations of disease include signs of end-organ damage such as hypercalcemia, renal insufficiency, anemia and/or osteolytic damage (2, 3). MM accounts for 1% of all cancers, with an incidence of 6-7 cases per 100,000 people, and is the second most common hematological malignant tumor (1, 4, 5). It is estimated that in 2021, a total of 34,920 new cases of the disease will be diagnosed and that 15,600 MM-related deaths will occur in the US (6).
In the past few decades, with the rapid development of new therapeutic approaches, such as proteasome inhibitors (PIs), immunomodulatory drugs (IMiDs), monoclonal antibodies (mABs), autologous stem cell transplantation (ASCT) and histone deacetylase (HDAC) inhibitors, the prognosis and outcome of patients with MM have been significantly improved (7–9). However, MM is still incurable to a large extent and has a high recurrence rate (10, 11). In this regard, new immunotherapies have been developed to combat MM.
Chimeric antigen receptor-engineered T cell (CAR-T cell) therapy has recently emerged with encouraging clinical results in MM (12, 13). The success of CAR-T cells targeting CD19 for chronic/acute lymphoblastic leukemia led to their FDA approval in 2017 (14–16). This has stimulated interest in extending CAR-T cell therapy to MM. CD19 and BCMA are commonly used in clinical trials at present, and novel targets are being developed (17–19). CD38 is highly and uniformly expressed in myeloma cells and is an ideal target antigen for MM (20). Although some CD38 CAR-T cells targeting MM have been reported (20, 21), clinical treatment with more novel CAR-T cells is needed to advance the treatment of hematological cancers. Due to the immunogenicity of the CAR transgene, the host immune response to the mouse-derived single-chain variable region (ScFv) leads to the poor persistence of CAR T cells, and the therapeutic effect is not long lasting (22). In CAR-T cell therapy, humanized ScFv has been applied as a strategy to reduce CAR immunogenicity and subsequently improve efficacy, but it does not effectively prevent the development of anti-IgE reactions that lead to allergic reactions (23, 24). Therefore, the use of human ScFv in the CAR structure may enhance the antitumor activity and persistence of CAR-T cells (23).
Herein, we report two human ScFv-derived, second-generation CAR-T cells targeting CD38. Subsequently, we found that these two kinds of CAR-T cells have notably effective cytokine secretion and specific tumor killing abilities, increased proliferation and expansion abilities in vitro, and a very robust xenograft tumor killing ability in vivo. Our novel CD38 CAR might be effective for cancer immunotherapy in the clinic.
The Phoenix-ECO cell line (CLR-3214) and PG13 cell line (TK-NIH3T3) were purchased from the American Tissue Culture Collection (ATCC, Manassas, VA, USA). Both cell lines were cultured in DMEM (Gibco, USA) with 10% fetal bovine serum (FBS, Gibco, USA) and used for retroviral vector packaging. The Daudi cell line (ml-cs-0509) was purchased from the American Tissue Culture Collection (ATCC, Manassas, VA, USA). The RPMI-gfp-luc, Raji-luc, and K562-hBCMA cell lines were kindly provided by Dr. Wu (China Agricultural University). All the tumor cell lines above were maintained in RPMI-1640 medium (Gibco, USA) with 10% FBS.
The CD38 CAR contains the following components: a signal peptide (SP) and myc taq upstream. The CD38 CAR antigen-targeting regions (ScFvs) contained heavy and light chain variables divided by a (G4S)3 linker, CD8 hinge-transmembrane domain, CD28 coactivation domain and CD3ζ intracellular signaling domain. All these CD38 CAR constructs were packaged into retroviral vectors.
The retroviral vector supernatants were generated by a two-step method using Phoenix-ECO and PG13 retroviral vector packaging cell lines in turn. The copy numbers of the vectors were analyzed by quantitative reverse transcription polymerase chain reaction (RT-qPCR). First, vector RNA was extracted from the vector-containing supernatants (QIAamp®Viral RNA Mini Kit, Cat#:52904), and then the vector RNA was reverse-transcribed into cDNA (QuantiNova Reverse Transcription Kit, Cat#:205410). After that, real-time PCR was carried out with the QuantStudio6 Flex system (Applied Biosystems), and the plasmid was continuously diluted to create a standard curve (QuantiNova SYBR ®Green PCR Kit, Cat#:75600) with the following formula: (DNA amount (ng)×6.022×1023)/(length (bp)×1×109×650). The titer of the recombinant CD38 CAR retroviral vector is reported in transforming units (TU)/mL. This experiment was repeated three times independently. The primer sequences are provided in Supplement Table 1.
All blood sampling from the donors was performed after informed consent was obtained, and the protocols were approved by the Beijing University of Chinese Medicine Medical Ethics Committee. Human peripheral blood mononuclear cells (PBMCs) were isolated from healthy human donors by Ficoll density gradient centrifugation (Lymphoprep, Stemcell, Canada). PBMCs were activated with 100 ng/mL soluble anti-CD3, clone OKT3 (Sino Biological, GMP-10977-H001) and 100 U/mL IL-2 (Sino Biological, GMP-11848-HNAE) and then cultured at 37°C in a 5% CO2 incubator for 48 hours. T cell growth medium was composed of AIM-V medium (Gibco, USA) with 10% FBS and 100 U/mL IL-2. Activated T cells were transduced using the spin inoculation method (25). The T cell transduction efficiency was measured by flow cytometry (BD LSRFortessa, USA), and the data were analyzed by FlowJo V10. Samples were directly stained with APC-conjugated anti-CD3 (Biolegend, Cat#:300312) antibody and PE-conjugated anti-myc (R&D, Cat#:9E10) antibody. The vector copy number integration-transduced primary T cells were determined by qPCR. The primer sequences are provided in Supplement Table 1. Expression of CD38 on CAR-T cell surfaces was stained with APC-conjugated anti-CD38 (Biolegend, Cat#: 102711) antibody and detected by flow cytometry.
To determine the lytic ability of CD38 CAR-T cells on CD38-positive tumor cells, CAR-T cells or untransduced T cells (Pan-T) were incubated with RPMI-gfp-luc or Raji-luc cells at different effector: target ratios (E:T ratios) for 12 hours. Luciferase activity was analyzed by a SpectraMax Series Multi-Mode Microplate Reader (model i3x; Molecular Devices). The following formula was used for analysis: % cell lysis = 1 – (experimental lysis – spontaneous lysis) × 100/(maximum lysis – spontaneous lysis). Each experiment was done in triplicate.
RPMI-gfp-luc cells were incubated with CAR-T cells or pan-T cells at an E:T ratio of 1:1. The cells were analyzed by an IncuCyte S3 Live-Cell analysis system (Sartorius, Germany). Images were captured every two hours for a total of 68 hours. Total integrated GFP intensity per well was assessed as a quantitative measure of live, GFP+ tumor cells. The values were normalized to the starting values. Each experiment was done in triplicate.
CAR-T cells or Pan-T cells were incubated with RPMI-gfp-luc, Raji-luc, Daudi cells or K562-hBCMA cells (K562-hBCMA cells expressing BCMA but not CD38 were used as a negative control) at different E:T ratios for 12 hours. Then, the cells were stained with BV421-conjugated anti-CD3, and Annexin V-Alexa Fluor 647 (BioFriend, Cat#: P04D12) staining was used for apoptotic cell detection. Data were collected by flow cytometry and analyzed by FlowJo. The apoptotic rate of tumor cells was calculated as the percentage of CD3-negative and annexin V-positive cells among total cells. Each experiment was done in triplicate.
To assess cytokine release by CAR-T cells, supernatants were harvested 12 hours after incubation with different types of tumor cells for assessment with a Cytometric Bead Array (CBA, Biolegend, Cat#: B314528) kit following the manufacturer’s protocol. Beads were analyzed by a flow cytometry assay. Each experiment was done in triplicate.
CAR-T cells or pan-T cells were labeled with carboxyfluorescein diacetate succinimidyl ester (CFSE, BD, Cat#: 565082) according to the manufacturer’s instructions to track CAR-T cell proliferation. Then, the labeled CAR-T cells were incubated with or without RPMI-gfp-luc cells at an E:T ratio of 1:1 without the addition of exogenous cytokine IL-2 for 72 hours. The proliferation of T cells was assessed by monitoring CFSE dilution. The cells were collected by flow cytometry, and data were analyzed by FlowJo.
NPG/Vst mice are members of the NOD-PrkdcscidIl2rgnull family. Female NPG/Vst mice with severe combined immunodeficiency were purchased from VITALSTART (Beijing, China). All mice were raised under specific pathogen-free conditions. The animal experiment program was approved by the Biomedical Research Ethics Committee of Beijing University of Chinese Medicine (BUCM-4-2020060101-2015).
NPG mice (6-8 weeks old) were administered intravenous (i.v.) inoculations of 2× 106 RPMI-gfp-luc cells. Fourteen days later, the transplanted mice were randomly divided into 4 groups (control group, pan-T group, CD38 CAR21-T group and CD38 CAR22-T group) with 5 mice in each group. On the same day, the mice received i.v. injections of 1x108/kg CAR T cells or Pan-T cells. The second injection was given seven days after the first CAR-T cell injection. From the third day after the first CAR-T cell injection (17 days after tumor inoculation), a multifunctional in vivo imager (MIIS, Molecular Devices) was used to monitor the tumor load once a week according to the manufacturer’s protocol. On the 16th day after tumor inoculation (the second day after the first injection of CAR-T cells), the peripheral blood of mice was collected, the plasma was separated, and the level of human cytokines was detected by a CBA kit. On the 35th day after tumor inoculation (21 days after CAR-T cell injection), 100 μL peripheral blood of mice was collected, and BV785-conjugated anti-CD3 (Biolegend, Cat#: 300472) staining was performed. Then, the expression of human CD3 in mouse blood was detected to assess the persistence of injected CAR-T cells in vivo by flow cytometry. All animals were weighed once a week. The experiments lasted up to 62 days. At the time of death of the mice, blood, liver, bone marrow and lysed erythrocytes were collected. The residual CD38-positive tumor cells were monitored by flow cytometry.
The data obtained were analyzed using the GraphPad Prism 8.0 software and expressed as arithmetic the mean ± SEM. Comparisons of two groups or data points were performed by Student’s t-test. All experiments were performed at least 3 times using independent donor cells to ascertain reproducibility. P-values < 0.05 were considered statistically significant.
To construct the CD38 CAR, we used the variable heavy and light chain sequences of two different CD38 antibodies as the ScFv sequences, CD8 as the hinge and transmembrane domains, CD28 as the costimulatory domain and CD3ζ as the intracellular activation domain (Figures 1A, B). Phoenix-ECO cells and PG13 cells were used to package and produce CD38 CAR retroviral vectors. Vector copy numbers were determined by RT-qPCR, and vector titers were calculated. The results showed that high titers of CD38 CAR retroviral particles were generated (Figure 1C). Activated human primary T cells were transduced with CD38 CAR retroviral vectors. The expression of CD38 CAR was detected by flow cytometry. The results showed that the two CD38 CAR retroviral vectors we packaged had high transduction efficiency (Figures 1D and S1A). The qPCR detection results further confirmed that the two CD38 CARs were integrated into the T cell genome (Figure 1E). CD38 expression was seen on the surface of 98.5% of T cells but disappeared after transduction (Figure 1F).
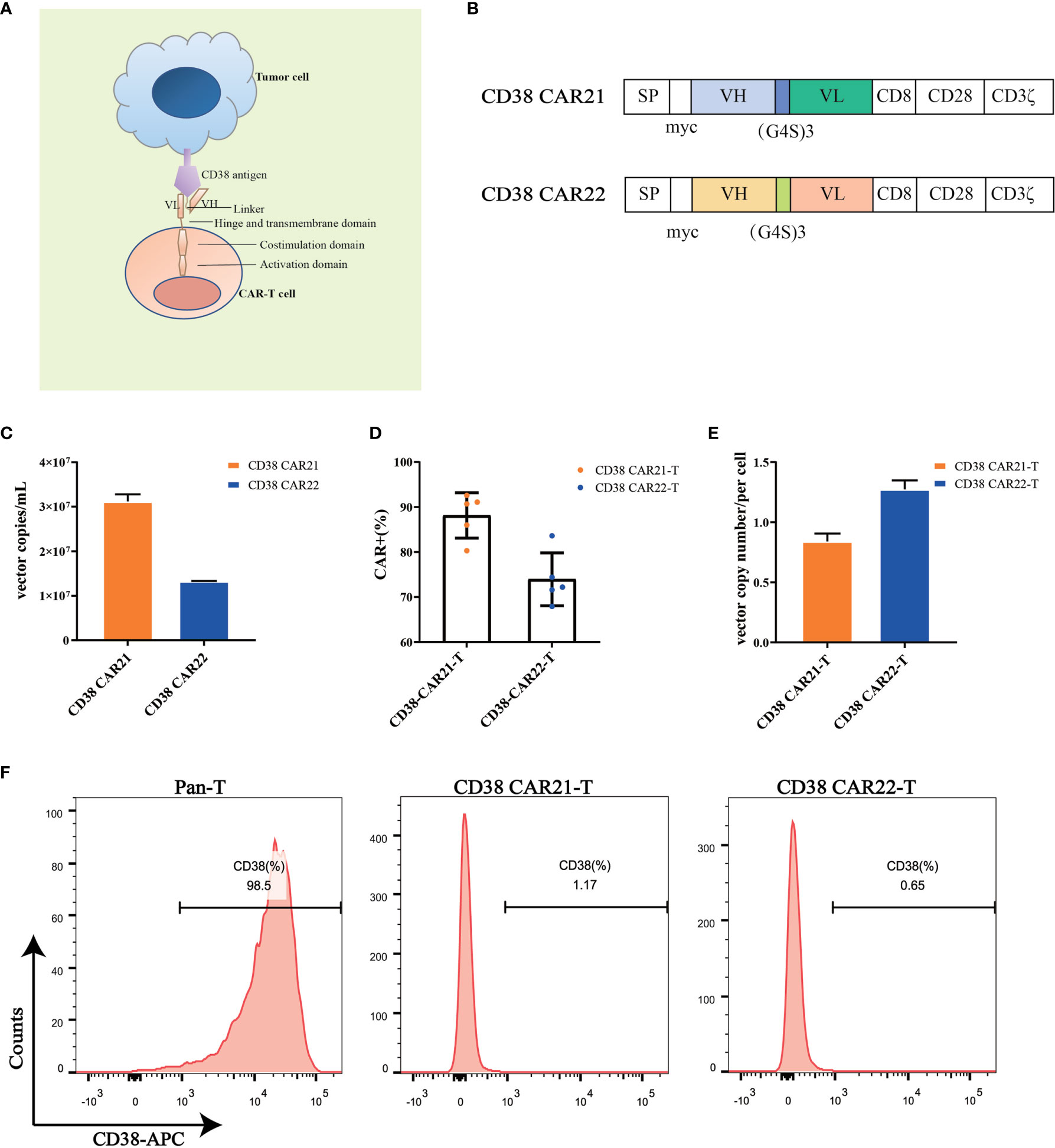
Figure 1 CD38 CAR construct and expression on primary T cells. (A, B) The construction of two CD38 CARs. The CD38-ScFv sequences were selected from two different CD38 antibody sequences (CAR21 and CAR22) with the CD8 hinge and transmembrane region, the CD28 cytoplasmic costimulatory region and the CD3ζ signaling domain. (C) The copy numbers of supernatant viral vectors produced by the PG13 cell line were detected by RT-qPCR. (D) The transduction efficiency of CD38 CAR in primary T cells from different donors (n= 5). The transduction efficiency was defined as the percentage of CD3-positive and myc-positive cells among total live cells. (E) The vector copy number of CD38 CAR integration per primary T cell was detected by RT-qPCR. (F) The expression of CD38 on the surface of human primary T cells transduced with the CD38 CAR was detected by flow cytometry.
Cytotoxicity is the most important feature of CAR-T cells and directly determines the antitumor activity of CAR-T cells. To determine the cytotoxicity of CD38 CAR-T cells on tumor cells, we established an in vitro tumor killing experiment in which CAR-T cells were incubated with different types of CD38-positive tumor cells at different E:T ratios. The results of CD38 expression on the tumor cells are shown in Figure S1B. First, the cytolytic effects of CD38 CAR-T cells on tumor cells were analyzed by luciferase assay. We found that compared with Pan-T cells, CD38 CAR-T cells showed a stronger cytolytic effect on RPMI-gfp-luc and Raji-luc cells, and this cytolytic ability was positively correlated with the E:T ratio (Figure 2A). Furthermore, we incubated CAR-T cells with RPMI-gfp-luc cells expressing green fluorescent protein at an E:T ratio of 1:1 for real-time monitoring and found that CD38 CAR-T cells had a robust killing effect after 2 hours (Figure 2B). These results indicated that our CD38 CAR-T cells have rapid and highly efficient effective antitumor capability.
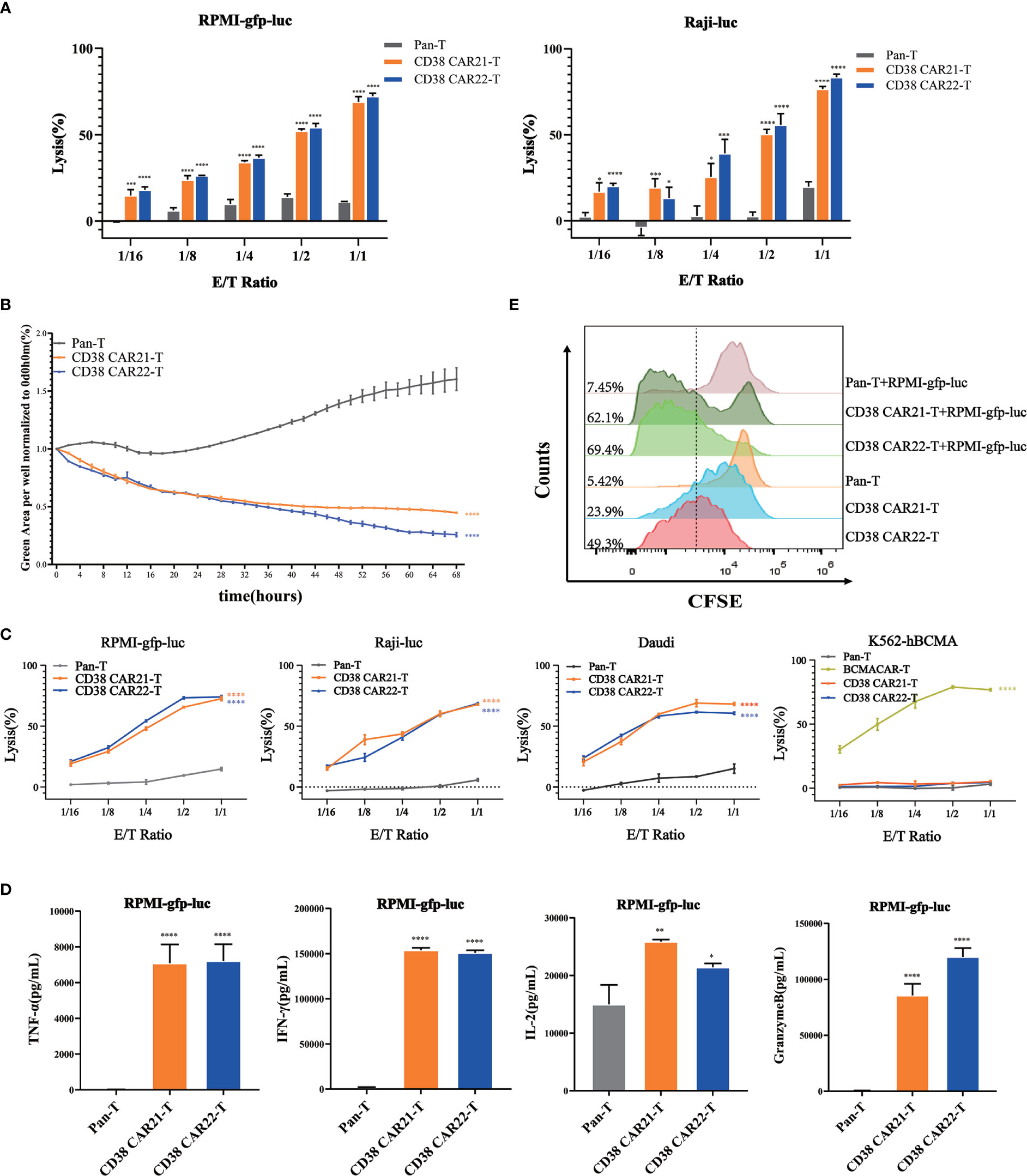
Figure 2 Cytotoxicity and proliferation of CD38 CAR-T cells in vitro. (A) The ability of CAR-T cells to kill tumor cells was measured by luciferase assay. CD38 CAR-T cells or pan-T cells were incubated with RPMI-gfp-luc cells or Raji-luc cells, and the luciferase signal was determined after 12 hours. (B) The ability of CAR-T cells to kill tumor cells was measured by an IncuCyte S3 system. RPMI-gfp-luc cells were incubated with CD38 CAR-T cells or pan-T cells at an E:T ratio of 1:1. Total integrated GFP intensity per well was assessed as a quantitative measure of live, GFP+ tumor cells. The values were normalized to the starting values. (C) The ability of CD38 CAR-T cells to kill tumor cells was determined by flow cytometry. CD38 CAR-T cells or pan-T cells were incubated with RPMI-gfp-luc cells, Raji-luc cells, Daudi cells or K562-hBCMA cells for 12 hours. (D) CD38 CAR-T cells were incubated with RPMI-gfp-luc cells for 12 hours, and the secretion of cytokines was measured with a flow cytometry-based CBA kit. The graph shows the secretion of TNFα, IFN-γ, IL-2 and granzyme B. (E) The proliferation of CD38 CAR-T cells. The proliferation ability was measured by the CFSE-based assay. Data were obtained from three replicates and are presented as the mean ± SEM (n=3). *indicates p value < 0.05; **< 0.01; ***< 0.005 and ****< 0.001, comparison of two groups was performed by Student’s t-test. TNF-α, tumor necrosis factor α; IFN-γ, interferon γ; IL-2, interleukin 2.
To confirm the antigen specificity of this antitumor effect of CD38 CAR-T cells, CD38 CAR-T cells were incubated with RPMI-gfp-luc, Raji-luc and Daudi cells expressing CD38 antigen. K562-hBCMA cells that did not express CD38 but expressed BCMA were used as controls. Samples were detected by flow cytometry. The results showed that CD38 CAR-T cells had a robust killing effect on CD38-positive tumor cells but no specific killing effect on CD38-negative K562-hBCMA cells. This result demonstrated the CD38 antigen specificity of the CD38 CAR-T cells against the tumor cells (Figure 2C).
The release of cytokines is a marker of CAR-T cell activation. CD38 CAR-T cells were incubated with RPMI-gfp-luc, Raji-luc and Daudi or K562-hBCMA cells, the cell culture medium was harvested, and cytokines such as TNF-α, IFN-γ, IL-2 and granzyme B were measured by a CBA kit. As shown in Figures 2D and S2, CD38 CAR-T cells secreted large amounts of TNF-α, IFN-γ, IL-2 and granzyme when incubated with CD38-positive tumor cells compared with pan-T cells, and there was no significant change in the killing effect of CD38 CAR-T cells on tumor cells that did not express CD38. The release of these cytokines indicates that CD38 CAR-T cells are effectively activated and further confirms that CD38 CAR-T cells have robust antitumor activity. Moreover, the antitumor activity of CD38 CAR T cells was specific for CD38 antigen.
The production of a robust and lasting antitumor immune response needs not only to trigger cytotoxicity and the production of cytokines but also to stimulate the proliferation of CAR-T cells. To evaluate the proliferation ability of the two kinds of CD38 CAR-T cells in vitro, we divided CD38 CAR-T cells into two groups: single culture and coculture with RPMI-gfp-luc. A CFSE-based assay was used to measure proliferation. The results showed that the green fluorescence signal of the two kinds of CD38 CAR-T cells shifted significantly to the left compared with that of pan-T cells, indicating that the two kinds of CD38 CAR-T cells expanded faster. Moreover, upon stimulation with RPMI-gfp-luc cells, CD38 CAR-T cells showed a better proliferation effect than CAR-T cells cultured alone (Figure 2E).
To evaluate the antitumor effect of CD38 CAR-T cells in vivo, we established tumor xenotransplantation models by i.v. injection of RPMI-gfp-luc tumor cells. A schema of the experimental protocol for this in vivo model is provided in Figure 3A. After 14 days, the transplanted mice were randomly divided into 4 groups (Figure S3A), and the mice received i.v. injections of 1x108/kg CAR-T cells or Pan-T cells. The second injection was given seven days after the first CAR-T cell injection. As shown in Figures 3B, C, the results of bioluminescence imaging in vivo showed that compared with the control group or Pan-T group, the tumor of the CD38 CAR-T cell groups was cleared completely after the second injection of CD38 CAR-T cells. At the end of the study, the CD38 antigen was virtually undetectable in the blood, liver and bone marrow (Figure S4), showing a strong antitumor ability in vivo until the end of the experiment, and no recurrence was observed. On the 16th day after tumor cell inoculation (the second day after the first injection of CAR-T cells), we found that the level of human IFN-γ in the blood of mice treated with CD38 CAR-T cells was significantly higher than that of mice in the Pan-T group, which indicated that CD38 CAR-T cells produced more IFN-γ and that CD38 CAR-T cells were effectively activated (Figure S3B). At 35 days after tumor inoculation, human T cells were still detected in the blood, and the number in the CAR-T group was higher than that in the control group (Figure S3C). This result demonstrated that CD38 CAR-T cell persistence was obviously increased in vivo compared with pan-T cell persistence, indicating that CAR-T cells could survive longer in vivo and have a more lasting antitumor effect. The survival rate of mice in the CD38 CAR-T groups was significantly higher than that in the control group and the Pan-T group, and the survival rate of CD38 CAR21-T cells was 100% until the end of the experiment (Figure 3D). In addition, the weight of mice treated with CD38 CAR-T cells was relatively stable compared with that of the pan-T group (Figure S3D).
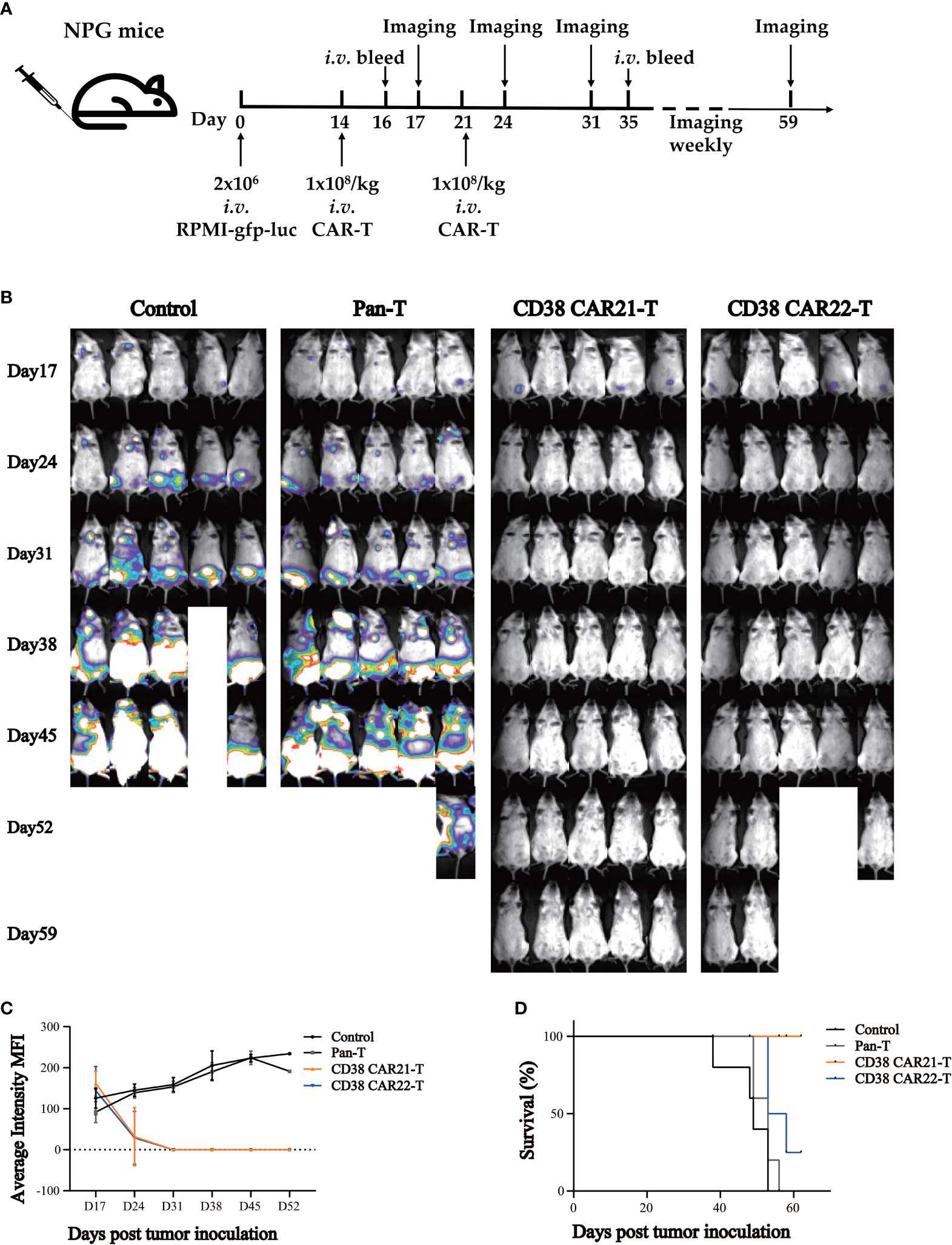
Figure 3 CD38 CAR-T cells exhibited significant antitumor activities in a mouse xenograft tumor model. (A) Experimental design indicating tumor cells, timeline, doses, and imaging time points. Mice were randomized after tumor engraftment. T cells from two independent donors were used (n=5). (B) Mice were assessed by bioluminescence imaging once a week starting 3 days after the first injection of CAR-T cells. (C) Tumor burden was monitored weekly and presented as the average intensity. (D) Survival analysis of mice after tumor cell inoculation.n= 5 mice per group.
MM is a malignant tumor mainly characterized by the malignant proliferation of plasma cells in bone marrow that secrete a large amount of monoclonal immunoglobulin (M protein) and eventually leads to organ damage (26, 27). MM is still incurable. Adoptive cellular immunotherapy is a promising method for the treatment of cancer. In recent years, immunotherapy based on CAR T cells has achieved remarkable success in the treatment of malignant hematological tumors, especially lymphoma (26, 28). With the success of CAR-T cells in the treatment of acute lymphoblastic leukemia, research on CAR-T therapy in MM has continuously advanced (8). The human CD38 antigen is a transmembrane glycoprotein that is highly expressed in MM cells. Although CD38 is also expressed on B cells, plasma cells, T cells, NK cells and myeloid progenitor cells, the expression level is much lower than that in MM cells (29). Anti-CD38 monoclonal antibodies such as daratumumab, have been approved by the FDA for the treatment of newly diagnosed MM (30) and relapsed/refractory MM (31). Compared with anti-CD38 monoclonal antibodies, CD38 CAR-T cells have substantially improved specificity and can induce a durable and efficient response (32). Although CD38 CAR-T cells targeting MM have been reported, there are still no approved CD38 CAR-T cell products for clinical use; thus, more novel CD38 CAR-T cell clinical treatments are needed to advance the treatment of hematological cancers. In this study, we demonstrated the great potential of using novel human ScFv-derived CAR-T cells targeting CD38 in the treatment of MM. MM cells express many markers; among them, CD38 molecules are uniformly and highly expressed on the surface of myeloma cells. Recently, many studies have suggested that CD38 may be an ideal target for antigen-specific adoptive cell therapy (20, 33, 34).
CARs mainly comprise single-strand variable regions as signal-binding domains and a linker that connects the heavy (VH) and light chain variable (LH) extracellular domains. In our study, we used a flexible linker (G4S)3 consisting of glycine and serine to increase the flexibility of target cell binding regions (35). At present, clinically used CAR-T cells mostly contain mouse-derived ScFv as the extracellular signal domain, but the host may produce an immune response to mouse-derived ScFv, resulting in a human anti-mouse antibody reaction, thus reducing the efficacy (36, 37). The ScFv sequences of two human CD38 antibodies screened by a previous phage display technique in our laboratory can reduce the immunogenicity of the CAR. The transmembrane region usually comes from CD8α, and the intracellular moiety contains the CD3ζ signaling chain. The costimulatory domain of second-generation CAR cells usually uses CD28 or 4-1BB, and CD28 promotes the secretion of cytokines by CAR-T cells and shows stronger antitumor ability (38–41). Third-generation CARs have two costimulatory domains to promote the proliferation and activation of CAR-T cells (42). At present, second-generation CARs are the most widely used in the clinic and the most effective (3). Therefore, in this experiment, we used the second-generation CAR structure, and the intracellular stimulation we used was CD28. We stably integrated CD38 CARs into the genome of mitotic T cells with a retroviral vector. We found that there was no expression of CD38 on the surface of successfully prepared CD38 CAR-T cells, but it did not affect the function of CD38 CAR-T cells, which was also described in other studies (39, 43). The loss of CD38 may be due to the “self-lysis” of CD38-positive T cell compartments (44).
To determine the antitumor ability of CD38 CAR-T cells in vitro, we selected different kinds of CD38-positive cells and used CD38-negative tumor cells as a control. In different experimental studies, we found that CD38 CAR-T cells have an efficient killing effect on CD38-positive tumor cells but not CD38-negative tumor cells. These results indicate that our CD38 CAR-T cells have a specific killing effect on tumor cells. The distribution of CD38 expression is rather extensive, and as such, normal cells expressing CD38 may be lysed by CD38 CAR-T cells; therefore, this situation cannot be ignored. In the cytokine secretion experiment, CD38 CAR-T cells secreted proinflammatory cytokines after being stimulated by tumor cells, which promoted the apoptosis of tumor cells. CD38 CAR-T cells could be activated by CD38-positive tumor cells but not by tumor cells that do not express CD38, which further confirms the killing specificity of this kind of CD38 CAR-T cell. These results are consistent with those of previous studies (34, 44, 45). We found that CAR-T cells proliferated faster than pan-T cells, and under the stimulation of tumor cells, the proliferation ability of CAR-T cells was further enhanced. It is possible to be due to the coactivation domain CD28. CD28 is an important co-stimulatory molecule for T lymphocytes, which could deliver signals that complement T cell receptor (TCR) in both qualitative and quantitative manners, thereby promoting T cell proliferation and survival (46).
Furthermore, we established NPG mouse xenograft tumor models to determine the antitumor ability of CD38 CAR-T cells in vivo. We found that IFN-γ cytokine secretion was significantly enhanced and that CD38 CAR-T cells were effectively activated two days after tail vein injection of CD38 CAR-T cells, which indicated strong antitumor ability. Moreover, there was no recurrence of tumors observed during the continuous observation period, and flow cytometry showed that CD38 CAR-T cells could persist in mice. This shows the persistence of the antitumor effect of CD38 CAR-T cells.
Overall, we successfully constructed second-generation human ScFv-derived CD38 CAR-T cells and assessed the in vitro functional activity, such as cytolysis and cytokine release, on CD38-positive tumor cells. These CAR T cells can significantly inhibit xenograft tumor growth in vivo, highlighting their antitumor effect. This research provides a solid foundation for further clinical study and illustrates that this CD38 CAR-T therapy has very good clinical potential.
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
The studies involving human participants were reviewed and approved by Beijing University of Chinese Medicine Medical Ethics Committee. The patients/participants provided their written informed consent to participate in this study. The animal study was reviewed and approved by Biomedical Research Ethics Committee of Beijing University of Chinese Medicine.
JW conceived the project and supervised the experiments. XL performed the experiments with the help of YF, FS, ZY, ZS, and BS. XL, TW, and ZY participated in the figure preparation and revised the manuscript. XL wrote the manuscript with revisions by JW and PW. All authors contributed to the article and approved the submitted version.
This work was supported by “Double First-Class” start-up funds from Beijing University of Chinese Medicine (to JW; No. 1000041510155).
The patent application was filed based on the work reported in this manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The authors thank GENERAL BIOSYSTEMS (www.generalbiol.com) for synthesizing the gene fragments. The authors thank Dr. Sen Wu from the College of Biological Sciences, China Agricultural University, for providing the tumor cell lines. The authors thank Boyao Nie from Beijing Vitalstar Biotechnology company for his help in preparing in vivo studies. The authors thank Sarah Conte, PhD, from American Journal Experts (http://www.aje.cn/pricing/) for editing the English text of a draft of this manuscript.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2021.703087/full#supplementary-material
1. Kumar SK, Rajkumar V, Kyle RA, Van Duin M, Sonneveld P, Mateos MV, et al. Multiple Myeloma. Nat Rev Dis Primers (2017) 3:17046. doi: 10.1038/nrdp.2017.46
2. Rajkumar SV, Dimopoulos MA, Palumbo A, Blade J, Merlini G, Mateos MV, et al. International Myeloma Working Group Updated Criteria for the Diagnosis of Multiple Myeloma. Lancet Oncol (2014) 15:e538–48. doi: 10.1016/s1470-2045(14)70442-5
3. Rodriguez-Lobato LG, Ganzetti M, Fernandez de Larrea C, Hudecek M, Einsele H, Danhof S. CAR T-Cells in Multiple Myeloma: State of the Art and Future Directions. Front Oncol (2020) 10:1243. doi: 10.3389/fonc.2020.01243
4. Willenbacher W, Seeber A, Steiner N, Willenbacher E, Gatalica Z, Swensen J, et al. Towards Molecular Profiling in Multiple Myeloma: A Literature Review and Early Indications of its Efficacy for Informing Treatment Strategies. Int J Mol Sci (2018) 19:2087. doi: 10.3390/ijms19072087
5. Latif A, Kapoor V, Sipra Q, Malik SU, Bilal J, Bin Riaz I, et al. Disease Milestones Through Bibliometric Analysis of the Top 100 Cited Articles in Multiple Myeloma. Cureus (2018) 10:e2438. doi: 10.7759/cureus.2438
6. Siegel RL, Miller KD, Fuchs HE, Jemal A. Cancer Statistics, 2021. CA Cancer J Clin (2021) 71:7–33. doi: 10.3322/caac.21654
7. Rajkumar S. Multiple Myeloma: Every Year a New Standard? Hematol Oncol (2019) 37(Suppl 1):62–5. doi: 10.1002/hon.2586
8. Sohail A, Mushtaq A, Iftikhar A, Warraich Z, Kurtin SE, Tenneti P, et al. Emerging Immune Targets for the Treatment of Multiple Myeloma. Immunotherapy (2018) 10:265–82. doi: 10.2217/imt-2017-0136
9. Afifi S, Michael A, Azimi M, Rodriguez M, Lendvai N, Landgren O. Role of Histone Deacetylase Inhibitors in Relapsed Refractory Multiple Myeloma: A Focus on Vorinostat and Panobinostat. Pharmacotherapy (2015) 35:1173–88. doi: 10.1002/phar.1671
10. Kumar SK, Callander NS, Hillengass J, Liedtke M, Kumar R. NCCN Guidelines Insights: Multiple Myeloma, Version 1.2020. J Natl Compr Cancer Netw (2019) 17:1154–65. doi: 10.6004/jnccn.2019.0049
11. Sonneveld P, Broijl A. Treatment of Relapsed and Refractory Multiple Myeloma. Haematologica (2016) 101:396–406. doi: 10.3324/haematol.2015.129189
12. Maude SL, Laetsch TW, Buechner J, Rives S, Boyer M, Bittencourt H, et al. Tisagenlecleucel in Children and Young Adults With B-Cell Lymphoblastic Leukemia. N Engl J Med (2018) 378:439–48. doi: 10.1056/NEJMoa1709866
13. Neelapu SS, Locke FL, Bartlett NL, Lekakis LJ, Miklos DB, Jacobson CA, et al. Axicabtagene Ciloleucel CAR T-Cell Therapy in Refractory Large B-Cell Lymphoma. N Engl J Med (2017) 377:2531–44. doi: 10.1056/NEJMoa1707447
14. Bcop A, Orozco P. Axicabtagene Ciloleucel: The First FDA-Approved CAR T-Cell Therapy for Relapsed/Refractory Large B-Cell Lymphoma. J Adv Pract Oncol (2019) 10:878–82. doi: 10.6004/jadpro.2019.10.8.9
15. Strohl WR, Naso M. Bispecific T-Cell Redirection Versus Chimeric Antigen Receptor (CAR)-T Cells as Approaches to Kill Cancer Cells. Antibodies (Basel) (2019) 8:41. doi: 10.3390/antib8030041
16. Zhang T, Cao L, Xie J, Shi N, Zhang Z, Luo Z, et al. Efficiency of CD19 Chimeric Antigen Receptor-Modified T Cells for Treatment of B Cell Malignancies in Phase I Clinical Trials: A Meta-Analysis. Oncotarget (2015) 6:33961–71. doi: 10.18632/oncotarget.5582
17. Berahovich R, Zhou H, Xu S, Wei Y, Guan J, Guan J, et al. CAR-T Cells Based on Novel BCMA Monoclonal Antibody Block Multiple Myeloma Cell Growth. Cancers (Basel) (2018) 10:323. doi: 10.3390/cancers10090323
18. Locke FL, Neelapu SS, Bartlett NL, Siddiqi T, Chavez JC, Hosing CM, et al. Phase 1 Results of ZUMA-1: A Multicenter Study of KTE-C19 Anti-CD19 CAR T Cell Therapy in Refractory Aggressive Lymphoma. Mol Ther (2017) 25:285–95. doi: 10.1016/j.ymthe.2016.10.020
19. Golubovskaya V, Zhou H, Li F, Valentine M, Sun J, Berahovich R, et al. Novel CD37, Humanized CD37 and Bi-Specific Humanized CD37-CD19 CAR-T Cells Specifically Target Lymphoma. Cancers (Basel) (2021) 13:981. doi: 10.3390/cancers13050981
20. Drent E, Groen RW, Noort WA, Themeli M, Lammerts van Bueren JJ, Parren PW, et al. Pre-Clinical Evaluation of CD38 Chimeric Antigen Receptor Engineered T Cells for the Treatment of Multiple Myeloma. Haematologica (2016) 101:616–25. doi: 10.3324/haematol.2015.137620
21. Drent E, Themeli M, Poels R, de Jong-Korlaar R, Yuan H, de Bruijn J, et al. A Rational Strategy for Reducing On-Target Off-Tumor Effects of CD38-Chimeric Antigen Receptors by Affinity Optimization. Mol Ther (2017) 25:1946–58. doi: 10.1016/j.ymthe.2017.04.024
22. Lamers CH, Willemsen R, van Elzakker P, van Steenbergen-Langeveld S, Broertjes M, Oosterwijk-Wakka J, et al. Immune Responses to Transgene and Retroviral Vector in Patients Treated With Ex Vivo-Engineered T Cells. Blood (2011) 117:72–82. doi: 10.1182/blood-2010-07-294520
23. Sommermeyer D, Hill T, Shamah SM, Salter AI, Chen Y, Mohler KM, et al. Fully Human CD19-Specific Chimeric Antigen Receptors for T-Cell Therapy. Leukemia (2017) 31:2191–9. doi: 10.1038/leu.2017.57
24. Lanitis E, Poussin M, Hagemann IS, Coukos G, Sandaltzopoulos R, Scholler N, et al. Redirected Antitumor Activity of Primary Human Lymphocytes Transduced With a Fully Human Anti-Mesothelin Chimeric Receptor. Mol Ther (2012) 20:633–43. doi: 10.1038/mt.2011.256
25. Mihara K, Yanagihara K, Takigahira M, Imai C, Kitanaka A, Takihara Y, et al. Activated T-Cell-Mediated Immunotherapy With a Chimeric Receptor Against CD38 in B-Cell Non-Hodgkin Lymphoma. J Immunother (2009) 32:737–43. doi: 10.1097/CJI.0b013e3181adaff1
26. Feinberg D, Paul B, Kang Y. The Promise of Chimeric Antigen Receptor (CAR) T Cell Therapy in Multiple Myeloma. Cell Immunol (2019) 345:103964. doi: 10.1016/j.cellimm.2019.103964
27. Gulla A, Anderson KC. Multiple Myeloma: The (R)Evolution of Current Therapy and a Glance Into Future. Haematologica (2020) 105:2358–67. doi: 10.3324/haematol.2020.247015
28. Davila ML, Riviere I, Wang X, Bartido S, Park J, Curran K, et al. Efficacy and Toxicity Management of 19-28z CAR T Cell Therapy in B Cell Acute Lymphoblastic Leukemia. Sci Transl Med (2014) 6:224ra25. doi: 10.1126/scitranslmed.3008226
29. de Weers M, Tai YT, van der Veer MS, Bakker JM, Vink T, Jacobs DC, et al. Daratumumab, a Novel Therapeutic Human CD38 Monoclonal Antibody, Induces Killing of Multiple Myeloma and Other Hematological Tumors. J Immunol (2011) 186:1840–8. doi: 10.4049/jimmunol.1003032
30. Mateos M-V, Dimopoulos MA, Cavo M, Suzuki K, Jakubowiak A, Knop S, et al. Daratumumab Plus Bortezomib, Melphalan, and Prednisone for Untreated Myeloma. New Engl J Med (2018) 378:519–29. doi: 10.1056/NEJMoa1714678
31. Dimopoulos MA, Oriol A, Nahi H, San-Miguel J, Bahlis NJ, Usmani SZ, et al. Daratumumab, Lenalidomide, and Dexamethasone for Multiple Myeloma. N Engl J Med (2016) 375:1319–31. doi: 10.1056/NEJMoa1607751
32. Ding L, Hu Y, Huang H. Novel Progresses of Chimeric Antigen Receptor (CAR) T Cell Therapy in Multiple Myeloma. Stem Cell Investig (2021) 8:1. doi: 10.21037/sci-2020-029
33. Drent E, Groen R, Noort W, Bueren J, Parren P, Kuball J, et al. CD38 Chimeric Antigen Receptor Engineered T Cells as Therapeutic Tools for Multiple Myeloma. Blood (2014) 124:4759. doi: 10.1182/blood.V124.21.4759.4759
34. Bhattacharyya J, Mihara K, Kitanaka A, Yanagihara K, Kubo T, Takei Y, et al. T-Cell Immunotherapy With a Chimeric Receptor Against CD38 Is Effective in Eradicating Chemotherapy-Resistant B-Cell Lymphoma Cells Overexpressing Survivin Induced by BMI-1. Blood Cancer J (2012) 2:e75. doi: 10.1038/bcj.2012.21
35. Rydzek J, Nerreter T, Peng H, Jutz S, Leitner J, Steinberger P, et al. Chimeric Antigen Receptor Library Screening Using a Novel NF-kappaB/NFAT Reporter Cell Platform. Mol Ther (2019) 27:287–99. doi: 10.1016/j.ymthe.2018.11.015
36. Dotti G, Gottschalk S, Savoldo B, Brenner MK. Design and Development of Therapies Using Chimeric Antigen Receptor-Expressing T Cells. Immunol Rev (2014) 257:107–26. doi: 10.1111/imr.12131
37. Turtle CJ, Hanafi LA, Berger C, Gooley TA, Cherian S, Hudecek M, et al. CD19 CAR-T Cells of Defined CD4+:CD8+ Composition in Adult B Cell ALL Patients. J Clin Invest (2016) 126:2123–38. doi: 10.1172/JCI85309
38. Tomuleasa C, Fuji S, Berce C, Onaciu A, Chira S, Petrushev B, et al. Chimeric Antigen Receptor T-Cells for the Treatment of B-Cell Acute Lymphoblastic Leukemia. Front Immunol (2018) 9:239. doi: 10.3389/fimmu.2018.00239
39. Rajabzadeh A, Rahbarizadeh F, Ahmadvand D, Kabir Salmani M, Hamidieh AA. A VHH-Based Anti-MUC1 Chimeric Antigen Receptor for Specific Retargeting of Human Primary T Cells to MUC1-Positive Cancer Cells. Cell J (2021) 22:502–13. doi: 10.22074/cellj.2021.6917
40. Li G, Boucher JC, Kotani H, Park K, Zhang Y, Shrestha B, et al. 4-1BB Enhancement of CAR T Function Requires NF-kappaB and TRAFs. JCI Insight (2018) 3:e121322. doi: 10.1172/jci.insight.121322
41. Textor A, Grunewald L, Anders K, Klaus A, Schwiebert S, Winkler A, et al. CD28 Co-Stimulus Achieves Superior CAR T Cell Effector Function Against Solid Tumors Than 4-1BB Co-Stimulus. Cancers (2021) 13:1050. doi: 10.3390/cancers13051050
42. Hombach AA, Rappl G, Abken H. Arming Cytokine-Induced Killer Cells With Chimeric Antigen Receptors: CD28 Outperforms Combined CD28-OX40 “Super-Stimulation”. Mol Ther (2013) 21:2268–77. doi: 10.1038/mt.2013.192
43. Mihara K, Bhattacharyya J, Kitanaka A, Yanagihara K, Kubo T, Takei Y, et al. T-Cell Immunotherapy With a Chimeric Receptor Against CD38 Is Effective in Eliminating Myeloma Cells. Leukemia (2012) 26:365–7. doi: 10.1038/leu.2011.205
44. Mihara K, Yanagihara K, Takigahira M, Ochiya T, Imai C, Kitanaka A, et al. Activated T Cell-Mediated Immunotherapy With a Chimeric Receptor Against CD38 in B-Cell Malignancies. Blood (2007) 110:2356. doi: 10.1182/blood.V110.11.2356.2356
45. Zhou R, Yazdanifar M, Roy LD, Whilding LM, Gavrill A, Maher J, et al. CAR T Cells Targeting the Tumor MUC1 Glycoprotein Reduce Triple-Negative Breast Cancer Growth. Front Immunol (2019) 10:1149. doi: 10.3389/fimmu.2019.01149
Keywords: chimeric antigen receptor, CAR-T, CD38, multiple myeloma, immunotherapy, xenograft
Citation: Li X, Feng Y, Shang F, Yu Z, Wang T, Zhang J, Song Z, Wang P, Shi B and Wang J (2021) Characterization of the Therapeutic Effects of Novel Chimeric Antigen Receptor T Cells Targeting CD38 on Multiple Myeloma. Front. Oncol. 11:703087. doi: 10.3389/fonc.2021.703087
Received: 30 April 2021; Accepted: 03 August 2021;
Published: 26 August 2021.
Edited by:
Qingxin Mu, University of Washington, United StatesReviewed by:
Caixia Li, The First Affiliated Hospital of Soochow University, ChinaCopyright © 2021 Li, Feng, Shang, Yu, Wang, Zhang, Song, Wang, Shi and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Bingjie Shi, c2hpYmluZ2ppZUBidWNtLmVkdS5jbg==; Jianxun Wang, Smlhbnh1bi5XYW5nQGJ1Y20uZWR1LmNu
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.