 Shengchao Xu
Shengchao Xu Xi Yan2†
Xi Yan2† Chengke Luo
Chengke Luo- 1Department of Neurosurgery, Xiangya Hospital of Central South University, Changsha, China
- 2Health Management Center, Xiangya Hospital of Central South University, Changsha, China
- 3Department of Microbiology, Xiangya School of Medicine, Central South University, Changsha, China
Background: Patient-derived orthotopic xenograft (PDOX) is a popular animal model for translational cancer research. Immunotherapy is a promising therapy against glioblastoma (GBM). However, the PDOX model is limited to evaluating immune-related events. Our study aims to establish GBM humanized PDOX (HPDOX) mice models to study the mechanism of anti-CTLA4 immunotherapy and immune-related adverse events (IRAEs).
Methods: HPDOX models were established by culturing GBM tissues and intracranially implanting them in NSG mice. Meanwhile, peripheral blood mononuclear cells (PBMCs) were separated from peripheral blood and of GBM patients and administrated in corresponding mice. The population of CD45+, CD3+, CD4+, CD8+, and regulatory T (Treg) cells was estimated in the peripheral blood or tumor.
Results: T cells derived from GBM patients were detected in HPDOX mice models. The application of anti-CTLA4 antibodies (ipilimumab and tremelimumab) significantly inhibited the growth of GBM xenografts in mice. Moreover, residual patient T cells were detected in the tumor microenvironment and peripheral blood of HPDOX mice and were significantly elevated by ipilimumab and tremelimumab. Additionally, Treg cells were decreased in mice with IRAEs. Lastly, the proportion of CD4+/CD8+ T cells dramatically increased after the administration of ipilimumab. And the degree of IRAEs may be related to CD56+ expression in HPDOX.
Conclusions: Our study established HPDOX mice models for investigating the mechanism and IRAEs of immunotherapies in GBM, which would offer a promising platform for evaluating the efficacy and IRAEs of novel therapies and exploring personalized therapeutic strategies.
Background
Glioblastoma (GBM) is one of the most malignant primary brain cancers. The median overall survival of GBM patients is only 14.6 months, despite the satisfactory surgery and concomitant chemoradiotherapy (1–3). Therefore, there is a clear urgent to reveal the mechanism of tumorigenesis and development and develop novel therapeutic agents against GBM. However, recent clinical trials without appropriate mice models in this area have resulted in dissatisfactory and inconsistent therapeutic effects. It cannot reflect the tumor microenvironment, principal histologic, and genetic characteristics of GBM. In this context, the development of accurate and reproducible animal models is essential.
The patient-derived orthotopic xenograft (PDOX) has attracted more and more attention to facilitate biologic studies, preclinical drug evaluation, and biomarker identification (4–8). Nevertheless, clinically relevant PDOX models are not fully capable of recapitulating patients’ immune systems, which impedes the evaluation of immunotherapy efficacy. In recent years, although the co-cultures combined GBM organoids with immune cells such as human peripheral blood mononuclear cells (PBMCs) have been of great concern, which could be an ideal platform for immunotherapy selection (9), since the tumor-derived spheres are still not in the same conditions such as hypoxia or immune microenvironment, as to those occur in intracranial. Besides, the co-culture conditions combined GBM organoids with immune cells are not the optimum one for each cell type according to compromise strategy (10, 11). Meanwhile, immune-related adverse events (IRAEs) arising under immunotherapy forcing us potentially without precedent to think of strategies to maintain the immune system. Therefore, revealing the mechanism of T cells from PBMCs in the action of IRAEs and anti-tumor immunity is essential to balancing the immune system in developing a cancer immunotherapy strategy.
Humanized PDOX models (HPDOX) were regarded as the next-generation PDOX. Although humanized mice were reported in few tumors such as myeloma and hepatocellular carcinoma, humanized GBM mice models were seldom studied in recent literature (12). Those models were essential to study the potential and limitations for differential immune-enhancing approaches, as well as contribute to refining the framework of emerging immunotherapy strategies and related IRAEs against GBM. Our study was conducted to establish humanized GBM mice models and investigate the efficacy and IRAEs of anti-CTLA4 immunotherapy. Our studies aimed to provide a platform to develop effective strategies to minimize immune therapeutic IRAEs without impeding anti-tumor immunity in the future.
Materials and Methods
Human GBM Tissue Specimens
Fresh GBM specimens were obtained from patients who received surgery in the Department of Neurosurgery, Xiangya Hospital from 2016 to 2020. All clinical samples were collected with informed consent obtained from the patients. All procedures were conducted following the Declaration of Helsinki (1964).
Peripheral Blood
All procedures were approved by the Ethics Committee of Xiangya Hospital. Peripheral blood (PB) specimens were collected from patients who received surgery in the Department of Neurosurgery, Xiangya Hospital from 2016 to 2020 with written informed consent obtained. PBMCs were isolated using Lymphoprep (Stem Cell Technologies) according to the manufacturer’s instructions.
T Cell Reconstitution and Anti-CTLA4 Antibodies
The reconstitution of CD4+ and CD8+ T cells of PB from mice was monitored every week. A total of 1 × 106 GBM patients’ PBMCs were implanted into sub-lethally irradiated (0.5 Gy) 4–6-week-old male NSG mice by tail vein. The anti-CTLA4 antibodies (ipilimumab, ipilimumab with N298A mutation, and tremelimumab) were generous gifts from Huntsman Cancer Institute, University of Utah, USA.
Generation of HPDOX Mice Model
Some 4–6-week-old male NSG (NOD.Cg−Prkdcscid Il2rgtm1Wjll/SzJ) mice were used in this study. All animal experiments were obtained and performed at the Laboratory Animal Center of the Central South University and all procedures were approved by the Ethics Committee of Xiangya Hospital following the Guide for the Care and Use of Laboratory Animals. PDOX was established from surgically resected specimens in NSG mice. Fresh surgical specimens were rinsed with Hank’s solution three times. Then the tumor tissue was cut into several 1–3 mm3 pieces. The tissue was incubated with accutase for 30 min at 37°C and dissociated into single cells. Cells were cultured in serum-free medium (Canada, Stemcell Technologies) in an incubator. Before transplantation, cells were digested and resuspended in the medium with a density of 1 × 108/ml. The injection was located at the skull 1–2 mm lateral and 1 mm anterior to the bregma. Each time, 5 μl of cell suspension was aspirated using the Hamilton syringe and slowly injected into the brain at a dept of 2–3 mm with a rate of 1 μl/min. After the completion of the injection, maintain the needle for 3 min before withdrawing to reduce the backflow of the injected cells spillover. After 14 days of PDOX established, the mice with criterion can be incorporated for further study: (a) The weight loss is no more than 10%; (b) Motor function is normal, without hypokinesis; (c) Hair’s clean and shiny, without ruffings; (d) The bowel and bladder functions are normal.; and (e) No signs of infection or any other illness. To estimate the tumor volume, the simple random sampling method was employed to select three mice from each group for sacrifice. The tumor volume was calculated according to the HE slides with the greatest cross-sectional area. Tumor volume was determined using the following equation: length (L) × width2 (W) × 0.5. Tumor volume of the three mice was within our expectation, whereas some mice were excluded because the mice failed to meet the standard as described previously. There is no significant difference in tumor volume at this time point (Figure S1, P >0.05). Afterward, the HPDOX mice model was established by injecting PBMCs from GBM patients at 4 h after irradiation (13). The antibodies were given twice a week after PBMCs transplantation. The mice in our study were monitored for up to 3 months for weight, health, or immune status. HPDOX suffered systemic IRAEs were assessed by the criterion of mice graft-versus-host disease (GVHD) clinical scoring system including weight loss, posture, activity, fur texture, and skin integrity (14). The mouse’s total score was estimated twice a week. Mice were sacrificed according to the following criteria: weight loss of 20–25%; tumor weight reached 10% of mice weight; appetite loss of more than 24 h; depression and hypothermia. The observation endpoint was defined as 104 days (14 days for model establishment and 3 months for observation).
Flow Cytometry
PB containing the anticoagulant sodium heparin was centrifuged at 1,500 rpm for 5 min at room temperature. Red blood cells were lysed by Red Blood Cell Lysis Buffer (Sigma), and the white blood cells were pelleted at 300g for 3 min. Flow cytometric analysis was performed using the LSR FORTESSA device (BD Biosciences, San Jose, CA, USA). The samples were incubated with the following antibodies to identify T and regulatory T (Treg) cells: anti-human CD3 FITC (BD Biosciences), anti-human CD4 Pacific Blue (BD Biosciences), anti-human CD8 APC (BD Biosciences), anti-human CD45 PE (BD Biosciences), IFNγ-APC (BioLegend), CD45RA- PE (BD Biosciences), CD45RO-APC-cy7 (BD Biosciences), anti-human FOXP3 PE (BD Biosciences), CFSE FITC (BD Biosciences), CD25 PE (BD Biosciences), CD127 PE-cy7 (BD Biosciences), CD56 PE (BD Biosciences), and HLA-DR PE-cy7 (BD Biosciences). The following controls were used: unstained cells and single-stained cells; and dead cells, which in conjunction with AutoComp software were used to set accurate compensation and data analysis. Cells were counted per sample, and the data were analyzed with FlowJo V10.
Statistical Analyses
Statistical analyses were conducted using GraphPad Prism v8.0. One-way ANOVA or unpaired two-tailed Student’s t-test was used to estimate the difference between two or more groups. Kaplan–Meier analysis was used to evaluate the survival difference between two groups. Two-sided p <0.05 was considered as statistical significance.
Results
Establishment of GBM HPDOX Mice Models
Establishment of GBM PDOX from patient’s tissues. The establishment of the PDOX model was conducted by injecting patients’ tumor-derived GBM cells into female immune-deficient nude mice. Candidate mice for PDOX models could be NSG, NOD-SCID, and nude. Herein we investigated in vitro culture of 26 PDOX generated and/or passaged in NSG mice. The workflow was shown in Figure 1, and details were described above. The demographic information of patients from whom PDOX was generated was summarized in Table 1. The age of patients ranged from 22 to 76 years old with a mean of 58 ± 12.05 years old, and 11 patients were women and 15 were men. The tumor volume varied from 4.88 to 115.33 cm3, with a mean of 48 ± 26.60 cm3. GBM cells from each specimen were injected into five mice, in which 12 specimens successfully inherited in mice. The overall engraftment rate of GBM PDOX in NSG mice was 46.15% (60/130). The total time of PDOX establishment ranged from 44 to 126 days, with a mean of 62 ± 17.5 days. Humanized GBM PDOX by transplanting the same patient’s PBMCs. Firstly, we established GBM PDOX mice. Two weeks later, the HPDOX models were established by engrafting the same patient’s PBMCs (Figure 1, bottom).
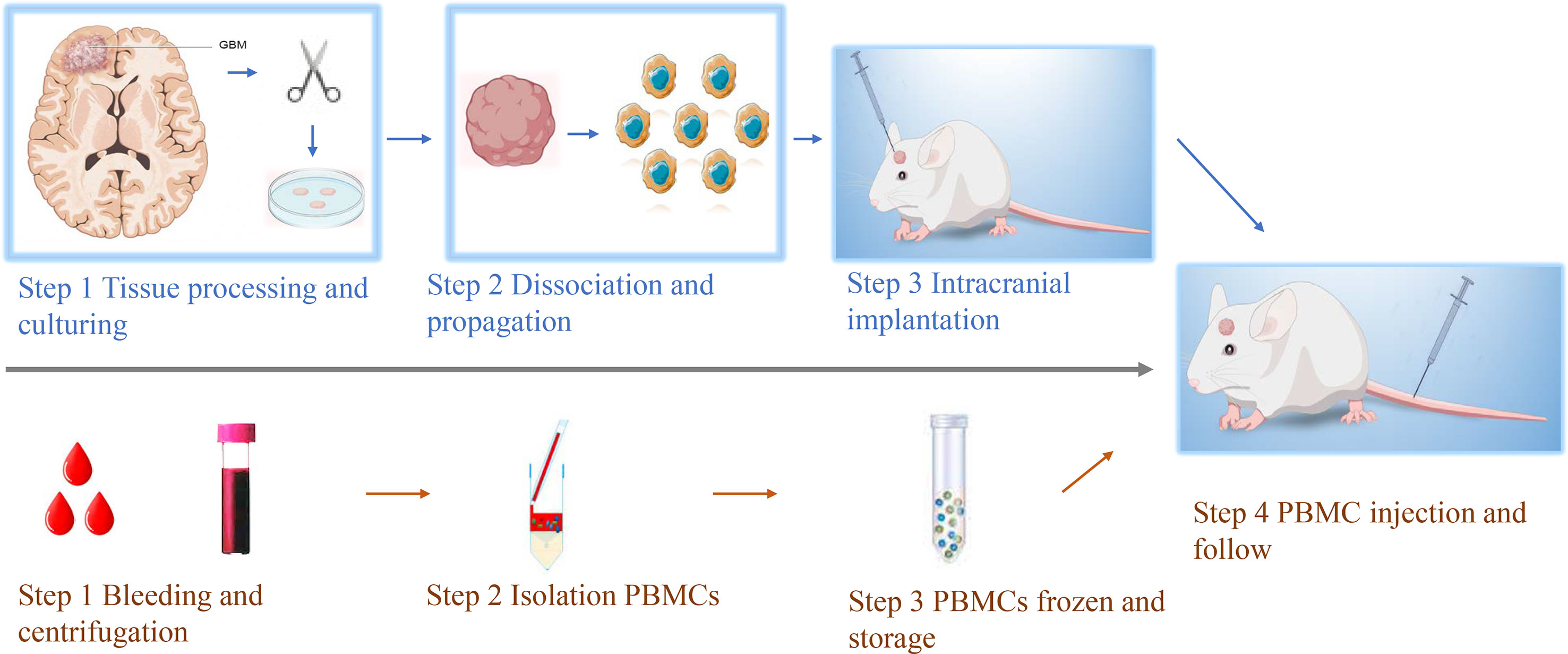
Figure 1 Workflow of establishing GBM HPDOX mice models. Top, the workflow of establishing the GBM PDOX model. Tumor specimens obtained from surgery were dissociated to be single tumor cells. Then tumor cells were cultured with proper conditions to enhance the formation of GBM neurospheres (Top step 1). After the obtainment of neurospheres could be tested (Top, steps 1 & 2). GBM neurospheres could be used to establish the PDOX model to study tumor biology or test novel medicine and instruments (Top, step 3). Bottom, the workflow of establishing patients’ PBMCs-derived humanized mouse models. The peripheral blood of GBM patients was collected and centrifugated (Bottom, step 1). The PBMCs were isolated and stored (Bottom, steps 2 & 3). After the establishment of the PDOX mice model for two weeks, the same patient’s PBMCs were injected into NSG mice for further study (step 4).
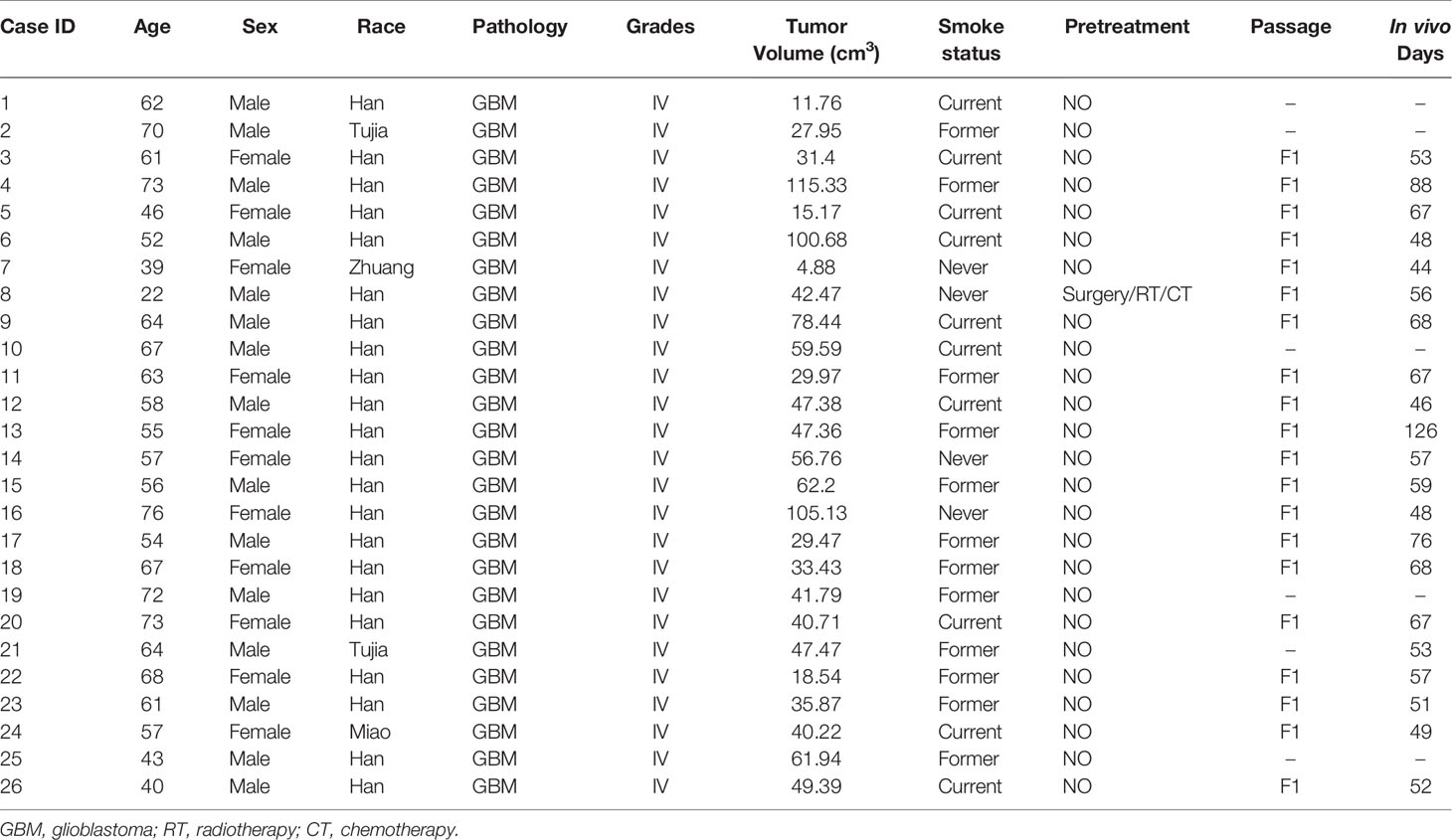
Table 1 The demographic and clinical information for patients of PDOX models.
Patients’ T Cells Reconstituted Well in GBM HPDOX Mice
Timepoint of the construction of HPDOX mice model. PBMCs were collected from GBM patients and the HPDOX model was constructed in PDOX mice bearing tumors derived from the same patients (Figure 2A). Flow cytometry of CD4+ and CD8+ T cells in HPDOX to explore the differentiated T cells within transplanted PBMCs (Figure 2B). The population of CD4+ (Figure 2C) and CD8+ (Figure 2D) T cells in GBM HPDOX mice. Our results showed both CD4+ and CD8+ T cells reconstituted well in GBM HPDOX mice.
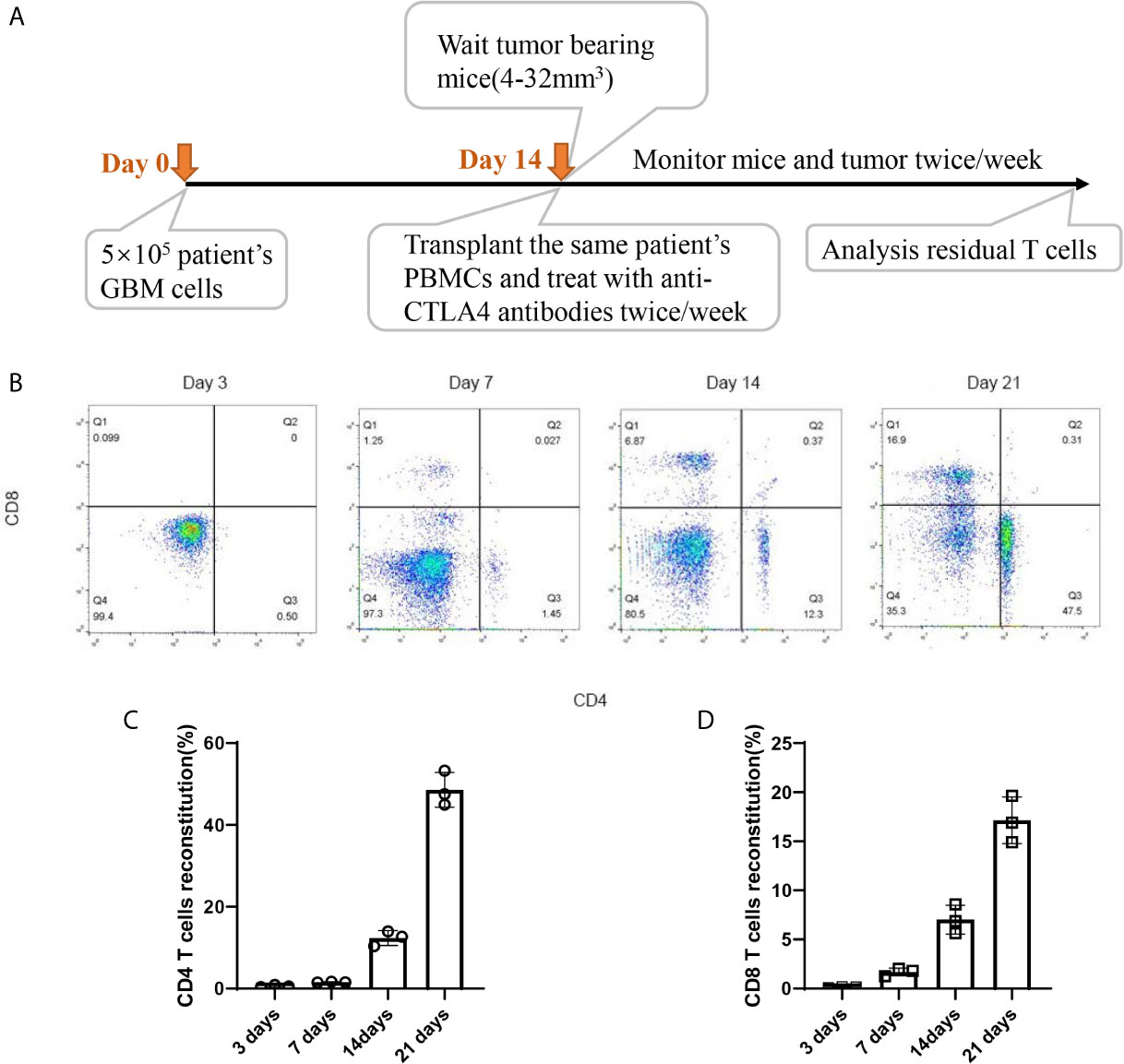
Figure 2 The reconstitution of T cells derived from GBM patients in HPDOX mice model. (A) Timepoint of the construction of HPDOX mice model. (B) Flow cytometry analysis of CD4+ and CD8+ T cells in NSG mice with transplanted PBMCs. (C, D) The population of CD4+ (C) and CD8+ (D) T cells in NSG mice with transplanted PBMCs. Each experiment was repeated at least three times, and three repeats were included each time.
Evaluation of Antitumor Effects of Anti-CTLA4 Antibodies in HPDOX Mice Model
GBM07 and GBM22 were selected as the representation since they had a high expression of CTLA4 (Table S1 and Figure 3A). After the construction of the HPDOX model, mice were treated with anti-CTLA4 antibody ipilimumab and tremelimumab twice weekly as the workflow described before. Results showed that tumor volume was significantly reduced after the application of ipilimumab and tremelimumab (P <0.05) (Figures 3B, C). After the intervention, residual human T cells (CD45+, CD3+) could be detected and their levels were significantly elevated in the tumor microenvironment and PB in HPDOX mice (P <0.05) (Figures 3D–G). Then we explored the population of the subgroups of the T cells. The activated T cells were presented with a higher expression of IFN-γ+ and CD25+. The population of the activated T cells was increased significantly after the treatment of anti-CTLA4 antibodies (P <0.01, Figures S2A–C). The exhausted T cells presented the lower expression of IFN-γ+ (Figure S2B). The exhausted T could differentiate into the activated T cells in certain conditions. The effector T cells are usually CD45RA positive, which was increased dramatically after treatment (P <0.001, Figures S2A, D). The memory T cells are CD45RO positive, which showed no significant difference between the groups (Figures S2A, E). The P value of the survival data of the HPDOX 07, and HPDOX 22 was 0.0082 and 0.0003 respectively, which indicated the significant difference between the treatment groups (Figures 3H, I). However, for the tremelimumab, the P value was 0.1118 and 0.0102 respectively, which indicated the significant difference between the treatment groups only in HPDOX 22 instead of HPDOX 07. These results indicated that CTLA4 played an inhibitory role in immune surveillance and HPDOX models could be appropriate approaches for revealing antitumor mechanisms and effects of anti-CTLA4 immunotherapy.
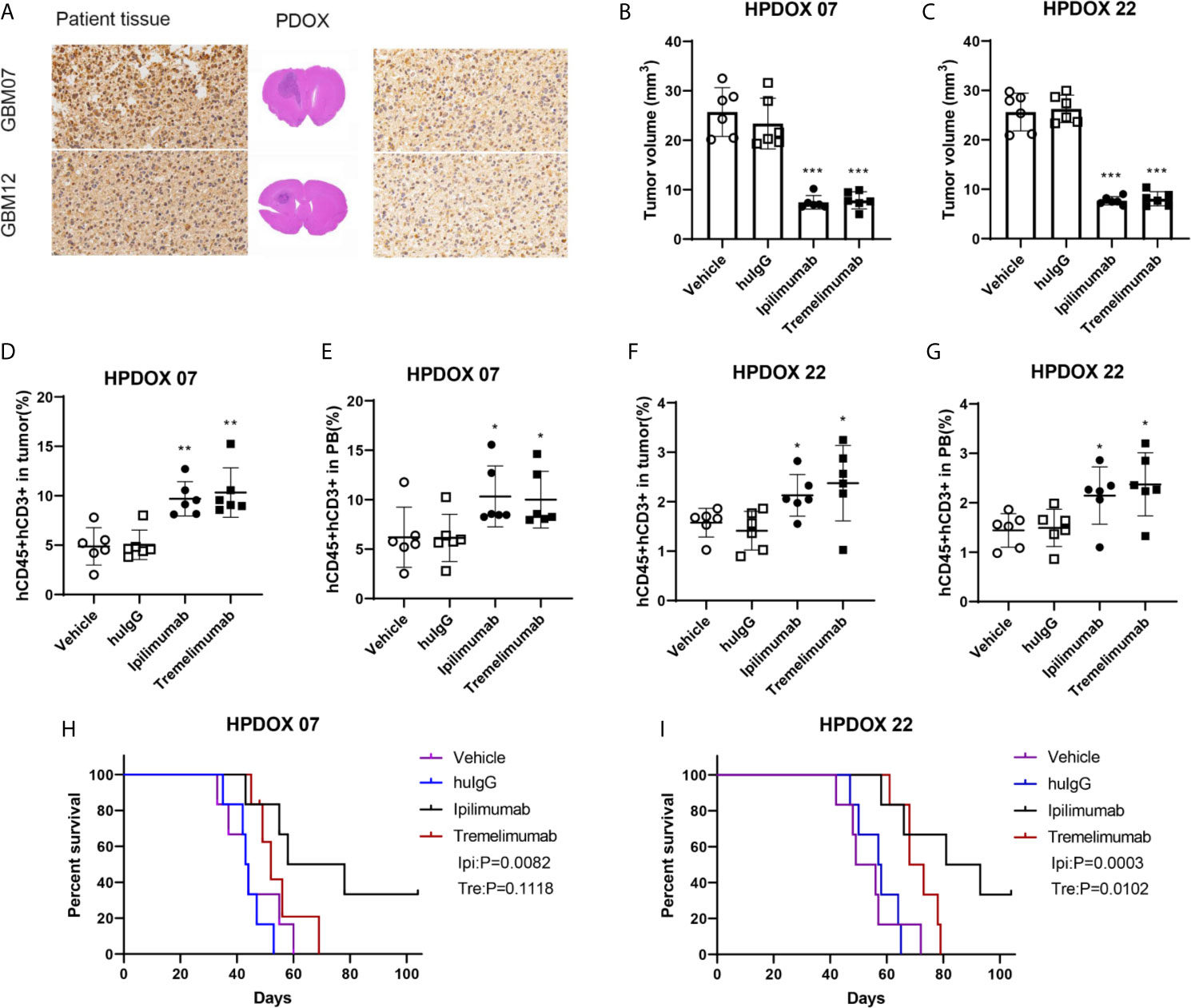
Figure 3 Evaluating the antitumor effect of anti-CTLA4 monoclonal antibodies in HPDOX. (A) IHC analyzes anti-CTLA4 in HPDOX 07, HPDOX 22; Scale bar: 20 μm (sides)/1,000 μm (middle). (B, C) Tumor volume of GBM xenograft in mice treated with Vehicle, huIgG, ipilimumab, and tremelimumab. (D–G) The population of T cells in GBM tissues (D, F) and peripheral blood (E, G) in mice treated with Vehicle, huIgG, ipilimumab, and tremelimumab. (H, I) The survival data of the HPDOX 07, and HPDOX 22 was presented after the treatment respectively. There were six mice in each group. Data were represented as mean ± SD. *P < 0.05, **P < 0.01, ***P < 0.001.
FOXP3+ and Ratio of CD4+/CD8+ T Cells Were Associated With IRAEs
To prolong the period for evaluation of the efficacy and IRAEs of immunotherapy, the detail of PBMCs transplantation is essential for the success of HPDOX (Table 2). The period can be prolonged to 47.90 ± 17.91 days engrafted with the decreased number (1 × 106 vs >1 × 107 normally) of autologous patients’ PBMCs (Figure S3 and Table 1). Further, we detected the effect of ipilimumab at different doses in HPDOX mice models. The construction of HPDOX was detailed as before and GBM26 was taken as an example. Ten mice in each group were administrated with a high dose (10 μg/g) and normal dose (3 μg/g) of ipilimumab respectively. IRAEs were detected in 7/10 in the high dose group, only 1/10 in the normal dose group within the next two months (Table 3). After the intervention, we found that CD4+ and CD8+T cells reconstituted well in GBM HPDOX26 mice (Figures 4A top and B), and the ratio of CD4+/CD8+ T cells was dramatically increased in mice with IRAEs (P <0.05) (Figure 4C). For the mice without IRAEs, the CD4/CD8 ratio after therapy was 2.34 ± 2.12, which is the normal range of human. But for the mice with IRAEs, the ratio shifts to 8.06 ± 2.69. The CD4/CD8 ratio increased because the percentage of CD4 T cells increased, while the percentage of CD4 T cells decreased in PB. Moreover, the FOXP3+ Treg cells were decreased in mice with IRAEs (P <0.05) (Figures 4A bottom and D). These results indicated that the promotion of CD4+ T cells and the suppression of CD8+ and FOXP3+ Treg cells were associated with the occurrence of IRAEs. The construction of the HPDOX mice model could provide a platform for assessing adverse events of immunotherapies.

Table 2 Establishing autologous GBM HPDOX.

Table 3 Evaluating the IRAEs under the treatment of ipilimumab.
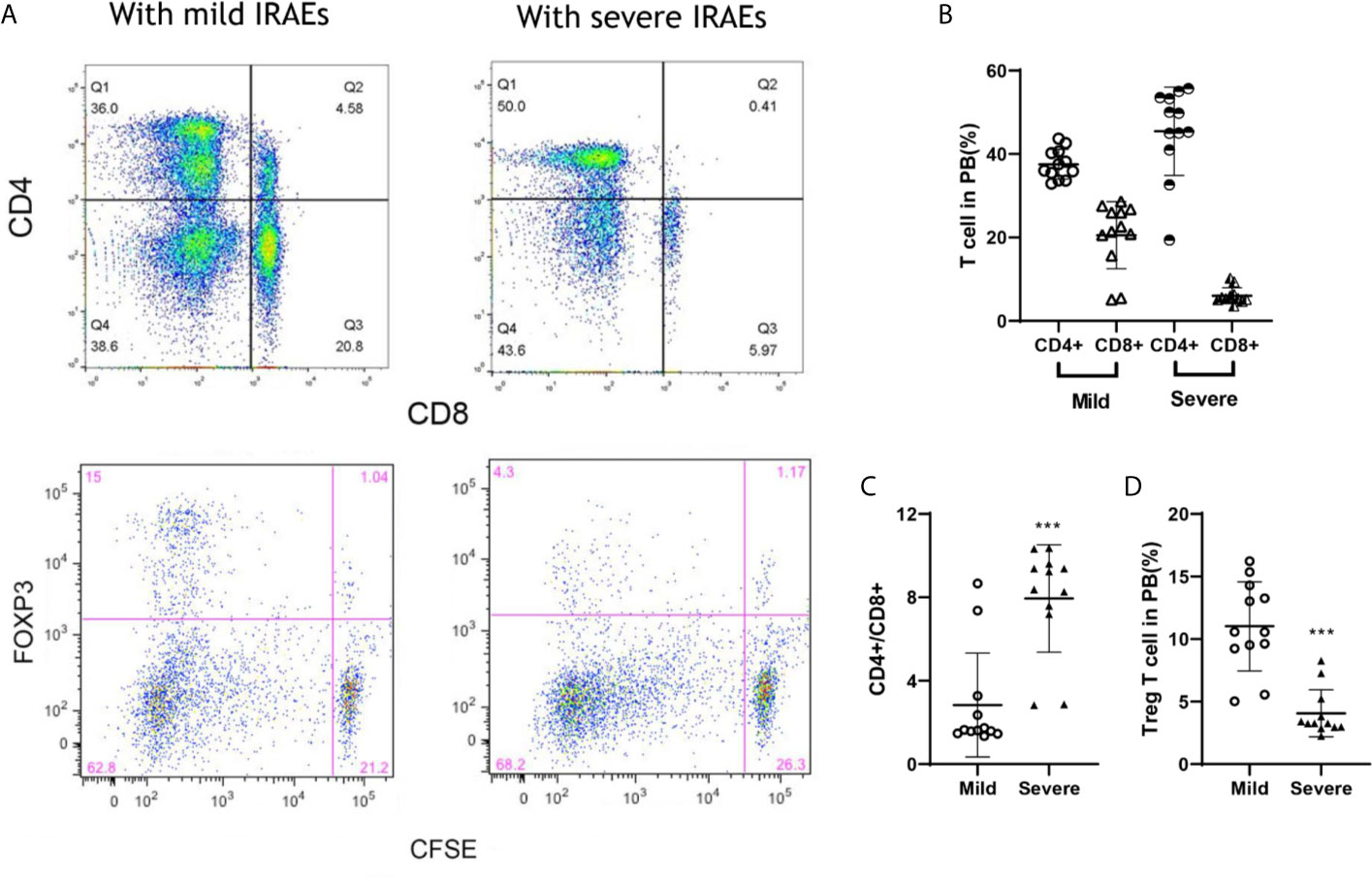
Figure 4 FOXP3+ and CD4+/CD8+ T cells were associated with IRAEs during anti-CTLA4 immunotherapy. (A) The population of CD4+, CD8+, and FOXP3+ T cells after the intervention of ipilimumab. (B, C) The population of T cells (B) and Treg cells (C) in peripheral blood of mice with IRAEs or not. (D) The ratio of CD4+/CD8+ T cells in peripheral blood of the HPDOX mice with IRAEs and not. There were twelve mice in each group. Data were represented as mean ± SD. ***P < 0.001.
The Degree of IRAEs in HPDOX May Be Related to CD56+ Cells
After the treatment of ipilimumab, the HPDOX with mild or severe IRAEs were accessed by flow cytometry to detect the possible mechanism of IRAEs. The whole blood was collected at the 7th and 35th days after the autologous PBMCs transplantation. As is shown in Figure 5, HPDOX suffered systemic IRAEs were assessed by the criterion of mouse GVHD clinical scoring system including weight loss, posture, activity, fur texture, and skin integrity (14). The CD4, CD8, CD25, CD127, CD56, and HLA-DR were detected to evaluate the expression of CD4, CD8 in T cells, the CD25+ CD127- Treg cells, CD56, and HLA-DR (Figures 5A, B). Those results showed, after the treatment of ipilimumab, there was no significant difference in the CD56 expression in the mild IRAEs group (Figure 5C), although increased in the severe IRAEs group. It indicates that the degree of IRAEs in HPDOX may be related to CD56+ cells. Similarly, the CD25+CD127- Treg cells were significantly decreased, while the HLA-DR expression was increased in both mild and severe IRAEs groups (Figures 5D, E).
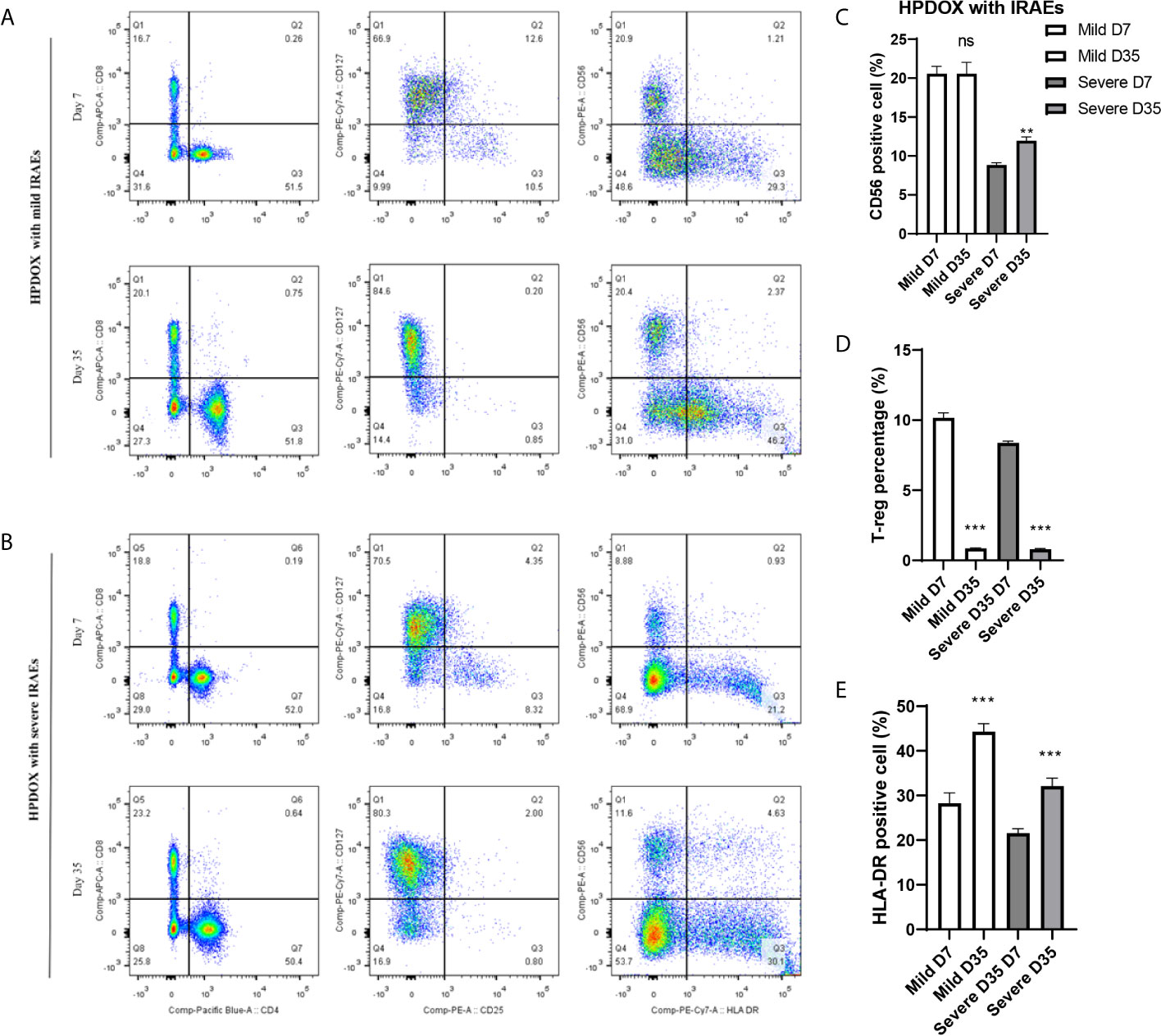
Figure 5 The degree of IRAEs may be related to CD56+ expression in HPDOX. (A, B) The population of CD4+, CD8+, the CD25+CD127− Treg cells, CD56, and HLA-DR in both mild and severe IRAEs groups after the intervention of ipilimumab. (C) There was no significant difference in the CD56 expression in the mild IRAEs group, although increased in the severe IRAEs group. (D, E) The CD25+CD127− Treg cells were significantly decreased, while the HLA-DR expression was increased in both mild and severe IRAEs groups. There were six mice in each group. Data were represented as mean ± SD. **P < 0.01, ***P < 0.001.
Discussion
During the past decades, various animal models have been developed to study brain tumor (15). Although established cell lines such as U87 and U251 have inherited most of the genetic and genomic features of GBM, they bear nothing in common with the actual patients’ GBM such as invasion histologic hallmarks (6). the development of severely immunocompromised mice has laid a solid foundation of PDOX, which has attracted more and more attention to facilitating biologic studies, preclinical drug evaluation, and biomarker identification (4–8). There are two main methods we have tried to obtain single cells for establishing the PDOX mice model: firstly, GBM tumor tissue right after they obtain from surgery is dissociated into single cells by treatment with Accutase™ solution to digest the extracellular material; secondly, the primary GBM cultures of tissue-derived cells which may acquire purer GBM cells but the resemblance disappeared with passages increasingly (6). The primary GBM culture conditions are also be used for the culture of GBM stem cells, which can enhance the success of produce those phenotypes from patients’ tissue in PDOX via few cells with enlarging tumor-initiating potential (4). Therefore, PDOX can reproduce biological features of GBM, such as brain invasion, microvascular proliferation, and anti-tumor therapy. Moreover, PDOX can simulate an appropriate microenvironment for cancer research compared with heterotopic extracranial implantation (16, 17). However, a limitation of PDOX should be addressed that it cannot be used in the researches on the immune of the microenvironment because of the requirement for immune-deficient nude mice as hosts (18).
Therefore, the HPDOX mice model was developed, which was a promising approach to facilitate the understanding of human immunity and the evaluation of the efficacy and IRAEs of immunotherapy in vivo. Stem-like cells derived HPDOX was reported with a low occurrence of IRAEs such as GVHD but represent few features of cancer in patients (19). HPDOX derived from patients’ PBMCs is the easiest way, which has limited application in following antigenic immune responses, but still is applied to access human immunosuppressive reagents (19). Although the novel mouse strains have been developed to inhibit the IRAEs such as GVHD by MHC complex-deficient mouse (20, 21), the immune cell differentiation and response could also be impeded. In this study, we established HPDOX mice models and evaluated the potential mechanism of anti-CTLA4 immunotherapy in GBM. Patients’ PBMCs could be engrafted into PDOX to reconstitute T cells for humanizing. Usually, the GVHD occurs 2–4 weeks after PBMC transfer. To prolong the period for evaluation of the efficacy and IRAEs of immunotherapy, the detail of PBMCs transplantation is essential for the success of HPDOX. The period can be prolonged after engrafted with the decreased number of autologous patients’ PBMCs. Specifically, IRAEs were assessed by weight loss, posture, activity, fur texture, and skin integrity. Although PBMCs derived humanized mice have been thoughted to be an appropriate platform to evaluate the efficacy of targeted therapy or immunotherapy, no previous literature has described its application in assessing the efficacy and IRAEs of immunotherapies in the GBM HPDOX mice model. The reconstitution of PBMCs to T cells is faster than stem-like cells, and it has higher veracity as a model (22). Similarly, our study revealed that the application of PBMCs could efficiently construct the HPDOX mice model to investigate the antitumor activities and IRAEs of anti-CTLA4 antibodies.
Immune checkpoint inhibitors (ICIs) enhance the anti-tumor immune response by blocking Treg-mediated immunosuppression. Why the T cells in the tumor microenvironment are few and non-sensitive still unrevealed. Recently, HPDOX has become a brand-new tool to assess cancer immunotherapy, which sets a robust foundation for cancer immune-related researches (23). In our study, we aim to demonstrate that HPDOX mice are appropriate to investigate the efficacy and IRAEs of anti-CTLA4 antibody therapies against GBM. Thus, this model can be used as a platform to evaluate whether patients benefit from certain targeted immunotherapy or not, which may provide a solid basis for clinical decisions. As one of the most common ICIs, anti-CTLA4 antibodies such as ipilimumab and tremelimumab have enhanced the anti-tumor immune response in both preclinical and clinical research and achieved unprecedented success. The achievement of ICIs mainly on account of two essential factors: 1) attenuate highly immunosuppressive tumor microenvironment by lowering frequencies of Treg cells; and 2) T effective cells are activated in tumor microenvironments (TME) by certain mechanisms, which play a critical role in anti-tumor in cellular immunity (24). Base on those points, anti-CTLA-4 antibodies might weaken their immunosuppressive effects via inhibiting the activated Treg cells.
Anti-CTLA4 antibodies block the CTLA4 molecules enhancing the anti-tumor immune response via inhibiting Treg-mediated immunosuppression in HPDOX. We observed the CTLA4 was blockaded by anti-CTLA4 antibodies and rescued the T cell exhaustion phenotype in the GBM HPDOX mouse model. Persistent exposure to high levels of antigen such as cancer or chronic infections may drive functional exhaustion of T cells (25). Recently, more and more researches focused on reversing T cell depletion abrogates the control of the proliferation of cancer. The critical role of CTLA4 in T cell exhaustion has been reported (26, 27). These findings warrant the clinical trial of CTLA4-targeted immunotherapy for GBM patients (NCT04606316). However, some patients do not benefit from anti-CTLA4 immunotherapy, which might because only a few T cells arrived in TME, which is not enough to reverse the immunosuppressive effects (28). In our study, ipilimumab, an anti-CTLA4 antibody that binds to CTLA4 specifically, showed the anti-GBM efficacy in inhibiting tumor growth via at least partly preserving T cells in the HPDOX model.
IRAEs showed a negative relationship with Treg numbers or percentages in the HPDOX model. This model may provide an emerging and promising tool to reveal the mechanism of clinical efficacy as well as IRAEs of immune-related therapies. IRAEs occurred in more than 90% of patients during the treatment with anti-CTLA4 antibody (29). Systemic administration of ICIs is usually not only influence by T cells in TME but also all T cells across the body. ICIs administrated by vein could induce IRAEs via unbalancing the T effect and Treg cells in normal tissues such as guts and skin. Those possible reasons reported for IRAEs during ICIs. On one side, Treg, expression of CTLA4, was inhibited by ADCC, which mediated by FcR expressing cells such as natural killer cells or macrophages in TME. On the other, T effective cells were activated and sustained via blocking the CTLA4 pathway. IRAEs could be induced by losing the functions or numbers of Treg, which are critical for maintaining tolerance (30). Based on those points, the dual roles of Treg are presented. On one hand, Treg cells can impede anti-tumor immunity to enhance immune evasion of tumor cells; On the other, Treg cells sustain an immune tolerance state and prevent from IRAEs. It reported that a negative relationship between Treg and IRAEs has been demonstrated in preclinical models, but not in GBM. Our study showed the patients’ PBMCs can co-exist in GBM HPDOX mice, which may be a useful platform for investigating the mechanism and role of the immune-related factors in IRAEs. In theory, it is a promising area to reform anti-CTLA4 antibodies and enhance its efficacy of cancer immunotherapy. To weaken ADCC and CDC effects, the N298A (Human IgG control) mutation was designed to prevent immune cells from IRAEs. However, our study showed N298A mutation did not affect the IRAEs of anti-CTLA4 antibodies, which might because patients’ natural killer cells or complement were not well restored in the HPDOX mice model (Figure S4 and Table S2). And the degree of IRAEs may be related to CD56+ expression in HPDOX.
The challenge of immunotherapy is to prevent IRAEs while preserving anti-tumor efficacy. In theory, the immunosuppressors or corticosteroids probably maintain immune tolerance in normal organs and tissues, which could also impede the antitumor efficacy. But they are not weakening the antitumor efficacy of ICIs. Interestingly, some clinical trials showed a positive relationship between the IRAEs and antitumor responses (31, 32). It’s a promising direction to study this mechanism in the future (33, 34). In the future, our study will also aim to make the current anti-CTLA4 immunotherapy more effective via inhibiting Treg cells and weaken their immunosuppressive effects. In the meanwhile, activating T effective cells by ICIs or vaccine in TME.
However, some important limitations should be considered. In the first place, in the tissues and plasma of mice, there are only low levels of human factors and cytokines resulted in a decreased number of myeloid cells and Treg cells. With the presence of GM-CSF, IL-6, IL-3, and M-CSF, the differentiation of T, B, and NK cells were more actively (35, 36). Secondary, although the novel mouse strains have been developed to inhibit the GVHD by mouse MHC complex, the immune cell types and responses in HPDOX still need to be further studied (12, 37). Thirdly, accessing the change in the transcriptome and epigenome of the patient tumor tissues is passaged in the HPDOX models (19, 38). Lastly, personalized medicine strategies will be needed to allow higher tumor infiltration and anti-tumor responses.
Conclusions
In conclusion, our study established HPDOX mice models for investigating the mechanism and IRAEs of immunotherapies in GBM, which would offer a promising platform for evaluating the efficacy and IRAEs of novel therapies and exploring personalized therapeutic strategies.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding author.
Ethics Statement
The studies involving human participants were reviewed and approved by the Ethics Committee of Xiangya Hospital. The patients/participants provided their written informed consent to participate in this study. The animal study was reviewed and approved by the Ethics Committee of Xiangya Hospital.
Author Contributions
CL conceived, designed, and supervised the study. SX and XY drafted the manuscript. SX and XY performed data analysis. SX, XY, and GD collected the data. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the National Natural Science Foundation of China (81902553); Natural Science Foundation of Hunan Province (2019JJ50942).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to show our gratitude to Prof. Chen (Huntsman Cancer Institute, University of Utah, USA) for providing CTLA4-specific antibodies (ipilimumab and tremelimumab).
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2021.692403/full#supplementary-material
Supplementary Figure 1 | The tumor volume before antibodies treatment. (A) The tumor volume of PDOX 07; (B) The tumor volume of PDOX 22. There were three mice in each group. Data were represented as mean ± SD. ns: P > 0.05.
Supplementary Figure 2 | The subgroups of T cells. (A–C) The activated T cells were presented with the higher expression of IFN-γ+ and CD25+. (B) The exhausted T cells presented the lower expression of IFN-γ+. (a & d) The effector T cells are CD45RA positive. (a & e) The memory T cells are CD45RO positive.
Supplementary Figure 3 | HPDOX with IRAEs such as graft-versus-host disease (GVHD). (left) The normal autologous GBM HPDOX without IRAES like GVHD. (right) The autologous GBM HPDOX mice suffered IRAES and/or GVHD. Specifically, IRAEs was assessed by weight and hair loss, posture, activity, fur texture and skin integrity.
Supplementary Figure 4 | Evaluating the antitumor effect of ipilimumab without and with the N298A mutation in HPDOX. (A, B) Tumor volume of GBM xenograft in mice treated with Vehicle, huIgG, ipilimumab, and ipilimumab with N298A. (C, E) The population of T cells in GBM tissues and (D, F) peripheral blood in mice treated with Vehicle, huIgG, ipilimumab, and ipilimumab with N298A. There were six mice in each group. Data were represented as mean ± SD. ns, no significant difference.
Abbreviations
PDOX, patient-derived orthotopic xenograft; GBM, glioblastoma; HPDOX, humanized patient-derived orthotopic xenograft; Treg, regulatory T; IRAEs, immune-related adverse events; PBMCs, peripheral blood mononuclear cells; PB, peripheral blood; GVHD, graft-versus-host disease; ICI, immune checkpoint inhibitors.
References
1. Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, et al. Radiotherapy Plus Concomitant and Adjuvant Temozolomide for Glioblastoma. N Engl J Med (2005) 352(10):987–96. doi: 10.1056/NEJMoa043330
2. Chinot OL, Wick W, Mason W, Henriksson R, Saran F, Nishikawa R, et al. Bevacizumab Plus Radiotherapy-Temozolomide for Newly Diagnosed Glioblastoma. N Engl J Med (2014) 370(8):709–22. doi: 10.1056/NEJMoa1308345
3. Gilbert MR, Dignam JJ, Armstrong TS, Wefel JS, Blumenthal DT, Vogelbaum MA, et al. A Randomized Trial of Bevacizumab for Newly Diagnosed Glioblastoma. N Engl J Med (2014) 370(8):699–708. doi: 10.1056/NEJMoa1308573
4. Singh SK, Hawkins C, Clarke ID, Squire JA, Bayani J, Hide T, et al. Identification of Human Brain Tumour Initiating Cells. Nature (2004) 432(7015):396–401. doi: 10.1038/nature03128
5. Bao S, Wu Q, McLendon RE, Hao Y, Shi Q, Hjelmeland AB, et al. Glioma Stem Cells Promote Radioresistance by Preferential Activation of the DNA Damage Response. Nature (2006) 444(7120):756–60. doi: 10.1038/nature05236
6. Lee J, Kotliarova S, Kotliarov Y, Li A, Su Q, Donin NM, et al. Tumor Stem Cells Derived From Glioblastomas Cultured in bFGF and EGF More Closely Mirror the Phenotype and Genotype of Primary Tumors Than do Serum-Cultured Cell Lines. Cancer Cell (2006) 9(5):391–403. doi: 10.1016/j.ccr.2006.03.030
7. Hidalgo M, Amant F, Biankin AV, Budinska E, Byrne AT, Caldas C, et al. Patient-Derived Xenograft Models: An Emerging Platform for Translational Cancer Research. Cancer Discov (2014) 4(9):998–1013. doi: 10.1158/2159-8290.CD-14-0001
8. Tentler JJ, Tan AC, Weekes CD, Jimeno A, Leong S, Pitts TM, et al. Patient-Derived Tumour Xenografts as Models for Oncology Drug Development. Nat Rev Clin Oncol (2012) 9(6):338–50. doi: 10.1038/nrclinonc.2012.61
9. Ye W, Luo C, Li C, Huang J, Liu F. Organoids to Study Immune Functions, Immunological Diseases and Immunotherapy. Cancer Lett (2020) 477:31–40. doi: 10.1016/j.canlet.2020.02.027
10. Klein E, Hau AC, Oudin A, Golebiewska A, Niclou SP. Glioblastoma Organoids: Pre-Clinical Applications and Challenges in the Context of Immunotherapy. Front Oncol (2020) 10:604121. doi: 10.3389/fonc.2020.604121
11. Bar-Ephraim YE, Kretzschmar K, Clevers H. Organoids in Immunological Research. Nat Rev Immunol (2020) 20(5):279–93. doi: 10.1038/s41577-019-0248-y
12. Byrne AT, Alferez DG, Amant F, Annibali D, Arribas J, Biankin AV, et al. Interrogating Open Issues in Cancer Precision Medicine With Patient-Derived Xenografts. Nat Rev Cancer (2017) 17(4):254–68. doi: 10.1038/nrc.2016.140
13. King MA, Covassin L, Brehm MA, Racki W, Pearson T, Leif J, et al. Human Peripheral Blood Leucocyte non-Obese Diabetic-Severe Combined Immunodeficiency Interleukin-2 Receptor Gamma Chain Gene Mouse Model of Xenogeneic Graft-Versus-Host-Like Disease and the Role of Host Major Histocompatibility Complex. Clin Exp Immunol (2009) 157(1):104–18. doi: 10.1111/j.1365-2249.2009.03933.x
14. Lai HY, Chou TY, Tzeng CH, Lee OK. Cytokine Profiles in Various Graft-Versus-Host Disease Target Organs Following Hematopoietic Stem Cell Transplantation. Cell Transplant (2012) 21(9):2033–45. doi: 10.3727/096368912X653110
15. Huszthy PC, Daphu I, Niclou SP, Stieber D, Nigro JM, Sakariassen PO, et al. In Vivo Models of Primary Brain Tumors: Pitfalls and Perspectives. Neuro Oncol (2012) 14(8):979–93. doi: 10.1093/neuonc/nos135
16. Jamal M, Rath BH, Tsang PS, Camphausen K, Tofilon PJ. The Brain Microenvironment Preferentially Enhances the Radioresistance of CD133(+) Glioblastoma Stem-Like Cells. Neoplasia (2012) 14(2):150–8. doi: 10.1593/neo.111794
17. Camphausen K, Purow B, Sproull M, Scott T, Ozawa T, Deen DF, et al. Orthotopic Growth of Human Glioma Cells Quantitatively and Qualitatively Influences Radiation-Induced Changes in Gene Expression. Cancer Res (2005) 65(22):10389–93. doi: 10.1158/0008-5472.CAN-05-1904
18. Oh T, Fakurnejad S, Sayegh ET, Clark AJ, Ivan ME, Sun MZ, et al. Immunocompetent Murine Models for the Study of Glioblastoma Immunotherapy. J Transl Med (2014) 12:107. doi: 10.1186/1479-5876-12-107
19. Stripecke R, Munz C, Schuringa JJ, Bissig KD, Soper B, Meeham T, et al. Innovations, Challenges, and Minimal Information for Standardization of Humanized Mice. EMBO Mol Med (2020) 12(7):e8662. doi: 10.15252/emmm.201708662
20. Yaguchi T, Kobayashi A, Inozume T, Morii K, Nagumo H, Nishio H, et al. Human PBMC-transferred Murine MHC Class I/II-deficient NOG Mice Enable Long-Term Evaluation of Human Immune Responses. Cell Mol Immunol (2018) 15(11):953–62. doi: 10.1038/cmi.2017.106
21. Brehm MA, Kenney LL, Wiles MV, Low BE, Tisch RM, Burzenski L, et al. Lack of Acute Xenogeneic Graft- Versus-Host Disease, But Retention of T-cell Function Following Engraftment of Human Peripheral Blood Mononuclear Cells in NSG Mice Deficient in MHC Class I and II Expression. FASEB J (2019) 33(3):3137–51. doi: 10.1096/fj.201800636R
22. Lin S, Huang G, Cheng L, Li Z, Xiao Y, Deng Q, et al. Establishment of Peripheral Blood Mononuclear Cell-Derived Humanized Lung Cancer Mouse Models for Studying Efficacy of PD-L1/PD-1 Targeted Immunotherapy. MAbs (2018) 10(8):1301–11. doi: 10.1080/19420862.2018.1518948
23. Jespersen H, Lindberg MF, Donia M, Soderberg EMV, Andersen R, Keller U, et al. Clinical Responses to Adoptive T-cell Transfer can be Modeled in an Autologous Immune-Humanized Mouse Model. Nat Commun (2017) 8(1):707. doi: 10.1038/s41467-017-00786-z
24. Tanaka A, Sakaguchi S. Regulatory T Cells in Cancer Immunotherapy. Cell Res (2017) 27(1):109–18. doi: 10.1038/cr.2016.151
25. Mueller SN, Ahmed R. High Antigen Levels are the Cause of T Cell Exhaustion During Chronic Viral Infection. Proc Natl Acad Sci U S A (2009) 106(21):8623–8. doi: 10.1073/pnas.0809818106
26. Zarour HM. Reversing T-cell Dysfunction and Exhaustion in Cancer. Clin Cancer Res (2016) 22(8):1856–64. doi: 10.1158/1078-0432.CCR-15-1849
27. Farhood B, Najafi M, Mortezaee K. CD8(+) Cytotoxic T Lymphocytes in Cancer Immunotherapy: A Review. J Cell Physiol (2019) 234(6):8509–21. doi: 10.1002/jcp.27782
28. Havel JJ, Chowell D, Chan TA. The Evolving Landscape of Biomarkers for Checkpoint Inhibitor Immunotherapy. Nat Rev Cancer (2019) 19(3):133–50. doi: 10.1038/s41568-019-0116-x
29. Michot JM, Bigenwald C, Champiat S, Collins M, Carbonnel F, Postel-Vinay S, et al. Immune-Related Adverse Events With Immune Checkpoint Blockade: A Comprehensive Review. Eur J Cancer (2016) 54:139–48. doi: 10.1016/j.ejca.2015.11.016
30. Kumar P, Saini S, Prabhakar BS. Cancer Immunotherapy With Check Point Inhibitor can Cause Autoimmune Adverse Events Due to Loss of Treg Homeostasis. Semin Cancer Biol (2020) 64:29–35. doi: 10.1016/j.semcancer.2019.01.006
31. Baik CS, Rubin EH, Forde PM, Mehnert JM, Collyar D, Butler MO, et al. Immuno-Oncology Clinical Trial Design: Limitations, Challenges, and Opportunities. Clin Cancer Res (2017) 23(17):4992–5002. doi: 10.1158/1078-0432.CCR-16-3066
32. Chang LS, Barroso-Sousa R, Tolaney SM, Hodi FS, Kaiser UB, Min L. Endocrine Toxicity of Cancer Immunotherapy Targeting Immune Checkpoints. Endocr Rev (2019) 40(1):17–65. doi: 10.1210/er.2018-00006
33. Sanmamed MF, Chen L. A Paradigm Shift in Cancer Immunotherapy: From Enhancement to Normalization. Cell (2018) 175(2):313–26. doi: 10.1016/j.cell.2018.09.035
34. Ye Q, Zhang Y, Cao Y, Wang X, Guo Y, Chen J, et al. Frenolicin B Targets Peroxiredoxin 1 and Glutaredoxin 3 to Trigger Ros/4e-Bp1-Mediated Antitumor Effects. Cell Chem Biol (2019) 26(3):366–77.e12. doi: 10.1016/j.chembiol.2018.11.013
35. Yu H, Borsotti C, Schickel JN, Zhu S, Strowig T, Eynon EE, et al. A Novel Humanized Mouse Model With Significant Improvement of Class-Switched, Antigen-Specific Antibody Production. Blood (2017) 129(8):959–69. doi: 10.1182/blood-2016-04-709584
36. Bryce PJ, Falahati R, Kenney LL, Leung J, Bebbington C, Tomasevic N, et al. Humanized Mouse Model of Mast Cell-Mediated Passive Cutaneous Anaphylaxis and Passive Systemic Anaphylaxis. J Allergy Clin Immunol (2016) 138(3):769–79. doi: 10.1016/j.jaci.2016.01.049
37. De La Rochere P, Guil-Luna S, Decaudin D, Azar G, Sidhu SS, Piaggio E. Humanized Mice for the Study of Immuno-Oncology. Trends Immunol (2018) 39(9):748–63. doi: 10.1016/j.it.2018.07.001
Keywords: glioblastoma, humanized patient-derived orthotopic xenograft, immune-related adverse events, anti-CTLA4 immunotherapy, regulatory T cells
Citation: Xu S, Yan X, Dai G and Luo C (2021) A Novel Mice Model for Studying the Efficacy and IRAEs of Anti-CTLA4 Targeted Immunotherapy. Front. Oncol. 11:692403. doi: 10.3389/fonc.2021.692403
Received: 08 April 2021; Accepted: 20 May 2021;
Published: 10 June 2021.
Edited by:
Haichang Li, The Ohio State University, United StatesReviewed by:
Shouheng Lin, Chinese Academy of Sciences (CAS), ChinaGamal Eldein Fathy Abd-Ellatef Abd-Elrahman, National Research Centre, Egypt
Copyright © 2021 Xu, Yan, Dai and Luo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Chengke Luo, Y2tfbHVvQGNzdS5lZHUuY24=
†These authors have contributed equally to this work