
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
SYSTEMATIC REVIEW article
Front. Oncol., 17 June 2021
Sec. Cancer Molecular Targets and Therapeutics
Volume 11 - 2021 | https://doi.org/10.3389/fonc.2021.677983
 Franz Sesti1
Franz Sesti1 Tiziana Feola1,2
Tiziana Feola1,2 Giulia Puliani1,3
Giulia Puliani1,3 Roberta Centello1Valentina Di Vito1Oreste Bagni4
Roberta Centello1Valentina Di Vito1Oreste Bagni4 Andrea Lenzi1
Andrea Lenzi1 Andrea M. Isidori1
Andrea M. Isidori1 Vito Cantisani5
Vito Cantisani5 Antongiulio Faggiano6
Antongiulio Faggiano6 Elisa Giannetta1*
Elisa Giannetta1*Background: Paragangliomas (PGLs) are neuroendocrine neoplasms arising from chromaffin cells of sympathetic or parasympathetic paraganglia. Systemic therapies have been used only in metastatic PGLs. Antiangiogenic agents, such as sunitinib, could be a viable therapeutic choice in the subgroup of patients with SDH-positive PGLs. We describe the case of a man with Familial Paraganglioma Syndrome type 1 (FPGL) related to a novel mutation in SDHD gene treated with sunitinib. Furthermore, we performed a systematic review of the literature aimed to address the following question: is sunitinib treatment effective in patients with advanced/progressive/metastatic PGL?
Methods: We performed a data search using MEDLINE, Cochrane Library, and Scopus between April 2019 and September 2020. We included studies reporting data on clinical or biological characteristics, or clinical outcomes of patients with PGLs treated with sunitinib.
Results: The search leaded to the selection of 25 publications. Data from case reports and case series showed that disease control rate (DCR = stable disease + partial response + complete response) was achieved in 34.7% of cases under sunitinib treatment. In 39% of patients DCR was followed by progressive disease (PD) or tumor relapse, 26.1% patients showed PD. Data from clinical trials showed that DCR was 83%, and the median progression free survival was 13.4 months.
Discussion: Data from the present literature review suggested that sunitinib could be a viable therapeutic option in advanced/progressive/metastatic inoperable PGLs. However, further trials on the efficacy of sunitinib in FPGL and sporadic PGL are needed.
Paragangliomas (PGLs) are neuroendocrine neoplasms (NENs) arising from chromaffin cells of sympathetic or parasympathetic paraganglia (1). A germline mutation is found in approximatively 40% of these tumors (2). Both in sporadic and inherited PGLs, it is possible to evidence two different pathogenetic pathways: alterations in proteins associated to Krebs cycle and hypoxia signaling (cluster I), and alteration in kinase signaling (cluster II) (3). Cluster I includes mutations in genes like von Hippel–Lindau (VHL), 2-oxoglutarate-dependent prolyl hydroxylase (PHD2), and succinate dehydrogenase (SDH), which is a component of the electron transport chain of the Krebs cycle and catalyzes the oxidation of succinate (4). Catalytic core of SDH is made of 2 subunits (SDHA, SDHB), anchored to the mitochondrial inner membrane through subunits SDHC and SDHD (5). Mutations in a subunit of SDH determine an accumulation of succinate, which causes an increase of hypoxia inducible factor (HIF) 1α for a reduction of its ubiquitination (6). The elevation in HIF1α leads to activation of the angiogenic pathway and alteration in cell metabolism (7).
SDHD mutations are responsible for Familial Paraganglioma Syndrome type 1 (FPGL1), inherited in autosomal dominant manner, characterized by head and neck parasympathetic PGLs (85% of cases), more rarely thoraco-abdominal sympathetic PGLs (20–25% of cases) and pheochromocytomas (10–25%) (8, 9). Some 157 mutations have been described in SDHD gene, including deletions and duplications (an updated list is available at https://databases.lovd.nl/shared/genes/SDHD) (10).
Malignant head and neck PGLs are extremely rare (8, 11). However, their local growth can determine compression and/or infiltration of the neighboring anatomical structures, causing dysphagia and cranial nerves palsy (12). Surgery represents the first-choice treatment for head and neck PGLs (13), but severe complications, including cranial nerve lesions and vascular damage can occur (14).
Systemic therapies have been used in metastatic PGLs only (15). Antiangiogenic agents, such as tyrosine kinase inhibitors (TKI), have demonstrated in a phase 2 trial to determine disease stability, as well as partial response in the subgroup of patients with SDH-positive PGLs (SDHA and SDHB) (16).
The effectiveness of sunitinib, a widely used TKI, in patients with advanced/progressive/metastatic PGL is still unclear. Starting from the observations in our clinical case, the present systematic review is aimed to address the following question: is sunitinib treatment effective in patients with advanced/progressive/metastatic PGL?
On September 2018, a 37-year-old man contacted the Neuroendocrine Tumor task force Unit (NETTARE) of the Policlinico Umberto I Hospital at “Sapienza” University for the appearance of bilateral later-cervical swellings three years before, diagnosed as PGLs. He smoked for 15 years. His personal history includes allergic asthma and coccygeal fistula surgically treated in 2003. The patient had a familial history for neoplasms on the father’s side: a grandmother with a non-better classified parathyroid neoplasm and a grandfather with a history of thyroid, bladder, and gastric cancer. On clinical examination left lateral cervical mass was fixed and firm, and no other relevant alterations were found. He reported dysphagia and dyspeptic symptoms without other disturbances; blood pressure and heart rate were normal.
The patient’s clinical history began in December 2015 when, for the appearance of bilateral laterocervical masses, he performed a magnetic resonance (MR) imaging of the neck that highlighted “the presence of two expansive lesions localized bilaterally in the vascular spaces of the neck, on the right of 45 × 25 × 70 mm and on the left of 48 × 44 × 53 mm, with an inhomogeneous architecture, and a marked and inhomogeneous contrast enhancement”. The MR angiography confirmed the vascular nature of the masses. The right lesion extended from the carotid bifurcation growing cranially up to the lacerated foramen, determining encasement of the internal and external carotid arteries, located both anteriorly. This formation exerted an evident mass effect in the retro-stylous vascular space, causing the lifting of the muscular tent of the upper constrictor and the partial distortion of the oropharyngeal air column. The left lesion surrounded the middle distal tract of the common carotid artery, determined encasement of two branches at the carotid bifurcation, but involved only the cervical tract of the internal carotid artery. Biochemical assessment showed negative urinary metanephrines (44 µg/24 h, range 20–345).
After two different multidisciplinary consultations, on September 2016 the patient underwent the removal of the right lesion after embolization, with histological examination compatible with PGL: “neoformation of 44 mm, consisting of solid round nests of monomorphic cells of medium-sized, immunoreactive for chromogranin A, synaptophysin, vimentin, negative for EMA, CAM5.2, AE1/AE3. At the periphery, cellular component of dendritic aspect, S100 positive. Ki67 index <2%”.
After surgery, the patient reported a lesion of the right hypoglossal nerve, developing right vocal cord hypo-mobility, paresis of the palatine veil on the right side, right hemilingual hypotrophy, and a complete deficit of tongue mobility. In March 2017, the left lesion was embolized.
During the follow-up the patient performed several morphological and functional imaging exams. The MR and computed tomography (CT) angiography confirmed the presence of residual tissue on the right side of 15 mm, whereas the embolized lesion on the left appeared slightly reduced in size compared to the pre-operative examination (40 × 43 × 53 mm), with presence of vital tissue. Both lesions showed significant uptake of radiotracer at 18F-fluoro-dihydroxyphenylalanine (18F-DOPA) positron emission tomography (PET)-CT with a SUVmax of 10.5 on the left and 5.66 on the right, and at 68Ga-DOTA-D-Phe1-Tyr3-Octreotate (68Ga-DOTATOC) PET-CT (Figures 1A, B). Moreover, the latter showed a further area of focal uptake in correspondence of the skull base on the right, adjacent to surgical clips.
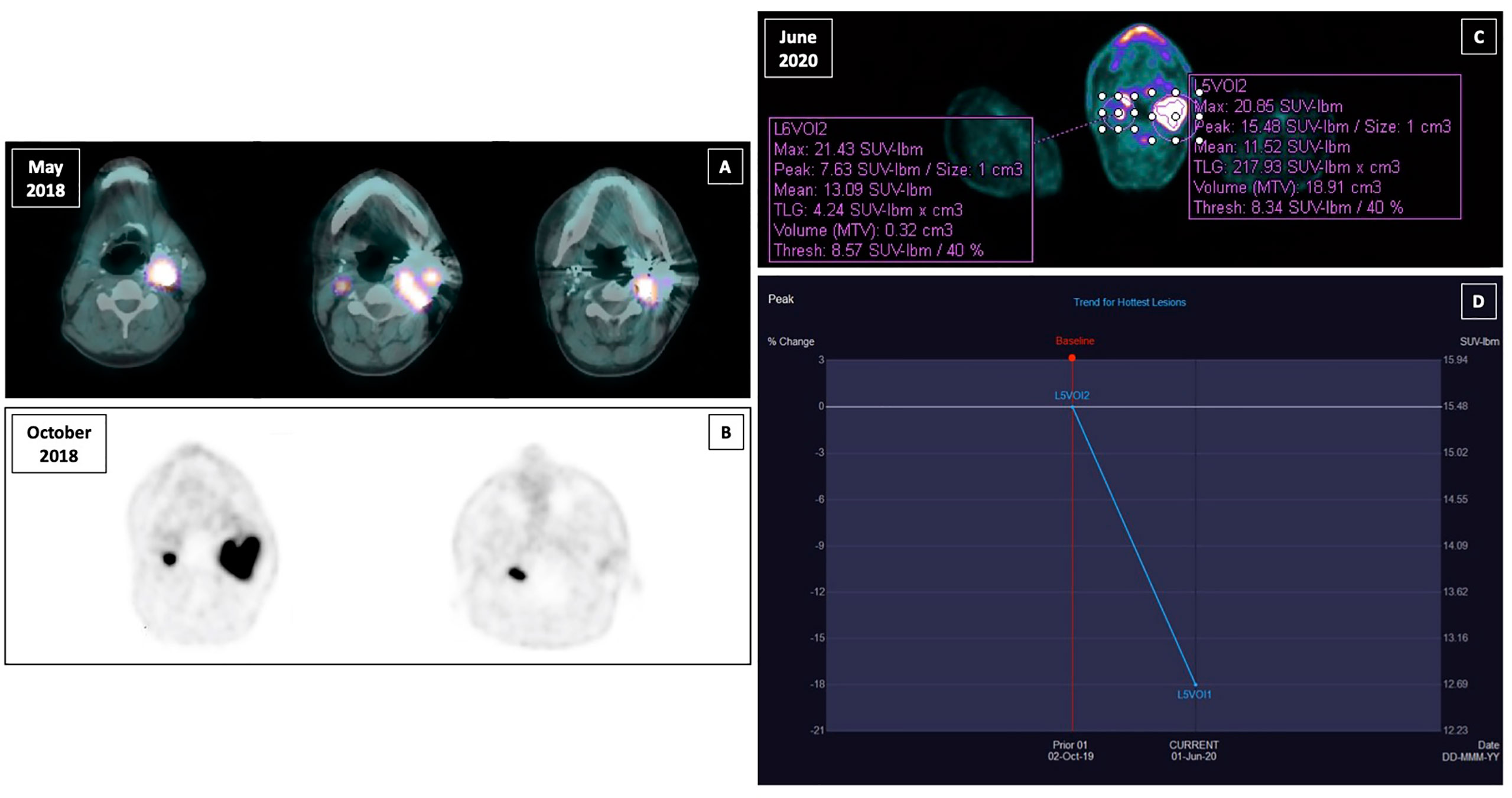
Figure 1 Functional imaging of right and left paraganglioma and response to sunitinib treatment assesed by PERCIST criteria. (A) 18F-fluoro-dihydroxyphenylalanine (18F-DOPA) positron emission tomography (PET)-computed tomography (CT) in May 2018; (B) 68Ga-DOTA-D-Phe1-Tyr3-Octreotate (68Ga-DOTATOC) PET-CT in October 2018; (C) 18F-fluorodeoxyglucose (18F-FDG) positron emission tomography (PET)-computed tomography (CT) with contrast medium performed after the third cycle of therapy in June 2020; (D) peak standardized uptake value corrected for lean body mass (SULpeak) trend for the hottest lesion between the two susequent 18F-FDG PET-CT (October 2020 and June 2020).
Considering the expression of somatostatin receptors, in December 2018 a therapy with somatostatin analogs (SSAs) was started (lanreotide 120 mg every 28 days subcutaneously). The patient experienced poor tolerance because of abdominal pain and acholic feces, so the dose was reduced to 90 mg every 28 days until the disappearance of the symptomatology. At 4-month MR angiography, tumor growth was observed (the right measured 22 vs. 15 mm; the left 69 × 46 × 50 vs. 40 × 43 × 53 mm). A 18F-fluorodeoxyglucose (FDG) PET-CT was performed according to our proposed follow-up algorithm (17), with the evidence of an intense metabolic activity in correspondence of the left voluminous expansive lesion (SUVmax 26.6) and the known right nodular lesion (SUVmax 27.4).
Given the morpho-functional features of the lesion (local aggressiveness, progression, high 18F-FDG uptake), the risk-effectiveness of the treatments’ strategies (high surgical risk, potential ineffectiveness of radiotherapy due to high tumor volume, poor efficacy and tolerance of SSA administration), and patient preferences, the NETTARE multidisciplinary board proposed a targeted therapy with a TKI: sunitinib.
In October 2019, sunitinib was started at a low dosage (25 mg daily orally), in order to minimize the potential toxicity, obtaining a relevant clinical response, with improvement of dysphagia and pain, and consequent improvement of quality of life. A good safety profile was reported. The follow-up with contrast-enhanced ultrasound (CEUS) of the neck showed a precocious size reduction of the lesions with onset of necrosis signals. The left lesion was decreased from 57 × 48 mm at baseline to 47 × 31 mm at 20 days and then was stable at 50 days, the right lesion was decreased from 13 mm at baseline to 9 mm at 20-day follow-up and then stable at 50-day follow-up. Near the end of the third cycle of sunitinib a 18F-FDG PET-CT with contrast medium was performed. The peak standardized uptake value corrected for lean body mass (SULpeak) of the left lesion was reduced by 18% (Figures 1C, D). Subsequently, a MR angiography performed at one-year follow-up showed the right lesion decreased to from 16 × 22 × 11 mm to 19 × 13 × 20 mm and left lesion decreased from 69 × 46 × 50 mm to 59 × 46 × 51 mm. At last MR follow-up in March 2021, right and left lesions were stable measuring respectively 18 × 16 × 13 mm and 60 × 44 × 47 mm. Thus, under sunitinib treatment the patient has been stable for 17 months (Figure 2).
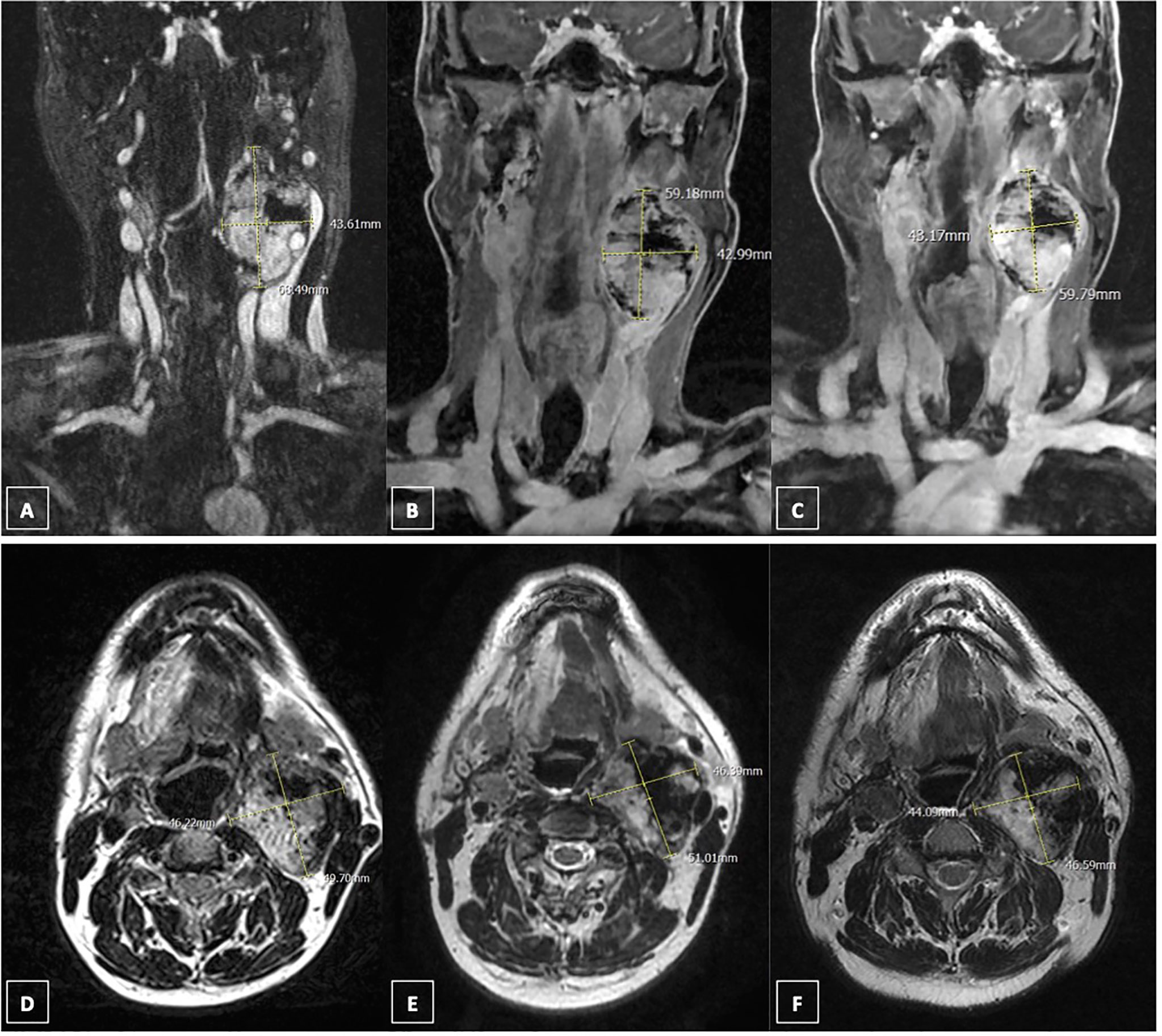
Figure 2 Response to sunitinib treatment assesed by magnetic resonance (MR). (A) coronal contrast-enhanced MR scan (fat-sat T1 sequence) of left paraganglioma in April 2019; (B) coronal contrast-enhanced MR scan (fat-sat T1 sequence) of left paraganglioma in July 2020; (C) coronal contrast-enhanced MR scan (fat-sat T1 sequence) of left paraganglioma in March 2021; (D) axial contrast-enhanced MR scan (T2 sequence) of left paraganglioma in April 2019; (E) axial contrast-enhanced MR scan (T2 sequence) of left paraganglioma in July 2020; (F) axial contrast-enhanced MR scan (T2 sequence) of left paraganglioma in March 2021.
Previously, the patient underwent a genetic consultation with the detection of a new mutation of SDHD gene c.16_28del p. Arg6PHEFS*5, that has never been described in literature, consistent with the diagnosis of FPGL1. A complete family history was collected in spite of the absence of other familial cases of PGL. At the genetic test among the first-degree relatives, the brother was negative, while the father was SDHD mutated. He underwent a neck and chest CT, that showed a lesion on the left carotid (max diameter 24 mm) as well as an enormous lesion in the mediastinum (>100 mm of maximal diameter), both with marked contrast enhancement. The 68Ga-DOTATOC PET-CT and 18F-FDG PET-CT were performed, highlighting uptake of both the radiotracers. The mediastinum lesion was removed after embolization by an expert team of thoracic surgeons of our Unit. The histological diagnosis was consistent with PGL: neoformation of 100 × 90 × 40 mm, associated to another neoformation of 15 mm with the same characteristics in proximity of the mail lesion. Both neoformations consisted of organoid nests of medium-sized cells with a large clear cytoplasm and a dispersed chromatin oval nucleus, which in the smaller lesion is hyperchromatic and irregular, surrounded by a thin continuous line of spindle cells with an elongated nucleus, and separated by a dense capillary vascular network. Immunochemistry was positive for synaptophysin, CD56, S100, GFAP, and weakly positive for chromogranin A. Ki67 index was 4%.
A systematic review was performed following a rigorous protocol based on the Cochrane Collaboration and PRISMA statements (18, 19), in order to address the following question: is sunitinib treatment effective in patients with advanced/progressive/metastatic PGL?
English-language original articles were independently searched by one author (FS) in several databases (MEDLINE, Cochrane Library, and Scopus) between April 2019 and September 2020. The following key words were used for study search: (sunitinib AND paraganglioma) OR (sunitinib AND paraganglion tumor) OR (sunitinib AND paraganglionic tumor) OR (sunitinib AND paraganglion neoplasm) OR (sunitinib AND paraganglionic neoplasm). Additional articles were identified by hand-searching reference lists of all the eligible articles retrieved.
The titles and the abstracts of all identified articles were independently screened by one reviewer (FS) to assess their relevance. Reviews, animal studies and non-original articles were excluded. Full texts of selected, potentially relevant, papers were further evaluated. For the purpose of this review suitability of studies was defined as eligible if reporting data on the clinical or biological characteristics, or clinical outcomes of patients with PGL treated with sunitinib. We therefore selected studies that met all the following eligibility criteria: (i) randomized-controlled trial, prospective or retrospective studies, case series or case reports; (ii) PGL diagnosis; and (iii) treatment with sunitinib.
One author (FS) independently extracted the following data from included publications: first author, year of publication, study design, age, sex, primary tumor site, mutation status, sunitinib schedule, radiographic response criteria, radiographic response, response duration, and toxicity. Table 1 summarizes these data.
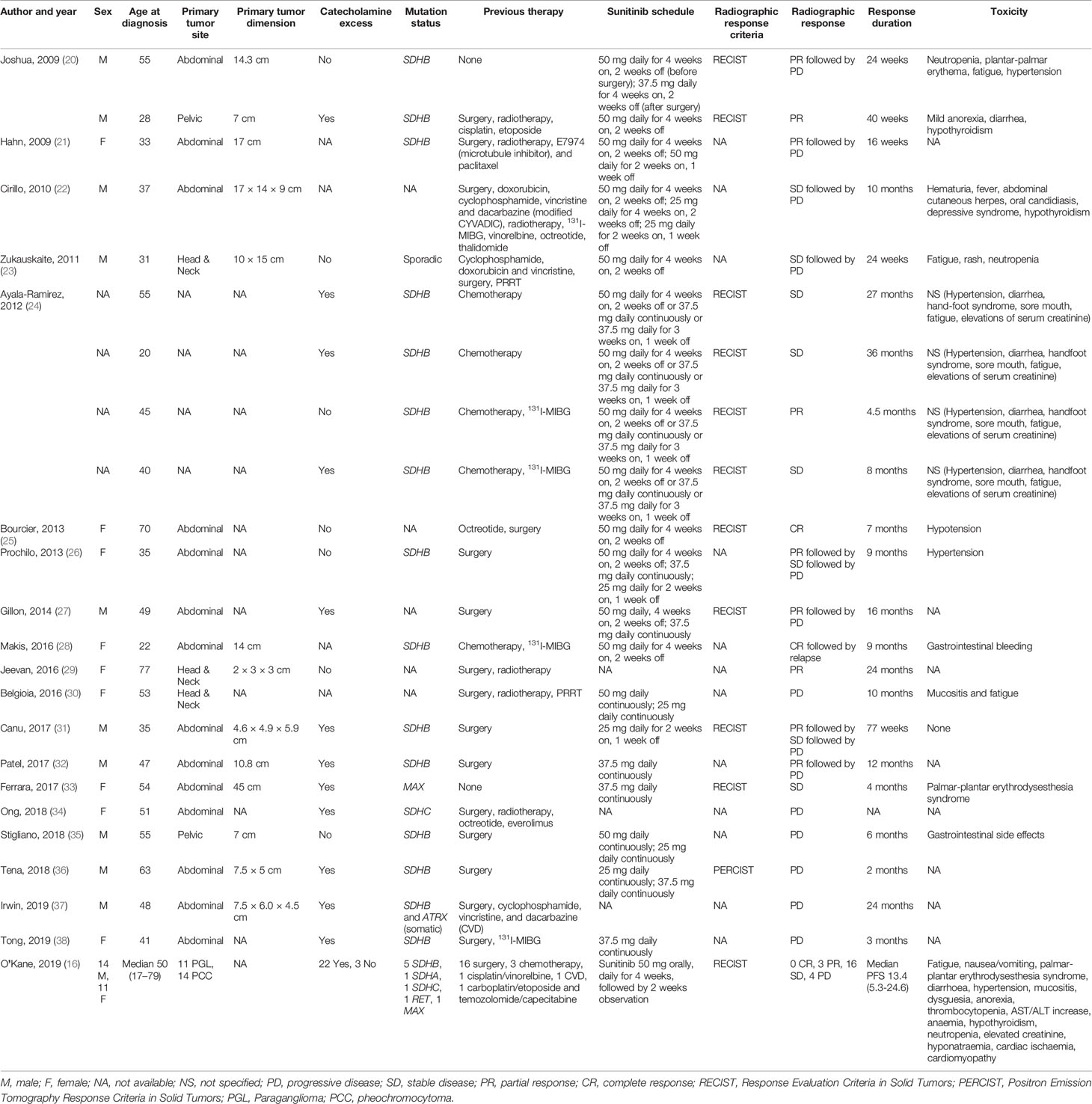
Table 1 Response to sunitinib in patients from 25 selected case reports, case series, and clinical trial.
Some 150 potentially relevant studies were identified, 116 were excluded on the basis of title and abstract screening. The main reasons for exclusion were not original studies, reviews, animal studies, duplicates, lack of group of interest (treatment with sunitinib). Of the 34 remaining publications, nine were excluded after full text assessment because they did not meet all the eligibility criteria. This process leaded to the selection of 25 publications (Figure 3) (16, 20–43).
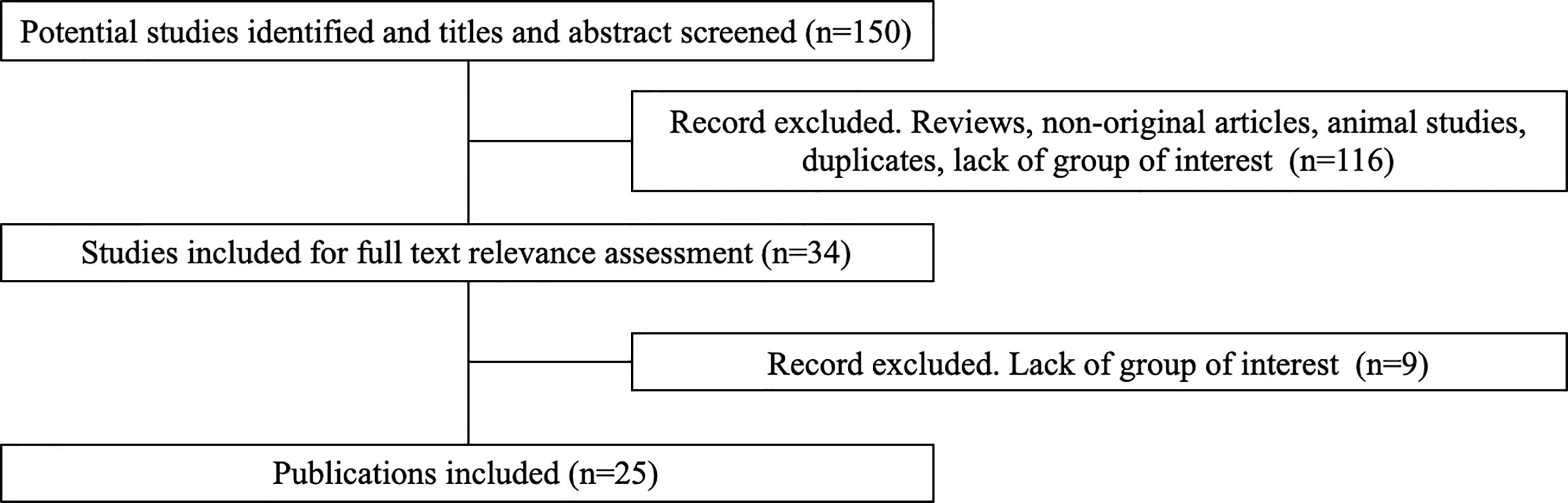
Figure 3 Flow-chart of the literature eligibility assessment process.
Data about objective response were available for 23 patients (nine women and 10 men, sex was not available for four patients) (Table 1). The patient age range was 20–77 years. Cumulative mean age, and cumulative median age were 45.4 and 47 years, respectively. In all cases disease status at baseline was represented by progressive or inoperable disease. Metastases were present in all patients. Some 14 patients had abdominal PGL, three head & neck PGL, and two pelvic PGL.
Mutation status was examined in 18 patients showing: SDHB (n = 15), MAX (n = 1), and SDHC (n = 1) mutations; one patient had sporadic PGL.
Treatment schedule included sunitinib regimens using 50, 37.5 or 25 mg. Six patients (26.1%) showed progressive disease (PD), four patients (17.4%) stable disease (SD), three patients (13%) partial response (PR), one (4.3%) patient complete response (CR), two patients (8.7%) SD followed by PD, six patients (26.1%) PR followed by PD, and one (4.3%) CR followed by relapse.
Regarding previous therapy, 16 patients underwent surgery before sunitinib treatment, 10 were treated with chemotherapy, six with radiotherapy, five with 131I MIBG, three with octreotide, two with PRRT, and one with everolimus. Only two patients started sunitinib as a first-line therapy. Data about objective response in this subset were available for two patients, one patient showed PR followed by PD, and one patient showed SD. In six cases sunitinib was used as first-line systemic therapy after surgery, four patients showed PR followed by PD, and two PD. In one case sunitinib was the first-line systemic therapy after surgery and radiotherapy, the patient showed PR. In any other case sunitinib was prescribed after one or more systemic therapies.
The only clinical trial available in the literature has been published by O’Kane et al. (16) who evaluated the effects of first-line sunitinib treatment (50 mg orally, daily for 4 weeks, followed by 2 weeks observation) in 25 patients (14 men and 11 women) with non-resectable or metastatic progressive PGL (11) or pheochromocytoma (14). Median age was 50 years (range: 17–79 years). Five patients were SDHB mutated, one SDHA, one SDHC, one RET, and one MAX. The disease control rate was 83% (95% CI: 56–93%), including three patients who achieved a PR, and 16 with SD. The median progression free survival (PFS) was 13.4 months (95% CI: 5.3–24.6) (16) (Table 1).
In the present case report, the patient presented bilateral neck PGLs, the right lesion has been removed surgically, however a permanent lesion of the right hypoglossal nerve has occurred. After surgery, the patient still complained dysphagia and pain related to the left PGL. Therefore, given the high surgical risk due to the dimension and the localization of the left lesion, and the patient’s preference, a medical therapy has been proposed.
Current guidelines are lacking therapeutic options for patients with inoperable neck PGLs (13). The established localized treatments include external beam radiotherapy (44), radiosurgery (45), and ablative therapy (46). Whereas systemic therapies include radionuclide therapy with 131I-metaiodobenzylguanidine (MIBG) (47) or peptide receptor radionuclide therapy (PRRT) (48), chemotherapy with cyclophosphamide, vincristine and dacarbazine (CVD) (49), or temozolomide (50). Among molecular targeted therapies the efficacy of TKI as lenvatinib (NCT03008369), cabozantinib (NCT02302833), nivolumab, and ipilimumab (NCT02834013) are currently being evaluated in clinical trials (51). Recently, data from a phase 2 clinical trial provided the rationale for pembrolizumab use in patients with advanced PGLs (52).
Somatostatin receptors (SSTRs) are expressed by PGLs (53, 54), as confirmed by studies investigating 68Ga PET-CT role in PGLs diagnosis, staging, and follow-up (17, 55). Therefore, in our case, considering the 68Ga DOTATOC uptake SSAs were administrated (lanreotide 120 mg every 28 days). Currently, the efficacy of SSAs in PGLs has not been studied in phase 2–3 clinical trials, nevertheless, in a prospective intervention study, one of four patients with progressive head and neck PGLs treated with octreotide showed a reduced tumor growth (56). In the present case, the SSA was reduced because of side effects, then discontinued because of tumor progression.
Sunitinib has been chosen as a second-line medical therapy after a multidisciplinary consult. Sunitinib is a multitargeted receptor TKI which exerts antiangiogenic and antitumor effects targeting platelet-derived growth factor receptor (PDGFR), vascular endothelial growth factor receptor (VEGFR), KIT, and FLT3 (57). The rationale derived from the high expression of HIF1α, HIF2α, VEGF, and VEGFR arising from HIF dysregulation and hypoxia-inducible target genes’ activation, due to germline mutations of SDHD (58). Indeed, the tumorigenesis of PGLs seems partly related to a pseudo-hypoxic drive (59). These data support the rationale of this antiangiogenic therapeutic agent in PGLs.
Recently, O’Kane et al. evaluated the effects of sunitinib treatment on 25 patients with progressive PGL or pheochromocytoma in a phase 2 clinical trial (16). A disease control rate (DCR = SD + PR + CR) of 83% (95% CI: 56–93%) was obtained (16). Notably, three patients who had a PR carried germline mutations of SDHA, SDHB, and RET. Moreover, in four other patients with germline SDH mutations (three SDHB and one SDHC) a prolonged SD was achieved. No patient with SDHD mutation was included in the study (16). In our review a significant radiographic DCR was achieved in 34.7% of cases (SD in 17.4%, PR in 13%, and CR in 4.3%). In 39% of patients DCR was followed by PD or tumor relapse. Furthermore, 26.1% patients showed PD.
The discordant results between O’Kane et al. trial and our systematic literature review could be related to the inhomogeneity of the two populations. Indeed, 13 of 23 patients included in our review received two or more lines of therapy before sunitinib, while in the SNIPP trial only one patient received two lines of therapy. Moreover, patients in the SNIPP trial had PGL or pheochromocytoma, while in our literature review patients with pheochromocytomas were not included in the analysis. Furthermore, patients in the two populations received different sunitinib schedules.
The role of sunitinib in these patients could soon become clearer thanks to the First International Randomized Study in Malignant Progressive Pheochromocytoma and Paraganglioma (FIRSTMAPPP) trial, which is investigating the efficacy of sunitinib, at a starting dose of 37.5 mg daily, on the PFS of patients with progressive malignant PGL or pheochromocytoma (NCT01371201). Estimated study completion date of FIRSTMAPPP trial is June 2021.
In our case, the tumor response was early assessed by CEUS, according to previous studies (17). After 20 days of sunitinib therapy a SD was observed, the left lesion was reduced by approximately 10 mm and the right by 4 mm. In the subsequent CEUS evaluation after 56 days both lesions showed stable dimensions. Furthermore, the objective tumor response was accompanied by a clinical improvement of dysphagia and pain, leading to a better quality of life. The treatment has been well tolerated who did not report any specific side effect.
A previous study showed that 18F-FDG PET could be a reliable technique to evaluate tumor objective response in sunitinib treated PGLs (24). Indeed, all five SDHB-mutated patients with sympathetic PGL who were studied with 18F-FDG PET showed a PR or a SD according to RECIST 1.1 criteria (24). In a SDHB knockout mouse model of PGL treated with sunitinib, 18F-FDG PET was able to detect a transient reduction of FDG uptake and total lesion glycolysis (TLG) during the first two weeks of treatment. However, both SUV and TLG increased after the third week. Metabolic resistance preceded tumor growth which was evident after four weeks of treatment. This evidence suggests that 18F-FDG PET could monitor precisely metabolic changes of PGL during an anti-angiogenic treatment and could possibly predict disease progression (60).
In the literature, to the best of our knowledge, among all patients with PGL treated with sunitinib, none was an SDHD mutation carrier, and none had a non-metastatic PGL (Table 1), making our patient the first case of SDHD-related benign PGL treated with sunitinib. Moreover, our patient showed a novel variant of SDHD gene, c.16_28del p. Arg6PHEFS*5, which is not described in the literature, neither present in genome (https://gnomad.broadinstitute.org) or in gene-specific databases (https://databases.lovd.nl/shared/genes/SDHD). The abovementioned variant is a small deletion of 13 nucleotides in exon 1 of SDHD gene, which determines a nucleotide sequence frameshift, resulting in a new aminoacidic sequence, starting from amino acid 6, with a premature stop after five amino acids. This variant has been classified as pathogenetic (class 5), according to American College of Medical Genetics and Genomics (61).
In the present paper we report the case of a patient affected by FPGL1 with locally-advanced bilateral neck PGLs. The peculiarity of the case lies in the fact that, to the best of our knowledge, this is the first reported patient with non-metastatic SDHD-related PGL treated with sunitinib. The treatment was safe and effective both in terms of tumor objective response and symptomatic relief. Moreover, both the patient and his father carry a novel mutation of SDHD gene, associated to the development of PGLs, never described in literature. On the basis of the reported pooled data from our systematic review, sunitinib could be a viable therapeutic option in advanced/progressive/metastatic PGLs, especially in patients with germline mutations. Further trials on the efficacy of sunitinib in FPGL and sporadic PGLs are needed.
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
FS is the first author for this case report and systematic review of literature. EG is the corresponding author that concepted and designed the study. TF, GP, RC, VV, OB, and CV contributed to the data collection, and manuscript, tables, figures preparation. CV, OB, AL, AI, AF, and EG revised critically this work. All authors contributed to the article and approved the submitted version.
Ministerial research project PRIN2017Z3N3YC.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We wish to thank Dr. Stefania Zovato of the Familial Cancer Clinic and Oncoendocrinology Veneto Institute of Oncology IOV- IRCCS for identifying the novel heterozygous SDHD gene variant in the patient; Dr. Giovanni Savarese, Dr. Raffaella Ruggiero and Dr. Luisa Circelli of the Genetic Ames Group, Napoli, Italy for identifying the novel heterozygous SDHD gene variant in father’s proband.
We also wish to thank the NETTARE Unit—NeuroEndocrine Tumor TAsk foRcE of “Sapienza” University of Rome, Italy, led by Prof. Andrea Lenzi, Prof. Andrea M. Isidori, Prof. Antongiulio Faggiano, and Prof. Elisa Giannetta, for integrating the patient’s multidisciplinary clinical, diagnostic and therapeutic management, and follow-up.
1. Lam AK. Update on Adrenal Tumours in 2017 World Health Organization (WHO) of Endocrine Tumours. Endocr Pathol (2017) 28(3):213–27. doi: 10.1007/s12022-017-9484-5
2. Khatami F, Mohammadamoli M, Tavangar SM. Genetic and Epigenetic Differences of Benign and Malignant Pheochromocytomas and Paragangliomas (Ppgls). Endocr Regul (2018) 52(1):41–54. doi: 10.2478/enr-2018-0006
3. Nolting S, Grossman AB. Signaling Pathways in Pheochromocytomas and Paragangliomas: Prospects for Future Therapies. Endocr Pathol (2012) 23(1):21–33. doi: 10.1007/s12022-012-9199-6
4. Fishbein L, Leshchiner I, Walter V, Danilova L, Robertson AG, Johnson AR, et al. Comprehensive Molecular Characterization of Pheochromocytoma and Paraganglioma. Cancer Cell (2017) 31(2):181–93. doi: 10.1016/j.ccell.2017.01.001
5. Selak MA, Armour SM, MacKenzie ED, Boulahbel H, Watson DG, Mansfield KD, et al. Succinate Links TCA Cycle Dysfunction to Oncogenesis by Inhibiting HIF-alpha Prolyl Hydroxylase. Cancer Cell (2005) 7(1):77–85. doi: 10.1016/j.ccr.2004.11.022
6. Pollard PJ, Wortham NC, Tomlinson IP. The TCA Cycle and Tumorigenesis: The Examples of Fumarate Hydratase and Succinate Dehydrogenase. Ann Med (2003) 35(8):632–9. doi: 10.1080/07853890310018458
7. Baysal BE, Maher ER. 15 YEARS of PARAGANGLIOMA: Genetics and Mechanism of Pheochromocytoma-Paraganglioma Syndromes Characterized by Germline SDHB and SDHD Mutations. Endocr Relat Cancer (2015) 22(4):T71–82. doi: 10.1530/ERC-15-0226
8. Benn DE, Robinson BG, Clifton-Bligh RJ. 15 YEARS of PARAGANGLIOMA: Clinical Manifestations of Paraganglioma Syndromes Types 1-5. Endocr Relat Cancer (2015) 22(4):T91–103. doi: 10.1530/ERC-15-0268
9. Baysal BE, Ferrell RE, Willett-Brozick JE, Lawrence EC, Myssiorek D, Bosch A, et al. Mutations in SDHD, a Mitochondrial Complex II Gene, in Hereditary Paraganglioma. Science (2000) 287(5454):848–51. doi: 10.1126/science.287.5454.848
10. Bayley JP, Devilee P, Taschner PE. The SDH Mutation Database: An Online Resource for Succinate Dehydrogenase Sequence Variants Involved in Pheochromocytoma, Paraganglioma and Mitochondrial Complex II Deficiency. BMC Med Genet (2005) 6:39. doi: 10.1186/1471-2350-6-39
11. Piccini V, Rapizzi E, Bacca A, Di Trapani G, Pulli R, Giache V, et al. Head and Neck Paragangliomas: Genetic Spectrum and Clinical Variability in 79 Consecutive Patients. Endocr Relat Cancer (2012) 19(2):149–55. doi: 10.1530/ERC-11-0369
12. Corssmit EP, Romijn JA. Clinical Management of Paragangliomas. Eur J Endocrinol (2014) 171(6):R231–43. doi: 10.1530/EJE-14-0396
13. Lenders JW, Duh QY, Eisenhofer G, Gimenez-Roqueplo AP, Grebe SK, Murad MH, et al. Pheochromocytoma and Paraganglioma: An Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab (2014) 99(6):1915–42. doi: 10.1210/jc.2014-1498
14. Luna-Ortiz K, Rascon-Ortiz M, Villavicencio-Valencia V, Granados-Garcia M, Herrera-Gomez A. Carotid Body Tumors: Review of a 20-Year Experience. Oral Oncol (2005) 41(1):56–61. doi: 10.1016/j.oraloncology.2004.06.006
15. Corssmit EPM, Snel M, Kapiteijn E. Malignant Pheochromocytoma and Paraganglioma: Management Options. Curr Opin Oncol (2020) 32(1):20–6. doi: 10.1097/CCO.0000000000000589
16. O’Kane GM, Ezzat S, Joshua AM, Bourdeau I, Leibowitz-Amit R, Olney HJ, et al. A Phase 2 Trial of Sunitinib in Patients With Progressive Paraganglioma or Pheochromocytoma: The SNIPP Trial. Br J Cancer (2019) 120(12):1113–9. doi: 10.1038/s41416-019-0474-x
17. Puliani G, Sesti F, Feola T, Di Leo N, Polti G, Verrico M, et al. Natural History and Management of Familial Paraganglioma Syndrome Type 1: Long-Term Data From a Large Family. J Clin Med (2020) 9(2):588. doi: 10.3390/jcm9020588
18. Moher D, Liberati A, Tetzlaff J, Altman DG, Group P. Preferred Reporting Items for Systematic Reviews and Meta-Analyses: The PRISMA Statement. BMJ (2009) 339:b2535. doi: 10.1136/bmj.b2535
19. Liberati A, Altman DG, Tetzlaff J, Mulrow C, Gotzsche PC, Ioannidis JP, et al. The PRISMA Statement for Reporting Systematic Reviews and Meta-Analyses of Studies That Evaluate Health Care Interventions: Explanation and Elaboration. J Clin Epidemiol (2009) 62(10):e1–34. doi: 10.1016/j.jclinepi.2009.06.006
20. Joshua AM, Ezzat S, Asa SL, Evans A, Broom R, Freeman M, et al. Rationale and Evidence for Sunitinib in the Treatment of Malignant Paraganglioma/Pheochromocytoma. J Clin Endocrinol Metab (2009) 94(1):5–9. doi: 10.1210/jc.2008-1836
21. Hahn NM, Reckova M, Cheng L, Baldridge LA, Cummings OW, Sweeney CJ. Patient With Malignant Paraganglioma Responding to the Multikinase Inhibitor Sunitinib Malate. J Clin Oncol (2009) 27(3):460–3. doi: 10.1200/JCO.2008.19.9380
22. Cirillo F. Metastatic Paraganglioma and Treatment With Sunitinib: A Case Report. Tumori (2010) 96(6):1022–7. doi: 10.1177/548.6526
23. Zukauskaite R, Hjorthgau K, Poulsen PL, Baerentzen S, Ladekarl M. Malignant Pheochromocytoma and Paraganglioma: Three Cases Illustrating the Use of Molecular Targeted Diagnostics and Therapy and Possible Role of New Drugs. Acta Oncol (2011) 50(8):1255–9. doi: 10.3109/0284186X.2011.584552
24. Ayala-Ramirez M, Chougnet CN, Habra MA, Palmer JL, Leboulleux S, Cabanillas ME, et al. Treatment With Sunitinib for Patients With Progressive Metastatic Pheochromocytomas and Sympathetic Paragangliomas. J Clin Endocrinol Metab (2012) 97(11):4040–50. doi: 10.1210/jc.2012-2356
25. Bourcier ME, Vinik AI. Sunitinib for the Treatment of Metastatic Paraganglioma and Vasoactive Intestinal Polypeptide-Producing Tumor (Vipoma). Pancreas (2013) 42(2):348–52. doi: 10.1097/MPA.0b013e31825c53fa
26. Prochilo T, Savelli G, Bertocchi P, Abeni C, Rota L, Rizzi A, et al. Targeting VEGF-VEGFR Pathway by Sunitinib in Peripheral Primitive Neuroectodermal Tumor, Paraganglioma and Epithelioid Hemangioendothelioma: Three Case Reports. Case Rep Oncol (2013) 6(1):90–7. doi: 10.1159/000348429
27. Gillon P, Godbert Y, Dupin C, Bubien V, Italiano A, Roubaud G. Long Clinical Benefit Achieved in Two Patients With Malignant Paraganglioma Treated by Metronomic Cyclophosphamide. Future Oncol (2014) 10(14):2121–5. doi: 10.2217/fon.14.171
28. Makis W, McCann K, McEwan AJ, Sawyer MB. Combined Treatment With 131i-MIBG and Sunitinib Induces Remission in a Patient With Metastatic Paraganglioma Due to Hereditary Paraganglioma-Pheochromocytoma Syndrome From an SDHB Mutation. Clin Nucl Med (2016) 41(3):204–6. doi: 10.1097/RLU.0000000000000973
29. Jeevan DS, Saleh M, LaBagnara M, Neil JA, Hillard VH. Malignant Carotid Body Tumor Presenting With Myelopathy: Case Report. J Neurosurg Spine (2016) 24(4):660–3. doi: 10.3171/2015.8.SPINE13483
30. Belgioia L, Pupillo F, Bacigalupo A, Corvo R. Long-Term Survival in a Patient With Head and Neck Paraganglioma Treated With Tailored Modalities for 20 Years: A Case Report. Tumori (2016) 102(Suppl. 2):S9–11. doi: 10.5301/tj.5000443
31. Canu L, Pradella S, Rapizzi E, Fucci R, Valeri A, Briganti V, et al. Sunitinib in the Therapy of Malignant Paragangliomas: Report on the Efficacy in a SDHB Mutation Carrier and Review of the Literature. Arch Endocrinol Metab (2017) 61(1):90–7. doi: 10.1590/2359-3997000000217
32. Patel S, Owen D, Schmidt CR, Kirschner LS, Phay J, Shirley L, et al. Favorable and Durable Response to Pazopanib in Metastatic Refractory Paraganglioma. J Oncol Pract (2017) 13(12):840–2. doi: 10.1200/JOP.2017.024984
33. Ferrara AM, Lombardi G, Pambuku A, Meringolo D, Bertorelle R, Nardin M, et al. Temozolomide Treatment of a Malignant Pheochromocytoma and an Unresectable MAX-related Paraganglioma. Anticancer Drugs (2018) 29(1):102–5. doi: 10.1097/CAD.0000000000000570
34. Ong RKS, Flores SK, Reddick RL, Dahia PLM, Shawa H. A Unique Case of Metastatic, Functional, Hereditary Paraganglioma Associated With an SDHC Germline Mutation. J Clin Endocrinol Metab (2018) 103(8):2802–6. doi: 10.1210/jc.2017-01302
35. Stigliano A, Lardo P, Cerquetti L, Aschelter AM, Matarazzo I, Capriotti G, et al. Treatment Responses to Antiangiogenetic Therapy and Chemotherapy in Nonsecreting Paraganglioma (PGL4) of Urinary Bladder With SDHB Mutation: A Case Report. Med (Baltimore) (2018) 97(30):e10904. doi: 10.1097/MD.0000000000010904
36. Tena I, Gupta G, Tajahuerce M, Benavent M, Cifrian M, Falcon A, et al. Successful Second-Line Metronomic Temozolomide in Metastatic Paraganglioma: Case Reports and Review of the Literature. Clin Med Insights Oncol (2018) 12:1179554918763367. doi: 10.1177/1179554918763367
37. Irwin T, Konnick EQ, Tretiakova MS. Malignant Intrarenal/Renal Pelvis Paraganglioma With Co-Occurring SDHB and ATRX Mutations. Endocr Pathol (2019) 30(4):270–5. doi: 10.1007/s12022-019-09594-1
38. Tong A, Li M, Cui Y, Ma X, Wang H, Li Y. Temozolomide Is a Potential Therapeutic Tool for Patients With Metastatic Pheochromocytoma/Paraganglioma-Case Report and Review of the Literature. Front Endocrinol (Lausanne) (2020) 11:61. doi: 10.3389/fendo.2020.00061
39. Ayala-Ramirez M, Habra MA, Busaidy N, Cote G, Rich T, Waguespack S, et al. A Catecholamine Crisis on Mount Kilimanjaro: A Hypoxia Effect? J Travel Med (2010) 17(6):424–6. doi: 10.1111/j.1708-8305.2010.00463.x
40. Nozieres C, Walter T, Joly MO, Giraud S, Scoazec JY, Borson-Chazot F, et al. A SDHB Malignant Paraganglioma With Dramatic Response to Temozolomide-Capecitabine. Eur J Endocrinol (2012) 166(6):1107–11. doi: 10.1530/EJE-11-1098
41. Narechania S, Bath A, Ghassemi L, Lokhande C, Haddad A, Yousuf AM, et al. Paraganglioma Presenting as Postpartum Fever of Unknown Origin. Case Rep Endocrinol (2015) 2015:864719. doi: 10.1155/2015/864719
42. Makis W, McCann K, McEwan AJ. The Challenges of Treating Paraganglioma Patients With (177)Lu-DOTATATE PRRT: Catecholamine Crises, Tumor Lysis Syndrome and the Need for Modification of Treatment Protocols. Nucl Med Mol Imaging (2015) 49(3):223–30. doi: 10.1007/s13139-015-0332-6
43. Bizzarri N, De Cian F, Di Domenico S, Centurioni MG, Mammoliti S, Ghirardi V, et al. Peritoneal Carcinomatosis From Ovarian Paraganglioma: Report of a Rare Case and Systematic Review of the Literature. J Obstet Gynaecol Res (2018) 44(9):1682–92. doi: 10.1111/jog.13713
44. van Hulsteijn LT, Corssmit EP, Coremans IE, Smit JW, Jansen JC, Dekkers OM. Regression and Local Control Rates After Radiotherapy for Jugulotympanic Paragangliomas: Systematic Review and Meta-Analysis. Radiother Oncol (2013) 106(2):161–8. doi: 10.1016/j.radonc.2012.11.002
45. Fatima N, Pollom E, Soltys S, Chang SD, Meola A. Stereotactic Radiosurgery for Head and Neck Paragangliomas: A Systematic Review and Meta-Analysis. Neurosurg Rev (2020) 44:741–52. doi: 10.1007/s10143-020-01292-5
46. Kohlenberg J, Welch B, Hamidi O, Callstrom M, Morris J, Sprung J, et al. Efficacy and Safety of Ablative Therapy in the Treatment of Patients With Metastatic Pheochromocytoma and Paraganglioma. Cancers (Basel) (2019) 11(2):195. doi: 10.3390/cancers11020195
47. van Hulsteijn LT, Niemeijer ND, Dekkers OM, Corssmit EP. (131)I-MIBG Therapy for Malignant Paraganglioma and Phaeochromocytoma: Systematic Review and Meta-Analysis. Clin Endocrinol (Oxf) (2014) 80(4):487–501. doi: 10.1111/cen.12341
48. Satapathy S, Mittal BR, Bhansali A. ‘Peptide Receptor Radionuclide Therapy in the Management of Advanced Pheochromocytoma and Paraganglioma: A Systematic Review and Meta-Analysis’. Clin Endocrinol (Oxf) (2019) 91(6):718–27. doi: 10.1111/cen.14106
49. Niemeijer ND, Alblas G, van Hulsteijn LT, Dekkers OM, Corssmit EP. Chemotherapy With Cyclophosphamide, Vincristine and Dacarbazine for Malignant Paraganglioma and Pheochromocytoma: Systematic Review and Meta-Analysis. Clin Endocrinol (Oxf) (2014) 81(5):642–51. doi: 10.1111/cen.12542
50. Hadoux J, Favier J, Scoazec JY, Leboulleux S, Al Ghuzlan A, Caramella C, et al. SDHB Mutations are Associated With Response to Temozolomide in Patients With Metastatic Pheochromocytoma or Paraganglioma. Int J Cancer (2014) 135(11):2711–20. doi: 10.1002/ijc.28913
51. Fanciulli G, Di Molfetta S, Dotto A, Florio T, Feola T, Rubino M, et al. Emerging Therapies in Pheochromocytoma and Paraganglioma: Immune Checkpoint Inhibitors in the Starting Blocks. J Clin Med (2020) 10(1):88. doi: 10.3390/jcm10010088
52. Naing A, Meric-Bernstam F, Stephen B, Karp DD, Hajjar J, Rodon Ahnert J, et al. Phase 2 Study of Pembrolizumab in Patients With Advanced Rare Cancers. J Immunother Cancer (2020) 8(1):e000347. doi: 10.1136/jitc-2019-000347
53. Leijon H, Remes S, Hagstrom J, Louhimo J, Maenpaa H, Schalin-Jantti C, et al. Variable Somatostatin Receptor Subtype Expression in 151 Primary Pheochromocytomas and Paragangliomas. Hum Pathol (2019) 86:66–75. doi: 10.1016/j.humpath.2018.11.020
54. Volante M, Brizzi MP, Faggiano A, La Rosa S, Rapa I, Ferrero A, et al. Somatostatin Receptor Type 2A Immunohistochemistry in Neuroendocrine Tumors: A Proposal of Scoring System Correlated With Somatostatin Receptor Scintigraphy. Mod Pathol (2007) 20(11):1172–82. doi: 10.1038/modpathol.3800954
55. Han S, Suh CH, Woo S, Kim YJ, Lee JJ. Performance of (68)Ga-DOTA-Conjugated Somatostatin Receptor-Targeting Peptide PET in Detection of Pheochromocytoma and Paraganglioma: A Systematic Review and Metaanalysis. J Nucl Med (2019) 60(3):369–76. doi: 10.2967/jnumed.118.211706
56. van Hulsteijn LT, van Duinen N, Verbist BM, Jansen JC, van der Klaauw AA, Smit JW, et al. Effects of Octreotide Therapy in Progressive Head and Neck Paragangliomas: Case Series. Head Neck (2013) 35(12):E391–6. doi: 10.1002/hed.23348
57. Cassol CA, Winer D, Liu W, Guo M, Ezzat S, Asa SL. Tyrosine Kinase Receptors as Molecular Targets in Pheochromocytomas and Paragangliomas. Mod Pathol (2014) 27(8):1050–62. doi: 10.1038/modpathol.2013.233
58. Gimenez-Roqueplo AP, Favier J, Rustin P, Mourad JJ, Plouin PF, Corvol P, et al. The R22X Mutation of the SDHD Gene in Hereditary Paraganglioma Abolishes the Enzymatic Activity of Complex II in the Mitochondrial Respiratory Chain and Activates the Hypoxia Pathway. Am J Hum Genet (2001) 69(6):1186–97. doi: 10.1086/324413
59. Pollard PJ, Briere JJ, Alam NA, Barwell J, Barclay E, Wortham NC, et al. Accumulation of Krebs Cycle Intermediates and Over-Expression of HIF1alpha in Tumours Which Result From Germline FH and SDH Mutations. Hum Mol Genet (2005) 14(15):2231–9. doi: 10.1093/hmg/ddi227
60. Facchin C, Perez-Liva M, Garofalakis A, Viel T, Certain A, Balvay D, et al. Concurrent Imaging of Vascularization and Metabolism in a Mouse Model of Paraganglioma Under Anti-Angiogenic Treatment. Theranostics (2020) 10(8):3518–32. doi: 10.7150/thno.40687
61. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and Guidelines for the Interpretation of Sequence Variants: A Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med (2015) 17(5):405–24. doi: 10.1038/gim.2015.30
Keywords: sunitinib, paraganglioma, Familial Paraganglioma Syndrome, succinate dehydrogenase, SDHD
Citation: Sesti F, Feola T, Puliani G, Centello R, Di Vito V, Bagni O, Lenzi A, Isidori AM, Cantisani V, Faggiano A and Giannetta E (2021) Sunitinib Treatment for Advanced Paraganglioma: Case Report of a Novel SDHD Gene Mutation Variant and Systematic Review of the Literature. Front. Oncol. 11:677983. doi: 10.3389/fonc.2021.677983
Received: 08 March 2021; Accepted: 25 May 2021;
Published: 17 June 2021.
Edited by:
Massimo Fantini, Precision Biologics, Inc., United StatesReviewed by:
Francoise Borson-Chazot, Université de Lyon, FranceCopyright © 2021 Sesti, Feola, Puliani, Centello, Di Vito, Bagni, Lenzi, Isidori, Cantisani, Faggiano and Giannetta. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Elisa Giannetta, ZWxpc2EuZ2lhbm5ldHRhQHVuaXJvbWExLml0
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.