 Chang Liu
Chang Liu Zhimin Fan
Zhimin Fan
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oncol., 03 September 2021
Sec. Cancer Metabolism
Volume 11 - 2021 | https://doi.org/10.3389/fonc.2021.698023
This article is part of the Research TopicTumor Metabolism in Cancer Therapy ResistanceView all 8 articles
Although chemotherapy can improve the overall survival and prognosis of cancer patients, chemoresistance remains an obstacle due to the diversity, heterogeneity, and adaptability to environmental alters in clinic. To determine more possibilities for cancer therapy, recent studies have begun to explore changes in the metabolism, especially glycolysis. The Warburg effect is a hallmark of cancer that refers to the preference of cancer cells to metabolize glucose anaerobically rather than aerobically, even under normoxia, which contributes to chemoresistance. However, the association between glycolysis and chemoresistance and molecular mechanisms of glycolysis-induced chemoresistance remains unclear. This review describes the mechanism of glycolysis-induced chemoresistance from the aspects of glycolysis process, signaling pathways, tumor microenvironment, and their interactions. The understanding of how glycolysis induces chemoresistance may provide new molecular targets and concepts for cancer therapy.
As a disease with a low cure rate, cancer is accompanied not only by abnormalities in proliferation, metastasis, and invasion but also by metabolic disorders (1, 2). In 1924, Otto Warburg first indicated that cancer utilizes glycolysis to provide adenosine triphosphate (ATP), nucleotide, lipid, and amino acid for the growth of cancer cells even under aerobic conditions; this phenomenon is called the Warburg effect (3). There is a significant difference in the usage of glucose between cancer and normal cells. Rapid proliferation of cancer cells and the abnormal structure and function of vascularization both lead to imbalance in the intake and consumption of oxygen, resulting in hypoxia, which drives cancer cells to choose glycolysis for energy supply (4, 5). At the same time, abnormally activated oncogene signaling pathways and the tumor microenvironment make cancer cells choose glycolysis as their primary energy source even under normoxia, which means pyruvate is mainly converted into lactate to play its role in energy source, rather than being incorporated into the tricarboxylic acid cycle (TCA cycle) (Figure 1) (6). Recently, more and more studies have proven that while being the energy source of cancer cells, glycolysis is also involved in the activation of oncogenes such as phosphatidylinositol 3-kinase (PI3K) and hypoxia inducible factor-1 alpha (HIF-1A) shift in the tumor microenvironment such as hypoxia and acidosis (7–10).
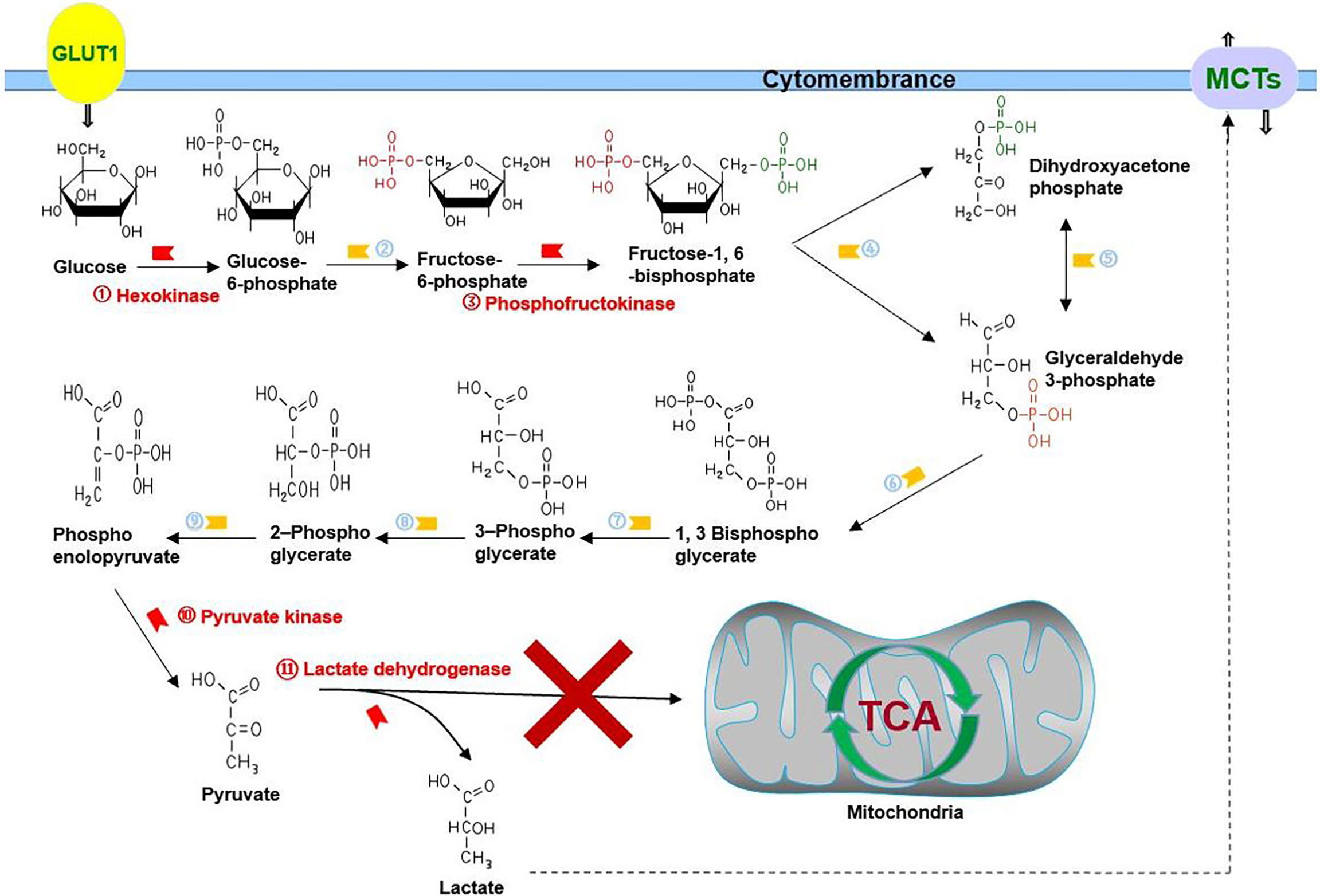
Figure 1 Glycolysis in cancer: cancer cells choose glycolysis as their primary energy source even under normoxia, which means pyruvate is mainly converted into lactate to play its role in energy source, rather than being incorporated into the TCA cycle. GLUT1 is responsible for transporting glucose, and MCTs are responsible for transporting lactate. ②, phosphohexose isomerase; ④, aldolase; ⑤, triose phosphate isomerase; ⑥, glyceraldehyde 3-phosphate dehydrogenase; ⑦, phosphoglycerate kinase; ⑧, phosphoglycerate mutase; ⑨, enolase.
Although recent years have seen a slight decline in cancer mortality, it remains an urgent national health problem and the second leading cause of death in the United States (11). Chemotherapy is one of the main treatments of cancer and is usually performed as neoadjuvant and adjuvant therapy (12, 13). Although chemotherapy can improve the overall survival and prognosis of cancer patients, chemoresistance remains a clinical obstacle that needs to be overcome due to the diversity, heterogeneity, and adaptability to environmental alters in clinics (14, 15). Chemoresistance is caused by multifactor interaction, and its mechanism can be summarized as mutation in drug targets and metabolism, apoptosis inhibition, activation of intracellular survival signaling pathways, enhanced deoxyribonucleic acid (DNA) repair, immune escape of cancer stem cells (CSCs), epigenetic alteration, and aberrant metabolism (16–18). A previous study on chemoresistance focused more on gene mutation and external factors. In recent years, cancer metabolism has become a new research hotspot (19, 20). Increasing studies have proven that glycolysis inhibition can be a novel method to improve chemoresistance (21–23).
Although the relationship between cancer metabolism and chemoresistance is clear, the causal relationship between them remains controversial. Therefore, systematically understanding the causal relationship between cancer metabolism and chemoresistance may provide new ideas for scientific research and clinical treatment. This review aimed to discusses the mechanism of glycolysis-induced chemoresistance from the aspects of glycolysis process, signaling pathways, and tumor microenvironment and their interactions, which will bring new insights for research and clinical therapy on chemoresistance.
Glucose transporter (GLUT) located on the cytomembrane is encoded by the SLC2 gene and divided into three categories and 14 subtypes, namely, Class 1 (GLUTs 1–4 and 14), Class 2 (GLUTs 5, 7, 9, and 11), and Class 3 (GLUTs 6, 8, 10, 12, and HMIT), which uptake glucose into the cytoplasm and participates in respiration, metabolism, and proliferation in cancer (24, 25).
GLUT1 has a high affinity for glucose and is highly presented in erythrocytes, endothelial cells, and cancer cells among the GLUT subtypes (26–30). Cancer cells depend on ATP contributed from aerobic glycolysis for survival, and often have an overexpression of GLUT1 for sufficient glucose uptake (25). Furthermore, overexpressed GLUT1 is significantly associated with poor differentiated cancers, positive lymph node metastasis, larger tumors, and worse overall survival and disease-free survival in cancer (31). Cancer is accompanied by an abnormal activation of PI3K, HIF-1A, RAS, MYC, and other pathways that activate nuclear factor kappa B subunit (NFκB) and mechanistic target of rapamycin kinase (mTOR) by facilitating GLUT1 overexpression and participate in cell proliferation, metastasis, and chemotherapy resistance (28, 30–32). Acetaldehyde dehydrogenase enhances stemness and paclitaxel resistance via GLUT in endometrial cancer (27); Ajuba, which belongs to the Ajuba LIM family, serves as adaptor proteins that have the ability to connect cell adhesion and nuclear signaling overexpression inhibits cisplatin efficiency via Yes‐associated protein (YAP)/GLUT1/B-cell lymphoma-extra-large (BCL-xL) in breast and gastric cancer (33); Wnt1-inducible signaling protein 1 inhibits mitochondrial activity and upregulates GLUT1 through the YAP1/GLUT1 pathway to enhance glycolysis and induces chemoresistance in laryngeal cancer, as well as in prostate, lung, colorectal, and breast cancer (34). A collaboration between GLUT1 inhibitors and chemotherapeutic drugs significantly facilitates apoptosis and chemosensitivity in breast cancer, oral squamous cell carcinoma, and laryngeal cancer (29, 32, 35), and mannose-conjugated platinum complexes are effective in cancer targeting mediated by GLUT1 (36). Resveratrol presents anticancer effects by inhibiting GLUT1 via the protein kinase B (AKT)/mTOR-dependent signaling pathway and targeting “classical” tumor-promoting pathways, such as PI3K/AKT, signal transducer and activator of transcription (STAT)3/5, and mitogen-activated protein kinase (MAPK), which enhance glycolysis via the upregulation of glycolytic enzymes and glucose transporters (37). As an inhibitor of glycolysis, 2-deoxyglucose (2-DG) competes with glucose to bind to GLUT1, and reverses chemoresistance in breast and prostate cancer (38–40). In summary, GLUT1 induces chemoresistance via itself or advocating other signaling pathways and contributes a new direction for clinical diagnosis, treatment, and prognosis of cancer.
GLUT3, which mainly presents in the nervous system, has a higher affinity for glucose than GLUT1 and exhibits the highest turnover rate among all GLUT family members (41, 42). GLUT3 is overexpressed in various cancer cells, such as glioblastoma (43), ovarian cancer (44), gastric cancer (45, 46), and non-small cell lung cancer (46), due to its high glycolytic efficiency. GLUT3 upregulation in glioblastoma ensures survival under restricted glucose conditions and increases cancer cell invasion that is not recapitulated by GLUT1 (43). Studies have reported that GLUT3 affects the neovascularization processes to counteract the antiangiogenic effect of temozolomide (TMZ) in glioblastoma (47). Tripartite motif 66 upregulates TMZ resistance via the C-MYC/GLUT3 signaling pathway in glioblastoma (48). DNA damage-inducible transcript 4 decreases TMZ efficacy in glioblastoma through GLUT3-mediated cancer stemness (49). YAP promotes the proliferation and migration of colorectal cancer cells via the GLUT3/Adenosine 5’-monophosphate (AMP)-activated protein kinase signaling pathway (50). Overexpression of YAP1 in gastric cancer cells can skew macrophage polarization to M2-like phenotype and induce GLUT3-depended glycolysis program, which further creates an immunosuppressive milieu to promote 5-fluorouracil (5-FU) resistance (45). Transcription factor 4 downregulation sensitizes melanoma cells to vemurafenib by inhibiting GLUT3-mediated glycolysis (51). Atorvastatin overcomes tyrosine kinase inhibitor (TKI) resistance via GLUT3 inhibition in non-small cell lung cancer (46). Melatonin promotes cisplatin-induced apoptosis via the downregulation of GLUT3 in hepatocellular carcinoma (52). GLUT3 can evolve as a new therapeutic target in the future; additionally, combined deletion of GLUT1 and GLUT3 may achieve better results (53).
GLUT12, which was first discovered in human breast cancer cell line michigan cancer foundation-7 (MCF-7), is limited to insulin-sensitive tissues, skeletal muscle, fat, and heart in normal human adult tissues (54, 55). Recent studies have found that GLUT12 is expressed in rhabdomyosarcomas, oligodendrogliomas, oligoastrocytomas, astrocytomas, and breast and prostate cancer (56, 57). Overexpression of GLUT12 in breast and prostate cancer is associated with cancer development and characteristic glycolytic metabolism observed in malignant cells (55, 58, 59). This effect may be mediated through P53, estradiol and epidermal growth factor (56, 60). GLUT12 could serve as a new therapeutic target due to its targeted expression on cancer cells. For example, microRNA let-7a-5p (miR let-7a-5p) inhibits the proliferation, migration, and invasion of triple-negative breast cancer via GLUT12 inhibition (60).
Hexokinases (HKs) are located in the cytoplasm phosphorylate intracellular glucose, which is the first rate-limiting step of glycolysis. There are four subtypes of HKs: HK1, HK2, HK3, and HK4, which are encoded by different genes on different chromosomes. HK1 generally exists in normal tissues, and HK2 is highly expressed and facilitates chemoresistance in various cancers (61–65).
HK2 transfers from the cytoplasm to the outer mitochondrial membrane and combines with voltage-dependent anion channel to display a series effects of anti-apoptosis and chemoresistance: (1) mitochondrially bound HK2(MitoHK-II) is in close proximity to the intramitochondrial ATP and consequently promotes glycolysis (66); (2) MitoHK-II inhibits apoptosis by precisely inhibiting or closing mitochondrial permeability transition pores (mPTPs), and then inhibiting the release of cytochrome c and other apoptotic factors (67); (3) MitoHK-II prevents the opening of mPTPs by inhibiting reactive oxygen species(ROS) accumulation and providing cellular protection against Ca2+ overload (61); (4) MitoHK-II competitively inhibits BCL2-associated X (Bax) binding to the mitochondria and transfers Bax back to the cytoplasm, thereby inhibiting apoptosis (62); and (5) when extracellular microenvironment is not conducive to the growth of cancer cells, such as during hypoxia, and in the presence of chemical drugs, HK1 ensures energy supply of glycolysis and HK2 inhibits the release of apoptotic factors via MitoHK-II (61, 68–70).
HK2 is also related to other cancer-associated factors. The PI3K/AKT/mTOR pathway facilitates the combination of HK2 and the outer mitochondrial membrane, which maintains a high metabolic rate, stemness, and promotes proliferation, invasion, metastasis, and chemoresistance of cancer (71, 72). In contrast, HK2 develops protective autophagy and inhibits cell apoptosis through the PI3K/AKT/mTOR pathway or the MAPK kinases (MEK)/extracellular regulated protein kinases (ERK) pathway of chemotherapeutic drugs (61), such as cisplatin in ovarian cancer (61, 73). P53 rebuilds the chemosensitivity of cisplatin by binding to the promoter region of HK2 in epithelial ovarian cancer (74). The long noncoding RNA-Suppressing Androgen Receptor in Renal Cell Carcinoma (lncRNA-SARCC) restores the sensitivity of osteosarcoma to cisplatin via miR-143 by targeting HK2 (75). MiR-125b recovers 5-FU and cisplatin sensitivity in various cancers by binding to HK2 mRNA (76–79). 3-Bromopyruvate (3-BrPA), a small molecule analog to lactate, is a potent inhibitor of HK2 and not only induces the cytotoxic effects of chloroethylnitrosoureas and reduces the synthesis of biomacromolecules required for DNA repair in gliomas (80) but also promotes cisplatin sensitivity in non-small-cell lung cancer overexpressing tripartite motif-containing 59 (TRIM 59), which results in a high glycolysis rate and cisplatin resistance via the regulation of phosphatase and tensin homolog deleted on chromosome ten (PTEN)/AKT/HK2 (81). HK2 induces chemoresistance by binding to the outer mitochondrial membrane or interacting with other cancer-associated factors, which can be a new target for cancer therapy.
Phosphofructokinase (PFK), located in the cytoplasm, is divided into two subtypess: PFK1 converts fructose 6-phosphate into fructose-1,6-bisphosphate, which is the second rate-limiting step in glycolysis, and PFK2, also called 6-phosphofructo-2-kinase/fructose-2,6-biphosphatase (PFKFB), converts fructose-6-phosphate to fructose-2,6-biphosphatase. PFKFB can regulate glycolysis via fructose-2,6-biphosphatase, which is recognized as an essential allosteric activator of PFK1 (82–84). PFKFB has four subtypes, namely, PFKFB1, PFKFB2, PFKFB3, and PFKFB4, of which PFKFB3 exhibits apical kinase activity and is overexpressed under various signals such as hypoxia, estrogen receptor, RAS activation, and P53 deletion in cancer, which promotes glycolysis flux in cancer metabolism (83, 85–87). Overexpression of PFKFB3 in cancer contributes to cyclin-dependent kinases, leading to the phosphorylation and degradation of Cip/Kip protein p27, thereby facilitating the cell cycle, enhancing cell proliferation, and inhibiting apoptosis (88). As a downstream component of vascular endothelial growth factor, PFKFB3 enhances angiogenesis and endothelial migration by regulating tube formation and directional migration of the filamentous and lamellar feet of the endothelium (83, 84, 89) and promotes blood vessel branching by inhibiting the pre-stalk activity of Notch signaling (90), thereby weakening the effect of antiangiogenic therapies and promoting the exchange of lactate between the cells in the tumor core and edge to meet their requirement of energy source (91).
PFKFB3 not only contributes to the proliferation, metastasis, and angiogenesis of cancer but also induces the resistance of liver cancer cells to sorafenib through the PFKFB3/HIF-1A positive feedback loop (92). Inhibition of PFKFB3 suppresses defensive autophagy induced by oxaliplatin and recovers cytotoxicity of oxaliplatin in colorectal cancer (93). Although cisplatin can induce PFKFB3 acetylation (K472) and hinder its nuclear localization signal activity, accumulation of PFKFB3 in the cytoplasm facilitates glycolysis to counteract the effects of cisplatin (94). Antiangiogenic therapies combined with the inhibition of PFKFB3 not only recover the normal vascular barrier function and blood perfusion but also result in metabolic changes in endothelial cells or vascular leakage, further impairing the delivery of chemotherapeutic drugs (85, 95). MiR-488 not only inhibits the proliferation and glycolysis of prostate cancer (96) but also inhibits oxaliplatin/5-FU resistance and glycolysis of colorectal cancer by targeting PFKFB3 (97). Liposomes co-loaded with PFKFB3 shRNA plasmid significantly upregulate the cytotoxicity of docetaxel in non-small cell lung cancer (98). Currently, increasing number of studies on molecular inhibitors of PFKFB3 are further exploring the possibility of clinical therapy, such as 3-(3-pyridinyl)-1-(4-pyridinyl)-2-propen-1-one (3PO), PFK, and PFK-158. PFK-158 has entered a phase 1 clinical trial (clinicaltrials.gov #NCT02044861) (84).
Pyruvate kinase (PK) detected in the cytoplasm produces pyruvate and ATP, which is the last rate-limiting step of glycolysis. PK encoded by PKM and PKLR gene is separated into four subtypes, namely, PKL, PKR, PKM1, and PKM2. PKL and PKR exist in the liver and erythrocytes, while PKM1 and PKM2 generally exist in normal tissues and cancer cells (99, 100). PKM2 is the major isoform in cancer, which can shuttle between the cytoplasm and nucleus, and engages in proliferation, anti-apoptosis, metastasis, chemoresistance and other processes in cancer (101–103). For example, lncRNA XIST/miR-137 axis induces glycolysis and 5-FU/cisplatin resistance in colorectal cancer by elevating the PKM2/PKM1 ratio (104).
The mechanism of PKM2-induced chemoresistance can be summarized in two aspects: PKM2 located in the cytoplasm facilitates glycolysis and metabolism, and when phosphorylated in the nucleus, PKM2 is displayed as a protein kinase regulating gene expression. PKM2 promotes not only glycolysis but also the production of glycolysis intermediates and enters the glycolysis branch pathway, such as the pentose phosphate pathway, which suppresses ROS accumulation and induces cisplatin resistance in esophageal squamous cancer (105). PKM2 can inhibit ROS accumulation and oxidative stress-induced apoptosis by binding to BCL2 protein on the mitochondrial membrane (100). Especially, miR‐122 inhibits docetaxel resistance of prostate and hepatocellular cancer and 5-FU resistance of colon cancer by targeting PKM2 (106–108). Exosomes derived from chemoresistant cancer cells can transfer ciRS-122 across the cells and facilitate glycolysis to reduce oxaliplatin sensitivity in chemosensitive cells by inhibiting miR-122 and upregulating PKM2 in colorectal cancer (109). On the other hand, studies have confirmed that the inhibition of PKM2 can increase the susceptibility of cancer cells overexpressing ATP-binding cassette (ABC) transporters to ATP depletion, thereby inhibiting glycolysis, inducing apoptosis, and increasing chemosensitivity (107–111). Phosphorylated PKM2 has three main functions: (1) PKM2 facilitates oncogene transcription and cancer proliferation by activating β-catenin, cyclin D1, and C-MyC (101, 112); (2) P53 and PKM2 in the nucleus can phosphorylate each other to form a cascade to protect against external stress (100); and (3) PKM2 can inactivate P53 by inhibiting P38-MAPK and induce gemcitabine resistance in pancreatic cancer (113).
Interestingly, although most studies have confirmed that the inhibition of PKM2 can significantly upregulate chemosensitivity (114, 115), some studies have suggested that the inhibition can induce chemoresistance (103, 116, 117). Thus, the ability of PKM2 to induce chemoresistance may be in accordance with the cell type, cycle, state, and so on, which needs more exploration in the future (118).
Lactate dehydrogenase (LDH), which is located in the cytoplasm, catalyzes the conversion of pyruvate to lactate, which is the end-product of glycolysis. LDH is composed of three monomeric subunits: LDHA, LDHB, and LDHC, which can constitute six kinds of tetrameric isoenzymes (119, 120). LDHC specifically exists in male germ cells (119), whereas LDHA and LDHB are mainly present in the skeleton muscles/liver and heart, respectively (121). In addition, LDHA is highly expressed and facilitates chemoresistance in various cancers (122–125).
The mechanism of LDHA-induced chemoresistance can be summarized as follows: First, as a direct target of the HIF-1A and C-MYC oncogenes (126), LDHA promotes biosynthesis and glycolysis, ensuring energy supply and proliferation of cancer cells. The peroxisome proliferator-activated receptor-c coactivator-1b promotes cell proliferation and tumor growth through LDHA-mediated glycolytic metabolism in multiple myeloma (127). Circular RNA circUBE2D2 accelerates glycolysis and sorafenib resistance via the miR-889-3p/LDHA axis in hepatocellular carcinoma (128). Family with sequence similarity 83 member D promotes glycolytic capacity and gemcitabine resistance through the Wnt/β-catenin/LDHA pathway in pancreatic adenocarcinoma (129). Second, LDHA is involved in cancer invasion and CSC phenotype through the acidic microenvironment maintained by lactate output (130). When LDHA is highly expressed, mesothelioma becomes more aggressive (131). LDHA is significantly related to octamer-binding transcription factor 4, which plays a key role in the self-renewal of embryonic stem cells in gastric cancer (132). Human coilin-interacting nuclear ATPase protein generates sufficient lactate to maintain an acidic microenvironment for invasion and CSC phenotype via LDHA in colorectal CSCs (133). Third, LDHA inhibits apoptosis by protecting the cancer cells from ROS damage and promoting the expression of antiapoptotic proteins (134–136). Catechin increases mitochondrial ROS, enhances apoptotic cell death, and reduces 5-FU resistance in gastric cancer via LDHA inhibition (137). LDHA inhibition results in increased mitochondrial pathway apoptosis via ROS production and elevated levels of Bax, cleaved poly (adenosine diphosphate-ribose) polymerase, cleaved caspase-9, cytoplasmic cytochrome C, and superoxide anion in breast cancer (138). Metformin facilitates apoptosis via LDHA inhibition in cholangiocarcinoma cells (139).
LDHA inhibition can significantly restore the sensitivity of chemotherapy drugs: LDHA knockdown sensitizes oral squamous cell carcinoma cells (122) and breast cancer cells (140) to Taxol and lung cancer cells to low doses of paclitaxel (141) via siRNA/shRNA. MiR‐34a re-sensitizes colon cancer cells to 5-FU (142), miR-329-3p sensitizes osteosarcoma cells to cisplatin (143), and miR-7 sensitizes gastric cancer cells to cisplatin (144) all via LDHA inhibition. Recently, increasing studies have begun to explore LDHA inhibitors, which can be divided into three categories, represented by oxamate, 3-dihydroxy-6-methyl-7-(phenylmethyl)-4-propylnaphthalene-1carboxylic acid(FX11), and N-hydroxyindoles (NHI) (145). As an analogue of pyruvate, oxamate inhibits LDHA by competing with substrates and overcomes cetuximab resistance in Ewing’s sarcoma (146). FX11 inhibits LDHA by competing with nicotinamide adenine dinucleotide (NADH) and induces oxidative stress and necrosis in human lymphoma and pancreatic cancer xenograft models (134). NHI competes with pyruvate and NADH and overcomes gemcitabine resistance in pancreatic cancer and hypoxic mesothelioma cells (147, 148).
Monocarboxylate transporters (MCTs), which are located on the cytomembrane, are encoded by the SLC16 gene and divided into 14 members that share the same basic structure, of which only the membrane-bound proton-coupled isoforms, MCT1, MCT2, MCT3, and MCT4, transport lactate through the plasma membrane (149, 150). MCT1 has a ubiquitous distribution, whereas MCT4 presents in highly glycolytic tissues (149). Both of them are highly expressed and responsible for the transportation of lactate in cancer cells (151), such as glioblastoma multiforme (152), head and neck cancer (153), and viral-driven lymphomas (154). MCT1 and MCT4 also play indirect roles in angiogenesis, invasion, malignant dissemination, and chemoresistance by regulating and interacting with CD147 (155–157).
MCT1 can transport lactate in both directions, and MCT4 mainly promotes the excretion of lactate from the cell (158), which induces chemoresistance; this can be summarized as five aspects: (1) lactate produced by cancer-associated fibroblasts (CAFs) is extruded through MCT4 and captured by cancer cells through MCT1, which promotes malignant proliferation and aggressiveness and reduces the effects of platinum-based chemotherapy in urothelial bladder cancer (159); (2) hypoxic cancer cells produce and transport lactate to oxygenated cancer cells adjacent to blood vessels via MCT1 and MCT4, which ensures the overall survival of the malignant glioma (160); (3) MCT1 and MCT4 avoid cell death due to intracellular acidification and maintain an acidic microenvironment by promoting lactate efflux in breast cancer (161), colorectal cancer (162) and glioblastomas (163); (4) MCT1 and MCT4 enhance lactate metabolism and inhibit ROS-dependent cellular apoptosis in colorectal cancer (164) and non-small cell lung cancer (165); and (5) MCT1-driven lactate import as a key process of the reverse Warburg effect favors stemness properties, which is a hallmark of chemoresistance in pancreatic adenocarcinoma (166) and glioblastoma (167).
MiR-124 sensitizes breast cancer cells to Taxol via MCT1 inhibition (168). Curcumin reverses chemoresistance in hepatic cancer cells via MCT1 inhibition (169). Co-inhibition of MCT1 and MCT4 can exert a better effect (170). A-cyano-4-hydroxy-cinnamic acid (ACCA), as a small-molecule inhibitor of MCTs, inhibits invasiveness and induces the necrosis of malignant glioma (171) and sensitizes colorectal cancer cells to cisplatin (172). In short, MCTs can also be a new target for further exploration. AZD3965 has been applied as a potent MCT1 inhibitor in various phase I/II clinical trials (173).
The PI3K/AKT signaling pathway typically activated in cancer is not only involved in cellular processes such as inflammation, autophagy, and tumor formation but also related to cancer metabolism (9, 20, 174). Activated AKT prevents the transport of pyruvate into the mitochondria for the TCA cycle and switches cancer metabolism from oxidative phosphorylation to aerobic glycolysis by triggering GLUTI expression, stimulating phosphofructokinase activity, phosphorylating HK2, and inhibiting PKM2 activity (175). Meanwhile, the PI3K/AKT pathway increases energy supply by regulating aerobic glycolysis, which enhances the ability of ABC transporters to excrete drugs (10). The AKT/mTOR signaling pathway maintains homeostasis of glycolysis, induces drug-resistant cells to overexpress C-MYC, directly stimulates glucose uptake, and enhances glycolysis (20, 176). Proteins and hormones in distinct cancer can utilize PI3K to promote glycolysis. For example, Ubiquitin-specific protease 6 N-terminal-like protein sustains chronic AKT phosphorylation and GLUT1 stability fueling aerobic glycolysis in breast cancer (177); TRIM32 promotes the growth of gastric cancer cells by enhancing AKT activity and GLUT1 expression (178). Studies have discussed that PI3K-induced glycolysis may be responsible for the formation of chemoresistant phenotypes of cancer cells (20), and the inhibition of glycolysis by interrupting the PI3K signaling pathway can automatically improve chemoresistance. Serine/threonine kinase 35 induces chemoresistance of colorectal cancer cells toward 5-FU, partially due to its role in inducing glycolytic process by regulating AKT (179). The overexpression of Helicobacter pylori-secreted Cytotoxin-associated gene A protein contributes to 5-FU resistance by enhancing glycolysis in gastric cancer via the activation of the AKT pathway (180); Copines-1 enhances oxaliplatin resistance of colorectal cancer cells by activating the AKT/GLUT1/HK2 signaling pathway (181). Downregulation of Krüppel-like factor 5 can inhibit hypoxia-induced cisplatin resistance in non-small-cell lung cancer, and its mechanism is via the inhibition of HIF-1α-dependent glycolysis through the inactivation of the PI3K/AKT/mTOR pathway (182). Knockdown of the transcription factor Forkhead box 6 can inhibit glycolysis of hepatocellular carcinoma cells and reduce their paclitaxel resistance via inhibiting the PI3K/AKT signaling pathway (183).
HIF-1 is a nucleoprotein secreted under hypoxia that acts as a transcription factor to regulate angiogenesis, endothelial cell migration (184), erythropoiesis (9), and innate immunity (185). HIF-1 induces the conversion from oxidative phosphorylation to aerobic glycolysis in cancer under normoxia (186). Abnormally stimulated HIF-1 functioning as a transcription factor inhibits mitochondrial activity and promotes glycolysis and cancer cell growth by facilitating the expression of glycolysis transporters and key enzymes such as GLUT1, HK2, FBP, PKM2, and LDHA (187–189). Especially, HIF-1 regulates oncogene expression (190); on the contrary, oncogene signaling pathways such as PI3K/AKT, MAPK/ERK, STAT3, and nuclear PKM2 can activate HIF-1 under normoxia (186). Sphingosine kinase 1 contributes to doxorubicin resistance and glycolysis of osteosarcoma by advocating HIF-1α expression (191). Human equilibrative nucleoside transporter 1 restores the chemosensitivity of gemcitabine by inhibiting glycolysis and glucose transport mediated by HIF-1α in pancreatic cancer (192). Glycolysis engages in chemoresistance induced by HIF-1 through different mechanisms: (1) HIF-1 switches metabolism from oxidative phosphorylation to glycolysis and leads to mitochondrial dysfunction; decreased accumulation of ROS elicits the inhibition of apoptosis, which disturbs the capability of chemotherapeutic drugs and facilitates chemoresistance (190, 193, 194); (2) Tumor-associated macrophages (TAMs) secrete vesicle-packaged HIF-1α-stabilizing IncRNA to inhibit HIF-1 degradation, promote glycolysis, and induce docetaxel resistance in breast cancer. Lactate production of glycolysis enhances HIF-1 expression through the ERK pathway, forming a positive feedback loop to induce chemoresistance (195) and (3) HIF-1 activates carbonic anhydrase IX (CAIX) to maintain normal intracellular pH in response to vinorelbine, thereby preventing cell apoptosis. As a transmembrane protein neutralizing intracellular acidosis, CAIX is induced by HIF-1 and is related to glycolysis in lung cancer (196). Chemotherapy combined with HIF-1 inhibition upregulates the sensitivity of chemotherapeutic drugs. For example, HIF-1 knockdown significantly improves chemosensitivity to cisplatin in prostate and ovarian cancer (197, 198). Baicalein deteriorates hypoxia-induced 5-FU resistance in gastric cancer by suppressing glycolysis and the PTEN/AKT/HIF-1 signaling pathway (199). Ascorbate combined with cisplatin increases ROS production and alters glycolysis and mitochondrial function by decreasing the HIF-1 activity, which further restores cisplatin sensitivity of osteosarcoma (200).
MYC is a group of oncogenes including C-MYC, L-MYC and N-MYC that is generally upregulated and amplificated in cancers (201). MYC functioning as a transcription factor directly upregulates GLUT, HK2, and PKM2 expression and inhibits mitochondrial respiration and activity (189). Especially, MYC upregulates genes that play an essential role in metabolic reorganization equally under normoxia and hypoxia (176, 202). Besides acting as a downstream factor of HIF1, C-MYC has a synergistic effect with HIF-1 on inducing glycolysis by promoting 3-phosphoinositide dependent kinase-1 and HK2 and inducing angiogenesis, leading to hypoxia adaptation, internal environment stability, and chemoresistance in cancer (202, 203). In addition, various proteins and molecules in cancer use C-MYC to promote glycolysis and induce chemoresistance: the epigenetic factor protein arginine methyltransferase 5 is an epigenetic enzyme that leads to increased C-MYC levels and subsequent enhancement of proliferation and glycolysis in pancreatic cancer (204); miR-155 positively regulates glucose metabolism via C-MYC in breast cancer (205); P21-activated kinase 2 (PAK2) utilizes the PAK2/C-MYC/PKM2 axis and induces camptothecin/etoposide resistance in head and neck carcinoma (206); gankyrin arrives glycolysis to promote tumorigenesis, metastasis, and sorafenib/regorafenib resistance by activating β-catenin/C-MYC signaling in human hepatocellular carcinoma (207); increased aerobic glycolysis mediates adriamycin resistance, which is related to excessive activation of the AKT/mTOR/C-MYC pathway in leukemia cells; oxamate rescues adriamycin sensitivity depending on the downregulation of glycolysis instead of P-glycoprotein (P-gp) (208).
The key process of glycolysis is inseparable from chemoresistance induced by oncogenes (Figure 2). On the one hand, the PI3K, HIF-1, and C-MYC signaling pathways can activate the expression of key glycolysis enzymes and transporters to ensure cancer metabolism and energy supply; on the other hand, the key enzymes and transporters of glycolysis can activate chemoresistance-related signaling pathways through their own or other protein mediators. The complementary synergistic effect of the key process of glycolysis and oncogene signaling pathway strives possibilities for the survival of cancer cells during chemotherapy.
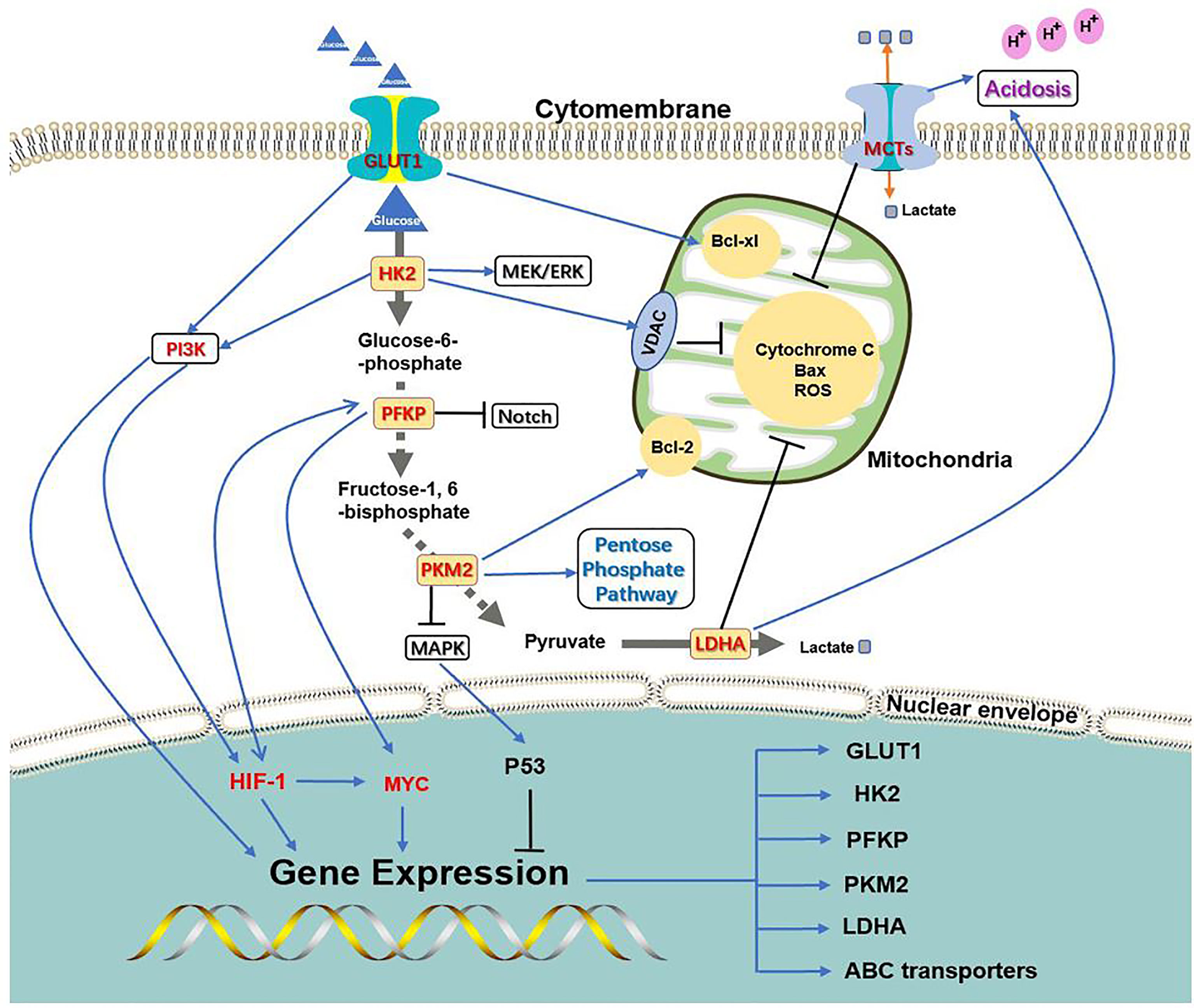
Figure 2 Association between glycolysis transporter, key enzymes, and PI3K, HIF-1, C-MYC signaling pathways. PI3K, HIF-1, and C-MYC signaling pathways can activate the expression of key glycolysis enzymes and transporters to ensure cancer metabolism and energy supply; at the same time, the key enzymes and transporters of glycolysis can activate chemoresistance-related signaling pathways through their own or other protein mediators.
In addition to gene mutation and metabolism change, cancer often impacts the surrounding microenvironment, of which the impact of metabolism on the microenvironment is more significant. With the transformation of metabolism, the microenvironment undergoes a series of adjustments such as hypoxia (209), acidosis (20), and stromal cell formation (210–212) to survival, which interact with glycolysis and induce chemoresistance in cancer (Figure 3).
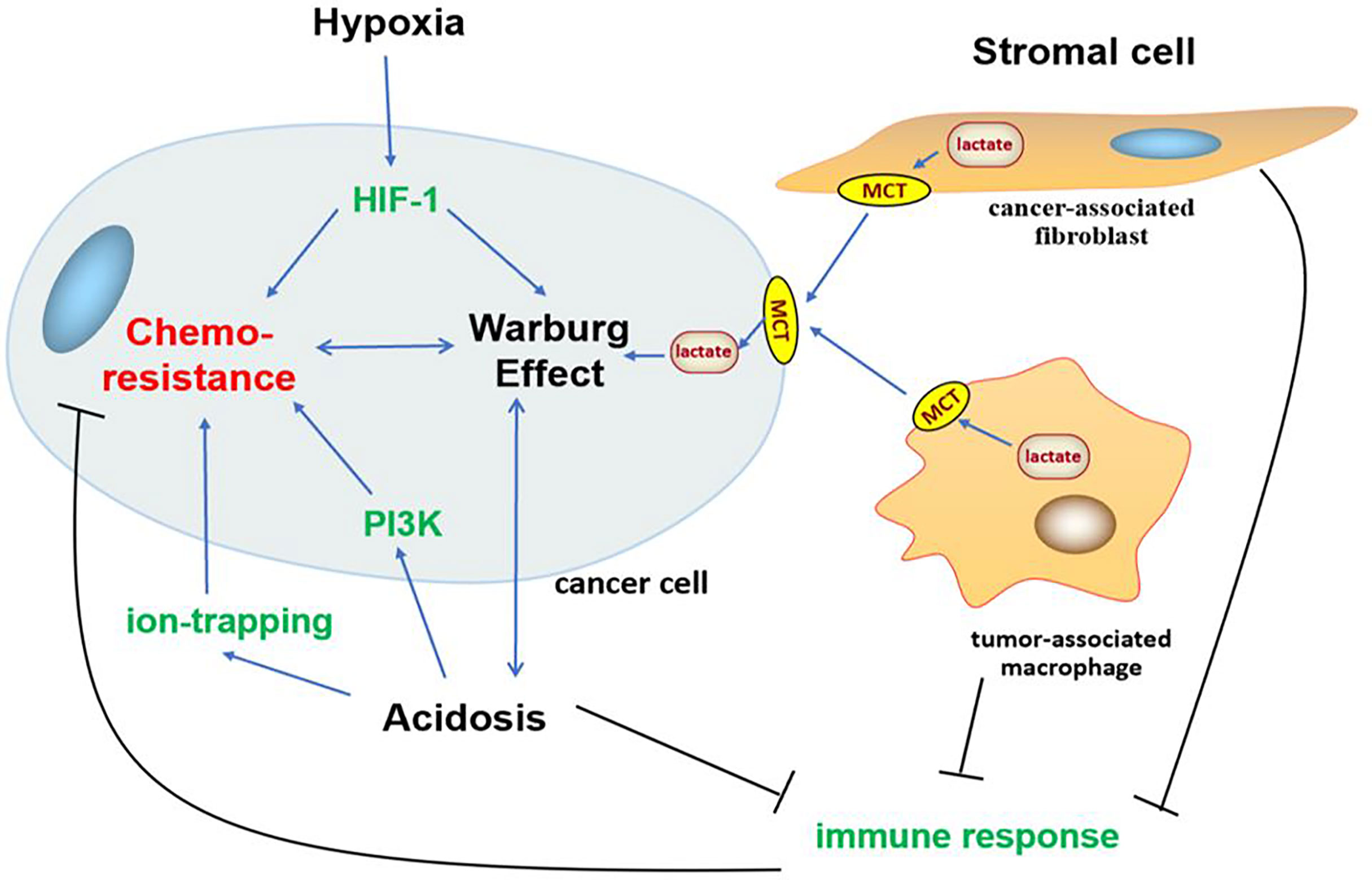
Figure 3 Association between Warburg effect-induced chemoresistance and tumor microenvironment. Tumor microenvironment undergoes a series of adjustments such as hypoxia, acidosis, and stromal cell formation to survival, which interact with glycolysis and oncogene PI3K, HIF-1 signaling pathways to induce chemoresistance in cancer.
Solid tumors are produced without the existing vascular system and can only exist by recruiting new blood vessels, which are always inadequate and dysfunctional. Rapid growth and proliferation usually lead to oxygen consumption and hypoxia in most tumor beds (209, 213). Hypoxia can promote glycolysis and stemness in hepatocellular cancer through ubiquitin-specific protease 22 (214), melanoma cancer via nodal signaling activity (215), and so on. Cancer triggers metabolism, angiogenesis, and erythropoiesis to counteract the disadvantages of hypoxia, of which HIF-1 is the central regulatory mechanism of hypoxia that acts by upregulating its downstream genes (216, 217). HIF-1 is the main transcription factor that induces the expression of almost all genes encoding glucose transporters and glycolytic key enzymes (218, 219), which allows hypoxic cancer cells to absorb glucose more efficiently, metabolize pyruvate to lactate, activate multi-drug resistance gene, and induce chemoresistance (220, 221). Hypoxia has synergistic effects with acidosis on inducing chemoresistance by upregulating the expression of fatty acid synthase and regulating lipid metabolism (209); it can induce acidosis by selecting glycolytic cells, and acidosis can further select cells with upregulated glycolysis and acidic resistance, thereby choosing cells with survival advantages (222). Both these negative factors facilitate the evolution of cancer and select cells with more survival advantages, which can retain genomic instability and a mutator phenotype, have sustained angiogenesis, and be resistant to apoptosis and chemotherapy (8, 222). For example, CAIX, a pH regulator under hypoxia, regulates the adaptation of hypoxia and acidosis by promoting glycolysis and stemness in breast cancer (223).
Aerobic glycolysis in cancer produces numerous amounts of lacttate, thereby cancer cells rapidly export lacttate via MCTs on the cytomembrane, which maintains intracellular acid–base balance and ensures aerobic glycolysis and lactate production to be continued; thus, cancer cells sustain a special pH gradient, which is more acidic extracellularly and more alkaline intracellularly (212, 224–226). Acidosis-induced chemoresistance can be summarized as follows: (1) Most chemotherapeutic drugs are either weak bases or weak acids; only few of them are zwitterions. Weakly basic drugs such as paclitaxel and vincristine will be neutralized and protonated under extracellular acidosis, making it difficult for them to pass through the cytomembrane and function. Even if they pass through the cytomembrane, they will be isolated into acidic vesicles of lysosomes and lose their efficacy. Although weakly acidic drugs increase their distribution in the interstitial fluid, they will become inactive before reaching the target due to intracellular alkalinity. This phenomenon is called the “ion trapping mechanism” (227–231). Meanwhile, extracellular acidosis facilitates P-gp, ABC subfamily B member 1 (ABCB1) and 2 (ABCB2) transporter, and intracellular acidic vesicles to remove drugs out of cancer cells, further inducing chemoresistance (224, 232–235). (2) Extracellular acidosis promotes chemoresistance signaling pathways by activating related proteins. For example, mild acidic stress not only facilitates unfolded protein response (UPR) but also triggers an adaptive UPR with progressive increase in glucose regulatory protein 78 expression, which reduces the cleavage of caspase 7 to induce sunitinib resistance in oral squamous cancer (212, 224); extracellular lactate functions as an agonist for G protein-coupled receptor 81 (GPR81) and promotes GPR81 upregulation of the PI3K/AKT/mTOR pathway to inhibit apoptosis, promote stem cell phenotype, inhibit immune response, and induce etoposide resistance in non-small-cell lung cancer (234, 236). (3) Lactate inhibits immune response in different ways: ① lactate directly inhibits the cytotoxicity of perforin and granzyme; ② high extracellular lacttate levels lead to the accumulation of endogenous lacttate in T cells, thereby reducing the secretion of pro-inflammatory cytokine; ③ lacttate indirectly weakens natural killer (NK) cell function by recruiting monocyte-derived dendritic cells (225); and (4) extracellular acidosis not only has synergistic effect with hypoxia but also facilitates glycolysis of stromal cells to produce lactate for fueling cancer cells, ensuring survival and proliferation and preventing apoptosis (166).
Stromal cells facilitate metabolism, invasion, metastasis, and chemoresistance in cancer by paracrine signaling, and the recruitment of immunosuppressive cells is the foundation of tumor microenvironment, in which CAFs and TAMs have representative functions (210, 211, 237). As cancer progresses, cancer cells not only harness neighboring cells recruiting glycolysis and glutaminolysis for both itself and for the neighboring cancer cells via MCT1 and MCT4, which is called the Reverse Warburg effect (91), but also promotes the differentiation of stromal cells into CAFs and TAMs to ensure survival advantage. For instance, cancer cells induce the CAFs phenotype by secreting microvesicle or extracellular vesicle, which supply energy and promote proliferation, migration, and resistance in nasopharyngea and oral squamous cancer (238, 239). Cancer cells diffuse excessive intracellular ROS into the extracellular space (240), which causes strong oxidative stress and facilitates the onset of CAFs phenotype in adjacent stromal cells, further eliminating ROS and providing nutrients in turn via MCTs (241). Studies have confirmed that CAFs promote a glycolytic switch, ROS elimination in chronic lymphocytic leukemia via a Notch/C-MYC signaling-dependent manner under hypoxia (242, 243). Ovarian cancer cells release cytokines that recruit and activate stromal fibroblasts and immune cells, thereby perpetuating an interstitial inflammatory state in the stroma that hinders the immune response and facilitates cancer survival and propagation (244). Although competition exists for oxygen and glucose between stromal and cancer cells, both CAFs and TAMs can fuel cancer cells via the Reverse Warburg effect under normoxia (210, 211). Meanwhile, TAMs not only secrete vesicle-packaged HIF-1α-stabilizing lncRNA to inhibit HIF-1 degradation, enhance glycolysis, and induce chemoresistance in breast cancer (195) but also inhibit T cell infiltration, resulting in decreased programmed death-ligand 1 expression in tumors, which compromises the tumor response to various anticancer therapies (245).
Aerobic glycolysis is an important hallmark that distinguishes cancer tissues from normal tissues; on the one hand, it interacts with oncogenes PI3K, HIF-1, and C-MYC for inducing chemoresistance by facilitating the overexpression of glucose transporters and key enzymes of glycolysis and resistant signaling pathways in different degrees, and on the other hand, it acts synergistically with hypoxia and acidosis for advocating oncogene signaling pathways and stromal cells on sustaining energy supply and immune escape in cancer. Lactate, a product of glycolysis, blocks the efficacy of chemotherapy drugs by maintaining a hypoxic, acidic cancer microenvironment. Although further studies are warranted to determine the exact mechanism of the Warburg effect-induced chemoresistance, studies on inhibitors targeting glycolysis transporters, key enzymes, and signaling pathways are undergoing or have entered clinical trials (Table 1). Therefore, the inhibition of aerobic glycolysis in cancer may be a new idea for chemotherapy, which provides a new possibility for clinical therapy.
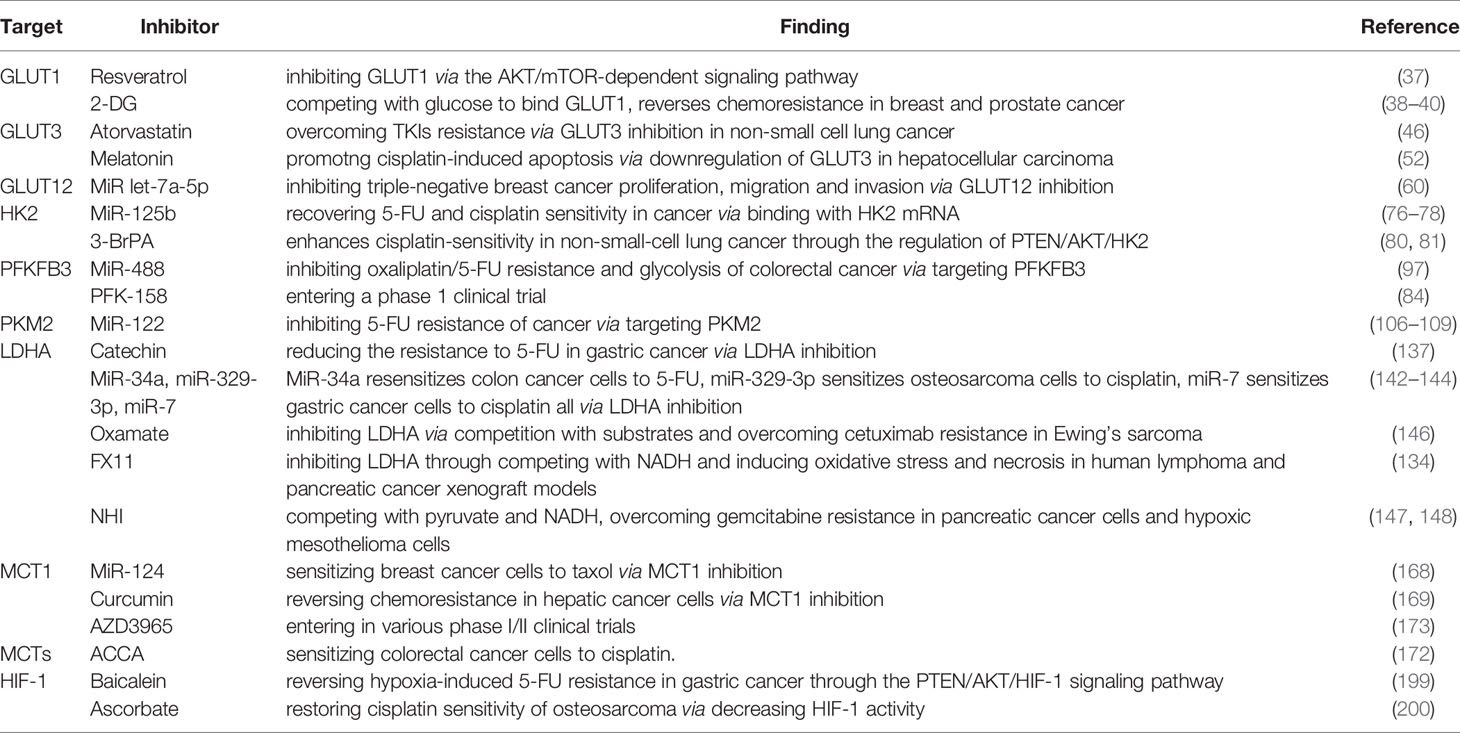
Table 1 A list of glycolytic inhibitors targeting transporters, key enzymes, and signaling pathway in the glucose metabolic pathway.
CL: Data curation, Writing - Original draft preparation. YJ: Writing - Review and Editing. ZF: Writing - Review and Editing. All authors contributed to the article and approved the submitted version.
National Natural Science Foundation of China (No. 82003173): translation fee 3000 RMB Jilin Province Department of Finance(JLSCZD2019-030) : publishing fee 6000 RMB.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Hanahan D, Weinberg RA. Hallmarks of Cancer: The Next Generation. Cell (2011) 144(5):646–74. doi: 10.1016/j.cell.2011.02.013
2. Icard P, Shulman S, Farhat D, Steyaert JM, Alifano M, Lincet H. How the Warburg Effect Supports Aggressiveness and Drug Resistance of Cancer Cells? Drug Resist Update (2018) 38:1–11. doi: 10.1016/j.drup.2018.03.001
4. Doktorova H, Hrabeta J, Khalil MA, Eckschlager T. Hypoxia-Induced Chemoresistance in Cancer Cells: The Role of Not Only HIF-1. BioMed Pap Med Fac. Univ. Palacky Olomouc Czech Repub (2015) 159(2):166–77. doi: 10.5507/bp.2015.025
5. Wouters BG, van den Beucken T, Magagnin MG, Lambin P, Koumenis C. Targeting Hypoxia Tolerance in Cancer. Drug Resist Update (2004) 7(1):25–40. doi: 10.1016/j.drup.2003.12.004
6. Warburg O, Wind F, Negelein E. The Metabolism of Tumors in the Body. J Gen Physiol (1927) 8(6):519–30. doi: 10.1085/jgp.8.6.519
7. Pillai SR, Damaghi M, Marunaka Y, Spugnini EP, Fais S, Gillies RJ. Causes, Consequences, and Therapy of Tumors Acidosis. Cancer Metastasis Rev (2019) 38(1-2):205–22. doi: 10.1007/s10555-019-09792-7
8. Vaupel P, Multhoff G. Fatal Alliance of Hypoxia-/HIF-1alpha-Driven Microenvironmental Traits Promoting Cancer Progression. Adv Exp Med Biol (2020) 1232:169–76. doi: 10.1007/978-3-030-34461-0_21
9. Abdel-Wahab AF, Mahmoud W, Al-Harizy RM. Targeting Glucose Metabolism to Suppress Cancer Progression: Prospective of Anti-Glycolytic Cancer Therapy. Pharmacol Res (2019) 150:104511. doi: 10.1016/j.phrs.2019.104511
10. Liu R, Chen Y, Liu G, Li C, Song Y, Cao Z, et al. PI3K/AKT Pathway as a Key Link Modulates the Multidrug Resistance of Cancers. Cell Death Dis (2020) 11(9):797. doi: 10.1038/s41419-020-02998-6
11. Weir HK, Thun MJ, Hankey BF, Ries LA, Howe HL, Wingo PA, et al. Annual Report to the Nation on the Status of Cancer, 1975-2000, Featuring the Uses of Surveillance Data for Cancer Prevention and Control. J Natl Cancer Inst (2003) 95(17):1276–99. doi: 10.1093/jnci/djg040
12. Tse T, Sehdev S, Seely J, Gravel DH, Clemons M, Cordeiro E, et al. Neoadjuvant Chemotherapy in Breast Cancer: Review of the Evidence and Conditions That Facilitated Its Use During the Global Pandemic. Curr Oncol (2021) 28(2):1338–47. doi: 10.3390/curroncol28020127
13. Klaiber U, Leonhardt CS, Strobel O, Tjaden C, Hackert T, Neoptolemos JP. Neoadjuvant and Adjuvant Chemotherapy in Pancreatic Cancer. Langenbecks Arch Surg (2018) 403(8):917–32. doi: 10.1007/s00423-018-1724-8
14. Zeng S, Pottler M, Lan B, Grutzmann R, Pilarsky C, Yang H. Chemoresistance in Pancreatic Cancer. Int J Mol Sci (2019) 20(18):4504–23. doi: 10.3390/ijms20184504
15. Das M, Law S. Role of Tumor Microenvironment in Cancer Stem Cell Chemoresistance and Recurrence. Int J Biochem Cell Biol (2018) 103:115–24. doi: 10.1016/j.biocel.2018.08.011
16. Ramos P, Bentires-Alj M. Mechanism-Based Cancer Therapy: Resistance to Therapy, Therapy for Resistance. Oncogene (2015) 34(28):3617–26. doi: 10.1038/onc.2014.314
17. Bosc C, Selak MA, Sarry JE. Resistance Is Futile: Targeting Mitochondrial Energetics and Metabolism to Overcome Drug Resistance in Cancer Treatment. Cell Metab (2017) 26(5):705–7. doi: 10.1016/j.cmet.2017.10.013
18. Pan ST, Li ZL, He ZX, Qiu JX, Zhou SF. Molecular Mechanisms for Tumour Resistance to Chemotherapy. Clin Exp Pharmacol Physiol (2016) 43(8):723–37. doi: 10.1111/1440-1681.12581
19. Zhao Y, Butler EB, Tan M. Targeting Cellular Metabolism to Improve Cancer Therapeutics. Cell Death Dis (2013) 4:e532. doi: 10.1038/cddis.2013.60
20. Ma L, Zong X. Metabolic Symbiosis in Chemoresistance: Refocusing the Role of Aerobic Glycolysis. Front Oncol (2020) 10:5. doi: 10.3389/fonc.2020.00005
21. Liu Y, Zhang Z, Wang J, Chen C, Tang X, Zhu J, et al. Metabolic Reprogramming Results in Abnormal Glycolysis in Gastric Cancer: A Review. Oncol. Targets Ther (2019) 12:1195–204. doi: 10.2147/OTT.S189687
22. Tekade RK, Sun X. The Warburg Effect and Glucose-Derived Cancer Theranostics. Drug Discovery Today (2017) 22(11):1637–53. doi: 10.1016/j.drudis.2017.08.003
23. Massari F, Ciccarese C, Santoni M, Iacovelli R, Mazzucchelli R, Piva F, et al. Metabolic Phenotype of Bladder Cancer. Cancer Treat Rev (2016) 45:46–57. doi: 10.1016/j.ctrv.2016.03.005
24. Mueckler M, Thorens B. The SLC2 (GLUT) Family of Membrane Transporters. Mol Aspects Med (2013) 34(2-3):121–38. doi: 10.1016/j.mam.2012.07.001
25. Barron CC, Bilan PJ, Tsakiridis T, Tsiani E. Facilitative Glucose Transporters: Implications for Cancer Detection, Prognosis and Treatment. Metabolism (2016) 65(2):124–39. doi: 10.1016/j.metabol.2015.10.007
26. Ding X, Liu J, Liu T, Ma Z, Wen D, Zhu J. miR-148b Inhibits Glycolysis in Gastric Cancer Through Targeting SLC2A1. Cancer Med (2017) 6(6):1301–10. doi: 10.1002/cam4.1008
27. Mori Y, Yamawaki K, Ishiguro T, Yoshihara K, Ueda H, Sato A, et al. ALDH-Dependent Glycolytic Activation Mediates Stemness and Paclitaxel Resistance in Patient-Derived Spheroid Models of Uterine Endometrial Cancer. Stem Cell Rep (2019) 13(4):730–46. doi: 10.1016/j.stemcr.2019.08.015
28. Xiao H, Wang J, Yan W, Cui Y, Chen Z, Gao X, et al. GLUT1 Regulates Cell Glycolysis and Proliferation in Prostate Cancer. Prostate (2018) 78(2):86–94. doi: 10.1002/pros.23448
29. Barbosa AM, Martel F. Targeting Glucose Transporters for Breast Cancer Therapy: The Effect of Natural and Synthetic Compounds. Cancers (Basel) (2020) 12(1):154–92. doi: 10.3390/cancers12010154
30. Gou Q, Dong C, Jin J, Liu Q, Lu W, Shi J, et al. PPARalpha Agonist Alleviates Tumor Growth and Chemo-Resistance Associated With the Inhibition of Glucose Metabolic Pathway. Eur J Pharmacol (2019) 863:172664. doi: 10.1016/j.ejphar.2019.172664
31. Yu M, Yongzhi H, Chen S, Luo X, Lin Y, Zhou Y, et al. The Prognostic Value of GLUT1 in Cancers: A Systematic Review and Meta-Analysis. Oncotarget (2017) 8(26):43356–67. doi: 10.18632/oncotarget.17445
32. Wang Y, Zhang X, Wang Z, Hu Q, Wu J, Li Y, et al. LncRNA-P23154 Promotes the Invasion-Metastasis Potential of Oral Squamous Cell Carcinoma by Regulating Glut1-Mediated Glycolysis. Cancer Lett (2018) 434:172–83. doi: 10.1016/j.canlet.2018.07.016
33. Li H, Fu L, Liu B, Lin X, Dong Q, Wang E. Ajuba Overexpression Regulates Mitochondrial Potential and Glucose Uptake Through YAP/Bcl-Xl/GLUT1 in Human Gastric Cancer. Gene (2019) 693:16–24. doi: 10.1016/j.gene.2019.01.018
34. Wang L, Sun J, Gao P, Su K, Wu H, Li J, et al. Wnt1-Inducible Signaling Protein 1 Regulates Laryngeal Squamous Cell Carcinoma Glycolysis and Chemoresistance via the YAP1/TEAD1/GLUT1 Pathway. J Cell Physiol (2019). doi: 10.1002/jcp.28253
35. Jiang T, Zhou ML, Fan J. Inhibition of GLUT-1 Expression and the PI3K/Akt Pathway to Enhance the Chemosensitivity of Laryngeal Carcinoma Cells In Vitro. Oncol. Targets Ther (2018) 11:7865–72. doi: 10.2147/OTT.S176818
36. Liu R, Li H, Gao X, Mi Q, Zhao H, Gao Q. Mannose-Conjugated Platinum Complexes Reveals Effective Tumor Targeting Mediated by Glucose Transporter 1. Biochem Biophys Res Commun (2017) 487(1):34–40. doi: 10.1016/j.bbrc.2017.04.004
37. Brockmueller A, Sameri S, Liskova A, Zhai K, Varghese E, Samuel SM, et al. Resveratrol’s Anti-Cancer Effects Through the Modulation of Tumor Glucose Metabolism. Cancers (Basel) (2021) 13(2):188–223. doi: 10.3390/cancers13020188
38. Ma S, Jia R, Li D, Shen B. Targeting Cellular Metabolism Chemosensitizes the Doxorubicin-Resistant Human Breast Adenocarcinoma Cells. BioMed Res Int (2015) 2015:453986. doi: 10.1155/2015/453986
39. Ben Sahra I, Laurent K, Giuliano S, Larbret F, Ponzio G, Gounon P, et al. Targeting Cancer Cell Metabolism: The Combination of Metformin and 2-Deoxyglucose Induces P53-Dependent Apoptosis in Prostate Cancer Cells. Cancer Res (2010) 70(6):2465–75. doi: 10.1158/0008-5472.Can-09-2782
40. Seo M, Crochet RB, Lee Y-H. Targeting Altered Metabolism—Emerging Cancer Therapeutic Strategies. Cancer Drug Design Discov (2014) . p:427–48. doi: 10.1016/B978-0-12-396521-9.00014-0
41. Simpson IA, Dwyer D, Malide D, Moley KH, Travis A, Vannucci SJ. The Facilitative Glucose Transporter GLUT3: 20 Years of Distinction. Am J Physiol Endocrinol Metab (2008) 295(2):E242–53. doi: 10.1152/ajpendo.90388.2008
42. Thorens B, Mueckler M. Glucose Transporters in the 21st Century. Am J Physiol Endocrinol Metab (2010) 298(2):E141–5. doi: 10.1152/ajpendo.00712.2009
43. Libby CJ, Gc S, Benavides GA, Fisher JL, Williford SE, Zhang S, et al. A Role for GLUT3 in Glioblastoma Cell Invasion That is Not Recapitulated by GLUT1. Cell Adh Migr (2021) 15(1):101–15. doi: 10.1080/19336918.2021.1903684
44. Ancey PB, Contat C, Meylan E. Glucose Transporters in Cancer - From Tumor Cells to the Tumor Microenvironment. FEBS J (2018) 285(16):2926–43. doi: 10.1111/febs.14577
45. He Z, Chen D, Wu J, Sui C, Deng X, Zhang P, et al. Yes Associated Protein 1 Promotes Resistance to 5-Fluorouracil in Gastric Cancer by Regulating GLUT3-Dependent Glycometabolism Reprogramming of Tumor-Associated Macrophages. Arch Biochem Biophys (2021) 702:108838. doi: 10.1016/j.abb.2021.108838
46. Ali A, Levantini E, Fhu CW, Teo JT, Clohessy JG, Goggi JL, et al. CAV1 - GLUT3 Signaling is Important for Cellular Energy and can be Targeted by Atorvastatin in Non-Small Cell Lung Cancer. Theranostics (2019) 9(21):6157–74. doi: 10.7150/thno.35805
47. Le Calve B, Rynkowski M, Le Mercier M, Bruyere C, Lonez C, Gras T, et al. Long-Term In Vitro Treatment of Human Glioblastoma Cells With Temozolomide Increases Resistance In Vivo Through Up-Regulation of GLUT Transporter and Aldo-Keto Reductase Enzyme AKR1C Expression. Neoplasia (N. Y. NY) (2010) 12(9):727–39. doi: 10.1593/neo.10526
48. Song Y, Meng L, Yu J, Cao Z, Sun J, Zhao H. TRIM66 Overexpression Promotes Glioma Progression and Regulates Glucose Uptake Through Cmyc/GLUT3 Signaling. Cancer Manag Res (2021) 13:5187–201. doi: 10.2147/CMAR.S293728
49. Ho KH, Chen PH, Chou CM, Shih CM, Lee YT, Cheng CH, et al. A Key Role of DNA Damage-Inducible Transcript 4 (DDIT4) Connects Autophagy and GLUT3-Mediated Stemness To Desensitize Temozolomide Efficacy in Glioblastomas. Neurotherapeutics (2020) 17(3):1212–27. doi: 10.1007/s13311-019-00826-0
50. Jiang L, Zhang J, Xu Q, Wang B, Yao Y, Sun L, et al. YAP Promotes the Proliferation and Migration of Colorectal Cancer Cells Through the Glut3/AMPK Signaling Pathway. Oncol Lett (2021) 21(4):312. doi: 10.3892/ol.2021.12573
51. Liu C, He S, Zhang J, Li S, Chen J, Han C. Silencing TCF4 Sensitizes Melanoma Cells to Vemurafenib Through Inhibiting GLUT3-Mediated Glycolysis. Oncol. Targets Ther (2020) 13:4905–15. doi: 10.2147/OTT.S245531
52. Mi L, Kuang H. Melatonin Regulates Cisplatin Resistance and Glucose Metabolism Through Hippo Signaling in Hepatocellular Carcinoma Cells. Cancer Manag Res (2020) 12:1863–74. doi: 10.2147/CMAR.S230466
53. Contat C, Ancey PB, Zangger N, Sabatino S, Pascual J, Escrig S, et al. Combined Deletion of Glut1 and Glut3 Impairs Lung Adenocarcinoma Growth. Elife (2020) 9:e53618. doi: 10.7554/eLife.53618
54. Macheda ML, Rogers S, Best JD. Molecular and Cellular Regulation of Glucose Transporter (GLUT) Proteins in Cancer. J Cell Physiol (2005) 202(3):654–62. doi: 10.1002/jcp.20166
55. Rogers S, Docherty SE, Slavin JL, Henderson MA, Best JD. Differential Expression of GLUT12 in Breast Cancer and Normal Breast Tissue. Cancer Lett (2003) 193(2):225–33. doi: 10.1016/s0304-3835(03)00010-7
56. Pujol-Gimenez J, de Heredia FP, Idoate MA, Airley R, Lostao MP, Evans AR. Could GLUT12 be a Potential Therapeutic Target in Cancer Treatment? A Preliminary Report. J Cancer (2015) 6(2):139–43. doi: 10.7150/jca.10429
57. White MA, Tsouko E, Lin CC, Rajapakshe K, Spencer JM, Wilkenfeld SR, et al. GLUT12 Promotes Prostate Cancer Cell Growth and is Regulated by Androgens and CaMKK2 Signaling. Endocr-Relat Cancer (2018) 25(4):453–69. doi: 10.1530/Erc-17-0051
58. Matsui C, Takatani-Nakase T, Maeda S, Nakase I, Takahashi K. Potential Roles of GLUT12 for Glucose Sensing and Cellular Migration in MCF-7 Human Breast Cancer Cells Under High Glucose Conditions. Anticancer Res (2017) 37(12):6715–22. doi: 10.21873/anticanres.12130
59. Chandler JD, Williams ED, Slavin JL, Best JD, Rogers S. Expression and Localization of GLUT1 and GLUT12 in Prostate Carcinoma. Cancer (2003) 97(8):2035–42. doi: 10.1002/cncr.11293
60. Shi Y, Zhang Y, Ran F, Liu J, Lin J, Hao X, et al. Let-7a-5p Inhibits Triple-Negative Breast Tumor Growth and Metastasis Through GLUT12-Mediated Warburg Effect. Cancer Lett (2020) 495:53–65. doi: 10.1016/j.canlet.2020.09.012
61. Roberts DJ, Miyamoto S. Hexokinase II Integrates Energy Metabolism and Cellular Protection: Akting on Mitochondria and TORCing to Autophagy. Cell Death Differ (2015) 22(2):248–57. doi: 10.1038/cdd.2014.173
62. Lee HJ, Li CF, Ruan D, He J, Montal ED, Lorenz S, et al. Non-Proteolytic Ubiquitination of Hexokinase 2 by HectH9 Controls Tumor Metabolism and Cancer Stem Cell Expansion. Nat Commun (2019) 10(1):2625. doi: 10.1038/s41467-019-10374-y
63. Wu Z, Wu J, Zhao Q, Fu S, Jin J. Emerging Roles of Aerobic Glycolysis in Breast Cancer. Clin Transl Oncol (2019) 22(5):631–46. doi: 10.1007/s12094-019-02187-8
64. Shi T, Ma Y, Cao L, Zhan S, Xu Y, Fu F, et al. B7-H3 Promotes Aerobic Glycolysis and Chemoresistance in Colorectal Cancer Cells by Regulating HK2. Cell Death Dis (2019) 10(4):308. doi: 10.1038/s41419-019-1549-6
65. Xiang XY, Kang JS, Yang XC, Su J, Wu Y, Yan XY, et al. SIRT3 Participates in Glucose Metabolism Interruption and Apoptosis Induced by BH3 Mimetic S1 in Ovarian Cancer Cells. Int J Oncol (2016) 49(2):773–84. doi: 10.3892/ijo.2016.3552
66. Bock FJ, Tait SWG. Mitochondria as Multifaceted Regulators of Cell Death. Nat Rev Mol Cell Biol (2020) 21(2):85–100. doi: 10.1038/s41580-019-0173-8
67. Vyssokikh MY, Brdiczka D. The Function of Complexes Between the Outer Mitochondrial Membrane Pore (VDAC) and the Adenine Nucleotide Translocase in Regulation of Energy Metabolism and Apoptosis. Acta Biochim Pol (2003) 50(2):389–404. doi: 10.18388/abp.2003_3693
68. Azoulay-Zohar H, Israelson A, Abu-Hamad S, Shoshan-Barmatz V. In Self-Defence: Hexokinase Promotes Voltage-Dependent Anion Channel Closure and Prevents Mitochondria-Mediated Apoptotic Cell Death. Biochem J (2004) 377(Pt 2):347–55. doi: 10.1042/BJ20031465
69. Krasnov GS, Dmitriev AA, Lakunina VA, Kirpiy AA, Kudryavtseva AV. Targeting VDAC-Bound Hexokinase II: A Promising Approach for Concomitant Anti-Cancer Therapy. Expert Opin Ther Targets (2013) 17(10):1221–33. doi: 10.1517/14728222.2013.833607
70. Zhang J, Chen G, Gao Y, Liang H. HOTAIR/miR-125 Axis-Mediated Hexokinase 2 Expression Promotes Chemoresistance in Human Glioblastoma. J Cell Mol Med (2020) 24(10):5707–17. doi: 10.1111/jcmm.15233
71. Liu CC, Chou KT, Hsu JW, Lin JH, Hsu TW, Yen DH, et al. High Metabolic Rate and Stem Cell Characteristics of Esophageal Cancer Stem-Like Cells Depend on the Hsp27-AKT-HK2 Pathway. Int J Cancer (2019) 145(8):2144–56. doi: 10.1002/ijc.32301
72. Deshmukh A, Deshpande K, Arfuso F, Newsholme P, Dharmarajan A. Cancer Stem Cell Metabolism: A Potential Target for Cancer Therapy. Mol Cancer (2016) 15(1):69. doi: 10.1186/s12943-016-0555-x
73. Zhang XY, Zhang M, Cong Q, Zhang MX, Zhang MY, Lu YY, et al. Hexokinase 2 Confers Resistance to Cisplatin in Ovarian Cancer Cells by Enhancing Cisplatin-Induced Autophagy. Int J Biochem Cell Biol (2018) 95:9–16. doi: 10.1016/j.biocel.2017.12.010
74. Han CY, Patten DA, Lee SG, Parks RJ, Chan DW, Harper ME, et al. P53 Promotes Chemoresponsiveness by Regulating Hexokinase II Gene Transcription and Metabolic Reprogramming in Epithelial Ovarian Cancer. Mol Carcinog (2019) 58(11):2161–74. doi: 10.1002/mc.23106
75. Wen JF, Jiang YQ, Li C, Dai XK, Wu T, Yin WZ. LncRNA-SARCC Sensitizes Osteosarcoma to Cisplatin Through the miR-143-Mediated Glycolysis Inhibition by Targeting Hexokinase 2. Cancer Biomark (2020) 28(2):231–46. doi: 10.3233/CBM-191181
76. Jiang JX, Gao S, Pan YZ, Yu C, Sun CY. Overexpression of microRNA-125b Sensitizes Human Hepatocellular Carcinoma Cells to 5-Fluorouracil Through Inhibition of Glycolysis by Targeting Hexokinase II. Mol Med Rep (2014) 10(2):995–1002. doi: 10.3892/mmr.2014.2271
77. Zhang Y, Liu Y, Xu X. Knockdown of LncRNA-UCA1 Suppresses Chemoresistance of Pediatric AML by Inhibiting Glycolysis Through the microRNA-125a/Hexokinase 2 Pathway. J Cell Biochem (2018) 119(7):6296–308. doi: 10.1002/jcb.26899
78. Shi H, Li K, Feng J, Liu G, Feng Y, Zhang X. LncRNA-DANCR Interferes With miR-125b-5p/HK2 Axis to Desensitize Colon Cancer Cells to Cisplatin Vis Activating Anaerobic Glycolysis. Front Oncol (2020) 10:1034. doi: 10.3389/fonc.2020.01034
79. Peng B, Theng PY, Le MTN. Essential Functions of miR-125b in Cancer. Cell Prolif (2021) 54(2):e12913. doi: 10.1111/cpr.12913
80. Sun X, Sun G, Huang Y, Zhang S, Tang X, Zhang N, et al. Glycolytic Inhibition by 3-Bromopyruvate Increases the Cytotoxic Effects of Chloroethylnitrosoureas to Human Glioma Cells and the DNA Interstrand Cross-Links Formation. Toxicology (2020) 435:152413. doi: 10.1016/j.tox.2020.152413
81. He R, Liu H. TRIM59 Knockdown Blocks Cisplatin Resistance in A549/DDP Cells Through Regulating PTEN/AKT/Hk2. Gene (2020) 747:144553. doi: 10.1016/j.gene.2020.144553
82. Minchenko OH, Tsuchihara K, Minchenko DO, Bikfalvi A, Esumi H. Mechanisms of Regulation of PFKFB Expression in Pancreatic and Gastric Cancer Cells. World J Gastroenterol (2014) 20(38):13705–17. doi: 10.3748/wjg.v20.i38.13705
83. Shi L, Pan H, Liu Z, Xie J, Han W. Roles of PFKFB3 in Cancer. Signal Transduct Target Ther (2017) 2:17044. doi: 10.1038/sigtrans.2017.44
84. Yi M, Ban Y, Tan Y, Xiong W, Li G, Xiang B. 6-Phosphofructo-2-Kinase/Fructose-2,6-Biphosphatase 3 and 4: A Pair of Valves for Fine-Tuning of Glucose Metabolism in Human Cancer. Mol Metab (2019) 20:1–13. doi: 10.1016/j.molmet.2018.11.013
85. Yang Q, Hou P. Targeting PFKFB3 in the Endothelium for Cancer Therapy. Trends Mol Med (2017) 23(3):197–200. doi: 10.1016/j.molmed.2017.01.008
86. Li HM, Yang JG, Liu ZJ, Wang WM, Yu ZL, Ren JG, et al. Blockage of Glycolysis by Targeting PFKFB3 Suppresses Tumor Growth and Metastasis in Head and Neck Squamous Cell Carcinoma. J Exp Clin Cancer Res (2017) 36(1):7. doi: 10.1186/s13046-016-0481-1
87. Imbert-Fernandez Y, Clem BF, O’Neal J, Kerr DA, Spaulding R, Lanceta L, et al. Estradiol Stimulates Glucose Metabolism via 6-Phosphofructo-2-Kinase (PFKFB3). J Biol Chem (2014) 289(13):9440–8. doi: 10.1074/jbc.M113.529990
88. Yalcin A, Clem BF, Imbert-Fernandez Y, Ozcan SC, Peker S, O’Neal J, et al. 6-Phosphofructo-2-Kinase (PFKFB3) Promotes Cell Cycle Progression and Suppresses Apoptosis via Cdk1-Mediated Phosphorylation of P27. Cell Death Dis (2014) 5:e1337. doi: 10.1038/cddis.2014.292
89. Trenti A, Tedesco S, Boscaro C, Ferri N, Cignarella A, Trevisi L, et al. The Glycolytic Enzyme PFKFB3 Is Involved in Estrogen-Mediated Angiogenesis via GPER1. J Pharmacol Exp Ther (2017) 361(3):398–407. doi: 10.1124/jpet.116.238212
90. De Bock K, Georgiadou M, Schoors S, Kuchnio A, Wong BW, Cantelmo AR, et al. Role of PFKFB3-Driven Glycolysis in Vessel Sprouting. Cell (2013) 154(3):651–63. doi: 10.1016/j.cell.2013.06.037
91. Alfarouk KO, Shayoub ME, Muddathir AK, Elhassan GO, Bashir AH. Evolution of Tumor Metabolism Might Reflect Carcinogenesis as a Reverse Evolution Process (Dismantling of Multicellularity). Cancers (Basel) (2011) 3(3):3002–17. doi: 10.3390/cancers3033002
92. Long Q, Zou X, Song Y, Duan Z, Liu L. PFKFB3/HIF-1alpha Feedback Loop Modulates Sorafenib Resistance in Hepatocellular Carcinoma Cells. Biochem Biophys Res Commun (2019) 513(3):642–50. doi: 10.1016/j.bbrc.2019.03.109
93. Yan S, Zhou N, Zhang D, Zhang K, Zheng W, Bao Y, et al. PFKFB3 Inhibition Attenuates Oxaliplatin-Induced Autophagy and Enhances Its Cytotoxicity in Colon Cancer Cells. Int J Mol Sci (2019) 20(21):5415–30. doi: 10.3390/ijms20215415
94. Li FL, Liu JP, Bao RX, Yan G, Feng X, Xu YP, et al. Acetylation Accumulates PFKFB3 in Cytoplasm to Promote Glycolysis and Protects Cells From Cisplatin-Induced Apoptosis. Nat Commun (2018) 9(1):508. doi: 10.1038/s41467-018-02950-5
95. Cantelmo AR, Conradi LC, Brajic A, Goveia J, Kalucka J, Pircher A, et al. Inhibition of the Glycolytic Activator PFKFB3 in Endothelium Induces Tumor Vessel Normalization, Impairs Metastasis, and Improves Chemotherapy. Cancer Cell (2016) 30(6):968–85. doi: 10.1016/j.ccell.2016.10.006
96. Wang J, Li X, Xiao Z, Wang Y, Han Y, Li J, et al. MicroRNA-488 Inhibits Proliferation and Glycolysis in Human Prostate Cancer Cells by Regulating PFKFB3. FEBS Open Bio (2019) 9(10):1798–807. doi: 10.1002/2211-5463.12718
97. Deng X, Li D, Ke X, Wang Q, Yan S, Xue Y, et al. Mir-488 Alleviates Chemoresistance and Glycolysis of Colorectal Cancer by Targeting PFKFB3. J Clin Lab Anal (2021) 35(1):e23578. doi: 10.1002/jcla.23578
98. Chowdhury N, Vhora I, Patel K, Doddapaneni R, Mondal A, Singh M. Liposomes Co-Loaded With 6-Phosphofructo-2-Kinase/Fructose-2, 6-Biphosphatase 3 (PFKFB3) shRNA Plasmid and Docetaxel for the Treatment of non-Small Cell Lung Cancer. Pharm Res (2017) 34(11):2371–84. doi: 10.1007/s11095-017-2244-x
99. Yang W, Lu Z. Pyruvate Kinase M2 at a Glance. J Cell Sci (2015) 128(9):1655–60. doi: 10.1242/jcs.166629
100. Zhang Z, Deng X, Liu Y, Liu Y, Sun L, Chen F. PKM2, Function and Expression and Regulation. Cell Biosci (2019) 9:52. doi: 10.1186/s13578-019-0317-8
101. Tamada M, Suematsu M, Saya H. Pyruvate Kinase M2: Multiple Faces for Conferring Benefits on Cancer Cells. Clin Cancer Res (2012) 18(20):5554–61. doi: 10.1158/1078-0432.CCR-12-0859
102. Goldberg MS, Sharp PA. Pyruvate Kinase M2-Specific siRNA Induces Apoptosis and Tumor Regression. J Exp Med (2012) 209(2):217–24. doi: 10.1084/jem.20111487
103. He Y, Wang Y, Liu H, Xu X, He S, Tang J, et al. Pyruvate Kinase Isoform M2 (PKM2) Participates in Multiple Myeloma Cell Proliferation, Adhesion and Chemoresistance. Leuk Res (2015) 39(12):1428–36. doi: 10.1016/j.leukres.2015.09.019
104. Zheng H, Zhang M, Ke X, Deng X, Li D, Wang Q, et al. LncRNA XIST/miR-137 Axis Strengthens Chemo-Resistance and Glycolysis of Colorectal Cancer Cells by Hindering Transformation From PKM2 to PKM1. Cancer Biomark (2020) 30(4):395–406. doi: 10.3233/CBM-201740
105. Fukuda S, Miyata H, Miyazaki Y, Makino T, Takahashi T, Kurokawa Y, et al. Pyruvate Kinase M2 Modulates Esophageal Squamous Cell Carcinoma Chemotherapy Response by Regulating the Pentose Phosphate Pathway. Ann Surg Oncol (2015) 22 Suppl 3:S1461–8. doi: 10.1245/s10434-015-4522-3
106. Zhu Z, Tang G, Yan J. MicroRNA-122 Regulates Docetaxel Resistance of Prostate Cancer Cells by Regulating PKM2. Exp Ther Med (2020) 20(6):247. doi: 10.3892/etm.2020.9377
107. Pan C, Wang X, Shi K, Zheng Y, Li J, Chen Y, et al. MiR-122 Reverses the Doxorubicin-Resistance in Hepatocellular Carcinoma Cells Through Regulating the Tumor Metabolism. PloS One (2016) 11(5):e0152090. doi: 10.1371/journal.pone.0152090
108. He J, Xie G, Tong J, Peng Y, Huang H, Li J, et al. Overexpression of microRNA-122 Re-Sensitizes 5-FU-Resistant Colon Cancer Cells to 5-FU Through the Inhibition of PKM2. Vitro Vivo Cell Biochem Biophys (2014) 70(2):1343–50. doi: 10.1007/s12013-014-0062-x
109. Wang X, Zhang H, Yang H, Bai M, Ning T, Deng T, et al. Exosome-Delivered circRNA Promotes Glycolysis to Induce Chemoresistance Through the miR-122-PKM2 Axis in Colorectal Cancer. Mol Oncol (2020) 14(3):539–55. doi: 10.1002/1878-0261.12629
110. van Niekerk G, Engelbrecht AM. Role of PKM2 in Directing the Metabolic Fate of Glucose in Cancer: A Potential Therapeutic Target. Cell Oncol (Dordr) (2018) 41(4):343–51. doi: 10.1007/s13402-018-0383-7
111. Kam Y, Das T, Tian H, Foroutan P, Ruiz E, Martinez G, et al. Sweat But No Gain: Inhibiting Proliferation of Multidrug Resistant Cancer Cells With “Ersatzdroges”. Int J Cancer (2015) 136(4):E188–96. doi: 10.1002/ijc.29158
112. Wang X, Zhang F, Wu XR. Inhibition of Pyruvate Kinase M2 Markedly Reduces Chemoresistance of Advanced Bladder Cancer to Cisplatin. Sci Rep (2017) 7:45983. doi: 10.1038/srep45983
113. Kim DJ, Park YS, Kang MG, You YM, Jung Y, Koo H, et al. Pyruvate Kinase Isoenzyme M2 is a Therapeutic Target of Gemcitabine-Resistant Pancreatic Cancer Cells. Exp Cell Res (2015) 336(1):119–29. doi: 10.1016/j.yexcr.2015.05.017
114. Qian Y, Bi L, Yang Y, Wang D. Effect of Pyruvate Kinase M2-Regulating Aerobic Glycolysis on Chemotherapy Resistance of Estrogen Receptor-Positive Breast Cancer. Anticancer Drugs (2018) 29(7):616–27. doi: 10.1097/CAD.0000000000000624
115. Guo W, Zhang Y, Chen T, Wang Y, Xue J, Zhang Y, et al. Efficacy of RNAi Targeting of Pyruvate Kinase M2 Combined With Cisplatin in a Lung Cancer Model. J Cancer Res Clin Oncol (2011) 137(1):65–72. doi: 10.1007/s00432-010-0860-5
116. Lin Y, Lv F, Liu F, Guo X, Fan Y, Gu F, et al. High Expression of Pyruvate Kinase M2 is Associated With Chemosensitivity to Epirubicin and 5-Fluorouracil in Breast Cancer. J Cancer (2015) 6(11):1130–9. doi: 10.7150/jca.12719
117. Yoo BC, Ku JL, Hong SH, Shin YK, Park SY, Kim HK, et al. Decreased Pyruvate Kinase M2 Activity Linked to Cisplatin Resistance in Human Gastric Carcinoma Cell Lines. Int J Cancer (2004) 108(4):532–9. doi: 10.1002/ijc.11604
118. Israelsen WJ, Vander Heiden MG. Pyruvate Kinase: Function, Regulation and Role in Cancer. Semin Cell Dev Biol (2015) 43:43–51. doi: 10.1016/j.semcdb.2015.08.004
119. Yu Y, Deck JA, Hunsaker LA, Deck LM, Royer RE, Goldberg E, et al. Selective Active Site Inhibitors of Human Lactate Dehydrogenases A4, B4, and C4. Biochem Pharmacol (2001) 62(1):81–9. doi: 10.1016/s0006-2952(01)00636-0
120. Van Wilpe S, Koornstra R, Den Brok M, De Groot JW, Blank C, De Vries J, et al. Lactate Dehydrogenase: A Marker of Diminished Antitumor Immunity. Oncoimmunology (2020) 9(1):1731942. doi: 10.1080/2162402X.2020.1731942
121. Granchi C, Bertini S, Macchia M, Minutolo F. Inhibitors of Lactate Dehydrogenase Isoforms and Their Therapeutic Potentials. Curr Med Chem (2010) 17(7):672–97. doi: 10.2174/092986710790416263
122. Feng L, LL E, Soloveiv MM, Wang DS, Zhang BO, Dong YW, et al. Synergistic Cytotoxicity of Cisplatin and Taxol in Overcoming Taxol Resistance Through the Inhibition of LDHA in Oral Squamous Cell Carcinoma. Oncol Lett (2015) 9(4):1827–32. doi: 10.3892/ol.2015.2931
123. Ooi AT, Gomperts BN. Molecular Pathways: Targeting Cellular Energy Metabolism in Cancer via Inhibition of SLC2A1 and LDHA. Clin Cancer Res (2015) 21(11):2440–4. doi: 10.1158/1078-0432.CCR-14-1209
124. Balinsky D, Platz CE, Lewis JW. Isozyme Patterns of Normal, Benign, and Malignant Human Breast Tissues. Cancer Res (1983) 43(12 Pt 1):5895–901.
125. Shi M, Cui J, Du J, Wei D, Jia Z, Zhang J, et al. A Novel KLF4/LDHA Signaling Pathway Regulates Aerobic Glycolysis in and Progression of Pancreatic Cancer. Clin Cancer Res (2014) 20(16):4370–80. doi: 10.1158/1078-0432.Ccr-14-0186
126. Lu H, Li X, Luo Z, Liu J, Fan Z. Cetuximab Reverses the Warburg Effect by Inhibiting HIF-1-Regulated LDH-A. Mol Cancer Ther (2013) 12(10):2187–99. doi: 10.1158/1535-7163.Mct-12-1245
127. Zhang H, Li L, Chen Q, Li M, Feng J, Sun Y, et al. PGC1beta Regulates Multiple Myeloma Tumor Growth Through LDHA-Mediated Glycolytic Metabolism. Mol Oncol (2018) 12(9):1579–95. doi: 10.1002/1878-0261.12363
128. Huang H, Peng J, Yi SJ, Ding CM, Ji W, Huang QS, et al. Circular RNA Circube2d2 Functions as an Oncogenic Factor in Hepatocellular Carcinoma Sorafenib Resistance and Glycolysis. Am J Transl Res (2021) 13(6):6076–+.
129. Hua YQ, Zhang K, Sheng J, Ning ZY, Li Y, Shi WD, et al. Fam83D Promotes Tumorigenesis and Gemcitabine Resistance of Pancreatic Adenocarcinoma Through the Wnt/beta-Catenin Pathway. Life Sci (2021) PII: S0024-3205(21)00190-9 119205. doi: 10.1016/j.lfs.2021.119205
130. Walenta S, Wetterling M, Lehrke M, Schwickert G, Sundfør K, Rofstad EK, et al. High Lactate Levels Predict Likelihood of Metastases, Tumor Recurrence, and Restricted Patient Survival in Human Cervical Cancers. Cancer Res (2000) 60(4):916–21.
131. Li Petri G, El Hassouni B, Sciarrillo R, Funel N, Mantini G, Zeeuw van der Laan EA, et al. Impact of Hypoxia on Chemoresistance of Mesothelioma Mediated by the Proton-Coupled Folate Transporter, and Preclinical Activity of New Anti-LDH-A Compounds. Br J Cancer (2020) 123(4):644–56. doi: 10.1038/s41416-020-0912-9
132. Zhang Y, Zhang X, Wang X, Gan L, Yu G, Chen Y, et al. Inhibition of LDH-A by Lentivirus-Mediated Small Interfering RNA Suppresses Intestinal-Type Gastric Cancer Tumorigenicity Through the Downregulation of Oct4. Cancer Lett (2012) 321(1):45–54. doi: 10.1016/j.canlet.2012.03.013
133. Ji Y, Yang C, Tang Z, Yang Y, Tian Y, Yao H, et al. Adenylate Kinase hCINAP Determines Self-Renewal of Colorectal Cancer Stem Cells by Facilitating LDHA Phosphorylation. Nat Commun (2017) 8:15308. doi: 10.1038/ncomms15308
134. Le A, Cooper CR, Gouw AM, Dinavahi R, Maitra A, Deck LM, et al. Inhibition of Lactate Dehydrogenase A Induces Oxidative Stress and Inhibits Tumor Progression. Proc Natl Acad Sci USA (2010) 107(5):2037–42. doi: 10.1073/pnas.0914433107
135. Wu H, Wang Y, Ying M, Jin C, Li J, Hu X. Lactate Dehydrogenases Amplify Reactive Oxygen Species in Cancer Cells in Response to Oxidative Stimuli. Signal Transduct Target Ther (2021) 6(1):242. doi: 10.1038/s41392-021-00595-3
136. Wang X, Xu L, Wu Q, Liu M, Tang F, Cai Y, et al. Inhibition of LDHA Deliver Potential Anticancer Performance in Renal Cell Carcinoma. Urologia Internationalis (2017) 99(2):237–44. doi: 10.1159/000445125
137. Han JH, Kim M, Kim HJ, Jang SB, Bae SJ, Lee IK, et al. Targeting Lactate Dehydrogenase A With Catechin Resensitizes SNU620/5FU Gastric Cancer Cells to 5-Fluorouracil. Int J Mol Sci (2021) 22(10):5406–25. doi: 10.3390/ijms22105406
138. Wang ZY, Loo TY, Shen JG, Wang N, Wang DM, Yang DP, et al. LDH-A Silencing Suppresses Breast Cancer Tumorigenicity Through Induction of Oxidative Stress Mediated Mitochondrial Pathway Apoptosis. Breast Cancer Res Treat (2012) 131(3):791–800. doi: 10.1007/s10549-011-1466-6
139. Tang D, Xu L, Zhang M, Dorfman RG, Pan Y, Zhou Q, et al. Metformin Facilitates BG45induced Apoptosis via an Antiwarburg Effect in Cholangiocarcinoma Cells. Oncol Rep (2018) 39(4):1957–65. doi: 10.3892/or.2018.6275
140. Zhou M, Zhao Y, Ding Y, Liu H, Liu Z, Fodstad O, et al. Warburg Effect in Chemosensitivity: Targeting Lactate Dehydrogenase-A Re-Sensitizes Taxol-Resistant Cancer Cells to Taxol. Mol Cancer (2010) 9:33. doi: 10.1186/1476-4598-9-33
141. Seth P, Grant A, Tang J, Vinogradov E, Wang X, Lenkinski R, et al. On-Target Inhibition of Tumor Fermentative Glycolysis as Visualized by Hyperpolarized Pyruvate. Neoplasia (N. Y. NY) (2011) 13(1):60–71. doi: 10.1593/neo.101020
142. Li X, Zhao H, Zhou X, Song L. Inhibition of Lactate Dehydrogenase A by microRNA-34a Resensitizes Colon Cancer Cells to 5-Fluorouracil. Mol Med Rep (2015) 11(1):577–82. doi: 10.3892/mmr.2014.2726
143. Li G, Li Y, Wang DY. Overexpression of miR-329-3p Sensitizes Osteosarcoma Cells to Cisplatin Through Suppression of Glucose Metabolism by Targeting LDHA. Cell Biol Int (2021) 45(4):766–74. doi: 10.1002/cbin.11476
144. Jin HF, Wang JF, Shao M, Zhou K, Ma X, Lv XP. Down-Regulation of miR-7 in Gastric Cancer Is Associated With Elevated LDH-A Expression and Chemoresistance to Cisplatin. Front Cell Dev Biol (2020) 8:555937. doi: 10.3389/fcell.2020.555937
145. Feng Y, Xiong Y, Qiao T, Li X, Jia L, Han Y. Lactate Dehydrogenase A: A Key Player in Carcinogenesis and Potential Target in Cancer Therapy. Cancer Med (2018) 7(12):6124–36. doi: 10.1002/cam4.1820
146. Fu J, Jiang H, Wu C, Jiang Y, Xiao L, Tian Y. Overcoming Cetuximab Resistance in Ewing’s Sarcoma by Inhibiting Lactate Dehydrogenase-A. Mol Med Rep (2016) 14(1):995–1001. doi: 10.3892/mmr.2016.5290
147. Maftouh M, Avan A, Sciarrillo R, Granchi C, Leon LG, Rani R, et al. Synergistic Interaction of Novel Lactate Dehydrogenase Inhibitors With Gemcitabine Against Pancreatic Cancer Cells in Hypoxia. Br J Cancer (2014) 110(1):172–82. doi: 10.1038/bjc.2013.681
148. Giovannetti E, Leon LG, Gomez VE, Zucali PA, Minutolo F, Peters GJ. A Specific Inhibitor of Lactate Dehydrogenase Overcame the Resistance Toward Gemcitabine in Hypoxic Mesothelioma Cells, and Modulated the Expression of the Human Equilibrative Transporter-1. Nucleosides Nucleotides Nucleic Acids (2016) 35(10-12):643–51. doi: 10.1080/15257770.2016.1149193
149. Halestrap AP. The SLC16 Gene Family - Structure, Role and Regulation in Health and Disease. Mol Aspects Med (2013) 34(2-3):337–49. doi: 10.1016/j.mam.2012.05.003
150. Halestrap AP. The Monocarboxylate Transporter Family–Structure and Functional Characterization. IUBMB Life (2012) 64(1):1–9. doi: 10.1002/iub.573
151. Bonatelli M, Fornari IF, Bernécule PN, Pinheiro LE, Costa RFA, Longatto-Filho A, et al. Expression of Glycolysis-Related Proteins in Cancer of Unknown Primary Origin. Front Oncol (2021) 11:682665. doi: 10.3389/fonc.2021.682665
152. Kubelt C, Peters S, Ahmeti H, Huhndorf M, Huber L, Cohrs G, et al. Intratumoral Distribution of Lactate and the Monocarboxylate Transporters 1 and 4 in Human Glioblastoma Multiforme and Their Relationships to Tumor Progression-Associated Markers. Int J Mol Sci (2020) 21(17):6254–73. doi: 10.3390/ijms21176254
153. Leu M, Kitz J, Pilavakis Y, Hakroush S, Wolff HA, Canis M, et al. Monocarboxylate Transporter-1 (MCT1) Protein Expression in Head and Neck Cancer Affects Clinical Outcome. Sci Rep (2021) 11(1):4578. doi: 10.1038/s41598-021-84019-w
154. Bonglack EN, Messinger JE, Cable JM, Ch’ng J, Parnell KM, Reinoso-Vizcaíno NM, et al. Monocarboxylate Transporter Antagonism Reveals Metabolic Vulnerabilities of Viral-Driven Lymphomas. Proc Natl Acad Sci USA (2021) 118(25):e2022495118. doi: 10.1073/pnas.2022495118
155. Afonso J, Santos LL, Miranda-Goncalves V, Morais A, Amaro T, Longatto-Filho A, et al. CD147 and MCT1-Potential Partners in Bladder Cancer Aggressiveness and Cisplatin Resistance. Mol Carcinog (2015) 54(11):1451–66. doi: 10.1002/mc.22222
156. Enerson BE, Drewes LR. Molecular Features, Regulation, and Function of Monocarboxylate Transporters: Implications for Drug Delivery. J Pharm Sci (2003) 92(8):1531–44. doi: 10.1002/jps.10389
157. Kirk P, Wilson MC, Heddle C, Brown MH, Barclay AN, Halestrap AP. CD147 is Tightly Associated With Lactate Transporters MCT1 and MCT4 and Facilitates Their Cell Surface Expression. EMBO J (2000) 19(15):3896–904. doi: 10.1093/emboj/19.15.3896
158. Felmlee MA, Jones RS, Rodriguez-Cruz V, Follman KE, Morris ME. Monocarboxylate Transporters (SLC16): Function, Regulation, and Role in Health and Disease. Pharmacol Rev (2020) 72(2):466–85. doi: 10.1124/pr.119.018762
159. Afonso J, Santos LL, Morais A, Amaro T, Longatto-Filho A, Baltazar F. Metabolic Coupling in Urothelial Bladder Cancer Compartments and Its Correlation to Tumor Aggressiveness. Cell Cycle (2016) 15(3):368–80. doi: 10.1080/15384101.2015.1121329
160. Reuss AM, Groos D, Buchfelder M, Savaskan N. The Acidic Brain-Glycolytic Switch in the Microenvironment of Malignant Glioma. Int J Mol Sci (2021) 22(11):5518–42. doi: 10.3390/ijms22115518
161. Tavares-Valente D, Sousa B, Schmitt F, Baltazar F, Queiros O. Disruption of pH Dynamics Suppresses Proliferation and Potentiates Doxorubicin Cytotoxicity in Breast Cancer Cells. Pharmaceutics (2021) 13(2):242–61. doi: 10.3390/pharmaceutics13020242
162. Li N, Kang Y, Wang L, Huff S, Tang R, Hui H, et al. ALKBH5 Regulates Anti-PD-1 Therapy Response by Modulating Lactate and Suppressive Immune Cell Accumulation in Tumor Microenvironment. Proc Natl Acad Sci USA (2020) 117(33):20159–70. doi: 10.1073/pnas.1918986117
163. Miranda-Goncalves V, Granja S, Martinho O, Honavar M, Pojo M, Costa BM, et al. Hypoxia-Mediated Upregulation of MCT1 Expression Supports the Glycolytic Phenotype of Glioblastomas. Oncotarget (2016) 7(29):46335–53. doi: 10.18632/oncotarget.10114
164. Li F, Zhan L, Dong Q, Wang Q, Wang Y, Li X, et al. Tumor-Derived Exosome-Educated Hepatic Stellate Cells Regulate Lactate Metabolism of Hypoxic Colorectal Tumor Cells via the IL-6/STAT3 Pathway to Confer Drug Resistance. Oncol. Targets Ther (2020) 13:7851–64. doi: 10.2147/OTT.S253485
165. Kuo TC, Huang KY, Yang SC, Wu S, Chung WC, Chang YL, et al. Monocarboxylate Transporter 4 Is a Therapeutic Target in Non-Small Cell Lung Cancer With Aerobic Glycolysis Preference. Mol Ther Oncolytics (2020) 18:189–201. doi: 10.1016/j.omto.2020.06.012
166. Sandforth L, Ammar N, Dinges LA, Rocken C, Arlt A, Sebens S, et al. Impact of the Monocarboxylate Transporter-1 (MCT1)-Mediated Cellular Import of Lactate on Stemness Properties of Human Pancreatic Adenocarcinoma Cells Dagger. Cancers (Basel) (2020) 12(3):581–601. doi: 10.3390/cancers12030581
167. Takada T, Takata K, Ashihara E. Inhibition of Monocarboxylate Transporter 1 Suppresses the Proliferation of Glioblastoma Stem Cells. J Physiol Sci (2016) 66(5):387–96. doi: 10.1007/s12576-016-0435-6
168. Hou L, Zhao Y, Song GQ, Ma YH, Jin XH, Jin SL, et al. Interfering Cellular Lactate Homeostasis Overcomes Taxol Resistance of Breast Cancer Cells Through the microRNA-124-Mediated Lactate Transporter (MCT1) Inhibition. Cancer Cell Int (2019) 19:193. doi: 10.1186/s12935-019-0904-0
169. Soni VK, Shukla D, Kumar A, Vishvakarma NK. Curcumin Circumvent Lactate-Induced Chemoresistance in Hepatic Cancer Cells Through Modulation of Hydroxycarboxylic Acid Receptor-1. Int J Biochem Cell Biol (2020) 123:105752. doi: 10.1016/j.biocel.2020.105752
170. Vander Linden C, Corbet C, Bastien E, Martherus R, Guilbaud C, Petit L, et al. Therapy-Induced DNA Methylation Inactivates MCT1 and Renders Tumor Cells Vulnerable to MCT4 Inhibition. Cell Rep (2021) 35(9):109202. doi: 10.1016/j.celrep.2021.109202
171. Colen CB, Shen Y, Ghoddoussi F, Yu P, Francis TB, Koch BJ, et al. Metabolic Targeting of Lactate Efflux by Malignant Glioma Inhibits Invasiveness and Induces Necrosis: An In Vivo Study. Neoplasia (N. Y. NY) (2011) 13(7):620–32. doi: 10.1593/neo.11134
172. Kumar A, Kant S, Singh SM. Targeting Monocarboxylate Transporter by Alpha-Cyano-4-Hydroxycinnamate Modulates Apoptosis and Cisplatin Resistance of Colo205 Cells: Implication of Altered Cell Survival Regulation. Apoptosis (2013) 18(12):1574–85. doi: 10.1007/s10495-013-0894-7
173. Bola BM, Chadwick AL, Michopoulos F, Blount KG, Telfer BA, Williams KJ, et al. Inhibition of Monocarboxylate Transporter-1 (MCT1) by AZD3965 Enhances Radiosensitivity by Reducing Lactate Transport. Mol Cancer Ther (2014) 13(12):2805–16. doi: 10.1158/1535-7163.Mct-13-1091
174. Howell JJ, Ricoult SJ, Ben-Sahra I, Manning BD. A Growing Role for mTOR in Promoting Anabolic Metabolism. Biochem Soc Trans (2013) 41(4):906–12. doi: 10.1042/BST20130041
175. Courtnay R, Ngo DC, Malik N, Ververis K, Tortorella SM, Karagiannis TC. Cancer Metabolism and the Warburg Effect: The Role of HIF-1 and PI3K. Mol Biol Rep (2015) 42(4):841–51. doi: 10.1007/s11033-015-3858-x
176. Zhang X, Ai Z, Chen J, Yi J, Liu Z, Zhao H, et al. Glycometabolic Adaptation Mediates the Insensitivity of Drug-Resistant K562/ADM Leukaemia Cells to Adriamycin via the AKT-mTOR/C-Myc Signalling Pathway. Mol Med Rep (2017) 15(4):1869–76. doi: 10.3892/mmr.2017.6189
177. Avanzato D, Pupo E, Ducano N, Isella C, Bertalot G, Luise C, et al. High USP6NL Levels in Breast Cancer Sustain Chronic AKT Phosphorylation and GLUT1 Stability Fueling Aerobic Glycolysis. Cancer Res (2018) 78(13):3432–44. doi: 10.1158/0008-5472.Can-17-3018
178. Wang J, Fang Y, Liu T. TRIM32 Promotes the Growth of Gastric Cancer Cells Through Enhancing AKT Activity and Glucose Transportation. BioMed Res Int (2020) 2020:4027627. doi: 10.1155/2020/4027627
179. Yang H, Zhu J, Wang G, Liu H, Zhou Y, Qian J. STK35 Is Ubiquitinated by NEDD4L and Promotes Glycolysis and Inhibits Apoptosis Through Regulating the AKT Signaling Pathway, Influencing Chemoresistance of Colorectal Cancer. Front Cell Dev Biol (2020) 8:582695. doi: 10.3389/fcell.2020.582695
180. Gao S, Song D, Liu Y, Yan H, Chen X. Helicobacter Pylori CagA Protein Attenuates 5-Fu Sensitivity of Gastric Cancer Cells Through Upregulating Cellular Glucose Metabolism. Oncol. Targets Ther (2020) 13:6339–49. doi: 10.2147/OTT.S230875
181. Wang Y, Pan S, He X, Wang Y, Huang H, Chen J, et al. CPNE1 Enhances Colorectal Cancer Cell Growth, Glycolysis, and Drug Resistance Through Regulating the AKT-GLUT1/HK2 Pathway. Oncol Targets Ther (2021) 14:699–710. doi: 10.2147/OTT.S284211
182. Gong T, Cui L, Wang H, Wang H, Han N. Knockdown of KLF5 Suppresses Hypoxia-Induced Resistance to Cisplatin in NSCLC Cells by Regulating HIF-1alpha-Dependent Glycolysis Through Inactivation of the PI3K/Akt/mTOR Pathway. J Transl Med (2018) 16(1):164. doi: 10.1186/s12967-018-1543-2
183. Yu X, Gao X, Mao X, Shi Z, Zhu B, Xie L, et al. Knockdown of FOXO6 Inhibits Glycolysis and Reduces Cell Resistance to Paclitaxel in HCC Cells via PI3K/Akt Signaling Pathway. Oncol. Targets Ther (2020) 13:1545–56. doi: 10.2147/OTT.S233031
184. George AL, Rajoria S, Suriano R, Mittleman A, Tiwari RK. Hypoxia and Estrogen are Functionally Equivalent in Breast Cancer-Endothelial Cell Interdependence. Mol Cancer (2012) 11:80. doi: 10.1186/1476-4598-11-80
185. Rius J, Guma M, Schachtrup C, Akassoglou K, Zinkernagel AS, Nizet V, et al. NF-kappaB Links Innate Immunity to the Hypoxic Response Through Transcriptional Regulation of HIF-1alpha. Nature (2008) 453(7196):807–11. doi: 10.1038/nature06905
186. Wang G, Wang JJ, Fu XL, Guang R, To ST. Advances in the Targeting of HIF-1alpha and Future Therapeutic Strategies for Glioblastoma Multiforme (Review). Oncol Rep (2017) 37(2):657–70. doi: 10.3892/or.2016.5309
187. Lu Y, Wang L, Ding W, Wang D, Wang X, Luo Q, et al. Ammonia Mediates Mitochondrial Uncoupling and Promotes Glycolysis via HIF-1 Activation in Human Breast Cancer MDA-MB-231cells. Biochem Biophys Res Commun (2019) 519(1):153–9. doi: 10.1016/j.bbrc.2019.08.152
188. Lim S, Liu H, Madeira da Silva L, Arora R, Liu Z, Phillips JB, et al. Immunoregulatory Protein B7-H3 Reprograms Glucose Metabolism in Cancer Cells by ROS-Mediated Stabilization of HIF1alpha. Cancer Res (2016) 76(8):2231–42. doi: 10.1158/0008-5472.CAN-15-1538
189. Marengo B, Garbarino O, Speciale A, Monteleone L, Traverso N, Domenicotti C, et al. and Metabolic Redox Changes in Cancer Cells: A Synergy Able to Induce Chemoresistance. Oxid Med Cell Longev (2019) 2019:7346492. doi: 10.1155/2019/7346492
190. Frolova O, Samudio I, Benito JM, Jacamo R, Kornblau SM, Markovic A, et al. Regulation of HIF-1alpha Signaling and Chemoresistance in Acute Lymphocytic Leukemia Under Hypoxic Conditions of the Bone Marrow Microenvironment. Cancer Biol Ther (2012) 13(10):858–70. doi: 10.4161/cbt.20838
191. Ren X, Su C. Sphingosine Kinase 1 Contributes to Doxorubicin Resistance and Glycolysis in Osteosarcoma. Mol Med Rep (2020) 22(3):2183–90. doi: 10.3892/mmr.2020.11295
192. Xi Y, Yuan P, Li T, Zhang M, Liu MF, Li B. Hent1 Reverses Chemoresistance by Regulating Glycolysis in Pancreatic Cancer. Cancer Lett (2020) 479:112–22. doi: 10.1016/j.canlet.2020.03.015
193. Zhang L, Yang H, Zhang W, Liang Z, Huang Q, Xu G, et al. Clk1-Regulated Aerobic Glycolysis is Involved in Glioma Chemoresistance. J Neurochem (2017) 142(4):574–88. doi: 10.1111/jnc.14096
194. Cao XP, Cao Y, Li WJ, Zhang HH, Zhu ZM. P4HA1/HIF1alpha Feedback Loop Drives the Glycolytic and Malignant Phenotypes of Pancreatic Cancer. Biochem Biophys Res Commun (2019) 516(3):606–12. doi: 10.1016/j.bbrc.2019.06.096
195. Chen F, Chen J, Yang L, Liu J, Zhang X, Zhang Y, et al. Extracellular Vesicle-Packaged HIF-1alpha-Stabilizing lncRNA From Tumour-Associated Macrophages Regulates Aerobic Glycolysis of Breast Cancer Cells. Nat Cell Biol (2019) 21(4):498–510. doi: 10.1038/s41556-019-0299-0
196. Sowa T, Menju T, Chen-Yoshikawa TF, Takahashi K, Nishikawa S, Nakanishi T, et al. Hypoxia-Inducible Factor 1 Promotes Chemoresistance of Lung Cancer by Inducing Carbonic Anhydrase IX Expression. Cancer Med (2017) 6(1):288–97. doi: 10.1002/cam4.991
197. Gu J, Li Y, Zeng J, Wang B, Ji K, Tang Y, et al. Knockdown of HIF-1alpha by siRNA-Expressing Plasmid Delivered by Attenuated Salmonella Enhances the Antitumor Effects of Cisplatin on Prostate Cancer. Sci Rep (2017) 7(1):7546. doi: 10.1038/s41598-017-07973-4
198. Ai Z, Lu Y, Qiu S, Fan Z. Overcoming Cisplatin Resistance of Ovarian Cancer Cells by Targeting HIF-1-Regulated Cancer Metabolism. Cancer Lett (2016) 373(1):36–44. doi: 10.1016/j.canlet.2016.01.009
199. Chen F, Zhuang M, Zhong C, Peng J, Wang X, Li J, et al. Baicalein Reverses Hypoxia-Induced 5-FU Resistance in Gastric Cancer AGS Cells Through Suppression of Glycolysis and the PTEN/Akt/HIF-1alpha Signaling Pathway. Oncol Rep (2015) 33(1):457–63. doi: 10.3892/or.2014.3550
200. Oka N, Komuro A, Amano H, Dash S, Honda M, Ota K, et al. Ascorbate Sensitizes Human Osteosarcoma Cells to the Cytostatic Effects of Cisplatin. Pharmacol Res Perspect (2020) 8(4):e00632. doi: 10.1002/prp2.632
201. Meyer N, Penn LZ. Reflecting on 25 Years With MYC. Nat Rev Cancer (2008) 8(12):976–90. doi: 10.1038/nrc2231
202. Gordan JD, Thompson CB, Simon MC. HIF and C-Myc: Sibling Rivals for Control of Cancer Cell Metabolism and Proliferation. Cancer Cell (2007) 12(2):108–13. doi: 10.1016/j.ccr.2007.07.006
203. Podar K, Anderson KC. A Therapeutic Role for Targeting C-Myc/Hif-1-Dependent Signaling Pathways. Cell Cycle (2010) 9(9):1722–8. doi: 10.4161/cc.9.9.11358
204. Qin Y, Hu Q, Xu J, Ji S, Dai W, Liu W, et al. PRMT5 Enhances Tumorigenicity and Glycolysis in Pancreatic Cancer via the FBW7/cMyc Axis. Cell Commun Signal (2019) 17(1):30. doi: 10.1186/s12964-019-0344-4
205. Kim S, Lee E, Jung J, Lee JW, Kim HJ, Kim J, et al. microRNA-155 Positively Regulates Glucose Metabolism via PIK3R1-FOXO3a-cMYC Axis in Breast Cancer. Oncogene (2018) 37(22):2982–91. doi: 10.1038/s41388-018-0124-4
206. Gupta A, Ajith A, Singh S, Panday RK, Samaiya A, Shukla S. PAK2-C-Myc-PKM2 Axis Plays an Essential Role in Head and Neck Oncogenesis via Regulating Warburg Effect. Cell Death Dis (2018) 9(8):825. doi: 10.1038/s41419-018-0887-0
207. Liu R, Li Y, Tian L, Shi H, Wang J, Liang Y, et al. Gankyrin Drives Metabolic Reprogramming to Promote Tumorigenesis, Metastasis and Drug Resistance Through Activating Beta-Catenin/C-Myc Signaling in Human Hepatocellular Carcinoma. Cancer Lett (2019) 443:34–46. doi: 10.1016/j.canlet.2018.11.030
208. Zhang X, Chen J, Ai Z, Zhang Z, Lin L, Wei H. Targeting Glycometabolic Reprogramming to Restore the Sensitivity of Leukemia Drug-Resistant K562/ADM Cells to Adriamycin. Life Sci (2018) 215:1–10. doi: 10.1016/j.lfs.2018.10.050
209. Kim JW, Gao P, Dang CV. Effects of Hypoxia on Tumor Metabolism. Cancer Metastasis Rev (2007) 26(2):291–8. doi: 10.1007/s10555-007-9060-4
210. Wanandi SI, Ningsih SS, Asikin H, Hosea R, Neolaka GMG. Metabolic Interplay Between Tumour Cells and Cancer-Associated Fibroblasts (CAFs) Under Hypoxia Versus Normoxia. Malays J Med Sci (2018) 25(3):7–16. doi: 10.21315/mjms2018.25.3.2
211. Vitale I, Manic G, Coussens LM, Kroemer G, Galluzzi L. Macrophages and Metabolism in the Tumor Microenvironment. Cell Metab (2019) 30(1):36–50. doi: 10.1016/j.cmet.2019.06.001
212. Visioli F, Wang Y, Alam GN, Ning Y, Rados PV, Nor JE, et al. Glucose-Regulated Protein 78 (Grp78) Confers Chemoresistance to Tumor Endothelial Cells Under Acidic Stress. PloS One (2014) 9(6):e101053. doi: 10.1371/journal.pone.0101053
213. Guo Q, Lan F, Yan X, Xiao Z, Wu Y, Zhang Q. Hypoxia Exposure Induced Cisplatin Resistance Partially via Activating P53 and Hypoxia Inducible Factor-1alpha in non-Small Cell Lung Cancer A549 Cells. Oncol Lett (2018) 16(1):801–8. doi: 10.3892/ol.2018.8767
214. Ling S, Shan Q, Zhan Q, Ye Q, Liu P, Xu S, et al. USP22 Promotes Hypoxia-Induced Hepatocellular Carcinoma Stemness by a HIF1alpha/USP22 Positive Feedback Loop Upon TP53 Inactivation. Gut (2019) 69(7):1322–34. doi: 10.1136/gutjnl-2019-319616
215. Li H, Chen J, Wang X, He M, Zhang Z, Cen Y. Nodal Induced by Hypoxia Exposure Contributes to Dacarbazine Resistance and the Maintenance of Stemness in Melanoma Cancer Stemlike Cells. Oncol Rep (2018) 39(6):2855–64. doi: 10.3892/or.2018.6387
216. Roma-Rodrigues C, Mendes R, Baptista PV, Fernandes AR. Targeting Tumor Microenvironment for Cancer Therapy. Int J Mol Sci (2019) 20(4):840–71. doi: 10.3390/ijms20040840
217. Liu L, Simon MC. Regulation of Transcription and Translation by Hypoxia. Cancer Biol Ther (2004) 3(6):492–7. doi: 10.4161/cbt.3.6.1010
218. Wang GL, Semenza GL. General Involvement of Hypoxia-Inducible Factor 1 in Transcriptional Response to Hypoxia. Proc Natl Acad Sci USA (1993) 90(9):4304–8. doi: 10.1073/pnas.90.9.4304
219. Semenza GL, Roth PH, Fang HM, Wang GL. Transcriptional Regulation of Genes Encoding Glycolytic Enzymes by Hypoxia-Inducible Factor 1. J Biol Chem (1994) 269(38):23757–63. doi: 10.1016/S0021-9258(17)31580-6
220. Thews O, Nowak M, Sauvant C, Gekle M. Hypoxia-Induced Extracellular Acidosis Increases P-Glycoprotein Activity and Chemoresistance in Tumors In Vivo via P38 Signaling Pathway. Adv Exp Med Biol (2011) 701:115–22. doi: 10.1007/978-1-4419-7756-4_16
221. Fahy L, Calvo J, Chabi S, Renou L, Le Maout C, Poglio S, et al. Hypoxia Favors Chemoresistance in T-ALL Through an HIF1alpha-Mediated Mtorc1 Inhibition Loop. Blood Adv (2021) 5(2):513–26. doi: 10.1182/bloodadvances.2020002832
222. Nunes SC. Tumor Microenvironment - Selective Pressures Boosting Cancer Progression. Adv Exp Med Biol (2020) 1219:35–49. doi: 10.1007/978-3-030-34025-4_2
223. Gibadulinova A, Bullova P, Strnad H, Pohlodek K, Jurkovicova D, Takacova M, et al. CAIX-Mediated Control of LIN28/let-7 Axis Contributes to Metabolic Adaptation of Breast Cancer Cells to Hypoxia. Int J Mol Sci (2020) 21(12):4299–317. doi: 10.3390/ijms21124299
224. da Silva VP, Mesquita CB, Nunes JS, de Bem Prunes B, Rados PV, Visioli F. Effects of Extracellular Acidity on Resistance to Chemotherapy Treatment: A Systematic Review. Med Oncol (2018) 35(12):161. doi: 10.1007/s12032-018-1214-4
225. Brown TP, Ganapathy V. Lactate/GPR81 Signaling and Proton Motive Force in Cancer: Role in Angiogenesis, Immune Escape, Nutrition, and Warburg Phenomenon. Pharmacol Ther (2020) 206:107451. doi: 10.1016/j.pharmthera.2019.107451
226. Huber V, De Milito A, Harguindey S, Reshkin SJ, Wahl ML, Rauch C, et al. Proton Dynamics in Cancer. J Transl Med (2010) 8:57. doi: 10.1186/1479-5876-8-57
227. Raghunand N, Mahoney BP, Gillies RJ. Tumor Acidity, Ion Trapping and Chemotherapeutics. Biochem Pharmacol (2003) 66(7):1219–29. doi: 10.1016/s0006-2952(03)00468-4
228. Raghunand N, Martinez-Zaguilan R, Wright SH, Gillies RJ. pH and Drug Resistance. II. Turnover of Acidic Vesicles and Resistance to Weakly Basic Chemotherapeutic Drugs. Biochem Pharmacol (1999) 57(9):1047–58. doi: 10.1016/s0006-2952(99)00021-0
229. Hrabeta J, Belhajova M, Subrtova H, Merlos Rodrigo MA, Heger Z, Eckschlager T. Drug Sequestration in Lysosomes as One of the Mechanisms of Chemoresistance of Cancer Cells and the Possibilities of Its Inhibition. Int J Mol Sci (2020) 21(12):4392–410. doi: 10.3390/ijms21124392
230. Mahoney BP, Raghunand N, Baggett B, Gillies RJ. Tumor Acidity, Ion Trapping and Chemotherapeutics. Biochem Pharmacol (2003) 66(7):1207–18. doi: 10.1016/s0006-2952(03)00467-2
231. Zhitomirsky B, Assaraf YG. Lysosomes as Mediators of Drug Resistance in Cancer. Drug Resist Update (2016) 24:23–33. doi: 10.1016/j.drup.2015.11.004
232. Federici C, Petrucci F, Caimi S, Cesolini A, Logozzi M, Borghi M, et al. Exosome Release and Low pH Belong to a Framework of Resistance of Human Melanoma Cells to Cisplatin. PloS One (2014) 9(2):e88193. doi: 10.1371/journal.pone.0088193
233. Thews O, Riemann A, Nowak M, Gekle M. Impact of Hypoxia-Related Tumor Acidosis on Cytotoxicity of Different Chemotherapeutic Drugs In Vitro and In Vivo. Adv Exp Med Biol (2014) 812:51–8. doi: 10.1007/978-1-4939-0620-8_7
234. Dong Q, Zhou C, Ren H, Zhang Z, Cheng F, Xiong Z, et al. Lactate-Induced MRP1 Expression Contributes to Metabolism-Based Etoposide Resistance in non-Small Cell Lung Cancer Cells. Cell Commun Signal (2020) 18(1):167. doi: 10.1186/s12964-020-00653-3
235. Alfarouk KO, Stock CM, Taylor S, Walsh M, Muddathir AK, Verduzco D, et al. Resistance to Cancer Chemotherapy: Failure in Drug Response From ADME to P-Gp. Cancer Cell Int (2015) 15:71. doi: 10.1186/s12935-015-0221-1
236. Vaupel P, Schmidberger H, Mayer A. The Warburg Effect: Essential Part of Metabolic Reprogramming and Central Contributor to Cancer Progression. Int J Radiat Biol (2019) 95(7):912–9. doi: 10.1080/09553002.2019.1589653
237. Son B, Lee S, Youn H, Kim E, Kim W, Youn B. The Role of Tumor Microenvironment in Therapeutic Resistance. Oncotarget (2017) 8(3):3933–45. doi: 10.18632/oncotarget.13907
238. Wu X, Zhou Z, Xu S, Liao C, Chen X, Li B, et al. Extracellular Vesicle Packaged LMP1-Activated Fibroblasts Promote Tumor Progression via Autophagy and Stroma-Tumor Metabolism Coupling. Cancer Lett (2020) 478:93–106. doi: 10.1016/j.canlet.2020.03.004
239. Jiang E, Xu Z, Wang M, Yan T, Huang C, Zhou X, et al. Tumoral Microvesicle-Activated Glycometabolic Reprogramming in Fibroblasts Promotes the Progression of Oral Squamous Cell Carcinoma. FASEB J (2019) 33(4):5690–703. doi: 10.1096/fj.201802226R
240. Ding L, Zhang W, Yang L, Pelicano H, Zhou K, Yin R, et al. Targeting the Autophagy in Bone Marrow Stromal Cells Overcomes Resistance to Vorinostat in Chronic Lymphocytic Leukemia. Oncol Targets Ther (2018) 11:5151–70. doi: 10.2147/OTT.S170392
241. Lisanti MP, Martinez-Outschoorn UE, Sotgia F. Oncogenes Induce the Cancer-Associated Fibroblast Phenotype: Metabolic Symbiosis and “Fibroblast Addiction” are New Therapeutic Targets for Drug Discovery. Cell Cycle (2013) 12(17):2723–32. doi: 10.4161/cc.25695
242. Aoki T, Shimada K, Sakamoto A, Sugimoto K, Morishita T, Kojima Y, et al. Emetine Elicits Apoptosis of Intractable B-Cell Lymphoma Cells With MYC Rearrangement Through Inhibition of Glycolytic Metabolism. Oncotarget (2017) 8(8):13085–98. doi: 10.18632/oncotarget.14393
243. Jitschin R, Braun M, Qorraj M, Saul D, Le Blanc K, Zenz T, et al. Stromal Cell-Mediated Glycolytic Switch in CLL Cells Involves Notch-C-Myc Signaling. Blood (2015) 125(22):3432–6. doi: 10.1182/blood-2014-10-607036
244. Thuwajit C, Ferraresi A, Titone R, Thuwajit P, Isidoro C. The Metabolic Cross-Talk Between Epithelial Cancer Cells and Stromal Fibroblasts in Ovarian Cancer Progression: Autophagy Plays a Role. Med Res Rev (2018) 38(4):1235–54. doi: 10.1002/med.21473
Keywords: chemoresistance, Warburg effect, tumor microenvironment, signaling pathway, transporters and key enzymes of glycolysis
Citation: Liu C, Jin Y and Fan Z (2021) The Mechanism of Warburg Effect-Induced Chemoresistance in Cancer. Front. Oncol. 11:698023. doi: 10.3389/fonc.2021.698023
Received: 20 April 2021; Accepted: 11 August 2021;
Published: 03 September 2021.
Edited by:
Sara Granja, University of Minho, PortugalReviewed by:
Khalid Omer Alfarouk, Alfarouk Biomedical Research LLC, United StatesCopyright © 2021 Liu, Jin and Fan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhimin Fan, ZmFuem1Aamx1LmVkdS5jbg==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.