 Xiao-Xin Sun
Xiao-Xin Sun Rosalie C. Sears
Rosalie C. Sears Mu-Shui Dai
Mu-Shui Dai- Department of Molecular & Medical Genetics, School of Medicine and the OHSU Knight Cancer Institute, Oregon Health & Science University, Portland, OR, United States
Deregulated MYC overexpression and activation contributes to tumor growth and progression. Given the short half-life and unstable nature of the MYC protein, it is not surprising that the oncoprotein is highly regulated via diverse posttranslational mechanisms. Among them, ubiquitination dynamically controls the levels and activity of MYC during normal cell growth and homeostasis, whereas the disturbance of the ubiquitination/deubiquitination balance enables unwanted MYC stabilization and activation. In addition, MYC is also regulated by SUMOylation which crosstalks with the ubiquitination pathway and controls MYC protein stability and activity. In this mini-review, we will summarize current updates regarding MYC ubiquitination and provide perspectives about these MYC regulators as potential therapeutic targets in cancer.
Introduction
The c-Myc oncoprotein (MYC thereafter) is a basic helix-loop-helix and leucine zipper (bHLH-LZ) transcription factor that regulates almost all aspects of cell biology by regulating gene transcription, including cell growth and proliferation, apoptosis and senescence, angiogenesis, metabolism, ribosome biogenesis, and stem cell homeostasis (1–4). MYC heterodimerizes with its partner protein MAX via its C-terminal bHLH-LZ domain and binds to the E-box element (CACGTG) at target gene promoters (5–7). The N-terminal transcription activation domain (TAD) recruits key transcription co-activators, chromatin modifiers, and mediators to promote the transcription-initiating complex formation and initiate transcription initiation (8, 9). MYC also promotes RNA polymerase II (RNAPII) pause-release upon recruiting the pTEF phosphorylation complex to phosphorylate Serine 2 of the C-terminal domain (CTD) of RNAPII (10). Early individual gene and profiling studies have identified a large number of MYC target genes (4, 11). Recent genome wide studies suggest that MYC might also be a global gene amplifier, promoting the transcription of most, if not all, actively transcribed genes (12–15). Currently, there are several models demonstrating the different modes of MYC transactivation function, including specific-gene regulation, global gene activation, and gene-specific affinity models (1, 3). New studies also suggest that MYC supports genome integrity by clearing stalled RNAPII and resolving transcription-replication conflicts (1, 16).
Since deregulated MYC overexpression contributes significantly to human cancers by regulating the expression of genes involved in almost all aspects of the cancer hallmarks (4, 17, 18), MYC levels and activity must be tightly controlled during normal homeostasis. Under normal physiological condition, MYC is an unstable protein with a half-life less than 15–30 min whereas in growth stimulated cells, MYC is transiently stabilized by shutting down proteasome degradation of MYC. This is controlled by a phosphorylation cascade involving two residues at the TAD: Thr (T) 58 and Ser (S) 62 (19–21). Upon growth stimulation, MYC is phosphorylated by RAS-induced kinases and cyclin-dependent kinases such as CDK2 at S62. Upon reduction in the growth signals, GSK3 is activated to phosphorylate T58, which requires prior S62 phosphorylation. Then, T58 phosphorylation promotes the recruitment of the proline isomerase PIN1 to catalyze the cis-to-trans isomerization of MYC at Pro (P) 63, followed by the recruitment of the phosphatase PP2A to dephosphorylate MYC at S62. T58 phosphorylated MYC is then targeted by the Fbw7 ubiquitin (Ub) E3 ligase for proteasome degradation (22–25). During the past two decades, more than a dozen ubiquitin ligases have been reported to regulate MYC stability and/or activity. In this mini-review, we briefly describe recent progress in understanding MYC control by the ubiquitin proteasome system including novel ubiquitin ligases and deubiquitinating enzymes and then focus on the perspectives of targeting these molecules for cancer therapy.
MYC Ubiquitin Ligases
To date, at least 18 Ub ligases have been discovered to mediate MYC ubiquitination, which regulates MYC protein stability and/or activity (Figure 1). While most of the Ub ligases, such as SCFFbw7, target MYC protein for degradation resulting in the inhibition of MYC activity, several other Ub ligases stabilize MYC. This can be done by antagonizing SCFFbw7-mediated MYC degradation, as in the case for SCFβ-TRCP, which ubiquitinates MYC via K33/K63/K48 mixed linkage, counteracting SCFFbw7-mediated K48-linked MYC ubiquitination and degradation (26). In another example, RNF4, a SUMO-targeted ubiquitin ligase (StubL), mediates K11- and K33-linked ubiquitination of MYC independently of SUMOylation, resulting in MYC stabilization (27). Also, HUWE1 mediates MYC ubiquitination by enhancing the recruitment of p300 and promoting MYC activity without targeting MYC for degradation (28). Yet, HUWE1 ubiquitinates and degrades Miz1, a protein that binds to MYC and accumulates at MYC-bound chromatin associated with repressed transcription (29). Thus, depletion of HUWE1 switches MYC from activating to repressive and suppresses MYC activity (29). On the other hand, SCFSkp2-mediated ubiquitination promotes MYC activity, but in this case it is coupled with targeting MYC for proteasome degradation (30, 31). We have reviewed most of the MYC Ub ligases, and the reader can refer to our previous publications (32, 33). Here, we describe newly identified MYC regulators and briefly discuss the role for the MYC ubiquitin ligases in the context of cell cycle and chromatin association.
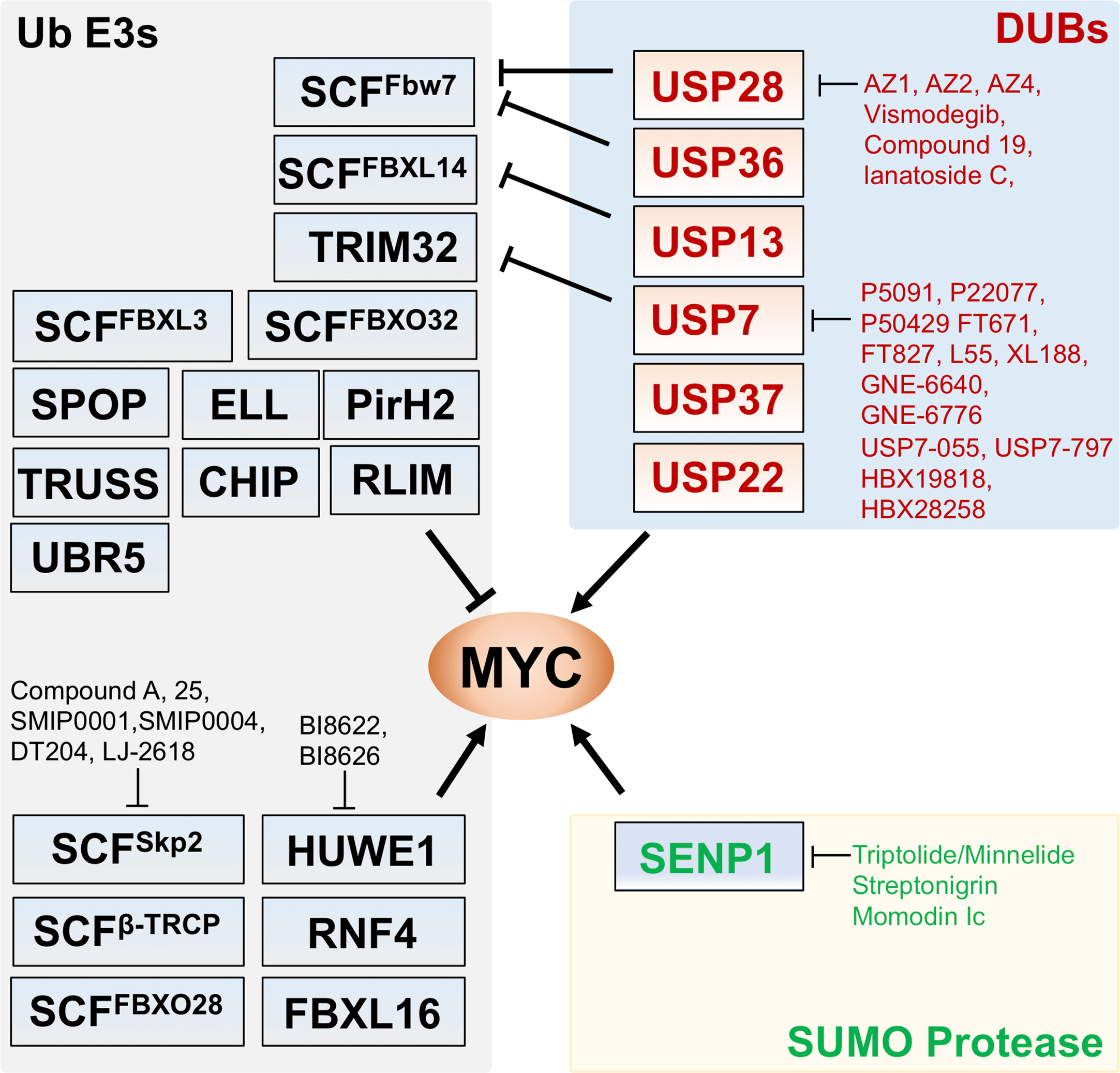
Figure 1 Regulation of MYC by Ub ligases, deubiquitinating enzymes (DUBs), and SUMO protease. Shown are the known Ub ligases (left) mediating MYC ubiquitination, DUBs (upper right) for MYC deubiquitination and SENP1 (lower right) that deSUMOylates and stabilizes MYC. The arrows indicate positive regulation of MYC activity whereas the bars indicate the inhibition. Small molecule inhibitors targeting the indicated Ub ligases, DUBs, and SENP1 are indicated.
Recently, the Westermarck group (34) showed that UBR5 ubiquitinates MYC and prevents cells from accumulating excess MYC protein. Interestingly, UBR5 suppresses MYC-dependent priming to therapy-induced apoptosis in cancer cells as it resets MYC to levels that are not enough to induce apoptosis, whereas in normal cells accumulated MYC triggers apoptosis. Indeed, MYC and UBR5 are often co-amplified in MYC-driven human cancers. Yet, UBR5 high expression (UBR5-high) cells dominate MYC-high cells at the single cell level in basal type breast cancers. This study suggests that UBR5-mediated MYC ubiquitination and degradation prevents the accumulation of too much MYC and thus benefits cancer cell survival (34). Therefore, UBR5 may promote tumor cell resistance to cancer therapy. It would be interesting to consider how UBR5 and other regulators of MYC that control MYC ubiquitination levels can tune MYC oncogenic potential.
The leucine-rich repeats (LRR) containing F-box protein FBXL16 has recently been shown to regulate MYC stability by antagonizing SCFFbw7. Interestingly, FBXL16 binds to Skp1 and PP2A but not Cul1, indicating that it may not form a SCF E3 ligase complex (35). FBXL16 binds to wild-type MYC and the T58A or S62A mutant with equal efficiency and does not compete with FBW7 for binding to MYC, but inhibits FBW7-mediated MYC ubiquitination in cells and in vitro. As both F-box and the LRR domains are required for FBXL16’s activity to counteract FBW7, it is possible that FBXL16 may form a complex with FBW7 and MYC where it directly suppresses FBW7 ligase activity. On another note, as FBXL16 also binds to PP2A and negatively affects its phosphatase activity (36), it would be interesting to test whether FBXL16 stabilizes MYC partially via inhibiting PP2A, thus suppressing MYC S62 dephosphorylation. Nevertheless, this less studied F-box protein functions to promote cancer cell growth and potentiates MYC oncogenic activity.
MYC Ubiquitination Dynamics
The identification of increasing numbers of Ub ligases targeting MYC suggests that these Ub ligases may control MYC levels and activity dynamically and coordinately and act in a cell context-dependent fashion. For example, FBXO32 is uniquely reported to function under starvation conditions and cell cycle exit, where it catalyzes K48-linked polyubiquitination of MYC at K326 leading to MYC degradation (37, 38). Several other MYC Ub ligases have been reported to function at specific cell cycle phases. TRUSS targets MYC for proteasomal degradation in G1 phase (39, 40). SKP2 expression is high at the G1/S transition and peaks at S phase, and SKP2 promotes S phase entry of MYC containing rat-1 cells, but not MYC null cells (30). In addition, overexpression of SKP2 correlates with the reduction of TRUSS in human cancers (40), suggesting an interplay of MYC Ub ligases in tightly controlling MYC stability during the cell cycle. Also, acting later in the cell cycle, FBXO28 is subjected to regulation by S- and G2/M-phase kinases, cyclin A-CDK2, and cyclin B-CDK1 that mediate phosphorylation of FBXO28 at S344 (41). This activates the Ub ligase activity of FBXO28 to ubiquitinate MYC at C-terminal lysines and recruit p300 co-activator for transactivation of a subset of target genes important for S- and G2/M-phases.
Genome-wide studies revealed wide-spread chromatin binding of MYC, including at high-affinity sites (E-box and CG island regions in the promoter and enhancer regions) bound by endogenous low levels of MYC and at low-affinity sites that are bound by overexpressed and deregulated levels of MYC through a mechanism called “invasion” (13–15, 42). In some experimental conditions, MYC is considered as a transcription amplifier to upregulate most, if not all, active transcribed genes. However, the DNA binding of MYC often does not match its transcription activity (3, 43). Also, MYC binding in certain binding sites with high-affinity promoters, such as ribosome biogenesis genes, can be saturated, and not increase with increasing MYC levels (44). These observations suggest additional control mechanisms between MYC DNA binding and transcriptional regulation.
Emerging evidences suggest that proper chromatin turnover of MYC is critical for its transactivation activity. This is initially observed by two studies showing that SKP2-mediated MYC ubiquitination and proteasome degradation increase MYC activity (30, 31). SKP2 binds to the MYC Box (MB) II which is also the binding motif for the TRAPP co-activator and is essential for MYC transactivation activity. The proteasome can be recruited by MYC and SKP2 to MYC target gene promoters such as the cyclin D2 promoter (31, 45). FBXO28 also binds to MB II and increases MYC activity by non-proteolytic ubiquitination of MYC (41). The role for MYC turnover in target gene transcription activation is further highlighted by a recent study (46) showing that MYC associates with the elongation factor complex PAF1 at promoters and ubiquitination and degradation releases this association. Upon MYC ubiquitination and degradation, the PAF1 complex can then be transferred to and bound to RNA Pol II to promote transcription elongation. Interestingly, PAF1C, a component of the PAF1 complex, interacts with the MBI and phosphorylation at T58 and S62 in MBI promotes PAF1C binding to MYC. Therefore, it would be interesting to examine whether and how MBI phosphorylation coordinates the recruitment of the PAF1 complex with Fbw7-mediated ubiquitination and degradation of MYC, and the role of other E3 ligases such as SKP2 that activate MYC transactivation in this dynamic process controlling transcription. The study also agrees with a recent work by Chen et al. (47) showing that depletion of PAF1 results in an increased release of paused Pol II and transcription elongation of many genes by recruiting the super elongation complex (SEC). Adding to the complexity is that ELL, a component of SEC that promotes transcription elongation, has recently been identified as a MYC Ub ligase. ELL ubiquitinates MYC and targets it for degradation and suppresses MYC-driven tumorigenesis (48). Whether this occurs at the chromatin and how ELL suppresses MYC activity remains to be addressed. Together, these observations are consistent with our studies indicating that MYC DNA binding and turnover are cyclic at promoters together with the cyclic binding of general transcription factors, mediators, and chromatin modifying enzymes as in the case of ERα turnover at promoters (49, 50).
MYC Deubiquitinating Enzymes (DUBs)
MYC ubiquitination can be removed by deubiquitination. Several deubiquitinating enzymes have been shown to deubiquitinate MYC and regulate its levels and activity. For example, USP28 deubiquitinates MYC via counteracting Fbw7α (51, 52), whereas USP36 deubiquitinates MYC in the nucleolus by counteracting Fbw7γ (33, 53). The role of USP36 in regulating MYC ubiquitination and stabilization was also reported in Drosophila, showing that the nucleolar isoform of Drosophila USP36 (dUSP36) deubiquitinates dMYC and regulates dMYC-dependent cell growth (54). USP22 and USP37 have been shown to deubiquitinate and stabilize MYC (55, 56). USP7 has been shown to deubiquitinate N-Myc (57) and antagonize TRIM32-mediated MYC ubiquitination (58). USP13 can antagonize FBXL14 by removing FBXL14-mediated MYC ubiquitination (59). The Otub6B isoform can regulate MYC levels but not its transcription, although it is unclear whether the isoform can directly mediate MYC deubiquitination (60). While USP36 regulates MYC deubiquitination in the nucleolus, many of the DUBs act on MYC in the nucleoplasm. It is interesting to know whether these DUBs counteract MYC ubiquitin ligases on chromatin to control MYC turnover during transcription and whether they crosstalk with other chromatin modifications, given that USP36 also deubiquitinates H2B (61).
Crosstalk of MYC SUMOylation With Ubiquitination
Recent studies have shown that MYC is also subject to SUMOylation (62–65). A unique feature about MYC SUMOylation is that it acts promiscuously with respect to the accepting lysines as mutating up to 10 lysines identified by mass spectrometry analysis still failed to abolish MYC SUMOylation (62). This might explain the early observations documenting MYC SUMOylation without a clear effect on MYC stability and activity (63, 65). We recently showed that the SUMO-specific protease SENP1 deSUMOylates MYC (66). Interestingly, SENP1-mediated deSUMOylation stabilizes and activates MYC, likely due to the indirect deubiquitination of MYC as MYC can be co-modified by both Ub and SUMO (66). This is also strongly supported by multiple recent proteomic studies showing ubiquitination of SUMO as well as SUMO-conjugation to multiple lysines of ubiquitin (67, 68). Similar regulation has been observed for HIF1α SUMOylation in that SENP1 deSUMOylation of HIF1α results in HIF1α stabilization as well (69). It is likely that SUMOylation actually regulates the stability of a large number of proteins via crosstalk with the Ub system. Nevertheless, the SUMO regulation of MYC adds another layer of complexity to the regulation of MYC protein stability and activity and provides another target in MYC-driven cancer cell growth. Given that SUMOylation plays a key role in transcription regulation (70, 71), it is interesting to understand how MYC SUMOylation/deSUMOylation plays a role in MYC turnover at the chromatin.
Targeting MYC Ubiquitination Pathway
MYC is commonly considered “undruggable” and direct targeting of MYC is very challenging because of its nucleus localization and the absence of active sites amendable to conventional small molecule ligand-binding (72–74), although this concept has evolved (75, 76). Recently, Omomyc, a peptide that competitively binds to E-box elements as a heterodimer with MAX or as a homodimer and suppresses the binding of MYC to E-boxes (77–79), has been shown to have therapeutic potential in vivo in various cancer models (77, 79, 80). Targeting MYC regulatory pathways, such as MYC ubiquitination and deubiquitination whose deregulation contributes to MYC stabilization in cancer, is another highly desirable approach. Several MYC Ub ligases, such as SKP2, HUWE1, and β-TRCP, promote MYC function and thus are promising therapeutic targets (Figure 1, Table 1). In particular, therapeutically targeting SKP2, which is overexpressed in a variety of cancers, has exciting potential to capitalize on MYC’s pro-apoptotic function. N-terminal ubiquitination of MYC by SKP2 increases MYC’s transactivation of pro-proliferative target genes while loss of SKP2 can not only lead to increased MYC levels but also increased apoptotic activity (30, 31, 107, 108). Inhibitors of SKP2 with anti-tumor properties include several natural products and recently identified small molecule compounds (83, 109). Small molecule compound A was found to inhibit SKP2 incorporation into the SCF complex, thus suppressing its ubiquitin ligase activity (81). SMIP0001 and SMIP0004 (82) have been shown to reduce SKP2 levels and thus induce p27 accumulation. Compound 25 and its analogs (83) disrupt the SKP2-SKP1 interaction in the SCF complex and inhibit the ligase activity and have been shown to suppress cancer cell growth. Chemical library screens identified a novel compound, designated as DT204, that reduces SKP2 binding to Cullin-1 and Commd1, and synergistically enhances BTZ-induced apoptosis (84). NSC689857 and NSC681152 disrupt the SKP2-Cks1 interaction (85), thus inhibiting p27 ubiquitination. It is interesting to examine whether these compounds that specifically inhibit p27 degradation also suppress MYC activity. A selenonucleoside called LJ-2618 downregulates the expression of SKP2 by promoting its degradation and induces G2/M cell cycle arrest in prostate cancer cells and xenograft tumor in vivo (86). Likewise, all-trans retinoic acid (ATRA) stimulates the ubiquitin-mediated degradation of SKP2 (87). This finding is intriguing because of the known effects of ATRA in stimulating cell differentiation, a consequence of MYC inhibition in some settings (110).
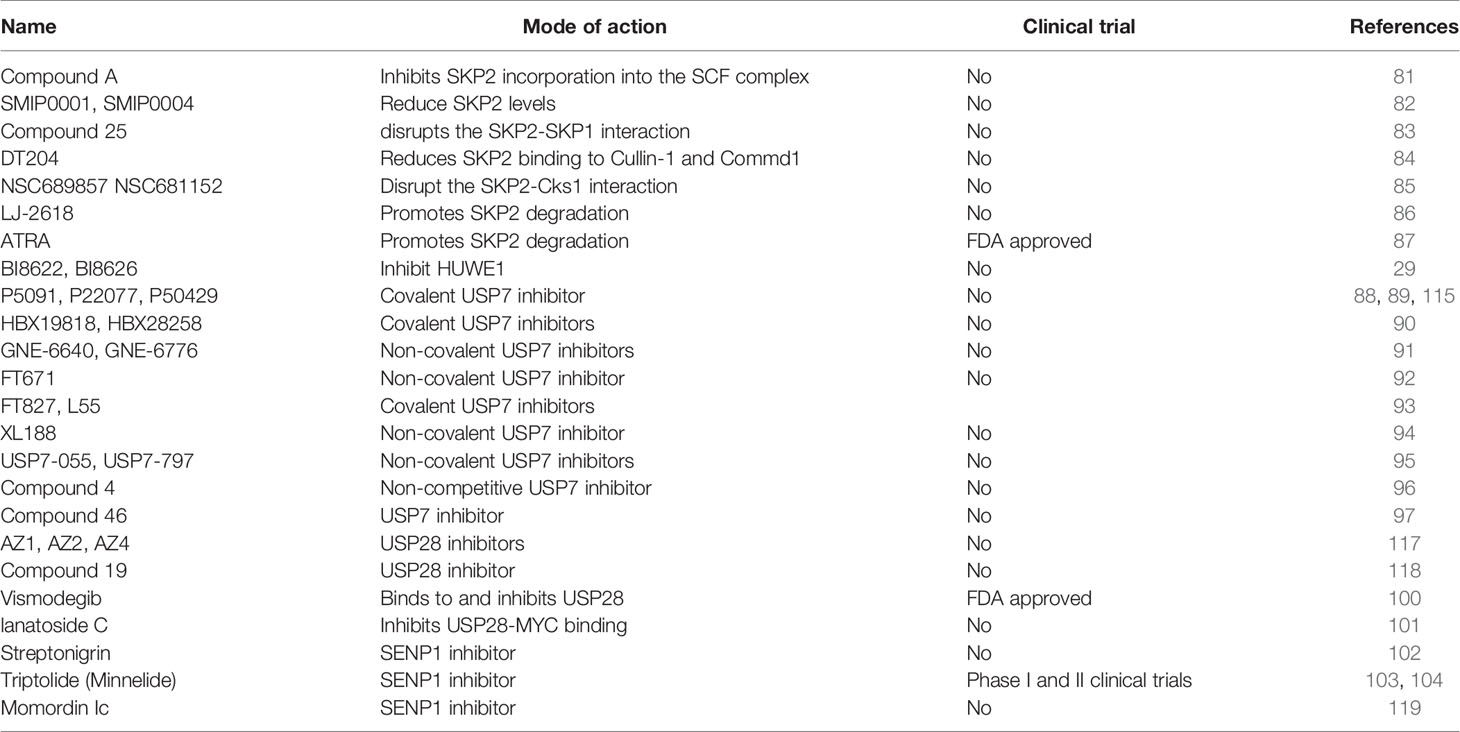
Table 1 Inhibitors targeting the MYC degradation pathway.
Targeting positive ubiquitination mediated by HUWE1 could also suppress MYC’s oncogenic activity. A recent high-throughput screening (HTS) has identified small molecule inhibitors of HUWE1, BI8622 and BI8626, that suppress transactivation of MYC target genes while increasing transrepression and the induction of apoptosis in colorectal cancer cells (29). Treatment of these compounds inhibits MYC-dependent transactivation in colorectal cancer cells but not in stem cells or normal colon epithelial cells, and this effect is associated with the role of HUWE1 inhibition in stabilizing Miz1 and Miz1-mediated suppression of MYC target genes (29). Also, HUWE1 is frequently deregulated in multiple myeloma (MM) and targeting HUWE1 with the small molecule inhibitors in combination with lenalidomide results in synergistic growth inhibition in MM cells in vitro and in vivo (111).
Many of the MYC DUBs positively regulate MYC stability and activity and could emerge as important cancer therapeutic targets as well. Studies have clearly indicated that different thresholds of MYC elicit different activities and that specific levels of MYC are required to maintain tumorigenesis (112, 113), supporting the idea of dropping MYC levels below these thresholds by strategies like inhibiting DUBs, such as USP28 or USP36 that antagonize FBW7-mediated MYC ubiquitination (52, 53) and USP7 that deubiquitinates N-MYC (57). Indeed, emerging studies have discovered a number of small molecule inhibitors for USP7 (114, 115) (Table 1). These USP7 inhibitors include trisubstituted thiophene P5091 (88) and its analogs P22077 (89) and P50429 (90), the acridine derivatives HBX19818 and HBX28258 (91), 2-Amino-4-ethylpyridine derivatives GNE-6640 and GNE-6776 (92), pyrazolo[3,4-d]pyrimidin-4-one-piperidine compounds FT671 and FT827 (93) and the derivative compound L55 (94), a Quinazolin-4-one derivative XL 188 (95), and recently reported novel chemical series including compounds USP7-055 and USP7-797 (96), compound 4 (97), and compound 46 (98). These USP7 inhibitors showed anti-proliferative effect in cancer cell lines and mouse xenograft models. For example, P5091 treatment induced multiple myeloma (MM) cell death, overcomes the resistance to Bortezomib, and inhibits MM xenograft tumor growth (88). Both P22077 and P50429 showed anti-proliferative effect in HCT116 cancer cells and mouse xenograft models (88, 116, 117). Treatment with GNE-6640 and GNE-6776 induced cancer cell death and increases cytotoxicity with chemotherapeutic agents (92). Most of these above studies focused on the role of USP7 in degrading MDM2 to induce p53 and p53-dependent anti-proliferative effects. Yet, effects on p53 mutant cancers are also evident (96). It would be beneficial to understand whether such a role also involves USP7 activity to deubiquitinate N-MYC especially in neuroblastoma with N-MYC amplification or antagonize TRIM32-mediated MYC ubiquitination (58) and whether USP7 deubiquitinates c-MYC as well.
Recently, a high throughput screening (HTS) identified the first set of benzylaminoethanol compounds AZ1, AZ2, and AZ4 that specifically inhibit USP28 (99). By inhibiting USP28, these compounds indeed reduce cellular MYC levels, induce apoptosis, and inhibit cell proliferation in a dose-dependent manner (99). AZ1 was also recently shown to markedly inhibit tumor cell growth and reduce tumor burden in an orthotopically transplanted lung tumor model in mice with well tolerance at doses up to 375 mg/kg (118). Liu et al. (100) reported a new compound 19, a [1,2,3]triazolo[4,5-d] pyrimidine derivative, that specifically inhibits USP28 and reduces gastric cancer cell proliferation and EMT with a better IC50 than that of AZ1. These effects are likely due to binding to USP28 and inducing USP28 degradation through ubiquitin-proteasome system (100). Vismodegib, a sonic hedgehog inhibitor used for the treatment of basal cell carcinoma, was recently shown to bind to USP28 and inhibit its DUB activity (101). Vismodegib exhibits selectivity towards USP28 and its evolutionally related USP25. Treatment of cancer cells with Vismodegib reduces the levels of c-Myc and Notch1 and suppresses cell growth (101). In addition, lanatoside C, a cardiac glycoside, has been shown to reduce MYC levels and suppress gastric cancer cell proliferation by inhibiting USP28 binding to MYC, thereby destabilizing MYC, although it remains to be determined whether lanatoside C directly targets USP28 (102).
Given that SENP1 positively regulates MYC levels and activity, SENP1 is an interesting cancer therapeutic target. Several SENP1 inhibitors have been reported (103–105). Streptonigrin (SN), a natural product isolated from Streptomyces flocculus, has been shown to inhibit SENP1 activity (IC50 = 0.518 µM towards SENP1, IC50 = 6.919 µM towards SENP2) (103). Triptolide, a small natural compound extracted from a Chinese herb Tripterygium wilfordii, has been shown to inhibit SENP1 expression (with IC50 = 0.0203 µM in PC-3 cells) (105) and destabilize MYC (104), yet the underlying mechanism is unknown. A water-soluble prodrug of Triptolide called Minnelide has been shown to exhibit promising anti-tumor effects in pancreatic and liver cancers (119). A phase II trial of Minnelide in patients with refractory pancreatic cancer (NCT03117920) was just completed with results pending. Also, a phase I clinical trial of Minnelide (NCT03129139) is ongoing in patients with advanced solid tumors. Also, Momordin Ic (Mc), a natural pentacyclic triterpenic compound, was shown to inhibit SENP1 activity with an IC50 of 15.37 µM in vitro (106). It is conceivable that by inhibiting MYC deSUMOylation, these SENP1 inhibitors could indirectly suppress MYC deubiquitination, thereby destabilizing MYC and exhibiting anti-proliferative effect in cancer cells.
Conclusion and Perspectives
Emerging evidence supports the dynamic MYC turnover by proteasome-mediated degradation and the complex crosstalk among different posttranslational modifications (PTMs) of MYC (ubiquitination, phosphorylation, acetylation, SUMOylation, and their reverse processes), resulting in the tight control of MYC transactivation activity, thus emphasizing the importance of MYC in the multi-step regulation of gene transcription and its deregulation in cancer. While several MYC Ub ligases such as the above mentioned SKP2 and HUWE1 have been actively explored as therapeutic targets, small molecule inhibitors suppressing other MYC ligases remain to be identified. This therapeutic strategy holds promise as, for example, knockdown of FBW7 is synthetically lethal to MYC-overexpressing cancer cells (120). Conversely, targeting UBR5 to accumulate MYC beyond the threshold levels that trigger cancer cell apoptosis in MYC-high cancers (34) is another example of inducing MYC synthetic lethality. Together, targeting these MYC posttranslational modifiers could yield potential cancer therapeutics, and additional research understanding the dynamic control processes and the effects of perturbing MYC levels in cancer will be important for their application. Future work would also be needed to further understand the crosstalk between MYC PTMs and the combined intervention of targeting multiple MYC PTMs. Further structure-biology studies and medicinal chemistry optimization could aid in improving target specificity and developing novel compounds. It is hopeful that certain compounds targeting the MYC ubiquitination-proteasome degradation pathways could ultimately move to clinic trials for treating MYC-dependent cancers. If successful, combinational therapies with other targeted therapies could also be desirable to treat advanced cancers, given that MYC crosstalk with many other oncogenic pathways such as RAS, mTOR, HIF signaling, and others.
Author Contributions
X-XS, YL, RS, M-SD wrote and edited the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by NIH grants R01 CA186241 to M-SD and RS, R01 GM130604 to M-SD, U54 CA209988 to RS, U01 CA224012 to RS, and R01 CA196228 to RS.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Baluapuri A, Wolf E, Eilers M. Target Gene-Independent Functions of MYC Oncoproteins. Nat Rev Mol Cell Biol (2020) 21:255–67. doi: 10.1038/s41580-020-0215-2
2. Bretones G, Delgado MD, Leon J. Myc and Cell Cycle Control. Biochim Biophys Acta (2015) 1849:506–16. doi: 10.1016/j.bbagrm.2014.03.013
3. Kress TR, Sabo A, Amati B. MYC: Connecting Selective Transcriptional Control to Global RNA Production. Nat Rev Cancer (2015) 15:593–607. doi: 10.1038/nrc3984
4. Meyer N, Penn LZ. Reflecting on 25 Years With MYC. Nat Rev Cancer (2008) 8:976–90. doi: 10.1038/nrc2231
5. Bieda M, Xu X, Singer MA, Green R, Farnham PJ. Unbiased Location Analysis of E2F1-Binding Sites Suggests a Widespread Role for E2F1 in the Human Genome. Genome Res (2006) 16:595–605. doi: 10.1101/gr.4887606
6. Fernandez PC, Frank SR, Wang L, Schroeder M, Liu S, Greene J, et al. Genomic Targets of the Human c-Myc Protein. Genes Dev (2003) 17:1115–29. doi: 10.1101/gad.1067003
7. Mao DY, Watson JD, Yan PS, Barsyte-Lovejoy D, Khosravi F, Wong WW, et al. Analysis of Myc Bound Loci Identified by CpG Island Arrays Shows That Max Is Essential for Myc-Dependent Repression. Curr Biol (2003) 13:882–6. doi: 10.1016/S0960-9822(03)00297-5
8. Adhikary S, Eilers M. Transcriptional Regulation and Transformation by Myc Proteins. Nat Rev Mol Cell Biol (2005) 6:635–45. doi: 10.1038/nrm1703
9. Tu WB, Helander S, Pilstal R, Hickman KA, Lourenco C, Jurisica I, et al. Myc and its Interactors Take Shape. Biochim Biophys Acta (2015) 1849:469–83. doi: 10.1016/j.bbagrm.2014.06.002
10. Rahl PB, Lin CY, Seila AC, Flynn RA, McCuine S, Burge CB, et al. c-Myc Regulates Transcriptional Pause Release. Cell (2010) 141:432–45. doi: 10.1016/j.cell.2010.03.030
11. Patel JH, Loboda AP, Showe MK, Showe LC, McMahon SB. Analysis of Genomic Targets Reveals Complex Functions of MYC. Nat Rev Cancer (2004) 4:562–8. doi: 10.1038/nrc1393
12. Lin CJ, Nasr Z, Premsrirut PK, Porco JA Jr, Hippo Y, Lowe SW, et al. Targeting Synthetic Lethal Interactions Between Myc and the eIF4F Complex Impedes Tumorigenesis. Cell Rep (2012) 1:325–33. doi: 10.1016/j.celrep.2012.02.010
13. Nie Z, Hu G, Wei G, Cui K, Yamane A, Resch W, et al. c-Myc is a Universal Amplifier of Expressed Genes in Lymphocytes and Embryonic Stem Cells. Cell (2012) 151:68–79. doi: 10.1016/j.cell.2012.08.033
14. Sabo A, Kress TR, Pelizzola M, de Pretis S, Gorski MM, Tesi A, et al. Selective Transcriptional Regulation by Myc in Cellular Growth Control and Lymphomagenesis. Nature (2014) 511:488–92. doi: 10.1038/nature13537
15. Walz S, Lorenzin F, Morton J, Wiese KE, von Eyss B, Herold S, et al. Activation and Repression by Oncogenic MYC Shape Tumour-Specific Gene Expression Profiles. Nature (2014) 511:483–7. doi: 10.1038/nature13473
16. Endres T, Solvie D, Heidelberger JB, Andrioletti V, Baluapuri A, Ade CP, et al. Ubiquitylation of MYC Couples Transcription Elongation With Double-Strand Break Repair at Active Promoters. Mol Cell (2021) 81:830–44.e813. doi: 10.1016/j.molcel.2020.12.035
18. Gabay M, Li Y, Felsher DW. MYC Activation Is a Hallmark of Cancer Initiation and Maintenance. Cold Spring Harbor Perspect Med (2014) 4:a014241. doi: 10.1101/cshperspect.a014241
19. Lutterbach B, Hann SR. Hierarchical Phosphorylation at N-terminal Transformation-Sensitive Sites in c-Myc Protein Is Regulated by Mitogens and in Mitosis. Mol Cell Biol (1994) 14:5510–22. doi: 10.1128/MCB.14.8.5510
20. Sears R, Nuckolls F, Haura E, Taya Y, Tamai K, Nevins JR. Multiple Ras-Dependent Phosphorylation Pathways Regulate Myc Protein Stability. Genes Dev (2000) 14:2501–14. doi: 10.1101/gad.836800
21. Hann SR. Role of Post-Translational Modifications in Regulating c-Myc Proteolysis, Transcriptional Activity and Biological Function. Semin Cancer Biol (2006) 16:288–302. doi: 10.1016/j.semcancer.2006.08.004
22. Moberg KH, Mukherjee A, Veraksa A, Artavanis-Tsakonas S, Hariharan IK. The Drosophila F Box Protein Archipelago Regulates dMyc Protein Levels In Vivo. Curr Biol (2004) 14:965–74. doi: 10.1016/j.cub.2004.04.040
23. Yada M, Hatakeyama S, Kamura T, Nishiyama M, Tsunematsu R, Imaki H, et al. Phosphorylation-Dependent Degradation of c-Myc Is Mediated by the F-Box Protein Fbw7. EMBO J (2004) 23:2116–25. doi: 10.1038/sj.emboj.7600217
24. Welcker M, Orian A, Jin J, Grim JE, Harper JW, Eisenman RN, et al. The Fbw7 Tumor Suppressor Regulates Glycogen Synthase Kinase 3 Phosphorylation-Dependent c-Myc Protein Degradation. Proc Natl Acad Sci USA (2004) 101:9085–90. doi: 10.1073/pnas.0402770101
25. Welcker M, Orian A, Grim JE, Eisenman RN, Clurman BE. A Nucleolar Isoform of the Fbw7 Ubiquitin Ligase Regulates c-Myc and Cell Size. Curr Biol (2004) 14:1852–7. doi: 10.1016/j.cub.2004.09.083
26. Popov N, Schulein C, Jaenicke LA, Eilers M. Ubiquitylation of the Amino Terminus of Myc by SCF(beta-TrCP) Antagonizes SCF(Fbw7)-mediated Turnover. Nat Cell Biol (2010) 12:973–81. doi: 10.1038/ncb2104
27. Thomas JJ, Abed M, Heuberger J, Novak R, Zohar Y, Beltran Lopez AP, et al. Rnf4-Dependent Oncogene Activation by Protein Stabilization. Cell Rep (2016) 16:3388–400. doi: 10.1016/j.celrep.2016.08.024
28. Adhikary S, Marinoni F, Hock A, Hulleman E, Popov N, Beier R, et al. The Ubiquitin Ligase HectH9 Regulates Transcriptional Activation by Myc and Is Essential for Tumor Cell Proliferation. Cell (2005) 123:409–21. doi: 10.1016/j.cell.2005.08.016
29. Peter S, Bultinck J, Myant K, Jaenicke LA, Walz S, Muller J, et al. Tumor Cell-Specific Inhibition of MYC Function Using Small Molecule Inhibitors of the HUWE1 Ubiquitin Ligase. EMBO Mol Med (2014) 6:1525–41. doi: 10.15252/emmm.201403927
30. Kim SY, Herbst A, Tworkowski KA, Salghetti SE, Tansey WP. Skp2 Regulates Myc Protein Stability and Activity. Mol Cell (2003) 11:1177–88. doi: 10.1016/S1097-2765(03)00173-4
31. von der Lehr N, Johansson S, Wu S, Bahram F, Castell A, Cetinkaya C, et al. The F-Box Protein Skp2 Participates in c-Myc Proteosomal Degradation and Acts as a Cofactor for c-Myc-Regulated Transcription. Mol Cell (2003) 11:1189–200. doi: 10.1016/S1097-2765(03)00193-X
32. Chen Y, Sun XX, Sears RC, Dai MS. Writing and Erasing MYC Ubiquitination and Sumoylation. Genes Dis (2019) 6:359–71. doi: 10.1016/j.gendis.2019.05.006
33. Sun XX, Sears RC, Dai MS. Deubiquitinating C-Myc: USP36 Steps Up in the Nucleolus. Cell Cycle (2015) 14:3786–93. doi: 10.1080/15384101.2015.1093713
34. Qiao X, Liu Y, Prada ML, Mohan AK, Gupta A, Jaiswal A, et al. Ubr5 Is Coamplified With MYC in Breast Tumors and Encodes an Ubiquitin Ligase That Limits MYC-Dependent Apoptosis. Cancer Res (2020) 80:1414–27. doi: 10.1158/0008-5472.CAN-19-1647
35. Morel M, Shah KN, Long W. The F-box Protein FBXL16 Up-Regulates the Stability of C-MYC Oncoprotein by Antagonizing the Activity of the F-Box Protein FBW7. J Biol Chem (2020) 295:7970–80. doi: 10.1074/jbc.RA120.012658
36. Honarpour N, Rose CM, Brumbaugh J, Anderson J, Graham RL, Sweredoski MJ, et al. F-Box Protein FBXL16 Binds PP2A-B55alpha and Regulates Differentiation of Embryonic Stem Cells Along the FLK1+ Lineage. Mol Cell Proteomics (2014) 13:780–91. doi: 10.1074/mcp.M113.031765
37. Mei Z, Zhang D, Hu B, Wang J, Shen X, Xiao W. Fbxo32 Targets c-Myc for Proteasomal Degradation and Inhibits c-Myc Activity. J Biol Chem (2015) 290:16202–14. doi: 10.1074/jbc.M115.645978
38. Chou JL, Su HY, Chen LY, Liao YP, Hartman-Frey C, Lai YH, et al. Promoter Hypermethylation of FBXO32, a Novel TGF-beta/SMAD4 Target Gene and Tumor Suppressor, Is Associated With Poor Prognosis in Human Ovarian Cancer. Lab Invest (2010) 90:414–25. doi: 10.1038/labinvest.2009.138
39. Choi SH, Wright JB, Gerber SA, Cole MD. Myc Protein is Stabilized by Suppression of a Novel E3 Ligase Complex in Cancer Cells. Genes Dev (2010) 24:1236–41. doi: 10.1101/gad.1920310
40. Jamal A, Swarnalatha M, Sultana S, Joshi P, Panda SK, Kumar V. The G1 Phase E3 Ubiquitin Ligase TRUSS That Gets Deregulated in Human Cancers Is a Novel Substrate of the S-Phase E3 Ubiquitin Ligase Skp2. Cell Cycle (2015) 14:2688–700. doi: 10.1080/15384101.2015.1056946
41. Cepeda D, Ng HF, Sharifi HR, Mahmoudi S, Cerrato VS, Fredlund E, et al. CDK-Mediated Activation of the SCF(FBXO) (28) Ubiquitin Ligase Promotes MYC-Driven Transcription and Tumourigenesis and Predicts Poor Survival in Breast Cancer. EMBO Mol Med (2013) 5:999–1018. doi: 10.1002/emmm.201202341
42. Lin CY, Loven J, Rahl PB, Paranal RM, Burge CB, Bradner JE, et al. Transcriptional Amplification in Tumor Cells With Elevated C-Myc. Cell (2012) 151:56–67. doi: 10.1016/j.cell.2012.08.026
43. Perna D, Faga G, Verrecchia A, Gorski MM, Barozzi I, Narang V, et al. Genome-Wide Mapping of Myc Binding and Gene Regulation in Serum-Stimulated Fibroblasts. Oncogene (2012) 31:1695–709. doi: 10.1038/onc.2011.359
44. Lorenzin F, Benary U, Baluapuri A, Walz S, Jung LA, von Eyss B, et al. Different Promoter Affinities Account for Specificity in MYC-dependent Gene Regulation. Elife (2016) 5:e15161. doi: 10.7554/eLife.15161
45. von der Lehr N, Johansson S, Larsson LG. Implication of the Ubiquitin/Proteasome System in Myc-Regulated Transcription. Cell Cycle (2003) 2:403–7. doi: 10.4161/cc.2.5.484
46. Jaenicke LA, von Eyss B, Carstensen A, Wolf E, Xu W, Greifenberg AK, et al. Ubiquitin-Dependent Turnover of MYC Antagonizes MYC/PAF1C Complex Accumulation to Drive Transcriptional Elongation. Mol Cell (2016) 61:54–67. doi: 10.1016/j.molcel.2015.11.007
47. Chen FX, Woodfin AR, Gardini A, Rickels RA, Marshall SA, Smith ER, et al. PAF1, a Molecular Regulator of Promoter-Proximal Pausing by RNA Polymerase Ii. Cell (2015) 162:1003–15. doi: 10.1016/j.cell.2015.07.042
48. Chen Y, Zhou C, Ji W, Mei Z, Hu B, Zhang W, et al. ELL Targets c-Myc for Proteasomal Degradation and Suppresses Tumour Growth. Nat Commun (2016) 7:11057. doi: 10.1038/ncomms11057
49. Metivier R, Penot G, Hubner MR, Reid G, Brand H, Kos M, et al. Estrogen Receptor-Alpha Directs Ordered, Cyclical, and Combinatorial Recruitment of Cofactors on a Natural Target Promoter. Cell (2003) 115:751–63. doi: 10.1016/S0092-8674(03)00934-6
50. Reid G, Hubner MR, Metivier R, Brand H, Denger S, Manu D, et al. Cyclic, Proteasome-Mediated Turnover of Unliganded and Liganded ERalpha on Responsive Promoters Is an Integral Feature of Estrogen Signaling. Mol Cell (2003) 11:695–707. doi: 10.1016/S1097-2765(03)00090-X
51. Diefenbacher ME, Chakraborty A, Blake SM, Mitter R, Popov N, Eilers M, et al. Usp28 Counteracts Fbw7 in Intestinal Homeostasis and Cancer. Cancer Res (2015) 75:1181–6. doi: 10.1158/0008-5472.CAN-14-1726
52. Popov N, Wanzel M, Madiredjo M, Zhang D, Beijersbergen R, Bernards R, et al. The Ubiquitin-Specific Protease USP28 is Required for MYC Stability. Nat Cell Biol (2007) 9:765–74. doi: 10.1038/ncb1601
53. Sun XX, He X, Yin L, Komada M, Sears RC, Dai MS. The Nucleolar Ubiquitin-Specific Protease USP36 Deubiquitinates and Stabilizes C-Myc. Proc Natl Acad Sci USA (2015) 112:3734–9. doi: 10.1073/pnas.1411713112
54. Thevenon D, Seffouh I, Pillet C, Crespo-Yanez X, Fauvarque MO, Taillebourg E. A Nucleolar Isoform of the Drosophila Ubiquitin Specific Protease Dusp36 Regulates Myc-Dependent Cell Growth. Front Cell Dev Biol (2020) 8:506. doi: 10.3389/fcell.2020.00506
55. Kim D, Hong A, Park HI, Shin WH, Yoo L, Jeon SJ, et al. Deubiquitinating Enzyme USP22 Positively Regulates c-Myc Stability and Tumorigenic Activity in Mammalian and Breast Cancer Cells. J Cell Physiol (2017) 232:3664–76. doi: 10.1002/jcp.25841
56. Pan J, Deng Q, Jiang C, Wang X, Niu T, Li H, et al. USP37 Directly Deubiquitinates and Stabilizes c-Myc in Lung Cancer. Oncogene (2015) 34:3957–67. doi: 10.1038/onc.2014.327
57. Tavana O, Li D, Dai C, Lopez G, Banerjee D, Kon N, et al. HAUSP Deubiquitinates and Stabilizes N-Myc in Neuroblastoma. Nat Med (2016) 22:1180–6. doi: 10.1038/nm.4180
58. Nicklas S, Hillje AL, Okawa S, Rudolph IM, Collmann FM, van Wuellen T, et al. A Complex of the Ubiquitin Ligase TRIM32 and the Deubiquitinase USP7 Balances the Level of c-Myc Ubiquitination and Thereby Determines Neural Stem Cell Fate Specification. Cell Death Differ (2019) 26:728–40. doi: 10.1038/s41418-018-0144-1
59. Fang X, Zhou W, Wu Q, Huang Z, Shi Y, Yang K, et al. Deubiquitinase USP13 Maintains Glioblastoma Stem Cells by Antagonizing FBXL14-Mediated Myc Ubiquitination. J Exp Med (2017) 214:245–67. doi: 10.1084/jem.20151673
60. Sobol A, Askonas C, Alani S, Weber MJ, Ananthanarayanan V, Osipo C, et al. Deubiquitinase OTUD6B Isoforms Are Important Regulators of Growth and Proliferation. Mol Cancer Res (2017) 15:117–27. doi: 10.1158/1541-7786.MCR-16-0281-T
61. DeVine T, Sears RC, Dai MS. The Ubiquitin-Specific Protease USP36 Is a Conserved Histone H2B Deubiquitinase. Biochem Biophys Res Commun (2018) 495:2363–8. doi: 10.1016/j.bbrc.2017.12.107
62. Gonzalez-Prieto R, Cuijpers SA, Kumar R, Hendriks IA, Vertegaal AC. c-Myc Is Targeted to the Proteasome for Degradation in a SUMOylation-Dependent Manner, Regulated by PIAS1, SENP7 and RNF4. Cell Cycle (2015) 14:1859–72. doi: 10.1080/15384101.2015.1040965
63. Kalkat M, Chan PK, Wasylishen AR, Srikumar T, Kim SS, Ponzielli R, et al. Identification of C-MYC SUMOylation by Mass Spectrometry. PloS One (2014) 9:e115337. doi: 10.1371/journal.pone.0115337
64. Rabellino A, Melegari M, Tompkins VS, Chen W, Van Ness BG, Teruya-Feldstein J, et al. Pias1 Promotes Lymphomagenesis Through MYC Upregulation. Cell Rep (2016) 15:2266–78. doi: 10.1016/j.celrep.2016.05.015
65. Sabo A, Doni M, Amati B. Sumoylation of Myc-family Proteins. PloS One (2014) 9:e91072. doi: 10.1371/journal.pone.0091072
66. Sun XX, Chen Y, Su Y, Wang X, Chauhan KM, Liang J, et al. SUMO Protease SENP1 deSUMOylates and Stabilizes C-Myc. Proc Natl Acad Sci USA (2018) 115:10983–8. doi: 10.1073/pnas.1802932115
67. Hendriks IA, Vertegaal AC. A Comprehensive Compilation of SUMO Proteomics. Nat Rev Mol Cell Biol (2016) 17:581–95. doi: 10.1038/nrm.2016.81
68. Lamoliatte F, McManus FP, Maarifi G, Chelbi-Alix MK, Thibault P. Uncovering the SUMOylation and Ubiquitylation Crosstalk in Human Cells Using Sequential Peptide Immunopurification. Nat Commun (2017) 8:14109. doi: 10.1038/ncomms14109
69. Cheng J, Kang X, Zhang S, Yeh ET. SUMO-Specific Protease 1 Is Essential for Stabilization of HIF1alpha During Hypoxia. Cell (2007) 131:584–95. doi: 10.1016/j.cell.2007.08.045
70. Chymkowitch P, Nguea PA, Enserink JM. SUMO-Regulated Transcription: Challenging the Dogma. Bioessays (2015) 37:1095–105. doi: 10.1002/bies.201500065
71. Cubenas-Potts C, Matunis MJ. SUMO: A Multifaceted Modifier of Chromatin Structure and Function. Dev Cell (2013) 24:1–12. doi: 10.1016/j.devcel.2012.11.020
72. Cermelli S, Jang IS, Bernard B, Grandori C. Synthetic Lethal Screens as a Means to Understand and Treat MYC-Driven Cancers. Cold Spring Harbor Perspect Med (2014) 4:a014209. doi: 10.1101/cshperspect.a014209
73. Delmore JE, Issa GC, Lemieux ME, Rahl PB, Shi J, Jacobs HM, et al. BET Bromodomain Inhibition as a Therapeutic Strategy to Target C-Myc. Cell (2011) 146:904–17. doi: 10.1016/j.cell.2011.08.017
74. Li B, Simon MC. Molecular Pathways: Targeting MYC-Induced Metabolic Reprogramming and Oncogenic Stress in Cancer. Clin Cancer Res (2013) 19:5835–41. doi: 10.1158/1078-0432.CCR-12-3629
75. Dang CV, Reddy EP, Shokat KM, Soucek L. Drugging the ‘Undruggable’ Cancer Targets. Nat Rev Cancer (2017) 17:502–8. doi: 10.1038/nrc.2017.36
76. Duffy MJ, Crown J. Drugging “Undruggable” Genes for Cancer Treatment: Are We Making Progress? Int J Cancer (2021) 148:8–17. doi: 10.1002/ijc.33197
77. Beaulieu ME, Jauset T, Masso-Valles D, Martinez-Martin S, Rahl P, Maltais L, et al. Intrinsic Cell-Penetrating Activity Propels Omomyc From Proof of Concept to Viable anti-MYC Therapy. Sci Transl Med (2019) 11:eaar5012. doi: 10.1126/scitranslmed.aar5012
78. Demma MJ, Mapelli C, Sun A, Bodea S, Ruprecht B, Javaid S, et al. Omomyc Reveals New Mechanisms to Inhibit the MYC Oncogene. Mol Cell Biol (2019) 39:e00248–19. doi: 10.1128/MCB.00248-19
79. Masso-Valles D, Soucek L. Blocking Myc to Treat Cancer: Reflecting on Two Decades of Omomyc. Cells (2020) 9:883. doi: 10.3390/cells9040883
80. Wang E, Sorolla A, Cunningham PT, Bogdawa HM, Beck S, Golden E, et al. Tumor Penetrating Peptides Inhibiting MYC as a Potent Targeted Therapeutic Strategy for Triple-Negative Breast Cancers. Oncogene (2019) 38:140–50. doi: 10.1038/s41388-018-0421-y
81. Chen Q, Xie W, Kuhn DJ, Voorhees PM, Lopez-Girona A, Mendy D, et al. Targeting the P27 E3 Ligase SCF(Skp2) Results in p27- and Skp2-Mediated Cell-Cycle Arrest and Activation of Autophagy. Blood (2008) 111:4690–9. doi: 10.1182/blood-2007-09-112904
82. Rico-Bautista E, Yang CC, Lu L, Roth GP, Wolf DA. Chemical Genetics Approach to Restoring p27Kip1 Reveals Novel Compounds With Antiproliferative Activity in Prostate Cancer Cells. BMC Biol (2010) 8:153. doi: 10.1186/1741-7007-8-153
83. Chan CH, Morrow JK, Li CF, Gao Y, Jin G, Moten A, et al. Pharmacological Inactivation of Skp2 SCF Ubiquitin Ligase Restricts Cancer Stem Cell Traits and Cancer Progression. Cell (2013) 154:556–68. doi: 10.1016/j.cell.2013.06.048
84. Malek E, Abdel-Malek MA, Jagannathan S, Vad N, Karns R, Jegga AG, et al. Pharmacogenomics and Chemical Library Screens Reveal a Novel SCF(SKP2) Inhibitor That Overcomes Bortezomib Resistance in Multiple Myeloma. Leukemia (2017) 31:645–53. doi: 10.1038/leu.2016.258
85. Ungermannova D, Lee J, Zhang G, Dallmann HG, McHenry CS, Liu X. High-Throughput Screening AlphaScreen Assay for Identification of Small-Molecule Inhibitors of Ubiquitin E3 Ligase Scfskp2-Cks1. J Biomol Screen (2013) 18:910–20. doi: 10.1177/1087057113485789
86. Byun WS, Jin M, Yu J, Kim WK, Song J, Chung HJ, et al. A Novel Selenonucleoside Suppresses Tumor Growth by Targeting Skp2 Degradation in Paclitaxel-Resistant Prostate Cancer. Biochem Pharmacol (2018) 158:84–94. doi: 10.1016/j.bcp.2018.10.002
87. Dow R, Hendley J, Pirkmaier A, Musgrove EA, Germain D. Retinoic Acid-Mediated Growth Arrest Requires Ubiquitylation and Degradation of the F-Box Protein Skp2. J Biol Chem (2001) 276:45945–51. doi: 10.1074/jbc.M103593200
88. Chauhan D, Tian Z, Nicholson B, Kumar KG, Zhou B, Carrasco R, et al. A Small Molecule Inhibitor of Ubiquitin-Specific Protease-7 Induces Apoptosis in Multiple Myeloma Cells and Overcomes Bortezomib Resistance. Cancer Cell (2012) 22:345–58. doi: 10.1016/j.ccr.2012.08.007
89. Altun M, Kramer HB, Willems LI, McDermott JL, Leach CA, Goldenberg SJ, et al. Activity-Based Chemical Proteomics Accelerates Inhibitor Development for Deubiquitylating Enzymes. Chem Biol (2011) 18:1401–12. doi: 10.1016/j.chembiol.2011.08.018
90. Pozhidaeva A, Valles G, Wang F, Wu J, Sterner DE, Nguyen P, et al. Usp7-Specific Inhibitors Target and Modify the Enzyme’s Active Site Via Distinct Chemical Mechanisms. Cell Chem Biol (2017) 24:1501–12.e1505. doi: 10.1016/j.chembiol.2017.09.004
91. Reverdy C, Conrath S, Lopez R, Planquette C, Atmanene C, Collura V, et al. Discovery of Specific Inhibitors of Human USP7/HAUSP Deubiquitinating Enzyme. Chem Biol (2012) 19:467–77. doi: 10.1016/j.chembiol.2012.02.007
92. Kategaya L, Di Lello P, Rouge L, Pastor R, Clark KR, Drummond J, et al. USP7 Small-Molecule Inhibitors Interfere With Ubiquitin Binding. Nature (2017) 550:534–8. doi: 10.1038/nature24006
93. Turnbull AP, Ioannidis S, Krajewski WW, Pinto-Fernandez A, Heride C, Martin ACL, et al. Molecular Basis of USP7 Inhibition by Selective Small-Molecule Inhibitors. Nature (2017) 550:481–6. doi: 10.1038/nature24451
94. Li M, Liu S, Chen H, Zhou X, Zhou J, Zhou S, et al. N-Benzylpiperidinol Derivatives as Novel USP7 Inhibitors: Structure-Activity Relationships and X-Ray Crystallographic Studies. Eur J Med Chem (2020) 199:112279. doi: 10.1016/j.ejmech.2020.112279
95. Lamberto I, Liu X, Seo HS, Schauer NJ, Iacob RE, Hu W, et al. Structure-Guided Development of a Potent and Selective Non-Covalent Active-Site Inhibitor of USP7. Cell Chem Biol (2017) 24:1490–500.e1411. doi: 10.1016/j.chembiol.2017.09.003
96. Ohol YM, Sun MT, Cutler G, Leger PR, Hu DX, Biannic B, et al. Novel, Selective Inhibitors of USP7 Uncover Multiple Mechanisms of Antitumor Activity In Vitro and In Vivo. Mol Cancer Ther (2020) 19:1970–80. doi: 10.1158/1535-7163.MCT-20-0184
97. Gavory G, O’Dowd CR, Helm MD, Flasz J, Arkoudis E, Dossang A, et al. Discovery and Characterization of Highly Potent and Selective Allosteric USP7 Inhibitors. Nat Chem Biol (2018) 14:118–25. doi: 10.1038/nchembio.2528
98. O’Dowd CR, Helm MD, Rountree JSS, Flasz JT, Arkoudis E, Miel H, et al. Identification and Structure-Guided Development of Pyrimidinone Based USP7 Inhibitors. ACS Med Chem Lett (2018) 9:238–43. doi: 10.1021/acsmedchemlett.7b00512
99. Wrigley JD, Gavory G, Simpson I, Preston M, Plant H, Bradley J, et al. Identification and Characterization of Dual Inhibitors of the USP25/28 Deubiquitinating Enzyme Subfamily. ACS Chem Biol (2017) 12:3113–25. doi: 10.1021/acschembio.7b00334
100. Liu Z, Zhao T, Li Z, Sun K, Fu Y, Cheng T, et al. Discovery of [1,2,3]Triazolo[4,5-D]Pyrimidine Derivatives as Highly Potent, Selective, and Cellularly Active USP28 Inhibitors. Acta Pharm Sin B (2020) 10:1476–91. doi: 10.1016/j.apsb.2019.12.008
101. Wang H, Meng Q, Ding Y, Xiong M, Zhu M, Yang Y, et al. USP28 and USP25 Are Downregulated by Vismodegib In Vitro and in Colorectal Cancer Cell Lines. FEBS J (2021) 288:1325–42. doi: 10.1111/febs.15461
102. Hu Y, Yu K, Wang G, Zhang D, Shi C, Ding Y, et al. Lanatoside C Inhibits Cell Proliferation and Induces Apoptosis Through Attenuating Wnt/beta-Catenin/c-Myc Signaling Pathway in Human Gastric Cancer Cell. Biochem Pharmacol (2018) 150:280–92. doi: 10.1016/j.bcp.2018.02.023
103. Ambaye N, Chen CH, Khanna S, Li YJ, Chen Y. Streptonigrin Inhibits SENP1 and Reduces the Protein Level of Hypoxia-Inducible Factor 1alpha (HIF1alpha) in Cells. Biochemistry (2018) 57:1807–13. doi: 10.1021/acs.biochem.7b00947
104. Han Y, Huang W, Liu J, Liu D, Cui Y, Huang R, et al. Triptolide Inhibits the AR Signaling Pathway to Suppress the Proliferation of Enzalutamide Resistant Prostate Cancer Cells. Theranostics (2017) 7:1914–27. doi: 10.7150/thno.17852
105. Huang W, He T, Chai C, Yang Y, Zheng Y, Zhou P, et al. Triptolide Inhibits the Proliferation of Prostate Cancer Cells and Down-Regulates SUMO-Specific Protease 1 Expression. PloS One (2012) 7:e37693. doi: 10.1371/journal.pone.0037693
106. Wu J, Lei H, Zhang J, Chen X, Tang C, Wang W, et al. Momordin Ic, a New Natural SENP1 Inhibitor, Inhibits Prostate Cancer Cell Proliferation. Oncotarget (2016) 7:58995–9005. doi: 10.18632/oncotarget.10636
107. Zhang Q, Spears E, Boone DN, Li Z, Gregory MA, Hann SR. Domain-Specific c-Myc Ubiquitylation Controls c-Myc Transcriptional and Apoptotic Activity. Proc Natl Acad Sci USA (2013) 110:978–83. doi: 10.1073/pnas.1208334110
108. Gstaiger M, Jordan R, Lim M, Catzavelos C, Mestan J, Slingerland J, et al. Skp2 is Oncogenic and Overexpressed in Human Cancers. Proc Natl Acad Sci USA (2001) 98:5043–8. doi: 10.1073/pnas.081474898
109. Huang HC, Lin CL, Lin JK. 1,2,3,4,6-Penta-O-Galloyl-Beta-D-Glucose, Quercetin, Curcumin and Lycopene Induce Cell-Cycle Arrest in MDA-MB-231 and BT474 Cells Through Downregulation of Skp2 Protein. J Agric Food Chem (2011) 59:6765–75. doi: 10.1021/jf201096v
110. Shachaf CM, Kopelman AM, Arvanitis C, Karlsson A, Beer S, Mandl S, et al. MYC Inactivation Uncovers Pluripotent Differentiation and Tumour Dormancy in Hepatocellular Cancer. Nature (2004) 431:1112–7. doi: 10.1038/nature03043
111. Crawford LJ, Campbell DC, Morgan JJ, Lawson MA, Down JM, Chauhan D, et al. The E3 Ligase HUWE1 Inhibition as a Therapeutic Strategy to Target MYC in Multiple Myeloma. Oncogene (2020) 39:5001–14. doi: 10.1038/s41388-020-1345-x
112. Shachaf CM, Gentles AJ, Elchuri S, Sahoo D, Soen Y, Sharpe O, et al. Genomic and Proteomic Analysis Reveals a Threshold Level of MYC Required for Tumor Maintenance. Cancer Res (2008) 68:5132–42. doi: 10.1158/0008-5472.CAN-07-6192
113. Murphy DJ, Junttila MR, Pouyet L, Karnezis A, Shchors K, Bui DA, et al. Distinct Thresholds Govern Myc’s Biological Output In Vivo. Cancer Cell (2008) 14:447–57. doi: 10.1016/j.ccr.2008.10.018
114. Pozhidaeva A, Bezsonova I. Usp7: Structure, Substrate Specificity, and Inhibition. DNA Repair (Amst) (2019) 76:30–9. doi: 10.1016/j.dnarep.2019.02.005
115. Li P, Liu HM. Recent Advances in the Development of Ubiquitin-Specific-Processing Protease 7 (USP7) Inhibitors. Eur J Med Chem (2020) 191:112107. doi: 10.1016/j.ejmech.2020.112107
116. Fan YH, Cheng J, Vasudevan SA, Dou J, Zhang H, Patel RH, et al. USP7 Inhibitor P22077 Inhibits Neuroblastoma Growth Via Inducing p53-Mediated Apoptosis. Cell Death Dis (2013) 4:e867. doi: 10.1038/cddis.2013.400
117. Weinstock J, Wu J, Cao P, Kingsbury WD, McDermott JL, Kodrasov MP, et al. Selective Dual Inhibitors of the Cancer-Related Deubiquitylating Proteases USP7 and USP47. ACS Med Chem Lett (2012) 3:789–92. doi: 10.1021/ml200276j
118. Prieto-Garcia C, Hartmann O, Reissland M, Braun F, Fischer T, Walz S, et al. Maintaining Protein Stability of Np63 Via USP28 Is Required by Squamous Cancer Cells. EMBO Mol Med (2020) 12:e11101. doi: 10.15252/emmm.201911101
119. Banerjee S, Saluja A. Minnelide, a Novel Drug for Pancreatic and Liver Cancer. Pancreatology (2015) 15:S39–43. doi: 10.1016/j.pan.2015.05.472
Keywords: MYC, protein stability, ubiquitination, deubiquitination, ubiquitin ligase, deubiquitinating enzyme, SUMOylation, SUMO-specific protease
Citation: Sun X-X, Li Y, Sears RC and Dai M-S (2021) Targeting the MYC Ubiquitination-Proteasome Degradation Pathway for Cancer Therapy. Front. Oncol. 11:679445. doi: 10.3389/fonc.2021.679445
Received: 11 March 2021; Accepted: 24 May 2021;
Published: 11 June 2021.
Edited by:
Alena Malyukova, Karolinska Institutet (KI), SwedenReviewed by:
Paul B. Fisher, Virginia Commonwealth University, United StatesPádraig D’Arcy, Linköping University, Sweden
Markus E. Diefenbacher, University of Wuerzburg, Germany
Copyright © 2021 Sun, Li, Sears and Dai. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mu-Shui Dai, ZGFpbUBvaHN1LmVkdQ==; Rosalie C. Sears, c2VhcnNyQG9oc3UuZWR1