
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Oncol., 09 August 2021
Sec. Radiation Oncology
Volume 11 - 2021 | https://doi.org/10.3389/fonc.2021.697308
This article is part of the Research TopicImpact of Radiotherapies in Pancreatic CancerView all 6 articles
 Bin Hu1†
Bin Hu1† Xiumei Ma1†Renhua Huang1†Zheng Wu1Jun Lu2
Xiumei Ma1†Renhua Huang1†Zheng Wu1Jun Lu2 Yuntao Guo3Jianmin Tang1Chunhui Ma4Jun Ma5*
Yuntao Guo3Jianmin Tang1Chunhui Ma4Jun Ma5* Lei Zhang1*Yongrui Bai1*
Lei Zhang1*Yongrui Bai1*Background: Pancreatic cancer (PC) is one of the most lethal human cancers, and radiation therapy (RT) is an important treating option. Many patients diagnosed with PC do not achieve objective responses because of the existence of intrinsic and acquired radioresistance. Therefore, biomarkers, which predict radiotherapy benefit in PC, are eagerly needed to be identified.
Methods: Whole-exome sequencing of six pancreatic ductal adenocarcinoma patients (PDAC) (three with a good response and three with a poor response) who had received radical surgery and then radiotherapy has been performed as standard of care treatment. Somatic and germline variants and the mutational signatures were analyzed with bioinformatics tools and public databases. Functional enrichment and pathway-based protein-protein interaction analyses were utilized to address the possibly mechanism in radioresistance. MTT, LDH, and colony formation assay were applied to evaluate cell growth and colony formation ability.
Results: In the present study, somatic mutations located in 441 genes were detected to be radiosensitivity-related loci. Seventeen genes, including the Smad protein family members (SMAD3 and SMAD4), were identified to influence the radiosensitivity in PDAC. The SMAD3 and SMAD4 genes mutate differently between radiosensitive and radioresistant PDAC patients. Mutation of SMAD3 potentiates the effects of ionizing radiation (IR) on cell growth and colony formation in PDAC cells, whereas mutation of SMAD4 had the opposite effects. SMAD3 and SMAD4 regulate the radiosensitivity of PDAC, at least in part, by P21 and FOXO3a, respectively.
Conclusions: These results indicate that mutations of SMAD3 and SMAD4 likely cause the difference of response to radiotherapy in PDAC, which might be considered as the biomarkers and potential targets for the radiotherapy of pancreatic cancer.
Pancreatic cancer (PC) is the fourth leading cause of tumor-related deaths presently, which is predicted to be the second most fatal cancer in 2030 (1, 2). In recent years, some treatment methods have been developed, and the comprehension for cancer biology is deepened. The survival rates of patients with cancer are improved by many new treatment modalities, such as targeted therapy, immunotherapy, and advanced radiological techniques. However, the prognosis of pancreatic cancer remains poor. Its overall survival rate is only around 10% for 1 year and less than 5% for 5 years (3). Pancreatic cancer patients that can be treated with surgical management only accounts for less than 20%. Therefore, the main treatment options for PC are still chemotherapy (CT) and radiation therapy (RT) (4). So far, 40% of cured patients has received radiotherapy (5). However, many PC patients failed to obtain objective therapeutic effect because of inherent or acquired radiation resistance.
Previous reports have proved that the efficiency of radiation therapy could be affected by a series of alterations concerning the tumor itself and the surrounding microenvironment, such as inhibition of apoptosis, increased DNA repair, inflammation, and hypoxia (6). Furthermore, lots of pathways, such as PI3K/AKT (7), Notch (8), Foxo (9), ATM/ATR (10), MEK/ERK (11), TGFβ (12, 13), Wnt (14), and genes, such as SMAD4 (15), MUC1 (16), RAD54 (17), have been identified to regulate the endogenous signals and contribute to resistance to radiation therapy (RI) in some cancers. Nevertheless, the molecular mechanisms of radioresistance in PC remain to be unravelled and in urgent need of further exploration.
To identify the critical mutations of affecting radiotherapy benefit in PC, whole exome sequencing was utilized in six pancreatic ductal adenocarcinoma (PDAC) patients (three with a good response and three with a poor response) who had received radical surgery. We aimed at finding novel therapeutic response predictive biomarkers and providing the basis for developing novel radiosensitizers.
Tumor and blood samples were collected from six Chinese patients diagnosed with PC. Briefly, radical surgery (total or partial gastrectomy) was performed on all six patients and the tumor tissues and blood samples were collected for analyzes. After resection, all patients received postoperative adjuvant chemotherapy (gemcitabine+tegafur, gimeracil, and oteracil porassium capsules), and then radiotherapy was further utilized for therapy. After radiotherapy, the patients continued to receive chemotherapy (tegafur, gimeracil, and oteracil porassium capsules). The original clinical and survival data of patients are shown in Table 1. We divided the patients into two group based on their responses to radiotherapy. Patients with no response to radiotherapy were considered as the radioresistant group (group N), and patients with complete response were considered as the sensitive group (group S). The studies were approved by the ethical committee of Ren Ji Hospital, Shanghai Jiao Tong University School of Medicine, and all patients signed informed consent in advance of their recruitment.
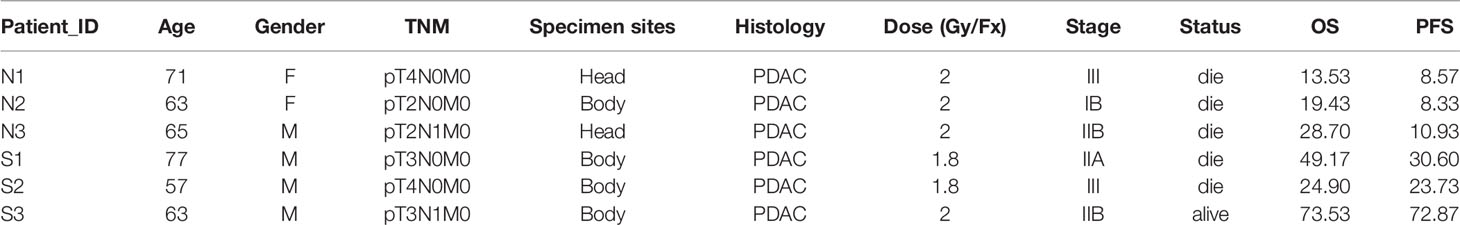
Table 1 Clinicopathological Characteristics of Six Primary Pancreatic Tumor Samples.
GenRead™ DNA FFPE Kit (Qiagen; Germany) was used to extract genomic DNA from formalin-fixed paraffin-embedded tissues and paired normal blood DNA. Then the genomic DNA was fragmented and hybridized following instructions on manufacturer’s protocol. These two methods were combined to verify the quality of isolated genomic DNA: 1) measuring DNA concentration using Qubit® DNA Assay Kit in Qubit® 2.0 Flurometer (Invitrogen, USA); 2) monitoring DNA degradation and contamination using 1% agarose gels. The Agilent SureSelect Clinical Research Exome Version 2 Probe set kit was applied to perform exome capture. Then, the Illumina HiSeq X platform was used to sequence the captured DNA as instructed in the manufacturer’s protocol, with a paired-end run of 2 × 151 bp in Nantong ZhongKe Medical Laboratory. HCS (HiSeq Control Software v2.2) and RTA (Real Time Analysis. v1.18) were used to analyze initial alignment and quality control. Illumina package bcl2fastq (v1.8.4) was applied to convert the binary base call (BCL) to a FASTQ format. Related sequencing data in this article have been uploaded to the GenBank Data Libraries with accession number as PRJNA684940.
Short reads (Raw data) in FASTQ format were obtained from fluorescence images on Hiseq platform by base calling. Fastp was used for quality control by discarding low quality paired reads, including reads that contain adapter contamination or more than 10% of uncertain bases in either one read or over 50% low-quality bases in either one read or less than 20 bases after trimming (18). Mapping results were obtained using Burrows-Wheeler Aligner (BWA) and stored in BAM format (19, 20). PCR duplicates of mapped reads were removed with Sambamba (21).
Germline mutations, including single-nucleotide polymorphisms (SNPs) and small insertions and deletions (InDels) of each exome, were detected using the HaplotypeCaller tool of Genome Analysis Toolkit (GATK) software following the protocol (22). To avoid false positives, these criteria were applied to filter.vcf outputs: SNP: “QD< 2.0 || FS > 60.0 || MQ< 40.0 || MQRankSum< -12.5 || ReadPosRankSum< -8.0”, InDel: “QD< 2.0 || FS > 200.0 || ReadPosRankSum< -20.0”. Annotation of filtered SNPs was conducted using ANNOVAR (23) and snpEFF (24). Only mutations that met the following criteria were considered for further analysis (1): the mutation locates in the exon or its flank region ( ± 2bp) (2); its allele frequency is less than 0.01 in GnomAD, 1,000 genomes and in house database (25, 26) (3); it is not a synonymous mutation (4); more than two softwares of SIFT, Polyphen2HDIV, Polyphen2HVAR, MutationTaster2, predict it to be harmful (27–30) (5). Its host gene is predicted to be associated with PC by phenolyzer software (31).
Mutect2 was used to detect somatic mutations, including single-nucleotide variants (SNVs), small insertions and deletions (InDels) (32). Filter Mutect Calls, a tool of GATK, was employed to assess the qualities of all potential variants and only the site with tag “PASS” in the “FILTER” filed were remained. With Vcf2maf (https://github.com/mskcc/vcf2maf) and VEP, vcf files were converted maf files and annotated somatic mutations (33). Cnvkit was used to identify the copy number variants (CNVs) (34).
The relative weights of mutational signatures of in each sample were calculated according to all somatic SNVs. The R package “deconstructSigs,” built on the basis of mutational signatures in COSMIC (http://cancer.sanger.ac.uk/cosmic/signatures), was applied to quantify the contribution of each signature for each tumor (35). The microsatellite instability (MSI) score was calculated withmsisensor2 (36).
The ABSOLUTE algorithm (37) was employed to assess the tumor purity and ploidy of all samples with the default parameters, which took segmented copy-number and MAF profiles as input. The dNdScv R package (38), which is a group of maximum-likelihood dN/dS methods, was used to quantify selection in cancer and somatic evolution in this study.
BxPC-3 cells were cultured in 1640 medium supplemented with 10% fetal bovine serum (FBS) (Gibco, Thermo Fisher Scientific, Inc, Waltham, MA, USA) and maintained in a humidified 5% CO2 atmosphere at 37˚C. WT (wild type) or Mut (a specific mutation) plasmids of SMAD3 and SMAD4 were purchased from Genechem (Shanghai, China). Briefly, the mutation plasmids were constructed as the mutation information found in the sample (exon2: c.220C>T for SMAD3 gene and exon9: c.1138A>T for SMAD4 gene) by chemical synthesis. To reduce the interference of endogenous expression of SMAD3 or SMAD4, lentiviruses containing SMAD3 or SMAD4 shRNA, purchased from Genechem, were first utilized to knockdown their expression before WT and Mut plasmids transfection in BxPC-3 cells. The siRNAs targeting P21 or FOXO3a were also purchased from Genechem (Shanghai, China).
BxPC-3 cells transfected with WT or Mut plasmid were seeded in 96-well plates. We then utilized Cell Counting Kit-8 (CCK-8) from MedChemExpress (Madison, WI, USA) and LDH assay kit (Promega, Madison, WI) to examine cell viability and cytotoxicity detection as the manufacturer’s instructions, respectively. The results were normalized against the Mock group from three independent experiments done in triplicate.
Cell colony formation detection was performed in a 6-well plate. Cells in different groups were seeded at optimal density. Fresh medium was changed every three days. After two weeks, we fixed the cells with methanol and then stained them with crystal violet. The pictures were photographed by a digital scanner. The surviving fraction (SF) was represented by calculating a ratio of the number of colonies formed and the product of the number of cells plated and the plating efficiency.
Protein extraction, protein determination, Western blot, and real-time PCR were performed as described previously (39).
Correlation between mutations and clinicohistological variables like overall survival time was measured by statistical R/Bioconductor packages. The significance of differences in data between the groups was determined by the Student’s t-test and Fisher’s exact test. With the clusterProfiler package, we performed KEGG pathway enrichment analyses for the significantly genes (40). Statistical significance was set as a P value of 0.05 or less.
Six cases of pancreatic ductal adenocarcinoma (PDAC) patients were involved in this study whose TNM stage ranges from T2 to T4. In our cohort, the mean age of PDAC patients was 66 years (range, 57–77 years; SD, 7.0) and four patients were male (66.7%). With a median follow-up of 17.3 months (SD 24.8), five patients had a dead status (83.3%). According to the response to radiotherapy with a dose of 1.8 to 2, three patients, with the overall survival was beyond 23 months, were considered as a good prognosis, whereas the other three patients were considered as a poor prognosis and died in 11 months (detail in Table 1).
Tumor tissues and paired blood samples of six individuals were collected, respectively to conduct the whole exome sequencing. After removing the low-quality, too short and adaptor containing reads, about 48.5 G bases per library were obtained, and the sequence depth ranged from 330X to 533X. Otherwise, the bases and depth of paired samples were 15.4 G and 149X, averagely. Then, all reads were mapped to the UCSC Homo sapiens genome (version 19), with the total mapping rate more than 99.5% in all samples. Clean data have the Q30 value more than 90% in our research and fits the need of following identification of somatic mutations (details in Supplemental Material S1).
A total of 2,380 somatic variations (median, 403.5; range, 74–735; SD, 241.3) were identified in the six pancreatic carcinomas samples, including 1,989 single nucleotide polymorphisms and 391 small insertions and deletions (Figure 1A). As expected, most variations are located in the exonic region (35.03%) and intronic area (44.33%) of the genomes (Figure 1B). We also found that cytosine (C) to thymine (T) transitions were the predominant nucleotide changes (Figure 1C). The somatic mutation profiles were analyzed to address the possible mechanism in the occurring and progressing of PDAC (Figure 1D). Signature 1, age, has the high weight in all six individuals, and signature 3 enriched in insensitive group members (COSMIC Signatures version 2).
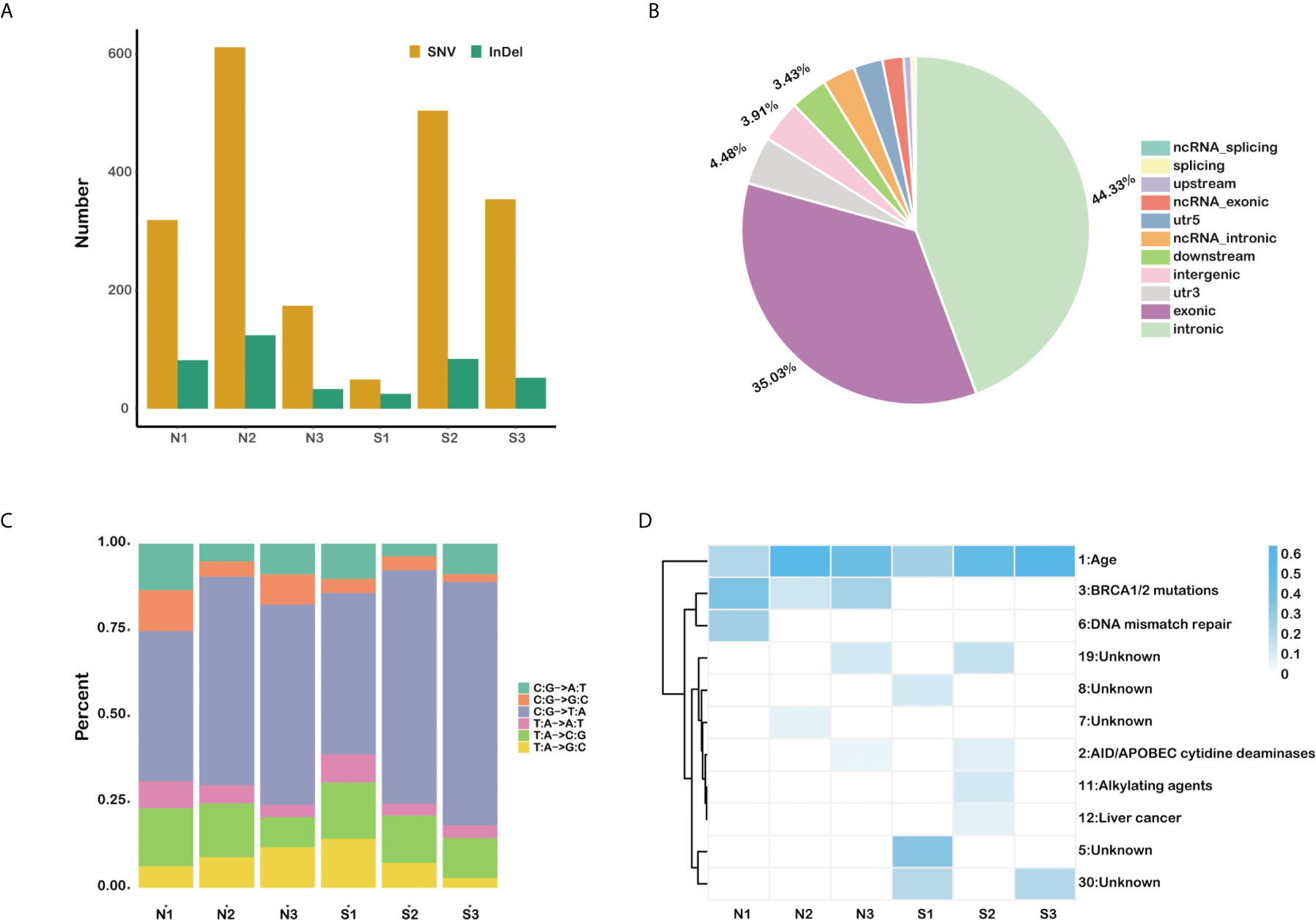
Figure 1 The somatic mutation profiles of six primary pancreatic tumor samples. (A) Total number of SNVs and InDels detected in each sample. (B) The statistics of somatic mutations in the genome. (C) The distribution of mutation type for each sample. (D) The mutation signature distribution based on COSMIC signature database.
It is known to all that tumor mutational burden and microsatellite instability were closely related with the response to immunotherapy in various types of cancers. To determine whether there are also correlations between tumor mutational burden and radiosensitivity in PDAC patients, we examined the number of all mutations in the target region as tumor mutation burden in this tumor type. As shown in Table 2, our results showed that there was no significant correlation between overall survival time (OS) and MSI (p=0.9194). Otherwise, the TMB was negatively correlated with the OS with a coefficient of -0.66 (p=0.175). To determine whether radiosensitivity was correlated with clonality, the purity and copy number alterations were then analyzed in this study. Our results indicated that there was no obvious association between OS and purity or ploidy (r = 0.029424 and r = 0.2, respectively) (Table 2). These results showed that the overall survival is not significantly correlated with the number of missense mutations, the instability of microsatellite, and clonality in PDAC patients treated with radiotherapy. The possibly influencing factors, age and tumor stage, were also analyzed (Table 2). However, the results showed that all these factors are not the core factors causing radioresistance in PDAC.
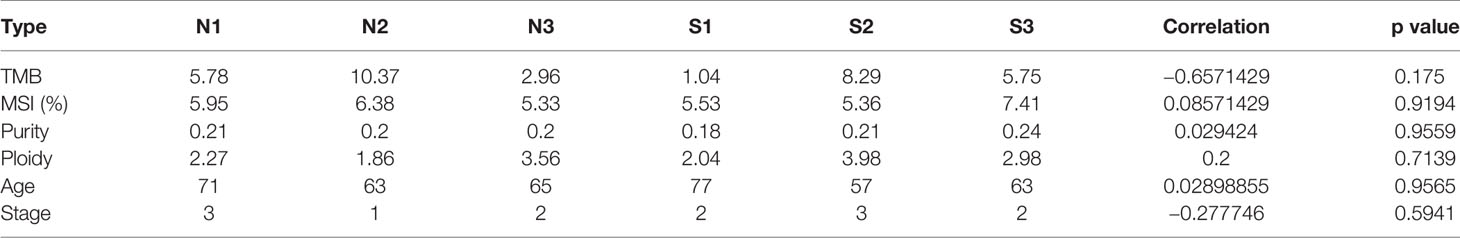
Table 2 Difference of TMB, MSI, and clonality between sensitive and insensitive groups.
Somatic mutations were filtered according to the criterions mentioned in the method to expose the candidate deleterious sites. 459 in 2301 variants remained, which is located in 441 genes, such as TP53, KRAS, PTEN, and so on (Figure 2A, detail in Supplemental Material S2). We next counted the number of variants for each gene in sensitive and insensitive members. Among them, 434 genes were only mutated in group N or group S, which were defined to be radiosensitivity-related genes. To address the possibly functions that these radiosensitivity-related genes took part in, Gene Ontology and KEGG enrichment analysis were conducted (Figure 2B). The most significant GO terms contained response to extracellular stimulus, mitotic cell cycle checkpoint, DNA catabolic process, and so on (detail in Supplemental Material S3). Pathway terms, like cell cycle, TGF-beta signaling pathway, and FoxO signaling pathway, were significantly enriched in our study, which might be closely related with radiosensitivity in PDAC (detail in Supplemental Material S4). As shown in Figures 2C, E, no significant differences on total number of genes and variation were observed between group N and group S. There are eight common genes in both groups, as well as 258 and 175 unique genes belonged to the group N and group S, respectively (Figure 2D). Moreover, two common mutations were shared in both groups, but 270 and 187 unique mutations belonged to the group N and group S, respectively (Figure 2F). Germline mutations were also analyzed in our study. All the mutations were filtered by the procedures mentioned in the method, and 754 sites in 639 genes remained (Details in Supplemental Material S5).
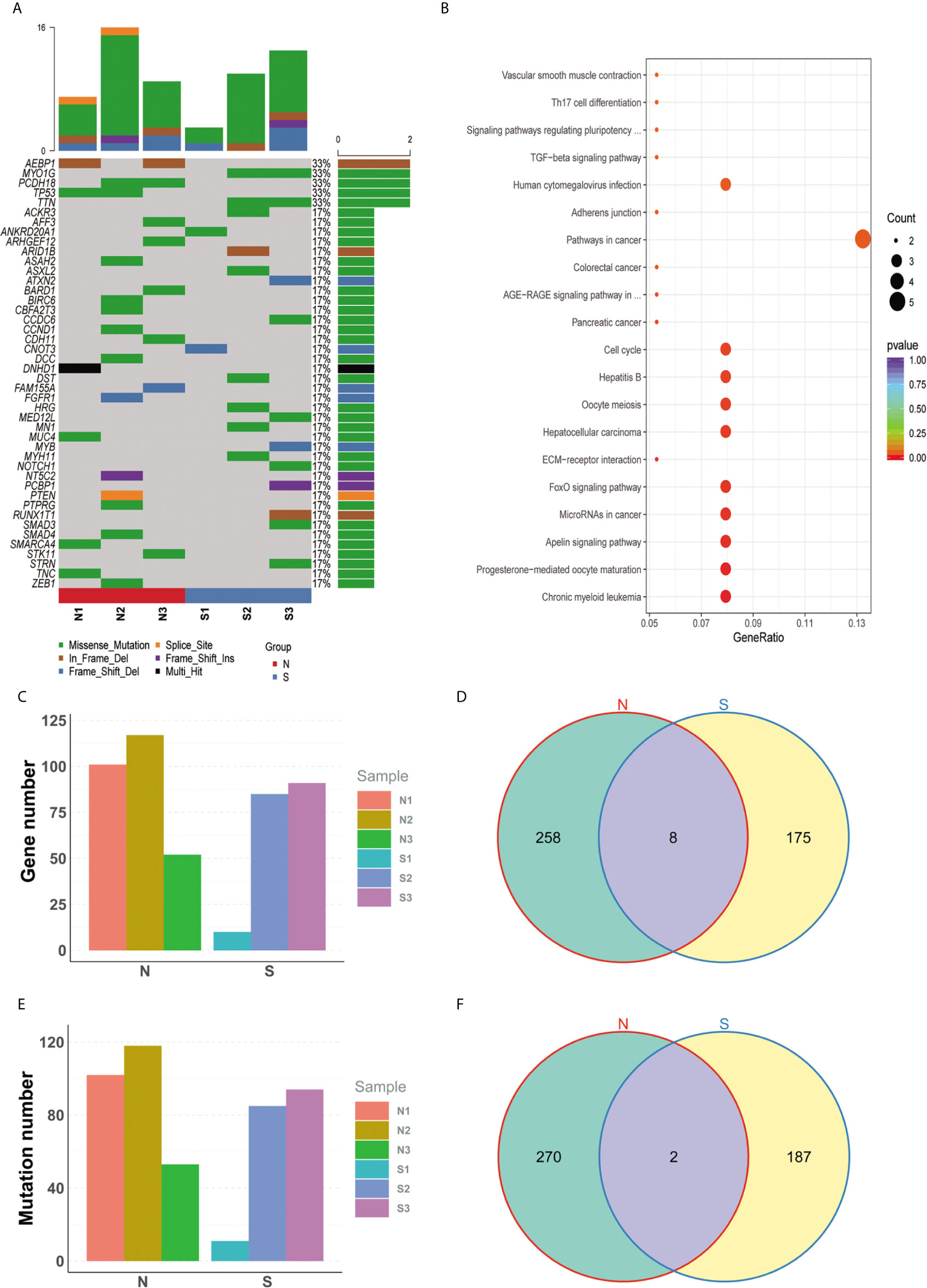
Figure 2 Specific mutated genes between group N and group S. (A) The oncoprint plot of all somatic mutations. (B) The bubble chart of kegg terms enriched for all diff-mutated genes. (C) No significant difference on total number of genes was observed between group N group and group S. (D) There are 8 common genes in both groups, as well as 258 and 175 unique genes belonged to the group N and group S, respectively. (E) No significant difference on total number of variations was observed between group N and group S. (F) 2 common mutations were shared in both groups, but 270 and 187 unique mutations belonged to the group N and group S, respectively.
Based on previous studies, 30 RI-related pathways were selected, including cell cycle, homologous recombination, nucleotide excision repair, and so on. Combined with the protein-protein interactions, a pathway-based protein-protein interaction (PPI) network was constructed with the RI-related genes (Table 3). Seventeen genes were involved. SMAD3, SMAD4, ARID1B, CRKL, and CASP10 played a center role in the network (Figure 3A). Moreover, the results of qPCR showed that the mRNA expression of these five genes was not affected by ionizing radiation in BxPC-3 cells (Figure 3B). It is worth noting that both SMAD3 and SMAD4 belong to the SMAD proteins family, which is a group of intracellular signal transducer proteins. The synonymous mutations of somatic mutations are listed in Supplemental Material S6.
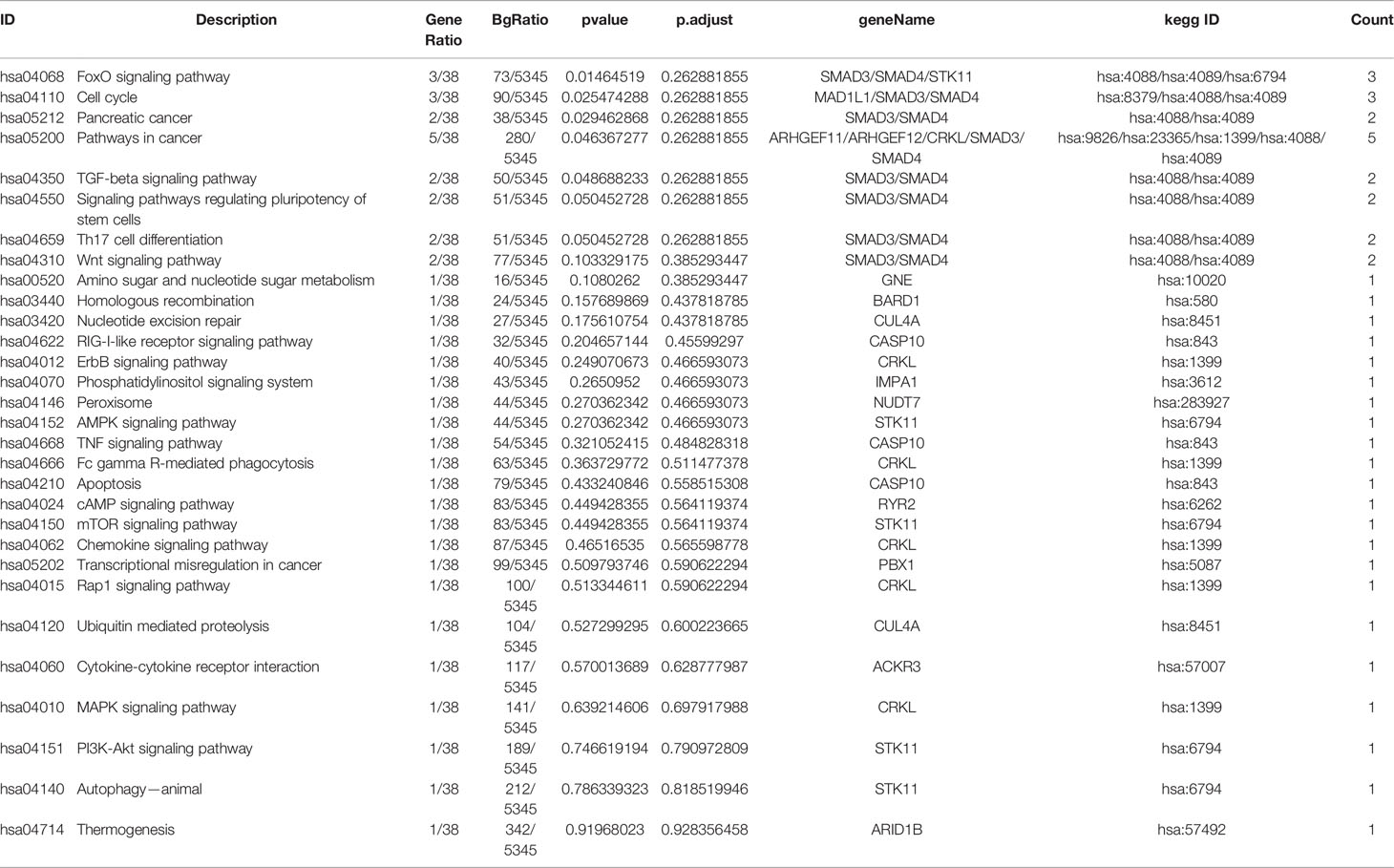
Table 3 RI Related Pathways of Somatic Mutation Related Genes.
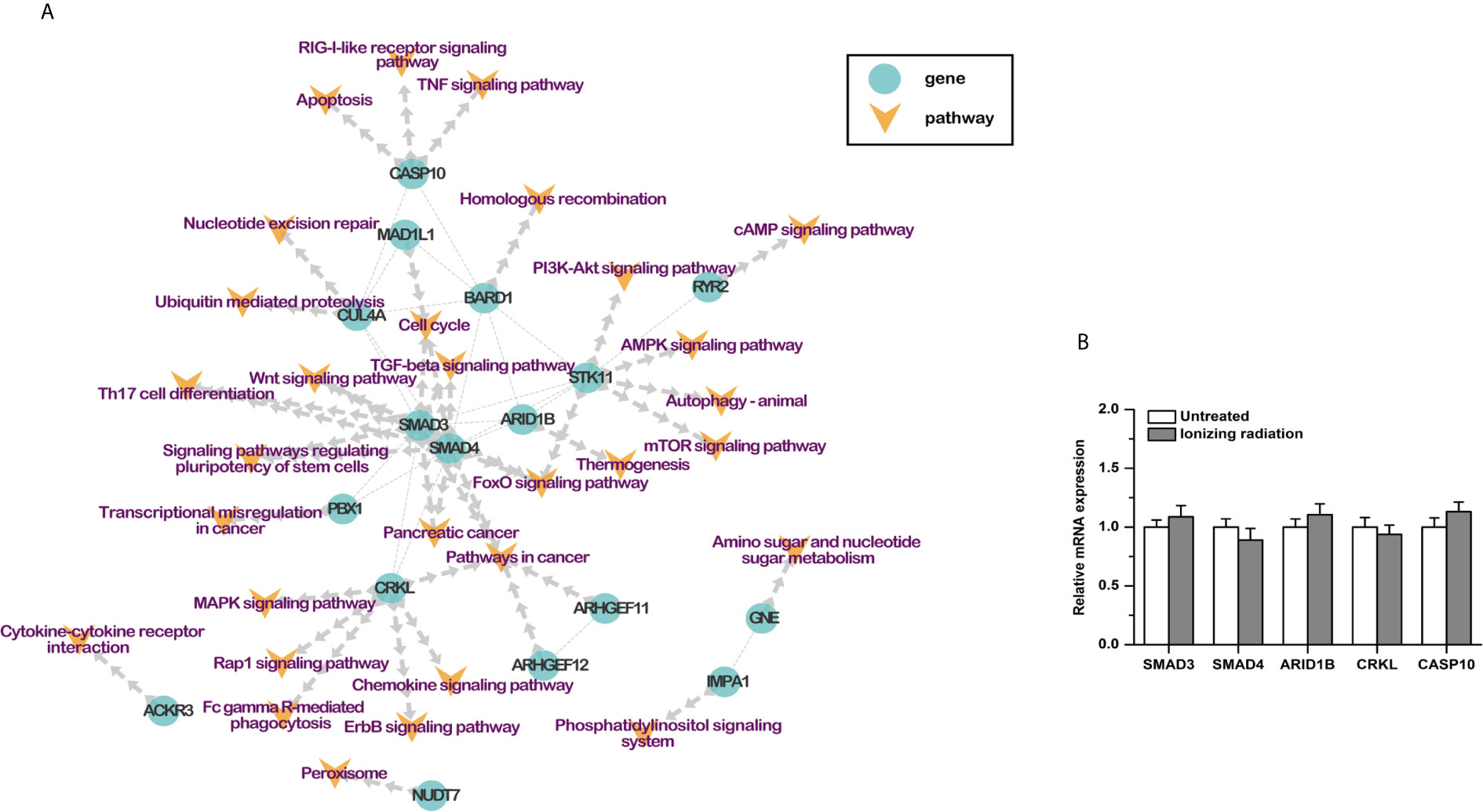
Figure 3 (A) Protein-protein interaction network of RI-related genes. (B) The mRNA expression of SMAD3, SMAD4, ARID1B, CRKL, and CASP10 was not affected by ionizing radiation (4Gy) in PDAC cells.
The ectopic wild-type (WT; without the variation) and mutant (Mut; with the variation) SMAD3 or SMAD4 cells were constructed in BxPC-3 cells (a PDAC cell line) to determine whether the mutation of SMAD3 or SMAD4 affected the radiosensitivity. As shown in Figures 4A, B, results of qPCR showed that there were no obvious differences on the expression of SMAD3 or SMAD4 between the WT and Mut cells, respectively. We then examined cell survival and death after ionizing radiation (IR) by utilizing CCK-8 and LDH release assay. The results showed that transfection of WT SMAD3 repressed the decreases of cell viability and the increase of LDH release induced by ionizing radiation (IR, 4Gy), whereas mutation of SMAD3 facilitated the effects of IR on cell survival and death (Figures 4C, D). Moreover, transfection of SMAD3 mutation significantly promoted IR-induced decreases of colony formation and surviving fraction in PDAC cells compared with the WT group (Figures 4E, F). On the contrary, the effects of IR on cell survival, cell death, colony formation, and surviving fraction were inhibited by the mutation of SMAD4 compared with the WT group (Figures 4G–J). These results indicate that SMAD3 mutation or SMAD4 mutation leads to the different response to radiotherapy in PDAC.
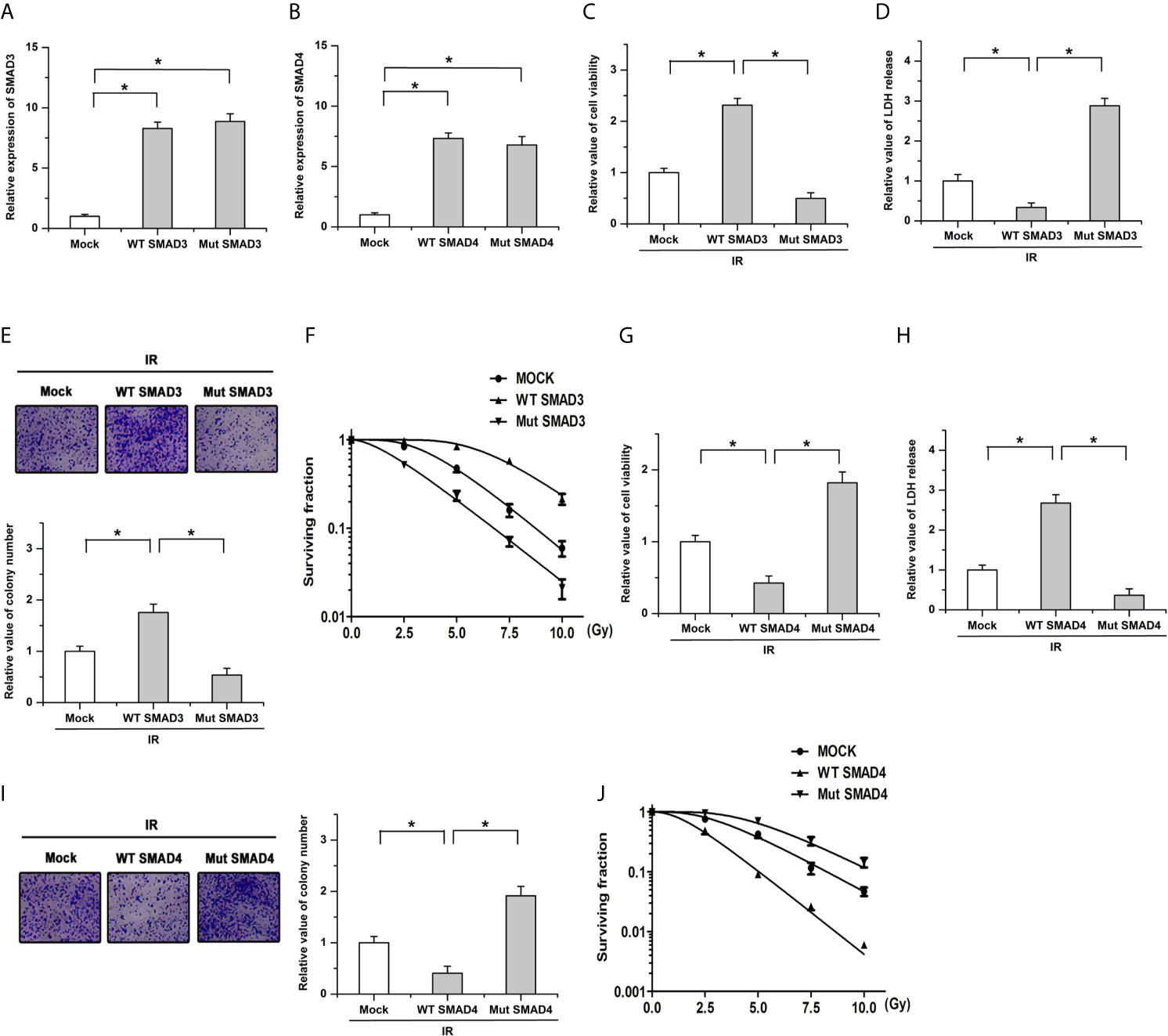
Figure 4 The radiosensitivity is affected by the mutation of SMAD3 or SMAD4 in PDAC. (A, B) Results of qPCR showed that there were no obvious differences on the expression of SMAD3 (A) or SMAD4 (B) between the WT and Mut cells, respectively. (C, D) The regulatory effects of IR on cell viability (C) and LDH release (D) were facilitated by the SMAD3 mutation. (E, F) The decreases of colony formation (E) and surviving fraction (F) induced by IR were enhanced by the mutation of SMAD3 compared with the WT group. (G, H) SMAD4 mutation mitigated the effects of IR on cell survival (G) and LDH release (H). (I, J) The decrease of colony formation (I) and surviving fraction (J) induced by IR were antagonized by SMAD4 mutation compared with the WT group. Cells infected with the empty vector were defined as the mock group. *P < 0.05.
Previous studies have indicated that SMAD3 directly binds to the promoter of cyclin-dependent kinase inhibitor 1 (P21) and SMAD4 could regulate the activity and expression of FOXO3a by forming a complex (41, 42). Based on the protein-protein interaction (PPI) network, SMAD3 and SMAD4 could regulate cell cycle and the FOXO signaling pathway, respectively. We then determined whether SMAD3 and SMAD4 regulated the radiosensitivity by P21 and FOXO3a in PDAC. Our results showed that SMAD3 overexpression facilitates the expression of P21, whereas the effects were abolished by the mutation of SMAD3 (Figure 5A). The inhibitory effects of SMAD3 on the decrease of cell viability and surviving fraction were attenuated by the knockdown of P21 (Figures 5B–D). On the other hand, the expression of FOXO3a was increased by transfection of WT SMAD4, whereas mutation of SMAD4 had no detectable effects on its expression (Figure 5E). Overexpression of SMAD4 mitigated cell viability and surviving fraction after IR treatment, which was attenuated by the knockdown of FOXO3a (Figures 5F–H). These results indicate that SMAD3 and SMAD4 regulate the radiosensitivity of PDAC, at least in part, by P21 and FOXO3a, respectively.
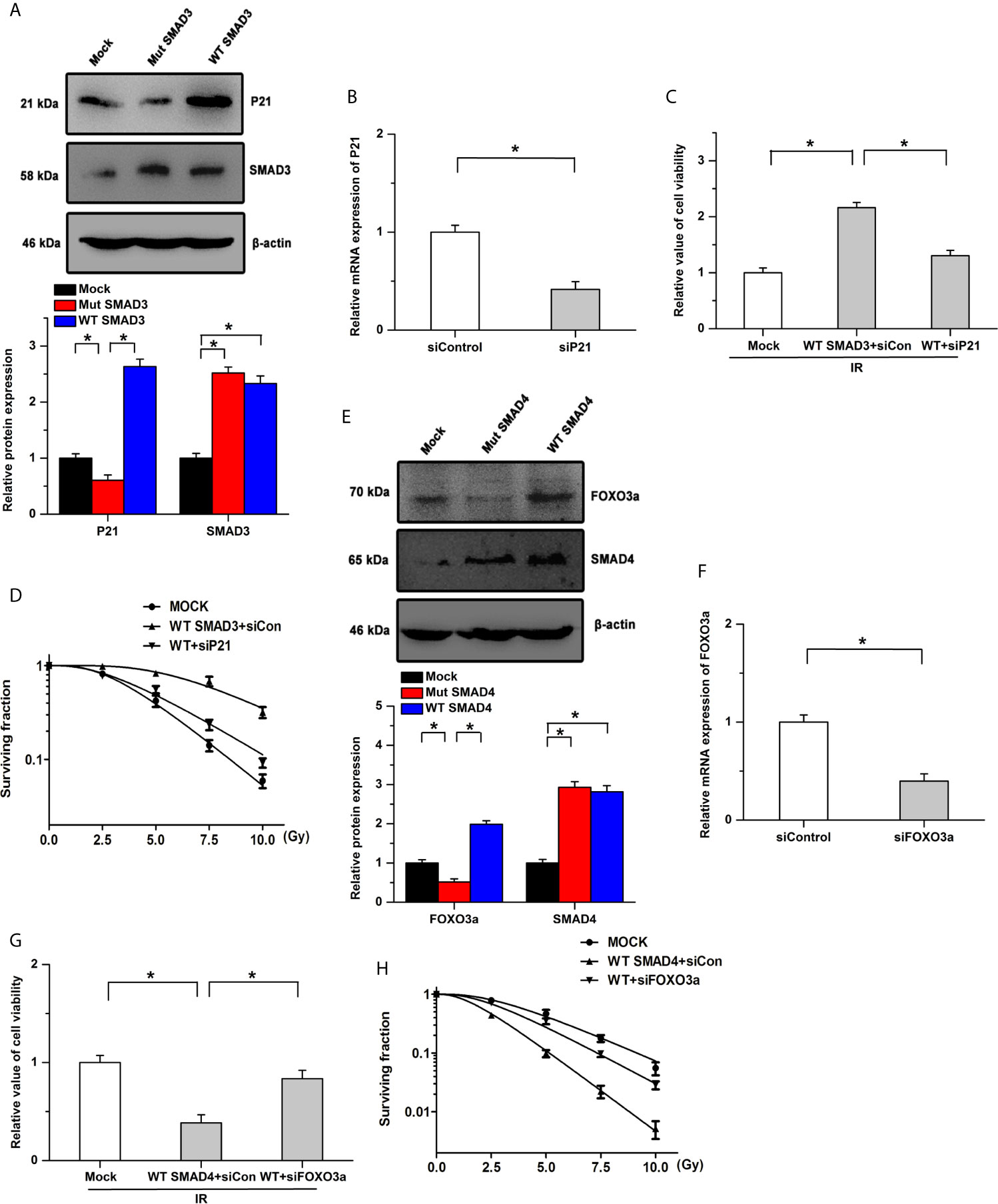
Figure 5 Effects of SMAD3 and SMAD4 on radiosensitivity are mediated by different signaling pathways. (A) Expression of P21 was significantly increased by transfection of WT SMAD3, while the effects were abolished by the mutation of SMAD3. (B) Knockdown efficiency of siP21 was verified by Real-time PCR. (C, D) SMAD3 inhibits IR-induced the decreases of cell viability (C) and surviving fraction (D), which were mitigated by P21 knockdown. WT indicates WT SMAD3, siCon indicates siControl. (E) Transfection of WT SMAD4, but not SMAD4 mutation, increased the expression of FOXO3a. (F) Real-time PCR was utilized to determine the knockdown efficiency. (G, H) Inhibitory effects of SMAD4 on the decreases of cell viability (G) and surviving fraction (H) induced by IR were attenuated by the knockdown of FOXO3a. WT indicates WT SMAD4, siCon indicates siControl. *P < 0.05.
Whole-exosome mutations profiles of PDAC were analyzed in this study. Somatic mutations locate in 441 genes were detected to be radiosensitivity-related loci. Functional enrichment and pathway-based protein-protein interaction analysis were used to address the possible mechanism involved in RI, 17 genes and 15 related pathways were identified to likely influence the radiosensitivity in PDAC. Furthermore, effects of IR on cell growth and colony formation were facilitated by the mutation of SMAD3, whereas SMAD4 mutation had the opposite effects in PDAC. The results indicate that SMAD3 and SMAD4 likely cause the difference of response to radiotherapy in PDAC.
Accumulating evidence has indicated that many mutational signatures exists in various types of tumors. It has been revealed that the unique combinations of mutation types resulted from different mutational processes (43). Consistent with most other cancer studies, all members enriched in the signature of age (signature 1), which is the result of an endogenous mutational process initiated by spontaneous deamination of 5-methylcytosine (44). Interestingly, all three insensitive group members exhibited an enrichment of the signature 3 (COSMIC Signatures version 2), which has been approved to be related with the failure of DNA double-strand break-repair by homologous recombination (45). However, germline and somatic BRCA1 and BRCA2 mutations, the proven failure causes of DNA double-strand break-repair, were not detected in our cohort, further studies would be done to explore the implicit mechanism.
As is known, the TMB and MSI score are reliable biomarkers of immunotherapy (46, 47). We analyzed the correlation between TMB, MSI, and OS and found that they were not obviously correlated, suggesting that the radiosensitivity might not be decided by these biomarkers. In the study of drug sensitivity, no significant association is found between OS and mutation burden or clonality in gastric cancer (48), whether it is the same in RI is still not studied. In this study, clonality, age, and tumor stage were also not the reason of RI in PDAC. All these results confirm that the mechanism in RI might be different with the other therapies.
The Smad protein family acts as the crucial mediators in the TGF-β signaling pathway, could transmit signals from the cell surface to the nucleus, and regulates gene activity and cell proliferation. As a member of Smad family, SMAD3 plays an important role in cancer progression, and its mutations status and DNA methylation level were related to the development of various kinds of cancers (49). Ken et al. found that increased expression of SMAD3 facilitates epithelial-mesenchymal transition and leads to poor prognosis in PDAC (50). In our study, SMAD3 was mutated in the radiosensitive group. Mutation of SMAD3 promoted the effects of IR on cell growth and colony formation in PDAC cells. SMAD4 also belongs to the Smad family same family and could form heteromeric complexes with other activated Smad proteins. The heteromeric complexes translocate and accumulate in the nucleus and then modulate the transcription of the target genes. It is reported that SMAD4 inhibits epithelial cell proliferation by acting as a tumor suppressor (51). The occurrences of pancreatic cancer, juvenile polyposis syndrome, and hereditary hemorrhagic telangiectasia syndrome are closed related with the mutations or deletions of SMAD4 (52–54). Previous studies have shown that resistance to radiotherapy likely resulted from the absence or low expression level of TGF-beta receptor or SMAD4 (15, 55). Wang F et al. have found that defective SMAD4 lead to the radioresistance by increasing persistently higher levels of ROS and promoting the autophagy caused by radiation in pancreatic cancer (55). In the present study, mutation of SMAD4 was found in the radioresistance group of PDAC, and SMAD4 acted as the central molecule in radioresistance. Mutation of SMAD4 mitigated the effects of IR on cell growth in PDAC, which is consistent with a previous study. However, further studies are still needed to elucidate the mechanism of SMAD3/SMAD4 on the response to radiotherapy in PDAC.
Furthermore, our results also showed that some PPI associated to SMAD genes might be associated with the radiosensitivity in PDAC. ARID1B, which encodes an AT-rich DNA interacting domain-containing protein, is a component of the SWI/SNF chromatin remodeling complex and may play a role in cell-cycle activation and influence DNA damage response. Yang L et al. found that depletion of ARID1A significantly enhances the radioresistance of pancreatic cancer cells and activates the PI3K/AKT signaling pathway (56). It is reported that depletion of ARID1B sensitizes colorectal cancer cells with ARID1A mutation to ionizing radiation (57). Inhibition of either ARID1A or ARID1B increases cellular sensitivity to ionizing radiation, and SWI/SNF factors are required for cellular resistance to ionizing radiation (58). CUL4A, belonging to the cullin family subunit of ubiquitin-protein ligases, inhibits the cellular repair capacity and the cell cycle checkpoint in response to DNA damage. Previous studies have shown that CUL4A overexpression renders MCF10A more sensitive to ionizing radiation, and knockdown of CUL4A facilitates the global genomic repair pathway and augments DNA damage response induced by ultraviolet (UV) irradiation (59, 60). Cellular sensitivity to ionizing radiation is decreased by CUL4A depletion by inhibiting the degradation of Chk1 (61). STK11 often serves as a tumor suppressor in some cancers. However, the potential functions of STK11 in response to IR are not always consistent. It is reported that STK11-activated autophagy enhances resistance to the combination of trametinib and radiation in KRAS-mutant NSCLC and promotes esophageal squamous cell carcinoma cell survival after radiation (62, 63). Conversely, inhibition of the STK11-SIK1 signaling pathway promotes epithelial-mesenchymal transition and radioresistance in NSCLC (64). In addition, CRKL, frequently unregulated in several malignant tumors, positively regulates the progression of cancers by promoting cell proliferation and metastasis (65). Knockdown of PBX1 significantly enhanced the radiosensitivity via STAT3 in esophageal squamous cancer (66). In our results, we found that mutations of CUL4A, STK11, and CRKL were acquired in the radioresistance group in PDAC, whereas mutation of ARID1B and PBX1 were found in the radiosensitive group. However, the concrete roles and corresponding mechanism of these genes in regulating the radiosensitivity of PDAC still need to be determined in further studies.
Meanwhile, this study had some limitations. Although we have identified that some key genes mutations might be associated with the radiosensitivity and proved that SMAD3 and SMAD4 affected radiosensitivity through different signaling pathways in PDAC in vitro, the number of patients used in our study is small, which may cause data bias in sequencing results. Further studies are needed to validate the effects of these key mutations on radiosensitivity of pancreatic cancer in more clinical samples and in vivo study. Moreover, it is necessary to determine whether the radiosensitivity is also affected by the mutations of SMAD3 or SMAD4 in other cancers, such as nasopharyngeal carcinoma and lung cancer.
In summary, our results have identified several genes mutations associated with the radiosensitivity in PDAC. Moreover, SMAD3 and SMAD4 genes mutate differently between radiosensitive and radioresistance patients with PDAC, implying that SMAD3 and SMAD4 participate in regulating the radiosensitivity in PDAC by different pathways. These results might provide some biomarkers and potential targets for the radiotherapy of PDAC patients in the future.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/genbank/, PRJNA684940.
The studies involving human participants were reviewed and approved by Renji Hospital. The patients/participants provided their written informed consent to participate in this study.
YB, JM and LZ designed this study. BH, RH, ZW, CM, YG and JL analyzed the data. LZ and CM wrote the manuscript. JT and XM collected the data. JM, XM and LZ revised the manuscript. All authors contributed to the article and approved the submitted version.
This study was sponsored by National Natural Science Foundation of China (grant no. 81802626 and 81972854), the Incubating Program for Clinical Research and Innovation of Renji Hospital (grant no. PYII-17-006), Science and Technology Commission of Shanghai Municipality (grant no. 21ZR1438500).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2021.697308/full#supplementary-material
1. Li D, Xie K, Wolff R, Abbruzzese JL. Pancreatic Cancer. Lancet (2004) 363:1049–57. doi: 10.1016/S0140-6736(04)15841-8
2. Rahib L, Smith BD, Aizenberg R, Rosenzweig AB, Fleshman JM, Matrisian LM. Projecting Cancer Incidence and Deaths to 2030: The Unexpected Burden of Thyroid, Liver, and Pancreas Cancers in the United States. Cancer Res (2014) 74:4006. doi: 10.1158/0008-5472.CAN-14-1642
3. Siegel R, Ma J, Zou Z, Jemal A. Cancer Statistics, 2014. CA Cancer J Clin (2014) 64:364. doi: 10.3322/caac.21208
4. Paulson AS, Tran Cao HS, Tempero MA, Lowy AM. Therapeutic Advances in Pancreatic Cancer. Gastroenterology (2013) 145:694. doi: 10.1053/j.gastro.2013.01.078
5. Sharma RA, Plummer R, Stock JK, Greenhalgh TA, Ataman O, Kelly S, et al. Clinical Development of New Drug-Radiotherapy Combinations. Nat Rev Clin Oncol (2016) 13:627–42. doi: 10.1038/nrclinonc.2016.79
6. Buckley AM, Lynam-Lennon N, O'Neill H, O'Sullivan J. Targeting Hallmarks of Cancer to Enhance Radiosensitivity in Gastrointestinal Cancers. Nat Rev Gastroenterol Hepatol (2020) 17:298–313. doi: 10.1038/s41575-019-0247-2
7. Park JH, Jung KH, Kim SJ, Fang Z, Yan HH, Son MK, et al. Radiosensitization of the PI3K Inhibitor HS-173 Through Reduction of DNA Damage Repair in Pancreatic Cancer. Oncotarget (2017) 8:112893–906. doi: 10.18632/oncotarget.22850
8. Bi YL, Min M, Shen W, Liu Y. Numb/Notch Signaling Pathway Modulation Enhances Human Pancreatic Cancer Cell Radiosensitivity. Tumour Biol (2016) 37:15145–55. doi: 10.1007/s13277-016-5311-8
9. Huang C, Chen D, Zhu H, Lv S, Li Q, Li G. LITAF Enhances Radiosensitivity of Human Glioma Cells Via the FoxO1 Pathway. Cell Mol Neurobiol (2019) 39:871–82. doi: 10.1007/s10571-019-00686-4
10. Ayars M, Eshleman J, Goggins M. Susceptibility of ATM-Deficient Pancreatic Cancer Cells to Radiation. Cell Cycle (2017) 16:991–8. doi: 10.1080/15384101.2017.1312236
11. Estrada-Bernal A, Chatterjee M, Haque SJ, Yang L, Morgan MA, Kotian S, et al. MEK Inhibitor GSK1120212-Mediated Radiosensitization of Pancreatic Cancer Cells Involves Inhibition of DNA Double-Strand Break Repair Pathways. Cell Cycle (2015) 14:3713–24. doi: 10.1080/15384101.2015.1104437
12. Chang VH, Tsai YC, Tsai YL, Peng SL, Chen SL, Chang TM, et al. Krüpple-Like Factor 10 Regulates Radio-Sensitivity of Pancreatic Cancer via UV Radiation Resistance-Associated Gene. Radiother Oncol (2017) 122:476–84. doi: 10.1016/j.radonc.2017.01.001
13. Meyer JE, Finnberg NK, Chen L, Cvetkovic D, Wang B, Zhou L, et al. Tissue TGF-β Expression Following Conventional Radiotherapy and Pulsed Low-Dose-Rate Radiation. Cell Cycle (2017) 16:1171–4. doi: 10.1080/15384101.2017.1317418
14. Xue J, Zhu W, Song J, Jiao Y, Luo J, Yu C, et al. Activation of Pparα by Clofibrate Sensitizes Pancreatic Cancer Cells to Radiation Through the Wnt/β-Catenin Pathway. Oncogene (2018) 37:953–62. doi: 10.1038/onc.2017.401
15. Dancea HC, Shareef MM, Ahmed MM. Role of Radiation-Induced TGF-Beta Signaling in Cancer Therapy. Mol Cell Pharmacol (2009) 1:44–56. doi: 10.4255/mcpharmacol.09.06
16. Gunda V, Souchek J, Abrego J, Shukla SK, Goode GD, Vernucci E, et al. MUC1-Mediated Metabolic Alterations Regulate Response to Radiotherapy in Pancreatic Cancer. Clin Cancer Res (2017) 23:5881–91. doi: 10.1158/1078-0432.CCR-17-1151
17. Wu Z, Jing S, Li Y, Gao Y, Yu S, Li Z, et al. The Effects of SAHA on Radiosensitivity in Pancreatic Cancer Cells by Inducing Apoptosis and Targeting RAD51. Biomed Pharmacother (2017) 89:705–10. doi: 10.1016/j.biopha.2017.02.067
18. Chen S, Zhou Y, Chen Y, Gu J. Fastp: An Ultra-Fast All-in-One FASTQ Preprocessor. Bioinformatics (2018) 34:i884–90. doi: 10.1093/bioinformatics/bty560
19. Li H, Durbin R. Fast and Accurate Short Read Alignment With Burrows-Wheeler Transform. Bioinformatics (2009) 25:1754–60. doi: 10.1093/bioinformatics/btp324
20. Karolchik D, Baertsch R, Diekhans M, Furey TS, Hinrichs A, Lu YT, et al. The UCSC Genome Browser Database. Nucleic Acids Res (2003) 31:51–4. doi: 10.1093/nar/gkg129
21. Tarasov A, Vilella AJ, Cuppen E, Nijman IJ, Prins P. Sambamba: Fast Processing of NGS Alignment Formats. Bioinformatics (2015) 31:2032–4. doi: 10.1093/bioinformatics/btv098
22. McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, et al. The Genome Analysis Toolkit: A MapReduce Framework for Analyzing Next-Generation DNA Sequencing Data. Genome Res (2010) 20:1297–303. doi: 10.1101/gr.107524.110
23. Wang K, Li M, Hakonarson H. ANNOVAR: Functional Annotation of Genetic Variants From High-Throughput Sequencing Data. Nucleic Acids Res (2010) 38:e164. doi: 10.1093/nar/gkq603
24. Cingolani P, Platts A, Wang le L, Coon M, Nguyen T, Wang L, et al. A Program for Annotating and Predicting the Effects of Single Nucleotide Polymorphisms, SnpEff: SNPs in the Genome of Drosophila Melanogaster Strain W1118; Iso-2; Iso-3. Fly (Austin) (2012) 6:80–92. doi: 10.4161/fly.19695
25. 1000 Genomes Project Consortium, Auton A, Brooks LD, Durbin RM, Garrison EP, Kang HM, et al. A Global Reference for Human Genetic Variation. Nature (2015) 526:68–74. doi: 10.1038/nature15393
26. Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, et al. Exome Aggregation Consortium. Analysis of Protein-Coding Genetic Variation in 60,706 Humans. Nature (2016) 536:285–91. doi: 10.1038/nature19057
27. Ng PC, Henikoff S. SIFT: Predicting Amino Acid Changes That Affect Protein Function. Nucleic Acids Res (2003) 31:3812–4. doi: 10.1093/nar/gkg509
28. Adzhubei I, Jordan DM, Sunyaev SR. Predicting Functional Effect of Human Missense Mutations Using PolyPhen-2. Curr Protoc Hum Genet (2013) 7:7.20. doi: 10.1002/0471142905.hg0720s76
29. Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, et al. A Method and Server for Predicting Damaging Missense Mutations. Nat Methods (2010) 7:248–9. doi: 10.1038/nmeth0410-248
30. Schwarz JM, Cooper DN, Schuelke M, Seelow D. MutationTaster2: Mutation Prediction for the Deep-Sequencing Age. Nat Methods (2014) 11:361–2. doi: 10.1038/nmeth.2890
31. Yang H, Robinson PN, Wang K. Phenolyzer: Phenotype-Based Prioritization of Candidate Genes for Human Diseases. Nat Methods (2015) 12:841–3. doi: 10.1038/nmeth.3484
32. Cibulskis K, Lawrence MS, Carter SL, Sivachenko A, Jaffe D, Sougnez C, et al. Sensitive Detection of Somatic Point Mutations in Impure and Heterogeneous Cancer Samples. Nat Biotechnol (2013) 31:213–9. doi: 10.1038/nbt.2514
33. McLaren W, Gil L, Hunt SE, Riat HS, Ritchie GR, Thormann A, et al. The Ensembl Variant Effect Predictor. Genome Biol (2016) 17:122. doi: 10.1186/s13059-016-0974-4
34. Talevich E, Shain AH, Botton T, Bastian BC. CNVkit: Genome-Wide Copy Number Detection and Visualization From Targeted DNA Sequencing. PLoS Comput Biol (2016) 12:e1004873. doi: 10.1371/journal.pcbi.1004873
35. Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SA, Behjati S, Biankin AV, et al. Signatures of Mutational Processes in Human Cancer. Nature (2013) 500:415–21. doi: 10.1038/nature12477
36. Niu B, Ye K, Zhang Q, Lu C, Xie M, McLellan MD, et al. MSIsensor: Microsatellite Instability Detection Using Paired Tumor-Normal Sequence Data. Bioinformatics (2014) 30:1015–6. doi: 10.1093/bioinformatics/btt755
37. Carter SL, Cibulskis K, Helman E, McKenna A, Shen H, Zack T, et al. Absolute Quantification of Somatic DNA Alterations in Human Cancer. Nat Biotechnol (2012) 30:413–21. doi: 10.1038/nbt.2203
38. Martincorena I, Raine KM, Gerstung M, Dawson KJ, Haase K, Van Loo P, et al. Universal Patterns of Selection in Cancer and Somatic Tissues. Cell (2017) 171:1029–41.e21. doi: 10.1016/j.cell.2017.09.042
39. Zhang L, Yuan C, Peng J, Zhou L, Jiang Y, Lin Y, et al. SHP-2-Mediated Upregulation of ZEB1 Is Important for PDGF-B-Induced Cell Proliferation and Metastatic Phenotype in Triple Negative Breast Cancer. Front Oncol (2020) 10:1230. doi: 10.3389/fonc.2020.01230
40. Yu G, Wang LG, Han Y, He QY. Clusterprofiler: An R Package for Comparing Biological Themes Among Gene Clusters. OMICS (2012) 16:284–7. doi: 10.1089/omi.2011.0118
41. Niu H, Huang Y, Yan L, Zhang L, Zhao M, Lu T, et al. Knockdown of SMAD3 Inhibits the Growth and Enhances the Radiosensitivity of Lung Adenocarcinoma via P21 In Vitro and In Vivo. Int J Biol Sci (2020) 16:1010–22. doi: 10.7150/ijbs.40173
42. Fu G, Peng C. Nodal Enhances the Activity of FoxO3a and its Synergistic Interaction With Smads to Regulate Cyclin G2 Transcription in Ovarian Cancer Cells. Oncogene (2011) 30:3953–66. doi: 10.1038/onc.2011.127
43. Helleday T, Eshtad S, Nik-Zainal S. Mechanisms Underlying Mutational Signatures in Human Cancers. Nat Rev Genet (2014) 15:585–98. doi: 10.1038/nrg3729
44. Alexandrov LB, Jones PH, Wedge DC, Sale JE, Campbell PJ, Nik-Zainal S, et al. Clock-Like Mutational Processes in Human Somatic Cells. Nat Genet (2015) 47:1402–7. doi: 10.1038/ng.3441
45. Alexandrov LB, Kim J, Haradhvala NJ, Huang MN, Tian Ng AW, Wu Y, et al. The Repertoire of Mutational Signatures in Human Cancer. Nature (2020) 578:94–101. doi: 10.1038/s41586-020-1943-3
46. Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, Havel JJ, et al. Cancer Immunology. Mutational Landscape Determines Sensitivity to PD-1 Blockade in non-Small Cell Lung Cancer. Science (2015) 348:124–8. doi: 10.1126/science.aaa1348
47. Dudley JC, Lin MT, Le DT, Eshleman JR. Microsatellite Instability as a Biomarker for PD-1 Blockade. Clin Cancer Res (2016) 22:813–20. doi: 10.1158/1078-0432.CCR-15-1678
48. Yang Q, Zhu C, Zhang Y, Wang Y, Wang Y, Zhu L, et al. Molecular Analysis of Gastric Cancer Identifies Genomic Markers of Drug Sensitivity in Asian Gastric Cancer. J Cancer (2018) 9:2973–80. doi: 10.7150/jca.25506
49. Ansar M, Wang CJ, Wang YH, Shen TH, Hung CS, Chang SC, et al. SMAD3 Hypomethylation as a Biomarker for Early Prediction of Colorectal Cancer. Int J Mol Sci (2020) 21:7395. doi: 10.3390/ijms21197395
50. Yamazaki K, Masugi Y, Effendi K, Tsujikawa H, Hiraoka N, Kitago M, et al. Upregulated SMAD3 Promotes Epithelial-Mesenchymal Transition and Predicts Poor Prognosis in Pancreatic Ductal Adenocarcinoma. Lab Invest (2014) 94:683–91. doi: 10.1038/labinvest.2014.53
52. Biankin AV, Waddell N, Kassahn KS, Gingras MC, Muthuswamy LB, Johns AL, et al. Pancreatic Cancer Genomes Reveal Aberrations in Axon Guidance Pathway Genes. Nature (2012) 491:399–405. doi: 10.1038/nature11547
53. Kinzler KW, Vogelstein B. Landscaping the Cancer Terrain. Science (1998) 280:1036–7. doi: 10.1126/science.280.5366.1036
54. Gallione CJ, Richards JA, Letteboer TG, Rushlow D, Prigoda NL, Leedom TP, et al. SMAD4 Mutations Found in Unselected HHT Patients. J Med Genet (2006) 43:793–7. doi: 10.1136/jmg.2006.041517
55. Wang F, Xia X, Yang C, Shen J, Mai J, Kim HC, et al. SMAD4 Gene Mutation Renders Pancreatic Cancer Resistance to Radiotherapy Through Promotion of Autophagy. Clin Cancer Res (2018) 24:3176–85. doi: 10.1158/1078-0432.CCR-17-3435
56. Yang L, Yang G, Ding Y, Dai Y, Xu S, Guo Q, et al. Inhibition of PI3K/AKT Signaling Pathway Radiosensitizes Pancreatic Cancer Cells With ARID1A Deficiency in Vitro. J Cancer (2018) 9:890–900. doi: 10.7150/jca.21306
57. Niedermaier B, Sak A, Zernickel E, Xu S, Groneberg M, Stuschke M. Targeting ARID1A-Mutant Colorectal Cancer: Depletion of ARID1B Increases Radiosensitivity and Modulates DNA Damage Response. Sci Rep (2019) 9:18207. doi: 10.1038/s41598-019-54757-z
58. Watanabe R, Ui A, Kanno S, Ogiwara H, Nagase T, Kohno T, et al. SWI/SNF Factors Required for Cellular Resistance to DNA Damage Include ARID1A and ARID1B and Show Interdependent Protein Stability. Cancer Res (2014) 74:2465–75. doi: 10.1158/0008-5472.CAN-13-3608
59. Gupta A, Yang LX, Chen LC. Study of the G2/M Cell Cycle Checkpoint in Irradiated Mammary Epithelial Cells Overexpressing Cul-4A Gene. Int J Radiat Oncol Biol Phys (2002) 52:822–30. doi: 10.1016/S0360-3016(01)02739-0
60. Liu L, Lee S, Zhang J, Peters SB, Hannah J, Zhang Y, et al. CUL4A Abrogation Augments DNA Damage Response and Protection Against Skin Carcinogenesis. Mol Cell (2009) 34:451–60. doi: 10.1016/j.molcel.2009.04.020
61. Kim KS, Heo JI, Choi KJ, Bae S. Enhancement of Cellular Radiation Sensitivity Through Degradation of Chk1 by the XIAP-XAF1 Complex. Cancer Biol Ther (2014) 15:1622–34. doi: 10.4161/15384047.2014.962305
62. Wang Y, Li N, Jiang W, Deng W, Ye R, Xu C, et al. Mutant LKB1 Confers Enhanced Radiosensitization in Combination With Trametinib in KRAS-Mutant Non-Small Cell Lung Cancer. Clin Cancer Res (2018) 24:5744–56. doi: 10.1158/1078-0432.CCR-18-1489
63. Lu C, Xie C. Radiation-Induced Autophagy Promotes Esophageal Squamous Cell Carcinoma Cell Survival via the LKB1 Pathway. Oncol Rep (2016) 35:3559–65. doi: 10.3892/or.2016.4753
64. Yao YH, Cui Y, Qiu XN, Zhang LZ, Zhang W, Li H, et al. Attenuated LKB1-SIK1 Signaling Promotes Epithelial-Mesenchymal Transition and Radioresistance of non-Small Cell Lung Cancer Cells. Chin J Cancer (2016) 35:50. doi: 10.1186/s40880-016-0113-3
65. Bian X, Liang Z, Feng A, Salgado E, Shim H. HDAC Inhibitor Suppresses Proliferation and Invasion of Breast Cancer Cells Through Regulation of miR-200c Targeting CRKL. Biochem Pharmacol (2018) 147:30–7. doi: 10.1016/j.bcp.2017.11.008
Keywords: pancreatic cancer, radiotherapy, radioresistance, biomarker, Smad
Citation: Hu B, Ma X, Huang R, Wu Z, Lu J, Guo Y, Tang J, Ma C, Ma J, Zhang L and Bai Y (2021) Identification of Key Genes Mutations Associated With the Radiosensitivity by Whole Exome Sequencing in Pancreatic Cancer. Front. Oncol. 11:697308. doi: 10.3389/fonc.2021.697308
Received: 19 April 2021; Accepted: 20 July 2021;
Published: 09 August 2021.
Edited by:
Ira Ida Skvortsova, Innsbruck Medical University, AustriaReviewed by:
Lin Zhu, Fudan University, ChinaCopyright © 2021 Hu, Ma, Huang, Wu, Lu, Guo, Tang, Ma, Ma, Zhang and Bai. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yongrui Bai, YmFpeW9uZ3J1aXpAMTYzLmNvbQ==; Lei Zhang, aHJibXV6bEAxMjYuY29t; Jun Ma, bWE4ODY2MTVAMTI2LmNvbQ==
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.