 Justin T. Avery
Justin T. Avery Ruowen Zhang
Ruowen Zhang Rebecca J. Boohaker
Rebecca J. Boohaker
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oncol., 25 May 2021
Sec. Molecular and Cellular Oncology
Volume 11 - 2021 | https://doi.org/10.3389/fonc.2021.673154
This article is part of the Research TopicThe Role of DNA Repair Pathways in Resistance to Chemotherapy and Radiotherapy in CancerView all 17 articles
GLI1 is a transcriptional effector at the terminal end of the Hedgehog signaling (Hh) pathway and is tightly regulated during embryonic development and tissue patterning/differentiation. GLI1 has low-level expression in differentiated tissues, however, in certain cancers, aberrant activation of GLI1 has been linked to the promotion of numerous hallmarks of cancer, such as proliferation, survival, angiogenesis, metastasis, metabolic rewiring, and chemotherapeutic resistance. All of these are driven, in part, by GLI1’s role in regulating cell cycle, DNA replication and DNA damage repair processes. The consequences of GLI1 oncogenic activity, specifically the activity surrounding DNA damage repair proteins, such as NBS1, and cell cycle proteins, such as CDK1, can be linked to tumorigenesis and chemoresistance. Therefore, understanding the underlying mechanisms driving GLI1 dysregulation can provide prognostic and diagnostic biomarkers to identify a patient population that would derive therapeutic benefit from either direct inhibition of GLI1 or targeted therapy towards proteins downstream of GLI1 regulation.
GLI1 is an effector transcriptional factor distal to both the canonical and non-canonical Hedgehog (Hh) signaling pathways. The Hh family of proteins contains three subfamilies: sonic hedgehog (SHh), desert hedgehog (DHh) and Indian hedgehog (IHh) (1). IHh and DHh are reported to be involved in normal tissue development, such as bone formation (2). SHh, first discovered in Drosophila, has been found to be highly conserved across many different vertebrate species including human, mouse, rat, frog, fish, and chicken, and is the most studied member of the hedgehog family (3). SHh plays a critical role in the embryonic development that is necessary for certain cell differentiation and maintenance of tissue polarity (4). Due to its conserved nature, and apparent critical functionality across organisms, SHh and the downstream pathway members have evolved to serve vastly diverse roles in both embryonic and non-embryonic cellular homeostasis. Herein, we focus specifically on our current understanding of SHh-GLI pathway and its clinical significance in human development and the consequences of its dysregulation in disease progression (5–8).
SHh-mediated transduction is initiated via extracellular SHh ligand binding to the 12-span transmembrane receptor, PATCHED-1 (PTCH-1) or the redundant receptor PTCH-2, in target cells (9, 10). In the absence of SHh, PTCH-1 and, redundantly, PTCH-2, catalytically inhibit downstream signaling activity with seven-transmembrane G-protein-coupled receptor, Smoothened (SMO) (11–13). Upon SHh binding to PTCH, the inhibitory interaction is terminated through internalization of PTCH, releasing SMO and allowing for phosphorylation to transduce signal into the cytoplasm (14). The resultant signal leads to the component dissociation of a large protein complex comprising of Sufu and GLIs in the cytoplasm, releasing the GLI transcription factors. Finally, the released GLI transcription factors translocate into the nucleus to execute transcriptional activation of specific target genes (15). Aberrant activation of the hedgehog pathway has been shown to promote oncogenic activities, such as metastasis, DNA damage repair, stem-ness, and chemotherapeutic resistance, in a variety of types of cancer (16–26).
There are two models for the over-activation of the Hh pathway in cancer: (a) ligand-dependent model: tumors are able to over-activate SHh-GLI pathway via autocrine signaling to produce high level of SHh ligands (18, 27–31). This can be observed in several epithelial originating tumors such as small cell lung cancer (SCLC), pancreatic, colon, and prostate cancer, and glioblastomas, and medulloblastomas. (b) Ligand-independent model: Clinical observations have found mutations of PTCH-1 and PTCH-2 in basal cell carcinomas and in medulloblastomas, resulting in dysregulated GLI signaling due to ineffective sequestration of SMO signaling, regardless of SHh ligand levels. Mutant PTCH often results in SMO constitutive activation, subsequently promoting cell transformation and tumorigenesis (32). Inactivation of PTCH-1 due to gene mutation has also been reported in trichoepitheliomas (33), esophageal squamous cell carcinomas (34), and transitional cell carcinomas of the bladder (35). In both models, the commonality is a failure to stifle SMO signal transduction.
Regardless of how SHh-GLI pathway is activated, all biological function of these upstream proteins such as SHh, PTCH and SMO depends on the transcriptional effectors at the distal end of the pathway: the GLI proteins. There are three GLI transcriptional proteins in this family, two which act as transcriptional activators (GLI1 and GLI2) and one transcriptional repressor (GLI3) (36). GLI1 was initially found to transcriptionally regulate specific target genes involved in mammalian development, such as patterning in the central nervous system, proliferation, differentiation, and survival (37). However, increased expression in terminally differentiated cells is a known oncogenic biomarker for numbers cancer subtypes (38–40), making it an ideal drug discovery target.
GLI1 (1106 amino acids; MW 117.9kDa) was originally identified as an amplified gene product in a malignant glioma (41) and was the first member described in the human GLI gene family. GLI1-DNA binding is mediated by five highly conserved tandem C2-H2 zinc finger (ZF) domains and a consensus histidine-cysteine linker sequence between zinc fingers (42). While ZF1-3 interacts with the phosphate backbone and contributes to binding stability and recruitment of co-regulatory factors, ZF4-5 regulates transcription, recognizing the consensus sequence 5'-GACCACCCA-3' in the promoter region of target genes. The two cytosine-pairs flanking the central adenine within the consensus site are critical for GLI binding, whereas the other positions can tolerate a certain degree of flexibility (43). In addition to the transcriptional ZF domain, the GLI proteins contain both nuclear export sequence (NES) and a nuclear localization signal (NLS), which facilitate the nucleo-cytoplasmic shuttling of GLI (44). GLI1 also contains a single SUFU-interacting site located at the N-terminus (SIN) (45), which is responsible for SUFU-mediated cytoplasmic retention of GLI1. The positioning of the SIN is unique to GLI1; GLI2 and GLI3 also have a SUFU-interacting site though it is located in the C-terminus (SIC) (45, 46). The GLI1 C-terminal region possesses a transactivation domain (TAD) which remodels chromatin and interacts with histone acetyltransferase (HAT), histone deacetylase (HDAC); SWI-SNF5; SWI/SNF-like Brg/Brm-associated factor; and the TFIID TATA box-binding protein-associated factor, TAFII31 (26). Like the SIN domain, all GLI proteins also possess a TAD, but GLI2 and GLI3 have an additional N-terminal repressor domain, which is lacking on GLI1. Therefore, GLI1 performs as a strong transcriptional activator (47), whereas full-length GLI2 is generally a weak activator since the fully activated form requires significant truncation of its N-terminus and C-terminus (48–51), and GLI3 has been reported as a strong repressor in most settings (52).
Two additional isoforms of GLI1, N-terminal deletion variant (GLI1ΔN) and truncated GLI1 (tGLI1), have been identified. GLI1ΔN is generation is the result of a 128-amino acid deletion on its N-terminus (47). This deletion results in loss of the lone critical suppressive SUFU-binding domain on the GLI1 protein sequence, while preserving the ZNF domains, NLS and NES, and the transactivation domain. As would be expected, this isoform of GLI1 functions as a constitutively active protein, with activity comparable to full-length GLI1 (GLI1FL) but surprisingly does not show a preferential expression in cancer tissues (53, 54). tGLI1 originates from a splicing of exon 3 and part of exon 4 of the GLI1 gene, resulting in the deletion of 41 amino acids (55). All functional domains are retained in tGLI1, and this isoform is observed specifically in tumor expression. It has been shown to regulate an additional set of target genes involved in EMT, invasion and metastasis (56). All three GLI1 isoforms (GLI1FL, GLI1ΔN, and tGLI1) could be activated by SHh ligand stimulation, but whether they induce differently transcriptional targets has not yet been determined.
Over-activation of Hh promotes the tumor microenvironment through pro-inflammatory mechanisms, angiogenesis, genome instability, mutation, resistance to cell death, energy imbalance, and is involved in invasion and metastasis (57, 58). Some studies, however, fail to observe a positive correlation between the Hh signaling pathway and the development/progression of cancer (59–61). For instance, Li discovered that SMO expression was not statistically correlated with CRC-specific or overall survival; the same results were reported by Stefanius, where no correlation between Hh and colorectal serrated adenocarcinomas was observed (62, 63). Our lab, like many others, observed a positive correlation between GLI1 expression and disease severity (64). We also demonstrated that both GLI1 and one of its transcriptional targets, NBS1, negatively correlate with CRC patient 5-year survival, driving chemotherapeutic resistance by overcoming FOLFOX induced DNA damage (standard of care treatment). The difference lies in the way GLI1 is activated—whether it be through canonical activation (PTCH/SMO) or non-canonical (RAS/RAF, etc) (Figure 1). Elevated levels of GLI1 in cancer are often driven by non-canonical pathways. As such, this explains why Vismodegib, the first SMO inhibitor to be approved by the FDA for the treatment of BCC (65), failed to demonstrate the effectiveness in clinical trials for the treatment of metastatic colorectal cancer where GLI expression is driven non-canonically (66). Therefore, it is important to determine how GLI1 is upregulated and its function in the initiation, progression, invasion and metastasis in order to develop a therapeutic target for new treatment schemes based on the inhibition, at different levels, of the Hh pathway (67–69).
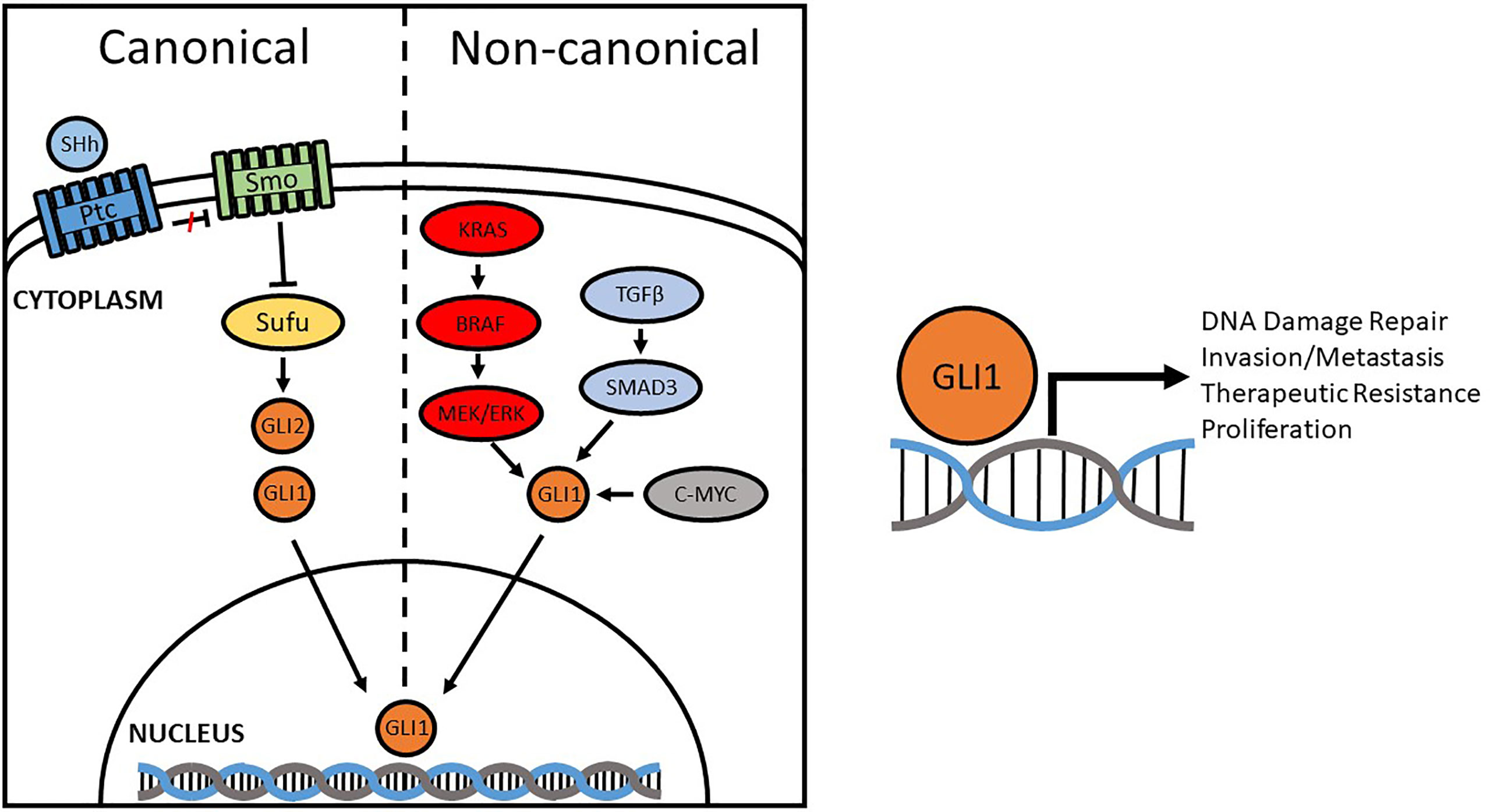
Figure 1 Canonical and Non-canonical activation of GLI1. Compounds originally designed to inhibit the Hedgehog pathway focused on canonical regulators, like SMO, but were found to be ineffective in some cancers due to non-canonical activation. Aberrant activation of GLI1 promotes DNA damage repair, invasion/metastasis, proliferation, and therapeutic resistance through transcriptional regulation of target genes.
The RAS-RAF-MEK-ERK pathway is the most common non-canonical mechanism involved both in transcriptional activation of GLI genes and in post-translational modifications of GLI-transcribed proteins. In colorectal cancer, constitutively activated mutant KRAS or altered stimulation of pathway components (mainly RAS, RAF, MEK) results in the hyperactivation of the mitogen-activated protein kinase (MAPK) extracellular signal-regulated kinase 1 and 2 (ERK1/2) and positively modulates tumor proliferation by increasing GLI1 transcriptional activity and expression of Hh target genes (54, 70, 71). This non-canonical activation pathway was confirmed in a study where EGF-induced stimulation of GLI is unaffected by SMO inhibition but was blocked by MEK1 inhibition (72). Similarly, the RAS-RAF pathway induces GLI1 and GLI2 transcriptional activity and increases mRNA and protein levels in a non-canonical manner in colon cancer cells (73). Pharmacological and genetic inhibition of GLI function is more effective in reducing tumor proliferation and inducing apoptosis than the inhibition of the canonical pathway at SMO level, suggesting that GLI activity is crucial for RAS/MEK-induced colon cancer proliferation (74, 75).
The PI3K-AKT-mTOR signaling pathway is another crucial non-canonical activator of GLI1, particularly evident in pancreatic cancers (76). Activation of PI3K-AKT signaling has been found to enhance GLI1 protein stability (77) since AKT is able to extend GLI proteins half-life in the cells by alleviating the inhibitory effect of PKA and facilitates nuclear translocation. Another mechanism of PI3K signaling activating GLI1 is via members of the ribosomal S6 kinase family (S6K/p70-S6K), which are the downstream effectors of the PI3K-AKT-mTOR axis. Activated S6K1 promotes GLI1 disassociation from SUFU by phosphorylating GLI1 at Serine residue at position 84, increasing GLI1 transcriptional activity (78). Additionally, p70-S6K2 has been shown to inhibit GSK3 by phosphorylating GLI1at Ser9, leading to decreases of GSK3b-mediated GLI1 degradation (79).
TGFβ is a multifunctioning cytokine that has been implicated in nearly all the key steps of tumorigenesis, tumor maintenance and advanced metastasis (80). In brief, TGFβ is secreted as a latent complex and sequestered in the extracellular matrix until activated. Once biologically available to its target cells, TGFβ binds its type 2 receptor (TGFBR2), leading to the recruitment of its type 1 receptor (TGFBR1) and subsequent downstream signaling resulting in nuclear localization of the SMAD2/3/4 complex (81). In advanced pancreatic cancer, TGFβ signaling contributes to a metastatic phenotype (82). GLI1 as an effector of TGFβ signaling as it interacts with SMAD proteins to induce a subset of TGFβ-inducible target genes, including BCL2, IL7, and Cyclin D1 (83). In the mouse model of PDAC, SMO-independent GLI1 activation promotes transformation and requires both TGFβ and KRAS signaling (84) where inhibition of TGFβ by TbRI antagonist SD208 significantly reduces tumor burden and increases infiltration of lymphocytes.
C-MYC, which is frequently over-amplified in colorectal cancer, has been confirmed to be another oncogene that activates GLI1 independently from Hh ligand-mediated signaling (85). C-MYC is a transcriptional activator of GLI1. C-MYC-GLI1 activated pathway could be blocked by small molecule inhibitors targeting either protein, downregulating GLI1 expression and, in turn, inducing cell apoptosis of colorectal cells. Similarly, aberrant expression of oncogenic EGFR, which is responsible for the over-activation of GLI1 through RAS-RAF-MEK pathway, promotes colorectal cancer metastasis and chemotherapeutic resistance. In triple negative breast cancer, elevated expression of GLI1 is driven by VEGF/NRP2 and α6β1 pathway results in an autocrine feedback loop with GLI1 enhancing the expression of NRP2 (86). Atypical protein kinase C iota/lambda (aPKC) has been identified as a novel regulator of GLI, and like the VEGF/NRP2 pathway, results in a positive feedback loop enhancing GLI1 overexpression in basal cell carcinoma (87) and has been also observed in drosophila (88). An interesting connection between GLI1 and p53 has also been reported because of loss of p53 results in aberrant GLI1 expression (89). Genetic mutations of aforementioned pathway genes have been shown to drive GLI1 expression in multiple types of cancer and cancer precursor diseases (90–95). An interesting GLI1 genetic translocation was first noted in 2004 when five pericytomas had an ACTB-GLI fusion transcript t(7;12) (96), with an additional three patients reported on 15 years later (97). Additional fusions were later observed with ACTB1/MALAT1/PTCH1-GLI1, which were associated with metastasis to the lung/lymph node in three of the patients (98).
While GLI1 and GLI2 are both transcriptional activators, GLI1 can be thought of as the primary effector of Hh signaling since GLI1 is a transcriptional target of GLI2, which may amplify Hh-induced, GLI2-mediated transcription of GLI1 target genes (99–102). As previously stated, GLI1 induced by Hh signaling is important in the regulation of cellular proliferation, stemness, cell fate determination, and cellular survival in a variety of organs (36, 103); however, its aberrant activation has been associated with many human cancers (104). For example, GLI1 is amplified in glioma (37), osteosarcoma, and rhabdomyosarcoma (105). Mutations in PTCH or SMO are also prevalent in basal cell carcinomas, medulloblastomas, and cancers of the esophagus and bladder (102), and sustained and activated Hh-Gli signaling has led to the development of medulloblastomas in PTCH heterozygous mice (106). Melanomas and carcinomas of the prostate have further demonstrated a need for elevated Hh-Gli signaling, since inhibition by cyclopamine (a SMO inhibitor) can result in reduction for these types of cancers (107, 108).
Although GLI1 plays a key role in canonically activated Hh cancers (103, 109), non-canonical oncogenic activation (CMYC, RAS/RAF, TGFβ, etc) is critical to address as well (110). For example, in gastrointestinal (GI) cancers, over-activation of GLI1 is driven by KRAS/BRAF mutation (102). It has recently been suggested that oncogenic GLI1 progresses during colon carcinogenesis (111, 112) and in metastatic disease (31), whereas in normal colonic tissue, Hh-GLI is strictly involved in differentiation (59, 113).
Colorectal cancer (CRC) is still one of the most common gastrointestinal cancers worldwide and results in approximately 33% mortality rate, despite several therapeutic advancements (114). The most important prognostic indicator is stage at diagnosis. The 5-year relative survival of patients diagnosed with CRC is 90% for patients with localized disease (non-metastatic), whereas clinical statistics shows less than 5% 5-year survival for metastatic CRC (115, 116). Therefore, oncogenic drivers of metastasis promote a significant problem to both CRC patients and clinicians (63, 117). The mechanism for CRC progression toward metastasis is multifactorial, with age, dietary habits, genetic alteration (mutational activation of oncogenes and inhibition of several tumor suppressor genes), intensity of epithelial-to-mesenchymal transformation (EMT), angiogenesis in tumor growth, and response to the therapeutic treatment all playing roles in the progression of disease (118, 119). Various gene mutations (KRAS, MYB, and BRAF) and gene abnormal amplification (CMYC and EGFR) have been associated with the molecular mechanisms underlying the development of CRC, all of which can result in non-canonical activation of GLI1 (120, 121). Another complication for studying and treating CRC is the heterogeneity of the disease. This heterogeneity is driven by the by pluripotent, self-renewing cancer stem cells (CSCs) which have unlimited self-renewal through symmetric cell division, and have the ability to give rise to progeny cells through asymmetric division, and an innate resistance to cytotoxic therapeutics (122). Additionally, may publications have implicated Wnt, Notch, Hh, and/or TGFβ signaling pathways in proliferation and maintenance of CSCs, and dysregulation of these pathways might cause the development of CRC (123–127). All of these pathways drive GLI1 expression, defining GLI1 as a cancer stem cell marker in multiple types of cancer, including colorectal (128–131).
Pancreatic ductal adenocarcinoma (PDAC) is one of the deadliest types of cancer in the United States, with a 5-year survival rate of less than 3.5% (132, 133). Removal of the tumor is the only potentially curative treatment to date, but this is not achievable for over 85% patients due to non-resectable cases like early-stage metastasis or complicated primary site (80). KRAS over-activation mutations play a major role in initiating the transformation from precursor lesions termed “pancreatic intraepithelial neo-plasias” (PanINs) to PDAC and promote cancer development and metastasis (134–136). In pancreatic cancer, KRAS mutations are present in 90% of cases (137). Multiple mutation types exist, with codon-13 (G13D) or -61 (Q61L or Q61H) occurring less frequently and 95% of KRAS mutations occurring at codon-12. Single-nucleotide mutations on codon-12 result in eight different amino acid substitutions, with G12D the predominant mutation (51%), G12V (30%), G12R (12%), G12C (2%), G12S (2%), G12A (2%), G12L/F (1%) (11, 14). These missense mutations enhance the level of GTP-bound active KRAS due to impairing intrinsic and GTPase-activating protein-mediated GTP hydrolysis, resulting in over-activating downstream signaling, increasing cell growth and survival, leading to neoplastic transformation (138–140). For patients with locally advanced and/or metastatic PDAC, a G12D KRAS mutation within the primary tumor is an independent prognostic factor that results in significantly decreased overall survival, including those within the subgroup that receive chemotherapy (141). Pancreatic cancer with activating mutations in KRAS or BRAF occur frequently, and oncogenic pathways like RAS/RAF/MEK/ERK, the PI3K-AKT-mTOR, and TGFβ signaling converge on the activation of GLI1, promoting cellular proliferation, tumor progression, chemotherapeutic resistance, and early metastasis (142, 143).
Glioblastoma multiforme (GBM) is the most aggressive and most common type of brain tumor. The standard of care for patients with GBM is maximum safe surgical resection followed by concurrent temozolamide (TMZ) and radiation therapy (144). TMZ is an alkylating agent that results in the transport of methyl groups to guanine and adenine, resulting in DNA damage and eventual cell cycle arrest and apoptosis. TMZ also acts as a radiation-sensitizer to enhance the DNA damage induced by the ionizing radiation. Individuals receiving this standard treatment have a median survival time between 12 and 15 months and have an average 5-year survival of 5% in the United States. Unfortunately, approximately 50% of patients do not respond to the standard of care regimen (145). Most of these cases are the result of overexpression of O6-methylguanine-DNA methyl-transferase (MGMT), a protein that directly counters the methyl damage caused by TMZ (146). In fact, hypomethylation of the MGMT promoter is a biomarker for aggressiveness of disease and poor response to therapy (147). GLI1 was recently identified as positive regulator of MGMT, having several putative binding sites in the MGMT promoter region (148). Aberrant activation of GLI family members has been linked to chemotherapeutic resistance to TMZ (69). Data set analysis from the Chinese Glioma Genome Atlas (CGGA) indicates that individuals with lower expression of GLI1 (149) have a statistically greater median survival when compared to GLI1 high-expressing patients. Several studies have examined the effect of GLI inhibition in GBM cells in vitro and found that treatment with GLI inhibitors, like GANT61, results in decreased expression of MGMT and re-sensitization to TMZ (148, 150, 151).
Neuroblastoma accounts for roughly 8% of all childhood malignancies and up to 15% of all pediatric cancer deaths (152). It is a heterogeneous solid tumor, and the heterogeneity is partially driven by the generation of extrachromosomal circular DNA (eccDNA) (153). eccDNA formation has been linked to the dysregulation of the double-stranded break (DSB) repair mechanism, specifically that which drives non-homologous end-joining (NHEJ) and is produced through R-loop defects or circularization of gene fragments (154). Oncogenic GLI1 drives R-loop formation, and treatment with GANT61 has been shown to decrease the generation of R-loop formation (155), likely additionally reducing the generation of eccDNAs.
EMT is considered to be an important feature in cancer development. This process allows the epithelial cells to undergo various biological changes, transforming them to a mesenchymal cell phenotype characterized by enhanced migration, invasiveness, and resistance to apoptosis. EMT markers, such as snail family of zinc-finger transcription factor 1 (SNAIL1), vimentin, and E-cadherin, are three of the primary factors that regulate the EMT transition. GLI1 can initiate cancer cell EMT by increasing expression of SNAIL1 and vimentin but decreasing E-cadherin, causing β-catenin to migrate into the nucleus and act as a transcription factor, inducing cell transformation (156, 157). Since β-catenin is an important member of the WNT signaling pathway, this results in cross-talk between WNT pathway and Hh pathway, resulting in GLI1 activation (158). Overexpression of GLI1 in colorectal cancer cells induces more invasive growth in organoid 3D cultures as well as in soft agar colony formation (159).
GLI1 activation has been linked to the DNA damage response (DDR) and promotes chemotherapeutic resistance. Recent studies have demonstrated that loss of either non-homologous end joining(NHEJ) gene DNA Ligase IV (Lig4), or genes involved in homologous recombination (HR) like X-ray cross complementation 2 (XRCC2), and breast cancer growth suppressor protein 2 (BRCA2), or (Lig4/XRCC2) in combination with p53 deficiency results in PTCH-1 downregulation and GLI1 activation (69, 160). DNA damaging agents, such as doxorubicin and cisplatin, induced concomitant expression of p53 and downregulation of GLI1 and its target genes (161). In response to damage, p53-induced cell cycle checkpoints prevents proliferation of damaged cells and provides sufficient time for repair, which is the opposite response that GLI1 promotes (69).
Specific inhibition of GLI1 induces extensive cell death while the inhibition of Hh signaling at the level of SMO did not in colorectal cancers (161). In HT29 cells, inhibition of GLI1 by siRNAs or GANT61 (a small molecule inhibitor) showed increased DNA damage and cell cycle arrest at G1–S and in early S-phase, resultant of down-regulation of cell cycle genes, such as E2F2, cyclin E2, Cdc25a, Cdk2 and cyclin A2, Cdc25c, cyclinB2, Cdc20, Cdc2. Inhibition of GLI1 induces serious DNA damage because it pauses DNA synthesis by impairing the ensemble of DNA licensing pre-complex and accumulates conflicts by head-to-head jam made by DNA and RNA synthesis machinery due to cell cycle arrest (155). Additionally, inhibition of GLI1 not only promotes cell cycle arrest it also impairs cell innate DNA damage response procedure. The DDR machinery is comprised of multiple sensors and repair enzymes that are deployed at various stages of the cell cycle to ensure the maintenance of chromosomal integrity and replicative fidelity. Numerous reports of overexpression of critical DDR component proteins in oncogenic environments indicate that chemo-resistance can arise due to over-activation of the MRE11, Rad50, NBS1 (MRN) complex. A critical component of the MRN complex is the Nijmegen breakage syndrome-1 (NBS1; p95, nibrin) protein, produced by NBS gene. Complexing with MRE11 and RAD50, NBS1 is the first factor to detect and bind to histone H2AX at the site of a DNA lesion which subsequently forms the multimeric MRN complex, initiating the process of DSBs repair (162–164). Overexpression of individual components of the MRN complex has been significantly associated with adverse clinical outcomes due to chemotherapeutic resistance. Therefore, induced novel therapeutic avenue would be to inhibit the DDR mechanism, allowing chemotherapeutic mechanisms that target DNA damage to work more effectively. The challenge, however, is to specifically eliminate DDR in cancer cells without affecting the normal and necessary functions of DDR in non-cancerous cells.
Ataxia-telangiectasia mutated (ATM) is a kinase that regulates a number of substrates, including the phosphorylation of NBS1, which is required to initiate and enhance NBS1’s DDR activity. As such, several programs have attempted to develop various ATM inhibitors aimed to inhibit DDR (165). Unfortunately, ATM itself is not a specific therapeutic target because of its multiple domained nature, critical kinase function in normal cellular processes, and essential role in the maintenance of chromosome integrity at all phases of the cell cycle (166). Some studies reported that the level of phosphorylated NBS1 (Ser343), which is regulated by its upstream kinase ATM/ATR, is a critical phosphorylation status thought to increases DNA damage response and promotes cell survival. To test this theory, our lab overexpressed wild type NBS1, domain-negative NBS1 (S343A), or phospho-mimic NBS1 (S343E) in HT29 cells. Overexpression of any NBS1 vector rescued ~25% of cells from apoptosis mediated by GLI inhibition. Surprisingly, the overexpression of S343E, S343A, or total NBS1 was not statistically different from one another, indicating that total levels of NBS1, elevated by GLI1 transcription, rather than the phosphorylation status, were responsible for protection from GLI inhibition-induced apoptosis (64). Since GLI1 is not typically expressed by differentiated cells, targeting oncogenic expression of GLI1 would result in fewer off-target effects and provide a specific therapeutic strategy.
Most of the efforts to-date have typically focused on targeting GLI inhibition through the canonical Hh pathway, targeting upstream regulators like SMO, and subsequently sequestering GLI1 in the cytoplasm. Five SMO inhibitors have been approved by the FDA for clinical trials: vismodegib (GDC-0449), sonidegib (NPV-LDE-225), saridegib (IPI-926), BMS-833923, glasdegib (PF-04449913), and taladegib (LY2940680) (167). Variable success using SMO inhibitors has been demonstrated across a variety of different cancer types in preclinical models (30, 31, 107, 168–171) and clinical models (172–177). This is due to the predominant dependence of certain types of human cancers on canonical Hh signaling, such as basal cell carcinoma (173, 177), and medulloblastoma (172). However, clinical trials in most solid tumors have failed, likely because of aforementioned non-canonical activation pathways (i.e., RAS-ERK, PI3K-AKT-mTORS6K1 signaling, p53 loss, epigenetic alterations, etc.). Therefore, direct targeting of GLI might represent a better choice to improve the antitumor activity of these drugs in such cases.
The library of GLI1 antagonists is not as extensive as that for SMO. The most commonly used small molecules are GANT58 and GANT61, which were identified in a cell-based GLI-dependent luciferase screening system (178). These two compounds belong to different chemical classes, with GANT61 being a hexahydropyrimidine derivative and GANT58 possessing a thiophene core with four pyridine rings. Compared to GANT58, GANT61 is more specific toward GLI proteins and effectively reduces GLI1 and GLI2 DNA-binding ability, inhibiting the Hh pathway with a half maximal effective concentration (EC50) of 5 μM in GLI1-expressing HEK293T cells (26). GANT61 binds to the GLI1 protein between ZF2 and ZF3, by interacting with Glu119 and Glu167, as demonstrated by in silico docking on the crystal structure of the ZF domain of GLI1 bound to DNA (119). Experimental analysis shows that mutation of the predicted binding sites significantly reduces GANT61-GLI binding affinity. The GANT61 binding site is different from the GLI DNA-binding region, and the inhibitor is not able to bind to other ZF transcription factors such as KLF4 or TFIIβ (26, 119). Unfortunately, GANT61 is not usable as a translational therapeutic as it is unstable and has poor PK properties (179).
Using GANT61 as an initial scaffold, Southern Research has recently developed a novel GLI1 inhibitor (SRI-38832) that has better PK properties and has shown efficacy in vivo (64). Additionally, there are several promising compounds showing the biological activity of GLI inhibition (180), arsenic trioxide (ATO), originally approved by the FDA for the treatment of acute promyelocytic leulemia, has been shown to inhibit GLI proteins by binding to GLI proteins and enhancing degradation (181). ATO is currently being tested in multiple clinical trials ranging from phase I to phase IV for either solid tumors and hematologic malignancies. However, recent reports indicate lack of efficacy against small cell lung cancer (182). Polyunsaturated fatty acids (PUFAs) have also been reported to repress GLI1 expression by stimulating GLI1 suppressor, nuclear factor of activated T cells 1 (NFATc1) expression (183). Glabrescione B (GlaB), an isoflavone naturally found in the seeds of Derris glabrescens, is able to bind the GLI1 ZF domain, thereby diminishing GLI1/DNA interaction (184). Leadiant Biosciences used Glabrescione B as their scaffold for generating a pool of compounds for GLI1 inhibition (185, 186). Computational modeling of the DNA/GLI1 protein interaction has also been used to develop an 8-hydroxyquinolines as a GLI1 inhibitors, with similar scaffolds as Lediant Bioscience’s compounds (187). Finally, the Hedgehog pathway inhibitors (HPIs) including HPI-1, HPI-2, HPI-3, and HPI-4, were identified with a high-throughput screening for compounds capable of abolishing the Hh target gene expression induced by the SMO agonist SAG (188). HPI-1 can suppress Hh pathway activation, likely through targeting a posttranslational modification of the GLI proteins and/or an interaction between the transcription factor and a co-factor (189). The detailed mechanisms of action have not yet been completely unraveled.
One specific problem often encountered is the non-specificity of developed compounds claiming to be specific for GLI1 (i.e., also inhibit GLI2 and decrease GLI2 protein/messenger expression). The homology of GLI1 and GLI2, along with the similarities in the promotor recognition sequence makes it difficult to design an inhibitor of one without inadvertently targeting the other. Computational modeling and structural biology (NMR; crystallography) can help to resolve the challenge of non-specificity.
GLI1 exists at the conjunction of multiple oncogenic pathways outside of the canonically understood hedgehog pathway. In the scope of oncogenesis, GLI1 activation is particularly dominant in subsets of a number of cancer types because parallel non-canonical pathways outside of hedgehog signaling influence GLI1 function. Additionally, the list of GLI1 transcriptional targets continues to expand, encompassing cell cycle regulators (Cdt1), DNA damage repair proteins (NBS1), and proliferation (FOXM1). In certain cancers, it promotes a dedifferentiation to a more stem-like phenotype. Because of GLI1’s regulatory fluidity, targeting upstream pathway members is often an exercise in futility, as seen by the failure of SMO inhibitors, for example. For this reason, GLI1 is a significant therapeutic target for the treatment of multiple cancer types.
Whether overexpressed due to canonical, non-canonical, or genetic mutation, elevated GLI1 expression drives several of the hallmarks of cancer including DNA damage repair, cell proliferation, and metastasis. Rather than target upstream regulators of GLI, targeting the distal effector provides the greatest potential for therapeutic benefit. Since GLI1 is canonically active in embryonic development, with minimal basal expression in differentiated cells, it 1) serves as a biomarker for de-differentiation in cancer cells, particularly those refractory to treatment and 2) provides a prominent target not readily expressed in most non-cancerous tissue. As such, by targeting the downstream effector (GLI1) rather than upstream activators, we can effectively inhibit the oncogenesis driven by aberrant GLI1 activation, and promote cancer-specific DNA damage. While many promising drug discovery campaigns are developing and looking for novel GLI1 inhibitors, more work needs to be done to develop a potent, specific inhibitory compound.
JA and RZ compiled the information and wrote the manuscript. RB reviewed and corrected the manuscript. JA and RZ have contributed equally to this work and share first authorship. All authors contributed to the article and approved the submitted version.
This work was supported by The National Institutes of Health [5RO1CA183921-05].
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Kumar S, Balczarek KA, Lai ZC. Evolution of the Hedgehog Gene Family. Genetics (1996) 142(3):965–72. doi: 10.1093/genetics/142.3.965
2. McMahon AP. More Surprises in the Hedgehog Signaling Pathway. Cell (2000) 100(2):185–8. doi: 10.1016/s0092-8674(00)81555-x
3. Ingham PW, Nakano Y, Seger C. Mechanisms and Functions of Hedgehog Signalling Across the Metazoa. Nat Rev Genet (2011) 12(6):393–406. doi: 10.1038/nrg2984
4. Mohler J. Requirements for Hedgehog, a Segmental Polarity Gene, in Patterning Larval and Adult Cuticle of Drosophila. Genetics (1988) 120(4):1061–72. doi: 10.1093/genetics/120.4.1061
5. Belloni E, Muenke M, Roessler E, Traverso G, Siegel-Bartelt J, Frumkin A, et al. Identification of Sonic Hedgehog as a Candidate Gene Responsible for Holoprosencephaly. Nat Genet (1996) 14(3):353–6. doi: 10.1038/ng1196-353
6. Villavicencio EH, Walterhouse DO, Iannaccone PM. The Sonic Hedgehog-Patched-Gli Pathway in Human Development and Disease. Am J Hum Genet (2000) 67(5):1047–54. doi: 10.1016/S0002-9297(07)62934-6
7. Gritli-Linde A, Bei M, Maas R, Zhang XM, Linde A, McMahon AP. Shh Signaling Within the Dental Epithelium is Necessary for Cell Proliferation, Growth and Polarization. Development (2002) 129(23):5323–37. doi: 10.1242/dev.00100
8. Collins RT, Cohen SM. A Genetic Screen in Drosophila for Identifying Novel Components of the Hedgehog Signaling Pathway. Genetics (2005) 170(1):173–84. doi: 10.1534/genetics.104.039420
9. Stone DM, Hynes M, Armanini M, Swanson TA, Gu Q, Johnson RL, et al. The Tumour-Suppressor Gene Patched Encodes a Candidate Receptor for Sonic Hedgehog. Nature (1996) 384(6605):129–34. doi: 10.1038/384129a0
10. Smyth I, Narang MA, Evans T, Heimann C, Nakamura Y, Chenevix-Trench G, et al. Isolation and Characterization of Human Patched 2 (PTCH2), a Putative Tumour Suppressor Gene Inbasal Cell Carcinoma and Medulloblastoma on Chromosome 1p32. Hum Mol Genet (1999) 8(2):291–7. doi: 10.1093/hmg/8.2.291
11. Murone M, Rosenthal A, de Sauvage FJ. Sonic Hedgehog Signaling by the Patched-Smoothened Receptor Complex. Curr Biol (1999) 9(2):76–84. doi: 10.1016/s0960-9822(99)80018-9
12. Taipale J, Cooper MK, Maiti T, Beachy PA. Patched Acts Catalytically to Suppress the Activity of Smoothened. Nature (2002) 418(6900):892–7. doi: 10.1038/nature00989
13. Casali A, Struhl G. Reading the Hedgehog Morphogen Gradient by Measuring the Ratio of Bound to Unbound Patched Protein. Nature (2004) 431(7004):76–80. doi: 10.1038/nature02835
14. Pasca di Magliano M, Hebrok M. Hedgehog Signalling in Cancer Formation and Maintenance. Nat Rev Cancer (2003) 3(12):903–11. doi: 10.1038/nrc1229
15. Hegde GV, Munger CM, Emanuel K, Joshi AD, Greiner TC, Weisenburger DD, et al. Targeting of Sonic hedgehog-GLI Signaling: A Potential Strategy to Improve Therapy for Mantle Cell Lymphoma. Mol Cancer Ther (2008) 7(6):1450–60. doi: 10.1158/1535-7163.MCT-07-2118
16. Goodrich LV, Milenkovic L, Higgins KM, Scott MP. Altered Neural Cell Fates and Medulloblastoma in Mouse Patched Mutants. Science (1997) 277(5329):1109–13. doi: 10.1126/science.277.5329.1109
17. Oro AE, Higgins KM, Hu Z, Bonifas JM, Epstein EH Jr., Scott MP. Basal Cell Carcinomas in Mice Overexpressing Sonic Hedgehog. Science (1997) 276(5313):817–21. doi: 10.1126/science.276.5313.817
18. Watkins DN, Berman DM, Burkholder SG, Wang B, Beachy PA, Baylin SB. Hedgehog Signalling Within Airway Epithelial Progenitors and in Small-Cell Lung Cancer. Nature (2003) 422(6929):313–7. doi: 10.1038/nature01493
19. Bowman RV, Yang IA, Semmler AB, Fong KM. Epigenetics of Lung Cancer. Respirology (2006) 11(4):355–65. doi: 10.1111/j.1440-1843.2006.00859.x
20. Clement V, Sanchez P, de Tribolet N, Radovanovic I, Ruiz i Altaba A. Hedgehog-GLI1 Signaling Regulates Human Glioma Growth, Cancer Stem Cell Self-Renewal, and Tumorigenicity. Curr Biol (2007) 17(2):165–72. doi: 10.1016/j.cub.2006.11.033
21. Dierks C, Beigi R, Guo GR, Zirlik K, Stegert MR, Manley P, et al. Expansion of Bcr-Abl-positive Leukemic Stem Cells is Dependent on Hedgehog Pathway Activation. Cancer Cell (2008) 14(3):238–49. doi: 10.1016/j.ccr.2008.08.003
22. Goel A, Boland CR. Epigenetics of Colorectal Cancer. Gastroenterology (2012) 1431442–1460(6):e1441. doi: 10.1053/j.gastro.2012.09.032
23. Seeber LM, van Diest PJ. Epigenetics in Ovarian Cancer. Methods Mol Biol (2012) 863:253–69. doi: 10.1007/978-1-61779-612-8_15
24. Ozen C, Yildiz G, Dagcan AT, Cevik D, Ors A, Keles U, et al. Genetics and Epigenetics of Liver Cancer. N Biotechnol (2013) 30(4):381–4. doi: 10.1016/j.nbt.2013.01.007
25. Rimkus TK, Carpenter RL, Qasem S, Chan M, Lo HW. Targeting the Sonic Hedgehog Signaling Pathway: Review of Smoothened and GLI Inhibitors. Cancers (Basel) (2016) 8(2):22. doi: 10.3390/cancers8020022
26. Mastrangelo E, Milani M. Role and Inhibition of GLI1 Protein in Cancer. Lung Cancer (Auckl) (2018) 9:35–43. doi: 10.2147/LCTT.S124483
27. Zurawel RH, Allen C, Chiappa S, Cato W, Biegel J, Cogen P, et al. Analysis of PTCH/SMO/SHH Pathway Genes in Medulloblastoma. Genes Chromosomes Cancer (2000) 27(1):44–51. doi: 10.1002/(sici)1098-2264(200001)27:1<44::aid-gcc6>3.0.co;2-v
28. Pola R, Ling LE, Silver M, Corbley MJ, Kearney M, Blake Pepinsky R, et al. The Morphogen Sonic Hedgehog is an Indirect Angiogenic Agent Upregulating Two Families of Angiogenic Growth Factors. Nat Med (2001) 7(6):706–11. doi: 10.1038/89083
29. Walsh PC. Hedgehog Signalling in Prostate Regeneration, Neoplasia and Metastasis. J Urol (2005) 173(4):1169. doi: 10.1097/01.ju.0000156734.69186.57
30. Stecca B, Mas C, Clement V, Zbinden M, Correa R, Piguet V, et al. Melanomas Require HEDGEHOG-GLI Signaling Regulated by Interactions Between GLI1 and the RAS-MEK/AKT Pathways. Proc Natl Acad Sci USA (2007) 104(14):5895–900. doi: 10.1073/pnas.0700776104
31. Varnat F, Duquet A, Malerba M, Zbinden M, Mas C, Gervaz P, et al. Human Colon Cancer Epithelial Cells Harbour Active HEDGEHOG-GLI Signalling That is Essential for Tumour Growth, Recurrence, Metastasis and Stem Cell Survival and Expansion. EMBO Mol Med (2009) 1(6-7):338–51. doi: 10.1002/emmm.200900039
32. Xie J, Murone M, Luoh SM, Ryan A, Gu Q, Zhang C, et al. Activating Smoothened Mutations in Sporadic Basal-Cell Carcinoma. Nature (1998) 391(6662):90–2. doi: 10.1038/34201
33. Vorechovsky I, Unden AB, Sandstedt B, Toftgard R, Stahle-Backdahl M. Trichoepitheliomas Contain Somatic Mutations in the Overexpressed PTCH Gene: Support for a Gatekeeper Mechanism in Skin Tumorigenesis. Cancer Res (1997) 57(21):4677–81.
34. Maesawa C, Tamura G, Iwaya T, Ogasawara S, Ishida K, Sato N, et al. Mutations in the Human Homologue of the Drosophila Patched Gene in Esophageal Squamous Cell Carcinoma. Genes Chromosomes Cancer (1998) 21(3):276–9. doi: 10.1002/(SICI)1098-2264(199803)21:3<276::AID-GCC15>3.0.CO;2-N
35. McGarvey TW, Maruta Y, Tomaszewski JE, Linnenbach AJ, Malkowicz SB. PTCH Gene Mutations in Invasive Transitional Cell Carcinoma of the Bladder. Oncogene (1998) 17(9):1167–72. doi: 10.1038/sj.onc.1202045
36. Ruiz i Altaba A, Mas C, Stecca B. The Gli Code: An Information Nexus Regulating Cell Fate, Stemness and Cancer. Trends Cell Biol (2007) 17(9):438–47. doi: 10.1016/j.tcb.2007.06.007
37. Kinzler KW, Bigner SH, Bigner DD, Trent JM, Law ML, O’Brien SJ, et al. Identification of an Amplified, Highly Expressed Gene in a Human Glioma. Science (1987) 236(4797):70–3. doi: 10.1126/science.3563490
38. Gupta S, Takebe N, Lorusso P. Targeting the Hedgehog Pathway in Cancer. Ther Adv Med Oncol (2010) 2(4):237–50. doi: 10.1177/1758834010366430
39. Skoda AM, Simovic D, Karin V, Kardum V, Vranic S, Serman L. The Role of the Hedgehog Signaling Pathway in Cancer: A Comprehensive Review. Bosn J Basic Med Sci (2018) 18(1):8–20. doi: 10.17305/bjbms.2018.2756
40. Yao Z, Han L, Chen Y, He F, Sun B, Kamar S, et al. Hedgehog Signalling in the Tumourigenesis and Metastasis of Osteosarcoma, and its Potential Value in the Clinical Therapy of Osteosarcoma. Cell Death Dis (2018) 9(6):701. doi: 10.1038/s41419-018-0647-1
41. Wong AJ, Bigner SH, Bigner DD, Kinzler KW, Hamilton SR, Vogelstein B. Increased Expression of the Epidermal Growth Factor Receptor Gene in Malignant Gliomas is Invariably Associated With Gene Amplification. Proc Natl Acad Sci USA (1987) 84(19):6899–903. doi: 10.1073/pnas.84.19.6899
42. Ruppert JM, Kinzler KW, Wong AJ, Bigner SH, Kao FT, Law ML, et al. The GLI-Kruppel Family of Human Genes. Mol Cell Biol (1988) 8(8):3104–13. doi: 10.1128/mcb.8.8.3104
43. Winklmayr M, Schmid C, Laner-Plamberger S, Kaser A, Aberger F, Eichberger T, et al. Non-Consensus GLI Binding Sites in Hedgehog Target Gene Regulation. BMC Mol Biol (2010) 11:2. doi: 10.1186/1471-2199-11-2
44. Bauer NC, Doetsch PW, Corbett AH. Mechanisms Regulating Protein Localization. Traffic (2015) 16(10):1039–61. doi: 10.1111/tra.12310
45. Han Y, Shi Q, Jiang J. Multisite Interaction With Sufu Regulates Ci/Gli Activity Through Distinct Mechanisms in Hh Signal Transduction. Proc Natl Acad Sci USA (2015) 112(20):6383–8. doi: 10.1073/pnas.1421628112
46. Han Y, Xiong Y, Shi X, Wu J, Zhao Y, Jiang J. Regulation of Gli Ciliary Localization and Hedgehog Signaling by the PY-NLS/karyopherin-beta2 Nuclear Import System. PloS Biol (2017) 15(8):e2002063. doi: 10.1371/journal.pbio.2002063
47. Carpenter RL, Lo HW. Hedgehog Pathway and GLI1 Isoforms in Human Cancer. Discovery Med (2012) 13(69):105–13.
48. Roessler E, Ermilov AN, Grange DK, Wang A, Grachtchouk M, Dlugosz AA, et al. A Previously Unidentified Amino-Terminal Domain Regulates Transcriptional Activity of Wild-Type and Disease-Associated Human GLI2. Hum Mol Genet (2005) 14(15):2181–8. doi: 10.1093/hmg/ddi222
49. Speek M, Njunkova O, Pata I, Valdre E, Kogerman P. A Potential Role of Alternative Splicing in the Regulation of the Transcriptional Activity of Human GLI2 in Gonadal Tissues. BMC Mol Biol (2006) 7:13. doi: 10.1186/1471-2199-7-13
50. Grachtchouk M, Pero J, Yang SH, Ermilov AN, Michael LE, Wang A, et al. Basal Cell Carcinomas in Mice Arise From Hair Follicle Stem Cells and Multiple Epithelial Progenitor Populations. J Clin Invest (2011) 121(5):1768–81. doi: 10.1172/JCI46307
51. Pantazi E, Gemenetzidis E, Trigiante G, Warnes G, Shan L, Mao X, et al. GLI2 Induces Genomic Instability in Human Keratinocytes by Inhibiting Apoptosis. Cell Death Dis (2014) 5:e1028. doi: 10.1038/cddis.2013.535
52. Tsanev R, Tiigimagi P, Michelson P, Metsis M, Osterlund T, Kogerman P. Identification of the Gene Transcription Repressor Domain of Gli3. FEBS Lett (2009) 583(1):224–8. doi: 10.1016/j.febslet.2008.12.010
53. Shimokawa T, Tostar U, Lauth M, Palaniswamy R, Kasper M, Toftgard R, et al. Novel Human Glioma-Associated Oncogene 1 (GLI1) Splice Variants Reveal Distinct Mechanisms in the Terminal Transduction of the Hedgehog Signal. J Biol Chem (2008) 283(21):14345–54. doi: 10.1074/jbc.M800299200
54. Pietrobono S, Gagliardi S, Stecca B. Non-Canonical Hedgehog Signaling Pathway in Cancer: Activation of GLI Transcription Factors Beyond Smoothened. Front Genet (2019) 10:556. doi: 10.3389/fgene.2019.00556
55. Lo HW, Zhu H, Cao X, Aldrich A, Ali-Osman F. A Novel Splice Variant of GLI1 That Promotes Glioblastoma Cell Migration and Invasion. Cancer Res (2009) 69(17):6790–8. doi: 10.1158/0008-5472.CAN-09-0886
56. Cao X, Geradts J, Dewhirst MW, Lo HW. Upregulation of VEGF-A and CD24 Gene Expression by the tGLI1 Transcription Factor Contributes to the Aggressive Behavior of Breast Cancer Cells. Oncogene (2012) 31(1):104–15. doi: 10.1038/onc.2011.219
57. Justilien V, Fields AP. Molecular Pathways: Novel Approaches for Improved Therapeutic Targeting of Hedgehog Signaling in Cancer Stem Cells. Clin Cancer Res (2015) 21(3):505–13. doi: 10.1158/1078-0432.CCR-14-0507
58. Hanna A, Shevde LA. Hedgehog Signaling: Modulation of Cancer Properies and Tumor Mircroenvironment. Mol Cancer (2016) 15:24. doi: 10.1186/s12943-016-0509-3
59. van den Brink GR, Bleuming SA, Hardwick JC, Schepman BL, Offerhaus GJ, Keller JJ, et al. Indian Hedgehog is an Antagonist of Wnt Signaling in Colonic Epithelial Cell Differentiation. Nat Genet (2004) 36(3):277–82. doi: 10.1038/ng1304
60. Taniguchi H, Yamamoto H, Akutsu N, Nosho K, Adachi Y, Imai K, et al. Transcriptional Silencing of Hedgehog-Interacting Protein by CpG Hypermethylation and Chromatic Structure in Human Gastrointestinal Cancer. J Pathol (2007) 213(2):131–9. doi: 10.1002/path.2216
61. Shi T, Mazumdar T, Devecchio J, Duan ZH, Agyeman A, Aziz M, et al. cDNA Microarray Gene Expression Profiling of Hedgehog Signaling Pathway Inhibition in Human Colon Cancer Cells. PloS One (2010) 5(10):E13054. doi: 10.1371/journal.pone.0013054
62. Stefanius K, Kantola T, Tuomisto A, Vahteristo P, Karttunen TJ, Aaltonen LA, et al. Downregulation of the Hedgehog Receptor PTCH1 in Colorectal Serrated Adenocarcinomas is Not Caused by PTCH1 Mutations. Virchows Arch (2011) 458(2):213–9. doi: 10.1007/s00428-010-1031-4
63. Li T, Liao X, Lochhead P, Morikawa T, Yamauchi M, Nishihara R, et al. SMO Expression in Colorectal Cancer: Associations With Clinical, Pathological, and Molecular Features. Ann Surg Oncol (2014) 21(13):4164–73. doi: 10.1245/s10434-014-3888-y
64. Zhang R, Ma J, Avery JT, Sambandam V, Nguyen TH, Xu B, et al. Gli1 Inhibitor Sri-38832 Attenuates Chemotherapeutic Resistance by Downregulating Nbs1 Transcription in BRAF(V600E) Colorectal Cancer. Front Oncol (2020) 10:241. doi: 10.3389/fonc.2020.00241
65. Dlugosz A, Agrawal S, Kirkpatrick P. Vismodegib. Nat Rev Drug Discov (2012) 11(6):437–8. doi: 10.1038/nrd3753
66. Berlin J, Bendell JC, Hart LL, Firdaus I, Gore I, Hermann RC, et al. A Randomized Phase II Trial of Vismodegib Versus Placebo With FOLFOX or FOLFIRI and Bevacizumab in Patients With Previously Untreated Metastatic Colorectal Cancer. Clin Cancer Res (2013) 19(1):258–67. doi: 10.1158/1078-0432.CCR-12-1800
67. Taipale J, Chen JK, Cooper MK, Wang B, Mann RK, Milenkovic L, et al. Effects of Oncogenic Mutations in Smoothened and Patched can be Reversed by Cyclopamine. Nature (2000) 406(6799):1005–9. doi: 10.1038/35023008
68. Das S, Samant RS, Shevde LA. Nonclassical Activation of Hedgehog Signaling Enhances Multidrug Resistance and Makes Cancer Cells Refractory to Smoothened-targeting Hedgehog Inhibition. J Biol Chem (2013) 288(17):11824–33. doi: 10.1074/jbc.M112.432302
69. Palle K, Mani C, Tripathi K, Athar M. Aberrant GLI1 Activation in DNA Damage Response, Carcinogenesis and Chemoresistance. Cancers (Basel) (2015) 7(4):2330–51. doi: 10.3390/cancers7040894
70. Samatar AA, Poulikakos PI. Targeting RAS-ERK Signalling in Cancer: Promises and Challenges. Nat Rev Drug Discov (2014) 13(12):928–42. doi: 10.1038/nrd4281
71. Rovida E, Stecca B. Mitogen-Activated Protein Kinases and Hedgehog-GLI Signaling in Cancer: A Crosstalk Providing Therapeutic Opportunities? Semin Cancer Biol (2015) 35:154–67. doi: 10.1016/j.semcancer.2015.08.003
72. Seto M, Ohta M, Asaoka Y, Ikenoue T, Tada M, Miyabayashi K, et al. Regulation of the Hedgehog Signaling by the Mitogen-Activated Protein Kinase Cascade in Gastric Cancer. Mol Carcinog (2009) 48(8):703–12. doi: 10.1002/mc.20516
73. Schnidar H, Eberl M, Klingler S, Mangelberger D, Kasper M, Hauser-Kronberger C, et al. Epidermal Growth Factor Receptor Signaling Synergizes With Hedgehog/GLI in Oncogenic Transformation Via Activation of the MEK/ERK/JUN Pathway. Cancer Res (2009) 69(4):1284–92. doi: 10.1158/0008-5472.CAN-08-2331
74. Schneider P, Bayo-Fina JM, Singh R, Kumar Dhanyamraju P, Holz P, Baier A, et al. Identification of a Novel Actin-Dependent Signal Transducing Module Allows for the Targeted Degradation of GLI1. Nat Commun (2015) 6:8023. doi: 10.1038/ncomms9023
75. Savona MR, Pollyea DA, Stock W, Oehler VG, Schroeder MA, Lancet J, et al. Phase Ib Study of Glasdegib, a Hedgehog Pathway Inhibitor, in Combination With Standard Chemotherapy in Patients With AML or High-Risk Mds. Clin Cancer Res (2018) 24(10):2294–303. doi: 10.1158/1078-0432.CCR-17-2824
76. Riobo NA, Lu K, Ai X, Haines GM, Emerson CP Jr. Phosphoinositide 3-Kinase and Akt are Essential for Sonic Hedgehog Signaling. Proc Natl Acad Sci U.S.A. (2006) 103(12):4505–10. doi: 10.1073/pnas.0504337103
77. Singh R, Dhanyamraju PK, Lauth M. DYRK1B Blocks Canonical and Promotes non-Canonical Hedgehog Signaling Through Activation of the mTOR/AKT Pathway. Oncotarget (2017) 8(1):833–45. doi: 10.18632/oncotarget.13662
78. Wang Y, Ding Q, Yen CJ, Xia W, Izzo JG, Lang JY, et al. The Crosstalk of mTOR/S6K1 and Hedgehog Pathways. Cancer Cell (2012) 21(3):374–87. doi: 10.1016/j.ccr.2011.12.028
79. Mizuarai S, Kawagishi A, Kotani H. Inhibition of p70S6K2 Down-Regulates Hedgehog/GLI Pathway in non-Small Cell Lung Cancer Cell Lines. Mol Cancer (2009) 8:44. doi: 10.1186/1476-4598-8-44
80. Jones S, Zhang X, Parsons DW, Lin JC, Leary RJ, Angenendt P, et al. Core Signaling Pathways in Human Pancreatic Cancers Revealed by Global Genomic Analyses. Science (2008) 321(5897):1801–6. doi: 10.1126/science.1164368
81. Principe DR, DeCant B, Mascarinas E, Wayne EA, Diaz AM, Akagi N, et al. Tgfbeta Signaling in the Pancreatic Tumor Microenvironment Promotes Fibrosis and Immune Evasion to Facilitate Tumorigenesis. Cancer Res (2016) 76(9):2525–39. doi: 10.1158/0008-5472.CAN-15-1293
82. Grusch M, Petz M, Metzner T, Ozturk D, Schneller D, Mikulits W. The Crosstalk of RAS With the TGF-beta Family During Carcinoma Progression and its Implications for Targeted Cancer Therapy. Curr Cancer Drug Targets (2010) 10(8):849–57. doi: 10.2174/156800910793357943
83. Nye MD, Almada LL, Fernandez-Barrena MG, Marks DL, Elsawa SF, Vrabel A, et al. The Transcription Factor GLI1 Interacts With SMAD Proteins to Modulate Transforming Growth Factor Beta-Induced Gene Expression in a p300/CREB-binding Protein-Associated Factor (PCAF)-Dependent Manner. J Biol Chem (2014) 289(22):15495–506. doi: 10.1074/jbc.M113.545194
84. Nolan-Stevaux O, Lau J, Truitt ML, Chu GC, Hebrok M, Fernandez-Zapico ME, et al. GLI1 is Regulated Through Smoothened-independent Mechanisms in Neoplastic Pancreatic Ducts and Mediates PDAC Cell Survival and Transformation. Genes Dev (2009) 23(1):24–36. doi: 10.1101/gad.1753809
85. Yoon JW, Gallant M, Lamm ML, Iannaccone S, Vieux KF, Proytcheva M, et al. Noncanonical Regulation of the Hedgehog Mediator GLI1 by c-MYC in Burkitt Lymphoma. Mol Cancer Res (2013) 11(6):604–15. doi: 10.1158/1541-7786.MCR-12-0441
86. Goel HL, Pursell B, Chang C, Shaw LM, Mao J, Simin K, et al. GLI1 Regulates a Novel neuropilin-2/alpha6beta1 Integrin Based Autocrine Pathway That Contributes to Breast Cancer Initiation. EMBO Mol Med (2013) 5(4):488–508. doi: 10.1002/emmm.201202078
87. Atwood SX, Li M, Lee A, Tang JY, Oro AE. GLI Activation by Atypical Protein Kinase C Iota/Lambda Regulates the Growth of Basal Cell Carcinomas. Nature (2013) 494(7438):484–8. doi: 10.1038/nature11889
88. Jiang K, Liu Y, Fan J, Epperly G, Gao T, Jiang J, et al. Hedgehog-regulated Atypical PKC Promotes Phosphorylation and Activation of Smoothened and Cubitus Interruptus in Drosophila. Proc Natl Acad Sci USA (2014) 111(45):E4842–50. doi: 10.1073/pnas.1417147111
89. Stecca B, Ruiz i Altaba A. A GLI1-p53 Inhibitory Loop Controls Neural Stem Cell and Tumour Cell Numbers. EMBO J (2009) 28(6):663–76. doi: 10.1038/emboj.2009.16
90. Sjoblom T, Jones S, Wood LD, Parsons DW, Lin J, Barber TD, et al. The Consensus Coding Sequences of Human Breast and Colorectal Cancers. Science (2006) 314(5797):268–74. doi: 10.1126/science.1133427
91. Lees CW, Zacharias WJ, Tremelling M, Noble CL, Nimmo ER, Tenesa A, et al. Analysis of Germline GLI1 Variation Implicates Hedgehog Signalling in the Regulation of Intestinal Inflammatory Pathways. PloS Med (2008) 5(12):e239. doi: 10.1371/journal.pmed.0050239
92. Liu JA, Lai FP, Gui HS, Sham MH, Tam PK, Garcia-Barcelo MM, et al. Identification of GLI Mutations in Patients With Hirschsprung Disease That Disrupt Enteric Nervous System Development in Mice. Gastroenterology (2015) 1491837-1848(7):e1835. doi: 10.1053/j.gastro.2015.07.060
93. Lucchesi C, Khalifa E, Laizet Y, Soubeyran I, Mathoulin-Pelissier S, Chomienne C, et al. Targetable Alterations in Adult Patients With Soft-Tissue Sarcomas: Insights for Personalized Therapy. JAMA Oncol (2018) 4(10):1398–404. doi: 10.1001/jamaoncol.2018.0723
94. Newell F, Kong Y, Wilmott JS, Johansson PA, Ferguson PM, Cui C, et al. Whole-Genome Landscape of Mucosal Melanoma Reveals Diverse Drivers and Therapeutic Targets. Nat Commun (2019) 10(1):3163. doi: 10.1038/s41467-019-11107-x
95. Wilmott JS, Johansson PA, Newell F, Waddell N, Ferguson P, Quek C, et al. Whole Genome Sequencing of Melanomas in Adolescent and Young Adults Reveals Distinct Mutation Landscapes and the Potential Role of Germline Variants in Disease Susceptibility. Int J Cancer (2019) 144(5):1049–60. doi: 10.1002/ijc.31791
96. Dahlen A, Mertens F, Mandahl N, Panagopoulos I. Molecular Genetic Characterization of the Genomic ACTB-GLI Fusion in Pericytoma With T(7;12). Biochem Biophys Res Commun (2004) 325(4):1318–23. doi: 10.1016/j.bbrc.2004.10.172
97. Kerr DA, Pinto A, Subhawong TK, Wilky BA, Schlumbrecht MP, Antonescu CR, et al. Pericytoma With T(7;12) and ACTB-GLI1 Fusion: Reevaluation of an Unusual Entity and its Relationship to the Spectrum of GLI1 Fusion-Related Neoplasms. Am J Surg Pathol (2019) 43(12):1682–92. doi: 10.1097/PAS.0000000000001360
98. Antonescu CR, Agaram NP, Sung YS, Zhang L, Swanson D, Dickson BC. A Distinct Malignant Epithelioid Neoplasm With Gli1 Gene Rearrangements, Frequent S100 Protein Expression, and Metastatic Potential: Expanding the Spectrum of Pathologic Entities With Actb/Malat1/Ptch1-GLI1 Fusions. Am J Surg Pathol (2018) 42(4):553–60. doi: 10.1097/PAS.0000000000001010
99. Bai CB, Auerbach W, Lee JS, Stephen D, Joyner AL. Gli2, But Not Gli1, is Required for Initial Shh Signaling and Ectopic Activation of the Shh Pathway. Development (2002) 129(20):4753–61. doi: 10.1242/dev.129.20.4753
100. Ikram MS, Neill GW, Regl G, Eichberger T, Frischauf AM, Aberger F, et al. GLI2 is Expressed in Normal Human Epidermis and BCC and Induces GLI1 Expression by Binding to its Promoter. J Invest Dermatol (2004) 122(6):1503–9. doi: 10.1111/j.0022-202X.2004.22612.x
101. Thiyagarajan S, Bhatia N, Reagan-Shaw S, Cozma D, Thomas-Tikhonenko A, Ahmad N, et al. Role of GLI2 Transcription Factor in Growth and Tumorigenicity of Prostate Cells. Cancer Res (2007) 67(22):10642–6. doi: 10.1158/0008-5472.CAN-07-2015
102. Katoh Y, Katoh M. Hedgehog Target Genes: Mechanisms of Carcinogenesis Induced by Aberrant Hedgehog Signaling Activation. Curr Mol Med (2009) 9(7):873–86. doi: 10.2174/156652409789105570
103. Kasper M, Schnidar H, Neill GW, Hanneder M, Klingler S, Blaas L, et al. Selective Modulation of Hedgehog/GLI Target Gene Expression by Epidermal Growth Factor Signaling in Human Keratinocytes. Mol Cell Biol (2006) 26(16):6283–98. doi: 10.1128/MCB.02317-05
104. Ruiz i Altaba A. Gli Proteins Encode Context-Dependent Positive and Negative Functions: Implications for Development and Disease. Development (1999) 126(14):3205–16. doi: 10.1242/dev.126.14.3205
105. Khatib ZA, Matsushime H, Valentine M, Shapiro DN, Sherr CJ, Look AT. Coamplification of the CDK4 Gene With MDM2 and GLI in Human Sarcomas. Cancer Res (1993) 53(22):5535–41.
106. Zurawel RH, Allen C, Wechsler-Reya R, Scott MP, Raffel C. Evidence That Haploinsufficiency of Ptch Leads to Medulloblastoma in Mice. Genes Chromosomes Cancer (2000) 28(1):77–81. doi: 10.1002/(SICI)1098-2264(200005)28:1<77::AID-GCC9>3.0.CO;2-Y
107. Sanchez P, Hernandez AM, Stecca B, Kahler AJ, DeGueme AM, Barrett A, et al. Inhibition of Prostate Cancer Proliferation by Interference With SONIC Hedgehog-GLI1 Signaling. Proc Natl Acad Sci USA (2004) 101(34):12561–6. doi: 10.1073/pnas.0404956101
108. Hu X, Lai D, Chen W, Zi S, Li J, Du P, et al. Differential Expression Profiles of the Hedgehog Signaling Pathway Between Microsatellite-Stable and Microsatellite-Unstable Colorectal Cancers. Mol Med Rep (2011) 4(5):873–7. doi: 10.3892/mmr.2011.529
109. Kasper M, Regl G, Frischauf AM, Aberger F. GLI Transcription Factors: Mediators of Oncogenic Hedgehog Signalling. Eur J Cancer (2006) 42(4):437–45. doi: 10.1016/j.ejca.2005.08.039
110. Eichberger T, Sander V, Schnidar H, Regl G, Kasper M, Schmid C, et al. Overlapping and Distinct Transcriptional Regulator Properties of the GLI1 and GLI2 Oncogenes. Genomics (2006) 87(5):616–32. doi: 10.1016/j.ygeno.2005.12.003
111. Yoshikawa K, Shimada M, Miyamoto H, Higashijima J, Miyatani T, Nishioka M, et al. Sonic Hedgehog Relates to Colorectal Carcinogenesis. J Gastroenterol (2009) 44(11):1113–7. doi: 10.1007/s00535-009-0110-2
112. Mazumdar T, DeVecchio J, Shi T, Jones J, Agyeman A, Houghton JA. Hedgehog Signaling Drives Cellular Survival in Human Colon Carcinoma Cells. Cancer Res (2011) 71(3):1092–102. doi: 10.1158/0008-5472.CAN-10-2315
113. Varnat F, Zacchetti G, Ruiz i Altaba A. Hedgehog Pathway Activity is Required for the Lethality and Intestinal Phenotypes of Mice With Hyperactive Wnt Signaling. Mech Dev (2010) 127(1-2):73–81. doi: 10.1016/j.mod.2009.10.005
114. Wu C, Zhu X, Liu W, Ruan T, Tao K. Hedgehog Signaling Pathway in Colorectal Cancer: Function, Mechanism, and Therapy. Onco Targets Ther (2017) 10:3249–59. doi: 10.2147/OTT.S139639
115. N IJ, Rijnierse A, de Wit N, Jonker-Termont D, Dekker J, Muller M, et al. Dietary Haem Stimulates Epithelial Cell Turnover by Downregulating Feedback Inhibitors of Proliferation in Murine Colon. Gut (2012) 61(7):1041–9. doi: 10.1136/gutjnl-2011-300239
116. Song J, Zhang J, Wang J, Wang J, Guo X, Dong W. Beta1 Integrin Mediates Colorectal Cancer Cell Proliferation and Migration Through Regulation of the Hedgehog Pathway. Tumour Biol (2015) 36(3):2013–21. doi: 10.1007/s13277-014-2808-x
117. Gulino A, Ferretti E, De Smaele E. Hedgehog Signalling in Colon Cancer and Stem Cells. EMBO Mol Med (2009) 1(6-7):300–2. doi: 10.1002/emmm.200900042
118. Chowdhury S, Pradhan RN, Sarkar RR. Structural and Logical Analysis of a Comprehensive Hedgehog Signaling Pathway to Identify Alternative Drug Targets for Glioma, Colon and Pancreatic Cancer. PloS One (2013) 8(7):e69132. doi: 10.1371/journal.pone.0069132
119. Agyeman A, Jha BK, Mazumdar T, Houghton JA. Mode and Specificity of Binding of the Small Molecule GANT61 to GLI Determines Inhibition of GLI-DNA Binding. Oncotarget (2014) 5(12):4492–503. doi: 10.18632/oncotarget.2046
120. Ciucci A, De Stefano I, Vellone VG, Lisi L, Bottoni C, Scambia G, et al. Expression of the Glioma-Associated Oncogene Homolog 1 (gli1) in Advanced Serous Ovarian Cancer is Associated With Unfavorable Overall Survival. PloS One (2013) 8(3):e60145. doi: 10.1371/journal.pone.0060145
121. Chung JH, Larsen AR, Chen E, Bunz F. A PTCH1 Homolog Transcriptionally Activated by p53 Suppresses Hedgehog Signaling. J Biol Chem (2014) 289(47):33020–31. doi: 10.1074/jbc.M114.597203
122. Ramasamy TS, Ayob AZ, Myint HH, Thiagarajah S, Amini F. Targeting Colorectal Cancer Stem Cells Using Curcumin and Curcumin Analogues: Insights Into the Mechanism of the Therapeutic Efficacy. Cancer Cell Int (2015) 15:96. doi: 10.1186/s12935-015-0241-x
123. Curtin JC, Lorenzi MV. Drug Discovery Approaches to Target Wnt Signaling in Cancer Stem Cells. Oncotarget (2010) 1(7):563–77. doi: 10.18632/oncotarget.191
124. Roy S, Majumdar AP. Signaling in Colon Cancer Stem Cells. J Mol Signal (2012) 7(1):11. doi: 10.1186/1750-2187-7-11
125. Ramirez A, Boulaiz H, Morata-Tarifa C, Peran M, Jimenez G, Picon-Ruiz M, et al. HER2-Signaling Pathway, JNK and ERKs Kinases, and Cancer Stem-Like Cells are Targets of Bozepinib Small Compound. Oncotarget (2014) 5(11):3590–606. doi: 10.18632/oncotarget.1962
126. So JY, Suh N. Targeting Cancer Stem Cells in Solid Tumors by Vitamin D. J Steroid Biochem Mol Biol (2015) 148:79–85. doi: 10.1016/j.jsbmb.2014.10.007
127. Zhang X, Zhang SS, Wei GJ, Deng ZM, Hu Y. Dysregulation of Hedgehog Signaling Pathway Related Components in the Evolution of Colonic Carcinogenesis. Int J Clin Exp Med (2015) 8(11):21379–85.
128. Miyazaki Y, Matsubara S, Ding Q, Tsukasa K, Yoshimitsu M, Kosai K, et al. Efficient Elimination of Pancreatic Cancer Stem Cells by Hedgehog/GLI Inhibitor GANT61 in Combination With mTOR Inhibition. Mol Cancer (2016) 15(1):49. doi: 10.1186/s12943-016-0534-2
129. Yang Z, Cui Y, Ni W, Kim S, Xuan Y. Gli1, a Potential Regulator of Esophageal Cancer Stem Cell, is Identified as an Independent Adverse Prognostic Factor in Esophageal Squamous Cell Carcinoma. J Cancer Res Clin Oncol (2017) 143(2):243–54. doi: 10.1007/s00432-016-2273-6
130. Lv L, Yang Z, Ma T, Xuan Y. Gli1, a Potential Cancer Stem Cell Marker, is Strongly Associated With Prognosis in Prostate Cancer. Int J Clin Exp Pathol (2018) 11(10):4957–66.
131. Yang Z, Zhang C, Qi W, Cui Y, Xuan Y. GLI1 Promotes Cancer Stemness Through Intracellular Signaling Pathway PI3K/Akt/NFkappaB in Colorectal Adenocarcinoma. Exp Cell Res (2018) 373(1-2):145–54. doi: 10.1016/j.yexcr.2018.10.006
132. Li D, Xie K, Wolff R, Abbruzzese JL. Pancreatic Cancer. Lancet (2004) 363(9414):1049–57. doi: 10.1016/S0140-6736(04)15841-8
133. Stathis A, Moore MJ. Advanced Pancreatic Carcinoma: Current Treatment and Future Challenges. Nat Rev Clin Oncol (2010) 7(3):163–72. doi: 10.1038/nrclinonc.2009.236
134. Grutzmann R, Pilarsky C, Ammerpohl O, Luttges J, Bohme A, Sipos B, et al. Gene Expression Profiling of Microdissected Pancreatic Ductal Carcinomas Using High-Density DNA Microarrays. Neoplasia (2004) 6(5):611–22. doi: 10.1593/neo.04295
135. Lim KH, Counter CM. Reduction in the Requirement of Oncogenic Ras Signaling to Activation of PI3K/AKT Pathway During Tumor Maintenance. Cancer Cell (2005) 8(5):381–92. doi: 10.1016/j.ccr.2005.10.014
136. Collins MA, Bednar F, Zhang Y, Brisset JC, Galban S, Galban CJ, et al. Oncogenic Kras is Required for Both the Initiation and Maintenance of Pancreatic Cancer in Mice. J Clin Invest (2012) 122(2):639–53. doi: 10.1172/JCI59227
137. Hahn SA, Schmiegel WH. Recent Discoveries in Cancer Genetics of Exocrine Pancreatic Neoplasia. Digestion (1998) 59(5):493–501. doi: 10.1159/000007526
138. Scheffzek K, Ahmadian MR, Kabsch W, Wiesmuller L, Lautwein A, Schmitz F, et al. The Ras-RasGAP Complex: Structural Basis for GTPase Activation and its Loss in Oncogenic Ras Mutants. Science (1997) 277(5324):333–8. doi: 10.1126/science.277.5324.333
139. Scheidig AJ, Burmester C, Goody RS. The Pre-Hydrolysis State of p21(ras) in Complex With GTP: New Insights Into the Role of Water Molecules in the GTP Hydrolysis Reaction of Ras-Like Proteins. Structure (1999) 7(11):1311–24. doi: 10.1016/s0969-2126(00)80021-0
140. Kim ST, Lim DH, Jang KT, Lim T, Lee J, Choi YL, et al. Impact of KRAS Mutations on Clinical Outcomes in Pancreatic Cancer Patients Treated With First-Line Gemcitabine-Based Chemotherapy. Mol Cancer Ther (2011) 10(10):1993–9. doi: 10.1158/1535-7163.MCT-11-0269
141. Kotoula V, Charalambous E, Biesmans B, Malousi A, Vrettou E, Fountzilas G, et al. Targeted KRAS Mutation Assessment on Patient Tumor Histologic Material in Real Time Diagnostics. PloS One (2009) 4(11):e7746. doi: 10.1371/journal.pone.0007746
142. Tuveson DA, Shaw AT, Willis NA, Silver DP, Jackson EL, Chang S, et al. Endogenous Oncogenic K-ras(G12D) Stimulates Proliferation and Widespread Neoplastic and Developmental Defects. Cancer Cell (2004) 5(4):375–87. doi: 10.1016/s1535-6108(04)00085-6
143. Ji Z, Mei FC, Xie J, Cheng X. Oncogenic KRAS Activates Hedgehog Signaling Pathway in Pancreatic Cancer Cells. J Biol Chem (2007) 282(19):14048–55. doi: 10.1074/jbc.M611089200
144. Fabian D, Guillermo Prieto Eibl MDP, Alnahhas I, Sebastian N, Giglio P, Puduvalli V, et al. Treatment of Glioblastoma (GBM) With the Addition of Tumor-Treating Fields (Ttf): A Review. Cancers (Basel) (2019) 11(2):174. doi: 10.3390/cancers11020174
145. Lee SY. Temozolomide Resistance in Glioblastoma Multiforme. Genes Dis (2016) 3(3):198–210. doi: 10.1016/j.gendis.2016.04.007
146. Jiapaer S, Furuta T, Tanaka S, Kitabayashi T, Nakada M. Potential Strategies Overcoming the Temozolomide Resistance for Glioblastoma. Neurol Med Chir (Tokyo) (2018) 58(10):405–21. doi: 10.2176/nmc.ra.2018-0141
147. Wick W, Weller M, van den Bent M, Sanson M, Weiler M, von Deimling A, et al. MGMT Testing–the Challenges for Biomarker-Based Glioma Treatment. Nat Rev Neurol (2014) 10(7):372–85. doi: 10.1038/nrneurol.2014.100
148. Wang K, Chen D, Qian Z, Cui D, Gao L, Lou M. Hedgehog/Gli1 Signaling Pathway Regulates MGMT Expression and Chemoresistance to Temozolomide in Human Glioblastoma. Cancer Cell Int (2017) 17:117. doi: 10.1186/s12935-017-0491-x
149. Yan W, Zhang W, You G, Zhang J, Han L, Bao Z, et al. Molecular Classification of Gliomas Based on Whole Genome Gene Expression: A Systematic Report of 225 Samples From the Chinese Glioma Cooperative Group. Neuro Oncol (2012) 14(12):1432–40. doi: 10.1093/neuonc/nos263
150. Li J, Cai J, Zhao S, Yao K, Sun Y, Li Y, et al. GANT61, a GLI Inhibitor, Sensitizes Glioma Cells to the Temozolomide Treatment. J Exp Clin Cancer Res (2016) 35(1):184. doi: 10.1186/s13046-016-0463-3
151. Melamed JR, Morgan JT, Ioele SA, Gleghorn JP, Sims-Mourtada J, Day ES. Investigating the Role of Hedgehog/GLI1 Signaling in Glioblastoma Cell Response to Temozolomide. Oncotarget (2018) 9(43):27000–15. doi: 10.18632/oncotarget.25467
152. Zafar A, Wang W, Liu G, Wang X, Xian W, McKeon F, et al. Molecular Targeting Therapies for Neuroblastoma: Progress and Challenges. Med Res Rev (2020) 41(2):961–1021. doi: 10.1002/med.21750
153. Koche RP, Rodriguez-Fos E, Helmsauer K, Burkert M, MacArthur IC, Maag J, et al. Extrachromosomal Circular DNA Drives Oncogenic Genome Remodeling in Neuroblastoma. Nat Genet (2020) 52(1):29–34. doi: 10.1038/s41588-019-0547-z
154. Ain Q, Schmeer C, Wengerodt D, Witte OW, Kretz A. Extrachromosomal Circular Dna: Current Knowledge and Implications for CNS Aging and Neurodegeneration. Int J Mol Sci (2020) 21(7):2477. doi: 10.3390/ijms21072477
155. Zhang R, Wu J, Ferrandon S, Glowacki KJ, Houghton JA. Targeting GLI by GANT61 Involves Mechanisms Dependent on Inhibition of Both Transcription and DNA Licensing. Oncotarget (2016) 7(49):80190–207. doi: 10.18632/oncotarget.13376
156. Li X, Deng W, Lobo-Ruppert SM, Ruppert JM. Gli1 Acts Through Snail and E-cadherin to Promote Nuclear Signaling by Beta-Catenin. Oncogene (2007) 26(31):4489–98. doi: 10.1038/sj.onc.1210241
157. Zheng X, Song T, Dou C, Jia Y, Liu Q. CtBP2 is an Independent Prognostic Marker That Promotes GLI1 Induced Epithelial-Mesenchymal Transition in Hepatocellular Carcinoma. Oncotarget (2015) 6(6):3752–69. doi: 10.18632/oncotarget.2915
158. Liao X, Siu MK, Au CW, Chan QK, Chan HY, Wong ES, et al. Aberrant Activation of Hedgehog Signaling Pathway Contributes to Endometrial Carcinogenesis Through Beta-Catenin. Mod Pathol (2009) 22(6):839–47. doi: 10.1038/modpathol.2009.45
159. Harrison W, Cochrane B, Neill G, Philpott M. The Oncogenic GLI Transcription Factors Facilitate Keratinocyte Survival and Transformation Upon Exposure to Genotoxic Agents. Oncogene (2014) 33(19):2432–40. doi: 10.1038/onc.2013.199
160. Frappart PO, Lee Y, Russell HR, Chalhoub N, Wang YD, Orii KE, et al. Recurrent Genomic Alterations Characterize Medulloblastoma Arising From DNA Double-Strand Break Repair Deficiency. Proc Natl Acad Sci USA (2009) 106(6):1880–5. doi: 10.1073/pnas.0806882106
161. Mazumdar T, Devecchio J, Agyeman A, Shi T, Houghton JA. Blocking Hedgehog Survival Signaling at the Level of the GLI Genes Induces DNA Damage and Extensive Cell Death in Human Colon Carcinoma Cells. Cancer Res (2011) 71(17):5904–14. doi: 10.1158/0008-5472.CAN-10-4173
162. Varon R, Vissinga C, Platzer M, Cerosaletti KM, Chrzanowska KH, Saar K, et al. Nibrin, a Novel DNA Double-Strand Break Repair Protein, is Mutated in Nijmegen Breakage Syndrome. Cell (1998) 93(3):467–76. doi: 10.1016/s0092-8674(00)81174-5
163. Kobayashi J, Antoccia A, Tauchi H, Matsuura S, Komatsu K. NBS1 and its Functional Role in the DNA Damage Response. DNA Repair (Amst) (2004) 3(8-9):855–61. doi: 10.1016/j.dnarep.2004.03.023
164. Antoccia A, Kobayashi J, Tauchi H, Matsuura S, Komatsu K. Nijmegen Breakage Syndrome and Functions of the Responsible Protein, NBS1. Genome Dyn (2006) 1:191–205. doi: 10.1159/000092508
165. Ronco C, Martin AR, Demange L, Benhida R. Atm, ATR, Chk1, CHK2 and WEE1 Inhibitors in Cancer and Cancer Stem Cells. Medchemcomm (2017) 8(2):295–319. doi: 10.1039/c6md00439c
166. Boohaker RJ, Xu B. The Versatile Functions of ATM Kinase. BioMed J (2014) 37(1):3–9. doi: 10.4103/2319-4170.125655
167. Pietrobono S, Stecca B. Targeting the Oncoprotein Smoothened by Small Molecules: Focus on Novel Acylguanidine Derivatives as Potent Smoothened Inhibitors. Cells (2018) 7(12):272. doi: 10.3390/cells7120272
168. Ecke I, Rosenberger A, Obenauer S, Dullin C, Aberger F, Kimmina S, et al. Cyclopamine Treatment of Full-Blown Hh/Ptch-associated RMS Partially Inhibits Hh/Ptch Signaling, But Not Tumor Growth. Mol Carcinog (2008) 47(5):361–72. doi: 10.1002/mc.20394
169. Robarge KD, Brunton SA, Castanedo GM, Cui Y, Dina MS, Goldsmith R, et al. Gdc-0449-a Potent Inhibitor of the Hedgehog Pathway. Bioorg Med Chem Lett (2009) 19(19):5576–81. doi: 10.1016/j.bmcl.2009.08.049
170. Sarangi A, Valadez JG, Rush S, Abel TW, Thompson RC, Cooper MK. Targeted Inhibition of the Hedgehog Pathway in Established Malignant Glioma Xenografts Enhances Survival. Oncogene (2009) 28(39):3468–76. doi: 10.1038/onc.2009.208
171. Mas C, Ruiz i Altaba A. Small Molecule Modulation of HH-GLI Signaling: Current Leads, Trials and Tribulations. Biochem Pharmacol (2010) 80(5):712–23. doi: 10.1016/j.bcp.2010.04.016
172. Rudin CM, Hann CL, Laterra J, Yauch RL, Callahan CA, Fu L, et al. Treatment of Medulloblastoma With Hedgehog Pathway Inhibitor GDC-0449. N Engl J Med (2009) 361(12):1173–8. doi: 10.1056/NEJMoa0902903
173. Von Hoff DD, LoRusso PM, Rudin CM, Reddy JC, Yauch RL, Tibes R, et al. Inhibition of the Hedgehog Pathway in Advanced Basal-Cell Carcinoma. N Engl J Med (2009) 361(12):1164–72. doi: 10.1056/NEJMoa0905360
174. De Smaele E, Ferretti E, Gulino A. Vismodegib, a Small-Molecule Inhibitor of the Hedgehog Pathway for the Treatment of Advanced Cancers. Curr Opin Investig Drugs (2010) 11(6):707–18.
175. Dierks C. Gdc-0449–targeting the Hedgehog Signaling Pathway. Recent Results Cancer Res (2010) 184:235–8. doi: 10.1007/978-3-642-01222-8_17
176. Low JA, de Sauvage FJ. Clinical Experience With Hedgehog Pathway Inhibitors. J Clin Oncol (2010) 28(36):5321–6. doi: 10.1200/JCO.2010.27.9943
177. LoRusso PM, Rudin CM, Reddy JC, Tibes R, Weiss GJ, Borad MJ, et al. Phase I Trial of Hedgehog Pathway Inhibitor Vismodegib (GDC-0449) in Patients With Refractory, Locally Advanced or Metastatic Solid Tumors. Clin Cancer Res (2011) 17(8):2502–11. doi: 10.1158/1078-0432.CCR-10-2745
178. Lauth M, Bergstrom A, Shimokawa T, Toftgard R. Inhibition of GLI-mediated Transcription and Tumor Cell Growth by Small-Molecule Antagonists. Proc Natl Acad Sci USA (2007) 104(20):8455–60. doi: 10.1073/pnas.0609699104
179. Calcaterra A, Iovine V, Botta B, Quaglio D, D’Acquarica I, Ciogli A, et al. Chemical, Computational and Functional Insights Into the Chemical Stability of the Hedgehog Pathway Inhibitor GANT61. J Enzyme Inhib Med Chem (2018) 33(1):349–58. doi: 10.1080/14756366.2017.1419221
180. Peer E, Tesanovic S, Aberger F. Next-Generation Hedgehog/GLI Pathway Inhibitors for Cancer Therapy. Cancers (Basel) (2019) 11(4):538. doi: 10.3390/cancers11040538
181. Beauchamp EM, Ringer L, Bulut G, Sajwan KP, Hall MD, Lee YC, et al. Arsenic Trioxide Inhibits Human Cancer Cell Growth and Tumor Development in Mice by Blocking Hedgehog/GLI Pathway. J Clin Invest (2011) 121(1):148–60. doi: 10.1172/JCI42874
182. Owonikoko TK, Zhang G, Kim HS, Stinson RM, Bechara R, Zhang C, et al. Patient-Derived Xenografts Faithfully Replicated Clinical Outcome in a Phase II Co-Clinical Trial of Arsenic Trioxide in Relapsed Small Cell Lung Cancer. J Transl Med (2016) 14(1):111. doi: 10.1186/s12967-016-0861-5
183. Comba A, Almada LL, Tolosa EJ, Iguchi E, Marks DL, Vara Messler M, et al. Nuclear Factor of Activated T Cells-Dependent Down-regulation of the Transcription Factor Glioma-associated Protein 1 (Gli1) Underlies the Growth Inhibitory Properties of Arachidonic Acid. J Biol Chem (2016) 291(4):1933–47. doi: 10.1074/jbc.M115.691972
184. Infante P, Mori M, Alfonsi R, Ghirga F, Aiello F, Toscano S, et al. Gli1/DNA Interaction is a Druggable Target for Hedgehog-dependent Tumors. EMBO J (2015) 34(2):200–17. doi: 10.15252/embj.201489213
185. Giannini G, Taddei M, Manetti F, Petricci E, Stecca B. Gli1 Inhibitors and Uses Thereof. (2017).
186. Manetti F, Stecca B, Santini R, Maresca L, Giannini G, Taddei M, et al. Pharmacophore-Based Virtual Screening for Identification of Negative Modulators of GLI1 as Potential Anticancer Agents. ACS Med Chem Lett (2020) 11(5):832–8. doi: 10.1021/acsmedchemlett.9b00639
187. Dash RC, Wen J, Zaino AM, Morel SR, Chau LQ, Wechsler-Reya RJ, et al. Structure-Based Virtual Screening Identifies an 8-Hydroxyquinoline as a Small Molecule GLI1 Inhibitor. Mol Ther Oncolytics (2021) 20:265–76. doi: 10.1016/j.omto.2021.01.004
188. Chen JK, Taipale J, Young KE, Maiti T, Beachy PA. Small Molecule Modulation of Smoothened Activity. Proc Natl Acad Sci USA (2002) 99(22):14071–6. doi: 10.1073/pnas.182542899
Keywords: hedgehog, GLI1, therapeutic resistance, DNA damage repair, cancer
Citation: Avery JT, Zhang R and Boohaker RJ (2021) GLI1: A Therapeutic Target for Cancer. Front. Oncol. 11:673154. doi: 10.3389/fonc.2021.673154
Received: 26 February 2021; Accepted: 30 April 2021;
Published: 25 May 2021.
Edited by:
José Díaz-Chávez, National Institute of Cancerology (INCAN), MexicoReviewed by:
Chinnadurai Mani, Texas Tech University Health Sciences Center, United StatesCopyright © 2021 Avery, Zhang and Boohaker. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rebecca J. Boohaker, UkJvb2hha2VyQHNvdXRoZXJucmVzZWFyY2gub3Jn
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.