
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Oncol. , 10 September 2021
Sec. Cancer Immunity and Immunotherapy
Volume 11 - 2021 | https://doi.org/10.3389/fonc.2021.664236
This article is part of the Research Topic Impact of the Glioma Microenvironment on Antitumor Immunity View all 14 articles
 Radhika Thokala1,2‡Zev A. Binder1,2‡Yibo Yin1,2†Logan Zhang1,2Jiasi Vicky Zhang1,2Daniel Y. Zhang2,3Michael C. Milone2,4Guo-li Ming3Hongjun Song2,5Donald M. O’Rourke1,2*
Radhika Thokala1,2‡Zev A. Binder1,2‡Yibo Yin1,2†Logan Zhang1,2Jiasi Vicky Zhang1,2Daniel Y. Zhang2,3Michael C. Milone2,4Guo-li Ming3Hongjun Song2,5Donald M. O’Rourke1,2*Tumor heterogeneity is a key reason for therapeutic failure and tumor recurrence in glioblastoma (GBM). Our chimeric antigen receptor (CAR) T cell (2173 CAR T cells) clinical trial (NCT02209376) against epidermal growth factor receptor (EGFR) variant III (EGFRvIII) demonstrated successful trafficking of T cells across the blood–brain barrier into GBM active tumor sites. However, CAR T cell infiltration was associated only with a selective loss of EGFRvIII+ tumor, demonstrating little to no effect on EGFRvIII- tumor cells. Post-CAR T-treated tumor specimens showed continued presence of EGFR amplification and oncogenic EGFR extracellular domain (ECD) missense mutations, despite loss of EGFRvIII. To address tumor escape, we generated an EGFR-specific CAR by fusing monoclonal antibody (mAb) 806 to a 4-1BB co-stimulatory domain. The resulting construct was compared to 2173 CAR T cells in GBM, using in vitro and in vivo models. 806 CAR T cells specifically lysed tumor cells and secreted cytokines in response to amplified EGFR, EGFRvIII, and EGFR-ECD mutations in U87MG cells, GBM neurosphere-derived cell lines, and patient-derived GBM organoids. 806 CAR T cells did not lyse fetal brain astrocytes or primary keratinocytes to a significant degree. They also exhibited superior antitumor activity in vivo when compared to 2173 CAR T cells. The broad specificity of 806 CAR T cells to EGFR alterations gives us the potential to target multiple clones within a tumor and reduce opportunities for tumor escape via antigen loss.
Chimeric antigen receptor (CAR) cells targeting pediatric B cell malignancies have shown unprecedented responses and were the first CAR T cell therapies to receive FDA approval, in 2017 (1–3). The successful application of this therapeutic technology in the treatment of solid tumors, including glioblastoma (GBM), remains a significant challenge; chief among them are tumor heterogeneity, immunosuppressive tumor microenvironment, and antigen escape (4, 5). Successful strategies for overcoming these obstacles are required to advance CAR T therapy in solid tumors.
Epidermal growth factor receptor (EGFR) was one of the first oncogenes identified in GBM and presents an attractive therapeutic target, given its extracellular nature and frequent alterations in GBM. Approximately 60% of GBM specimens contain a mutation, rearrangement, splicing alteration, and/or amplification of EGFR (6). EGFR overexpression, mediated through focal amplification of the EGFR locus as double minute chromosomes, has long been recognized as the most common EGFR alteration, present in 60% of GBM patients (7, 8). Tumor-specific EGFR variant III (EGFRvIII), resulting from deletion of exon 2–7 of wild-type EGFR (wtEGFR), is present in 30% of GBM patients (9). In addition, oncogenic missense mutations EGFRA289D/T/V, EGFRR108G/K, and EGFRG598V have been identified in 12%–13% of cases in the extracellular domain (ECD) of EGFR, independent of EGFRvIII. Missense mutations and EGFRvIII often co-occur with EGFR amplification and activate EGFR receptor independent of its ligand (10). Several of the missense mutations have been shown to have a negative effect on patient survival, driving tumor proliferation and invasion (11).
Our first-in-man CAR T clinical trial (NCT02209376) against EGFRvIII in recurrent GBM demonstrated the safety of a peripheral infusion of CAR T cells and resulted in successful trafficking of the CAR T cells to active tumor sites, across the blood–brain barrier (12). After treatment, CAR T cells infiltrated the GBM tumors rapidly, proliferated in situ, and persisted over a prolonged period of time. However, CAR T cell infiltration was associated only with a selective loss of EGFRvIII+ GBM cells. Importantly, post-CAR T-treated tumor specimens showed the continued presence of EGFR amplification and missense mutations, despite the decrease in EGFRvIII target antigen. Persistence of EGFR amplification and ECD missense mutations in the context of loss of EGFRvIII expression suggested that tumor heterogeneity played an essential role for tumor recurrence and continued regrowth.
mAb806, originally raised against EGFRvIII, recognizes a conformationally exposed epitope of wtEGFR when it is overexpressed on tumor cells. The same epitope is not exposed in EGFR expressed on normal non-overexpressing cells (13, 14). ABT-414, an antibody–drug conjugate composed of a humanized mAb806 (ABT-806), showed early efficacy in phase I/II clinical trials with no apparent skin toxicity in treated GBM patients (15). However, a recent Phase III trial was terminated when an interim analysis failed to demonstrate a survival benefit over placebo (16). mAb806 showed an increased binding affinity for not only EGFRvIII but also EGFR ECD mutations and a low affinity for wtEGFR (11). These findings suggest that mAb806 is a viable therapeutic option for tumors harboring EGFR alterations in addition to EGFRvIII.
In the present study, we have developed EGFR-specific CAR T cells derived from the single-chain fragment variable region (scFv) of 806 mAb, using our standard 4-1BB-ζ construct (17). We then compared 806 CAR T activity with EGFRvIII-specific CAR T cells (2173 CAR T), currently in clinic, for specificity against oncogenic EGFR alterations, including amplified EGFR, EGFRvIII, and extracellular mutations in vitro and in vivo.
806 scFvs were swapped with scFv of our standard CD19-BB-ζ lentiviral vector described previously to generate 806-BB-ζ CAR (17, 18). Briefly, the nucleotide coding sequences of 806 or C225 scFv with the huCD8 leader were synthesized by GeneArt (Thermo Fisher Scientific, Waltham, MA) with 5′ Xba1 and 3′Nhe1 and ligated to Xba1 and Nhe1 sites of CD19-BB-ζ car construct. The C225-BB-ζ CAR was obtained from Dr. Avery Posey’s lab at the University of Pennsylvania. The 2173-BB-ζ CAR T construct was obtained from Dr. Laura Johnson’s Lab at the University of Pennsylvania (19, 20).
Human primary total T cells (CD4 and CD8) were isolated from normal healthy donors following leukapheresis by negative selection using RosetteSep kits (STEMCELL Technologies, Vancouver, CA, Canada). All specimens were collected with protocol approved by the University Review Board, and written informed consent was obtained from each donor. T cells were cultured in RPMI 1640 (Thermo Fisher Scientific) supplemented with 10% fetal bovine serum (FBS) (VWR, Radnor, PA, USA), 10 mM HEPES (Thermo Fisher Scientific), 100 U/mL penicillin (Thermo Fisher Scientific), and 100 g/ml streptomycin sulfate (Thermo Fisher Scientific) and stimulated with magnetic beads coated with anti-CD3/anti-CD28 (Thermo Fisher Scientific) at the 1:3 T cell-to-bead ratio. Approximately 24 h after activation, T cells were transduced with lentiviral vectors encoding the CAR transgene at an MOI of 3 to 6. On day 5, beads were removed and thereafter cells were counted and fed every 2 days, supplemented with IL 2 150 U/ml until they were either used for functional assays or cryopreserved for future use.
The human cell line U87MG was purchased from the American Type Culture Collection (ATCC) and maintained in MEM (Richter’s modification) (Thermo Fisher Scientific) with components GlutaMAX-1 (Thermo Fisher Scientific), HEPES pyruvate, and penicillin/streptomycin supplemented with 10% FBS. Primary human keratinocytes were purchased from the Dermatology Core Facility at the University of Pennsylvania. K562 cells were purchased from ATCC and maintained in RPMI media (Invitrogen, Carlsbad, CA, USA) supplemented with 10% FBS, 20 mM HEPES, and 1% penicillin/streptomycin. Primary astrocytes were purchased (ScienCell Research Laboratories, Carlsbad, CA, USA) and cultured according to the manufacturer’s instructions. The cells from early passages were used for cytotoxicity and cytokine experiments. GSC cell lines were cultured in DMEM F-12 media (Sigma-Aldrich, St. Louis, MO, USA) supplemented with 2% B27 without vitamin A (Thermo Fisher Scientific), 20 mM HEPES, and penicillin/streptomycin.
To produce the overexpressing EGFR cell line (designated as U87MG-EGFR), the lentivirus co-expressing wtEGFR and Cyan Fluorescent Protein (CFP) under the control of the EF-1α promoter was transduced into the U87MG cell line. On post-transduction day 4, cells were sorted on an Influx cell sorter (BD, Franklin Lakes, NJ, USA) on the basis of high EGFR expression and subsequently expanded. The lentivirus co-expressing CFP and EGFR mutants EGFRR108K/G, EGFRA289D/T/V, or EGFRvIII was transduced into U87MG-EGFR, GSC5077 neurosphere cells (21), and K562 cell lines. CFP-positive cells were sorted by fluorescence-activated cell sorting (FACS). For luciferase killing assays and in vivo tracking studies, U87MG and U87MG-EGFR mutant cell lines were transduced with lentivirus click beetle green (CBG) luciferase and green fluorescent protein (GFP). Anti-GFP-positive cells were sorted by FACS.
CAR T cells and K562 targets expressing EGFR and its variants were cocultured in 1:2 ratio in the R10 medium in a 96-well plate, in triplicate. Plates were incubated at 37°C with 5% CO2. After 48 h, supernatants were collected and cytokine levels were assessed by ELISA kit (R&D Systems, Minneapolis, MN, USA) for IFN-γ, TNF-α, and IL2 production, according to the manufacturer’s instructions.
The cytolytic efficacy of CAR T cells against K562 cells was evaluated by 4-h chromium release assays using E:T ratios of 5:1, 2.5:1, and 1:1. 51Cr-labeled target cells were incubated with CAR T cells in complete medium or 0.1% Triton X-100, to determine spontaneous and maximum 51Cr release respectively, in a V-bottomed 96-well plate. The mean percentage of specific cytolysis of triplicate wells was calculated from the release of 51Cr using a TopCount NXT (Perkin-Elmer Life and Analytical Sciences, Inc., Waltham, MA) as:
Data was reported as mean ± SD.
CBG+ target cell lines (U87 variants and GSC5077 variants) were cocultured with CAR T cells at E:T ratios of 10:1, 5:1, and 2.5:1, for 24 h at 37°C. One hundred microliters of the mixture was transferred to a 96-well black luminometer plate, 100 μl of 66 μg/ml D-luciferin (GoldBio, St. Louis, MO, USA) was added, and the luminescence was immediately determined. Results were reported as percent killing based on luciferase activity in wells with tumor cells alone.
To assess CD107a degranulation, we plated 1 × 105 T cells and 5 × 105 stimulator target cells per well in round-bottom 96-well plates, to a final volume of 200 μl in complete R10 medium, in triplicates. The CD107a-PE antibody (BD) was added into each well and incubated at 37°C for 4 h, along with surface staining for CD8 (BioLegend, San Diego, CA, USA) and CD3 and then analyzed by flow cytometry.
For CAR detection, cells were stained with biotinylated protein L (GenScript, Piscataway, NJ, USA), goat anti-mouse IgG, and anti-human IgG (Jackson ImmunoResearch Laboratories, West Grove, PA), followed by streptavidin-conjugated allophycocyanin (APC) (BD). The surface expression of EGFR and its mutants was detected by CFP and APC-conjugated cetuximab antibody (Novus Biologicals, Centennial, CO, USA). EGFRvIII expression was detected by anti-EGFRvIII antibody, clone DH8.3 (Santa Cruz Biotechnology, Dallas, TX, USA). Flow analysis done by LSRFortessa (BD) and data were analyzed by FlowJo software (BD).
All mouse experiments were conducted according to Institutional Animal Care and Use Committee (IACUC)–approved protocols. NSG mice were injected with 2.5 × 105 U87MG-EGFR/EGFRvIII/Luc+ tumors subcutaneously in 100 μl of PBS on day 0, seven animals per cohort. Tumor progression was evaluated by luminescence emission on an IVIS Lumina III In Vivo Imaging System (Caliper Life Sciences, Hopkinton, MA, USA) after intraperitoneal D-luciferin injection according to the manufacturer’s instructions (GoldBio). Tumor size was measured by calipers in two dimensions and approximated to volume using the following calculation:
Seven days after tumor implantation, mice were treated with 3 × 106 CAR T cells intravenously via the tail vein, in 100 μl of PBS. Survival was followed over time until predetermined IACUC-approved endpoints were reached.
GBM organoids (GBOs) were established from primary patient tissue, under a University of Pennsylvania Institutional Review Board-approved protocol and with patient written informed consent, and cocultured with CAR T cells as described previously (22, 23). GBOs were fixed and stained after coculture, using anti-CD3 (BioLegend), anti-cleaved caspase 3 (Cell Signaling Technology, Danvers, MA), anti-EGFR (Thermo Fisher Scientific), anti-EGFRvIII (Cell Signaling Technology), and DAPI (Sigma). To control for tumor heterogeneity, four GBOs per condition were used. Mutational data and variant allele fractions (VAF) were obtained from the Center for Personalized Diagnostics at the University of Pennsylvania, as described previously (24).
All in vitro experiments were performed at least in triplicate. GraphPad Prism 6 software (GraphPad Software, San Diego, CA, USA) was used for statistical analyses. Data were presented as mean ± standard deviation. The differences between means were tested by appropriate tests. For the mouse experiments, changes in tumor radiance from baseline at each time point were calculated and compared between groups using the t-test or Wilcoxon rank-sum test, as appropriate. Survival determined from the time of T cell injection was analyzed by the Kaplan–Meier method, and differences in survival between groups were compared by the log-rank Mantel–Cox test.
In the present study, we have generated CARs that target EGFR and EGFR mutants by fusing the scFv derived from mAb806 to a second-generation CAR construct containing 4-1BB-CD3ζ signaling 806 CAR, the design of which is shown schematically in Figure 1A. The EGFRvIII-specific 4-1BB-CD3ζ-based 2173 CAR used in our clinical trials (NCT02209376 and NCT03726515) was generated for comparative evaluation with 806 CAR. 4-1BB-based cetuximab (C225) and CD19 CARs were used as positive and negative controls. Lentiviral vectors encoding CARs were transduced into a mixture of CD4 and CD8 T cells, and surface expression was confirmed by flow cytometry (Figure 1B). We next turned to generating target-positive tumor cell lines, expressing the mutations EGFRR108K/G, EGFRA289D/T/V, EGFRG598V, and EGFRvIII, for testing of our CAR constructs (Figure 1C). In order to more faithfully model the EGFR mutations, which are almost always co-expressed with amplified wtEGFR, we transduced the GBM cell line U87MG and patient-derived glioma stem cell line GSC5077 (21), both of which express low levels of wtEGFR, with a lentiviral vector encoding wtEGFR (Figure 1D) (resultant lines referred to as U87MG-EGFR and GSC5077-EGFR), as well as K562 chronic myelogenous leukemia (CML) cells that lack endogenous expression of EGFR, with wtEGFR (Figure 1E). U87MG-EGFR, GSC5077-EGFR, and K562 cells were also transduced with EGFRvIII lentivirus and expression was then analyzed by an EGFRvIII-specific antibody (Figure 1F). A lentiviral vector co-expressing CFP and the targeted EGFR extracellular mutants (Figure 1D) was transduced into U87MG-EGFR, GSC5077-EGFR, and K562 cells. The resulting CFP-positive cells were sorted by fluorescence-activated cell sorting to obtain a positively transduced cell population (Figure 1G).
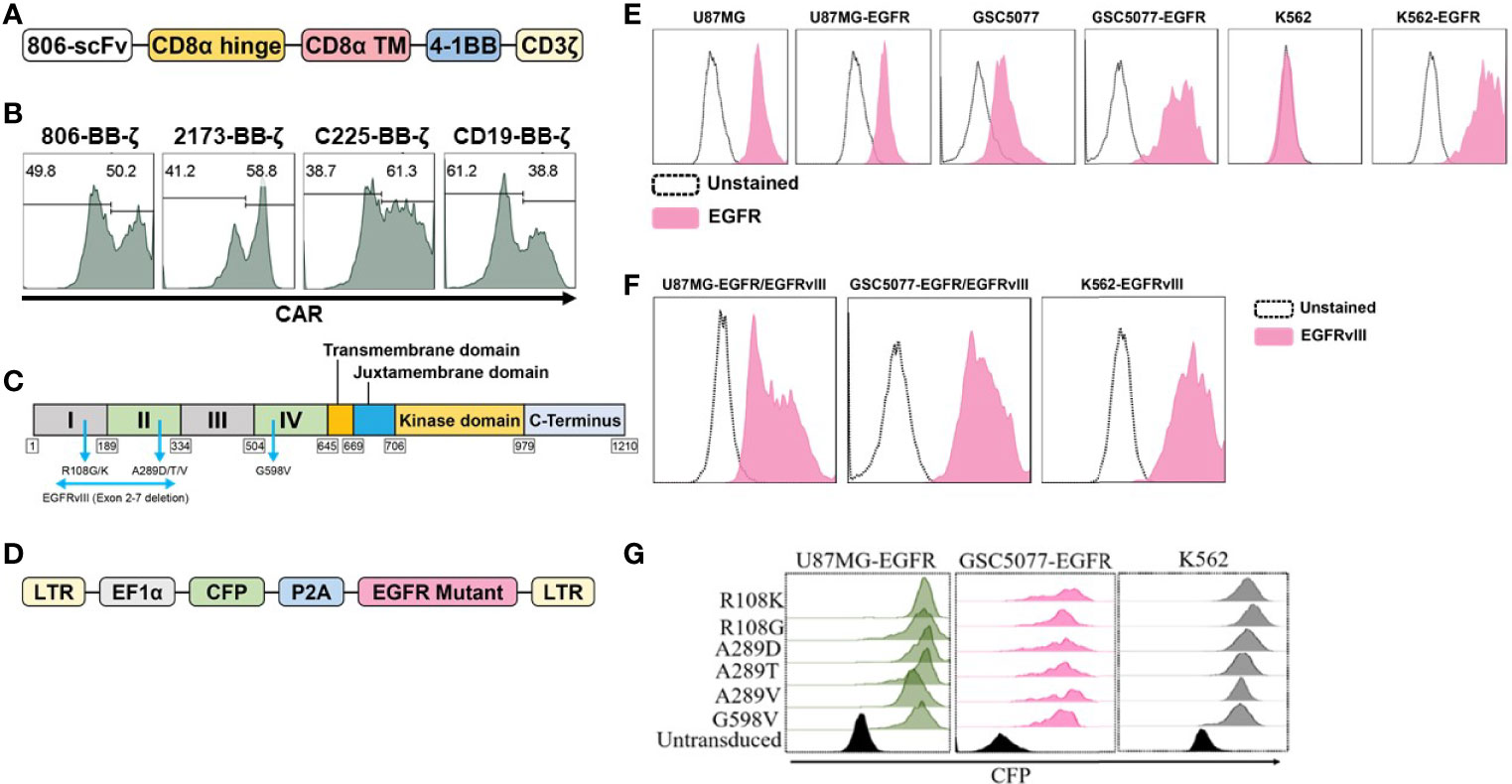
Figure 1 Construction and expression of 806 CAR and EGFR mutant cell lines. (A) Schematic diagram of vector map of 806 CAR containing the 4-1BB co-stimulatory domain. (B) CAR surface expression in primary human CD4+ and CD8+ T cells. Human T cells were simulated for 24 h with anti-CD3/anti-CD28 T-cell activating beads and transduced with CAR transgenes, and CAR expression was analyzed by flow cytometry using biotinylated goat-anti-mouse (806, C225, and CD19 CARs) and goat-anti human F(ab)2 fragment-specific antibodies (2173 CARs) followed by secondary staining with streptavidin-APC. (C) Schematic showing targeted missense mutations in the extracellular domain of EGFR, EGFRR108K/G, EGFRA289D/T/V, EGFRG598V, and splice variant EGFRvIII. (D) Schematic of lentiviral vector co-expressing CFP and wtEGFR or EGFR mutant. (E) Flow-based analysis of endogenous and ectopically expressed EGFR in U87MG, GSC5077, and K562 cell lines using the cetuximab antibody. (F) U87MG, U87MG-EGFR, and GSC5077-EGFR expression of EGFRvIII. (G) U87MG-EGFR, GSC5077-EGFR, and K562 cell lines were transduced with a lentiviral vector co-expressing CFP and indicated EGFR missense mutations and sorted by CFP expression using fluorescent-activated cell sorting.
To determine the specificity of the 806 and 2173 CARs for overexpressed wtEGFR, EGFRvIII, and the EGFR-ECD mutants, 2173 and 806 EGFR BB-ζ CAR T cells were cocultured with U87MG-EGFR and GSC5077-EGFR cell lines expressing EGFRvIII and extracellular mutants EGFRR108K/G, EGFRA289D/T/V, and EGFRG598V, in 24-h bioluminescence-luciferase based killing assays (Figures 2A, B). While 2173 CAR T cells demonstrated specificity for EGFRvIII alone, 806 CAR T cells efficiently lysed all targets and exhibited similar cytolytic potential as C225 CAR T (Figures 2A, B). Notably, 806 CAR T cells were able to kill U87MG cells, despite expressing only low levels of wtEGFR, at an equal level when compared to overexpressed wtEGFR and EGFRvIII. Since U87MG-EGFR mutants expressed endogenous and ectopic EGFR, we could not distinguish if the 806 scFv-binding specificity was restricted to the mutant or wtEGFR. To test the exclusive specificity to the mutants, we cocultured 806 and 2173 CAR T cells with the CML cell line K562, transduced to express wtEGFR, EGFRvIII, or EGFR mutants, as K562 does not have any endogenous EGFR (Figure 2B). 806 CAR T cells did not lyse untransduced K562 cells, confirming the lack of EGFR on the parental line. The 806 CAR T cells selectively targeted K562 cells expressing EGFR, EGFRvIII, or EGFR-ECD mutants and demonstrated similar efficacy as C225 CAR T cells. 2173 CAR T cells lysed K562-EGFRvIII cells but did not show any activity against either wtEGFR or the ECD mutants, as expected (Figure 2B). T cell activation was assessed by induction of surface CD107a expression after coculture of CAR T cells with target-expressing cells (Figure 2C). Antigen-specific effector cytokine production was assessed by coculturing K562 target cells transduced with EGFR and its variants with CAR T cells. The resulting supernatants were analyzed for IFN-γ, TNF-α, and IL2 production (Figure 2D). Untransduced K562 and Nalm6 cells were used as negative controls. 806 and C225 CAR T cells produced similar levels of CD107 degranulation (Figure 2C) and IFN-γ, TNF-α, and IL2 (Figure 2D) in response to EGFRvIII, EGFR-ECD mutants, and EGFR overexpressing cells, while 2173 CAR T cells responded to EGFRvIII alone.
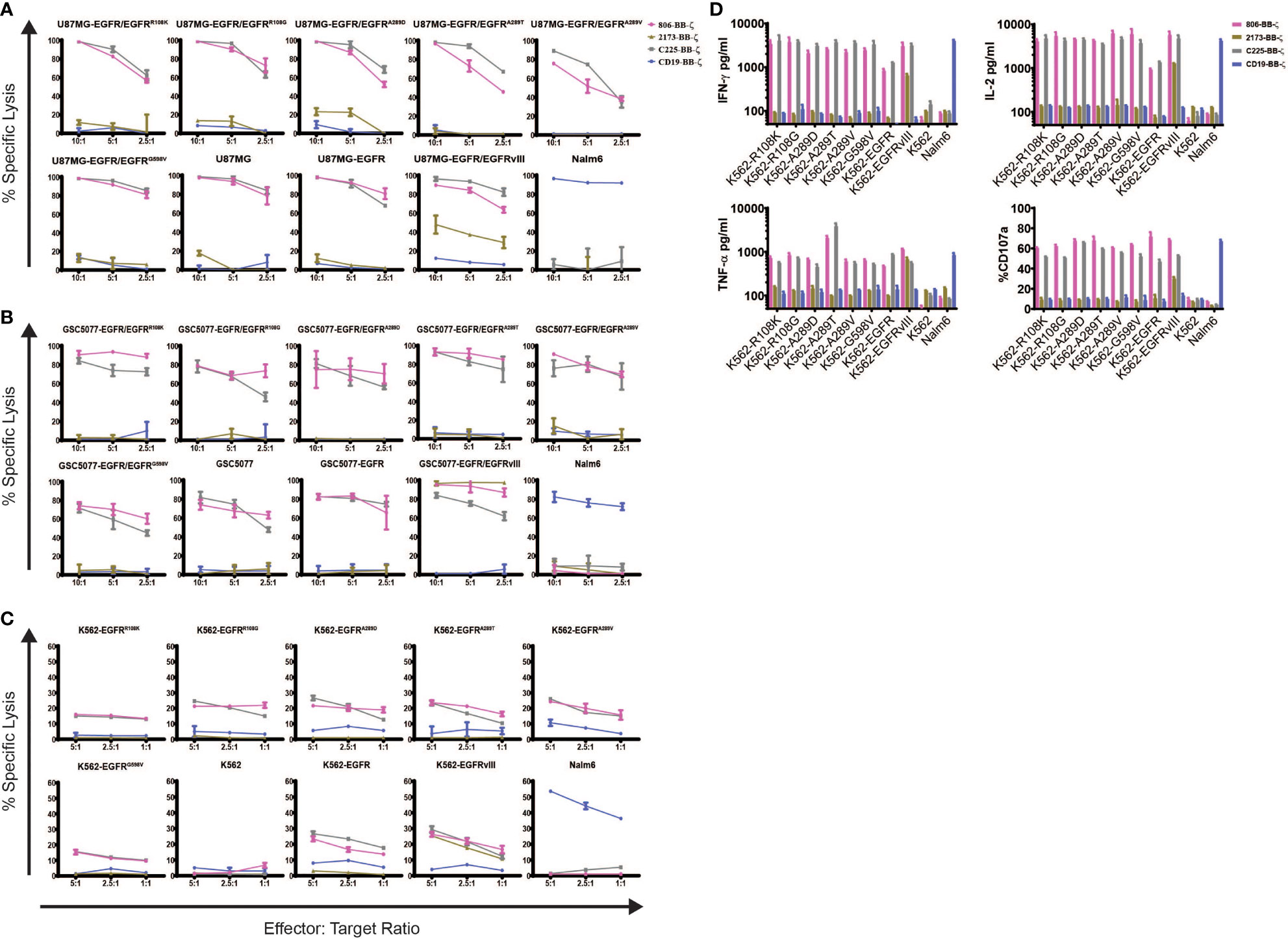
Figure 2 In vitro characterization of 806 EGFR CAR T cells. Antigen-specific cytolytic activity of 806 and 2173 CAR T cells against cell lines expressing EGFR and its variants. (A) U87MG-EGFR and GSC5077-EGFR cell lines expressing EGFRvIII, EGFRR108K/G, EGFRA289D/T/V, and EGFRG598V mutant variants were stably transduced with Click Beetle Green (CBG) and cocultured with CAR T cells at indicated effector-to-target ratios for 24 h. One representative experiment from three normal donors is shown. Samples were performed in triplicates in three replicative experiments. C225-BB-ζ CAR, and CD19-BB-ζ CAR were used as positive and negative controls, respectively. (B) Antigen-specific cytolytic activity of 806 and 2173 CAR T cells in EGFR and its variants expressed in K562 cells in a 4-h chromium release assay at indicated effector-to-target ratios. (C) K562 cells expressing wtEGFR, EGFRvIII, or EGFR-mutants were cocultured with 806 CART cells for 48 h. IFN-γ, TNF-α, and IL2 secretion was measured in the supernatant by ELISA. Bar charts represent results from a single experiment, and values represent the average ± SD of triplicates. (D) CD107a upregulation on CAR T cells stimulated with K562 cells expressing wtEGFR, EGFRvIII, or EGFR-mutants for 4 h. The percentage of CD107a expression was quantified on CD3 cells (values represent the average of ± SD of two repeated experiments).
Having confirmed the function of the 806 CARs, we next sought to compare the reactivity of 806 and 2173 CAR T cells in response to endogenous levels of EGFR in normal cells, in vitro. We cultured primary human keratinocytes and astrocytes, as those cell types express wtEGFR (Figure 3A) and used them to stimulate CAR T cells. We observed production of IFN-γ by C225 CAR T cells, in response to EGFR presented by either astrocytes or keratinocytes, as well as U87MG-EGFR (Figure 3B). In contrast, 2173 CAR T cells produced IFN-γ in response to EGFRvIII antigen alone. 806 CAR T cells exhibited low or no cytotoxicity when cocultured with astrocytes or keratinocytes (Figure 3C), with corresponding low IFN-γ production (Figure 3B).
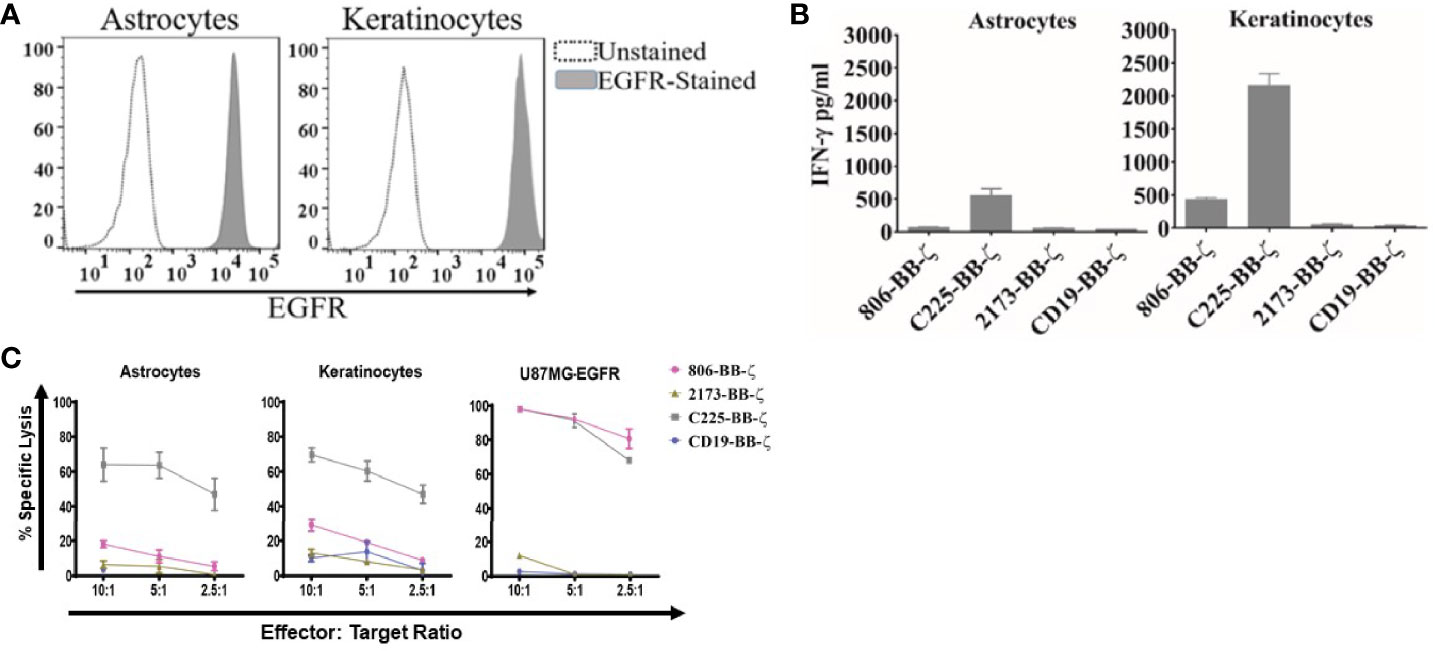
Figure 3 Antitumor efficacy of 806 CAR T cells in primary astrocytes and keratinocytes. (A) Surface expression of EGFR assessed by flow cytometry on human primary astrocytes and keratinocytes using EGFR-specific cetuximab antibody. (B) Primary astrocytes and keratinocytes were cocultured with 806 CAR T cells at indicated ratios in a 4-h chromium assay, and results are representative of a single experiment showing the average ± SD of triplicates. (C) Levels of IFN-γ measured in supernatants by ELISA 24 h after coculturing 806 and 2173 CAR T cells with primary astrocytes and keratinocytes at an effector-to-target ratio of 1:1. Results are representative of a single experiment with the average ± SD of triplicates.
Having compared the antigen-specific effector function of 806 CAR with 2173 CARs, we next sought to confirm its in vivo antitumor effects, using immunodeficient NSG mice bearing human GBM tumors (Figure 4A). On Day 0, U87MG-EGFR/EGFRvIII tumors were implanted subcutaneously, and on Day 5, tumor engraftment was confirmed by bioluminescence imaging (BLI). On Day 7, a single dose of 3 × 106 CAR-positive T cells were infused intravenously (n = 7 per cohort). Total bioluminescence (Figure 4B) and individual bioluminescence (Figure 4C) were assessed in 806 and 2173 CAR T cell-treated groups. Animals in the negative control cohort, receiving CD19 CAR T cells, demonstrated rapid tumor growth, with all mice reaching a predetermined humane experimental endpoint by 42 days after initial tumor engraftment. To be noted, all mice in the CD19, 2173, and 806 groups reached experimental endpoint by day 42, 63, and 91, respectively (Figure 4A).
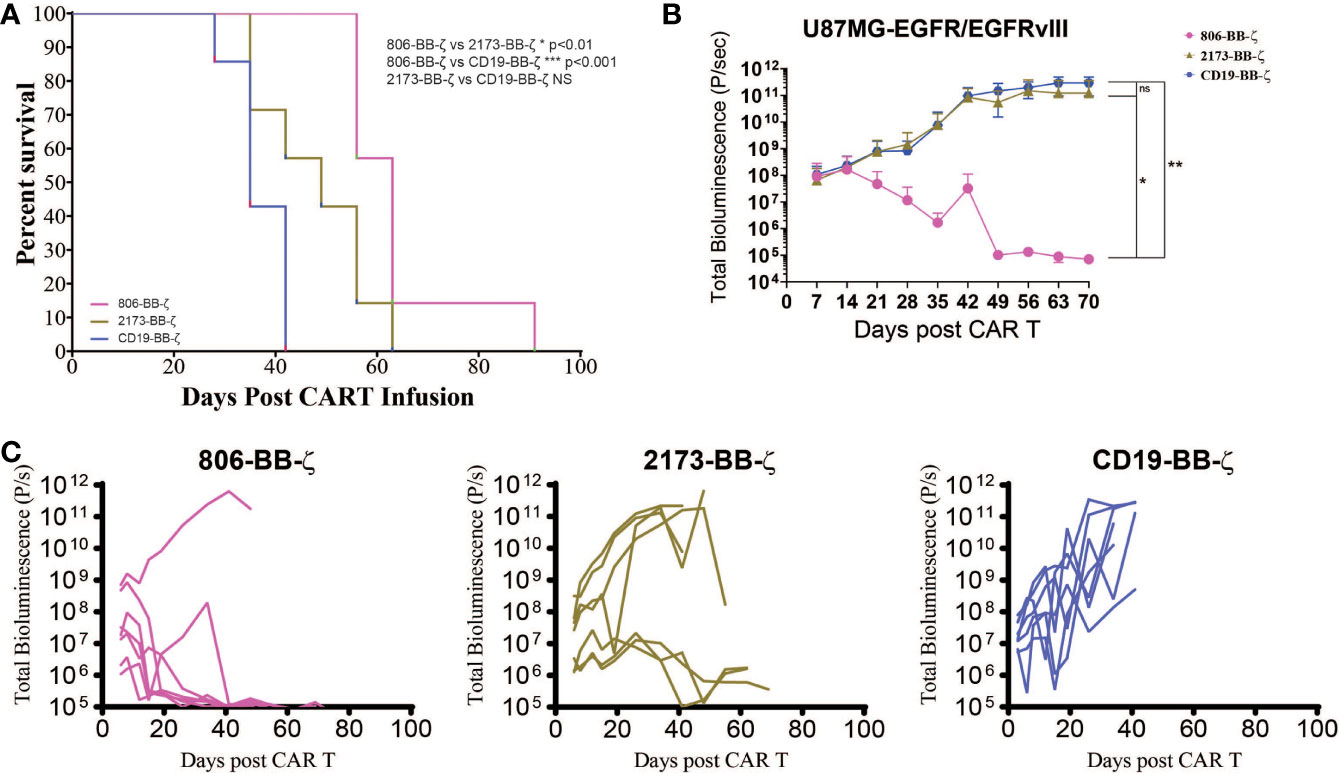
Figure 4 In vivo antitumor effect of 806 CAR T cells in NSG mice bearing U87MG-EGFR/EGFRvIII+ xenografts. Seven days after 250,000 U87MG-EGFR/EGFRvIII cells were subcutaneously implanted into mice, 3 × 106 T cells were injected intravenously with indicated CAR constructs. (A) Survival based on time to endpoint was plotted using a Kaplan–Meier curve and statistically significant differences between CAR groups were determined using the log-rank Mantel-Cox test. Tumor burden was assessed by bioluminescent imaging. Bars indicate means ± SD (n = 7 mice per group). Tumor burden was quantified as total flux (B) and in individual mice (C) in units of photons/second. Bars indicate means± SD (n = 7 mice). P = photons. ns, p > 0.05; *p ≤ 0.05; **p ≤ 0.01.
Given the ability of the 806 CAR to target EGFR alterations beyond EGFRvIII, we turned to patient-derived GBM organoids (GBOs) to demonstrate activity in a heterogeneous model previously characterized to be of high fidelity to human tumors (22, 23). GBOs retain the originating tumor heterogeneity to a high degree out beyond 12 weeks of culturing and maintain the expression of endogenous EGFR and its alterations, providing a valuable model platform for testing therapies aimed at addressing tumor escape. The GBOs selected for coculture experiments contained multiple EGFR mutations (Figure 5A). GBO 9057 had EGFR copy number gain, EGFRvIII, and two missense mutations, EGFRG598V and EGFRC595Y. The missense mutation was found to have a VAF of 24%, while EGFRvIII was identified in less than 10% of the reads, based on next-generation sequencing (NGS). GBO 9066 had EGFR copy number gain, EGFRA289V, and EGFRG598V. Both EGFRA289V and EGFRG598V had a VAF of less than 15%, making determination of co-occurrence impossible through NGS.
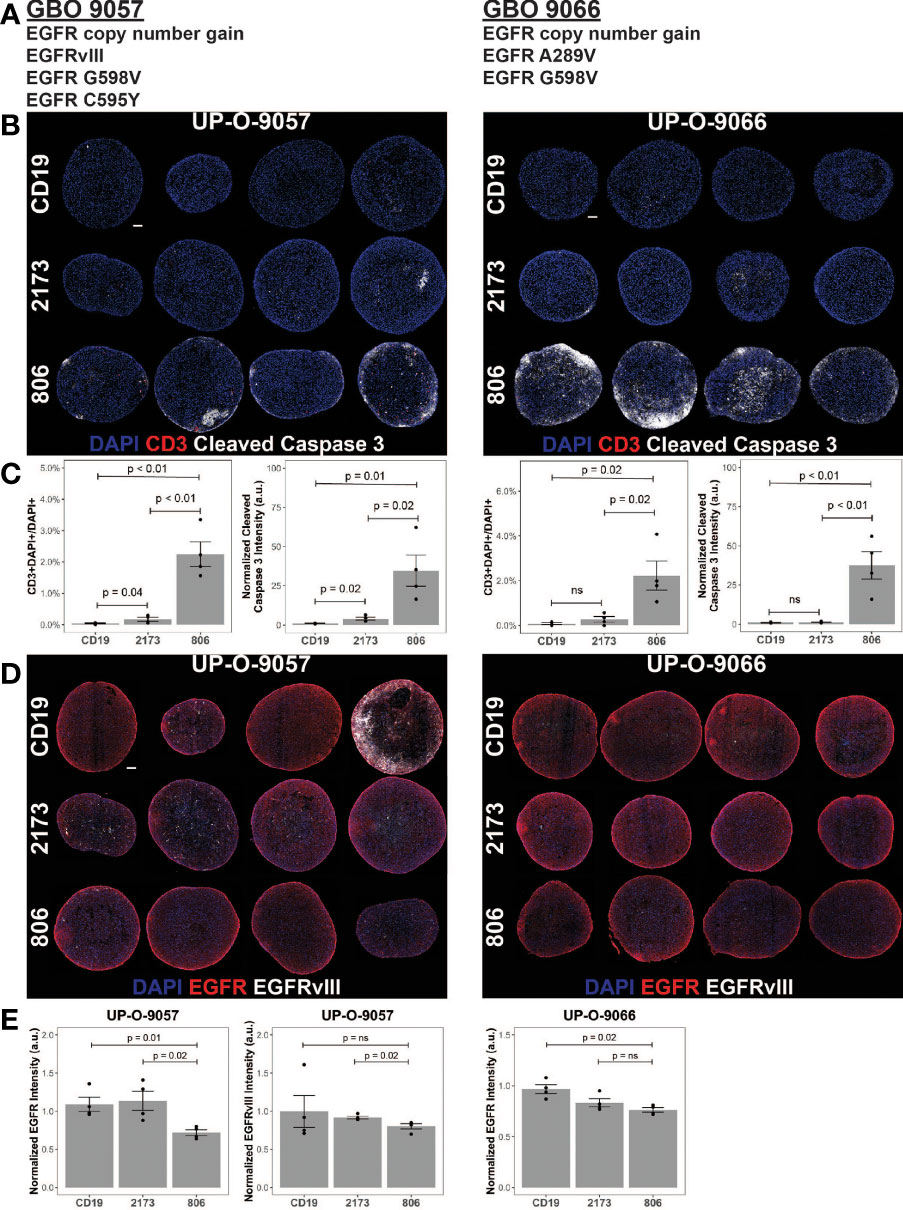
Figure 5 806 CAR T activity in heterogeneous GBOs highlights cross-reactivity of 806 binder against oncogenic EGFRs. CAR T coculture with GBOs was used to demonstrate anti-EGFR activity. (A) EGFR alterations identified in each GBO line. (B) Immunofluorescence images of CAR T cells engrafted GBOs, for four organoids per condition, 9057 (left) and 9066 (right). Blue = DAPI; red = CD3+; white = cleaved caspase 3+ (CC3+), scale bar = 100 µm. (C) Quantification of CD3+ cells (left) and CC3+ cells (right) showing antitumor activity from the 806 CAR T cells. (D) Immunofluorescence images of CAR T cell targets in GBOs, for 9057 (left) and 9066 (right). Blue = DAPI; red = EGFR+; white = EGFRvIII+, scale bar = 100 µm. (E) Quantification of EGFR+ (left) and EGFRvIII+ signals (right) showing antitumor activity from the 806 CAR T cells. Error bars are ± standard error. ns, p > 0.05.
GBOs were cocultured with 806, 2173, and CD19 CAR T cells, at a 1:10 E:T ratio, for 72 h before fixation and evaluation. CAR T cell infiltration, as quantified by CD3 staining, was more significant in the 806 CAR T cell population than either the 2173 or CD19 CAR T cell population (Figure 5B). Cleaved caspase 3 (CC3) was used as a measure of cell death and antitumor activity. As with the CD3+ cell infiltration, the 806 CAR coculture resulted in higher CC3 levels than either the 2173 or CD19 CAR cocultures (Figure 5C). These results highlighted the broad cross-reactivity of the 806 CAR in a heterogeneous, high-fidelity GBM model. wtEGFR staining in both GBO lines provided additional evidence of the cross-reactive nature of the 806 CAR (Figure 5D). Staining intensity, normalized to CD19 CAR-treated GBOs, showed consistent decreases in 806 CAR-treated GBOs, to a greater degree than the 2173 CAR-treated GBOs (Figure 5E).
We have shown broad cross-reactivity of 806 CAR T cells to EGFR mutant proteins resulting in enhanced anti-GBM tumor killing, along with a low on-target, off-tumor effect against both astrocytes and keratinocytes that express wild-type EGFR. Importantly, 806 CAR T cells are able to more effectively control tumor growth in a wtEGFR/EGFRvIII model. 806 CAR T cells also demonstrate greater killing in GBOs with heterogeneity of endogenous EGFR and EGFR mutants, confirming its potential to more effectively treat GBM tumors by limiting the impact of tumor escape due to antigen loss.
With regard to the CAR T trial in recurrent GBM (12), the demonstrated tumor recurrence was likely due to the exclusive specificity of the scFv employed in the trial. The 2173 construct was chosen for its selective binding to a novel glycine residue formed at the exon 2–7 deletion in EGFRvIII and for a lack of cross-reactivity to wtEGFR (20). However, the binding affinity and target repertoire were of secondary importance. Given the co-occurrence of amplified wtEGFR with EGFRvIII and most ECD missense mutations (11), there is a clinically relevant rationale for targeting multiple EGFR alterations in the GBM population (25). Dual targeting of EGFR and EGFRvIII by CAR T and NK cells has been demonstrated in recent studies using scFvs specific for both antigens (26–29). Our work expands on that, as 806 CAR T cells were able to lyse GBM (U87MG, GSC5077) and non-GBM (K562) cell lines modified to express not only wtEGFR and EGFRvIII but also EGFR extracellular mutations. In comparison, 2173 CAR T cells exhibited specificity for EGFRvIII alone (12, 20). While 806 CAR treated animals did eventually reach experimental endpoints, their loss of weight, patchy hair, and red eyes were suggestive of the development of graft-vs-host disease and not tumor growth. This hypothesis was supported by a lack of palpable or visible tumor at autopsy.
GBM tumors are significantly heterogeneous, both intratumorally (30, 31) and intertumorally (6). Intratumorally, there are mixed cytological subtypes, exhibiting regional differences in gene expression, key genetic mutations, and chromosomal alterations. This polyclonal nature contributes to therapeutic resistance and tumor escape (32). To address intratumoral heterogeneity, relevant targeted therapies would ideally be able to target larger tumor cell populations within the entire tumor bulk. Given the co-occurrence of wtEGFR amplification seen with EGFR mutations and splice variants (24), the cross-reactive EGFR-targeting 806 scFv should provide greater tumor cell coverage, resulting in better tumor control. The potential for broader tumor control was demonstrated through the high-fidelity, heterogeneous GBO model (22). GBOs have retained the originating tumor heterogeneity to a high degree, as assessed by both mRNA and protein levels. These “mini-tumors” provided the opportunity to test 806 CAR T cells against a target-heterogeneous tumor and showcase its ability to exert antitumor activity against a greater portion of the tumor than the compared 2173 CAR and CD19 CAR T cells. While the VAFs associated with the originating tumors of the GBOS allow for hypothesizing of independent EGFR mutant tumor populations, one caveat is that the NGS methods used do not allow for concrete determination of subpopulations. The data were subject to bias from tumor viability and number of reads of the sample. Additionally, the heterogeneity of the EGFR variants on amplified alleles is complicated by the mechanisms of amplification of EGFR. GBMs frequently harbor double minutes, extrachromosomal sequences of DNA that are acentric and lead to asymmetric distribution to daughter cells (33). This causes increased cell-to-cell heterogeneity of EGFR alterations in GBM.
Intertumoral variation, from patient to patient, reduces the applicable population for targeted therapies. However, there are gene families frequently found altered across GBM (6). In particular, EGFR amplification is found in up to 60% of GBMs. Concurrently with amplification, 30%–40% of GBM tumors express the constitutively active mutant variant, EGFRvIII (34). Combined with the intratumoral expression of EGFR variants, these data suggest that targeting the EGFR family of tumor-specific alterations may successfully address both inter- and intratumoral heterogeneity.
Several EGFRvIII-targeted agents are currently in development or in clinical trials for the treatment of GBM. Although the preclinical data from experimental studies evaluating these therapies have been promising, their efficacy in the clinic has yet to be conclusively demonstrated (35–37). In a vaccination approach to target the EGFRvIII in GBM patients, a phase III trial for newly diagnosed glioblastoma failed to show overall efficacy despite 60%–80% of recurrent tumors showing complete loss of EGFRvIII positive cells (38). Additional trials targeting EGFRvIII demonstrated similar loss of EGFRvIII concurrent with tumor recurrence (12, 39, 40). Similarly, the EGFRvIII-targeting CAR T trial illustrated the continued presence of EGFR amplification and oncogenic EGFR ECD missense mutations despite EGFRvIII antigen loss in posttreatment tumor specimens (12). These results confirm the need to target multiple EGFR alterations simultaneously.
Despite preclinical efficacy, the success of wtEGFR targeting mAbs cetuximab and panitumumab has been associated with on-target, off-tumor toxicity in other tumor types, due to their significant binding to EGFR expressed on normal tissues (41, 42). Their clinical activity in GBM has yet to be successfully demonstrated in large-scale studies. Coculture of 806 CAR T cells with basal physiologic EGFR-expressing normal tissue cell lines did not lead to significant cell killing in our work. Previous work has suggested that the 806 epitope is exposed on both mutated EGFR (EGFRvIII, EGFRR108G/K, EGFRA289D/T/V) as well as amplified wtEGFR found on tumors, but not accessible on wtEGFR found on normal tissue (43). The wtEGFR differences have been proposed to be due to different posttranslational mannose modifications and kinetics of EGFR trafficking in tumors compared to normal tissue (44). Multiple clinical trials with humanized mAb 806 conjugated to a microtubule inhibitor (ABT-414) have demonstrated only low levels of cutaneous toxicity (45–47). The therapeutic window of CAR T cells for tumor-associated antigens relies on the quantitative difference between antigen-overexpressing tumor and antigen-low normal tissue. Preclinical studies targeting EGFR and erbB2 with affinity-lowered CAR T cells have demonstrated potent antitumor effects against high antigen density while sparing low antigen density normal tissue (48–50). The demonstrated cross-reactivity of 806 CAR T cells for EGFR alterations, including amplified wtEGFR, EGFRvIII, and ECD missense mutations, suggests that 806 CAR T cells may be a more efficacious therapeutic strategy to achieve tumor control and prevent tumor escape via target antigen loss.
The original contributions presented in the study are included in the article/supplementary material. Further inquiries can be directed to the corresponding author.
The studies involving human participants were reviewed and approved by the University of Pennsylvania Institutional Review Board. The patients/participants provided their written informed consent to participate in this study. The animal study was reviewed and approved by University of Pennsylvania Institutional Animal Care and Use Committee.
RT and ZB conceived and carried out the experiments, with contributions from YY, LZ, JZ, and DZ for specific assays. MM, GLM, HS, and DO’R supervised the project. RT and ZB wrote the manuscript. All authors contributed to the article and approved the submitted version.
The described work was funded by the GBM Translational Center of Excellence, the Templeton Family Initiative in Neuro-Oncology, The Maria and Gabriele Troiano Brain Cancer Immunotherapy Fund, and NIH (R35NS116843 to HS and R35NS097370 to GLM).
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The described work involves patent applications owned by the University of Pennsylvania. MM is an inventor on multiple issued and pending patents related to CAR T cell technology used in this study. These patents are assigned to the University of Pennsylvania and have been licensed to third parties for which royalties have or may be received.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The authors thank the Human Immunology Core at the University of Pennsylvania for providing leukocytes for the described work, the Stem Cell and Xenograft Core at the University of Pennsylvania for assistance with the animal work, and the Small Animal Imaging Facility at the University of Pennsylvania for the bioluminescence imaging.
1. Castellarin M, Watanabe K, June CH, Kloss CC, Posey AD Jr. Driving Cars to the Clinic for Solid Tumors. Gene Ther (2018) 25(3):165–75. doi: 10.1038/s41434-018-0007-x
2. Fesnak AD, June CH, Levine BL. Engineered T Cells: The Promise and Challenges of Cancer Immunotherapy. Nat Rev Cancer (2016) 16(9):566–81. doi: 10.1038/nrc.2016.97
3. Porter DL, Levine BL, Kalos M, Bagg A, June CH. Chimeric Antigen Receptor-Modified T Cells in Chronic Lymphoid Leukemia. N Engl J Med (2011) 365(8):725–33. doi: 10.1056/NEJMoa1103849
4. Jackson C, Ruzevick J, Phallen J, Belcaid Z, Lim M. Challenges in Immunotherapy Presented by the Glioblastoma Multiforme Microenvironment. Clin Dev Immunol (2011) 2011:732413. doi: 10.1155/2011/732413
5. Nduom EK, Weller M, Heimberger AB. Immunosuppressive Mechanisms in Glioblastoma. Neuro Oncol (2015) 17(Suppl 7):vii9–vii14. doi: 10.1093/neuonc/nov151
6. Brennan CW, Verhaak RG, McKenna A, Campos B, Noushmehr H, Salama SR, et al. The Somatic Genomic Landscape of Glioblastoma. Cell (2013) 155(2):462–77. doi: 10.1016/j.cell.2013.09.034
7. Cominelli M, Grisanti S, Mazzoleni S, Branca C, Buttolo L, Furlan D, et al. EGFR Amplified and Overexpressing Glioblastomas and Association With Better Response to Adjuvant Metronomic Temozolomide. J Natl Cancer Inst (2015) 107(5). doi: 10.1093/jnci/djv041
8. Lopez-Gines C, Gil-Benso R, Ferrer-Luna R, Benito R, Serna E, Gonzalez-Darder J, et al. New Pattern of EGFR Amplification in Glioblastoma and the Relationship of Gene Copy Number With Gene Expression Profile. Mod Pathol (2010) 23(6):856–65. doi: 10.1038/modpathol.2010.62
9. Nishikawa R, Ji XD, Harmon RC, Lazar CS, Gill GN, Cavenee WK, et al. A Mutant Epidermal Growth Factor Receptor Common in Human Glioma Confers Enhanced Tumorigenicity. Proc Natl Acad Sci USA (1994) 91(16):7727–31. doi: 10.1073/pnas.91.16.7727
10. Lee JC, Vivanco I, Beroukhim R, Huang JH, Feng WL, DeBiasi RM, et al. Epidermal Growth Factor Receptor Activation in Glioblastoma Through Novel Missense Mutations in the Extracellular Domain. PloS Med (2006) 3(12):e485. doi: 10.1371/journal.pmed.0030485
11. Binder ZA, Thorne AH, Bakas S, Wileyto EP, Bilello M, Akbari H, et al. Epidermal Growth Factor Receptor Extracellular Domain Mutations in Glioblastoma Present Opportunities for Clinical Imaging and Therapeutic Development. Cancer Cell (2018) 34(1):163–77.e7. doi: 10.1016/j.ccell.2018.06.006
12. O’Rourke DM, Nasrallah MP, Desai A, Melenhorst JJ, Mansfield K, Morrissette JJD, et al. A Single Dose of Peripherally Infused EGFRvIII-Directed CAR T Cells Mediates Antigen Loss and Induces Adaptive Resistance in Patients With Recurrent Glioblastoma. Sci Transl Med (2017) 9(399). doi: 10.1126/scitranslmed.aaa0984
13. Gan HK, Burgess AW, Clayton AH, Scott AM. Targeting of a Conformationally Exposed, Tumor-Specific Epitope of EGFR as a Strategy for Cancer Therapy. Cancer Res (2012) 72(12):2924–30. doi: 10.1158/0008-5472.CAN-11-3898
14. Panousis C, Rayzman VM, Johns TG, Renner C, Liu Z, Cartwright G, et al. Engineering and Characterisation of Chimeric Monoclonal Antibody 806 (Ch806) for Targeted Immunotherapy of Tumours Expressing De2-7 EGFR or Amplified EGFR. Br J Cancer (2005) 92(6):1069–77. doi: 10.1038/sj.bjc.6602470
15. Phillips AC, Boghaert ER, Vaidya KS, Mitten MJ, Norvell S, Falls HD, et al. ABT-414, an Antibody-Drug Conjugate Targeting a Tumor-Selective EGFR Epitope. Mol Cancer Ther (2016) 15(4):661–9. doi: 10.1158/1535-7163.MCT-15-0901
16. Lassman A, Pugh S, Wang T, Aldape K, Gan H, Preusser M, et al. ACTR-21. A Randomized, Double-Blind, Placebo-Controlled Phase 3 Trial of Depatuxizumab Mafooditin (ABT-414) in Epidermal Growth Factor Receptor (EGFR) Amplified (AMP) Newly Diagnosed Glioblastosma (nGBM). Neuro-Oncology (2019) 21(Supplement_6):vi17–vi. doi: 10.1093/neuonc/noz175.064
17. Milone MC, Fish JD, Carpenito C, Carroll RG, Binder GK, Teachey D, et al. Chimeric Receptors Containing CD137 Signal Transduction Domains Mediate Enhanced Survival of T Cells and Increased Antileukemic Efficacy In Vivo. Mol Ther (2009) 17(8):1453–64. doi: 10.1038/mt.2009.83
18. Imai C, Mihara K, Andreansky M, Nicholson IC, Pui CH, Geiger TL, et al. Chimeric Receptors With 4-1BB Signaling Capacity Provoke Potent Cytotoxicity Against Acute Lymphoblastic Leukemia. Leukemia (2004) 18(4):676–84. doi: 10.1038/sj.leu.2403302
19. Carpenito C, Milone MC, Hassan R, Simonet JC, Lakhal M, Suhoski MM, et al. Control of Large, Established Tumor Xenografts With Genetically Retargeted Human T Cells Containing CD28 and CD137 Domains. Proc Natl Acad Sci USA (2009) 106(9):3360–5. doi: 10.1073/pnas.0813101106
20. Johnson LA, Scholler J, Ohkuri T, Kosaka A, Patel PR, McGettigan SE, et al. Rational Development and Characterization of Humanized Anti-EGFR Variant III Chimeric Antigen Receptor T Cells for Glioblastoma. Sci Transl Med (2015) 7(275):275ra22. doi: 10.1126/scitranslmed.aaa4963
21. Yin Y, Boesteanu AC, Binder ZA, Xu C, Reid RA, Rodriguez JL, et al. Checkpoint Blockade Reverses Anergy in IL-13ralpha2 Humanized scFv-Based CAR T Cells to Treat Murine and Canine Gliomas. Mol Ther Oncolytics (2018) 11:20–38. doi: 10.1016/j.omto.2018.08.002
22. Jacob F, Salinas RD, Zhang DY, Nguyen PTT, Schnoll JG, Wong SZH, et al. A Patient-Derived Glioblastoma Organoid Model and Biobank Recapitulates Inter- and Intra-Tumoral Heterogeneity. Cell (2020) 180(1):188–204.e22. doi: 10.1016/j.cell.2019.11.036
23. Jacob F, Ming GL, Song H. Generation and Biobanking of Patient-Derived Glioblastoma Organoids and Their Application in CAR T Cell Testing. Nat Protoc (2020) 15(12):4000–33. doi: 10.1038/s41596-020-0402-9
24. Nasrallah MP, Binder ZA, Oldridge DA, Zhao J, Lieberman DB, Roth JJ, et al. Molecular Neuropathology in Practice: Clinical Profiling and Integrative Analysis of Molecular Alterations in Glioblastoma. Acad Pathol (2019) 6:2374289519848353. doi: 10.1177/2374289519848353
25. Reilly EB, Phillips AC, Buchanan FG, Kingsbury G, Zhang Y, Meulbroek JA, et al. Characterization of ABT-806, a Humanized Tumor-Specific Anti-EGFR Monoclonal Antibody. Mol Cancer Ther (2015) 14(5):1141–51. doi: 10.1158/1535-7163.MCT-14-0820
26. Genssler S, Burger MC, Zhang C, Oelsner S, Mildenberger I, Wagner M, et al. Dual Targeting of Glioblastoma With Chimeric Antigen Receptor-Engineered Natural Killer Cells Overcomes Heterogeneity of Target Antigen Expression and Enhances Antitumor Activity and Survival. Oncoimmunology (2016) 5(4):e1119354. doi: 10.1080/2162402X.2015.1119354
27. Han J, Chu J, Keung Chan W, Zhang J, Wang Y, Cohen JB, et al. CAR-Engineered NK Cells Targeting Wild-Type EGFR and EGFRvIII Enhance Killing of Glioblastoma and Patient-Derived Glioblastoma Stem Cells. Sci Rep (2015) 5:11483. doi: 10.1038/srep11483
28. Jiang H, Gao H, Kong J, Song B, Wang P, Shi B, et al. Selective Targeting of Glioblastoma With EGFRvIII/EGFR Bitargeted Chimeric Antigen Receptor T Cell. Cancer Immunol Res (2018) 6(11):1314–26. doi: 10.1158/2326-6066.CIR-18-0044
29. Ravanpay AC, Gust J, Johnson AJ, Rolczynski LS, Cecchini M, Chang CA, et al. EGFR806-CAR T Cells Selectively Target a Tumor-Restricted EGFR Epitope in Glioblastoma. Oncotarget (2019) 10(66):7080–95. doi: 10.18632/oncotarget.27389
30. Darmanis S, Sloan SA, Croote D, Mignardi M, Chernikova S, Samghababi P, et al. Single-Cell RNA-Seq Analysis of Infiltrating Neoplastic Cells at the Migrating Front of Human Glioblastoma. Cell Rep (2017) 21(5):1399–410. doi: 10.1016/j.celrep.2017.10.030
31. Patel AP, Tirosh I, Trombetta JJ, Shalek AK, Gillespie SM, Wakimoto H, et al. Single-Cell RNA-Seq Highlights Intratumoral Heterogeneity in Primary Glioblastoma. Science (2014) 344(6190):1396–401. doi: 10.1126/science.1254257
32. Skaga E, Kulesskiy E, Fayzullin A, Sandberg CJ, Potdar S, Kyttala A, et al. Intertumoral Heterogeneity in Patient-Specific Drug Sensitivities in Treatment-Naive Glioblastoma. BMC Cancer (2019) 19(1):628. doi: 10.1186/s12885-019-5861-4
33. Bailey C, Shoura MJ, Mischel PS, Swanton C. Extrachromosomal DNA-Relieving Heredity Constraints, Accelerating Tumour Evolution. Ann Oncol (2020) 31(7):884–93. doi: 10.1016/j.annonc.2020.03.303
34. Gan HK, Kaye AH, Luwor RB. The EGFRvIII Variant in Glioblastoma Multiforme. J Clin Neurosci (2009) 16(6):748–54. doi: 10.1016/j.jocn.2008.12.005
35. Choi BD, Archer GE, Mitchell DA, Heimberger AB, McLendon RE, Bigner DD, et al. EGFRvIII-Targeted Vaccination Therapy of Malignant Glioma. Brain Pathol (2009) 19(4):713–23. doi: 10.1111/j.1750-3639.2009.00318.x
36. Karpel-Massler G, Schmidt U, Unterberg A, Halatsch ME. Therapeutic Inhibition of the Epidermal Growth Factor Receptor in High-Grade Gliomas: Where do We Stand? Mol Cancer Res (2009) 7(7):1000–12. doi: 10.1158/1541-7786.MCR-08-0479
37. Rosenthal M, Curry R, Reardon DA, Rasmussen E, Upreti VV, Damore MA, et al. Safety, Tolerability, and Pharmacokinetics of Anti-EGFRvIII Antibody-Drug Conjugate AMG 595 in Patients With Recurrent Malignant Glioma Expressing EGFRvIII. Cancer Chemother Pharmacol (2019) 84(2):327–36. doi: 10.1007/s00280-019-03879-2
38. Heimberger AB, Sampson JH. The PEPvIII-KLH (CDX-110) Vaccine in Glioblastoma Multiforme Patients. Expert Opin Biol Ther (2009) 9(8):1087–98. doi: 10.1517/14712590903124346
39. Weller M, Butowski N, Tran DD, Recht LD, Lim M, Hirte H, et al. Rindopepimut With Temozolomide for Patients With Newly Diagnosed, EGFRvIII-Expressing Glioblastoma (ACT IV): A Randomised, Double-Blind, International Phase 3 Trial. Lancet Oncol (2017) 18(10):1373–85. doi: 10.1016/S1470-2045(17)30517-X
40. Schuster J, Lai RK, Recht LD, Reardon DA, Paleologos NA, Groves MD, et al. Multicenter Trial of Rindopepimut (CDX-110) in Newly Diagnosed Glioblastoma: The ACT III Study. Neuro Oncol (2015) 17(6):854–61. doi: 10.1093/neuonc/nou348
41. Saltz LB, Meropol NJ, Loehrer PJ, Needle MN, Kopit J, Mayer RJ. Phase II Trial of Cetuximab in Patients With Refractory Colorectal Cancer That Expresses the Epidermal Growth Factor Receptor. J Clin Oncol (2004) 22(7):1201–8. doi: 10.1200/JCO.2004.10.182
42. Holch JW, Held S, Stintzing S, Fischer von Weikersthal L, Decker T, Kiani A, et al. Relation of Cetuximab-Induced Skin Toxicity and Early Tumor Shrinkage in Metastatic Colorectal Cancer Patients: Results of the Randomized Phase 3 Trial FIRE-3 (AIO Krk0306). Ann Oncol (2020) 31(1):72–8. doi: 10.1016/j.annonc.2019.10.001
43. Orellana L, Thorne AH, Lema R, Gustavsson J, Parisian AD, Hospital A, et al. Oncogenic Mutations at the EGFR Ectodomain Structurally Converge to Remove a Steric Hindrance on a Kinase-Coupled Cryptic Epitope. Proc Natl Acad Sci USA (2019) 116(20):10009–18. doi: 10.1073/pnas.1821442116
44. Johns TG, Mellman I, Cartwright GA, Ritter G, Old LJ, Burgess AW, et al. The Antitumor Monoclonal Antibody 806 Recognizes a High-Mannose Form of the EGF Receptor That Reaches the Cell Surface When Cells Over-Express the Receptor. FASEB J (2005) 19(7):780–2. doi: 10.1096/fj.04-1766fje
45. Cleary JM, Reardon DA, Azad N, Gandhi L, Shapiro GI, Chaves J, et al. A Phase 1 Study of ABT-806 in Subjects With Advanced Solid Tumors. Invest New Drugs (2015) 33(3):671–8. doi: 10.1007/s10637-015-0234-6
46. Reardon DA, Lassman AB, van den Bent M, Kumthekar P, Merrell R, Scott AM, et al. Efficacy and Safety Results of ABT-414 in Combination With Radiation and Temozolomide in Newly Diagnosed Glioblastoma. Neuro Oncol (2017) 19(7):965–75. doi: 10.1093/neuonc/now257
47. van den Bent M, Gan HK, Lassman AB, Kumthekar P, Merrell R, Butowski N, et al. Efficacy of Depatuxizumab Mafodotin (ABT-414) Monotherapy in Patients With EGFR-Amplified, Recurrent Glioblastoma: Results From a Multi-Center, International Study. Cancer Chemother Pharmacol (2017) 80(6):1209–17. doi: 10.1007/s00280-017-3451-1
48. Caruso HG, Hurton LV, Najjar A, Rushworth D, Ang S, Olivares S, et al. Tuning Sensitivity of CAR to EGFR Density Limits Recognition of Normal Tissue While Maintaining Potent Antitumor Activity. Cancer Res (2015) 75(17):3505–18. doi: 10.1158/0008-5472.CAN-15-0139
49. Liu X, Jiang S, Fang C, Yang S, Olalere D, Pequignot EC, et al. Affinity-Tuned ErbB2 or EGFR Chimeric Antigen Receptor T Cells Exhibit an Increased Therapeutic Index Against Tumors in Mice. Cancer Res (2015) 75(17):3596–607. doi: 10.1158/0008-5472.CAN-15-0159
Keywords: GBM, glioma, immunotherapy, CAR T cells, adoptive T cell therapy, EGFR
Citation: Thokala R, Binder ZA, Yin Y, Zhang L, Zhang JV, Zhang DY, Milone MC, Ming GL, Song H and O’Rourke DM (2021) High-Affinity Chimeric Antigen Receptor With Cross-Reactive scFv to Clinically Relevant EGFR Oncogenic Isoforms. Front. Oncol. 11:664236. doi: 10.3389/fonc.2021.664236
Received: 04 February 2021; Accepted: 18 August 2021;
Published: 10 September 2021.
Edited by:
Payal Watchmaker, University of California, San Francisco, United StatesReviewed by:
Giedre Krenciute, St. Jude Children’s Research Hospital, United StatesCopyright © 2021 Thokala, Binder, Yin, Zhang, Zhang, Zhang, Milone, Ming, Song and O’Rourke. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Donald M. O’Rourke, donald.orourke@pennmedicine.upenn.edu
†Present address: Yibo Yin, Department of Neurosurgery, First Affiliated Hospital of Harbin Medical University, Harbin, China
‡These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.