 Francesca Mancini
Francesca Mancini Ludovica Giorgini
Ludovica Giorgini Emanuela Teveroni
Emanuela Teveroni Alfredo Pontecorvi
Alfredo Pontecorvi Fabiola Moretti
Fabiola Moretti- 1Research Unit on Human Reproduction, International Scientific Institute Paul VI, Fondazione Policlinico A. Gemelli, IRCCS, Rome, Italy
- 2Institute of Biochemistry and Cell Biology, National Research Council of Italy, Monterotondo, Italy
- 3Catholic University of the Sacred Heart of Rome, Fondazione Policlinico A. Gemelli, IRCCS, Rome, Italy
Sex profoundly affects cancer incidence and susceptibility to therapy, with sex hormones highly contributing to this disparity. Various studies and omics data suggest a relationship between sex and the oncosuppressor p53 circuitry, including its regulators MDM2 and MDM4. Association of this network with genetic variation underlies sex-related altered cancer risk, age of onset, and cancer sensitivity to therapy. Moreover, sex-related factors, mainly estrogenic hormones, can affect the levels and/or function of the p53 network both in hormone-dependent and independent cancer. Despite this evidence, preclinical and clinical studies aimed to evaluate p53 targeted therapy rarely consider sex and related factors. This review summarizes the studies reporting the relationship between sex and the p53 circuitry, including its associated regulators, MDM2 and MDM4, with particular emphasis on estrogenic hormones. Moreover, we reviewed the evaluation of sex/hormone in preclinical studies and clinical trials employing p53-target therapies, and discuss how patients’ sex and hormonal status could impact these therapeutic approaches.
Introduction
Cancer statistics reveal sex (meant as biological factors) differences in incidence, therapy response, and mortality of many cancers (1). The majority of these tumors, excluding sex-related prostate ovary and breast cancer, present a higher incidence, increased invasive property, and cancer death in males compared to female, even after correction for environmental exposures and risk habits, as smoking and alcohol consumption, more common among men (2). Primarily, genetic factors residing on sex chromosomes contribute to these disparities (3). Indeed, the X chromosome contains various genes involved in oncogenesis (4). Since a percentage of genes is not silenced in inactivated X-chromosome, their higher expression levels in females compared to males may underlie cancer differences (4, 5).
Additionally, an important factor contributing to sexual differentiation is the circulating sex hormones with a strong relevance of estrogen (6, 7). Women have a higher risk of developing lung cancer upon smoking than men. Various studies suggest that the interaction between tobacco carcinogens and endogenous and exogenous sex steroids may be relevant (8). Nonetheless, this disparity persists among adults aged 85 and older, thus beyond the women’s reproductive age (9). The levels of intra-tissue sex hormones determined by local production of the estrogen (intracrinology) are assuming great importance in many hormone-related tumors (10, 11). Finally, epigenetic modification of DNA is different in the two sexes with DNA methylation enhanced in various organs of experimental feminine rodents. Since cancer is linked to epigenetic dysregulation, these differences play an important function, too [reviewed in (5)].
All these factors can variously affect molecular pathways involved in oncogenesis. The p53 pathway is one of the most relevant in tumor development and therapy response, and p53 is a crucial oncosuppressor in both humans and rodents (12, 13). TP53 gene is mutated in about 50% of human cancers, while the protein is inactivated in tumors bearing wild-type p53 (wt-p53). One of the most frequent inactivation mechanisms of wt-p53 is the overexpression of its negative regulators, MDM2 and MDM4 (also MDMX). These two proteins form a heterodimer that controls p53 activity and levels. In addition, the two proteins function singularly towards p53 with different outcomes depending on the tissue (14, 15). Given its relevance, re-activation of wt-p53 oncosuppressive activity is a field of intense study. In tumors retaining wt-p53, most of the approaches target the inhibitory proteins MDM2/MDM4, either singularly or in combination [reviewed in (16, 17)]. These approaches apply mainly to solid tumors as myeloma, melanoma, and liposarcoma, although some trials have also been applied to acute myeloid leukemia (AML) (for details, see Therapies Directed to wt-p53 Re-Activation). Unfortunately, at present, none of these therapeutic approaches have reached the patient’s bed.
Over time, different studies have demonstrated crosstalk between p53/MDM2/MDM4 and sex. Although p53 and its regulators MDM2 and MDM4 genes reside on autosomes, genes associated with p53 circuitry reside on the X chromosome, affecting its function in a sex-related manner. Moreover, genetic variations in the p53/MDM2/MDM4 genes (in the promoter, 3′UTR region, introns, or coding sequence) are responsive to hormone status, affecting p53/MDM4/MDM2 levels and function. Despite all these data, sex differences are not consistently evaluated in preclinical and clinical trials targeting the p53 circuitry.
In this review, we summarize data regarding sex-related factors associated with wt-p53, MDM2, and MDM4. Particularly, we recapitulate the sex-related factors that can affect p53 function by acting on p53 itself or its regulators MDM2 and MDM4. Moreover, we will review sex differences in therapeutic approaches aimed to reactivate wt-p53 and discuss how the consideration of sex could affect the results of these approaches.
Effects of Sex on p53 Circuitry
p53
Given the importance of the oncosuppressor p53 in counteracting cancer development, many studies investigated the association of p53 status with sex-related genetic factors and the potential effect of sex-related factors on p53 function.
Genetic Factors
A direct association of p53 status with sex-associated cancer difference has been reported in various tumors. In lung cancer, p53 mutations are more frequent in females than in males [reviewed in (8)]. Also, females carrying mutant p53 have an increased risk of developing adrenocortical carcinoma (ACC), suggesting that the p53 oncosuppressive activity impacts ACC development more strongly in females than in males (18). Since ACC risk is also increased in females of pediatric age (19), factors other than sex hormones likely affect p53 function in ACC in a sex-related way (5). At present, these factors have not been identified.
In exon 4 of the p53 gene, the SNP-R72P is present (Table 1). The R72 variant possesses higher ability to induce apoptosis compared to P72 [reviewed in (20)]. A hospital-based case–control study of hepatocellular carcinoma (HCC) development in a Turkish population demonstrated that the P72 homozygote (p53Pro/Pro) is associated with increased HCC risk in males but not in females (21). A similar association of the variant alleles (P/R + P/P) has been found with a particular squamous cell carcinoma (Kangri cancer) in Indian male subjects (22). These studies suggest that the penetrance/efficacy of the p53Pro/Pro variant is different among male and female subjects. Which factors, genetic or hormonal, alter the p53Pro/Pro activity is currently unknown. It has been proposed that biallelic expression of X-linked tumor-suppressor genes in females explains a portion of the reduced cancer incidence in females compared to males across various tumor types (4). Recently, Haupt and collaborators reported X-linked genes associated with the p53 network. Starting from bio-informatic analyses, they showed that in many non-reproductive cancer histotypes, p53 mutation is more frequent in males than in females with a concomitant lower survival rate (23). Then, they identified X-linked genes encoding for proteins essential for genomic fidelity that are connected to p53. Due to the chromosome X inactivation, females are protected from these gene germline mutations, while males are exposed to a higher risk because they have only a single copy of the X chromosome. The underlying hypothesis is that this link brings to enhanced selection of p53 inactivation in men. This phenomenon is not evident in hormone-dependent tumors since in male breast cancer the frequency of p53 inactivation is reduced compared to females (24). Molecular proof of the ability of these X-linked genes to confer p53-mediated increased protection from cancer in the female gender has not yet been provided.
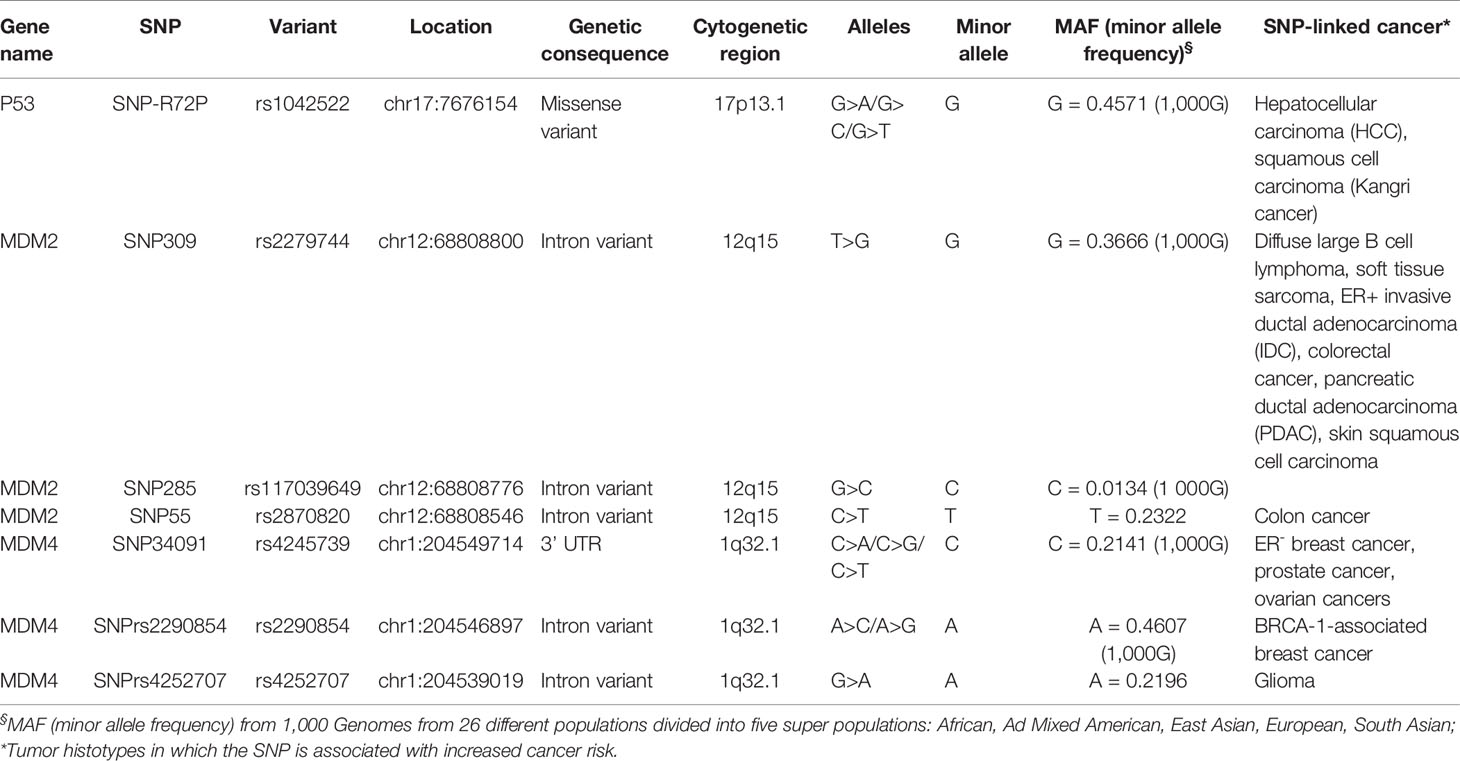
Table 1 Summary of p53, MDM2, MDM4 SNPs relevant to cancer in a sex/hormone-related way.
Hormone Activity
Many studies investigated the effect of sex hormones, especially estrogen, on p53 function (25). The picture deriving from these studies is very complex, in some cases reporting opposite results. An important factor that may contribute to this inconsistency is the type of estrogen receptor involved. Indeed, estrogen activity is mediated by the membrane-bound G protein-coupled estrogen receptor 1 (GPER or GP3R0) and the nuclear estrogen receptors (ERs) α and β (ERα, ERβ), with ERβ often exhibiting opposite activity to ERα. Hormone-stimulated nuclear ERs translocate into the nucleus and direct transcription as homo- and heterodimers or as partners of other transcription factors (indirect genomic signaling) (26). Additionally, the two nuclear receptor genes (named ESR1 and ESR2 corresponding to ERα and ERβ, respectively) originate alternative forms that can interact with the full-length receptors and repress their function. Therefore, the studies performed by stimulation with 17β-estradiol (E2, the primary estrogenic hormone that interacts with all estrogen receptors) without characterization of the receptor and the ERα and ERβ isoforms present in the system can be variously interpreted. An additional factor that adds complexity to the interpretation of estrogen activity towards p53 is the positive regulation of MDM2 levels by estrogens (see relative paragraph MDM2). Since MDM2 is a negative regulator of p53, the fine-tuning of MDM2 and p53 by estrogens can result in different outcomes.
Since ERα is a critical therapeutic target in hormone-responsive breast and endometrial cancers, many data described in literature investigated the interplay between p53 and ERα. Particularly, since p53 mutation is not common in estrogen-responsive breast cancer, accounting for about 20% of tumors (27, 28), many studies focused on this tissue.
Two main and opposite estrogenic hormone activities towards p53 have been described: a positive activity at different levels and a repressive activity mainly related to p53-transcriptional function.
Cooperative Estrogen-p53 Activity
As concerns the mechanism of cooperation, there are various studies from Olivier’s group (29–31). In cell lines derived from breast cancer MCF7 cells, they demonstrated that estrogen through endogenous ERα increases p53 levels and enhances p53-mediated response to DNA damage. They further showed that focal adhesion kinase (FAK), a critical regulator of adhesion and motility, is downregulated by p53 in response to E2 (31). These studies have been further confirmed by Berger and colleagues, who demonstrated that the p53 promoter contains four ERα responsive elements (ERE) (32). Accordingly, knockdown of ERα leads to decreased p53 mRNA and protein levels and its targets, MDM2 and p21. This results in increased colony formation in an estrogen-free medium upon a cytostatic dose (100–400 nM) of doxorubicin (32). Of note, these authors demonstrate that the ERβ receptor is not involved since its exogenous expression does not alter p53 levels and activity. In support of this data, Klaus and colleagues analyzed the radiation-responsiveness of the mammary epithelium in ovariectomized mice upon E2 and progesterone (P) treatment (33). These hormones activate the p53 response to radiation in terms of increased p21. Also, by comparing the mammary epithelium of BALB/c mice with different p53 status (Trp53+/+, Trp53+/−, Trp53−/−), they demonstrated that E + P upregulated p53 nuclear levels and apoptosis upon ionizing radiation, also in the haploinsufficient background (BALB/c Trp53+/−) (34). Interestingly, parity acted similarly and delayed the onset of spontaneous mammary tumors in these mice, confirming a protective role of hormones towards breast cancer development. Similar data were obtained by Sivaraman using the rat model (35). Importantly, epidemiologic studies show that women with full-term pregnancy have a significantly reduced risk of developing ER+ breast cancer (36). In agreement with these studies, Kupperwasser and colleagues reported that mare serum gonadotropin (PMSG) and human gonadotropin (hCG) treatment increases p53 nuclear fraction leading to enhanced mammary gland apoptosis following ionizing radiation (37). Interestingly, a recent proteome analysis of MCF-7 cells demonstrated that estrogen modulates cyto-nuclear shuttling; in response to estrogen, dynamic subcellular redistribution of proteins is the major phenomenon compared to the alteration of protein levels (38). Overall, this data strongly supports that estrogen enhances the oncosuppressive function of p53 in the breast tissue. Also, in another normal epithelial context as Young Adult Mouse Colon cells (YAMC), estrogen induces p53 downstream targets as PUMA, Bcl-2-associated X protein (Bax), and Noxa, and sensitizes cells to p53-mediated apoptosis (39), extending the cooperative function of p53 and estrogen in another epithelium. The good prognosis of ERα+/wt-p53 breast cancer can also be related to a cooperative activity between these two factors. To integrate this hypothesis, p53 inhibits ERα transcriptional activity on synthetic estrogen-responsive elements (40), suggesting a tumor-suppressive function of p53 towards ERα in hormone-activated signaling pathways.
This data raises a question about the consequences on p53 of anti-estrogenic therapies in breast cancer (41). Since these drugs antagonize ER function, they should reduce p53 oncosuppressive activity. The observation that anti-estrogenic therapy displays partial agonist activity in a gene-specific and tissue-specific manner partly solves this issue (42). In this regard, Olivier’s group demonstrated that 4-hydroxy-tamoxifen (OHT), a selective estrogen receptor modulator (SERM), suppresses cell proliferation more effectively in breast cancer cell lines bearing wild-type p53 compared to cells with mutated p53. Furthermore, p53 expression levels have been reported as a positive prognostic factor for OHT treatment (43). Conversely, the activity of fulvestrant (ICI 182,780), a selective estrogen receptor degrader (SERD) that acts by inducing degradation of nuclear estrogen receptors is independent of p53 status (30). This data suggests that the p53-estrogen crosstalk is differently affected by estrogen, OHT, and fulvestrant, and supports the efficacy of anti-estrogenic therapies in wt-p53 breast cancer.
By considering hepatic tissue, Pok and colleagues showed that testosterone positively regulates hepatocyte cell cycle regulators and reduces p53 and p21, while E2 plays the opposite effect (44). Accordingly, in liver cancer cell lines, E2, via ERα, activates the transcription of p53 and its target miR-23a, promoting p53-dependent apoptosis and, in turn, inhibiting HCC development (45). Since men are more susceptible than women to hepatocellular carcinoma (HCC) at age <60 years (46), this data supports a protective role of estrogenic hormones in liver cancer risk and highlights a possible mechanism by which sex hormones contribute to establishing the male prevalence of hepatocarcinoma.
Few studies analyzed the effects of ERβ on p53. In colon cancer cell lines, ERβ overexpression enhances p53 levels and activity through p14ARF-mediated downregulation of MDM2. Of note, this cell outcome was observed in many but not all colon cancer cell lines analyzed. The reason for these results remains unexplained (47). Similarly, in the human colon metastatic LoVo cell line, overexpression of ERβ enhances p53-mediated apoptosis in an estrogen-dependent manner (48). Overall, these studies indicate a proapoptotic function and anti-oncogenic activity of ERβ towards p53 although in tissues other than the breast.
Antagonistic Estrogen-p53 Activity
Opposite to this view, Das’s group reported that ERα inhibits p53 function on some transcriptional targets (49). The model proposed by these authors is that ERα and p53 cooperatively bind on the promoters of some p53-targets genes at whose levels ERα represses p53 activity. In most of these studies, cell outcome is not reported, so they are not entirely comparable to previous studies. One limitation of these studies is that they are often based on ERα overexpression. Indeed, in MCF7 without ERα overexpression, endogenous ERα does not affect p53 transcriptional function following E2 or fulvestrant treatments (50). A further explanation can derive from studies of Brown’s group (51). They demonstrated in MCF7 that E2 and OHT reduce the apoptosis induced by cytotoxic high dose of doxorubicin (10 μM) whereas fulvestrant is inefficacious. Using genome-wide approaches, they reported the modulation of a subset of p53 and ER target genes, but not changes of p53 levels and its binding to these gene promoters. This data suggests that different p53 targets can be variously regulated upon specific estrogen treatments, leading to different cell outcomes. Also, Lewandowski and collaborators reported an antagonistic activity of estrogen towards p53. In MCF7 cells, E2, through ERα, mediates the relocalization of p53 from the nucleus to the cytoplasm, inhibiting its transcriptional activity as revealed by decreased p21 levels. This, in turn, results in reduced sensitivity of MCF7 to TNF-mediated cell death while ERβ behaves oppositely, antagonizing cytoplasmic relocalization of p53 (52). Interestingly, a study from Bargonetti’s group demonstrated that in MCF7, estrogen-induced cell proliferation and downregulation of p21 are p53-independent but MDM2 dependent (53). Therefore, E2-induced p21 modulation as a marker of p53 activity can be misleading. Moreover, the different crosstalk between ERα and p53 could also be ascribed to specific treatments, such as cytostatic (32) vs. cytotoxic (49) doses of doxorubicin, γ-irradiation (33), or TNFα (51).
MDM2
MDM2 protein is involved in a negative feedback loop with p53, by which p53 activates transcription of the MDM2 oncogene, which in turn inhibits p53 activity. This loop is essential to maintain both proteins at moderate levels and reset cell behavior after p53 activation. Due to their intertwined role, an unbalanced expression or activity of MDM2 is involved in cancer, and many studies highlighted sex-related factors leading to unbalanced MDM2 (15). Additionally, MDM2 regulates targets other than p53, which also have relevance to cancer (54).
Genetic Factors
The expression of the MDM2 gene is probably the best example of sex-mediated regulation of p53 circuitry in cancer. An initial report from Blaydes’s group demonstrated the activation of the MDM2 P2 promoter by the AP1-ETS transcription factors in an ERα dependent manner (55). Subsequently, Bond and colleagues identified the SNP309 T/G within this promoter (56) (Table 1). The SNP309G variant extends the length of the DNA binding site for specificity protein 1 (Sp1), increasing the affinity for this transcriptional factor. As a result, Sp1 increases MDM2 levels, leading to an attenuation of the oncosuppressive p53 activity. This SNP is indeed associated with an early age of cancer diagnosis. Since Sp1 is a co-transcriptional factor of ERs, the authors demonstrated that SNP309 accelerates the age of onset of various cancer types (diffuse large B cell lymphoma, soft tissue sarcoma, invasive ductal breast carcinoma, IDC)—in female but not in male patients (57). Accordingly, this sex difference is associated with ER+ but not ER− invasive ductal breast carcinoma and is more evident in non-menopausal women. Similar data were reported for colorectal cancer, in which female SNP309G carriers were diagnosed with cancer earlier than those carrying the wild-type gene (58). Other studies evidenced the relevance of this SNP in a p53-independent way due to the ubiquitin ligase activity of MDM2 towards other targets (53, 59, 60). Overall, this data underlies the role of the estrogen-mediated pathway on MDM2 function through SNP309. At odds with this data, some studies did not find an association between SNP309 and estrogen status on cancer risk [reviewed in (61)]. Although, in many cases, the authors did not take into account the sex and the hormone levels, a resolving study from Lønning’s group defined the presence of the additional SNP285G>C, which antagonizes the Sp1 binding to SNP309 (Table 1) (62). The presence of this SNP reduces the risk of both ovarian and breast cancers, highlighting the relevance of MDM2 fine-tuning for cancer development.
Further complexity has been recently added by Lozano’s group, who demonstrated that MDM2 SNP309G exhibits tissue-specific regulation and different impacts on cancer risk (63). Accordingly, Grochola and colleagues showed that in pancreatic ductal adenocarcinoma (PDAC), the SNP309G is associated with earlier onset in men but not in women. They attributed this effect to the function of Sp1 as a coactivator of androgen receptors present in PDAC (64).
In 2015, Kato’s group identified an additional SNP in MDM2-P2 promoter, the SNP55 (rs2870820, C/T) (65). Both SNP55T and SNP55C bind Sp1, whereas only the C allelic variant creates an additional consensus sequence for the transcriptional factor NF-kB. The NF-kB p50/p50 homodimer interferes with Sp1 transcriptional activity, as demonstrated by Hirano and colleagues (66). In the context of MDM2, this results in transcriptional repression of the gene (65). Therefore, this SNP further contributes to fine-tuning MDM2 levels. Subsequently, Helwa and colleagues reported that women with SNP55TT or SNP55TC genotype have a higher risk of colon cancer, particularly left-sided colon cancer, than those with SNP55CC genotype (66). Conversely, this SNP does not seem to affect breast, lung, prostate, and endometrial cancer risk (66). In a recent study, the same group analyzed the impact of the combination of all three SNPs and reported that the SNP55T allele variant is associated with a lower risk of endometrial cancer in women carrying the SNP285G and SNP309T. At the same time, this haplotype is not correlated with the risk of ovarian cancer (67).
These results collectively validate the MDM2 SNPs as important cancer modifiers by attenuating the cell-protective activity of p53 or p53-independent pathways. To date, the characterization of these SNPs in the application of MDM2-target therapies has not been reported.
Hormone Activity
Besides the effect of the hormone on MDM2 transcription through SNPs, other studies evidenced a regulation of MDM2 by the estrogenic pathway at the protein levels. ERα stabilizes MDM2 since the use of fulvestrant significantly reduces the MDM2 half-life. Particularly, fulvestrant decreased basal expression of MDM2 through increased protein turnover in the absence of E2 (68). This in turn, increases cell apoptosis and sensitivity of MCF7 breast cancer cells to chemotherapic drugs, doxorubicin, paclitaxel, and etoposide (68). Accordingly, high levels of MDM2 are detected in ERα+ breast carcinoma (69). In a reciprocal fashion, Cavailles’s group demonstrated MDM2 activity towards ERα: MDM2 interacts with ERα and p53 and induces ERα degradation through its ubiquitin ligase activity in a ligand-independent manner (70). Conversely, in a p53-independent way, Bargonetti’s group reported the ability of MDM2 to facilitate the estrogen-mediated activation of cell proliferation (53), suggesting different activities of MDM2 dependent on p53 background.
The overall positive effects of estrogenic hormones on MDM2, at the mRNA and protein levels, suggest that anti-estrogenic therapies in breast cancer could synergize with MDM2-targeted drugs.
MDM4
MDM4 is a double-faced p53 regulator: it cooperates with MDM2 in inhibiting p53, thus behaving as an oncogenic factor. Conversely, under DNA damage conditions, it cooperates with p53 and promotes cell apoptosis (71, 72). MDM4 also possesses a p53-independent function by suppressing the mTOR-mediated pathway (73, 74). Of relevance, the proapoptotic activities reside on the cytoplasmic fraction of MDM4 (75, 76). Accordingly, most tumors show high levels of MDM4 in the nuclear compartment (77). Wide-genome studies reported the association of specific SNPs in the MDM4 gene with hormone-mediated cancer and estrogen receptor-negative breast tumors, suggesting that the presence of ERs may select for a particular MDM4 gene status.
Genetic Factors
Despite the description of various SNPs in the MDM4 gene (78), the majority of data focused on the SNP 34091 (A > C) in the 3′UTR region of human MDM4 (Table 1). Data collected from the Collaborative Oncological Gene-environment (COGS) showed a significant association of this SNP with hormone-dependent cancers (79–81). This SNP located 32 bp downstream of the stop codon should create an illegitimate binding site for miR-887 (80, 82) and has-miR-191, a miRNA often expressed in tumor tissues, leading to downregulation of MDM4 in the MDM4-C variant (83). Unexpectedly, the SNP34091C variant is associated with increased risk of breast cancer, high-grade ovarian cancer (HGSOC), and prostate cancer suggesting a specific sensitivity of these hormone-dependent cancers to presumably low MDM4 levels (79, 84). SNP34091C is associated with increased cancer risk only in ER-negative breast cancer, suggesting that the presence of ERs interferes with MDM4 activity (79, 85). Additionally, in ER-negative breast cancer and HGSOC, the presence of this SNP is not correlated to the status of p53, suggesting that ERs interfere with MDM4 p53-independent activities. This data is in agreement with the cytoplasmic proapoptotic function of MDM4. The ER ability to re-localize MDM4 in the nucleus would abrogate the MDM4 cytoplasmic anti-oncogenic function. Accordingly, many human tumors express nuclear MDM4 (77). Other studies on ovarian cancer reported contradictory results. Wynendaele reported an association of the AA haplotype with reduced overall survival of ovarian carcinoma. Conversely, Gansmo reported an association of C variant with HGSOC (84). A possible explanation raised by these last authors is that in HGSOC, p53 is mutated in almost 90% of tumors. Therefore, the effect of this variant is mediated via pathways other than p53 (84).
The association between hormone-related pathways and this SNP is supported by the observation that it does not alter colon- and lung-cancer risk in a large population-based control study (86). Of merit, this study distinguished male and female patients compared to male and female controls. Association with prostate cancer was not found, although some authors reported a trend of association of the C allele with higher prostate cancer aggressiveness (87).
At odds with the previous study, other authors reported a reduced cancer risk for the SNP34091C variant in esophageal squamous cell carcinoma and non-Hodgkin lymphoma (88, 89). Information on ERs status in these studies is not available and cannot be entirely compared to previous results. Additionally, the different geographic populations (Caucasian vs. Chinese) may underlie this discrepancy. Indeed, the frequency of the MDM4-SNP34091 is different between these two populations (90). Finally, other miRNAs may affect SNP function (91).
Additional SNPs have been identified in other cancer types. Couch and colleagues reported the association of the minor allele of MDM4 SNP rs2290854 with breast cancer risk in mutant BRCA1 carriers, suggesting that this MDM4 variant can be a modifying factor for breast cancer in this mutant background (92) (Table 1). Also, in this case, this SNP is associated with ER− but not ER+ breast cancer. Moreover, two recent papers analyzed the sex-related association of various MDM4 SNP with glioma in the European and Chinese populations (93, 94). The authors evidenced the association of the A allele of a novel MDM4 SNP (rs4252707) (Table 1) with the increased risk of this tumor. However, although the higher frequency of glioma in males, they did not find an association with sex.
Hormone Activity
Lozano’s group was the first to demonstrate a dissimilar activity of overexpressed MDM4 between male and female mice (95). Her group reported a higher incidence of multiple tumors and a decreased animal survival in Mdm4 transgenic p53-null males but not in females, indicating sexual dimorphism of Mdm4 activity, at least in rodents. Using the same animal model, our group demonstrated that in a wt-p53 background, Mdm4 promotes tumor development following DNA damage in a sex-independent way, indicating that Mdm4 oncogenic properties are not affected by sex. In contrast, there is increased chemotherapy sensitivity in Mdm4-overexpressing male but not in female mice. Molecular analysis demonstrated that E2 re-localizes MDM4 in the nucleus, antagonizing the cytoplasmic MDM4-mediated DNA damage response (96). Noteworthy, treatment of animals with fulvestrant rescues MDM4-mediated proapoptotic activity and increases tumor sensitivity to chemotherapy. Accordingly, MDM4 nuclear localization and intra-tumor estrogen availability correlate with decreased platinum sensitivity and apoptosis and predict poor disease-free survival in human HGSOC. A specular finding was reported by Das’s group (97). They showed that ERα is a positive regulator of MDM4 oncogenic activity, and treatment of tumor cells with ER inhibitors (fulvestrant or OHT) reduces MDM4 protein levels. Overall, this data indicates that sex and/or ERα regulate MDM4 activity. Depending on p53 background, they can result in opposite outcomes. The assessment of sex/hormone status in MDM4-target therapy could confirm the relevance of these factors in drug efficacy and suggest the potential usefulness of combinatorial treatments.
Therapies Directed to wt-p53 Re-Activation
In the last decades, different therapeutic approaches were developed to reactivate wt-p53 functions in cancer. Peptides and small molecule compounds were described to target the critical inhibitory binding of MDM2 and MDM4 to p53 or to stimulate p53 by acting on the protein itself [reviewed in (16, 98, 99)].
Compounds in the p53 network that entered clinical trials are summarized in Table 2. Among them, the majority refers to phase I studies that evaluate safety and tolerability, with few or no results about the efficacy of the therapies. Despite the low number of patients usually enrolled in phase I, none of these trials reported the hormone/gender status in their evaluations. Here, we briefly reviewed the active clinical trials suggesting the potential role of sex/hormone.
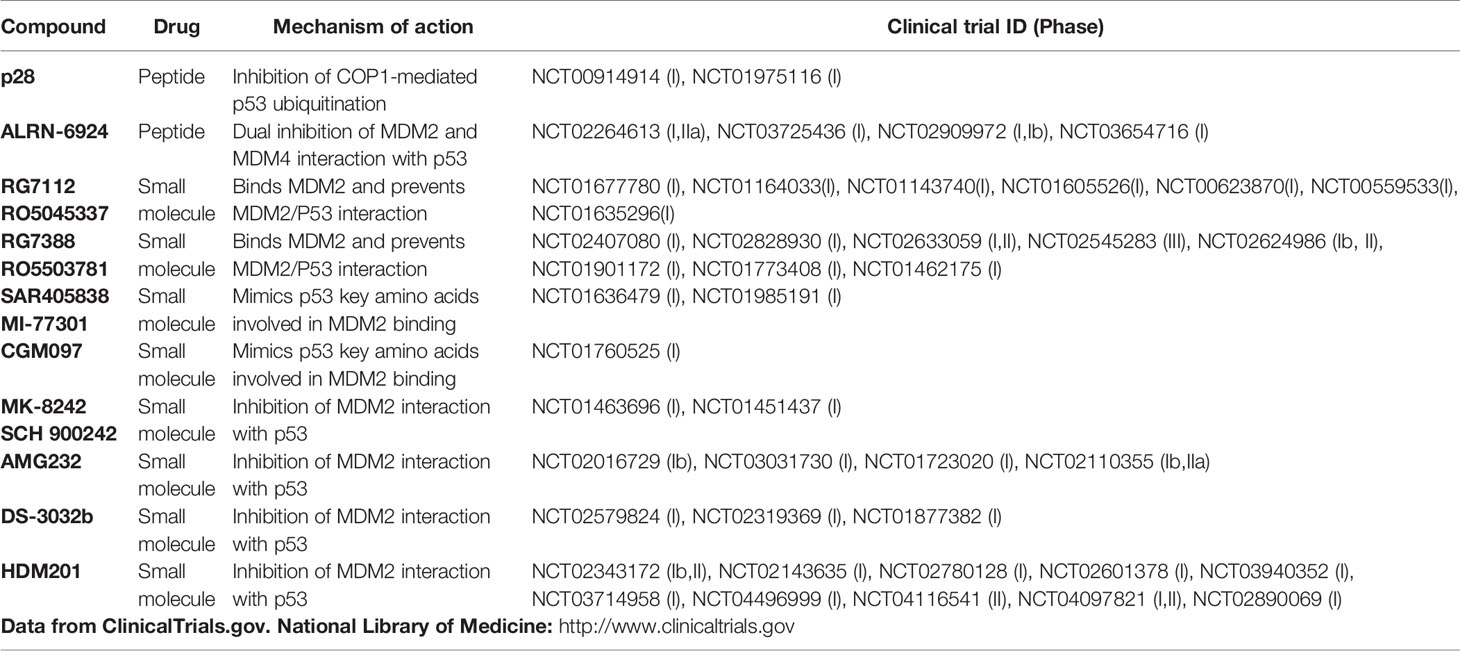
Table 2 Strategies for wt-p53 re-activation, which entered clinical trial phases.
P28 is a peptide derived from the Azurin, a Pseudomonas aeruginosa redox protein that exerts an antiproliferative activity towards cancer cells by inhibiting COP-1 mediated ubiquitination of p53, thus in an MDM2/MDM4 independent way (100, 101). In a Phase I study (NCT00914914), p28 proved preliminary evidence of anti-tumor activity in patients with melanoma and colon cancer (Table 2). Although no results regarding gender are displayed (102), P28 efficacy towards these non-hormone mediated cancers might suggest that COP-1 mediated regulation of p53 is not affected by hormone status. Accordingly, a second Phase I study NCT01975116 established safety in children with recurrent or refractory central nervous system cancer (CNS) (103). Comparing this peptide efficacy in males and females could highlight the potential effects of genetic factor/s on p53 function.
ALRN-6924 is a peptide that targets both MDM2 and MDM4 and prevents their binding to p53 (104). Phase I studies evaluated safety in acute myeloid leukemia (AML), myelodysplastic syndrome (MDS), and solid cancers (Table 2). Two recent trials are recruiting patients to evaluate the safety and efficacy of ALRN-6924 in combination with drugs used in chemotherapy as Cytarabine for patients with leukemia (NCT03654716) or Paclitaxel for those with breast cancer (NCT03725436). The results of this last trial could be of interest to evaluate the relevance of ERs in p53 circuitry since the inclusion criteria are breast cancer carrying wt-p53 and positive for ERs.
RG7112 and RG7388, two derivatives of cis-imidazoline molecule known as “nutlin”, are under testing in clinical trials (Table 2). RG7112, despite promising results in terms of p53 activation, was dropped because of significant toxicity (105). RG7388, known as Idasanutlin, is a nutlin analog with a higher affinity and specificity for MDM2. This compound underwent several clinical trials, including phase II and phase III trials in combination with chemotherapy or novel therapies as monoclonal antibodies and other new anticancer small molecules (Table 2). Based on the relevance of ERα in MDM2 levels, the efficacy of this drug could be increased in those tumors previously shown more sensitive to SNP variations.
Spirooxindole-based compound mimics the p53 key amino acids that bind MDM2. MI-77301 from Sanofi (106) and the substituted dihydroisoquinolinone derivative CGM097 from Novartis, entered in phase I clinical trials (Table 2). For both compounds, the anti-tumor activity has been verified in preclinical studies (107), whereas no results have been reported from clinical trials. Of interest, p53 status is not sufficient to predict CGM097 sensitivity in a panel of 477 cell lines from the Cancer Cell Line Encyclopedia (CCLE). In the 13 gene signature predicting CGM097 response, MDM2 levels are the most significant predictor (108). Given the relevance of ER/hormone status in affecting MDM2 levels, the assessment of hormone status could be relevant in the analysis of the efficacy of these compounds in the related clinical trials.
MK-8242 from Merck is a small-molecules that inhibits MDM2 interaction with p53 and can induce growth arrest at very low concentration (109). In the NCT01463696 trial, three of 47 patients with liposarcoma showed a partial response, and 31 patients stable disease (110) (Table 2). Since 60% of these patients are males, it would be interesting to re-evaluate the results by separate analysis of patients based on sex/hormone status.
AMG232 by Amgen is a piperidinone-derived compound that acts as a potent inhibitor of the MDM2−p53 complex and shows high anti-tumor activity in xenograft models (111). This compound underwent several clinical trials, including phases I and II, as single or combination treatments for solid tumors, AML, myeloma, and melanoma (Table 2), but its effects have never been evaluated in the light of sex/hormones.
DS3032b developed by Daiichi Sankyo showed stable disease in 77% of patients with solid tumors (112) and reduced bone marrow blasts after the first cycle in half patients and complete remission in two patients with hematological malignancies.
HDM201 developed by Novartis is an imidazopyrrolidinone analog that inhibits the P53–MDM2 interaction with high efficiency. It was able to induce p53 dependent apoptosis and tumor regression in xenograft tumor models (113). Many clinical studies are ongoing in patients with wild-type p53 tumors of different histotypes such as AML, solid tumors, and multiple myeloma (Table 2). Some results of clinical benefit from these trials have been reported: approximately 25–30% of patients had a partial response or stable disease, although the tumor histotypes are not specified (114).
Many of the tumors under these clinical trials—including myeloma and lymphoma—show sex differences, with male prevalence. Evaluating these drugs in terms of sex and/or hormone status could give valuable information for more personalized medicine.
Discussion
Although sex is an important factor in determining cancer development, progression, and sensitivity to therapy, sex-based studies of cancer biology and treatment are still largely insufficient, and the factors driving the sex-related cancer disparity remain to be clarified. Even after recommendations from NIH and other funding agencies to consider sex and gender at all levels of biomedical research, animal studies and clinical trials that distinguish gender populations are few (115). P53-target therapies do not make an exception, as demonstrated by Table 2. None of those clinical trials reported separate results for men and women or considered the hormonal status of patients. Still, many studies demonstrate that p53 activity is affected by sex-related genetic and/or hormone determinants. This review reflects the abundance and complexity of the sex-related molecular factors that affect p53 response in human tumors. Based on the data here reviewed, nowadays, it is difficult to predict which genetic or hormonal factors could contribute to defining a more personalized application of single or combinatorial treatments.
For this reason, evaluation of sex/hormones in preclinical studies and clinical trials could help clarify the factors that finally affect p53 function in vivo and could guide future molecular studies besides drive a more appropriate and successful application of these therapies. Indeed, “false” negative or positive results could be due to the confounding effects of mixed backgrounds. As stated by Clayton and Collins, “inadequate inclusion of female cells and animals in experiments and inadequate analysis of data by sex may well contribute to the troubling rise of reproducibility in preclinical biomedical research” (115).
Inclusion of sex/hormone status in the analysis of p53 data could open the possibility of combinatorial treatments with anti-hormone or other target therapies. Based on the beneficial effect of SERM on tumors with wt-p53 and possible depressing activity of MDM2 levels, a combinatorial treatment of SERM therapy with p53-reactivating drugs in breast cancer could be hypothesized. Accordingly, Rozeboom and colleagues recently suggested a new clinical trial based on triple therapy with a BCL2 inhibitor (venetoclax), an anti-estrogen (tamoxifen/fulvestrant), and an MDM2 inhibitor (AMG-232/MI-77301) in the ER+/WT TP53 metastatic breast cancer setting (116). Finally, the ascertainment of sex/hormone and p53 crosstalk could guide different drug dosages and improve safety-toxicity drug features. This is particularly relevant given the more active immune response in females than males and the well-known required lower doses of heart disease drugs in females compared to men (117, 118).
Author Contributions
FMa, LG, ET, and FMo wrote the manuscript. AP revised the manuscript. FMo conceived and reviewed the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This study was supported by grant from Italian Association for Cancer Research (AIRC) under IG 2018—ID. 21814 project—P.I. Moretti Fabiola.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Dr. Donato Civitareale for critical reading and useful suggestion.
References
1. Siegel RL, Miller KD, Fuchs HE, Jemal A. Cancer Statistics, 2021. CA: A Cancer J Clin (2021) 71(1):7–33. doi: 10.3322/caac.21654
2. Wisnivesky JP, Halm EA. Sex Differences in Lung Cancer Survival: Do Tumors Behave Differently in Elderly Women? J Clin Oncol Off J Am Soc Clin Oncol (2007) 25(13):1705–12. doi: 10.1200/JCO.2006.08.1455
3. Lopes-Ramos CM, Quackenbush J, DeMeo DL. Genome-Wide Sex and Gender Differences in Cancer. Front Oncol (2020) 10:2486–503. doi: 10.3389/fonc.2020.597788
4. Dunford A, Weinstock DM, Savova V, Schumacher SE, Cleary JP, Yoda A, et al. Tumor-Suppressor Genes That Escape From X-Inactivation Contribute to Cancer Sex Bias. Nat Genet (2017) 49(1):10–6. doi: 10.1038/ng.3726
5. Rubin JB, Lagas JS, Broestl L, Sponagel J, Rockwell N, Rhee G, et al. Sex Differences in Cancer Mechanisms. Biol Sex Dif (2020) 11(1):1–29. doi: 10.1186/s13293-020-00291-x
6. Clocchiatti A, Cora E, Zhang Y, Dotto GP. Sexual Dimorphism in Cancer. Nat Rev Cancer (2016) 16(5):330–9. doi: 10.1038/nrc.2016.30
7. Thomas C, Gustafsson JA. The Different Roles of ER Subtypes in Cancer Biology and Therapy. Nat Rev Cancer (2011) 11(8):597–608. doi: 10.1038/nrc3093
8. Stapelfeld C, Dammann C, Maser E. Sex-Specificity in Lung Cancer Risk. Int J Cancer (2020) 146(9):2376–82. doi: 10.1002/ijc.32716
9. DeSantis CE, Miller KD, Dale W, Mohile SG, Cohen HJ, Leach CR, et al. Cancer Statistics for Adults Aged 85 Years and Older, 2019. CA: A Cancer J Clin (2019) 69(6):452–67. doi: 10.3322/caac.21577
10. Konings G, Brentjens L, Delvoux B, Linnanen T, Cornel K, Koskimies P, et al. Intracrine Regulation of Estrogen and Other Sex Steroid Levels in Endometrium and Non-Gynecological Tissues; Pathology, Physiology, and Drug Discover. Front Pharmacol (2018) 9:940. doi: 10.3389/fphar.2018.00940
11. Labrie F. Intracrinology and Menopause: The Science Describing the Cell-Specific Intracellular Formation of Estrogens and Androgens From DHEA and Their Strictly Local Action and Inactivation in Peripheral Tissues. Menopause (New York NY) (2019) 26(2):220–4. doi: 10.1097/GME.0000000000001177
12. Wasylishen AR, Lozano G. Attenuating the P53 Pathway in Human Cancers: Many Means to the Same End. Cold Spring Harb Perspect Med (2016) 6(8):a026211. doi: 10.1101/cshperspect.a026211
13. Joerger AC, Fersht AR. The P53 Pathway: Origins, Inactivation in Cancer, and Emerging Therapeutic Approache. Annu Rev Biochem (2016) 85:375–404. doi: 10.1146/annurev-biochem-060815-014710
14. Zhang Y, Xiong S, Li Q, Hu S, Tashakori M, Van Pelt C, et al. Tissue-Specific and Age-Dependent Effects of Global MDM2 Loss. J Pathol (2014) 233(4):380–91. doi: 10.1002/path.4368
15. Karni-Schmidt O, Lokshin M, Prives C. The Roles of MDM2 and MDMX in Cance. Annu Rev Pathol (2016) 11:617–44. doi: 10.1146/annurev-pathol-012414-040349
16. Teveroni E, Luca R, Pellegrino M, Ciolli G, Pontecorvi A, Moretti F. Peptides and Peptidomimetics in the P53/MDM2/MDM4 Circuitry - A Patent Review. Expert Opin Ther Patents (2016) 26(12):1417–29. doi: 10.1080/13543776.2017.1233179
17. Sanz G, Singh M, Peuget S, Selivanova G. Inhibition of P53 Inhibitors: Progress, Challenges and Perspectives. J Mol Cell Biol (2019) 11(7):586–99. doi: 10.1093/jmcb/mjz075
18. Audenet F, Méjean A, Chartier-Kastler E, Rouprêt M. Adrenal Tumours Are More Predominant in Females Regardless of Their Histological Subtype: A Review. World J Urol (2013) 31(5):1037–43. doi: 10.1007/s00345-012-1011-1
19. Ribeiro RC, Pinto EM, Zambetti GP. Familial Predisposition to Adrenocortical Tumors: Clinical and Biological Features and Management Strategies. Best Pract Res Clin Endocrinol Metab (2010) 24(3):477–90. doi: 10.1016/j.beem.2010.03.002
20. Whibley C, Pharoah PD, Hollstein M. P53 Polymorphisms: Cancer Implications. Nat Rev Cancer (2009) 9(2):95–107. doi: 10.1038/nrc2584
21. Sümbül AT, Akkız H, Bayram S, Bekar A, Akgöllü E, Sandıkçı M. P53 Codon 72 Polymorphism Is Associated With Susceptibility to Hepatocellular Carcinoma in the Turkish Population: A Case-Control Study. Mol Biol Rep (2012) 39(2):1639–47. doi: 10.1007/s11033-011-0903-2
22. Pandith AA, Khan NP, Rashid N, Azad N, Zaroo I, Hafiz A, et al. Impact of Codon 72 Arg > Pro Single Nucleotide Polymorphism in TP53 Gene in the Risk of Kangri Cancer: A Case Control Study in Kashmi. Tumour Biol J Int Soc Oncodev Biol Med (2012) 33(4):927–33. doi: 10.1007/s13277-012-0318-2
23. Haupt S, Caramia F, Herschtal A, Soussi T, Lozano G, Chen H, et al. Identification of Cancer Sex-Disparity in the Functional Integrity of P53 and Its X Chromosome Network. Nat Commun (2019) 10(1):5385–96. doi: 10.1038/s41467-019-13266-3
24. Moelans CB, de Ligt J, van der Groep P, Prins P, Besselink NJM, Hoogstraat M, et al. The Molecular Genetic Make-Up of Male Breast Cancer. Endocrine-Related Cancer (2019) 26(10):779–94. doi: 10.1530/ERC-19-0278
25. Berger C, Qian Y, Chen X. The P53-Estrogen Receptor Loop in Cancer. Curr Mol Med (2013) 13(8):1229–40. doi: 10.2174/15665240113139990065
26. Fuentes N, Silveyra P. Estrogen Receptor Signaling Mechanisms. Adv Protein Chem Struct Biol (2019) 116:135–70. doi: 10.1016/bs.apcsb.2019.01.001
27. Welcome - IARC TP53 Database 2016 . Available at: http://p53.iarc.fr/.
28. Pharoah PD, Day NE, Caldas C. Somatic Mutations in the P53 Gene and Prognosis in Breast Cancer: A Meta-Analysis. Br J Cancer (1999) 80(12):1968–73. doi: 10.1038/sj.bjc.6690628
29. Fernández-Cuesta L, Anaganti S, Hainaut P, Olivier M. Estrogen Levels Act as a Rheostat on P53 Levels and Modulate P53-Dependent Responses in Breast Cancer Cell Lines. Breast Cancer Res Treat (2011) 125(1):35–42. doi: 10.1007/s10549-010-0819-x
30. Fernandez-Cuesta L, Anaganti S, Hainaut P, Olivier M. P53 Status Influences Response to Tamoxifen But Not to Fulvestrant in Breast Cancer Cell Lines. Int J Cancer (2011) 128(8):1813–21. doi: 10.1002/ijc.25512
31. Anaganti S, Fernández-Cuesta L, Langerød A, Hainaut P, Olivier M. P53-Dependent Repression of Focal Adhesion Kinase in Response to Estradiol in Breast Cancer Cell-Lines. Cancer Letters (2011) 300(2):215–24. doi: 10.1016/j.canlet.2010.10.008
32. Berger CE, Qian Y, Liu G, Chen H, Chen X. P53, a Target of Estrogen Receptor (ER) α, Modulates DNA Damage-Induced Growth Suppression in ER-Positive Breast Cancer Cells. J Biol Chem (2012) 287(36):30117–27. doi: 10.1074/jbc.M112.367326
33. Becker KA, Lu S, Dickinson ES, Dunphy KA, Mathews L, Schneider SS, et al. Estrogen and Progesterone Regulate Radiation-Induced P53 Activity in Mammary Epithelium Through TGF-Beta-Dependent Pathways. Oncogene (2005) 24(42):6345–53. doi: 10.1038/sj.onc.1208787
34. Dunphy KA, Blackburn AC, Yan H, O'Connell LR, Jerry DJ. Estrogen and Progesterone Induce Persistent Increases in P53-Dependent Apoptosis and Suppress Mammary Tumors in BALB/c-Trp53+/- Mice. Breast Cancer Res BCR (2008) 10(3):R43. doi: 10.1186/bcr2094
35. Sivaraman L, Conneely OM, Medina D, O'Malley BW. P53 Is a Potential Mediator of Pregnancy and Hormone-Induced Resistance to Mammary Carcinogenesis. Proc Natl Acad Sci USA (2001) 98(22):12379–84. doi: 10.1073/pnas.221459098
36. Fortner RT, Sisti J, Chai B, Collins LC, Rosner B, Hankinson SE, et al. Parity, Breastfeeding, and Breast Cancer Risk by Hormone Receptor Status and Molecular Phenotype: Results From the Nurses' Health Studie. Breast Cancer Res BCR (2019) 21(1):40. doi: 10.1186/s13058-019-1119-y
37. Kuperwasser C, Pinkas J, Hurlbut GD, Naber SP, Jerry DJ. Cytoplasmic Sequestration and Functional Repression of P53 in the Mammary Epithelium Is Reversed by Hormonal Treatment. Cancer Res (2000) 60(10):2723–29.
38. Pinto G, Alhaiek AA, Amadi S, Qattan AT, Crawford M, Radulovic M, et al. Systematic Nucleo-Cytoplasmic Trafficking of Proteins Following Exposure of MCF7 Breast Cancer Cells to Estradiol. J Proteome Res (2014) 13(2):1112–27. doi: 10.1021/pr4012359
39. Weige CC, Allred KF, Armstrong CM, Allred CD. P53 Mediates Estradiol Induced Activation of Apoptosis and DNA Repair in Non-Malignant Colonocytes. J Steroid Biochem Mol Biol (2012) 128(3-5):113–20. doi: 10.1016/j.jsbmb.2011.10.010
40. Yu CL, Driggers P, Barrera-Hernandez G, Nunez SB, Segars JH, Cheng S. The Tumor Suppressor P53 Is a Negative Regulator of Estrogen Receptor Signaling Pathways. Biochem Biophys Res Commun (1997) 239(2):617–20. doi: 10.1006/bbrc.1997.7522
41. Lewis-Wambi JS, Jordan VC. Estrogen Regulation of Apoptosis: How Can One Hormone Stimulate and Inhibit? Breast Cancer Res BCR (2009) 11(3):206. doi: 10.1186/bcr2255
42. Traboulsi T, El EM, Gleason JL, Mader S. Antiestrogens: Structure-Activity Relationships and Use in Breast Cancer Treatment. J Mol Endocrinol (2017) 58(1):R15–31. doi: 10.1530/JME-16-0024
43. Coates AS, Millar EK, O'Toole SA, Molloy TJ, Viale G, Goldhirsch A, et al. Prognostic Interaction Between Expression of P53 and Estrogen Receptor in Patients With Node-Negative Breast Cancer: Results From IBCSG Trials VIII and IX. Breast Cancer Res BCR (2012) 14(6):R143. doi: 10.1186/bcr3348
44. Pok S, Barn VA, Wong HJ, Blackburn AC, Board P, Farrell GC, et al. Testosterone Regulation of Cyclin E Kinase: A Key Factor in Determining Gender Differences in Hepatocarcinogenesis. J Gastroenterol Hepatol (2016) 31(6):1210–9. doi: 10.1111/jgh.13232
45. Huang FY, Wong DK, Seto WK, Lai CL, Yuen MF. Estradiol Induces Apoptosis via Activation of miRNA-23a and P53: Implication for Gender Difference in Liver Cancer Development. Oncotarget (2015) 6(33):34941–52. doi: 10.18632/oncotarget.5472
46. El-Serag HB. Hepatocellular Carcinoma. N Engl J Med (2011) 365(12):1118–27. doi: 10.1056/NEJMra1001683
47. Hartman J, Edvardsson K, Lindberg K, Zhao C, Williams C, Ström A, et al. Tumor Repressive Functions of Estrogen Receptor Beta in SW480 Colon Cancer Cells. Cancer Res (2009) 69(15):6100–6. doi: 10.1158/0008-5472.CAN-09-0506
48. Hsu HH, Cheng SF, Wu CC, Chu CH, Weng YJ, Lin CS, et al. Apoptotic Effects of Over-Expressed Estrogen Receptor-Beta on LoVo Colon Cancer Cell Is Mediated by P53 Signalings in a Ligand-Dependent Manner. Chin J Physiol (2006) 49(2):110–6.
49. Liu W, Konduri SD, Bansal S, Nayak BK, Rajasekaran SA, Karuppayil SM, et al. Estrogen Receptor-Alpha Binds P53 Tumor Suppressor Protein Directly and Represses Its Function. J Biol Chem (2006) 281(15):9837–40. doi: 10.1074/jbc.C600001200
50. Sayeed A, Konduri SD, Liu W, Bansal S, Li F, Das GM. Estrogen Receptor Alpha Inhibits P53-Mediated Transcriptional Repression: Implications for the Regulation of Apoptosis. Cancer Res (2007) 67(16):7746–55. doi: 10.1158/0008-5472.CAN-06-3724
51. Bailey ST, Shin H, Westerling T, Liu XS, Brown M. Estrogen Receptor Prevents P53-Dependent Apoptosis in Breast Cancer. Proc Natl Acad Sci USA (2012) 109(44):18060–5. doi: 10.1073/pnas.1018858109
52. Lewandowski SA, Thiery J, Jalil A, Leclercq G, Szczylik C, Chouaib S. Opposite Effects of Estrogen Receptors Alpha and Beta on MCF-7 Sensitivity to the Cytotoxic Action of TNF and P53 Activity. Oncogene (2005) 24(30):4789–98. doi: 10.1038/sj.onc.1208595
53. Brekman A, Singh KE, Polotskaia A, Kundu N, Bargonetti J. A P53-Independent Role of Mdm2 in Estrogen-Mediated Activation of Breast Cancer Cell Proliferation. Breast Cancer Res BCR (2011) 13(1):R3. doi: 10.1186/bcr2804
54. Hu L, Zhang H, Bergholz J, Sun S, Xiao ZX. MDM2/MDMX: Master Negative Regulators for P53 and RB. Mol Cell Oncol (2016) 3(2):e1106635. doi: 10.1080/23723556.2015.1106635
55. Phelps M, Darley M, Primrose JN, Blaydes JP. P53-Independent Activation of the Hdm2-P2 Promoter Through Multiple Transcription Factor Response Elements Results in Elevated Hdm2 Expression in Estrogen Receptor Alpha-Positive Breast Cancer Cells. Cancer Res (2003) 63(10):2616–23.
56. Bond GL, Hu W, Bond EE, Robins H, Lutzker SG, Arva NC, et al. A Single Nucleotide Polymorphism in the MDM2 Promoter Attenuates the P53 Tumor Suppressor Pathway and Accelerates Tumor Formation in Humans. Cell (2004) 119(5):591–602. doi: 10.1016/j.cell.2004.11.022
57. Bond GL, Hirshfield KM, Kirchhoff T, Alexe G, Bond EE, Robins H, et al. MDM2 SNP309 Accelerates Tumor Formation in a Gender-Specific and Hormone-Dependent Manner. Cancer Res (2006) 66(10):5104–10. doi: 10.1158/0008-5472.CAN-06-0180
58. Alhopuro P, Ylisaukko-Oja SK, Koskinen WJ, Bono P, Arola J, Järvinen HJ, et al. The MDM2 Promoter Polymorphism SNP309–>G and the Risk of Uterine Leiomyosarcoma, Colorectal Cancer, and Squamous Cell Carcinoma of the Head and Neck. J Med Genet (2005) 42(9):694–8. doi: 10.1136/jmg.2005.031260
59. Nayak MS, Yang JM, Hait WN. Effect of a Single Nucleotide Polymorphism in the Murine Double Minute 2 Promoter (SNP309) on the Sensitivity to Topoisomerase II-Targeting Drugs. Cancer Res (2007) 67(12):5831–9. doi: 10.1158/0008-5472.CAN-06-4533
60. Kundu N, Brekman A, Kim JY, Xiao G, Gao C, Bargonetti J. Estrogen-Activated MDM2 Disrupts Mammary Tissue Architecture Through a P53-Independent Pathway. Oncotarget (2017) 8(29):47916–30. doi: 10.18632/oncotarget.18147
61. Bond GL, Levine AJ. A Single Nucleotide Polymorphism in the P53 Pathway Interacts With Gender, Environmental Stresses and Tumor Genetics to Influence Cancer in Humans. Oncogene (2007) 26(9):1317–23. doi: 10.1038/sj.onc.1210199
62. Knappskog S, Bjørnslett M, Myklebust LM, Huijts PE, Vreeswijk MP, Edvardsen H, et al. The MDM2 Promoter SNP285C/309G Haplotype Diminishes Sp1 Transcription Factor Binding and Reduces Risk for Breast and Ovarian Cancer in Caucasian. Cancer Cell (2011) 19(2):273–82. doi: 10.1016/j.ccr.2010.12.019
63. Ortiz GJ, Li Y, Post SM, Pant V, Xiong S, Larsson CA, et al. Contrasting Effects of an Mdm2 Functional Polymorphism on Tumor Phenotypes. Oncogene (2018) 37(3):332–40. doi: 10.1038/onc.2017.344
64. Grochola LF, Müller TH, Bond GL, Taubert H, Udelnow A, Würl P. MDM2 SNP309 Associates With Accelerated Pancreatic Adenocarcinoma Formation. Pancreas (2010) 39(1):76–80. doi: 10.1097/MPA.0b013e3181b9f105
65. Okamoto K, Tsunematsu R, Tahira T, Sonoda K, Asanoma K, Yagi H, et al. SNP55, a New Functional Polymorphism of MDM2-P2 Promoter, Contributes to Allele-Specific Expression of MDM2 in Endometrial Cancers. BMC Med Genet (2015) 16:67. doi: 10.1186/s12881-015-0216-8
66. Helwa R, Gansmo LB, Romundstad P, Hveem K, Vatten L, Ryan BM, et al. MDM2 Promoter SNP55 (Rs2870820) Affects Risk of Colon Cancer But Not Breast-, Lung-, or Prostate Cancer. Sci Rep (2016) 6:33153. doi: 10.1038/srep33153
67. Helwa R, Gansmo LB, Bjørnslett M, Halle MK, Werner HMJ, Romundstad P, et al. Impact of MDM2 Promoter SNP55 (Rs2870820) on Risk of Endometrial and Ovarian Cancer. Biomarkers (2021) 26(4):302–8. doi: 10.1080/1354750X.2021.1891291
68. Dolfi SC, Jäger AV, Medina DJ, Haffty BG, Yang JM, Hirshfield KM. Fulvestrant Treatment Alters MDM2 Protein Turnover and Sensitivity of Human Breast Carcinoma Cells to Chemotherapeutic Drugs. Cancer Lett (2014) 350(1-2):52–60. doi: 10.1016/j.canlet.2014.04.009
69. Sheikh MS, Shao ZM, Hussain A, Fontana JA. The P53-Binding Protein MDM2 Gene is Differentially Expressed in Human Breast Carcinoma. Cancer Res (1993) 53(14):3226–8.
70. Duong V, Boulle N, Daujat S, Chauvet J, Bonnet S, Neel H, et al. Differential Regulation of Estrogen Receptor Alpha Turnover and Transactivation by Mdm2 and Stress-Inducing Agents. Cancer Res (2007) 67(11):5513–21. doi: 10.1158/0008-5472.CAN-07-0967
71. Mancini F, Di Conza G, Monti O, Macchiarulo A, Pellicciari R, Pontecorvi A, et al. Puzzling Over MDM4-P53 Network. Int J Biochem Cell Biol (2010) 42(7):1080–3. doi: 10.1016/j.biocel.2010.04.010
72. Chen SH, Forrester W, Lahav G. Schedule-Dependent Interaction Between Anticancer Treatments. Science (2016) 351(6278):1204–8. doi: 10.1126/science.aac5610
73. Mancini F, Teveroni E, Di Conza G, Monteleone V, Arisi I, Pellegrino M, et al. MDM4 Actively Restrains Cytoplasmic Mtorc1 by Sensing Nutrient Availability. Mol Cancer (2017) 16:13. doi: 10.1186/s12943-017-0626-7
74. Kon N, Wang D, Li T, Jiang L, Qiang L, Gu W. Inhibition of Mdmx (Mdm4) In Vivo Induces Anti-Obesity Effects. Oncotarget (2018) 9(7):7282–97. doi: 10.18632/oncotarget.23837
75. Mancini F, Moretti F. Mitochondrial MDM4 (MDMX) An Unpredicted Role in the P53-Mediated Intrinsic Apoptotic Pathway. Cell Cycle (2009) 8(23):3854–9. doi: 10.4161/cc.8.23.10089
76. Mancini F, Pieroni L, Monteleone V, Luca R, Fici L, Luca E, et al. MDM4/HIPK2/p53 Cytoplasmic Assembly Uncovers Coordinated Repression of Molecules With Anti-Apoptotic Activity During Early DNA Damage Response. Oncogene (2016) 35(2):228–40. doi: 10.1038/onc.2015.76
77. Gembarska A, Luciani F, Fedele C, Russell EA, Dewaele M, Villar S, et al. MDM4 Is a Key Therapeutic Target in Cutaneous Melanoma. Nat Med (2012) 18(8):1239–47. doi: 10.1038/nm.2863
78. Atwal GS, Kirchhoff T, Bond EE, Montagna M, Menin C, Bertorelle R, et al. Altered Tumor Formation and Evolutionary Selection of Genetic Variants in the Human MDM4 Oncogene. Proc Natl Acad Sci USA (2009) 106(25):10236–41. doi: 10.1073/pnas.0901298106
79. Garcia-Closas M, Couch FJ, Lindstrom S, Michailidou K, Schmidt MK, Brook MN, et al. Genome-Wide Association Studies Identify Four ER Negative-Specific Breast Cancer Risk Loci. Nat Genet (2013) 45(4):392–8, 398e1-2. doi: 10.1038/ng.2561
80. Eeles RA, Olama AA, Benlloch S, Saunders EJ, Leongamornlert DA, Tymrakiewicz M, et al. Identification of 23 New Prostate Cancer Susceptibility Loci Using the iCOGS Custom Genotyping Array. Nat Genet (2013) 45(4):385–91, 391e1–2. doi: 10.1038/ng.2560
81. Sakoda LC, Jorgenson E, Witte JS. Turning of COGS Moves Forward Findings for Hormonally Mediated Cancers. Nat Genet (2013) 45(4):345–8. doi: 10.1038/ng.2587
82. Stegeman S, Moya L, Selth LA, Spurdle AB, Clements JA, Batra J. A Genetic Variant of MDM4 Influences Regulation by Multiple microRNAs in Prostate Cancer. Endocr Relat Cancer (2015) 22(2):265–76. doi: 10.1530/ERC-15-0013
83. Wynendaele J, Bohnke A, Leucci E, Nielsen SJ, Lambertz I, Hammer S, et al. An Illegitimate microRNA Target Site Within the 3' UTR of MDM4 Affects Ovarian Cancer Progression and Chemosensitivity. Cancer Res (2010) 70(23):9641–9. doi: 10.1158/0008-5472.CAN-10-0527
84. Gansmo LB, Bjornslett M, Halle MK, Salvesen HB, Dorum A, Birkeland E, et al. The MDM4 SNP34091 (Rs4245739) C-Allele Is Associated With Increased Risk of Ovarian-But Not Endometrial Cancer. Tumour Biol (2016) 37(8):10697–702. doi: 10.1007/s13277-016-4940-2
85. Milne RL, Kuchenbaecker KB, Michailidou K, Beesley J, Kar S, Lindstrom S, et al. Identification of Ten Variants Associated With Risk of Estrogen-Receptor-Negative Breast Cancer. Nat Genet (2017) 49(12):1767–78. doi: 10.1038/ng.3785
86. Gansmo LB, Romundstad P, Birkeland E, Hveem K, Vatten L, Knappskog S, et al. MDM4 SNP34091 (Rs4245739) and Its Effect on Breast-, Colon-, Lung-, and Prostate Cancer Risk. Cancer Med (2015) 4(12): 1901–7. doi: 10.1002/cam4.555
87. Kotarac N, Dobrijevic Z, Matijasevic S, Savic-Pavicevic D, Brajuskovic G. Association of KLK3, VAMP8 and MDM4 Genetic Variants Within microRNA Binding Sites With Prostate Cancer: Evidence From Serbian Populatio. Pathol Oncol Res POR (2020) 26(4):2409–23. doi: 10.1007/s12253-020-00839-7
88. Fan C, Wei J, Yuan C, Wang X, Jiang C, Zhou C, et al. The Functional TP53 Rs1042522 and MDM4 Rs4245739 Genetic Variants Contribute to Non-Hodgkin Lymphoma Risk. PloS One (2014) 9(9):e107047. doi: 10.1371/journal.pone.0107047
89. Zhou L, Zhang X, Li Z, Zhou C, Li M, Tang X, et al. Association of a Genetic Variation in a miR-191 Binding Site in MDM4 With Risk of Esophageal Squamous Cell Carcinoma. PloS One (2013) 8(5):e64331. doi: 10.1371/journal.pone.0064331
90. Abecasis GR, Auton A, Brooks LD, DePristo MA, Durbin RM, Handsaker RE, et al. An Integrated Map of Genetic Variation From 1,092 Human Genomes. Nature (2012) 491(7422):56–65. doi: 10.1038/nature11632
91. Anwar SL, Wulaningsih W, Watkins J. Profile of the Breast Cancer Susceptibility Marker Rs4245739 Identifies a Role for miRNAs. Cancer Biol Med (2017) 14(4):387–95. doi: 10.20892/j.issn.2095-3941.2017.0050
92. Couch FJ, Wang X, McGuffog L, Lee A, Olswold C, Kuchenbaecker KB, et al. Genome-Wide Association Study in BRCA1 Mutation Carriers Identifies Novel Loci Associated With Breast and Ovarian Cancer Risk. PloS Genet (2013) 9(3):e1003212. doi: 10.1371/journal.pgen.1003212
93. Sun P, Yan F, Fang W, Zhao J, Chen H, Ma X, et al. MDM4 Contributes to the Increased Risk of Glioma Susceptibility in Han Chinese Population. Sci Rep (2018) 8(1). doi: 10.1038/s41598-018-29468-6
94. Melin BS, Barnholtz-Sloan JS, Wrensch MR, Johansen C, Il'yasova D, Kinnersley B, et al. Genome-Wide Association Study of Glioma Subtypes Identifies Specific Differences in Genetic Susceptibility to Glioblastoma and Non-Glioblastoma Tumors. Nat Genet (2017) 49(5):789–4. doi: 10.1038/ng.3823
95. Xiong S, Pant V, Zhang Y, Aryal NK, You MJ, Kusewitt D, et al. The P53 Inhibitor Mdm4 Cooperates With Multiple Genetic Lesions in Tumourigenesis. J Pathol (2017) 241(4):501–10. doi: 10.1002/path.4854
96. Luca R, di Blasio G, Gallo D, Monteleone V, Manni I, Fici L, et al. Estrogens Counteract Platinum-Chemosensitivity by Modifying the Subcellular Localization of MDM4. Cancers (Basel) (2019) 11(9):1349. doi: 10.3390/cancers11091349
97. Swetzig WM, Wang J, Das GM. Estrogen Receptor Alpha (Erα/ESR1) Mediates the P53-Independent Overexpression of MDM4/MDMX and MDM2 in Human Breast Cancer. Oncotarget (2016) 7(13):16049–69. doi: 10.18632/oncotarget.7533
98. Skalniak L, Surmiak E, Holak TA. A Therapeutic Patent Overview of MDM2/X-Targeted Therapies (2014-2018). Expert Opin Ther Patents (2019) 29(3):151–70. doi: 10.1080/13543776.2019.1582645
99. Duffy MJ, Synnott NC, O'Grady S, Crown J. Targeting P53 for the Treatment of Cancer. Semin Cancer Biol (2020) S1044-579X(20). doi: 10.1016/j.semcancer.2020.07.005
100. Yamada T, Goto M, Punj V, Zaborina O, Chen ML, Kimbara K, et al. Bacterial Redox Protein Azurin, Tumor Suppressor Protein P53, and Regression of Cancer. Proc Natl Acad Sci USA (2002) 99(22):14098–103. doi: 10.1073/pnas.222539699
101. Punj V, Bhattacharyya S, Saint-Dic D, Vasu C, Cunningham EA, Graves J, et al. Bacterial Cupredoxin Azurin as an Inducer of Apoptosis and Regression in Human Breast Cancer. Oncogene (2004) 23(13):2367–78. doi: 10.1038/sj.onc.1207376
102. Warso MA, Richards JM, Mehta D, Christov K, Schaeffer C, Rae Bressler L, et al. A First-in-Class, First-in-Human, Phase I Trial of P28, a Non-HDM2-Mediated Peptide Inhibitor of P53 Ubiquitination in Patients With Advanced Solid Tumours. Br J Cancer (2013) 108(5):1061–70. doi: 10.1038/bjc.2013.74
103. Lulla RR, Goldman S, Yamada T, Beattie CW, Bressler L, Pacini M, et al. Phase 1 Trial of P28 (NSC745104), a Non-HDM2-Mediated Peptide Inhibitor of P53 Ubiquitination in Pediatric Patients With Recurrent or Progressive Central Nervous System Tumors: A Pediatric Brain Tumor Consortium Studu. Neuro Oncol (2016) 18(9):1319–25. doi: 10.1093/neuonc/now047
104. Carvajal LA, Neriah DB, Senecal A, Benard L, Thiruthuvanathan V, Yatsenko T, et al. Dual Inhibition of MDMX and MDM2 as a Therapeutic Strategy in Leukemia. Sci Trans Med (2018) 10(436):eaao3003. doi: 10.1126/scitranslmed.aao3003
105. Andreeff M, Kelly KR, Yee K, Assouline S, Strair R, Popplewell L, et al. Results of the Phase I Trial of RG7112, a Small-Molecule MDM2 Antagonist in Leukemia. Clin Cancer Res Off J Am Assoc Cancer Res (2016) 22(4):868–76. doi: 10.1158/1078-0432.CCR-15-0481
106. Wang S, Sun W, Zhao Y, McEachern D, Meaux I, Barrière C, et al. SAR405838: An Optimized Inhibitor of MDM2-P53 Interaction That Induces Complete and Durable Tumor Regression. Cancer Res (2014) 74(20):5855–65. doi: 10.1158/0008-5472.CAN-14-0799
107. Holzer P, Masuya K, Furet P, Kallen J, Valat-Stachyra T, Ferretti S, et al. Discovery of a Dihydroisoquinolinone Derivative (NVP-CGM097): A Highly Potent and Selective MDM2 Inhibitor Undergoing Phase 1 Clinical Trials in P53wt Tumor. J Medicinal Chem (2015) 58(16):6348–58. doi: 10.1021/acs.jmedchem.5b00810
108. Jeay S, Gaulis S, Ferretti S, Bitter H, Ito M, Valat T, et al. A Distinct P53 Target Gene Set Predicts for Response to the Selective P53-HDM2 Inhibitor NVP-Cgm097. eLife (2015) 4:e06498. doi: 10.7554/eLife.06498
109. Ravandi F, Gojo I, Patnaik MM, Minden MD, Kantarjian H, Johnson-Levonas AO, et al. A Phase I Trial of the Human Double Minute 2 Inhibitor (MK-8242) in Patients With Refractory/Recurrent Acute Myelogenous Leukemia (AM). Leukemia Res (2016) 48:92–100. doi: 10.1016/j.leukres.2016.07.004
110. Wagner AJ, Banerji U, Mahipal A, Somaiah N, Hirsch H, Fancourt C, et al. Phase I Trial of the Human Double Minute 2 Inhibitor MK-8242 in Patients With Advanced Solid Tumor. J Clin Oncol Off J Am Soc Clin Oncol (2017) 35(12):1304–11. doi: 10.1200/JCO.2016.70.7117
111. Sun D, Li Z, Rew Y, Gribble M, Bartberger MD, Beck HP, et al. Discovery of AMG 232, a Potent, Selective, and Orally Bioavailable MDM2-P53 Inhibitor in Clinical Development. J Medicinal Chem (2014) 57(4):1454–72. doi: 10.1021/jm401753e.
112. DiNardo CD, Rosenthal J, Andreeff M, Zernovak O, Kumar P, Gajee R, et al. Phase 1 Dose Escalation Study of MDM2 Inhibitor DS-3032b in Patients With Hematological Malignancies - Preliminary Results | Blood | American Society of Hematology. Blood (2016) 128:593. doi: 10.1182/blood.V128.22.593.593
113. Ferretti S, Rebmann R, Berger M, Santacroce F, Albrecht G, Pollehn K, et al. Insights Into the Mechanism of Action of NVP-HDM201, a Differentiated and Versatile Next-Generation Small-Molecule Inhibitor of Mdm2, Under Evaluation in Phase I Clinical Trials. Cancer Res (2016) 76(14):1224. doi: 10.1158/1538-7445.AM2016-1224
114. Hyman D, Chatterjee M, Langenberg MHG, Lin CC, Suarez C, Tai D, et al. Dose- and Regimen-Finding Phase I Study of NVP-HDM201 in Patients (Pts) With TP53 Wild-Type (Wt) Advanced Tumors. Eur J Cancer (2016) 69:S128–S9. doi: 10.1016/S0959-8049(16)32982-3
115. Clayton JA, Collins FS. Policy: NIH to Balance Sex in Cell and Animal Studies. Nature (2014) 509(7500):282–3. doi: 10.1038/509282a
116. Rozeboom B, Dey N, De P. ER+ Metastatic Breast Cancer: Past, Present, and a Prescription for an Apoptosis-Targeted Future. Am J Cancer Res (2019) 9(12):2821–31.
117. Mauvais-Jarvis F, Bairey M N, Barnes PJ, Brinton RD, Carrero JJ, DeMeo DL, et al. Sex and Gender: Modifiers of Health, Disease, and Medicine. Lancet (Lond Eng) (2020) 396(10250):565–82. doi: 10.1016/S0140-6736(20)31561-0
Keywords: cancer therapy, p53, sex, estrogen, MDM2, MDM4
Citation: Mancini F, Giorgini L, Teveroni E, Pontecorvi A and Moretti F (2021) Role of Sex in the Therapeutic Targeting of p53 Circuitry. Front. Oncol. 11:698946. doi: 10.3389/fonc.2021.698946
Received: 22 April 2021; Accepted: 16 June 2021;
Published: 08 July 2021.
Edited by:
Aniello Cerrato, Istituto per l’Endocrinologia e l’oncologia “Gaetano Salvatore (CNR), ItalyReviewed by:
Lawrence Panasci, Segal Cancer Centre, CanadaFrancesca Maffei, University of Bologna, Italy
Copyright © 2021 Mancini, Giorgini, Teveroni, Pontecorvi and Moretti. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Fabiola Moretti, ZmFiaW9sYS5tb3JldHRpQGNuci5pdA==; Francesca Mancini, bWFuY2luaS5jaGljY2FAZ21haWwuY29t
†These authors share first authorship
‡These authors share last authorship