
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Oncol. , 30 April 2021
Sec. Gastrointestinal Cancers
Volume 11 - 2021 | https://doi.org/10.3389/fonc.2021.664927
 Qun Zhao1*†
Qun Zhao1*† Jian Guo1†Xinran Cheng1Yingying Liao2Yun Bi1Yingxia Gong1Xudong Zhang1Yang Guo1Xianhui Wang1Wei Yu3Shu Jin4
Jian Guo1†Xinran Cheng1Yingying Liao2Yun Bi1Yingxia Gong1Xudong Zhang1Yang Guo1Xianhui Wang1Wei Yu3Shu Jin4 Yan Tan1
Yan Tan1 Xianjun Yu1*
Xianjun Yu1*Receptor-interacting protein 3 (RIPK3), a member of the family of serine/threonine protein kinases, emerged as a critical regulator of necroptosis. Downregulated expression of RIPK3 is correlated with poor prognosis in multiple tumor types. Here, we show that RIPK3 is involved in the progression of spontaneous intestinal tumorigenesis. As a clinical correlate, reduced expression of RIPK3 is positively associated with histological grade, lymphatic metastasis and poor prognosis in CRC patients. RIPK3-deficient (Ripk3-/-) mice exhibit increased tumor formation in Apcmin/+ spontaneous intestinal tumorigenesis. Apcmin/+Ripk3-/- tumors promote hyperactivation of IL-6/STAT3 signaling, which exacerbates proliferation and inhibits apoptosis. Blocking IL-6 signaling suppressed tumor formation and reduced STAT3 activation in Apcmin/+Ripk3-/- mice. Thus, our results reveal that RIPK3 is a tumor suppressor in spontaneous intestinal tumorigenesis, and implicate targeting the IL-6/STAT3 signaling axis as a potential therapeutic strategy for intestinal tumor patients with reduced RIPK3.
Colorectal cancer (CRC) is one of the most common cancers worldwide, and the number of deaths is rising in developing countries (1, 2). An increased frequency of CRC patients is usually diagnosed at an advance stage (3). Histologically, over 95% of CRC cases are adenocarcinomas, while squamous cell carcinomas, lymphomas and sarcomas are rarer (4). Increasing evidence reveals that excessive neoplastic transformation of colonic epithelial cells have the ability to initiate and develop CRC (5, 6). Moreover, CRCs arise in intestinal epithelial cells upon loss of tumor suppressor genes such as adenomatous polyposis coli (APC) (7). Thus, investigation of the molecular mechanisms in CRC tumorigenesis is urgently needed to prevent and treat CRC.
Inflammation is a dynamic and complex process to prevent overwhelming dampens and maintain tissue homeostasis (8, 9). Accumulation of the inflammatory environment drives intestinal cell proliferation and tissue regeneration through activation of pro-oncogene factors such as signal transducer and activator of transcription 3 (STAT3) (10, 11). Elevated levels of interleukin 6 (IL-6) are linked with chronic intestinal inflammation and tumorigenesis (12, 13). The role of IL-6/STAT3 signaling in intestinal cell proliferation and tumorigenesis is well established (14–16). A better understanding of the regulation of IL-6/STAT3 activation would facilitate the development of novel approaches for CRC therapy.
Necroptosis is a form of necrotic-regulated cell death upon extracellular and intracellular stimuli in certain pathologies (17–19). Receptor-interacting protein kinase 1 (RIPK1) and RIPK3 are critical regulatory molecules during necroptosis (20–22). RIPK1 interacts with RIPK3 via the RIP homotypic interaction motif (RHIM) domains and then forms the necrosome (23). RIPK1-RIPK3 necrosome complex results in the recruitment and phosphorylation of mixed-lineage kinase domain-like (MLKL), which leads to impair membrane integrity and drive necroptosis (24, 25). Several studies have shown that RIPK3 is involved in several inflammatory diseases, including intestinal inflammation, acute pancreatitis and skin inflammatory diseases (23, 26, 27). In fact, RIPK3 also exhibits a necroptosis-independent function in intestinal inflammation and tumorigenesis (28, 29). Reduced RIPK3 expression has been shown in human CRC tumors, and low expression of RIPK3 is significantly correlated with poor progression (30, 31). In contrast, overexpression of RIPK3 suppresses CRC cell proliferation, migration and invasion (32). In particular, loss of RIPK3 is susceptible to intestinal tumorigenesis in inflammation-associated colon cancer models (31, 33). However, colitis-associated tumorigenesis represents only 1% of colorectal cancer, and sporadic intestinal tumors are more prevalent (34). Thus, it is necessary to address the function of RIPK3 in the progression of spontaneous intestinal tumorigenesis.
In the present study, we revealed a tumor suppressor role for RIPK3 in spontaneous intestinal tumorigenesis. Expression analyses of RIPK3 expression showed that RIPK3 is significantly downregulated in CRC tumors, which predicted a poor prognosis in CRC. The absence of RIPK3 exhibits dramatically increased tumor numbers in Apcmin/+ mice through the hyperactivation of IL-6/STAT3 signals. Anti-IL-6R antibody therapy suppressed STAT3 activation and attenuated tumor burden in Apcmin/+Ripk3-/- mice. Our findings highlight that reduced RIPK3 predicts a more aggressive disease and worse outcome in CRC.
All the mice were on a C57BL/6 background and maintained in a specific pathogen-free (SPF) facility. Ripk3-/- mice and Apcmin/+ mice have been described previously (29, 35). Ripk3-/- mice were crossed with Apcmin/+ mice to generate Apcmin/+Ripk3+/- and Ripk3+/- mice. Then, Apcmin/+Ripk3+/- mice were crossed with Ripk3+/- mice to generate Apcmin/+ and Apcmin/+Ripk3-/- mice. This study was conducted according the guidelines of the Institutional Animal Care and Use Committee of the Hubei University of Medicine.
Clinical scores were calculated as described in our previous studies (35). The disease activity index (DAI) was quantified with a clinical score assessment based on stool consistency and fecal blood. Briefly, stool scores were determined as follows: 0 = well-formed pellets; 1 = semi-formed stools in the anus and 2 = liquid stools adhered in the anus. The bleeding scores were determined as follows: 0 = no observed blood; 1= visible blood traces in stool; and 2= gross bleeding in rectal tissue.
Tumor load was determined according to a published protocol (36). Tumor load was calculated based on tumor numbers and tumor diameter.
Isolated intestinal tissues were homogenized using a Mini-Rotor (Thermo Scientific) and lysed in complete radioimmunoprecipitation assay (RIPA) buffer containing Roche’s cOmplete™ protease inhibitor cocktail. The protein concentrations were quantified by a quantitative BCA protein kit (P0010S; Beyotime Biotechnology, Shanghai, China). The proteins were separated by SDS-PAGE and detected using chemiluminescent substrate (Thermo Scientific). The following primary antibodies were used: rabbit anti-RIPK3 (#ab152130, Abcam; 1:1000), mouse anti-STAT3 (#9139, CST; 1:1000), rabbit anti-phospho-STAT3 (Tyr705) (#9145, CST; 1:1000), rabbit anti-PCNA (#13110, CST; 1:1000), rabbit anti-Cleaved-Caspase-3 (#9661, CST; 1:1000), and rabbit anti-Cyclin D1 (#2978, CST; 1:1000), and rabbit anti-C-myc (#9402, CST; 1:1000), and anti-GAPDH (#5174, CST; 1:5000).
Intestinal tissues were homogenized in TRIzol reagent (Life Technologies) to obtain RNA. RNA was reverse-transcribed to complementary DNA using HiScript III RT SuperMix for qPCR (gDNA wiper) (# R323, Vazyme Biotech Co.,Ltd), and the levels of genes were measured using quantitative RT-PCR using SYBR Premix Ex Taq™ (#RR420, TAKARA) according to the manufacturer’s instructions. mRNA quantities were normalized against that of GAPDH.
Intestines were cleared and then cut into pieces. The colon crypt cells were collected following shaking after incubation in cold PBS with 3 mM EDTA/1.5 mM DTT.
The intestinal tissues were collected and then homogenized in RIPA buffer containing Roche’s cOmplete™ protease inhibitor cocktail for 30 mins on ice. The protein concentration of IL-6 was measured by enzyme linked immunosorbent assay (ELISA) (eBioscience) (37).
IL-6 signaling was antagonized with the IL-6R antibody tocilizumab (#A2012, Selleckchem; Houston, TX, USA). Four-week-old Apcmin/+Ripk3-/- mice were intraperitoneally injected with 4 mg/kg tocilizumab weekly. After 10 weeks of treatment, all mice were euthanized, and the intestinal tissues were collected for further analysis.
The data are represented as the mean ± standard error of the mean (SEM) and were analyzed by GraphPad Prism software. Significant significance was determined by 2-tailed Student’s t-test, one-way ANOVA, Pearson’s correlation coefficients test, or log-rank test. P values below 0.05 were statistically significant.
To determine the role of RIPK3 in human CRC, we analyzed the levels of RIPK3 expression in containing 41 normal intestinal specimens and 286 CRC tumor samples from the Cancer Genome Atlas (TCGA) transcriptome database. CRC tumor samples showed significantly decreased expression of RIPK3 relative to normal intestinal tissue samples (Figure 1A). We next examined the levels of RIPK3 in different stages of CRC tumor progression. The results revealed that RIPK3 expression was higher in normal intestinal controls than in four CRC stages (Figure 1B). Interestingly, RIPK3 expression was lower in metastatic tumors than in normal counterparts (Figure 1C). More importantly, overall survival was significantly higher in patients with high RIPK3 expression than in patients with reduced RIPK3 expression in colorectal cancer (Figure 1D). These data indicate reduced RIPK3 expression in human CRC and suggest a suppressor role of RIPK3 in colorectal tumorigenesis.
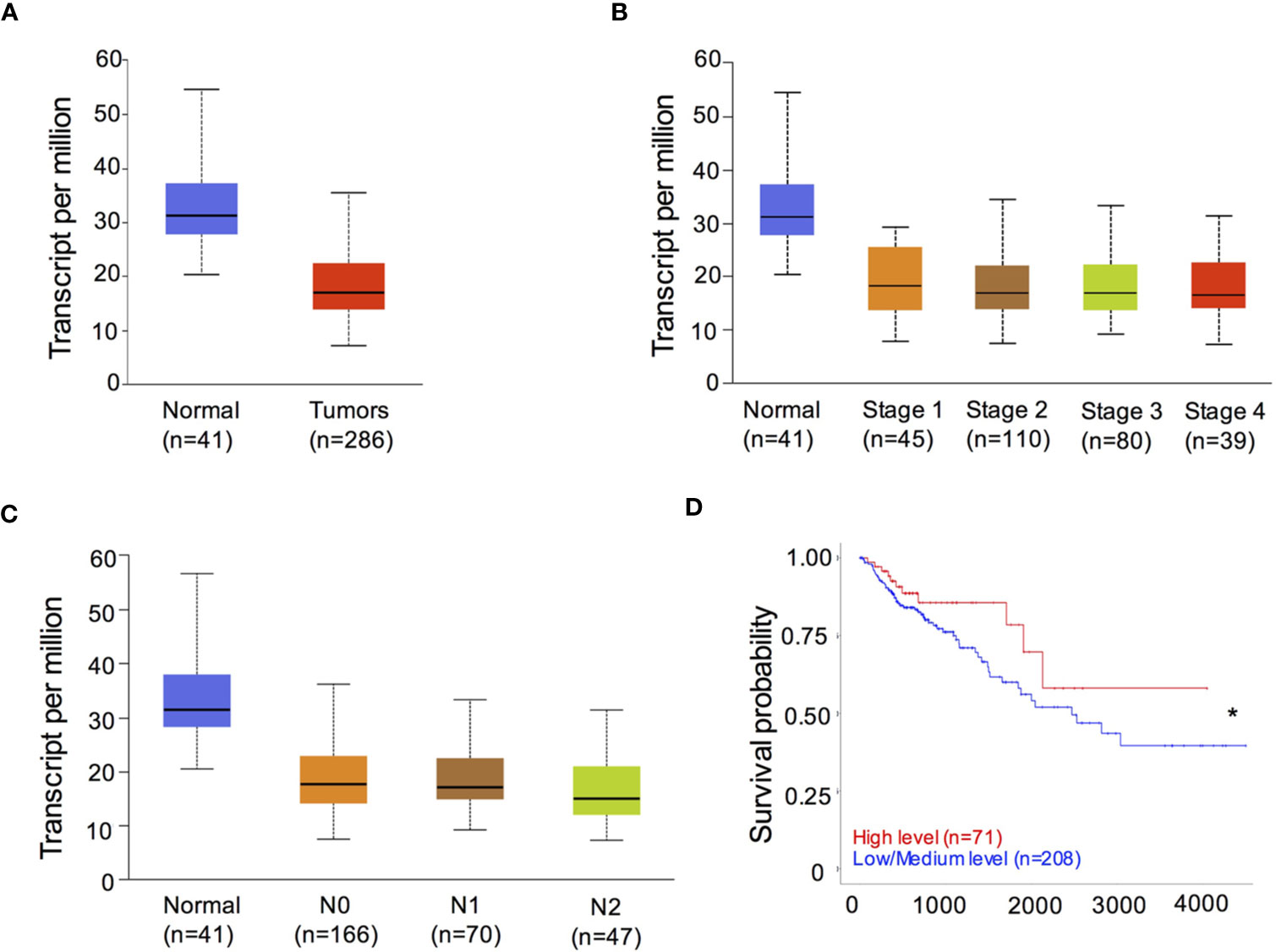
Figure 1 RIPK3 expression is reduced in colorectal cancer (CRC) patients. (A) The levels of RIPK3 mRNA expression in colorectal cancer (CRC) and normal colorectal (NC) tissues in the TCGA colorectal database. n, sample numbers. (B) The levels of RIPK3 mRNA expression in different stages of CRC progression. (C) The levels of RIPK3 mRNA expression in metastatic tumors and normal counterparts. (D) Kaplan- Meier analyses of RIPK3 expression in colorectal cancer patients. *p < 0.05.
Mutations of the WT Apc allele (Apcmin/+) are found in ~85% of intestinal tumors and promote intestinal tumorigenesis. Apcmin/+ mice are a suitable mouse model for the study of sporadic intestinal tumorigenesis. We hypothesized that RIPK3 deletion might increase the tumor burden in Apcmin/+ mice during intestinal tumorigenesis. To address the role of RIPK3 in intestinal tumorigenesis, Apcmin/+Ripk3-/- mice were obtained by crossing Apcmin/+ mice with Ripk3-/- mice. Tumor progression in Apcmin/+Ripk3-/- mice and those of Apcmin/+ mice was analyzed. The results showed significantly more lesions in the Apcmin/+Ripk3-/- mice than in Apcmin/+ mice (Figure 2A). Moreover, Apcmin/+Ripk3-/- mice exhibited increased tumor numbers and tumor loads compared with in Apcmin/+ mice (Figures 2B, C). Of note, the size of the tumor distribution in Apcmin/+Ripk3-/- mice tended to be larger than Apcmin/+ mice (Figure 2D). H&E staining also confirmed that there were larger polyps in Apcmin/+Ripk3-/- intestines than in Apcmin/+ littermate controls (Figure 2E). Accordingly, anemia and thymus atrophy were significantly exacerbated in Apcmin/+Ripk3-/- mice compared to the Apcmin/+ littermate controls (Figures 2F, G). Analyses of Kaplan-Meier survival found that the survival time of the Apcmin/+Ripk3-/- mice was dramatically shorter, with a median survival of only 132 days relative to the median survival of the Apcmin/+ mice, which was 195 days (Figure 2H).
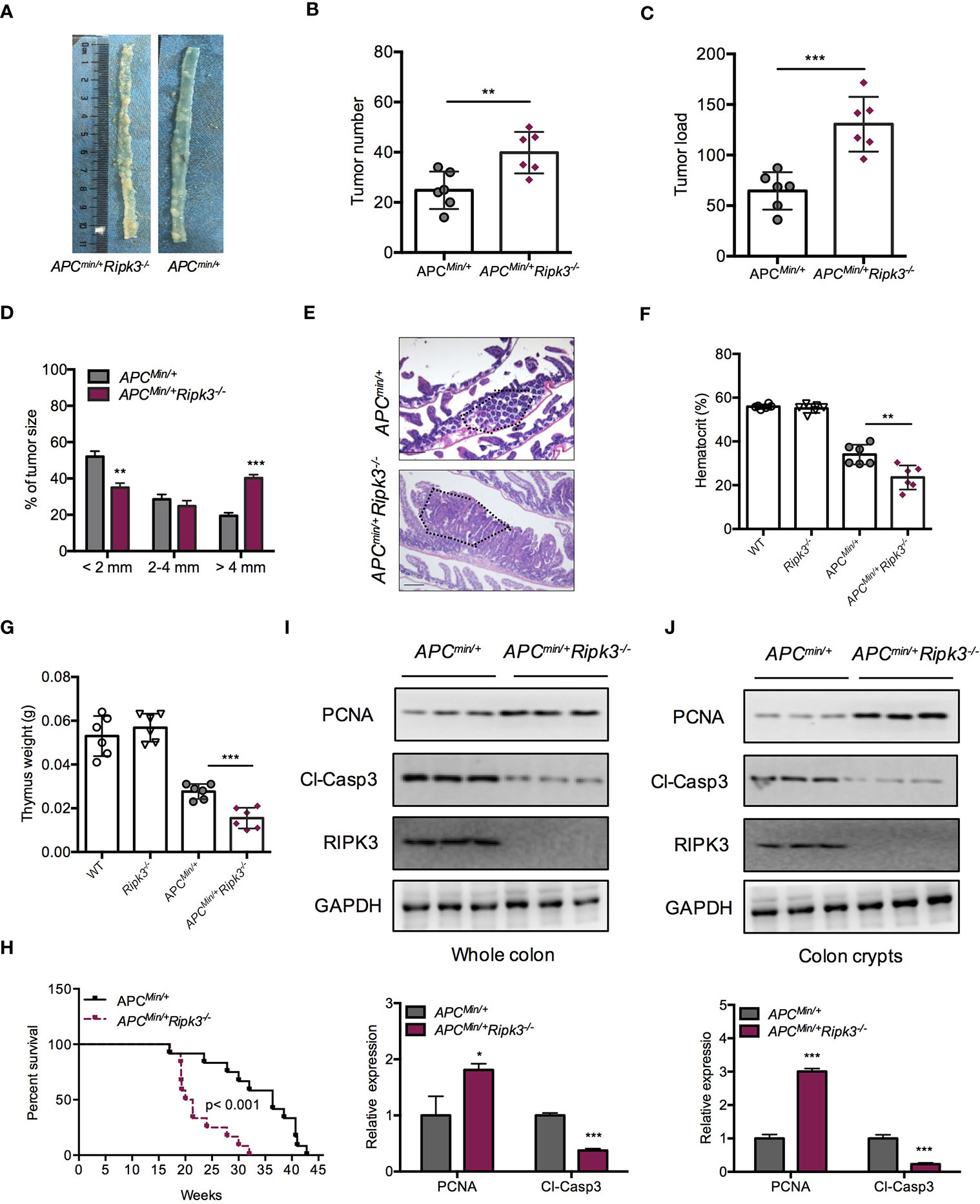
Figure 2 Loss of RIPK3 aggravates tumor burden in ApcMin/+ mice. (A) Polyps in representative intestines from Apcmin/+ and Apcmin/+Ripk3-/- mice. (B, C) Number of polyps formed (B) and tumor load (C) in 16-week-old Apcmin/+ and Apcmin/+Ripk3-/- intestines. (D) Distribution of tumor size in 16-week-old Apcmin/+ and Apcmin/+Ripk3-/- mice. (E) Images of the H&E-stained intestines from Apcmin/+ and Apcmin/+Ripk3-/- mice. (F, G) Hematocrit (F) and thymus weight (G) of 16-week-old WT, Ripk3-/-, Apcmin/+ and Apcmin/+Ripk3-/- mice. (H) Survival of Apcmin/+ and Apcmin/+Ripk3-/- mice as indicated. *p < 0.05, **p < 0.01 and ***p < 0.001 versus Apcmin/+ mice. (I, J) Whole intestines and colonic crypts were isolated from 16-week-old Apcmin/+ and Apcmin/+Ripk3-/- mice. PCNA and cleavage of caspase-3 were analyzed by western blotting. *p < 0.05 and ***p < 0.001 versus Apcmin/+ mice.
Since loss of RIPK3 resulted in higher tumor burdens in the Apcmin/+ model, we hypothesized that RIPK3 inhibited tumor progression through regulating cellular proliferation. Compared with Apcmin/+ mice, Apcmin/+Ripk3-/- tumors and colon crypt showed increased PCNA and decreased cleavage of caspase-3 proteins (Figures 2I, J). However, there were no significant difference in proliferation and apoptosis was found in WT and Ripk3-/- intestines (Figure S1), suggesting that loss of RIPK3 accelerates intestinal tumorigenesis by promoting proliferation and preventing apoptosis. These results demonstrate that loss of RIPK3 aggravates intestinal tumorigenesis in Apcmin/+ mice.
Activation of the STAT3 signaling pathway is associated with colorectal carcinogenesis and progression. In Apcmin/+ mice, hyperactivation of STAT3 accelerates intestinal tumorigenesis (38). To evaluate the status of STAT3 in intestinal epithelial cells and tumors from Apcmin/+Ripk3-/- mice, we analyzed protein expression in intestinal tumors and colon crypt cells. The results indicated that the levels of pSTAT3 were higher in Apcmin/+Ripk3-/- tumors than in Apcmin/+ tumors, WT and Ripk3-/- intestines (Figure 3A). We also found higher levels of pSTAT3 in Apcmin/+Ripk3-/- colon crypts than in control mice (Figure 3B). As tumors from Apcmin/+Ripk3-/- mice display activation of STAT3 signaling, we determined the reason for STAT3 hyperactivation in Apcmin/+Ripk3-/- mice. IL-6 maintains persistent activation of STAT3 signaling in CRC (16). We found significantly increased levels of IL-6 mRNA and protein in Apcmin/+Ripk3-/- tumors (Figures 3C, D). Aberrant activation of STAT3 promotes intestinal tumorigenesis by upregulating STAT3 target genes, which are involved in cell survival and proliferation (39). The results showed that the expression of STAT3 target genes (Cyclin D1, C-myc and Survivin) was increased in Apcmin/+Ripk3-/- tumors compared with Apcmin/+ tumors (Figure 3E). Consistently, we found higher levels of these target genes in Apcmin/+Ripk3-/- colon crypts than in Apcmin/+ mice (Figure 3F). Similarly, qRT-PCR results also showed significantly increased expression of these genes in Apcmin/+Ripk3-/- tumors and colon crypts (Figures 3G, H). However, no remarkable increase in expression of STAT3 target genes was observed in either the intestines of the WT or Ripk3-/- intestines (Figure S2). These results indicate that IL-6/STAT3 signaling is activated in Apcmin/+Ripk3-/- mice during intestinal tumorigenesis.
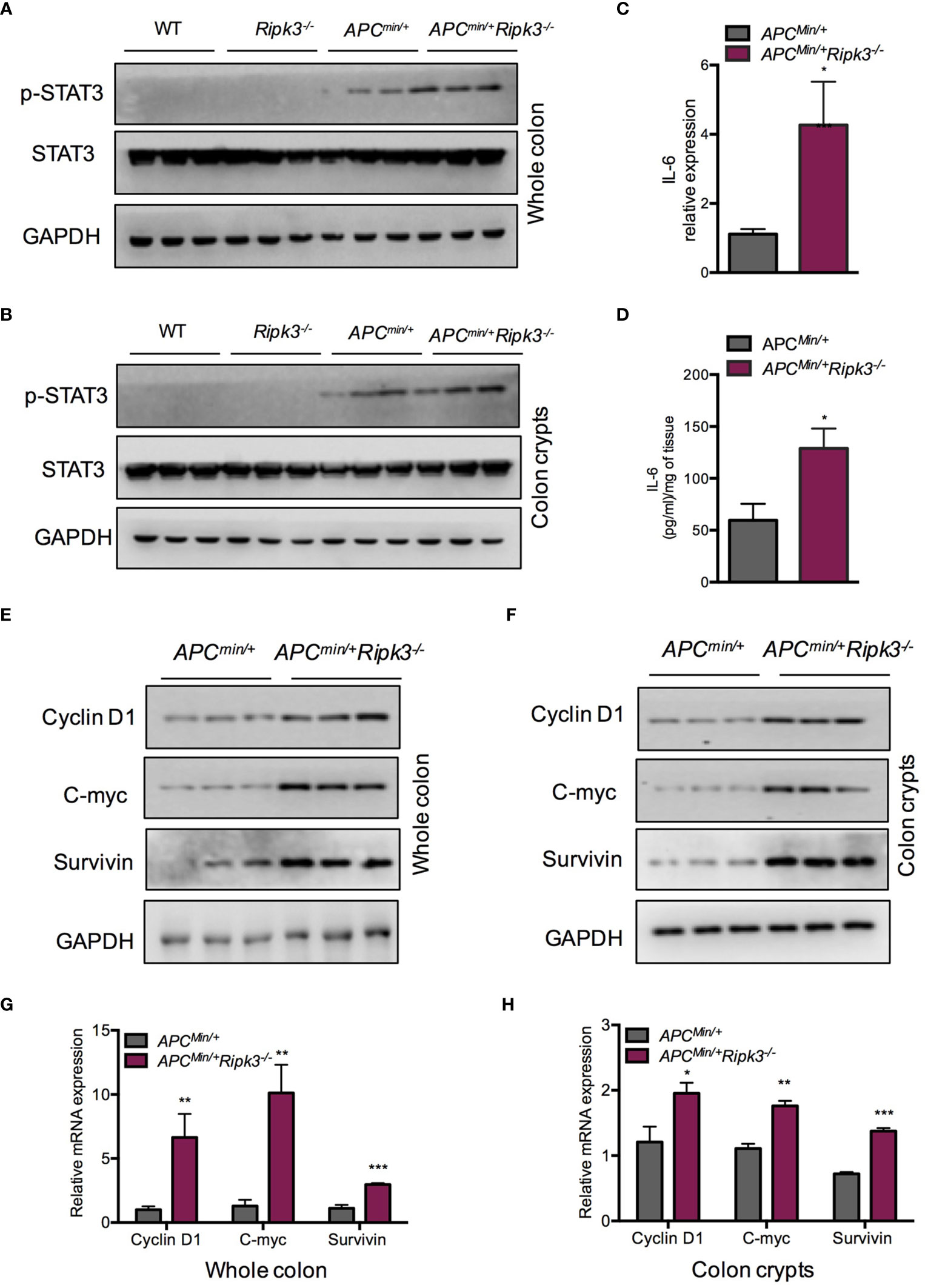
Figure 3 Loss of RIPK3 activates STAT3 signals in Apcmin/+ mice. (A) The expression of pSTAT3 in 6-week-old Apcmin/+ and Apcmin/+Ripk3-/- intestinal tumors. (B) The levels of pSTAT3 in Apcmin/+ and Apcmin/+Ripk3-/- colonic crypts. (C) Quantitative mRNA expression of IL-6 in Apcmin/+ and Apcmin/+Ripk3-/- intestinal tumors. (D) The protein levels of IL-6 in Apcmin/+ and Apcmin/+Ripk3-/- intestinal tumors as determined by ELISA. (E) The expression of STAT3 target genes in Apcmin/+ and Apcmin/+Ripk3-/- intestinal tumors. (F) The expression of STAT3 target genes in Apcmin/+ and Apcmin/+Ripk3-/- colonic crypts. (G, H) Quantitative mRNA expression of STAT3 target genes in Apcmin/+ and Apcmin/+Ripk3-/- intestinal tumors and colonic crypts. *p < 0.05, **p < 0.01, ***p < 0.001 versus Apcmin/+ mice.
IL-6 is required for homeostasis of intestinal crypts (40, 41), and tumor formation is suppressed in the absence of IL-6 in the Apcmin/+ model (38, 42). To confirm the role of IL-6/STAT3 signaling in Apcmin/+Ripk3-/- mice, we investigated the effect of IL-6 using an anti-IL-6 receptor (anti-IL-6R) antibody. We injected the anti-IL-6R antibody into Apcmin/+Ripk3-/- mice for 10 weeks (Figure 4A). Treatment with anti-IL-6R antibody resulted in a lower clinical score in Apcmin/+Ripk3-/- mice compared to the untreated (UT) mice (Figure 4B). After IL-6R antibody treatment for 10 weeks, the anti-IL6-R-treated Apcmin/+Ripk3-/- mice showed fewer intestinal polyps than UT Apcmin/+Ripk3-/- mice (Figure 4C). As expected, anti-IL-6R antibody therapy alleviated anemia and thymus atrophy in Apcmin/+Ripk3-/- mice (Figures 4D, E). To elucidate whether STAT3 activation changes in anti-IL-6R-treated Apcmin/+Ripk3-/- mice, intestinal polyps were isolated and analyzed by western blotting. Anti-IL-6R-treated Apcmin/+Ripk3-/- intestines had decreased levels of p-STAT3 compared to UT Apcmin/+Ripk3-/- mice (Figure 4F). Consistently, the expression of STAT3 target genes Cyclin D1, C-myc and Survivin, was markedly suppressed in anti-IL-6R-treated Apcmin/+Ripk3-/- tumors (Figures 4G, H). These results demonstrate that increased IL-6/STAT3 signaling plays a causative role in the increased tumor burden in Apcmin/+Ripk3-/- mice.
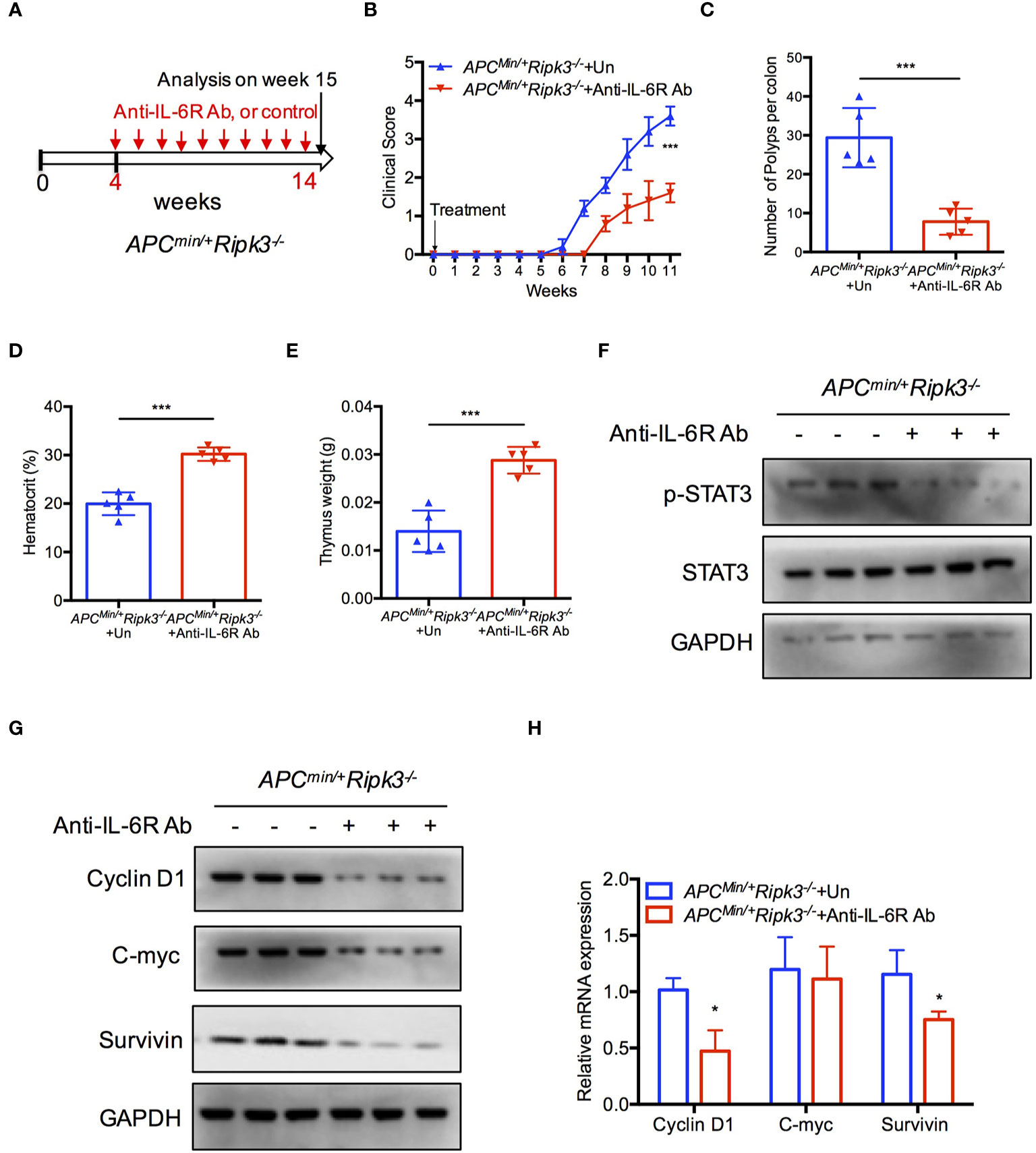
Figure 4 Inhibition of IL-6 blocks tumor burden in Apcmin/+Ripk3-/- mice. (A) Scheme for anti-IL6R antibody treatment in Apcmin/+Ripk3-/- mice. (B) Four-week-old Apcmin/+Ripk3-/- mice were injected with anti-IL6R antibody with at a dosage of 4 mg/kg weekly for 10 weeks. The clinical scores were monitored. (C) Numbers of intestinal tumors in Un- or anti-IL6R-treated Apcmin/+Ripk3-/- mice. (D, E) Hematocrit (D) and thymus weight (E) of Apcmin/+Ripk3-/- mice after anti-IL6R therapy for 10 weeks. (F) Intestinal tumors were collected from PBS and anti-IL6R-treated Apcmin/+Ripk3-/- mice. The levels of p-STAT3 were determined by western blotting. (G, H) The expression of STAT3 target genes expression in PBS and anti-IL6R-treated Apcmin/+Ripk3-/- intestinal tumors. *p < 0.05, ***p < 0.001 versus Un-treated Apcmin/+Ripk3-/- mice.
In this study, we found that loss of RIPK3 promoted the initiation and progression of spontaneous intestinal tumors. RIPK3 is reduced in human CRC patient cohorts and reduced RIPK3 is associated with poor prognosis in CRC. In the Apcmin/+ model, loss of RIPK3 elevated IL-6 levels, in turn hyperactivating the STAT3 signaling pathway and contributing to accelerated intestinal tumorigenesis. Notably, blocking IL-6 with a neutralizing IL-6 receptor antibody effectively attenuated tumor burden and STAT3 hyperactivation in Apcmin/+Ripk3-/- mice. Our results highlight that RIPK3 plays a suppressive role in intestinal tumorigenesis by inactivating the IL-6/STAT3 signaling axis.
Necroptosis is involved in intestinal inflammation in mice and human IBD patients (43, 44). Abnormal RIPK3 expression is increased in Crohn’s disease (CD) and ulcerative colitis (UC) patients (44, 45). RIPK3 has a necroptosis-independent function by inhibiting inflammatory responses in DSS-induced acute colitis (28). In some types of tumors, reduced expression of RIPK3 appears to occur commonly and was found to correlate with reduced overall survival and poor prognosis (32, 46, 47). Previous studies have shown that RIPK3 suppresses inflammation-associated CRC (31, 33), while conflicting results suggest that RIPK3 promotes colitis-associated colorectal cancer (48). Recent work has examined the lack of an overt phenotype in Ripk3-/- mice in an inflammation-associated CRC model (49). These studies present conflicting results that might be due to differences in experimental conditions, commensal microflora and genetic background of mice. Thus, the results of a previous study indicate that the role of RIPK3 in the initiation and progression of CRC is still not clear. In addition, sporadic intestinal tumors are the majority of colon cancers, and the importance of RIRP3 needs to be addressed in sporadic intestinal tumors. Here, we took advantage of genetically engineered mouse models (GEMs) and established that RIRP3 deficiency drove increased tumor burden during intestinal tumorigenesis in Apcmin/+ mice. The present study revealed that the survival time was shorter in Apcmin/+ mice in the absence of RIRP3, which is consistent with reduced RIPK3 correlated with poor clinical outcomes in CRC. Consistently, we found reduced RIPK3 expression in human CRC compared with normal tissues using the TCGA database. RIPK3 expression was significantly decreased in four CRC stages versus in healthy controls. Importantly, significantly lower expression of RIPK3 predicted poor overall survival in CRC patients. In summary, these data reveal that RIPK3 exerts tumor-suppressive roles in intestinal tumorigenesis.
Intestinal tumors originate from epithelial cells through activation of multiple key signaling pathways for cell growth, differentiation, and survival. Compensatory proliferation and widespread apoptosis are known to contribute to promoting colorectal carcinogenesis. Here, our results indicated that Apcmin/+Ripk3-/- tumors exhibited higher proliferative rates and reduced apoptotic compared to Apcmin/+ tumors. Notably, untransformed colon tissue and colon crypts of Apcmin/+Ripk3-/- mice also exhibited higher proliferation and lower apoptosis. Thus, cooperation with increased proliferation and decreased apoptosis contributes to accelerated intestinal tumors in Apcmin/+Ripk3-/- mice.
Inflammatory cytokines stimulate the activation of STAT3 signaling, which in turn contributes to promoted epithelial cell survival and resistance to apoptosis (5). STAT3 is critical for intestinal regeneration and tumorigenesis by regulating survival, cell cycle progression, and inflammation (50). Hyperactivation of STAT3 in epithelial cells has been linked to the development of CRC. Constitutively activated STAT3 in mice is resistant to intestinal tumor development (14). In contrast, IEC-specific STAT3-deficient mice exhibit decreased intestinal tumors (16). In this study, we observed that loss of RIPK3 promoted the activation of STAT3, thereby accelerating intestinal tumorigenesis in Apcmin/+ mice. Moreover, RIPK3-deficient mice exhibited increased expression of STAT3 target genes, including Cyclin D1, C-myc and Survivin in the Apcmin/+ model. Enhanced levels of IL-6 play a key role in STAT3 signaling in tumor formation and the development of CRC (42, 51, 52). Indeed, the levels of IL-6 were significantly upregulated in Apcmin/+Ripk3-/- tumors. However, blocking IL-6 signals by anti-IL-6R antibody therapy was sufficient to suppress tumor formation in anti-IL6-R-treated Apcmin/+Ripk3-/- mice. As expected, decreased levels of pSTAT3 and target genes were observed in anti-IL-6R-treated Apcmin/+Ripk3-/- mice and correlated positively with decreased tumor numbers. Altogether, these data suggest that increased disease and polyp burden in Apcmin/+Ripk3-/- mice are most likely mediated by hyperactivation of the IL-6/STAT3 signaling axis. The contribution of the cell-specific function of RIPK3 in the suppression of CRC needs to be further investigated using RIPK3 conditional knockout mice in future studies.
In summary, we report that RIPK3 exerts a suppressive role in intestinal tumorigenesis by suppressing the IL-6/STAT3 signaling axis. Mechanistic analysis provides evidence to further understand the role of RIPK3 in intestinal tumorigenesis and identify that increasing RIPK3 may be considered as a potential therapeutic strategy for CRC therapy.
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding authors.
The animal study was reviewed and approved by Hubei University of Medicine. Written informed consent was obtained from the owners for the participation of their animals in this study.
XY and QZ conceived the study and designed the experiments. XY, JG, XC, YB, YG, XZ and QZ performed experiments. XY, JG, XC, YL, YG, XW, WY, SJ, YT and QZ analyzed data. XY and QZ wrote the manuscript. All authors contributed to the article and approved the submitted version.
Our study was funded by the National Natural Science Foundation of China (81902852, 81502548), the Natural Science Foundation of Hubei Provincial Department of Education (D20202101), the Foundation of Health Commission of Hubei Province (WJ2019M053), the China Postdoctoral Science Foundation (2020M670220), the Biomedical Research Foundation, Hubei University of Medicine (HBMUPI201809), Faculty Development Grants from Hubei University of Medicine (2018QDJZR06), Open Project of Hubei Key Laboratory of Embryonic Stem Cell Research of Hubei University of Medicine (2020ESOF008), and Innovative Research Program for Graduates of Hubei University of Medicine (YC2020005), and the Student’s Platform for Innovation and Entrepreneurship Training Program (202010929001).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2021.664927/full#supplementary-material
1. Siegel RL, Miller KD, Sauer AG, Fedewa SA, Butterly LF, Anderson JC, et al. Colorectal cancer statistics, 2020. CA Cancer J Clin (2020) 70:145–64. doi: 10.3322/caac.21601
2. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2020. CA Cancer J Clin (2020) 70:7–30. doi: 10.3322/caac.21590
3. Bailey CE, Hu CY, You N, Bednarski BK, Rodriguez-Bigas MA, Skibber JM, et al. Increasing disparities in the age-related incidences of colon and rectal cancers in the United States, 1975-2010. JAMA Surg (2015) 150:17–22. doi: 10.1001/jamasurg.2014.1756
4. Fleming M, Ravula S, Tatishchev SF, Wang HL. Colorectal carcinoma: Pathologic aspects. J Gastrointest Oncol (2012) 3:153–73. doi: 10.3978/j.issn.2078-6891.2012.030
5. Grivennikov SI, Greten FR, Karin M. Immunity, inflammation, and cancer. Cell (2010) 140(6):883–99. doi: 10.1016/j.cell.2010.01.025
6. West NR, McCuaig S, Franchini F, Powrie F. Emerging cytokine networks in colorectal cancer. Nat Rev Immunol (2015) 15:615–29. doi: 10.1038/nri3896
7. Kinzler KW, Vogelstein B. Lessons from hereditary colorectal cancer. Cell (1996) 87:159–70. doi: 10.1016/s0092-8674(00)81333-1
8. Lasry A, Zinger A, Ben-Neriah Y. Inflammatory networks underlying colorectal cancer. Nat Immunol (2016) 17:230–40. doi: 10.1038/ni.3384
9. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell (2011) 144:646–74. doi: 10.1016/j.cell.2011.02.013
10. Li J, Huang L, Zhao H, Yan Y, Lu J. The Role of Interleukins in Colorectal Cancer. Int J Biol Sci (2020) 16:2323–39. doi: 10.7150/ijbs.46651
11. O’Shea JJ, Holland SM, Staudt LM. JAKs and STATs in immunity, immunodeficiency, and cancer. N Engl J Med (2013) 368:161–70. doi: 10.1056/NEJMra1202117
12. Becker C, Fantini MC, Wirtz S, Nikolaev A, Lehr HA, Galle PR, et al. IL-6 signaling promotes tumor growth in colorectal cancer. Cell Cycle (2005) 4:217–20. doi: 10.4161/cc.4.2.1413
13. Waldner MJ, Foersch S, Neurath MF. Interleukin-6–a key regulator of colorectal cancer development. Int J Biol Sci (2012) 8:1248–53. doi: 10.7150/ijbs.4614
14. Bollrath J, Phesse TJ, von Burstin VA, Putoczki T, Bennecke M, Bateman T, et al. gp130-mediated Stat3 activation in enterocytes regulates cell survival and cell-cycle progression during colitis-associated tumorigenesis. Cancer Cell (2009) 15:91–102. doi: 10.1016/j.ccr.2009.01.002
15. Corvinus FM, Orth C, Moriggl R, Tsareva SA, Wagner S, Pfitzner EB, et al. Persistent STAT3 activation in colon cancer is associated with enhanced cell proliferation and tumor growth. Neoplasia (2005) 7:545–55. doi: 10.1593/neo.04571
16. Grivennikov S, Karin E, Terzic J, Mucida D, Yu G-Y, Vallabhapurapu S, et al. IL-6 and Stat3 are required for survival of intestinal epithelial cells and development of colitis-associated cancer. Cancer Cell (2009) 15:103–13. doi: 10.1016/j.ccr.2009.01.001
17. Imtiyaz HZ, Rosenberg S, Zhang Y, Rahman ZSM, Hou Y-J, Manser T, et al. The Fas-associated death domain protein is required in apoptosis and TLR-induced proliferative responses in B cells. J Immunol (2006) 176:6852–61. doi: 10.4049/jimmunol.176.11.6852
18. Feoktistova M, Geserick P, Kellert B, Dimitrova DP, Langlais C, Hupe M, et al. cIAPs block Ripoptosome formation, a RIP1/caspase-8 containing intracellular cell death complex differentially regulated by cFLIP isoforms. Mol Cell (2011) 43:449–63. doi: 10.1016/j.molcel.2011.06.011
19. Upton JW, Kaiser WJ, Mocarski ES. DAI/ZBP1/DLM-1 complexes with RIP3 to mediate virus-induced programmed necrosis that is targeted by murine cytomegalovirus vIRA. Cell Host Microbe (2012) 11:290–7. doi: 10.1016/j.chom.2019.09.004
20. Chan FK-M, Shisler J, Bixby JG, Felices M, Zheng L, Appel M, et al. A role for tumor necrosis factor receptor-2 and receptor-interacting protein in programmed necrosis and antiviral responses. J Biol Chem (2003) 278:51613–21. doi: 10.1074/jbc.M305633200
21. Holler N, Zaru R, Micheau O, Thome M, Attinger A, Valitutti S, et al. Fas triggers an alternative, caspase-8-independent cell death pathway using the kinase RIP as effector molecule. Nat Immunol (2000) 1:489–95. doi: 10.1038/82732
22. Cho YS, Challa S, Moquin D, Genga R, Ray TD, Guildford M, et al. Phosphorylation-driven assembly of the RIP1-RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell (2009) 137:1112–23. doi: 10.1016/j.cell.2009.05.037
23. He S, Wang L, Miao L, Wang T, Du F, Zhao L, et al. Receptor interacting protein kinase-3 determines cellular necrotic response to TNF-alpha. Cell (2009) 137:1100–11. doi: 10.1016/j.cell.2009.05.021
24. Sun L, Wang H, Wang Z, He S, Chen S, Liao D, et al. Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell (2012) 148:213–27. doi: 10.1016/j.cell.2011.11.031
25. Yoon S, Kovalenko A, Bogdanov K, Wallach D. MLKL, the protein that mediates necroptosis, also regulates endosomal trafficking and extracellular vesicle generation. Immunity (2017) 47:51–65. doi: 10.1016/j.immuni.2017.06.001
26. Pasparakis M, Vandenabeele P. Necroptosis and its role in inflammation. Nature (2015) 517:311–20. doi: 10.1038/nature14191
27. Gunther C, Neumann H, Neurath MF, Becker C. Apoptosis, necrosis and necroptosis: cell death regulation in the intestinal epithelium. Gut (2013) 62:1062–71. doi: 10.1136/gutjnl-2011-301364
28. Moriwaki K, Balaji S, McQuade T, Malhotra N, Kang J, Chan FK-M. The necroptosis adaptor RIPK3 promotes injury-induced cytokine expression and tissue repair. Immunity (2014) 41:567–78. doi: 10.1016/j.immuni.2014.09.016
29. Zhao Q, Yu X, Zhang H, Liu Y, Zhang X, Wu X, et al. RIPK3 mediates necroptosis during embryonic development and postnatal inflammation in fadd-deficient mice. Cell Rep (2017) 19:798–808. doi: 10.1016/j.celrep.2017.04.011
30. Najafov A, Chen H, Yuan J. Necroptosis and Cancer. Trends Cancer (2017) 3:294–301. doi: 10.1016/j.trecan.2017.03.002
31. Bozec D, Iuga AC, Roda G, Dahan S, Yeretssian G. Critical function of the necroptosis adaptor RIPK3 in protecting from intestinal tumorigenesis. Oncotarget (2016) 7:46384–400. doi: 10.18632/oncotarget.10135
32. Feng X, Song Q, Yu A, Tang H, Peng Z, Wang X. Receptor-interacting protein kinase 3 is a predictor of survival and plays a tumor suppressive role in colorectal cancer. Neoplasma (2015) 62:592–601. doi: 10.4149/neo_2015_071
33. Yan G, Zhao H, Zhang Q, Zhou Y, Wu L, Lei J, et al. A RIPK3-PGE2 circuit mediates myeloid-derived suppressor cell-potentiated colorectal carcinogenesis. Cancer Res (2018) 78:5586–99. doi: 10.1158/0008-5472.CAN-17-3962
34. Yashiro M. Ulcerative colitis-associated colorectal cancer. World J Gastroenterol (2014) 20:16389–97. doi: 10.3748/wjg.v20.i44.16389
35. Zhao Q, Bi Y, Zhong J, Ren ZT, Liu YX, Jia JJ, et al. Pristimerin suppresses colorectal cancer through inhibiting inflammatory responses and Wnt/beta-catenin signaling. Toxicol Appl Pharmacol (2020) 386:114813. doi: 10.1016/j.taap.2019.114813
36. Neufert C, Becker C, Neurath MF. An inducible mouse model of colon carcinogenesis for the analysis of sporadic and inflammation-driven tumor progression. Nat Protoc (2007) 2:1998–2004. doi: 10.1038/nprot.2007.279
37. Zhao Q, Yu X, Li M, Liu Y, Han Y, Zhang X, et al. MLKL attenuates colon inflammation and colitis-tumorigenesis via suppression of inflammatory responses. Cancer Lett (2019) 459:100–11. doi: 10.1016/j.canlet.2019.05.034
38. Schmidt S, Schumacher N, Schwarz J, Tangermann S, Kenner L, Schlederer M, et al. ADAM17 is required for EGF-R-induced intestinal tumors via IL-6 trans-signaling. J Exp Med (2018) 215:1205–25. doi: 10.1084/jem.20171696
39. Zhao Q, Bi Y, Zhong J, Li X, Guo J, Liu Y-X, et al. 10,11-dehydrocurvularin exerts antitumor effect against human breast cancer by suppressing STAT3 activation. Acta Pharmacol Sin (2020). doi: 10.1038/s41401-020-0499-y
40. Aden K, Breuer A, Rehman A, Geese H, Tran F, Sommer J, et al. Classic IL-6R signalling is dispensable for intestinal epithelial proliferation and repair. Oncogenesis (2016) 5:e270. doi: 10.1038/oncsis.2016.71
41. Jeffery V, Goldson AJ, Dainty JR, Chieppa M, Sobolewski A. IL-6 Signaling Regulates Small Intestinal Crypt Homeostasis. J Immunol (2017) 199:304–11. doi: 10.4049/jimmunol.1600960
42. Baltgalvis KA, Berger FG, Pena MMO, Davis JM, Muga SJ, Carson JA. Interleukin-6 and cachexia in ApcMin/+ mice. Am J Physiol Regul Integr Comp Physiol (2008) 294:R393–401. doi: 10.1152/ajpregu.00716.2007
43. Welz P-S, Wullaert A, Vlantis K, Kondylis V, Fernandez-Majada V, Ermolaeva M, et al. FADD prevents RIP3-mediated epithelial cell necrosis and chronic intestinal inflammation. Nature (2011) 477:330–4. doi: 10.1038/nature10273
44. Pierdomenico M, Negroni A, Stronati L, Vitali R, Prete E, Bertin J, et al. Necroptosis is active in children with inflammatory bowel disease and contributes to heighten intestinal inflammation. Am J Gastroenterol (2014) 109:279–87. doi: 10.1038/ajg.2013.403
45. Wu T, Dai Y, Xue L, Sheng Y, Xu L, Xue Y. Expression of receptor interacting protein 3 and mixed lineage kinase domain-like protein-key proteins in necroptosis is upregulated in ulcerative colitis. Ann Palliat Med (2019) 8:483–9. doi: 10.21037/apm.2019.07.04
46. Nugues AL, El Bouazzati H, Hetuin D, Berthon C, Loyens A, Bertrand E, et al. RIP3 is downregulated in human myeloid leukemia cells and modulates apoptosis and caspase-mediated p65/RelA cleavage. Cell Death Dis (2014) 5:e1384. doi: 10.1038/cddis.2014.347
47. Koo G-B, Morgan MJ, Lee D-G, Kim W-J, Yoon J-H, Koo JS, et al. Methylation-dependent loss of RIP3 expression in cancer represses programmed necrosis in response to chemotherapeutics. Cell Res (2015) 25:707–25. doi: 10.1038/cr.2015.56
48. Liu Z-Y, Zheng M, Li Y-M, Fan X-Y, Wang J-C, Li Z-C, et al. RIP3 promotes colitis-associated colorectal cancer by controlling tumor cell proliferation and CXCL1-induced immune suppression. Theranostics (2019) 9:3659–73. doi: 10.7150/thno.32126
49. Alvarez-Diaz S, Preaudet A, Samson AL, Nguyen PM, Fung KY, Garnham AL, et al. Necroptosis is dispensable for the development of inflammation-associated or sporadic colon cancer in mice. Cell Death Differ (2020). doi: 10.1038/s41418-020-00673-z
50. Rigby RJ, Simmons JG, Greenhalgh CJ, Alexander WS, Lund PK. Suppressor of cytokine signaling 3 (SOCS3) limits damage-induced crypt hyper-proliferation and inflammation-associated tumorigenesis in the colon. Oncogene (2007) 26:4833–41. doi: 10.1038/sj.onc.1210286
51. Wang K, Kim MK, Di Caro G, Wong J, Shalapour S, Wan J, et al. Interleukin-17 receptor a signaling in transformed enterocytes promotes early colorectal tumorigenesis. Immunity (2014) 41:1052–63. doi: 10.1016/j.immuni.2014.11.009
Keywords: RIPK3, intestinal tumorigenesis, APC mutant mouse model, STAT3 (signal transducer and activator of transcription 3), IL-6 receptor antibody
Citation: Zhao Q, Guo J, Cheng X, Liao Y, Bi Y, Gong Y, Zhang X, Guo Y, Wang X, Yu W, Jin S, Tan Y and Yu X (2021) RIPK3 Suppresses the Progression of Spontaneous Intestinal Tumorigenesis. Front. Oncol. 11:664927. doi: 10.3389/fonc.2021.664927
Received: 06 February 2021; Accepted: 23 March 2021;
Published: 30 April 2021.
Edited by:
Zexian Liu, Sun Yat-sen University Cancer Center (SYSUCC), ChinaReviewed by:
Mengtao Cao, Shenzhen Second People’s Hospital, ChinaCopyright © 2021 Zhao, Guo, Cheng, Liao, Bi, Gong, Zhang, Guo, Wang, Yu, Jin, Tan and Yu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xianjun Yu, eGp5dUBoYm11LmVkdS5jbg==; Qun Zhao, emhhb3F1bkBoYm11LmVkdS5jbg==
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.