 Yan-Qi Li
Yan-Qi Li Zhi Zheng
Zhi Zheng Quan-Xing Liu
Quan-Xing Liu Xiao Lu
Xiao Lu Jiao Zhang
Jiao Zhang Hong Zheng
Hong Zheng Ji-Gang Dai
Ji-Gang Dai
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oncol. , 29 April 2021
Sec. Pharmacology of Anti-Cancer Drugs
Volume 11 - 2021 | https://doi.org/10.3389/fonc.2021.670804
This article is part of the Research Topic Investigating Drugs used Off-Label in Various Cancers View all 12 articles
Drug repositioning is a strategy for identifying new antitumor drugs; this strategy allows existing and approved clinical drugs to be innovatively repurposed to treat tumors. Based on the similarities between parasitic diseases and cancer, recent studies aimed to investigate the efficacy of existing antiparasitic drugs in cancer. In this review, we selected two antihelminthic drugs (macrolides and benzimidazoles) and two antiprotozoal drugs (artemisinin and its derivatives, and quinolines) and summarized the research progresses made to date on the role of these drugs in cancer. Overall, these drugs regulate tumor growth via multiple targets, pathways, and modes of action. These antiparasitic drugs are good candidates for comprehensive, in-depth analyses of tumor occurrence and development. In-depth studies may improve the current tumor diagnoses and treatment regimens. However, for clinical application, current investigations are still insufficient, warranting more comprehensive analyses.
There exists a close connection between parasitic infections and cancer (1–3). Helminth infections are widespread the world over, and the causative parasites are thought to be responsible for causing cancer in humans (4). Thus far, Schistosoma haematobium, Clonorchis sinensis, and Opisthorchis viverrini have been recognized as clear biological carcinogens (1). The specific carcinogenic mechanism is not yet clear, and the metabolites of catechol estrogens and parasite-derived oxysterols may play an important role (5). Unlike worms, protozoa have not been identified as biological carcinogens; however, certain characteristics of protozoa are similar to those of cancer. In a manner similar to the immune evasion strategies employed by cancers, Trypanosoma cruzi and Leishmania parasites leverage the immune mechanisms to persist in the body and establish a chronic infection (6). Although malaria is the most widespread parasitic disease in the world, it does not seem to be carcinogenic (2). However, the incidence of malaria is positively correlated with mortality in most cancers, with the exception of colorectal, lung, gastric, and several other types of cancer whose mortalities exhibit an inverse correlation with malaria (7, 8). Thus, the relationship between malaria and cancer is worth exploring.
Cancer is the second leading cause of death worldwide and is a major burden of disease (9, 10). However, with proper treatment, many cancers can be cured. Drugs are essential in the treatment of tumors but are often not as effective as required because of drug resistance and low specificity (11). Despite the emergence of highly specific monoclonal antibody drugs, the drugs are unsuitable for a wide range of clinical applications because they have strict requirements for the target (12). Therefore, the development of new antitumor drugs is still urgently needed. However, owing to the similarities between parasitic diseases and cancer as well as the successful clinical administration of antiparasitic drugs for years, it seems feasible to repurpose existing antiparasitic drugs into antitumor drugs. In fact, some antiparasitic and antitumor drugs share the same target, a variety of drugs targeting CDKs, TGR enzyme, tubulin/microtubule system have been confirmed to have dual effects on anti-parasites and anti-cancer (13).
Research on the repurposing antiparasitic drugs for tumors has gradually gained popularity. However, many studies have reported contradictory results. We selected two well-researched antihelminthic drugs and two antiprotozoal drugs and summarized the corresponding research progress to provide direction for further exploration into the repurposing of antiparasitic drugs as antitumor drugs.
Macrolides are antiparasitic drugs with dual functions in vitro and in vivo. The mechanism of macrolides is mainly to increase the concentration of the inhibitory transmitter GABA and enhance the permeability of the nerve membrane to chloride ions, causing neuromuscular paralysis and death (14). Macrolides have long been used to kill nematodes and consist of two main categories: avermectins and milbemycins. Apart from the antiparasitic effects, macrolide drugs also show different levels of anticancer activity (Figure 1).
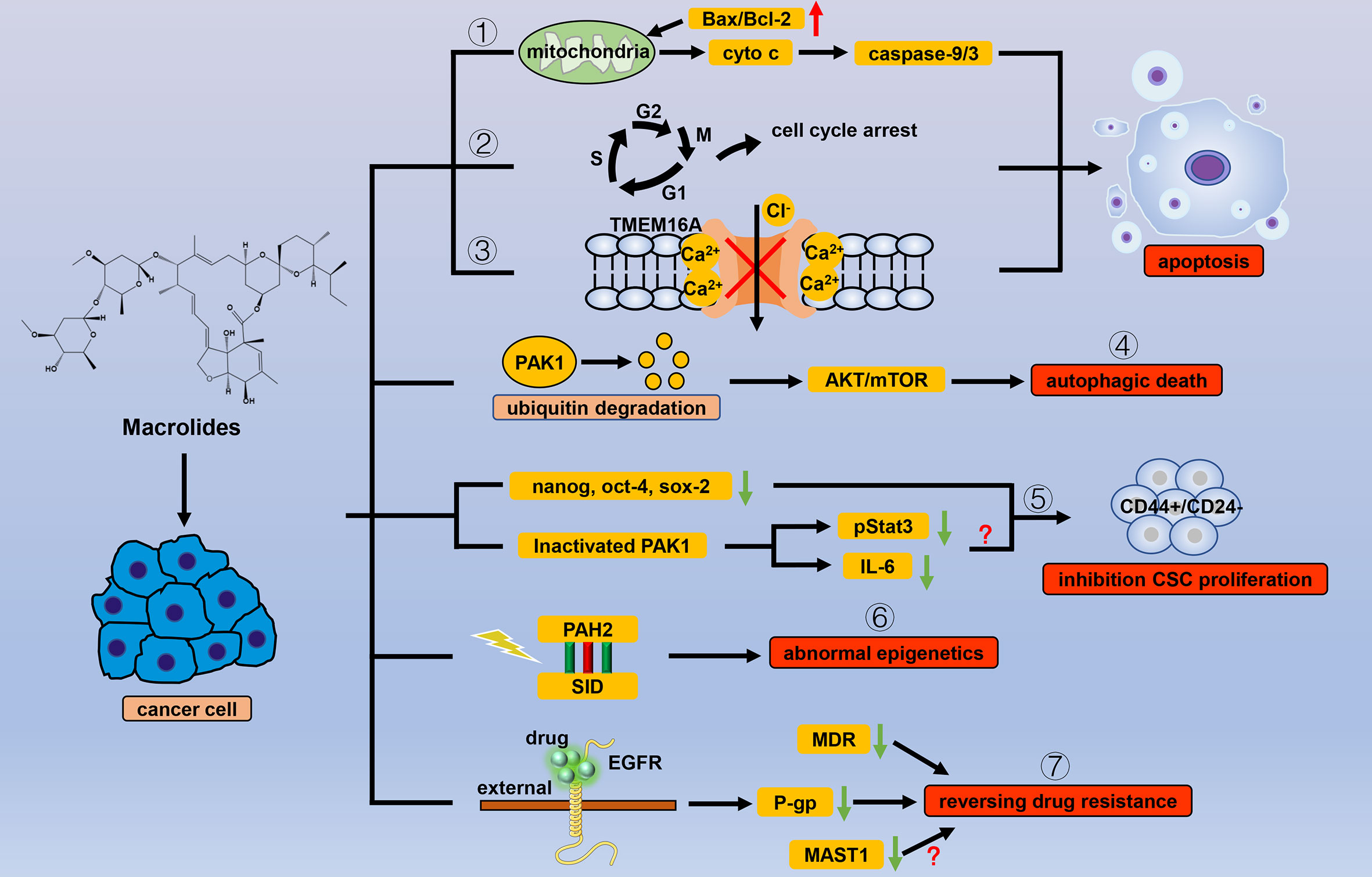
Figure 1 Efficacy of macrolide antiparasitic drugs in cancer. Apoptosis is the chief mechanism used by macrolide drugs to kill cancer cells. Macrolides trigger apoptosis through the (1) mitochondrial pathway, (2) cell cycle arrest, and (3) inhibition of the current of Ca2+ ion-activated Cl- channels. Other than apoptosis, macrolides can cause autophagic death of cancer cells by (4) degrading PAK1. When used for cancer cells, macrolides show selectivity for CSCs by (5) inhibiting stem cell genes and inactivating PAK1. Macrolides also reverse the abnormal epigenetics of tumor cells through (6) the combination of PAH2 and SID domain. By binding to the extracellular segment of EGFR, macrolides can (7) inhibit the transcription of P-gp, thereby reversing tumor resistance in which MDR and MASTA1 are also involved.
Commonly used avermectins include avermectin (AVM), ivermectin (IVM), doramectin (DRM), eprinomectin (EPM), and selamectin (SLM). Although all these avermectins show anticancer activity, studies on IVM are more comprehensive than those on other drugs. IVM can regulate the natural progression of tumors via multiple pathways.
Apoptosis is an important mechanism used by IVMs to kill cancer cells. Eukaryotic translation initiation factor 4A isoform 3 (EIF4A3) is an RNA-binding protein involved in the splicing modulation of BCL2L1/Bcl-X and is considered to be closely associated with apoptosis. SILAC-based quantitative proteomic analysis revealed that IVM inhibited the expression of EIF4A3 and 116 EIF4A3-binding mRNAs (15). In LA795 cells, IVM analogs (IVM, EPM, and SLM) can significantly inhibit currents mediated by the transmembrane member 16A (TMEM16A), an endogenous Ca2+-activated Cl- channel closely related to tumorigenesis, thereby inducing apoptosis (16). In addition, IVMs utilize the well-studied pathway of mitochondrial apoptosis to exert their anticancer activity. When acting on HeLa cells, IVM can increase the ratio of Bax/Bcl-2 and induce release of mitochondrial cytochrome c into the cytoplasm, thus stimulating caspase-9/-3-mediated apoptosis (17). In chronic myeloid leukemia and renal cell carcinoma cells, IVM can induce apoptosis by inducing mitochondrial dysfunction (18).
Cell cycle arrest can easily lead to apoptosis. Several studies have revealed that in cancer cells, IVM arrests the cell cycle in different phases by regulating the expression of proteins that control the cell cycle, thereby inducing apoptosis (19–22). Especially in epithelial ovarian cancer, IVM induces apoptosis through multiphasic cell cycle arrest, and exhibits KPNB1-dependent antitumor effects (22). The cell cycle is also the target of many chemotherapeutic drugs, and combinatorial treatment with IVM and clinical drugs is worth investigating. In fact, in multiple in vivo and in vitro experiments, IVM significantly enhanced the efficacy of various drugs, including cisplatin and tamoxifen, but this result still needs clinical verification (20, 23).
Autophagy regulation is another important mechanism underlying IVM action. A study on breast cancer revealed that when IVM was applied to cancer cells, no obvious apoptosis was observed before 24 h of treatment, but the inhibition of growth of these cancer cells during the 24 h was evident. Later in that study, autophagy flux increased during the first 24 h of IVM treatment, and the anticancer effect during this period was reversed when IVM was used to treat cancer cells Beclin 1 or Atg5 knockdown (24). Current studies argue that IVM degrades PAK1 in cancer cells through the ubiquitination pathway, thereby inactivating the AKT-mTOR pathway, which is the key negative regulatory pathway of autophagy (24, 25). Although mechanisms used by IVM require more detailed exploration, it is encouraging that more studies indicate that induction of autophagy may be used as a method of synergistic treatment in clinical tumor chemotherapy, highlighting the potential of IVM as a clinical adjuvant drug (26, 27).
In addition to the anticancer mechanism, the selective functional characteristics of IVMs are also noteworthy. IVMs exhibit more pronounced toxicity toward cancer cells than toward non-cancer cells, which may be related to higher mitochondrial biogenesis in cancer cells (28). More importantly, when acting on cancer cells, IVM still exhibits different levels of cytotoxicity in different cancer cell subgroups. The CD44+/CD24- subpopulation of breast cancer cells have been previously reported to possess stem/progenitor cell properties (29, 30). IVM preferentially inhibits the viability of CD44 +/CD24- subpopulation cells (cancer stem cells (CSCs)) and reduces the expression of stemness genes (NANOG, POU5F1, and SOX2) (31). Current research points out that this may be related to the ability of IVMs to inactivate p21-activated kinase (PAK1), thereby reducing the levels of pStat3 and extracellular IL-6 and inhibiting the formation of CSCs (32). Research on the specific effect of IVM on CSCs is still limited, and many other knowledge gaps exist that require further research. In general, the selective nature of IVMs shows that it is almost non-toxic to non-cancer cells but can effectively inhibit the growth of cancer cells, demonstrating its unique potential as an anticancer drug.
IVMs have also shown anticancer capabilities in many fields other than these mentioned above. Although research on these aspects is rare, it has broadened the scope of exploration of IVMs. The latest research showed that IVMs could reverse tumor resistance. IVM at a low dose that does not produce evident cytotoxicity can bind to the extracellular domain of EGFR, which inhibits the activation of EGFR and its downstream signaling cascade ERK/Akt/NF-κB, thus inhibiting the transcription factor NF-κB and leading to reduction in P-glycoprotein (P-gp) transcription (33). Moreover, in triple-negative breast cancer (TNBC), the targeted disruption of the Sin3 (a master transcriptional scaffold and corepressor that plays an essential role in the regulation of gene transcription and maintenance of chromatin structure) complex by introducing a Sin3 interaction domain (SID) decoy that interferes with PAH2 binding by sequestering SID-containing partner proteins reverted the silencing of genes involved in cell growth and differentiation (34–36). Interestingly, IVM and SLM can be used as small molecule inhibitors of SID peptides that play a similar role to that of Sin3 disruption, indicating that AVMs can also exert anticancer effects by regulating the abnormal epigenetics of tumors (37). Furthermore, the activation of WNT-TCF signaling is implicated in multiple diseases, but there are no WNT-TCF antagonists in clinical use. However, SLM and IVM can reduce the expression of target proteins in this pathway by mimicking dnTCF, further demonstrating the application potential of these drugs (38).
The milbemycin family comprises a series of 16-membered macrolide antibiotics that contain a highly characteristic spiroketal group that can be produced by several Streptomyces, these antibiotics have strong biological activities and are used as highly selective and potent broad-spectrum antiparasitic agents (39–41). At present, research on milbemycins in cancer was relatively rare, and milbemycins have been found to play an important role in reversing tumor drug resistance. Milbemycins can restore the sensitivity of cancer cells toward chemotherapy drugs by reducing the expression of MDR1 or P-gp, and its concentration has no obvious cytotoxic effect on cancer cells (42, 43). Cisplatin (DDP) is one of the most widely used chemotherapeutic drugs and is considered the first-line treatment for many cancers, but drug resistance limits its therapeutic potential. A recent study found that serine/threonine kinase 1 (MAST1) was a major driver of DDP resistance in human cancers (44). Encouragingly, in multidrug and cisplatin-resistant human lung adenocarcinoma (A549/DDP) cells, a milbemycin compound isolated from Streptomyces sp. FJS31-2, named VM48130, reduced the expression of multiple resistance-related genes, including MAST1, to reverse resistance, which further demonstrated the potential of milbemycin as an adjuvant drug in clinical chemotherapy (45). The anticancer mechanisms of these compounds also include other aspects. For example, moxidectin effectively inhibited the proliferation of rat C6 and human U251 glioma cells. Mitochondria-related apoptotic pathways, cell cycle arrest, and autophagy induced by the AKT/mTOR signaling pathway in cancer cells are all considered to be involved in this process, but the specific mechanism remains to be explored (46, 47).
Benzimidazole is a broad-spectrum antiparasitic drug with a structure similar to that of purines and is mainly used in clinics for nematodes. Benzimidazoles include albendazole (ABZ), flubendazole (FLU), fenbendazole (FBZ), oxibendazole (OBZ), and febantel (FBT). In general, these drugs mainly exert their antiparasitic effects by interfering with sugar metabolism, affecting adenosine triphosphate (ATP) production, and binding to tubulin to affect the cell cycle (48, 49). These biological processes are also critical in cancer, and it seems inevitable that such drugs are effective in tumor treatment. Correspondingly, many studies have shown that benzimidazoles have prominent anticancer activity (Figure 2).
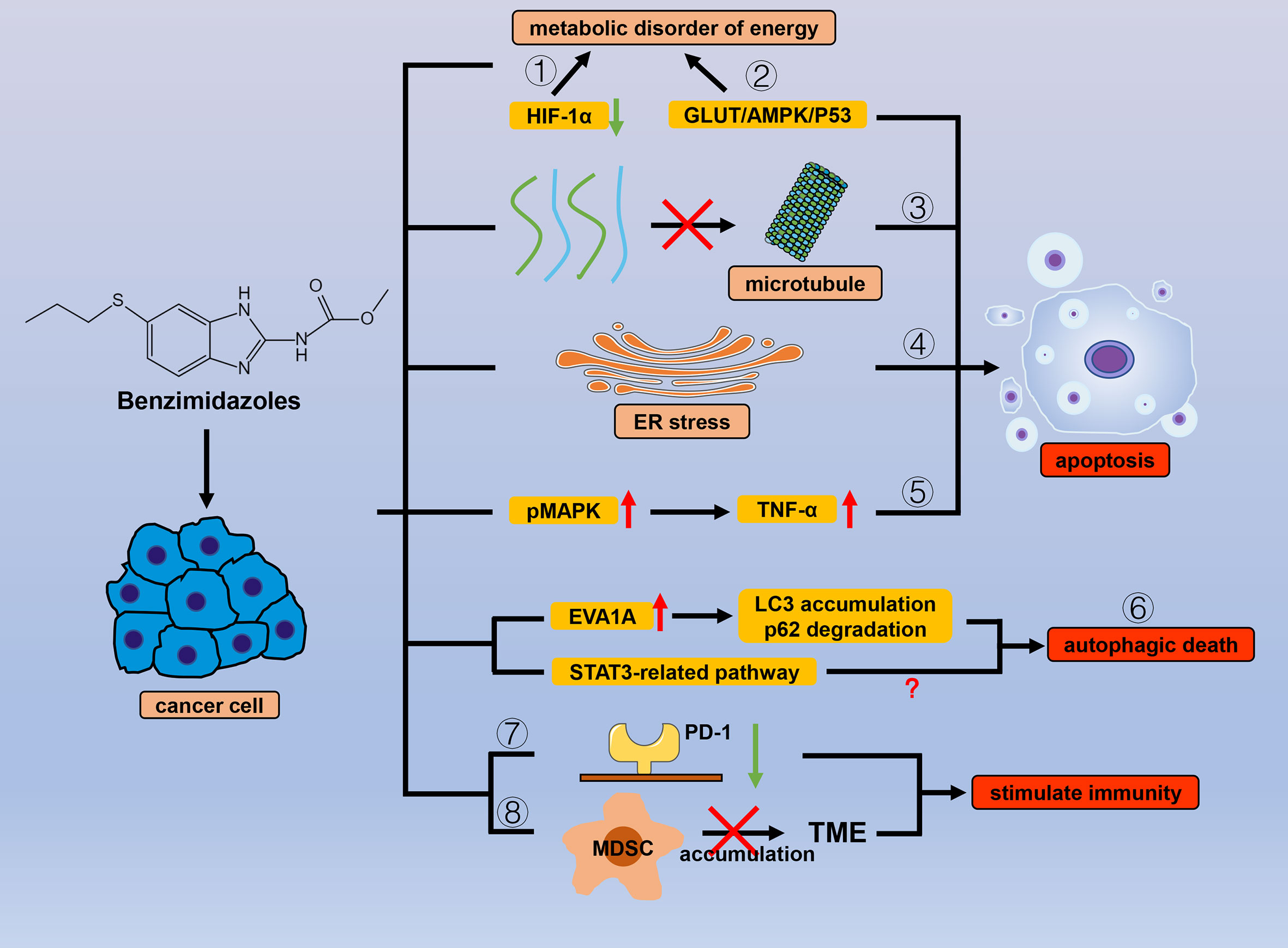
Figure 2 Efficacy of benzimidazole antiparasitic drugs in cancer. Benzimidazoles can cause energy metabolism disorders and reduce cancer cell tolerance to hypoxic environments by (1) inhibiting the expression of HIF-1α and (2) inhibiting sugar intake through the GLUT/AMPK/P53 pathway. Apart from sugar metabolism, benzimidazoles also induce apoptosis by (3) inhibiting microtubule polymerization and (4) ER stress, and (5) promoting MAPK phosphorylation. By affecting numerous autophagy-related proteins (LC3, P62, and EVA1A) and downstream signals related to STAT3, benzimidazole can also cause (6) autophagic cancer cell death. (7) Benzimidazoles reduce the expression of PD-1 and (8) inhibit the accumulation of MDSC in the TME to stimulate antitumor immunity.
Based on the antiparasitic mechanism, studies have found that ABZ can inhibit glucose uptake through the GLUT1/AMPK/P53 signaling pathway, thereby disrupting sugar metabolism in cancer cells and inducing cell apoptosis (50). ABZ, a microtubule-targeting agent (MTA), induces apoptosis by disrupting microtubule formation and causes mitotic arrest in tumors (51, 52). ABZ causes bundles of short microtubules to form along the edges of cells rather than covering the entire cell, leading to a series of biological reactions (52). MTAs are a class of drugs currently used in chemotherapy. A synergistic antiproliferative effect is observed upon using combinatorial therapy involving low concentrations of ABZ, colchicine, and ABZ plus 2-methoxyestradiol (2ME) (53). However, recent studies have revealed an interesting mechanism underlying the anticancer activity of ABZs, i.e., targeting the microtubules. In K562 cells, when compared with paclitaxel and other MTAs, ABZ treatment significantly increased the number of cells arrested at the G2/M phase in a short time, and ABZs did not immediately activate apoptosis. Subsequently, ABZs could upregulate SIRT3 expression, which is believed to regulate SOD2 activity to clear mitochondrial reactive oxygen species (ROS). They further speculated that this ability allowed ABZs to protect cancer cells from cytotoxicity in the short-term, but when SIRT3 expression was further reduced, this unique ABZ mechanism was no longer effective (54, 55).
Whether it be inhibiting the glucose metabolism pathway or targeting microtubules to exert anticancer activity, triggering cancer cell apoptosis is the common end result. In fact, ABZs can induce apoptosis in other ways. According to a study on cutaneous squamous cell carcinoma, ABZs increase apoptosis-related signals by inducing endoplasmic reticulum (ER) stress, and pretreatment with the ER stress inhibitor 4-PBA attenuates ABZ-induced apoptosis (56). Furthermore, in human leukemia U937 cells, ABZs increase MAPK phosphorylation and upregulate TNF-α expression, thus inducing apoptosis. The same pathway is seemingly involved in the ABZ-induced death of HL-60 cells (57).
Moreover, most cancer cells generate ATP using accelerated glycolysis rates, and glucose is converted to lactate instead of being metabolized by oxidative phosphorylation, even when oxygen is abundant (58, 59). Current research indicates that hypoxia-inducible factor-1α (HIF-1α) plays a critical role in this process. In general, under hypoxic conditions, HIF-1α maintains the survival requirements of cancer cells by regulating the expression of a series of glycolytic enzymes and can also bind to the vascular endothelial cell growth factor (VEGF) gene promoter to induce VEGF expression and angiogenesis (60, 61). Therefore, HIF-1α is becoming an increasingly attractive therapeutic target in the treatment of cancer. It is encouraging that ABZs significantly inhibited the expression of HIF-1α in non-small cell lung cancer and ovarian cancer; however, ABZ treatment did not affect the HIF-1α mRNA level, suggesting that other unknown regulatory pathways may be involved in this process (62, 63).
Research institutions have already carried out phase I clinical trials of oral ABZ to treat advanced cancer patients to detect its maximum tolerated dose. Results from the 36 patients with refractory solid tumors enrolled in the study showed that the recommended dose for further study was 1,200 mg twice daily for 14 days in a 21-day cycle, with myelosuppression being the main dose-limiting toxicity (64). Although no patients achieved partial or complete response according to the RECIST study’s criteria, 4 out of 24 patients with assessable tumor markers (16%) demonstrated a decrease in tumor markers of more than 50%. In contrast, another patient had a significant decrease in tumor markers and a prolonged period of stable disease. Overall, as research continues to progress, new anticancer drugs based on ABZ can be expected.
Similar to ABZ, FLU induces monopolar spindle formation by inhibiting tubulin polymerization, inhibiting proliferation and migration, ultimately triggering apoptosis in a variety of cancer cell lines (65–69). However, more distinctive is that autophagy seems to play an important role in the anticancer activity of FLU. Using molecular docking simulation technology to screen numerous small molecule drugs approved by the Food and Drug Administration (FDA), FLU was found to have the highest antitumor activity and the ability to target autophagy-related gene (ATG) 4 B. Molecular dynamics simulation revealed that FLU bound with high affinity to ATG4B protein, and that it could induce autophagy and exhibit an antiproliferative effect on TNBC cancer cells (70). The latest research, however, has proposed another possible mechanism for FLU’s anticancer activity on TNBC cells. FLU treatment promotes autophagy by upregulating the expression of Eva-1 homolog A (EVA1A), a protein involved in autophagy and apoptosis-induced cell death (71). EVA1A knockdown, results in the partial inhibition of LC3 puncta accumulation, p62 degradation, and LC3 lipidation in TNBC cells (72). FLU also promotes autophagy in other malignant cell lines, such as A549 and H460 (73), through the regulation of the signal transducer and activator of transcription 3 (STAT3)-related pathway to induce apoptosis in human colorectal cancer cells, but the specific mechanism remains unclear (74).
Moreover, FLU has clinical value because it is potentially involved in tumor immunotherapy and molecular targeted drug resistance. Programmed cell death protein-1 (PD-1) and programmed cell death-ligand 1 (PD-L1) are immune system regulators that play a role in dampening the immune response to cancer cells, and PD-1 inhibitors have already changed the paradigm of cancer treatment in many cancers (75, 76). However, all available PD-1/PD-L1 treatments are antibodies that require intravenous infusion, resulting in exorbitant costs. PD-1/PD-L1 treatments can also have unpredictable and/or poor response in second-line treatment; therefore, finding a small molecule inhibitor is more convenient (77). A study on melanoma showed that FLU could inhibit the expression of PD-1 in cancer cells and the accumulation of myeloid-derived suppressor cells (MDSCs) in the tumor microenvironment, indicating its ability to elicit the host’s antitumor immunity, but the specific mechanism remains to be explored (78). In general, this suggests huge potential application of FLU in tumor immunotherapy. Apart from this, trastuzumab provides significant clinical benefit for HER2-positive breast cancers, but nearly 70% of patients experience primary or acquired resistance, which dramatically limits the therapeutic effect (79). Encouragingly, FLU significantly reduced p95HER2 expression and the phosphorylation level of HER2, HER3, and AKT, preventing the hetero-dimerization of HER2/HER3 in trastuzumab-resistant cells (80), which play an important role in trastuzumab resistance (81–83). Combination therapy with FLU seems to be a possible solution to improve the efficacy of trastuzumab.
Artemisinin (ARS) is a 1,2,-trioxane from the Chinese medicinal plant Sweet Wormwood, and since its antimalarial effect was discovered, research on ARS has been continuously focused on. A variety of ARS and its derivatives (ARTs), including dihydroartemisinin (DHA), artemether (ARM), artesunate (ART), and artemisitene (ATT), have emerged because of the advancements of drug modification and synthesis technology. ARS-based combination therapies are established standard treatments for malaria worldwide (84–87). It is currently considered that the heme-irons released by Plasmodium-attacking red blood cells can cleave the endoperoxide bridge of ARS via a Fe (II) Fenton-type reaction, and that free radical intermediates kill the Plasmodia (88, 89). Other pathways are also involved in ARTs antimalarial response (90, 91), but comprehensive research on antimalarial mechanisms remains necessary. Interestingly, as with other natural products, antimalarial properties are not the only benefits of ARTs, and ARTs have shown application value in many diseases, including cancer (Figure 3).
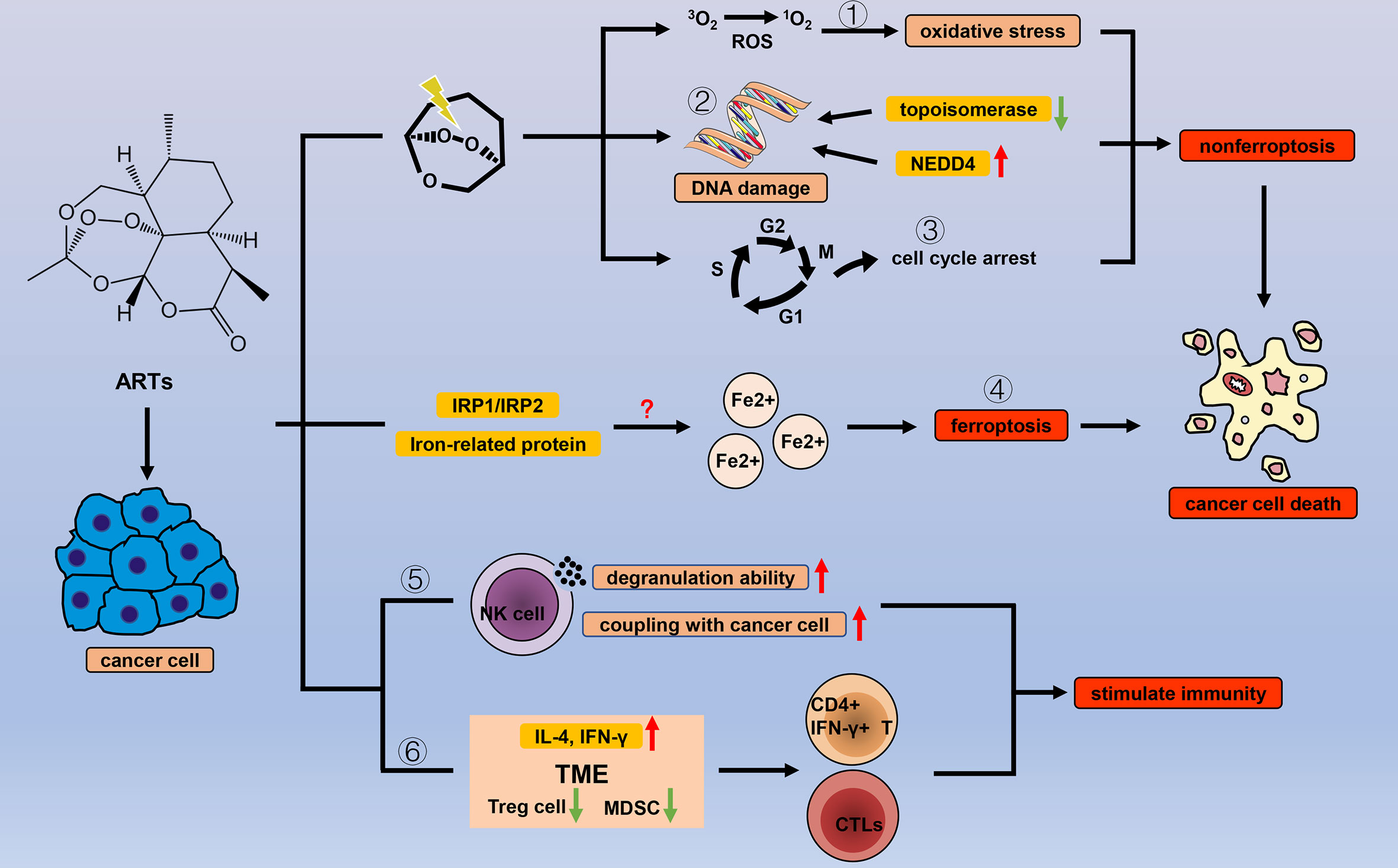
Figure 3 Efficacy of ARTs in cancer. With the help of endogenous peroxides, ARTs can cause oxidative stress by (1) increasing the concentration of ROS, (2) DNA damage, and (3) cell cycle arrest to cause cancer cell death. In addition to the common types of cell death, ARTs increase the concentration of unstable iron ions in cancer cells by (4) regulating a variety of iron-related proteins and IRP1/IRP2, thereby triggering ferroptosis. ARTs strengthen the cancer-killing effect of NK cells by (5) enhancing their degranulation ability and increasing the connection between NK cells and cancer cells. ARTs (6) reduce negative regulation factors (Treg cells and MDSCs) and increase IL-4 and IFN-γ in TME to stimulate T cell immune response.
Long-term studies on the anticancer mechanism of ARTs have shown that the endoperoxide moiety is essential for its biological activity, and ARTs without the endoperoxide moiety were inactive (92, 93). In general, ARTs mainly exert anticancer effects in three ways with the endoperoxide moiety. (i) The cleavage of the endoperoxide moiety leads to the formation of ROS and oxidative stress in the tumor. Excessive production of ROS causes death by damaging cellular components, including DNA, proteins, and lipids (94); a notable feature of ARTs is that they can spontaneously generate a large amount of ROS in a heme-dependent manner (95–97). Many studies have indicated that ARTs increase the expression of cleaved caspase-3 and PARP in a variety of cancer cells by producing excessive amounts of ROS, thus inducing apoptosis in cancer cells (98–100). Further, excessive amounts of ROS may trigger an ER stress response in cancer cells (99), but the specific mechanism is not clear. Interestingly, increasing the concentration of ferrous ions and oxygen in the tumor environment to further increase the concentration of ROS has been shown to enhance the anticancer activity of ARTs (101, 102), which provides a possible strategy for the development of new anticancer drugs based on ARTs.
(ii) ARTs rely on excessive amounts of ROS to cause DNA damage in cancer cells. In the alkaline comet assay, both ARS and ART caused significant DNA damage, and the fold changes of OTM and tail DNA significantly increased (103). In addition, molecular docking indicated that various ARTs might induce DNA damage in cancer cells by inhibiting topoisomerase 1 (104), an enzyme that resolves the topological stress in genomic DNA by preventing double-stranded breaks in the DNA during cell proliferation (105). Subsequent studies involved in-depth exploration of the specific mechanism of ARTs. ATT can increase the expression of E3 ligase NEDD4, resulting in the destabilization of c-Myc protein, thereby inhibiting the expression of DNA topoisomerases. Importantly, neither a decrease in the concentration of NEDD4 protein nor DNA damage has been observed in non-cancer cells (106). DNA damage inducers such as cisplatin and doxorubicin have become the first-line cancer treatment (107, 108). However, these drugs lack strict selectivity, and are toxic to non-cancer cells. The high selectivity of ATT as a DNA damage inducer suggests that ARTs may be a potential alternative to cisplatin and doxorubicin.
(iii) ARTs induce cycle arrest to trigger cancer cell death. This anticancer effect of ARTs is closely related to excessive amounts of ROS and oxidative DNA lesions that affect cell integrity, leading to perturbations in DNA replication and cell division mechanisms. More specifically, CDK4 encodes a cyclin-dependent serine-threonine kinase in response to mitogenic or proliferation-promoting stimuli and interacts with cyclin D1 to phosphorylate the tumor suppressor protein Rb (109, 110). DHA can regulate the cyclin D1-CDK4-Rb pathway by inhibiting the expression of CDK4 to trigger cycle arrest (111). In cisplatin-resistant human breast carcinoma cells, ARS exerts anticancer activity by targeting multiple key cell cycle-related proteins, including cyclin-B1, cyclin D1, and cyclin E, to trigger cycle block (112). In general, with the help of the endoperoxide moiety, ARTs can kill cancer cells in a variety of ways. Moreover, these pathways are closely interconnected.
Ferroptosis, a new type of programmed cell death that is iron-dependent and differs from apoptosis, cell necrosis, and autophagy, has recently become a research hotspot in cancer (113–115). Interestingly, ART anticancer activity is also based on ferroptosis. Current research shows two main mechanisms of ferroptosis. In the first mechanism, the expression of the core enzyme GPX4 in the antioxidant system is reduced or inactivated, depending on the level of intracellular GSH (116). In the second mechanism, unstable iron ions accumulate in cancer cells (100). The accumulation of unstable iron ions may be the main mechanism by which ART-induced ferroptosis. The anticancer effects of ARTs are related to many iron-related proteins, including transferrin (TF), transferrin receptors 1 and 2 (TFRC, TFR2), ceruloplasmin (CP), and lactoferrin (LTF) (117). Furthermore, in DHA-induced ferroptosis, intracellular GSH levels are not affected. However, when DHA is combined with deferoxamine (DFO), an iron chelator, DHA-induced ferroptosis is completely inhibited. Further, DHA may inhibit the translation activity of a series of ferritin-related genes by maintaining the binding of iron regulatory protein-1 (IRP1) and IRP2 to iron-responsive element sequences, thus greatly increasing the concentration of unstable iron ions in cancer cells (118). However, counterintuitively, ARTs also strangely induce a negative feedback pathway in ferroptosis. ARTs can significantly increase the expression of GRP78, an antiferroptosis glucose-regulated protein, and knockdown of GRP78-enhanced ART-induced ferroptosis in AsPC-1 and PaTU8988 cells (119). DHA increases the expression of HSPA5, a negative regulator of DHA-induced ferroptosis, by activating GPX4 in glioma cells (120). It is worth considering that the induction of ferroptosis activity by ARTs could be enhanced by inhibiting the negative feedback pathway. In summary, the specific mechanism by which ARTs induce ferroptosis is still unclear, and more research is warranted. In addition, except for ferroptosis, other types of programmed cell death modes, such as apoptosis and autophagy, are also important for ARTs to kill cancer cells, and many excellent research results have been published (Table 1).
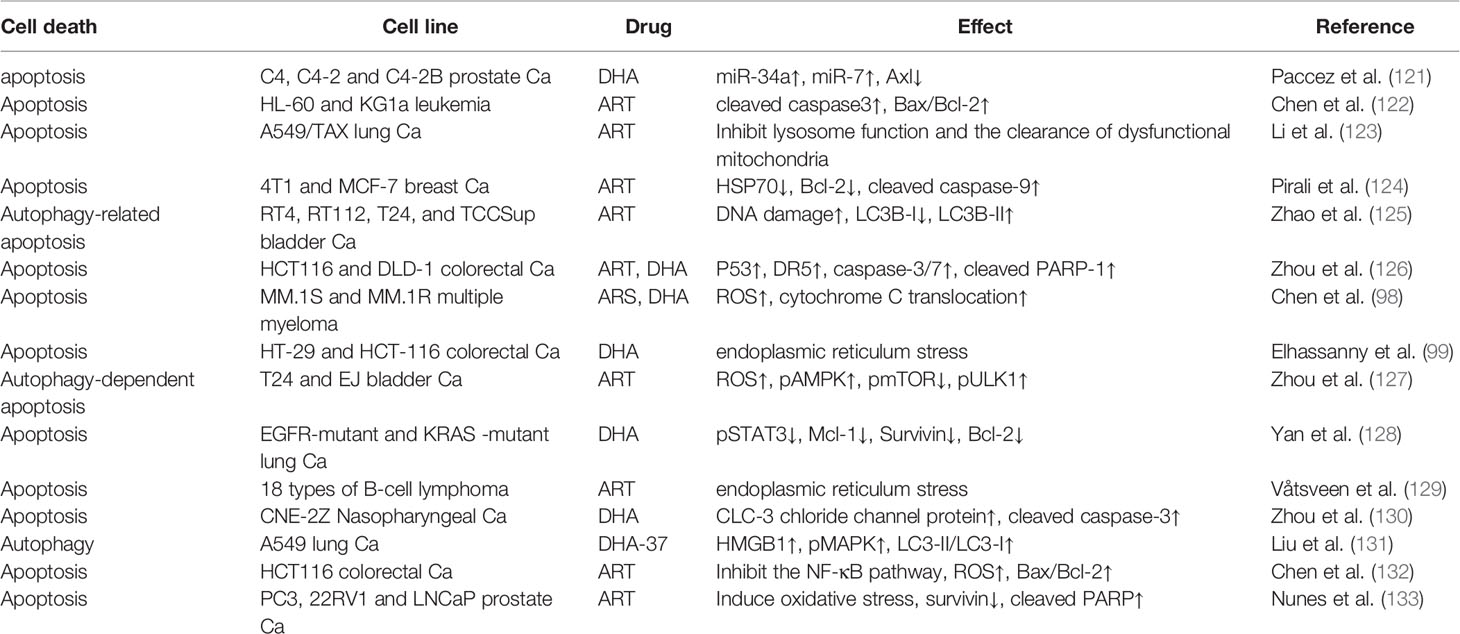
Table 1 Non- ferroptosis cancer cell death induced by ARTs.
The regulation of the immune system seems to be one of the antitumor mechanisms of ARTs. This idea was first supported when ARS was directly applied to natural killer (NK) cells, and the degranulation ability of NK cells was enhanced to effectively kill cancer cells. Combination treatment with the degranulation inhibitor concanamycin A completely reversed these effects of ARS. ARS does not change the expression of activated receptors on the surface of NK cells to enhance their degranulation ability, but it could induce the activation of downstream signaling molecules (134). Subsequently, ARS can also increase the coupling between tumor and NK cells, but the expression of the main ligands of NK receptors is not affected; the specific mechanism needs to be further explored (135). Moreover, except for the innate immune system, ARTs can also regulate specific immune systems. For example, ARM can reduce Treg cells and increase IL-4 and IFN-γ in the tumor microenvironment (136). More directly, treatment with ARS in a 4T1 breast cancer model significantly reduced the number of MDSCs and Treg cells in mice and significantly increased CD4+ IFN-γ+ T cells and cytotoxic T lymphocytes (CTLs) (137). Although obvious immune regulation can be observed, few studies have investigated immune system regulation by ARTs, and the exact underlying mechanism remains unclear; however, it is still a promising research direction.
As studies on ARTs in cancer have increased, so too have the number of case reports and related clinical trials that support the potential role of ARTs in cancer treatment. There were two uveal melanoma cases in which compassionate treatment with ART was administered after ineffective standard chemotherapy (138). One patient received fotemustine plus ART and reached a temporary response, although the tumor progressed under prior fotemustine therapy alone, and the patient died 23 months after being diagnosed with stage 4 disease. The second patient reached disease stabilization after administration of dacarbazine and ART, and the survival time of the patient greatly exceeded that of the median survival time for patients with uveal melanoma, which is 2 to 5 months. Furthermore, the results of a phase I clinical study on intravenous ART in patients with advanced solid tumor malignancies were recently published (139). Nineteen patients were enrolled in the study and had various cancers. In the study, dose-limiting toxicities were observed in one of six patients at dose levels of 12 mg/kg (neutropenic fever) and 18 mg/kg (grade 3 hypersensitivity reaction on C1D1); both patients treated with 25 mg/kg experienced dose-limiting toxicities (one patient had grade 3 nausea/vomiting, and the other experienced neutropenic infection, grade 3 ALT elevation, and grade 4 ALT elevation). They also observed a disease control rate of 27% (4 out of 16). In summary, although there are issues to be resolved, ARTs have great potential as anticancer drugs.
Similar to ARTs, the design and synthesis of quinoline drugs, which are characterized by a quinoline ring, has been researched for application as antimalarial drugs. Quinolines exert their antimalarial effects during the blood or liver stages of the life cycle of the parasite (140), but different drugs have different mechanisms (141, 142). With the progress in research, an increasing number of quinoline drugs have shown therapeutic effects in other diseases, including cancer (Figure 4).
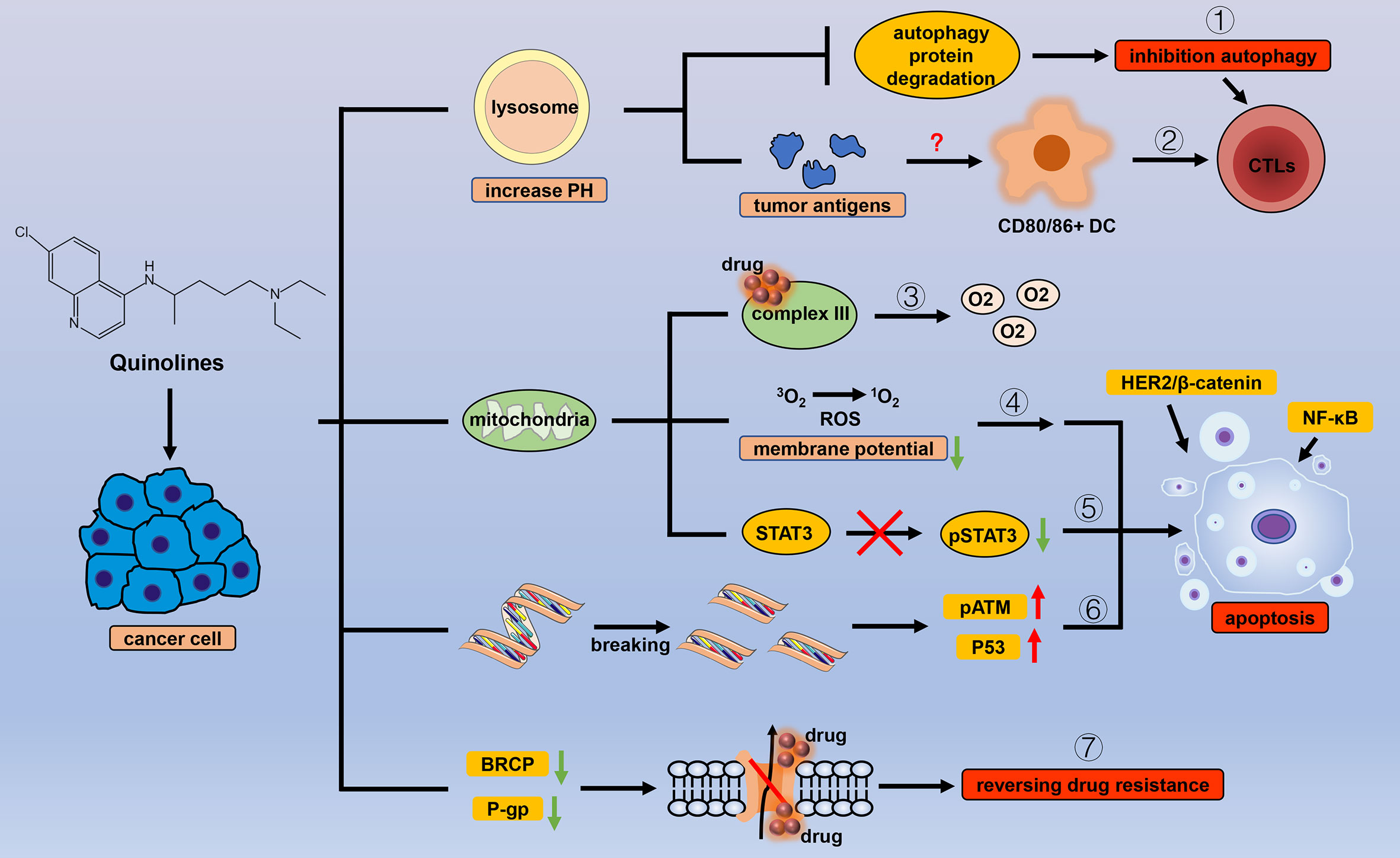
Figure 4 Efficacy of quinoline antiparasitic drugs in cancer. Lysosomes are an important target of quinoline drugs, and quinolines increase the pH of the lysosome, thereby triggering a variety of cascade reactions. Quinolines can (1) inhibit the degradation of autophagy proteins to block autophagy and (2) stimulate antitumor immune responses by increasing tumor antigens. Mitochondria are also a target for quinolines. Quinolines increase the oxygen concentration in cancer cells by (3) inhibiting the related processes of mitochondrial complex III and cause apoptosis by (4) inducing oxidative stress with the change in mitochondrial membrane potential, (5) inhibiting the phosphorylation level of STAT3 in mitochondria, and (6) introducing double-strand breaks in DNA in cancer cells. (7) Quinolines inhibit the drug delivery mediated by P-gp and BRCP.
Chloroquine (CQ), a 4-aminoquinoline, has been used as an antimalarial drug for many years and is often recommended to be co-administered with primaquine to prevent recurrence of Plasmodium vivax (143). CQ is currently considered a protonated, weakly basic drug that increases the pH and accumulates in the food vacuole of parasites, thereby interfering with the degradation of host red blood cell hemoglobin, and preventing the growth of malaria parasite (144). The exact mechanism requires further investigation. Similar to other antiparasitic drugs, CQ has also shown potential in the treatment of cancers and other diseases (145, 146).
As lysosomotropic agents, CQ increases the pH of the lysosome from 4.5 to 6, such a change in pH is not conducive for the activity of lysosomal enzymes. This mode of action is known as a lysosomotropic effect (147, 148). Therefore, CQ can affect various biological processes by acting on receptors, enzymes, and transcriptional factors, which would determine the therapeutic effects in cancer (149) (1). Inhibit autophagy to induce anticancer effects. As an autophagy inhibitor approved by the FDA. At present, this particular function has been studied quite thoroughly. CQ blocks the final step of the autophagy process by impeding the degradation of autophagic proteins such as light chain 3B-II (LC3B-II) (150, 151). Thus, CQ prevents the production and recycling of important nutrients and metabolites, causing tumor cell damage and death. Further, the inhibition of the final stage of autophagy increases cytotoxic effects in cancer cells by promoting cell apoptosis and cell cycle arrest (152) (2). Regulate immunity to prevent tumor cells from escaping. Successive studies have shown that when ultra-low concentrations of chemotherapeutics are used on tumor cells, the expression of some genes related to inflammation, immunity, and tumor antigens in the cell increases, thereby increasing the cell’s ability to use CTLs to induce immunogenic death of tumor cells (153–157). A recent study reported for the first time the effect of low concentrations of CQ on the immunogenicity of tumor cells (158). In that study, when HCT-116 colon cancer cells were treated with CQ and 5-fluorouracil in combination, their cell lysates significantly induced the maturation of dendritic cells, and the expression of surface markers, including CD80 and CD86, was significantly increased. Additionally, dendritic cells increased the production of CTLs and triggered tumor cell death. Subsequently, they detected an increase in tumor-associated carcinoembryonic antigen family gene expression in the treated cancer cells, but the specific mechanism still needs to be explored, which may be related to the inhibition of autophagy by CQ. CQ has also been found to directly affect the function and differentiation of various immune cells by altering the pH of the lysosome and has been described in detail in this review (149).
CQ also has multiple functions in tumors by regulating a variety of key signaling molecules. Platinum drugs are recognized as the mainstay drugs for the treatment of epithelial ovarian cancer (159). However, drug-resistant cancer cells can survive the DNA damage induced by anticancer drugs through DNA repair pathways or bypassing cell cycle checkpoints (160, 161); however, CQ can upregulate the expression of p21WAF1/CIP1 to prevent this phenomenon and reverse the drug resistance of cancer cells. This function seems to depend on autophagy inhibition, but the connection is not yet clear (162). Many other functions of CQ depend on its inhibitory effect on autophagy. However, CQ can reduce the expression of CXC chemokine receptor 4 (CXCR4) by inhibiting STAT3 expression, thereby reducing the stemness of esophageal squamous cell carcinoma cells. This process is independent of autophagy, and the expression of key molecules, ATG7 and BECN1, in the autophagy pathway does not change (163), suggesting that CQ has other effects that do not depend on autophagy inhibition, but these have not yet been elucidated. In addition, CQ can regulate the expression of signaling molecules, including NF-κB and p53, to exert an anticancer effect (164).
As an autophagy inhibitor approved by the FDA, CQ has received widespread attention, and the possibilities of its clinical application have been extensively studied. To date, many clinical trials using CQ alone or in combination to treat cancer have been conducted (165, 166). Most findings indicate that the combination of CQ with other drugs was well tolerated, and the maximum tolerated dose increases compared with using CQ alone. However, in these trials, no significant differences between the treatment and control group and no significant improvement in overall survival were observed. This may also be related to the small sample size used in the phase I clinical trials; thus, the efficacy of CQ should be further evaluated and explored in a larger sample.
Atovaquone (ATV), a hydroxy 1,4-naphthoquinoline, is a homolog of coenzyme Q. Current research shows that ATV’s antimalarial site is the mitochondrial complex III (141). Compared with non-cancer cells, cancer cells rely more heavily on mitochondrial functions to generate ATP for growth and survival (167, 168). Naturally, respiratory energy metabolism, especially oxidative phosphorylation, has become a focus of research. ATV, an inhibitor of the mitochondrial complex III, can significantly reduce the oxygen consumption rate and increase the concentration of oxygen in cancer cells (169). In clinical practice, hypoxia is a major problem in cancer treatment (170). The current novel photodynamic therapy technology mainly relies on ROS to kill cancer cells, but the hypoxic tumor environment limits its therapeutic value (171, 172); thus, combinations with ATV may be a solution. Many studies have also revealed that ATV can indeed improve the efficacy of radiotherapy, chemotherapy, and immunotherapy by reducing the rate of oxygen consumption (173, 174).
The reduction in mitochondrial function greatly inhibits the proliferation of CSCs, which mainly rely on mitochondrial respiration rather than glycolysis. The decrease in mitochondrial membrane potential and the increase in ROS levels lead to apoptosis in CSCs; thus, ATV selectively acts on CSCs (175). More specifically, ATV can inhibit the phosphorylation of mitochondrial STAT3 but not of nuclear STAT3. The inhibition of STAT3 phosphorylation is not accompanied by changes in JAK, Src, or MEK, indicating that this function of ATV is independent of JAK/Src/MEK (176). ATV can also reduce the expression of a variety of STAT3 target genes, thereby exerting anticancer effects in tumors (177, 178). There is still much to be discovered regarding mitochondria and related functions, an aspect that has received much attention in the context of cancer treatment (179).
Mitochondrial respiratory function is not the only target of ATVs. ATVs can degrade HER2 and β-catenin in a proteasome-dependent manner, thereby inhibiting the activation of HER2/β-catenin and triggering apoptosis in cancer cells; however, application of the proteasome inhibitor MG-132 eliminates this effect of ATV (180). In addition, ATV can also introduce double-stranded breaks in DNA, thereby upregulating phosphorylated ATM and p53 to trigger cycle arrest and apoptosis in cancer cells (181). ATV is also an inhibitor of BRCP and P-gp-mediated drug delivery (182), indicating the broader application value of ATV, but its specific mechanism remains to be elucidated.
People are increasingly aware of the value of drug repositioning with the urgent clinical situation that large number of new anticancer drugs have appeared unignorable drug resistance after short-term clinical use (183, 184). Encouragingly, numerous clinically approved non-cancer drugs have shown antitumor activity in vitro, in vivo, and even clinically, and antiparasitic drugs account for a large portion of these repurposed drugs (185).
Based on the current research, the application of antiparasitic drugs in anticancer therapy is multitargeted and multimodal (Table 2). Macrolide drugs induce cell cycle arrest, apoptosis, and autophagic death in cancer cells by regulating multiple targets and multiple signaling pathways, including WNT-TCF and PAK1-AKT-mTOR, and also play a role in reversing tumor resistance. Benzimidazole drugs induce cancer cell death by inhibiting sugar metabolism and interfering with the formation of microtubules. In particular, benzimidazoles can also inhibit the expression of HIF-1α protein to inhibit the stress behavior of cancer cells under hypoxic environments, and by changing the tumor microenvironment to stimulate the host’s antitumor immunity, benzimidazoles may also exert anticancer effects. Endoperoxide is very important for the anticancer activity of ARTs. Specifically, an endoperoxide moiety can produce a large amount of ROS in tumor cells and trigger a series of biological processes that are lethal to cancer cells. Interestingly, ARTs increase the concentration of unstable iron ions in cancer cells by regulating various ferritin and related pathways to induce ferroptosis, a new programmed cell death mode (115, 186), which may be worthwhile to study further. Similar to clinical antimalarial drugs, quinoline drugs also exert anticancer effects. CQ, a lysosomotropic agent, affects various biological pathways in cancer cells by changing the pH of lysosomes. Autophagy is currently studied more thoroughly than other mechanisms, but CQ also increases the immunogenicity of cancer cells. In addition, as a competitive inhibitor of the mitochondrial complex III, ATV can significantly affect the oxidative phosphorylation of cancer cells to change the hypoxic environment of tumor cells and greatly improve the efficacy of various clinical treatment methods. At present, the application of antiparasitic drugs in tumor treatment is not only at the stage of theoretical experimentation, but several clinical trials have already been carried out to analyzed the feasibility of specific drugs.

Table 2 Antitumor mechanism of antiparasitic drugs.
In summary, antiparasitic drugs are involved in almost all aspects of tumors, including cell cycle, apoptosis, autophagy, ferroptosis, stress, energy homeostasis, immunity, and drug resistance. Besides, when used in combination with existing clinical tumor drugs, many antiparasitic drugs show significant synergistic effects (187–189). However, according to current research results, antiparasitic drugs are still far from being repurposed into antitumor drugs that can be widely used in clinical practice. The current research on the anticancer mechanisms of antiparasitic drugs is still not comprehensive enough, and more thorough research is needed. In addition, antiparasitic and tumor therapy have two different application environments, and many problems remain to be solved. The first problem is drug delivery; the external microenvironment of the parasite-infected foci is relatively normal, whereas the growth of tumors mainly depends on glycolysis, which leads to an acidic external microenvironment. Whether this microenvironment affects the delivery of drugs needs further exploration. The second problem is drug concentration. Different drug concentrations cause different dominant effects. The concentration that has the best anticancer effect and does not cause side effects in the human body needs to be established. The third problem is that although antiparasitic drugs have many advantages, we cannot rule out they might promote tumor growth. Studies have shown that CQ-induced stress in cancer cells can activate NF-κB, thereby conferring transcriptional and phenotypic plasticity to cells, resulting in the reprogramming of cells and allows tumor cells to escape cell death induced by either drug therapy or the immune system (190, 191). Fortunately, for the first two issues, many studies have conducted in-depth study. Increasing evidence shows that nanotechnology-based drug delivery methods yield better therapeutic effects at lower concentrations and might be clinically implemented in the near future (192–198). Successive preclinical studies and clinical trials have clarified in detail the therapeutic effects of different drug concentrations and various possible side effects. However, there are very few studies have investigated the cancer-promoting effects of antiparasitic drugs. However, this may be very important for us to fully understand the role of antiparasitic drugs in tumors. After understanding these cancer-promoting effects, with the help of modern drug modification and improvement technologies (199–202), it can greatly accelerate the real application of antiparasitic drugs in clinical cancer treatment. More problems are likely to be encountered as research and practice progress. Nevertheless, it is undeniable that antiparasitic drugs indeed have great potential for development as broad-spectrum, clinically applicable antitumor drugs.
J-GD and HZ contributed substantially to the design of this review and gave the approval of the final version for publishing. DZ and JZ prepared the table. Q-XL and XL prepared the figures. Y-QL and ZZ wrote the manuscript. All authors contributed to the article and approved the submitted version.
This work was supported by grants from the National Natural Science Foundation of China (No. 81972190), Natural Science Foundation of Chongqing (cstc2018jcyjAX0178) and the Scientific Research Project of Undergraduate, the Army (Third Military) Medical University (2019XBK48).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Arora N, Kaur R, Anjum F, Tripathi S, Mishra A, Kumar R, et al. Neglected Agent Eminent Disease: Linking Human Helminthic Infection, Inflammation, and Malignancy. Front Cell Infect Microbiol (2019) 9:402. doi: 10.3389/fcimb.2019.00402
2. van Tong H, Brindley PJ, Meyer CG, Velavan TP. Parasite Infection, Carcinogenesis and Human Malignancy. Ebiomedicine (2017) 15:12–23. doi: 10.1016/j.ebiom.2016.11.034
3. Pastille E, Frede A, McSorley HJ, Gräb J, Adamczyk A, Kollenda S, et al. Intestinal Helminth Infection Drives Carcinogenesis in Colitis-Associated Colon Cancer. PloS Pathog (2017) 13(9):e1006649. doi: 10.1371/journal.ppat.1006649
4. Moormann AM, Bailey JA. Malaria - How This Parasitic Infection Aids and Abets EBV-associated Burkitt Lymphomagenesis. Curr Opin Virol (2016) 20:78–84. doi: 10.1016/j.coviro.2016.09.006
5. Brindley PJ, Costa J, Sripa B. Why Does Infection With Some Helminths Cause Cancer? Trends Cancer (2015) 1(3):174–82. doi: 10.1016/j.trecan.2015.08.011
6. Ouaissi A, Ouaissi M. Molecular Basis of Trypanosoma Cruzi and Leishmania Interaction With Their Host(s): Exploitation of Immune and Defense Mechanisms by the Parasite Leading to Persistence and Chronicity, Features Reminiscent of Immune System Evasion Strategies in Cancer Diseases. Arch Immunol Ther Exp (Warsz) (2005) 53(2):102–14.
7. Qin L, Chen C, Chen L, Xue R, Ou-Yang M, Zhou C, et al. Worldwide Malaria Incidence and Cancer Mortality are Inversely Associated. Infect Agent Cancer (2017) 12:14. doi: 10.1186/s13027-017-0117-x
8. Lehrer S. Association Between Malaria Incidence and All Cancer Mortality in Fifty U.S. States and the District of Columbia. Anticancer Res (2010) 30(4):1371–3.
9. Gao F, Zhang X, Wang T, Xiao J. Quinolone Hybrids and Their Anti-Cancer Activities: An Overview. Eur J Med Chem (2019) 165:59–79. doi: 10.1016/j.ejmech.2019.01.017
10. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global Cancer Statistics 2018: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J Clin (2018) 68(6):394–424. doi: 10.3322/caac.21492
11. Xu Z, Zhao SJ, Liu Y. 1,2,3-Triazole-containing Hybrids as Potential Anticancer Agents: Current Developments, Action Mechanisms and Structure-Activity Relationships. Eur J Med Chem (2019) 183:111700. doi: 10.1016/j.ejmech.2019.111700
12. Feng M, Xiong G, Cao Z, Yang G, Zheng S, Song X, et al. Pd-1/Pd-L1 and Immunotherapy for Pancreatic Cancer. Cancer Lett (2017) 407:57–65. doi: 10.1016/j.canlet.2017.08.006
13. Dorosti Z, Yousefi M, Sharafi SM, Darani HY. Mutual Action of Anticancer and Antiparasitic Drugs: Are There Any Shared Targets? Future Oncol (2014) 10(15):2529–39. doi: 10.2217/fon.14.65
14. Geary TG. Ivermectin 20 Years on: Maturation of a Wonder Drug. Trends Parasitol (2005) 21(11):530–2. doi: 10.1016/j.pt.2005.08.014
15. Li N, Zhan X. Anti-Parasite Drug Ivermectin can Suppress Ovarian Cancer by Regulating lncRNA-EIF4A3-mRNA Axes. Epma J (2020) 11(2):289–309. doi: 10.1007/s13167-020-00209-y
16. Zhang X, Zhang G, Zhai W, Zhao Z, Wang S, Yi J. Inhibition of TMEM16A Ca(2+)-Activated Cl(-) Channels by Avermectins is Essential for Their Anticancer Effects. Pharmacol Res (2020) 156:104763. doi: 10.1016/j.phrs.2020.104763
17. Zhang P, Zhang Y, Liu K, Liu B, Xu W, Gao J, et al. Ivermectin Induces Cell Cycle Arrest and Apoptosis of HeLa Cells Via Mitochondrial Pathway. Cell Prolif (2019) 52(2):e12543. doi: 10.1111/cpr.12543
18. Wang J, Xu Y, Wan H, Hu J. Antibiotic Ivermectin Selectively Induces Apoptosis in Chronic Myeloid Leukemia Through Inducing Mitochondrial Dysfunction and Oxidative Stress. Biochem Biophys Res Commun (2018) 497(1):241–7. doi: 10.1016/j.bbrc.2018.02.063
19. Juarez M, Schcolnik-Cabrera A, Dominguez-Gomez G, Chavez-Blanco A, Diaz-Chavez J, Duenas-Gonzalez A. Antitumor Effects of Ivermectin At Clinically Feasible Concentrations Support its Clinical Development as a Repositioned Cancer Drug. Cancer Chemother Pharmacol (2020) 85(6):1153–63. doi: 10.1007/s00280-020-04041-z
20. Zhang X, Qin T, Zhu Z, Hong F, Xu Y, Zhang X, et al. Ivermectin Augments the In Vitro and In Vivo Efficacy of Cisplatin in Epithelial Ovarian Cancer by Suppressing Akt/mTOR Signaling. Am J Med Sci (2020) 359(2):123–9. doi: 10.1016/j.amjms.2019.11.001
21. Intuyod K, Hahnvajanawong C, Pinlaor P, Pinlaor S. Anti-Parasitic Drug Ivermectin Exhibits Potent Anticancer Activity Against Gemcitabine-Resistant Cholangiocarcinoma In Vitro. Anticancer Res (2019) 39(9):4837–43. doi: 10.21873/anticanres.13669
22. Kodama M, Kodama T, Newberg JY, Katayama H, Kobayashi M, Hanash SM, et al. In Vivo Loss-of-Function Screens Identify KPNB1 as a New Druggable Oncogene in Epithelial Ovarian Cancer. Proc Natl Acad Sci USA (2017) 114(35):E7301–10. doi: 10.1073/pnas.1705441114
23. Lokich E, Singh RK, Han A, Romano N, Yano N, Kim K, et al. HE4 Expression is Associated With Hormonal Elements and Mediated by Importin-Dependent Nuclear Translocation. Sci Rep (2014) 4:5500. doi: 10.1038/srep05500
24. Dou Q, Chen HN, Wang K, Yuan K, Lei Y, Li K, et al. Ivermectin Induces Cytostatic Autophagy by Blocking the PAK1/Akt Axis in Breast Cancer. Cancer Res (2016) 76(15):4457–69. doi: 10.1158/0008-5472.CAN-15-2887
25. Wang K, Gao W, Dou Q, Chen H, Li Q, Nice EC, et al. Ivermectin Induces PAK1-mediated Cytostatic Autophagy in Breast Cancer. Autophagy (2016) 12(12):2498–9. doi: 10.1080/15548627.2016.1231494
26. Wei W, Hardin H, Luo QY. Targeting Autophagy in Thyroid Cancers. Endocr Relat Cancer (2019) 26(4):R181–94. doi: 10.1530/ERC-18-0502
27. Peng PL, Kuo WH, Tseng HC, Chou FP. Synergistic Tumor-Killing Effect of Radiation and Berberine Combined Treatment in Lung Cancer: The Contribution of Autophagic Cell Death. Int J Radiat Oncol Biol Phys (2008) 70(2):529–42. doi: 10.1016/j.ijrobp.2007.08.034
28. Zhu M, Li Y, Zhou Z. Antibiotic Ivermectin Preferentially Targets Renal Cancer Through Inducing Mitochondrial Dysfunction and Oxidative Damage. Biochem Biophys Res Commun (2017) 492(3):373–8. doi: 10.1016/j.bbrc.2017.08.097
29. Tanaka H, Nakamura M, Kameda C, Kubo M, Sato N, Kuroki S, et al. The Hedgehog Signaling Pathway Plays an Essential Role in Maintaining the CD44+CD24-/low Subpopulation and the Side Population of Breast Cancer Cells. Anticancer Res (2009) 29(6):2147–57.
30. Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Prospective Identification of Tumorigenic Breast Cancer Cells. Proc Natl Acad Sci USA (2003) 100(7):3983–8. doi: 10.1073/pnas.0530291100
31. Dominguez-Gomez G, Chavez-Blanco A, Medina-Franco JL, Saldivar-Gonzalez F, Flores-Torrontegui Y, Juarez M, et al. Ivermectin as an Inhibitor of Cancer Stem−Like Cells. Mol Med Rep (2018) 17(2):3397–403. doi: 10.3892/mmr.2017.8231
32. Kim JH, Choi HS, Kim SL, Lee DS. The PAK1-Stat3 Signaling Pathway Activates IL-6 Gene Transcription and Human Breast Cancer Stem Cell Formation. Cancers (Basel) (2019) 11(10):1527. doi: 10.3390/cancers11101527
33. Jiang L, Wang P, Sun YJ, Wu YJ. Ivermectin Reverses the Drug Resistance in Cancer Cells Through EGFR/ERK/Akt/NF-κb Pathway. J Exp Clin Cancer Res (2019) 38(1):265. doi: 10.1186/s13046-019-1251-7
34. Farias EF, Petrie K, Leibovitch B, Murtagh J, Chornet MB, Schenk T, et al. Interference With Sin3 Function Induces Epigenetic Reprogramming and Differentiation in Breast Cancer Cells. Proc Natl Acad Sci USA (2010) 107(26):11811–6. doi: 10.1073/pnas.1006737107
35. Ellison-Zelski SJ, Solodin NM, Alarid ET. Repression of ESR1 Through Actions of Estrogen Receptor Alpha and Sin3A At the Proximal Promoter. Mol Cell Biol (2009) 29(18):4949–58. doi: 10.1128/MCB.00383-09
36. Ellison-Zelski SJ, Alarid ET. Maximum Growth and Survival of Estrogen Receptor-Alpha Positive Breast Cancer Cells Requires the Sin3A Transcriptional Repressor. Mol Cancer (2010) 9:263. doi: 10.1186/1476-4598-9-263
37. Kwon YJ, Petrie K, Leibovitch BA, Zeng L, Mezei M, Howell L, et al. Selective Inhibition of SIN3 Corepressor With Avermectins as a Novel Therapeutic Strategy in Triple-Negative Breast Cancer. Mol Cancer Ther (2015) 14(8):1824–36. doi: 10.1158/1535-7163.MCT-14-0980-T
38. Melotti A, Mas C, Kuciak M, Lorente-Trigos A, Borges I, Ruiz IAA. The River Blindness Drug Ivermectin and Related Macrocyclic Lactones Inhibit WNT-TCF Pathway Responses in Human Cancer. EMBO Mol Med (2014) 6(10):1263–78. doi: 10.15252/emmm.201404084
39. Prichard RK. Is Anthelmintic Resistance a Concern for Heartworm Control? What can We Learn From the Human Filariasis Control Programs? Vet Parasitol (2005) 133(2-3):243–53. doi: 10.1016/j.vetpar.2005.04.008
40. Wang XJ, Wang M, Wang JD, Jiang L, Wang JJ, Xiang WS. Isolation and Identification of Novel Macrocyclic Lactones From Streptomyces Avermitilis NEAU1069 With Acaricidal and Nematocidal Activity. J Agric Food Chem (2010) 58(5):2710–4. doi: 10.1021/jf902496d
41. Xiang WS, Wang JD, Wang XJ, Zhang J. Two New Beta-Class Milbemycins From Streptomyces Bingchenggensis: Fermentation, Isolation, Structure Elucidation and Biological Properties. J Antibiot (Tokyo) (2007) 60(6):351–6. doi: 10.1038/ja.2007.47
42. Gao A, Liang H, Wang X, Zhang X, Jing M, Zhang J, et al. Reversal Effects of Two New Milbemycin Compounds on Multidrug Resistance in MCF-7/adr Cells In Vitro. Eur J Pharmacol (2011) 659(2-3):108–13. doi: 10.1016/j.ejphar.2011.03.023
43. Xiang W, Gao A, Liang H, Li C, Gao J, Wang Q, et al. Reversal of P-glycoprotein-mediated Multidrug Resistance In Vitro by Milbemycin Compounds in Adriamycin-Resistant Human Breast Carcinoma (MCF-7/adr) Cells. Toxicol Vitro (2010) 24(6):1474–81. doi: 10.1016/j.tiv.2010.07.020
44. Jin L, Chun J, Pan C, Li D, Lin R, Alesi GN, et al. Mast1 Drives Cisplatin Resistance in Human Cancers by Rewiring Craf-Independent MEK Activation. Cancer Cell (2018) 34(2):315–30. doi: 10.1016/j.ccell.2018.06.012
45. Li XQ, Yue CW, Xu WH, Lü YH, Huang YJ, Tian P, et al. A Milbemycin Compound Isolated From Streptomyces Sp. FJS31-2 With Cytotoxicity and Reversal of Cisplatin Resistance Activity in A549/DDP Cells. BioMed Pharmacother (2020) 128:110322. doi: 10.1016/j.biopha.2020.110322
46. Liu J, Liang H, Khilji S, Li H, Song D, Chen C, et al. Moxidectin Induces Cytostatic Autophagic Cell Death of Glioma Cells Through Inhibiting the AKT/mTOR Signalling Pathway. J Cancer (2020) 11(19):5802–11. doi: 10.7150/jca.46697
47. Song D, Liang H, Qu B, Li Y, Liu J, Chen C, et al. Moxidectin Inhibits Glioma Cell Viability by Inducing G0/G1 cell Cycle Arrest and Apoptosis. Oncol Rep (2018) 40(3):1348–58. doi: 10.3892/or.2018.6561
48. Lacey E. The Role of the Cytoskeletal Protein, Tubulin, in the Mode of Action and Mechanism of Drug Resistance to Benzimidazoles. Int J Parasitol (1988) 18(7):885–936. doi: 10.1016/0020-7519(88)90175-0
49. Valdez J, Cedillo R, Hernández-Campos A, Yépez L, Hernández-Luis F, Navarrete-Vázquez G, et al. Synthesis and Antiparasitic Activity of 1H-Benzimidazole Derivatives. Bioorg Med Chem Lett (2002) 12(16):2221–4. doi: 10.1016/s0960-894x(02)00346-3
50. Liu H, Sun H, Zhang B, Liu S, Deng S, Weng Z, et al. (18)F-Fdg PET Imaging for Monitoring the Early Anti-Tumor Effect of Albendazole on Triple-Negative Breast Cancer. Breast Cancer-Tokyo (2020) 27(3):372–80. doi: 10.1007/s12282-019-01027-5
51. Zhang X, Zhao J, Gao X, Pei D, Gao C. Anthelmintic Drug Albendazole Arrests Human Gastric Cancer Cells At the Mitotic Phase and Induces Apoptosis. Exp Ther Med (2017) 13(2):595–603. doi: 10.3892/etm.2016.3992
52. Chu SW, Badar S, Morris DL, Pourgholami MH. Potent Inhibition of Tubulin Polymerisation and Proliferation of Paclitaxel-Resistant 1A9PTX22 Human Ovarian Cancer Cells by Albendazole. Anticancer Res (2009) 29(10):3791–6.
53. Ehteda A, Galettis P, Pillai K, Morris DL. Combination of Albendazole and 2-Methoxyestradiol Significantly Improves the Survival of HCT-116 Tumor-Bearing Nude Mice. BMC Cancer (2013) 13:86. doi: 10.1186/1471-2407-13-86
54. Wang LJ, Liou LR, Shi YJ, Chiou JT, Lee YC, Huang CH, et al. Albendazole-Induced SIRT3 Upregulation Protects Human Leukemia K562 Cells From the Cytotoxicity of MCL1 Suppression. Int J Mol Sci (2020) 21(11):3907. doi: 10.3390/ijms21113907
55. Chen Y, Zhang J, Lin Y, Lei Q, Guan KL, Zhao S, et al. Tumour Suppressor SIRT3 Deacetylates and Activates Manganese Superoxide Dismutase to Scavenge ROS. EMBO Rep (2011) 12(6):534–41. doi: 10.1038/embor.2011.65
56. Zhang QL, Lian DD, Zhu MJ, Li XM, Lee JK, Yoon TJ, et al. Antitumor Effect of Albendazole on Cutaneous Squamous Cell Carcinoma (Scc) Cells. BioMed Res Int (2019) 2019:3689517. doi: 10.1155/2019/3689517
57. Wang LJ, Lee YC, Huang CH, Shi YJ, Chen YJ, Pei SN, et al. Non-Mitotic Effect of Albendazole Triggers Apoptosis of Human Leukemia Cells Via SIRT3/ROS/p38 MAPK/TTP Axis-Mediated TNF-α Upregulation. Biochem Pharmacol (2019) 162:154–68. doi: 10.1016/j.bcp.2018.11.003
58. Kim JW, Dang CV. Cancer’s Molecular Sweet Tooth and the Warburg Effect. Cancer Res (2006) 66(18):8927–30. doi: 10.1158/0008-5472.CAN-06-1501
59. Alfarouk KO, Muddathir AK, Shayoub ME. Tumor Acidity as Evolutionary Spite. Cancers (Basel) (2011) 3(1):408–14. doi: 10.3390/cancers3010408
60. Cairns RA, Harris IS, Mak TW. Regulation of Cancer Cell Metabolism. Nat Rev Cancer (2011) 11(2):85–95. doi: 10.1038/nrc2981
61. Ahluwalia A, Tarnawski AS. Critical Role of Hypoxia Sensor–HIF-1α in VEGF Gene Activation. Implications for Angiogenesis and Tissue Injury Healing. Curr Med Chem (2012) 19(1):90–7. doi: 10.2174/092986712803413944
62. Zhou F, Du J, Wang J. Albendazole Inhibits HIF-1α-Dependent Glycolysis and VEGF Expression in non-Small Cell Lung Cancer Cells. Mol Cell Biochem (2017) 428(1-2):171–8. doi: 10.1007/s11010-016-2927-3
63. Pourgholami MH, Cai ZY, Badar S, Wangoo K, Poruchynsky MS, Morris DL. Potent Inhibition of Tumoral Hypoxia-Inducible Factor 1alpha by Albendazole. BMC Cancer (2010) 10:143. doi: 10.1186/1471-2407-10-143
64. Pourgholami MH, Szwajcer M, Chin M, Liauw W, Seef J, Galettis P, et al. Phase I Clinical Trial to Determine Maximum Tolerated Dose of Oral Albendazole in Patients With Advanced Cancer. Cancer Chemother Pharmacol (2010) 65(3):597–605. doi: 10.1007/s00280-009-1157-8
65. Hanušová V, Skálová L, Králová V, Matoušková P. The Effect of Flubendazole on Adhesion and Migration in SW480 and SW620 Colon Cancer Cells. Anticancer Agents Med Chem (2018) 18(6):837–46. doi: 10.2174/1871520618666171213141911
66. Kralova V, Hanušová V, Caltová K, Špaček P, Hochmalová M, Skálová L, et al. Flubendazole and Mebendazole Impair Migration and Epithelial to Mesenchymal Transition in Oral Cell Lines. Chem Biol Interact (2018) 293:124–32. doi: 10.1016/j.cbi.2018.07.026
67. Čáňová K, Rozkydalová L, Vokurková D, Rudolf E. Flubendazole Induces Mitotic Catastrophe and Apoptosis in Melanoma Cells. Toxicol Vitro (2018) 46:313–22. doi: 10.1016/j.tiv.2017.10.025
68. Králová V, Hanušová V, Rudolf E, Čáňová K, Skálová L. Flubendazole Induces Mitotic Catastrophe and Senescence in Colon Cancer Cells In Vitro. J Pharm Pharmacol (2016) 68(2):208–18. doi: 10.1111/jphp.12503
69. Hou ZJ, Luo X, Zhang W, Peng F, Cui B, Wu SJ, et al. Flubendazole, FDA-approved Anthelmintic, Targets Breast Cancer Stem-Like Cells. Oncotarget (2015) 6(8):6326–40. doi: 10.18632/oncotarget.3436
70. Zhang L, Guo M, Li J, Zheng Y, Zhang S, Xie T, et al. Systems Biology-Based Discovery of a Potential Atg4B Agonist (Flubendazole) That Induces Autophagy in Breast Cancer. Mol Biosyst (2015) 11(11):2860–6. doi: 10.1039/c5mb00466g
71. Hu J, Li G, Qu L, Li N, Liu W, Xia D, et al. TMEM166/EVA1A Interacts With ATG16L1 and Induces Autophagosome Formation and Cell Death. Cell Death Dis (2016) 7(8):e2323. doi: 10.1038/cddis.2016.230
72. Zhen Y, Zhao R, Wang M, Jiang X, Gao F, Fu L, et al. Flubendazole Elicits Anti-Cancer Effects Via Targeting EVA1A-modulated Autophagy and Apoptosis in Triple-negative Breast Cancer. Theranostics (2020) 10(18):8080–97. doi: 10.7150/thno.43473
73. Dong T, Lu Z, Li J, Liu Y, Wen J. [Flubendazole Inhibits the Proliferation of A549 and H460 Cells and Promotes Autophagy]. Zhongguo Fei Ai Za Zhi (2020) 23(5):306–13. doi: 10.3779/j.issn.1009-3419.2020.104.17
74. Lin S, Yang L, Yao Y, Xu L, Xiang Y, Zhao H, et al. Flubendazole Demonstrates Valid Antitumor Effects by Inhibiting STAT3 and Activating Autophagy. J Exp Clin Cancer Res (2019) 38(1):293. doi: 10.1186/s13046-019-1303-z
75. Dolan DE, Gupta S. PD-1 Pathway Inhibitors: Changing the Landscape of Cancer Immunotherapy. Cancer Control (2014) 21(3):231–7. doi: 10.1177/107327481402100308
76. Gong J, Chehrazi-Raffle A, Reddi S, Salgia R. Development of PD-1 and PD-L1 Inhibitors as a Form of Cancer Immunotherapy: A Comprehensive Review of Registration Trials and Future Considerations. J Immunother Cancer (2018) 6(1):8. doi: 10.1186/s40425-018-0316-z
77. Li K, Tian H. Development of Small-Molecule Immune Checkpoint Inhibitors of PD-1/PD-L1 as a New Therapeutic Strategy for Tumour Immunotherapy. J Drug Target (2019) 27(3):244–56. doi: 10.1080/1061186X.2018.1440400
78. Li Y, Acharya G, Elahy M, Xin H, Khachigian LM. The Anthelmintic Flubendazole Blocks Human Melanoma Growth and Metastasis and Suppresses Programmed Cell Death Protein-1 and Myeloid-Derived Suppressor Cell Accumulation. Cancer Lett (2019) 459:268–76. doi: 10.1016/j.canlet.2019.05.026
79. Slamon DJ, Leyland-Jones B, Shak S, Fuchs H, Paton V, Bajamonde A, et al. Use of Chemotherapy Plus a Monoclonal Antibody Against HER2 for Metastatic Breast Cancer That Overexpresses HER2. N Engl J Med (2001) 344(11):783–92. doi: 10.1056/NEJM200103153441101
80. Kim YJ, Sung D, Oh E, Cho Y, Cho TM, Farrand L, et al. Flubendazole Overcomes Trastuzumab Resistance by Targeting Cancer Stem-Like Properties and HER2 Signaling in HER2-positive Breast Cancer. Cancer Lett (2018) 412:118–30. doi: 10.1016/j.canlet.2017.10.020
81. Sáez R, Molina MA, Ramsey EE, Rojo F, Keenan EJ, Albanell J, et al. p95HER-2 Predicts Worse Outcome in Patients With HER-2-positive Breast Cancer. Clin Cancer Res (2006) 12(2):424–31. doi: 10.1158/1078-0432.CCR-05-1807
82. Watanabe S, Yonesaka K, Tanizaki J, Nonagase Y, Takegawa N, Haratani K, et al. Targeting of the HER2/HER3 Signaling Axis Overcomes Ligand-Mediated Resistance to Trastuzumab in HER2-positive Breast Cancer. Cancer Med (2019) 8(3):1258–68. doi: 10.1002/cam4.1995
83. Parra-Palau JL, Morancho B, Peg V, Escorihuela M, Scaltriti M, Vicario R, et al. Effect of p95HER2/611CTF on the Response to Trastuzumab and Chemotherapy. J Natl Cancer Inst (2014) 106(11):dju291. doi: 10.1093/jnci/dju291
84. Visser BJ, Wieten RW, Kroon D, Nagel IM, Bélard S, van Vugt M, et al. Efficacy and Safety of Artemisinin Combination Therapy (ACT) for non-Falciparum Malaria: A Systematic Review. Malar J (2014) 13:463. doi: 10.1186/1475-2875-13-463
85. Naing C, Whittaker MA, Mak JW, Aung K. A Systematic Review of the Efficacy of a Single Dose Artemisinin-Naphthoquine in Treating Uncomplicated Malaria. Malar J (2015) 14:392. doi: 10.1186/s12936-015-0919-5
86. Dahal P, D’Alessandro U, Dorsey G, Guerin PJ, Nsanzabana C, Price RN, et al. Clinical Determinants of Early Parasitological Response to ACTs in African Patients With Uncomplicated Falciparum Malaria: A Literature Review and Meta-Analysis of Individual Patient Data. BMC Med (2015) 13:212. doi: 10.1186/s12916-015-0445-x
87. Yakasai AM, Hamza M, Dalhat MM, Bello M, Gadanya MA, Yaqub ZM, et al. Adherence to Artemisinin-Based Combination Therapy for the Treatment of Uncomplicated Malaria: A Systematic Review and Meta-Analysis. J Trop Med (2015) 2015:189232. doi: 10.1155/2015/189232
88. Cumming JN, Ploypradith P, Posner GH. Antimalarial Activity of Artemisinin (Qinghaosu) and Related Trioxanes: Mechanism(s) of Action. Adv Pharmacol (1997) 37:253–97. doi: 10.1016/s1054-3589(08)60952-7
89. O’Neill PM, Posner GH. A Medicinal Chemistry Perspective on Artemisinin and Related Endoperoxides. J Med Chem (2004) 47(12):2945–64. doi: 10.1021/jm030571c
90. Shandilya A, Chacko S, Jayaram B, Ghosh I. A Plausible Mechanism for the Antimalarial Activity of Artemisinin: A Computational Approach. Sci Rep (2013) 3:2513. doi: 10.1038/srep02513
91. Lisewski AM, Quiros JP, Ng CL, Adikesavan AK, Miura K, Putluri N, et al. Supergenomic Network Compression and the Discovery of EXP1 as a Glutathione Transferase Inhibited by Artesunate. Cell (2014) 158(4):916–28. doi: 10.1016/j.cell.2014.07.011
92. Zheng GQ. Cytotoxic Terpenoids and Flavonoids From Artemisia Annua. Planta Med (1994) 60(1):54–7. doi: 10.1055/s-2006-959408
93. Beekman AC, Wierenga PK, Woerdenbag HJ, Van Uden W, Pras N, Konings AW, et al. Artemisinin-Derived Sesquiterpene Lactones as Potential Antitumour Compounds: Cytotoxic Action Against Bone Marrow and Tumour Cells. Planta Med (1998) 64(7):615–9. doi: 10.1055/s-2006-957533
94. Trachootham D, Alexandre J, Huang P. Targeting Cancer Cells by ROS-mediated Mechanisms: A Radical Therapeutic Approach? Nat Rev Drug Discovery (2009) 8(7):579–91. doi: 10.1038/nrd2803
95. Mercer AE, Copple IM, Maggs JL, O’Neill PM, Park BK. The Role of Heme and the Mitochondrion in the Chemical and Molecular Mechanisms of Mammalian Cell Death Induced by the Artemisinin Antimalarials. J Biol Chem (2011) 286(2):987–96. doi: 10.1074/jbc.M110.144188
96. Zhang CJ, Wang J, Zhang J, Lee YM, Feng G, Lim TK, et al. Mechanism-Guided Design and Synthesis of a Mitochondria-Targeting Artemisinin Analogue With Enhanced Anticancer Activity. Angew Chem Int Ed Engl (2016) 55(44):13770–4. doi: 10.1002/anie.201607303
97. Wang J, Zhang J, Shi Y, Xu C, Zhang C, Wong YK, et al. Mechanistic Investigation of the Specific Anticancer Property of Artemisinin and Its Combination With Aminolevulinic Acid for Enhanced Anticolorectal Cancer Activity. ACS Cent Sci (2017) 3(7):743–50. doi: 10.1021/acscentsci.7b00156
98. Chen Y, Li R, Zhu Y, Zhong S, Qian J, Yang D, et al. Dihydroartemisinin Induces Growth Arrest and Overcomes Dexamethasone Resistance in Multiple Myeloma. Front Oncol (2020) 10:767. doi: 10.3389/fonc.2020.00767
99. Elhassanny A, Soliman E, Marie M, McGuire P, Gul W, ElSohly M, et al. Heme-Dependent ER Stress Apoptosis: A Mechanism for the Selective Toxicity of the Dihydroartemisinin, NSC735847, in Colorectal Cancer Cells. Front Oncol (2020) 10:965. doi: 10.3389/fonc.2020.00965
100. Zhu S, Yu Q, Huo C, Li Y, He L, Ran B, et al. Ferroptosis: A Novel Mechanism of Artemisinin and its Derivatives in Cancer Therapy. Curr Med Chem (2020) 28(2):329–45. doi: 10.2174/0929867327666200121124404
101. Guo S, Yao X, Jiang Q, Wang K, Zhang Y, Peng H, et al. Dihydroartemisinin-Loaded Magnetic Nanoparticles for Enhanced Chemodynamic Therapy. Front Pharmacol (2020) 11:226. doi: 10.3389/fphar.2020.00226
102. Luo Y, Sun X, Huang L, Yan J, Yu BY, Tian J. Artemisinin-Based Smart Nanomedicines With Self-Supply of Ferrous Ion to Enhance Oxidative Stress for Specific and Efficient Cancer Treatment. ACS Appl Mater Interfaces (2019) 11(33):29490–7. doi: 10.1021/acsami.9b07390
103. Li X, Gu S, Sun D, Dai H, Chen H, Zhang Z. The Selectivity of Artemisinin-Based Drugs on Human Lung Normal and Cancer Cells. Environ Toxicol Pharmacol (2018) 57:86–94. doi: 10.1016/j.etap.2017.12.004
104. Kadioglu O, Chan A, Cong LQA, Wong V, Colligs V, Wecklein S, et al. Artemisinin Derivatives Target Topoisomerase 1 and Cause Dna Damage in Silico and In Vitro. Front Pharmacol (2017) 8:711. doi: 10.3389/fphar.2017.00711
105. Bollimpelli VS, Dholaniya PS, Kondapi AK. Topoisomerase Iiβ and its Role in Different Biological Contexts. Arch Biochem Biophys (2017) 633:78–84. doi: 10.1016/j.abb.2017.06.021
106. Chen J, Li W, Cui K, Ji K, Xu S, Xu Y. Artemisitene Suppresses Tumorigenesis by Inducing DNA Damage Through Deregulating c-Myc-topoisomerase Pathway. Oncogene (2018) 37(37):5079–87. doi: 10.1038/s41388-018-0331-z
107. Nitiss JL. Targeting DNA Topoisomerase II in Cancer Chemotherapy. Nat Rev Cancer (2009) 9(5):338–50. doi: 10.1038/nrc2607
108. Zhang P, Wang H, Rowe PS, Hu B, Wang Y. MEPE/OF45 as a New Target for Sensitizing Human Tumour Cells to DNA Damage Inducers. Br J Cancer (2010) 102(5):862–6. doi: 10.1038/sj.bjc.6605572
109. Harbour JW, Luo RX, Dei SA, Postigo AA, Dean DC. Cdk Phosphorylation Triggers Sequential Intramolecular Interactions That Progressively Block Rb Functions as Cells Move Through G1. Cell (1999) 98(6):859–69. doi: 10.1016/s0092-8674(00)81519-6
110. Musgrove EA, Caldon CE, Barraclough J, Stone A, Sutherland RL. Cyclin D as a Therapeutic Target in Cancer. Nat Rev Cancer (2011) 11(8):558–72. doi: 10.1038/nrc3090
111. Fan HN, Zhu MY, Peng SQ, Zhu JS, Zhang J, Qu GQ. Dihydroartemisinin Inhibits the Growth and Invasion of Gastric Cancer Cells by Regulating Cyclin D1-CDK4-Rb Signaling. Pathol Res Pract (2020) 216(2):152795. doi: 10.1016/j.prp.2019.152795
112. Guan X, Guan Y. Artemisinin Induces Selective and Potent Anticancer Effects in Drug Resistant Breast Cancer Cells by Inducing Cellular Apoptosis and Autophagy and G2/M Cell Cycle Arrest. J Buon (2020) 25(3):1330–6.
113. Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, et al. Ferroptosis: An Iron-Dependent Form of Nonapoptotic Cell Death. Cell (2012) 149(5):1060–72. doi: 10.1016/j.cell.2012.03.042
114. Gao M, Monian P, Quadri N, Ramasamy R, Jiang X. Glutaminolysis and Transferrin Regulate Ferroptosis. Mol Cell (2015) 59(2):298–308. doi: 10.1016/j.molcel.2015.06.011
115. Stockwell BR, Friedmann AJ, Bayir H, Bush AI, Conrad M, Dixon SJ, et al. Ferroptosis: A Regulated Cell Death Nexus Linking Metabolism, Redox Biology, and Disease. Cell (2017) 171(2):273–85. doi: 10.1016/j.cell.2017.09.021
116. Yang WS, SriRamaratnam R, Welsch ME, Shimada K, Skouta R, Viswanathan VS, et al. Regulation of Ferroptotic Cancer Cell Death by GPX4. Cell (2014) 156(1-2):317–31. doi: 10.1016/j.cell.2013.12.010
117. Ooko E, Saeed ME, Kadioglu O, Sarvi S, Colak M, Elmasaoudi K, et al. Artemisinin Derivatives Induce Iron-Dependent Cell Death (Ferroptosis) in Tumor Cells. Phytomedicine (2015) 22(11):1045–54. doi: 10.1016/j.phymed.2015.08.002
118. Chen GQ, Benthani FA, Wu J, Liang D, Bian ZX, Jiang X. Artemisinin Compounds Sensitize Cancer Cells to Ferroptosis by Regulating Iron Homeostasis. Cell Death Differ (2020) 27(1):242–54. doi: 10.1038/s41418-019-0352-3
119. Wang K, Zhang Z, Wang M, Cao X, Qi J, Wang D, et al. Role of GRP78 Inhibiting Artesunate-Induced Ferroptosis in KRAS Mutant Pancreatic Cancer Cells. Drug Des Devel Ther (2019) 13:2135–44. doi: 10.2147/DDDT.S199459
120. Chen Y, Mi Y, Zhang X, Ma Q, Song Y, Zhang L, et al. Dihydroartemisinin-Induced Unfolded Protein Response Feedback Attenuates Ferroptosis Via PERK/ATF4/HSPA5 Pathway in Glioma Cells. J Exp Clin Cancer Res (2019) 38(1):402. doi: 10.1186/s13046-019-1413-7
121. Paccez JD, Duncan K, Sekar D, Correa RG, Wang Y, Gu X, et al. Dihydroartemisinin Inhibits Prostate Cancer Via JARID2/miR-7/miR-34a-dependent Downregulation of Axl. Oncogenesis (2019) 8(3):14. doi: 10.1038/s41389-019-0122-6
122. Chen S, Gan S, Han L, Li X, Xie X, Zou D, et al. Artesunate Induces Apoptosis and Inhibits the Proliferation, Stemness, and Tumorigenesis of Leukemia. Ann Transl Med (2020) 8(12):767. doi: 10.21037/atm-20-4558
123. Li Z, Zhu YT, Xiang M, Qiu JL, Luo SQ, Lin F. Enhanced Lysosomal Function is Critical for Paclitaxel Resistance in Cancer Cells: Reversed by Artesunate. Acta Pharmacol Sin (2020) 42(4):624–32. doi: 10.1038/s41401-020-0445-z
124. Pirali M, Taheri M, Zarei S, Majidi M, Ghafouri H. Artesunate, as a HSP70 Atpase Activity Inhibitor, Induces Apoptosis in Breast Cancer Cells. Int J Biol Macromol (2020) 164:3369–75. doi: 10.1016/j.ijbiomac.2020.08.198
125. Zhao F, Vakhrusheva O, Markowitsch SD, Slade KS, Tsaur I, Cinatl JJ, et al. Artesunate Impairs Growth in Cisplatin-Resistant Bladder Cancer Cells by Cell Cycle Arrest, Apoptosis and Autophagy Induction. Cells-Basel (2020) 9(12):2643. doi: 10.3390/cells9122643
126. Zhou X, Zijlstra SN, Soto-Gamez A, Setroikromo R, Quax WJ. Artemisinin Derivatives Stimulate DR5-Specific Trail-Induced Apoptosis by Regulating Wildtype P53. Cancers (Basel) (2020) 12(9):2514 doi: 10.3390/cancers12092514
127. Zhou X, Chen Y, Wang F, Wu H, Zhang Y, Liu J, et al. Artesunate Induces Autophagy Dependent Apoptosis Through Upregulating ROS and Activating AMPK-mTOR-ULK1 Axis in Human Bladder Cancer Cells. Chem Biol Interact (2020) 331:109273. doi: 10.1016/j.cbi.2020.109273
128. Yan X, Li P, Zhan Y, Qi M, Liu J, An Z, et al. Dihydroartemisinin Suppresses STAT3 Signaling and Mcl-1 and Survivin Expression to Potentiate ABT-263-induced Apoptosis in Non-small Cell Lung Cancer Cells Harboring EGFR or RAS Mutation. Biochem Pharmacol (2018) 150:72–85. doi: 10.1016/j.bcp.2018.01.031
129. Våtsveen TK, Myhre MR, Steen CB, Wälchli S, Lingjærde OC, Bai B, et al. Artesunate Shows Potent Anti-Tumor Activity in B-cell Lymphoma. J Hematol Oncol (2018) 11(1):23. doi: 10.1186/s13045-018-0561-0
130. Zhou C, Tang X, Xu J, Wang J, Yang Y, Chen Y, et al. Opening of the CLC-3 Chloride Channel Induced by Dihydroartemisinin Contributed to Early Apoptotic Events in Human Poorly Differentiated Nasopharyngeal Carcinoma Cells. J Cell Biochem (2018) 119(11):9560–72. doi: 10.1002/jcb.27274
131. Liu X, Wu J, Fan M, Shen C, Dai W, Bao Y, et al. Novel Dihydroartemisinin Derivative DHA-37 Induces Autophagic Cell Death Through Upregulation of HMGB1 in A549 Cells. Cell Death Dis (2018) 9(11):1048. doi: 10.1038/s41419-018-1006-y
132. Chen X, Wong YK, Lim TK, Lim WH, Lin Q, Wang J, et al. Artesunate Activates the Intrinsic Apoptosis of HCT116 Cells Through the Suppression of Fatty Acid Synthesis and the NF-κb Pathway. Molecules (2017) 22(8):1272. doi: 10.3390/molecules22081272
133. Nunes JJ, Pandey SK, Yadav A, Goel S, Ateeq B. Targeting NF-Kappa B Signaling by Artesunate Restores Sensitivity of Castrate-Resistant Prostate Cancer Cells to Antiandrogens. Neoplasia (2017) 19(4):333–45. doi: 10.1016/j.neo.2017.02.002
134. Houh YK, Kim KE, Park S, Hur DY, Kim S, Kim D, et al. The Effects of Artemisinin on the Cytolytic Activity of Natural Killer (Nk) Cells. Int J Mol Sci (2017) 18(7):1600. doi: 10.3390/ijms18071600
135. Lu Z, Bi J, Wan X. Artemisinin Sensitizes Tumor Cells to NK Cell-Mediated Cytolysis. Biochem Biophys Res Commun (2020) 524(2):418–23. doi: 10.1016/j.bbrc.2020.01.094
136. Farsam V, Hassan ZM, Zavaran-Hosseini A, Noori S, Mahdavi M, Ranjbar M. Antitumor and Immunomodulatory Properties of Artemether and its Ability to Reduce CD4+ CD25+ Foxp3+ T Reg Cells In Vivo. Int Immunopharmacol (2011) 11(11):1802–8. doi: 10.1016/j.intimp.2011.07.008
137. Cao Y, Feng YH, Gao LW, Li XY, Jin QX, Wang YY, et al. Artemisinin Enhances the Anti-Tumor Immune Response in 4T1 Breast Cancer Cells In Vitro and In Vivo. Int Immunopharmacol (2019) 70:110–6. doi: 10.1016/j.intimp.2019.01.041
138. Berger TG, Dieckmann D, Efferth T, Schultz ES, Funk JO, Baur A, et al. Artesunate in the Treatment of Metastatic Uveal Melanoma–First Experiences. Oncol Rep (2005) 14(6):1599–603. doi: 10.3892/or.14.6.1599
139. Deeken JF, Wang H, Hartley M, Cheema AK, Smaglo B, Hwang JJ, et al. A Phase I Study of Intravenous Artesunate in Patients With Advanced Solid Tumor Malignancies. Cancer Chemother Pharmacol (2018) 81(3):587–96. doi: 10.1007/s00280-018-3533-8
140. Milner DJ. Malaria Pathogenesis. Cold Spring Harb Perspect Med (2018) 8(1):a025569. doi: 10.1101/cshperspect.a025569
141. Nixon GL, Moss DM, Shone AE, Lalloo DG, Fisher N, O’Neill PM, et al. Antimalarial Pharmacology and Therapeutics of Atovaquone. J Antimicrob Chemother (2013) 68(5):977–85. doi: 10.1093/jac/dks504
142. Watkins WM. Pharmacology and Pharmacokinetics of New Antimalarials. Med Trop (Mars) (1995) 55(4 Suppl):33–6.
143. Commons RJ, Simpson JA, Thriemer K, Humphreys GS, Abreha T, Alemu SG, et al. The Effect of Chloroquine Dose and Primaquine on Plasmodium Vivax Recurrence: A WorldWide Antimalarial Resistance Network Systematic Review and Individual Patient Pooled Meta-Analysis. Lancet Infect Dis (2018) 18(9):1025–34. doi: 10.1016/S1473-3099(18)30348-7
144. Skrzypek R, Callaghan R. The “Pushmi-Pullyu” of Resistance to Chloroquine in Malaria. Essays Biochem (2017) 61(1):167–75. doi: 10.1042/EBC20160060
145. Thomé R, Lopes SC, Costa FT, Verinaud L. Chloroquine: Modes of Action of an Undervalued Drug. Immunol Lett (2013) 153(1-2):50–7. doi: 10.1016/j.imlet.2013.07.004
146. Aguiar A, Murce E, Cortopassi WA, Pimentel AS, Almeida M, Barros D, et al. Chloroquine Analogs as Antimalarial Candidates With Potent In Vitro and In Vivo Activity. Int J Parasitol Drugs Drug Resist (2018) 8(3):459–64. doi: 10.1016/j.ijpddr.2018.10.002
147. Pasquier B. Autophagy Inhibitors. Cell Mol Life Sci (2016) 73(5):985–1001. doi: 10.1007/s00018-015-2104-y
148. Mindell JA. Lysosomal Acidification Mechanisms. Annu Rev Physiol (2012) 74:69–86. doi: 10.1146/annurev-physiol-012110-142317
149. Martinez GP, Zabaleta ME, Di Giulio C, Charris JE, Mijares MR. The Role of Chloroquine and Hydroxychloroquine in Immune Regulation and Diseases. Curr Pharm Des (2020) 26(35):4467–85. doi: 10.2174/1381612826666200707132920
150. Amaravadi RK, Lippincott-Schwartz J, Yin XM, Weiss WA, Takebe N, Timmer W, et al. Principles and Current Strategies for Targeting Autophagy for Cancer Treatment. Clin Cancer Res (2011) 17(4):654–66. doi: 10.1158/1078-0432.CCR-10-2634
151. Cortegiani A, Ingoglia G, Ippolito M, Giarratano A, Einav S. A Systematic Review on the Efficacy and Safety of Chloroquine for the Treatment of COVID-19. J Crit Care (2020) 57:279–83. doi: 10.1016/j.jcrc.2020.03.005
152. Pascolo S. Time to Use a Dose of Chloroquine as an Adjuvant to Anti-Cancer Chemotherapies. Eur J Pharmacol (2016) 771:139–44. doi: 10.1016/j.ejphar.2015.12.017
153. Kaneno R, Shurin GV, Kaneno FM, Naiditch H, Luo J, Shurin MR. Chemotherapeutic Agents in Low Noncytotoxic Concentrations Increase Immunogenicity of Human Colon Cancer Cells. Cell Oncol (Dordr) (2011) 34(2):97–106. doi: 10.1007/s13402-010-0005-5
154. Zitvogel L, Apetoh L, Ghiringhelli F, Kroemer G. Immunological Aspects of Cancer Chemotherapy. Nat Rev Immunol (2008) 8(1):59–73. doi: 10.1038/nri2216
155. Zitvogel L, Kroemer G. Anticancer Immunochemotherapy Using Adjuvants With Direct Cytotoxic Effects. J Clin Invest (2009) 119(8):2127–30. doi: 10.1172/JCI39991
156. Vanmeerbeek I, Sprooten J, De Ruysscher D, Tejpar S, Vandenberghe P, Fucikova J, et al. Trial Watch: Chemotherapy-Induced Immunogenic Cell Death in Immuno-Oncology. Oncoimmunology (2020) 9(1):1703449. doi: 10.1080/2162402X.2019.1703449
157. Kaneno R, Shurin GV, Tourkova IL, Shurin MR. Chemomodulation of Human Dendritic Cell Function by Antineoplastic Agents in Low Noncytotoxic Concentrations. J Transl Med (2009) 7:58. doi: 10.1186/1479-5876-7-58
158. Zamame RJ, Romagnoli GG, Falasco BF, Gorgulho CM, Sanzochi FC, Dos SD, et al. Blocking Drug-Induced Autophagy With Chloroquine in HCT-116 Colon Cancer Cells Enhances DC Maturation and T Cell Responses Induced by Tumor Cell Lysate. Int Immunopharmacol (2020) 84:106495. doi: 10.1016/j.intimp.2020.106495
159. Kim S, Han Y, Kim SI, Kim HS, Kim SJ, Song YS. Tumor Evolution and Chemoresistance in Ovarian Cancer. NPJ Precis Oncol (2018) 2:20. doi: 10.1038/s41698-018-0063-0
160. Sakthivel KM, Hariharan S. Regulatory Players of DNA Damage Repair Mechanisms: Role in Cancer Chemoresistance. BioMed Pharmacother (2017) 93:1238–45. doi: 10.1016/j.biopha.2017.07.035
161. Nagel ZD, Kitange GJ, Gupta SK, Joughin BA, Chaim IA, Mazzucato P, et al. Dna Repair Capacity in Multiple Pathways Predicts Chemoresistance in Glioblastoma Multiforme. Cancer Res (2017) 77(1):198–206. doi: 10.1158/0008-5472.CAN-16-1151
162. Hwang JR, Kim WY, Cho YJ, Ryu JY, Choi JJ, Jeong SY, et al. Chloroquine Reverses Chemoresistance Via Upregulation of p21(WAF1/CIP1) and Autophagy Inhibition in Ovarian Cancer. Cell Death Dis (2020) 11(12):1034. doi: 10.1038/s41419-020-03242-x
163. Yue D, Zhang D, Shi X, Liu S, Li A, Wang D, et al. Chloroquine Inhibits Stemness of Esophageal Squamous Cell Carcinoma Cells Through Targeting Cxcr4-STAT3 Pathway. Front Oncol (2020) 10:311. doi: 10.3389/fonc.2020.00311
164. Varisli L, Cen O, Vlahopoulos S. Dissecting Pharmacological Effects of Chloroquine in Cancer Treatment: Interference With Inflammatory Signaling Pathways. Immunology (2020) 159(3):257–78. doi: 10.1111/imm.13160
165. Compter I, Eekers D, Hoeben A, Rouschop K, Reymen B, Ackermans L, et al. Chloroquine Combined With Concurrent Radiotherapy and Temozolomide for Newly Diagnosed Glioblastoma: A Phase IB Trial. Autophagy (2020), 1–9. doi: 10.1080/15548627.2020.1816343
166. Zhou W, Wang H, Yang Y, Chen ZS, Zou C, Zhang J. Chloroquine Against Malaria, Cancers and Viral Diseases. Drug Discovery Today (2020) 25(11):2012–22. doi: 10.1016/j.drudis.2020.09.010
167. Lagadinou ED, Sach A, Callahan K, Rossi RM, Neering SJ, Minhajuddin M, et al. BCL-2 Inhibition Targets Oxidative Phosphorylation and Selectively Eradicates Quiescent Human Leukemia Stem Cells. Cell Stem Cell (2013) 12(3):329–41. doi: 10.1016/j.stem.2012.12.013
168. Miranda S, Foncea R, Guerrero J, Leighton F. Oxidative Stress and Upregulation of Mitochondrial Biogenesis Genes in Mitochondrial DNA-depleted HeLa Cells. Biochem Biophys Res Commun (1999) 258(1):44–9. doi: 10.1006/bbrc.1999.0580
169. Tian S, Chen H, Tan W. Targeting Mitochondrial Respiration as a Therapeutic Strategy for Cervical Cancer. Biochem Biophys Res Commun (2018) 499(4):1019–24. doi: 10.1016/j.bbrc.2018.04.042
170. Wang D, Xue B, Ohulchanskyy TY, Liu Y, Yakovliev A, Ziniuk R, et al. Inhibiting Tumor Oxygen Metabolism and Simultaneously Generating Oxygen by Intelligent Upconversion Nanotherapeutics for Enhanced Photodynamic Therapy. Biomaterials (2020) 251:120088. doi: 10.1016/j.biomaterials.2020.120088
171. Agostinis P, Berg K, Cengel KA, Foster TH, Girotti AW, Gollnick SO, et al. Photodynamic Therapy of Cancer: An Update. CA Cancer J Clin (2011) 61(4):250–81. doi: 10.3322/caac.20114
172. Li XY, Tan LC, Dong LW, Zhang WQ, Shen XX, Lu X, et al. Susceptibility and Resistance Mechanisms During Photodynamic Therapy of Melanoma. Front Oncol (2020) 10:597. doi: 10.3389/fonc.2020.00597
173. Chen D, Sun X, Zhang X, Cao J. Targeting Mitochondria by Anthelmintic Drug Atovaquone Sensitizes Renal Cell Carcinoma to Chemotherapy and Immunotherapy. J Biochem Mol Toxicol (2018) 32(9):e22195. doi: 10.1002/jbt.22195
174. Ashton TM, Fokas E, Kunz-Schughart LA, Folkes LK, Anbalagan S, Huether M, et al. The Anti-Malarial Atovaquone Increases Radiosensitivity by Alleviating Tumour Hypoxia. Nat Commun (2016) 7:12308. doi: 10.1038/ncomms12308
175. Fiorillo M, Lamb R, Tanowitz HB, Mutti L, Krstic-Demonacos M, Cappello AR, et al. Repurposing Atovaquone: Targeting Mitochondrial Complex III and OXPHOS to Eradicate Cancer Stem Cells. Oncotarget (2016) 7(23):34084–99. doi: 10.18632/oncotarget.9122
176. Lv Z, Yan X, Lu L, Su C, He Y. Atovaquone Enhances Doxorubicin’s Efficacy Via Inhibiting Mitochondrial Respiration and STAT3 in Aggressive Thyroid Cancer. J Bioenerg Biomembr (2018) 50(4):263–70. doi: 10.1007/s10863-018-9755-y
177. Takabe H, Warnken ZN, Zhang Y, Davis DA, Smyth H, Kuhn JG, et al. A Repurposed Drug for Brain Cancer: Enhanced Atovaquone Amorphous Solid Dispersion by Combining a Spontaneously Emulsifying Component With a Polymer Carrier. Pharmaceutics (2018) 10(2):60. doi: 10.3390/pharmaceutics10020060
178. Xiang M, Kim H, Ho VT, Walker SR, Bar-Natan M, Anahtar M, et al. Gene Expression-Based Discovery of Atovaquone as a STAT3 Inhibitor and Anticancer Agent. Blood (2016) 128(14):1845–53. doi: 10.1182/blood-2015-07-660506
179. Ashton TM, McKenna WG, Kunz-Schughart LA, Higgins GS. Oxidative Phosphorylation as an Emerging Target in Cancer Therapy. Clin Cancer Res (2018) 24(11):2482–90. doi: 10.1158/1078-0432.CCR-17-3070
180. Gupta N, Srivastava SK. Atovaquone: An Antiprotozoal Drug Suppresses Primary and Resistant Breast Tumor Growth by Inhibiting Her2/β-Catenin Signaling. Mol Cancer Ther (2019) 18(10):1708–20. doi: 10.1158/1535-7163.MCT-18-1286
181. Gao X, Liu X, Shan W, Liu Q, Wang C, Zheng J, et al. Anti-Malarial Atovaquone Exhibits Anti-Tumor Effects by Inducing DNA Damage in Hepatocellular Carcinoma. Am J Cancer Res (2018) 8(9):1697–711.
182. Rijpma SR, van den Heuvel JJ, van der Velden M, Sauerwein RW, Russel FG, Koenderink JB. Atovaquone and Quinine Anti-Malarials Inhibit ATP Binding Cassette Transporter Activity. Malar J (2014) 13:359. doi: 10.1186/1475-2875-13-359
183. Ashburn TT, Thor KB. Drug Repositioning: Identifying and Developing New Uses for Existing Drugs. Nat Rev Drug Discovery (2004) 3(8):673–83. doi: 10.1038/nrd1468
184. Langedijk J, Mantel-Teeuwisse AK, Slijkerman DS, Schutjens MH. Drug Repositioning and Repurposing: Terminology and Definitions in Literature. Drug Discovery Today (2015) 20(8):1027–34. doi: 10.1016/j.drudis.2015.05.001
185. Juarez M, Schcolnik-Cabrera A, Dueñas-Gonzalez A. The Multitargeted Drug Ivermectin: From an Antiparasitic Agent to a Repositioned Cancer Drug. Am J Cancer Res (2018) 8(2):317–31.
186. Hirschhorn T, Stockwell BR. The Development of the Concept of Ferroptosis. Free Radic Biol Med (2019) 133:130–43. doi: 10.1016/j.freeradbiomed.2018.09.043
187. Ayyagari VN, Hsieh TJ, Diaz-Sylvester PL, Brard L. Evaluation of the Cytotoxicity of the Bithionol - Cisplatin Combination in a Panel of Human Ovarian Cancer Cell Lines. BMC Cancer (2017) 17(1):49. doi: 10.1186/s12885-016-3034-2
188. Gao A, Wang X, Xiang W, Liang H, Gao J, Yan Y. Reversal of P-glycoprotein-mediated Multidrug Resistance In Vitro by Doramectin and Nemadectin. J Pharm Pharmacol (2010) 62(3):393–9. doi: 10.1211/jpp.62.03.0016
189. Wu ZH, Lu MK, Hu LY, Li X. Praziquantel Synergistically Enhances Paclitaxel Efficacy to Inhibit Cancer Cell Growth. PloS One (2012) 7(12):e51721. doi: 10.1371/journal.pone.0051721
190. Moschovi M, Critselis E, Cen O, Adamaki M, Lambrou GI, Chrousos GP, et al. Drugs Acting on Homeostasis: Challenging Cancer Cell Adaptation. Expert Rev Anticancer Ther (2015) 15(12):1405–17. doi: 10.1586/14737140.2015.1095095
191. Vlahopoulos SA. Aberrant Control of NF-κb in Cancer Permits Transcriptional and Phenotypic Plasticity, to Curtail Dependence on Host Tissue: Molecular Mode. Cancer Biol Med (2017) 14(3):254–70. doi: 10.20892/j.issn.2095-3941.2017.0029
192. Hettiarachchi G, Samanta SK, Falcinelli S, Zhang B, Moncelet D, Isaacs L, et al. Acyclic Cucurbit[n]uril-Type Molecular Container Enables Systemic Delivery of Effective Doses of Albendazole for Treatment of SK-OV-3 Xenograft Tumors. Mol Pharm (2016) 13(3):809–18. doi: 10.1021/acs.molpharmaceut.5b00723
193. Akbarian A, Ebtekar M, Pakravan N, Hassan ZM. Folate Receptor Alpha Targeted Delivery of Artemether to Breast Cancer Cells With Folate-Decorated Human Serum Albumin Nanoparticles. Int J Biol Macromol (2020) 152:90–101. doi: 10.1016/j.ijbiomac.2020.02.106
194. Yang B, Shi J. Developing New Cancer Nanomedicines by Repurposing Old Drugs. Angew Chem Int Ed Engl (2020) 59(49):21829–38. doi: 10.1002/anie.202004317
195. Chen J, Jiang Z, Xu W, Sun T, Zhuang X, Ding J, et al. Spatiotemporally Targeted Nanomedicine Overcomes Hypoxia-Induced Drug Resistance of Tumor Cells After Disrupting Neovasculature. Nano Lett (2020) 20(8):6191–8. doi: 10.1021/acs.nanolett.0c02515
196. Wei L, Chen J, Ding J. Sequentially Stimuli-Responsive Anticancer Nanomedicines. Nanomed (Lond) (2021) 16(4):261–4. doi: 10.2217/nnm-2021-0019
197. Feng X, Xu W, Xu X, Li G, Ding J, Chen X. Cystine Proportion Regulates Fate of Polypeptide Nanogel as Nanocarrier for Chemotherapeutics. Sci China Chem (2021) 64(2):293–301. doi: 10.1007/s11426-020-9884-6
198. Ding J, Chen J, Gao L, Jiang Z, Zhang Y, Li M, et al. Engineered Nanomedicines With Enhanced Tumor Penetration. Nano Today (2019) 29:100800. doi: 10.1016/j.nantod.2019.100800
199. Zheng P, Ding B, Jiang Z, Xu W, Li G, Ding J, et al. Ultrasound-Augmented Mitochondrial Calcium Ion Overload by Calcium Nanomodulator to Induce Immunogenic Cell Death. Nano Lett (2021) 21(5):2088–93. doi: 10.1021/acs.nanolett.0c04778
200. Ma W, Chen Q, Xu W, Yu M, Yang Y, Zou B, et al. Self-Targeting Visualizable Hyaluronate Nanogel for Synchronized Intracellular Release of Doxorubicin and Cisplatin in Combating Multidrug-Resistant Breast Cancer. Nano Res (2021) 14(3):846–57. doi: 10.1007/s12274-020-3124-y
201. Liu J, Li Z, Zhao D, Feng X, Wang C, Li D, et al. Immunogenic Cell Death-Inducing Chemotherapeutic Nanoformulations Potentiate Combination Chemoimmunotherapy. Mater Design (2021) 202:109465. doi: 10.1016/j.matdes.2021.109465
Keywords: drug repositioning, antiparasitic drugs, macrolides, benzimidazoles, artemisinin, quinolines, autophagy, ferroptosis
Citation: Li Y-Q, Zheng Z, Liu Q-X, Lu X, Zhou D, Zhang J, Zheng H and Dai J-G (2021) Repositioning of Antiparasitic Drugs for Tumor Treatment. Front. Oncol. 11:670804. doi: 10.3389/fonc.2021.670804
Received: 22 February 2021; Accepted: 13 April 2021;
Published: 29 April 2021.
Edited by:
Christian Celia, University of Studies G. d’Annunzio Chieti and Pescara, ItalyReviewed by:
Patrícia Peres De Oliveira, Universidade Federal de São João del Rei, BrazilCopyright © 2021 Li, Zheng, Liu, Lu, Zhou, Zhang, Zheng and Dai. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ji-Gang Dai, ZGFpamlnYW5nQHRtbXUuZWR1LmNu; Hong Zheng, emllY29lQHRtbXUuZWR1LmNu
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.