
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Oncol. , 03 May 2021
Sec. Molecular and Cellular Oncology
Volume 11 - 2021 | https://doi.org/10.3389/fonc.2021.658151
This article is part of the Research Topic lncRNAs in Cancer Metastasis and Therapy Resistance View all 41 articles
 Chung-Hsien Shih1Li-Ling Chuang2,3Mong-Hsun Tsai4,5Li-Han Chen6Eric Y. Chuang5,7,8
Chung-Hsien Shih1Li-Ling Chuang2,3Mong-Hsun Tsai4,5Li-Han Chen6Eric Y. Chuang5,7,8 Tzu-Pin Lu5,9
Tzu-Pin Lu5,9 Liang-Chuan Lai1,5*
Liang-Chuan Lai1,5*Hypoxia, a common process during tumor growth, can lead to tumor aggressiveness and is tightly associated with poor prognosis. Long noncoding RNAs (lncRNAs) are long ribonucleotides (>200 bases) with limited ability to translate proteins, and are known to affect many aspects of cellular function. One of their regulatory mechanisms is to function as a sponge for microRNA (miRNA) to modulate its biological functions. Previously, MALAT1 was identified as a hypoxia-induced lncRNA. However, the regulatory mechanism and functions of MALAT1 in breast cancer are still unclear. Therefore, we explored whether MALAT1 can regulate the functions of breast cancer cells through miRNAs. Our results showed the expression levels of MALAT1 were significantly up-regulated under hypoxia and regulated by HIF-1α and HIF-2α. Next, in contrast to previous reports, nuclear and cytoplasmic fractionation assays and fluorescence in situ hybridization indicated that MALAT1 was mainly located in the cytoplasm. Therefore, the labeling of MALAT1 as a nuclear marker should be done with the caveat. Furthermore, expression levels of miRNAs and RNA immunoprecipitation using antibody against AGO2 showed that MALAT1 functioned as a sponge of miRNA miR-3064-5p. Lastly, functional assays revealed that MALAT1 could promote cellular migration and proliferation of breast cancer cells. Our findings provide evidence that hypoxia-responsive long non-coding MALAT1 could be transcriptionally activated by HIF-1α and HIF-2α, act as a miRNA sponge of miR-3064-5p, and promote tumor growth and migration in breast cancer cells. These data suggest that MALAT1 may be a candidate for therapeutic targeting of breast cancer progression.
Several studies have confirmed that the tumor microenvironment promotes cancer progression in many ways, especially via therapeutic resistance. Hypoxia is a common feature of malignant tumors (1). It has been described as a complicated incident of the tumor microenvironment that promotes tumor aggressiveness and metastasis (2, 3), and is strongly associated with poor prognosis (4, 5). Hypoxia is harmful to cancer cells, but it drives their adaptation, thereby promoting malignant progression (6, 7). In response to hypoxia, cancer cells exhibit modified expression of numerous genes regulated by hypoxia-inducible factors (HIFs), the major components of hypoxia signaling pathways (8). Most of the HIF-dependent responses rely on changes in the expression of genes associated with angiogenesis, proliferation, epithelial to mesenchymal transition, and metastasis (9). These changes allow malignant cells to survive the harsh hypoxic environment. However, the details of how hypoxia leads to tumor progression remain to be identified.
Long noncoding RNAs (lncRNAs) are transcripts which are longer than 200 nucleotides but have limited protein-coding capacity (10). Emerging evidence has shown that lncRNAs are a critical factor for both normal development and tumorigenesis (11, 12), and that they participate in epigenetic regulation of gene expression (13, 14). In recent studies, lncRNAs such as metastasis-associated lung adenocarcinoma transcript 1 (MALAT1) have been shown to participate in cancer progression. MALAT1 was initially identified as being up-regulated in primary human non-small cell lung cancer cells with higher metastasis ability (15), and was associated with metastasis and survival of cancer cells (14). Later, it was observed to have aberrant expression levels in many cancers (16, 17), and to be involved in post−transcriptionally modified primary transcripts and regulated gene expression (18). Although hypoxia-inducible factor was a major regulator of the non-coding and coding transcriptome in hypoxia (19), the regulatory mechanism of MALAT1 in breast cancer cells remains to be clarified. In addition to lncRNA, microRNAs (miRNAs), a class of small non-coding RNA transcripts (~22 nucleotides), also regulate the gene expression levels by binding to the 3'-untranslated regions (3'-UTRs) of target mRNAs (20, 21). Many studies have reported that miRNAs are differentially expressed in hypoxia and related to various aspects of cancer pathogenesis and progression, such as cell differentiation, proliferation, migration, invasion, apoptosis, and drug resistance (22–28). Some studies have reported the interaction between lncRNA and miRNA, specifically that lncRNA can be competing endogenous RNA by acting as a sponge for miRNA (29, 30).
Previously, we used next-generation sequencing (NGS) technology to identify oxygen-responsive lncRNAs in MCF-7 breast cancer cells and identified MALAT1 as one of the top five up-regulated lncRNAs under hypoxia. However, the regulatory mechanism and function of MALAT1 in breast cancer are not known. Therefore, the purpose of this study was to explore the regulatory mechanisms and functions of MALAT1 in breast cancer cells. Expression levels of MALAT1 in MCF-7 under HIF-1A or HIF-2A overexpression were examined by quantitative RT-PCR (qPCR). Endogenous expression levels of MALAT1 in MCF-7 cells grown at different oxygen concentrations were examined by qPCR. Luciferase reporter assays verified the direct interaction between HIF-1α or HIF-2α and the putative hypoxia response elements in the MALAT1 promoter. To confirm the distribution of MALAT1 in breast cancer cells, nuclear-cytoplasmic RNA fractionation assays and RNA fluorescence in situ hybridization (FISH) were performed. To identify miRNAs affected by MALAT1, NGS was performed in MALAT1-knockdown cells under hypoxia, followed by RNA immunoprecipitation (RIP) assays using antibody against AGO2 protein, the essential component of the miRNA-induced silencing complex, and by qPCR. Furthermore, a series of functional assays of MALAT1 were performed. The results indicate a role for MALAT1 as a sponge for miRNA, which increases the metastatic potential of MCF-7 breast cancer cells.
MCF-7 and MDA-MB-231 breast cancer cells and HEK293T human embryonic kidney cells were maintained in Dulbecco’s Modified Eagle Medium (DMEM) (GIBCO, Carlsbad, CA, USA). All were supplemented with 10% fetal bovine serum (HyClone, Logan, UT, USA) and 1% antibiotics (penicillin-streptomycin) (GIBCO). Human mammary epithelial cell line MCF-10A was maintained in Dulbecco’s Modified Eagle Medium: Nutrient Mixture F-12 (DMEM/F12) (GIBCO) containing horse serum (5%), epidermal growth factor (20 ng/ml), hydrocortisone (0.5 mg/ml) (Sigma, Saint Louis, MO, USA), cholera toxin (100 ng/ml) (Sigma), insulin (10 μg/ml) (Sigma) and 1% antibiotics (penicillin-streptomycin) (GIBCO). Cells were incubated at 37°C in a humidified incubator with 5% CO2. In some experiments, cells were treated with 300 μM cobalt (II) chloride (Sigma) to mimic hypoxic conditions or cultured in a hypoxia chamber (Ruskinn Technology, Bridgend, UK) filled with a gas mixture of 0.5% O2, 5% CO2 and 94.5% N2 for 24 h.
To overexpress MALAT1, the expression plasmid pCMV-MALAT1 was kindly provided by Dr. Yi-Ching Wang, National Cheng Kung University, Taiwan. To overexpress HIF-1α and HIF-2α under normoxic conditions, pcDNA3-HIF-1α-P402A/P564A (Addgene plasmid #18955) and pcDNA3-HIF-2α-P405A/P531A (Addgene plasmid #18956) were bought from Addgene (Cambridge, MA, USA). The mutations produce proteins that resist O2-regulated prolyl hydroxylation in the oxygen-dependent degradation domain and are thus stable under normoxia. To determine promoter activity by luciferase assay, the luciferase expression plasmid pGL3 was purchased from the Biomedical Resource Core of the 1st Core Facility Lab, National Taiwan University (NTU) College of Medicine (Taipei, Taiwan). Briefly, the NDRG1-OT1 promoter region encompassing -1 ~ -2,000 bp relative to the transcription start site of MALAT1 was amplified from human genomic DNA by PCR and subcloned into the pGL3-basic vector to create the pGL3-MALAT1 promoter plasmid.
To determine the binding activity of miR-3064-5p on MALAT1, luciferase expression plasmids with mutations at the binding sites pmiR-GLO-MALAT1 S1 (1,279 - 1,302 bp) and pmiR-GLO-MALAT1 S2 (7,837 - 7,860 bp) were purchased from the Biomedical Resource Core of the 1st Core Facility Lab (NTU).
Lentiviral plasmids pLKO_TRC005_shMALAT1 #1 and pLKO_TRC005_shMALAT1 #2 encoded short hairpin RNA (shRNA) against MALAT1 (GeneID: 378938). The oligonucleotides synthesized for these shRNAs were as follows: shMALAT1 #1: 5'- GAG CGA AAG GAT GCC CAT CCG CCC TTT TTG AAT TCT AGA TCT TGA GAC AAA-3' (sense), 5'-GAG CGA AAG GAT GCC CAT CCG CCC CCG GTA CCT CGT CC-3' (antisense); shMALAT1 #2: 5'-GAG AGA GGG AAG CTC GTT AGT GCC TTT TTG AAT TCT AGA TCT TGA GAC AAA-3' (sense), 5'-GAG AGA GGG AAG CTC GTT AGT GCC CCG GTA CCT CGT CC-3' (antisense).
MCF-7 and MDA-MB-231 cells were transfected with pCMV-MALAT1 or pEYFP-N1 empty vector using jetPRIME transfection reagent (Polyplus-transfection, SA) according to the manufacturer’s instructions. Four hours later, the transfection medium was replaced with fresh medium containing serum. After 24 h, cells were checked for RNA expression by qPCR.
The lentiviral vectors were co-transfected with packaging plasmids psPAX2 and pMD2G (Addgene) into HEK293T cells. Infectious lentivirus was harvested at 24, 36, and 48 h after transfection, and filtered through 0.45 μm PVDF filters. MDA-MB-231 cells were infected with concentrated virus, and the culture medium was replaced after 24 h of incubation. Then, cells were selected by treating with puromycin for 2 days. The expression levels of MALAT1 in cells were validated by qPCR.
One of the HIF core binding motifs (hypoxia response element, HRE), located at -235 to -231 bp relative to the transcription start site of MALAT1, was identified. The seed region of miR-3064-5p was located at 1,295 ~ 1,301 and 7,853 ~ 7,859 bp of MALAT1. The mutations of the HRE in the pGL3-MALAT1 promoter plasmid and the miR-3064-5p binding site mutations of pmiR-GLO-MALAT1 were introduced by Biomedical Resource Core of the 1st Core Facility Lab (NTU). In addition, the mutated sequences were validated by sequencing.
To determine the effects of HIF-1α and HIF-2α on the MALAT1 promoter construct, HEK293T cells were seeded in 24-well plates at a density of 5×104 cells/well. After 24 h, cells were transfected with 100 ng wild-type or mutant HRE firefly luciferase reporter construct, and 2 ng Renilla luciferase plasmid (pGL3 [hRluc/TK], kindly provided by Dr. Meng-Chun Hu, NTU) using jetPRIME (Polyplus-transfection) reagent. Also, cells were transfected with 50 ng of pcDNA3-HIF1α-P402A/P564A or 100 ng of pcDNA3-HIF2α-P405A/P531ApcDNA3. After 24 h, cells were lysed in cell lysis buffer (92.8 mM K2HPO4, 9.2 mM KH2PO4 and 0.2% Triton X-100 in ddH2O), and the luciferase activity was measured using the Dual-Glo luciferase reporter assay system (Promega, Fitchburg, WI, USA) and normalized to Renilla luciferase activity.
To determine the effect of miR-3064-5p on the MALAT1 reporter construct, HEK293 cells were co-transfected with 0.025 nmol of miR-3064-5p mimic and 100 ng of the reporter vector containing the wild-type MALAT1 S1 or S2 or the mutant MALAT1 S1 or S2. After 48 h, the cells were collected, and the luciferase activities were measured using the Dual-Glo luciferase reporter assay system (Promega).
Total RNA was isolated using NucleoZOL reagent (Machery-Nagel, Düren, Germany) according to the manufacturer’s instructions. One μg of total RNA was reverse-transcribed using the High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Carlsbad, CA, USA). For reverse transcription of miRNA, SuperScript IV Reverse Transcriptase (Invitrogen, Carlsbad, CA, USA) was used with the primers listed in Table 1. Per the manufacturer’s instructions, 2.5% of each reaction was used as the template for qPCR with 5× HOT FIREPol® EvaGreen® qPCR Mix Plus (OmicsBio, New Taipei City, Taiwan), and the reactions were performed on a StepOnePlus Real-Time PCR System (Thermo Fisher, Waltham, MA, USA). The primer pairs used for detection of cDNAs are listed in Table 1. At last, the relative gene expression levels were normalized to 18S rRNA or U6 using the 2-ΔΔCt method.
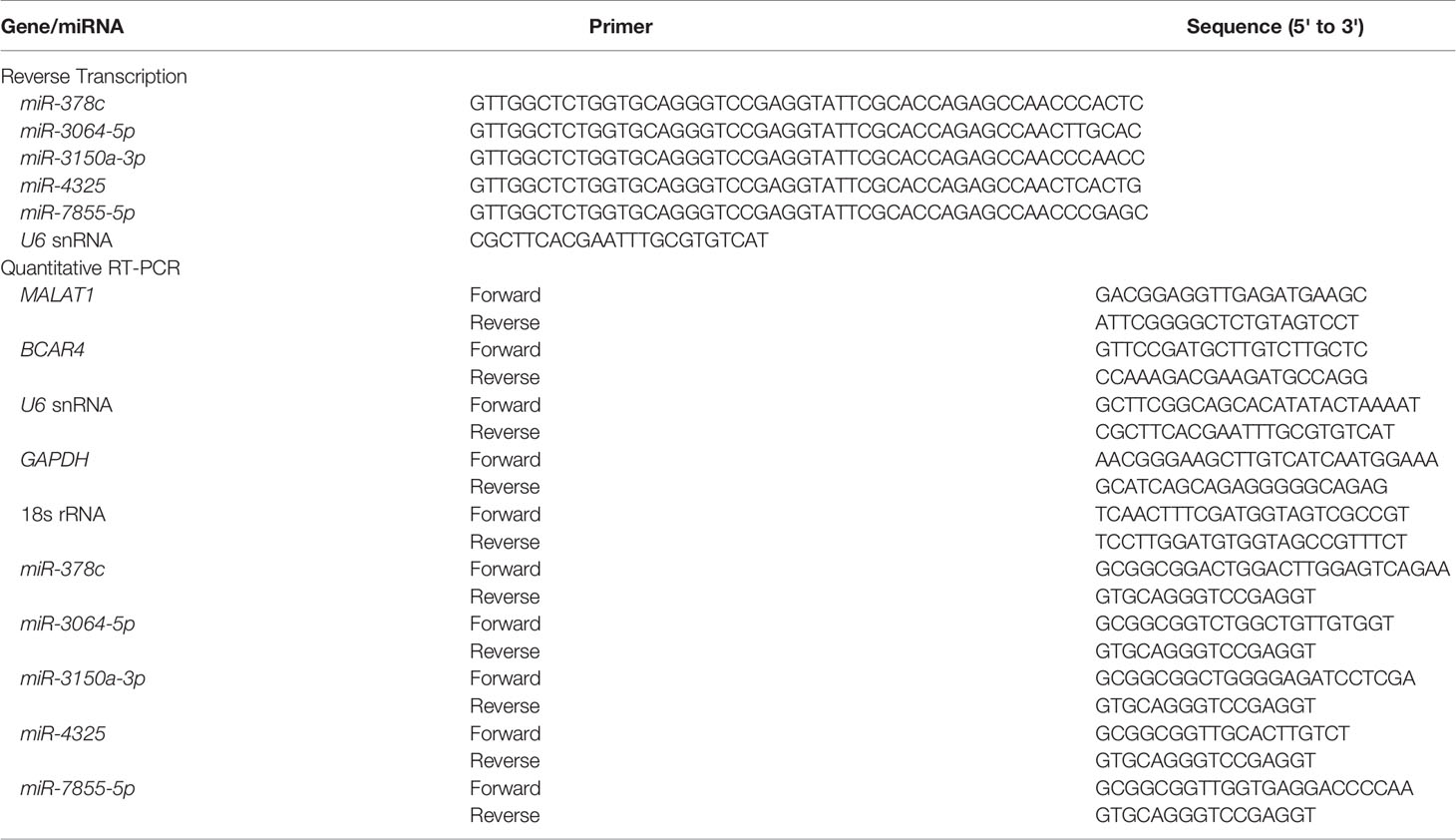
Table 1 The primers for reverse transcription and quantitative RT-PCR.
To determine the subcellular localization of RNA, fractionation of nuclear and cytoplasmic RNA was performed using the Cytoplasmic & Nuclear RNA Purification Kit (Norgen Biotek, Thorold, ON, Canada). Cells were first lysed with Lysis Buffer J (Norgen Biotek), and the lysate was separated by centrifugation, after which the supernatant contained the cytoplasmic RNA and the pellet contained the nuclear RNA. Buffer SK (Norgen Biotek) and ethanol were then added to the cytoplasmic and nuclear fractions, and the solution was loaded onto a spin-column to collect RNA. The bound RNA was then washed with Wash Solution A (Norgen Biotek), and the purified RNA was eluted with Elution Buffer E (Norgen Biotek). The isolated RNA was subsequently reverse-transcribed, and the relative expression level was measured by qPCR. The pairs of primers used are listed in Table 1.
For FISH, the MALAT1 hybridization protocol was followed from a previous publication (31). Briefly, cells were seeded onto an autoclaved glass chamber slide at a density of 3 × 105 cells/well and incubated overnight. Cells were fixed by fixation buffer and incubated for 10 min at room temperature. For permeabilizing the cells, each well was soaked in 70% ethanol at 4°C overnight. Cells were then hybridized by fluorescein probes (tgaaccaaagctgcactgtg; Protech, Taiwan) labeled at the 5' end with a final concentration of 4 μM in hybridization buffer, and incubated at 37°C overnight. The next day, the hybridization buffer was removed, and cells were washed in phosphate-buffered saline (PBS) 3 times. Finally, the slide was mounted on a DAPI Fluoromount-G (SouthernBiotech). Images were acquired using a Zeiss LSM880 confocal microscope (Carl Zeiss AG, Gina, Germany).
MCF-7 cells were seeded in a six-well plate at a density of 800 cells/well. MDA-MB-231 cells were seeded in a six-well plate at a density of 500 cells/well. After 2 weeks of incubation, cells were fixed with 600 μl 75% methanol/25% acetate (Sigma) for 10 min and washed by PBS followed by staining with 0.1% crystal violet for another 10 min. Colonies with cell numbers greater than 50 were quantified using ImageJ 1.8.0 (National Institutes of Health).
Cells were seeded on an Ibidi Culture-Insert (Ibidi, Martinsried, Germany) at a density of 2.5 × 104 cells/reservoir and incubated overnight. The inserts were carefully removed with sterile tweezers to create a cell-free gap. The ability of cells to migrate into the gap was captured by microphotography at indicated time points and quantified using ImageJ 1.8.0 (National Institutes of Health).
Cells were seeded at a density of 5 × 104 cells/100 μl in a 96-well plate. After seeding 12 h, 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay solution (Sigma, St. Louis, MO, USA) (1 ml/well of a 5 mg/ml solution in PBS) was added to each well and incubated for 1 h. After incubating for 1 h, medium with MTT solution was removed and 100 μl dimethyl sulfoxide (Sigma) was added to each well to dissolve the converted purple formazan. The absorbance of formazan was measured at 570 nm using an enzyme-linked immunosorbent assay (ELISA) reader (Bio Tek, Winooski, VT, USA).
Cells were harvested by trypsinization and fixed with cold 75% ethanol at -20°C overnight. Cells were washed with PBS and resuspended in propidium iodide (PI) (Life Technologies, Carlsbad, CA, USA) solution (20 μg PI/ml, 100% Triton-X 1μl/ml, 20ng RNase/ml in PBS) for 10 min on ice. The suspension was analyzed with a Beckman Coulter FC500 cytometer (Beckman, Brea, CA, USA).
To validate the interaction between RNA and RNA binding proteins, the Magna RIP Kit (Millipore) was used. Before lysis, cells were washed with cold PBS, and samples were harvested with cell scrapers. Then, cells were lysed in RIP Lysis Buffer (Millipore) with RNase inhibitor and protease inhibitor cocktail (Millipore), and the magnetic beads for immunoprecipitation were prepared according to the manufacturer’s instructions. The RNA binding protein-RNA complexes were immunoprecipitated with premade magnetic beads at 4°C with overnight agitation. After washing the beads with ice-cold RIP Wash Buffer (Millipore), the RNA binding proteins were digested with proteinase K at 55°C for 30 min with shaking. The purified RNA was isolated with TRIzol reagent (Ambion, Thermo Fisher) and reverse-transcribed, and the relative gene expression level was measured by qPCR. The pairs of primers used are listed in Table 1.
Statistical analysis was carried out using Microsoft Excel to assess differences between experimental groups. All results were reported as means ± SDs for at least 3 independent experiments. Statistical significance was analyzed by Student’s t test and expressed as a P value. P values lower than 0.05 were considered to indicate statistical significance.
To investigate endogenous expression levels of MALAT1, we first determined the expression of MALAT1 in several breast cell lines, including MCF-10A (non-cancerous mammary gland epithelial cell), MCF-7 (luminal A cancer), and MDA-MB-231 (triple negative cancer) (Figure 1A). The expression levels of MALAT1 in MDA-MB-231 cells were significantly (P < 0.05) higher than those in MCF-10A and MCF-7 cells. Also, the relative expression levels of MALAT1 in MCF-7 cells under hypoxia or CoCl2 treatment were examined by qPCR. The relative expression of MALAT1 in MDA-MB-231 cells (Figure 1B) and MCF-7 cells (Figure 1C) under hypoxia were significantly (P < 0.05) up-regulated. MALAT1 was similarly up-regulated in MCF-7 cells treated with CoCl2, which mimics hypoxia (Figure 1D).
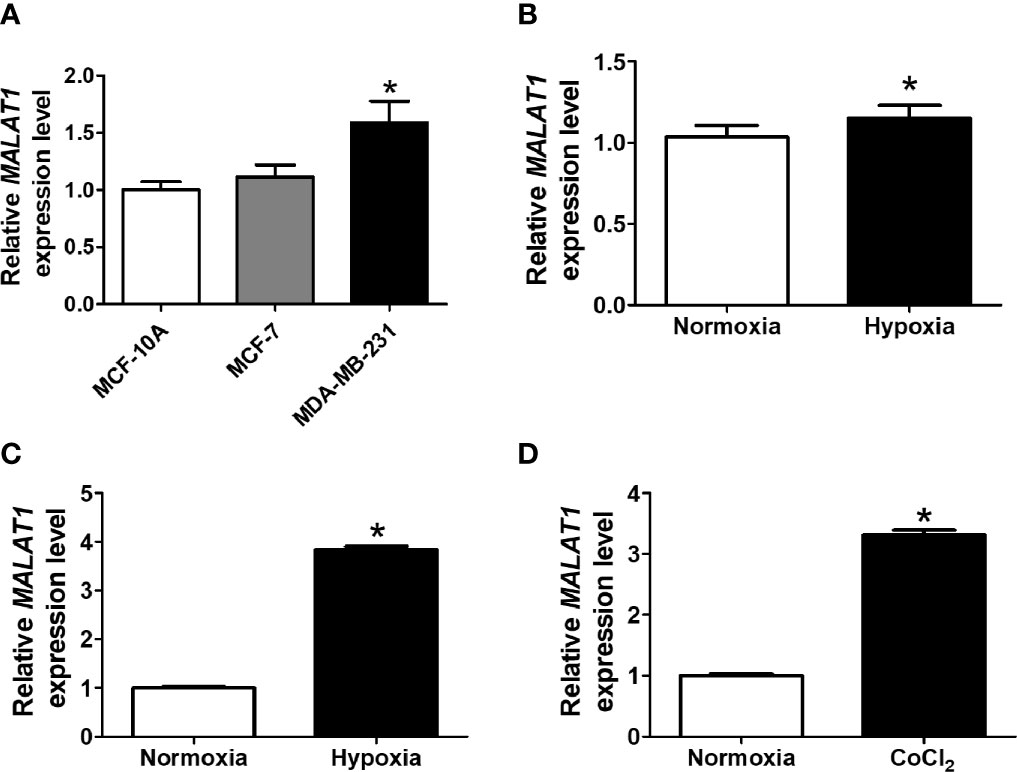
Figure 1 MALAT1 is up-regulated under hypoxia in breast cancer cells. (A) Relative endogenous expression levels of MALAT1 in MCF-10A, MCF-7, and MDA-MB-231 cells. The expression levels were measured by qPCR. (B, C) Relative expression levels of MALAT1 in MDA-MB-231 (B) and MCF-7 (C) cells under hypoxia. (D) Relative expression levels of MALAT1 in MCF-7 cells treated with CoCl2. Data shown are the means ± SDs (n=3). *P < 0.05.
Since MALAT1 was significantly up-regulated under hypoxia, we hypothesized that this effect was triggered by HIFs. In order to mimic HIF1A or HIF2A overexpression, we transfected a degradation-resistant HIF1A mutant (pcDNA3-HIF-1α-P402A/P564A) (Figure 2A) or HIF2A mutant (pcDNA3-HIF-2α-P405A/P531A) (Figure 2B) into MCF-7 cells under normoxia to observe the effects of HIF-1α and HIF-2α on MALAT1 expression. The RNA expression levels of MALAT1 were increased significantly (P < 0.05) when either HIF1A or HIF2A was overexpressed (Figures 2C, D). To further investigate how HIF-1A or HIF2A increased MALAT1 expression, the promoter sequence analysis revealed that there was one putative HRE ([A/G]CGTG) located at -235 to -231 bp relative to the transcription start site of MALAT1. Therefore, the promoter region of MALAT1 (-2,000 to -1 bp) was inserted into the pGL3-basic vector carrying the firefly luciferase gene. In addition, to validate the putative HRE site, the HRE sequences were mutated from GCGTG to TGTAT (Figure 2E). Overexpression of HIF-1A (Figure 2F) or HIF-2A (Figure 2G) both increased the luciferase activity, and the HRE site mutation significantly (P < 0.05) decreased both luciferase activities (Figures 2F, G). These results indicate that both HIF-1α and HIF-2α up-regulate the transcriptional levels of MALAT1 by binding to the HRE in its promoter. Furthermore, when MALAT1 was knocked down by shRNA, the expression levels of HIF-1A and HIF-2A were significantly up-regulated, suggesting the negative feedback of MALAT1 on HIF-1A and HIF-2A (Figure 2H).
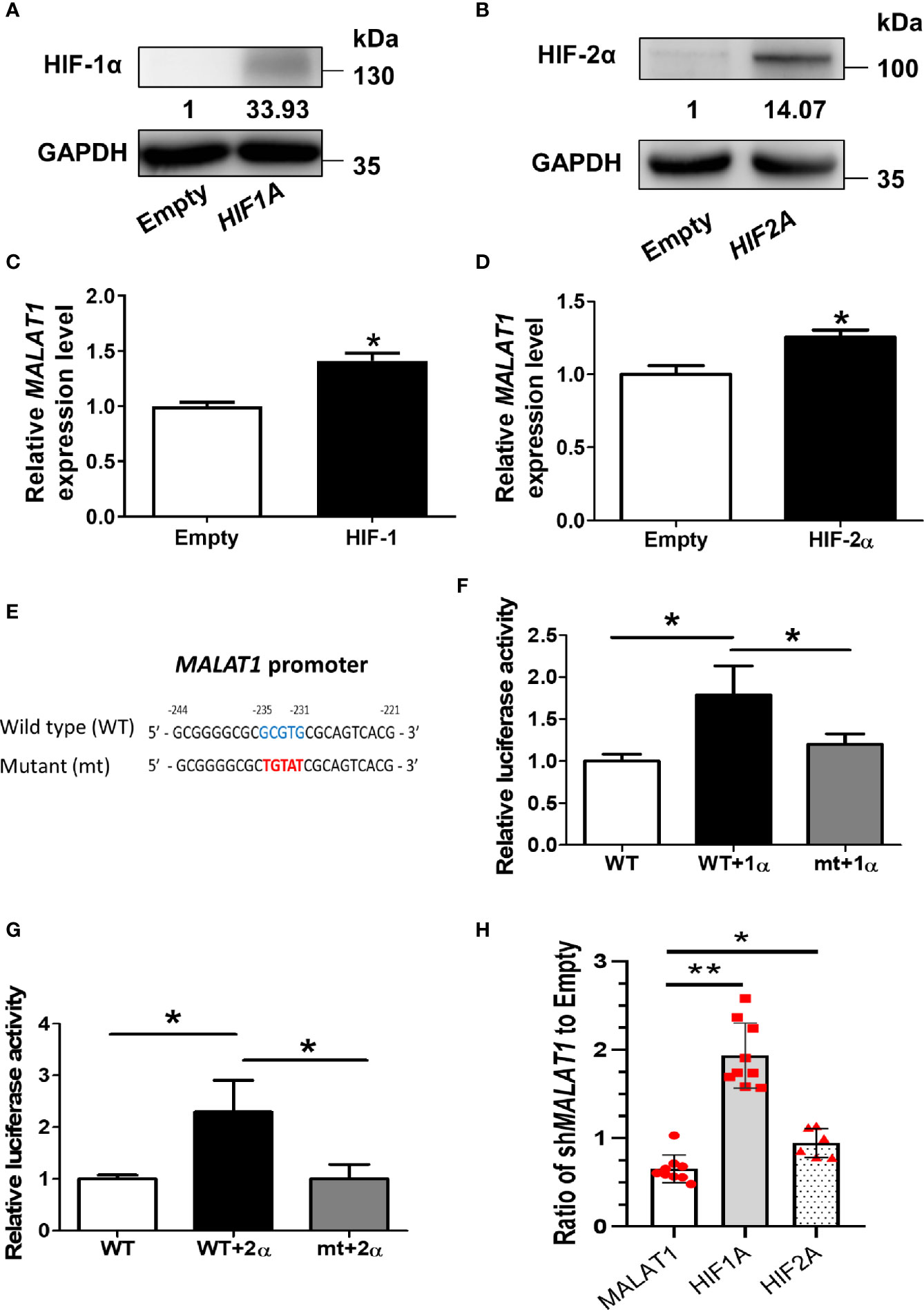
Figure 2 HIF-1α and HIF-2α up-regulate the transcriptional levels of MALAT1. (A) Western blot analysis of HIF-1α in MCF-7 cells over-expressing HIF-1α-P402A/P564A under normoxia. (B) Western blot analysis of HIF-2α in MCF-7 cells over-expressing HIF-2α-P405A/P531A under normoxia. (C, D) Relative expression levels of MALAT1 in MCF-7 cells overexpressing HIF-1α-P402A/P564A (C) or HIF-2α-P405A/P531A (D). The expression levels were measured by qPCR. (E) Schematic diagram of the putative HRE ([A/G]CGTG; -235 ~ -231 bp) in the promoter region (-2,000 ~ -1 bp) of MALAT1. (F, G) Luciferase reporter assays of wild-type (WT) and mutant (mt) MALAT1 promoters in HEK-293T cells overexpressing HIF-1α-P402A/P564A (F) or HIF-2α-P405A/P531A (G). HEK-293T cells were transfected with HIF-1α or HIF-2α expressing plasmids, firefly luciferase plasmids, and Renilla luciferase vectors. The relative firefly luciferase activity was measured and normalized to Renilla luciferase activity. All data shown are the means ± SDs (n = 3). *P < 0.05. (H) Relative expression levels of MALAT1, HIF1A, and HIF2A in MCF-7 cells transduced with shMALAT1.The expression levels were measured by qPCR. Loading control: 18S rRNA. **P < 0.01. *P < 0.05.
To determine whether MALAT1 could serve as a miRNA sponge to modulate cell functions, we first investigated the distribution of MALAT1 in MCF-7 cells and MDA-MB-231 cells under normoxia (Figures 3A, B) and hypoxia (Figures 3E, F). The positive control for nuclear function was BCAR4, and for cytoplasmic function was GAPDH. Surprisingly, MALAT1 was mainly distributed in the cytoplasm, not the nucleus. To confirm this phenomenon, we also investigated the distribution of MALAT1 in lung cancer cell lines A549 and H1299 under normoxia, because MALAT1 was reported to be a highly abundant nuclear transcript in these cells (Figures 3C, D) (18, 32, 33). Yet, similar results were observed in lung cancer cells. Nuclear-cytoplasmic RNA fractionation assays indicated that MALAT1 was mainly located in the cytoplasm of MCF-7 cells and MDA-MB-231 cells either under normoxia or hypoxia. Furthermore, RNA FISH was used to determine the location of MALAT1 (Figure 3G). These results also showed that MALAT1 is mainly located in the cytoplasm in MCF-7 cells and MDA-MB-231 cells.
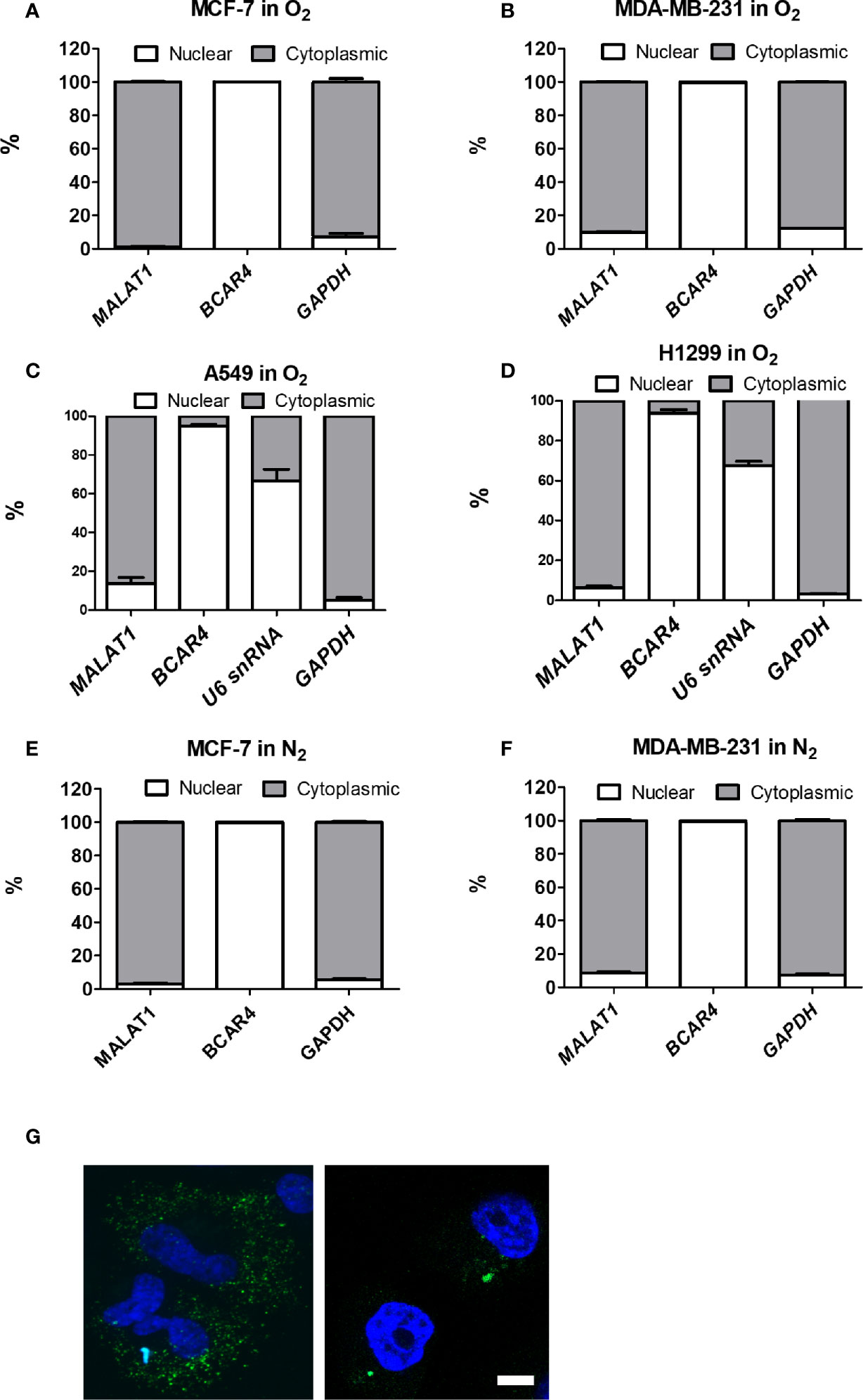
Figure 3 MALAT1 is located primarily in the cytoplasm of MCF-7 and MDA-MB-231 cells under normoxia or hypoxia. (A–D) Cytoplasmic and nuclear distribution of MALAT1 in breast cancer cells [MCF-7 (A), MDA-MB-231 (B)] and lung cancer cells [A549 (C), H1299 (D)] cells under normoxia. (E, F) Cytoplasmic and nuclear distribution of MALAT1 in MCF-7 (E) and MDA-MB-231 (F) cells under hypoxia. Relative abundance of RNA was normalized to the total amount of RNA and detected by qPCR. BCAR4 and U6 snRNA: nuclear marker. GAPDH: cytoplasmic marker. Data shown are the means ± SDs (n=3). (G) RNA FISH of MALAT1. Cell nuclei were stained by Hoechst staining (blue). MALAT1 was hybridized with MALAT1-FITC probes (green) in breast cancer cell lines and detected by a Zeiss LSM880 microscope. Magnification:1,000×; Scale bar: 5 μm.
In our previous research, five differentially expressed miRNAs, including miR-378c, miR-3150a-3p, miR-3064-5p, miR-4325, and miR-7855-5p, were significantly up-regulated using NGS when MALAT1 was knocked down under hypoxia (data not shown). We hypothesized that MALAT1 may serve as a sponge to these miRNAs. To test our hypothesis, MALAT1 was first silenced in MDA-MB-231 cells by two shRNAs targeted to different sites of MALAT1. MALAT1 expression levels were significantly (P < 0.05) down-regulated when MALAT1 was knocked down under hypoxia (Figure 4A). Next, the expression levels of these five miRNAs were validated by qPCR when MALAT1 was knocked down under hypoxia (Figure 4B). The results showed that the expression levels of miR-3064-5p, miR-3150, miR-4325, and miR-7855-5p were significantly (P < 0.05) up-regulated when MALAT1 was knocked down under hypoxia. Furthermore, the expression levels of miR-3064-5p, miR-3150, miR-4325, and miR-7855-5p were significantly down-regulated when MALAT1 was overexpressed (Figures 4C, D). These results indicate that the expression levels of MALAT1 are negatively correlated with those of miR-3064-5p, miR-3150, miR-4325, and miR-7855-5p.
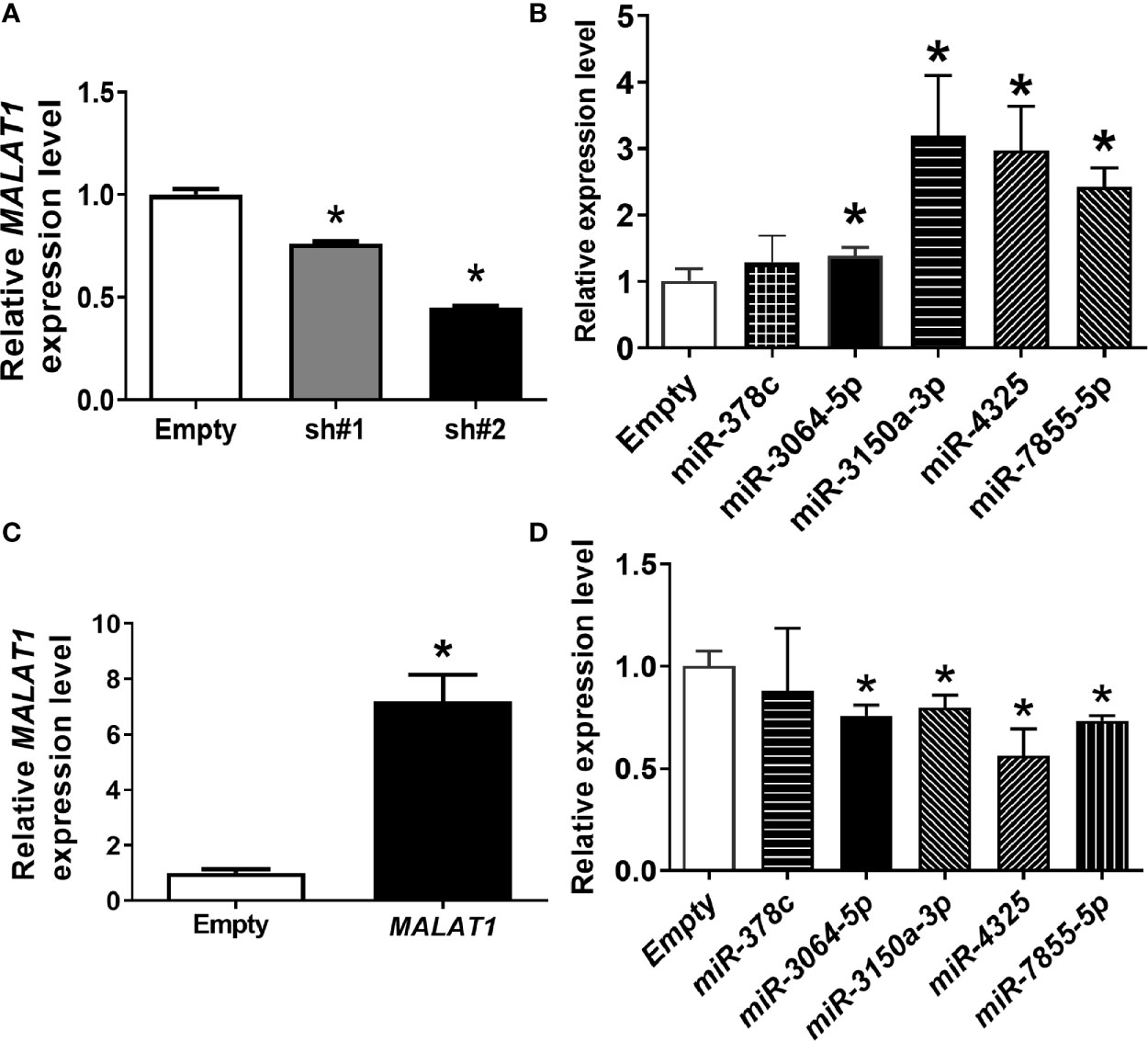
Figure 4 The inverse relationship between expression levels of miRNAs and MALAT1. (A) Relative expression levels of MALAT1 in MDA-MB-231 cells were reduced with shRNA against MALAT1 under hypoxia. The expression levels were measured by qPCR. Loading control: 18S rRNA. (B) Relative expression levels of miRNAs in MDA-MB-231 cells transfected with shRNA against MALAT1. (C) Relative expression levels of MALAT1 in MCF-7 cells overexpressing MALAT1. Cells were transfected with pCMV-MALAT1 and the expression levels were detected by qPCR. (D) Relative expression levels of miRNAs in MCF-7 cells transfected with pCMV-MALAT1. Data shown are the means ± SDs (n=3). *P < 0.05.
To validate that MALAT1 physically bound to miRNAs and served as a miRNA sponge in breast cancer, miR-3064-5p was chosen for further experiments, because miR-3064-5p has been reported to inhibit cell proliferation and invasion in ovarian cancer and to suppress angiogenesis in hepatocellular carcinoma (34, 35). First, since argonaute2 (AGO2) protein is an essential component of the miRNA-induced silencing complex (miRISC), RIP assays using anti-AGO2 antibody were performed. As shown in Figures 5A, B, MALAT1 and miR-3064-5p were both significantly (P < 0.05) enriched with AGO2 immunoprecipitation compared with IgG control group. Next, sequence analysis of MALAT1 revealed two putative binding sites for miR-3064-5p, located at 1,279~1,302 bp (Site 1) and 7,837~7,860 bp (Site 2) relative to the transcription start site. Therefore, wild-type or mutant versions of the regions of MALAT1 (1,048~1,547 or 7,607~8,106 bp) containing the putative binding sites of miR-3064-5p were inserted into the pmiR-GLO vector (Figure 5C), and luciferase reporter assays were performed. Overexpression of miR-3064-5p mimic decreased the MALAT1 promoter-mediated luciferase activity, but the luciferase activity was rescued only by the site 2 mutation, not the site 1 mutation (Figures 5D, E). Taken together, these results indicate that miR-3064-5p regulates MALAT1 directly by binding at 7,837~7,860 bp of MALAT1.
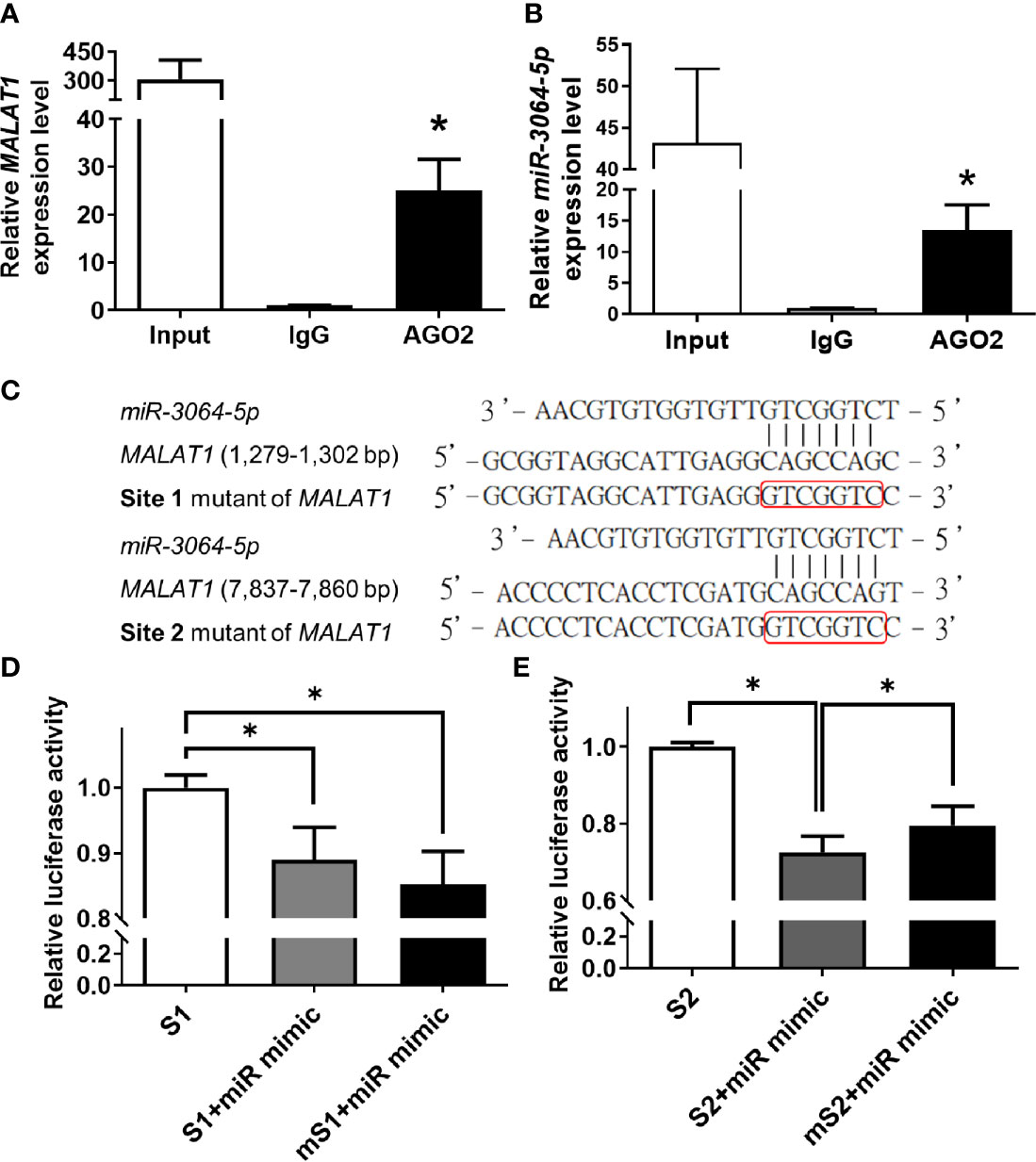
Figure 5 MALAT1 binds to miR-3064-5p directly. (A, B) RIP using antibody against AGO2. The relative RNA levels of MALAT1 (A) and miR-3064-5p (B) were quantified and normalized to the IgG group using qPCR. Input: positive control; IgG: negative control. (C) Schematic representation of firefly reporter constructs containing the fragments of Site 1 (1,279~1,302 bp) and Site 2 (7,837~7,860 bp) of MALAT1, and mutants with mutation at the binding sites of miR-3064-5p. (D, E) Luciferase reporter assays of MALAT1 fragment with wild-type or mutant (m) Site 1 (D) or Site 2 (E) in cells overexpressing miR-3064-5p mimic. HEK-293T cells were transfected with miR-3064-5p mimic and firefly/Renilla plasmids. The relative firefly luciferase activity was measured and normalized to Renilla luciferase activity. Data shown are the means ± SDs (n=3). *P < 0.05.
Since more expression of MALAT1 was observed in MDA-MB-231 cells, we studied the functional effects of MALAT1 knockdown on cell proliferation, cell migration, colony formation, and cell cycle distribution in MDA-MB-231 cells. The effect of MALAT1 on cell proliferation was examined by MTT assays. MDA-MB-231 cells with MALAT1 knockdown had significantly (P < 0.05) decreased cell proliferation (Figure 6A). Next, the effects of MALAT1 on cell migration were examined by wound healing assays. MDA-MB-231 cells with MALAT1 knockdown had significantly (P < 0.05) decreased cell migration (Figure 6B). Long term effects of MALAT1 on cell proliferation were observed through colony formation assays. MDA-MB-231 cells with MALAT1 knockdown had significantly decreased colony numbers (Figure 6C). Lastly, the effects of MALAT1 on cell cycle distribution were examined by flow cytometry. In MALAT1 knockdown cells, the percentage of cells in G1 phase significantly increased as compared to the empty vector controls (Figure 6D). Conversely, the percentage of cells in the S and G2/M phases decreased. Taken together, these data show that MDA-MB-231 cells with MALAT1 knockdown had significantly decreased ability to multiply, migrate, and colonize, suggesting that breast tumor malignancy could be mediated by MALAT1.
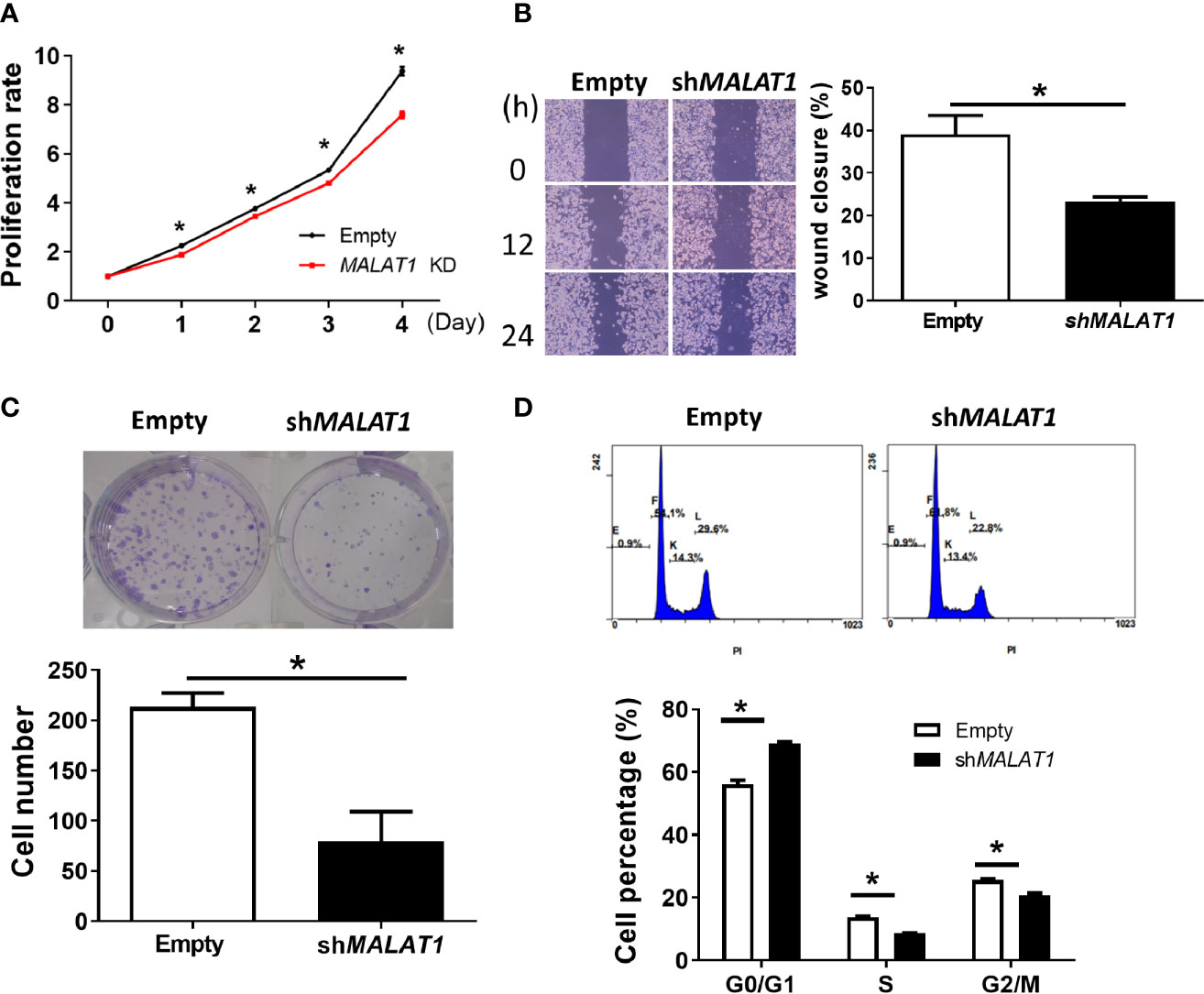
Figure 6 Knockdown of MALAT1 decreases proliferation and metastasis in MDA-MB-231 cells. (A) Measurement of cell proliferation using MTT assays. Cell growth was measured in MDA-MB-231 cells transduced with lentivirus which expresses shRNA against MALAT1. The proliferation rate was normalized to day zero. (B) Wound healing assay. Left: Representative pictures of wound healing assays. After 24 h of transduction, wound healing was photographed at 0, 12, and 24 h. Right: Migration ability was quantified as reduction in wound size at 24 h. (C) Colony formation assay. Top: Representative pictures of colony formation assays. Colonies with cell numbers more than fifty were counted. Bottom: Quantification of results. (D) Cell cycle distribution by flow cytometry. Top: Representative diagrams of flow cytometry. After 48 h of transfection, cells were harvested and stained with PI. Bottom: Quantification of results as the percentage of cells in each phase of the cell cycle. All data shown are the means ± SDs (n = 3). *P < 0.05.
Finally, considering that the endogenous expression levels of MALAT1 in MCF-7 cells were lower than in MDA-MB-231 cells, we also studied the functional effects of overexpressing MALAT1 in MCF-7 cells. MCF-7 cells with MALAT1 overexpression had significantly increased cell proliferation (Figure 7A), cell migration (Figure 7B), and ability to form colonies (Figure 7C). However, in MALAT1 overexpressing cells, the distribution of cells in each phase of the cell cycle showed no significant differences as compared to empty controls (Figure 7D). These data confirm that MALAT1 overexpression leads to characteristics associated increased tumor malignancy in MCF-7 cells.
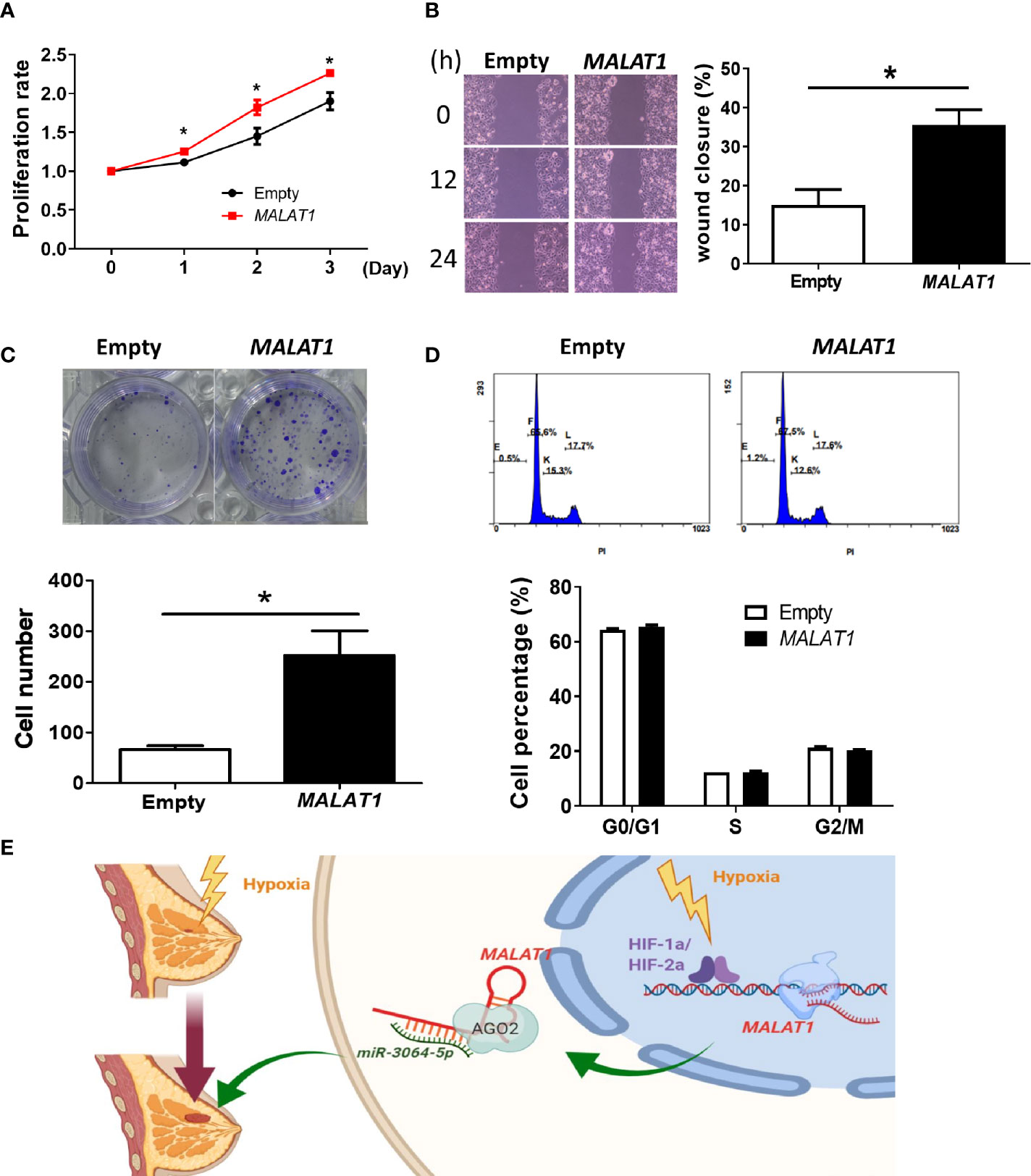
Figure 7 Overexpression of MALAT1 promotes proliferation and migration in MCF-7 cells. (A) Measurement of cell proliferation using MTT assays. Cell growth was measured in MCF-7 cells overexpressing MALAT1. The proliferation rate was normalized to day zero. (B) Wound healing assay. Left: Representative pictures of wound healing assays. After 24 h of transfection, wound healing was photographed at 0, 12, and 24 h. Right: Migration ability was quantified as reduction in wound size at 24 h. (C) Colony formation assay. Top: Representative pictures of colony formation assays. Colonies with cell numbers more than fifty were counted. Bottom: Quantification of results. (D) Cell cycle distribution by flow cytometry. Top: Representative diagrams of flow cytometry. After 48 h of transfection, cells were harvested and stained with PI. Bottom: Quantification of results as the percentage of cells in each phase of the cell cycle. (E) A proposed model for illustrating the regulatory mechanisms and function of hypoxia-induced lncRNA MALAT1 in breast cancer cells. All data shown are the means ± SDs (n = 3). *P < 0.05.
Taken together, these data suggested that hypoxia-responsive long non-coding MALAT1 could be transcriptionally activated by HIF-1α and HIF-2α, act as a miRNA sponge of miR-3064-5p, and promote tumor growth and migration in breast cancer cells (Figure 7E).
In this study, we demonstrated that MALAT1 was up-regulated under hypoxia in breast cancer cells. Luciferase reporter assays showed that HIF-1A and HIF-2A both increased the transcriptional activity of MALAT1. Next, the nuclear and cytoplasmic fractionation assays and FISH indicated that MALAT1 was mainly located in the cytoplasm. Four hypoxia-responsive miRNAs, including miR-3064-5p, miR-3150, miR-4325, and miR-7855-5p, had reverse relationships with the expression of MALAT1. In addition, RIP assays using antibody against AGO2 showed that MALAT1 served as a miRNA sponge for miR-3064-5p. Lastly, functional assays revealed that MALAT1 could promote breast cancer cell aggressiveness, by increasing proliferation and migration and altering cell cycle distribution.
LncRNAs are known to play a crucial role in carcinogenesis (36). For example, the lncRNA PRLB promotes tumorigenesis through regulating the miR-4766-5p/SIRT1 axis (37). LncRNA HIFCAR/MIR31HG was found to be a HIF-1α co-activator that promoted oral cancer progression (38). LncRNA UCA1 promoted proliferation, migration, and immune escape and inhibited cell apoptosis in gastric cancer (39). Our results revealed that the endogenous expression levels of MALAT1 in MDA-MB-231 metastatic breast cancer cells were higher than in normal MCF-10A breast epithelial cells and in less aggressive MCF-7 breast cancer cells (Figure 1A), suggesting that MALAT1 plays a role in the oncogenic characteristics in breast cancer. Another study reported similar results, that MALAT1 expression levels were significantly higher in tumor tissues as compared with adjacent noncancerous tissues (40).
So far, some lncRNAs have been confirmed to respond to hypoxia in several malignant tumors and to regulate gene expression to adjust to microenvironments deficient in oxygen (41–44). Recent RNA-seq results showed that >100 lncRNAs, including H19, MIR210HG, and MALAT1, were up-regulated in human umbilical vein endothelial cells under hypoxia (45). We found that MALAT1 was also up-regulated in hypoxia in breast cancer cells (Figure 1). Similarly, MALAT1 was reported to be induced in hypoxia and to regulate polypyrimidine tract-binding protein (PTB)-associated splicing factor transcriptionally in A549 lung cancer cells (46). In hypoxia, HIF-1 is known to function as an oxygen-regulated transcriptional activator that is expressed ubiquitously and plays essential roles in mammalian development, physiology, and disease pathogenesis (47–49). Unlike the ubiquitously expressed HIF-1α, HIF-2α is mainly expressed in endothelial cells (50). Some genes are regulated only by HIF-2α and not HIF-1α in cancer. In our results, ectopic expression of both HIF-1α or HIF-2α increased MALAT1 expression (Figures 2C, D). Furthermore, promoter analysis revealed that there is one putative HRE in the MALAT1 promoter (Figure 2E). Our results revealed that both HIF-1α and HIF-2α could up-regulate expression of MALAT1 by binding to its promoter. Several studies have revealed evidence that MALAT1 promotes arsenite-induced glycolysis in human hepatic L-02 cells through HIF-1α stabilization (51). Our results suggest that HIF-2α could also regulate cell functions by modulating the expression of noncoding RNAs, such miRNA and lncRNA. This is consistent with the finding that the HIF-2α/MALAT1/miR-216b axis up-regulated autophagy to promote multi-drug resistance in hepatocellular carcinoma cells (52).
Furthermore, the negative feedback loop of MALAT1 on HIF-1A and HIF-2A was discovered in this study (Figure 2H). MALAT1 was reported to increase HIF-1α expression by blocking the ubiquitin-proteasome pathway in arsenite-induced glycolysis (53). Also, a positive feedback loop between MALAT1 and HIF-2α was discovered in arsenite induced hepatocellular carcinomas (54). Our results extend the current understanding regarding the reciprocal regulation between MALAT1 and HIF-1A as well as MALAT1 and HIF-2A.
Initially, MALAT1 was identified as being up-regulated in primary human non-small cell lung cancer cells with heightened metastatic potential (15). Also early in its history, MALAT1 was found to be abundant in neurons and to modulate synaptogenesis by regulating gene expression in cultured hippocampal neurons (55). In one study, MALAT1 was called noncoding nuclear-enriched abundant transcript 2 (NEAT2), indicating its nuclear abundance in several cancer cell lines, and its role in alternative splicing regulation (46). For a long time, MALAT1 was considered to be a nuclear marker in certain cancer cell lines (18, 32, 33), especially cancers with aggressive metastatic tumors (15), and it has been shown to be involved in proliferation and invasion of lung cancer cells (56) and cervical cancer cells (57). However, the results of our nuclear-cytoplasmic RNA fractionation assays indicated that MALAT1 was mainly located in the cytoplasm of MCF-7, MDA-MB-231, A549, and H1299 cells, under either normoxia or hypoxia (Figure 3). These results are consistent with recent studies showing that MALAT1 was located in the cytoplasm in human hepatocellular carcinoma cells, monocytes, and human pulmonary microvascular endothelial cells (29, 30, 35). Therefore, the labeling of MALAT1 as a nuclear marker should be done with the caveat that this status is dependent on the cell type.
LncRNAs and miRNAs can work cooperatively to mediate gene expression via post-transcriptional mechanisms. Since MALAT1 was mainly in the cytoplasm of breast cancer cells, we hypothesized that MALAT1 may serve as a miRNA sponge. In this study, we showed that four hypoxia-responsive miRNAs (miR-3064-5p, miR-3150a-3p, miR-4325, and miR-7855-5p) had negative correlation with the expression MALAT1 (Figure 4). Furthermore, MALAT1 was enriched in RIP assays using antibody against AGO2 (Figure 6A). The results of luciferase reporter assays also indicated that miR-3064-5p regulated MALAT1 directly by binding at 7,837~7,860 bp relative to the transcription start site of MALAT1. However, mutation of this site on MALAT1 did not fully rescue the luciferase activity (Figure 5E), suggesting that miR-3064-5p may target other sites of MALAT1. Other references have also reported that MALAT1 could serve as a miRNA sponge. For example, MALAT1 functioned as a competing endogenous RNA (ceRNA) by sponging miR-3064-5p, which alleviated the suppressive effect on angiogenesis in human hepatocellular carcinoma via the FOXA1/CD24/Src pathway (35). MALAT1 targeted miR-150-5p to exacerbate acute respiratory distress syndrome by upregulating ICAM-1 expression (29). MALAT1 bound miR-23a to suppress inflammation in septic mice (30).
In our experiments, MALAT1 was shown to promote cell proliferation and migration in MFC7 and MDA-MB-231 cells (Figures 6, 7). When MALAT1 was knocked down in MDA-MB-231 cells, the percentage of G1 phase of cell cycle significantly increased, indicating that silencing of MALAT1 resulted in G1 arrest. However, the downstream genes of MALAT1/miRNA-3064-5p that influence the functions of breast cancer cells are still unknown. In the future, we can use predictive tools to investigate the target genes of miRNA-3064-5p and validate these genes experimentally. Also, an animal model is needed to confirm the role of the MALAT1/miR-3064-5p pathway in breast cancer. Since the suppressive role of Malat1 on metastatic ability of breast cancer in mouse has been reported (58), more experiments in animal studies and clinical trials are still warranted to explore the MALAT1 pathway as a therapeutic target for breast cancer.
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
The study was approved by the Biosafety Committee of College of Medicine, National Taiwan University [BG1050086].
C-HS and L-CL conceived and designed the experiments. C-HS performed the experiments. C-HS, L-LC, and T-PL analyzed the data. L-HC, M-HT, and EC contributed reagents, materials, and/or analysis tools. C-HS and L-CL wrote the paper. All authors contributed to the article and approved the submitted version.
This work was supported by a grant from the Ministry of Science and Technology [MOST 109-2320-B-002-016-MY3]. These funding sources had no role in the design of this study and will not have any role during its execution, analyses, interpretation of the data, or decision to submit results.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank Melissa Stauffer for editorial assistance. We also benefited from technical assistance from the Biomedical Resource Core at the 1st Core Facility Lab, NTU College of Medicine.
1. Shao C, Yang F, Miao S, Liu W, Wang C, Shu Y, et al. Role of Hypoxia-Induced Exosomes in Tumor Biology. Mol Cancer (2018) 17:120. doi: 10.1186/s12943-018-0869-y
2. Harris AL. Hypoxia–a Key Regulatory Factor in Tumour Growth. Nat Rev Cancer (2002) 2:38–47. doi: 10.1038/nrc704
3. Gillies RJ, Verduzco D, Gatenby RA. Evolutionary Dynamics of Carcinogenesis and Why Targeted Therapy Does Not Work. Nat Rev Cancer (2012) 12:487–93. doi: 10.1038/nrc3298
4. Muz B, de la Puente P, Azab F, Azab AK. The Role of Hypoxia in Cancer Progression, Angiogenesis, Metastasis, and Resistance to Therapy. Hypoxia (Auckl) (2015) 3:83–92. doi: 10.2147/HP.S93413
5. Masson N, Ratcliffe PJ. Hypoxia Signaling Pathways in Cancer Metabolism: The Importance of Co-Selecting Interconnected Physiological Pathways. Cancer Metab (2014) 2:3. doi: 10.1186/2049-3002-2-3
6. Knowles HJ, Harris AL. Hypoxia and Oxidative Stress in Breast Cancer. Hypoxia and Tumourigenesis. Breast Cancer Res (2001) 3:318–22. doi: 10.1186/bcr314
7. Jiang Y, Wu GH, He GD, Zhuang QL, Xi QL, Zhang B, et al. The Effect of Silencing HIF-1alpha Gene in BxPC-3 Cell Line on Glycolysis-Related Gene Expression, Cell Growth, Invasion, and Apoptosis. Nutr Cancer (2015) 67:1314–23. doi: 10.1080/01635581.2015.1085584
8. Semenza GL. Hypoxia-Inducible Factors in Physiology and Medicine. Cell (2012) 148:399–408. doi: 10.1016/j.cell.2012.01.021
9. Semenza GL. Targeting HIF-1 for Cancer Therapy. Nat Rev Cancer (2003) 3:721–32. doi: 10.1038/nrc1187
10. Lin C, Yang L. Long Noncoding RNA in Cancer: Wiring Signaling Circuitry. Trends Cell Biol (2018) 28:287–301. doi: 10.1016/j.tcb.2017.11.008
11. Terracciano D, Terreri S, de Nigris F, Costa V, Calin GA, Cimmino A. The Role of a New Class of Long Noncoding RNAs Transcribed From Ultraconserved Regions in Cancer. Biochim Biophys Acta Rev Cancer (2017) 1868:449–55. doi: 10.1016/j.bbcan.2017.09.001
12. Kornienko AE, Guenzl PM, Barlow DP, Pauler FM. Gene Regulation by the Act of Long non-Coding RNA Transcription. BMC Biol (2013) 11:59. doi: 10.1186/1741-7007-11-59
13. Mercer TR, Dinger ME, Mattick JS. Long non-Coding RNAs: Insights Into Functions. Nat Rev Genet (2009) 10:155–9. doi: 10.1038/nrg2521
14. Wilusz JE, Sunwoo H, Spector DL. Long Noncoding RNAs: Functional Surprises From the RNA World. Genes Dev (2009) 23:1494–504. doi: 10.1101/gad.1800909
15. Ji P, Diederichs S, Wang W, Böing S, Metzger R, Schneider PM, et al. Malat-1, a Novel Noncoding RNA, and Thymosin Beta4 Predict Metastasis and Survival in Early-Stage non-Small Cell Lung Cancer. Oncogene (2003) 22:8031–41. doi: 10.1038/sj.onc.1206928
16. Schmidt LH, Spieker T, Koschmieder S, Schäffers S, Humberg J, Jungen D, et al. The Long Noncoding MALAT-1 RNA Indicates a Poor Prognosis in non-Small Cell Lung Cancer and Induces Migration and Tumor Growth. J Thorac Oncol (2011) 6:1984–92. doi: 10.1097/JTO.0b013e3182307eac
17. Ying L, Chen Q, Wang Y, Zhou Z, Huang Y, Qiu F. Upregulated MALAT-1 Contributes to Bladder Cancer Cell Migration by Inducing Epithelial-to-Mesenchymal Transition. Mol Biosyst (2012) 8:2289–94. doi: 10.1039/c2mb25070e
18. Tripathi V, Ellis JD, Shen Z, Song DY, Pan Q, Watt AT, et al. The Nuclear-Retained Noncoding RNA MALAT1 Regulates Alternative Splicing by Modulating SR Splicing Factor Phosphorylation. Mol Cell (2010) 39:925–38. doi: 10.1016/j.molcel.2010.08.011
19. Choudhry H, Schödel J, Oikonomopoulos S, Camps C, Grampp S, Harris AL, et al. Extensive Regulation of the non-Coding Transcriptome by Hypoxia: Role of HIF in Releasing Paused Rnapol2. EMBO Rep (2014) 15:70–6. doi: 10.1002/embr.201337642
20. Lou W, Liu J, Gao Y, Zhong G, Chen D, Shen J, et al. MicroRNAs in Cancer Metastasis and Angiogenesis. Oncotarget (2017) 8:115787–802. doi: 10.18632/oncotarget.23115
21. Mahdian-Shakib A, Dorostkar R, Tat M, Hashemzadeh MS, Saidi N. Differential Role of microRNAs in Prognosis, Diagnosis, and Therapy of Ovarian Cancer. BioMed Pharmacother (2016) 84:592–600. doi: 10.1016/j.biopha.2016.09.087
22. Giovannetti E, Erozenci A, Smit J, Danesi R, Peters GJ. Molecular Mechanisms Underlying the Role of microRNAs (miRNAs) in Anticancer Drug Resistance and Implications for Clinical Practice. Crit Rev Oncol Hematol (2012) 81:103–22. doi: 10.1016/j.critrevonc.2011.03.010
23. Kumar MS, Armenteros-Monterroso E, East P, Chakravorty P, Matthews N, Winslow MM, et al. HMGA2 Functions as a Competing Endogenous RNA to Promote Lung Cancer Progression. Nature (2014) 505:212–7. doi: 10.1038/nature12785
24. Khan S, Ayub H, Khan T, Wahid F. MicroRNA Biogenesis, Gene Silencing Mechanisms and Role in Breast, Ovarian and Prostate Cancer. Biochimie (2019) 167:12–24. doi: 10.1016/j.biochi.2019.09.001
25. Kim YW, Kim EY, Jeon D, Liu JL, Kim HS, Choi JW, et al. Differential microRNA Expression Signatures and Cell Type-Specific Association With Taxol Resistance in Ovarian Cancer Cells. Drug Des Devel Ther (2014) 8:293–314. doi: 10.2147/DDDT.S51969
26. Mulrane L, McGee SF, Gallagher WM, O’Connor DP. miRNA Dysregulation in Breast Cancer. Cancer Res (2013) 73:6554–62. doi: 10.1158/0008-5472.CAN-13-1841
27. Chan YC, Khanna S, Roy S, Sen CK. miR-200b Targets Ets-1 and is Down-Regulated by Hypoxia to Induce Angiogenic Response of Endothelial Cells. J Biol Chem (2011) 286:2047–56. doi: 10.1074/jbc.M110.158790
28. Lei Z, Li B, Yang Z, Fang H, Zhang GM, Feng ZH, et al. Regulation of HIF-1alpha and VEGF by miR-20b Tunes Tumor Cells to Adapt to the Alteration of Oxygen Concentration. PloS One (2009) 4:e7629. doi: 10.1371/journal.pone.0007629
29. Yao MY, Zhang WH, Ma WT, Liu QH, Xing LH, Zhao GF. Long non-Coding RNA MALAT1 Exacerbates Acute Respiratory Distress Syndrome by Upregulating ICAM-1 Expression Via microRNA-150-5p Downregulation. Aging (Albany NY) (2020) 12:6570–85. doi: 10.18632/aging.102953
30. Xie W, Chen L, Chen L, Kou Q. Silencing of Long non-Coding RNA MALAT1 Suppresses Inflammation in Septic Mice: Role of microRNA-23a in the Down-Regulation of MCEMP1 Expression. Inflammation Res (2020) 69:179–90. doi: 10.1007/s00011-019-01306-z
31. Dunagin M, Cabili MN, Rinn J, Raj A. Visualization of lncRNA by Single-Molecule Fluorescence in Situ Hybridization. Methods Mol Biol (2015) 1262:3–19. doi: 10.1007/978-1-4939-2253-6_1
32. Hutchinson JN, Ensminger AW, Clemson CM, Lynch CR, Lawrence JB, Chess A. A Screen for Nuclear Transcripts Identifies Two Linked Noncoding RNAs Associated With SC35 Splicing Domains. BMC Genomics (2007) 8:1–16. doi: 10.1186/1471-2164-8-39
33. Bernard D, Prasanth KV, Tripathi V, Colasse S, Nakamura T, Xuan Z, et al. A Long Nuclear-Retained non-Coding RNA Regulates Synaptogenesis by Modulating Gene Expression. EMBO J (2010) 29:3082–93. doi: 10.1038/emboj.2010.199
34. Bai L, Wang H, Wang AH, Zhang LY, Bai J. MicroRNA-532 and microRNA-3064 Inhibit Cell Proliferation and Invasion by Acting as Direct Regulators of Human Telomerase Reverse Transcriptase in Ovarian Cancer. PloS One (2017) 12:e0173912. doi: 10.1371/journal.pone.0173912
35. Zhang P, Ha M, Li L, Huang X, Liu C. MicroRNA-3064-5p Sponged by MALAT1 Suppresses Angiogenesis in Human Hepatocellular Carcinoma by Targeting the FOXA1/CD24/Src Pathway. FASEB J (2020) 34:66–81. doi: 10.1096/fj.201901834R
36. Bhan A, Soleimani M, Mandal SS. Long Noncoding RNA and Cancer: A New Paradigm. Cancer Res (2017) 77:3965–81. doi: 10.1158/0008-5472.CAN-16-2634
37. Liang Y, Song X, Li Y, Sang Y, Zhang N, Zhang H, et al. A Novel Long non-Coding RNA-PRLB Acts as a Tumor Promoter Through Regulating miR-4766-5p/SIRT1 Axis in Breast Cancer. Cell Death Dis (2018) 9:563. doi: 10.1038/s41419-018-0582-1
38. Shih JW, Chiang WF, Wu ATH, Wu MH, Wang LY, Yu YL, et al. Long Noncoding RNA LncHIFCAR/MIR31HG is a HIF-1α Co-Activator Driving Oral Cancer Progression. Nat Commun (2017) 8:15874. doi: 10.1038/ncomms15874
39. Wang CJ, Zhu CC, Xu J, Wang M, Zhao WY, Liu Q, et al. The Lncrna UCA1 Promotes Proliferation, Migration, Immune Escape and Inhibits Apoptosis in Gastric Cancer by Sponging Anti-Tumor Mirnas. Mol Cancer (2019) 18:115. doi: 10.1186/s12943-019-1032-0
40. Huang XJ, Xia Y, He GF, Zheng LL, Cai YP, Yin Y, et al. MALAT1 Promotes Angiogenesis of Breast Cancer. Oncol Rep (2018) 40:2683–9. doi: 10.3892/or.2018.6705
41. Deng SJ, Chen HY, Ye Z, Deng SC, Zhu S, Zeng Z, et al. Hypoxia-Induced LncRNA-BX111 Promotes Metastasis and Progression of Pancreatic Cancer Through Regulating ZEB1 Transcription. Oncogene (2018) 37:5811–28. doi: 10.1038/s41388-018-0382-1
42. Fan H, Li J, Wang J, Hu Z. Long Non-Coding Rnas (Lncrnas) Tumor-Suppressive Role of lncRNA on Chromosome 8p12 (Tslnc8) Inhibits Tumor Metastasis and Promotes Apoptosis by Regulating Interleukin 6 (Il-6)/Signal Transducer and Activator of Transcription 3 (Stat3)/Hypoxia-Inducible Factor 1-Alpha (HIF-1α) Signaling Pathway in Non-Small Cell Lung Cancer. Med Sci Monit (2019) 25:7624–33. doi: 10.12659/MSM.917565
43. Xue M, Chen W, Xiang A, Wang R, Chen H, Pan J, et al. Hypoxic Exosomes Facilitate Bladder Tumor Growth and Development Through Transferring Long non-Coding RNA-UCA1. Mol Cancer (2017) 16:143. doi: 10.1186/s12943-017-0714-8
44. Zhang J, Jin HY, Wu Y, Zheng ZC, Guo S, Wang Y, et al. Hypoxia-Induced LncRNA PCGEM1 Promotes Invasion and Metastasis of Gastric Cancer Through Regulating SNAI1. Clin Transl Oncol (2019) 21:1142–51. doi: 10.1007/s12094-019-02035-9
45. Voellenkle C, Garcia-Manteiga JM, Pedrotti S, Perfetti A, De Toma I, Da Silva D, et al. Implication of Long Noncoding RNAs in the Endothelial Cell Response to Hypoxia Revealed by RNA-Sequencing. Sci Rep (2016) 6:24141. doi: 10.1038/srep24141
46. Hu L, Tang J, Huang X, Zhang T, Feng X. Hypoxia Exposure Upregulates MALAT-1 and Regulates the Transcriptional Activity of PTB-associated Splicing Factor in A549 Lung Adenocarcinoma Cells. Oncol Lett (2018) 16:294–300. doi: 10.3892/ol.2018.8637
47. Semenza GL. Hydroxylation of HIF-1: Oxygen Sensing At the Molecular Level. Physiol (Bethesda) (2004) 19:176–82. doi: 10.1152/physiol.00001.2004
48. Brahimi-Horn MC, Pouysségur J. Harnessing the Hypoxia-Inducible Factor in Cancer and Ischemic Disease. Biochem Pharmacol (2007) 73:450–7. doi: 10.1016/j.bcp.2006.10.013
49. Manalo DJ, Rowan A, Lavoie T, Natarajan L, Kelly BD, Ye SQ, et al. Transcriptional Regulation of Vascular Endothelial Cell Responses to Hypoxia by HIF-1. Blood (2005) 105:659–69. doi: 10.1182/blood-2004-07-2958
50. Krämer A, Green J, Pollard J Jr, Tugendreich S. Causal Analysis Approaches in Ingenuity Pathway Analysis. Bioinformatics (2014) 30:523–30. doi: 10.1093/bioinformatics/btt703
51. Luo F, Liu X, Ling M, Lu L, Shi L, Lu X, et al. The Lncrna MALAT1, Acting Through HIF-1α Stabilization, Enhances Arsenite-Induced Glycolysis in Human Hepatic L-02 Cells. Biochim Biophys Acta (2016) 1862:1685–95. doi: 10.1016/j.bbadis.2016.06.004
52. Yuan P, Cao W, Zang Q, Li G, Guo X, Fan J. The Hif-2α-MALAT1-miR-216b Axis Regulates Multi-Drug Resistance of Hepatocellular Carcinoma Cells Via Modulating Autophagy. Biochem Biophys Res Commun (2016) 478:1067–73. doi: 10.1016/j.bbrc.2016.08.065
53. Luo F, Liu X, Ling M, Lu L, Shi L, Lu X, et al. The Lncrna MALAT1, Acting Through HIF-1alpha Stabilization, Enhances Arsenite-Induced Glycolysis in Human Hepatic L-02 Cells. Biochim Biophys Acta (2016) 1862:1685–95. doi: 10.1016/j.bbadis.2016.06.004
54. Luo F, Sun B, Li H, Xu Y, Liu Y, Liu X, et al. A Malat1/Hif-2α Feedback Loop Contributes to Arsenite Carcinogenesis. Oncotarget (2016) 7:5769. doi: 10.18632/oncotarget.6806
55. Bernard D, Prasanth KV, Tripathi V, Colasse S, Nakamura T, Xuan Z, et al. A Long Nuclear-Retained non-Coding RNA Regulates Synaptogenesis by Modulating Gene Expression. EMBO J (2010) 29:3082–93. doi: 10.1038/emboj.2010.199
56. Tano K, Mizuno R, Okada T, Rakwal R, Shibato J, Masuo Y, et al. MALAT-1 Enhances Cell Motility of Lung Adenocarcinoma Cells by Influencing the Expression of Motility-Related Genes. FEBS Lett (2010) 584:4575–80. doi: 10.1016/j.febslet.2010.10.008
57. Guo F, Li Y, Liu Y, Wang J, Li Y, Li G. Inhibition of Metastasis-Associated Lung Adenocarcinoma Transcript 1 in CaSki Human Cervical Cancer Cells Suppresses Cell Proliferation and Invasion. Acta Biochim Biophys Sin (Shanghai) (2010) 42:224–9. doi: 10.1093/abbs/gmq008
Keywords: hypoxia, long non-coding RNA, MALAT1, breast cancer, hypoxia inducible factor-1α, microRNA, miR-3064-5p
Citation: Shih C-H, Chuang L-L, Tsai M-H, Chen L-H, Chuang EY, Lu T-P and Lai L-C (2021) Hypoxia-Induced MALAT1 Promotes the Proliferation and Migration of Breast Cancer Cells by Sponging MiR-3064-5p. Front. Oncol. 11:658151. doi: 10.3389/fonc.2021.658151
Received: 25 January 2021; Accepted: 12 April 2021;
Published: 03 May 2021.
Edited by:
Aamir Ahmad, University of Alabama at Birmingham, United StatesReviewed by:
Hani Choudhry, King Abdulaziz University, Saudi ArabiaCopyright © 2021 Shih, Chuang, Tsai, Chen, Chuang, Lu and Lai. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Liang-Chuan Lai, bGxhaUBudHUuZWR1LnR3
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.