 Isabella Giacomini1†
Isabella Giacomini1† Federico Gianfanti1,2†
Federico Gianfanti1,2† Maria Andrea Desbats2
Maria Andrea Desbats2 Genny Orso1
Genny Orso1 Massimiliano Berretta3Tommaso Prayer-Galetti4
Massimiliano Berretta3Tommaso Prayer-Galetti4 Eugenio Ragazzi1*
Eugenio Ragazzi1* Veronica Cocetta1
Veronica Cocetta1- 1Department of Pharmaceutical and Pharmacological Sciences, University of Padova, Padova, Italy
- 2Veneto Institute of Molecular Medicine, VIMM, Padova, Italy
- 3Department of Clinical and Experimental Medicine, University of Messina, Messina, Italy
- 4Department of Surgery, Oncology and Gastroenterology - Urology, University of Padova, Padova, Italy
Cholesterol is a ubiquitous sterol with many biological functions, which are crucial for proper cellular signaling and physiology. Indeed, cholesterol is essential in maintaining membrane physical properties, while its metabolism is involved in bile acid production and steroid hormone biosynthesis. Additionally, isoprenoids metabolites of the mevalonate pathway support protein-prenylation and dolichol, ubiquinone and the heme a biosynthesis. Cancer cells rely on cholesterol to satisfy their increased nutrient demands and to support their uncontrolled growth, thus promoting tumor development and progression. Indeed, transformed cells reprogram cholesterol metabolism either by increasing its uptake and de novo biosynthesis, or deregulating the efflux. Alternatively, tumor can efficiently accumulate cholesterol into lipid droplets and deeply modify the activity of key cholesterol homeostasis regulators. In light of these considerations, altered pathways of cholesterol metabolism might represent intriguing pharmacological targets for the development of exploitable strategies in the context of cancer therapy. Thus, this work aims to discuss the emerging evidence of in vitro and in vivo studies, as well as clinical trials, on the role of cholesterol pathways in the treatment of cancer, starting from already available cholesterol-lowering drugs (statins or fibrates), and moving towards novel potential pharmacological inhibitors or selective target modulators.
Introduction
In the last decades, the study of metabolic reprogramming has been revealed as one of the hallmarks of cancer and chemotherapy resistance. It has been demonstrated that cancer cells change their metabolism, increasing glucose demand, glutamine or lipid synthesis, exploiting the pentose phosphate pathway or altering their mitochondrial function, in order to support a higher proliferation rate leading to tumor progression and chemotherapy resistance (1–10). Among these altered pathways, cholesterol metabolic reprogramming has acquired a pivotal role in the field of cancer research.
The discovery of cholesterol dates back to the second half of the eighteenth century, when Poulletier de la Salle isolated for the first time this molecule from human gallstone and bile. Since then, a huge number of researches were undertaken, which eventually led to the understanding of key molecular events of cholesterol biology, such as transport in blood and cellular metabolism. Nowadays, this peculiar lipid is still extensively studied for its involvement in several pathophysiological processes (11, 12). Cholesterol is a ubiquitous sterol found in vertebrate organisms with a plethora of biological functions that are essential for proper cellular growth and activity (13, 14). Due to its alicyclic nature, cholesterol is highly hydrophobic and resides predominantly within the phospholipidic bilayer of cell membranes, where it preserves the barrier function by modulating permeability, fluidity and rigidity (15, 16). In this setting, cholesterol preferentially interacts with the saturated acyl chains of adjacent sphingolipids and glycophosphatidylinositol-anchored proteins of the outer leaflet, forming small ordered and tightly packed microdomains, physically separated from the shorter and unsaturated phospholipids of the bilayer (16–18). These assemblies, usually called lipid rafts, are involved in several biological processes, such as biosynthetic and endocytic vesicular trafficking (19), ceramide-mediated apoptosis (20), host-pathogen interactions (pathogen binding and uptake) (21), cytoskeletal dynamics and rearrangement, cellular polarization (22) and signal transduction (IgE signaling, T-cell antigen receptor signaling, Ras signaling, Hedgehog signaling) (23). Although the most known role of cholesterol as a structural and functional component of cellular membranes is unquestionable (15, 16, 18), it also represents the precursor of bile acids, and its oxidation allows the biosynthesis of steroid hormones in steroid-producing tissues. In addition, the isoprenoid intermediates of the mevalonate pathway can be diverted toward the biosynthesis of dolichol, ubiquinone and the side tail of heme a (24, 25), or exploited as substrates for protein-prenylation (26). Lastly, cholesterol has also been found to interact with a large variety of proteins, including receptors, enzymes, etc. by both covalent and non-covalent binding, thus regulating protein stability, localization, and activity. These interactions indicate cholesterol as an important element in the regulation of many biochemical pathways, through the control of protein localization and activity (27).
Due to the crucial role played by this sterol in several physiological settings, disruption of cholesterol homeostasis and metabolic reprogramming may be responsible for the development of cardiovascular disorders and is implicated in the pathogenesis of diabetes, Alzheimer disease and many types of cancer (28–32). Intracellular and systemic cholesterol concentrations are tightly regulated by the balance between de novo biosynthesis, uptake, efflux, and storage, and metabolic alterations in lipid/cholesterol pathways have been shown to modulate cancer cells’ sensitivity to chemotherapeutic agents. The dependence of cancer cells on aberrant lipid and cholesterol metabolism could point to these pathways as an attractive target to treat cancer as well as to sensitize them to anticancer therapies (33). Many cholesterol-lowering drugs are approved and used for the treatment of hypercholesterolemia and for the control of pathologies and metabolic disorders. This work focuses on the correlation between cholesterol metabolism and cancer, considering the importance of these pathways in sustaining cell growth, invasion or migration. Furthermore, starting from the relatively recent findings on the role of sterol in tumor progression and chemotherapy response, we will consider how the pharmacological targeting of increased cholesterol metabolism pathways could represent a promising approach for cancer treatment.
Cholesterol Metabolism
Cholesterol metabolism in humans is complex. Cholesterol is either supplied from the diet (exogenous) or synthesized de novo (~70% of total body cholesterol, endogenous). Here below we provide a brief section on the main aspects related to cholesterol metabolism, introductory to understanding the reprogramming aspect observed in cancer cells. For a more accurate description of the fine regulation of cellular processes involving cholesterol, we refer to several specific reviews (34–37).
Cholesterol Biosynthesis
The biosynthetic cascade which leads to cholesterol production (Figure 1) occurs virtually in every mammalian cell, with liver and intestine being the anatomical sites responsible for more than 50% of total cholesterol biosynthesis (38, 39). This process is orchestrated by more than 20 enzymes which are distributed between the cytosol and the endoplasmic reticulum (ER) (40). The first step is catalyzed by the cytoplasmatic enzyme acetylacetyl-CoA thiolase which allows the condensation of two acetyl-CoA molecules to obtain acetylacetyl-CoA. In the second reaction, the enzyme 3-hydroxy-3-methylglutaryl-CoA synthase (HMGCS) allows the introduction of the third molecule of acetyl-CoA for the formation of the branched-chain molecule 3-hydroxy-3-methylglutaryl-CoA, which is then reduced to mevalonate in the first rate-limiting step of cholesterol biosynthesis by 3-hydroxy-3-methylglutaryl-CoA reductase (HMGCR). Afterwards, mevalonate undergoes two subsequent phosphorylations performed by mevalonate kinase (MVK) and phosphomevalonate kinase (PMVK) and an ATP-dependent decarboxylation which eventually yields the isoprenoid precursor isopentyl pyrophosphate (IPP). This intermediate is converted into its isomer dimethylallyl pyrophosphate (DMAPP) in a reversible reaction catalyzed by isopentenyl pyrophosphate isomerase. The condensation of one molecule of IPP with one molecule of DMAPP allows the formation of geranyl pyrophosphate (GPP), which is in turn combined with another IPP molecule by the enzyme farnesyl diphosphate synthase to yield farnesyl pyrophosphate (FPP), a key isoprenoid. At this point, the mevalonate pathway diverts toward the formation of either non-sterol isoprenoids, such as geranylgeranyl pyrophosphate (GGPP), or sterols, through the head-to-head condensation of two FPP molecules, mediated by squalene synthase, which gives rise to squalene. Intracellular accumulation of non-sterol products is required for post-translational modification processes (N-glycosylation and Cys-prenylation) of diverse proteins that play important roles in cellular growth and signal transduction (24, 41). On the other hand, squalene epoxidase (SQLE), the other rate-limiting enzyme of cholesterol biosynthesis, converts squalene into its epoxydic form 2,3-epoxysqualene, which is then cyclized to lanosterol by the enzyme lanosterol synthase. The last phase of cholesterol biosynthesis involves 19 oxygen-based reactions which include demethylations, double-bond reductions, and double bond replacements. In this context, lanosterol enters the Bloch branch or the Kandutsch–Russell pathway and is processed through the formation of several intermediates which yields desmosterol and 7-dehydrocholesterol, the direct precursors of cholesterol (42–44). Recently, the existence of a third hybrid pathway has also been suggested for the conversion of lanosterol into cholesterol (45).
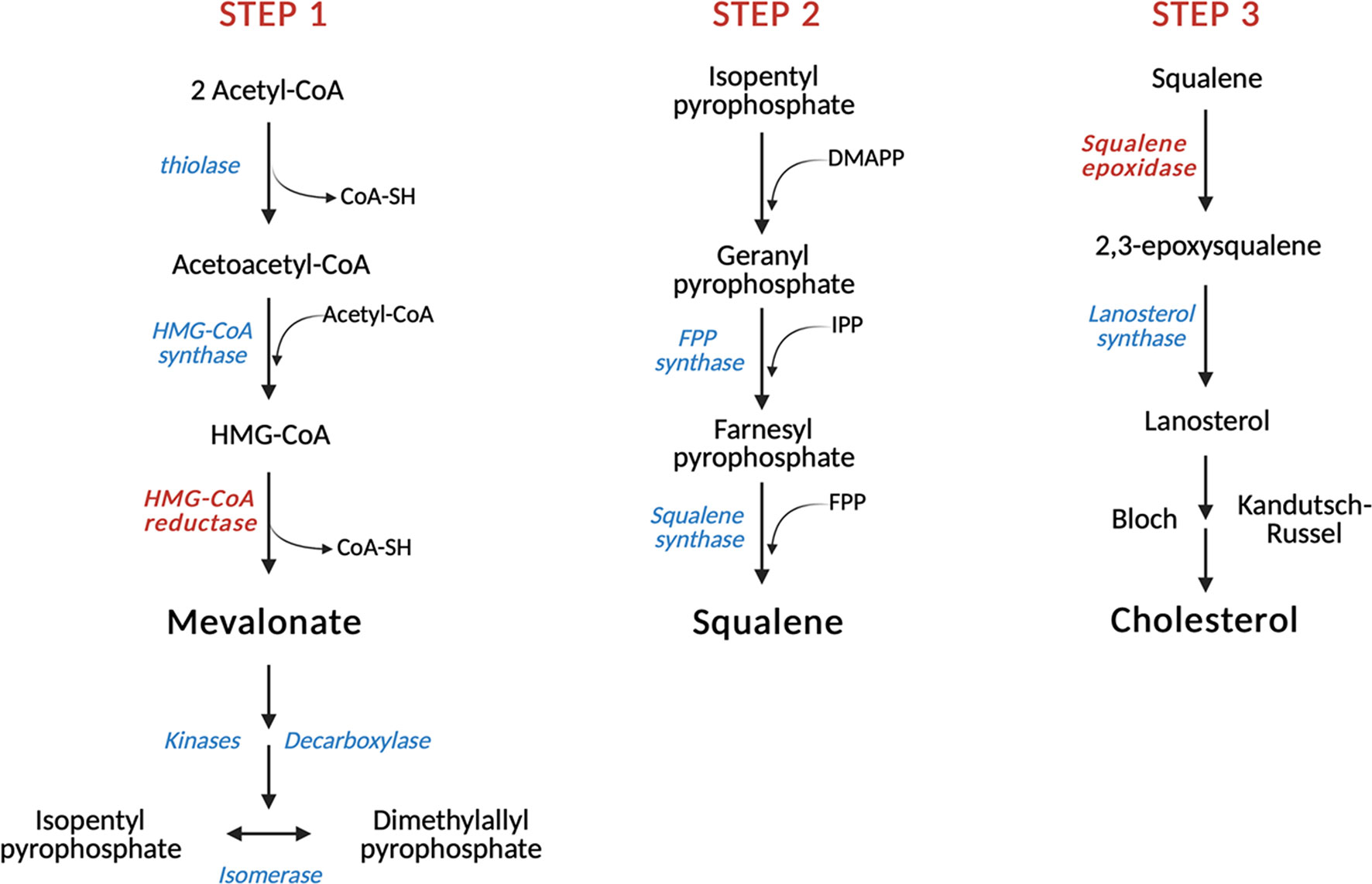
Figure 1 Schematic representation of cholesterol biosynthesis. In the first step of cholesterol biosynthesis, three molecules of acetyl-CoA condense to form HMG-CoA, which is then reduced to mevalonate by the first step-limiting enzyme HMG-CoA reductase (HMGCR). Subsequent reactions allow the conversion of mevalonate into FPP, an isoprenoid that gives rise to squalene in a reaction catalyzed by squalene synthase (SQS). Squalene is then converted by the second rate-limiting enzyme squalene epoxidase (SQLE) into its epoxidic form, which is eventually cyclized to lanosterol by the enzyme lanosterol synthase. Further oxygen-based reactions lead to the formation of cholesterol. Red: rate-limiting enzymes. HMG-CoA, 3-hydroxy-3-methylglutaryl-CoA; IPP, Isopentyl pyrophosphate; DMAPP, Dimethylallyl pyrophosphate; FPP, Farnesyl pyrophosphate.
Cholesterol Uptake and Efflux
The dietary intake of cholesterol is extremely important to ensure the maintenance of its homeostasis (46, 47). In the small intestinal lumen, dietary sterols are solubilized into micelles by bile acids and adsorbed in a process facilitated by the Niemann–Pick C1-like-1 (NPC1L1) protein, which is localized in the apical membrane of enterocytes and allows cholesterol uptake in a clathrin-mediated endocytosis fashion (48, 49). Once inside the enterocyte, cholesterol is mainly converted to cholesteryl esters by the ER enzyme acyl-coenzymeA cholesterol acetyltransferase 2 (ACAT2) and then packed into nascent chylomicrons, together with dietary triglycerides and apolipoprotein B-48 (50, 51). Through the lymphatic system, chylomicrons are poured into the bloodstream and metabolized by the endothelial enzyme lipoprotein lipase, which hydrolyzes the triglycerides contained in the core to yield chylomicron remnants. The released fatty acids are used by peripheral tissues including muscles and adipose tissue either for storage or oxidation, while dietary cholesterol is delivered to the liver by chylomicron remnants (52, 53). Hepatic cholesterol and triglycerides are coupled to apolipoprotein B and incorporated into VLDL particles, which are secreted into the blood and hydrolyzed by plasma lipases to yield IDL. IDLs are further converted into LDLs, particles rich in cholesterol and cholesteryl esters which are captured by LDL receptor-expressing tissues, including the liver and other extrahepatic tissues (54, 55). On the other hand, LDL is driven towards the lysosomal compartment where lysosomal lipases hydrolyze the cholesteryl esters stored in the core to cholesterol, which eventually exits from the lysosome lumen aided by the coordinated action of NPC1, NPC2 and LAMP2 and reaches other cellular organelles, mostly via non-vesicular transport mediated by sterol transfer proteins (STPs) (56–58). Cholesterol elimination also significantly impacts cellular homeostasis. Therefore, the excess of cellular cholesterol in peripheral tissues has to be stored as less-toxic cholesteryl esters in lipid droplets or disposed and moved towards the liver for recycling or excretion, by a process usually referred to as reverse cholesterol transport (59, 60). Cholesterol removal from extrahepatic cells is driven by HDL particles, which accumulate and transport cholesteryl esters to the liver, the adrenal glands, and the gonads (61). Cholesteryl esters are then converted to free cholesterol by cholesteryl ester hydrolase (CEH) for either steroid hormones synthesis in steroid-producing organs or cholesterol excretion and bile acids synthesis in the liver (62, 63). Cellular cholesterol efflux is controlled by four regulatory proteins belonging to the ATP-binding cassette (ABC) transporter superfamily, namely ABCA1, ABCG1, ABCG5 and ABCG8. ABCA1 mediates the transport of cholesterol and phospholipids to lipid-free apolipoprotein A-I (apo A-I) in the blood allowing the generation of nascent discoidal HDL particles, which are converted into globular and mature HDLs under the action of lecithin:cholesterol acyl transferase (LCAT) by accepting further cholesterol from ABCG1 (63, 64).
Cholesterol Storage
Intracellular cholesterol excess is usually esterified by the ER enzyme acyl coenzyme A cholesterol acetyltransferase (ACAT), which catalyzes the transfer of a fatty acyl group to cholesterol (65). Indeed, ACAT-produced cholesteryl esters can be easily stored into lipid droplets preventing free-cholesterol lipotoxicity (66). Cholesterol esterification is also involved in lipoprotein and steroid hormone production, as well as in chylomicrons assembly for cholesterol absorption (67). Two ACAT isoenzymes have been identified in mammals so far, consistent with their different tissue distribution. ACAT1 is widely expressed throughout the body, suggesting its involvement in maintaining cholesterol homeostasis, while ACAT2 expression is exclusive in enterocytes and hepatocytes, where it contributes to lipoprotein biosynthesis and assembly (68, 69).
Regulation of Cholesterol Homeostasis
In order to ensure the maintenance of cellular and systemic cholesterol homeostasis, mammalian cells must carefully orchestrate the set of molecular pathways involved in cholesterol biosynthesis, uptake, storage and efflux (70, 71). This is accomplished by sterol-sensitive systems, which couple variations in cellular sterol levels with adaptive responses. Particularly, three adaptive factors are considered as key regulators of cholesterol homeostasis, namely sterol regulatory element-binding protein-2 (SREBP2), liver X receptors (LXRs) and nuclear factor erythroid 2 related factor-1 (NRF1) (40). SREBP2 belongs to the basic-helix-loop-helix-leucine zipper (bHLH-Zip) family of transcription factors and lies within the ER membrane associated with SREBP-cleavage activating protein (SCAP) through its C-terminal portion (72). The N-terminal transcription factor portion, usually referred to as nuclear SREBP2 (nSREBP2), undergoes dimerization and is then imported inside the nucleus, where binds to sterol responsive elements (SREs) in the promoter regions of target genes, inducing their transcription (73, 74). Conversely, when ER-membrane cholesterol levels increase above the threshold, the sterol sensitive domain (SSD) of SCAP binds to cholesterol and SCAP switches to an open conformation promoting its interaction with insulin-induced gene 1 (INSIG1) protein (75). nSREBP2 binds to and induces the transcription of HMGCR and SQLE genes, which encode for the two rate-limiting enzymes of cholesterol biosynthesis, increasing sterols intracellular levels (76, 77). HMGCR levels are also regulated either by direct interaction with ER-sterols through its SSD (INSIG1-mediated ubiquitination) or by covalent modification (AMPK-mediated phosphorylation) (78, 79). SREBP2 activation also increases the expression of NPC1L1 and LDLR genes, two master regulators of cholesterol intestinal absorption and cholesterol intake by peripheral cells, respectively (80, 81). Moreover, a SRE motif is contained upstream of SREBP2 gene, suggesting that nSREBP2 promotes the activation of its own gene (feed-forward mechanism) (82). Under increasing cholesterol levels, the ER preserves cellular homeostasis by recruiting the adaptive factor NRF1 (61). NRF1 resides within the ER-membrane but is rapidly activated by proteolysis, released from the ER and translocated into the nucleus, where it regulates the transcription of its target genes by binding to anti-oxidant response elements (AREs) (83). Particularly, when NRF1 is activated and enters the nucleus, it represses the transcriptional activity of LXR, which promotes cholesterol excretion, export and storage, while inhibiting de novo biosynthesis. Differentially to SREBP2 and NRF1, LXRs are nuclear receptors which, upon heterodimerization with the retinoid X receptor-α (RXR α), bind to LXR responsive elements (LXRE) and regulate the expression of several genes involved in lipid homeostasis (84). Once activated, LXRs promotes the activation of genes involved in bile acids production (CYP7A1), cholesterol excretion (ABCG5, ABCG8) and reverse cholesterol transport (ABCA1, ABCG1) (85–87). Moreover, LXRs impair cholesterol intestinal absorption by down-regulating NPC1L1 expression and inhibit cholesterol cellular uptake by promoting IDOL-mediated LDLR degradation (88–90). Overall, LXRs activity prevents lipotoxicity induced by intracellular accumulation of sterols.
Cholesterol Lowering Drugs
Since cholesterol plays a key role in many cellular processes, disruption of cholesterol homeostasis is linked to the onset of several diseases, including metabolic disorders, atherosclerosis, cancer, etc. Several therapeutic classes of drugs are currently used to treat hypercholesterolemia (Table 1) and to prevent associated cardiovascular diseases (110). Statins are the first-line treatment of hypercholesterolemia and they have an important role in the prevention of cardiovascular diseases. Statins are competitive inhibitors of 3-hydroxy-3-methylglutaryl-CoA reductase (HMGCR), the enzyme responsible for the reduction of HMG-CoA into mevalonate (91). This specific block causes effects on cholesterol metabolism, such as diminished plasma triglycerides, enhanced HDL, and upregulation of LDL receptor (LDLR) expression, which leads to increased LDL uptake in hepatocytes and decreased blood LDL content (92). Fibrates are another therapeutic class of drugs prescribed to treat hypercholesterolemia (98, 99). They are agonists of the transcription factor PPARα, that once activated, translocates in the nucleus, heterodimerizes with the retinoid X receptor (RXR) and binds to peroxisome proliferator response elements (PPREs) starting the gene’s transcription (100). The effects on lipoprotein metabolism and cellular cholesterol homeostasis are decreased hepatic synthesis and decreased serum levels of triglycerides, reduced synthesis of VLDL, increased HDL cholesterol, and regulation in fatty acid synthesis and uptake, such as regulation of FAT or CD-36 (98, 99). Other therapeutic classes of cholesterol-lowering drugs are represented by selective cholesterol absorption inhibitors, such as ezetimibe; resins, such as cholestyramine, colestipol and colesevelam, which are bile acid sequestrants (103); apolipoprotein B synthesis inhibitors, such as mipomersen (105); microsomal transfer protein inhibitors, such as lomitapide (105). A new promising therapeutic class of cholesterol-lowering drugs is represented by PCSK9 inhibitors. PCSK9 is predominantly produced in hepatocytes, where it decreases LDLR number. When PCSK9 binds LDLR there is a consequent block of LDLR in an open conformation and its recycling is blocked. Then, LDLR is degraded by lysosomes (109). Another recently approved cholesterol-lowering drug is bempedoic acid (8-hydroxy-2,2,14,14-tetramethylpentadecanedioic acid) (97), acting as ATP citrate lyase inhibitor, an enzyme upstream from 3-hydroxy-3-methylglutaryl-CoA. Since the focus of this review is the repositioning of cholesterol-lowering drugs in oncology, we refer to Table 1 for a schematic explanation of the mechanisms of action, effects on cholesterol metabolism, and possible side effects of the drugs.
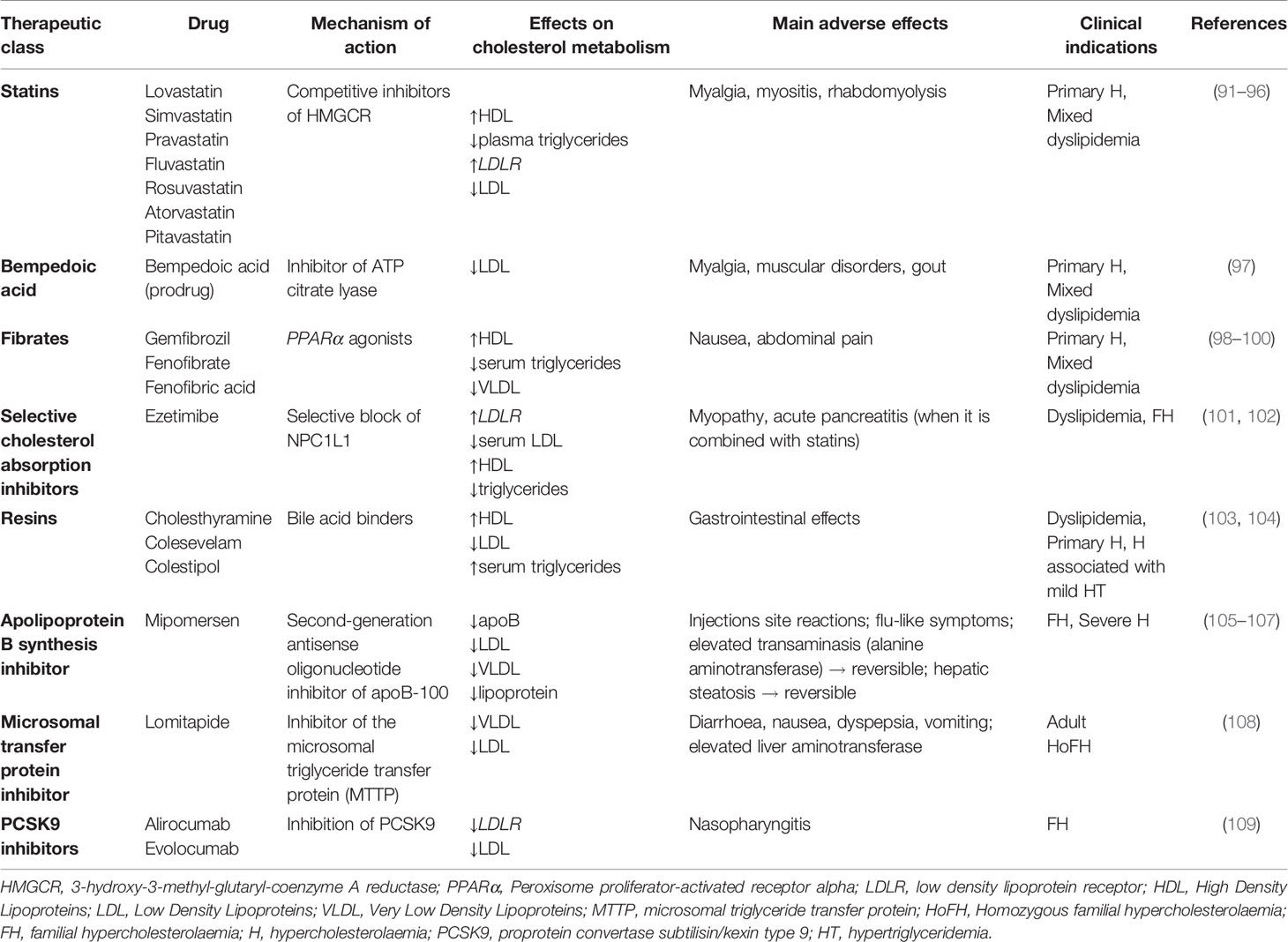
Table 1 Current cholesterol-lowering drugs and relative mechanism of action, main effects on cholesterol metabolism, adverse effects and therapeutic indications.
Cholesterol Metabolic Reprogramming in Cancer: Pharmacological Targeting
Cancer cells are highly proliferative and therefore strongly dependent on cholesterol to satisfy their increasing demand of substrates for membrane biosynthesis (111). Accordingly, cholesterol is generally beneficial for cancer growth and development, as it promotes oncogenic signaling and evasion of apoptosis, as well as cell migration and invasion (112–115). Notably, cancer cells increase their cholesterol demand by enhancing de novo biosynthesis or uptake, by altering the cholesterol efflux, or by increasing its storage, as will be described in the following sections. Also, cholesterol homeostasis is largely compromised in cancer development and progression, as will be discussed below. In line with this, the differential cholesterol requirements of tumors provide novel therapeutic strategies for the treatment of several malignancies. As above mentioned, the purpose of this review is to sum up the main alterations in cholesterol-related metabolic pathways observed in in vitro and in vivo cancer models. The current scientific evidence highlights the reprogramming of lipid/cholesterol pathways in many cancers, thus suggesting intriguing targets exploitable for a combined therapy with conventional chemotherapeutic agents in the fight against cancer. In the following sections we will describe the main alterations observed in cancer cells in the context of cholesterol metabolism, pointing out the more interesting targets identified since now. The identification of specific targets has opened the possibility to exploit them for a pharmacological approach by using cholesterol lowering/modulating drugs. Here we will review the current literature focused on the use of cholesterol targeting drugs in the context of cancer treatment. Results from in vitro and in vivo studies have allowed the translation into clinical trials of some drugs that are discussed in the following sections, highlighting the potential of this combined approach for cancer treatment (Table 2).
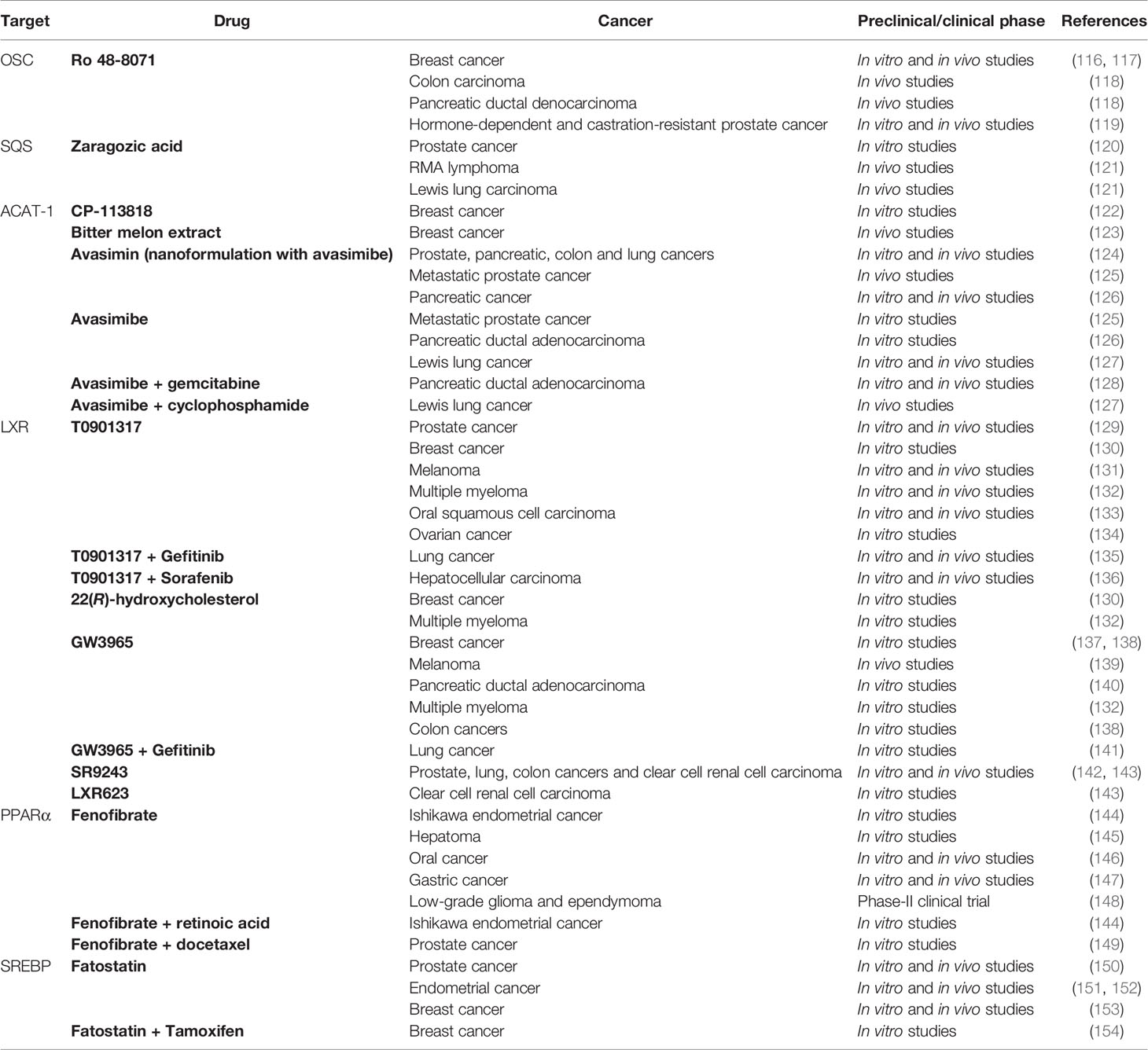
Table 2 Sum up of drugs acting on cholesterol metabolism investigated as potential treatment in cancer therapy.
Enhanced Cholesterol de novo Biosynthesis
Many cancers upregulate de novo cholesterol biosynthesis, thereby fueling the oncogenic machinery and sustaining tumor progression (155). Aberrant cholesterol biosynthetic program can be considered as a hallmark of transformed cancer cells and has been correlated with lower overall patient survival in melanoma, acute myeloid leukemia and sarcoma (32). Consistently, in breast cancer cholesterol biosynthesis-related genes are considered reliable prognostic factors associated with shorter relapse-free survival (156). Cholesterol biogenesis is carried on through the mevalonate pathway (Figure 2), which leads to the production of farnesyl pyrophosphate (FPP), responsible for the formation of either the non-sterol isoprenoid geranylgeranyl pyrophosphate (GGPP) or squalene. The first rate-limiting enzyme HMGCR is overexpressed in many tumors, such as prostate cancer, gastric cancer and colon cancer (157–159). Indeed, the accumulation of non-sterol isoprenoids mediates several oncogenic activities by post-translationally modifying key proteins directly involved in the expression of oncogenes, cytoskeletal organization and cell survival/proliferation (24, 160). This process is collectively known as protein prenylation and allows the covalent attachment of lipid moieties to small oncogenic G proteins, thereby promoting their activation and transforming function (26). GTP-binding proteins Rho, Rac, Rab, Rap, Ras (Ras, Rho, Rab superfamily of GTPases) are all dependent from farnesylation and geranylgeranylation to exert their tumorigenic activities, which eventually promote cell cycle progression and cellular survival, as well as tumor cells motility, migration and metastasis (41, 161, 162). Moreover, isoprenoids are involved in ubiquinone biogenesis. Ubiquinone (CoQ) is a redox active lipid that functions as electron carrier in the mitochondrial respiratory chain. CoQ sustains p53-deficient colon cancer cells growth and development by promoting de novo pyrimidine synthesis and maintaining the integrity of the electron transfer chain even under nutrients starvation and oxygen restriction (163). Differently from steroidogenic healthy tissues, HMGCR activity in tumors is refractory to sterol-mediated negative feedback regulation (36, 164). Therefore, HMGCR altered regulation allows the accumulation of isoprenoids even in cholesterol-enriched conditions, thereby sustaining the production of non-sterol mevalonate intermediates essential for the establishment of tumor malignant phenotype (41). The alternative branch of mevalonate pathway diverts towards the formation of sterols through the activity of squalene synthase (SQS), which gives rise to squalene. In lung cancer patients, SQS is frequently overexpressed and associated with poor prognosis and tumor metastasis. Indeed, the enhanced expression of SQS induces cholesterol biosynthesis, which in turn sustains Tumor Necrosis Factor Receptor 1 (TNFR1) accumulation into lipid rafts and subsequent NF-κB and MMP1 activation (165). Squalene epoxidase (SQLE) converts squalene into squalene-2,3-epoxide and represents the other rate-limiting enzyme in sterol biogenesis. SQLE activity is dysregulated in many tumors, such as breast, lung and colorectal cancer (166–168). Colorectal tumors are characterized by higher SQLE expression levels when compared with healthy tissues, which sustain tumor development by promoting extracellular signal-regulated kinase 1/2 (ERK1/2) oncogenic activity (169). Similarly, in breast cancer SQLE is frequently amplified at the gene level and strongly overexpressed in more aggressive and undifferentiated tumors, thereby demonstrating its oncogenic potential (170). On the other hand, a subset of tumors presents SQLE downregulation and subsequent cholesterol auxotrophy. Lymphoma SQLE-deficient cells accumulate squalene, which modifies cellular membranes and lipid droplets composition, thereby protecting neoplastic cells from the oxidative damage and ferroptosis (171, 172). In breast cancer cells, NAD(P)H-dependent steroid dehydrogenase-like protein (NSDHL) and sterol-C4-methyl oxidase (SC4MOL), two enzymes of the Kandutsch-Russell pathway, are overexpressed and translocate to the plasma membrane. Here, they promote metastasis development by modulating lipid rafts’ sterol composition (173, 174). Another key post-squalenic enzyme is oxidosqualene cyclase (OSC), which mediates 2,3-oxidosqualene cyclization into lanosterol (175). In metastatic mouse models of human colorectal and pancreatic cancer, OSC promotes tumor neovascularization and metastatic potential. Consistently, OSC inhibitors hamper endothelial cell migration and promote cell apoptosis, thereby inhibiting tumor-associated angiogenesis and dissemination to distal organs (116, 118). Additionally, OSC plays an important role in cell self-renewal and its expression is increased in breast cancer stem cells (156, 176). In conclusion, the mevalonate pathway is oncogenic at many levels and frequently dysregulated in several cancers.
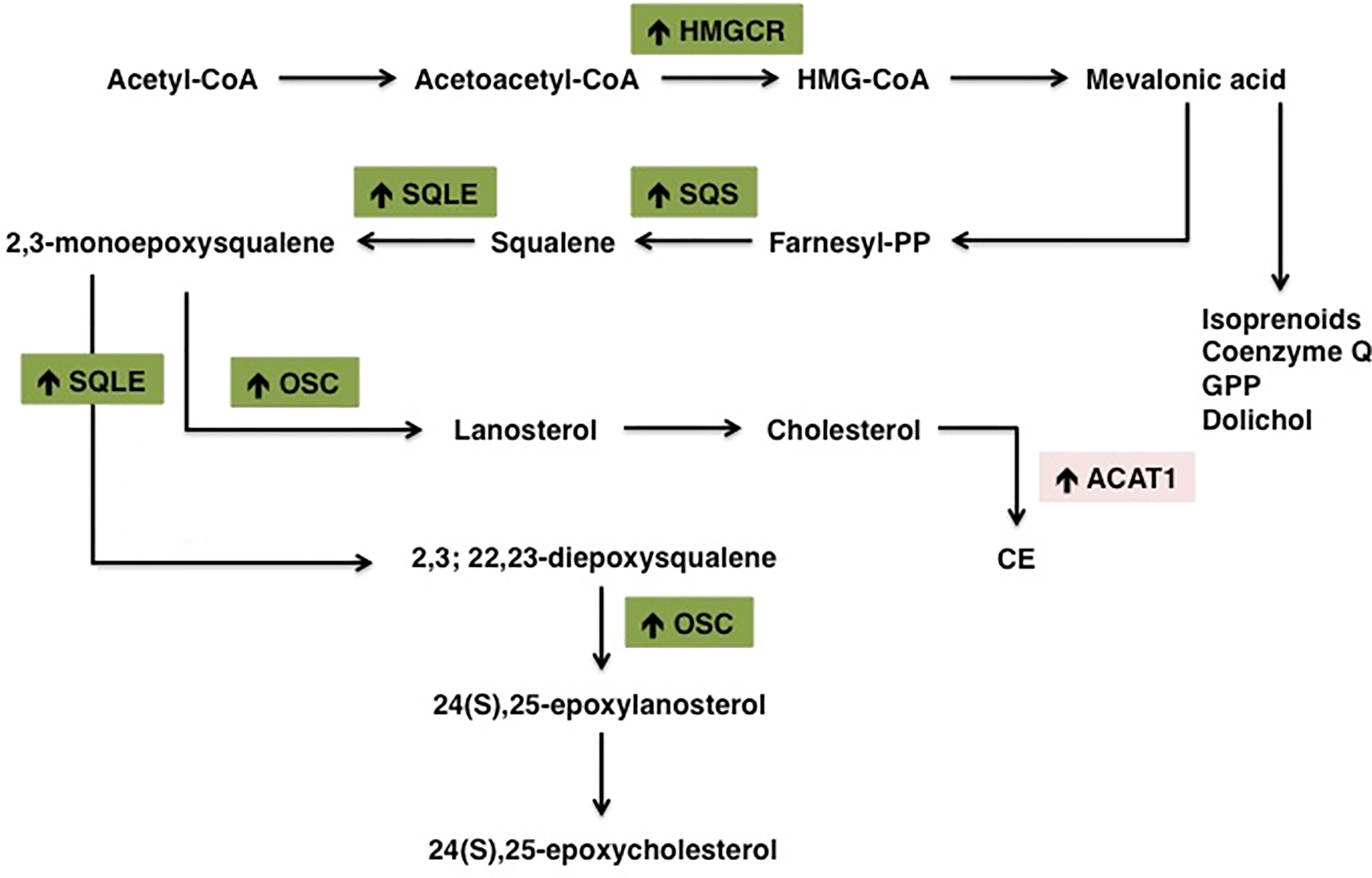
Figure 2 Schematic representation of the main alterations in cholesterol metabolism pathway in tumors. Cancer cells are highly proliferative and therefore strongly dependent on cholesterol to sustain the high demand of substrates for membrane biosynthesis. Cancer cells increase their cholesterol demand by enhancing de novo biosynthesis (or exogenous uptake). Increased/overexpressed enzymes in cholesterol biosynthesis pathway are indicated with (↑). HMGCR, 3-hydroxy-3-methylglutaryl-CoA reductase; SQS, Squalene synthase; SQLE, Squalene epoxidase; ACAT1, Acetyl-CoA Acetyltransferase 1.
Pharmacological Modulation of HMGCR
As mentioned, HMGCR is the rate-limiting enzyme of the mevalonate pathway, which produces cholesterol. Considering the overexpression of this enzyme in many tumors, targeting HMGCR could be a good strategy for cancer clinical therapy. As stated, statins are the commonest pharmacological inhibitors of HMGCR and the repositioning of these drugs in the cancer field is well studied and established. Statins exerts antitumor activities with different molecular mechanisms, such as reducing cell proliferation or tumor cell survival (177), suppressing angiogenesis or causing apoptosis, and reducing tumor growth and metastasis (178–180). The efficacy of these drugs has been evaluated both in monotherapy and in combination therapy with standard chemotherapeutic agents. In vitro studies evidenced that statins are able to inhibit cell proliferation and viability, or causing apoptosis in different human cancer cell lines, such as breast, ovarian and prostate tumor cells. In particular, it has been demonstrated that the open-ring conformation of statins is responsible for the inhibition of HMGCR and apoptosis induction (181). In vivo studies evidenced that statins reduce tumor growth and arrest metastasis progression (113, 182–186). Terzi and co-workers (187) evaluated the effect of two statins, atorvastatin and simvastatin, combined with the standard chemotherapeutic agent, bortezomib, in human multiple myeloma. The results showed that statins are able to improve the effectiveness of bortezomib and reduce the adverse effects (187). The combined treatment of pivastatin with gemcitabine synergically reduced cell proliferation of MIA PaCa-2 cells inducing cell cycle arrest. Moreover, the same combination reduced the tumor growth in in vivo xenograft models (188). A new formulation of atorvastatin was developed in order to cross the blood-brain barrier and target the glioblastoma tumor cells. This new nanoformulation was cytotoxic in mouse and human glioblastoma cells, and was able to reduce the growth in a three-dimensional (3D) tumor model (189). The anticancer role of statin treatment in combination with conventional anticancer drugs, has been tested in clinical trials for the treatment of different types of tumors, such as breast, prostate, ovarian or lung cancers, bringing to light controversial effects of the association (Table 3 summarizes some clinical trials on breast, ovarian and prostate cancer). Different clinical trials demonstrated that statins are able to reduce tumor progression and enhance the survival rate of patients with breast cancer (194). Farooqui reviewed different randomized controlled trials and concluded that the addition of statins to standard chemotherapy is not able to enhance the survival in patients with advanced cancers and a prognosis of less than 2 years (195). A Swedish study, instead, concluded that statins use for 6 months in patients with multiple myeloma enhanced the survival rate of both men and women (196). In another study, atorvastatin was administered in patients with prostate cancer for 27 days before prostatectomy. In this case, drug administration was not able to decrease tumor proliferation with respect to placebo, however, a longer administration of atorvastatin showed beneficial effects (197). In light of this result, it could be interesting to define the specific chronic drug administration before surgery. In fact, other clinical trials evidenced that a chronic therapy of 6 months before surgery resulted to be more efficient compared with a 3 months therapy in reducing tumors (198). It is thus evident that the current knowledge obtained from several in vitro and in vivo studies in different types of tumors confirmed and deepened the molecular mechanisms of statins as anticancer drugs. Since statins were already approved for hypercholesterolemia treatment, their repositioning in the oncological field has benefited from an easier and faster translation into clinic. However, clinical trials evaluating the antitumor effect of these drugs are still few. Monotherapy studies highlight the potential of this class of drugs in cancer prevention, but the pharmacological differences among all statins, as well as the heterogeneity of tested tumors, lead to a lack of proven positive outcomes. In addition, it has been demonstrated the impossibility to administer high doses because of their adverse effects (199). At the same time, relatively few clinical trials take into consideration the combined therapy with standard chemotherapeutic agents, a strategy allowing low dose administration and lower toxicity of the single agents. Thus, further studies evaluating the beneficial effect of statins in combination with conventional chemotherapy have to be conducted in order to assess their potential in cancer therapy.
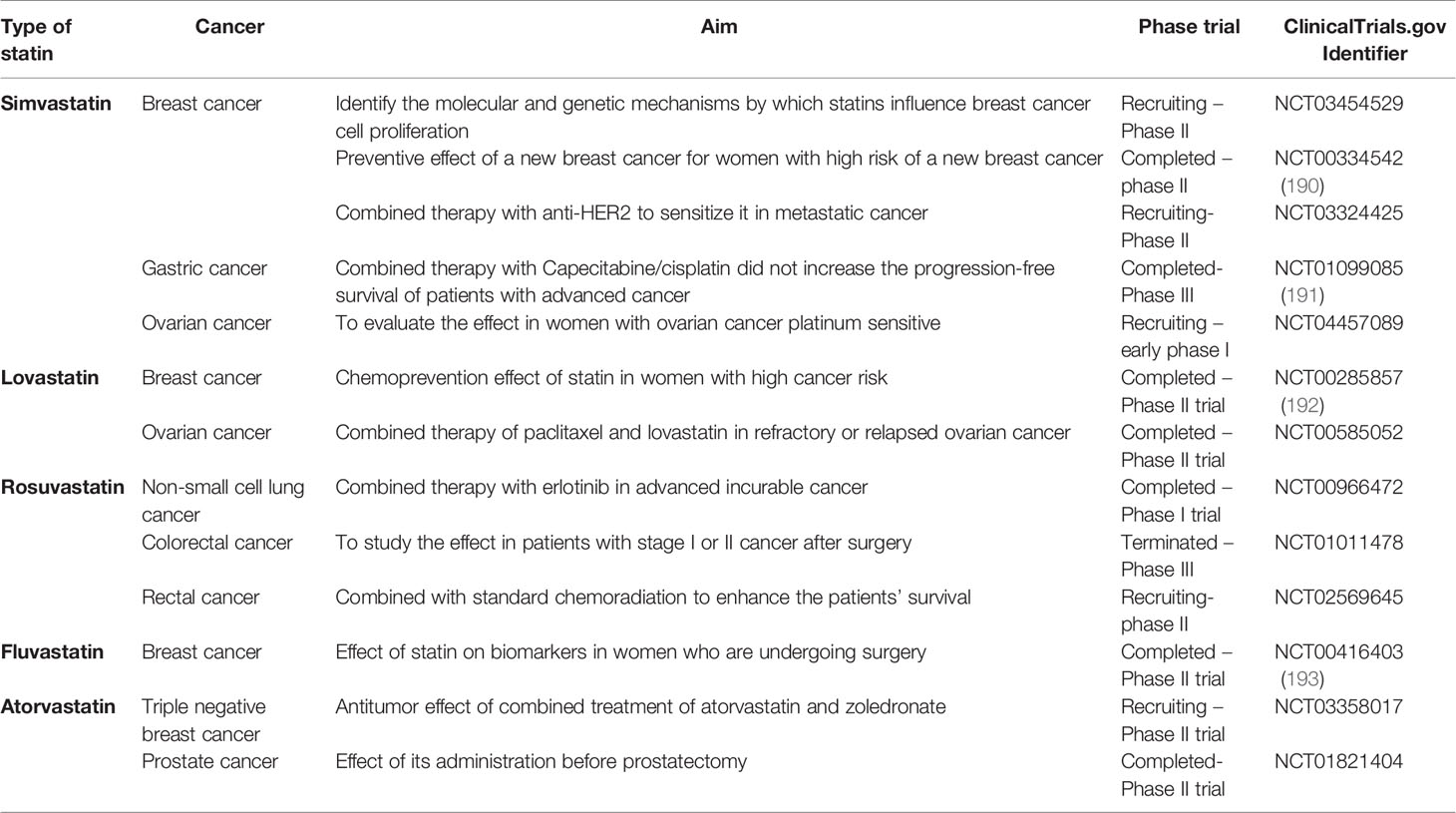
Table 3 Statins currently under evaluation in clinical trials for cancer treatment.
Pharmacological Modulation of OSC
2,3-Oxidosqualene cyclase (OSC) is the enzyme that catalyzes the conversion of 2, 3-monoepoxysqualene into lanosterol acting downstream of HMGCR (116). Since lanosterol is the precursor of cholesterol, the inhibition of OSC causes decreased cholesterol synthesis (175), but unlike statins the LDL catabolism is not affected. This suggests that OSC inhibitors act in a different way (200, 201). Staedler et al. (175) demonstrated the antitumor effect of OSC inhibitors in human glioblastoma and brain-derived endothelial cells. Moreover, the combined treatment with OSC inhibitors and atorvastatin showed an increased antitumor effect in human glioblastoma cells with respect to monotherapy (175). Among the OSC inhibitors, Ro 48-8071 ([4’ -[6-(Allylmethylamino)hexyloxy]-4-bromo-2’ - fluorobenzophenone fumarate]) emerged for its potential antitumor effect. Grinter and coworkers (117) demonstrated that this molecule was able to inhibit cell proliferation in BT-474 human breast cancer cells (117). Ro 48-8071 decreased cell viability of ERα-positive human breast cancer cells (BT-474 and MCF-7), without affecting normal AG11132A cells. Moreover, this compound proved efficacy in reducing tumor growth in in vivo mouse xenograft model injected with BT-474 cells, without showing toxicity at doses administered (116). Maione et al. (118) demonstrated the antitumor effect of Ro 48-8071 in murine models of intestine and pancreas cancers. In fact, the role of this compound was investigated in mouse xenograft models injected with human colon carcinoma HCT116 cells, and pancreatic ductal adenocarcinoma HPAF-II cells. Results evidenced that Ro 48-8071 was able to weaken angiogenesis and inhibit tumor growth in the two mouse models previously mentioned (118). Hyder and coworkers (119) investigated the effect of Ro 48-8071 on cell viability and apoptosis in different lines of human prostate cancer cells. Data showed that this compound reduced cell viability in hormone-dependent LNCaP, castration-resistant PC-3 and DU145 prostate cancer cells, also causing apoptosis (119). Since several studies demonstrated the antiproliferative role of ER-β (202–205), castration-resistant prostate cancer cells were treated with the combination of Ro 48-8071 and ER-β agonist diarylpropionitrile showing enhanced activity in inhibiting cell viability. In vivo treatment with Ro 48-8071 was able to suppress the growth of prostate cancer PC-3 cell xenografts in mice (119).
Pharmacological Modulation of SQS
Squalene synthase (SQS) is an enzyme that catalyzes the conversion of FPP into squalene, the precursor of cholesterol (206). Since squalene synthase is the enzyme responsible for the first committed step in cholesterol production, its targeting results of interest in clinical therapy (207). Zaragozic acids, natural products obtained from fungi, are pharmacological inhibitors of SQS. Brusselmans et al. (120) demonstrated that the expression of squalene synthase was enhanced in LNCaP prostate cancer cells following androgen stimulation. Thus, the inhibition of the enzyme by downregulation or by treatment with zaragozic acid A, was able to cause both the arrest in growth and the induction of cytotoxicity in prostate cancer cells (120). Lanterna and coworkers (121) tested two different isoforms of zaragozic acid, A and B, in mouse models of RMA lymphoma and LLC Lewis lung carcinoma and results confirmed the ability of zaragozic acid in reducing tumor growth, without showing adverse effects (121).
Enhanced Cholesterol Uptake
An alternative strategy exploited by cancer cells to promote sterol-mediated proliferation is to increase the uptake of exogenous cholesterol. NPC1L1 is a critical sterol transporter, essential for cholesterol intestinal uptake. In colorectal cancer, NPC1L1 promotes colitis-associated tumorigenesis by inducing cholesterol absorption and increasing its plasmatic levels (208). Malignant cells capture and internalize cholesterol through the activity of LDLRs. Indeed, LDLRs expression levels are increased in many cancers, including glioblastoma and leukemia, as well as in pancreatic and lung cancers (209). Also, higher levels of LDLRs negatively correlate with the survival of patients affected by pancreatic adenocarcinoma (210). HER2-positive and triple-negative breast tumors characterized by higher LDLRs intratumoral levels are associated with poorer prognosis, suggesting an important contribution of LDL cholesterol in breast cancer progression (211). LDLRs promote tumor development and progression, by modulating cancer cell invasive and migratory potential, as well as adhesivity and plasticity. Indeed, LDLRs foster epithelial-to-mesenchymal transition (EMT), the secretion of metalloproteinase MMP-9 and the activation of Wnt/β-catenin oncogenic signaling (212, 213). On the other hand, HDL cholesterol is accumulated by steroidogenic organs in a process mediated by scavenger receptor type B class 1 (SRB1). SRB1 is overexpressed in diverse human malignancies, such as prostate, breast, ovarian, and colorectal cancers (214). In lung adenocarcinoma, SRB1 represents an independent prognostic factor and its expression positively correlates with malignant tumor behavior and impaired overall survival (215). Consistently, cancer patients present lower levels of HDL cholesterol compared to healthy subjects, suggesting that cancer cells exploit HDL cholesterol from peripheral tissues to sustain their malignant phenotype by picking it up in a SRB1-mediated fashion (36). Indeed, high levels of SRB1 characterize highly undifferentiated and metastatic prostate tumors, which are usually associated with androgen independence. Accordingly, SRB1-mediated cholesterol supplying might provide sterol precursors, thereby promoting tumor self-production of androgens and the development of castration-resistant phenotypes (216). Currently, there are not pharmacological inhibitors targeting LDLR and SRB1. However, increasing evidence highlighted the correlation between high-cholesterol diet and increased tumor growth and development of metastasis (217–219). Dietary cholesterol, that represents only the 30% of total cholesterol in the human body is delivered to liver through chylomicrons, which are hydrolyzed to fatty acids and stored in adipose tissue (220). Once cholesterol arrives at hepatocytes, it is removed either in form of free cholesterol or converted in bile acids. Excess of cholesterol is converted into cholesterol esters and stored in hepatocytes (221). Accumulating evidence, based on metabolic mechanisms, highlights that tissues enriched of stored fatty acids could be more prone to be related with increased cancer risk (222, 223). Pelton and coworkers (224) demonstrated that high-cholesterol diet enhances the tumor growth of human breast cancer injected in a mouse model. In light of this consideration, the administration of low-cholesterol diet or ezetimibe slightly decreased the growth of tumors by a reduction of cholesterol levels (224). Moreover, it has been shown that the inhibition of the xanthine oxidase with a pharmacological inhibitor was able to reduce both tumor growth and metastasis in breast mouse model fed with high-cholesterol diet (225). Thus, it appears that a low-cholesterol diet could be a promising strategy to counteract tumor growth. Furthermore, combining the low-cholesterol diet with anticancer drugs could become an exploitable field in clinical therapy.
Dysregulated Cholesterol Efflux
In physiological conditions, the excess of cellular cholesterol is removed from peripheral tissues through a process controlled by ATP binding cassette transporters, mainly ABCA1. In general, malignant cells show decreased levels of ABCA1, thereby promoting intracellular cholesterol storage. Indeed, ABCA1 deregulation leads to cholesterol accumulation in the mitochondrial compartment, which in turn supports the malignant transformation (32, 155). Moreover, higher levels of intracellular cholesterol directly affect the lipid composition of plasma membranes, as well as their physical properties: cholesterol enrichment increases the phospholipids’ degree of order in the bilayer while reducing its permeability, thereby promoting tumor resistance to membrane-active anticancer drugs (226). Peroxisome proliferator-activated receptors (PPAR) α and γ play a pivotal role in modulating both intracellular and extracellular cholesterol fluctuations (227, 228). Indeed, PPARα and PPARγ activation promotes LXR-mediated ABCA1 expression, thus inducing cholesterol efflux to the lipid-poor apolipoprotein A-I (229). Also, PPARα blocks cholesterol biosynthesis by inhibiting sterol regulatory element-binding protein 2 (SREBP-2) activity (230). Accordingly, PPARα and PPARγ are considered tumor suppressor genes which inhibit tumor progression (231). On the other hand, cholesterol integration within the plasma-membrane reduces malignant cell migration and metastatic potential. Specifically, increased plasma membrane-associated cholesterol reduces the fluidity of the bilayer, resulting in restricted cell motility and EMT, a critical event for the development of tumor metastasis (232). The variation in cholesterol content, in fact, affects membrane fluidity, permeability and rigidity, thus impacting several processes, such as invasion, migration or tumor development, growth and metastasis (233). In particular, different studies reported that increased cholesterol percentage deriving from higher de novo biosynthesis leads to enhanced rigidity and decreased fluidity, thus contributing to decreased cell mobility (234), or to altered membrane permeability, which is involved in altered cancer cell response to drug treatments (226). In line with this, variations in intracellular cholesterol levels mediated by ABCA1 overexpression drive the onset of EMT and the promotion of tumor invasiveness, whilst human solid tumors at advanced stages are characterized by high levels of ABCA1 expression (235). Therefore, metastatic cancer cells substantially reduce their cholesterol levels in the plasma membrane compartment by overexpressing ABCA1, which eventually mediates cholesterol efflux (232). Conversely, the development of primary tumors requires pro-oncogenic and survival-stimulatory signaling pathways, which are dependent or modulated by lipid rafts (114). Accumulations of cholesterol in lipid rafts induce the aberrant activation of tyrosine kinase receptors, such as IGF1 and HER2, as well as PI3K/AKT-mediated tumorigenic signaling (234, 236, 237).
Pharmacological Modulation of PPARα
Saidi and coworkers (144) tested fenofibrate, which is an agonist of PPARα, in Ishikawa endometrial cancer cells. The results showed inhibition of cell viability and apoptosis induction. Moreover, the combined use of fenofibrate and retinoic acid, which is an agonist of retinoid-X-receptor (RXR), enhanced the inhibition of cell proliferation (144). The mechanism of action of fenofibrate still remains unclear. In fact, fenofibrate reduced the cell proliferation of human hepatoma cells through the inhibition of Akt phosphorylation and not through a PPARα-dependent mechanism (145). Jan et al. (146) proposed metabolic reprogramming as the mechanism underpinning the anticancer effect of fenofibrate. In particular, this drug caused the reduction of oral cancer cell proliferation and activated the glycolysis pathway. Moreover, in vivo administration of fenofibrate in mice reduced the tumor growth (146). The antitumor effect of fenofibrate through the reprogramming of cancer metabolism is also confirmed in gastric carcinoma. In fact, the use of this drug reduced both in vitro cancer cell proliferation and in vivo tumor growth. In addition, Chen and coworkers (238) demonstrated that treating cells with fenofibrate causes mitochondrial dysfunction due to its accumulation too, suggesting also the PPARα involvement in mitochondria reprogramming (238). Thus, targeting PPARα could be an interesting tool for cancer treatment. Moreover, fenofibrate was also tested in combination with chemotherapeutic agents, suggesting that using the combined therapy could be a strategy to overcome drug resistance. In fact, treating prostate resistant cancer cells with fenofibrate is able to resensitize them to docetaxel (149).
Enhanced Cholesterol Storage
The overload of free cholesterol inside the cell is extremely toxic (239). Therefore, in physiological conditions, the excess of free cholesterol is avoided by producing its esterified form, namely cholesteryl ester (240). Cholesteryl esters can be readily stored into lipid droplets, thus preventing the lipotoxic potential of free cholesterol (241). The accumulation of intracellular cholesterol is strongly oncogenic and represents a common hallmark of cancer (242, 243). For instance, the intracellular levels of cholesteryl esters and lipid droplets are substantially increased in breast cancer, leukemia, and glioblastoma (244–246). In colorectal cancer, lipid droplet-enriched malignant stem cells are characterized by increased clonogenic and tumorigenic potentials (247). Consistently, the cholesteryl ester-producing enzyme acetyltransferase ACAT1 is upregulated in many cancers, including hepatocellular carcinoma, castration-resistant prostate cancer, and pancreatic cancer, whilst its expression positively correlates with reduced overall survival and recurrence-free survival in adrenocortical carcinoma (126, 248–250). ACAT1 overexpression and cholesteryl esters enrichment play a dual role in promoting cancer progression. Higher levels of cholesterol esterification lead to decreased contents of free cholesterol, thereby protecting malignant cells from ER stress and apoptosis (126, 251). On the other hand, esterified cholesterol represents an intracellular source of cholesterol, which can be exploited by cancer cells when needed to fuel the malignant phenotype. Consistently, the cholesteryl ester-metabolizing enzyme lysosomal acid lipase (LAL) is upregulated in tumor tissues, thus providing malignant cells with ready-to-use free cholesterol (243, 252). PTEN deficiency drives cholesteryl esters accumulation in pancreatic cancer through the activation of the downstream PI3K/Akt/mTOR/SREBP signaling pathway; increased content of esterified cholesterol promotes tumorigenesis and metastatic potential (126). Similarly, cholesterol reservoirs are enriched in advanced and metastatic human prostate cancer, while nearly absent in healthy prostate, benign prostatic hyperplasia, and prostatic intraepithelial neoplasia. Accumulation of cholesteryl esters is triggered by PTEN loss, which in turn induces the expression of SREBP and LDLR via PI3K/AKT/mTOR, thus promoting ACAT1-mediated cholesterol storage in lipid droplets (243). Thanks to cholesterol esterification and subsequent accumulation, prostate cancer cells reduce the intracellular levels of free cholesterol, thereby avoiding free cholesterol lipotoxicity and maintaining SREPB-induced cholesterol biogenesis and uptake (62, 253). Moreover, increased contents of esterified cholesterol might fuel the development of castration-refractory prostate tumors by providing androgen precursors for de novo steroidogenesis (125, 254, 255). The excess of lipid droplets accumulated in tumor cells is the leading cause of enhanced cell proliferation and the responsible of cancer aggressiveness. Thus, it appears that targeting enhanced cholesterol storage could be an interesting tool in cancer therapy (113).
Pharmacological Modulation of ACAT-1
ACAT-1 is overexpressed in two ER- lines of human breast cancer, MDA-MB-231, and MDA-MB-436. Higher expression of this enzyme could be related to a higher cell proliferation rate (122). Treating cells with CP-113818, which is an ACAT-1 inhibitor, caused a reduction in cell proliferation and migration, suggesting the correlation mentioned above (122). Bitter melon extract, a natural ACAT-1 inhibitor, exerts antitumor effects towards breast cancer cells. Shim and coworkers (123) fed orthotopic mice models of MDA-MB-231 cells with this compound showing a reduction in tumor growth through cholesterol metabolism modulation (123). It has also been demonstrated that ACAT-1 is a metabolic “tumor promoter”, since it is overexpressed in human breast cancer cells leading to tumor formation and lung metastasis (256). Cancer cells use this mitochondrial enzyme to recycle ketone acids into acetyl-CoA enhancing the ATP production. Ozsvari and coworkers (257) investigated ACAT-1 as a therapeutic target and the use of in silico drug design identified mitoketoscins. These molecules belong to a new therapeutic class of drugs that inhibits mitochondrial functions and ACAT-1 (257). Lo et al. (258) demonstrated that ACAT-1 is overexpressed in MES-SA/Dx doxorubicin-resistant uterine sarcoma cancer cells compared to the sensitive counterpart, suggesting a correlation with drug resistance. Thus, the ACAT-1 knock-down caused a decrease in cell viability, showing an important role of this enzyme in the onset of drug resistance (258). Lee and coworkers (124) developed avasimin, which is a nanoformulation containing avasimibe, an ACAT-1 inhibitor. They tested the formulation in different human cancer cell lines, showing that avasimin was able to reduce lipid droplets accumulation in PC3 prostate cancer cells. Concerning the effect on cell viability the nanoformulation was used to treat human PC3, MIA-PaCa2 pancreatic cancer cells, A549 lung cancer cells, and HCT116 colon cancer cells, showing a reduction in cell viability in all cell lines. Moreover, they evaluated in vivo the avasimin effect in PC3 and HCT116 cell xenograft mouse model. The results evidenced a decrease in both tumor growths after the avasimin intravenous treatment (124). Li et al. (126) demonstrated an overexpression of ACAT-1 in MIA PaCa-2 and PANC-1 human pancreatic cancer cells compared to normal cells. Treating cells with avasimibe or genetic silencing of ACAT-1 caused the block of cholesterol esterification that led to a reduction in cell invasion and migration. Results showed a higher sensitivity to the ACAT-1 inhibition of cancer cells compared to the normal counterpart. A xenograft mouse model injected with MIA PaCa-2 cells was treated with avasimibe and results showed a reduction in tumor growth, decreased metastatic lesions in lymph nodes and in liver compared to untreated mice (126). Moreover, it has been suggested an important role of cholesteryl ester in the development of metastasis. Thus, Lee and coworkers (125) tested avasimibe in PC-3M metastatic prostate cancer cell lines derived from PC-3 xenografts liver metastasis. The treatment showed a decrease in cell migration rate. In addition, when they treated PC-3M xenograft mice with avasimin a reduction in tumor growth and metastasis development were observed. Taken together, these data suggested an implication of cholesteryl ester in the development of metastasis in prostate cancer. ACAT-1 inhibition compromised Wnt/β-catenin signaling consequently overcoming metastasis formation (125). Li et al. (128) demonstrated a correlation between cholesterol metabolism and gemcitabine resistance, since it was found a higher accumulation of cholesteryl ester in gemcitabine-resistant pancreatic ductal adenocarcinoma cells compared to the sensitive counterpart. In addition, it has been demonstrated that Akt is implicated in cholesteryl ester accumulation. Treatment of resistant cells with avasimibe, an ACAT-1 inhibitor, caused a reduction in cell proliferation. Moreover, the combined treatment with gemcitabine and avasimibe showed synergic effect in vitro and resulted in decreasing tumor growth in in vivo xenograft mouse model injected with Mia PaCa-2 cells. Avasimibe treatment downregulates Akt contributing to resensitization of resistant cells (128). It has been demonstrated that the upregulation of ACAT-1 is implicated in the development of metastasis in LLC Lewis lung cancer. Treatment of LLC cells with avasimibe caused a decrease in cell proliferation and migration. Moreover, avasimibe alone or in combination with cyclophosphamide was able to reduce both tumor growth and metastasis formation in xenograft mouse model (127).
Oncogenic Signaling and Cholesterol Homeostasis
In physiological conditions, cholesterol homeostasis is maintained by sterol-sensitive systems, mainly SREBP2 and Liver X receptors (LXR). Oncogenic potential gaining and tumor suppressor activity loss in cancer cells deeply affect cholesterol metabolism. As a general rule, oncogenic pathways induce cholesterol biosynthesis and uptake, thus promoting increased intracellular levels of sterols, while tumor suppressor pathways lead to cholesterol lowering inside the cells (34). Indeed, the oncogenic MYC induces cholesterol biosynthesis by upregulating HMGCR expression, which is essential during oesophageal squamous cell carcinoma malignant transformation (259, 260). Similarly, aberrant EGFR oncogenic signaling is involved in SCAP-mediated SREBP-2 activation, thus promoting LDLR expression and subsequent cholesterol uptake (261, 262). In human hepatocellular carcinomas, the pro-oncogenic activity of c-FOS mediates LXRα downregulation, which leads to cholesterol retention and production of tumorigenic oxysterols (263). Oxysterols are oxygenated cholesterol metabolites which target and modulate the activity of many nuclear receptors, including LXRs, retinoid-related orphan receptors (RORs), as well as the Hedgehog signaling pathway (264, 265). Among them, 27-hydroxycholesterol (27HC) is an endogenous selective estrogen receptor modulator involved in breast and prostate cancers progression (266). 27HC promotes cell proliferation through p53 inactivation, as well as cell migratory potential via Signal Transducer and Activator of Transcription-3 (STAT-3)-mediated MMP9 activation and subsequent EMT induction (267, 268). 27HC is also involved in tumor angiogenesis by inducing VEGF activation through ERα signaling or reactive oxygen species-mediated STAT-3 recruiting (269). Consistently, advanced breast cancers upregulate CYP27A1 while decreasing the expression of CYP7B1, thereby promoting 27HC accumulation (31, 270). Higher levels of intracellular cholesterol in cancer cells are determined by aberrant HMGCR activity, due to disrupted sterol-controlled feedback regulation or SREBP-mediated overexpression (271–273). In hypoxic tumor microenvironments, SREBPs and their downstream genes are strongly upregulated and support cell survival and tumor growth (274). The activity of SREBPs is promoted by many oncogenic signaling pathways, including PI3K/Akt and Ras/ERK, which eventually induce cholesterol biosynthesis and uptake, while inhibiting its ABCA1-mediated efflux (275–277). In line with this, tumor suppressors genes p53 and PTEN increase cholesterol clearance by increasing ABCA1 activity, while reducing cholesterol absorption and accumulation (126, 278). Indeed, PTEN and p53 loss induce PI3K/Akt signaling, thereby promoting LDLR-induced cholesterol uptake and subsequent formation of cholesteryl esters (243, 279, 280).
Pharmacological Modulation of LXR
Liver X receptors (LXR) are nuclear receptors involved in cholesterol metabolism. Targeting LXR could be a good strategy because its activation is able to modulate the cholesterol pathway. The consequence is decreased cholesterol levels into cells, causing limited cancer cell proliferation. As already explained above, LXR can be activated by endogenous ligands, such as oxysterols but also by agonists, such as T0901317 (130). Treating LNCaP human prostate cancer cells with LXR agonist T0901317 caused cell death through apoptosis. Moreover, the treatment with T0901317 in a xenograft mouse model injected with LNCaP cells was able to reduce tumor growth (129). It is known that increased levels of cholesterol activate Akt enhancing its phosphorylation besides improving tumor progression (281). Pommier and coworkers investigated the effect of LXR activation on cholesterol metabolism. T0901317 treatment was able to increase the expression of LXR target gene Abcg1 and consequently causing a higher cholesterol efflux. Moreover, overexpression of Abcg1 modulates reverse cholesterol transport causing cholesterol exhaustion in rafts and the inactivation of Akt signaling pathway (129). T0901317 showed anticancer properties also in ovarian cancer. In fact, treatment of ovarian cancer cells with this compound was able to inhibit cell proliferation and cause apoptosis (134). It was demonstrated that MCF-7 human breast cancer cells express LXR. Thus, treating these cells with the LXR synthetic agonist, T0901317, and the natural one, 22(R)-hydroxycholesterol, resulted in a reduction of cell proliferation besides both agonists caused cell death through apoptosis. In addition, T0901317 treatment was able to decrease intracellular cholesterol and LXR activation increased the expression of its target gene Abcg1in MCF-7 cells (130). Furthermore, it has been demonstrated that the combined therapy of T0901317 and gefitinib, an anticancer drug, was able to reduce cell and tumor growth both in vitro and in vivo in a lung cancer model (135). This agonist combined with sorafenib enhanced the antitumor effect of the chemotherapeutic agent in hepatocellular carcinoma. In fact, the activation of LXR blocks two pathways, MET and EGFR, avoiding their availability for lipid rafts and consequently enhancing the efflux of cholesterol (136). MCF-7, T-47D, SK-BR-3, or MDA-MB-231 human breast cancer cell lines, which are genetically different, expressed both LXR isoforms, LXR-α and LXR-β. Treating these cell lines with GW3965 LXR ligand caused a reduction in proliferation. Nguyen-Vu and coworkers correlated decreased cell proliferation with the downregulation of genes involved in cell growth. For example, they showed that the downregulation of E2F2, which is a transcription factor, caused a reduced proliferation of MCF-7 and T-47D ER+ cancer cells (137). GW3965 treatment inhibited cell proliferation in both human MCF-7 breast and SW480 colon cancer cell lines. Investigating the molecular mechanisms underlying this anti-proliferative effect, Hassan and coworkers (138) demonstrated that the activation of LXR caused the decrease of Akt phosphorylation leading to its inactivation (138). The expression of the isoform LXR-β was assessed in three different human pancreatic ductal adenocarcinoma cell lines, BxPC-3 and MIA-PaCa-2 and PANC-1. Treating cells with GW3965 increased the expression of the LXR target gene Abca1. Moreover, cell proliferation of human pancreatic ductal adenocarcinoma cell lines decreased after treatment with LXR agonist and the cell cycle was blocked (140).
The agonist GW3965 in combination with the standard chemotherapeutic agent gefitinib demonstrated synergic effect in resensitization of gefitinib-resistant lung cancer cells (141). Pencheva and her group (139) hypothesized that targeting LXR could be a strategy to block metastasis progression in melanoma. Treating melanoma cells with LXR agonists, GW3965 or T0901317, did not cause an impact on cell proliferation but affected cell invasion. In particular, the use of LXR agonists was able to block lung metastasis development and reduce brain metastasis progression in mouse melanoma models. In addition, it was also demonstrated that oral or diet administration of GW3965 to dacarbazine-resistant mice was able to strongly reduce melanoma tumor growth and that the combined treatment of LXR agonist with dacarbazine has proven to be more active compared to GW3965 alone. Moreover, the same agonist was able to reduce tumor growth in mouse melanoma models resistant to vemurafenib, and again, the combined treatment of GW3965 with vemurafenib had a higher effect compared to LXR alone (139). Zhang and coworkers (131) demonstrated that both LXR isoforms are expressed in murine B16F10 melanoma cells. When LXR is activated through the agonist T0901317 there was a decrease in melanoma cell proliferation and apoptosis through caspase-3 activation. Moreover, the treatment of mouse melanoma models with T0901317 reduced tumor growth. In order to confirm the involvement of LXR signaling in melanoma anti-tumor activity, the LXR target genes Abca1 and SREBF1 were checked confirming their increased expression in mice treated with T0901317 (131). Human multiple myeloma cells expressed both LXR isoforms, LXR-α and LXR-β. Treating cells with LXR ligand, 22(R)-hydroxycholesterol and the two agonists, GW3965 and T0901317, strongly increased the expression of two target genes, Abca1 and Abcg1, while slightly the one of the target gene SREBP-1c (132). The Hedgehog (Hh) signaling pathway is a regulator of proliferation, differentiation and it has been linked to carcinogenesis (282). Agarwal and coworkers showed that activating LXR represents a strategy to inhibit Hh signaling pathway in human multiple myeloma cells. Moreover, they showed that treating cells with LXR agonists was able to inhibit clonogenic growth both in vitro and in vivo (132). SR9243 is a specific inverse agonist of LXR. Flaveny et al. (142) demonstrated that treatment of prostate, lung and colon cancer cells with SR9243 decreases cell proliferation, causes cell death through apoptosis and reduces tumor growth in mouse xenografts. Moreover, a combined treatment of SR9243 with cisplatin or 5’-fluorouracil sensitized cells to chemotherapeutic drugs. In particular, SR9243 treatment caused down-regulation of GCK1, PFK2, PFK1, and LDH Warburg genes, and decreased expression of FASN, SREBP1-c, and SCD1 lipogenic gene both in in vitro and in vivo colon xenograft models (142). Similar results regarding lipid metabolism were obtained in clear renal cell carcinoma both in vitro and in vivo (143). SR9243 was also able to reduce the expression of LXR target gene Abca1 involved in cholesterol transport (142). In addition, Wu et al. (143) tested the LXR agonist LXR623 in clear cell renal cell carcinoma demonstrating its role in decreasing cell proliferation and causing apoptosis. Considering that LXR is a transcription factor able to regulate the expression of different target genes, including those related to glycolysis and lipogenesis, targeting this receptor could represent a promising approach in cancer treatment. Kaneko and coworkers (133) demonstrated that LXR-α was expressed in human oral squamous cell carcinoma. Thus, activating LXR with T0901317 resulted in a reduction in cancer cell viability through the induction of the target gene Abca1. Moreover, SAS cells were injected in SCID mice and then they were treated with T0901317. The results evidenced a reduction in tumor growth after treatment (133).
The LXR agonist GW3965 was also used to target LDLR in glioblastoma. In fact, this pharmacological approach both inhibits the uptake of exogenous LDL and enhances the cholesterol excision from cells. Treating cancer cells with this drug induced apoptosis in vitro and reduced in vivo tumor growth (283).
Pharmacological Modulation of SREBP
Fatostatin is a non-sterol diarylthiazole derivative and a specific inhibitor of SREBP. The mechanism of action of this drug consists in binding the SREBP cleavage activating protein (SCAP), and consequently blocking cholesterol biosynthesis (150, 151). Targeting SREBP could be a new pharmacological approach for cancer treatment. Fatostatin showed antitumor effect in both androgen-responsive and androgen-nonresponsive prostate cancer cells by the in vitro inhibition of cell proliferation and cell cycle arrest. In addition, it was able to reduce in vivo tumor growth (150). Gholkar and coworkers (284) investigated the mechanism underlying the antitumor effect of fatostatin in different types of tumors, such as human breast and cervix cells, showing its ability to block the tubulin polymerization and arrest cells in mitosis (284). Fatostatin reduced cell viability in endometrial cancer (151, 152) and decreased the tumor growth in xenograft mice enhancing their survival rate (151). ER-positive breast cancer cells treated with fatostatin showed decreased cell viability and higher lipid accumulation. In particular, increased ceramides’ levels are strictly related to apoptosis. The xenograft volume decreased after treatment with fatostatin (153). Moreover, the combined treatment with tamoxifen resulted synergic in reducing both in vitro cell proliferation and in vivo tumor growth in breast cancer (154).
Conclusions
This review examined the most relevant aspects of the metabolic reprogramming that have been observed in cancer focusing on cholesterol metabolism. Understanding the metabolic vulnerabilities of tumor tissues can help in the identification of new therapeutic targets in order to develop a better cancer treatment. The large amount of literature of the last decades provided overwhelming evidence of lipid and cholesterol metabolism alterations in cancer. High levels of cholesterol are essential to sustain fast tumor cell proliferation and the complex role of cholesterol in cancer development, progression, and susceptibility to chemotherapy is firmly established. Intracellular cholesterol levels can be regulated by de novo synthesis, reduced degradation, increased uptake or storage. This review summarizes the current knowledge regarding the alteration in all these aspects of cholesterol metabolism, highlighting the molecular targets and the possible pharmacological approaches that are currently under investigation (Figure 3 and Table 2). Despite a large amount of in vitro and in vivo evidence suggesting the use of cholesterol-related drugs against cancer, the clinical translation is still limited (Table 3). To date, only statins and fenofibrate have resulted in clinical trials for cancer therapy showing promising results. Besides the treatment with a single agent, a common therapeutic strategy is the drug combination, which can affect simultaneously different pathways in cancers; thus, it is of interest to underline that the combination of conventional chemotherapeutic drugs with cholesterol-lowering agents is under investigation showing encouraging results.
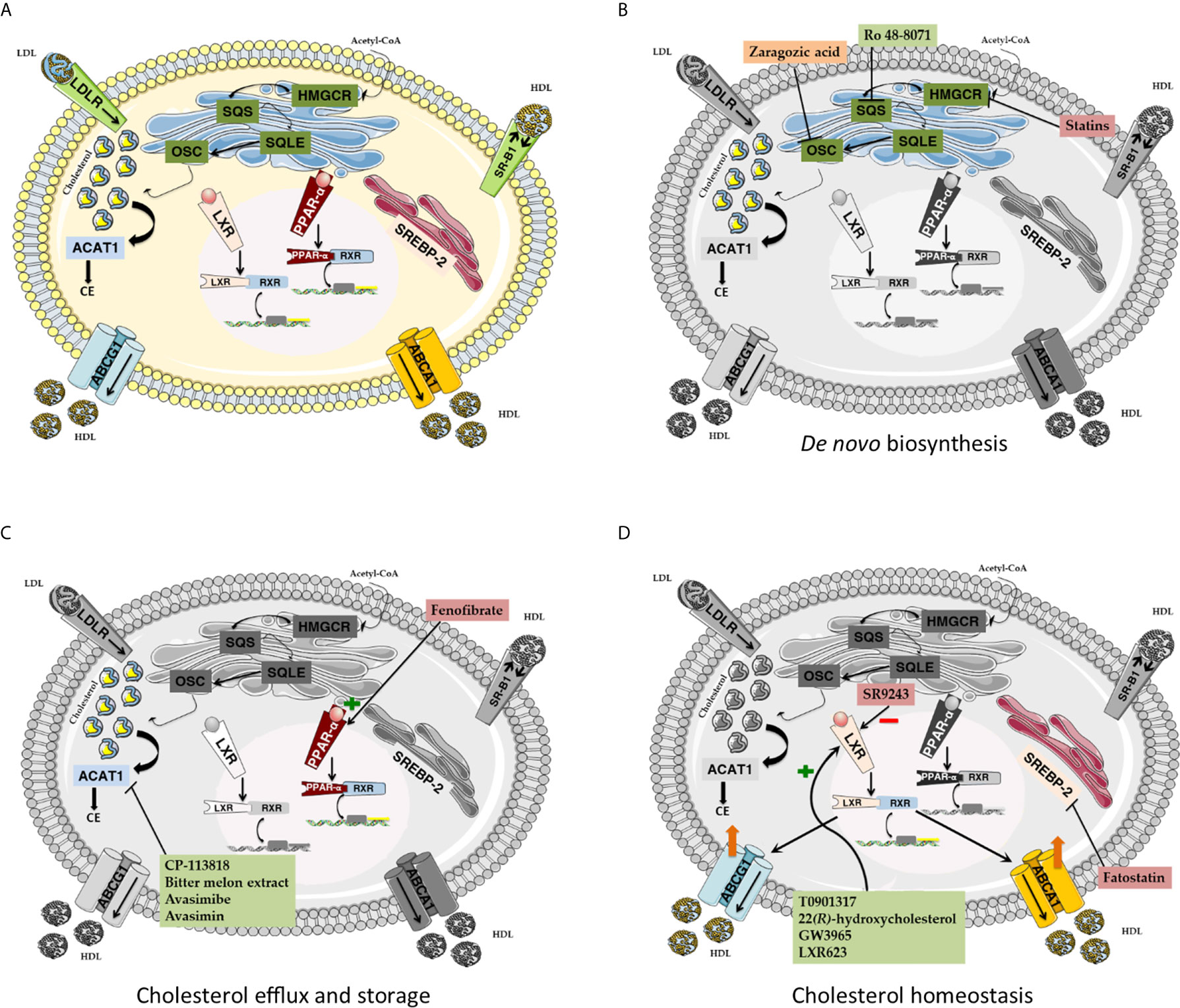
Figure 3 (A) Cholesterol metabolism. De novo cholesterol biosynthesis mainly relies on the activity of four key enzymes. HMGCR catalyzes the formation of mevalonate. Mevalonate is essential for farnesyl pyrophosphate biosynthesis, which is in turn exploited by SQS for squalene production. SQL converts squalene into its epoxydic form, which is eventually cyclized to lanosterol by OSC. Lastly, lanosterol is converted to cholesterol. HDL particles collect extrahepatic cholesterol and allow its cellular uptake by interacting with SR-B1. Alternatively, LDL-associated cholesterol can be captured and internalized in coated endocytic vesicles in a LDLR-mediated fashion. Intracellular cholesterol excess is converted into cholesteryl esters by ACAT1 and stored into lipid droplets. Cellular cholesterol efflux is mainly controlled by ABCA1 and ABCG1, two regulatory proteins belonging to the ATP-binding cassette transporter superfamily. Cellular cholesterol homeostasis is maintained by sterol-sensitive systems, such as SREBP2 and LXR. SREBP2-mediated adaptative response promotes cholesterol biosynthesis and uptake. Conversely, LXR promotes cholesterol excretion while impairing its uptake and production. PPARα activation promotes LXR-mediated ABCA1 expression and blocks cholesterol biosynthesis by inhibiting SREBP2. (B) Pharmacological targeting of de novo cholesterol biosynthesis pathway. Statins target and inhibit the activity of the rate-limiting enzyme HMGCR. Ro 48-8071 and Zaragozic acid act downstream of the mevalonate pathway, by inhibiting the activity of SQS and OSC, respectively. (C) Pharmacological targeting of cholesterol efflux and storage. The PPARα agonist Fenofibrate promotes PPARα-RXR interaction, thereby activating the PPARα signaling cascade. Both synthetic (CP-113818, Avasimibe, Avasimin) and natural (Bitter melon extract) inhibitors of ACAT-1 block cholesterol esterification and intracellular overload. (D) Pharmacological targeting of cholesterol homeostasis. LXR agonists, such as GW3965, T0901317, 22(R)-hydroxycholesterol and LXR623, can activate LXR signaling cascade, leading to increased cholesterol efflux and reduced cholesterol uptake. HMGCR, 3-hydroxy-3-methylglutaryl-CoA reductase; SQS, Squalene synthase; SQLE, Squalene epoxidase; OSC, 2,3-oxidosqualene cyclase; SR B1, scavenger receptor type B class 1; LDLR, LDL receptor; ACAT1, Acetyl-CoA Acetyltransferase 1. PPAR- α, peroxisome proliferator-activated receptor; LXR, liver X receptor; SREBP-2, sterol regulatory element-binding protein 2; ABCA1, ATP Binding Cassette Subfamily A Member 1; ABCG1, ATP Binding Cassette Subfamily G Member 1; HDL, High-density Lipoprotein; LDL, Low-density Lipoprotein.
A better understanding of the metabolic dependencies of tumors also provides new hints for therapeutic strategies in cancer therapy. Novel studies are focusing on the exploitation of lipid/cholesterol metabolic vulnerabilities of cancer to develop new drug delivery systems and strategies. Recent works make use of engineered lipids or adipocytes to deliver anticancer drugs to the tumors (285, 286). Moreover, LDL and HDL particles have been proposed as delivery systems for anticancer drugs. A work by Sobot et al. (287) proposed a chemical linkage of gemcitabine to squalene-moiety (the lipid precursors of cholesterol biosynthesis) assembled in nanoparticles. They showed that LDLR levels positively correlated with nanoparticle uptake and cytotoxic effect in cancer cells and in tumor-bearing mice (287). Mooberry and coworkers, instead, tested a formulation of paclitaxel encapsulated in synthetic/reconstituted high-density lipoprotein (rHDL). The increased uptake of anticancer drug is mediated by SR-B1, which is overexpressed in prostate cancer cells (288). Thus, conjugation of cholesterol moieties with anticancer drugs is an attractive approach (289) for cancer therapy, which can also improve the chemotherapy efficacy and reduce the cytotoxicity to normal cells.
The repurposing of cholesterol-lowering drugs for cancer therapy might be a promising approach to selectively affect cancer cells, highly dependent from cholesterol and to eventually improve the efficacy of conventional chemotherapy by affecting different signaling/metabolic pathways. A deep elucidation of cholesterol-linked metabolic vulnerabilities in cancers may offer new opportunities to develop new drug delivery strategies, allowing a more selective targeting of cancer cells, thus improving the quality of cancer therapy in patients.
Author Contributions
VC and ER conceived the review. IG and FG wrote the literature review. VC, MB, GO, TP-G, MD, and ER supervised and edited the review. All authors contributed to the article and approved the submitted version.
Funding
IG, FG, VC, GO, and ER are supported by the University of Padova.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer AC declared a shared affiliation with the authors to the handling editor at the time of review.
Acknowledgments
The authors thank Dr. Andrea Pagetta for graphical support.
References
1. Yoshida GJ. Metabolic Reprogramming: The Emerging Concept and Associated Therapeutic Strategies. J Exp Clin Cancer Res (2015) 34:111. doi: 10.1186/s13046-015-0221-y
2. Ward PS, Thompson CB. Metabolic Reprogramming: A Cancer Hallmark Even Warburg Did Not Anticipate. Cancer Cell (2012) 21:297–308. doi: 10.1016/j.ccr.2012.02.014
3. Cocetta V, Ragazzi E, Montopoli M. Mitochondrial Involvement in Cisplatin Resistance. Int J Mol Sci (2019) 20:3384. doi: 10.3390/ijms20143384
4. Zaal EA, Berkers CR. The Influence of Metabolism on Drug Response in Cancer. Front Oncol (2018) 8:500. doi: 10.3389/fonc.2018.00500
5. Desbats MA, Giacomini I, Prayer-Galetti T, Montopoli M. Metabolic Plasticity in Chemotherapy Resistance. Front Oncol (2020) 10:281. doi: 10.3389/fonc.2020.00281
6. Fendt S-M, Frezza C, Erez A. Targeting Metabolic Plasticity and Flexibility Dynamics for Cancer Therapy. Cancer Discov (2020) 10:1797–807. doi: 10.1158/2159-8290.CD-20-0844
7. Giacomini I, Ragazzi E, Pasut G, Montopoli M. The Pentose Phosphate Pathway and Its Involvement in Cisplatin Resistance. Int J Mol Sci (2020) 21:937. doi: 10.3390/ijms21030937
8. Morandi A, Indraccolo S. Linking Metabolic Reprogramming to Therapy Resistance in Cancer. Biochim Biophys Acta BBA - Rev Cancer (2017) 1868:1–6. doi: 10.1016/j.bbcan.2016.12.004
9. Cocetta V, Ragazzi E, Montopoli M. Links Between Cancer Metabolism and Cisplatin Resistance. Int Rev Cell Mol Biol (2020) 354:107–64. doi: 10.1016/bs.ircmb.2020.01.005
10. Phan LM, Yeung S-CJ, Lee M-H. Cancer Metabolic Reprogramming: Importance, Main Features, and Potentials for Precise Targeted Anti-Cancer Therapies. Cancer Biol Med (2014) 11:1–19. doi: 10.7497/j.issn.2095-3941.2014.01.001
11. Olson RE. Discovery of the Lipoproteins, Their Role in Fat Transport and Their Significance as Risk Factors. J Nutr (1998) 128:439S–43S. doi: 10.1093/jn/128.2.439S
12. Schekman R. Discovery of the Cellular and Molecular Basis of Cholesterol Control. Proc Natl Acad Sci (2013) 110:14833–6. doi: 10.1073/pnas.1312967110
13. Simons K, Ikonen E. How Cells Handle Cholesterol. Science (2000) 290:1721–6. doi: 10.1126/science.290.5497.1721
14. Farese RV, Herz J. Cholesterol Metabolism and Embryogenesis. Trends Genet (1998) 14:115–20. doi: 10.1016/S0168-9525(97)01377-2
15. Goldstein JL, Brown MS. History of Discovery : The LDL Receptor. Arterioscler Thromb (2010) 29:431–8. doi: 10.1161/ATVBAHA.108.179564.History
16. Munro S. Lipid Rafts: Elusive or Illusive? Cell (2003) 115:377–88. doi: 10.1016/S0092-8674(03)00882-1
17. Simons K, Ehehalt R. Cholesterol, Lipid Rafts, and Disease. J Clin Invest (2002) 110:597–603. doi: 10.1172/JCI0216390
18. Sezgin E, Levental I, Mayor S, Eggeling C. The Mystery of Membrane Organization: Composition, Regulation and Roles of Lipid Rafts. Nat Rev Mol Cell Biol (2017) 18:361–74. doi: 10.1038/nrm.2017.16
19. Simons K, Ikonen E. Functional Rafts in Cell Membranes. Nature (1997) 387:569–72. doi: 10.1038/42408
20. Kolesnick R. The Therapeutic Potential of Modulating the Ceramide/Sphingomyelin Pathway. J Clin Invest (2002) 110:3–8. doi: 10.1172/JCI16127
21. van der Goot FG, Harder T. Raft Membrane Domains: From a Liquid-Ordered Membrane Phase to a Site of Pathogen Attack. Semin Immunol (2001) 13:89–97. doi: 10.1006/smim.2000.0300
22. Head BP, Patel HH, Insel PA. Interaction of Membrane/Lipid Rafts With the Cytoskeleton: Impact on Signaling and Function. Biochim Biophys Acta (2014) 1838:532–45. doi: 10.1016/j.bbamem.2013.07.018
23. Simons K, Toomre D. Lipid Rafts and Signal Transduction. Nat Rev Mol Cell Biol (2000) 1:31–9. doi: 10.1038/35036052
24. Buhaescu I, Izzedine H. Mevalonate Pathway: A Review of Clinical and Therapeutical Implications. Clin Biochem (2007) 40:575–84. doi: 10.1016/j.clinbiochem.2007.03.016
25. Shi Z, Ruvkun G. The Mevalonate Pathway Regulates microRNA Activity in Caenorhabditis Elegans. Proc Natl Acad Sci USA (2012) 109:4568–73. doi: 10.1073/pnas.1202421109
26. Berndt N, Hamilton AD, Sebti SM. Targeting Protein Prenylation for Cancer Therapy. Nat Rev Cancer (2011) 11:775–91. doi: 10.1038/nrc3151
27. Hulce JJ, Cognetta AB, Niphakis MJ, Tully SE, Cravatt BF. Proteome-Wide Mapping of Cholesterol-Interacting Proteins in Mammalian Cells. Nat Methods (2013) . 10:259–64. doi: 10.1038/nmeth.2368
28. Martins IJ, Hone E, Foster JK, Sünram-Lea SI, Gnjec A, Fuller SJ, et al. Apolipoprotein E, Cholesterol Metabolism, Diabetes, and the Convergence of Risk Factors for Alzheimer’s Disease and Cardiovascular Disease. Mol Psychiatry (2006) 11:721–36. doi: 10.1038/sj.mp.4001854
29. Van Der Kant R, Goldstein LSB, Ossenkoppele R. Amyloid-β-Independent Regulators of Tau Pathology in Alzheimer Disease. Nat Rev Neurosci (2020) 21:21–35. doi: 10.1038/s41583-019-0240-3
30. Haase CL, Tybjærg-Hansen A, Nordestgaard BG, Frikke-Schmidt R. HDL Cholesterol and Risk of Type 2 Diabetes: A Mendelian Randomization Study. Diabetes (2015) 64:3328–33. doi: 10.2337/db14-1603
31. Silvente-Poirot S, Poirot M. Cholesterol and Cancer, in the Balance. Science (2014) 343:1445–6. doi: 10.1126/science.1252787
32. Kuzu OF, Noory MA, Robertson GP. The Role of Cholesterol in Cancer. Cancer Res (2016) 76:2063–70. doi: 10.1158/0008-5472.CAN-15-2613
33. Germain N, Dhayer M, Boileau M, Fovez Q, Kluza J, Marchetti P. Lipid Metabolism and Resistance to Anticancer Treatment. Biology (2020) 9:474. doi: 10.3390/biology9120474
34. Huang B, Song B, Xu C. Cholesterol Metabolism in Cancer: Mechanisms and Therapeutic Opportunities. Nat Metab (2020) 2:132–41. doi: 10.1038/s42255-020-0174-0
35. Ding X, Zhang W, Li S, Yang H. The Role of Cholesterol Metabolism in Cancer. Am J Cancer Res (2019) 9:219–27.
36. Cruz PMR, Mo H, McConathy WJ, Sabnis N, Lacko AG. The Role of Cholesterol Metabolism and Cholesterol Transport in Carcinogenesis: A Review of Scientific Findings, Relevant to Future Cancer Therapeutics. Front Pharmacol (2013) 4:119. doi: 10.3389/fphar.2013.00119
37. Xu H, Zhou S, Tang Q, Xia H, Bi F. Cholesterol Metabolism: New Functions and Therapeutic Approaches in Cancer. Biochim Biophys Acta Rev Cancer (2020) 1874:188394. doi: 10.1016/j.bbcan.2020.188394
38. Repa JJ, Mangelsdorf DJ. The Role of Orphan Nuclear Receptors in the Regulation of Cholesterol Homeostasis. Annu Rev Cell Dev Biol (2000) 16:459–81. doi: 10.1146/annurev.cellbio.16.1.459
39. Kruit JK, Groen AK, van Berkel TJ, Kuipers F. Emerging Roles of the Intestine in Control of Cholesterol Metabolism. World J Gastroenterol (2006) 12:6429–39. doi: 10.3748/wjg.v12.i40.6429
40. Luo J, Yang H, Song B-L. Mechanisms and Regulation of Cholesterol Homeostasis. Nat Rev Mol Cell Biol (2020) 21:225–45. doi: 10.1038/s41580-019-0190-7
41. Mo H, Elson CE. Studies of the Isoprenoid-Mediated Inhibition of Mevalonate Synthesis Applied to Cancer Chemotherapy and Chemoprevention. Exp Biol Med (2004) 229:567–85. doi: 10.1177/153537020422900701
42. Ikonen E. Cellular Cholesterol Trafficking and Compartmentalization. Nat Rev Mol Cell Biol (2008) 9:125–38. doi: 10.1038/nrm2336
43. Singh P, Saxena R, Srinivas G, Pande G, Chattopadhyay A. Cholesterol Biosynthesis and Homeostasis in Regulation of the Cell Cycle. PloS One (2013) 8:e58833. doi: 10.1371/journal.pone.0058833
44. Kandutsch AA, Russell AE. Preputial Gland Tumor Sterols. 3. A Metabolic Pathway From Lanosterol to Cholesterol. J Biol Chem (1960) 235:2256–61. doi: 10.1016/S0021-9258(18)64608-3
45. Ačimovič J, Goyal S, Košir R, Goličnik M, Perše M, Belič A, et al. Cytochrome P450 Metabolism of the Post-Lanosterol Intermediates Explains Enigmas of Cholesterol Synthesis. Sci Rep (2016) 6:28462. doi: 10.1038/srep28462
46. Jin U, Park SJ, Park SM. Cholesterol Metabolism in the Brain and Its Association With Parkinson’s Disease. Exp Neurobiol (2019) 28:554–67. doi: 10.5607/en.2019.28.5.554
47. Goedeke L, Fernández-Hernando C. Regulation of Cholesterol Homeostasis. Cell Mol Life Sci (2012) 69:915–30. doi: 10.1007/s00018-011-0857-5
48. Jia L, Betters JL, Yu L. Niemann-Pick C1-Like 1 (NPC1L1) Protein in Intestinal and Hepatic Cholesterol Transport. Annu Rev Physiol (2011) 73:239–59. doi: 10.1146/annurev-physiol-012110-142233
49. Betters JL, Yu L. NPC1L1 and Cholesterol Transport. FEBS Lett (2010) 584:2740–7. doi: 10.1016/j.febslet.2010.03.030
50. Nguyen TM, Sawyer JK, Kelley KL, Davis MA, Rudel LL. Cholesterol Esterification by ACAT2 is Essential for Efficient Intestinal Cholesterol Absorption: Evidence From Thoracic Lymph Duct Cannulation. J Lipid Res (2012) 53:95–104. doi: 10.1194/jlr.M018820
51. Ko CW, Qu J, Black DD, Tso P. Regulation of Intestinal Lipid Metabolism: Current Concepts and Relevance to Disease. Nat Rev Gastroenterol Hepatol (2020) 17:169–83. doi: 10.1038/s41575-019-0250-7
52. Ginsberg HN. Lipoprotein Physiology. Endocrinol Metab Clin North Am (1998) 27:503–19. doi: 10.1016/s0889-8529(05)70023-2
53. Ramasamy I. Recent Advances in Physiological Lipoprotein Metabolism. Clin Chem Lab Med (2014) 52:1695–727. doi: 10.1515/cclm-2013-0358
54. Holmes MV, Ala-Korpela M. What Is “LDL Cholesterol”? Nat Rev Cardiol (2019) 16:197–8. doi: 10.1038/s41569-019-0157-6
55. Go GW, Mani A. Low-Density Lipoprotein Receptor (LDLR) Family Orchestrates Cholesterol Homeostasis. Yale J Biol Med (2012) 85:19–28.
56. Luo X, Cheng C, Tan Z, Li N, Tang M, Yang L, et al. Emerging Roles of Lipid Metabolism in Cancer Metastasis. Mol Cancer (2017) 16:76. doi: 10.1186/s12943-017-0646-3
57. Kwon HJ, Abi-Mosleh L, Wang ML, Deisenhofer J, Goldstein JL, Brown MS, et al. Structure of N-Terminal Domain of NPC1 Reveals Distinct Subdomains for Binding and Transfer of Cholesterol. Cell (2009) 137:1213–24. doi: 10.1016/j.cell.2009.03.049
58. Li J, Pfeffer SR. Lysosomal Membrane Glycoproteins Bind Cholesterol and Contribute to Lysosomal Cholesterol Export. eLife (2016) 5:e21635. doi: 10.7554/eLife.21635
59. Ouimet M, Barrett TJ, Fisher EA. HDL and Reverse Cholesterol Transport. Circ Res (2019) 124:1505–18. doi: 10.1161/CIRCRESAHA.119.312617
60. Brufau G, Groen AK, Kuipers F. Reverse Cholesterol Transport Revisited: Contribution of Biliary Versus Intestinal Cholesterol Excretion. Arterioscler Thromb Vasc Biol (2011) 31:1726–33. doi: 10.1161/ATVBAHA.108.181206
61. Röhrl C, Stangl H. HDL Endocytosis and Resecretion. Biochim Biophys Acta (2013) 1831:1626–33. doi: 10.1016/j.bbalip.2013.07.014
62. Chang TY, Chang CCY, Ohgami N, Yamauchi Y. Cholesterol Sensing, Trafficking, and Esterification. Annu Rev Cell Dev Biol (2006) 22:129–57. doi: 10.1146/annurev.cellbio.22.010305.104656
63. Gelissen IC, Harris M, Rye KA, Quinn C, Brown AJ, Kockx M, et al. ABCA1 and ABCG1 Synergize to Mediate Cholesterol Export to Apoa-I. Arterioscler Thromb Vasc Biol (2006) 26:534–40. doi: 10.1161/01.ATV.0000200082.58536.e1
64. Ossoli A, Pavanello C, Calabresi L. High-Density Lipoprotein, Lecithin: Cholesterol Acyltransferase, and Atherosclerosis. Endocrinol Metab (2016) 31:223. doi: 10.3803/EnM.2016.31.2.223
65. Chang TY, Li BL, Chang CCY, Urano Y. Acyl-Coenzyme A:Cholesterol Acyltransferases. Am J Physiol Endocrinol Metab (2009) 297:E1–9. doi: 10.1152/ajpendo.90926.2008
66. Petan T, Jarc E, Jusović M. Lipid Droplets in Cancer: Guardians of Fat in a Stressful World. Molecules (2018) 23:1941. doi: 10.3390/molecules23081941
67. Miyazaki A, Sakashita N, Lee O, Takahashi K, Horiuchi S, Hakamata H, et al. Expression of ACAT-1 Protein in Human Atherosclerotic Lesions and Cultured Human Monocytes-Macrophages. Arterioscler Thromb Vasc Biol (1998) 18:1568–74. doi: 10.1161/01.ATV.18.10.1568
68. Chang CCY, Sakashita N, Ornvold K, Lee O, Chang ET, Dong R, et al. Immunological Quantitation and Localization of ACAT-1 and ACAT-2 in Human Liver and Small Intestine. J Biol Chem (2000) 275:28083–92. doi: 10.1074/jbc.M003927200
69. Joyce CW, Shelness GS, Davis MA, Lee RG, Skinner K, Anderson RA, et al. ACAT1 and ACAT2 Membrane Topology Segregates a Serine Residue Essential for Activity to Opposite Sides of the Endoplasmic Reticulum Membrane. Mol Biol Cell (2000) 11:3675–87. doi: 10.1091/mbc.11.11.3675
70. Steck TL, Lange Y. Cell Cholesterol Homeostasis: Mediation by Active Cholesterol. Trends Cell Biol (2010) 20:680–7. doi: 10.1016/j.tcb.2010.08.007
71. Wong J, Quinn CM, Brown AJ. SREBP-2 Positively Regulates Transcription of the Cholesterol Efflux Gene, ABCA1, by Generating Oxysterol Ligands for LXR. Biochem J (2006) 400:485–91. doi: 10.1042/BJ20060914
72. Brown MS, Radhakrishnan A, Goldstein JL. Retrospective on Cholesterol Homeostasis: The Central Role of Scap. Annu Rev Biochem (2018) 87:783–807. doi: 10.1146/annurev-biochem-062917-011852
73. Nagoshi E, Yoneda Y. Dimerization of Sterol Regulatory Element-Binding Protein 2 Via the Helix-Loop-Helix-Leucine Zipper Domain is a Prerequisite for Its Nuclear Localization Mediated by Importin β. Mol Cell Biol (2001) 21:2779–89. doi: 10.1128/MCB.21.8.2779-2789.2001
74. Zhao X, Yang F. Regulation of SREBP-Mediated Gene Expression. Acta Biophys Sin (2012) 28:287. doi: 10.3724/SP.J.1260.2012.20034
75. Radhakrishnan A, Sun LP, Kwon HJ, Brown MS, Goldstein JL. Direct Binding of Cholesterol to the Purified Membrane Region of SCAP: Mechanism for a Sterol-Sensing Domain. Mol Cell (2004) 15:259–68. doi: 10.1016/j.molcel.2004.06.019
76. Howe V, Sharpe LJ, Prabhu AV, Brown AJ. New Insights Into Cellular Cholesterol Acquisition: Promoter Analysis of Human HMGCR and SQLE , Two Key Control Enzymes in Cholesterol Synthesis. Biochim Biophys Acta Mol Cell Biol Lipids (2017) 1862:647–57. doi: 10.1016/j.bbalip.2017.03.009
77. Horton JD, Goldstein JL, Brown MS. SREBPs: Activators of the Complete Program of Cholesterol and Fatty Acid Synthesis in the Liver. J Clin Invest (2002) 109:1125–31. doi: 10.1172/JCI0215593
78. Theesfeld CL, Pourmand D, Davis T, Garza RM, Hampton RY. The Sterol-Sensing Domain (SSD) Directly Mediates Signal-Regulated Endoplasmic Reticulum-Associated Degradation (ERAD) of 3-Hydroxy-3-Methylglutaryl (HMG)-CoA Reductase Isozyme Hmg2. J Biol Chem (2011) 286:26298–307. doi: 10.1074/jbc.M111.244798
79. Clarke PR, Hardie DG. Regulation of HMG-CoA Reductase: Identification of the Site Phosphorylated by the AMP-Activated Protein Kinase In Vitro and in Intact Rat Liver. EMBO J (1990) 9:2439–46. doi: 10.1002/j.1460-2075.1990.tb07420.x
80. Alrefai WA, Annaba F, Sarwar Z, Dwivedi A, Saksena S, Singla A, et al. Modulation of Human Niemann-Pick C1-Like 1 Gene Expression by Sterol: Role of Sterol Regulatory Element Binding Protein 2. Am J Physiol Gastrointest Liver Physiol (2007) 292:G369–376. doi: 10.1152/ajpgi.00306.2006
81. Kotzka J, Müller-Wieland D, Roth G, Kremer L, Munck M, Schürmann S, et al. Sterol Regulatory Element Binding Proteins (SREBP)-1a and SREBP-2 are Linked to the MAP-kinase Cascade. J Lipid Res (2000) 41:99–108. doi: 10.1016/S0022-2275(20)32079-4
82. Sato R, Inoue J, Kawabe Y, Kodama T, Takano T, Maeda M. Sterol-Dependent Transcriptional Regulation of Sterol Regulatory Element-binding Protein-2. J Biol Chem (1996) 271:26461–4. doi: 10.1074/jbc.271.43.26461
83. Radhakrishnan SK, den Besten W, Deshaies RJ. p97-dependent Retrotranslocation and Proteolytic Processing Govern Formation of Active Nrf1 Upon Proteasome Inhibition. eLife (2014) 3:e01856. doi: 10.7554/eLife.01856
84. Wang B, Tontonoz P. Liver X Receptors in Lipid Signalling and Membrane Homeostasis. Nat Rev Endocrinol (2018) 14:452–63. doi: 10.1038/s41574-018-0037-x
85. Peet DJ, Turley SD, Ma W, Janowski BA, Lobaccaro JM, Hammer RE, et al. Cholesterol and Bile Acid Metabolism Are Impaired in Mice Lacking the Nuclear Oxysterol Receptor LXR Alpha. Cell (1998) 93:693–704. doi: 10.1016/s0092-8674(00)81432-4
86. Yvan-Charvet L, Wang N, Tall AR. Role of HDL, ABCA1, and ABCG1 Transporters in Cholesterol Efflux and Immune Responses. Arterioscler Thromb Vasc Biol (2010) 30:139–43. doi: 10.1161/ATVBAHA.108.179283
87. Venkateswaran A, Laffitte BA, Joseph SB, Mak PA, Wilpitz DC, Edwards PA, et al. Control of Cellular Cholesterol Efflux by the Nuclear Oxysterol Receptor LXR Alpha. Proc Natl Acad Sci USA (2000) 97:12097–102. doi: 10.1073/pnas.200367697
88. Duval C, Touche V, Tailleux A, Fruchart JC, Fievet C, Clavey V, et al. Niemann–Pick C1 Like 1 Gene Expression is Down-Regulated by LXR Activators in the Intestine. Biochem Biophys Res Commun (2006) 340:1259–63. doi: 10.1016/j.bbrc.2005.12.137
89. Zelcer N, Hong C, Boyadjian R, Tontonoz P. LXR Regulates Cholesterol Uptake Through Idol-Dependent Ubiquitination of the LDL Receptor. Science (2009) 325:100–4. doi: 10.1126/science.1168974
90. Zhang L, Reue K, Fong LG, Young SG, Tontonoz P. Feedback Regulation of Cholesterol Uptake by the LXR–IDOL–LDLR Axis. Arterioscler Thromb Vasc Biol (2012) 32:2541–6. doi: 10.1161/ATVBAHA.112.250571
91. Stancu C, Sima A. Statins: Mechanism of Action and Effects. J Cell Mol Med (2001) 5:378–87. doi: 10.1111/j.1582-4934.2001.tb00172.x
92. Bedi O, Dhawan V, Sharma PL, Kumar P. Pleiotropic Effects of Statins: New Therapeutic Targets in Drug Design. Naunyn Schmiedebergs Arch Pharmacol (2016) 389:695–712. doi: 10.1007/s00210-016-1252-4
93. Endo A. A Historical Perspective on the Discovery of Statins. Proc Jpn Acad Ser B (2010) 86:484–93. doi: 10.2183/pjab.86.484
94. Davies JT, Delfino SF, Feinberg CE, Johnson MF, Nappi VL, Olinger JT, et al. Current and Emerging Uses of Statins in Clinical Therapeutics: A Review. Lipid Insights (2016) 9:13–29. doi: 10.4137/LPI.S37450
95. Ramkumar S, Raghunath A, Raghunath S. Statin Therapy: Review of Safety and Potential Side Effects. Acta Cardiol Sin (2016) 32:631–9. doi: 10.6515/ACS20160611A
96. Liu A, Wu Q, Guo J, Ares I, Rodríguez J-L, Martínez-Larrañaga MR, et al. Statins: Adverse Reactions, Oxidative Stress and Metabolic Interactions. Pharmacol Ther (2019) 195:54–84. doi: 10.1016/j.pharmthera.2018.10.004
97. Ray KK, Bays HE, Catapano AL, Lalwani ND, Bloedon LT, Sterling LR, et al. Clear Harmony Trial. Safety and Efficacy of Bempedoic Acid to Reduce LDL Cholesterol. N Engl J Med (2019) 380:1022–32. doi: 10.1056/NEJMoa1803917
98. Fazio S, Linton MF. The Role of Fibrates in Managing Hyperlipidemia: Mechanisms of Action and Clinical Efficacy. Curr Atheroscler Rep (2004) 6:148–57. doi: 10.1007/s11883-004-0104-8
99. Backes JM, Gibson CA, Ruisinger JF, Moriarty PM. Fibrates: What Have We Learned in the Past 40 Years? Pharmacotherapy (2007) 27:412–24. doi: 10.1592/phco.27.3.412
100. Laganà A, Vitale S, Nigro A, Sofo V, Salmeri F, Rossetti P, et al. Pleiotropic Actions of Peroxisome Proliferator-Activated Receptors (PPARs) in Dysregulated Metabolic Homeostasis, Inflammation and Cancer: Current Evidence and Future Perspectives. Int J Mol Sci (2016) 17:999. doi: 10.3390/ijms1707099933
101. Phan BA, Dayspring TD, Toth PP. Ezetimibe Therapy: Mechanism of Action and Clinical Update. Vasc Health Risk Manag (2012) 8:415–27. doi: 10.2147/VHRM.S33664
102. Florentin M, Liberopoulos EN, Elisaf MS. Ezetimibe-Associated Adverse Effects: What the Clinician Needs to Know: Ezetimibe and Side Effects. Int J Clin Pract (2007) 62:88–96. doi: 10.1111/j.1742-1241.2007.01592.x
103. Corsini A, Windler E, Farnier M. Colesevelam Hydrochloride: Usefulness of a Specifically Engineered Bile Acid Sequestrant for Lowering LDL-Cholesterol. Eur J Cardiovasc Prev Rehabil (2009) 16:1–9. doi: 10.1097/HJR.0b013e32831215db
104. Scaldaferri F, Pizzoferrato M, Ponziani FR, Gasbarrini G, Gasbarrini A. Use and Indications of Cholestyramine and Bile Acid Sequestrants. Intern Emerg Med (2013) 8:205–10. doi: 10.1007/s11739-011-0653-0
105. Gouni-Berthold I, Berthold HK. Mipomersen and Lomitapide: Two New Drugs for the Treatment of Homozygous Familial Hypercholesterolemia. Atheroscler Suppl (2015) 18:28–34. doi: 10.1016/j.atherosclerosissup.2015.02.005
106. Agarwala A, Jones P, Nambi V. The Role of Antisense Oligonucleotide Therapy in Patients With Familial Hypercholesterolemia: Risks, Benefits, and Management Recommendations. Curr Atheroscler Rep (2015) 17:467. doi: 10.1007/s11883-014-0467-4
107. Parhofer K. Mipomersen: Evidence-Based Review of its Potential in the Treatment of Homozygous and Severe Heterozygous Familial Hypercholesterolemia. Core Evid (2012) 7:29–38. doi: 10.2147/CE.S25239
108. Goulooze SC, Cohen AF, Rissmann R. Lomitapide: New Drug Mechanisms: Lomitapide. Br J Clin Pharmacol (2015) 80:179–81. doi: 10.1111/bcp.12612
109. Chaudhary R, Garg J, Shah N, Sumner A. PCSK9 Inhibitors: A New Era of Lipid Lowering Therapy. World J Cardiol (2017) 9:76. doi: 10.4330/wjc.v9.i2.76
110. Knopp RH. Drug Treatment of Lipid Disorders. N Engl J Med (1999) 341:498–511. doi: 10.1056/NEJM199908123410707
111. Gorin A, Gabitova L, Astsaturov I. Regulation of Cholesterol Biosynthesis and Cancer Signaling. Curr Opin Pharmacol (2012) 12:710–6. doi: 10.1016/j.coph.2012.06.011
112. Kuzu OF, Gowda R, Noory MA, Robertson GP. Modulating Cancer Cell Survival by Targeting Intracellular Cholesterol Transport. Br J Cancer (2017) 117:513–24. doi: 10.1038/bjc.2017.200
113. Gu L, Saha ST, Thomas J, Kaur M. Targeting Cellular Cholesterol for Anticancer Therapy. FEBS J (2019) 286:4192–208. doi: 10.1111/febs.15018
114. Li YC, Park MJ, Ye S-K, Kim C-W, Kim Y-N. Elevated Levels of Cholesterol-Rich Lipid Rafts in Cancer Cells Are Correlated With Apoptosis Sensitivity Induced by Cholesterol-Depleting Agents. Am J Pathol (2006) 168:1107–18. doi: 10.2353/ajpath.2006.050959
115. Liu Z, Liu X, Liu S, Cao Q. Cholesterol Promotes the Migration and Invasion of Renal Carcinoma Cells by Regulating the KLF5/miR-27a/FBXW7 Pathway. Biochem Biophys Res Commun (2018) 502:69–75. doi: 10.1016/j.bbrc.2018.05.122
116. Liang Y, Besch-Williford C, Aebi JD, Mafuvadze B, Cook MT, Zou X, et al. Cholesterol Biosynthesis Inhibitors as Potent Novel Anti-Cancer Agents: Suppression of Hormone-Dependent Breast Cancer by the Oxidosqualene Cyclase Inhibitor RO 48-8071. Breast Cancer Res Treat (2014) 146:51–62. doi: 10.1007/s10549-014-2996-5
117. Grinter SZ, Liang Y, Huang S-Y, Hyder SM, Zou X. An Inverse Docking Approach for Identifying New Potential Anti-Cancer Targets. J Mol Graph Model (2011) 29:795–9. doi: 10.1016/j.jmgm.2011.01.002
118. Maione F, Oliaro-Bosso S, Meda C, Di Nicolantonio F, Bussolino F, Balliano G, et al. The Cholesterol Biosynthesis Enzyme Oxidosqualene Cyclase is a New Target to Impair Tumour Angiogenesis and Metastasis Dissemination. Sci Rep (2015) 5:9054. doi: 10.1038/srep09054
119. Liang Y, Mafuvadze B, Aebi JD, Hyder SM. Cholesterol Biosynthesis Inhibitor RO 48-8071 Suppresses Growth of Hormone-Dependent and Castration-Resistant Prostate Cancer Cells. OncoTargets Ther (2016) 9:3223–32. doi: 10.2147/OTT.S105725
120. Brusselmans K, Timmermans L, Van de Sande T, Van Veldhoven PP, Guan G, Shechter I, et al. Squalene Synthase, a Determinant of Raft-Associated Cholesterol and Modulator of Cancer Cell Proliferation. J Biol Chem (2007) 282:18777–85. doi: 10.1074/jbc.M611763200
121. Lanterna C, Musumeci A, Raccosta L, Corna G, Moresco M, Maggioni D, et al. The Administration of Drugs Inhibiting Cholesterol/Oxysterol Synthesis is Safe and Increases the Efficacy of Immunotherapeutic Regimens in Tumor-Bearing Mice. Cancer Immunol Immunother (2016) 65:1303–15. doi: 10.1007/s00262-016-1884-8
122. Antalis CJ, Arnold T, Rasool T, Lee B, Buhman KK, Siddiqui RA. High ACAT1 Expression in Estrogen Receptor Negative Basal-Like Breast Cancer Cells Is Associated With LDL-induced Proliferation. Breast Cancer Res Treat (2010) 122:661–70. doi: 10.1007/s10549-009-0594-8
123. Shim SH, Sur S, Steele R, Albert CJ, Huang C, Ford DA, et al. Disrupting Cholesterol Esterification by Bitter Melon Suppresses Triple-Negative Breast Cancer Cell Growth. Mol Carcinog (2018) 57:1599–607. doi: 10.1002/mc.22882
124. Lee SSY, Li J, Tai JN, Ratliff TL, Park K, Cheng JX. Avasimibe Encapsulated in Human Serum Albumin Blocks Cholesterol Esterification for Selective Cancer Treatment. ACS Nano (2015) 9:2420–32. doi: 10.1021/nn504025a
125. Lee HJ, Li J, Vickman RE, Li J, Liu R, Durkes AC, et al. Cholesterol Esterification Inhibition Suppresses Prostate Cancer Metastasis by Impairing the Wnt/β-Catenin Pathway. Mol Cancer Res (2018) 16:974–85. doi: 10.1158/1541-7786.MCR-17-0665
126. Li J, Gu D, Lee SSY, Song B, Bandyopadhyay S, Chen S, et al. Abrogating Cholesterol Esterification Suppresses Growth and Metastasis of Pancreatic Cancer. Oncogene (2016) 35:6378–88. doi: 10.1038/onc.2016.168
127. Bi M, Qiao X, Zhang H, Wu H, Gao Z, Zhou H, et al. Effect of Inhibiting ACAT−1 Expression on the Growth and Metastasis of Lewis Lung Carcinoma. Oncol Lett (2019) 18:1548–56. doi: 10.3892/ol.2019.10427
128. Li J, Qu X, Tian J, Zhang JT, Cheng JX. Cholesterol Esterification Inhibition and Gemcitabine Synergistically Suppress Pancreatic Ductal Adenocarcinoma Proliferation. PloS One (2018) 13:e0193318. doi: 10.1371/journal.pone.0193318
129. Pommier AJC, Alves G, Viennois E, Bernard S, Communal Y, Sion B, et al. Liver X Receptor Activation Downregulates AKT Survival Signaling in Lipid Rafts and Induces Apoptosis of Prostate Cancer Cells. Oncogene (2010) 29:2712–23. doi: 10.1038/onc.2010.30
130. Roz AE, Bard JM, Huvelin JM, Nazih H. LXR Agonists and ABCG1-dependent Cholesterol Efflux in MCF-7 Breast Cancer Cells: Relation to Proliferation and Apoptosis. Anticancer Res (2012) 32:3007–13.
131. Zhang W, Jiang H, Zhang J, Zhang Y, Liu A, Zhao Y, et al. Liver X Receptor Activation Induces Apoptosis of Melanoma Cell Through Caspase Pathway. Cancer Cell Int (2014) 14:16. doi: 10.1186/1475-2867-14-16
132. Agarwal JR, Wang Q, Tanno T, Rasheed Z, Merchant A, Ghosh N, et al. Activation of Liver X Receptors Inhibits Hedgehog Signaling, Clonogenic Growth, and Self-Renewal in Multiple Myeloma. Mol Cancer Ther (2014) 13:1873–81. doi: 10.1158/1535-7163.MCT-13-0997
133. Kaneko T, Kanno C, Ichikawa-Tomikawa N, Kashiwagi K, Yaginuma N, Ohkoshi C, et al. Liver X Receptor Reduces Proliferation of Human Oral Cancer Cells by Promoting Cholesterol Efflux Via Up-Regulation of ABCA1 Expression. Oncotarget (2015) 6:33345–57. doi: 10.18632/oncotarget.5428
134. Rough JJ, Monroy MA, Yerrum S, Daly JM. Anti-Proliferative Effect of LXR Agonist T0901317 in Ovarian Carcinoma Cells. J Ovarian Res (2010) 3:13. doi: 10.1186/1757-2215-3-13
135. Lou R, Cao H, Dong S, Shi C, Xu X, Ma R, et al. Liver X Receptor Agonist T0901317 Inhibits the Migration and Invasion of Non-Small-Cell Lung Cancer Cells In Vivo and In Vitro. Anticancer Drugs (2019) 30:495–500. doi: 10.1097/CAD.0000000000000758
136. Shao W, Zhu W, Lin J, Luo M, Lin Z, Lu L, et al. Liver X Receptor Agonism Sensitizes a Subset of Hepatocellular Carcinoma to Sorafenib by Dual-Inhibiting MET and EGFR. Neoplasia (2020) 22:1–9. doi: 10.1016/j.neo.2019.08.002
137. Nguyen-Vu T, Vedin LL, Liu K, Jonsson P, Lin JZ, Candelaria NR, et al. Liver × Receptor Ligands Disrupt Breast Cancer Cell Proliferation Through an E2F-Mediated Mechanism. Breast Cancer Res (2013) 15:R51. doi: 10.1186/bcr3443
138. Hassan TS, Paniccia A, Russo V, Steffensen KR. LXR Inhibits Proliferation of Human Breast Cancer Cells Through the PI3K-Akt Pathway. Nucl Recept Res (2015) 2:1–10. doi: 10.11131/2015/101154
139. Pencheva N, Buss CG, Posada J, Merghoub T, Tavazoie SF. Broad-Spectrum Therapeutic Suppression of Metastatic Melanoma Through Nuclear Hormone Receptor Activation. Cell (2014) 156:986–1001. doi: 10.1016/j.cell.2014.01.038
140. Candelaria NR, Addanki S, Zheng J, Nguyen-Vu T, Karaboga H, Dey P, et al. Antiproliferative Effects and Mechanisms of Liver X Receptor Ligands in Pancreatic Ductal Adenocarcinoma Cells. PloS One (2014) 9:e106289. doi: 10.1371/journal.pone.0106289
141. Wang Q, Shen B, Qin X, Liu S, Feng J. Akt/mTOR and AMPK Signaling Pathways are Responsible for Liver X Receptor Agonist GW3965-Enhanced Gefitinib Sensitivity in Non-Small Cell Lung Cancer Cell Lines. Transl Cancer Res (2019) 8:66–76. doi: 10.21037/tcr.2018.12.34
142. Flaveny CA, Griffett K, El-Gendy BE-DM, Kazantzis M, Sengupta M, Amelio AL, et al. Broad Anti-tumor Activity of a Small Molecule That Selectively Targets the Warburg Effect and Lipogenesis. Cancer Cell (2015) 28:42–56. doi: 10.1016/j.ccell.2015.05.007
143. Wu G, Wang Q, Xu Y, Li J, Zhang H, Qi G, et al. Targeting the Transcription Factor Receptor LXR to Treat Clear Cell Renal Cell Carcinoma: Agonist or Inverse Agonist? Cell Death Dis (2019) 10:416. doi: 10.1038/s41419-019-1654-6
144. Saidi SA, Holland CM, Charnock-Jones DS, Smith SK. In Vitro and In Vivo Effects of the PPAR-Alpha Agonists Fenofibrate and Retinoic Acid in Endometrial Cancer. Mol Cancer (2006) 5:13. doi: 10.1186/1476-4598-5-13
145. Yamasaki D, Kawabe N, Nakamura H, Tachibana K, Ishimoto K, Tanaka T, et al. Fenofibrate Suppresses Growth of the Human Hepatocellular Carcinoma Cell Via PPARα-Independent Mechanisms. Eur J Cell Biol (2011) 90:657–64. doi: 10.1016/j.ejcb.2011.02.005
146. Jan CI, Tsai MH, Chiu CF, Huang YP, Liu CJ, Chang NW. Fenofibrate Suppresses Oral Tumorigenesis Via Reprogramming Metabolic Processes: Potential Drug Repurposing for Oral Cancer. Int J Biol Sci (2016) 12:786–98. doi: 10.7150/ijbs.13851
147. Chen X, Chen S, Yu D. Metabolic Reprogramming of Chemoresistant Cancer Cells and the Potential Significance of Metabolic Regulation in the Reversal of Cancer Chemoresistance. Metabolites (2020) 10:289. doi: 10.3390/metabo10070289
148. Robison NJ, Campigotto F, Chi SN, Manley PE, Turner CD, Zimmerman MA, et al. A Phase II Trial of a Multi-Agent Oral Antiangiogenic (Metronomic) Regimen in Children With Recurrent or Progressive Cancer. Pediatr Blood Cancer (2014) 61:636–42. doi: 10.1002/pbc.24794
149. Luty M, Piwowarczyk K, Łabędź-Masłowska A, Wróbel T, Szczygieł M, Catapano J, et al. Fenofibrate Augments the Sensitivity of Drug-Resistant Prostate Cancer Cells to Docetaxel. Cancers (2019) 11:77. doi: 10.3390/cancers11010077
150. Li X, Chen YT, Hu P, Huang WC. Fatostatin Displays High Antitumor Activity in Prostate Cancer by Blocking Srebp-Regulated Metabolic Pathways and Androgen Receptor Signaling. Mol Cancer Ther (2014) 13:855–66. doi: 10.1158/1535-7163.MCT-13-0797
151. Yao L, Chen S, Li W. Fatostatin Inhibits the Development of Endometrial Carcinoma in Endometrial Carcinoma Cells and a Xenograft Model by Targeting Lipid Metabolism. Arch Biochem Biophys (2020) 684:108327. doi: 10.1016/j.abb.2020.108327
152. Gao S, Shi Z, Li X, Li W, Wang Y, Liu Z, et al. Fatostatin Suppresses Growth and Enhances Apoptosis by Blocking SREBP-Regulated Metabolic Pathways in Endometrial Carcinoma. Oncol Rep (2018) 39:1919–29. doi: 10.3892/or.2018.6265
153. Brovkovych V, Izhar Y, Danes JM, Dubrovskyi O, Sakallioglu IT, Morrow LM, et al. Fatostatin Induces Pro- and Anti-Apoptotic Lipid Accumulation in Breast Cancer. Oncogenesis (2018) 7:66. doi: 10.1038/s41389-018-0076-0
154. Liu Y, Zhang N, Zhang H, Wang L, Duan Y, Wang X, et al. Fatostatin in Combination With Tamoxifen Induces Synergistic Inhibition in ER-Positive Breast Cancer. Drug Des Devel Ther (2020) 14:3535–45. doi: 10.2147/DDDT.S253876
155. Smith B, Land H. Anticancer Activity of the Cholesterol Exporter ABCA1 Gene. Cell Rep (2012) 2:580–90. doi: 10.1016/j.celrep.2012.08.011
156. Ehmsen S, Pedersen MH, Wang G, Terp MG, Arslanagic A, Hood BL, et al. Increased Cholesterol Biosynthesis Is a Key Characteristic of Breast Cancer Stem Cells Influencing Patient Outcome. Cell Rep (2019) 27:3927–38. doi: 10.1016/j.celrep.2019.05.104
157. Chushi L, Wei W, Kangkang X, Yongzeng F, Ning X, Xiaolei C. HMGCR is Up-Regulated in Gastric Cancer and Promotes the Growth and Migration of the Cancer Cells. Gene (2016) 587:42–7. doi: 10.1016/j.gene.2016.04.029
158. Ashida S, Kawada C, Inoue K. Stromal Regulation of Prostate Cancer Cell Growth by Mevalonate Pathway Enzymes HMGCS1 and HMGCR. Oncol Lett (2017) 14:6533–42. doi: 10.3892/ol.2017.7025
159. Wong W-LW, Dimitroulakos J, Minden M, Penn L. HMG-CoA Reductase Inhibitors and the Malignant Cell: The Statin Family of Drugs as Triggers of Tumor-Specific Apoptosis. Leukemia (2002) 16:508–19. doi: 10.1038/sj.leu.2402476
160. Goldstein JL, Brown MS. Regulation of the Mevalonate Pathway. Nature (1990) 343:425–30. doi: 10.1038/343425a0
161. Bonetti PO, Lerman LO, Napoli C, Lerman A. Statin Effects Beyond Lipid Lowering—are They Clinically Relevant? Eur Heart J (2003) 24:225–48. doi: 10.1016/S0195-668X(02)00419-0
162. Gimple RC, Wang X. RAS: Striking at the Core of the Oncogenic Circuitry. Front Oncol (2019) 9:965. doi: 10.3389/fonc.2019.00965
163. Kaymak I, Maier CR, Schmitz W, Campbell AD, Dankworth B, Ade CP, et al. Mevalonate Pathway Provides Ubiquinone to Maintain Pyrimidine Synthesis and Survival in P53-Deficient Cancer Cells Exposed to Metabolic Stress. Cancer Res (2020) 80:189–203. doi: 10.1158/0008-5472.CAN-19-0650
165. Yang YF, Jan YH, Liu YP, Yang CJ, Su CY, Chang YC, et al. Squalene Synthase Induces Tumor Necrosis Factor Receptor 1 Enrichment in Lipid Rafts to Promote Lung Cancer Metastasis. Am J Respir Crit Care Med (2014) 190:675–87. doi: 10.1164/rccm.201404-0714OC
166. Parris TZ, Kovács A, Hajizadeh S, Nemes S, Semaan M, Levin M, et al. Frequent MYC Coamplification and DNA Hypomethylation of Multiple Genes on 8q in 8p11-p12-amplified Breast Carcinomas. Oncogenesis (2014) 3:e95. doi: 10.1038/oncsis.2014.8
167. Liu Y, Sun W, Zhang K, Zheng H, Ma Y, Lin D, et al. Identification of Genes Differentially Expressed in Human Primary Lung Squamous Cell Carcinoma. Lung Cancer (2007) 56:307–17. doi: 10.1016/j.lungcan.2007.01.016
168. Yuen HF, McCrudden CM, Huang YH, Tham JM, Zhang X, Zeng Q, et al. TAZ Expression as a Prognostic Indicator in Colorectal Cancer. PloS One (2013) 8:e54211. doi: 10.1371/journal.pone.0054211
169. Sui Z, Zhou J, Cheng Z, Lu P. Squalene Epoxidase (SQLE) Promotes the Growth and Migration of the Hepatocellular Carcinoma Cells. Tumor Biol (2015) 36:6173–9. doi: 10.1007/s13277-015-3301-x
170. Brown DN, Caffa I, Cirmena G, Piras D, Garuti A, Gallo M, et al. Squalene Epoxidase is a Bona Fide Oncogene by Amplification With Clinical Relevance in Breast Cancer. Sci Rep (2016) 6:19435. doi: 10.1038/srep19435
171. Garcia-Bermudez J, Baudrier L, Bayraktar EC, Shen Y, La K, Guarecuco R, et al. Squalene Accumulation in Cholesterol Auxotrophic Lymphomas Prevents Oxidative Cell Death. Nature (2019) 567:118–22. doi: 10.1038/s41586-019-0945-5
172. Viswanathan VS, Ryan MJ, Dhruv HD, Gill S, Eichhoff OM, Seashore-Ludlow B, et al. Dependency of a Therapy-Resistant State of Cancer Cells on a Lipid Peroxidase Pathway. Nature (2017) 547:453–7. doi: 10.1038/nature23007
173. Murai T. Cholesterol Lowering: Role in Cancer Prevention and Treatment. Biol Chem (2014) 396:1–11. doi: 10.1515/hsz-2014-0194
174. Xue T, Zhang Y, Zhang L, Yao L, Hu X, Xu LX. Proteomic Analysis of Two Metabolic Proteins With Potential to Translocate to Plasma Membrane Associated With Tumor Metastasis Development and Drug Targets. J Proteome Res (2013) 12:1754–63. doi: 10.1021/pr301100r
175. Staedler D, Chapuis-Bernasconi C, Dehmlow H, Fischer H, Juillerat-Jeanneret L, Aebi JD. Cytotoxic Effects of Combination of Oxidosqualene Cyclase Inhibitors With Atorvastatin in Human Cancer Cells. J Med Chem (2012) 55:4990–5002. doi: 10.1021/jm300256z
176. Mejia-Pous C, Damiola F, Gandrillon O. Cholesterol Synthesis-Related Enzyme Oxidosqualene Cyclase is Required to Maintain Self-Renewal in Primary Erythroid Progenitors. Cell Prolif (2011) 44:441–52. doi: 10.1111/j.1365-2184.2011.00771.x
177. Beckwitt CH, Shiraha K, Wells A. Lipophilic Statins Limit Cancer Cell Growth and Survival, Via Involvement of Akt Signaling. PloS One (2018) 13:e0197422. doi: 10.1371/journal.pone.0197422
178. Hindler K, Cleeland CS, Rivera E, Collard CD. The Role of Statins in Cancer Therapy. Oncologist (2006) 11:306–15. doi: 10.1634/theoncologist.11-3-306
179. Pisanti S, Picardi P, Ciaglia E, D’Alessandro A, Bifulco M. Novel Prospects of Statins as Therapeutic Agents in Cancer. Pharmacol Res (2014) 88:84–98. doi: 10.1016/j.phrs.2014.06.013
180. Gizzo S, Quaranta M, Nardelli GB, Noventa M. Lipophilic Statins as Anticancer Agents: Molecular Targeted Actions and Proposal in Advanced Gynaecological Malignancies. Curr Drug Targets (2015) 16:1142–59. doi: 10.2174/1389450116666150330113239
181. Fatehi Hassanabad A. Current Perspectives on Statins as Potential Anti-Cancer Therapeutics: Clinical Outcomes and Underlying Molecular Mechanisms. Transl Lung Cancer Res (2019) 8:692–9. doi: 10.21037/tlcr.2019.09.08
182. Osmak M. Statins and Cancer: Current and Future Prospects. Cancer Lett (2012) 324:1–12. doi: 10.1016/j.canlet.2012.04.011
183. Ahmadi M, Amiri S, Pecic S, Machaj F, Rosik J, Łos MJ, et al. Pleiotropic Effects of Statins: A Focus on Cancer. Biochim Biophys Acta BBA - Mol Basis Dis (2020) 1866:165968. doi: 10.1016/j.bbadis.2020.165968
184. Altwairgi AK. Statins Are Potential Anticancerous Agents (Review). Oncol Rep (2015) 33:1019–39. doi: 10.3892/or.2015.3741
185. Di Bello E, Zwergel C, Mai A, Valente S. The Innovative Potential of Statins in Cancer: New Targets for New Therapies. Front Chem (2020) 8:516. doi: 10.3389/fchem.2020.00516
186. Matusewicz L, Meissner J, Toporkiewicz M, Sikorski AF. The Effect of Statins on Cancer Cells—Review. Tumor Biol (2015) 36:4889–904. doi: 10.1007/s13277-015-3551-7
187. Terzi H, Altun A, Şencan M. In Vitro Comparison of the Cytotoxic Effects of Statins on U266 Myeloma Cell Line. Indian J Med Res (2019) 150:630–4. doi: 10.4103/ijmr.IJMR_672_18
188. Chen YH, Chen YC, Lin CC, Hsieh YP, Hsu CS, Hsieh MC. Synergistic Anticancer Effects of Gemcitabine With Pitavastatin on Pancreatic Cancer Cell Line MIA PaCa-2 In Vitro and In Vivo. Cancer Manag Res (2020) 12:4645–65. doi: 10.2147/CMAR.S247876
189. Lübtow MM, Oerter S, Quader S, Jeanclos E, Cubukova A, Krafft M, et al. In Vitro Blood–Brain Barrier Permeability and Cytotoxicity of an Atorvastatin-Loaded Nanoformulation Against Glioblastoma in 2D and 3D Models. Mol Pharm (2020) 17:1835–47. doi: 10.1021/acs.molpharmaceut.9b01117
190. Higgins MJ, Prowell TM, Blackford AL, Byrne C, Khouri NF, Slater SA, et al. A Short-Term Biomarker Modulation Study of Simvastatin in Women at Increased Risk of a New Breast Cancer. Breast Cancer Res Treat (2012) 131:915–24. doi: 10.1007/s10549-011-1858-7
191. Kim ST, Kang JH, Lee J, Park SH, Park JO, Park YS, et al. Simvastatin Plus Capecitabine–Cisplatin Versus Placebo Plus Capecitabine–Cisplatin in Patients With Previously Untreated Advanced Gastric Cancer: A Double-Blind Randomised Phase 3 Study. Eur J Cancer (2014) 50:2822–30. doi: 10.1016/j.ejca.2014.08.005
192. Vinayak S, Schwartz EJ, Jensen K, Lipson J, Alli E, McPherson L, et al. A Clinical Trial of Lovastatin for Modification of Biomarkers Associated With Breast Cancer Risk. Breast Cancer Res Treat (2013) 142:389–98. doi: 10.1007/s10549-013-2739-z
193. Garwood ER, Kumar AS, Baehner FL, Moore DH, Au A, Hylton N, et al. Fluvastatin Reduces Proliferation and Increases Apoptosis in Women With High Grade Breast Cancer. Breast Cancer Res Treat (2010) 119:137–44. doi: 10.1007/s10549-009-0507-x
194. Beckwitt CH, Brufsky A, Oltvai ZN, Wells A. Statin Drugs to Reduce Breast Cancer Recurrence and Mortality. Breast Cancer Res (2018) 20:144. doi: 10.1186/s13058-018-1066-z
195. Farooqi MAM, Malhotra N, Mukherjee SD, Sanger S, Dhesy-Thind SK, Ellis P, et al. Statin Therapy in the Treatment of Active Cancer: A Systematic Review and Meta-Analysis of Randomized Controlled Trials. PloS One (2018) 13:e0209486. doi: 10.1371/journal.pone.0209486
196. Brånvall E, Ekberg S, Eloranta S, Wästerlid T, Birmann BM, Smedby KE. Statin Use is Associated With Improved Survival in Multiple Myeloma: A Swedish Population-Based Study of 4315 Patients. Am J Hematol (2020) 95:652–61. doi: 10.1002/ajh.25778
197. Murtola TJ, Syvälä H, Tolonen T, Helminen M, Riikonen J, Koskimäki J, et al. Atorvastatin Versus Placebo for Prostate Cancer Before Radical Prostatectomy—A Randomized, Double-blind, Placebo-Controlled Clinical Trial. Eur Urol (2018) 74:697–701. doi: 10.1016/j.eururo.2018.06.037
198. Bono AV, Pagano F, Montironi R, Zattoni F, Manganelli A, Selvaggi FP, et al. Effect of Complete Androgen Blockade on Pathologic Stage and Resection Margin Status of Prostate Cancer: Progress Pathology Report of the Italian PROSIT Study. Urology (2001) 57:117–21. doi: 10.1016/S0090-4295(00)00866-9
199. Matusewicz L, Czogalla A, Sikorski AF. Attempts to Use Statins in Cancer Therapy: An Update. Tumor Biol (2020) 42:101042832094176. doi: 10.1177/1010428320941760
200. Charlton-Menys V, Durrington PN. Squalene Synthase Inhibitors: Clinical Pharmacology and Cholesterol-Lowering Potential. Drugs (2007) 67:11–6. doi: 10.2165/00003495-200767010-00002
201. Cenedella RJ, Jacob R, Borchman D, Tang D, Neely AR, Samadi A, et al. Direct Perturbation of Lens Membrane Structure may Contribute to Cataracts Caused by U18666A, an Oxidosqualene Cyclase Inhibitor. J Lipid Res (2004) 45:1232–41. doi: 10.1194/jlr.M300469-JLR200
202. Dey P, Barros RPA, Warner M, Ström A, Gustafsson J-Å. Insight Into the Mechanisms of Action of Estrogen Receptor β in the Breast, Prostate, Colon, and CNS. J Mol Endocrinol (2013) 51:T61–74. doi: 10.1530/JME-13-0150101
203. Dey P, Ström A, Gustafsson J-Å. Estrogen Receptor β Upregulates FOXO3a and Causes Induction of Apoptosis Through PUMA in Prostate Cancer. Oncogene (2014) 33:4213–25. doi: 10.1038/onc.2013.384
204. Singh V, Sharma V, Verma V, Pandey D, Yadav SK, Maikhuri JP, et al. Apigenin Manipulates the Ubiquitin–Proteasome System to Rescue Estrogen Receptor-β From Degradation and Induce Apoptosis in Prostate Cancer Cells. Eur J Nutr (2015) 54:1255–67. doi: 10.1007/s00394-014-0803-z
205. Pravettoni A, Mornati O, Martini PGV, Marino M, Colciago A, Celotti F, et al. Estrogen Receptor Beta (ERbeta) and Inhibition of Prostate Cancer Cell Proliferation: Studies on the Possible Mechanism of Action in DU145 Cells. Mol Cell Endocrinol (2007) 263:46–54. doi: 10.1016/j.mce.2006.08.008
206. Pandit J, Danley DE, Schulte GK, Mazzalupo S, Pauly TA, Hayward CM, et al. Crystal Structure of Human Squalene Synthase: A Key Enzyme in Cholesterol Biosynthesis. J Biol Chem (2000) 275:30610–7. doi: 10.1074/jbc.M004132200
207. Bergstrom JD, Dufresne C, Bills GF, Nallin-Omstead M, Byrne K. Discovery, Biosynthesis, and Mechanism of Action of the Zaragozic Acids: Potent Inhibitors of Squalene Synthase. Annu Rev Microbiol (1995) 49:607–39. doi: 10.1146/annurev.mi.49.100195.003135
208. He J, Shin H, Wei X, Kadegowda AKG, Chen R, Xie SK. NPC1L1 Knockout Protects Against Colitis-Associated Tumorigenesis in Mice. BMC Cancer (2015) 15:189. doi: 10.1186/s12885-015-1230-0
209. Huang J, Li L, Lian J, Schauer S, Vesely PW, Kratky D, et al. Tumor-Induced Hyperlipidemia Contributes to Tumor Growth. Cell Rep (2016) 15:336–48. doi: 10.1016/j.celrep.2016.03.020
210. Gonias SL, Karimi-Mostowfi N, Murray SS, Mantuano E, Gilder AS. Expression of LDL Receptor-Related Proteins (LRPs) in Common Solid Malignancies Correlates With Patient Survival. PloS One (2017) 12:e0186649. doi: 10.1371/journal.pone.0186649
211. Gallagher EJ, Zelenko Z, Neel BA, Antoniou IM, Rajan L, Kase N, et al. Elevated Tumor LDLR Expression Accelerates LDL Cholesterol-Mediated Breast Cancer Growth in Mouse Models of Hyperlipidemia. Oncogene (2017) 36:6462–71. doi: 10.1038/onc.2017.247
212. Campion O, Al Khalifa T, Langlois B, Thevenard-Devy J, Salesse S, Savary K, et al. Contribution of the Low-Density Lipoprotein Receptor Family to Breast Cancer Progression. Front Oncol (2020) 10:882. doi: 10.3389/fonc.2020.00882
213. Roslan Z, Muhamad M, Selvaratnam L, Ab-Rahim S. The Roles of Low-Density Lipoprotein Receptor-Related Proteins 5, 6, and 8 in Cancer: A Review. J Oncol (2019) 2019:4536302. doi: 10.1155/2019/4536302
214. Mooberry LK, Sabnis NA, Panchoo M, Nagarajan B, Lacko AG. Targeting the SR-B1 Receptor as a Gateway for Cancer Therapy and Imaging. Front Pharmacol (2016) 7:466. doi: 10.3389/fphar.2016.00466
215. Feng H, Wang M, Wu C, Yu J, Wang D, Ma J, et al. High Scavenger Receptor Class B Type I Expression is Related to Tumor Aggressiveness and Poor Prognosis in Lung Adenocarcinoma: A STROBE Compliant Article. Medicine (Baltimore) (2018) 97:e0203. doi: 10.1097/MD.0000000000010203
216. Schörghofer D, Kinslechner K, Preitschopf A, Schütz B, Röhrl C, Hengstschläger M, et al. The HDL Receptor SR-BI is Associated With Human Prostate Cancer Progression and Plays a Possible Role In Establishing Androgen Independence. Reprod Biol Endocrinol (2015) 13:88. doi: 10.1186/s12958-015-0087-z
217. Wang C, Li P, Xuan J, Zhu C, Liu J, Shan L, et al. Cholesterol Enhances Colorectal Cancer Progression Via ROS Elevation and MAPK Signaling Pathway Activation. Cell Physiol Biochem (2017) 42:729–42. doi: 10.1159/000477890
218. Llaverias G, Danilo C, Mercier I, Daumer K, Capozza F, Williams TM, et al. Role of Cholesterol in the Development and Progression of Breast Cancer. Am J Pathol (2011) 178:402–12. doi: 10.1016/j.ajpath.2010.11.005
219. Riscal R, Skuli N, Simon MC. Even Cancer Cells Watch Their Cholesterol! Mol Cell (2019) 76:220–31. doi: 10.1016/j.molcel.2019.09.008
220. Kapourchali FR, Surendiran G, Goulet A, Moghadasian MH. The Role of Dietary Cholesterol in Lipoprotein Metabolism and Related Metabolic Abnormalities: A Mini-Review. Crit Rev Food Sci Nutr (2016) 56:2408–15. doi: 10.1080/10408398.2013.842887
221. Püschel GP, Henkel J. Dietary Cholesterol Does Not Break Your Heart But Kills Your Liver. Porto BioMed J (2018) 3:e12. doi: 10.1016/j.pbj.0000000000000012
222. Quail DF, Dannenberg AJ. The Obese Adipose Tissue Microenvironment in Cancer Development and Progression. Nat Rev Endocrinol (2019) 15:139–54. doi: 10.1038/s41574-018-0126-x
223. Cedó L, Reddy ST, Mato E, Blanco-Vaca F, Escolà-Gil JC. HDL and LDL: Potential New Players in Breast Cancer Development. J Clin Med (2019) 8:853. doi: 10.3390/jcm8060853
224. Pelton K, Coticchia CM, Curatolo AS, Schaffner CP, Zurakowski D, Solomon KR, et al. Hypercholesterolemia Induces Angiogenesis and Accelerates Growth of Breast Tumors In Vivo. Am J Pathol (2014) 184:2099–110. doi: 10.1016/j.ajpath.2014.03.006
225. Oh SH, Choi SY, Choi HJ, Ryu HM, Kim YJ, Jung HY, et al. The Emerging Role of Xanthine Oxidase Inhibition for Suppression of Breast Cancer Cell Migration and Metastasis Associated With Hypercholesterolemia. FASEB J (2019) 33:7301–14. doi: 10.1096/fj.201802415RR
226. Heilos D, Röhrl C, Pirker C, Englinger B, Baier D, Mohr T, et al. Altered Membrane Rigidity Via Enhanced Endogenous Cholesterol Synthesis Drives Cancer Cell Resistance to Destruxins. Oncotarget (2018) 9:25661–80. doi: 10.18632/oncotarget.25432
227. Han T, Lv Y, Wang S, Hu T, Hong H, Fu Z. PPARγ Overexpression Regulates Cholesterol Metabolism in Human L02 Hepatocytes. J Pharmacol Sci (2019) 139:1–8. doi: 10.1016/j.jphs.2018.09.013
228. Tachibana K, Yamasaki D, Ishimoto K, Doi T. The Role of PPARs in Cancer. PPAR Res (2008) 2008:102737. doi: 10.1155/2008/102737
229. Chinetti G, Lestavel S, Bocher V, Remaley AT, Neve B, Torra IP, et al. PPAR-α and PPAR-γ Activators Induce Cholesterol Removal From Human Macrophage Foam Cells Through Stimulation of the ABCA1 Pathway. Nat Med (2001) 7:53–8. doi: 10.1038/83348
230. Grabacka M, Reiss K. Anticancer Properties of PPARα-Effects on Cellular Metabolism and Inflammation. PPAR Res (2008) 2008:930705. doi: 10.1155/2008/930705
231. Gou Q, Gong X, Jin J, Shi J, Hou Y. Peroxisome Proliferator-Activated Receptors (PPARs) Are Potential Drug Targets for Cancer Therapy. Oncotarget (2017) 8:60704–9. doi: 10.18632/oncotarget.19610
232. Zhao W, Prijic S, Urban BC, Tisza MJ, Zuo Y, Li L, et al. Candidate Antimetastasis Drugs Suppress the Metastatic Capacity of Breast Cancer Cells by Reducing Membrane Fluidity. Cancer Res (2016) 76:2037–49. doi: 10.1158/0008-5472.CAN-15-1970
233. Preta G. New Insights Into Targeting Membrane Lipids for Cancer Therapy. Front Cell Dev Biol (2020) 8:571237. doi: 10.3389/fcell.2020.571237
234. Zhang J, Li Q, Wu Y, Wang D, Xu L, Zhang Y, et al. Cholesterol Content in Cell Membrane Maintains Surface Levels of ErbB2 and Confers a Therapeutic Vulnerability in ErbB2-Positive Breast Cancer. Cell Commun Signal (2019) 17:15. doi: 10.1186/s12964-019-0328-4
235. Aguirre-Portolés C, Feliu J, Reglero G, Ramírez de Molina A. ABCA1 Overexpression Worsens Colorectal Cancer Prognosis by Facilitating Tumour Growth and Caveolin-1-Dependent Invasiveness, and These Effects can be Ameliorated Using the BET Inhibitor Apabetalone. Mol Oncol (2018) 12:1735–52. doi: 10.1002/1878-0261.12367
236. Koundouros N, Poulogiannis G. Reprogramming of Fatty Acid Metabolism in Cancer. Br J Cancer (2020) 122:4–22. doi: 10.1038/s41416-019-0650-z
237. Adam RM, Mukhopadhyay NK, Kim J, Di Vizio D, Cinar B, Boucher K, et al. Cholesterol Sensitivity of Endogenous and Myristoylated Akt. Cancer Res (2007) 67:6238–46. doi: 10.1158/0008-5472.CAN-07-0288
238. Chen L, Peng J, Wang Y, Jiang H, Wang W, Dai J, et al. Fenofibrate-Induced Mitochondrial Dysfunction and Metabolic Reprogramming Reversal: The Anti-Tumor Effects in Gastric Carcinoma Cells Mediated by the PPAR Pathway. Am J Transl Res (2020) 12:428–46.
239. Ertunc ME, Hotamisligil GS. Lipid Signaling and Lipotoxicity in Metaflammation: Indications for Metabolic Disease Pathogenesis and Treatment. J Lipid Res (2016) 57:2099–114. doi: 10.1194/jlr.R066514
240. Wang YJ, Bian Y, Luo J, Lu M, Xiong Y, Guo SY, et al. Cholesterol and Fatty Acids Regulate Cysteine Ubiquitylation of ACAT2 Through Competitive Oxidation. Nat Cell Biol (2017) 19:808–19. doi: 10.1038/ncb3551
241. Jarc E, Petan T. Lipid Droplets and the Management of Cellular Stress. Yale J Biol Med (2019) 92:435–52.
242. Qiu B, Ackerman D, Sanchez DJ, Li B, Ochocki JD, Grazioli A, et al. HIF2α-Dependent Lipid Storage Promotes Endoplasmic Reticulum Homeostasis in Clear-Cell Renal Cell Carcinoma. Cancer Discov (2015) 5:652–67. doi: 10.1158/2159-8290.CD-14-1507
243. Yue S, Li J, Lee SY, Lee HJ, Shao T, Song B, et al. Cholesteryl Ester Accumulation Induced by PTEN Loss and PI3K/AKT Activation Underlies Human Prostate Cancer Aggressiveness. Cell Metab (2014) 19:393–406. doi: 10.1016/j.cmet.2014.01.019
244. de Gonzalo-Calvo D, López-Vilaró L, Nasarre L, Perez-Olabarria M, Vázquez T, Escuin D, et al. Intratumor Cholesteryl Ester Accumulation is Associated With Human Breast Cancer Proliferation and Aggressive Potential: A Molecular and Clinicopathological Study. BMC Cancer (2015) 15:460. doi: 10.1186/s12885-015-1469-5
245. Mulas MF, Abete C, Pulisci D, Pani A, Massidda B, Dessì S, et al. Cholesterol Esters as Growth Regulators of Lymphocytic Leukaemia Cells. Cell Prolif (2011) 44:360–71. doi: 10.1111/j.1365-2184.2011.00758.x
246. Bemlih S, Poirier M-D, El Andaloussi A. Acyl-Coenzyme A: Cholesterol Acyltransferase Inhibitor Avasimibe Affect Survival and Proliferation of Glioma Tumor Cell Lines. Cancer Biol Ther (2010) 9:1025–32. doi: 10.4161/cbt.9.12.11875
247. Tirinato L, Liberale C, Di Franco S, Candeloro P, Benfante A, La Rocca R, et al. Lipid Droplets: A New Player in Colorectal Cancer Stem Cells Unveiled by Spectroscopic Imaging. Stem Cells (2015) 33:35–44. doi: 10.1002/stem.1837
248. Jiang Y, Sun A, Zhao Y, Ying W, Sun H, Yang X, et al. Proteomics Identifies New Therapeutic Targets of Early-Stage Hepatocellular Carcinoma. Nature (2019) 567:257–61. doi: 10.1038/s41586-019-0987-8
249. Saraon P, Cretu D, Musrap N, Karagiannis GS, Batruch I, Drabovich AP, et al. Quantitative Proteomics Reveals That Enzymes of the Ketogenic Pathway are Associated With Prostate Cancer Progression. Mol Cell Proteomics (2013) 12:1589–601. doi: 10.1074/mcp.M112.023887
250. Lacombe AMF, Soares IC, Mariani BM de P, Nishi MY, Bezerra-Neto JE, Charchar H da S, et al. Sterol O-Acyl Transferase 1 as a Prognostic Marker of Adrenocortical Carcinoma. Cancers (2020) 12:247. doi: 10.3390/cancers12010247
251. Warner GJ, Stoudt G, Bamberger M, Johnson WJ, Rothblat GH. Cell Toxicity Induced by Inhibition of Acyl Coenzyme A:Cholesterol Acyltransferase and Accumulation of Unesterified Cholesterol. J Biol Chem (1995) 270:5772–8. doi: 10.1074/jbc.270.11.5772
252. Wang J, Tan M, Ge J, Zhang P, Zhong J, Tao L, et al. Lysosomal Acid Lipase Promotes Cholesterol Ester Metabolism and Drives Clear Cell Renal Cell Carcinoma Progression. Cell Prolif (2018) 51:e12452. doi: 10.1111/cpr.12452
253. Chen Y, Hughes-Fulford M. Human Prostate Cancer Cells Lack Feedback Regulation of Low-Density Lipoprotein Receptor and its Regulator, SREBP2. Int J Cancer (2001) 91:41–5. doi: 10.1002/1097-0215(20010101)91:1<41::AID-IJC1009>3.0.CO;2-2
254. Locke JA, Guns ES, Lubik AA, Adomat HH, Hendy SC, Wood CA, et al. Androgen Levels Increase by Intratumoral De Novo Steroidogenesis During Progression of Castration-Resistant Prostate Cancer. Cancer Res (2008) 68:6407–15. doi: 10.1158/0008-5472.CAN-07-5997
255. Leon CG, Locke JA, Adomat HH, Etinger SL, Twiddy AL, Neumann RD, et al. Alterations in Cholesterol Regulation Contribute to the Production of Intratumoral Androgens During Progression to Castration-Resistant Prostate Cancer in a Mouse Xenograft Model. Prostate (2010) 70:390–400. doi: 10.1002/pros.21072
256. Martinez-Outschoorn UE, Lin Z, Whitaker-Menezes D, Howell A, Sotgia F, Lisanti MP. Ketone Body Utilization Drives Tumor Growth and Metastasis. Cell Cycle (2012) 11:3964–71. doi: 10.4161/cc.22137
257. Ozsvari B, Sotgia F, Simmons K, Trowbridge R, Foster R, Lisanti MP. Mitoketoscins: Novel Mitochondrial Inhibitors for Targeting Ketone Metabolism in Cancer Stem Cells (CSCs). Oncotarget (2017) 8:78340–50. doi: 10.18632/oncotarget.21259
258. Lo YW, Lin ST, Chang SJ, Chan CH, Lyu KW, Chang JF, et al. Mitochondrial Proteomics With siRNA Knockdown to Reveal ACAT1 and MDH2 in the Development of Doxorubicin-Resistant Uterine Cancer. J Cell Mol Med (2015) 19:744–59. doi: 10.1111/jcmm.12388
259. Dong Y, Tu R, Liu H, Qing G. Regulation of Cancer Cell Metabolism: Oncogenic MYC in the Driver’s Seat. Signal Transduct Target Ther (2020) 5:124. doi: 10.1038/s41392-020-00235-2
260. Zhong C, Fan L, Yao F, Shi J, Fang W, Zhao H. HMGCR is Necessary for the Tumorigenecity of Esophageal Squamous Cell Carcinoma and Is Regulated by Myc. Tumor Biol (2014) 35:4123–9. doi: 10.1007/s13277-013-1539-8
261. Cheng C, Geng F, Cheng X, Guo D. Lipid Metabolism Reprogramming and its Potential Targets in Cancer. Cancer Commun Lond Engl (2018) 38:27. doi: 10.1186/s40880-018-0301-4
262. Haskins JW, Zhang S, Means RE, Kelleher JK, Cline GW, Canfrán-Duque A, et al. Neuregulin-Activated ERBB4 Induces the SREBP-2 Cholesterol Biosynthetic Pathway and Increases Low-Density Lipoprotein Uptake. Sci Signal (2015) 8:ra111. doi: 10.1126/scisignal.aac5124
263. Bakiri L, Hamacher R, Graña O, Guío-Carrión A, Campos-Olivas R, Martinez L, et al. Liver Carcinogenesis by FOS-Dependent Inflammation and Cholesterol Dysregulation. J Exp Med (2017) 214:1387–409. doi: 10.1084/jem.20160935
264. Kloudova A, Guengerich PF, Soucek P. The Role of Oxysterols in Human Cancer. Trends Endocrinol Metab (2017) 28:485–96. doi: 10.1016/j.tem.2017.03.002
265. Olkkonen VM, Béaslas O, Nissilä E. Oxysterols and Their Cellular Effectors. Biomolecules (2012) 2:76–103. doi: 10.3390/biom2010076
266. He S, Nelson ER. 27-Hydroxycholesterol, an Endogenous Selective Estrogen Receptor Modulator. Maturitas (2017) 104:29–35. doi: 10.1016/j.maturitas.2017.07.014
267. Raza S, Ohm JE, Dhasarathy A, Schommer J, Roche C, Hammer KDP, et al. The Cholesterol Metabolite 27-Hydroxycholesterol Regulates p53 Activity and Increases Cell Proliferation Via MDM2 in Breast Cancer Cells. Mol Cell Biochem (2015) 410:187–95. doi: 10.1007/s11010-015-2551-7
268. Shen Z, Zhu D, Liu J, Chen J, Liu Y, Hu C, et al. 27-Hydroxycholesterol Induces Invasion and Migration of Breast Cancer Cells by Increasing MMP9 and Generating EMT Through Activation of STAT-3. Environ Toxicol Pharmacol (2017) 51:1–8. doi: 10.1016/j.etap.2017.02.001
269. Zhu D, Shen Z, Liu J, Chen J, Liu Y, Hu C, et al. The ROS-mediated Activation of STAT-3/VEGF Signaling is Involved in the 27-Hydroxycholesterol-Induced Angiogenesis in Human Breast Cancer Cells. Toxicol Lett (2016) 264:79–86. doi: 10.1016/j.toxlet.2016.11.006
270. Revilla G, Pons M de P, Baila-Rueda L, García-León A, Santos D, Cenarro A, et al. Cholesterol and 27-Hydroxycholesterol Promote Thyroid Carcinoma Aggressiveness. Sci Rep (2019) 9:10260. doi: 10.1038/s41598-019-46727-2
271. Clendening JW, Pandyra A, Boutros PC, El Ghamrasni S, Khosravi F, Trentin GA, et al. Dysregulation of the Mevalonate Pathway Promotes Transformation. Proc Natl Acad Sci (2010) 107:15051–6. doi: 10.1073/pnas.0910258107
272. Xue L, Qi H, Zhang H, Ding L, Huang Q, Zhao D, et al. Targeting SREBP-2-Regulated Mevalonate Metabolism for Cancer Therapy. Front Oncol (2020) 10:1510. doi: 10.3389/fonc.2020.01510
273. Zhong C, Fan L, Li Z, Yao F, Zhao H. SREBP2 is Upregulated in Esophageal Squamous Cell Carcinoma and Co−Operates With C−Myc to Regulate HMGCR Expression. Mol Med Rep (2019) 20:3003–10. doi: 10.3892/mmr.2019.10577
274. Lewis CA, Brault C, Peck B, Bensaad K, Griffiths B, Mitter R, et al. SREBP Maintains Lipid Biosynthesis and Viability of Cancer Cells Under Lipid- and Oxygen-Deprived Conditions and Defines a Gene Signature Associated With Poor Survival in Glioblastoma Multiforme. Oncogene (2015) 34:5128–40. doi: 10.1038/onc.2014.439
275. Porstmann T, Santos CR, Griffiths B, Cully M, Wu M, Leevers S, et al. SREBP Activity is Regulated by mTORC1 and Contributes to Akt-Dependent Cell Growth. Cell Metab (2008) 8:224–36. doi: 10.1016/j.cmet.2008.07.007
276. Ricoult SJH, Yecies JL, Ben-Sahra I, Manning BD. Oncogenic PI3K and K-Ras Stimulate De Novo Lipid Synthesis Through mTORC1 and SREBP. Oncogene (2016) 35:1250–60. doi: 10.1038/onc.2015.179
277. Dong F, Mo Z, Eid W, Courtney KC, Zha X. Akt Inhibition Promotes ABCA1-Mediated Cholesterol Efflux to ApoA-I Through Suppressing mTORC1. PloS One (2014) 9:e113789. doi: 10.1371/journal.pone.0113789
278. Moon SH, Huang CH, Houlihan SL, Regunath K, Freed-Pastor WA, Morris JP 4th, et al. P53 Represses the Mevalonate Pathway to Mediate Tumor Suppression. Cell (2019) 176:564–80.e19. doi: 10.1016/j.cell.2018.11.011
279. Peck B, Schulze A. Lipid Metabolism at the Nexus of Diet and Tumor Microenvironment. Trends Cancer (2019) 5:693–703. doi: 10.1016/j.trecan.2019.09.007
280. Ingallina E, Sorrentino G, Bertolio R, Lisek K, Zannini A, Azzolin L, et al. Mechanical Cues Control Mutant p53 Stability Through a Mevalonate–RhoA Axis. Nat Cell Biol (2018) 20:28–35. doi: 10.1038/s41556-017-0009-8
281. Zhuang L, Kim J, Adam RM, Solomon KR, Freeman MR. Cholesterol Targeting Alters Lipid Raft Composition and Cell Survival in Prostate Cancer Cells and Xenografts. J Clin Invest (2005) 115:959–68. doi: 10.1172/JCI200519935
282. Jia Y, Wang Y, Xie J. The Hedgehog Pathway: Role in Cell Differentiation, Polarity and Proliferation. Arch Toxicol (2015) 89:179–91. doi: 10.1007/s00204-014-1433-1
283. Guo D, Reinitz F, Youssef M, Hong C, Nathanson D, Akhavan D, et al. An LXR Agonist Promotes Glioblastoma Cell Death Through Inhibition of an EGFR/AKT/SREBP-1/LDLR-Dependent Pathway. Cancer Discov (2011) 1:442–56. doi: 10.1158/2159-8290.CD-11-0102
284. Gholkar AA, Cheung K, Williams KJ, Lo YC, Hamideh SA, Nnebe C, et al. Fatostatin Inhibits Cancer Cell Proliferation by Affecting Mitotic Microtubule Spindle Assembly and Cell Division. J Biol Chem (2016) 291:17001–8. doi: 10.1074/jbc.C116.737346
285. Wen D, Wang J, Van Den Driessche G, Chen Q, Zhang Y, Chen G, et al. Adipocytes as Anticancer Drug Delivery Depot. Matter (2019) 1:1203–14. doi: 10.1016/j.matt.2019.08.007
286. Shen H, Shi S, Zhang Z, Gong T, Sun X. Coating Solid Lipid Nanoparticles With Hyaluronic Acid Enhances Antitumor Activity Against Melanoma Stem-Like Cells. Theranostics (2015) 5:755–71. doi: 10.7150/thno.10804
287. Sobot D, Mura S, Rouquette M, Vukosavljevic B, Cayre F, Buchy E, et al. Circulating Lipoproteins: A Trojan Horse Guiding Squalenoylated Drugs to LDL-Accumulating Cancer Cells. Mol Ther (2017) 25:1596–605. doi: 10.1016/j.ymthe.2017.05.016
288. Mooberry LK, Nair M, Paranjape S, McConathy WJ, Lacko AG. Receptor Mediated Uptake of Paclitaxel From a Synthetic High Density Lipoprotein Nanocarrier. J Drug Target (2010) 18:53–8. doi: 10.3109/10611860903156419
Keywords: cholesterol, cancer, metabolic reprogramming, cancer therapy, pharmacological targeting, pharmacological modulation, metabolic targeting agents
Citation: Giacomini I, Gianfanti F, Desbats MA, Orso G, Berretta M, Prayer-Galetti T, Ragazzi E and Cocetta V (2021) Cholesterol Metabolic Reprogramming in Cancer and Its Pharmacological Modulation as Therapeutic Strategy. Front. Oncol. 11:682911. doi: 10.3389/fonc.2021.682911
Received: 19 March 2021; Accepted: 06 May 2021;
Published: 24 May 2021.
Edited by:
Miriam Martini, University of Turin, ItalyReviewed by:
Alessandro Carrer, Veneto Institute of Molecular Medicine (VIMM), ItalyPaola Defilippi, University of Turin, Italy
Paolo E. Porporato, University of Turin, Italy
Copyright © 2021 Giacomini, Gianfanti, Desbats, Orso, Berretta, Prayer-Galetti, Ragazzi and Cocetta. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Eugenio Ragazzi, ZXVnZW5pby5yYWdhenppQHVuaXBkLml0
†These authors have contributed equally to this work and share first authorship