
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oncol. , 13 July 2021
Sec. Cancer Metabolism
Volume 11 - 2021 | https://doi.org/10.3389/fonc.2021.697894
This article is part of the Research Topic Tumor Metabolism in Cancer Therapy Resistance View all 8 articles
 Guofeng Ma1,2†
Guofeng Ma1,2† Chun Li3†Zhilei Zhang1,2
Chun Li3†Zhilei Zhang1,2 Ye Liang2Zhijuan Liang2Yuanbin Chen2
Ye Liang2Zhijuan Liang2Yuanbin Chen2 Liping Wang2Dan Li2Manqin Zeng4Wenhong Shan5*Haitao Niu1,2*
Liping Wang2Dan Li2Manqin Zeng4Wenhong Shan5*Haitao Niu1,2*Immunotherapy, especially PD-1/PD-L1 checkpoint blockade immunotherapy, has led tumor therapy into a new era. However, the vast majority of patients do not benefit from immunotherapy. One possible reason for this lack of response is that the association between tumors, immune cells and metabolic reprogramming in the tumor microenvironment affect tumor immune escape. Generally, the limited amount of metabolites in the tumor microenvironment leads to nutritional competition between tumors and immune cells. Metabolism regulates tumor cell expression of PD-L1, and the PD-1/PD-L1 immune checkpoint regulates the metabolism of tumor and T cells, which suggests that targeted tumor metabolism may have a synergistic therapeutic effect together with immunotherapy. However, the targeting of different metabolic pathways in different tumors may have different effects on tumor immune escape. Herein, we discuss the influence of glucose metabolism and glutamine metabolism on tumor immune escape and describe the theoretical basis for strategies targeting glucose or glutamine metabolism in combination with PD-1/PD-L1 checkpoint blockade immunotherapy.
The traditional method of cancer treatment is surgical removal, followed by two different types of treatment: radiotherapy plus chemotherapy and targeted therapy that inhibit tumor angiogenesis and oncogenic signaling. However, these therapies are usually effective for only early-stage cancers and usually cannot cure advanced-stage cancers and have substantial side effects. Research on resistance to radiotherapy and chemotherapy usually focuses on tumor cell-intrinsic effects, such as cell cycle arrest and cell death caused by DNA damage, the switching of survival signalling pathways and the activation of new signaling pathways that lead to tumor resistance (1, 2). Research on resistance to targeted drugs mainly focuses on gene mutations. Gene mutations usually occur after the administration of targeted agents, such that the targeted drugs cannot bind to the target tumor molecules. The reprogramming of biological systems is also a reason for drug resistance (3). Therefore, methods to improve treatment mainly focus on the activation of new signaling pathways and the inherent genetic heterogeneity of tumor cells. However, these methods still cannot solve the problem of tumor drug resistance. The present study further examines tumor immunotherapy and metabolism in the tumor microenvironment (TME) in order to find a better way to treat tumors.
The study of the cancer-immunity cycle led to the development of checkpoint immunotherapy (4, 5). The PD-1/PD-L1 pathway is one of the core pathways involved in tumor immune escape, and it received the most attention in recent years. PD-L1 and PD-1 are important inhibitory costimulatory molecules that regulate the immune response of T cells, and these cells are also known as immune checkpoint molecules (6). PD-1 is expressed on activated T cells, and its ligands PD-L1 and PD-L2 are expressed on immune cells and tumors. PD-1 binds to PD-L1 on tumor cells and drives T cells into a dysfunctional state (7). Pembrolizumab and Nivolumab are two antibodies that inhibit PD-1 interactions with its ligands, and these agents are approved by the FDA for the treatment of non-small cell lung cancer, advanced melanoma, and renal cell carcinoma (8, 9). Although the development of PD-1/PD-L1 checkpoint immunotherapy has greatly promoted the progress of tumor treatment strategies, most patients do not respond to treatment or have off-target effects in clinical trials. These shortcomings may be attributed to genetic mutations (10), the status of immune infiltration in the tumor (11) and the expression of PD-L1 by tumors and immune cells (12, 13). Therefore, it is necessary to design a more reasonable combination therapy strategy to improve the effect of checkpoint immunotherapy.
Metabolic reprogramming is a hallmark of malignant tumors. Different stages of tumor progression are accompanied with different types of metabolic reprogramming. These different metabolic phenotypes may provide strategies for the targeted metabolic treatment of tumors (14). The Warburg effect is a typical example of metabolic reprogramming that is controlled by oncogenes. Under aerobic conditions, tumor cells still rely on the conversion of glucose to lactic acid for energy, which provides sufficient energy for tumor proliferation and produces a specifically acidic TME that can inhibit the function of T cells, and it is more conducive to tumor progression (15, 16). Glutamine is another important nutrient involved in tumor progression that can regulate tumor energy production, signal transduction and redox homeostasis. Glutamine transporter variants such as SLC1A5 have been shown to promote tumor metabolic reprogramming (17). The main problem facing tumor metabolism treatment strategies is how to specifically target tumor metabolism without affecting normal cell metabolism and inhibiting the function of antitumor immune effect cells in the TME. In addition, owing to the diversity of cell metabolism pathways, there are numerous metabolic bypass pathways, and the compensation of these pathways also limits the effect of targeting metabolism. Therefore, it is necessary to further study tumor metabolism pathways to understand the relationship between tumor metabolism and tumor immunity in the TME, and design more reasonable combination immunotherapies and targeted metabolic therapy strategies to treat tumors more effectively.
In this review, we discuss two strategies targeting tumor glucose metabolism or glutamine metabolism and explore how the targeting of glucose or glutamine metabolism affects tumor immune escape via the regulation of tumor PD-L1 expression and the function of T cells in addition to their direct effects on the tumor’s own biological activity. In this way, we further reveal the mechanism underlying strategies that combine targeted glucose or glutamine metabolic with PD-1/PD-L1 checkpoint blockade immunotherapy and provide a strong rationale for these strategies in the treatment of tumors.
The difference in metabolism between tumor and normal tissues suggests a reprogramming of tumor metabolism. Unlike normal cells, tumor cells can use large amounts of glucose to produce lactic acid through glycolysis in the cytoplasm even in the presence of high oxygen. In addition, this kind of high glycolytic flux produces large amounts of ATP, with a correspondingly low rate of oxidative phosphorylation (OXPHOS) in mitochondria. This phenomenon is called the Warburg effect (15, 16, 18). The further study of tumor glucose metabolism revealed that tumors often represent a mosaic of tumor cells with different metabolic characteristics. Some tumors rely more on oxygen, and other tumors are more prone to glycolysis (19). For example, metabolic heterogeneity of glucose metabolism exists within and between human lung tumors, human clear cell renal cell carcinomas (ccRCCs), high-grade serous ovarian and pancreatic cancer (20–23). However, the Warburg effect is not only important for energy purposes but also to provide building blocks for synthesis of macromolecules for tumors that rely on this effect, and it may be used as a marker of tumor malignancy (24–28). Traditionally, the Warburg effect enables tumors to obtain the large amount of energy that is needed for rapid proliferation, which promotes tumor growth and metastasis (29, 30). Recent studies have shown that the Warburg effect also played an important role in the tumor immune escape mechanism (31–33).
The Warburg effect is the main energy source of some tumors, and this reliance causes tumor cells to consume a large amount of glucose to survive. Imperfect blood vessel development in solid tumors leads to a limited supply of nutrients for tumors. Therefore, glucose in the TME is often lacking (33–35). PD-L1 is a negative immunoregulator that is regulated by glycolysis in many tumors. In vitro experiments found that a reduction in glucose content in the culture medium upregulated the expression of PD-L1 in renal cancer cells via the EGFR/ERK/C-Jun pathway (36). The expression of PD-L1 correlated with the uptake of 18F-FDG in lung adenocarcinoma (37). Pyruvate kinase is the key enzyme in the last step of glycolysis. The isoenzyme pyruvate kinase isozyme type M2 (PKM2) has been shown to promote the expression of PD-L1 in tumor and immune cells. The use of the PKM2 activator TEPP-46 to increase the conversion of phosphoenolpyruvate to pyruvate reduces the expression of PD-L1 in a murine CT26 colon carcinoma model and in tumors (38). TCGA database analysis also proved that glycolytic activity was related to active immune characteristics in various cancers, and in vitro experimental studies have proved that glycolysis can increase the expression of PD-L1 in breast cancer, osteosarcoma and ovarian cancer (39). Therefore, tumors with glucose-deficient TME regulate the expression of PD-L1 via glycolysis, which may cause tumor immune escape (Figure 1).
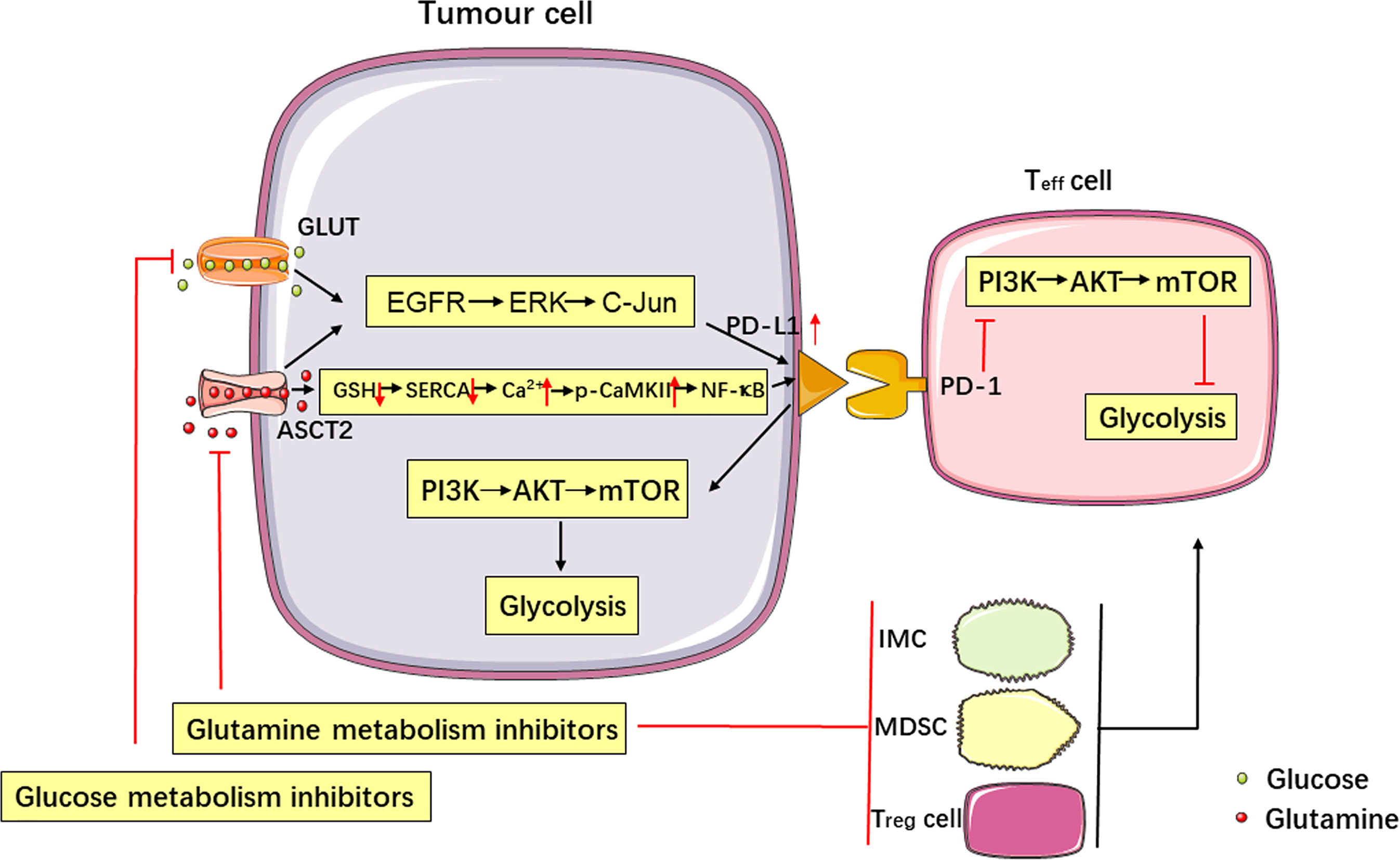
Figure 1 Metabolism affects tumor immune escape, and the PD-1/PD-L1 immune checkpoint regulates metabolic pathways. Glucose and glutamine metabolism upregulate the expression of PD-L1 in tumor cells via the EGFR/ERK/C-Jun pathway. Inhibition of glutamine use in tumor cells increases PD-L1 expression by reducing the levels of GSH, inhibiting the SERCA activation, and increasing cytosolic Ca2+ levels and CaMKII phosphorylation, which further activates the downstream NF-κB signalling pathway. Targeting glutamine metabolism can inhibit the production of immune cells negatively affecting the immune response (IMCs, MDSCs and Treg cells) and upregulate the function of Teff cells, thereby enhancing the antitumor immune response. Activation of the PD-L1/PD-1 signalling pathway promotes aerobic glycolysis (i.e., the Warburg effect) in tumor cells, inhibits glucose metabolism in Teff cells by stimulating the PI3K-AKT-mTOR signalling pathway, and produces synergistic inhibition of the antitumor response.
In recent years, PD-1/PD-L1 immune checkpoint blockade (ICB) therapy has made significant progress in the treatment of tumors. This type of therapy is based on the interaction between PD-L1 and PD-1, which inhibits the activation and proliferation of T cells and mediates tumor immune escape. Increasing evidence shows that the interaction of PD-L1 and PD-1 regulates the glucose metabolism of tumors and T cells, which affect the nutrient competition between these two cell types in the TME. The interaction of PD-1 with PD-L1 or PD-L2 on T cells impairs aerobic glycolysis via inhibition of the PI3K-AKT-mTOR pathway (40). The expression of PD-L1 on cancer cells drives activation of the PI3K-Akt-mTOR pathway to stimulate aerobic glycolysis, increases glucose uptake and enhance T cell competition for glucose (41–43). Therefore, the PD-1/PD-L1 axis can synergistically promote tumor immune escape via the upregulation of tumor cell glycolysis and inhibiting of T cell glycolysis (Figure 1).
The different glucose metabolism modes of tumors and T cells suggest competition between these types of cells for glucose use in the TME. The Warburg effect supports the rapid growth of tumors and the demand for macromolecules, which provides an external advantage for tumor cells, accelerates the consumption of glucose in the TME, and limits the glucose uptake of tumor-infiltrating T cells, which results in dysfunction (44, 45). For example, the effector function of CD4+ and CD8 + Teff cells was reduced under low-glucose conditions, and the production of interferon-γ (IFN-γ), intermediate-17 (IL-17) and granzyme B was inhibited (46–48). The production of an intermediate of T cell glycolysis, phosphoenolpyruvate (PEP), was inhibited, which interfered with the signal transduction of the calcium-dependent transcription factor nuclear factor of activated T cells (NFAT) (42). These results indicate that glucose metabolism directly controls the activation of T cells, and the limited use of glucose by T cells in the TME damages their functions and reduce immune responses. Primary ovarian cancer cells in a coculture system suppressed the expression of the methyltransferase EZH2 by maintaining high expression of the microRNAs miR-101 and miR-26a and imposing glucose restriction on T cells, which inhibited T cell function (49). Increasing the glycolytic ability of mouse sarcoma cells led to the inhibition of CD8+ T cell function in a coculture system (41). The function of CD4+ T cells was inhibited in a mouse model of melanoma overexpressing HK2. Overexpression of the glycolytic enzyme PEP carboxykinase in tumor-specific CD4+ T cells enhanced the antitumor effect (42). These results indicate that the function of effector T cells in the TME is related to the glycolytic activity of tumors, and tumors cause T cell metabolism disorders via glucose competition and effects on tumor immune escape (Figure 2).
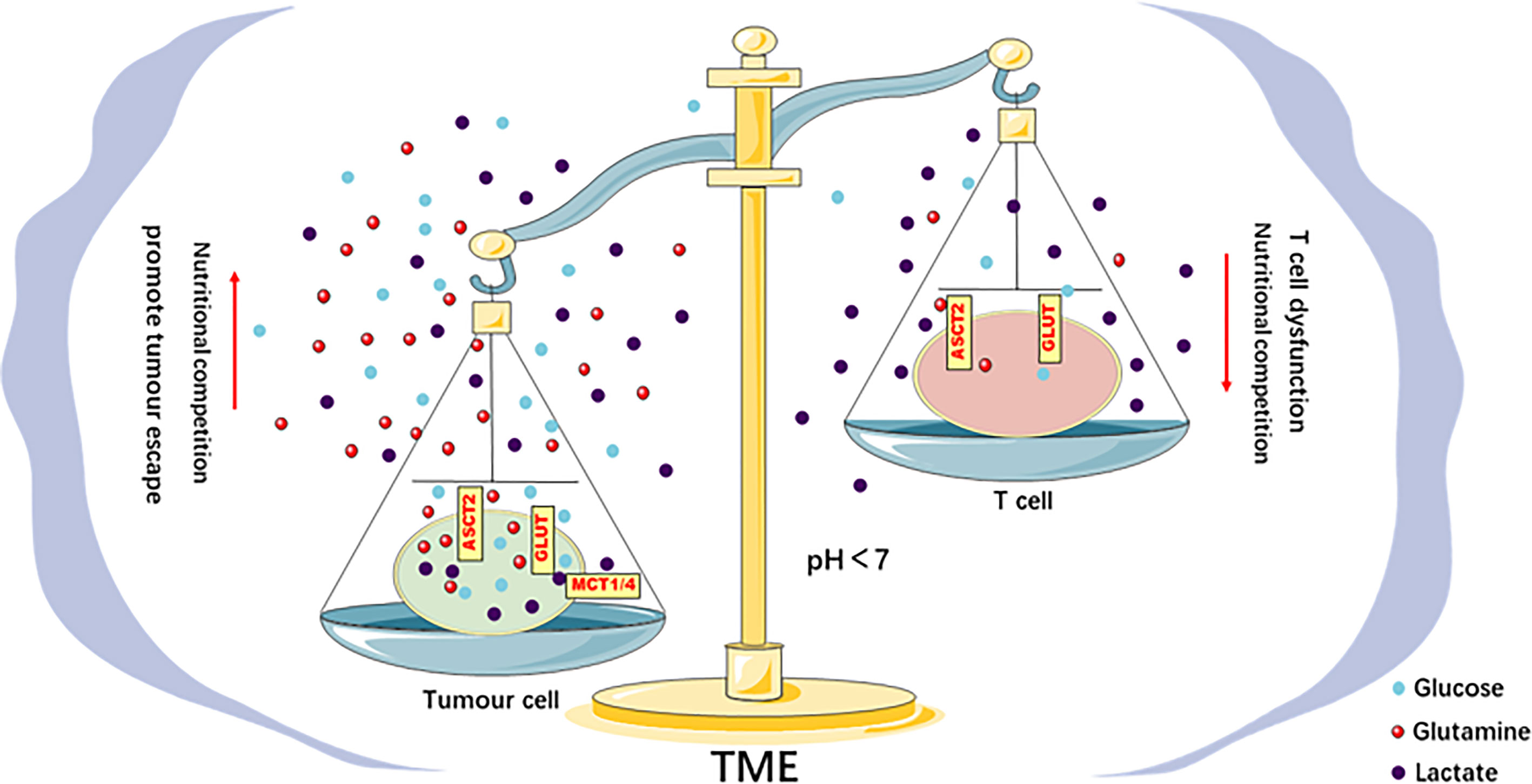
Figure 2 Metabolic competition in the TME drives tumor progression. There is competition for glucose and glutamine between tumor and Teff cells in the TME. This competition leads to the limited use of energy materials by Teff cells and impairs their function, which promotes immune escape. The acidic TME caused by the lactic acid produced by the tumor Warburg effect inhibits the function of Teff cells, impairs the antitumor immune response, and promotes tumor progression.
The Warburg effect of tumor cells produces a large amount of lactic acid. Lactic acid plays a different role in tumor cells than it does in T cells. Lactic acid establishes metabolic coupling between tumor and nonmalignant cells or between tumor cells to maintain tumor growth (50). Genetically modified mouse models of lung and pancreatic cancer found that circulating lactic acid was used to generate energy and induce the release of glucose to promote tumor growth (51). In contrast, the acidic TME formed by the large amount of lactic acid produced by tumor glycolysis inhibited the proliferation, survival, cytotoxicity and cytokine production of T cells (52, 53). In vitro experiments found that the activation process of mouse CD8+ T cells was dysfunctional under high-lactate and high-H+ conditions, and MAP kinase signal transduction during the activation of human effector CD8+ T cells was seriously damaged (53, 54). Under acidic pH conditions, PSGL-1 can act as a selective receptor for VISTA to inhibit the activity of T cells, which ultimately leads to immune escape of tumor cells (55). The low lactate levels produced by mouse melanoma cells in an LDHA knockout mouse melanoma model mediated a strong tumor exclusion response (53). These results indicate that the acidic TME mediated by the Warburg effect is beneficial to tumor progression but inhibits the function of T cells, which may cause tumor immune escape (Figure 2).
In conclusion, tumor glucose metabolism can affect tumor immune escape via regulation of the expression of PD-L1 in tumors and its effects on the function of T cells in TME via different pathways. Therefore, the influence of tumor glucose metabolism on tumor immune escape should be considered for interventions.
Glutamine is one of the most abundant amino acids in the human body, and it is an indispensable energy source for tumor survival and progression. Many tumor cells rely on glutamine for survival. Glutamine metabolism provides energy for tumor cells by producing ATP and participating in the tricarboxylic acid cycle. Glutamine also provides raw materials for the synthesis of macromolecular substances in tumor cells, such as nucleotides and hexosamine (56). Notably, glutamine metabolism is involved in tumor proliferation and metastasis (57, 58). Previous studies have demonstrated a correlation between tumor progression and glutamine metabolism. Tumor cells rely on glutamine metabolism for energy and the synthesis of macromolecules even when aerobic glycolysis provides sufficient energy (59). Glutamine metabolism provides high levels of NADPH for tumor cells to maintain the redox state and ensure the survival of tumor cells (60). Recent studies have shown that glutamine metabolism also affected tumor immune escape.
Glutamine metabolism maintains tumor proliferation and progression, and the targeting of glutamine metabolism can effectively inhibit tumor growth. Notably, recent studies have shown that targeted glutamine metabolism affected tumor immune escape via different mechanisms during tumor growth inhibition. Glutamine deprivation in the culture medium upregulated the expression of PD-L1 on renal cancer cells via the EGFR/ERK/C-Jun pathway (61). The inhibition of glutamine use in lung and colon tumors increased the expression of PD-L1 by reducing GSH levels. The reduction in GSH levels inhibited sarcoplasmic reticulum Ca2+-ATPase (SERCA) activation and increased cytosolic Ca2+ levels and CaMKII phosphorylation, which further activated the downstream nuclear factor-kappa B (NF-κB) signalling pathway to promote the expression of PD-L1 (62). However, researchers found that glutamine deprivation in renal cancer cells can weaken an immunosuppressive TME. Glutamine deprivation induced M2 macrophages to secrete IL-23 via activation of the hypoxia-inducible factor 1 alpha (HIF1α) pathway. IL-23 inhibits the proliferation and activation of Treg cells, which upregulates the proliferation and activity of CD4+ and CD8+ Teff cells, to enhance the antitumor immune response (63). The use of a small-molecule inhibitor of glutamine metabolism demonstrated that blockade of glutamine metabolism inhibited the generation and recruitment of myeloid-derived suppressor cells (MDSCs) via inhibition of the production of colony-stimulating factor 3 (CSF3), which enhanced the function of T cells in TME (64). Another study showed that glutamine metabolism can regulate the production of immature myeloid cells (IMCs) with a highly immunosuppressive effect. Blockade of glutamine metabolism improved the therapeutic effect of anti-PD-L1 agents in an ICB therapy-resistant mouse model, which indicates that glutamine metabolism can regulate the antitumor immune response (65). All of these data indicate the complexity of targeting glutamine metabolism to regulate the immune response because these strategies may simultaneously cause tumor immune escape via upregulation of the expression of PD-L1 in tumors and inhibition of tumor immune escape by enhancing the function of T cells (Figure 1).
Some tumor cells require a high consumption of glutamine, in addition to glucose, to meet tumor progression needs. Therefore, there is also competition for glutamine between tumors and T cells in the TME. As mentioned above, the glutamine metabolic pathway in tumor cells affects the proliferation and activation of T cells, and glutamine deprivation inhibits T cell proliferation and cytokine production (66, 67). Studies have shown that the MAPK/ERK pathway upregulates glutamine uptake by T cells in the process of T cell activation. The MAPK/ERK pathway also upregulates glutamine uptake and use by tumor cells (67, 68). Therefore, the MAPK/ERK pathway may play an important role in the competition for glutamine between T cells and tumor cells. Triple-negative breast cancer cells competitively deprive glutamine in the TME and limits the use of glutamine by tumor-infiltrating T cells, which affects the function of T cells and damages antitumor immune responses. The concentration of glutamine in the tumor interstitium was increased in a tumor model with specific GLS loss, and the activity of intratumoral T cells was increased. Limiting glutamine in the TME also differentially affected tumors and T cells. For example, the glutamine transporter inhibitor V-9302 selectively inhibits glutamine uptake by tumor, but not CD8+ T, cells. CD8+ T cells can regulate glutamine metabolism by upregulating glutamine transporter ATB0,+/Slc6a14 (69). The glutamine antagonist 6-diazo-5-oxo-L-norleucine (DON) and its precursor, JHU-083, inhibit tumor metabolism and have a strong antitumor effect. In contrast, the antitumor effects of CD8+T cells may be increased via metabolic reprogramming strategies that upregulate glycolysis and oxidative metabolism (70). These data show that tumor cells competitively use glutamine, which limits the use of glutamine by T cells and affects T cell function. The targeting of glutamine metabolism can inhibit tumor cell proliferation and relieves the restriction of glutamine use by tumor cells, which enhances the function of T cells (Figure 2).
In summary, tumor glutamine metabolism can regulate the function of T cells and affects tumor immune escape by regulating the expression of tumor PD-L1, the activation of regulatory immune cells and nutritional competition. Therefore, it is necessary to fully consider the effects of the upregulation of tumor PD-L1 expression and enhancing the function of T cells on tumor immune escape when intervening in tumor glutamine metabolism.
Immunotherapy produced fundamental changes to cancer treatment. Immune checkpoint inhibitors (ICIs), especially PD-1/PD-L1 ICIs, achieved amazing therapeutic effects in a variety of tumor types. However, there are a large number of people in whom ICIs are ineffective or who gradually develop drug resistance during the process of treatment. Therefore, solving the problem of drug resistance in immunotherapy has become a hot area of research. Previous studies attributed the failure of ICI therapy to different types of T cell production disorders or dysfunctions, such as insufficient antitumor T cell production, insufficient tumor-specific T cell function, and impaired T cell immune memory function (71). Increasing the generation and function of Teff cells and reducing the generation and function of Treg cells have become key goals for solving the problem of drug resistance in immunotherapy. As mentioned above, the different effects of glucose and glutamine metabolism on tumor and T cells determine the effect of metabolic interactions between tumor and T cells on tumor immune escape. Glucose metabolism and glutamine metabolism not only affect tumor proliferation and immune effects but also have different effects on different types of T cells. Therefore, strategies targeting glucose or glutamine metabolism in combination with PD-1/PD-L1 checkpoint blockade immunotherapy should enhance the power of cancer immunotherapy.
The Warburg effect provides an abundance of energy and intermediates for tumor metabolism to maintain tumor progression. Tumor cells also regulate the function of T cells via glucose competition, lactic acid acidification of the TME and direct regulation of PD-L1 expression. The PD-1/PD-L1 axis upregulates tumor cell glycolysis and simultaneously inhibits T cell glycolysis. Current strategies targeting glucose metabolism for the treatment of tumors mainly target rate-limiting enzymes of glycolysis, such as hexokinase 2 (HK2), PKM2, phosphofructokinase-2/fructose-2,6-bisphosphatase 3 (PFKFB3) and lactate dehydrogenase (LDHA) (38, 72–74). The relationship between tumor glucose metabolism and the tumor immune escape provides a theoretical basis for strategies targeting glucose metabolism in combination with PD-1/PD-L1 checkpoint blockade immunotherapy for tumor treatment. Therefore, targeted glucose metabolism in combination with PD-1/PD-L1 checkpoint blockade immunotherapy for tumor treatment will further improve the therapeutic effect. This is mainly based on the following ideas: ① The targeting of glucose metabolism may limit the use of glucose by tumors and upregulate glucose in the TME, which is more conducive to the function of Teff cells. Therefore, the concurrent use of PD-1/PD-L1 ICBs may further improve the function of T cells. ② The targeting of glucose metabolism inhibits tumor cell production of lactic acid, improves the acidic TME, and weakens the inhibitory effect on the function of effector T cells. The combined use of PD-1/PD-L1 ICBs may further enhance the function of T cells. ③ The targeting of glucose metabolism may upregulate the expression of PD-L1 in tumor cells, and the simultaneous use of PD-L1 monoclonal antibody may improve the body’s antitumor immune response and produce synergistic anticancer effects. ④ The interaction of PD-1 and PD-L1 can not only inhibit the activation and proliferation of T cells but also upregulate the glycolysis of tumor cells and inhibits the glycolysis of T cells. The targeting of glucose metabolism in combination with PD-1/PD-L1 ICBs can relieve this effect and improve the body’s antitumor immune response (Figure 3).
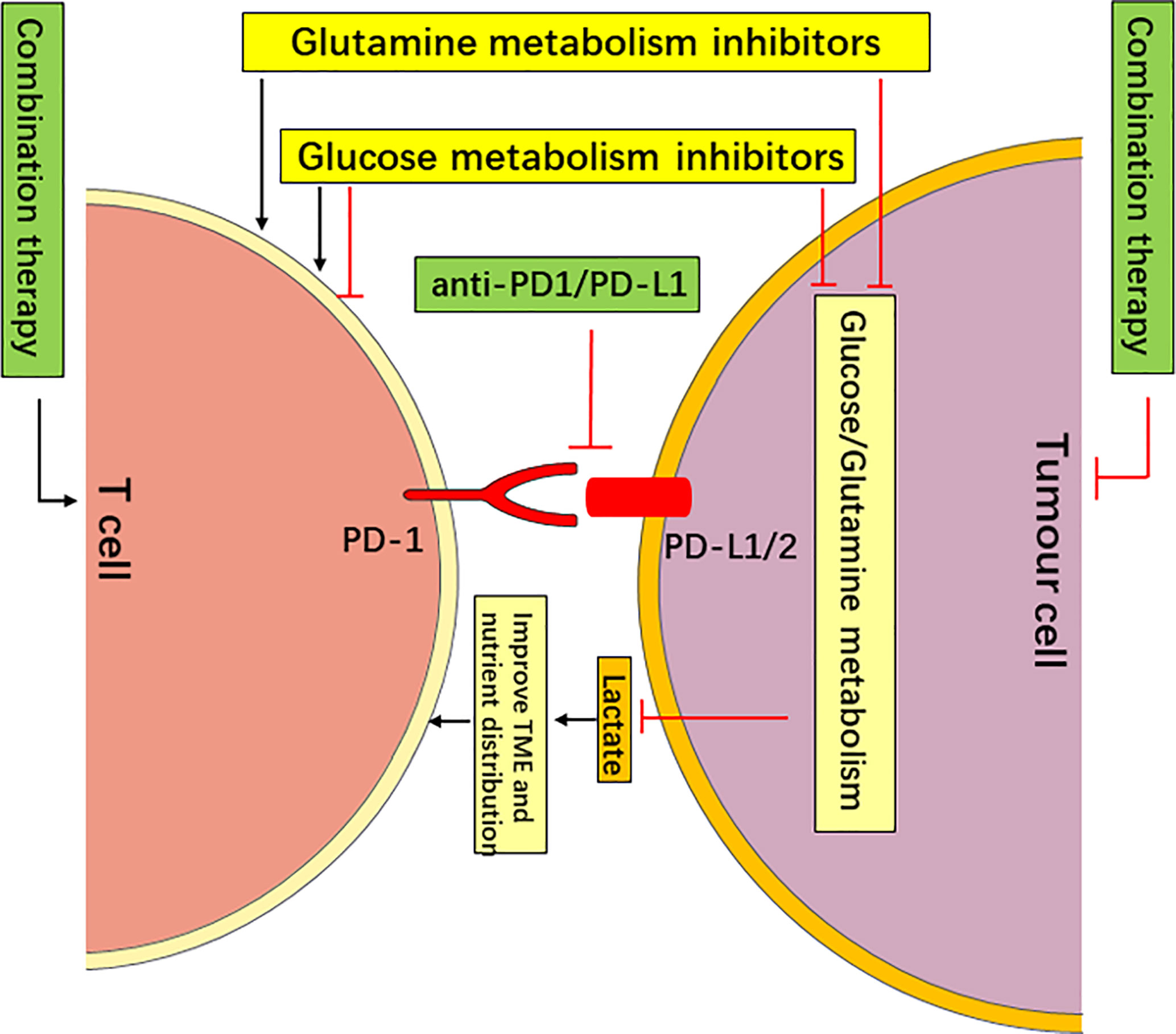
Figure 3 Targeting Metabolism in Combination with PD-1/PD-L1 Checkpoint Blockade Immunotherapy May Have a Synergistic Anticancer effect. The targeting of glucose or glutamine metabolism has an antitumor effect by starving tumors, and it improves the nutrient distribution and acidic TME, which is more conducive to the function of T cells. The targeting of glutamine metabolism may improve the function of T cells via metabolic reprogramming (such as DON and V-9302). However, the targeting of glucose metabolism may enhance or inhibit the function of different types of T cells (such as 2-DG). Therefore, the targeting of glucose or glutamine metabolism in combination with PD-1/PD-L1 checkpoint blockade immunotherapy may have a synergistic anti-cancer effect.
Although none of these combination therapies are approved for clinical treatment, the results of relevant experiments have proven the superiority of combination therapy. For example, metformin, which affects glucose metabolism and levels, reduced the hypoxic state of xenograft tumors, which rendered the tumors responsive to PD-1 blockade and enhancing the function of immune cells (75). PFKFB3 promoted the glycolysis of tumors and the production of lactic acid (76), and inhibition of PFKFB3 eliminated the Warburg effect, inhibit tumor progression and metastasis (77), and improved the therapeutic response to antibodies targeting the inhibitory immune checkpoint receptor in a mouse B16 melanoma model (78). LDHA is a key catalytic enzyme for aerobic glycolysis. Inhibition of LDHA reduced tumor growth in a xenograft model (79, 80), and researchers have proven that low levels of LDHA were associated with better anti-PD-1 antibody therapeutic responses in patients with melanoma (81). These experimental results support that the targeting of glycolysis enhances the effect of anti-PD-1/PD-L1 immunotherapy. Several current combinatory therapies are being tested in clinical trials (Table 1). However, the current challenge is how to target the glucose metabolism of tumor cells without affecting the glucose metabolism of immune and normal cells. In fact, it has been proved that inhibiting of glycolytic enzymes or use of the competitive glucose analogue 2-DG can support the formation of long-term memory CD8+ T cells, but it also inhibits the proliferation and function of tumor-infiltrating effector immune cells (82–85).Current unnatural sugar metabolism labeling technology achieved a preferential labeling of cancer cells and targeted delivery (86). The combination of imaging technology and isotope-labeling technology provides a “visualization” solution for targeted tumor metabolism. However, the clinical application of these therapies requires more time, and more research is needed to solve these problems in the future.
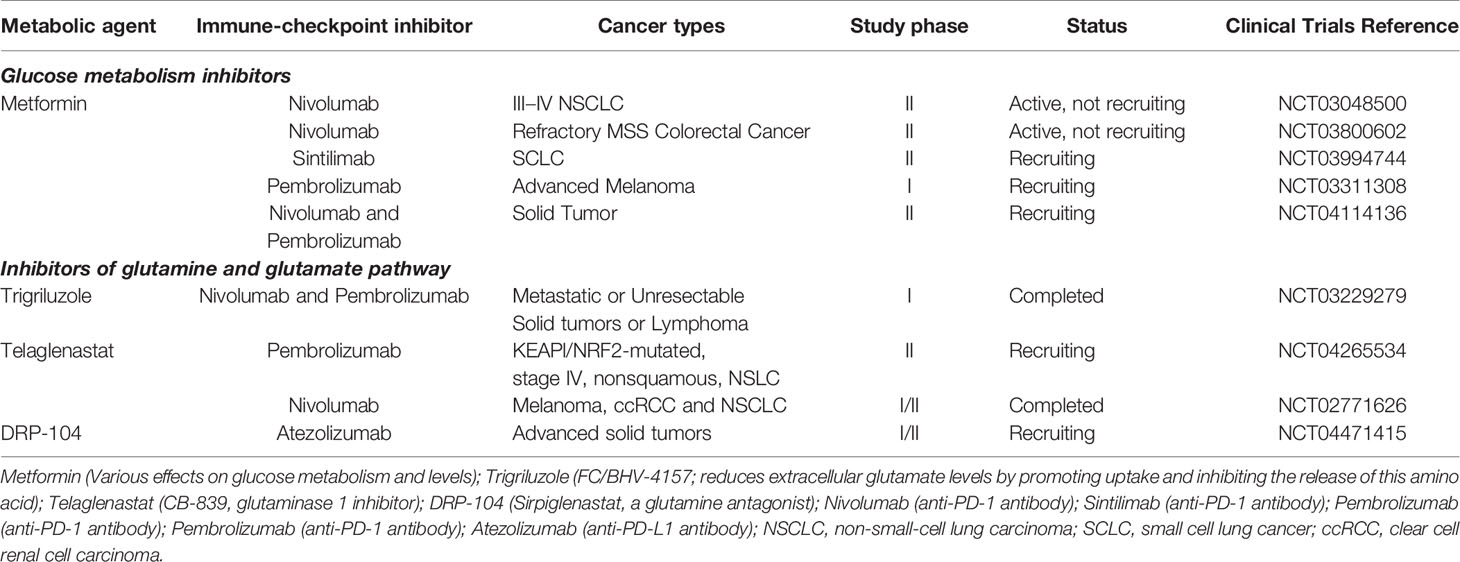
Table 1 Currently ongoing trials of glucose and glutamine metabolic interventions combined with immune checkpoint inhibitors.
Glutamine metabolism meets the metabolic needs of rapidly proliferating tumor cells. Similar to the glucose competition in the TME, there is also glutamine competition between tumor and immune cells, and glutamine metabolism directly affects tumor immune escape. A large number of basic and clinical experiments have shown that targeting of tumor glutamine metabolism can effectively inhibit tumor growth. Current strategies targeting glutamine metabolism mainly focus on key enzymes of glutamine metabolism, such as glutaminase (GLS), the glutamine transporter SLC1A5 and glutamate dehydrogenase 1 (GLUD1) (87–89). The relationship between tumor glutamine metabolism and immune escape provides a theoretical basis for the targeting of glutamine metabolism in combination with PD-1/PD-L1 immune checkpoint blockers for tumor treatment. Therefore, targeted of glutamine metabolism in combination with anti-PD-1/PD-L1 for tumor treatment will further improve the therapeutic effect, which is mainly based on the following ideas. ① The targeting of glutamine metabolism to inhibit the use of glutamine by tumors relieves the tumor’s restriction on the use of glutamine by T cells in the TME, which enhances the function of T cells. Combining with PD-1/PD-L1 checkpoint blockade immunotherapy may further improve the antitumor immune response. ② The targeting of glutamine metabolism directly affects the immune escape of tumors via different mechanisms, such as regulation of the generation of negative immune cells, improving the efficacy of anti-PD-L1 therapy and direct upregulation of the function of Teff cells in TME. Therefore, combinations with PD-1/PD-L1 immune checkpoint inhibitor may further improve the antitumor effect. ③ Glutamine metabolism directly regulates PD-L1 expression in tumor cells and inhibits the function of Teff cells. Combinations with anti-PD-L1 monoclonal antibodies can relieve this effect and improve the body’s antitumor immune response (Figure 3).
Several compounds targeting glutamine metabolism have been developed for antitumor therapy. The glutamine antagonist DON and its precursor JHU-083 inhibit tumor cell proliferation via inhibition of the activity of a variety of enzymes that are required for tumor glutamine metabolism. JHU-083 has been proven to delay tumor growth and promotes the production of durable and highly active antitumor T cells. The combination of JHU-083 with PD-1 immune checkpoint inhibitors enhanced the antitumor effects (70). These results indicate the potential of targeting glutamine metabolism in combination with PD-1/PD-L1 checkpoint blockade immunotherapy for tumor treatment. Although no such combination therapy is available for clinical treatment, some related combination therapies are in clinical trials, including the combination of the GLS inhibitor CB-839 and a PD-1 ICI for cancer treatment (Table 1). CB-839 treatment also enhanced CTL-mediated antitumor responses in mouse models (90), which may explain the synergistic effect between CB-839 and PD-1 inhibitors. Renal cancer has been proven to constitute a glutamine-dependent tumor (91). The targeting of glutamine may be a potential treatment for renal cancer. However, one study found that deprivation of glutamine in the culture medium upregulate the expression of PD-L1 in renal cancers (61). In addition, it has been proved that targeting of glutamine metabolism can upregulate the expression of PD-L1 in lung cancer and colon cancer (62). Therefore, the targeting of glutamine metabolism in combination with PD-L1-targeted ICIs may produce a more powerful therapeutic effect in the future. Although the targeting of glutamine metabolism in combination with PD-1/PD-L1 immune checkpoint therapy showed promising results, similar to strategies targeting glucose metabolism, it is necessary to determine how to achieve maximum killing of cancer cells while minimizing the negative impact on normal and immune cells. Different drugs that target glutamine metabolism may have different effects on immune cells in TME. For example, JHU-083 and V-9302 inhibit glutamine metabolism in Teff cells, but T cells regulate their own metabolism via different mechanisms without affecting their antitumor function (69, 70). Therefore, in addition to the combination of imaging technology and isotope labeling technology to solve the above mentioned problems, the development of more reasonable drugs that target glutamine metabolism without affecting or enhancing the function of Teff cells on the basis of the difference in glutamine metabolism between tumor and Teff cells is the focus of future research.
Immunotherapy, especially PD-1/PD-L1 checkpoint blockade immunotherapy, have ushered in a new era of cancer treatment. Although immunotherapy is unique in its ability to achieve long-term and complete responses, most patients do not benefit from treatment. Therefore, it is necessary to examine new treatment strategies to further improve immunotherapy efficacy, such as the combination of immunotherapy with targeted therapy (92). The in-depth study of tumor and immune cell glucose and glutamine metabolism produced increasing evidence indicating that the metabolic interaction between the tumor cells and immune cells may be related to a poor response to immunotherapy. Therefore, the targeting of tumor glucose or glutamine metabolism in combination with PD-1/PD-L1 ICIs may provide new treatment opportunities for patients with tumors. However, we need to pay more attention to which types of tumors may benefit from combination therapy. Siska and colleagues classified tumors into four metabolic types using the metabolic-tumor-stroma score. Targeting glycolysis might be essential to allow an effective immune response in a mixed tumor type with glycolysis and OXPHIS (MeTS3) and a highly glycolytic Warburg type (MeTS4), which may benefit from combination therapy (19). There is no reliable method to judge glutamine metabolism; thus, more in-depth research is needed.
In this review, we consider the combination of targeted glucose or glutamine metabolic therapy and PD-1/PD-L1 checkpoint blockade immunotherapy for the treatment of tumors. These strategies are mainly based on the mutual regulatory relationship between the metabolism of cancer and immune cells in the TME. We discussed the metabolic competition between tumor and immune cells in the TME, the differences in metabolic reprogramming between these cells, the different effects of metabolism on tumor, Teff and Treg cells, and the effect of targeting tumor glucose or glutamine metabolism in the TME on the tumor immune response. In future studies, the relationship between metabolic reprogramming and tumor immune escape should be fully considered to optimize therapy and avoid problems, such as off-target immunity, drug resistance, and possible metastasis, and the ineffectiveness of targeted metabolic therapy. The targeting of glucose metabolism and glutamine metabolism may directly regulate the expression of PD-L1 in tumor cells (36, 61), which modulate the effects of targeted metabolic therapy because it may cause tumor immune escape. Therefore, the combined use of targeted glucose or glutamine metabolic therapy and PD-L1 ICIs may induce a synergistic effect. However, the interaction between metabolism and tumor immune escape has additional effects. The intricate relationship between metabolism and immune escape reflects the difficulty of targeting the metabolic adaptations of tumor cells without affecting tumor clearance by Teff cells. Therefore, it is necessary to fully examine the metabolic mechanism of tumor immune escape, the metabolic requirements of immune cells, and the relationship between PD-1/PD-L1 immune checkpoints and metabolism to evaluate the impact of targeted glucose or glutamine metabolic therapy on immune checkpoints and the impact of immune checkpoint treatment on tumor metabolism. Designing a more reasonable combination treatment plan based on the metabolic crosstalk between tumor and immune cells can avoid treatment failure and increase the effectiveness of PD-1/PD-L1 checkpoint blockade immunotherapy in more cancers.
GM, CL, HN, and WS collected articles and designed the manuscript. GM, CL, and ZZ wrote the manuscript. YL and ZL prepared tables and figures. YC, LW, DL, and MZ revised and approved the manuscript. All authors contributed to the article and approved the submitted version.
This study was financially supported by the National Natural Science Foundation of China (82071750, 81772713, 81472411), Taishan Scholar Program of Shandong Province (tsqn20161077), Major Science and technology innovation project of Shandong Province (2019JZZY021002), Key projects of Qingdao Science and Technology Program (18-6-1-64-nsh), Research and Development Program of Shandong Province (2018GSF118197).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Holohan C, Van Schaeybroeck S, Longley DB, Johnston PG. Cancer Drug Resistance: An Evolving Paradigm. Nat Rev Cancer (2013) 13(10):714–26. doi: 10.1038/nrc3599
2. Advani SJ, Camargo MF, Seguin L, Mielgo A, Anand S, Hicks AM, et al. Kinase-Independent Role for CRAF-Driving Tumour Radioresistance via CHK2. Nat Commun (2015) 6:8154. doi: 10.1038/ncomms9154
3. Boumahdi S, de Sauvage FJ. The Great Escape: Tumour Cell Plasticity in Resistance to Targeted Therapy. Nat Rev Drug Discov (2020) 19(1):39–56. doi: 10.1038/s41573-019-0044-1
4. Muller WA. Localized Signals That Regulate Transendothelial Migration. Curr Opin Immunol (2016) 38:24–9. doi: 10.1016/j.coi.2015.10.006
5. Slaney CY, Kershaw MH, Darcy PK. Trafficking of T Cells Into Tumors. Cancer Res (2014) 74(24):7168. doi: 10.1158/0008-5472.CAN-14-2458
6. Topalian SL, Drake CG, Pardoll DM. Immune Checkpoint Blockade: A Common Denominator Approach to Cancer Therapy. Cancer Cell (2015) 27(4):450–61. doi: 10.1016/j.ccell.2015.03.001
7. Pauken KE, Wherry EJ. Overcoming T Cell Exhaustion in Infection and Cancer. Trends Immunol (2015) 36(4):265–76. doi: 10.1016/j.it.2015.02.008
8. Gunturi A, McDermott DF. Nivolumab for the Treatment of Cancer. Expert Opin Investig Drugs (2015) 24(2):253–60. doi: 10.1517/13543784.2015.991819
9. Khoja L, Butler MO, Kang SP, Ebbinghaus S, Joshua AM. Pembrolizumab. J Immunother Cancer (2015) 3:36. doi: 10.1186/s40425-015-0078-9
10. Wellenstein MD, Visser KED. Cancer-Cell-Intrinsic Mechanisms Shaping the Tumor Immune Landscape - ScienceDirect. Immunity (2018) 48(3):399–416. doi: 10.1016/j.immuni.2018.03.004
11. Coussens LM. Neutralizing Tumor-Promoting Chronic Inflammation: A Magic Bullet? Science (2013) 339(6127):286–91. doi: 10.1126/science.1232227
12. Herbst RS, Soria JC, Kowanetz M, Fine GD, Hamid O, Gordon MS, et al. Predictive Correlates of Response to the Anti-PD-L1 Antibody MPDL3280A in Cancer Patients. Nature (2014) 515(7528):563–7. doi: 10.1038/nature14011
13. Tumeh PC, Harview CL, Yearley JH, Shintaku IP, Taylor EJ, Robert L, et al. PD-1 Blockade Induces Responses by Inhibiting Adaptive Immune Resistance. Nature (2014) 515(7528):568–71. doi: 10.1038/nature13954
14. Faubert B, Solmonson A, DeBerardinis RJ. Metabolic Reprogramming and Cancer Progression. Science (2020) 368(6487):eaaw5473. doi: 10.1126/science.aaw5473
15. Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg Effect: The Metabolic Requirements of Cell Proliferation. Science (2009) 324(5930):1029–33. doi: 10.1126/science.1160809
16. McCarty MF, Whitaker J. Manipulating Tumor Acidification as a Cancer Treatment Strategy. Altern Med Rev (2010) 15(3):264–72. doi: 10.1111/j.1365-2036.2010.04338.x
17. Yoo HC, Park SJ, Nam M, Kang J, Kim K, Yeo JH, et al. A Variant of SLC1A5 Is a Mitochondrial Glutamine Transporter for Metabolic Reprogramming in Cancer Cells. Cell Metab (2020) 31(2):267–83.e12. doi: 10.1016/j.cmet.2019.11.020
18. Chang CH, Curtis JD, Maggi LB Jr., Faubert B, Villarino AV, O’Sullivan D, et al. Posttranscriptional Control of T Cell Effector Function by Aerobic Glycolysis. Cell (2013) 153(6):1239–51. doi: 10.1016/j.cell.2013.05.016
19. Siska PJ, Singer K, Evert K, Renner K. The Immunological Warburg Effect: Can a Metabolic-Tumor-Stroma Score (MeTS) Guide Cancer Immunotherapy? Immunol Rev (2020) 295(1):187–202. doi: 10.1111/imr.12846
20. Hensley CT, Faubert B, Yuan Q, Lev-Cohain N, Jin E, Kim J, et al. Metabolic Heterogeneity in Human Lung Tumors. Cell (2016) 164(4):681–94. doi: 10.1016/j.cell.2015.12.034
21. Courtney KD, Bezwada D, Mashimo T, Pichumani K, Vemireddy V, Funk AM, et al. Isotope Tracing of Human Clear Cell Renal Cell Carcinomas Demonstrates Suppressed Glucose Oxidation In Vivo. Cell Metab (2018) 28(5):793–800.e2. doi: 10.1016/j.cmet.2018.07.020
22. Brooks SA, Khandani AH, Fielding JR, Lin W, Sills T, Lee Y, et al. Alternate Metabolic Programs Define Regional Variation of Relevant Biological Features in Renal Cell Carcinoma Progression. Clin Cancer Res (2016) 22(12):2950–9. doi: 10.1158/1078-0432.ccr-15-2115
23. Daemen A, Peterson D, Sahu N, McCord R, Du X, Liu B, et al. Metabolite Profiling Stratifies Pancreatic Ductal Adenocarcinomas Into Subtypes With Distinct Sensitivities to Metabolic Inhibitors. Proc Natl Acad Sci USA (2015) 112(32):E4410–7. doi: 10.1073/pnas.1501605112
24. Jiang B. Aerobic Glycolysis and High Level of Lactate in Cancer Metabolism and Microenvironment. Genes Dis (2017) 4(1):25–7. doi: 10.1016/j.gendis.2017.02.003
25. Deberardinis RJ, Sayed N, Ditsworth D, Thompson CB. Brick by Brick: Metabolism and Tumor Cell Growth. Curr Opin Genet Dev (2008) 18(1):54–61. doi: 10.1016/j.gde.2008.02.003
26. Bonatelli M, Silva ECA, Cárcano FM, Zaia MG, Lopes LF, Scapulatempo-Neto C, et al. The Warburg Effect Is Associated With Tumor Aggressiveness in Testicular Germ Cell Tumors. Front Endocrinol (2019) 10:417. doi: 10.3389/fendo.2019.00417
27. Hanahan D, Weinberg RA. Hallmarks of Cancer: The Next Generation. Cell (2011) 144(5):646–74. doi: 10.1016/j.cell.2011.02.013
29. Hsu PP, Sabatini DM. Cancer Cell Metabolism: Warburg and Beyond. Cell (2008) 134(5):703–7. doi: 10.1016/j.cell.2008.08.021
30. Locasale JW, Cantley LC. Altered Metabolism in Cancer. BMC Biol (2010) 8:88. doi: 10.1186/1741-7007-8-88
31. Li X, Wenes M, Romero P, Huang SC. Navigating Metabolic Pathways to Enhance Antitumour Immunity and Immunotherapy. Nat Rev Clin Oncol (2019) 16: (7):425–41. doi: 10.1038/s41571-019-0203-7
32. Cerezo M, Rocchi S. Cancer Cell Metabolic Reprogramming: A Keystone for the Response to Immunotherapy. Cell Death Dis (2020) 11(11):964. doi: 10.1038/s41419-020-03175-5
33. Bader JE, Voss K, Rathmell JC. Targeting Metabolism to Improve the Tumor Microenvironment for Cancer Immunotherapy. Mol Cell (2020) 78(6):1019–33. doi: 10.1016/j.molcel.2020.05.034
34. Siska PJ, Beckermann KE, Mason FM, Andrejeva G, Greenplate AR, Sendor AB, et al. Mitochondrial Dysregulation and Glycolytic Insufficiency Functionally Impair CD8 T Cells Infiltrating Human Renal Cell Carcinoma. JCI Insight (2017) 2(12):1–13. doi: 10.1172/jci.insight.93411
35. Sullivan MR, Danai LV, Lewis CA. Quantification of Microenvironmental Metabolites in Murine Cancers Reveals Determinants of Tumor Nutrient Availability. Elife (2019) 8:e44235. doi: 10.7554/eLife.44235
36. Yu Y, Liang Y, Li D, Wang L, Liang Z, Chen Y, et al. Glucose Metabolism Involved in PD-L1-Mediated Immune Escape in the Malignant Kidney Tumour Microenvironment. Cell Death Discov (2021) 7(1):15. doi: 10.1038/s41420-021-00401-7
37. Cui Y, Li X, Du B, Diao Y, Li Y. PD-L1 in Lung Adenocarcinoma: Insights Into the Role of (18)F-FDG PET/CT. Cancer Manage Res (2020) 12:6385–95. doi: 10.2147/cmar.s256871
38. Palsson-McDermott EM, Dyck L, Zasłona Z, Menon D, McGettrick AF, Mills KHG, et al. Pyruvate Kinase M2 Is Required for the Expression of the Immune Checkpoint PD-L1 in Immune Cells and Tumors. Front Immunol (2017) 8:1300. doi: 10.3389/fimmu.2017.01300
39. Jiang Z, Liu Z, Li M, Chen C, Wang X. Increased Glycolysis Correlates With Elevated Immune Activity in Tumor Immune Microenvironment. EBioMedicine (2019) 42:431–42. doi: 10.1016/j.ebiom.2019.03.068
40. Freemerman AJ, Johnson AR, Sacks GN, Milner JJ, Kirk EL, Troester MA, et al. Metabolic Reprogramming of Macrophages: Glucose Transporter 1 (GLUT1)-Mediated Glucose Metabolism Drives a Proinflammatory Phenotype. J Biol Chem (2014) 289(11):7884–96. doi: 10.1074/jbc.M113.522037
41. Chang CH, Qiu J, O’Sullivan D, Buck MD, Noguchi T, Curtis JD, et al. Metabolic Competition in the Tumor Microenvironment Is a Driver of Cancer Progression. Cell (2015) 162(6):1229–41. doi: 10.1016/j.cell.2015.08.016
42. Ho PC, Bihuniak JD, Macintyre AN, Staron M, Liu X, Amezquita R, et al. Phosphoenolpyruvate Is a Metabolic Checkpoint of Anti-Tumor T Cell Responses. Cell (2015) 162(6):1217–28. doi: 10.1016/j.cell.2015.08.012
43. Lim S, Liu H, Madeira da Silva L, Arora R, Liu Z, Phillips JB, et al. Immunoregulatory Protein B7-H3 Reprograms Glucose Metabolism in Cancer Cells by ROS-Mediated Stabilization of HIF1α. Cancer Res (2016) 76(8):2231–42. doi: 10.1158/0008-5472.can-15-1538
44. Shi LZ, Wang R, Huang G, Vogel P, Neale G, Green DR, et al. HIF1?-Dependent Glycolytic Pathway Orchestrates a Metabolic Checkpoint for the Differentiation of TH17 and Treg Cells. J Exp Med (2011) 208(7):1367–76. doi: 10.1084/jem.20110278
45. Dang EV, Barbi J, Yang HY, Jinasena D, Yu H, Zheng Y, et al. Control of T(H)17/T(reg) Balance by Hypoxia-Inducible Factor 1. Cell (2011) 146(5):772–84. doi: 10.1016/j.cell.2011.07.033
46. Cham CM, Gajewski TF. Glucose Availability Regulates IFN-γ Production and P70s6 Kinase Activation in CD8+ Effector T Cells. J Immunol (2005) 174(8):4670–7. doi: 10.4049/jimmunol.174.8.4670
47. Cham CM, Driessens G, O’Keefe JP, Gajewski TF. Glucose Deprivation Inhibits Multiple Key Gene Expression Events and Effector Functions in CD8 T Cells. Eur J Immunol (2008) 38(9):2438–50. doi: 10.1002/eji.200838289
48. Blagih J, Coulombe F, Vincent EE, Dupuy F, Galicia-Vázquez G, Yurchenko E, et al. The Energy Sensor AMPK Regulates T Cell Metabolic Adaptation and Effector Responses In Vivo. Immunity (2015) 42(1):41–54. doi: 10.1016/j.immuni.2014.12.030
49. Zhao E, Maj T, Kryczek I, Li W, Wu K, Zhao L, et al. Cancer Mediates Effector T Cell Dysfunction by Targeting microRNAs and EZH2 Via Glycolysis Restriction. Nat Immunol (2016) 17(1):95–103. doi: 10.1038/ni.3313
50. Ippolito L, Morandi A, Giannoni E, Chiarugi P. Lactate: A Metabolic Driver in the Tumour Landscape. Trends Biochem Sci (2019) 44(2):153–66. doi: 10.1016/j.tibs.2018.10.011
51. Hui S, Ghergurovich JM, Morscher RJ, Jang C, Teng X, Lu W, et al. Glucose Feeds the TCA Cycle Via Circulating Lactate. Nature (2017) 551(7678):115–8. doi: 10.1038/nature24057
52. Fischer K, Hoffmann P, Voelkl S, Meidenbauer N, Ammer J, Edinger M, et al. Inhibitory Effect of Tumor Cell-Derived Lactic Acid on Human T Cells. Blood (2007) 109(9):3812–9. doi: 10.1182/blood-2006-07-035972
53. Brand A, Singer K, Koehl GE, Kolitzus M, Schoenhammer G, Thiel A, et al. LDHA-Associated Lactic Acid Production Blunts Tumor Immunosurveillance by T and NK Cells. Cell Metab (2016) 24(5):657–71. doi: 10.1016/j.cmet.2016.08.011
54. Mendler AN, Hu B, Prinz PU, Kreutz M, Gottfried E, Noessner E. Tumor Lactic Acidosis Suppresses CTL Function by Inhibition of P38 and JNK/c-Jun Activation. Int J Cancer (2012) 131(3):633–40. doi: 10.1002/ijc.26410
55. Johnston RJ, Su LJ, Pinckney J, Critton D, Boyer E, Krishnakumar A, et al. VISTA Is an Acidic pH-Selective Ligand for PSGL-1. Nature (2019) 574(7779):565–70. doi: 10.1038/s41586-019-1674-5
56. Daye D, Wellen KE. Metabolic Reprogramming in Cancer: Unraveling the Role of Glutamine in Tumorigenesis. Semin Cell Dev Biol (2012) 23(4):362–9. doi: 10.1016/j.semcdb.2012.02.002
57. Son J, Lyssiotis CA, Ying H, Wang X, Hua S, Ligorio M, et al. Glutamine Supports Pancreatic Cancer Growth Through a KRAS-Regulated Metabolic Pathway. Nature (2013) 496(7443):101–5. doi: 10.1038/nature12040
58. Zhang J, Pavlova NN, Thompson CB. Cancer Cell Metabolism: The Essential Role of the Nonessential Amino Acid, Glutamine. EMBO J (2017) 36: (10):1302–15. doi: 10.15252/embj.201696151
59. Medina MA, Sánchez-Jiménez F, Márquez J, Rodríguez Quesada A, Núñez de Castro I. Relevance of Glutamine Metabolism to Tumor Cell Growth. Mol Cell Biochem (1992) 113(1):1–15. doi: 10.1007/bf00230880
60. DeBerardinis RJ, Mancuso A, Daikhin E, Nissim I, Yudkoff M, Wehrli S, et al. Beyond Aerobic Glycolysis: Transformed Cells can Engage in Glutamine Metabolism That Exceeds the Requirement for Protein and Nucleotide Synthesis. Proc Natl Acad Sci USA (2007) 104(49):19345–50. doi: 10.1073/pnas.0709747104
61. Ma G, Liang Y, Chen Y, Wang L, Li D, Liang Z, et al. Glutamine Deprivation Induces PD-L1 Expression Via Activation of EGFR/ERK/c-Jun Signaling in Renal Cancer. Mol Cancer Res (2020) 18(2):324–39. doi: 10.1158/1541-7786.mcr-19-0517
62. Byun JK, Park M, Lee S, Yun JW, Lee J, Kim JS, et al. Inhibition of Glutamine Utilization Synergizes With Immune Checkpoint Inhibitor to Promote Antitumor Immunity. Mol Cell (2020) 80(4):592–606.e8. doi: 10.1016/j.molcel.2020.10.015
63. Fu Q, Xu L, Wang Y, Jiang Q, Liu Z, Zhang J, et al. Tumor-Associated Macrophage-Derived Interleukin-23 Interlinks Kidney Cancer Glutamine Addiction With Immune Evasion. Eur Urol (2019) 75(5):752–63. doi: 10.1016/j.eururo.2018.09.030
64. Oh MH, Sun IH, Zhao L, Leone RD, Sun IM, Xu W, et al. Targeting Glutamine Metabolism Enhances Tumor-Specific Immunity by Modulating Suppressive Myeloid Cells. J Clin Invest (2020) 130(7):3865–84. doi: 10.1172/jci131859
65. Wu WC, Sun HW. Immunosuppressive Immature Myeloid Cell Generation Is Controlled by Glutamine Metabolism in Human Cancer. Cancer Immunol Res (2019) 7: (10):1605–18. doi: 10.1158/2326-6066.cir-18-0902
66. Wang R, Dillon CP, Shi LZ, Milasta S, Carter R, Finkelstein D, et al. The Transcription Factor Myc Controls Metabolic Reprogramming Upon T Lymphocyte Activation. Immunity (2011) 35(6):871–82. doi: 10.1016/j.immuni.2011.09.021
67. Carr EL, Kelman A, Wu GS, Gopaul R, Senkevitch E, Aghvanyan A, et al. Glutamine Uptake and Metabolism Are Coordinately Regulated by ERK/MAPK During T Lymphocyte Activation. J Immunol (Baltimore Md: 1950) (2010) 185(2):1037–44. doi: 10.4049/jimmunol.0903586
68. Yang R, Li X, Wu Y, Zhang G, Liu X, Li Y, et al. EGFR Activates GDH1 Transcription to Promote Glutamine Metabolism Through MEK/ERK/ELK1 Pathway in Glioblastoma. Oncogene (2020) 39: (14):2975–86. doi: 10.1038/s41388-020-1199-2
69. Edwards DN, Ngwa VM, Raybuck AL, Wang S, Hwang Y, Kim LC, et al. Selective Glutamine Metabolism Inhibition in Tumor Cells Improves Antitumor T Lymphocyte Activity in Triple-Negative Breast Cancer. J Clin Invest (2021) 131(4):140100–15. doi: 10.1172/jci140100
70. Leone RD, Zhao L. Glutamine Blockade Induces Divergent Metabolic Programs to Overcome Tumor Immune Evasion. Science (2019) 366: (6468):1013–21. doi: 10.1126/science.aav2588
71. Jenkins RW, Barbie DA, Flaherty KT. Mechanisms of Resistance to Immune Checkpoint Inhibitors. Br J Cancer (2018) 118(1):9–16. doi: 10.1038/bjc.2017.434
72. DeWaal D, Nogueira V, Terry AR. Hexokinase-2 Depletion Inhibits Glycolysis and Induces Oxidative Phosphorylation in Hepatocellular Carcinoma and Sensitizes to Metformin. Nat Commun (2018) 9: (1):446. doi: 10.1038/s41467-017-02733-4
73. Brooke DG, van Dam EM, Watts CK, Khoury A, Dziadek MA, Brooks H, et al. Targeting the Warburg Effect in Cancer; Relationships for 2-Arylpyridazinones as Inhibitors of the Key Glycolytic Enzyme 6-Phosphofructo-2-Kinase/2,6-Bisphosphatase 3 (PFKFB3). Bioorg Med Chem (2014) 22(3):1029–39. doi: 10.1016/j.bmc.2013.12.041
74. Doherty JR, Cleveland JL. Targeting Lactate Metabolism for Cancer Therapeutics. J Clin Invest (2013) 123(9):3685–92. doi: 10.1172/jci69741
75. Scharping NE, Menk AV, Whetstone RD, Zeng X, Delgoffe GM. Efficacy of PD-1 Blockade Is Potentiated by Metformin-Induced Reduction of Tumor Hypoxia. Cancer Immunol Res (2017) 5(1):9–16. doi: 10.1158/2326-6066.cir-16-0103
76. Li FL, Liu JP, Bao RX, Yan G, Feng X, Xu YP, et al. Acetylation Accumulates PFKFB3 in Cytoplasm to Promote Glycolysis and Protects Cells From Cisplatin-Induced Apoptosis. Nat Commun (2018) 9(1):508. doi: 10.1038/s41467-018-02950-5
77. Li HM, Yang JG, Liu ZJ, Wang WM, Yu ZL, Ren JG, et al. Blockage of Glycolysis by Targeting PFKFB3 Suppresses Tumor Growth and Metastasis in Head and Neck Squamous Cell Carcinoma. J Exp Clin Cancer Res (2017) 36(1):7. doi: 10.1186/s13046-016-0481-1
78. Chesney JA, Telang S, Yaddanapudi K, Grewal JS. Targeting 6-Phosphofructo-2-Kinase (PFKFB3) as an Immunotherapeutic Strategy. J Clin Oncol (2016) 34(15_suppl):e14548–e. doi: 10.1200/JCO.2016.34.15_suppl.e14548
79. Yang M, Ma C, Liu S, Shao Q, Gao W, Song B, et al. HIF-Dependent Induction of Adenosine Receptor A2b Skews Human Dendritic Cells to a Th2-Stimulating Phenotype Under Hypoxia. Immunol Cell Biol (2010) 88(2):165–71. doi: 10.1038/icb.2009.77
80. Deck LM, Royer RE, Chamblee BB, Hernandez VM, Malone RR, Torres JE, et al. Selective Inhibitors of Human Lactate Dehydrogenases and Lactate Dehydrogenase From the Malarial Parasite Plasmodium Falciparum. J Med Chem (1998) 41(20):3879–87. doi: 10.1021/jm980334n
81. Weide B, Martens A, Hassel JC, Berking C, Postow MA, Bisschop K, et al. Baseline Biomarkers for Outcome of Melanoma Patients Treated With Pembrolizumab. Clin Cancer Res (2016) 22(22):5487–96. doi: 10.1158/1078-0432.ccr-16-0127
82. Bonuccelli G, Whitaker-Menezes D, Castello-Cros R, Pavlides S, Pestell RG, Fatatis A, et al. The Reverse Warburg Effect: Glycolysis Inhibitors Prevent the Tumor Promoting Effects of Caveolin-1 Deficient Cancer Associated Fibroblasts. Cell Cycle (2010) 9(10):1960–71. doi: 10.4161/cc.9.10.11601
83. Kouidhi S, Ben Ayed F, Benammar Elgaaied A. Targeting Tumor Metabolism: A New Challenge to Improve Immunotherapy. Front Immunol (2018) 9:353. doi: 10.3389/fimmu.2018.00353
84. Sukumar M, Liu J, Ji Y, Subramanian M, Crompton JG, Yu Z, et al. Inhibiting Glycolytic Metabolism Enhances CD8+ T Cell Memory and Antitumor Function. J Clin Invest (2013) 123(10):4479–88. doi: 10.1172/jci69589
85. Zhang D, Li J, Wang F, Hu J, Wang S, Sun Y. 2-Deoxy-D-Glucose Targeting of Glucose Metabolism in Cancer Cells as a Potential Therapy. Cancer Lett (2014) 355(2):176–83. doi: 10.1016/j.canlet.2014.09.003
86. Wang H, Mooney DJ. Metabolic Glycan Labelling for Cancer-Targeted Therapy. Cancer Lett (2020) 12: (12):1102–14. doi: 10.1038/s41557-020-00587-w
87. Biancur DE, Paulo JA. Compensatory Metabolic Networks in Pancreatic Cancers Upon Perturbation of Glutamine Metabolism. Nat Commun (2017) 8:15965. doi: 10.1038/ncomms15965
88. Schulte ML, Fu A, Zhao P, Li J, Geng L, Smith ST, et al. Pharmacological Blockade of ASCT2-Dependent Glutamine Transport Leads to Antitumor Efficacy in Preclinical Models. Nat Med (2018) 24: (2):194–202. doi: 10.1038/nm.4464
89. Jin L, Li D, Alesi GN, Fan J, Kang HB, Lu Z, et al. Glutamate Dehydrogenase 1 Signals Through Antioxidant Glutathione Peroxidase 1 to Regulate Redox Homeostasis and Tumor Growth. Cancer Cell (2015) 27(2):257–70. doi: 10.1016/j.ccell.2014.12.006
90. Johnson MO, Wolf MM, Madden MZ, Andrejeva G, Sugiura A, Contreras DC, et al. Distinct Regulation of Th17 and Th1 Cell Differentiation by Glutaminase-Dependent Metabolism. Cell (2018) 175(7):1780–95.e19. doi: 10.1016/j.cell.2018.10.001
91. Wettersten HI, Hakimi AA, Morin D, Bianchi C, Johnstone ME, Donohoe DR, et al. Grade-Dependent Metabolic Reprogramming in Kidney Cancer Revealed by Combined Proteomics and Metabolomics Analysis. Cancer Res (2015) 75(12):2541–52. doi: 10.1158/0008-5472.can-14-1703
Keywords: immunotherapy, PD-1/PD-L1 immune checkpoint, glucose metabolism, glutamine metabolism, combination therapy, tumor microenvironment
Citation: Ma G, Li C, Zhang Z, Liang Y, Liang Z, Chen Y, Wang L, Li D, Zeng M, Shan W and Niu H (2021) Targeted Glucose or Glutamine Metabolic Therapy Combined With PD-1/PD-L1 Checkpoint Blockade Immunotherapy for the Treatment of Tumors - Mechanisms and Strategies. Front. Oncol. 11:697894. doi: 10.3389/fonc.2021.697894
Received: 20 April 2021; Accepted: 21 June 2021;
Published: 13 July 2021.
Edited by:
Céline Pinheiro, Barretos Cancer Hospital, BrazilReviewed by:
Peter J. Siska, University Medical Center Regensburg, GermanyCopyright © 2021 Ma, Li, Zhang, Liang, Liang, Chen, Wang, Li, Zeng, Shan and Niu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Wenhong Shan, c3doeHNndHRAMTI2LmNvbQ==; Haitao Niu, bml1aHQwNTMyQDEyNi5jb20=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.