 Rihan Hai1,2
Rihan Hai1,2 Guang Shu
Guang Shu Gang Yin
Gang Yin
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oncol. , 29 July 2021
Sec. Pharmacology of Anti-Cancer Drugs
Volume 11 - 2021 | https://doi.org/10.3389/fonc.2021.700947
Over decades of studies, accumulating evidence has suggested that epigenetic dysregulation is a hallmark of tumours. Post-translational modifications of histones are involved in tumour pathogenesis and development mainly by influencing a broad range of physiological processes. Histone deacetylases (HDACs) and histone acetyltransferases (HATs) are pivotal epigenetic modulators that regulate dynamic processes in the acetylation of histones at lysine residues, thereby influencing transcription of oncogenes and tumour suppressor genes. Moreover, HDACs mediate the deacetylation process of many nonhistone proteins and thus orchestrate a host of pathological processes, such as tumour pathogenesis. In this review, we elucidate the functions of HDACs in cancer.
Epigenetics refers to molecular processes that regulate gene expression without altering the DNA sequence, including diverse molecular modifications of DNA and chromatin, such as histone acetylation and DNA methylation (1, 2). Epigenetics can turn on/off gene expression and thus plays a crucial role in tumorigenesis and cancer progression (3). Abnormal epigenetic alterations and destroyed epigenetic integrity are common characteristics of tumour cells (4). Among all epigenetic mechanisms, histone modifications have been shown to be important in carcinogenesis.
Histone modifications are catalysed by specific enzyme complexes through ATP-consuming processes, which in turn impact gene transcription, duplication, repair and recombination (4). Acetylation and methylation are involved in the regulation of amino-terminal (N-terminal) tail domains of core histones and affect the post-translational crosstalk between DNA and histones by recruiting proteins and transcription factors (5).
Histone acetylation involves reversible alterations of the N-terminal lysine on histones and is subsequently linked to different cellular processes and disease development (6, 7). Histone acetylation is accomplished by two enzymes: histone acetyltransferases (HATs) and histone deacetylases (HDACs or KDACs), which add or remove acetyl groups (8) (Figure 1). The existence of acetylated lysine in the histone tail causes the chromatin to be more open, allowing initiation of gene transcription. In contrast, deacetylation enhances the ionic interactions between histones, which have a positive charge, and DNA, which has a negative charge. Deacetylation is related to compacted chromatin, which is not conducive to gene transcription (9). These modifications show strong relationships to gene expression in normal and cancerous cells (10).
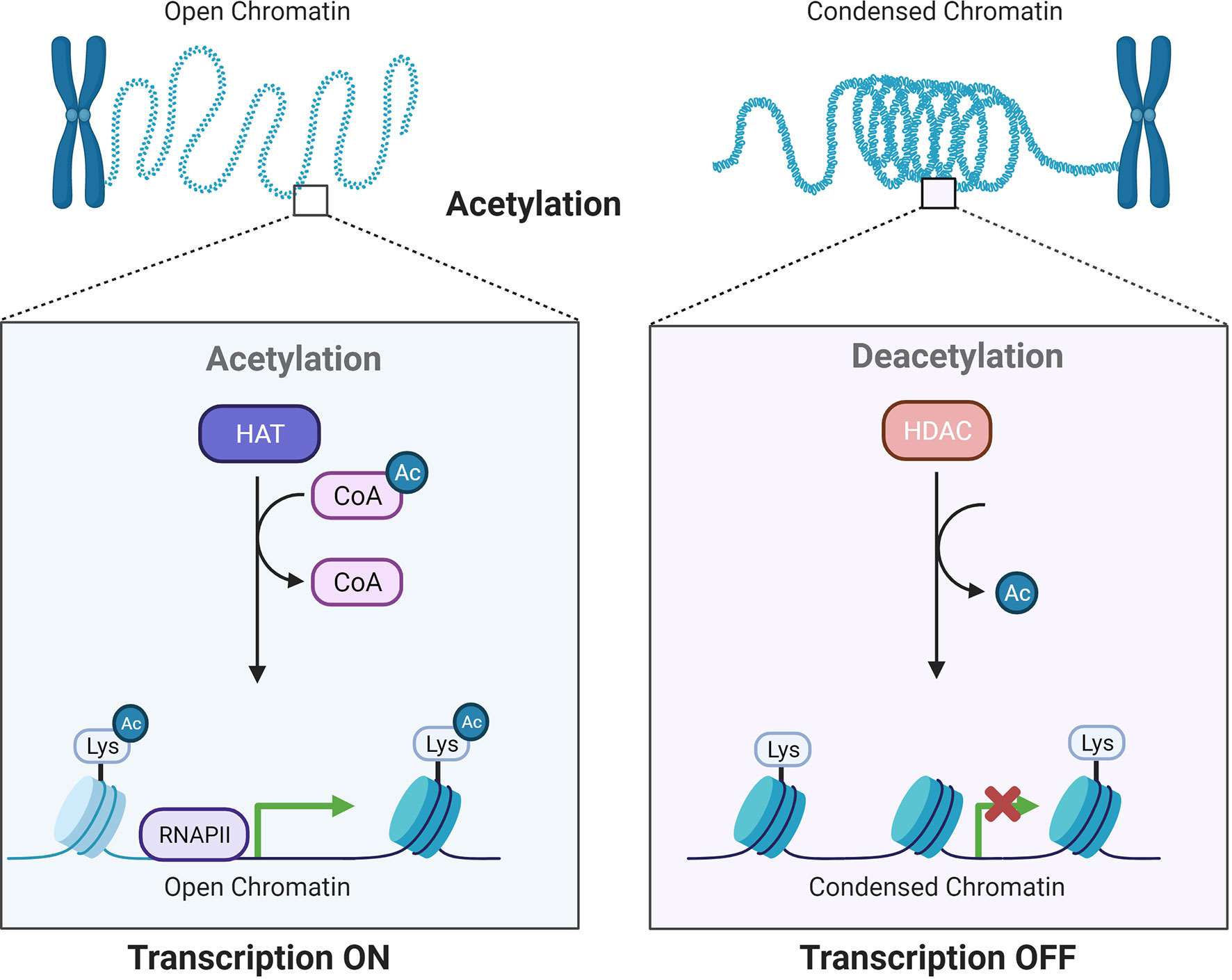
Figure 1 Acetylation by HATs and deacetylation by HDACs influence gene transcriptional activity. HATs and HDACs add or remove acetyl groups at the N−terminus lysine, which leads to an open or condensed state of the chromatin.
HDACs act as gene silencing complexes, and studies have suggested that HDACs suppress gene expression through transcription factors such as E2F1. Furthermore, evidence indicates that HDACs can eliminate acetylation of nonhistone proteins (11). HDACs inhibit the process by which T cells recognize and destroy tumour cells (12).
Human HDACs can be classified into four classes with 18 members (Figure 2). Class I and II HDACs were found to show high similarities in catalytic sites. Except for HDAC8, HDAC isoforms appear to combine with more proteins or more HDAC isoforms in multiprotein complexes (13, 14). Many types of HDACs are derived from nucleotide polymorphisms and optional splicing. Various isoforms of HDAC9 have been identified (15).

Figure 2 The domain structure of human HDACs. The basic domain structure of eighteen HDAC members. Class I, II, and IV HDACs contain Zn2+-dependent domains, while SIRTs contain NAD+-dependent domains. Nuclear localization sequences and zinc finger motifs in different HDACs are also shown.
These enzymes have been identified in the nucleus. The expression of class I HDACs is elevated in cancer cells. HDAC1 mainly shows oncogenic activity, but a dual role of HDAC1 was found in different stages of acute promyelocytic leukaemia (APL) (16). HDAC1 functions as a tumour suppressor by restraining the activity of PML-RAR in the preleukaemic stage of APL. Moreover, it exerts tumour-promoting activity in established tumour cells (16). This finding reveals the importance of identifying the role of HDACs at different stages in different cancer cell types for optimal treatment, such as personalized design of inhibitors.
HDAC1 and HDAC2 are involved in the deacetylation of p53, which results in enhanced regulation of target genes by p53 (17, 18). The interaction between HDAC3 and cancer-associated genes influences the angiogenic and carcinogenic potential of tumours and the efficacy of antitumour drug treatment (19). Furthermore, HDAC3 can catalyse the deacetylation of the Notch1 intracellular domain (NICD1), thereby promoting NICD1 protein stability, which is involved in the progression of T cell acute lymphoblastic leukaemia (ALL) and chronic lymphocytic leukaemia (CLL) (20). In addition, HDAC3 inhibits NF-κB lysine acetylation, thereby exerting a proinflammatory effect (21).
When the expression of HDAC1 is decreased, the levels of HDAC2 and HDAC3 are increased. However, highly expressed HDAC2 and HDAC3 are unable to compensate for the lack of HDAC1 (22).
HDAC8 is overexpressed in diverse cancer tissues, including colon, breast, lung, pancreatic, and liver cancers and childhood neuroblastoma (23). The distal region of HDAC8 allosterically regulates the activity of this enzyme (24). In addition, oxidoreduction reactions can modulate the activity of HDAC8 (25).
Homologous proteins of class I HDAC can form stable multiprotein complexes with other proteins. Over the years, several HDAC1/2 corepressor complexes, such as switch-independent protein 3A (SIN3A), mitotic deacetylase (MiDAC), nucleosome remodelling and deacetylase (NuRD), mesoderm induction early response (MIER), corepressor of REST (CoREST), and arginine-glutamic acid dipeptide repeats (RERE), have been identified (26, 27).
The structures of SIN3A and NuRD demonstrated the diversity of protein complex components, which are related to their function and context-specific cellular activity (28).
The SIN3 complex is composed of SIN3A/B, HDAC1/2, suppressor of defective silencing 3 (SDS3), and SIN3-associated protein p30 (SAP30) (27). Except for SIN3A/B, all corepressor proteins possess an ELM2–SANT domain. The SIN3A complex has more enzymatic activities based on its functional molecules (29, 30). The SIN3 complexes can also form a dimer (31). The acetylation of STAT3 and its interaction with SIN3A inhibit the expression of tumour suppressor genes (32). Nevertheless, the interaction pattern with the above modulators is still unclear.
Recently, PHF23, an H3K4me3 reader, was shown to directly bind the SIN3-HDAC complex and repress its deacetylation activity. Thus, the PHF23-SIN3-HDAC (PSH) complex consequently enhances the activation of downstream tumour suppressor genes (33). Aberrant PSH levels have a stimulatory role in chromosome 17p-deleted tumours.
The NuRD complex consists of chromodomain helicase DNA-binding proteins (CHD3/CHD4/CHD5), HDAC1/HDAC2, metastasis associated proteins (MTA1/MTA2/MTA3), methyl domain binding proteins (MBD2/MBD3), retinoblastoma binding proteins (RBBP4/RBBP7), and GATA zinc finger domain proteins (GATAD2B/GATAD2A) (34, 35), and all NuRD components showed high expression in tumour cells (36).
The NuRD complex is formed by six main protein subunits, which have some functional differences (37). The NuRD complex modulates the process by which different cells read DNA, which can trigger pluripotency in stem cells and is involved in the transformation of adult cells to induced pluripotent stem cells (37, 38). The NuRD complex plays important roles in diverse malignant phenotypes in hepatocellular carcinoma (HCC) cells (36).
Class II HDACs have different effects in different tissues (39). HDAC4, HDAC5 and HDAC7 alter cell differentiation on the basis of certain signals and thus lead to changes in the gene expression status. HDAC4, HDAC5, HDAC7, and HDAC9 (class IIa members) are encoded by various genes (40). The catalytic domains of HDAC6 and HDAC10 (class IIb members) show similarity with those of HDAC9. Furthermore, HDAC9 was reported to have splice variants (26). In addition, phosphorylation of class IIa enzymes is associated with their localization and activities in the nucleus (41). Compared to that of other HDACs, the histone deacetylating activity of class IIa HDACs is low. However, class IIa HDACs exert enzymatic activity when forming protein complexes with SMRT/N-CoR (42). A recent study reported that class IIa HDACs are directly involved in lung vascular barrier disruption (43). Class IIa HDACs may also regulate the endothelial cell barrier through deacetylation, since HDAC7 is closely related to cytoskeletal processes (43).
Class IIa HDACs repress transcriptional processes in diverse tissues. In addition to acting as transcription repressors, class II HDACs are associated with autophagic progression and cytoskeleton microtubules (44). For example, HDAC6 can act as a α-tubulin deacetylase and participate in multiple cytoplasmic pathways related to microtubules (45, 46), revealing that it may be an important therapeutic for treating Alzheimer’s disease and cancer (47). HDAC4 and HDAC9 contain different common genomic binding sites. However, HDAC4 binds additional sites that may escape modulation by HDAC9 (48). HDAC9 inhibits cardiomyocyte hypertrophy and skeletal muscle differentiation. HDAC4 suppresses the activity of Runx2, thereby repressing chondrocyte hypertrophy and endochondral bone formation (49). The expression of HDAC5 and lysine-specific demethylase 1 (LSD1) was elevated in breast cancer, and HDAC5 enhanced the LSD1 protein stability and reduced the nuclear level of H3K4me1/me2 in breast cancer cells (50).
To date, seven Sirtuins have been identified. Sirtuins contain catalytic domains, which function in a nicotinamide adenine dinucleotide (NAD+)-dependent manner in transcriptional processes. Sirtuins have also been reported to have lysine defatty-acylase activity (51).
SIRT1 was shown to have a dual role in tumour cell growth (52, 53). This molecule acts as a positive regulator of proteins involved in tumour suppressor pathways or DNA damage repair (54). SIRT1 also has a negative effect on tumorigenesis since it can decrease oncogene transcriptional activity through interaction and deacetylation of c-MYC (52, 55). SIRT2 was reported to exert tumour repressing effects. The deletion of SIRT2 disrupted the checkpoints of the cell cycle and led to enhanced tumorigenesis (56, 57). In addition to mitosis, SIRT2 can regulate genome integrity (58). SIRT3 was reported to modulate transcription factors in breast cancers (59). In lung cancer cells, SIRT4 suppressed cell proliferation, invasion and migration (60). In liver cancer, SIRT5 enhanced tumour cell proliferation (61). In retinoblastoma, SIRT6 was shown to be a cancer suppressor protein and function by restraining the metabolism of cells (62). SIRT6 can stabilize the normal genome of neighbouring cells (63). Moreover, SIRT7 was observed to target H3 histones with high specificity and recruit RNA polymerase I (64, 65).
Located in the N-terminal tail, the catalytic domain of HDAC11 is very similar to that of HDAC3 and HDAC8 (66). Similar to Sirtuins, HDAC11 also possesses acylase activity. Determination of defatty-acylase activity is considered an alternative method of identifying HDACs, and zinc-dependent HDACs showed notable differences in activity (67, 68). HDAC11 participates in oligodendrocyte progression and promotes oligodendrocyte differentiation (69). This molecule is also involved in immune responses by decreasing IL-10 levels (68). A recent study found that HDAC11 can promote the malignant phenotypes of JAK2-driven myeloproliferative neoplasms (70).
HDACs are involved in various stages of cancer (71). Highly expressed HDACs are usually associated with terminal illness and inferior outcomes of patients. For instance, upregulation of HDAC1, HDAC2, and HDAC3 expression correlated with worse survival in patients with gastric and ovarian tumours (72), and elevated levels of HDAC8 expression in neuroblastoma were related to advanced disease and negative outcomes (73, 74).
Notably, the HDAC expression level may not be a prerequisite of their functions in cancer, since abnormal activities of HDACs in cancer progression are common (75). Furthermore, some HDAC families serve as subunits of large protein complexes and can promote carcinogenesis (76, 77).
Research has shown that histone deacetylase activity is not generally necessary for gene expression. The acetyltransferase activity of these molecules cannot recruit p300/CBP and transcription factors but can facilitate the recruitment of TFIID and RNAPII at virtually all enhancers and enhancer-regulated genes (78). Histone acetylation promotes transcription of paused genes through release of Pol II into elongation (79). Research has shown that global histone acetylation depends on ongoing transcription. For instance, Wang and his colleagues have found that in K562 cells, transcription inhibition leads to rapid loss of H3K27ac from enhancers and promoters. Hos2 histone deacetylase primarily interacts with genes with high genome-wide activity and catalyses deacetylation of lysines in the H3 and H4 histone tails (80). Therefore, whether HDACs have context-specific rather than general functions in modulation of gene expression requires further exploration.
HDAC inhibitors (HDACis) were shown to exert antitumour effects through various mechanisms in several cancer cell lines (81). Different HDACis show distinctive mechanisms in influencing cell growth, apoptosis, migration, and angiogenesis (82, 83). HDACis can be classified into four main types according to their structures: short-chain fatty acids, hydroxamic acids, cyclic peptides, and benzamides (84). In recent years, four HDACis have been officially approved by the FDA for clinical therapy of T-cell lymphoma (TCL) and multiple myeloma: vorinostat, romidepsin, belinostat and panobinostat (11, 85). For example, panobinostat causes G2/M cell cycle arrest and apoptosis due to suppression of HDAC3 and HDAC6 and thus leads to degradation of Aurora A and B kinases (86). Panobinostat also enhanced the expression of CDH1 and restrained epithelial-mesenchymal transition (EMT) in triple-negative breast cancer (87). Entinostat leads to low Bcl-XL expression and promotes the apoptotic process of tumours. Entinostat was also shown to cause G1 cell cycle arrest by upregulating p21 expression (88).
Generally, the anticancer activity of HDACis indicates the tumour-promoting effects of HDACs. However, genetic inactivation of HDACs might play a tumorigenic role. HDAC1 somatic mutations and HDAC4 homozygous deletions were observed in 8.3% of dedifferentiated liposarcomas (89) and 4% of melanomas (90). In addition, HDAC2 has been shown to exert an anticancer effect in vitro and in vivo. Class II HDACs may also serve as cancer suppressors in specific cellular settings. HDAC6 showed low expression in liver cancer cells and liver transplantation patients. Furthermore, the status of HDAC6 is related to poor prognosis of disease. For class III HDACs, a SIRT6 mutation, which is a loss-of-function mutation, was observed in tumour cells and promoted tumour formation (91).
HDACs are involved in tumour pathogenesis and progression by deacetylating histone and nonhistone proteins that participate in the modulation of multiple tumorigenic pathways (92) (Figures 3, 4 and Table 1).
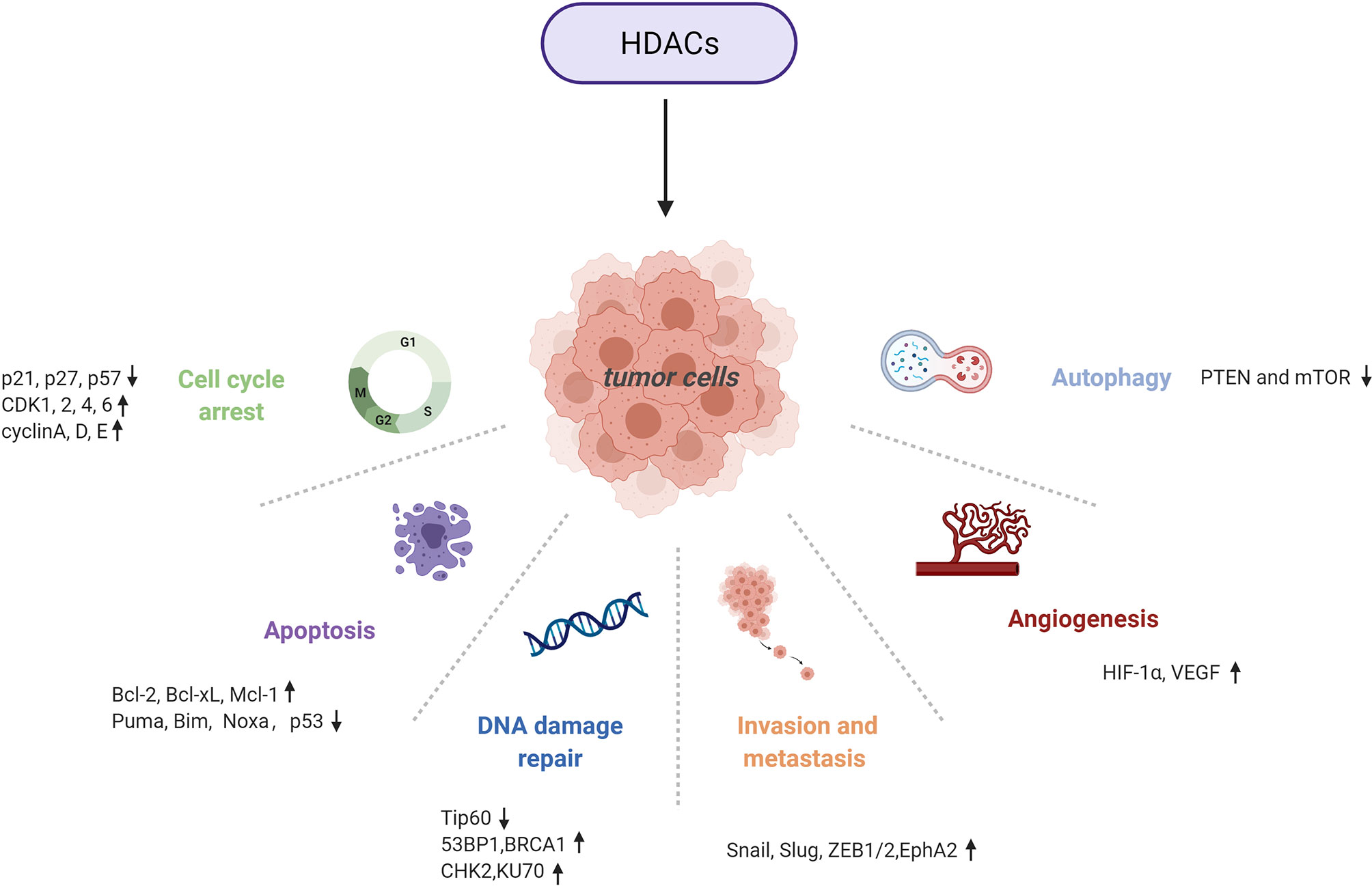
Figure 3 Multiple tumorigenic pathways activated by HDACs. An overview of HDAC-involved tumorigenic processes, including the cell cycle, apoptosis, DNA damage repair, metastasis, angiogenesis, and autophagy.
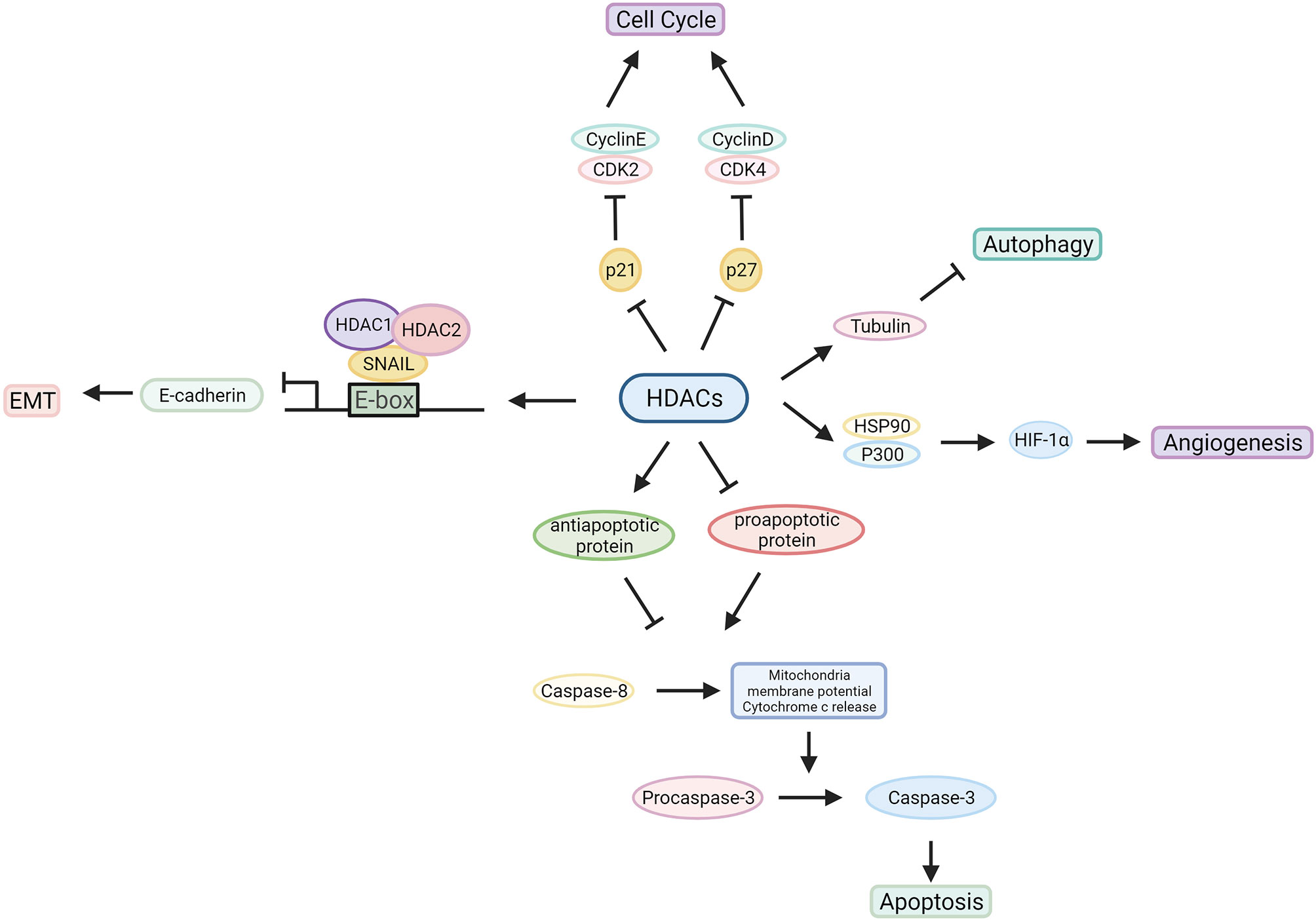
Figure 4 Characterized mechanism of HDACs in cancer development. HDACs influence the tumorigenic process by regulating oncogene and tumour suppressor gene expression or interacting with related proteins.
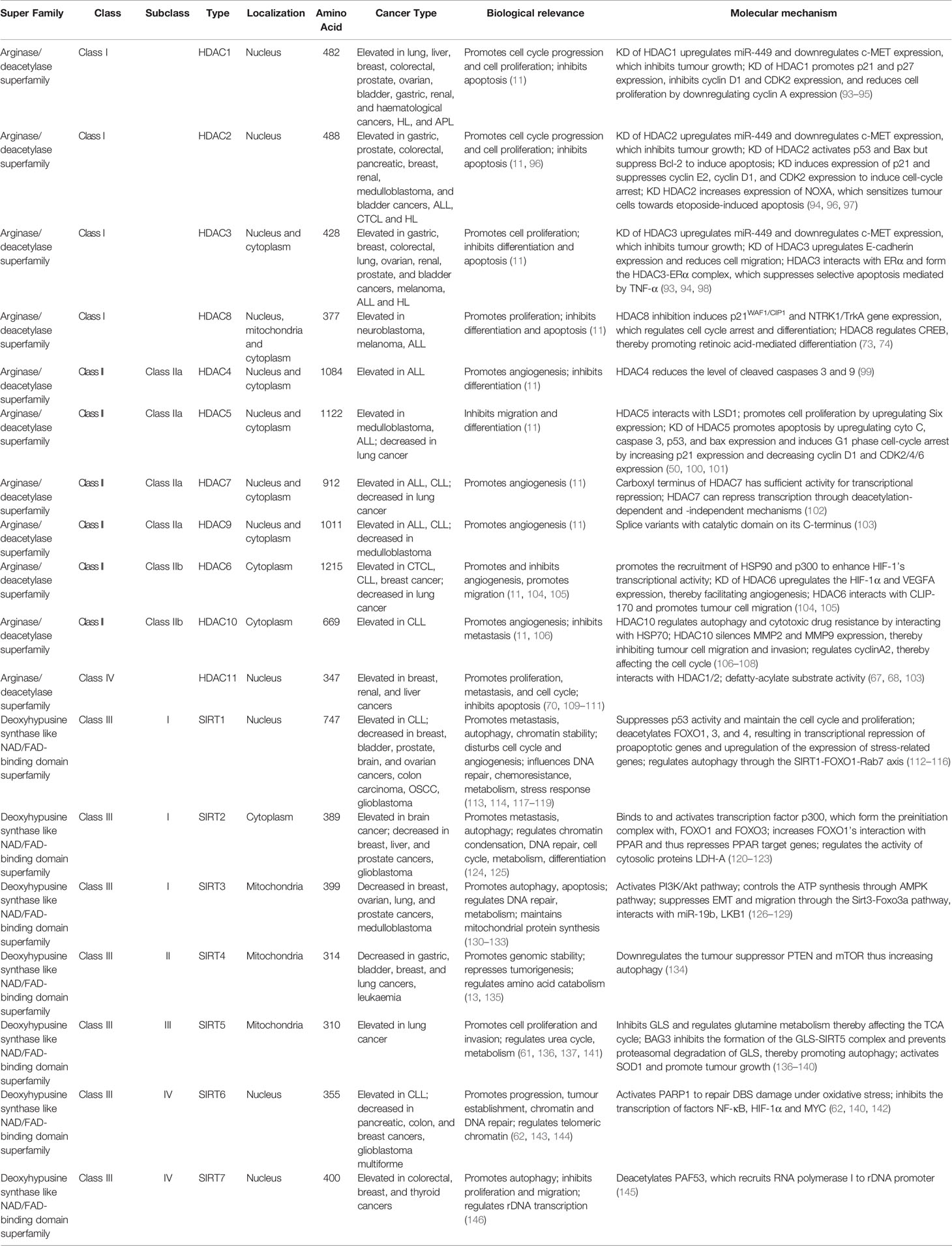
Table 1 Summary of histone deacetylases.
HDACs facilitate the stage-specific development of cancers; for example, HDAC1 reduces cell cycle suppressors by interacting with Rb and influencing E2F1 activity (9). Therefore, HDACis can impede the transition from G1 to S phase by recurring Rb activity via dephosphorylation and inhibition of E2F1 activities (147). HDAC inhibition also exerts anticancer effects by blocking the cell cycle, which is achieved by inducing high expression of cyclin-dependent kinase (CDK) inhibitors or low expression of cyclins and CDKs (148). HDAC1 and HDAC2 can bind to genes such as p21WAF1/CIP1 and p27KIP1, resulting in suppression of their expression (149). Silencing p21 and p27 (CDK inhibitors) led to facilitated cell proliferation. Thus, inhibition of HDAC1 and HDAC2 promotes cell cycle arrest at G1 phase (148).
Along with HDAC1’s effect on the G1/S transition, it is also involved in the G2/M transition. Knocking down HDAC1 in tumour cells partially contributed to G2/M phase arrest (150). Furthermore, in adult neural stem/progenitor cells, HDAC3 was reported to modulate the G2/M transition through alteration of CDK1 expression (151). In addition, HDAC3 contributes to cell progression and proliferation by modulating the signal transducer and activator of transcription3 signalling pathway in liver cancers (152). Moreover, HDAC10 participates in G2/M transition by regulating the transcription of cyclin A2 protein depending on let-7 and HMGA2 (153). HDAC10 plays a crucial role in unchecked cell progression, since deletion of HDAC10 induces blockade at mitotic entry and thus restrains cell proliferation in human lung cancers (154). High expression of Sp1 induced by HDAC1/2/6 facilitates cell division of malignant cells by enhancing BMI1 and hTERT and, more importantly, G2/M progression (155). Furthermore, an HDAC6 inhibitor can lead to G2/M arrest in temozolomide-resistant cells (155).
Members of class III and class IV HDACs also play roles in cell cycle regulation. SIRT1 can disturb the cell cycle by acting on p53 and blocking all p53-dependent pathways (156). HDAC11 was shown to have a negative effect on E2F7 and E2F8, cell cycle suppressors, thus contributing to cancer cell survival within lymph nodes (109).
HDACis can block the cell cycle at G1/S and G2/M phases, which is similar to the gene knockdown results, confirming multiple effects of HDACs during the cell cycle. These results support the promising role of HDACs as targets for treating aberrant tumour cell growth and proliferation.
HDACs participate in the extrinsic and intrinsic pathways of apoptosis. For the extrinsic pathway, HDACs can block TRAIL- or TGF-β-mediated pathways (157). For the intrinsic pathway, the HDAC family alters pro- and antiapoptotic proteins. HDACs can inhibit proapoptotic Bcl-2 proteins, such as NOXA and BAX (158, 159), via direct acetylation or by nonhistone protein KU70 modification (160). Moreover, HDACs can promote apoptosis in glioblastoma and other cancer types (161).
HDACi treatment can promote cell apoptosis by the extrinsic/intrinsic pathway and by improving the sensitivity of tumour cells (162). In addition, some HDACis have been assessed in preclinical cancer models. Vorinostat and panobinostat, which are nonselective HDACis, inhibited FLIP expression in a c-MYC-mediated manner (163). By suppressing HDAC1 and HDAC2, HDACis impeded the growth of prostate cancer (164). HDACis successfully reduced the level of FLIP and enhanced caspase-8 activity in non-small cell lung cancer (NSCLC). Under HDACis, cells are sensitive to activators such as TRAIL, thereby promoting apoptosis (165). Furthermore, HDAC2 depletion could help sensitize pancreatic cancer cells to TRAIL-induced apoptosis by upregulating the expression of TRAIL receptor DR5 (TRAIL-R2) (166). Regarding the intrinsic pathway, HDAC inhibition could promote apoptosis by downregulating the expression of Bcl-2 proteins, such as Bcl-2, Bcl-xL and Mcl-1 (161), while upregulating the expression of proapoptotic proteins (167), which include Puma, Bim, and Noxa (168).
Professor Wang found that HDAC1, HDAC2 and HDAC3 are related to microcystin-leucine arginine (MC-LR)-induced apoptosis in SD rat testicular cells. Specifically, HDACs were activated by MC-LR and subsequently reduced the acetylation state of normal testicular cells, resulting in cell cycle abnormalities and consequently cell apoptosis. These researchers also reported that TSA could re-establish MC-LR-induced apoptosis and cell cycle arrest (169).
Overexpression of HDAC1, HDAC2 and HDAC8 has been demonstrated to downregulate the expression of p53, a tumour suppressor gene that participates in cell apoptosis, thus resulting in the inhibition of apoptosis (170–172). Lnc-Ip53, a lncRNA that can be transactivated by p53, was reported to interact with HDAC1 and p300, thereby restraining p53 acetylation, reducing p53 activity and subsequently inhibiting apoptosis (173). HDACis, like TSA, stabilized the acetylation caused by p53 and increased PUMA expression by promoting the binding of p53 to the PUMA promoter, thus abrogating resistance to DNA damage-induced cell death in renal cancer (155, 174). Along with HDAC1, HDAC2 and HDAC8 (50), HDAC3 can interact with oestrogen receptor-α (ERα) and form the HDAC3-ERα complex, which suppressed selective apoptosis mediated by TNF-α in MCF-7 cells, and the process is dependent on caspase-7 (98).
Other publications also reported the function of p53 in HDACi-mediated apoptosis. The HDACi-induced process includes activating p53, whereas p53 was not found to be a prerequisite for anticancer processes (175). A majority of studies have shown that HDACis function without p53 since their anticancer effects do not fluctuate with cell p53 status (176). Despite this, other studies have concluded that p53 plays a crucial role in inducing tumour cells in response to HDACi treatment. To test this hypothesis, scientists have generated isogenic HCT-116 colon tumour cells with different p53 statuses. Vorinostat, apicidin, and VPA can function efficiently regardless of the level of p53. Nevertheless, entinostat is influenced by p53 status to a certain degree (175). This molecule can inhibit [FADD]-like interleukin-1 β-converting enzyme inhibitory protein [FLIP(L)] and thus promote p53-induced apoptosis (177).
Moreover, HDAC1 and HDAC8 simultaneously repress BMF. The HDAC8 inhibitor methylselenopyruvate was found to promote the transcription of BMF and apoptosis induced by BMF in colon cancer cells (178). Recently, HDAC11 was shown to be important for the proliferation of oncogenic JAK2-driven myeloproliferative neoplasms, and downregulation of HDAC11 expression could promote apoptosis in human leukaemia (70).
A growing body of research has shown that HDACs are involved in DNA damage repair (DDR) responses. HDACs serve as protective agents against DNA damage due to their necessary role in remodelling chromatin and balancing the acetylation of proteins associated with DNA damage (179).
HDAC1, along with HDAC2, binds to DNA damage regions to deacetylate histone proteins at H3K56 and H4K16 and accelerates nonhomologous end joining (NHEJ) pathways, thereby promoting double-strand break (DSB) repair (180). For HDAC3, inactivating HDAC3 leads to genomic instability, and HDAC3 deficiency in the liver contributes to HCC (181). More specifically, HDAC3 deacetylase acts on and stabilizes Tip60, thereby reducing apoptosis triggered by DNA damage (182). Furthermore, HDAC inhibition blocks DSB repair and sensitizes tumour cells to ionizing radiation and DNA damage agents.
HDAC3 was also reported to participate in nucleotide excision repair (NER). HDAC3 acts on H3K14 after ultraviolet irradiation and promotes XPC recruitment to DNA-damaged sites, thereby exerting a positive effect on the global genomic NER (183).
Several class II HDACs are involved in DNA damage repair. For example, HDAC4 and 53BP1 colocalize in nuclear foci after DSBs. Knocking down HDAC4 resulted in low levels of 53BP1 and inactivation of the DNA damage-induced G2 checkpoint (184). HDAC9 and HDAC10 were shown to play roles in homologous recombination (153, 185). Furthermore, Doctor Zhang reported that the DNA mismatch repair protein MutS protein homologue 2 (MSH2) is associated with class IIb HDACs. Specifically, HDAC6 functions as an MSH2 inhibitor via deacetylation and ubiquitination (186).
Sirtuins interact with many DDR proteins, such as Ku70, NBS1, hMOF, WRN, APE1, XPA, PARP-1, TopBP1, and KAP1, thus regulating several DDR pathways (187). In tumour cells, SIRT1 was reported to prevent p53 acetylation and thus promote cell survival after DNA damage (156, 188). Therefore, knocking down SIRT1 exerts positive feedback in cancer treatment, revealing that SIRT1 is an potential target for cancer treatment. However, when cancer cells suffer severe damage, SIRT1 is repressed due to phosphorylation at Ser682 by the DNA damage-responsive kinase HIPK2 (189). This process results in cell death under severe DNA damage. SIRT1 plays a crucial role in supporting genome integrity and stability (190). Thus, enhancing SIRT1 in this manner may be a potential therapeutic strategy.
SIRT6 also plays a notable role in DNA repair. This molecule was first reported to abrogate genome instability through the alteration of DNA base excision repair. SIRT6 can be directly recruited to DNA damage sites and enhance mono-ADP-ribosylation of PARP1, thereby facilitating DSB repair (143), which is achieved through phosphorylation of SIRT6 by JNK (191). To suppress genomic instability, SIRT6 recruits and deacetylates the ISWI-chromatin remodeler SNF2H at histone H3K56 (192). A recent study showed that inhibition of HDAC8 or SIRT6 induces DNA repair deficiencies in homologous recombination and NHEJ pathways in leukaemia-initiating cells, and such DNA repair deficiencies are synergistic with nicotinamide phosphoribosyl transferase (NAMPT) targeting (193). Overall, we found that some class III HDAC members, due to their deacetylase activity, are essential in the DNA damage response.
EMT is an important step in tumour cell invasion and metastasis. Many studies have shown that HDACs can regulate EMT in diverse cancer types. The most important feature of EMT is downregulated E-cadherin (encoded by CDH1) expression. Some CDH1 transcriptional inhibitors, such as Snail, Slug, Twist, and ZEB1/2, have been identified (194–196).
HDACs bind to the CDH1 promoter and deacetylate H3 and H4 histones. In pancreatic cancer, the Snail/HDAC1/2 complex was shown to repress CDH1, causing a reduction in E-cadherin expression and subsequently inducing EMT (197). The complex mentioned above was also found to can interact with EZH2 and cause CDH1 silencing (198). Additional studies reported that ZEB1 induces the binding of HDACs with the CHD1 promoter in human pancreatic cancers (199). Furthermore, ZEB1 and HDACs can modify the splicing of CDH1 exon 11. Therefore, the reduction in CDH1 expression is a comprehensive outcome caused by transcriptional inhibition and abnormal splicing (200). Mocetinostat, an HDACi, efficiently reduced the expression of ZEB1 in pancreatic tumour cells (87), revealing a therapeutic effect of class I HDACis in treating EMT and metastasis of cancers.
A recent study demonstrated that class I HDACs are related to maspin repression, which is often detected in prostate cancer. Inhibiting class I HDACs facilitated maspin re-expression, thus suppressing the proliferation and migration of prostate tumour cells (201). In addition, in colorectal cancers, HDAC3 was recruited to Runx 2 and repressed metastasis (202).
HDAC7 was reported to enhance EphA2 expression by downregulating miR-4465 expressing, showing a positive effect on tumour proliferation, migration, and invasion in nasopharyngeal carcinoma (NPC) (203). HDAC11 can upregulate the expression of RRM2, a gene associated with promigratory and metastatic phenotypes in diverse cancers, thus promoting metastasis of tumour cells (109).
The function of SIRT1 in the EMT process is associated with tumour types. In prostate cancer cells, SIRT1 and ZEB1 simultaneously bind to CDH1, thereby silencing transcription, which results in metastasis (204). In addition, the MPP8-SIRT1 interaction is considered to play a crucial role in CDH1 repression in prostate cancer cells (205). In melanoma, SIRT1 can deacetylate Beclin-1, thereby leading to E-cadherin in autophagy, and a low level of E-cadherin results in an enhanced EMT process (206). In colorectal cancer, EMT is activated by SIRT1 by upregulating Fra-1 expression (207). However, SIRT1 was suggested to reduce metastasis through deacetylation of Smad4 and repression of TGF-β-triggered signalling. The latter could affect matrix metalloproteinase-7 (MMP-7) in breast and oral cancer cells (208, 209). SIRT2 was reported to deacetylate Slug and stabilize its protein to enhance EMT (210). Thus, Sirtuins play important roles in metastasis.
Angiogenesis is critical in tumour growth and metastasis. Hypoxia or a hypoxic microenvironment enables the initial stage of angiogenesis, and its process is mainly controlled by hypoxia-inducible factor 1α (HIF-1α). HDACs such as HDAC1 function explicitly as deacetylase enzymes of HIF-1α, thereby preventing HIF-1α degradation. Abnormal expression of HDAC1 leads to overexpression of HIF-1α and VEGF in tumours, which subsequently promotes angiogenesis (211). Consistent with this conclusion, HDACis can lead to HIF-1 and VEGF degradation and repression (211). HDAC4, HDAC6, HDAC10, and SIRTs show similar mechanisms, as they promote angiogenesis by enhancing VEGF, EGF, and HIF 1α levels (157).
HDAC4, HDAC5, and HDAC6 act as mediators of HIF-1 activity by promoting the recruitment of obligatory cofactors, such as HSP90 and p300, thereby enhancing its transcriptional activity (212). Moreover, SIRT1 was found to play an opposite role, since it deacetylases HIF-1α and prevents HIF-1α from interacting with p300, resulting in a reduction in HIF-1α activity. SIRT1 inhibition in the hypoxic microenvironment maintains high HIF-1 activity (213).
However, HDAC5 and HDAC6 also have antiangiogenic roles since they can deacetylate HIF-1a chaperones, namely, HSP70 and HSP90. Suppressing HDAC5 and HDAC6 causes substantial acetylation of these proteins, and thus, HIF-1 is in an immature form and easily degrades (214). In endothelial cells, HDAC5 inhibits the expression of proangiogenic genes, including FGF2 or Slit2 (215). HDAC5 can suppress cysteine-rich angiogenic inducer 61 (CYR-61), which is an antifibrotic and proangiogenic mediator, to repress angiogenesis (216). HDAC6 also has a positive effect on angiogenesis through deacetylation of the actin-remodelling protein cortactin and thus induces migration and sprouting in endothelial cells (217).
Overall, HDACs function by regulating various pro- and antiangiogenic proteins in angiogenesis, revealing that they may be promising targets for cancer therapy. HDAC inhibition is considered to exert antiangiogenic effects by downregulating the expression of proangiogenic genes.
Autophagy exerts a dual role in tumorigenesis. Autophagy can eliminate damaged subcellular fractions, thereby preventing the transformation of normal cells into tumour cells (218). Therefore, the deletion of autophagic proteins was found to facilitate tumorigenesis. However, autophagy also promotes survival of cancer cells under metabolic stress, which may result in resistance to anticancer treatment (219).
Many HDAC enzymes show dual effects in the autophagic process. Class I type HDACs are thought to be conducive to autophagic flux in mice (220). Knocking down HDAC1 and HDAC2 was reported to impede autophagic flux (221). HDAC2 can directly function on the SNARE domain of syntaxin 17 (STX17) during autophagy. Deacetylated STX17 interacts with SNAP29 and HOPS, thus promoting the fusion of autophagosomes with lysosomes (222).
HDAC4 and HDAC5 are considered to alter autophagic flux while functioning as positive regulators of tumour cell growth. HDAC6 is needed to clear misfolded proteins by inducing autophagy (66) and enhances autophagy through its relationship with microtubule proteins (223). HDAC6 possesses a ubiquitin-binding domain, which is involved in responding to cytotoxic protein aggregates (224). In neurodegenerative diseases and cerebral ischaemia, HDAC6 plays an intermediary role between autophagy and the ubiquitin proteasome system (UPS). Specifically, autophagy can be strongly promoted and serve a compensatory role for HDAC6 under UPS damage (225, 226). HDAC6 plays an essential role in ubiquitin-selective quality control (QC) autophagy, rather than starvation-induced autophagy (227), in HDAC6 knockout mouse embryonic fibroblasts (MEFs). QC autophagy is reasonably distinct from starvation-induced autophagy due to the participation of ubiquitinated substrates and ubiquitin-binding HDAC6 and p62 (228). Furthermore, mitochondria have a selective method of elimination similar to autophagy. Specifically, parkin-mediated mitochondrial ubiquitination can also recruit the autophagic components HDAC6 and p62, which facilitate autophagic initiation and progression in impaired mitochondria (228). Despite p62’s passive role in autophagy as a substrate receptor, a recent study indicated that it can also act positively as an autophagic suppressor by promoting HDAC6 expression and subsequently recruiting deacetylated α-tubulin and unstable microtubules, resulting in dysfunctional autophagic flux, in prostate cancer cells (229). In neuroblastoma, loss of HDAC10 caused autophagosome/lysosome fusion blockade and autophagic flux arrest, contributing to enhanced cell sensitivity to chemotherapy (106). HDAC10 can also deacetylate HSP70 protein families, which may be linked to autophagy-mediated cell survival (106). Thus, class IIb HDACs are likely to primarily modulate autophagic flux by influencing autophagosome-autolysosome fusion.
Sirtuins are also involved in the regulation of the autophagic process. SIRT1 showed different effects in different cell lines and has a dual function in autophagy (230). SIRT1 is required to trigger starvation-induced autophagy since it affects Atg5, Atg7, Atg8, and LC3, essential members of the autophagic process (231, 232). Moreover, forkhead box O3 (FOXO3) can be deacetylated by SIRT1 and subsequently upregulates the expression of numerous autophagic genes. Additional studies have revealed that FOXK1/2, counterbalancing factors of FOXO3, recruit SIN3A-HDAC complexes to interfere with the acetylation of histone H4 and the expression of crucial autophagic genes (233). In addition, SIRT1 was shown to influence the PI3K/Beclin 1 and mTOR pathways in embryonic stem cells (ESCs), which in turn affects oxidative stress-induced autophagy (234). Unlike SIRT1, SIRT3 and SIRT5 were reported to participate in not only late but also early autophagic regulation (230). SIRT3 can act on mitochondria and trigger clearance under oxidative stress or starvation conditions (235). SIRT3 can also regulate the phosphorylation of activated protein kinase (AMPK) to enhance autophagy (236, 237). SIRT5 deacetylates lactate dehydrogenase B (LDHB) and increases its enzymatic activity. Protons (H) generated by LDHB contribute to autophagy in cancer cells (238). SIRT5 was also reported to participate in ammonia-induced autophagy, which is achieved through alteration of glutamine metabolism (136). SIRT2 dissociates from FOXO1 under stress conditions and drives the latter into a hyperacetylated state, which facilitates the autophagic process (136). SIRT6 was also reported to induce autophagy by restricting the transcription of the transcriptional repressor Nkx3.2, thus enhancing GATA5 expression (239).
In summary, the HDAC context-dependent functions in autophagic processes contribute to cancer treatment by targeted therapy.
HDAC3 is involved in lipopolysaccharide (LPS)-directed cytokine secretion in monocytes and M1 macrophages. HDAC3 disrupts the process by which inflammation-activated M1 macrophages mediate LPS tolerance (240).
Inhibition of HDAC mediates tumour-associated macrophages to specify the antitumour phenotype, resulting in decreased immune suppression and increased antitumour immune responses (241). Retinoic acid-related orphan receptor α (RORα)/HDAC-directed inhibition of NF-κB signalling modulates cholesterol metabolism in CD8+ T cells, which may provide a new therapeutic target in cancers such as colon tumours (242). Research has shown that suppression of HDACs is involved in restoration of IFN signalling, leading to enhancement of long-term antitumour immunity and repression of prostate tumour growth in bone (243). Moreover, Tcf1-instrinsic HDAC activity participates in suppressing excessive CTLA4 induction in protein immunization-elicited T cells, therefore protecting B-cell functions (244).
Suppression of HDACs is involved in regulating infiltrating macrophages and repressing the trafficking of myeloid-derived suppressor cells into tumours, thereby enhancing T cell activation in the tumour microenvironment (241). Suppression of HDACs plays an antitumour role in many cancers, such as pancreatic tumours, colorectal tumours and NSCLC, which can mediate tumour microenvironment changes, therefore enhancing the antitumour function of anti-PD-1 antibodies (245). Inhibition of HDACs can promote the expression of antigen-presenting machinery genes and CTL infiltration. Research has shown that HDACs are involved in regulating the tumour microenvironment, therefore regulating the immunotherapy response (246). Anne and her colleagues found that HDAC is involved in the transition of the tumour microenvironment from “cold” to “hot”, thereby inhibiting immune checkpoint blockade therapies (247).
Over the years, HDACs have been intensively investigated. To date, HDACs have been declared a key driver of cancers by modulating the dynamic acetylation of histone and nonhistone cellular substrates. As important regulators of histone acetylation clearance, HDACs exhibit abnormal expression and functions in cancer, indicating they are promising targets. Nevertheless, the precise mechanism of HDACs as tumorigenic agents is still worth studying. In many types of tumours knocking out HDACs was shown to lead to cell cycle arrest or apoptosis. In addition, the antitumour effect of HDACs was detected in a specific cellular setting. Selective HDACis serve as effective tools in elucidating the roles of HDACs. Moreover, additional studies are needed to systematically determine how each HDAC functions in specific conditions.
RH wrote the manuscript. LH completed the English revision. GY provided constructive feedback and guidance. GS completed critical revisions and proofread the manuscript. All authors contributed to the article and approved the submitted version.
This work was supported by the National Natural Science Foundation of China under Grant Number 81572900, The Fundamental Research Funds for the Central Universities of Central South University under Grant Number 2020105330104, and the National Key R&D Program of China, Stem Cell, and Translation Research under Grant Number 2016YFA0102000.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
AMPK, activated protein kinase; APL, acute promyelocytic leukaemia; Bcl-2, B-cell lymphoma-2; CDK, cyclin-dependent kinase; CLL, chronic lymphocytic leukaemia; CREB, cAMP-response element-binding protein; CTCL, cutaneous T cell lymphoma; DSB, DNA double-strand break; E2F, Early 2 factor; ELM2, egl-27 and MTA1 homology domain 2; ERα, oestrogen receptor-α; FOXO, forkhead box O; GLS, mitochondrial glutaminase; HATs, histone acetyltransferases; HCC, hepatocellular carcinoma; HDAC, histone deacetylases; HIF-1, hypoxia-inducible factors 1; HL, Hodgkin’s lymphoma; ISWI, imitation switch; LDH, lactate dehydrogenase; LSD1, lysine-specific demethylase 1 proteins; MC-LR, microcystin-leucine arginine; MMPs, matrix metalloproteinases; MPNs, myeloproliferative neoplasms; MPP8, M-phase phosphoprotein 8; MSH2,MutS protein homologue 2; NER, nucleotide excision repair; NHEJ, nonhomologous end joining pathway; NICD1, Notch1 intracellular domain; NuRD, nucleosome remodelling and deacetylase; OSCC, oral squamous cell carcinoma; PHF23, plant homeodomain finger protein 23; PML-RAR, promyelocytic leukaemia-retinoic acid receptor; Rb, retinoblastoma protein; SANT, Swi3, Ada2, NCoR and TFIIIB domain; SDS3, suppressor of defectivesilencing3; SIN3A, switch-independent protein 3A; STX17, syntaxin 17; T-ALL, T cell acute lymphoblastic leukaemia; UPS, ubiquitin proteasome system; XPC, xeroderma pigmentosum complementation group C.
1. Mirzaei H, Ghorbani S, Khanizadeh S, Namdari H, Faghihloo E, Akbari A. Histone Deacetylases in Virus-Associated Cancers. Rev Med Virol (2020) 30(1):e2085. doi: 10.1002/rmv.2085
2. He L, Li H, Wu A, Peng Y, Shu G, Yin G. Functions of N6-Methyladenosine and Its Role in Cancer. Mol Cancer (2019) 18(1):176. doi: 10.1186/s12943-019-1109-9
3. Fyodorov DV, Zhou BR, Skoultchi AI, Bai Y. Emerging Roles of Linker Histones in Regulating Chromatin Structure and Function. Nat Rev Mol Cell Biol (2018) 19(3):192–206. doi: 10.1038/nrm.2017.94
4. Shanmugam MK, Arfuso F, Arumugam S, Chinnathambi A, Jinsong B, Warrier S, et al. Role of Novel Histone Modifications in Cancer. Oncotarget (2018) 9(13):11414–26. doi: 10.18632/oncotarget.23356
5. Ellmeier W, Seiser C. Histone Deacetylase Function in CD4(+) T Cells. Nat Rev Immunol (2018) 18(10):617–34. doi: 10.1038/s41577-018-0037-z
6. Adeegbe DO, Liu Y, Lizotte PH, Kamihara Y, Aref AR, Almonte C, et al. Synergistic Immunostimulatory Effects and Therapeutic Benefit of Combined Histone Deacetylase and Bromodomain Inhibition in Non-Small Cell Lung Cancer. Cancer Discov (2017) 7(8):852–67. doi: 10.1158/2159-8290.CD-16-1020
7. Gil J, Ramirez-Torres A, Encarnacion-Guevara S. Lysine Acetylation and Cancer: A Proteomics Perspective. J Proteomics (2017) 150:297–309. doi: 10.1016/j.jprot.2016.10.003
8. Fullwiley D, Gibbon S. Wellcome Trust–Funded Monographs and Book Chapters Genomics in Emerging and Developing Economies. In: Gibbon S, Prainsack B, Hilgartner S, Lamoreaux J, editors. Handbook of Genomics, Health and Society. London, UK: Routledge (2018). p. 228–37. © 2018 selection and editorial matter Sahra Gibbon, Barbara Prainsack, Stephen Hilgartner and Janelle Lamoreaux, individual chapters, the contributors.
9. Draney C, Austin MC, Leifer AH, Smith CJ, Kener KB, Aitken TJ, et al. HDAC1 Overexpression Enhances β-Cell Proliferation by Down-Regulating Cdkn1b/P27. Biochem J (2018) 475(24):3997–4010. doi: 10.1042/bcj20180465
10. Barneda-Zahonero B, Parra M. Histone Deacetylases and Cancer. Mol Oncol (2012) 6(6):579–89. doi: 10.1016/j.molonc.2012.07.003
11. Yoon S, Eom GH. HDAC and HDAC Inhibitor: From Cancer to Cardiovascular Diseases. Chonnam Med J (2016) 52(1):1–11. doi: 10.4068/cmj.2016.52.1.1
12. Burke B, Eden C, Perez C, Belshoff A, Hart S, Plaza-Rojas L, et al. Inhibition of Histone Deacetylase (HDAC) Enhances Checkpoint Blockade Efficacy by Rendering Bladder Cancer Cells Visible for T Cell-Mediated Destruction. Front Oncol (2020) 10:699. doi: 10.3389/fonc.2020.00699
13. Aventaggiato M, Vernucci E, Barreca F, Russo MA, Tafani M. Sirtuins' Control of Autophagy and Mitophagy in Cancer. Pharmacol Ther (2021) 221:107748. doi: 10.1016/j.pharmthera.2020.107748
14. Yang XJ, Seto E. HATs and HDACs: From Structure, Function and Regulation to Novel Strategies for Therapy and Prevention. Oncogene (2007) 26(37):5310–8. doi: 10.1038/sj.onc.1210599
15. Petrie K, Guidez F, Howell L, Healy L, Waxman S, Greaves M, et al. The Histone Deacetylase 9 Gene Encodes Multiple Protein Isoforms. J Biol Chem (2003) 278(18):16059–72. doi: 10.1074/jbc.M212935200
16. Santoro F, Botrugno OA, Dal Zuffo R, Pallavicini I, Matthews GM, Cluse L, et al. A Dual Role for Hdac1: Oncosuppressor in Tumorigenesis, Oncogene in Tumor Maintenance. Blood (2013) 121(17):3459–68. doi: 10.1182/blood-2012-10-461988
17. Lagger S, Meunier D, Mikula M, Brunmeir R, Schlederer M, Artaker M, et al. Crucial Function of Histone Deacetylase 1 for Differentiation of Teratomas in Mice and Humans. EMBO J (2010) 29(23):3992–4007. doi: 10.1038/emboj.2010.264
18. Wilting RH, Yanover E, Heideman MR, Jacobs H, Horner J, van der Torre J, et al. Overlapping Functions of Hdac1 and Hdac2 in Cell Cycle Regulation and Haematopoiesis. EMBO J (2010) 29(15):2586–97. doi: 10.1038/emboj.2010.136
19. Kwon Y, Kim Y, Jung HS, Jeoung D. Role of HDAC3-miRNA-CAGE Network in Anti-Cancer Drug-Resistance. Int J Mol Sci (2018) 20(1):51. doi: 10.3390/ijms20010051
20. Ferrante F, Giaimo BD, Bartkuhn M, Zimmermann T, Close V, Mertens D, et al. HDAC3 Functions as a Positive Regulator in Notch Signal Transduction. Nucleic Acids Res (2020) 48(7):3496–512. doi: 10.1093/nar/gkaa088
21. Leus NG, Zwinderman MR, Dekker FJ. Histone Deacetylase 3 (HDAC 3) as Emerging Drug Target in NF-κb-Mediated Inflammation. Curr Opin Chem Biol (2016) 33:160–8. doi: 10.1016/j.cbpa.2016.06.019
22. Lagger G, O'Carroll D, Rembold M, Khier H, Tischler J, Weitzer G, et al. Essential Function of Histone Deacetylase 1 in Proliferation Control and CDK Inhibitor Repression. EMBO J (2002) 21(11):2672–81. doi: 10.1093/emboj/21.11.2672
23. Vanaja GR, Ramulu HG, Kalle AM. Overexpressed HDAC8 in Cervical Cancer Cells Shows Functional Redundancy of Tubulin Deacetylation With HDAC6. Cell Commun Signal (2018) 16(1):20. doi: 10.1186/s12964-018-0231-4
24. Werbeck ND, Shukla VK, Kunze MBA, Yalinca H, Pritchard RB, Siemons L, et al. A Distal Regulatory Region of a Class I Human Histone Deacetylase. Nat Commun (2020) 11(1):3841. doi: 10.1038/s41467-020-17610-w
25. Jänsch N, Meyners C, Muth M, Kopranovic A, Witt O, Oehme I, et al. The Enzyme Activity of Histone Deacetylase 8 Is Modulated by a Redox-Switch. Redox Biol (2019) 20:60–7. doi: 10.1016/j.redox.2018.09.013
26. Seto E, Yoshida M. Erasers of Histone Acetylation: The Histone Deacetylase Enzymes. Cold Spring Harb Perspect Biol (2014) 6(4):a018713. doi: 10.1101/cshperspect.a018713
27. Millard CJ, Watson PJ, Fairall L, Schwabe JWR. Targeting Class I Histone Deacetylases in a "Complex" Environment. Trends Pharmacol Sci (2017) 38(4):363–77. doi: 10.1016/j.tips.2016.12.006
28. Kelly RD, Cowley SM. The Physiological Roles of Histone Deacetylase (HDAC) 1 and 2: Complex Co-Stars With Multiple Leading Parts. Biochem Soc Trans (2013) 41(3):741–9. doi: 10.1042/bst20130010
29. Sif S, Saurin AJ, Imbalzano AN, Kingston RE. Purification and Characterization of Msin3a-Containing Brg1 and Hbrm Chromatin Remodeling Complexes. Genes Dev (2001) 15(5):603–18. doi: 10.1101/gad.872801
30. Deplus R, Delatte B, Schwinn MK, Defrance M, Méndez J, Murphy N, et al. TET2 and TET3 Regulate GlcNAcylation and H3K4 Methylation Through OGT and SET1/COMPASS. EMBO J (2013) 32(5):645–55. doi: 10.1038/emboj.2012.357
31. Clark MD, Marcum R, Graveline R, Chan CW, Xie T, Chen Z, et al. Structural Insights Into the Assembly of the Histone Deacetylase-Associated Sin3L/Rpd3L Corepressor Complex. Proc Natl Acad Sci USA (2015) 112(28):E3669–78. doi: 10.1073/pnas.1504021112
32. Gambi G, Di Simone E, Basso V, Ricci L, Wang R, Verma A, et al. The Transcriptional Regulator Sin3A Contributes to the Oncogenic Potential of STAT3. Cancer Res (2019) 79(12):3076–87. doi: 10.1158/0008-5472.Can-18-0359
33. Chen M, Chen X, Li S, Pan X, Gong Y, Zheng J, et al. An Epigenetic Mechanism Underlying Chromosome 17p Deletion-Driven Tumorigenesis. Cancer Discov (2020) 11(1):194–207. doi: 10.1158/2159-8290.Cd-20-0336
34. Pierson TM, Otero MG, Grand K, Choi A, Graham JM Jr., Young JI, et al. The NuRD Complex and Macrocephaly Associated Neurodevelopmental Disorders. Am J Med Genet C Semin Med Genet (2019) 181(4):548–56. doi: 10.1002/ajmg.c.31752
35. Millard CJ, Varma N, Saleh A, Morris K, Watson PJ, Bottrill AR, et al. The Structure of the Core NuRD Repression Complex Provides Insights Into Its Interaction With Chromatin. Elife (2016) 5:e13941. doi: 10.7554/eLife.13941
36. Shao S, Cao H, Wang Z, Zhou D, Wu C, Wang S, et al. CHD4/NuRD Complex Regulates Complement Gene Expression and Correlates With CD8 T Cell Infiltration in Human Hepatocellular Carcinoma. Clin Epigenet (2020) 12(1):31. doi: 10.1186/s13148-020-00827-3
37. Dos SRL, Tosti L, Radzisheuskaya A, Caballero IM, Kaji K, Hendrich B, et al. MBD3/NuRD Facilitates Induction of Pluripotency in a Context-Dependent Manner. Cell Stem Cell (2014) 15(3):392. doi: 10.1016/j.stem.2014.08.005
38. Mor N, Rais Y, Sheban D, Peles S, Aguilera-Castrejon A, Zviran A, et al. Neutralizing Gatad2a-Chd4-Mbd3/NuRD Complex Facilitates Deterministic Induction of Naive Pluripotency. Cell Stem Cell (2018) 23(3):412–25.e10. doi: 10.1016/j.stem.2018.07.004
39. Haberland M, Montgomery RL, Olson EN. The Many Roles of Histone Deacetylases in Development and Physiology: Implications for Disease and Therapy. Nat Rev Genet (2009) 10(1):32–42. doi: 10.1038/nrg2485
40. Witt O, Deubzer HE, Milde T, Oehme I. HDAC Family: What Are the Cancer Relevant Targets? Cancer Lett (2009) 277(1):8–21. doi: 10.1016/j.canlet.2008.08.016
41. Bahl S, Seto E. Regulation of Histone Deacetylase Activities and Functions by Phosphorylation and Its Physiological Relevance. Cell Mol Life Sci (2020) 78(2):427–45. doi: 10.1007/s00018-020-03599-4
42. Fischle W, Dequiedt F, Hendzel MJ, Guenther MG, Lazar MA, Voelter W, et al. Enzymatic Activity Associated With Class II HDACs Is Dependent on a Multiprotein Complex Containing HDAC3 and SMRT/N-CoR. Mol Cell (2002) 9(1):45–57. doi: 10.1016/s1097-2765(01)00429-4
43. Kovacs-Kasa A, Kovacs L, Cherian-Shaw M, Patel V, Meadows ML, Fulton DJ, et al. Inhibition of Class IIa HDACs Improves Endothelial Barrier Function in Endotoxin-Induced Acute Lung Injury. J Cell Physiol (2021) 236(4):2893–905. doi: 10.1002/jcp.30053
44. Lahm A, Paolini C, Pallaoro M, Nardi MC, Jones P, Neddermann P, et al. Unraveling the Hidden Catalytic Activity of Vertebrate Class IIa Histone Deacetylases. Proc Natl Acad Sci USA (2007) 104(44):17335–40. doi: 10.1073/pnas.0706487104
45. Hai Y, Christianson DW. Histone Deacetylase 6 Structure and Molecular Basis of Catalysis and Inhibition. Nat Chem Biol (2016) 12(9):741–7. doi: 10.1038/nchembio.2134
46. Gawel JM, Shouksmith AE, Raouf YS, Nawar N, Toutah K, Bukhari S, et al. PTG-0861: A Novel HDAC6-Selective Inhibitor as a Therapeutic Strategy in Acute Myeloid Leukaemia. Eur J Med Chem (2020) 201:112411. doi: 10.1016/j.ejmech.2020.112411
47. Valenzuela-Fernandez A, Cabrero JR, Serrador JM, Sanchez-Madrid F. HDAC6: A Key Regulator of Cytoskeleton, Cell Migration and Cell-Cell Interactions. Trends Cell Biol (2008) 18(6):291–7. doi: 10.1016/j.tcb.2008.04.003
48. Di Giorgio E, Dalla E, Franforte E, Paluvai H, Minisini M, Trevisanut M, et al. Different Class IIa HDACs Repressive Complexes Regulate Specific Epigenetic Responses Related to Cell Survival in Leiomyosarcoma Cells. Nucleic Acids Res (2020) 48(2):646–64. doi: 10.1093/nar/gkz1120
49. Vega RB, Matsuda K, Oh J, Barbosa AC, Yang X, Meadows E, et al. Histone Deacetylase 4 Controls Chondrocyte Hypertrophy During Skeletogenesis. Cell (2004) 119(4):555–66. doi: 10.1016/j.cell.2004.10.024
50. Cao C, Vasilatos SN, Bhargava R, Fine JL, Oesterreich S, Davidson NE, et al. Functional Interaction of Histone Deacetylase 5 (HDAC5) and Lysine-Specific Demethylase 1 (LSD1) Promotes Breast Cancer Progression. Oncogene (2017) 36(1):133–45. doi: 10.1038/onc.2016.186
51. Kawaguchi M, Ieda N, Nakagawa H. Development of Peptide-Based Sirtuin Defatty-Acylase Inhibitors Identified by the Fluorescence Probe, SFP3, That Can Efficiently Measure Defatty-Acylase Activity of Sirtuin. J Med Chem (2019) 62(11):5434–52. doi: 10.1021/acs.jmedchem.9b00315
52. Carafa V, Altucci L, Nebbioso A. Dual Tumor Suppressor and Tumor Promoter Action of Sirtuins in Determining Malignant Phenotype. Front Pharmacol (2019) 10:38. doi: 10.3389/fphar.2019.00038
53. Dai H, Sinclair DA, Ellis JL, Steegborn C. Sirtuin Activators and Inhibitors: Promises, Achievements, and Challenges. Pharmacol Ther (2018) 188:140–54. doi: 10.1016/j.pharmthera.2018.03.004
54. Farghali H, Kemelo MK, Canova NK. SIRT1 Modulators in Experimentally Induced Liver Injury. Oxid Med Cell Longev (2019) 2019:8765954. doi: 10.1155/2019/8765954
55. Yuan J, Minter-Dykhouse K, Lou Z. A C-Myc-SIRT1 Feedback Loop Regulates Cell Growth and Transformation. J Cell Biol (2009) 185(2):203–11. doi: 10.1083/jcb.200809167
56. Chen J, Chan AW, To KF, Chen W, Zhang Z, Ren J, et al. SIRT2 Overexpression in Hepatocellular Carcinoma Mediates Epithelial to Mesenchymal Transition by Protein Kinase B/glycogen Synthase Kinase-3beta/Beta-Catenin Signaling. Hepatology (2013) 57(6):2287–98. doi: 10.1002/hep.26278
57. McGlynn LM, Zino S, MacDonald AI, Curle J, Reilly JE, Mohammed ZM, et al. SIRT2: Tumour Suppressor or Tumour Promoter in Operable Breast Cancer? Eur J Cancer (2014) 50(2):290–301. doi: 10.1016/j.ejca.2013.10.005
58. Kim HS, Vassilopoulos A, Wang RH, Lahusen T, Xiao Z, Xu X, et al. SIRT2 Maintains Genome Integrity and Suppresses Tumorigenesis Through Regulating APC/C Activity. Cancer Cell (2011) 20(4):487–99. doi: 10.1016/j.ccr.2011.09.004
59. Toubai T, Tamaki H, Peltier DC, Rossi C, Oravecz-Wilson K, Liu C, et al. Mitochondrial Deacetylase SIRT3 Plays an Important Role in Donor T Cell Responses After Experimental Allogeneic Hematopoietic Transplantation. J Immunol (2018) 201(11):3443–55. doi: 10.4049/jimmunol.1800148
60. Fu L, Dong Q, He J, Wang X, Xing J, Wang E, et al. SIRT4 Inhibits Malignancy Progression of NSCLCs, Through Mitochondrial Dynamics Mediated by the ERK-Drp1 Pathway. Oncogene (2017) 36(19):2724–36. doi: 10.1038/onc.2016.425
61. Bringman-Rodenbarger LR, Guo AH, Lyssiotis CA, Lombard DB. Emerging Roles for SIRT5 in Metabolism and Cancer. Antioxid Redox Signal (2018) 28(8):677–90. doi: 10.1089/ars.2017.7264
62. Sebastian C, Zwaans BM, Silberman DM, Gymrek M, Goren A, Zhong L, et al. The Histone Deacetylase SIRT6 Is a Tumor Suppressor That Controls Cancer Metabolism. Cell (2012) 151(6):1185–99. doi: 10.1016/j.cell.2012.10.047
63. Klein MA, Denu JM. Biological and Catalytic Functions of Sirtuin 6 as Targets for Small-Molecule Modulators. J Biol Chem (2020) 295(32):11021–041. doi: 10.1074/jbc.REV120.011438
64. Chen S, Blank MF, Iyer A, Huang B, Wang L, Grummt I, et al. SIRT7-Dependent Deacetylation of the U3-55k Protein Controls pre-rRNA Processing. Nat Commun (2016) 7:10734. doi: 10.1038/ncomms10734
65. Kim JK, Noh JH, Jung KH, Eun JW, Bae HJ, Kim MG, et al. Sirtuin7 Oncogenic Potential in Human Hepatocellular Carcinoma and Its Regulation by the Tumor Suppressors MiR-125a-5p and MiR-125b. Hepatology (2013) 57(3):1055–67. doi: 10.1002/hep.26101
66. Yang XJ, Seto E. The Rpd3/Hda1 Family of Lysine Deacetylases: From Bacteria and Yeast to Mice and Men. Nat Rev Mol Cell Biol (2008) 9(3):206–18. doi: 10.1038/nrm2346
67. Bagchi RA, Ferguson BS, Stratton MS, Hu T, Cavasin MA, Sun L, et al. HDAC11 Suppresses the Thermogenic Program of Adipose Tissue via BRD2. JCI Insight (2018) 3(15):e120159. doi: 10.1172/jci.insight.120159
68. Cao J, Sun L, Aramsangtienchai P, Spiegelman NA, Zhang X, Huang W, et al. HDAC11 Regulates Type I Interferon Signaling Through Defatty-Acylation of SHMT2. Proc Natl Acad Sci USA (2019) 116(12):5487–92. doi: 10.1073/pnas.1815365116
69. Jagielska A, Lowe AL, Makhija E, Wroblewska L, Guck J, Franklin RJM, et al. Mechanical Strain Promotes Oligodendrocyte Differentiation by Global Changes of Gene Expression. Front Cell Neurosci (2017) 11:93. doi: 10.3389/fncel.2017.00093
70. Yue L, Sharma V, Horvat NP, Akuffo AA, Beatty MS, Murdun C, et al. HDAC11 Deficiency Disrupts Oncogene-Induced Hematopoiesis in Myeloproliferative Neoplasms. Blood (2020) 135(3):191–207. doi: 10.1182/blood.2019895326
71. Wang P, Wang Z, Liu J. Role of HDACs in Normal and Malignant Hematopoiesis. Mol Cancer (2020) 19(1):5. doi: 10.1186/s12943-019-1127-7
72. Weichert W, Denkert C, Noske A, Darb-Esfahani S, Dietel M, Kalloger SE, et al. Expression of Class I Histone Deacetylases Indicates Poor Prognosis in Endometrioid Subtypes of Ovarian and Endometrial Carcinomas. Neoplasia (2008) 10(9):1021–7. doi: 10.1593/neo.08474
73. Oehme I, Deubzer HE, Wegener D, Pickert D, Linke JP, Hero B, et al. Histone Deacetylase 8 in Neuroblastoma Tumorigenesis. Clin Cancer Res (2009) 15(1):91–9. doi: 10.1158/1078-0432.CCR-08-0684
74. Rettig I, Koeneke E, Trippel F, Mueller WC, Burhenne J, Kopp-Schneider A, et al. Selective Inhibition of HDAC8 Decreases Neuroblastoma Growth In Vitro and In Vivo and Enhances Retinoic Acid-Mediated Differentiation. Cell Death Dis (2015) 6:e1657. doi: 10.1038/cddis.2015.24
75. West AC, Johnstone RW. New and Emerging HDAC Inhibitors for Cancer Treatment. J Clin Invest (2014) 124(1):30–9. doi: 10.1172/JCI69738
76. Hug BA, Lazar MA. ETO Interacting Proteins. Oncogene (2004) 23(24):4270–4. doi: 10.1038/sj.onc.1207674
77. Falkenberg KJ, Johnstone RW. Histone Deacetylases and Their Inhibitors in Cancer, Neurological Diseases and Immune Disorders. Nat Rev Drug Discov (2014) 13(9):673–91. doi: 10.1038/nrd4360
78. Narita T, Ito S, Higashijima Y, Chu WK, Neumann K, Walter J, et al. Enhancers Are Activated by P300/CBP Activity-Dependent PIC Assembly, RNAPII Recruitment, and Pause Release. Mol Cell (2021) 81(10):2166–82. doi: 10.1016/j.molcel.2021.03.008
79. Vaid R, Wen J, Mannervik M. Release of Promoter-Proximal Paused Pol II in Response to Histone Deacetylase Inhibition. Nucleic Acids Res (2020) 48(9):4877–90. doi: 10.1093/nar/gkaa234
80. Wang A, Kurdistani SK, Grunstein M. Requirement of Hos2 Histone Deacetylase for Gene Activity in Yeast. Science (2002) 298(5597):1412–4. doi: 10.1126/science.1077790
81. Slaughter MJ, Shanle EK, Khan A, Chua KF, Hong T, Boxer LD, et al. HDAC Inhibition Results in Widespread Alteration of the Histone Acetylation Landscape and BRD4 Targeting to Gene Bodies. Cell Rep (2021) 34(3):108638. doi: 10.1016/j.celrep.2020.108638
82. Bao L, Diao H, Dong N, Su X, Wang B, Mo Q, et al. Histone Deacetylase Inhibitor Induces Cell Apoptosis and Cycle Arrest in Lung Cancer Cells via Mitochondrial Injury and P53 Up-Acetylation. Cell Biol Toxicol (2016) 32(6):469–82. doi: 10.1007/s10565-016-9347-8
83. Wawruszak A, Kalafut J, Okon E, Czapinski J, Halasa M, Przybyszewska A, et al. Histone Deacetylase Inhibitors and Phenotypical Transformation of Cancer Cells. Cancers (Basel) (2019) 11(2):148. doi: 10.3390/cancers11020148
84. Qiu X, Xiao X, Li N, Li Y. Histone Deacetylases Inhibitors (HDACis) as Novel Therapeutic Application in Various Clinical Diseases. Prog Neuropsychopharmacol Biol Psychiatry (2017) 72:60–72. doi: 10.1016/j.pnpbp.2016.09.002
85. Bailey H, Stenehjem DD, Sharma S. Panobinostat for the Treatment of Multiple Myeloma: The Evidence to Date. J Blood Med (2015) 6:269–76. doi: 10.2147/jbm.S69140
86. Cha TL, Chuang MJ, Wu ST, Sun GH, Chang SY, Yu DS, et al. Dual Degradation of Aurora A and B Kinases by the Histone Deacetylase Inhibitor LBH589 Induces G2-M Arrest and Apoptosis of Renal Cancer Cells. Clin Cancer Res (2009) 15(3):840–50. doi: 10.1158/1078-0432.CCR-08-1918
87. Meidhof S, Brabletz S, Lehmann W, Preca BT, Mock K, Ruh M, et al. ZEB1-Associated Drug Resistance in Cancer Cells Is Reversed by the Class I HDAC Inhibitor Mocetinostat. EMBO Mol Med (2015) 7(6):831–47. doi: 10.15252/emmm.201404396
88. Frys S, Simons Z, Hu Q, Barth MJ, Gu JJ, Mavis C, et al. Entinostat, a Novel Histone Deacetylase Inhibitor Is Active in B-Cell Lymphoma and Enhances the Anti-Tumour Activity of Rituximab and Chemotherapy Agents. Br J Haematol (2015) 169(4):506–19. doi: 10.1111/bjh.13318
89. Stark M, Hayward N. Genome-Wide Loss of Heterozygosity and Copy Number Analysis in Melanoma Using High-Density Single-Nucleotide Polymorphism Arrays. Cancer Res (2007) 67(6):2632–42. doi: 10.1158/0008-5472.CAN-06-4152
90. Taylor BS, DeCarolis PL, Angeles CV, Brenet F, Schultz N, Antonescu CR, et al. Frequent Alterations and Epigenetic Silencing of Differentiation Pathway Genes in Structurally Rearranged Liposarcomas. Cancer Discovery (2011) 1(7):587–97. doi: 10.1158/2159-8290.CD-11-0181
91. Kugel S, Feldman JL, Klein MA, Silberman DM, Sebastian C, Mermel C, et al. Identification of and Molecular Basis for SIRT6 Loss-Of-Function Point Mutations in Cancer. Cell Rep (2015) 13(3):479–88. doi: 10.1016/j.celrep.2015.09.022
92. Singh AK, Bishayee A, Pandey AK. Targeting Histone Deacetylases With Natural and Synthetic Agents: An Emerging Anticancer Strategy. Nutrients (2018) 10(6):731. doi: 10.3390/nu10060731
93. Hayashi A, Horiuchi A, Kikuchi N, Hayashi T, Fuseya C, Suzuki A, et al. Type-Specific Roles of Histone Deacetylase (HDAC) Overexpression in Ovarian Carcinoma: HDAC1 Enhances Cell Proliferation and HDAC3 Stimulates Cell Migration With Downregulation of E-Cadherin. Int J Cancer (2010) 127(6):1332–46. doi: 10.1002/ijc.25151
94. Buurman R, Gürlevik E, Schäffer V, Eilers M, Sandbothe M, Kreipe H, et al. Histone Deacetylases Activate Hepatocyte Growth Factor Signaling by Repressing microRNA-449 in Hepatocellular Carcinoma Cells. Gastroenterology (2012) 143(3):811–20.e15. doi: 10.1053/j.gastro.2012.05.033
95. Xie HJ, Noh JH, Kim JK, Jung KH, Eun JW, Bae HJ, et al. HDAC1 Inactivation Induces Mitotic Defect and Caspase-Independent Autophagic Cell Death in Liver Cancer. PLoS One (2012) 7(4):e34265. doi: 10.1371/journal.pone.0034265
96. Fritsche P, Seidler B, Schuler S, Schnieke A, Gottlicher M, Schmid RM, et al. HDAC2 Mediates Therapeutic Resistance of Pancreatic Cancer Cells via the BH3-Only Protein NOXA. Gut (2009) 58(10):1399–409. doi: 10.1136/gut.2009.180711
97. Jung KH, Noh JH, Kim JK, Eun JW, Bae HJ, Xie HJ, et al. HDAC2 Overexpression Confers Oncogenic Potential to Human Lung Cancer Cells by Deregulating Expression of Apoptosis and Cell Cycle Proteins. J Cell Biochem (2012) 113(6):2167–77. doi: 10.1002/jcb.24090
98. Park SH, Kim H, Kwak S, Jeong JH, Lee J, Hwang JT, et al. HDAC3-ERalpha Selectively Regulates TNF-Alpha-Induced Apoptotic Cell Death in MCF-7 Human Breast Cancer Cells via the P53 Signaling Pathway. Cells (2020) 9(5):1280. doi: 10.3390/cells9051280
99. Colarossi L, Memeo L, Colarossi C, Aiello E, Iuppa A, Espina V, et al. Inhibition of Histone Deacetylase 4 Increases Cytotoxicity of Docetaxel in Gastric Cancer Cells. Proteomics Clin Appl (2014) 8(11-12):924–31. doi: 10.1002/prca.201400058
100. Feng GW, Dong LD, Shang WJ, Pang XL, Li JF, Liu L, et al. HDAC5 Promotes Cell Proliferation in Human Hepatocellular Carcinoma by Up-Regulating Six1 Expression. Eur Rev Med Pharmacol Sci (2014) 18(6):811–6.
101. Fan J, Lou B, Chen W, Zhang J, Lin S, Lv FF, et al. Down-Regulation of HDAC5 Inhibits Growth of Human Hepatocellular Carcinoma by Induction of Apoptosis and Cell Cycle Arrest. Tumour Biol (2014) 35(11):11523–32. doi: 10.1007/s13277-014-2358-2
102. Pham L, Kaiser B, Romsa A, Schwarz T, Gopalakrishnan R, Jensen ED, et al. HDAC3 and HDAC7 Have Opposite Effects on Osteoclast Differentiation. J Biol Chem (2011) 286(14):12056–65. doi: 10.1074/jbc.M110.216853
103. Hassell KN. Histone Deacetylases and Their Inhibitors in Cancer Epigenetics. Diseases (2019) 7(4):57. doi: 10.3390/diseases7040057
104. Li D, Sun X, Zhang L, Yan B, Xie S, Liu R, et al. Histone Deacetylase 6 and Cytoplasmic Linker Protein 170 Function Together to Regulate the Motility of Pancreatic Cancer Cells. Protein Cell (2014) 5(3):214–23. doi: 10.1007/s13238-013-0010-3
105. Lv Z, Weng X, Du C, Zhang C, Xiao H, Cai X, et al. Downregulation of HDAC6 Promotes Angiogenesis in Hepatocellular Carcinoma Cells and Predicts Poor Prognosis in Liver Transplantation Patients. Mol Carcinog (2016) 55(5):1024–33. doi: 10.1002/mc.22345
106. Oehme I, Linke JP, Bock BC, Milde T, Lodrini M, Hartenstein B, et al. Histone Deacetylase 10 Promotes Autophagy-Mediated Cell Survival. Proc Natl Acad Sci USA (2013) 110(28):E2592–601. doi: 10.1073/pnas.1300113110
107. Song C, Zhu S, Wu C, Kang J. Histone Deacetylase (HDAC) 10 Suppresses Cervical Cancer Metastasis Through Inhibition of Matrix Metalloproteinase (MMP) 2 and 9 Expression. J Biol Chem (2013) 288(39):28021–33. doi: 10.1074/jbc.M113.498758
108. Li Y, Peng L, Seto E. Histone Deacetylase 10 Regulates the Cell Cycle G2/M Phase Transition via a Novel Let-7-HMGA2-Cyclin A2 Pathway. Mol Cell Biol (2015) 35(20):3547–65. doi: 10.1128/MCB.00400-15
109. Leslie PL, Chao YL, Tsai YH, Ghosh SK, Porrello A, Van Swearingen AED, et al. Histone Deacetylase 11 Inhibition Promotes Breast Cancer Metastasis From Lymph Nodes. Nat Commun (2019) 10(1):4192. doi: 10.1038/s41467-019-12222-5
110. Deubzer HE, Schier MC, Oehme I, Lodrini M, Haendler B, Sommer A, et al. HDAC11 Is a Novel Drug Target in Carcinomas. Int J Cancer (2013) 132(9):2200–8. doi: 10.1002/ijc.27876
111. Buglio D, Khaskhely NM, Voo KS, Martinez-Valdez H, Liu YJ, Younes A. HDAC11 Plays an Essential Role in Regulating OX40 Ligand Expression in Hodgkin Lymphoma. Blood (2011) 117(10):2910–7. doi: 10.1182/blood-2010-08-303701
112. Vaziri H, Dessain SK, Ng EE, Imai SI, Frye RA, Pandita TK, et al. Hsir2(SIRT1) Functions as an NAD-Dependent P53 Deacetylase. Cell (2001) 107(2):149–59. doi: 10.1016/s0092-8674(01)00527-x
113. Brunet A, Sweeney LB, Sturgill JF, Chua KF, Greer PL, Lin Y, et al. Stress-Dependent Regulation of FOXO Transcription Factors by the SIRT1 Deacetylase. Science (2004) 303(5666):2011–5. doi: 10.1126/science.1094637
114. Yang Q, Wang B, Zang W, Wang X, Liu Z, Li W, et al. Resveratrol Inhibits the Growth of Gastric Cancer by Inducing G1 Phase Arrest and Senescence in a Sirt1-Dependent Manner. PLoS One (2013) 8(11):e70627. doi: 10.1371/journal.pone.0070627
115. Qiu G, Li X, Che X, Wei C, He S, Lu J, et al. SIRT1 Is a Regulator of Autophagy: Implications in Gastric Cancer Progression and Treatment. FEBS Lett (2015) 589(16):2034–42. doi: 10.1016/j.febslet.2015.05.042
116. Qiu G, Li X, Wei C, Che X, He S, Lu J, et al. The Prognostic Role of SIRT1-Autophagy Axis in Gastric Cancer. Dis Markers (2016) 2016:6869415. doi: 10.1155/2016/6869415
117. Dali-Youcef N, Lagouge M, Froelich S, Koehl C, Schoonjans K, Auwerx J. Sirtuins: The 'Magnificent Seven', Function, Metabolism and Longevity. Ann Med (2007) 39(5):335–45. doi: 10.1080/07853890701408194
118. Jing H, Lin H. Sirtuins in Epigenetic Regulation. Chem Rev (2015) 115(6):2350–75. doi: 10.1021/cr500457h
119. Yu XJ, Guo XZ, Li C, Chong Y, Song TN, Pang JF, et al. SIRT1-ZEB1-Positive Feedback Promotes Epithelial-Mesenchymal Transition Process and Metastasis of Osteosarcoma. J Cell Biochem (2019) 120(3):3727–35. doi: 10.1002/jcb.27653
120. Rack JG, VanLinden MR, Lutter T, Aasland R, Ziegler M. Constitutive Nuclear Localization of an Alternatively Spliced Sirtuin-2 Isoform. J Mol Biol (2014) 426(8):1677–91. doi: 10.1016/j.jmb.2013.10.027
121. Wang F, Nguyen M, Qin FX, Tong Q. SIRT2 Deacetylates FOXO3a in Response to Oxidative Stress and Caloric Restriction. Aging Cell (2007) 6(4):505–14. doi: 10.1111/j.1474-9726.2007.00304.x
122. Zhao Y, Wang L, Yang J, Zhang P, Ma K, Zhou J, et al. Anti-Neoplastic Activity of the Cytosolic FoxO1 Results From Autophagic Cell Death. Autophagy (2010) 6(7):988–90. doi: 10.4161/auto.6.7.13289
123. Zhao Y, Yang H, Wang X, Zhang R, Wang C, Guo Z. Sirtuin-3 (SIRT3) Expression Is Associated With Overall Survival in Esophageal Cancer. Ann Diagn Pathol (2013) 17(6):483–5. doi: 10.1016/j.anndiagpath.2013.06.001
124. Jing E, Gesta S, Kahn CR. SIRT2 Regulates Adipocyte Differentiation Through FoxO1 Acetylation/Deacetylation. Cell Metab (2007) 6(2):105–14. doi: 10.1016/j.cmet.2007.07.003
125. Wang Y, Yang J, Hong T, Chen X, Cui L. SIRT2: Controversy and Multiple Roles in Disease and Physiology. Ageing Res Rev (2019) 55:100961. doi: 10.1016/j.arr.2019.100961
126. Jiang HK, Miao Y, Wang YH, Zhao M, Feng ZH, Yu XJ, et al. Aerobic Interval Training Protects Against Myocardial Infarction-Induced Oxidative Injury by Enhancing Antioxidase System and Mitochondrial Biosynthesis. Clin Exp Pharmacol Physiol (2014) 41(3):192–201. doi: 10.1111/1440-1681.12211
127. Zheng Y, Shi B, Ma M, Wu X, Lin X. The Novel Relationship Between Sirt3 and Autophagy in Myocardial Ischemia-Reperfusion. J Cell Physiol (2019) 234(5):5488–95. doi: 10.1002/jcp.27329
128. Katta S, Karnewar S, Panuganti D, Jerald MK, Sastry BKS, Kotamraju S. Mitochondria-Targeted Esculetin Inhibits PAI-1 Levels by Modulating STAT3 Activation and miR-19b via SIRT3: Role in Acute Coronary Artery Syndrome. J Cell Physiol (2018) 233(1):214–25. doi: 10.1002/jcp.25865
129. Hawley SA, Boudeau J, Reid JL, Mustard KJ, Udd L, Makela TP, et al. Complexes Between the LKB1 Tumor Suppressor, STRAD Alpha/Beta and MO25 Alpha/Beta Are Upstream Kinases in the AMP-Activated Protein Kinase Cascade. J Biol (2003) 2(4):28. doi: 10.1186/1475-4924-2-28
130. Alhazzazi TY, Kamarajan P, Verdin E, Kapila YL. SIRT3 and Cancer: Tumor Promoter or Suppressor? Biochim Biophys Acta (2011) 1816(1):80–8. doi: 10.1016/j.bbcan.2011.04.004
131. Chen J, Wang A, Chen Q. SirT3 and P53 Deacetylation in Aging and Cancer. J Cell Physiol (2017) 232(9):2308–11. doi: 10.1002/jcp.25669
132. Alhazzazi TY, Kamarajan P, Joo N, Huang JY, Verdin E, D'Silva NJ, et al. Sirtuin-3 (SIRT3), a Novel Potential Therapeutic Target for Oral Cancer. Cancer (2011) 117(8):1670–8. doi: 10.1002/cncr.25676
133. Lombard DB, Alt FW, Cheng HL, Bunkenborg J, Streeper RS, Mostoslavsky R, et al. Mammalian Sir2 Homolog SIRT3 Regulates Global Mitochondrial Lysine Acetylation. Mol Cell Biol (2007) 27(24):8807–14. doi: 10.1128/MCB.01636-07
134. Liu M, Wang Z, Ren M, Yang X, Liu B, Qi H, et al. SIRT4 Regulates PTEN Stability Through IDE in Response to Cellular Stresses. FASEB J (2019) 33(4):5535–47. doi: 10.1096/fj.201801987R
135. Csibi A, Fendt SM, Li C, Poulogiannis G, Choo AY, Chapski DJ, et al. The Mtorc1 Pathway Stimulates Glutamine Metabolism and Cell Proliferation by Repressing SIRT4. Cell (2013) 153(4):840–54. doi: 10.1016/j.cell.2013.04.023
136. Polletta L, Vernucci E, Carnevale I, Arcangeli T, Rotili D, Palmerio S, et al. SIRT5 Regulation of Ammonia-Induced Autophagy and Mitophagy. Autophagy (2015) 11(2):253–70. doi: 10.1080/15548627.2015.1009778
137. Lin ZF, Xu HB, Wang JY, Lin Q, Ruan Z, Liu FB, et al. SIRT5 Desuccinylates and Activates SOD1 to Eliminate ROS. Biochem Biophys Res Commun (2013) 441(1):191–5. doi: 10.1016/j.bbrc.2013.10.033
138. Greene KS, Lukey MJ, Wang X, Blank B, Druso JE, Lin MJ, et al. SIRT5 Stabilizes Mitochondrial Glutaminase and Supports Breast Cancer Tumorigenesis. Proc Natl Acad Sci USA (2019) 116(52):26625–32. doi: 10.1073/pnas.1911954116
139. Zhao S, Wang JM, Yan J, Zhang DL, Liu BQ, Jiang JY, et al. BAG3 Promotes Autophagy and Glutaminolysis via Stabilizing Glutaminase. Cell Death Dis (2019) 10(4):284. doi: 10.1038/s41419-019-1504-6
140. Zhong L, D'Urso A, Toiber D, Sebastian C, Henry RE, Vadysirisack DD, et al. The Histone Deacetylase Sirt6 Regulates Glucose Homeostasis via Hif1alpha. Cell (2010) 140(2):280–93. doi: 10.1016/j.cell.2009.12.041
141. Chang L, Xi L, Liu Y, Liu R, Wu Z, Jian Z. SIRT5 Promotes Cell Proliferation and Invasion in Hepatocellular Carcinoma by Targeting E2F1. Mol Med Rep (2018) 17(1):342–9. doi: 10.3892/mmr.2017.7875
142. Kawahara TL, Michishita E, Adler AS, Damian M, Berber E, Lin M, et al. SIRT6 Links Histone H3 Lysine 9 Deacetylation to NF-kappaB-Dependent Gene Expression and Organismal Life Span. Cell (2009) 136(1):62–74. doi: 10.1016/j.cell.2008.10.052
143. Mao Z, Hine C, Tian X, Van Meter M, Au M, Vaidya A, et al. SIRT6 Promotes DNA Repair Under Stress by Activating PARP1. Science (2011) 332(6036):1443–6. doi: 10.1126/science.1202723
144. Michishita E, McCord RA, Berber E, Kioi M, Padilla-Nash H, Damian M, et al. SIRT6 Is a Histone H3 Lysine 9 Deacetylase That Modulates Telomeric Chromatin. Nature (2008) 452(7186):492–6. doi: 10.1038/nature06736
145. Chen S, Seiler J, Santiago-Reichelt M, Felbel K, Grummt I, Voit R. Repression of RNA Polymerase I Upon Stress Is Caused by Inhibition of RNA-Dependent Deacetylation of PAF53 by SIRT7. Mol Cell (2013) 52(3):303–13. doi: 10.1016/j.molcel.2013.10.010
146. Jiang Y, Han Z, Wang Y, Hao W. Depletion of SIRT7 Sensitizes Human Non-Small Cell Lung Cancer Cells to Gemcitabine Therapy by Inhibiting Autophagy. Biochem Biophys Res Commun (2018) 506(1):266–71. doi: 10.1016/j.bbrc.2018.10.089
147. Li Y, Seto E. HDACs and HDAC Inhibitors in Cancer Development and Therapy. Cold Spring Harb Perspect Med (2016) 6(10):a026831. doi: 10.1101/cshperspect.a026831
148. Zhao H, Wang Y, Yang C, Zhou J, Wang L, Yi K, et al. EGFR-vIII Downregulated H2AZK4/7AC Though the PI3K/AKT-HDAC2 Axis to Regulate Cell Cycle Progression. Clin Transl Med (2020) 9(1):10. doi: 10.1186/s40169-020-0260-7
149. Yamaguchi T, Cubizolles F, Zhang Y, Reichert N, Kohler H, Seiser C, et al. Histone Deacetylases 1 and 2 Act in Concert to Promote the G1-To-S Progression. Genes Dev (2010) 24(5):455–69. doi: 10.1101/gad.552310
150. Wei D, Lu T, Ma D, Yu K, Zhang T, Xiong J, et al. Synergistic Activity of Imatinib and AR-42 Against Chronic Myeloid Leukemia Cells Mainly Through HDAC1 Inhibition. Life Sci (2018) 211:224–37. doi: 10.1016/j.lfs.2018.09.040
151. Jiang Y, Hsieh J. HDAC3 Controls Gap 2/Mitosis Progression in Adult Neural Stem/Progenitor Cells by Regulating CDK1 Levels. Proc Natl Acad Sci USA (2014) 111(37):13541–6. doi: 10.1073/pnas.1411939111
152. Lu XF, Cao XY, Zhu YJ, Wu ZR, Zhuang X, Shao MY, et al. Histone Deacetylase 3 Promotes Liver Regeneration and Liver Cancer Cells Proliferation Through Signal Transducer and Activator of Transcription 3 Signaling Pathway. Cell Death Dis (2018) 9(3):398. doi: 10.1038/s41419-018-0428-x
153. Liu X, Wang Y, Zhang R, Jin T, Qu L, Jin Q, et al. HDAC10 Is Positively Associated With PD-L1 Expression and Poor Prognosis in Patients With NSCLC. Front Oncol (2020) 10:485. doi: 10.3389/fonc.2020.00485
154. Li Y, Zhang X, Zhu S, Dejene EA, Peng W, Sepulveda A, et al. HDAC10 Regulates Cancer Stem-Like Cell Properties in KRAS-Driven Lung Adenocarcinoma. Cancer Res (2020) 80(16):3265–78. doi: 10.1158/0008-5472.CAN-19-3613
155. Yang WB, Hsu CC, Hsu TI, Liou JP, Chang KY, Chen PY, et al. Increased Activation of HDAC1/2/6 and Sp1 Underlies Therapeutic Resistance and Tumor Growth in Glioblastoma. Neuro Oncol (2020) 22(10):1439–51. doi: 10.1093/neuonc/noaa103
156. Lin XL, Li K, Yang Z, Chen B, Zhang T. Dulcitol Suppresses Proliferation and Migration of Hepatocellular Carcinoma via Regulating SIRT1/p53 Pathway. Phytomedicine (2020) 66:153112. doi: 10.1016/j.phymed.2019.153112
157. Patra S, Panigrahi DP, Praharaj PP, Bhol CS, Mahapatra KK, Mishra SR, et al. Dysregulation of Histone Deacetylases in Carcinogenesis and Tumor Progression: A Possible Link to Apoptosis and Autophagy. Cell Mol Life Sci (2019) 76(17):3263–82. doi: 10.1007/s00018-019-03098-1
158. Gong P, Li K, Li Y, Liu D, Zhao L, Jing Y. HDAC and Ku70 Axis- an Effective Target for Apoptosis Induction by a New 2-Cyano-3-Oxo-1,9-Dien Glycyrrhetinic Acid Analogue. Cell Death Dis (2018) 9(6):623. doi: 10.1038/s41419-018-0602-1
159. Subramanian C, Opipari AW Jr., Bian X, Castle VP, Kwok RP. Ku70 Acetylation Mediates Neuroblastoma Cell Death Induced by Histone Deacetylase Inhibitors. Proc Natl Acad Sci USA (2005) 102(13):4842–7. doi: 10.1073/pnas.0408351102
160. Cohen HY, Lavu S, Bitterman KJ, Hekking B, Imahiyerobo TA, Miller C, et al. Acetylation of the C Terminus of Ku70 by CBP and PCAF Controls Bax-Mediated Apoptosis. Mol Cell (2004) 13(5):627–38. doi: 10.1016/s1097-2765(04)00094-2
161. Nguyen TTT, Zhang Y, Shang E, Shu C, Quinzii CM, Westhoff MA, et al. Inhibition of HDAC1/2 Along With TRAP1 Causes Synthetic Lethality in Glioblastoma Model Systems. Cells (2020) 9(7):1661. doi: 10.3390/cells9071661
162. Ding J, Liu J, Zhang Z, Guo J, Cheng M, Wan Y, et al. Design, Synthesis and Biological Evaluation of Coumarin-Based N-Hydroxycinnamamide Derivatives as Novel Histone Deacetylase Inhibitors With Anticancer Activities. Bioorg Chem (2020) 101:104023. doi: 10.1016/j.bioorg.2020.104023
163. Bangert A, Cristofanon S, Eckhardt I, Abhari BA, Kolodziej S, Hacker S, et al. Histone Deacetylase Inhibitors Sensitize Glioblastoma Cells to TRAIL-Induced Apoptosis by C-Myc-Mediated Downregulation of cFLIP. Oncogene (2012) 31(44):4677–88. doi: 10.1038/onc.2011.614
164. Jang YG, Ko EB, Choi KC. Gallic Acid, a Phenolic Acid, Hinders the Progression of Prostate Cancer by Inhibition of Histone Deacetylase 1 and 2 Expression. J Nutr Biochem (2020) 84:108444. doi: 10.1016/j.jnutbio.2020.108444
165. Riley JS, Hutchinson R, McArt DG, Crawford N, Holohan C, Paul I, et al. Prognostic and Therapeutic Relevance of FLIP and Procaspase-8 Overexpression in Non-Small Cell Lung Cancer. Cell Death Dis (2013) 4:e951. doi: 10.1038/cddis.2013.481
166. Schuler S, Fritsche P, Diersch S, Arlt A, Schmid RM, Saur D, et al. HDAC2 Attenuates TRAIL-Induced Apoptosis of Pancreatic Cancer Cells. Mol Cancer (2010) 9:80. doi: 10.1186/1476-4598-9-80
167. Zhang J, Zhong Q. Histone Deacetylase Inhibitors and Cell Death. Cell Mol Life Sci (2014) 71(20):3885–901. doi: 10.1007/s00018-014-1656-6
168. Liu T, Wu Z, He Y, Xiao Y, Xia C. Single and Dual Target Inhibitors Based on Bcl-2: Promising Anti-Tumor Agents for Cancer Therapy. Eur J Med Chem (2020) 201:112446. doi: 10.1016/j.ejmech.2020.112446
169. Wang Y, Liu H, Liu X, Zhang X, Wu J, Yuan L, et al. Histone Acetylation Plays an Important Role in MC-LR-Induced Apoptosis and Cycle Disorder in SD Rat Testicular Cells. Chemosphere (2020) 241:125073. doi: 10.1016/j.chemosphere.2019.125073
170. Chen R, Zhang M, Zhou Y, Guo W, Yi M, Zhang Z, et al. The Application of Histone Deacetylases Inhibitors in Glioblastoma. J Exp Clin Cancer Res (2020) 39(1):138. doi: 10.1186/s13046-020-01643-6
171. Jang YG, Hwang KA, Choi KC. Rosmarinic Acid, a Component of Rosemary Tea, Induced the Cell Cycle Arrest and Apoptosis Through Modulation of HDAC2 Expression in Prostate Cancer Cell Lines. Nutrients (2018) 10(11). doi: 10.3390/nu10111784
172. Yu X, Li H, Zhu M, Hu P, Liu X, Qing Y, et al. Involvement of P53 Acetylation in Growth Suppression of Cutaneous T-Cell Lymphomas Induced by HDAC Inhibition. J Invest Dermatol (2020) 140(10):2009–22. doi: 10.1016/j.jid.2019.12.041
173. Zhang LZ, Yang JE, Luo YW, Liu FT, Yuan YF, Zhuang SM. A P53/lnc-Ip53 Negative Feedback Loop Regulates Tumor Growth and Chemoresistance. Adv Sci (Weinh) (2020) 7(21):2001364. doi: 10.1002/advs.202001364
174. Zhou X, Tolstov Y, Arslan A, Roth W, Grullich C, Pahernik S, et al. Harnessing the P53-PUMA Axis to Overcome DNA Damage Resistance in Renal Cell Carcinoma. Neoplasia (2014) 16(12):1028–35. doi: 10.1016/j.neo.2014.09.012
175. Sonnemann J, Marx C, Becker S, Wittig S, Palani CD, Kramer OH, et al. P53-Dependent and P53-Independent Anticancer Effects of Different Histone Deacetylase Inhibitors. Br J Cancer (2014) 110(3):656–67. doi: 10.1038/bjc.2013.742
176. Ellis L, Bots M, Lindemann RK, Bolden JE, Newbold A, Cluse LA, et al. The Histone Deacetylase Inhibitors LAQ824 and LBH589 Do Not Require Death Receptor Signaling or a Functional Apoptosome to Mediate Tumor Cell Death or Therapeutic Efficacy. Blood (2009) 114(2):380–93. doi: 10.1182/blood-2008-10-182758
177. Lees A, McIntyre AJ, Crawford NT, Falcone F, McCann C, Holohan C, et al. The Pseudo-Caspase FLIP(L) Regulates Cell Fate Following P53 Activation. Proc Natl Acad Sci USA (2020) 117(30):17808–19. doi: 10.1073/pnas.2001520117
178. Kang Y, Nian H, Rajendran P, Kim E, Dashwood WM, Pinto JT, et al. HDAC8 and STAT3 Repress BMF Gene Activity in Colon Cancer Cells. Cell Death Dis (2014) 5:e1476. doi: 10.1038/cddis.2014.422
179. Kolbinger FR, Koeneke E, Ridinger J, Heimburg T, Muller M, Bayer T, et al. The HDAC6/8/10 Inhibitor TH34 Induces DNA Damage-Mediated Cell Death in Human High-Grade Neuroblastoma Cell Lines. Arch Toxicol (2018) 92(8):2649–64. doi: 10.1007/s00204-018-2234-8
180. Miller KM, Tjeertes JV, Coates J, Legube G, Polo SE, Britton S, et al. Human HDAC1 and HDAC2 Function in the DNA-Damage Response to Promote DNA Nonhomologous End-Joining. Nat Struct Mol Biol (2010) 17(9):1144–51. doi: 10.1038/nsmb.1899
181. Bhaskara S, Knutson SK, Jiang G, Chandrasekharan MB, Wilson AJ, Zheng S, et al. Hdac3 Is Essential for the Maintenance of Chromatin Structure and Genome Stability. Cancer Cell (2010) 18(5):436–47. doi: 10.1016/j.ccr.2010.10.022
182. Yi J, Huang X, Yang Y, Zhu WG, Gu W, Luo J. Regulation of Histone Acetyltransferase TIP60 Function by Histone Deacetylase 3. J Biol Chem (2014) 289(49):33878–86. doi: 10.1074/jbc.M114.575266
183. Nishimoto K, Niida H, Uchida C, Ohhata T, Kitagawa K, Motegi A, et al. HDAC3 Is Required for XPC Recruitment and Nucleotide Excision Repair of DNA Damage Induced by UV Irradiation. Mol Cancer Res (2020) 18(9):1367–78. doi: 10.1158/1541-7786.MCR-20-0214
184. Kao GD, McKenna WG, Guenther MG, Muschel RJ, Lazar MA, Yen TJ. Histone Deacetylase 4 Interacts With 53BP1 to Mediate the DNA Damage Response. J Cell Biol (2003) 160(7):1017–27. doi: 10.1083/jcb.200209065
185. Yang Q, Yang Y, Zhou N, Tang K, Lau WB, Lau B, et al. Epigenetics in Ovarian Cancer: Premise, Properties, and Perspectives. Mol Cancer (2018) 17(1):109. doi: 10.1186/s12943-018-0855-4
186. Zhang M, Xiang S, Joo HY, Wang L, Williams KA, Liu W, et al. HDAC6 Deacetylates and Ubiquitinates MSH2 to Maintain Proper Levels of MutSalpha. Mol Cell (2014) 55(1):31–46. doi: 10.1016/j.molcel.2014.04.028
187. Lagunas-Rangel FA. Current Role of Mammalian Sirtuins in DNA Repair. DNA Repair (Amst) (2019) 80:85–92. doi: 10.1016/j.dnarep.2019.06.009
188. Luo J, Nikolaev AY, Imai S, Chen D, Su F, Shiloh A, et al. Negative Control of P53 by Sir2alpha Promotes Cell Survival Under Stress. Cell (2001) 107(2):137–48. doi: 10.1016/s0092-8674(01)00524-4
189. Conrad E, Polonio-Vallon T, Meister M, Matt S, Bitomsky N, Herbel C, et al. HIPK2 Restricts SIRT1 Activity Upon Severe DNA Damage by a Phosphorylation-Controlled Mechanism. Cell Death Differ (2016) 23(1):110–22. doi: 10.1038/cdd.2015.75
190. Wang RH, Sengupta K, Li C, Kim HS, Cao L, Xiao C, et al. Impaired DNA Damage Response, Genome Instability, and Tumorigenesis in SIRT1 Mutant Mice. Cancer Cell (2008) 14(4):312–23. doi: 10.1016/j.ccr.2008.09.001
191. Van Meter M, Simon M, Tombline G, May A, Morello TD, Hubbard BP, et al. JNK Phosphorylates SIRT6 to Stimulate DNA Double-Strand Break Repair in Response to Oxidative Stress by Recruiting PARP1 to DNA Breaks. Cell Rep (2016) 16(10):2641–50. doi: 10.1016/j.celrep.2016.08.006
192. Toiber D, Erdel F, Bouazoune K, Silberman DM, Zhong L, Mulligan P, et al. SIRT6 Recruits SNF2H to DNA Break Sites, Preventing Genomic Instability Through Chromatin Remodeling. Mol Cell (2013) 51(4):454–68. doi: 10.1016/j.molcel.2013.06.018
193. Zhang P, Brinton LT, Williams K, Sher S, Orwick S, Lai TH, et al. Targeting DNA Damage Repair Functions of Two Histone Deacetylases, HDAC8 and SIRT6, Sensitizes Acute Myeloid Leukemia to NAMPT Inhibition. Clin Cancer Res (2021) 27(8):2352–66. doi: 10.1158/1078-0432.CCR-20-3724
194. Cho MH, Park JH, Choi HJ, Park MK, Won HY, Park YJ, et al. DOT1L Cooperates With the C-Myc-P300 Complex to Epigenetically Derepress CDH1 Transcription Factors in Breast Cancer Progression. Nat Commun (2015) 6:7821. doi: 10.1038/ncomms8821
195. Kim J, Kang J, Kang YL, Woo J, Kim Y, Huh J, et al. Ketohexokinase-A Acts as a Nuclear Protein Kinase That Mediates Fructose-Induced Metastasis in Breast Cancer. Nat Commun (2020) 11(1):5436. doi: 10.1038/s41467-020-19263-1
196. Zhang Q, Agoston AT, Pham TH, Zhang W, Zhang X, Huo X, et al. Acidic Bile Salts Induce Epithelial to Mesenchymal Transition via VEGF Signaling in Non-Neoplastic Barrett's Cells. Gastroenterology (2019) 156(1):130–44.e10. doi: 10.1053/j.gastro.2018.09.046
197. von Burstin J, Eser S, Paul MC, Seidler B, Brandl M, Messer M, et al. E-Cadherin Regulates Metastasis of Pancreatic Cancer In Vivo and Is Suppressed by a SNAIL/HDAC1/HDAC2 Repressor Complex. Gastroenterology (2009) 137(1):361–71, 71.e1-5. doi: 10.1053/j.gastro.2009.04.004
198. Tong ZT, Cai MY, Wang XG, Kong LL, Mai SJ, Liu YH, et al. EZH2 Supports Nasopharyngeal Carcinoma Cell Aggressiveness by Forming a Co-Repressor Complex With HDAC1/HDAC2 and Snail to Inhibit E-Cadherin. Oncogene (2012) 31(5):583–94. doi: 10.1038/onc.2011.254
199. Aghdassi A, Sendler M, Guenther A, Mayerle J, Behn CO, Heidecke CD, et al. Recruitment of Histone Deacetylases HDAC1 and HDAC2 by the Transcriptional Repressor ZEB1 Downregulates E-Cadherin Expression in Pancreatic Cancer. Gut (2012) 61(3):439–48. doi: 10.1136/gutjnl-2011-300060
200. Liao W, Jordaan G, Srivastava MK, Dubinett S, Sharma S, Sharma S. Effect of Epigenetic Histone Modifications on E-Cadherin Splicing and Expression in Lung Cancer. Am J Cancer Res (2013) 3(4):374–89.
201. Shankar E, Pandey M, Verma S, Abbas A, Candamo M, Kanwal R, et al. Role of Class I Histone Deacetylases in the Regulation of Maspin Expression in Prostate Cancer. Mol Carcinog (2020) 59(8):955–66. doi: 10.1002/mc.23214
202. Pathania R, Ramachandran S, Mariappan G, Thakur P, Shi H, Choi JH, et al. Combined Inhibition of DNMT and HDAC Blocks the Tumorigenicity of Cancer Stem-Like Cells and Attenuates Mammary Tumor Growth. Cancer Res (2016) 76(11):3224–35. doi: 10.1158/0008-5472.CAN-15-2249
203. Li QG, Xiao T, Zhu W, Yu ZZ, Huang XP, Yi H, et al. HDAC7 Promotes the Oncogenicity of Nasopharyngeal Carcinoma Cells by miR-4465-EphA2 Signaling Axis. Cell Death Dis (2020) 11(5):322. doi: 10.1038/s41419-020-2521-1
204. Byles V, Zhu L, Lovaas JD, Chmilewski LK, Wang J, Faller DV, et al. SIRT1 Induces EMT by Cooperating With EMT Transcription Factors and Enhances Prostate Cancer Cell Migration and Metastasis. Oncogene (2012) 31(43):4619–29. doi: 10.1038/onc.2011.612
205. Sun L, Kokura K, Izumi V, Koomen JM, Seto E, Chen J, et al. MPP8 and SIRT1 Crosstalk in E-Cadherin Gene Silencing and Epithelial-Mesenchymal Transition. EMBO Rep (2015) 16(6):689–99. doi: 10.15252/embr.201439792
206. Sun T, Jiao L, Wang Y, Yu Y, Ming L. SIRT1 Induces Epithelial-Mesenchymal Transition by Promoting Autophagic Degradation of E-Cadherin in Melanoma Cells. Cell Death Dis (2018) 9(2):136. doi: 10.1038/s41419-017-0167-4
207. Cheng F, Su L, Yao C, Liu L, Shen J, Liu C, et al. SIRT1 Promotes Epithelial-Mesenchymal Transition and Metastasis in Colorectal Cancer by Regulating Fra-1 Expression. Cancer Lett (2016) 375(2):274–83. doi: 10.1016/j.canlet.2016.03.010
208. Simic P, Williams EO, Bell EL, Gong JJ, Bonkowski M, Guarente L. SIRT1 Suppresses the Epithelial-to-Mesenchymal Transition in Cancer Metastasis and Organ Fibrosis. Cell Rep (2013) 3(4):1175–86. doi: 10.1016/j.celrep.2013.03.019
209. Chen IC, Chiang WF, Huang HH, Chen PF, Shen YY, Chiang HC. Role of SIRT1 in Regulation of Epithelial-to-Mesenchymal Transition in Oral Squamous Cell Carcinoma Metastasis. Mol Cancer (2014) 13:254. doi: 10.1186/1476-4598-13-254
210. Zhou W, Ni TK, Wronski A, Glass B, Skibinski A, Beck A, et al. The SIRT2 Deacetylase Stabilizes Slug to Control Malignancy of Basal-Like Breast Cancer. Cell Rep (2016) 17(5):1302–17. doi: 10.1016/j.celrep.2016.10.006
211. Hrgovic I, Doll M, Pinter A, Kaufmann R, Kippenberger S, Meissner M. Histone Deacetylase Inhibitors Interfere With Angiogenesis by Decreasing Endothelial VEGFR-2 Protein Half-Life in Part via a VE-Cadherin-Dependent Mechanism. Exp Dermatol (2017) 26(2):194–201. doi: 10.1111/exd.13159
212. Schoepflin ZR, Shapiro IM, Risbud MV. Class I and IIa HDACs Mediate HIF-1alpha Stability Through PHD2-Dependent Mechanism, While HDAC6, A Class IIb Member, Promotes HIF-1alpha Transcriptional Activity in Nucleus Pulposus Cells of the Intervertebral Disc. J Bone Miner Res (2016) 31(6):1287–99. doi: 10.1002/jbmr.2787
213. Lim JH, Lee YM, Chun YS, Chen J, Kim JE, Park JW. Sirtuin 1 Modulates Cellular Responses to Hypoxia by Deacetylating Hypoxia-Inducible Factor 1alpha. Mol Cell (2010) 38(6):864–78. doi: 10.1016/j.molcel.2010.05.023
214. Chen S, Yin C, Lao T, Liang D, He D, Wang C, et al. AMPK-HDAC5 Pathway Facilitates Nuclear Accumulation of HIF-1alpha and Functional Activation of HIF-1 by Deacetylating Hsp70 in the Cytosol. Cell Cycle (2015) 14(15):2520–36. doi: 10.1080/15384101.2015.1055426
215. Urbich C, Rossig L, Kaluza D, Potente M, Boeckel JN, Knau A, et al. HDAC5 Is a Repressor of Angiogenesis and Determines the Angiogenic Gene Expression Pattern of Endothelial Cells. Blood (2009) 113(22):5669–79. doi: 10.1182/blood-2009-01-196485
216. Tsou PS, Khanna D, Sawalha AH. Identification of Cysteine-Rich Angiogenic Inducer 61 as a Potential Antifibrotic and Proangiogenic Mediator in Scleroderma. Arthritis Rheumatol (2019) 71(8):1350–9. doi: 10.1002/art.40890
217. Kaluza D, Kroll J, Gesierich S, Yao TP, Boon RA, Hergenreider E, et al. Class IIb HDAC6 Regulates Endothelial Cell Migration and Angiogenesis by Deacetylation of Cortactin. EMBO J (2011) 30(20):4142–56. doi: 10.1038/emboj.2011.298
218. Cecconi F, Levine B. The Role of Autophagy in Mammalian Development: Cell Makeover Rather Than Cell Death. Dev Cell (2008) 15(3):344–57. doi: 10.1016/j.devcel.2008.08.012
219. Degenhardt K, Mathew R, Beaudoin B, Bray K, Anderson D, Chen G, et al. Autophagy Promotes Tumor Cell Survival and Restricts Necrosis, Inflammation, and Tumorigenesis. Cancer Cell (2006) 10(1):51–64. doi: 10.1016/j.ccr.2006.06.001
220. Wu W, Luo M, Li K, Dai Y, Yi H, Zhong Y, et al. Cholesterol Derivatives Induce Dephosphorylation of the Histone Deacetylases Rpd3/HDAC1 to Upregulate Autophagy. Autophagy (2020) 17(2):512–28. doi: 10.1080/15548627.2020.1725376
221. Moresi V, Carrer M, Grueter CE, Rifki OF, Shelton JM, Richardson JA, et al. Histone Deacetylases 1 and 2 Regulate Autophagy Flux and Skeletal Muscle Homeostasis in Mice. Proc Natl Acad Sci USA (2012) 109(5):1649–54. doi: 10.1073/pnas.1121159109
222. Shen Q, Shi Y, Liu J, Su H, Huang J, Zhang Y, et al. Acetylation of STX17 (Syntaxin 17) Controls Autophagosome Maturation. Autophagy (2020) 17(5):1157–69. doi: 10.1080/15548627.2020.1752471
223. Majora M, Sondenheimer K, Knechten M, Uthe I, Esser C, Schiavi A, et al. HDAC Inhibition Improves Autophagic and Lysosomal Function to Prevent Loss of Subcutaneous Fat in a Mouse Model of Cockayne Syndrome. Sci Transl Med (2018) 10(456):eaam7510. doi: 10.1126/scitranslmed.aam7510
224. Boyault C, Zhang Y, Fritah S, Caron C, Gilquin B, Kwon SH, et al. HDAC6 Controls Major Cell Response Pathways to Cytotoxic Accumulation of Protein Aggregates. Genes Dev (2007) 21(17):2172–81. doi: 10.1101/gad.436407
225. Pandey UB, Nie Z, Batlevi Y, McCray BA, Ritson GP, Nedelsky NB, et al. HDAC6 Rescues Neurodegeneration and Provides an Essential Link Between Autophagy and the UPS. Nature (2007) 447(7146):859–63. doi: 10.1038/nature05853
226. Liu X, Yamashita T, Shang J, Shi X, Morihara R, Huang Y, et al. Molecular Switching From Ubiquitin-Proteasome to Autophagy Pathways in Mice Stroke Model. J Cereb Blood Flow Metab (2020) 40(1):214–24. doi: 10.1177/0271678X18810617
227. Lee JY, Koga H, Kawaguchi Y, Tang W, Wong E, Gao YS, et al. HDAC6 Controls Autophagosome Maturation Essential for Ubiquitin-Selective Quality-Control Autophagy. EMBO J (2010) 29(5):969–80. doi: 10.1038/emboj.2009.405
228. Lee JY, Nagano Y, Taylor JP, Lim KL, Yao TP. Disease-Causing Mutations in Parkin Impair Mitochondrial Ubiquitination, Aggregation, and HDAC6-Dependent Mitophagy. J Cell Biol (2010) 189(4):671–9. doi: 10.1083/jcb.201001039
229. Jiang X, Huang Y, Liang X, Jiang F, He Y, Li T, et al. Metastatic Prostate Cancer-Associated P62 Inhibits Autophagy Flux and Promotes Epithelial to Mesenchymal Transition by Sustaining the Level of HDAC6. Prostate (2018) 78(6):426–34. doi: 10.1002/pros.23487
230. Garva R, Thepmalee C, Yasamut U, Sudsaward S, Guazzelli A, Rajendran R, et al. Sirtuin Family Members Selectively Regulate Autophagy in Osteosarcoma and Mesothelioma Cells in Response to Cellular Stress. Front Oncol (2019) 9:949. doi: 10.3389/fonc.2019.00949
231. Lee IH, Cao L, Mostoslavsky R, Lombard DB, Liu J, Bruns NE, et al. A Role for the NAD-Dependent Deacetylase Sirt1 in the Regulation of Autophagy. Proc Natl Acad Sci USA (2008) 105(9):3374–9. doi: 10.1073/pnas.0712145105
232. Huang R, Xu Y, Wan W, Shou X, Qian J, You Z, et al. Deacetylation of Nuclear LC3 Drives Autophagy Initiation Under Starvation. Mol Cell (2015) 57(3):456–66. doi: 10.1016/j.molcel.2014.12.013
233. Bowman CJ, Ayer DE, Dynlacht BD. Foxk Proteins Repress the Initiation of Starvation-Induced Atrophy and Autophagy Programs. Nat Cell Biol (2014) 16(12):1202–14. doi: 10.1038/ncb3062
234. Ou X, Lee MR, Huang X, Messina-Graham S, Broxmeyer HE. SIRT1 Positively Regulates Autophagy and Mitochondria Function in Embryonic Stem Cells Under Oxidative Stress. Stem Cells (2014) 32(5):1183–94. doi: 10.1002/stem.1641
235. Tseng AH, Shieh SS, Wang DL. SIRT3 Deacetylates FOXO3 to Protect Mitochondria Against Oxidative Damage. Free Radic Biol Med (2013) 63:222–34. doi: 10.1016/j.freeradbiomed.2013.05.002
236. Zhang M, Deng YN, Zhang JY, Liu J, Li YB, Su H, et al. SIRT3 Protects Rotenone-Induced Injury in SH-SY5Y Cells by Promoting Autophagy Through the LKB1-AMPK-mTOR Pathway. Aging Dis (2018) 9(2):273–86. doi: 10.14336/AD.2017.0517
237. Han D, Jiang L, Gu X, Huang S, Pang J, Wu Y, et al. SIRT3 Deficiency Is Resistant to Autophagy-Dependent Ferroptosis by Inhibiting the AMPK/mTOR Pathway and Promoting GPX4 Levels. J Cell Physiol (2020) 235(11):8839–51. doi: 10.1002/jcp.29727
238. Shi L, Yan H, An S, Shen M, Jia W, Zhang R, et al. SIRT5-Mediated Deacetylation of LDHB Promotes Autophagy and Tumorigenesis in Colorectal Cancer. Mol Oncol (2019) 13(2):358–75. doi: 10.1002/1878-0261.12408
239. Guo J, Wang Z, Wu J, Liu M, Li M, Sun Y, et al. Endothelial SIRT6 Is Vital to Prevent Hypertension and Associated Cardiorenal Injury Through Targeting Nkx3.2-GATA5 Signaling. Circ Res (2019) 124(10):1448–61. doi: 10.1161/CIRCRESAHA.118.314032
240. Ghiboub M, Zhao J, Li Yim AYF, Schilderink R, Verseijden C, van Hamersveld PHP, et al. HDAC3 Mediates the Inflammatory Response and LPS Tolerance in Human Monocytes and Macrophages. Front Immunol (2020) 11:550769. doi: 10.3389/fimmu.2020.550769
241. Li X, Su X, Liu R, Pan Y, Fang J, Cao L, et al. HDAC Inhibition Potentiates Anti-Tumor Activity of Macrophages and Enhances Anti-PD-L1-Mediated Tumor Suppression. Oncogene (2021) 40(10):1836–50. doi: 10.1038/s41388-020-01636-x
242. Lee IK, Song H, Kim H, Kim IS, Tran NL, Kim SH, et al. Rorα Regulates Cholesterol Metabolism of CD8(+) T Cells for Anticancer Immunity. Cancers (Basel) (2020) 12(7):1733. doi: 10.3390/cancers12071733
243. Owen KL, Gearing LJ, Zanker DJ, Brockwell NK, Khoo WH, Roden DL, et al. Prostate Cancer Cell-Intrinsic Interferon Signaling Regulates Dormancy and Metastatic Outgrowth in Bone. EMBO Rep (2020) 21(6):e50162. doi: 10.15252/embr.202050162
244. Li F, Zhao X, Zhang Y, Shao P, Ma X, Paradee WJ, et al. T(FH) Cells Depend on Tcf1-Intrinsic HDAC Activity to Suppress CTLA4 and Guard B-Cell Help Function. Proc Natl Acad Sci USA (2021) 118(2):e2014562118. doi: 10.1073/pnas.2014562118
245. Kim YD, Park SM, Ha HC, Lee AR, Won H, Cha H, et al. HDAC Inhibitor, CG-745, Enhances the Anti-Cancer Effect of Anti-PD-1 Immune Checkpoint Inhibitor by Modulation of the Immune Microenvironment. J Cancer (2020) 11(14):4059–72. doi: 10.7150/jca.44622
246. Bretz AC, Parnitzke U, Kronthaler K, Dreker T, Bartz R, Hermann F, et al. Domatinostat Favors the Immunotherapy Response by Modulating the Tumor Immune Microenvironment (TIME). J Immunother Cancer (2019) 7(1):294. doi: 10.1186/s40425-019-0745-3
247. Knox T, Sahakian E, Banik D, Hadley M, Palmer E, Noonepalle S, et al. Selective HDAC6 Inhibitors Improve Anti-PD-1 Immune Checkpoint Blockade Therapy by Decreasing the Anti-Inflammatory Phenotype of Macrophages and Down-Regulation of Immunosuppressive Proteins in Tumor Cells. Sci Rep (2019) 9(1):6136. doi: 10.1038/s41598-019-42237-3
Keywords: epigenetics, protein acetylation, histone deacetylases, cancer, tumorigenesis
Citation: Hai R, He L, Shu G and Yin G (2021) Characterization of Histone Deacetylase Mechanisms in Cancer Development. Front. Oncol. 11:700947. doi: 10.3389/fonc.2021.700947
Received: 27 April 2021; Accepted: 05 July 2021;
Published: 29 July 2021.
Edited by:
Jiang-Jiang Qin, Institute of Cancer and Basic Medicine (CAS), ChinaReviewed by:
Carole Seidel, Institut National de Recherche et de Sécurité (INRS), FranceCopyright © 2021 Hai, He, Shu and Yin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Gang Yin, Z2FuZ3lpbkBjc3UuZWR1LmNu
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.