
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oncol., 23 June 2021
Sec. Cancer Immunity and Immunotherapy
Volume 11 - 2021 | https://doi.org/10.3389/fonc.2021.660039
This article is part of the Research TopicUnderstanding the Interplay Between the Tumor Immune Microenvironment and Genetic Alterations in Thoracic MalignanciesView all 13 articles
 Stefanie Hiltbrunner1,2†
Stefanie Hiltbrunner1,2† Laura Mannarino3†
Laura Mannarino3† Michaela B. Kirschner4
Michaela B. Kirschner4 Isabelle Opitz4
Isabelle Opitz4 Angelica Rigutto1,2
Angelica Rigutto1,2 Alexander Laure1,2
Alexander Laure1,2 Michela Lia5
Michela Lia5 Paolo Nozza6Antonio Maconi7
Paolo Nozza6Antonio Maconi7 Sergio Marchini3
Sergio Marchini3 Maurizio D’Incalci3
Maurizio D’Incalci3 Alessandra Curioni-Fontecedro1,2‡
Alessandra Curioni-Fontecedro1,2‡ Federica Grosso5,8*‡
Federica Grosso5,8*‡Malignant pleural mesothelioma (MPM) is a rare and fatal disease of the pleural lining. Up to 80% of the MPM cases are linked to asbestos exposure. Even though its use has been banned in the industrialized countries, the cases continue to increase. MPM is a lethal cancer, with very little survival improvements in the last years, mirroring very limited therapeutic advances. Platinum-based chemotherapy in combination with pemetrexed and surgery are the standard of care, but prognosis is still unacceptably poor with median overall survival of approximately 12 months. The genomic landscape of MPM has been widely characterized showing a low mutational burden and the impairment of tumor suppressor genes. Among them, BAP1 and BLM are present as a germline inactivation in a small subset of patients and increases predisposition to tumorigenesis. Other studies have demonstrated a high frequency of mutations in DNA repair genes. Many therapy approaches targeting these alterations have emerged and are under evaluation in the clinic. High-throughput technologies have allowed the detection of more complex molecular events, like chromotripsis and revealed different transcriptional programs for each histological subtype. Transcriptional analysis has also paved the way to the study of tumor-infiltrating cells, thus shedding lights on the crosstalk between tumor cells and the microenvironment. The tumor microenvironment of MPM is indeed crucial for the pathogenesis and outcome of this disease; it is characterized by an inflammatory response to asbestos exposure, involving a variety of chemokines and suppressive immune cells such as M2-like macrophages and regulatory T cells. Another important feature of MPM is the dysregulation of microRNA expression, being frequently linked to cancer development and drug resistance. This review will give a detailed overview of all the above mentioned features of MPM in order to improve the understanding of this disease and the development of new therapeutic strategies.
Malignant pleural mesothelioma (MPM) is an aggressive malignancy of the pleural lining with limited treatment options. It is strongly associated with exposure to fibrous material such as asbestos. Due to the long latency period of up to 40 years and the ongoing use of asbestos in developing countries, the cases are still rising. Patients with MPM have a very short median overall survival of around 12 months after diagnosis and are treated with a combination of surgery, radiotherapy and chemotherapy. Pharmacological treatment has not changed for years consisting in the combination of cisplatin with pemetrexed and/or bevacizumab in some cases. Studies performed so far deciphered the genomic, transcriptional and epigenomic landscape of MPM, highlighting a complex and not yet known scenario. Very recently, the combination of two immune checkpoint inhibitors showed an improvement in overall survival compared to standard chemotherapy in first line. Nevertheless, current therapies have not improved, there is no second line therapy available and inclusion into clinical trials is currently the best option. The tumor microenvironment (TME) of mesothelioma consists of a wide variety of innate and adaptive immune cell subtypes, stromal and endothelial cells and has been characterized as a highly inflammatory TME favoring treatment with immune checkpoint inhibitors. On the other hand, mesothelioma is considered a non-immunogenic cancer due to a low tumor mutational burden and paucity of activated T cells. Thus, the understanding of the crosstalk and interactions of immune, stromal and tumor cells is of major importance for the development of novel therapies and the discovery of new therapeutic targets.
Inhaled mineral fibers traveling to the visceral pleura and deposition of mineral fibers in the pleural lining leads to a permanent innate stimuli with subsequent chronic inflammation, production of oxygen radicals and necrotic cell death of mesothelial cells. Asbestos fibers are biopersistent and non-degradable, which plays an important role in their carcinogenic potential (1). Mesothelial cells exposed to asbestos fibers secrete C-C chemokine ligand 2 (CCL2), which attracts macrophages to the site (2). Reactive-oxygen species induce DNA damage and mutations in mesothelial cells (3) leading to necrotic cell death and to the production and release of damage-associated molecular patterns (DAMPs) including High Mobility Group Box 1 protein (HMGB1). HMGB1 is translocated from the nucleus to the cytoplasm and secreted into the extracellular space, where it can bind to its receptors TLR2, TLR4 and receptor for Advanced Glycation Endproducts (RAGE). The binding of HMGB1 to mesothelial cells enhances their proliferation and migration capacity. The release of HMGB1 also promotes autophagy, allowing a higher fraction of mesothelial cells to survive asbestos exposure. HMGB1 silencing was shown to inhibit autophagy and to increase asbestos-induced mesothelial cell death, thereby decreasing asbestos induced transformation (4) (Figure 1). The importance of HMGB1 in cancerous transformation was also studied in a mouse mesothelioma model, where the investigators demonstrated that inhibition of HMGB1 binding to its receptors led to decreased tumor growth (5, 6) again pointing out the importance of this mediator in MPM progression. In addition, serum concentrations of HMGB1 are also significantly higher in mesothelioma patients compared to healthy controls, indicating its significance in tumor development (7).
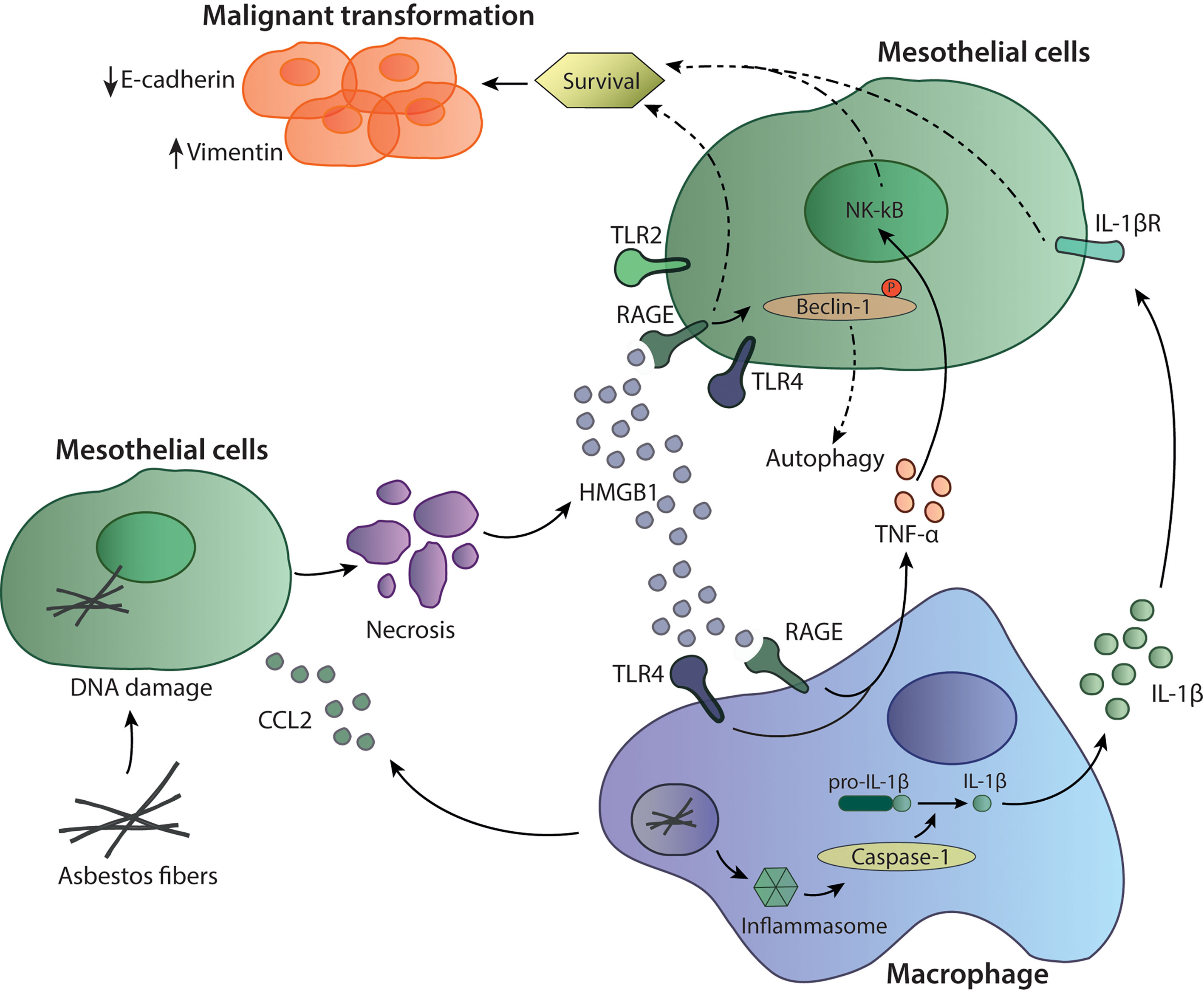
Figure 1 Mechanisms of asbestos-induced carcinogenesis. Asbestos fibers reach the mesothelial cells where they can induce cell death and the release of inflammatory mediators such as HMGB1 and CCL2. The recruited macrophages are activated through HMGB1 binding to TLR4 and RAGE to induce TNF-α or by inflammasome activation through asbestos fibers. Activation of caspase-1 and cleavage of pro-IL-1β to the active form IL-1β can lead to further survival signals in mesothelial cells. HMGB1 can also bind to TLR4 and RAGE expressed on mesothelial cells supporting survival of those cells.
Recruited macrophages phagocyte asbestos fibers leading to secretion of proinflammatory mediators such as TNF-α, supporting carcinogenesis and cancer cells survival (8). On the other hand, asbestos itself can also activate the inflammasome, a multiprotein complex part of the innate immune system, leading to activation of caspase-1 and cleavage of pro-IL-1β to IL-1β (9). IL-1β released by tumor-associated macrophages (TAMs) and its binding to IL-1R on mesothelial cells can be part of the malignant phenotype inducing cell survival and proliferation (10). Furthermore, production of TNF-α and IL-1β by macrophages can also be induced through the inflammatory environment and the presence of extracellular HMGB1, which protects mesothelial cells from asbestos-induced cell death (11, 12). TNF-α released by macrophages signals through NF-κB in mesothelial cells and supports their survival to asbestos exposure (13). Thus, TNF-α and IL-1β are important players in the transformation of non-tumorigenic mesothelial cells (14). Interestingly, HMGB1 was also shown to play an important role in epithelial to mesenchymal transition (EMT) as it led to upregulation of the EMT markers vimentin and α-smooth muscle actin (12). HMGB1 can also reduce expression of E-cadherins, an epithelial marker and upregulates mesenchymal markers promoting EMT (12). Altogether, this indicates the importance of HMGB1, TNF-α and IL-1β in mediating mesothelioma malignant transformation and progression (Figure 1).
The mesothelioma tumor microenvironment consists of a complex structure of stromal cells, immune cells and vasculature. All of these components result in a heterogeneous plethora of possible mesothelioma phenotypes, making this disease very difficult to treat. The immune compartment is characterized by the presence of many regulatory and inhibitory cells such as regulatory T cells, type 2 macrophages and myeloid-derived suppressor cells (MDSC). Immune infiltrates also include T and B cells, NK cells, dendritic cells (DC) and neutrophils.
Innate lymphoid cells (ILC) such as natural killer (NK) cells can be found in mesothelioma tumors, however, in a very small proportion (15, 16). NK cells are innate immune cells and belong to the innate lymphoid cell family (17), with high cytotoxic capacity and without the need for antigen-specific stimulation (18). NK cells are often characterized to have impaired effector functions in different solid tumors due to local immunosuppressive microenvironment leading to hampering of effector functions (19). However, little is known about the role of NK cells in mesothelioma or their possible engagement for mesothelioma therapy.
MPM tumors are infiltrated with NK cells shown by mRNA expression analysis of specific NK cell makers, which were even higher expressed in MPM compared to other cancers. However, the presence of the NK cell markers was not linked to better overall survival (20).
Similar results were obtained with staining for CD56 by immunohistochemistry (IHC) of a tissue-microarray in both epithelioid and non-epithelioid subgroups (16). In addition, different studies report different results about the expression of inhibitory or activating molecules on NK cells. A study by Nishimura et al. described that NK cells isolated from the blood of mesothelioma patients had lower cytotoxic activity compared to NK cells from healthy individuals and showed a reduced expression of the activating receptor NKp46 but normal levels of another activating receptor NKG2D (21). Another study described a higher frequency of CD56bright and a lower frequency of CD56dim NK cells in mesothelioma patients compared to healthy controls. Interestingly, treatment with anti-CTLA-4 immune checkpoint inhibitor changed this ratio from a higher frequency of CD56dim to a more physiological level of healthy controls (20).
Pleural effusion of MPM patients is often used to study the presence and function of different immune cells. NK cells isolated from pleural effusion show high expression of the immune checkpoint molecules T cell immunoglobulin and mucin-domain containing-3 (TIM-3) and lymphocyte activation gene-3 (LAG-3), whereas both molecules are expressed to the highest levels on NK cells and to a lesser extent on CD4 and CD8 T cells (22). Interestingly, LAG-3 is not expressed on MPM tumor cells (23). Here, the investigators claim that effusions are more often present in an inflammatory context, which could influence the expression of suppressive immune checkpoint molecules as they have shown that the early activation marker CD69 is significantly correlated with the expression of TIM-3. Another explanation could be that matching pleural effusion and tumor tissue do not reflect each other’s immune cell composition (24). Nevertheless, differences in effusion and tumor samples could also be due to different analysis methods (22). Furthermore, NK cells in pleural effusion from MPM patients are functional and produce high amounts of TNF-α and INF-γ upon stimulation (25) but have also an impaired expression of perforin, which can be restored by IL-2 stimulation in vitro. However, incubation of NK cells with pleural effusion completely abrogated the activation status of the NK cells, indicating the presence of inhibitory cytokines in the pleural effusion (26) (Figure 2). Similar results were obtained in a study performed by Vacca et al., they also described NK cells from pleural effusion from different cancer patients (including mesothelioma patients) as functionally capable to produce cytokines, perforin and granzyme A and B and to perform cytotoxic functions upon in vitro stimulation with IL-2. Furthermore, NK cells express normal levels of activating receptor including NKp30, NKp44, NKG2D, and DNAM‐1 after stimulation. This suggests a possibility for reactivation of NK cells and no expression of an anergic phenotype as described in other studies (27). However, the functional capacities of NK cells in human tumors were not investigated and it is currently unclear if they are in a state of exhaustion or can perform effector functions normally. A mouse syngenic mesothelioma model using the AE17 cell line, reveals that depletion of NK cells with an anti-asialo GM1 antibody did not influence tumor growth (28). Current data about NK cells in MPM tumors does not correlate to overall survival, nevertheless, more data is needed to understand their functional effector capacity and their exhaustion profile intratumorally and the possibility to target them with therapeutic approaches.
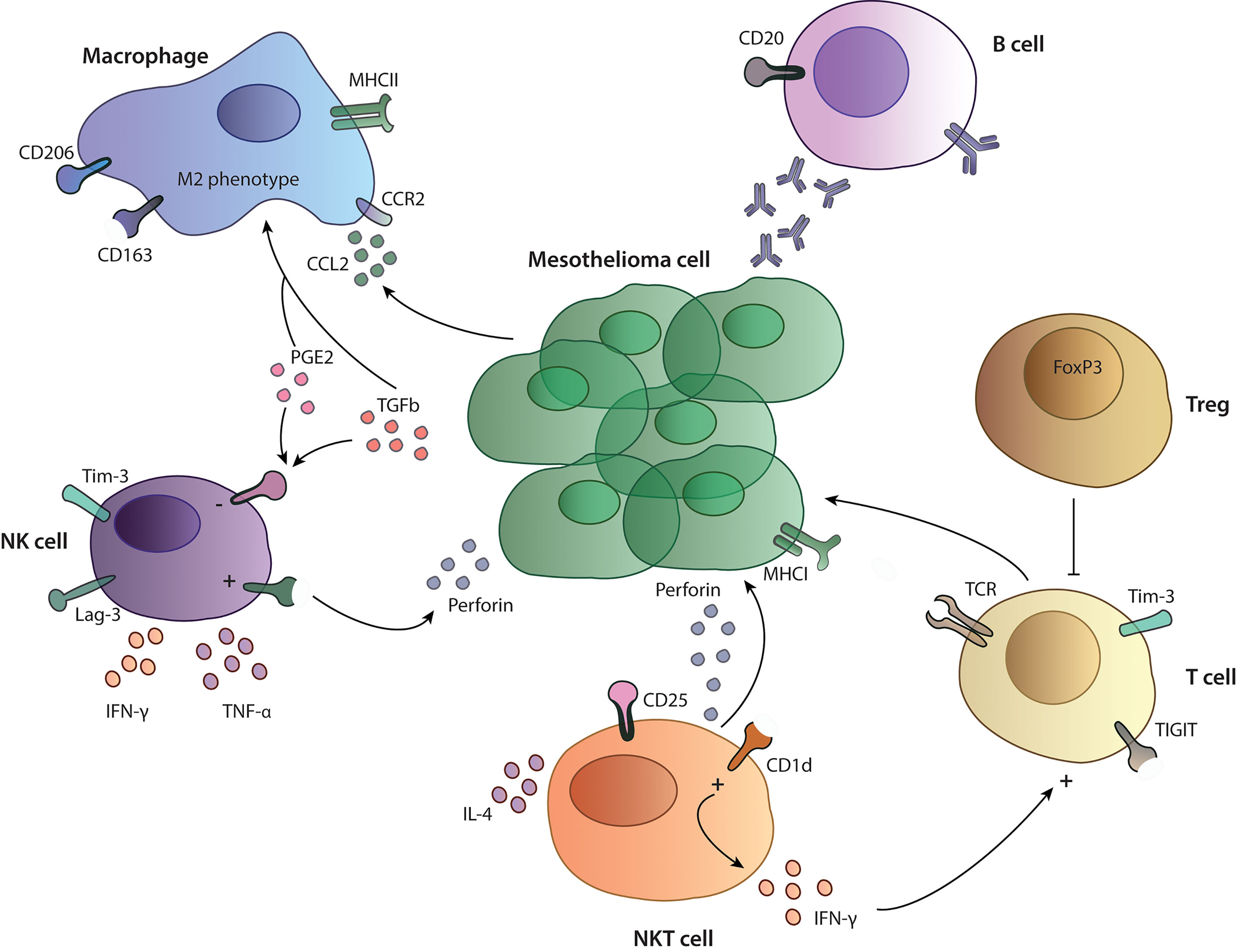
Figure 2 Tumor microenvironment in mesothelioma. Overview on the functionality and interactions of different immune cells studied in MPM patients. NK cells and T cells express inhibitory receptors such as TIM-3, LAG-3 and TIGIT and are influenced by a suppressive cytokines (PGE2, TGF-β) and the presence of Treg cells in performing their cytotoxic functions. Macrophages show a M2-like phenotype with expression of CD206 and CD163 on their surface. B cells in the TME produce specific antibodies against cancer cells, participating in the anti-tumor immune response.
Natural killer T (NKT) cells are a distinct population of T cells recognizing glycolipids presented on the non-classical class I-like molecule CD1d in contrast to normal T cells, which recognize peptide fragments presented on MHC molecules (29, 30). NKT cells have lytic activity, but their main function lies in the production and secretion of a wide variety of cytokines. Upon activation, they can produce high amounts of Th1 or Th2 cytokines, which can lead to bystander activation of NK cells, CD8 T cells and dendritic cells.
Little is known about the function of NKT cells in the tumor microenvironment of patients with MPM. Altomare et al. investigated the presence of NKT cells in the blood of MPM patients. Here, they showed that MPM patients have a higher frequency of circulating NKT cells compared to healthy volunteers, whereas there were no differences in their ability to produce IFN-γ and IL-4 (31) (Figure 2). NKT cells have been mainly studied in the context of mesothelioma mouse models and as a therapeutic target, since they can easily be activated by artificial glycolipids. In the pleural effusion of a MPM mouse model, NKT cells are present, express high levels of the activation marker CD25 and produce large amounts of INF-γ. In the same model, activation of NKT cells through administration of glycolipids led to prolonged survival in the treated groups (32, 33), indicating that these cells have an anti-tumor phenotype and can activate other cytotoxic cells. More studies are needed in order to understand the possibility to use them as a therapeutic target in MPM.
Macrophages are innate immune cells specialized in phagocytosis, engulfing and digestion of invading organisms and cell debris and play an important role in tissue homeostasis. Monocytes are recruited from the blood to the TME through locally produced chemokines and become TAMs and, in patients’ blood, increased amounts of circulating monocytes and a low lymphocyte to monocyte ratio have been reported to negatively correlate with overall survival (34, 35). TAMs can be divided into two subset depending on their function and marker expression. M1 macrophages are proinflammatory macrophages and have strong capacity to kill invading pathogens and contribute as well to tissue destruction. M2 macrophages are important mediators in tissue remodeling, allergic diseases and angiogenesis. Nevertheless, macrophages are a functionally diverse and plastic group and can reverse their polarization from M2 to M1 depending on the chemokine environment (36–38).
Various cytokines can induce differentiation of monocytes and macrophages into TAMs, such as CCL2, C–C chemokine ligand 4 (CCL4), C–C chemokine ligand 5 (CCL5) and C-X-C motif chemokine ligand 12 (CXCL12) secreted by MPM cells (39, 40). CCL2, the most studied TAM-associated chemokine, recruits immune cells such as T cells, macrophages and dendritic cells through binding to CCR2 on the cells to the inflammatory site (41). CCL2 is upregulated in pleural effusion and serum from MPM patients compared to benign pleural effusion or pleural effusion from other malignancies and serum from healthy volunteers, respectively (42–44). Interestingly, CCL2 levels also correlate with the tumor stage, indicating an important role of macrophages in disease progression (42). Other chemokine receptors like CXCR1, CXCR4, CCR5, and CCR7 are infrequently expressed on mesothelioma cells isolated from pleural effusion of MPM patients (45). Therefore, those chemokine pathways might only play a role in a subset of MPM patients.
Activation of the colony stimulating factor-1 receptor (CSF-1R) through M-CSF or IL-34 can induce differentiation of monocytes to macrophages (46). Incubation of monocytes with pleural effusion from MPM patients or supernatant from MPM cell cultures resulted in a CD14midCD163high M2 immunosuppressive macrophage phenotype (47) (Figure 2). It has been shown that pleural effusions from MPM patients contain M-CSF and that they can induce differentiation of monocytes to M2-like macrophages in a CSF-1R dependent manner (48). M2 macrophages themselves can induce proliferation of MPM cells and induce treatment resistance to chemotherapies (47). Another study from Cioce et al. showed that autocrine CSF-1R signaling through AKT and β-catenin is a crucial signaling pathway for chemotherapy resistance and survival (49). Thus, the CSF1R/AKT axis represents an interesting target for further therapeutic development.
Other important factors for the differentiation of monocytes and macrophages like prostaglandin E2 (PGE2) and Transforming Growth Factor β (TGF-β) are present in pleural effusion and in the supernatant of MPM tumor cell lines (50, 51). PGE2 is an immunosuppressive factor in the TME and can induce a suppressive phenotype in macrophages with high suppressive capacity on T cell proliferation (52). The human monocytic cell line THP-1 activated with lipopolysaccharide (LPS), developed an immunosuppressive phenotype when co-cultured with the MPM cell line Mero84. An increased production of immunosuppressive cytokines like PGE2 and IL-10 was produced by macrophages with a shift towards M2 phenotype (53).
A very close interplay has been described between the production of TGF-β and priorities of macrophages in several settings. TGF-β is a critical cytokine in tissue homeostasis and can have pleiotropic functions in cancer. It can inhibit proliferation of cancer cells but also induce tumor progression and metastasis, thereby function as a tumor promoting cytokine (54). TGF-β concentrations in pleural effusions are significantly higher in MPM patients compared to those from primary lung cancer patients and they correlate with disease stage and tumor volume (55–58). Patients with high TGF-β concentrations in pleural effusions have significantly shorter survival, however, circulating serum TGF-β concentrations do not have a predictive value (58).
Upon production of the above mentioned chemokines, monocytes then become TAMs. TAMs have been widely described to express a pro-tumoral M2 phenotype, but recent studies suggest that they might have M1 and M2 properties at the same time (38, 59). M2 macrophages are considered to promote tumor growth, proliferation and invasiveness. Increased TAM levels correlate with poor survival, bad prognosis and increased metastasis potential in different tumors (60–63). In the human MPM tumor microenvironment, TAMs account for the majority of tumor infiltrating cells with about 25-40% of total immune infiltrates (23, 64). TAMs in MPM express an immunosuppressive M2 phenotype with high levels of the surface molecules CD163, CD206 and Interleukin 4 receptor α. Independent on the histological subtype, MPM is generally heavily infiltrated with macrophages without any correlation with tumor stage, but interestingly with survival in the non-epithelioid group (34) (Table 1). This could be due to presence of more immunosuppressive cytokines supporting the pro-tumoral role of infiltrated macrophages. A study by Marcq et al. showed a correlation between CD68+ macrophages with the presence of CD4+FoxP3+ regulatory T cells, accounting for a downregulation of the adaptive immune response and support of an immunosuppressive TME, which could explain partially the difference between the prognostic differences in survival (23). Nevertheless, the prognostic value of macrophages and in particular M2 macrophages has led to divergent conclusions, depending on the different studies and datasets, while certain studies report a significance others do not (16, 34, 65–67) (Table 1). In order to further dissect the role of these cells in MPM, an orthotopic mouse model of MPM was used, where a tumor promoting effect of macrophages was described: mice with a high tumor burden had higher numbers of macrophages/monocytes in the pleural effusion as well as higher percentages of M2 suppressive macrophages (71). In another orthotopic, syngenic murine peritoneal mesothelioma model, the tumor burden, measured by tumor growth rates, invasiveness and number of metastasis was significantly reduced when macrophages were depleted in these mice (72). Both studies indicate TAMs as a negative prognostic factor for tumor progression in mice.
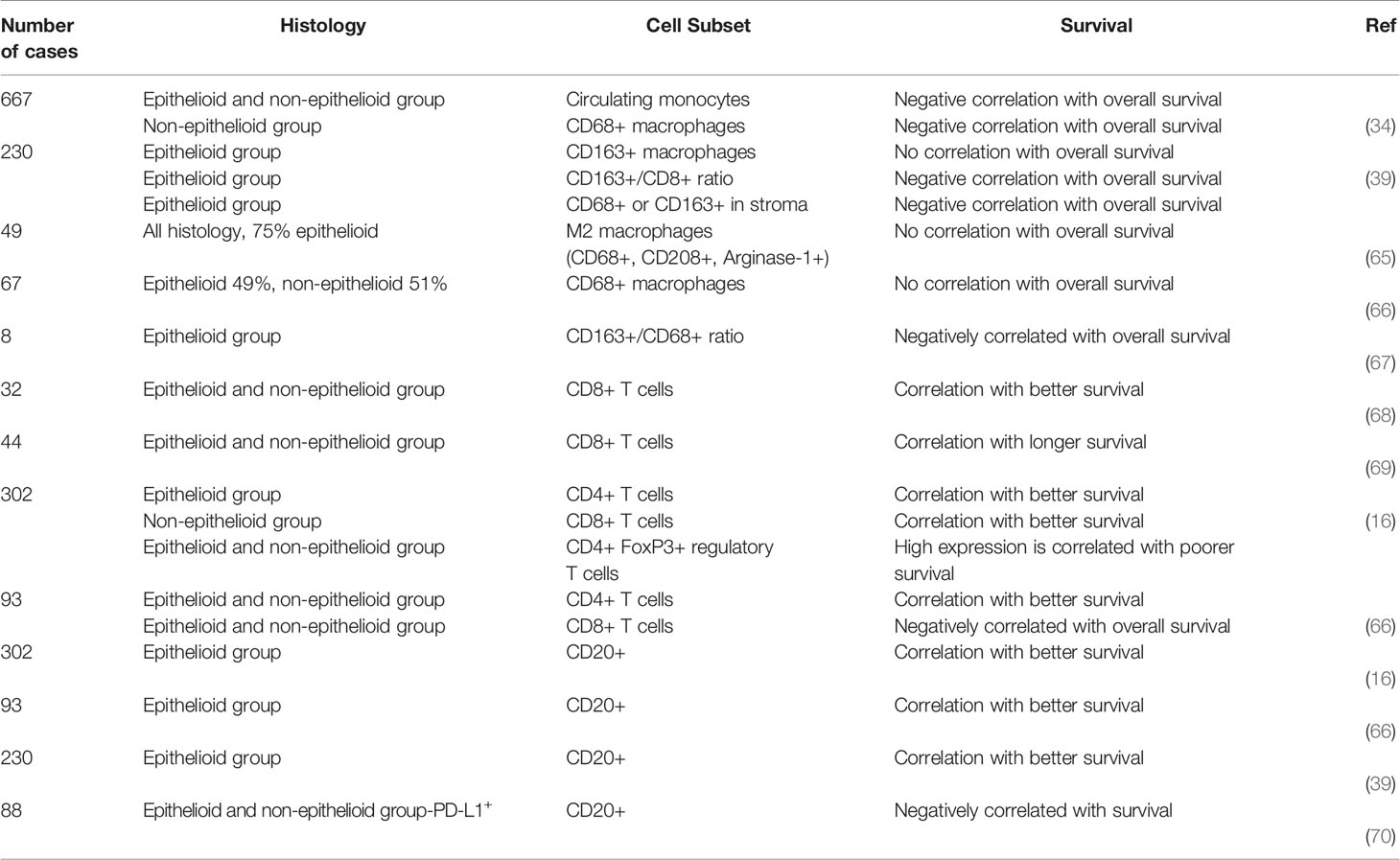
Table 1 Summary of publications of overall survival correlated with immune infiltrates.
In conclusion, MPM seems to alter the myeloid cell differentiation program by tumor-derived factors, which contributes to tumor suppression and a pro-tumoral immune response. TAMs and monocytes could be a potential target to alter this effect and induce an anti-tumoral immune response.
Myeloid-derived suppressor cells (MDSC) represent a pathological status of monocytes and neutrophils and are present in different pathological conditions. MDSC represent a small proportion of tumor infiltrating cells in MPM, below 10%, but with important pro-tumoral functions (65). MDSC promote tumor development and progression through different mechanisms, they have the ability to suppress T cells, remodel the TME, support EMT and angiogenesis (73, 74). MDSC are a heterogeneous group of myeloid cells but can be roughly divided in two groups, the granulocytic (Gr-MDSC) and the monocytic (M-MDSC) subset (74).
In MPM, the two populations of MDSCs can be found in the TME; the granulocytic like subtype expressing CD15high/CD33low and the monocytic like subtype expressing CD15low/CD33high (15, 67, 75, 76). Gr-MDSCs are recruited to the tumor site through G-CSF and GM-CSF released by MPM tumor cells and further differentiate into an immunosuppressive phenotype within the tumor (75, 77, 78). In MPM, MDSCs inhibit proliferation of CD8+ T cells through secretion of immunosuppressive molecules like reactive oxygen species (ROS), nitric oxide (NO) and kynurenine (75, 79, 80). High amounts of tumor infiltrating MDSCs significantly decrease progression free survival and overall survival in MPM patients (65).
Targeting MDSC in murine model of mesothelioma leads to reduced numbers of intra-tumoral MDSC with reduced capability to produce ROS and to reduce tumor growth (81). Targeting MDSC in MPM patients might represent a way to reduce intra-tumoral immune suppression and enhance immunotherapy regimes in future.
T cells play an important role in anti-cancer immunity in solid tumors and overall survival is closely linked to the presence of tumor infiltrating lymphocytes (TILs) across different tumor types (82). The approval of immune checkpoint inhibitors targeting CTLA-4 or the PD-1/PD-L1 interaction and their enormous clinical success in different malignancies (83, 84) further points out the importance of T cells in controlling cancer cells. In MPM CD3+ T cells are highly abundant in the TME and the presence of CD8+ TILs is a favorable marker for prognosis (68, 69). However, 60 to 80% of the cases analyzed are usually MPM tumors with an epithelioid subtype, and only few cases with the most aggressive sarcomatoid form are included. Sarcomatoid tumors show fewer CD4+ and CD8+ T cells in the tumor and are characterized by a loss of Th1 features such as T-bet (marker for Th1 polarization) and granzyme B expression, which are required for an efficacious anti-tumor immune response. In addition, the sarcomatoid subtype does not express HLA class I molecules leading to escape of T cell mediated cytotoxicity (17). Another study compared the epithelioid group with a non-epithelioid group (sarcomatoid and biphasic) and linked immune markers with outcome: high CD4+ counts in the epithelioid subset was associated with better prognosis, in contrary, in the non-epithelioid subsets, high CD8+ counts were associated with better prognosis, in both, high expression of FoxP3 was correlated to poorer survival (16, 66) (Table 1). Infiltration of T cells also varies between PD-L1 high and low expressing tumors. PD-L1 high tumors have more CD45+ cells infiltrated compared to PD-L1 low tumors, and significantly more CD3+ cells including CD4+ and CD8+ and regulatory T cells and express more co-inhibitory receptors such as TIM-3. Nevertheless, there is also a huge variability between patients, which could account for the differences in responses to immunotherapies (15, 85). Thus, the presence of TILs and the expression of PD-L1 are not sufficient to predict responses to such therapies, but rather the whole immune-context, including the presence of suppressive cells and inhibitory receptors might predict outcome. Other important factors for response to immunotherapy in MPM patients are the effector functions of TILs. On the one hand, cytotoxic T cells in MPM express more T cell immunoglobulin and ITIM domain (TIGIT) and TIM-3 compared to T cells from health lung tissue and had a minor ability to produce IFN-γ upon stimulation (86) (Figure 2). On the other hand, patients with MPM do have approximately double the amount of regulatory T cells (Tregs) cells in the periphery compared to healthy control. Tregs are important in sustaining peripheral tolerance and preventing autoimmune disease by suppressing other cells. The balance between effector T and B cells and Tregs is crucial for the quality and magnitude of the immune response. Nevertheless, the presence of Tregs can also block required anti-tumor immune responses (87) and their presence in tumors is associated with poorer prognosis in different malignancies (88). In a study from Klampatsa et al. around 12.8% of all CD4+ T cells in the MPM tissue are positive for FoxP3 compared to 2.2% from healthy lung tissue. In addition, there were significantly more Tregs in the tumor compared to the blood (86, 89). In a murine MPM model Tregs were also shown to be crucial for cancer progression, depleting Tregs with an anti-CD25 antibody led to reduced tumor growth (90).
Immune checkpoints have drawn attention during the last years due to the development of antibodies blocking the interaction of PD-1 and PD-L1 and its extraordinary success in cancer therapy. The PD-L1/PD-1 axis leads to inactivation of T cells. In the context of cancer, T cells are continuously exposed to tumor antigens, which leads to a state of dysfunctionality and unresponsiveness called exhaustion. Blocking the PD-L1/PD-1 axis with therapeutic antibodies reactivates T cells against cancer. PD-L1 is expressed in mesothelioma tumors, however, the positivity rate very much depends on the study, the cohort and the assay performed for analysis (23, 91–93). A recent overview analysis of four different antibodies used to stain for PD-L1 gave an incoherent picture over the different assays. The use of reliable antibodies and standardization of staining methods are important features in order to receive comparable studies (94). In MPM, high PD-L1 expression is associated with histology and is higher expressed in sarcomatoid/biphasic subtypes (66, 92, 95). In the recently published PROMISE-MESO trial, 48.9% of the patient had less than 1% PD-L1 expression, 28.2% had 1-20% and 18.5% more than 20% PD-L1 expression (96). Another study described 73% to be positive (> 1%) and 27% negative (<1%) (97). Inaguma et al. describe 33% of MPM to be positive and 67% negative. In addition, high PD-L1 expression is negatively correlated with overall survival (85, 95, 98). Importantly, PD-L1 expression is not only restricted to TME but also on tumor cells of the pleural effusions (99); therefore, further investigations with detailed analysis of abundance and localization of expression are warranted.
Recent studies have identified a wide range of other immune checkpoint molecules beside PD1/PD-L1, which could be suitable for cancer treatment, in particular TIM-3, LAG-3, TIGIT and V-domain Ig suppressor of T cell activation (VISTA). TIM-3 plays a major role in controlling the function of NK and T cells. Upregulation of TIM-3 on peripheral immune cells and its cognate ligand galectin-9 on tumor cells inhibits immune responses. Galectin-9 is expressed on MPM tumors where it can suppress T cell response (65). Furthermore, a higher number of TIM-3+ cells in peripheral NK and T cell populations correlate with a poor prognosis in many solid tumor types (20). In MPM, T cells in pleural effusion express inhibitory molecules such as PD-1, TIM-3, LAG-3 and have a higher diversity of TCR clones compared to blood of the same patient (65, 100). Interestingly, the LAG-3 gene is higher expressed on mesothelioma tumors compared to lung adenocarcinoma, while PD-L1 gene is higher expressed is lung adenocarcinoma (101). Understanding differences in the TME of various solid tumors can open up new options for more personalized immunotherapeutic approaches. A recent study defines a subgroup of patients co-expressing inhibitory molecules TIM-3, PD-L1, CTLA-4 and LAG-3 where this expression is associated with a shorter survival (102). Another immune checkpoint molecule, VISTA, is a negative regulator of T cell activation and it is highly expressed on myeloid cells. Similar to PD-L1, VISTA can support the conversion of naïve T cells to FoxP3+ regulatory T cells (103, 104). In MPM, VISTA is highly expressed on the epithelioid subtype but to a lesser extent on more aggressive subtypes. Interestingly, it is also expressed on normal and reactive mesothelium (105). In contrary to PD-L1 patients with high VISTA expression had a better overall survival, moreover, patients with concurrent high expression of VISTA and VEGFR2 survive almost five times longer compared to patients with low expression (102, 106).
B cells are essential cells of the adaptive immune system and function as antigen presenting cells (APC) thereby contributing to T cell activation, differentiation and polarization. B cells also play an important role in promoting the formation of tumor-associated tertiary lymphoid structures (TLS), areas for B cell maturation and isotype switching (107). The presence of B cells in the tumor can be a prognostic factor in different malignancies (108). In MPM, only few studies addressed the function and importance of B cells in the tumor and in pleural effusion. Krishnan et al. showed high levels of tumor-specific antibodies in murine models of mesothelioma treated with immunotherapy compared with untreated controls. In addition, disease eradication in all treated animals and complete failure of the treatment in B cell-deficient mice have been demonstrated (109). These findings line up with previous preclinical data, which showed increased levels of IgM and IgG after anti-CD40 antibody treatment and during tumor regression in mice (110). This data suggests that antibodies generated upon treatment play an important role in the tumor immunity and are essential for tumor responses (Figure 2). Besides high levels of antibodies, Jackman et al. showed an increased percent of B cells in the tumor as well as in the secondary lymphoid organs. In contrast, the number of T cells and of the other cells of the immune system (e.g., macrophages, NK, granulocytes) remained low (110). In a previous study, high antibody titers against four tumor-associated antigens (GeneX, THBS-2, STUB-1 and IFT88) were identified in the sera of MPM patients. In particular, high levels of antibodies against two of those antigens, GeneX and THBS-2, were detected in almost all MPM patients, with a decrease after surgical resection (111). These findings represent another example of the existence of a specific humoral immune response in MPM patients: antibodies produced by tumor infiltrating B cells can be used as a tumor marker for diagnosis and follow up. The association of high B cell numbers in the tumor with survival has been recently described. Different studies showed that high counts of CD20+ cells in patients with epithelioid mesothelioma positively correlated with survival, however, this was not the case in the non-epithelioid subgroup (16, 39, 66) (Table 1). In contrast, another study identified elevated B cell numbers in the sarcomatoid subgroup (70). In addition, in PD-L1neg MPMs B cells are considered a good prognostic factor, whereas in PD-L1+ MPMs, CD20+ infiltrates are associated with a poorer outcome (70) (Table 1). Patil et al. classified MPM cases into three different subgroups, based on immune profiles: one subgroup showed higher expression of B cell markers and antigen presentation-related genes compared with the other ones (91). This indicates that B cell infiltration is not a constant feature in all MPM cases and cannot be considered as a hallmark as, sometimes it is rarely detectable (76). Nevertheless, in some patients increased levels of B cells constitute a window of opportunity to develop novel immunotherapies and to identify novel MPM targeting receptors. Through bulk RNA-Seq data analysis from MPM tissue, the BCR sequence can be identified to generate candidate antibodies binding to MPM target cells. Another new approach to produce high-affinity antibodies involves the isolation of memory B cells from peripheral blood of the donors, when immortalized, such B cells stably secrete monoclonal antigen-specific antibodies, which could be used as further therapeutic agents (112). More research is needed to better understand the role of B cells in MPM but based on these premises, this cell population can be considered an important candidate for the development of new therapies.
As previously described, the role of external agents in inducing mesothelioma is mainly attributable to the chronic inflammation guided by HMGB1, NF-kB and the PIK3CA pathway. Meanwhile, clinical studies of large cohorts of individuals and the improvement of genome wide sequencing technologies have helped the identification of an increased number of oncological diseases associated with germline mutations (113). These genes mainly encode for tumor suppressor proteins involved in cell cycle regulation, apoptosis and DNA repair pathways; being involved in cancer development, they are called cancer susceptibility genes (113). The identification of these genes have paved the way for the development of targeted therapeutic approaches as well as for cancer prevention and surveillance (114, 115). The most common inherited cancer risk factor associated with MPM is the aberration of BRCA1 Associated Protein 1 (BAP1), a deubiquitinating enzyme located on chromosome 3p21.1 acting as tumor suppressor gene (116, 117). BAP1 is involved in different biological pathways, such as DNA replication, apoptosis, regulation of gene transcription, deubiquitation of histones and DNA repair (116, 117). Germline defects of BAP1 are responsible for the BAP1-tumor predisposition syndrome (BAP1-TPDS) including the occurrence of renal cell carcinoma, uveal melanoma, cholangiocarcinoma and mesothelioma. High frequency of germline mutations of BAP1 were demonstrated to cause mesothelioma in 2001, when an epidemic spread of cases was reported in a village in Cappadocia (113, 118). These results were further confirmed by other groups: in 2018, Betti et al. reported a frequency of 7.7% of pathogenic germline variants in a cohort of 39 patients (119), while Pastorino et al. reported a frequency of 30.7% in a cohort of 52 patients affected by malignant mesothelioma (120).
BAP1 alterations occur in one mutant allele and are inherited as autosomal dominant mutations: a study of germline-mutated mesothelioma showed 43.1% of relatives carried the same mutation of their probands (120). The peculiarity of mesotheliomas carrying BAP1 defects is a higher frequency in non- or low-exposure to asbestos as reported by Pastorino et al (120). These alterations are more frequently detected in young adults with MPM: 4% in patients older than 75 years and 20% in the ones of 55 years of age or younger (121). Among the three histotypes of MPM, such as epithelioid, sarcomatoid, and biphasic, BAP1-germline mutations are found more frequently in the epithelioid and this correlates with a better prognosis (113, 115).
Today, the assessment of BAP1 status has become part of the diagnostic routine of mesothelioma allowing to distinguish between benign and malignant mesothelial cells and to identify biphasic mesothelioma (116). However, the implementation of sequencing technologies with more extensive studies will uncover molecular features associated with mesothelioma onset. Interestingly, a recent study has revealed that BAP1 is not the only cancer-susceptibility-gene predisposing to mesothelioma. Bononi et al. have shown that heterozygous mutations in the Bloom syndrome gene (BLM), a gene involved in the DNA repair, promote the development of mesothelioma and the risk is further increased by exposure to asbestos (122).
These results suggest the importance of early detection of cancer risk factors in the population via tailored screening programs in order to prevent cancer development.
The DNA damage response (DDR) consists of a complex network of genes that respond to different types of DNA damages, such as Double Strand Breaks (DSBs) and Single Strand Breaks (SSBs), being organized in pathways as homologous recombination (HR), non-homologous end-joining (NHEJ), mismatch repair (MMR) and nucleotide excision repair (NER). Defects in one or more pathways lead to genomic instability and promote tumorigenesis and cancer progression (123, 124).
DDR has been an attractive therapeutic target upon the discovery of synthetic lethality, which occurs when the inefficiency of a DNA repair system causes the recruitment of other DNA damage pathways. This concept has first been applied to HR genes like BRCA1/2 due to their interaction with the poly(ADP-ribose) polymerase (PARP), a family of proteins that are activated in the presence of DNA damage and stabilize the replication machinery during repair (123). Thus, a new category of drugs called PARP-inhibitors (PARPi) have been developed to target the rescue of DNA repair pathway and lead to genomic instability and cell death (123). Among the most studied PARPi there are talazoparib, rucaparib, niraparib, olaparib and veliparib; although being first intended to target tumor lacking functional BRCA1/2, their action has been expanded to a larger category of Homologous Recombination Deficiency (HRD) tumors, that comprise other HR genes like ATM, ATR, RAD51, BARD1 (124).
Evidence reported in several works has shown a high percentage of germline mutations in MPM occurring in DNA repairing genes. Betti et al. tested a panel of 94 cancer predisposing genes and found mutations in PALB2, BRCA1/2, FANC1, ATM, SLX4, FANCC, FANCF, PMS1 and XPC covering almost the 10% of all tested patients (118). Most of these genes were involved in specific DNA repair mechanisms like HR, MMR and NER. A similar result was further confirmed by Panou et al. that reported an improved survival in patients with MPM bearing DNA repairing defects (121). A study by Guo et al. addressed the role of DNA repair genes in the pathogenesis of MPM and identified mutations in novel target genes like MSH3, BARD1 and RECQL4 that have not been previously described (125). A recent review by Fuso Nerini et al. confirmed that considering different studies performed on MPM, the DNA repair pathways are among the most frequently affected (124).
In this context, the use of PARPi in mesothelioma has been encouraged, however preliminary data do not allow a clear conclusion of PARPi effectiveness (124) (Table 2). In fact, while some studies have shown combinatorial treatment with cisplatin and olaparib is effective in mesothelioma cells with a defective HR (137), another study has demonstrated that olaparib has limited anti-tumor activity also in BAP1 mutated patients (NCT03531840). Other clinical trials are still ongoing (NCT03207347, NCT03654833) and will help clarify the effects of PARPi in mesothelioma.
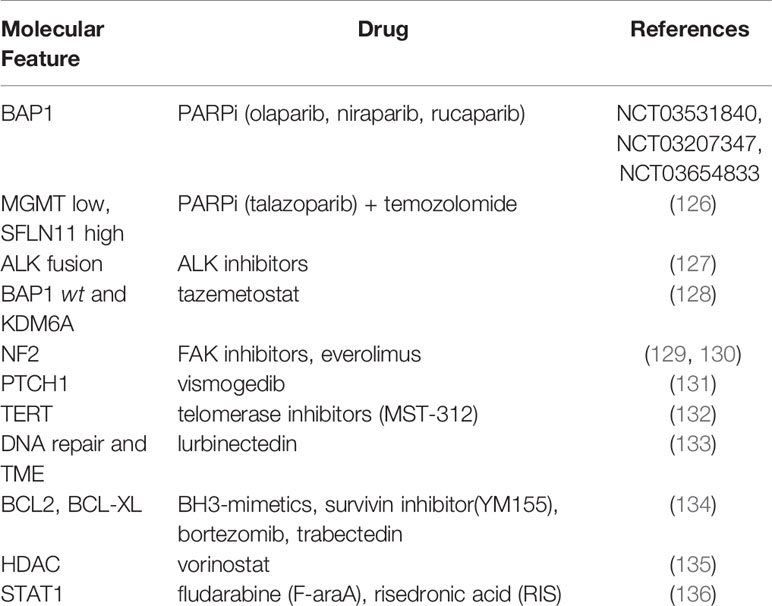
Table 2 Druggable targets in mesothelioma.
One of the main concern about DNA damage and mesothelioma has been the assessment of the role of BAP1 in sensing cells to PARPi. The role of BAP1 in the DDR is due to its interaction with BRCA1 and BARD1, however this association is far to be completely understood and need further investigation (116). A recent study exploited the sensitivity to PARPi in patient-derived mesothelioma cells (126). They showed that response to PARPi is independent on BAP1 mutational status. Conversely, they demonstrated that PARPi sensitivity, especially to talazoparib in combination with temozolomide, is mainly related to the combination of low expression levels of O-6-Methylguanine-DNA methyltransferase (MGMT) and high expression levels of Schlafen 11 (SLFN11) (126) (Table 2). Overall, this evidence suggests that targeting the DDR in MPM is still an attractive strategy, most of all in a context of combined therapy; however, more preclinical studies are needed to exploit other combinations and unravel molecular mechanisms and drug-interactions that could lead to improved patients outcome.
The -omics field in the study of cancer pathology has evolved rapidly in the last decades. The improvement of high-throughput technologies and computational approaches have made a big step forward in cancer characterization and drug-response investigation becoming crucial in the context of translational research.
Genomic and transcriptomic studies have improved the molecular characterization of MPM and set new hypothesis for therapeutic approaches. The first genomic studies from Bueno et al. in 2016 analyzed 211 transcriptomes and 216 whole exomes of mesotheliomas, while Hmeljak et al. in 2018 analyzed 74 samples from The Cancer Genome Atlas (TCGA) by the integration of the exome and the transcriptome (105, 138). Both studies confirmed frequent mutations in the CDKN2A, NF2, TP53, LATS2, and SETD2 genes and a low mutational burden with less than two non-synonymous mutations per megabase (Mb). Bueno et al. identified other genomic aberrations such as gene fusions and splice alterations in the most relevant genes like NF2, BAP1 and SETD2 (138). Both studies assessed a somatic copy-number alteration (SCNA) landscape with more copy losses than amplifications that included BAP1, NF2, CDKN2B, LATS2, as a further confirmation that MPM development is driven primarily by loss of tumor suppressor genes than by activation of classic oncogenic drivers (105, 138).
The pivotal role of BAP1 in mesothelioma is confirmed also at the somatic level, since 60% of cases present a second hit (120), even if the percentage could be even higher since studies performed so far have used next generation sequencing (NGS) approaches that lacked the identification of large deletions, while assessment from different platforms, like IHC and multiplex ligation–dependent probe amplification (MLPA) have increased detection perfomances (116). Somatic BAP1 mutations preferentially affect the epithelioid subtype and correlate to better prognosis (116).
Recently, a work by Zhang et al. has depicted a detailed picture of MPM genomic features. Indeed, the study of intratumor heterogeneity of MPM through an exome sequencing approach has shown that most MPMs follow a linear evolution with BAP1 being the most frequent ancestral mutation and NF2 arising mainly as a late event. Moreover, a minority of patients presented a branched evolution that was associated with a higher tumor lymphocyte infiltration and antigen burden, suggesting a possible sensitivity to immunotherapy (139).
Aberrant copy number alterations in CDKN2A and p16 arm identified with sequencing approaches were confirmed in other studies through fluorescent in-situ hybridization (FISH) and IHC and they were associated with higher asbestos fiber exposure (140).
Somatic mutations in CDKN2A, NF2, BAP1 were also reported in cases of malignant peritoneal mesothelioma, with CDKN2A less frequent as compared to pleural mesothelioma (141). Interestingly, the same work reported gene fusions such as EWSR1-ATF1 and FUS-ATF1 and ALK rearrangements that are hardly found in pleural mesothelioma and seem to be specific for young women as compared to ALK-wild type patients but might respond to targeted treatment (127, 141, 142) (Table 2). Two recent studies have reported novel somatic mutations in RDX and MXRA5 genes, independently (143, 144). In Torricelli et al. RDX and MXRA5 are present in 42% and 40.6% of the cohort, respectively and authors stated that MXRA5 is specific for the biphasic histotype together with NOD2 (143). The same genes were also described in the RAMES study where RDX and MXRA5 represented the 42% and 23% of MPMs, respectively, however in this case MXRA5 was identified in both epithelioid and non-epithelioid histotypes (144). Moreover, the same gene is reported to be significantly correlated to longer survival rate in a cohort of epithelioid only MPMs (145). Therefore, although the correlation of MXRA5 to histopathologic or clinical features needs further interpretation, more studies on this gene and RDX are warranted.
The extensive work of the genomic studies presented so far have defined various genetic features of mesothelioma, but, to date, they have no role in patient stratification and treatment. Interestingly, more insights in MPM molecular characterization have emerged from transcriptomic studies. The first molecular classification in epithelioid, sarcomatoid, and biphasic, was proposed in 2016 with the identification of 400 most variable genes within the groups, 189 upregulated and 241 downregulated, which also correlated with survival (138). Specifically, the epithelioid subtype presented up-regulation of UPK3B, ELMO3, CLDN15 while LOXL2 and VIM were up-regulated in the sarcomatoid subtype, thus showing a key difference in EMT regulation in the two groups. Using a data-integration approach Hmeljak et al. came to the same histotype classification, however, they stressed a relevant issue: since MPM transcriptomic can be used for histotype stratification, it is possible to use it even further for prognosis within a single histotype. For example, although the epithelioid histotype has a better survival rate than sarcomatoid, even within this subgroup it is possible to identify different clinical courses. Guided from this hypothesis, a cluster of patients with epithelioid subtype with poor prognosis was identified and associated with higher AURKA mRNA expression (105). Previously, the association of AURKA expression to a worse prognosis was detected by Borchert et al (137). This evidence suggests that for a further improvement in MPM therapeutic approaches it is essential to consider integrated data analysis, such as the combination of genomic and transcriptomic features.
As previously stated, mesothelioma presents as a tumor with low mutational burden with a median of 23 mutations per biopsy specimen (146) with ~1.2 mutations per Mb (105, 138). This finding identifies mesothelioma as an atypical tumor, since it is known that exposure to carcinogens and environmental pollution characterize tumors with a highly compromised genomic landscape and high mutational burden. However, latest development of sequencing technologies have revealed hidden aspects in cancer malignancies that have not been previously investigated, such as chromotripsis. The word chromotripsis derives from “chromo” which stands for chromosome and “tripsi” which means breaking into small pieces (147) and refers to a mutational phenomenon of DNA breakage from a single event that spreads into hundreds of catastrophic chromosomal damages (148). Accumulated DNA damages lead to the formation of micronuclei, usually containing single chromosome, that are disrupted during cell cycle and spread genetic material in the cells (146). Pieces of chromosomes can be included in the nuclei during mitosis, and this generates chromosome rearrangements and fusions (149) (Figure 3). Recent studies have addressed this topic in malignant mesothelioma. With the intent to provide reliable cell line models of mesothelioma, Oey et al. have characterized the genome of tumors and tumor-derived cell lines through whole genome sequencing (151). Here, the authors have identified recurrent events of high chromosomal instability like chromoanagenesis and chromotripsis. Interestingly, inter- and intra-chromosomal rearrangements affected genes like CDKN2A, one of the most frequently mutated in mesothelioma and KDM6A, a gene that has been associated with sensitivity to enhancer of zeste homolog 2 (EZH2) inhibitors, like tazemetostat (151, 153). This finding might have important implications, as BAP1-lacking mesotheliomas have demonstrated sensitivity to EZH2 inhibitors (128) (Table 2).
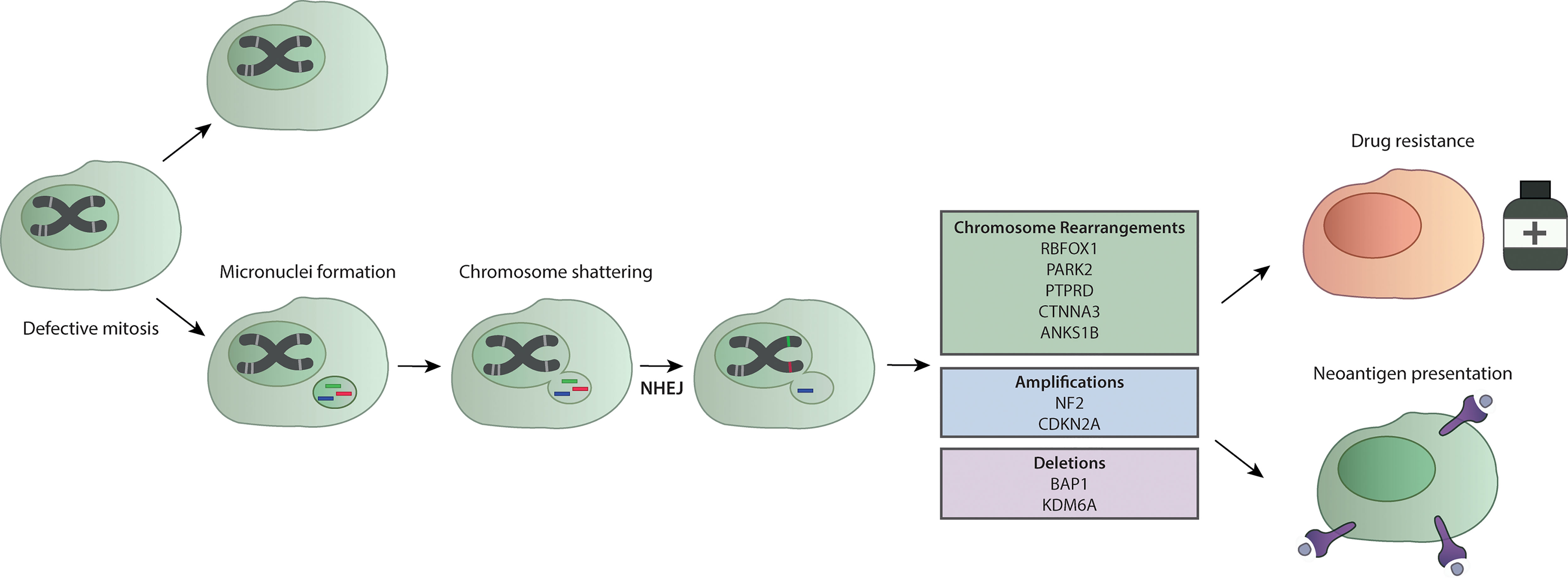
Figure 3 Chromotripsis in mesothelioma. A defective mitosis process leads to the formation of micronuclei. Further replication leads to chromosome shattering and the addition of genetic material through the NHEJ. This process forms chromosome rearrangements that in MPM interest genes like RBFOX1, PARK2, PTPRD, CTNNA3 and ANK51B, and are linked to amplification of key MPM genes like NF2 and CDKN2A, or the loss of genetic material of BAP1 and KDM6A (113, 150, 151). Amplification of oncogenes due to chromotripsis has been recently associated with drug resistance (152). The extensive chromosomal breakage upon chromotripsis events leads to neoantigen presentation that can make cells more sensitive to immunotherapy (150).
A more recent study addressed chromotripsis in mesothelioma through a new approach (150) called Mate-pair sequencing (Mpseq). Mpseq generates larger sequencing fragments and can detect chromosomal rearrangements and large insertion/deletion (150). Mansfield et al. found rearrangements that lead to CDKN2A, BAP1 and NF2 chromosomal instabilities. In particular, they have evaluated the MESO cohort of TCGA and identified chromotripsis events in 69% of patients, mostly occurring in tumor suppressor genes and non-coding DNA (150).
Results obtained in other cancers have shown that chromotripsis contributes to oncogene amplification, thus promoting cancer progression (152, 154). Among them, those harboring telomerase reverse transcriptase (TERT) gains have shown higher prevalence for chromothripsis (149) and this can be linked to mesothelioma with TERT-impairment that present poorer prognosis. This evidence suggests that chromotripsis is a specific genomic feature of mesothelioma and that future studies should include investigations of this complex event, especially for therapeutic developments. To this, chromotripsis has been recently associated with the development of drug-resistance in cancer through a mechanism that involves the repairing systems of the DNA via PARP and the NHEJ pathway (152). In addition, the notion of mesothelioma as a low mutated tumor should be reconsidered since most studies have defined the mutational burden in relation to only nucleotide changes, however the previously discussed findings suggest to consider genomic and structural rearrangements as well. Indeed, an increased expression of neoantigens resulting from these catastrophic events in mesothelioma correlate with clonal expansion of tumor-infiltrating T cells (150) suggesting a possible role in response to immunotherapy (Figure 3). All together, these discoveries indicate that the group of MPM presenting chromotripsis could benefit from combinatorial drug treatment, including PARPi or immunotherapy, that are worth being further exploited.
Following BAP1, the second most frequent mutated gene in mesothelioma is the neurofibromatosis type 2 (NF2) which encodes the protein merlin (138). This protein plays a role in the Hippo and the mTOR pathways, other than being involved in EMT (155). In mesothelioma, NF2 is found mostly as a biallelic inactivation. As reported by Sato et al. preclinical in vivo studies have shown the central role of NF2 in sensitizing tissue to asbestos and developing mesothelioma (129). In other studies, conducted on patients cohort, it has emerged that impairment of the NF2 gene is more frequent in the sarcomatoid subtype rather than the epithelioid and this correlates with worst prognosis (129, 138). To date, NF2 alterations represent a possible target for treatment such as drugs aiming at interfering with its function in extracellular matrix signal transduction. Up to date, studies are mainly based on preclinical models, cell lines or xenograft; however, promising results suggest further developments. To this, the initial studies investigating FAK inhibitors as VS-4718, or YAP and mTOR inhibitors in MPM lead to positive results in preclinical studies but not in clinical trials (129) (Table 2). Others showed the antagonistic relationship between FAK and Wnt pathways in malignant mesothelioma: dysregulated Wnt signaling is associated with invasion and resistance to apoptosis, while FAK signaling promotes invasion and EMT (130). The most interesting outcome has been reached with the development of K-975, a small molecule that inhibits the transcriptional enhanced associate domain (TEAD) protein that belongs to the Hippo pathway. K-975 showed a potent inhibitory effect on the proliferation of MPM cell lines, with a greater activity on NF2-non-expressing cells (156). Overall, these studies suggest that NF2 could be considered as a potential target for MPM warranting further investigations.
Recently, the involvement of the Hedgehog pathway in MPM has been investigated. Here, in a patient diagnosed with epithelioid MPM, a mutation in PTCH1 was identified (131). This gene is involved in the Hedgehog pathway and is druggable by vismodegib, already approved for the treatment of basal cell carcinoma (BCC) (Table 2). Despite the patient underwent several lines of treatment, vismodegib led to a very good partial response which lasted for over two years. The lack of genomic testing throughout the course of chemotherapy does not allow understanding whether the PTCH1 mutation was acquired under pharmacological pressure; still, this result suggests an interesting novel target in MPM.
Pirker et al. investigated the role of TERT promoter in mesothelioma (132). They found that mutations in TERT are prevalently associated with non-epithelioid subtype and to poor survival. In accordance with this evidence, TERT mutations are mutually exclusive with BAP1, which is more frequent in the epithelioid subtype and correlates with better prognosis, as previously discussed. An interesting aspect is that cell lines derived from tumors with mutated TERT present a more stable genome, e.g., in the number of gains or losses, in comparison to wild type ones, with a specific imbalance in chromosomes 1, 5q, 9p, 7, 14, and 20. These evidences brought to test telomerase inhibitors like MST-312 MPM, however with moderate effects (132) (Table 2).
BCL-2 is a family protein involved in tumorigenesis of different cancer types. It consists of BCL-2, BCL-XL, BCL-W, MCL-1 and BFL-1 proteins. In mesothelioma, overexpression of BCL-2 was found in 20% of cell lines and in 24% of a tumor samples, while the expression of BCL-XL and MCL-1 was identified as a general feature of mesothelioma, suggesting their critical role as pro-survival factors (134). As BCL-2 proteins act directly on the apoptosis pathway downstream from TP53, this makes its targeting an interesting therapeutic approach (134). Preclinical studies have been performed in mesothelioma cell lines using direct or indirect targeting of BCL2 and BCL-XL: both in cell lines and in vivo models this treatment was able to induce an apoptotic effect. This has paved the way to the development of BH3-mimetics a novel class of compound that mimic the interaction of BCL-2 protein through their BH3 domain. In mesothelioma, BH3-mimetics have been used in combination with YM155, a survivin inhibitor, or bortezomib, with an increased apoptotic effect in comparison to controls (Table 2). These drugs have also been used in combination with trabectedin, a marine compound, showing that BCL-2 mRNA expression inversely correlated with response to the treatment (134).
The marine-derived drug trabectedin has been already tested in MPM, although without any efficacy. In fact, the ATREUS trial, where this drug was administered as second line therapy in epithelioid MPM and as a first or second line in non-epithelioid subtype, showed poor efficacy of trabectedin and high liver toxicity that did not justify further use of this drug (157). However, lurbinectedin, an analogue of trabectedin, has shown promising results in a phase II clinical trial, where it was administered as a second or third line therapy in MPM patients (133) (Table 2). Lurbinectedin was efficacious independently on the MPM histotype and previous treatment. Further investigations to understand which patients might benefit from this drug are needed; however, the possible dual effect of lurbinectedin on both tumor cells and microenvironment might open new therapeutic venues for this disease (158).
In a recent work Dell’Anno et al. have used a drug repositioning approach to screen five MPM cell lines with 1170 FDA-approved drugs (136). They identified fludarabine (F-araA) and risedronic acid (RIS) as effective in MPM through a mechanism of inhibition of STAT1 expression and nucleic acids synthesis. Although promising, these results are yet limited by the use of only five cell lines without molecular characterization of these cells. Further studies including diverse and deeply characterized models are needed to successfully develop treatments for MPM.
In the past 20 years it has become increasingly clear that microRNAs are important players in the regulation of physiological processes within the cells, and that their dysregulation can be a major driver of malignant transformation and tumor progression (159, 160). Following the first description of a direct link between the loss of expression of a specific microRNA and cancer in 2002 (161), many studies followed investigating both the oncogenic and tumor suppressive potential of microRNAs. Hence, to date we know of many microRNAs that are involved in several processes linked to cancer development and progression, from regulation of cell proliferation and cell cycle processes to EMT and involvement in cancer immune escape (160, 162–164).
While still somewhat understudied in MPM, the studies available thus far, have shown that microRNAs play an important role in the biology of MPM, and represent valuable biomarker candidates (165, 166).
Considering that MPM is a diseases characterized by the loss of tumor suppressors, it is not surprising that this is also reflected on microRNA level with the majority of dysregulated microRNAs being lost, while only a limited number has been shown to be upregulated in MPM (165–167). The first study investigating dysregulation of microRNAs in MPM was performed by Guled et al. in 2009, who could show differential expression of microRNAs in MPM tissue compared to normal pericardial mesothelium, but also differential expression between the different histological subtypes of MPM (168). This study was shortly after followed by a study from Busacca et al. on MPM cell lines compared to immortalized mesothelial cells, which showed that the microRNAs with the greatest differential expression between MPM and normal cells, also discriminated between histopathological subtypes when investigated in tumor samples (169).
While the dysregulated microRNAs identified in these early studies were not functionally investigated, the obtained data strongly hinted towards a relevant role for microRNAs in MPM biology. The first studies providing functional data on dysregulated microRNAs, investigated the effect of re-expression of miR-29c-5p (miR-29c*) (170) and miR-31 (171), and in both cases, re-expression using microRNA mimics resulted in reduced proliferation and invasion of MPM cell lines, supporting a tumor suppressive role of these microRNAs. Following these initial studies, further tumor suppressor microRNAs were investigated in MPM such as miR-145 (172) and miR-205 (173), both of which are likely to alter EMT via targeting OCT4 and ZEB1/2 respectively. One of most comprehensively investigated tumor suppressive microRNAs in MPM is probably the miR-15~107 super family, and here in particular the family member miR-16. Reid et al. have shown that the members of this microRNA superfamily are quite consistently lost in MPM tissue compared to normal pleura, as well as in MPM cell lines (174). In vitro analyses then revealed that re-expression of miR-16-5p resulted in reduced cell proliferation and colony forming ability, as well as induction of apoptosis and a G0/G1 cell cycle arrest in MPM cell lines, but not in the non-malignant mesothelial cell line MeT-5A. These observed effects were most likely brought about by the downregulation of miR-16-5p target genes BCL2 and CCND1. In addition, re-expression of miR-16 resulted in sensitization of cells to pemetrexed and gemcitabine, suggesting an additional role of the miR-15~107 family in response to antimetabolite chemotherapy. Most importantly, mouse experiments, in which miR-16 was systemically delivered to the tumor cells using (EnGenic) minicells resulted in significant tumor growth inhibition in vivo. Based on these data, a phase I clinical trial was performed in MPM and non-small cell lung cancer (NSCLC) patients, which apart from reaching its goal of proving safety of the miR-16 replacement therapy approach also showed in one patient a remarkable metabolic and radiological response to the treatment (175, 176). In a more recent study, the same group has now shown that miR-16-5p is a regulator of PD-L1 expression, hence also linking this tumor suppressor to response to immune checkpoint inhibition (177). While miR-16-5p is the most comprehensively investigated tumor, suppressive microRNA linked to MPM biology, other microRNAs such as miR-193a-3p (178), miR-137-5p (179), miR-126 (180, 181), miR-34b/c (182, 183), and miR-215-5p (184) have also promising anti-proliferative and anti-tumor activity, when re-expressed in vitro or in vivo.
Compared to the tumor suppressive microRNAs, which have been investigated relatively frequently in MPM, studies of oncogenic microRNAs are much rarer, also due to the fact that not many microRNAs have been found to be consistently overexpressed in MPM tumor tissue. One example however are miR-182-5p and miR-183-5p, which inhibition using microRNA inhibitors results in reduced proliferation and invasion (185).
Taken together, the available expression and functional studies highlight that microRNA dysregulation, in particular the loss of tumor suppressive microRNAs, is likely to represent an important contributor to MPM biology, and therefore to the development and progression of this devastating disease. Considering the encouraging data obtained from in vivo microRNAs replacement studies and early clinical trials, additional research efforts in this area are certainly warranted. Especially also in light of the recent development of mRNA-based vaccines against COVID-19, the field of RNA-based vaccines has significantly advanced, possibly also opening additional avenues for delivery of microRNAs to cancer cells in order to replace lost tumor suppressors.
In addition to their important role in contributing to MPM biology, microRNAs have in recent years also been investigated for their biomarker potential both in tumor tissue and in blood (165, 166). Regarding the diagnostic value of tumor microRNA expression, the first published study by Gee et al. performed microRNA profiling in MPM tissue and pleural metastases from lung adenocarcinomas (186). This study identified in particular low expression of members of the miR-220 family as potential diagnostic factors for differentiating MPM and pleural metastases. A subsequent study by Benjamin et al., then investigated microRNA expression in a larger set of MPM and adenocarcinomas of the lung or pleura, and identified in addition to members of the miR-200 family, also members of the miR-192 family as potential diagnostic markers (187). Based on this data, a microRNAs expression signature was generated and independently validated in a small set of patients. The signature consisting of miR-192, -193-5p and -200c showed high accuracy (95%) in discriminating MPM from adenocarcinomas, and was subsequently marketed by Rosetta Genomics. While additional candidates such as miR-126, -143, -145 and -652 have been proposed (188, 189), none of the microRNAs or diagnostic signatures described here has been followed up extensively, hence none of them is in routine clinical use.
Similarly, several studies have been undertaken investigating the prognostic value of microRNAs in MPM. Among the most promising candidates is the tumor suppressive microRNA miR-29c-5p, which expression is not only associated with the histological subtype (higher in epithelioid), but also significantly associated with survival (170). Similarly, the tumor suppressor miR-31 has recently also been proposed to hold prognostic potential, with lower expression being associated with longer survival (190). Furthermore, in a study investigating specimens from patients undergoing extrapleural pneumonectomy, a signature consisting of 6 microRNAs, the so-called miR-Score, was identified, which was able to separate patients with good and poor prognosis with an accuracy of 87% (191). However, similar to the diagnostic candidates, independent validation studies are lacking.
An attractive alternative to microRNA expression in tissue samples is the presence of microRNAs in the circulation. Other than longer RNA species, microRNAs show remarkable stability in the blood of patients due to the fact that they are mainly bound to lipoproteins or encapsulated into extracellular vesicles. With this in mind, a small number of studies has been published proposing for example low levels of miR-103 in whole blood (192, 193), low levels of miR-126 (181, 188, 194) in serum, low level of miR-132 in plasma (195) or high levels of miR-625-3p (196) in plasma or serum to be associated with the presence of MPM, and allowed to discriminate those patients from healthy controls and asbestos-exposed individuals. While these studies again provide first evidence of the biomarker potential of microRNAs in the circulation, large independent validation studies undertaken by independent research groups are usually missing, and none of the candidates is yet ready for routine clinical application.
In addition to the above mentioned specific alterations of DNA or RNA in MPM, great interest is now being paid to the epigenome, that includes post-translational modifications that ultimately impact on gene expression not encoded by the DNA (197). Epigenetics modifications play an important role in the regulation of gene expression and include DNA methylation, histone modifications and chromatin remodeling. Dysfunction of these mechanism have been linked to tumorigenesis, cancer progression and metastasis (198, 199).
Due to the lack of druggable molecular targets, the level of investigation in MPM disease has moved so far. DNA methylation associated with tumor-suppressor genes and mechanisms involving histone modifications have been described and linked to MPM phenotypes and histological subtypes (198). DNA methylation provokes gene silencing by adding a methyl group to the fifth carbon of the cytosine base, with a process that mainly occurs in promoter regions, thus modifying the expression of the associated genes, or on repetitive DNA elements, such as LINE1, causing chromatin modifications (200). Although DNA methylation patterns are fairly stable markers of differentiated tissues that regulate specific gene expression, changes in the methylation profile can occur due to aging, exposure to environmental stimuli and chronic inflammation (200, 201). In particular, accumulation levels of DNA methylation has been associated with higher cancer risk and cancer onset (202). The investigation of the DNA methylation profiles can be carried on at the tumor level, otherwise it is possible to search specific markers in the circulating peripheral blood. Studies conducted in malignant mesothelioma tumors have identified hypermethylated regions in TMEM30B, KAZALD1 and MAPK13 (203). DNA methyltransferases (DNMT1, DNMT3a and DNMT3b) were hypermethylated in mesothelioma cells in comparison to normal pleura, a result that was further confirmed in the TCGA cohort (203). More recently, through genome-wide methylation array technology Guarrera et al. analyzed the methylome of 163 MPM cases whose exposure to asbestos was previously assessed in comparison to control samples (202). Here, the authors identified differential profiles of DNA methylation, 98% of which comprised hypomethylated single-CpG. These genomic regions were mainly associated with genes involved in immune systems processes. These profiles were not histotype-specific, a part from the couple CXCR6 and FYCO1 which had lower methylation level in biphasic mesotheliomas than epithelioid. However, the striking result was the most significant hypomethylated CpG in 7p22.2 associated with the Forkhead Box K1 (FOXK1), a transcription factor involved in pathways like development and metabolism, and, most of all, directly interacting with BAP1. It has been suggested that the dephosporylation of FOXK1 transactivated CCL-2 gene and promotes the activation of TAMs (204). These recent evidences push to go further in the investigation of these markers, especially considering their involvement in the immune system processes that make them a likely target for immunotherapy.
The identification of DNA methylation as a marker in blood has also been explored so far. The most studied gene is mesothelin encoding the mesothelin-related peptide (SMRP) which is generally methylated in the normal pleura, while it is modified in MPM. However, it has a low sensitivity to be considered as a good marker for MPM (204). This led Santarelli et al. to identify a new marker for asbestos-exposed mesotheliomas (194). Thrombomodulin (TM) expression is silenced in malignant mesothelioma through a mechanism that involved the methylation of TM promoter by the recruitment of PARP1. Since the methylation of TM promoter has been associated with survival and given the role of PARP1 in the methylation mechanism, this marker may be of interest for further investigation for therapeutic development. In 2019, Cugliari et al. analyzed the peripheral blood of 159 MPMs and identified the CpG dinucleotide cg03546163 region associated with the gene FKBP5 as a significant marker for prognosis (205). This is very interesting as FKBP5 increases chemosensitivity to the AKT pathway, which is druggable in mesothelioma as previously described.
Other epigenetic modifications involve the acetylation and methylation of histones. A study by Goto et al. has identified a high expression of state of histone H3 lysine methylation (H3K27me3) mark (206). Interestingly, high expression marks of H3K27me3 have been associated with overexpression of EZH2 (207). This last has been found as a marker of poor prognosis in mesothelioma (207). Moreover, preclinical studies have shown that loss of function in BAP1 make cells sensitive to the inhibitors of EZH2. Among them, tazemetostat, a first-in-class small-molecule inhibitor of EZH2 received approval from the FDA in January 2020 for the treatment of locally advanced or metastatic sarcoma (207), while an ongoing clinical trial named NCT02860286 has shown antitumor activity of tazemetostat in a cohort of 74 patients lacking BAP1 (128). However, the use of drugs targeting the epigenome has already been attempted in mesothelioma with negative results. In fact, in the phase III VANTAGE-014 trial the efficacy of vorinostat, a histone deacetylase inhibitor, was tested against placebo with no improvement in overall survival (135) (Table 2). As claimed by Garassino et al., the main biases of this study were the random selection of patients irrespective of clinicopathological features and the rapid development into a phase III trial (208). These results do not undermine the use of drugs targeting the epigenome, instead they underline the necessity to improve patients’ stratification previous to enrollment into clinical trials. In summary, the investigation of potentially reversible modifications like the epigenetics markers seem to be relevant in MPM, and, when identified through liquid biopsy, they could represent a novel and promising approach for diagnosis and monitoring of cancer progression.
In this review, we have covered multiple aspects of mesothelioma microenvironment that have played and will play an important role for immunotherapeutic approaches. To this, targeting the TME with anti-PD1 (nivolumab) and anti-CTLA-4 (ipilimumab) has revealed as the most effective strategy in this disease with few therapeutic options (209). In addition, genomic and transcriptomic have allowed the identification of druggable features, currently under evaluation alone or in combination with immunotherapies. Moreover, microRNAs expression has shown a role in a better understanding of the biology of MPM, and promising preliminary data suggests a possible application in the clinic for diagnosis and monitoring as epigenetic studies.
In conclusion, a comprehensive knowledge of MPM biological aspects is crucial for a deeper understanding of such complex disease and for the improvement of patients’ outcome.
SH and LM: conceptualization, original draft preparation, and writing. AC-F and FG: conceptualization, original draft preparation, writing, and supervision. MK: conceptualization and writing. All the authors: review and editing. All authors contributed to the article and approved the submitted version.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We would like to thank Fondazione Buzzi and Stiftung für angewandte Krebsforschung.
ADCC, antibody-dependent cellular cytotoxicity; APC, antigen presenting cells; BAP1-TPDS,BAP1-tumor predisposition syndromes; BLM, Bloom syndrome gene; CCL2, C-C chemokine ligand 2; CCL4, C–C chemokine ligand 4; CCL5, C–C chemokine ligand 5; CSF-1R, colony stimulating factor-1 receptor; CXCL12, C-X-C Motif Chemokine Ligand 12 ; DAMPs, damage-associated molecular patterns; DC, dendritic cells; DDR, DNA damage response; DSBs, Double Strand Breaks; EMT, epithelial to mesenchymal transition; EZH2, enhancer of zeste homolog 2; FISH, fluorescent in-situ hybridization; FOXK1, Forkhead Box K1; Gr-MDSC, granulocytic myeloid-derived suppressor cells; H3K27me3, histone H3 lysine methylation; HMGB1, high mobility group protein B1; HR, Homologous recombination; HRD, Homologous Recombination Deficiency; IHC, immunohistochemistry; ILC, Innate lymphoid cells; LAG-3, lymphocyte activation gene-3; M-MDSC, monocytic myeloid-derived suppressor cells; MDSC, myeloid-derived suppressor cells; MGMT, O6-methylguanine-DNA methyltransferase; MLPA, multiplex ligation–dependent probe amplification; MMR, Mismatch Repair; MPM, malignant pleural mesothelioma; Mpseq, Mate-pair sequencing; NER, Nucleotide Excision Repair; NF2, neurofibromatosis type 2; NFKB, Nuclear Factor Kappa B; NGS, next generation sequencing; NHEJ, non-homologous end-joining; NK, natural killer; NKT, Natural killer T; NO, nitric oxide; NSCLC, non-small cell lung cancer; PARP, poly(ADP-ribose) polymerase; PARPi, PARP-inhibitors; PGE2, prostaglandin E2; RAGE, receptor for Advanced Glycation Endproducts; ROS, reactive oxygen species; SCNA, somatic copy-number alteration; SLFN11, Schlafen 11; SMRP, mesothelin-related peptide; SSBs, Single Strand Breaks; TAMs, tumor-associated macrophages; TCGA, The Cancer Genome Atlas; TEAD, transcriptional enhanced associate domain; TERT, telomerase reverse transcriptase; TGF-β, Transforming Growth Factor β; TIGIT, T cell immunoglobulin and ITIM domain; TILs, tumor infiltrating lymphocytes; TIM-3, T cell immunoglobulin and mucin-domain containing-3; TLS, tertiary lymphoid structures; TM, Thrombomodulin; TME, tumor microenvironment; Treg, Regulatory T cells; VISTA, V-domain Ig suppressor of T cell activation; αGC, glycolipid α-galactosylceramide
ADCC: antibody-dependent cellular cytotoxicity
APC: antigen presenting cells
BAP1-TPDS: BAP1-tumor predisposition syndromes
BLM: Bloom syndrome gene
CCL2: C-C chemokine ligand 2
CCL4: C–C chemokine ligand 4
CCL5: C–C chemokine ligand 5
CSF-1R: colony stimulating factor-1 receptor
CXCL12: C-X-C Motif Chemokine Ligand 12
DAMPs: damage-associated molecular patterns
DC: dendritic cells
DDR: DNA damage response
DSBs: Double Strand Breaks
EMT: epithelial to mesenchymal transition
EZH2: enhancer of zeste homolog 2
FISH: fluorescent in-situ hybridization
FOXK1: Forkhead Box K1
Gr-MDSC: granulocytic myeloid-derived suppressor cells
H3K27me3: histone H3 lysine methylation
HMGB1: high mobility group protein B1
HR: Homologous recombination
HRD: Homologous Recombination Deficiency
IHC: immunohistochemistry
ILC: Innate lymphoid cells
LAG-3: lymphocyte activation gene-3
M-MDSC: monocytic myeloid-derived suppressor cells
MDSC: myeloid-derived suppressor cells
MGMT: O6-methylguanine-DNA methyltransferase
MLPA: multiplex ligation–dependent probe amplification
MMR: Mismatch Repair
MPM: malignant pleural mesothelioma
Mpseq: Mate-pair sequencing
NER: Nucleotide Excision Repair
NF2: neurofibromatosis type 2
NFKB: Nuclear Factor Kappa B
NGS: next generation sequencing
NHEJ: non-homologous end-joining
NK: natural killer
NKT: Natural killer T
NO: nitric oxide
NSCLC: non-small cell lung cancer
PARP: poly(ADP-ribose) polymerase
PARPi: PARP-inhibitors
PGE2: prostaglandin E2
RAGE: receptor for Advanced Glycation Endproducts
ROS: reactive oxygen species
SCNA: somatic copy-number alteration
SLFN11: Schlafen 11
SMRP: mesothelin-related peptide
SSBs: Single Strand Breaks
TAMs: tumor-associated macrophages
TCGA: The Cancer Genome Atlas
TEAD: transcriptional enhanced associate domain
TERT: telomerase reverse transcriptase
TGF-β: Transforming Growth Factor β
TIGIT: T cell immunoglobulin and ITIM domain
TILs: tumor infiltrating lymphocytes
TIM-3: T cell immunoglobulin and mucin-domain containing-3
TLS: tertiary lymphoid structures
TM: Thrombomodulin
TME: tumor microenvironment
Treg: Regulatory T cells
VISTA: V-domain Ig suppressor of T cell activation
αGC: glycolipid α-galactosylceramide
1. Donaldson K, Murphy FA, Duffin R, Poland CA. Asbestos, Carbon Nanotubes and the Pleural Mesothelium: A Review of the Hypothesis Regarding the Role of Long Fibre Retention in the Parietal Pleura, Inflammation and Mesothelioma. Part Fibre Toxicol (2010) 7:5. doi: 10.1186/1743-8977-7-5
2. Tanaka S, Choe N, Iwagaki A, Hemenway DR, Kagan E. Asbestos Exposure Induces MCP-1 Secretion by Pleural Mesothelial Cells. Exp Lung Res (2000) 26:241–55. doi: 10.1080/019021400404528
3. Xu A, Wu LJ, Santella RM, Hei TK. Role of Oxyradicals in Mutagenicity and DNA Damage Induced by Crocidolite Asbestos in Mammalian Cells. Cancer Res (1999) 59:5922–6.
4. Xue J, Patergnani S, Giorgi C, Suarez J, Goto K, Bononi A, et al. Asbestos Induces Mesothelial Cell Transformation Via HMGB1-Driven Autophagy. Proc Natl Acad Sci USA (2020) 117:25543–52. doi: 10.1073/pnas.2007622117
5. Jube S, Rivera ZS, Bianchi ME, Powers A, Wang E, Pagano I, et al. Cancer Cell Secretion of the DAMP Protein HMGB1 Supports Progression in Malignant Mesothelioma. Cancer Res (2012) 72:3290–301. doi: 10.1158/0008-5472.CAN-11-3481
6. Mukherjee A, Vasquez KM. Targeting Chromosomal Architectural HMGB Proteins Could be the Next Frontier in Cancer Therapy. Cancer Res (2020) 80:2075–82. doi: 10.1158/0008-5472.CAN-19-3066
7. Napolitano A, Antoine DJ, Pellegrini L, Baumann F, Pagano I, Pastorino S, et al. HMGB1 and Its Hyperacetylated Isoform are Sensitive and Specific Serum Biomarkers to Detect Asbestos Exposure and to Identify Mesothelioma Patients. Clin Cancer Res (2016) 22:3087–96. doi: 10.1158/1078-0432.CCR-15-1130
8. Padmore T, Stark C, Turkevich LA, Champion JA. Quantitative Analysis of the Role of Fiber Length on Phagocytosis and Inflammatory Response by Alveolar Macrophages. Biochim Biophys Acta Gen Subj (2017) 1861:58–67. doi: 10.1016/j.bbagen.2016.09.031
9. Wen H, Ting JP-Y, O’Neill LAJ. A Role for the NLRP3 Inflammasome in Metabolic Diseases–did Warburg Miss Inflammation? Nat Immunol (2012) 13:352–7. doi: 10.1038/ni.2228
10. Horio D, Minami T, Kitai H, Ishigaki H, Higashiguchi Y, Kondo N, et al. Tumor-Associated Macrophage-Derived Inflammatory Cytokine Enhances Malignant Potential of Malignant Pleural Mesothelioma. Cancer Sci (2020) 111:2895–906. doi: 10.1111/cas.14523
11. Yang H, Rivera Z, Jube S, Nasu M, Bertino P, Goparaju C, et al. Programmed Necrosis Induced by Asbestos in Human Mesothelial Cells Causes High-Mobility Group Box 1 Protein Release and Resultant Inflammation. Proc Natl Acad Sci USA (2010) 107:12611–6. doi: 10.1073/pnas.1006542107
12. Qi F, Okimoto G, Jube S, Napolitano A, Pass HI, Laczko R, et al. Continuous Exposure to Chrysotile Asbestos Can Cause Transformation of Human Mesothelial Cells Via HMGB1 and TNF-α Signaling. Am J Pathol (2013) 183:1654–66. doi: 10.1016/j.ajpath.2013.07.029
13. Yang H, Bocchetta M, Kroczynska B, Elmishad AG, Chen Y, Liu Z, et al. TNF-Alpha Inhibits Asbestos-Induced Cytotoxicity Via a NF-KappaB-Dependent Pathway, A Possible Mechanism for Asbestos-Induced Oncogenesis. Proc Natl Acad Sci USA (2006) 103:10397–402. doi: 10.1073/pnas.0604008103
14. Wang Y, Faux SP, Hallden G, Kirn DH, Houghton CE, Lemoine NR, et al. Interleukin-1beta and Tumour Necrosis Factor-Alpha Promote the Transformation of Human Immortalised Mesothelial Cells by Erionite. Int J Oncol (2004) 25:173–8. doi: 10.3892/ijo.25.1.173
15. Awad MM, Jones RE, Liu H, Lizotte PH, Ivanova EV, Kulkarni M, et al. Cytotoxic T Cells in PD-L1-Positive Malignant Pleural Mesotheliomas Are Counterbalanced by Distinct Immunosuppressive Factors. Cancer Immunol Res (2016) 4:1038–48. doi: 10.1158/2326-6066.CIR-16-0171
16. Chee SJ, Lopez M, Mellows T, Gankande S, Moutasim KA, Harris S, et al. Evaluating the Effect of Immune Cells on the Outcome of Patients With Mesothelioma. Br J Cancer (2017) 117:1341–8. doi: 10.1038/bjc.2017.269
17. Tazzari M, Brich S, Tuccitto A, Bozzi F, Beretta V, Spagnuolo RD, et al. Complex Immune Contextures Characterise Malignant Peritoneal Mesothelioma: Loss of Adaptive Immunological Signature in the More Aggressive Histological Types. J Immunol Res (2018) 2018:5804230. doi: 10.1155/2018/5804230
18. Vivier E, Raulet DH, Moretta A, Caligiuri MA, Zitvogel L, Lanier LL, et al. Innate or Adaptive Immunity? The Example of Natural Killer Cells. Science (2011) 331:44–9. doi: 10.1126/science.1198687
19. Platonova S, Cherfils-Vicini J, Damotte D, Crozet L, Vieillard V, Validire P, et al. Profound Coordinated Alterations of Intratumoral NK Cell Phenotype and Function in Lung Carcinoma. Cancer Res (2011) 71:5412–22. doi: 10.1158/0008-5472.CAN-10-4179
20. Sottile R, Tannazi M, Johansson MH, Cristiani CM, Calabró L, Ventura V, et al. NK- and T-Cell Subsets in Malignant Mesothelioma Patients: Baseline Pattern and Changes in the Context of Anti-CTLA-4 Therapy. Int J Cancer (2019) 145:2238–48. doi: 10.1002/ijc.32363
21. Nishimura Y, Kumagai-Takei N, Matsuzaki H, Lee S, Maeda M, Kishimoto T, et al. Functional Alteration of Natural Killer Cells and Cytotoxic T Lymphocytes Upon Asbestos Exposure and in Malignant Mesothelioma Patients. BioMed Res Int (2015) 2015:238431. doi: 10.1155/2015/238431
22. Marcq E, Waele JD, Audenaerde JV, Lion E, Santermans E, Hens N, et al. Abundant Expression of TIM-3, LAG-3, PD-1 and PD-L1 as Immunotherapy Checkpoint Targets in Effusions of Mesothelioma Patients. Oncotarget (2017) 8:89722–35. doi: 10.18632/oncotarget.21113
23. Marcq E, Siozopoulou V, De Waele J, van Audenaerde J, Zwaenepoel K, Santermans E, et al. Prognostic and Predictive Aspects of the Tumor Immune Microenvironment and Immune Checkpoints in Malignant Pleural Mesothelioma. Oncoimmunology (2017) 6:e1261241. doi: 10.1080/2162402X.2016.1261241
24. Lievense LA, Bezemer K, Cornelissen R, Kaijen-Lambers MEH, Hegmans JPJJ, Aerts JGJV. Precision Immunotherapy; Dynamics in the Cellular Profile of Pleural Effusions in Malignant Mesothelioma Patients. Lung Cancer (2017) 107:36–40. doi: 10.1016/j.lungcan.2016.04.015
25. Tumino N, Martini S, Munari E, Scordamaglia F, Besi F, Mariotti FR, et al. Presence of Innate Lymphoid Cells in Pleural Effusions of Primary and Metastatic Tumors: Functional Analysis and Expression of PD-1 Receptor. Int J Cancer (2019) 145:1660–8. doi: 10.1002/ijc.32262
26. Bosi A, Zanellato S, Bassani B, Albini A, Musco A, Cattoni M, et al. Natural Killer Cells From Malignant Pleural Effusion Are Endowed With a Decidual-Like Proangiogenic Polarization. J Immunol Res (2018) 2018:2438598. doi: 10.1155/2018/2438598
27. Vacca P, Martini S, Garelli V, Passalacqua G, Moretta L, Mingari MC. NK Cells From Malignant Pleural Effusions are Not Anergic But Produce Cytokines and Display Strong Antitumor Activity on Short-Term IL-2 Activation. Eur J Immunol (2013) 43:550–61. doi: 10.1002/eji.201242783
28. Jackaman C, Lansley S, Allan JE, Robinson BWS, Nelson DJ. IL-2/CD40-Driven NK Cells Install and Maintain Potency in the Anti-Mesothelioma Effector/Memory Phase. Int Immunol (2012) 24:357–68. doi: 10.1093/intimm/dxs005
29. Rossjohn J, Pellicci DG, Patel O, Gapin L, Godfrey DI. Recognition of CD1d-Restricted Antigens by Natural Killer T Cells. Nat Rev Immunol (2012) 12:845–57. doi: 10.1038/nri3328
30. Bendelac A, Savage PB, Teyton L. The Biology of NKT Cells. Annu Rev Immunol (2007) 25:297–336. doi: 10.1146/annurev.immunol.25.022106.141711
31. Altomare E, Fallarini S, Biaggi G, Gattoni E, Botta M, Lombardi G. Increased Frequency of Circulating Invariant Natural Killer T Cells in Malignant Pleural Mesothelioma Patients. Cancer Biol Ther (2012) 13:702–11. doi: 10.4161/cbt.20553
32. Tagawa T, Wu L, Anraku M, Yun Z, Rey-McIntyre K, de Perrot M. Antitumor Impact of Interferon-γ Producing CD1d-Restricted NKT Cells in Murine Malignant Mesothelioma. J Immunother (2013) 36:391–9. doi: 10.1097/CJI.0b013e3182a801f2
33. Wu L, Yun Z, Tagawa T, de la Maza L, Wu MO, Yu J, et al. Activation of CD1d-restricted Natural Killer T Cells can Inhibit Cancer Cell Proliferation During Chemotherapy by Promoting the Immune Responses in Murine Mesothelioma. Cancer Immunol Immunother (2014) 63:1285–96. doi: 10.1007/s00262-014-1597-9
34. Burt BM, Rodig SJ, Tilleman TR, Elbardissi AW, Bueno R, Sugarbaker DJ. Circulating and Tumor-Infiltrating Myeloid Cells Predict Survival in Human Pleural Mesothelioma. Cancer (2011) 117:5234–44. doi: 10.1002/cncr.26143
35. Tanrikulu AC, Abakay A, Komek H, Abakay O. Prognostic Value of the Lymphocyte-to-Monocyte Ratio and Other Inflammatory Markers in Malignant Pleural Mesothelioma. Environ Health Prev Med (2016) 21:304–11. doi: 10.1007/s12199-016-0530-6
36. Davis MJ, Tsang TM, Qiu Y, Dayrit JK, Freij JB, Huffnagle GB, et al. Macrophage M1/M2 Polarization Dynamically Adapts to Changes in Cytokine Microenvironments in Cryptococcus Neoformans Infection. mBio (2013) 4:e00264–00213. doi: 10.1128/mBio.00264-13
37. Goerdt S, Orfanos CE. Other Functions, Other Genes: Alternative Activation of Antigen-Presenting Cells. Immunity (1999) 10:137–42. doi: 10.1016/s1074-7613(00)80014-x
38. Mantovani A, Sozzani S, Locati M, Allavena P, Sica A. Macrophage Polarization: Tumor-Associated Macrophages as a Paradigm for Polarized M2 Mononuclear Phagocytes. Trends Immunol (2002) 23:549–55. doi: 10.1016/s1471-4906(02)02302-5
39. Ujiie H, Kadota K, Nitadori J-I, Aerts JG, Woo KM, Sima CS, et al. The Tumoral and Stromal Immune Microenvironment in Malignant Pleural Mesothelioma: A Comprehensive Analysis Reveals Prognostic Immune Markers. Oncoimmunology (2015) 4:e1009285. doi: 10.1080/2162402X.2015.1009285
40. Fujii M, Toyoda T, Nakanishi H, Yatabe Y, Sato A, Matsudaira Y, et al. TGF-β Synergizes With Defects in the Hippo Pathway to Stimulate Human Malignant Mesothelioma Growth. J Exp Med (2012) 209:479–94. doi: 10.1084/jem.20111653
41. Gschwandtner M, Derler R, Midwood KS. More Than Just Attractive: How CCL2 Influences Myeloid Cell Behavior Beyond Chemotaxis. Front Immunol (2019) 10:2759. doi: 10.3389/fimmu.2019.02759
42. Kishimoto T, Fujimoto N, Ebara T, Omori T, Oguri T, Niimi A, et al. Serum Levels of the Chemokine CCL2 are Elevated in Malignant Pleural Mesothelioma Patients. BMC Cancer (2019) 19:1204. doi: 10.1186/s12885-019-6419-1
43. Gueugnon F, Leclercq S, Blanquart C, Sagan C, Cellerin L, Padieu M, et al. Identification of Novel Markers for the Diagnosis of Malignant Pleural Mesothelioma. Am J Pathol (2011) 178:1033–42. doi: 10.1016/j.ajpath.2010.12.014
44. Blanquart C, Gueugnon F, Nguyen J-M, Roulois D, Cellerin L, Sagan C, et al. CCL2, Galectin-3, and SMRP Combination Improves the Diagnosis of Mesothelioma in Pleural Effusions. J Thorac Oncol (2012) 7:883–9. doi: 10.1097/JTO.0b013e31824c9272
45. Davidson B, Dong HP, Holth A, Berner A, Risberg B. Chemokine Receptors are Infrequently Expressed in Malignant and Benign Mesothelial Cells. Am J Clin Pathol (2007) 127:752–9. doi: 10.1309/LN2075V7C8K31CH8
46. Boulakirba S, Pfeifer A, Mhaidly R, Obba S, Goulard M, Schmitt T, et al. IL-34 and CSF-1 Display an Equivalent Macrophage Differentiation Ability But a Different Polarization Potential. Sci Rep (2018) 8:256. doi: 10.1038/s41598-017-18433-4
47. Chéné A-L, d’Almeida S, Blondy T, Tabiasco J, Deshayes S, Fonteneau J-F, et al. Pleural Effusions From Patients With Mesothelioma Induce Recruitment of Monocytes and Their Differentiation Into M2 Macrophages. J Thorac Oncol (2016) 11:1765–73. doi: 10.1016/j.jtho.2016.06.022
48. Blondy T, d’Almeida SM, Briolay T, Tabiasco J, Meiller C, Chéné A-L, et al. Involvement of the M-CSF/IL-34/CSF-1R Pathway in Malignant Pleural Mesothelioma. J Immunother Cancer (2020) 8:e000182. doi: 10.1136/jitc-2019-000182
49. Cioce M, Canino C, Goparaju C, Yang H, Carbone M, Pass HI. Autocrine CSF-1R Signaling Drives Mesothelioma Chemoresistance Via AKT Activation. Cell Death Dis (2014) 5:e1167. doi: 10.1038/cddis.2014.136
50. Zhang F, Wang H, Wang X, Jiang G, Liu H, Zhang G, et al. TGF-β Induces M2-like Macrophage Polarization Via SNAIL-Mediated Suppression of a Pro-Inflammatory Phenotype. Oncotarget (2016) 7:52294–306. doi: 10.18632/oncotarget.10561
51. Gratchev A. TGF-β Signalling in Tumour Associated Macrophages. Immunobiology (2017) 222:75–81. doi: 10.1016/j.imbio.2015.11.016
52. Lievense LA, Cornelissen R, Bezemer K, Kaijen-Lambers MEH, Hegmans JPJJ, Aerts JGJV. Pleural Effusion of Patients With Malignant Mesothelioma Induces Macrophage-Mediated T Cell Suppression. J Thorac Oncol (2016) 11:1755–64. doi: 10.1016/j.jtho.2016.06.021
53. Izzi V, Chiurchiù V, D’Aquilio F, Palumbo C, Tresoldi I, Modesti A, et al. Differential Effects of Malignant Mesothelioma Cells on THP-1 Monocytes and Macrophages. Int J Oncol (2009) 34:543–50. doi: 10.3892/ijo_00000180
54. Seoane J, Gomis RR. Tgf-β Family Signaling in Tumor Suppression and Cancer Progression. Cold Spring Harb Perspect Biol (2017) 9(12):a022277. doi: 10.1101/cshperspect.a022277
55. Kumar-Singh S, Weyler J, Martin MJ, Vermeulen PB, Van Marck E. Angiogenic Cytokines in Mesothelioma: A Study of VEGF, FGF-1 and -2, and TGF Beta Expression. J Pathol (1999) 189:72–8. doi: 10.1002/(SICI)1096-9896(199909)189:1<72::AID-PATH401>3.0.CO;2-0
56. DeLong P, Carroll RG, Henry AC, Tanaka T, Ahmad S, Leibowitz MS, et al. Regulatory T Cells and Cytokines in Malignant Pleural Effusions Secondary to Mesothelioma and Carcinoma. Cancer Biol Ther (2005) 4:342–6. doi: 10.4161/cbt.4.3.1644
57. Maeda J, Ueki N, Ohkawa T, Iwahashi N, Nakano T, Hada T, et al. Transforming Growth Factor-Beta 1 (TGF-Beta 1)- and Beta 2-Like Activities in Malignant Pleural Effusions Caused by Malignant Mesothelioma or Primary Lung Cancer. Clin Exp Immunol (1994) 98:319–22. doi: 10.1111/j.1365-2249.1994.tb06144.x
58. Stockhammer P, Ploenes T, Theegarten D, Schuler M, Maier S, Aigner C, et al. Detection of TGF-β in Pleural Effusions for Diagnosis and Prognostic Stratification of Malignant Pleural Mesothelioma. Lung Cancer (2020) 139:124–32. doi: 10.1016/j.lungcan.2019.11.013
59. Solinas G, Germano G, Mantovani A, Allavena P. Tumor-Associated Macrophages (TAM) as Major Players of the Cancer-Related Inflammation. J Leukoc Biol (2009) 86:1065–73. doi: 10.1189/jlb.0609385
60. Pollard JW. Trophic Macrophages in Development and Disease. Nat Rev Immunol (2009) 9:259–70. doi: 10.1038/nri2528
61. Bingle L, Brown NJ, Lewis CE. The Role of Tumour-Associated Macrophages in Tumour Progression: Implications for New Anticancer Therapies. J Pathol (2002) 196:254–65. doi: 10.1002/path.1027
62. Tsutsui S, Yasuda K, Suzuki K, Tahara K, Higashi H, Era S. Macrophage Infiltration and Its Prognostic Implications in Breast Cancer: The Relationship With VEGF Expression and Microvessel Density. Oncol Rep (2005) 14:425–31. doi: 10.3892/or.14.2.425
63. Condeelis J, Pollard JW. Macrophages: Obligate Partners for Tumor Cell Migration, Invasion, and Metastasis. Cell (2006) 124:263–6. doi: 10.1016/j.cell.2006.01.007
64. Minnema-Luiting J, Vroman H, Aerts J, Cornelissen R. Heterogeneity in Immune Cell Content in Malignant Pleural Mesothelioma. Int J Mol Sci (2018) 19(4):1041. doi: 10.3390/ijms19041041
65. Salaroglio IC, Kopecka J, Napoli F, Pradotto M, Maletta F, Costardi L, et al. Potential Diagnostic and Prognostic Role of Microenvironment in Malignant Pleural Mesothelioma. J Thorac Oncol (2019) 14:1458–71. doi: 10.1016/j.jtho.2019.03.029
66. Pasello G, Zago G, Lunardi F, Urso L, Kern I, Vlacic G, et al. Malignant Pleural Mesothelioma Immune Microenvironment and Checkpoint Expression: Correlation With Clinical-Pathological Features and Intratumor Heterogeneity Over Time. Ann Oncol (2018) 29:1258–65. doi: 10.1093/annonc/mdy086
67. Cornelissen R, Lievense LA, Maat AP, Hendriks RW, Hoogsteden HC, Bogers AJ, et al. Ratio of Intratumoral Macrophage Phenotypes is a Prognostic Factor in Epithelioid Malignant Pleural Mesothelioma. PloS One (2014) 9:e106742. doi: 10.1371/journal.pone.0106742
68. Anraku M, Cunningham KS, Yun Z, Tsao M-S, Zhang L, Keshavjee S, et al. Impact of Tumor-Infiltrating T Cells on Survival in Patients With Malignant Pleural Mesothelioma. J Thorac Cardiovasc Surg (2008) 135:823–9. doi: 10.1016/j.jtcvs.2007.10.026
69. Yamada N, Oizumi S, Kikuchi E, Shinagawa N, Konishi-Sakakibara J, Ishimine A, et al. CD8+ Tumor-Infiltrating Lymphocytes Predict Favorable Prognosis in Malignant Pleural Mesothelioma After Resection. Cancer Immunol Immunother (2010) 59:1543–9. doi: 10.1007/s00262-010-0881-6
70. Fusco N, Vaira V, Righi I, Sajjadi E, Venetis K, Lopez G, et al. Characterization of the Immune Microenvironment in Malignant Pleural Mesothelioma Reveals Prognostic Subgroups of Patients. Lung Cancer (2020) 150:53–61. doi: 10.1016/j.lungcan.2020.09.026
71. Colin DJ, Cottet-Dumoulin D, Faivre A, Germain S, Triponez F, Serre-Beinier V. Experimental Model of Human Malignant Mesothelioma in Athymic Mice. Int J Mol Sci (2018) 19(7):1881. doi: 10.3390/ijms19071881
72. Miselis NR, Wu ZJ, Van Rooijen N, Kane AB. Targeting Tumor-Associated Macrophages in An Orthotopic Murine Model of Diffuse Malignant Mesothelioma. Mol Cancer Ther (2008) 7:788–99. doi: 10.1158/1535-7163.MCT-07-0579
73. Gao D, Joshi N, Choi H, Ryu S, Hahn M, Catena R, et al. Myeloid Progenitor Cells in the Premetastatic Lung Promote Metastases by Inducing Mesenchymal to Epithelial Transition. Cancer Res (2012) 72:1384–94. doi: 10.1158/0008-5472.CAN-11-2905
74. Umansky V, Blattner C, Gebhardt C, Utikal J. The Role of Myeloid-Derived Suppressor Cells (MDSC) in Cancer Progression. Vaccines (Basel) (2016) 4(4):36. doi: 10.3390/vaccines4040036
75. Khanna S, Graef S, Mussai F, Thomas A, Wali N, Yenidunya BG, et al. Tumor-Derived GM-CSF Promotes Granulocyte Immunosuppression in Mesothelioma Patients. Clin Cancer Res (2018) 24:2859–72. doi: 10.1158/1078-0432.CCR-17-3757
76. Hegmans JPJJ, Hemmes A, Hammad H, Boon L, Hoogsteden HC, Lambrecht BN. Mesothelioma Environment Comprises Cytokines and T-Regulatory Cells That Suppress Immune Responses. Eur Respir J (2006) 27:1086–95. doi: 10.1183/09031936.06.00135305
77. Rikimaru T, Ichikawa Y, Ogawa Y, Higuchi E, Kinosita M, Oizumi K, et al. Production of Granulocyte Colony-Stimulating Factor by Malignant Mesothelioma. Eur Respir J (1995) 8:183–4. doi: 10.1183/09031936.95.08010183
78. Fujiwara A, Higashiyama M, Kanou T, Okami J, Tokunaga T, Tomita Y, et al. Granulocyte-Colony Stimulating Factor (G-CSF) Producing Malignant Pleural Mesothelioma: Report of a Case. Thorac Cancer (2015) 6:105–9. doi: 10.1111/1759-7714.12140
79. Jackaman C, Yeoh TL, Acuil ML, Gardner JK, Nelson DJ. Murine Mesothelioma Induces Locally-Proliferating IL-10(+)TNF-α(+)CD206(-)CX3CR1(+) M3 Macrophages That Can Be Selectively Depleted by Chemotherapy or Immunotherapy. Oncoimmunology (2016) 5:e1173299. doi: 10.1080/2162402X.2016.1173299
80. Zhao Y, Wu T, Shao S, Shi B, Zhao Y. Phenotype, Development, and Biological Function of Myeloid-Derived Suppressor Cells. Oncoimmunology (2016) 5:e1004983. doi: 10.1080/2162402X.2015.1004983
81. Veltman JD, Lambers MEH, van Nimwegen M, Hendriks RW, Hoogsteden HC, Aerts JGJV, et al. COX-2 Inhibition Improves Immunotherapy and is Associated With Decreased Numbers of Myeloid-Derived Suppressor Cells in Mesothelioma. Celecoxib Influences MDSC Function. BMC Cancer (2010) 10:464. doi: 10.1186/1471-2407-10-464
82. Fridman WH, Pagès F, Sautès-Fridman C, Galon J. The Immune Contexture in Human Tumours: Impact on Clinical Outcome. Nat Rev Cancer (2012) 12:298–306. doi: 10.1038/nrc3245
83. Borghaei H, Paz-Ares L, Horn L, Spigel DR, Steins M, Ready NE, et al. Nivolumab Versus Docetaxel in Advanced Nonsquamous Non-Small-Cell Lung Cancer. N Engl J Med (2015) 373:1627–39. doi: 10.1056/NEJMoa1507643
84. Brahmer J, Reckamp KL, Baas P, Crinò L, Eberhardt WEE, Poddubskaya E, et al. Nivolumab Versus Docetaxel in Advanced Squamous-Cell Non-Small-Cell Lung Cancer. N Engl J Med (2015) 373:123–35. doi: 10.1056/NEJMoa1504627
85. Thapa B, Salcedo A, Lin X, Walkiewicz M, Murone C, Ameratunga M, et al. The Immune Microenvironment, Genome-Wide Copy Number Aberrations, and Survival in Mesothelioma. J Thorac Oncol (2017) 12:850–9. doi: 10.1016/j.jtho.2017.02.013
86. Klampatsa A, O’Brien SM, Thompson JC, Rao AS, Stadanlick JE, Martinez MC, et al. Phenotypic and Functional Analysis of Malignant Mesothelioma Tumor-Infiltrating Lymphocytes. Oncoimmunology (2019) 8:e1638211. doi: 10.1080/2162402X.2019.1638211
87. Sakaguchi S, Yamaguchi T, Nomura T, Ono M. Regulatory T Cells and Immune Tolerance. Cell (2008) 133:775–87. doi: 10.1016/j.cell.2008.05.009
88. Shang B, Liu Y, Jiang S, Liu Y. Prognostic Value of Tumor-Infiltrating FoxP3+ Regulatory T Cells in Cancers: A Systematic Review and Meta-Analysis. Sci Rep (2015) 5:15179. doi: 10.1038/srep15179
89. McCoy MJ, Nowak AK, van der Most RG, Dick IM, Lake RA. Peripheral CD8(+) T Cell Proliferation is Prognostic for Patients With Advanced Thoracic Malignancies. Cancer Immunol Immunother (2013) 62:529–39. doi: 10.1007/s00262-012-1360-z
90. Needham DJ, Lee JX, Beilharz MW. Intra-Tumoural Regulatory T Cells: A Potential New Target in Cancer Immunotherapy. Biochem Biophys Res Commun (2006) 343:684–91. doi: 10.1016/j.bbrc.2006.03.018
91. Patil NS, Righi L, Koeppen H, Zou W, Izzo S, Grosso F, et al. Molecular and Histopathological Characterization of the Tumor Immune Microenvironment in Advanced Stage of Malignant Pleural Mesothelioma. J Thorac Oncol (2018) 13:124–33. doi: 10.1016/j.jtho.2017.09.1968
92. Cedrés S, Ponce-Aix S, Zugazagoitia J, Sansano I, Enguita A, Navarro-Mendivil A, et al. Analysis of Expression of Programmed Cell Death 1 Ligand 1 (PD-L1) in Malignant Pleural Mesothelioma (MPM). PloS One (2015) 10:e0121071. doi: 10.1371/journal.pone.0121071
93. Inaguma S, Lasota J, Wang Z, Czapiewski P, Langfort R, Rys J, et al. Expression of ALCAM (CD166) and PD-L1 (CD274) Independently Predicts Shorter Survival in Malignant Pleural Mesothelioma. Hum Pathol (2018) 71:1–7. doi: 10.1016/j.humpath.2017.04.032
94. Watanabe T, Okuda K, Murase T, Moriyama S, Haneda H, Kawano O, et al. Four Immunohistochemical Assays to Measure the PD-L1 Expression in Malignant Pleural Mesothelioma. Oncotarget (2018) 9:20769–80. doi: 10.18632/oncotarget.25100
95. Mansfield AS, Roden AC, Peikert T, Sheinin YM, Harrington SM, Krco CJ, et al. B7-H1 Expression in Malignant Pleural Mesothelioma is Associated With Sarcomatoid Histology and Poor Prognosis. J Thorac Oncol (2014) 9:1036–40. doi: 10.1097/JTO.0000000000000177
96. Popat S, Curioni-Fontecedro A, Dafni U, Shah R, O’Brien M, Pope A, et al. A Multicentre Randomised Phase III Trial Comparing Pembrolizumab Versus Single-Agent Chemotherapy for Advanced Pre-Treated Malignant Pleural Mesothelioma: The European Thoracic Oncology Platform (ETOP 9-15) PROMISE-meso Trial. Ann Oncol (2020) 31:1734–45. doi: 10.1016/j.annonc.2020.09.009
97. Losi L, Bertolini F, Guaitoli G, Fabbiani L, Banchelli F, Ambrosini-Spaltro A, et al. Role of Evaluating Tumor−Infiltrating Lymphocytes, Programmed Death−1 Ligand 1 and Mismatch Repair Proteins Expression in Malignant Mesothelioma. Int J Oncol (2019) 55:1157–64. doi: 10.3892/ijo.2019.4883
98. Jin L, Gu W, Li X, Xie L, Wang L, Chen Z. PD-L1 and Prognosis in Patients With Malignant Pleural Mesothelioma: A Meta-Analysis and Bioinformatics Study. Ther Adv Med Oncol (2020) 12:1758835920962362. doi: 10.1177/1758835920962362
99. Khanna S, Thomas A, Abate-Daga D, Zhang J, Morrow B, Steinberg SM, et al. Malignant Mesothelioma Effusions Are Infiltrated By CD3+ T Cells Highly Expressing PD-L1 and the PD-L1+ Tumor Cells Within These Effusions Are Susceptible to ADCC by the Anti-PD-L1 Antibody Avelumab. J Thorac Oncol (2016) 11:1993–2005. doi: 10.1016/j.jtho.2016.07.033
100. Chee J, Watson MW, Chopra A, Nguyen B, Cook AM, Creaney J, et al. Tumour Associated Lymphocytes in the Pleural Effusions of Patients With Mesothelioma Express High Levels of Inhibitory Receptors. BMC Res Notes (2018) 11(1):864. doi: 10.1186/s13104-018-3953-x
101. Wu C, Mairinger F, Casanova R, Batavia AA, Leblond A-L, Soltermann A. Prognostic Immune Cell Profiling of Malignant Pleural Effusion Patients by Computerized Immunohistochemical and Transcriptional Analysis. Cancers (Basel) (2019) 11(12):1953. doi: 10.3390/cancers11121953
102. Alcala N, Mangiante L, Le-Stang N, Gustafson CE, Boyault S, Damiola F, et al. Redefining Malignant Pleural Mesothelioma Types as a Continuum Uncovers Immune-Vascular Interactions. EBioMedicine (2019) 48:191–202. doi: 10.1016/j.ebiom.2019.09.003
103. Lines JL, Pantazi E, Mak J, Sempere LF, Wang L, O’Connell S, et al. VISTA is An Immune Checkpoint Molecule for Human T Cells. Cancer Res (2014) 74:1924–32. doi: 10.1158/0008-5472.CAN-13-1504
104. Wang L, Pino-Lagos K, de Vries VC, Guleria I, Sayegh MH, Noelle RJ. Programmed Death 1 Ligand Signaling Regulates the Generation of Adaptive Foxp3+CD4+ Regulatory T Cells. Proc Natl Acad Sci USA (2008) 105:9331–6. doi: 10.1073/pnas.0710441105
105. Hmeljak J, Sanchez-Vega F, Hoadley KA, Shih J, Stewart C, Heiman D, et al. Integrative Molecular Characterization of Malignant Pleural Mesothelioma. Cancer Discovery (2018) 8:1548–65. doi: 10.1158/2159-8290.CD-18-0804
106. Muller S, Victoria Lai W, Adusumilli PS, Desmeules P, Frosina D, Jungbluth A, et al. V-Domain Ig-containing Suppressor of T-Cell Activation (VISTA), a Potentially Targetable Immune Checkpoint Molecule, Is Highly Expressed in Epithelioid Malignant Pleural Mesothelioma. Mod Pathol (2020) 33:303–11. doi: 10.1038/s41379-019-0364-z
107. Sautès-Fridman C, Petitprez F, Calderaro J, Fridman WH. Tertiary Lymphoid Structures in the Era of Cancer Immunotherapy. Nat Rev Cancer (2019) 19:307–25. doi: 10.1038/s41568-019-0144-6
108. Ladányi A, Kiss J, Mohos A, Somlai B, Liszkay G, Gilde K, et al. Prognostic Impact of B-Cell Density in Cutaneous Melanoma. Cancer Immunol Immunother (2011) 60:1729–38. doi: 10.1007/s00262-011-1071-x
109. Krishnan S, Bakker E, Lee C, Kissick HT, Ireland DJ, Beilharz MW. Successful Combined Intratumoral Immunotherapy of Established Murine Mesotheliomas Requires B-cell Involvement. J Interferon Cytokine Res (2015) 35:100–7. doi: 10.1089/jir.2014.0054
110. Jackaman C, Cornwall S, Graham PT, Nelson DJ. CD40-Activated B Cells Contribute to Mesothelioma Tumor Regression. Immunol Cell Biol (2011) 89:255–67. doi: 10.1038/icb.2010.88
111. Shigematsu Y, Hanagiri T, Kuroda K, Baba T, Mizukami M, Ichiki Y, et al. Malignant Mesothelioma-Associated Antigens Recognized by Tumor-Infiltrating B Cells and the Clinical Significance of the Antibody Titers. Cancer Sci (2009) 100:1326–34. doi: 10.1111/j.1349-7006.2009.01181.x
112. Nicolini F, Bocchini M, Angeli D, Bronte G, Delmonte A, Crinò L, et al. Fully Human Antibodies for Malignant Pleural Mesothelioma Targeting. Cancers (Basel) (2020) 12(4):915. doi: 10.3390/cancers12040915
113. Carbone M, Adusumilli PS, Alexander HR, Baas P, Bardelli F, Bononi A, et al. Mesothelioma: Scientific Clues for Prevention, Diagnosis, and Therapy. CA Cancer J Clin (2019) 69:402–29. doi: 10.3322/caac.21572
114. McGee RB, Nichols KE. Introduction to Cancer Genetic Susceptibility Syndromes. Hematol Am Soc Hematol Educ Program (2016) 2016:293–301. doi: 10.1182/asheducation-2016.1.293
115. Pastorino L, Andreotti V, Dalmasso B, Vanni I, Ciccarese G, Mandalà M, et al. Insights Into Genetic Susceptibility to Melanoma by Gene Panel Testing: Potential Pathogenic Variants in ACD, Atm, BAP1, and POT1. Cancers (2020) 12:1007. doi: 10.3390/cancers12041007
116. Carbone M, Harbour JW, Brugarolas J, Bononi A, Pagano I, Dey A, et al. Biological Mechanisms and Clinical Significance of BAP1 Mutations in Human Cancer. Cancer Discovery (2020) 10:1103–20. doi: 10.1158/2159-8290.CD-19-1220
117. Hong JH, Chong ST, Lee P-H, Tan J, Heng HL, Ishak NDB, et al. Functional Characterisation Guides Classification of Novel BAP1 Germline Variants. NPJ Genomic Med (2020) 5:1–10. doi: 10.1038/s41525-020-00157-6
118. Betti M, Casalone E, Ferrante D, Aspesi A, Morleo G, Biasi A, et al. Germline Mutations in DNA Repair Genes Predispose Asbestos-Exposed Patients to Malignant Pleural Mesothelioma. Cancer Lett (2017) 405:38–45. doi: 10.1016/j.canlet.2017.06.028
119. Betti M, Aspesi A, Ferrante D, Sculco M, Righi L, Mirabelli D, et al. Sensitivity to Asbestos Is Increased in Patients With Mesothelioma and Pathogenic Germline Variants in BAP1 or Other DNA Repair Genes. Genes Chromosomes Cancer (2018) 57:573–83. doi: 10.1002/gcc.22670
120. Pastorino S, Yoshikawa Y, Pass HI, Emi M, Nasu M, Pagano I, et al. A Subset of Mesotheliomas With Improved Survival Occurring in Carriers of BAP1 and Other Germline Mutations. J Clin Oncol (2018) 36(35):JCO2018790352. doi: 10.1200/JCO.2018.79.0352
121. Panou V, Gadiraju M, Wolin A, Weipert CM, Skarda E, Husain AN, et al. Frequency of Germline Mutations in Cancer Susceptibility Genes in Malignant Mesothelioma. J Clin Oncol (2018) 36:2863–71. doi: 10.1200/JCO.2018.78.5204
122. Bononi A, Goto K, Ak G, Yoshikawa Y, Emi M, Pastorino S, et al. Heterozygous Germline BLM Mutations Increase Susceptibility to Asbestos and Mesothelioma. PNAS (2020) 117:33466–73. doi: 10.1073/pnas.2019652117
123. Pilié PG, Tang C, Mills GB, Yap TA. State-of-the-Art Strategies for Targeting the DNA Damage Response in Cancer. Nat Rev Clin Oncol (2019) 16:81–104. doi: 10.1038/s41571-018-0114-z
124. Fuso Nerini I, Roca E, Mannarino L, Grosso F, Frapolli R, D’Incalci M. Is DNA Repair a Potential Target for Effective Therapies Against Malignant Mesothelioma? Cancer Treat Rev (2020) 90:102101. doi: 10.1016/j.ctrv.2020.102101
125. Guo R, DuBoff M, Jayakumaran G, Kris MG, Ladanyi M, Robson ME, et al. Novel Germline Mutations in DNA Damage Repair in Patients With Malignant Pleural Mesotheliomas. J Thorac Oncol (2020) 15:655–60. doi: 10.1016/j.jtho.2019.12.111
126. Rathkey D, Khanal M, Murai J, Zhang J, Sengupta M, Jiang Q, et al. Sensitivity of Mesothelioma Cells to PARP Inhibitors Is Not Dependent on BAP1 But Is Enhanced by Temozolomide in Cells With High-Schlafen 11 and Low-O6-Methylguanine-DNA Methyltransferase Expression. J Thorac Oncol (2020) 15:843–59. doi: 10.1016/j.jtho.2020.01.012
127. Hung YP, Dong F, Watkins JC, Nardi V, Bueno R, Dal Cin P, et al. Identification of ALK Rearrangements in Malignant Peritoneal Mesothelioma. JAMA Oncol (2018) 4:235–8. doi: 10.1001/jamaoncol.2017.2918
128. Zauderer MG, Szlosarek PW, Le Moulec S, Popat S, Taylor P, Planchard D, et al. Safety and Efficacy of Tazemetostat, an Enhancer of Zeste-Homolog 2 Inhibitor, in Patients With Relapsed or Refractory Malignant Mesothelioma. JCO (2020) 38:9058–8. doi: 10.1200/JCO.2020.38.15_suppl.9058
129. Sato T, Sekido Y. NF2/Merlin Inactivation and Potential Therapeutic Targets in Mesothelioma. Int J Mol Sci (2018) 19(4):988. doi: 10.3390/ijms19040988
130. Wörthmüller J, Rüegg C. The Crosstalk Between FAK and Wnt Signaling Pathways in Cancer and Its Therapeutic Implication. Int J Mol Sci (2020) 21:9107. doi: 10.3390/ijms21239107
131. Popat S, Sharma B, MacMahon S, Nicholson AG, Sharma RK, Schuster K, et al. Durable Response to Vismodegib in PTCH1 F1147fs Mutant Relapsed Malignant Pleural Mesothelioma: Implications for Mesothelioma Drug Treatment. JCO Precis Oncol (2021) 5:39–43. doi: 10.1200/PO.20.00260
132. Pirker C, Bilecz A, Grusch M, Mohr T, Heidenreich B, Laszlo V, et al. Telomerase Reverse Transcriptase Promoter Mutations Identify a Genomically Defined and Highly Aggressive Human Pleural Mesothelioma Subgroup. Clin Cancer Res (2020) 26:3819–30. doi: 10.1158/1078-0432.CCR-19-3573
133. Metaxas Y, Früh M, Eboulet EI, Grosso F, Pless M, Zucali PA, et al. Lurbinectedin as Second- or Third-Line Palliative Therapy in Malignant Pleural Mesothelioma: An International, Multi-Centre, Single-Arm, Phase II Trial (SAKK 17/16). Ann Oncol (2020) 31:495–500. doi: 10.1016/j.annonc.2019.12.009
134. Arulananda S, Lee EF, Fairlie WD, John T. The Role of BCL-2 Family Proteins and Therapeutic Potential of BH3-Mimetics in Malignant Pleural Mesothelioma. Expert Rev Anticancer Ther (2020) 21(4):413–24. doi: 10.1080/14737140.2021.1856660
135. Krug LM, Kindler HL, Calvert H, Manegold C, Tsao AS, Fennell D, et al. Vorinostat in Patients With Advanced Malignant Pleural Mesothelioma Who Have Progressed on Previous Chemotherapy (VANTAGE-014): A Phase 3, Double-Blind, Randomised, Placebo-Controlled Trial. Lancet Oncol (2015) 16:447–56. doi: 10.1016/S1470-2045(15)70056-2
136. Dell’Anno I, Martin SA, Barbarino M, Melani A, Silvestri R, Bottaro M, et al. Drug-Repositioning Screening Identified Fludarabine and Risedronic Acid as Potential Therapeutic Compounds for Malignant Pleural Mesothelioma. Invest New Drugs (2020) 39(3):644–57. doi: 10.1007/s10637-020-01040-y
137. Borchert S, Wessolly M, Schmeller J, Mairinger E, Kollmeier J, Hager T, et al. Gene Expression Profiling of Homologous Recombination Repair Pathway Indicates Susceptibility for Olaparib Treatment in Malignant Pleural Mesothelioma In Vitro. BMC Cancer (2019) 19:108. doi: 10.1186/s12885-019-5314-0
138. Bueno R, Stawiski EW, Goldstein LD, Durinck S, De Rienzo A, Modrusan Z, et al. Comprehensive Genomic Analysis of Malignant Pleural Mesothelioma Identifies Recurrent Mutations, Gene Fusions and Splicing Alterations. Nat Genet (2016) 48:407–16. doi: 10.1038/ng.3520
139. Zhang M, Luo J-L, Sun Q, Harber J, Dawson AG, Nakas A, et al. Clonal Architecture in Mesothelioma Is Prognostic and Shapes the Tumour Microenvironment. Nat Commun (2021) 12:1751. doi: 10.1038/s41467-021-21798-w
140. Kettunen E, Savukoski S, Salmenkivi K, Böhling T, Vanhala E, Kuosma E, et al. CDKN2A Copy Number and p16 Expression in Malignant Pleural Mesothelioma in Relation to Asbestos Exposure. BMC Cancer (2019) 19:507. doi: 10.1186/s12885-019-5652-y
141. Hung YP, Dong F, Torre M, Crum CP, Bueno R, Chirieac LR. Molecular Characterization of Diffuse Malignant Peritoneal Mesothelioma. Modern Pathol (2020) 33:2269–79. doi: 10.1038/s41379-020-0588-y
142. Rüschoff JH, Gradhand E, Kahraman A, Rees H, Ferguson JL, Curioni-Fontecedro A, et al. STRN-ALK Rearranged Malignant Peritoneal Mesothelioma With Dramatic Response Following Ceritinib Treatment. JCO Precis Oncol (2019) 3:PO.19.00048. doi: 10.1200/PO.19.00048
143. Torricelli F, Lococo F, Di Stefano TS, Lorenzini E, Piana S, Valli R, et al. Deep Sequencing Analysis Identified a Specific Subset of Mutations Distinctive of Biphasic Malignant Pleural Mesothelioma. Cancers (2020) 12:2454. doi: 10.3390/cancers12092454
144. Pagano M, Ceresoli LG, Zucali PA, Pasello G, Garassino M, Grosso F, et al. Mutational Profile of Malignant Pleural Mesothelioma (MPM) in the Phase Ii RAMES Study. Cancers (2020) 12:2948. doi: 10.3390/cancers12102948
145. Torricelli F, Saxena A, Nuamah R, Neat M, Harling L, Ng W, et al. Genomic Analysis in Short- and Long-Term Patients With Malignant Pleura Mesothelioma Treated With Palliative Chemotherapy. Eur J Cancer (2020) 132:104–11. doi: 10.1016/j.ejca.2020.03.002
146. Carbone M, Yang H, Gaudino G. Does Chromothripsis Make Mesothelioma an Immunogenic Cancer? J Thorac Oncol (2019) 14:157–9. doi: 10.1016/j.jtho.2018.11.006
147. Stephens PJ, Greenman CD, Fu B, Yang F, Bignell GR, Mudie LJ, et al. Massive Genomic Rearrangement Acquired in a Single Catastrophic Event During Cancer Development. Cell (2011) 144:27–40. doi: 10.1016/j.cell.2010.11.055
148. Tubio JMC, Estivill X. Cancer: When Catastrophe Strikes a Cell. Nature (2011) 470:476–7. doi: 10.1038/470476a
149. Voronina N, Wong JKL, Hübschmann D, Hlevnjak M, Uhrig S, Heilig CE, et al. The Landscape of Chromothripsis Across Adult Cancer Types. Nat Commun (2020) 11:2320. doi: 10.1038/s41467-020-16134-7
150. Mansfield AS, Peikert T, Vasmatzis G. Chromosomal Rearrangements and Their Neoantigenic Potential in Mesothelioma. Transl Lung Cancer Res (2020) 9:S92–9. doi: 10.21037/tlcr.2019.11.12
151. Oey H, Daniels M, Relan V, Chee TM, Davidson MR, Yang IA, et al. Whole-Genome Sequencing of Human Malignant Mesothelioma Tumours and Cell Lines. Carcinogenesis (2019) 40:724–34. doi: 10.1093/carcin/bgz066
152. Shoshani O, Brunner SF, Yaeger R, Ly P, Nechemia-Arbely Y, Kim DH, et al. Chromothripsis Drives the Evolution of Gene Amplification in Cancer. Nature (2020) 591(7848):137–41. doi: 10.1038/s41586-020-03064-z
153. van Haaften G, Dalgliesh GL, Davies H, Chen L, Bignell G, Greenman C, et al. Somatic Mutations of the Histone H3K27 Demethylase Gene UTX in Human Cancer. Nat Genet (2009) 41:521–3. doi: 10.1038/ng.349
154. Cortés-Ciriano I, Lee JJ-K, Xi R, Jain D, Jung YL, Yang L, et al. Comprehensive Analysis of Chromothripsis in 2,658 Human Cancers Using Whole-Genome Sequencing. Nat Genet (2020) 52:331–41. doi: 10.1038/s41588-019-0576-7
155. Wadowski B, De Rienzo A, Bueno R. The Molecular Basis of Malignant Pleural Mesothelioma. Thorac Surg Clin (2020) 30:383–93. doi: 10.1016/j.thorsurg.2020.08.005
156. Kaneda A, Seike T, Danjo T, Nakajima T, Otsubo N, Yamaguchi D, et al. The Novel Potent TEAD Inhibitor, K-975, Inhibits YAP1/TAZ-TEAD Protein-Protein Interactions and Exerts an Anti-Tumor Effect on Malignant Pleural Mesothelioma. Am J Cancer Res (2020) 10:4399–415.
157. Cortinovis D, Grosso F, Carlucci L, Zucali PA, Pasello G, Tiseo M, et al. Trabectedin in Malignant Pleural Mesothelioma: Results From the Multicentre, Single Arm, Phase II Atreus Study. Clin Lung Cancer (2020) S1525-7304(20)30222-9. doi: 10.1016/j.cllc.2020.06.028
158. Barreca M, Spanò V, Montalbano A, Cueto M, Díaz Marrero AR, Deniz I, et al. Marine Anticancer Agents: An Overview With a Particular Focus on Their Chemical Classes. Mar Drugs (2020) 18(12):619. doi: 10.3390/md18120619
159. Garzon R, Calin GA, Croce CM. MicroRNAs in Cancer. Annu Rev Med (2009) 60:167–79. doi: 10.1146/annurev.med.59.053006.104707
160. Peng Y, Croce CM. The Role of MicroRNAs in Human Cancer. Signal Transduct Target Ther (2016) 1:15004. doi: 10.1038/sigtrans.2015.4
161. Calin GA, Dumitru CD, Shimizu M, Bichi R, Zupo S, Noch E, et al. Frequent Deletions and Down-Regulation of Micro- RNA Genes miR15 and miR16 at 13q14 in Chronic Lymphocytic Leukemia. Proc Natl Acad Sci USA (2002) 99:15524–9. doi: 10.1073/pnas.242606799
162. Behl T, Kumar C, Makkar R, Gupta A, Sachdeva M. Intercalating the Role of MicroRNAs in Cancer: As Enemy or Protector. Asian Pac J Cancer Prev (2020) 21:593–8. doi: 10.31557/APJCP.2020.21.3.593
163. Yi M, Xu L, Jiao Y, Luo S, Li A, Wu K. The Role of Cancer-Derived microRNAs in Cancer Immune Escape. J Hematol Oncol (2020) 13:25. doi: 10.1186/s13045-020-00848-8
164. Si W, Shen J, Zheng H, Fan W. The Role and Mechanisms of Action of microRNAs in Cancer Drug Resistance. Clin Epigenet (2019) 11:25. doi: 10.1186/s13148-018-0587-8
165. Reid G. MicroRNAs in Mesothelioma: From Tumour Suppressors and Biomarkers to Therapeutic Targets. J Thorac Dis (2015) 7:1031–40. doi: 10.3978/j.issn.2072-1439.2015.04.56
166. Birnie KA, Prêle CM, Thompson PJ, Badrian B, Mutsaers SE. Targeting microRNA to Improve Diagnostic and Therapeutic Approaches for Malignant Mesothelioma. Oncotarget (2017) 8:78193–207. doi: 10.18632/oncotarget.20409
167. Reid G, Johnson TG, van Zandwijk N. Manipulating microRNAs for the Treatment of Malignant Pleural Mesothelioma: Past, Present and Future. Front Oncol (2020) 10:105. doi: 10.3389/fonc.2020.00105
168. Guled M, Lahti L, Lindholm PM, Salmenkivi K, Bagwan I, Nicholson AG, et al. CDKN2A, NF2, and JUN Are Dysregulated Among Other Genes by miRNAs in Malignant Mesothelioma -a miRNA Microarray Analysis. Genes Chromosomes Cancer (2009) 48:615–23. doi: 10.1002/gcc.20669
169. Busacca S, Germano S, De Cecco L, Rinaldi M, Comoglio F, Favero F, et al. MicroRNA Signature of Malignant Mesothelioma With Potential Diagnostic and Prognostic Implications. Am J Respir Cell Mol Biol (2010) 42:312–9. doi: 10.1165/rcmb.2009-0060OC
170. Pass HI, Goparaju C, Ivanov S, Donington J, Carbone M, Hoshen M, et al. hsa-miR-29c* Is Linked to the Prognosis of Malignant Pleural Mesothelioma. Cancer Res (2010) 70:1916–24. doi: 10.1158/0008-5472.CAN-09-3993
171. Ivanov SV, Goparaju CMV, Lopez P, Zavadil J, Toren-Haritan G, Rosenwald S, et al. Pro-Tumorigenic Effects of miR-31 Loss in Mesothelioma. J Biol Chem (2010) 285:22809–17. doi: 10.1074/jbc.M110.100354
172. Cioce M, Ganci F, Canu V, Sacconi A, Mori F, Canino C, et al. Protumorigenic Effects of mir-145 Loss in Malignant Pleural Mesothelioma. Oncogene (2014) 33:5319–31. doi: 10.1038/onc.2013.476
173. Fassina A, Cappellesso R, Guzzardo V, Dalla Via L, Piccolo S, Ventura L, et al. Epithelial-Mesenchymal Transition in Malignant Mesothelioma. Mod Pathol (2012) 25:86–99. doi: 10.1038/modpathol.2011.144
174. Reid G, Pel ME, Kirschner MB, Cheng YY, Mugridge N, Weiss J, et al. Restoring Expression of miR-16: A Novel Approach to Therapy for Malignant Pleural Mesothelioma. Ann Oncol (2013) 24:3128–35. doi: 10.1093/annonc/mdt412
175. Kao SC, Fulham M, Wong K, Cooper W, Brahmbhatt H, MacDiarmid J, et al. A Significant Metabolic and Radiological Response After a Novel Targeted MicroRNA-based Treatment Approach in Malignant Pleural Mesothelioma. Am J Respir Crit Care Med (2015) 191:1467–9. doi: 10.1164/rccm.201503-0461LE
176. van Zandwijk N, Pavlakis N, Kao SC, Linton A, Boyer MJ, Clarke S, et al. Safety and Activity of microRNA-loaded Minicells in Patients With Recurrent Malignant Pleural Mesothelioma: A First-in-Man, Phase 1, Open-Label, Dose-Escalation Study. Lancet Oncol (2017) 18:1386–96. doi: 10.1016/S1470-2045(17)30621-6
177. Kao SC, Cheng YY, Williams M, Kirschner MB, Madore J, Lum T, et al. Tumor Suppressor Micrornas Contribute to the Regulation of PD-L1 Expression in Malignant Pleural Mesothelioma. J Thorac Oncol (2017) 12:1421–33. doi: 10.1016/j.jtho.2017.05.024
178. Williams M, Kirschner MB, Cheng YY, Hanh J, Weiss J, Mugridge N, et al. miR-193a-3p is a Potential Tumor Suppressor in Malignant Pleural Mesothelioma. Oncotarget (2015) 6:23480–95. doi: 10.18632/oncotarget.4346
179. Johnson TG, Schelch K, Cheng YY, Williams M, Sarun KH, Kirschner MB, et al. Dysregulated Expression of the MicroRNA miR-137 and Its Target YBX1 Contribute to the Invasive Characteristics of Malignant Pleural Mesothelioma. J Thorac Oncol (2018) 13:258–72. doi: 10.1016/j.jtho.2017.10.016
180. Tomasetti M, Re M, Monaco F, Gaetani S, Rubini C, Bertini A, et al. MiR-126 in Intestinal-Type Sinonasal Adenocarcinomas: Exosomal Transfer of MiR-126 Promotes Anti-Tumour Responses. BMC Cancer (2018) 18:896. doi: 10.1186/s12885-018-4801-z
181. Tomasetti M, Staffolani S, Nocchi L, Neuzil J, Strafella E, Manzella N, et al. Clinical Significance of Circulating miR-126 Quantification in Malignant Mesothelioma Patients. Clin Biochem (2012) 45:575–81. doi: 10.1016/j.clinbiochem.2012.02.009
182. Kubo T, Toyooka S, Tsukuda K, Sakaguchi M, Fukazawa T, Soh J, et al. Epigenetic Silencing of microRNA-34b/c Plays an Important Role in the Pathogenesis of Malignant Pleural Mesothelioma. Clin Cancer Res (2011) 17:4965–74. doi: 10.1158/1078-0432.CCR-10-3040
183. Ueno T, Toyooka S, Fukazawa T, Kubo T, Soh J, Asano H, et al. Preclinical Evaluation of microRNA-34b/c Delivery for Malignant Pleural Mesothelioma. Acta Med Okayama (2014) 68:23–6. doi: 10.18926/AMO/52140
184. Singh A, Bhattacharyya N, Srivastava A, Pruett N, Ripley RT, Schrump DS, et al. Microrna-215-5p Treatment Suppresses Mesothelioma Progression Via the MDM2-p53-Signaling Axis. Mol Ther (2019) 27:1665–80. doi: 10.1016/j.ymthe.2019.05.020
185. Suzuki R, Amatya VJ, Kushitani K, Kai Y, Kambara T, Takeshima Y. miR-182 and Mir-183 Promote Cell Proliferation and Invasion by Targeting FOXO1 in Mesothelioma. Front Oncol (2018) 8:446. doi: 10.3389/fonc.2018.00446
186. Gee GV, Koestler DC, Christensen BC, Sugarbaker DJ, Ugolini D, Ivaldi GP, et al. Downregulated microRNAs in the Differential Diagnosis of Malignant Pleural Mesothelioma. Int J Cancer (2010) 127:2859–69. doi: 10.1002/ijc.25285
187. Benjamin H, Lebanony D, Rosenwald S, Cohen L, Gibori H, Barabash N, et al. A Diagnostic Assay Based on microRNA Expression Accurately Identifies Malignant Pleural Mesothelioma. J Mol Diagn (2010) 12:771–9. doi: 10.2353/jmoldx.2010.090169
188. Santarelli L, Strafella E, Staffolani S, Amati M, Emanuelli M, Sartini D, et al. Association of MiR-126 With Soluble Mesothelin-Related Peptides, a Marker for Malignant Mesothelioma. PloS One (2011) 6:e18232. doi: 10.1371/journal.pone.0018232
189. Andersen M, Grauslund M, Ravn J, Sørensen JB, Andersen CB, Santoni-Rugiu E. Diagnostic Potential of miR-126, miR-143, miR-145, and miR-652 in Malignant Pleural Mesothelioma. J Mol Diagn (2014) 16:418–30. doi: 10.1016/j.jmoldx.2014.03.002
190. Matsumoto S, Nabeshima K, Hamasaki M, Shibuta T, Umemura T. Upregulation of microRNA-31 Associates With a Poor Prognosis of Malignant Pleural Mesothelioma With Sarcomatoid Component. Med Oncol (2014) 31:303. doi: 10.1007/s12032-014-0303-2
191. Kirschner MB, Cheng YY, Armstrong NJ, Lin RCY, Kao SC, Linton A, et al. MiR-score: A Novel 6-microRNA Signature That Predicts Survival Outcomes in Patients With Malignant Pleural Mesothelioma. Mol Oncol (2015) 9:715–26. doi: 10.1016/j.molonc.2014.11.007
192. Weber DG, Casjens S, Johnen G, Bryk O, Raiko I, Pesch B, et al. Combination of MiR-103a-3p and Mesothelin Improves the Biomarker Performance of Malignant Mesothelioma Diagnosis. PloS One (2014) 9:e114483. doi: 10.1371/journal.pone.0114483
193. Weber DG, Johnen G, Bryk O, Jöckel K-H, Brüning T. Identification of miRNA-103 in the Cellular Fraction of Human Peripheral Blood as a Potential Biomarker for Malignant Mesothelioma–A Pilot Study. PloS One (2012) 7:e30221. doi: 10.1371/journal.pone.0030221
194. Santarelli L, Staffolani S, Strafella E, Nocchi L, Manzella N, Grossi P, et al. Combined Circulating Epigenetic Markers to Improve Mesothelin Performance in the Diagnosis of Malignant Mesothelioma. Lung Cancer (2015) 90:457–64. doi: 10.1016/j.lungcan.2015.09.021
195. Weber DG, Gawrych K, Casjens S, Brik A, Lehnert M, Taeger D, et al. Circulating miR-132-3p as a Candidate Diagnostic Biomarker for Malignant Mesothelioma. Dis Markers (2017) 2017:9280170. doi: 10.1155/2017/9280170
196. Kirschner MB, Cheng YY, Badrian B, Kao SC, Creaney J, Edelman JJB, et al. Increased Circulating miR-625-3p: A Potential Biomarker for Patients With Malignant Pleural Mesothelioma. J Thorac Oncol (2012) 7:1184–91. doi: 10.1097/JTO.0b013e3182572e83
197. Jin B, Li Y, Robertson KD. DNA Methylation. Genes Cancer (2011) 2:607–17. doi: 10.1177/1947601910393957
198. Yoshikawa Y, Kuribayashi K, Minami T, Ohmuraya M, Kijima T. Epigenetic Alterations and Biomarkers for Immune Checkpoint Inhibitors–Current Standards and Future Perspectives in Malignant Pleural Mesothelioma Treatment. Front Oncol (2020) 10:554570. doi: 10.3389/fonc.2020.554570
199. Park JW, Han J-W. Targeting Epigenetics for Cancer Therapy. Arch Pharm Res (2019) 42:159–70. doi: 10.1007/s12272-019-01126-z
200. Ferrari L, Carugno M, Mensi C, Pesatori AC. Circulating Epigenetic Biomarkers in Malignant Pleural Mesothelioma: State of the Art and Critical Evaluation. Front Oncol (2020) 10:445. doi: 10.3389/fonc.2020.00445
201. Takeshima H, Ushijima T. Accumulation of Genetic and Epigenetic Alterations in Normal Cells and Cancer Risk. NPJ Precis Oncol (2019) 3:1–8. doi: 10.1038/s41698-019-0079-0
202. Guarrera S, Viberti C, Cugliari G, Allione A, Casalone E, Betti M, et al. Peripheral Blood DNA Methylation as Potential Biomarker of Malignant Pleural Mesothelioma in Asbestos-Exposed Subjects. J Thorac Oncol (2019) 14:527–39. doi: 10.1016/j.jtho.2018.10.163
203. McLoughlin KC, Kaufman AS, Schrump DS. Targeting the Epigenome in Malignant Pleural Mesothelioma. Transl Lung Cancer Res (2017) 6:350–65. doi: 10.21037/tlcr.2017.06.06
204. Nakatsumi H, Matsumoto M, Nakayama KI. Noncanonical Pathway for Regulation of CCL2 Expression by An mTORC1-FOXK1 Axis Promotes Recruitment of Tumor-Associated Macrophages. Cell Rep (2017) 21:2471–86. doi: 10.1016/j.celrep.2017.11.014
205. Cugliari G, Catalano C, Guarrera S, Allione A, Casalone E, Russo A, et al. Dna Methylation of FKBP5 as Predictor of Overall Survival in Malignant Pleural Mesothelioma. Cancers (Basel) (2020) 12(11):3470. doi: 10.3390/cancers12113470
206. Goto Y, Shinjo K, Kondo Y, Shen L, Toyota M, Suzuki H, et al. Epigenetic Profiles Distinguish Malignant Pleural Mesothelioma From Lung Adenocarcinoma. Cancer Res (2009) 69:9073–82. doi: 10.1158/0008-5472.CAN-09-1595
207. Cakiroglu E, Senturk S. Genomics and Functional Genomics of Malignant Pleural Mesothelioma. Int J Mol Sci (2020) 21(17):6342. doi: 10.3390/ijms21176342
208. Garassino MC, Marsoni S. A Lesson From Vorinostat in Pleural Mesothelioma. Lancet Oncol (2015) 16:359–60. doi: 10.1016/S1470-2045(15)70084-7
Keywords: mesothelioma, tumor microenvironment, genetic alterations, immunotherapy, targeted therapy
Citation: Hiltbrunner S, Mannarino L, Kirschner MB, Opitz I, Rigutto A, Laure A, Lia M, Nozza P, Maconi A, Marchini S, D’Incalci M, Curioni-Fontecedro A and Grosso F (2021) Tumor Immune Microenvironment and Genetic Alterations in Mesothelioma. Front. Oncol. 11:660039. doi: 10.3389/fonc.2021.660039
Received: 28 January 2021; Accepted: 25 May 2021;
Published: 23 June 2021.
Edited by:
Giulia Pasello, Veneto Institute of Oncology (IRCCS), ItalyReviewed by:
Luana Calabro’, Siena University Hospital, ItalyCopyright © 2021 Hiltbrunner, Mannarino, Kirschner, Opitz, Rigutto, Laure, Lia, Nozza, Maconi, Marchini, D’Incalci, Curioni-Fontecedro and Grosso. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Federica Grosso, ZmVkZXJpY2EuZ3Jvc3NvQG9zcGVkYWxlLmFsLml0
†These authors share first authorship
‡These authors share last authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.