 Xun Wu1†
Xun Wu1† J. Ernesto Fajardo-Despaigne1†
J. Ernesto Fajardo-Despaigne1† Christine Zhang1Vishala Neppalli2Versha Banerji3,4James B. Johnston3,4
Christine Zhang1Vishala Neppalli2Versha Banerji3,4James B. Johnston3,4 Spencer B. Gibson1,3,4
Spencer B. Gibson1,3,4 Aaron J. Marshall1,3,4,5*
Aaron J. Marshall1,3,4,5*- 1Department of Immunology, Max Rady College of Medicine, Rady Faculty of Health Sciences, University of Manitoba, Winnipeg, MB, Canada
- 2Hematopathology Laboratory, Shared Health Manitoba, Winnipeg, MB, Canada
- 3Research Institute in Oncology and Hematology, CancerCare Manitoba, Winnipeg, MB, Canada
- 4Department of Internal Medicine, Max Rady College of Medicine, Rady Faculty of Health Sciences, University of Manitoba, Winnipeg, MB, Canada
- 5Department of Biochemistry and Medical Genetics, Max Rady College of Medicine, Rady Faculty of Health Sciences, University of Manitoba, Winnipeg, MB, Canada
Follicular helper T cells (TFH) have specialized properties in promoting normal B cell activation but their role in chronic lymphocytic leukemia (CLL) is unknown. We find that TFH cells are elevated in CLL patients and are phenotypically abnormal, expressing higher levels of PD-1, TIGIT, CD40L, IFNγ and IL-21, and exhibiting abnormal composition of TFH1, TFH2 and TFH17 subsets. Frequencies of CD4-positive T cells expressing TFH1 markers and IL-21 were positively correlated with patient lymphocyte counts and RAI stage, suggesting that accumulation of abnormal TFH cells is concomitant with expansion of the leukemic B cell clone. Treatment with ibrutinib led to normalization of TFH frequencies and phenotype. TFH cells identified in CLL bone marrow display elevated expression of several functional markers compared to blood TFH cells. CLL T cell-B cell co-culture experiments revealed a correlation of patient TFH frequencies with functional ability of their CD4-positive T cells to promote CLL proliferation. Conversely, CLL cells can preferentially activate the TFH cell subset in co-culture. Together our results indicate that CLL development is associated with expansion of abnormal TFH populations that produce elevated levels of cytokines and costimulatory molecules which may help support CLL proliferation.
Highlights
1. Follicular helper T cells with altered cytokine and receptor profiles are progressively expanded in CLL and normalized upon treatment.
2. CLL B cells can preferentially activate follicular helper T cells, promoting CD4+ T cell-driven CLL B cell proliferation in vitro.
Introduction
Monoclonal B cell lymphocytosis (MBL) and chronic lymphocytic leukemia (CLL) are lymphoproliferative disorders characterized by the presence of abnormal numbers of CD5+ monoclonal B lymphocytes in the blood or tissues (1). MBL is the precursor to CLL, with approximately 1% of high-count cases requiring therapy each year following progressing to CLL (2). The clinical course of CLL patients is heterogeneous and prognostic markers have been developed to predict which patients may have aggressive disease (3). Independent prognostic markers for CLL include Rai stage, age, IGVH mutational status, β2-microglobulin level and TP53 loss-of-function (1, 3), which are used to calculate the International Prognostic Index (4). Within tissue microenvironments, CLL B cells come into close contact with other cells such as stromal cells, which provide signals that promote survival and drug resistance (5). The lymphoid tissue environment also promotes activation of B cell antigen receptor (BCR) signaling pathways and CLL proliferation (6).
Although CLL has historically been characterized as a disease of enhanced cell survival, active signaling and proliferation within lymphoid tissue is now appreciated to be an important factor determining disease prognosis (7, 8). Within bone marrow, spleen and lymph nodes, CLL proliferation occurs in “proliferation centers” where CLL cells interact directly with T cells, myeloid cells and stromal cells and display markers of active signaling (9). CLL patients showing highly active proliferation centers exhibit aggressive disease and poor prognosis (10). Inhibitors of Bruton’s Tyrosine Kinase (BTK) have proven to be efficacious in treating CLL via interrupting BCR signaling as well as the supportive cell:cell interactions within the lymphoid tissue microenvironment (11). BTK inhibitor treatment of CLL patients frequently results in a transient increase in circulating malignant cells after treatment, concomitant with dramatic loss of leukemic cells from lymph nodes (12), suggesting that these treatments trigger a rapid dissolution of proliferation centers.
Autologous human T cells were found to be required for CLL proliferation in a mouse xenograft model (13), suggesting they play an essential role distinct from stromal cells. Normal B cell follicles, as well as germinal centers containing activated B cells, are known to depend on a specialized subset of CD4+ T cells called follicular helper T cells (TFH). These CD4+ T cells express the chemokine receptor CXCR5 (14, 15), that allows them to migrate toward its ligand CXCL13, the B cell follicle chemokine made by follicular dendritic cells (16, 17). Normal TFH cells produce a unique spectrum of cytokines and costimulatory molecules and provide essential co-stimulatory signals to sustain B cell survival and proliferation within germinal centers (18). Functionally distinct TFH subpopulations have been identified based on their differential expression of CXCR3 and CCR6 (19, 20). Abnormalities in TFH populations have been observed in a number autoimmune diseases, where considerable evidence implicates them as drivers of pathological B cell responses (19). While substantial evidence indicates that T cell populations are altered in CLL (21–23), a full assessment of TFH populations across the spectrum of MBL and CLL has not previously been reported.
Here we report a comprehensive assessment of TFH populations and associations of their frequency and phenotypes with CLL biomarkers, clinical stage and immune dysfunction. We find evidence that CLL TFH exhibit an increased functional capacity to produce co-stimulatory receptors and cytokines linked to CLL survival and proliferation and skewing to a TFH1-like phenotype in advanced stage patients. Finally, we find that CLL cells can preferentially activate TFH cells in vitro and observe an association of TFH frequencies with the ability of activated CD4+ T cells to trigger CLL proliferation. These results define alterations in TFH phenotype and function in CLL and indicate a potential role for these cells as part of the dysfunctional immune microenvironment in this disease.
Methods
Patient Samples and Clinical Biomarkers
Peripheral blood and bone marrow aspirates were obtained from CLL patients attending the CLL clinic at CancerCare Manitoba. Informed consent of patients and control subjects was obtained under a protocol approved the Research Ethics Board at the University of Manitoba. Rai staging was determined using standard clinical criteria. Clinical biomarkers including CD38, IgM, IgG, IgA and lymphocyte count were determined using standard protocols and obtained from the Manitoba Tumor Bank and CAISIS database. Mononuclear cells were isolated using Ficoll-Paque density gradient and cryopreserved in 10% DMSO medium prior to analysis. Lymph node biopsies were formalin-fixed and paraffin-embedded prior to sectioning.
T Cell Phenotyping
For assessment of TFH subpopulations, peripheral blood or bone marrow mononuclear cells were stained for the markers CD3, CD4, CD14, CD19, CXCR5, PD-1, ICOS, CD45RA, CCR7, CXCR3, CCR6, TIGIT (antibody details in Supplementary Table 1) and LIVE/DEAD™ Fixable Aqua viability dye (Invitrogen™) at room temperature for 30 minutes. Stained cells were run on a Beckman Coulter Cytoflex instrument. TFH populations were quantified as percent of the singlet, CD4+, Dump (CD19/CD14/LiveDead) negative population as illustrated in Supplementary Figure 1.
Production of Costimulatory Molecules and Cytokines
For assessment of cytokine production, cryopreserved peripheral blood mononuclear cells (PBMC) were cultured overnight and then stimulated for 6 hours with 50ng/ml PMA and 1μg/ml ionomycin (Selleck Chemicals), with 10μg/ml Brefeldin A (Selleck) added for the last 4 hours. Cells were then cell surface stained for CD3, CD14, CD19, CXCR5, CCR6, CXCR3 as described above, fixed and permeabilized using eBioscience™ Fixation/Permeabilization buffer and stained intracellularly for CD4, IFNγ, IL-21 and CD40L at room temperature for 45 minutes. Intracellular CD4 staining was utilized to improve discrimination of CD4+ cells, as cell surface CD4 is downmodulated upon treatment with PMA+ionomycin. Antibody details are provided in Supplementary Table 1.
Immunofluorescence Microscopy
Lymph node tissue sections were deparaffinized, rehydrated and boiled for 20 minutes in Target Retrieval Solution, Citrate pH 6.1 (Agilent). After washing, serial tissue sections were blocked with 1% BSA and 2% FBS in PBS followed by staining with unconjugated primary antibodies at 4°C overnight. After washing, sections were incubated with secondary antibodies for 4 hours at room temperature with shaking, washed and then stained with directly conjugated primary antibodies at 4°C overnight. Following a final wash, sections were air dried and mounted with ProLong™ Gold antifade reagent (Invitrogen) and kept at -20°C until analysis. Antibody details are provided in Supplementary Table 2. Images were captured with a CSU-X1M5000 spinning disc confocal microscope (Carl Zeiss) equipped with 405/488/561/635nm lasers.
CD4 T Cell: CLL B Cell Co-Culture Assay
Autologous CD4 T cells and CLL B cells were purified from PBMC using EasySep™ Human CD4+ T Cell Isolation Kit and EasySep™ Human B Cell Enrichment Kit II without CD43 Depletion (both STEMCELL Technologies), respectively. Purified CD4 T cells were suspended at 1x10^6 cells/mL in RPMI 1640 media (GE Healthcare) with 10% FBS (Life Technologies). T cells were cultured overnight in 24-well plates with/without addition of ImmunoCult™ Human CD3/CD28/CD2 T Cell Activator (STEMCELL Technologies) at 25 μL cocktail/mL of cells. B cells were cultured overnight in U-bottom 96-well plates at 1x10^6 cells/well, in the presence of sCD40L+IL-4 (both 50ng/mL; R&D Systems). After 14-16 hours incubation, cells were washed to remove stimuli. B cells were stained with carboxyfluorescein succinimidyl ester (CFSE) (Sigma-Aldrich) at 0.3 μM in PBS for 5min at room temperature, then washed with culture media. T cells (with or without pre-activation) were co-cultured together with CFSE-labelled autologous CLL-B cells in U-bottom 96-well plates, using 2x10^5 T and 1x10^6 B cells per well. T and B cells alone were included as controls. Starting at day 2 of co-culture, 100 μL of culture medium was gently taken out and fresh medium added to each well daily. At indicated times, wells were harvested and flow cytometry analyses were carried out using the panel detailed in Supplementary Table 1. Briefly, cells were stained for CD4, CD19, CXCR5, CXCR3, CCR6, CCR7, CD69, CD25, CD134/OX40, PD-1, CD38 and LIVE/DEAD™ Fixable Aqua viability dye (Invitrogen™). Following wash, cells were fixed and permeabilized as above and stained for Ki-67 at room temperature for 45 min.
Data Analysis and Statistics
Flow data were analyzed by FlowJo® V10 (FlowJo, LLC). Statistical analysis was performed with GraphPad Prism (GraphPad Software Inc). Confocal images were processed by ImageJ (V1.47). Box and whisker plots illustrate the median, interquartile range and 10-90% percentile values. Statistical tests used are indicated in figure legends and differences were considered to be statistically significant at values of *(p<0.05), **(p<0.01), ***(p<0.001) and ****(p<0.0001).
Results
Follicular Helper T Cells Are Expanded and Phenotypically Distinct in CLL Patients
Peripheral blood mononuclear cells collected from CLL patients, MBL patients or age-matched controls were analyzed by multicolor flow cytometry to assess T follicular helper (TFH) cell populations. Gating on CD4+CXCR5+CD19-CD14- live lymphocytes (Figure S1) revealed a significant elevation in both TFH frequencies and overall TFH numbers in CLL but not MBL patients (Figure 1A). CLL TFH express higher levels of TFH-associated activation markers PD-1 and ICOS than corresponding non-TFH CD4 T cells and more PD-1 than control TFH (Figure 1B). Compared to control TFH, CLL TFH populations contain a higher proportion of CD45RA-/CCR7- effector memory cells and fewer CD45RA-CCR7+ central memory cells (Figure S2). Within the TFH population we further examined TFH1, TFH2 and TFH17 subset composition based on expression of chemokine receptors CXCR3 and CCR6 (24) and found that CLL patients demonstrate significant skewing towards the CXCR3+CCR6- TFH1 population (Figure 1C). The increased TFH1 skewing in CLL patients was accompanied by significantly reduced frequencies of the TFH2 population, whereas no significant change in TFH17 cells was observed (Figure 1C). Together these results indicate that TFH cells are expanded and phenotypically altered in CLL.
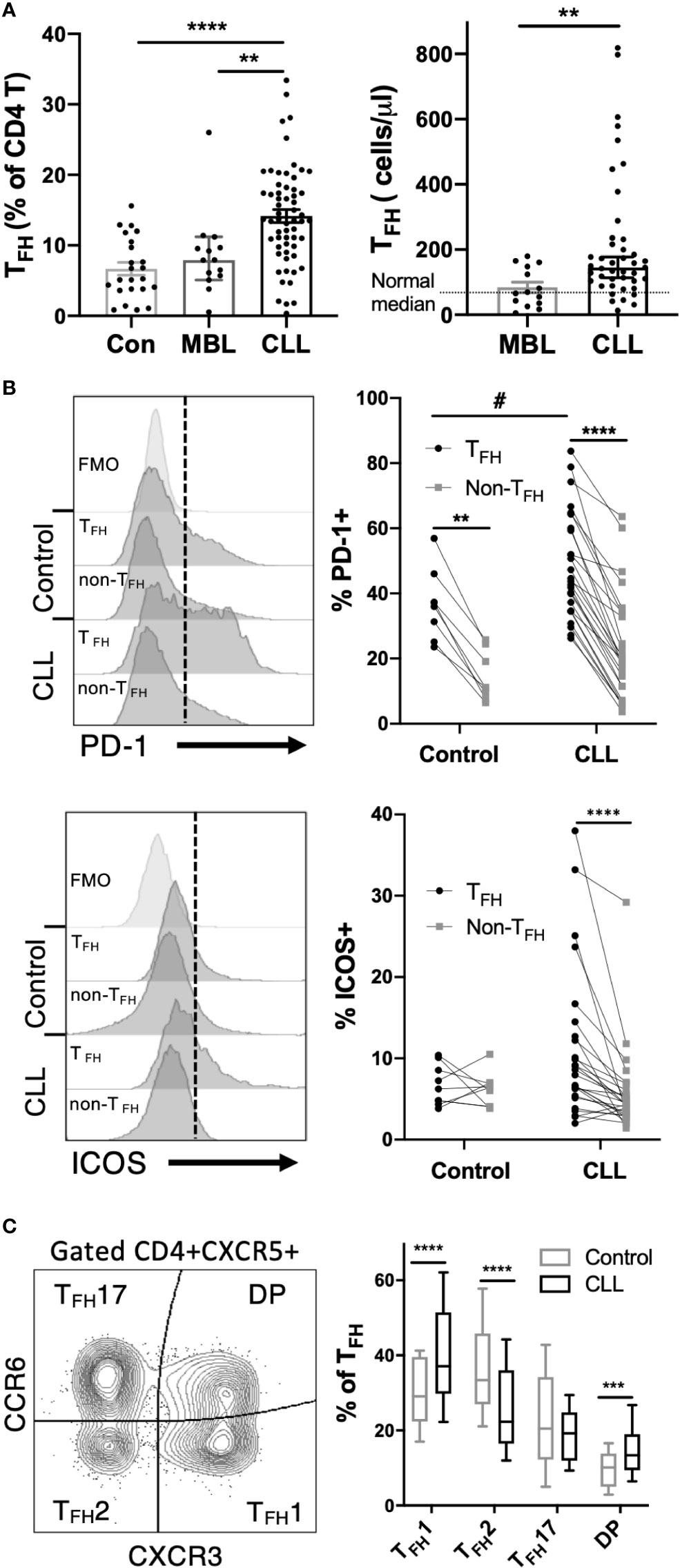
Figure 1 TFH cells are expanded and phenotypically altered in CLL patients. Peripheral blood mononuclear cells (PBMC) were isolated from CLL and MBL patients or control donors and analyzed by flow cytometry. For complete gating strategy see Supplementary Data. (A) TFH frequency and absolute numbers are significantly increased in CLL patients. N=23 for controls, 14 for MBL and 61 for CLL. All graphs illustrate the individual patient values, median, and 95% confidence interval. Mann-Whitney U test, **(p<0.01), ****(p<0.0001). (B) Expression of PD-1 or ICOS on TFH and non-TFH CD4+ T cell populations. TFH were gated as CD4+CXCR5+CD45RA- cells. Individual patients or control values are connected by lines. Histograms labeled FMO show fluorescence minus one staining controls. *denotes significance by Wilcoxon paired t test, ****(p<0.0001). #denotes significance by Mann-Whitney U test, #(p<0.05). (C) Proportions of TFH1, TFH2 and TFH17 sub-populations as determined by CCR6 and CXCR3 expression. Mann-Whitney U test, ***(p<0.001), ****(p<0.0001).
Association of Follicular Helper T Cells With Disease Burden
Within the spectrum of CLL patients in the cohort analyzed, we examined whether TFH frequencies were associated with clinical parameters or established biomarkers of disease. Strikingly, we found that both frequency of TFH among CD4+ T cells and TFH1 skewing were positively correlated with blood lymphocyte counts (Figure 2A). TFH expression of PD-1 or ICOS were also positively correlated with lymphocyte count (Figure S3). Frequencies of TFH and skewing to TFH1 were significantly elevated in high risk CLL (Rai 3-4) relative to low risk CLL (Rai 0) and CD38+ CLL patients showed more TFH1 skewing than CD38- patients (Figure 2B). The latter is consistent with a report that CD38 expression is driven by the TH1 cytokine IFNγ25. Interestingly, TFH frequency was inversely correlated with plasma IgM and IgG levels (Figure 2C). Together these results suggest that TFH accumulation and selective skewing to a TFH1 phenotype occurs in parallel with expansion of the leukemic B cell clone and decline in normal B cell function.
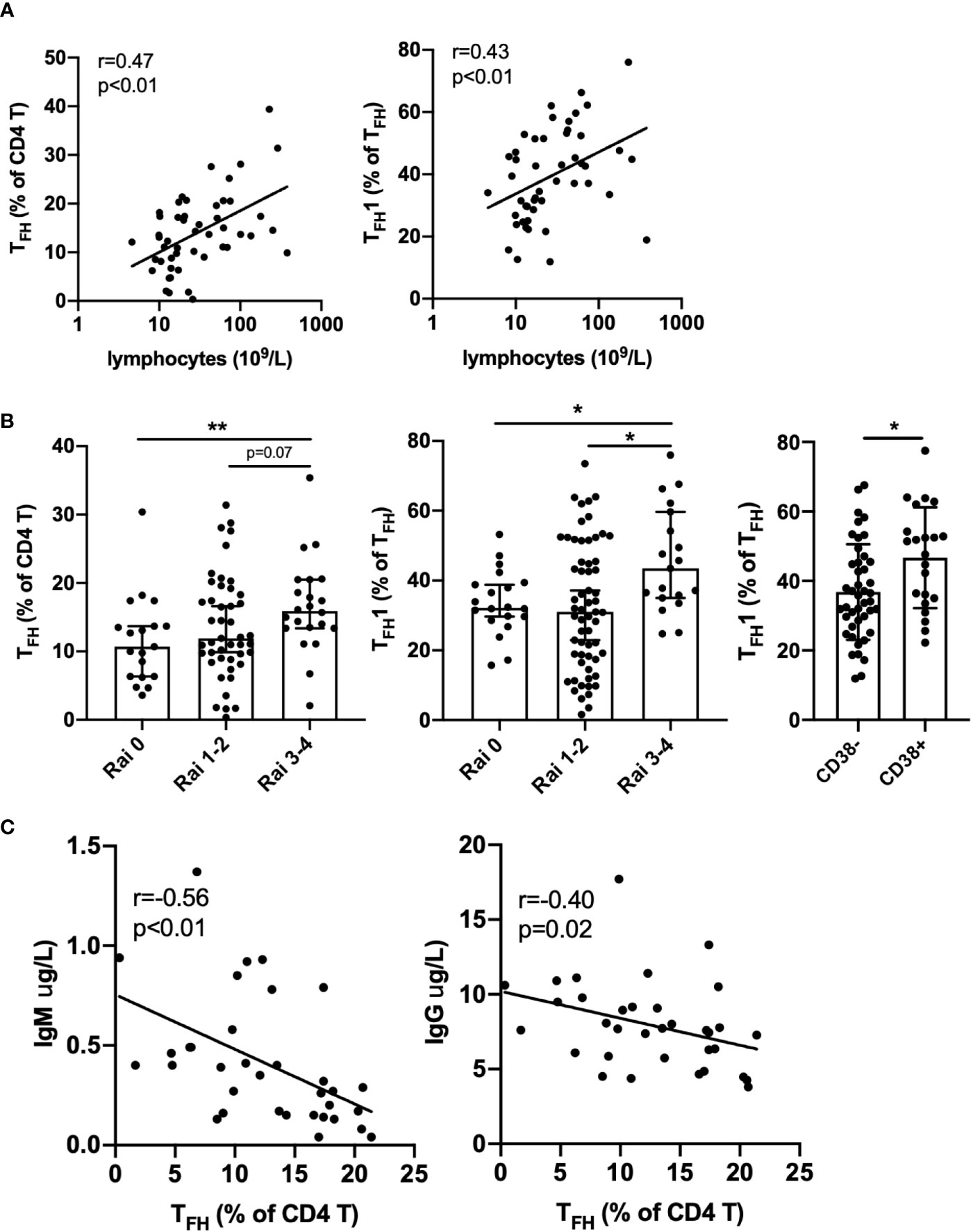
Figure 2 Association of TFH with disease burden and immune deficiency in CLL patients. (A) TFH frequency and TFH1 skewing are positively correlated with lymphocyte count. (B) TFH frequency or TFH1 skewing are elevated in advanced Rai stage patients and TFH1 frequency is increased in CD38 positive versus CD38 negative patients. All graphs illustrate the individual patient values, median, and 95% confidence interval. (C) TFH frequency is negatively correlated with plasma IgM and IgG levels. Significance was determined by Spearman correlation (A, C) or Mann-Whitney test, *(p<0.05), **(p<0.01) (B).
Impact of Ibrutinib Treatment on TFH Populations
To determine how CLL treatment impacts TFH populations, we examined patients during the first year of ibrutinib treatment. Ibrutinib treatment led to a gradual decline in the frequency of TFH cells over time, in parallel with a decline in total lymphocyte count (Figure 3A). In one patient who exhibited prolonged lymphocytosis after treatment, there was a concurrent transient increase in the TFH population prior to normalization. Notably, TFH composition changed post-ibrutinib treatment, with patients exhibiting a gradual re-balancing of TFH1, TFH2, and TFH17 subsets (Figure 3A). Significant reductions in both TFH frequencies and TFH1 skewing were observed after 40 weeks of treatment (Figure 3B). This was accompanied by increased frequencies of CXCR3-CCR6- TFH2-like cells, while TFH17 frequencies were not altered (Figure 3C). These results suggest that ibrutinib treatment can normalize TFH subsets concomitant with reduction in disease burden.
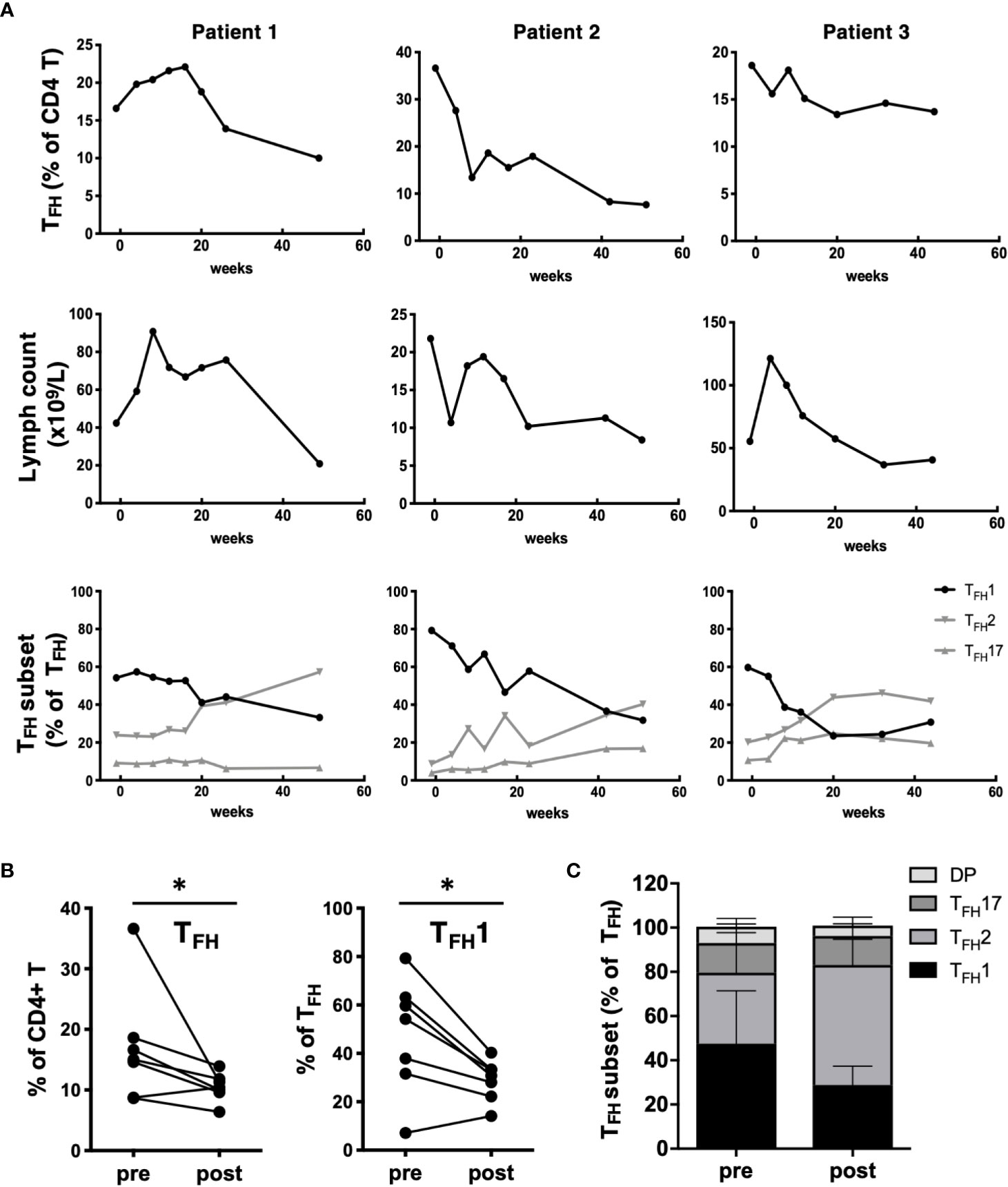
Figure 3 Impact of ibrutinib treatment on TFH populations. (A) PBMC samples were collected from CLL patients prior to starting treatment with ibrutinib and at multiple time points over a one-year follow-up. Changes in TFH frequencies, lymphocyte counts or TFH subset frequencies for 3 representative patients over time after ibrutinib treatment for three representative patients are shown. (B) Graphs showing pre/post treatment (>40 wk) frequencies of TFH or TFH1 subsets, with individual patient data connected by a line (*p<0.05, Wilcoxon test). (C) Stacked bar graph summarizing the average composition of four TFH subpopulations pre and post ibrutinib treatment (N=7).
CLL TFH Cells Produce High Levels of CD40L, TIGIT, IFNγ and IL-21
We further examined expression of costimulatory molecules and cytokines by CLL TFH cells. We found that TFH express higher levels of CD40L than non-TFH in both CLL patients and controls, however CLL TFH exhibit a strikingly elevated expression of CD40L (approximately 5-fold on average) relative to control TFH (Figure 4A). In addition, we found that CLL TFH express high levels of TIGIT (Figure 4A), an inhibitory immunoreceptor previously thought to function in T:B cell interactions (25, 26). We examined the ability of CLL TFH to produce the canonical Type 1 cytokine IFNγ and the TFH-associated cytokine IL-21 (Figure 4B). Remarkably, we observed a substantial increase in both the frequency IL-21 producing and IL-21/IFNγ double-producing cells in CLL patients, with the CLL TFH population containing significantly more IL-21 and double-producing cells than control TFH cells (Figure 4B). TFH1 cells produced significantly more IL-21 and IFNγ than other TFH subsets, but interestingly produced slightly less CD40L (Figure S4). We further assessed whether levels of CD40L, TIGIT or IL-21 expression by TFH are associated with disease burden or stage. Interestingly, while CD40L and TIGIT expression did not show strong associations, IL-21 expression by TFH was significantly associated with lymphocyte count and Rai stage (Figure 4C). These results indicate that CLL TFH cells produce abnormally high levels of costimulatory molecules and cytokines known to stimulate CLL survival and proliferation and the expression of IL-21 by these cells is associated with adverse biomarkers and disease burden.
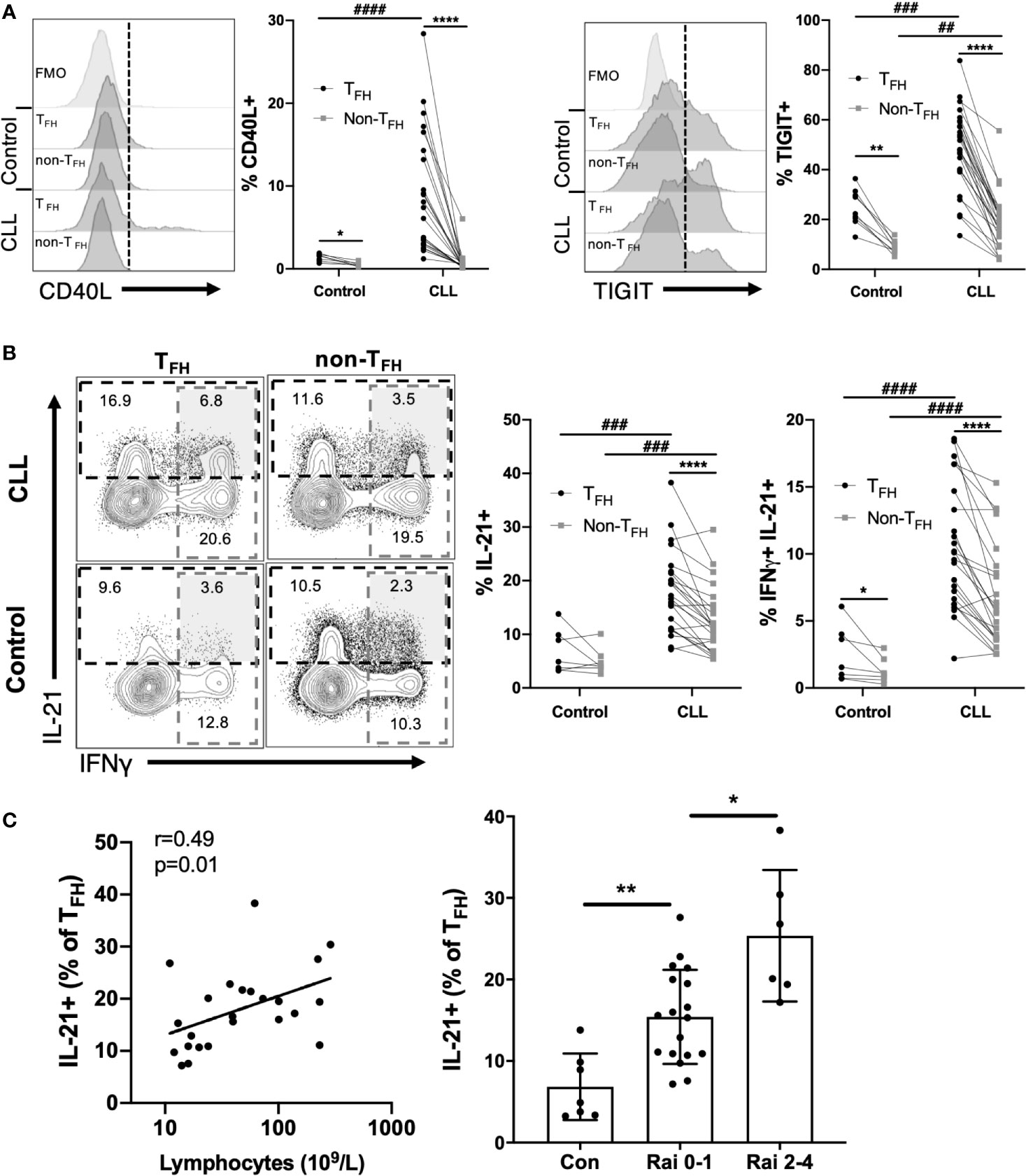
Figure 4 CLL TFH cells express abnormally high levels of CD40L, TIGIT, IFNγ and IL-21. (A) Expression of CD40L or TIGIT within TFH and non-TFH CD4+ T cell populations from controls and CLL patients. Left panels show representative staining histograms within the indicated cell populations. Histograms labeled FMO show fluorescence minus one staining controls. Right graphs show percent CD40L or TIGIT positivity among TFH or non-TFH populations, with individual subjects connected by lines. (B) Intracellular staining of IFNγ and IL-21 in TFH or non-TFH cell populations. Representative flow plots illustrate gating (left panels). Gated IL-21+ or IFNγ+/IL-21+ cell frequencies within the indicated groups (right panels). In (A, B) *denotes significance by Wilcoxon paired t test, ****(p<0.0001); #denotes significance by Mann-Whitney U test, ##(p<0.01), ###(p<0.001), ####(p<0.0001). (C) TFH cell expression of IL-21 is associated with lymphocyte count and Rai stage. Left panel, Spearman correlation; Right panel, Mann-Whitney U test, *(p<0.05), **(p<0.01).
Activated TFH1-Like Cells Are Present in CLL Lymphoid Tissues
CLL cells are present at varying levels in the bone marrow and lymph nodes, and signals present in these microenvironments are thought to drive CLL proliferation. To investigate whether circulating TFH cells may represent the counterpart of TFH populations present within lymphoid tissues, paired blood and bone marrow samples from CLL patients were analyzed. We found a significant correlation between blood and marrow TFH and TFH1 cell frequencies from the same patients (Figure 5A). Interestingly, TFH populations in bone marrow showed increased activation status relative to those in peripheral blood of the same patients, expressing significantly more IL-21, IFNγ and IL-21/IFNγ double-producing cells (Figure 5B). While levels of PD-1 was also elevated, CD40L and TIGIT were similar in bone marrow and peripheral blood TFH (Figure 5C). We further examined lymph node tissue sections from CLL patients using immunofluorescent staining and could identify T cells present within proliferation centers expressing the TFH1 markers CD3, CD4, CXCR5, CXCR3 and PD-1 (Figure 6). These results suggest that TFH cells are present in both circulation and within lymphoid tissues, with the latter showing similar skewing to TFH1 and a highly activated phenotype.
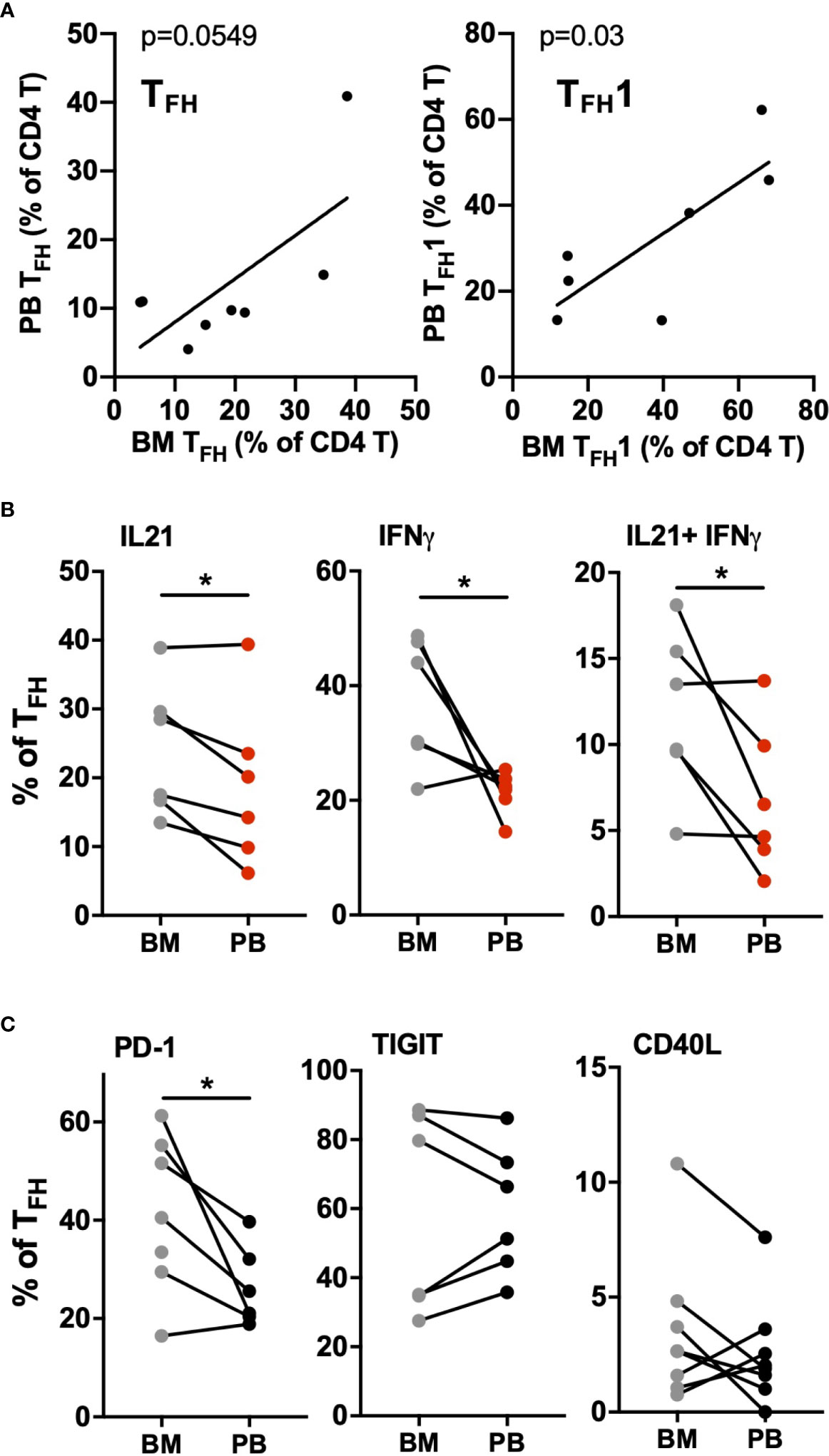
Figure 5 CLL bone marrow contains high levels of activated TFH1 cells. Mononuclear cells isolated from bone marrow aspirates (BM) and peripheral blood samples (PB) of the same patients were analyzed by flow cytometry. (A) Correlation between TFH (left) or TFH1 (right) frequencies in marrow versus matched blood samples (B) Comparison of cytokine production in marrow versus blood TFH populations. (C) Comparison of activation/costimulatory markers in marrow versus blood TFH populations. * denotes significance by Wilcoxon paired t test, *(p<0.05).
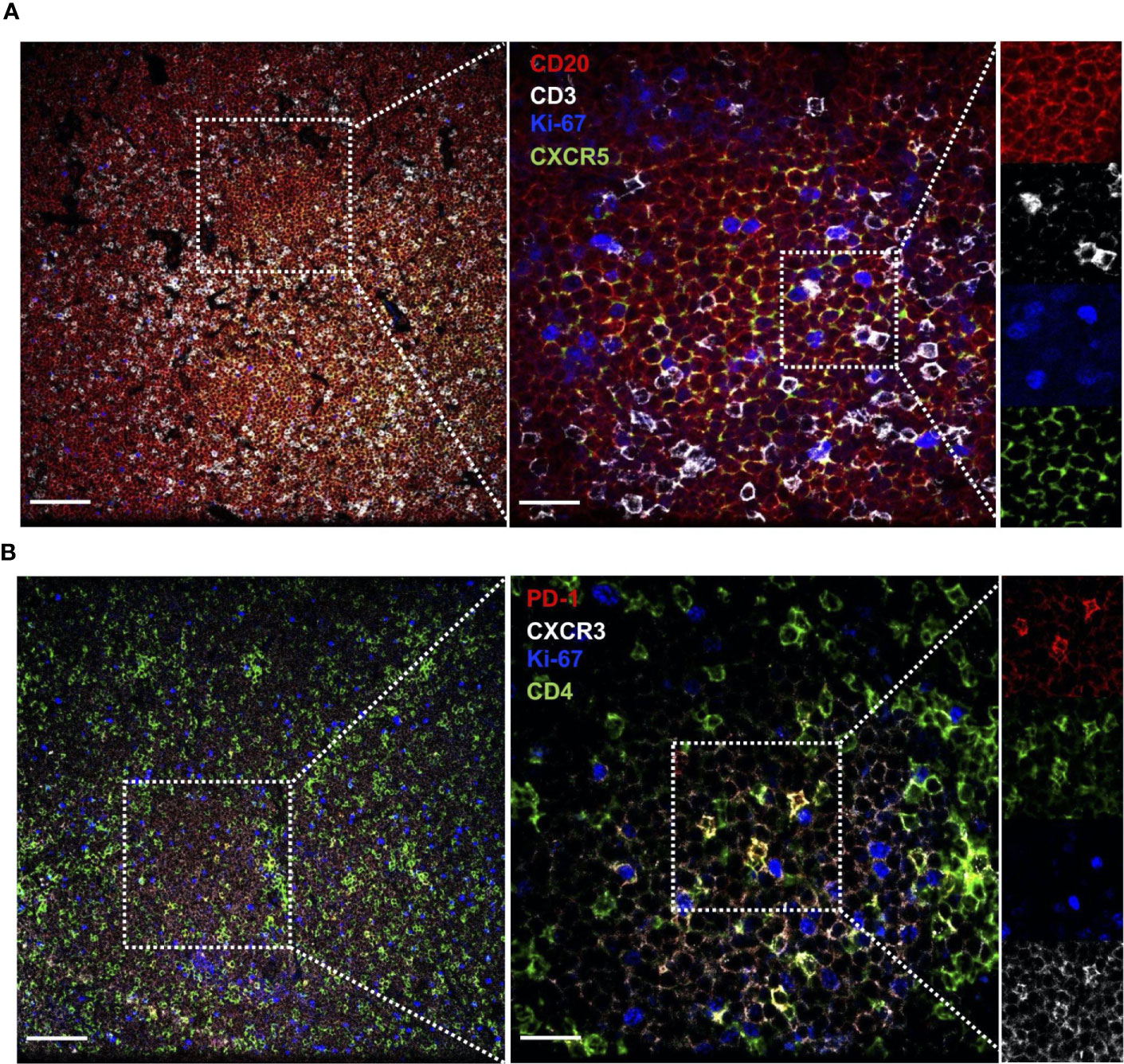
Figure 6 CLL lymph nodes contains cells expressing activated TFH1 markers. Lymph node sections from CLL patients were stained with the indicated labelled antibodies and imaged by confocal microscopy. (A) Section stained for CLL B cell marker CD20, T cell marker CD3, proliferation marker Ki67 and CXCR5 (expressed on CLL B cells and TFH). (B) Section stained for activation marker PD-1, T cell marker CD4, proliferation marker Ki67 and CXCR3 (expressed at low levels on CLL B cells and high levels on TFH1 cells). Several magnifications are shown (Left panel scale bar = 100µm, right panel scale bar = 25µm) to illustrate T cell presence within CLL B cell clusters containing proliferating cells, and close contact between CLL cells and T cells expressing CD3, CD4, CXCR5, PD-1 and CXCR3.
Autologous Activated CD4 T Cells Can Promote CLL B Cell Survival, Activation and Proliferation
We developed an in vitro system to study CLL B cell interaction with autologous CD4+ T cells. CD4 T cells were isolated from patient PBMC, pre-cultured overnight with a T cell-activation cocktail or medium alone, then washed and co-cultured with B cells isolated from the same patient that were labeled with cell division tracking dye CFSE. Resulting CLL-B cell division, and expression of activation markers by B and T cells, were assessed after 2 and 6 days of co-culture. At day 2, a significant increase in CLL B cell expression of CD69 and nuclear proliferation antigen Ki-67 was observed in the presence of activated CD4 T cells, compared to co-cultures with non-activated T cells or B cells alone (Figure 7A). Activated CD4 T:B cell co-culture also promoted an increase in CD25 activation marker expression among B cells and increased CLL B cell survival at this time point, whereas increased CD38 expression or cell division (as assessed by CFSE dilution) were not observed at day 2 (Figure S5A). After 6 days of co-culture, CLL-B cell division was observed (Figure 7B) and most divided cells also expressed Ki-67 (Figure S5B). At day 6, CLL B cells also exhibited significant upregulation of the activation marker CD25 (Figure 7B) as well as CD38 and CD69 (Figure S5C) when co-cultured with activated CD4 T cells. Notably, there was a positive correlation between patient TFH frequency and the frequency of divided, Ki-67+ and CD25+ CLL B cells observed in co-cultures (Figure 7C), consistent with a potential role for TFH cells in driving CLL-B cell activation and proliferation.
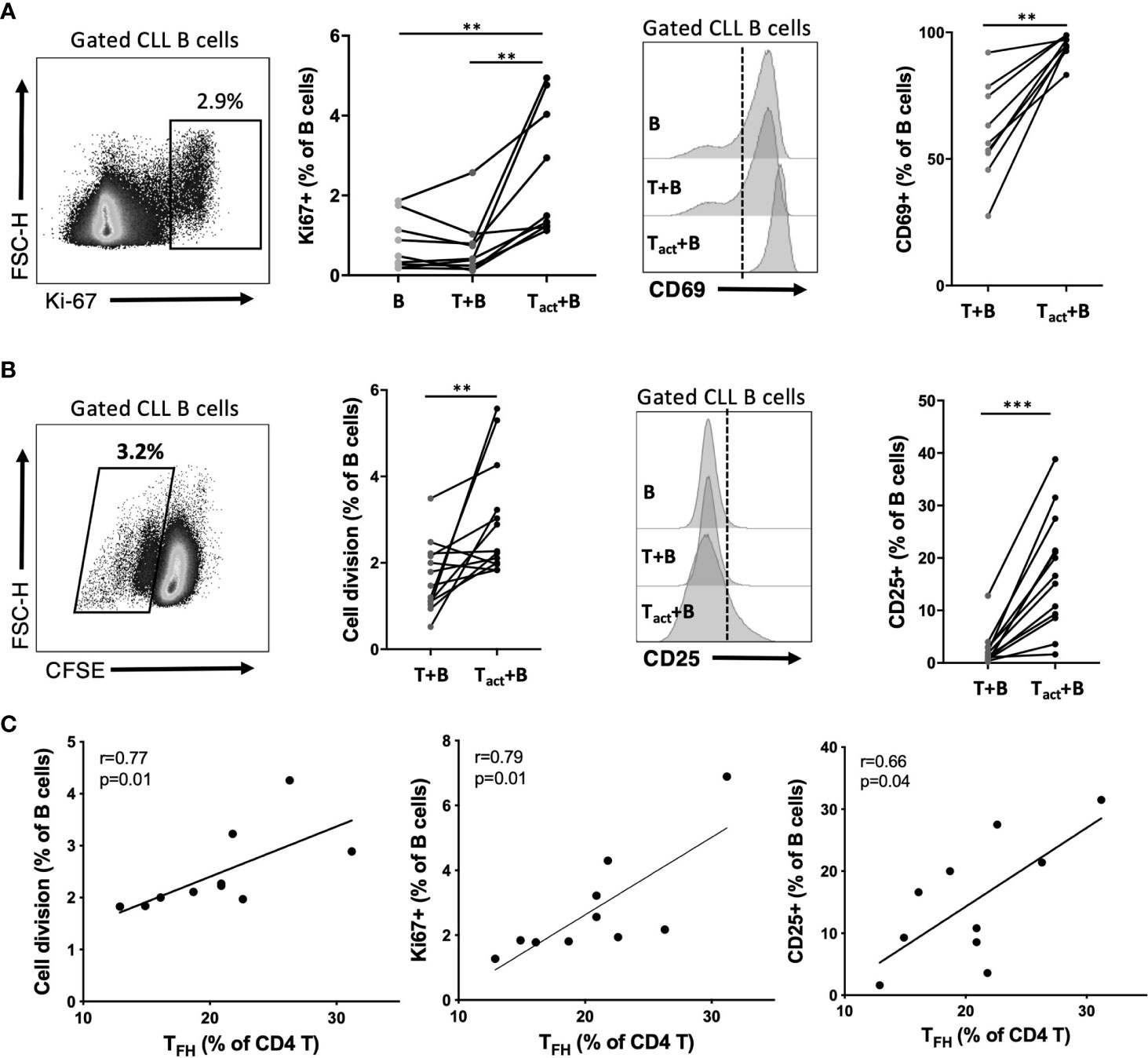
Figure 7 Impact of activated autologous CD4+ T cells on CLL activation and proliferation. Purified CLL cells were co-cultured with purified autologous CD4+ T cells from the same PBMC sample. Prior to co-culture, CLL cells were labeled with CFSE and T cells were briefly exposed to an activation cocktail (Tact) or kept in medium alone (T). After 2 or 6 days of co-culture, cells were analyzed by flow cytometry to determine cell division and expression of activation markers. B cells were gated as live CD19+CD4- cells. (A) Expression of nuclear proliferation antigen Ki67 or activation marker CD69 at day 2 of culture. (B) CLL cell division assessment based on CFSE dilution and expression of activation marker CD25 at day 6 of culture. In (A, B) *denotes significance by Wilcoxon paired t test, **(p<0.01), ***(p<0.001). (C) Correlation of observed B cell division, Ki67 expression or CD25 expression with the frequency of TFH within CD4 T cells for each patient sample cultured (Spearman correlation).
CLL Cells Activate TFH Cells in Co-Culture
During CLL:T cell interactions, CLL-B cells can also impact CD4+ T cell activation (27), but it is unclear whether particular cell subsets are preferentially targeted. In order to assess CLL B cell-dependent activation of CD4+ T cell subsets in co-culture, CD4 T cells untreated with activation cocktail were assessed for their expression of various activation markers in the presence or absence of autologous CLL cells. It was found that the presence of CLL B cells led to an increase in expression of activation markers in a small fraction (less than 15%) of autologous CD4 T cells (Figure 8A). This upregulation was significant for CD25, CD69 and PD-1 within 2 days of co-culture (Figure 8A). The frequency of CD4 T cells co-expressing both CD25 and OX40, associated with antigen-specific T cell recall responses (28), was also increased in the presence of the B cells at both timepoints (Figure 8A). To determine whether TFH cells were preferentially activated by CLL-B cells, we assessed activation marker expression on TFH and non-TFH fractions. The presence of B cells provoked an increase in CD69+ cells in both populations, however the fold increase in expression was significantly higher for TFH (Figure 8B). In addition to CD69, TFH cells showed greater fold increases in expression of CD25, CD25/OX40 and PD-1 than non-TFH cells when co-cultured with CLL (Figure 8B). TFH cell expression of activation markers CD25 and OX40 increased between day 2 and day 6 of culture, while no further increase in CD69 or PD-1 were observed after day 2 (Figure 8C). We noted that while the patient-specific frequencies of TFH cells were very stable over the first two days of culture, they began to decline by day 6 in cultures without B cells but were well maintained in the presence of CLL B cells (Figure 8D). Taken together, these results indicate that in this co-culture system, CLL-B cells can promote TFH cell activation and maintenance.
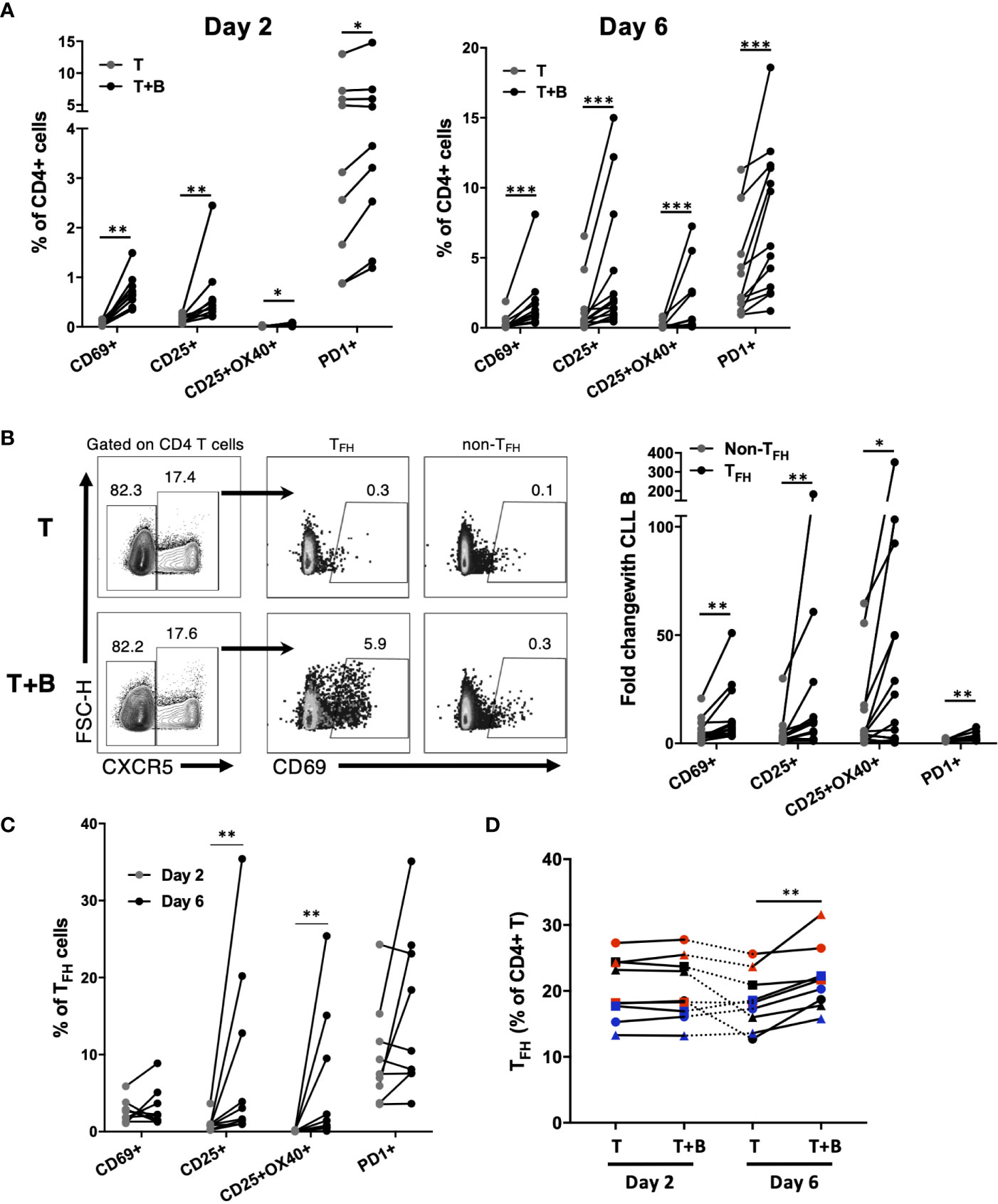
Figure 8 Impact of CLL B cells on activation of autologous CD4+ T cell subsets. Purified CLL cells were co-cultured with purified autologous CD4+ T cells that were not pre-activated. (A) Expression of activation markers on CD4+ T cells after 2 days or 6 days of co-culture. (B) Example flow cytometry analysis showing expression of CD69 on TFH or non-TFH CD4+ T cells. Data in right panel is expressed as fold change in activation marker expression among TFH or non-TFH (calculated as percent expression in T+B cell co-culture/percent expression in T cell only culture). (C) CD25 and OX40 expression increase on TFH cells co-cultured with CLL B cells between day 2 and day 6. (D) Patient-specific frequencies of TFH are maintained in CLL B cell co-culture. Individual patients are connected by lines. *denotes significance by Wilcoxon paired t test, *(p<0.05), **(p<0.01), ***(p<0.001).
Discussion
Abnormalities in T cell subsets and function in CLL have been reported in a number of studies, including changes in CD4/CD8 ratios, expansion of the Treg population, loss of naïve and increased memory populations, and increased expression of exhaustion markers (23, 29, 30). Here we report an in-depth analysis of follicular helper T cell subsets and functional status as well as their association with disease progression, immune status and ibrutinib treatment. Our results are partially consistent with studies from Asian and UK patients which also observed elevated TFH frequencies in CLL (31, 32). Our study reveals that elevation in TFH cells is first observable in Rai 0 CLL patients, whereas no increase was apparent in the MBL group. These previous studies reported lack of association with IgVH mutation status (31, 32) and we also found no association with either IgVH mutation status or ZAP-70 status (data not shown). Our study demonstrates a significant positive correlation between TFH frequencies and total lymphocyte counts, indicating that increasing acquisition of the TFH phenotype among CD4+ T cells occurs in tandem with expansion of the malignant B cell clone. A previous study found increased TFH frequencies in Binet C versus A/B patients (32), consistent with our observed trends of higher TFH frequencies, in the Rai 3-4 group. Surprisingly, we further find that expanded TFH populations in advanced stage CLL exhibit significant TFH1 skewing. Strikingly, increased expression of PD-1, ICOS and IL-21 by TFH cells were associated with disease burden, and CLL TFH cells were also found to over-express other stimulatory molecules such as IFNγ, TIGIT and CD40L.
Together our findings indicate that CLL TFH express more PD-1 than either non-TFH CD4+ T cells from CLL patients or TFH from controls. PD-1 is a marker for chronically-activated or exhausted T cells, and was previously reported to be elevated in CLL CD4+ T cells (33). Our results are consistent with this finding, and further define the CD4 sub-populations abnormally expressing this marker. In the context of the TFH literature, the PD-1+ TFH phenotype which we describe here (PD-1+CXCR5+CCR7low) is not associated with functional exhaustion but rather has been previously associated with recent TFH activation in tissues and enhanced functional capacity for providing B cell help (34, 35). However, we found evidence that TFH frequencies are negatively correlated with IgM and IgG levels. This finding is consistent with these TFH cells being dysfunctional in relation to their normal supportive roles for antibody responses; or alternatively it may reflect the association of TFH expansion with expansion of the CLL clone, and concomitant disruption of normal B cell niches within tissues.
Our results indicate that ibrutinib can normalize TFH frequencies and activation gradually over several months of treatment. These results are partially consistent with previous studies which found that ibrutinib can reduce T cell expression of PD-1 as well as memory markers (36, 37). The effects of ibrutinib on T cells may be due to inhibition of the kinase ITK expressed in T cells, since treatment with acalabrutib, which does not inhibit ITK, showed lesser impact on T cells (37). Patients with BTK mutations were found to have severe deficiency in TFH cells (38), suggesting the possibility that BTK itself may be required for TFH development and/or maintenance. Alternatively, ibrutinib may indirectly affect TFH cell populations via depletion of the CLL B cell clone and/or its other effects on the immune microenvironment.
Previous studies have provided considerable evidence that blood TFH are clonally related to TFH in lymphoid tissues. We found cells expressing markers of activated TFH cells within proliferation centers in lymph nodes and in bone marrow, suggesting they are in direct contact with CLL B cells within these microenvironments. TFH cells present in the bone marrow expressed a more activated phenotype than peripheral blood TFH, including highly elevated PD-1 and IL-21/IFNγ expression in some patients, indicating that lymphoid tissues are likely their primary site of activation. Given that IFNγ can impair hematopoietic stem cell function and contribute to bone marrow failure (39), it is possible that the presence of these activated T cells in marrow may directly participate in disruption of marrow function in advanced stage disease.
CLL TFH cells exhibit elevated expression of B cell stimulatory molecules CD40L and IL-21. Numerous studies have shown that the culture with CD40L-expressing T or stromal cell lines promotes CLL-B cell proliferation (40–42). The addition of IL-21 to CD40L-stimulated CLL-B cells increases the frequency of divided cells and the average number of divisions (31, 42). Our study provides evidence that TFH cells are the predominant T cell subset producing these CLL stimulatory factors in vivo. Additionally, we observed that CLL TFH cells expressed high levels of TIGIT, molecule that has been linked to CLL-B cell survival (43). TFH1 cells are reported to be relatively poor helpers for normal B cell responses (24), however it is possible that the abnormal TFH1-like cells in CLL are adapted to support malignant CLL B cells. A study using a mouse xenograft model found that T cells driving CLL cell proliferation had a TH1 phenotype (44), indicating the potential of these cells to serve as supportive cells in the microenvironment. Some studies have implicated IFNγ as a supportive cytokine for human CLL cells (27, 45). Together, our data demonstrate that abnormal TFH present in CLL patients over-produce multiple factors that could potentially support CLL survival and proliferation in tissues.
Our in vitro co-culture studies revealed evidence that CLL cells can preferentially activate TFH and that TFH expansion is associated with ability of activated CD4+ T cells to trigger CLL proliferation in vitro. Activated autologous CD4 T cells promoted CLL cell activation marker expression and division in line with other studies showing that the expression of activation markers by CLL cells is increased after 2-3 days of co-culture with T cells (27, 45), while CLL division is only observed after 4 days (27, 40, 46). To our knowledge, this is the first study to report this functional activity of purified autologous CD4 T cells in CLL, without addition of other factors such as stromal cells or cytokines. We observe that reactivation of CD4+ T cells using anti-CD3/28 is required to observe affects on CLL B cells, which may reflect that blood TFH, like blood CLL cells, are relatively quiescent compared to their counterparts in lymphoid tissues. Notably, the frequency of TFH cells within the CD4 T cell pool positively correlated with CLL-B cell division; however we were unable to purify sufficient numbers of TFH from CLL patient blood to carry out co-culture studies. Thus, while our data are consistent with a role of TFH cells in driving CLL proliferation, it remains possible that increased frequency of TFH could be associated with other alterations in CD4 T cell or autologous CLL cell populations that are critical for proliferation under these conditions.
Notably, the reciprocal activation of TFH cells by CLL cells in co-culture included induction of CD25/OX40 double positive TFH cells. Co-expression of these activation markers has been shown to be dependent on antigen-specific stimulation (28). Previous studies have raised the possibility of antigen-specific cognate interactions between CLL and CD4+ T cells leading to oligoclonal expansions (47), however the phenotype of these oligoclonal T cells has not been determined. One study found that CLL : CD4 T cell interactions in vitro can be abrogated by an anti-pan-MHC II antibody (27), consistent with cognate interaction. In addition to induction of activation marker expression on TFH, we found that CLL cells could maintain TFH frequencies during in vitro culture, resulting in significantly higher TFH frequencies in the presence of CLL after 6 days. These results suggest that direct bi-directional interactions between CLL cells and abnormal TFH cells is a significant feature of the dysfunctional CLL immune microenvironment.
Together, our findings suggest that alterations in TFH frequencies, activation status, subset distribution and costimulatory molecule expression can serve as alternative biomarkers in CLL reflective of lymphoid tissue involvement and disease progression. TFH cells may represent significant sources of CLL stimulatory molecules and play roles in disrupting normal immune function and promoting CLL proliferation.
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics Statement
The studies involving human participants were reviewed and approved by Research Ethics Board at the University of Manitoba. The patients/participants provided their written informed consent to participate in this study.
Author Contributions
XW and JF performed research, analyzed data, performed statistical analyses and wrote the manuscript. CZ performed research and analyzed data. SG, VN, VB and JJ designed research and collected CLL patient samples. AM designed research, analyzed and interpreted data, supervised trainees and wrote the manuscript. All authors contributed to the article and approved the submitted version.
Funding
XW and JF received graduate studentship funding from Research Manitoba. This study was supported by the Manitoba Tumour Bank, a member of the Canadian Tissue Repository Network, funded in part by the CancerCare Manitoba Foundation and the Canadian Institutes of Health Research. This study was supported by grants from the Leukemia and Lymphoma Society of Canada and Research Manitoba.
Conflict of Interest
VB and JJ: Consultant and research funding from Astra Zeneca, Abbvie, Janssen and Gilead unrelated to this work. VB: Received fees from BIOGEN for compounds unrelated to this work. SG: Research funding from Gilead, Abbvie and Janssen unrelated to this work.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors thank Mandy Squires for help with the patient database, Laurie Lange and Yun Li for blood sample biobanking and retrieval, Dr. Jie Li for preparing lymph node sections, Donna Hewitt and Linda Davidson with help obtaining healthy blood donor sample and Ahmed Ali and Sen Hou for advice and technical support.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2021.674492/full#supplementary-material
References
1. Hallek M, Cheson BD, Catovsky D, Caligaris-Cappio F, Dighiero G, Dohner H, et al. Iwcll Guidelines for Diagnosis, Indications for Treatment, Response Assessment, and Supportive Management of CLL. Blood (2018) 131(25):2745–60. doi: 10.1182/blood-2017-09-806398
2. Strati P, Shanafelt TD. Monoclonal B-Cell Lymphocytosis and Early-Stage Chronic Lymphocytic Leukemia: Diagnosis, Natural History, and Risk Stratification. Blood (2015) 126(4):454–62. doi: 10.1182/blood-2015-02-585059
3. Shanafelt TD, Geyer SM, Kay NE. Prognosis At Diagnosis: Integrating Molecular Biologic Insights Into Clinical Practice for Patients With CLL. Blood (2004) 103(4):1202–10. doi: 10.1182/blood-2003-07-2281
4. International CLLIPIwg. An International Prognostic Index for Patients With Chronic Lymphocytic Leukaemia (CLL-IPI): A Meta-Analysis of Individual Patient Data. Lancet Oncol (2016) 17(6):779–90. doi: 10.1016/S1470-2045(16)30029-8
5. Burger JA, Ghia P, Rosenwald A, Caligaris-Cappio F. The Microenvironment in Mature B-Cell Malignancies: A Target for New Treatment Strategies. Blood (2009) 114(16):3367–75. doi: 10.1182/blood-2009-06-225326
6. Herishanu Y, Perez-Galan P, Liu D, Biancotto A, Pittaluga S, Vire B, et al. The Lymph Node Microenvironment Promotes B-Cell Receptor Signaling, NF-Kappab Activation, and Tumor Proliferation in Chronic Lymphocytic Leukemia. Blood (2011) 117(2):563–74. doi: 10.1182/blood-2010-05-284984
7. Murphy EJ, Neuberg DS, Rassenti LZ, Hayes G, Redd R, Emson C, et al. Leukemia-Cell Proliferation and Disease Progression in Patients With Early Stage Chronic Lymphocytic Leukemia. Leukemia (2017) 31(6):1348–54. doi: 10.1038/leu.2017.34
8. Burger JA, Li KW, Keating MJ, Sivina M, Amer AM, Garg N, et al. Leukemia Cell Proliferation and Death in Chronic Lymphocytic Leukemia Patients on Therapy With the BTK Inhibitor Ibrutinib. JCI Insight (2017) 2(2):e89904. doi: 10.1172/jci.insight.89904
9. Herreros B, Rodriguez-Pinilla SM, Pajares R, Martinez-Gonzalez MA, Ramos R, Munoz I, et al. Proliferation Centers in Chronic Lymphocytic Leukemia: The Niche Where NF-Kappab Activation Takes Place. Leukemia (2010) 24(4):872–6. doi: 10.1038/leu.2009.285
10. Gine E, Martinez A, Villamor N, Lopez-Guillermo A, Camos M, Martinez D, et al. Expanded and Highly Active Proliferation Centers Identify a Histological Subtype of Chronic Lymphocytic Leukemia (“Accelerated” Chronic Lymphocytic Leukemia) With Aggressive Clinical Behavior. Haematologica (2010) 95(9):1526–33. doi: 10.3324/haematol.2010.022277
11. Burger JA, Tedeschi A, Barr PM, Robak T, Owen C, Ghia P, et al. Ibrutinib as Initial Therapy for Patients With Chronic Lymphocytic Leukemia. N Engl J Med (2015) 373(25):2425–37. doi: 10.1056/NEJMoa1509388
12. Herman SE, Niemann CU, Farooqui M, Jones J, Mustafa RZ, Lipsky A, et al. Ibrutinib-Induced Lymphocytosis in Patients With Chronic Lymphocytic Leukemia: Correlative Analyses From a Phase II Study. Leukemia (2014) 28(11):2188–96. doi: 10.1038/leu.2014.122
13. Bagnara D, Kaufman MS, Calissano C, Marsilio S, Patten PE, Simone R, et al. A Novel Adoptive Transfer Model of Chronic Lymphocytic Leukemia Suggests a Key Role for T Lymphocytes in the Disease. Blood (2011) 117(20):5463–72. doi: 10.1182/blood-2010-12-324210
14. Breitfeld D, Ohl L, Kremmer E, Ellwart J, Sallusto F, Lipp M, et al. Follicular B Helper T Cells Express CXC Chemokine Receptor 5, Localize to B Cell Follicles, and Support Immunoglobulin Production. J Exp Med (2000) 192(11):1545–52. doi: 10.1084/jem.192.11.1545
15. Schaerli P, Willimann K, Lang AB, Lipp M, Loetscher P, Moser B. CXC Chemokine Receptor 5 Expression Defines Follicular Homing T Cells With B Cell Helper Function. J Exp Med (2000) 192(11):1553–62. doi: 10.1084/jem.192.11.1553
16. Vinuesa CG, Linterman MA, Goodnow CC, Randall KL. T Cells and Follicular Dendritic Cells in Germinal Center B-Cell Formation and Selection. Immunol Rev (2010) 237(1):72–89. doi: 10.1111/j.1600-065X.2010.00937.x
17. Cyster JG, Ansel KM, Reif K, Ekland EH, Hyman PL, Tang HL, et al. Follicular Stromal Cells and Lymphocyte Homing to Follicles. Immunol Rev (2000) 176:181–93. doi: 10.1034/j.1600-065X.2000.00618.x
18. Crotty S. Follicular Helper CD4 T Cells (TFH). Annu Rev Immunol (2011) 29:621–63. doi: 10.1146/annurev-immunol-031210-101400
19. Ueno H, Banchereau J, Vinuesa CG. Pathophysiology of T Follicular Helper Cells in Humans and Mice. Nat Immunol (2015) 16:142–52. doi: 10.1038/ni.3054
20. Schmitt N, Bentebibel S-E, Ueno H. Phenotype and Functions of Memory Tfh Cells in Human Blood. Trends Immunol (2014) 35:436–42. doi: 10.1016/j.it.2014.06.002
21. Gorgun G, Holderried TA, Zahrieh D, Neuberg D, Gribben JG. Chronic Lymphocytic Leukemia Cells Induce Changes in Gene Expression of CD4 and CD8 T Cells. J Clin Invest (2005) 115(7):1797–805. doi: 10.1172/JCI24176
22. Ramsay AG, Clear AJ, Fatah R, Gribben JG. Multiple Inhibitory Ligands Induce Impaired T-Cell Immunologic Synapse Function in Chronic Lymphocytic Leukemia That Can Be Blocked With Lenalidomide: Establishing a Reversible Immune Evasion Mechanism in Human Cancer. Blood (2012) 120(7):1412–21. doi: 10.1182/blood-2012-02-411678
23. Forconi F, Moss P. Perturbation of the Normal Immune System in Patients With CLL. Blood (2015) 126(5):573–81. doi: 10.1182/blood-2015-03-567388
24. Morita R, Schmitt N, Bentebibel S-E, Ranganathan R, Bourdery L, Zurawski G, et al. Human Blood CXCR5+CD4+ T Cells are Counterparts of T Follicular Cells and Contain Specific Subsets That Differentially Support Antibody Secretion. Immunity (2011) 34:108–21. doi: 10.1016/j.immuni.2010.12.012
25. Godefroy E, Zhong H, Pham P, Friedman D, Yazdanbakhsh K. TIGIT-Positive Circulating Follicular Helper T Cells Display Robust B-Cell Help Functions: Potential Role in Sickle Cell Alloimmunization. Haematologica (2015) 100(11):1415–25. doi: 10.3324/haematol.2015.132738
26. Yu X, Harden K, Gonzalez LC, Francesco M, Chiang E, Irving B, et al. The Surface Protein TIGIT Suppresses T Cell Activation by Promoting the Generation of Mature Immunoregulatory Dendritic Cells. Nat Immunol (2009) 10(1):48–57. doi: 10.1038/ni.1674
27. Os A, Burgler S, Ribes AP, Funderud A, Wang D, Thompson KM, et al. Chronic Lymphocytic Leukemia Cells are Activated and Proliferate in Response to Specific T Helper Cells. Cell Rep (2013) 4(3):566–77. doi: 10.1016/j.celrep.2013.07.011
28. Havenar-Daughton C, Reiss SM, Carnathan DG, Wu JE, Kendric K, Torrents de la Pena A, et al. Cytokine-Independent Detection of Antigen-Specific Germinal Center T Follicular Helper Cells in Immunized Nonhuman Primates Using a Live Cell Activation-Induced Marker Technique. J Immunol (2016) 197(3):994–1002. doi: 10.4049/jimmunol.1600320
29. Riches JC, Davies JK, McClanahan F, Fatah R, Iqbal S, Agrawal S, et al. T Cells From CLL Patients Exhibit Features of T-Cell Exhaustion But Retain Capacity for Cytokine Production. Blood (2013) 121(9):1612–21. doi: 10.1182/blood-2012-09-457531
30. Rissiek A, Schulze C, Bacher U, Schieferdecker A, Thiele B, Jacholkowski A, et al. Multidimensional Scaling Analysis Identifies Pathological and Prognostically Relevant Profiles of Circulating T-Cells in Chronic Lymphocytic Leukemia. Int J Cancer (2014) 135(10):2370–9. doi: 10.1002/ijc.28884
31. Ahearne MJ, Willimott S, Pinon L, Kennedy DB, Miall F, Dyer MJ, et al. Enhancement of CD154/IL4 Proliferation by the T Follicular Helper (Tfh) Cytokine, IL21 and Increased Numbers of Circulating Cells Resembling Tfh Cells in Chronic Lymphocytic Leukaemia. Br J Haematol (2013) 162(3):360–70. doi: 10.1111/bjh.12401
32. Cha Z, Zang Y, Guo H, Rechlic JR, Olasnova LM, Gu H, et al. Association of Peripheral CD4+ CXCR5+ T Cells With Chronic Lymphocytic Leukemia. Tumour Biol (2013) 34(6):3579–85. doi: 10.1007/s13277-013-0937-2
33. Palma M, Gentilcore G, Heimersson K, Mozaffari F, Nasman-Glaser B, Young E, et al. T Cells in Chronic Lymphocytic Leukemia Display Dysregulated Expression of Immune Checkpoints and Activation Markers. Haematologica (2017) 102(3):562–72. doi: 10.3324/haematol.2016.151100
34. Locci M, Havenar-Daughton C, Landais E, Wu J, Kroenke MA, Arlehamn CL, et al. Human Circulating PD-1+CXCR3-CXCR5+ Memory Tfh Cells are Highly Functional and Correlate With Broadly Neutralizing HIV Antibody Responses. Immunity (2013) 39(4):758–69. doi: 10.1016/j.immuni.2013.08.031
35. He J, Tsai LM, Leong YA, Hu X, Ma CS, Chevalier N, et al. Circulating Precursor CCR7(Lo)PD-1(Hi) CXCR5(+) CD4(+) T Cells Indicate Tfh Cell Activity and Promote Antibody Responses Upon Antigen Reexposure. Immunity (2013) 39(4):770–81. doi: 10.1016/j.immuni.2013.09.007
36. Kondo K, Shaim H, Thompson PA, et al. Ibrutinib Modulates the Immunosuppressive CLL Microenvironment Through STAT3-Mediated Suppression of Regulatory B-Cell Function and Inhibition of the PD-1/PD-L1 Pathway. Leukemia (2018) 32(4):960–70. doi: 10.1038/leu.2017.304
37. Long M, Beckwith K, Do P, Mundy BL, Gordon A, Lehman AM, et al. Ibrutinib Treatment Improves T Cell Number and Function in CLL Patients. J Clin Invest (2017) 127:3052–64. doi: 10.1172/JCI89756
38. Ma CS, Wong N, Rao G, Avery DT, Torpy J, Hambridge T, et al. Monogenic Mutations Differentially Affect the Quantity and Quality of T Follicular Helper Cells in Patients With Human Primary Immunodeficiencies. J Allergy Clin Immunol (2015) 136(4):993–1006.e1001 doi: 10.1016/j.jaci.2015.05.036.
39. Morales-Mantilla DE, King KY. The Role of Interferon-Gamma in Hematopoietic Stem Cell Development, Homeostasis, and Disease. Curr Stem Cell Rep (2018) 4(3):264–71. doi: 10.1007/s40778-018-0139-3
40. Asslaber D, Grossinger EM, Girbl T, Hofbauer SW, Egle A, Weiss L, et al. Mimicking the Microenvironment in Chronic Lymphocytic Leukaemia - Where Does the Journey Go? Br J Haematol (2013) 160(5):711–4. doi: 10.1111/bjh.12151
41. Hamilton E, Pearce L, Morgan L, Robinson S, Ware V, Brennan P, et al. Mimicking the Tumour Microenvironment: Three Different Co-Culture Systems Induce a Similar Phenotype But Distinct Proliferative Signals in Primary Chronic Lymphocytic Leukaemia Cells. Br J Haematol (2012) 158(5):589–99. doi: 10.1111/j.1365-2141.2012.09191.x
42. Pascutti MF, Jak M, Tromp JM, Derks IA, Remmerswaal EB, Thijssen R, et al. IL-21 and CD40L Signals From Autologous T Cells Can Induce Antigen-Independent Proliferation of CLL Cells. Blood (2013) 122(17):3010–9. doi: 10.1182/blood-2012-11-467670
43. Catakovic K, Gassner FJ, Ratswohl C, Zaborsky N, Rebhandl S, Schubert M, et al. TIGIT Expressing CD4+T Cells Represent a Tumor-Supportive T Cell Subset in Chronic Lymphocytic Leukemia. Oncoimmunology (2017) 7(1):e1371399. doi: 10.1080/2162402X.2017.1371399
44. Patten PE, Ferrer G, Chen SS, Simone R, Marsilio S, Yan XJ, et al. Chronic Lymphocytic Leukemia Cells Diversify and Differentiate In Vivo Via a Nonclassical Th1-Dependent, Bcl-6-Deficient Process. JCI Insight (2016) 1(4). doi: 10.1172/jci.insight.86288
45. Burgler S, Gimeno A, Parente-Ribes A, Wang D, Os A, Devereux S, et al. Chronic Lymphocytic Leukemia Cells Express CD38 in Response to Th1 Cell-Derived IFN-Gamma by a T-Bet-Dependent Mechanism. J Immunol (2015) 194(2):827–35. doi: 10.4049/jimmunol.1401350
46. Patten PE, Buggins AG, Richards J, Wotherspoon A, Salisbury J, Mufti GJ, et al. CD38 Expression in Chronic Lymphocytic Leukemia is Regulated by the Tumor Microenvironment. Blood (2008) 111(10):5173–81. doi: 10.1182/blood-2007-08-108605
47. Zaborsky N, Holler C, Geisberger R, Asslaber D, Gassner FJ, Egger V, et al. B-Cell Receptor Usage Correlates With the Sensitivity to CD40 Stimulation and the Occurrence of CD4+ T-Cell Clonality in Chronic Lymphocytic Leukemia. Haematologica (2015) 100(8):e307–310. doi: 10.3324/haematol.2015.124719
Keywords: chronic lymphocytic leukemia (CLL), T follicular helper (Tfh) cell, interleukin 21 (IL-21), ibrutinib, TIGIT, coculture assay, bone marrow
Citation: Wu X, Fajardo-Despaigne JE, Zhang C, Neppalli V, Banerji V, Johnston JB, Gibson SB and Marshall AJ (2021) Altered T Follicular Helper Cell Subsets and Function in Chronic Lymphocytic Leukemia. Front. Oncol. 11:674492. doi: 10.3389/fonc.2021.674492
Received: 01 March 2021; Accepted: 06 April 2021;
Published: 28 April 2021.
Edited by:
Dimitar G. Efremov, International Centre for Genetic Engineering and Biotechnology, ItalyReviewed by:
Martina Seiffert, German Cancer Research Center, GermanyAngelika Muchowicz, Medical University of Warsaw, Poland
Copyright © 2021 Wu, Fajardo-Despaigne, Zhang, Neppalli, Banerji, Johnston, Gibson and Marshall. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Aaron J. Marshall, YWFyb24ubWFyc2hhbGxAdW1hbml0b2JhLmNh
†These authors have contributed equally to this work