 Tianyi Wu
Tianyi Wu Lizhao Wu*
Lizhao Wu*- Department of Pathophysiology, College of Basic Medical Sciences, China Medical University, Shenyang, China
Gastric cancer is the most common malignant tumor in the digestive tract, with very high morbidity and mortality in developing countries. The pathogenesis of gastric cancer is a complex biological process mediated by abnormal regulation of proto-oncogenes and tumor suppressor genes. Although there have been some in-depth studies on gastric cancer at the molecular level, the specific mechanism has not been fully elucidated. RB family proteins (including RB, p130, and p107) are involved in cell cycle regulation, a process that largely depends on members of the E2F gene family that encode transcriptional activators and repressors. In gastric cancer, inactivation of the RB-E2F pathway serves as a core transcriptional mechanism that drives cell cycle progression, and is regulated by cyclins, cyclin-dependent kinases, cyclin-dependent kinase inhibitors, p53, Helicobacter pylori and some other upstream molecules. The E2F proteins are encoded by eight genes (i.e. E2F1 to E2F8), each of which may play a specific role in gastric cancer. Interestingly, a single E2F such as E2F1 can activate or repress transcription, and enhance or inhibit cell proliferation, depending on the cell environment. Thus, the function of the E2F transcription factor family is very complex and needs further exploration. Importantly, the presence of H. pylori in stomach mucosa may affect the RB and p53 tumor suppressor systems, thereby promoting the occurrence of gastric cancer. This review aims to summarize recent research progress on important roles of the complex RB-E2F signaling network in the development and effective treatment of gastric cancer.
Introduction
Gastric cancer (GC) is a common type of gastrointestinal cancers. Worldwide it is the fifth most frequently diagnosed cancer and the third leading cause of cancer death (1). Although activation of proto-oncogenes and inactivation of tumor suppressor genes are considered as driving forces for GC, the pathogenesis of GC is a complex biological process mediated by both abnormal regulation of multiple genes and environmental insults (2). In recent years, the incidence of GC in western countries has been reduced, but it is still a serious public health problem in developing countries (1). Risk factors include Helicobacter pylori (H. pylori) infection, pickled food, smoking, obesity, chronic gastritis, and iron deficiency (3, 4). The most commonly used classification of GC is the two-category classification based on Lauren’s criteria: intestinal type and diffuse type, which are different not only morphologically, but also clinically and epidemiologically (5). The intestinal type is highly differentiated with a distinct premalignant state during cancer development, whereas the diffuse type is poorly differentiated lacking obvious premalignant lesions (5).
It is well known that tumorigenesis is a complex biological process usually mediated by polygenic mutations. The retinoblastoma (RB) gene (i.e. RB1) is the first tumor suppressor gene cloned in humans by positional cloning (6). It plays an important role in cell cycle regulation by regulating the adenoviral early region 2 binding factor (E2F) transcription factor family (7–10). The RB-E2F pathway not only regulates the cell cycle, but is also regulated by the cell cycle (10). In essence, it links the cell cycle to the transcriptional machinery, and plays a major role in the control of cell growth, apoptosis and differentiation, biological processes that are implicated in cancer development (9, 11).
The role of RB family proteins in GC was last reviewed in 2010 (12). Although much progress has since been made in understanding how the RB-E2F pathway is involved in the pathogenesis of GC, the specific role of E2F family members and the RB-E2F pathway in GC has not been systematically reviewed since a review article on the role of E2Fs in cancers of digestive system was published in 2013 (13). In this review, we will discuss research progress on the role of RB and E2F family members as well as their major upstream regulators in the initiation, progression and prognosis of GC. In addition, we will also summarize major research findings on how H. pylori infection impacts the development of GC by functionally disrupting the RB and p53 tumor suppressor systems. Finally, we will discuss major clinical implications of this research progress in effective treatment of GC.
Gastric Cancer
Intestinal gastric cancer (IGC) is thought to be initiated primarily by H. pylori infection, with higher incidence in older men in high-risk areas (14, 15). Well differentiated and poorly differentiated gastric adenocarcinomas usually harbor different genetic changes, with well-differentiated being more frequently associated with changes in important cancer-related genes such as RB and PTEN (16). IGC has a relatively clear development process that is called metaplasia-neoplasia-carcinoma sequence or Correa’s cascade, from atrophic gastritis to intestinal metaplasia (IM) to dysplasia and then to IGC (17). IM is a recognized premalignant lesion of gastric mucosa, defined as the replacement of gastric mucosa by epithelial cells with intestinal morphology, and is associated with an increased risk of GC (18, 19). IM can be either complete (with the large-intestine phenotype) or incomplete (with the small-intestine phenotype), with the latter more frequently associated with malignant transformation (18). In a 10-year prospective study published in 2018, it was found that IM cells had both genetic and epigenetic mutations that differed from GC cells (20). For example, TP53 and ARID1A, which are involved in the regulation of the RB-E2F pathway, are the most frequently mutated genes in GC, but are rarely mutated in IM (20). However, the exact mechanism of different genomic and epigenetic alterations between IM and GC and their application value in the prevention of GC still need to be explored (20).
Diffuse gastric cancer (DGC) usually results from pangastritis, has no atrophy and occurs mainly in younger female patients in low-risk areas (14, 15). DGC is poorly differentiated with stronger metastasis and invasiveness, and is often associated with CDH1 deficiency (21). By exploring the co-expression network of GC-related genes, an integrative functional genomics study group revealed differences between the two major subtypes of GC in transcriptional and epigenetic regulations as well as in stem cell characteristics (22). IGC was believed to be more affected by E2F-mediated transcription (22). Considering different characteristics of the two subtypes, development of subtype-specific targeted treatment strategies for GC deserves more attention.
In addition to the aforementioned Lauren’s classification, GC can also be divided into four molecular subtypes based on analyzing the data from The Cancer Genome Atlas (TCGA) project (23). The first subtype is Epstein–Barr virus (EBV)-associated GC, accounting for about 10% of GC (24). The association between EBV and GC was first recognized in 1990 (25). EBV has been shown to induce the nuclear export of E2F4 and E2F5 to prevent cell cycle arrest, an action that may have implications for the pathogenesis of GC (26). The second subtype is microsatellite unstable GC, accounting for 15–20% of GC. The hallmark of this subtype is microsatellite instability (MSI), accompanied with increased gene mutation rates (23). The intracranial histological heterogeneity of GC with MSI was associated with progressive frameshift mutations of TGF- receptor type II and E2F-4 (27). High levels of MSI were more common in IGC and in the antrum, with better differentiation and more lymphoid infiltration (28). The other two molecular subtypes of GC are genomic stable (GS) GC and GC with chromosomal instability (CIN) (23), which includes poorly differentiated endocrine carcinomas that are often accompanied with the inactivation of p53- and RB-related pathways (29). Interestingly, a similar study based on gene expression profiling identified three subtypes of gastric adenocarcinoma: proliferative, metabolic, and mesenchymal, with the proliferative subtype being often associated with the activation of E2F-mediated pathway (30). Since patients with different subtypes likely have different clinical characteristics and molecular basis, they may benefit from different treatments. Abnormalities of key components in the RB-E2F pathway identified in patients with GC are summarized in Figure 1 and Table 1.
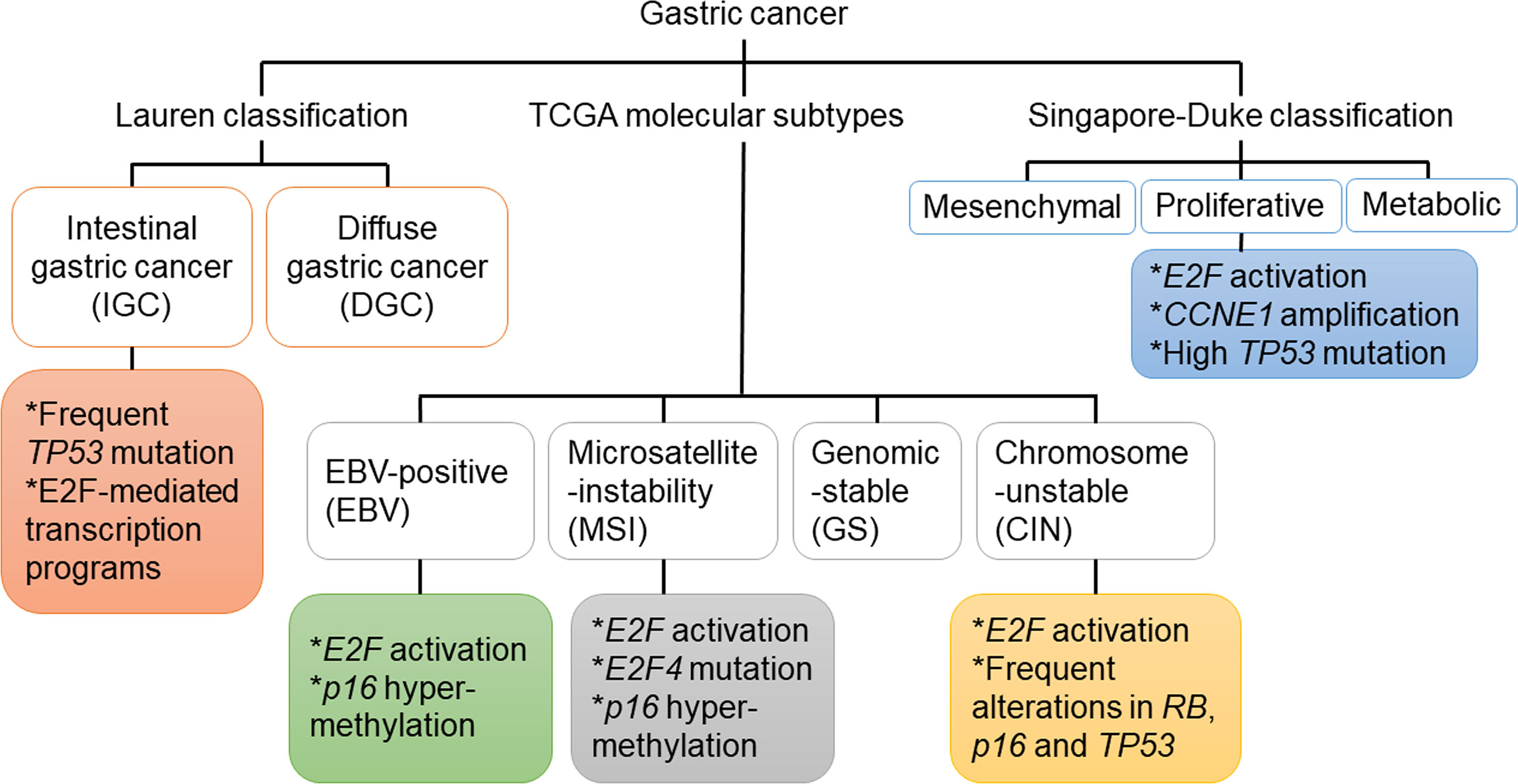
Figure 1 Genetic and epigenetic mutations of genes in key components of the RB-E2F pathway that were identified in different subtypes of gastric cancer.
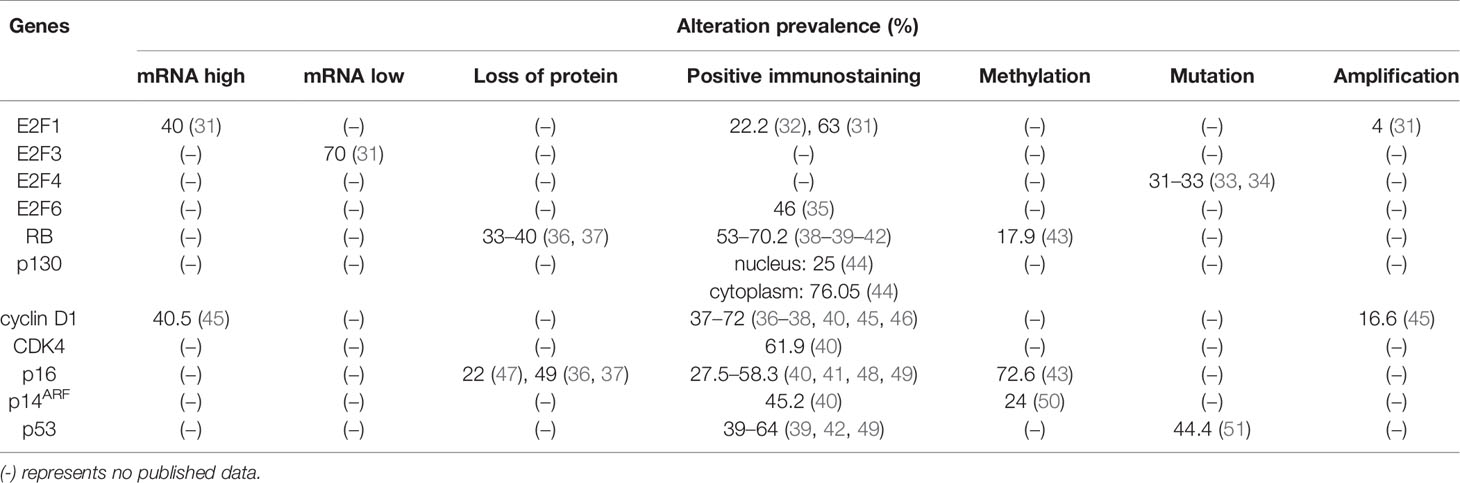
Table 1 Abnormalities in key components of the RB-E2F pathway in patients with GC.
The RB Family
The RB family consists of three members in humans, which are collectively referred to as “pocket proteins” and are involved in the regulation of the cell cycle (52). They are also involved in many biological processes such as proliferation, differentiation, senescence, apoptosis, gene regulation, and interact with many other cellular proteins (53, 54). The eponymous member of the pocket protein gene family is RB1 or RB, which was named from an inherited eye tumor called retinoblastoma (55). The RB gene was mapped on chromosome 13q14.2 (6). RB is widely distributed in various tissues and interacts with a large number of transcription factors and chromatin-remodeling proteins, allowing itself to bind to transcription factors and to modify chromatin structure (56). In addition to regulating the cell cycle, RB has also been shown to inhibit apoptosis (57). Consistent with an important role of RB in tumorigenesis, loss of function of RB has been associated with the development of many human cancers (58–64).
The second member of the family is p130, which was cloned in 1993 and mapped on chromosome 16q12.2 (65, 66). The third member of the family is p107, which was mapped on 20q11.2 (67). Interestingly, the three pocket proteins have overlapping and interdependent functions (68). In both quiescent and p53 activation conditions, RB and p130 can cooperate to repress G1/S genes, a process that RB plays a predominant role (69, 70). In the absence of RB and p130, p107 can also repress G1/S genes (69). Under the condition of DNA damage, p130 and p107 can cooperate to repress the G2/M genes and thus block cell cycle entry into mitosis (69). In general, when DNA damage leads to p53 activation, RB, p130 and p107 cooperatively repress G1/S genes while p130 and p107 cooperatively repress G2/M genes (69). In mice, pocket proteins have overlapping functions in suppressing the development of various types of tumors. For example, RB and p107 worked together to suppress the development of retinoblastoma (71, 72), head and neck cancers (73), and spontaneous skin tumors (74). In addition, RB and p130 worked together to suppress the development of retinoblastoma (75, 76).
The E2F Family
The E2F family of transcription factors includes 10 members, encoded by eight different genes, E2F1–E2F8 (9). E2F3 consists of two isoforms, E2F3a and E2F3b, derived from two different promoters (77). Members of the E2F family have both distinct and overlapping functions, and are important for various biological processes such as cell cycle control, cellular proliferation and apoptosis (78, 79). E2F1–6 are canonical E2Fs, which form heterodimers with dimerization partner (DP) proteins (80). E2F7 and E2F8 are atypical E2Fs, which do not bind to DP but have two DNA binding domains (9). All E2F members can bind to DNA in a sequence-specific manner to initiate transcriptional activation or repression of target genes (80). E2F1-3a are transcriptional activators, whereas E2F3b-8 are transcriptional repressors (9). However, it is worth noting that E2F3b can also act as a transcriptional activator (81, 82), even though its expression pattern during the cell cycle is similar to that of a canonical E2F repressor (77). The functional specificity of E2F-DP complex is determined by the E2F subunit, but in the absence of DP, E2Fs become non-functional (78).
In quiescent cells, pocket proteins can bind to E2F-DP heterodimers to repress E2F target genes. It is worth noting that different pocket proteins preferentially bind to different E2F transcription factors (83). RB binds to E2F1, E2F2, and E2F3 to form the repressive RB-E2F complex, while p107 and p130 bind to E2F4 and E2F5 to form the repressive DREAM (DP, RB-like, E2F and MuvB) complex (10). E2F6, E2F7 and E2F8 are not bound by pocket proteins (9). In G0 and early G1 phases, hypophosphorylated RB, which is in an activated state, binds to the pocket domain of E2F1-3 and inhibits E2F-mediated target gene activation, thereby blocking cell cycle progression at the G1/S transition (84). In addition, E2F4 and E2F5 can form complexes with p107 and p130 to mediate gene repression (84). When cells receive growth stimuli, activation of cyclin dependent kinases (CDKs) leads to the phosphorylation of pocket proteins and collapse of the previously formed RB-E2F complexes and DREAM complexes (70). The subsequent release of E2F1-3 from those complexes can activate target genes required for cell cycle entry (10, 52).
Pocket Proteins in Gastric Cancer
Various studies showed that RB plays important roles in the various aspects of GC. However, earlier studies focused on evaluations of its protein levels in various contexts of GC appeared to yield seemingly conflicting results. For example, compared with non-neoplastic tissues, tumors could have higher (38) or lower (48) levels of RB. In addition, altered RB protein levels were more frequent in less-invasive GC than in advanced invasive GC (85). In univariate and multivariate analyses, positive RB expression was found to be significantly correlated with the presence of lymph node metastasis (39). Nevertheless, another study showed that the expression of RB in lymph node metastasis was lower than that of the corresponding primary tumor (36). These inconsistent data may be related to the fact that RB function is largely dependent on its posttranslational regulation (i.e. phosphorylation). Therefore, defining the precise role of RB in various processes of GC likely requires evaluation of its phosphorylation status instead of just its mRNA or protein levels. In addition, since RB function is manifested at least in part through limiting activities of activator E2Fs, evaluating the RB status in GC patient samples will benefit from simultaneously evaluating the status of activator E2Fs. It is interesting to note that DNA methylation of the RB gene promoter was found in significantly more GC samples (17.9%) than in normal samples (5.5%) (43), suggesting that RB methylation may also play a role in GC.
Although little is known about the precise role of p107 in GC, cellular localization of p130 seems to play an important role in some aspects of GC. For example, high levels of nuclear localization of p130 were significantly correlated with lower grade GC, whereas high levels of cytoplasmic localization of p130 were significantly correlated with IGC (49). Besides, p130 was localized in the cytoplasm in DGC but in the nucleus in normal cells, further supporting an important role of its nuclear delocalization in the development of GC (44). However, no correlation has been found between cytoplasmic localization of p130 and tumor grade or survival of DGC. Although the functional consequence of p130 nuclear delocalization on the development of GC is currently unclear, it is plausible that such delocalization promotes the development of GC through inhibiting the function of p130 as a transcriptional modulator. Further investigations are needed to experimentally determine the precise role of p130 nuclear delocalization in GC and its underlying mechanisms.
Since the 2010 review, two significant advances have been made in the understanding of pocket proteins in GC. The first interesting and important finding was that p130 was primarily localized in the nucleus in normal cells but was mainly localized in the cytoplasm in DGC cells (44). Future studies should be directed to understanding the precise role of p130 subcellular localization in GC, the nucleo-cytoplasmic shuttling mechanisms, and whether p130 nuclear delocalization facilitates GC development by impairing p130-mediated transcriptional repression. In addition, it has been found that besides RB phosphorylation, RB promoter methylation may also play a role in the development of GC (43), highlighting the importance of epigenetic regulation of pocket proteins in GC. It would be interesting to know whether RB promoter methylation levels are different among various subtypes of GC, or among different stages of GC development.
The E2F Family in Gastric Cancer
Among E2F family members, E2F1 is so far the most widely studied in tumors, including GC. It is interesting to note that in vitro different levels of E2F1 had different effects on cell fate: low levels of E2F1 could promote cell cycle progression, medium levels of E2F1 could cause cell cycle arrest, and high levels of E2F1 could lead to cell apoptosis (86). Several earlier studies using either transgenic mouse models or in vitro systems showed that the role of E2F1 in tumorigenesis was pleiotropic, manifested by the fact that it might either promote or suppress tumorigenesis, depending on dominant signaling pathways and cell types (87–91). In GC, E2F1 gene amplification was rare, but its overexpression was detected in about 40% of patients (31). Gene expression microarray data and bioinformatic analysis of public datasets also showed that E2F1 was up-regulated in GC (92, 93). In addition, high mRNA levels of E2F1 were related to poor survival (93). In order to better understand the role of E2F1 in biological processes of GC, various research groups investigated effects of E2F1 overexpression or knockdown on the tumorigenicity of GC cells. For instance, overexpression of E2F1 in MGC-803 GC cell line led to significantly increased levels of apoptosis but significantly reduced levels of cellular proliferation and invasiveness, consistent with the tumor suppressor function of E2F1 (94). In addition, overexpression of E2F1 suppressed tumor growth and promoted tumor cell apoptosis in nude mice implanted with E2F1-overexpressing MGC-803 cells (95). Furthermore, adenovirus-mediated overexpression of E2F1 in AGS and SNU-1 GC cell lines induced apoptosis and reduced cell survival rate (96). On the other hand, E2F1 downregulation by intratumor-injection of E2F1 shRNA in nude mice engrafted with MGC-803 cells inhibited tumor growth and promoted apoptosis, accompanied by up-regulation of PTEN, Caspase-3 and Caspase-9 (97). In addition, in cisplatin-resistant SGC7901/DDP cells, shRNA-mediated E2F1 downregulation blocked cell cycle progression, promoted apoptosis, and increased the sensitivity of cells to several chemotherapeutic drugs, suggesting that E2F1 served as an oncogene and promoted multidrug resistance in GC (98). Although the dual role of E2F1 in GC is likely context dependent, mechanisms underlying the seemingly inconsistent results are provided from the existing literature. For example, E2F1 is considered as a proto-oncogene, and elevated E2F1 levels are sufficient to drive cell proliferation and cell cycle progression (99, 100), which can also explain E2F1 overexpression corresponding to poor prognosis (101). On the other hand, the tumor suppressive effect of E2F1 can be explained by E2F1-mediated apoptosis and growth arrest (102–104). E2F1 can inhibit the degradation of p53 by inducing the expression of p14ARF, leading to increased apoptosis and cell cycle arrest (105). In addition, E2F1 can also induce p53 independent of p14ARF (103). This also explains why RB-negative tumors tend to be p53 negative, probably to avoid the negative E2F1-p14ARF feedback (106). Interestingly, E2F1 protein levels could also reflect the sensitivity of GC patients to adjuvant chemoradiotherapy after radical gastrectomy. For example, among postoperative patients receiving adjuvant chemoradiotherapy, the E2F1 immuno-positive group had a higher survival rate than the E2F1 immuno-negative group (32). The immunopositivity of E2F1 might be used as an indicator for good response for adjuvant chemoradiotherapy and radiotherapy after surgery (107). A key determinant of the efficacy of anticancer therapies is the ability of cancer cells to undergo apoptosis in response to DNA damage factors (108). The success of radiotherapy or chemotherapy is at least partly due to the fact that cancer cells are more likely than normal cells to die when induced by DNA damage. Under the condition of DNA damage, E2F1 induces apoptosis through activation of various cell death pathways, which may explain the higher sensitivity of samples with high E2F expression to radiotherapy and chemotherapy (109).
Given the important role of E2F1 in GC, E2F1 has been considered as a potential therapeutic target for GC patients (93). However, since E2F1 activity is also important for normal cellular proliferation, therapeutically targeting E2F1 may have significant side effects on normal tissues that are capable of proliferating. In addition, due to the highly overlapping and compensatory effects of E2F activators (78), simple targeted intervention of E2F1 may lead to compensational upregulation of other two E2F activators, making such a therapy less effective. Therefore, E2F1 targeted therapy may require simultaneously targeting the other two E2F activators to achieve a better clinical outcome. Furthermore, the bidirectional effect of E2F1 on GC suggests that success on the targeted therapy is likely dependent on a clear understanding of the predominant oncogenic pathways involved in individual patients.
There are also several studies on E2F4 in GC. E2F4 mutation was found to be a common and an early event in the occurrence of GC, and might occur in the process of precancerous lesions such as IM and dysplasia (33, 34). E2F4 mutation in gastrointestinal tumors might not be random as it appeared frequently in a microsatellite region at exon 7 with a serine-encoding trinucleotide repeat sequence (33, 110). In addition, E2F4 frameshift mutation was associated with differentiation grades of GC as frameshift mutation of the microsatellite regions encoding serine repeats might inhibit the formation of RB-E2F4 complex and reduce the level of differentiation (27). Furthermore, a study of MSI suggested that E2F4 might be involved in the transformation of gastric adenocarcinoma into squamous cell carcinoma (111). Interestingly, by establishing an E2F-related transcriptional regulatory network, a research group found that target genes regulated by E2F1 and E2F4 showed a large number of differential expressions in GC, indicating that E2F1 and E2F4 might play important roles in tumorigenesis of GC (92). It was found that E2F4 mRNA levels increased with the degree of tumor invasion and malignancy (92). Bioinformatic analysis of a Gene Expression Profiling Interactive Analysis (GEPIA2) dataset representing 408 GC samples and 211 normal tissues showed that there was no difference in average expression levels of E2F4 between GC samples and normal tissues, but bioinformatics analysis using a completely different and consolidated Gene Expression Omnibus (GEO) dataset representing a much larger sample size (i.e. up to 876 GC samples) showed that patients with relatively high E2F4 expression had worse survival than those with relatively low E2F4 expression (93). As a member of the DREAM complex, E2F4 can repress many cell cycle genes (112), which are common markers of proliferation that can stratify most cancers, including GC (101). Although both high expression of E2F4 in advanced GC and its correlation with poor prognosis are seemingly contradictory to the repressive role of E2F4 in cell cycle control, there are existing studies supporting an oncogenic role of E2F4. For example, E2F4 induced proliferation and promoted the development of skin tumors in a keratin 5 promoter-driven E2F4 transgenic mice (113). In addition, E2F4 reduced apoptosis in cardiac myocytes (114). However, more investigations should be done to explore the precise role and underlying cellular and molecular mechanisms of E2F4 in GC initiation and progression, and to determine whether E2F4 overexpression is associated with a specific subtype of GC.
Compared to E2F1 and E2F4, other E2F family members have been much less studied regarding their potential roles in GC. Gene expression microarray data showed that mRNA levels of E2F2 in GC samples were increased compared with those in normal samples (92). Using Northern blot technique to analyze 30 GC samples and their corresponding non-neoplastic mucosa, a Japanese research group found that mRNA levels of E2F3 were lower in 70% of GC samples than in normal controls (31). In contrast, bioinformatic analysis of RNA sequencing data from a GEPIA2 dataset representing a much larger sample size (i.e. 408 GC samples and 211 normal gastric tissues) showed that expression levels of E2F3, along with E2F2, E2F5, E2F7 and E2F8, were significantly higher in GC samples than those in normal tissues (93). These conflicting results on E2F3 expression levels from the two studies may be due to differences in patients’ genetic background (i.e. mostly Japanese vs. mostly Caucasians and African Americans) and/or techniques (i.e. Northern blot vs. RNA sequencing) used to evaluate E2F3 expression levels. Moreover, high levels of E2F2, E2F3, E2F5, E2F6, E2F7 and E2F8 were related to better survivals (93). E2F6 was localized in the nucleus, and was at high levels in gastric adenocarcinoma without lymph node metastasis (35). Similarly, univariate analysis showed that the expression of E2F6 was negatively correlated with lymph node metastasis, suggesting that E2F6 might suppress the metastasis of GC (35). Thus it is clear that considerably more study is warranted to investigate the role and mechanism of E2F2, E2F3, E2F5, E2F6, E2F7 and E2F8 in GC. For example, it would be interesting to know whether all or some of the aforementioned upregulated E2F factors (93) are coordinately overexpressed in GC samples.
Since the summary of the role of E2F transcription factors in digestive tract malignancies in 2013, much progress has been made in understanding the roles of E2F family members in GC. There have been more data to explain the bidirectional effect of E2F1 on GC. In addition, the relationship between E2F1 and better chemoradiotherapeutic response in GC has been established. It is worth pointing out that the bidirectional effect of E2F1 and its effect on chemoradiotherapeutic sensitivity have also been found in many other tumors (109, 115). Furthermore, the application of bioinformatics has facilitated our understanding of GC-specific genetic alterations in various E2F members as well as their prognostic and other clinical implications.
Upstream Regulators of the RB-E2F Pathway in Gastric Cancer
Many upstream regulators of the RB-E2F pathway also play important roles in GC. The activities of RB and other pocket proteins are mainly regulated by phosphorylation through CDKs, which are in turn regulated by cyclins and cyclin-dependent kinase inhibitors (CKIs) (116, 117). Therefore, cyclins, CDKs, and CKIs as well as any molecules that regulate these three types of proteins may be involved in the pathogenesis of GC.
The cyclin D1 protein was almost undetectable in normal gastric mucosa, but was elevated in about half of GC cases, indicating that overexpression of cyclin D1 might be an early event in the process of tumorigenesis in GC (45, 46). The p16 gene, also known as p16INK4a, is located on chromosome 9p21 (118) and encodes for a protein that is an inhibitor of CDK4 (119, 120). As a CKI, p16 is able to competitively block the cyclin D1-CDK4 complex by binding to CDK4, an action that inhibits CDK4-mediated RB phosphorylation and prevents cell cycle progression from G1 to S phase (118). Loss of p16 function leads to an abnormal increase in cyclin D1-CDK4 complex activity, resulting in sustained RB phosphorylation (118). At the same time, phosphorylation of RB in G1 phase results in increased expression of p16 to limit CDK4 activity (118). This negative feedback loop of p16 and RB is critical for normal cell cycle control to protect cells from abnormal cellular proliferation. Therefore, deregulation of key components in the feedback loop is likely associated with the development of GC. For example, various p16 abnormalities have been identified in GC patient samples. An early study showed that about 50% of GC samples were detected with the loss of p16 expression (36). Interestingly, the expression of p16 in distal gastric carcinomas was higher than that in gastric cardia carcinomas (40). In GC, abnormal methylation of CpG islands in the promoter region of p16 downregulated p16 (47). Methylation of p16 was present in about 70% of GC samples, while there was almost no p16 methylation in normal samples (43). In addition, methylation of p16 was found in both IGC and DGC, but had no significant correlation with either tumor staging or histology (50). It is worth noting that hypermethylation of p16 significantly increased in MSI-high GC (121). Furthermore, p16 hypermethylation is also very common in EBV-associated GC, and may even be one of the important causes of EBV-associated GC (122, 123). The expression levels of p16 and RB were not only altered in GC, but also negatively correlated (41, 124, 125).
Several other upstream regulators in the RB-E2F pathway have also been implicated in GC. For example, transforming growth factor-beta1 (TGF-β1) inhibited GC cell growth by upregulating its downstream target p21, thereby blocking p130 phosphorylation and preventing aberrant cell cycle progression by downmodulating CDK activities (126). In addition, the tumor suppressor function of periostin was achieved by its induction of RB phosphorylation and the subsequent release of E2F1, which activated its target gene p14ARF, leading to the inactivation of MDM2 and the consequential reduced ubiquitination of p53 and E-cadherin (127). Moreover, SPIN1 could form a positive feedback loop with E2F1 to promote the development of GC (128). Furthermore, ATAD2 knockdown in GC cells led to reduced levels of cyclin D1, cyclin E, E2F1 and RB phosphorylation, thus inhibiting proliferation and cell cycle progression (129). Interestingly, decrease of intracellular chloride ion concentration could increase the level of p21 and reduce the phosphorylation of CDK2 and RB (130). This effect led to cell cycle arrest and inhibited the growth of tumor cells, providing us with a new therapeutic strategy (130). The fact that many upstream molecules of the RB-E2F pathway have a large proportion of genetic and epigenetic alterations in GC (Figure 1 and Table 1) suggests that in addition to the downstream effectors of RB such as activator E2Fs, its upstream regulators such as p16 and cyclin D1 also play important roles in GC, with the specific regulation network shown in Figure 2. Therefore, understanding the status of the upstream regulators of RB will not only further help us better understand the functional role of RB in various process of GC, but may also provide additional insights on the diagnosis, prognosis and effective treatment of GC.
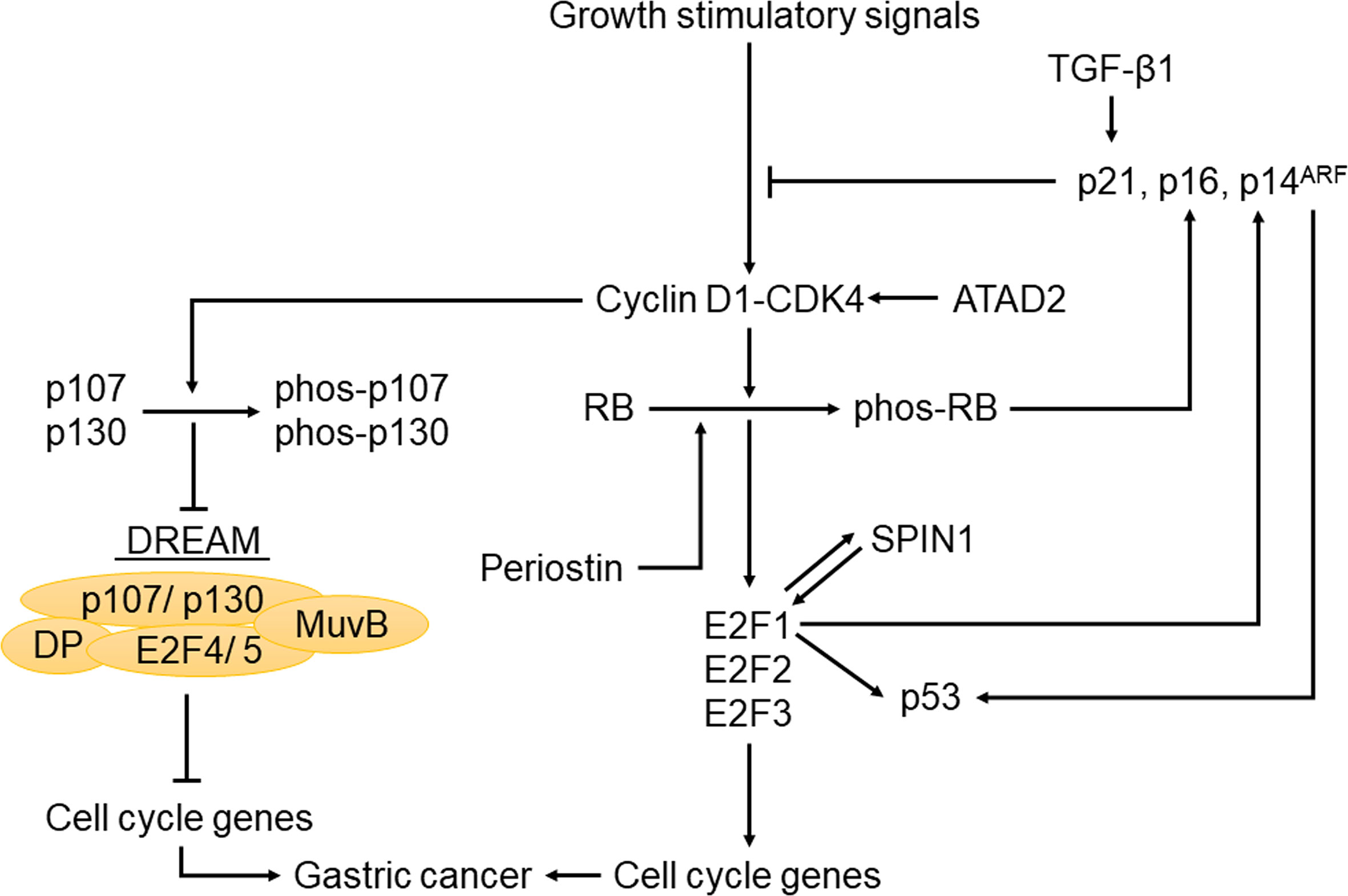
Figure 2 The regulatory network of the RB-E2F pathway in gastric cancer. When cells receive growth stimuli, activation of CDKs leads to the phosphorylation of pocket proteins, collapse of the previously formed RB-E2F complexes and DREAM complexes. The subsequent release of E2F1-3 can activate target genes required for cell cycle entry. Meanwhile, this pathway is also regulated by upstream and downstream molecules in gastric cancer, such as CKIs, p53, TGF-β1, ATAD2 and SPIN1.
Helicobacter pylori and the RB and p53 Tumor Suppressor Systems
The p53 tumor suppressor gene, also known as TP53, was first discovered in 1979 (131). In cells under non-stressed condition, p53 is usually present in small amounts (132). However, in the case of stress, such as hypoxia, DNA damage, proto-oncogene activation, radiotherapy and chemotherapy, p53 protein is stabilized to initiate a damage response cascade (132). If the damage cannot be repaired in time, p53 would induce apoptosis by binding to the apoptosis stimulating proteins of p53 (ASPP) (133, 134). Like RB, the p53 tumor suppressor also controls cell cycle but through independent and interrelated pathways (135). Therefore, it is not surprising that alterations of TP53 and RB are common events in human GC. It was reported that TP53 gene mutations were found in about 50% of GC cases (51).
H. pylori is a Gram-negative bacterium that was found in stomach mucosa, and is an important risk factor for GC, equivalent to a type I carcinogen (136). About half of the population in this world has H. pylori infection, and infection rates in Asian countries are generally higher than those in western countries (137). H. pylori could cause abnormal DNA methylation and inflammation, which increased the risk of GC (138). However, there was no significant difference in the rates of H. pylori infection either between IGC and DGC, or between proximal and distal tumors (139). Interestingly, high levels of RB methylation in H. pylori-positive individuals might increase the risk of GC (140). The proportions of RB tumor suppressor and the p53 tumor suppressor pathway abnormalities in H. pylori-infected GC were higher than that in non-H. pylori-infected GC (42). It was reported that H. pylori infection might first activate C-MYC and BCL-2 in IM, and then inactivate the RB and p53 tumor suppressor pathways in dysplasia, causing a severe imbalance of proliferation and apoptosis in precancerous lesions, leading to the occurrence of GC (42).
The pathogenicity of H. pylori is mainly due to its flagellum, lipopolysaccharide, vacuolar toxin VacA, and cytotoxin-related gene pathogenicity island (cagPAI) (141–143). VacA could generate a protective intracellular reservoir where H. pylori survives by usurping lysosomal and autophagy pathways. Besides, it was found that gastric epithelial cell apoptosis induced by VacA did not require RB regulation, and occurred whether or not p53 was expressed (144). The most important and widely studied virulence factor of H. pylori strains is cytotoxin-associated protein (CagA). CagA is introduced into gastric epithelial cells by the type IV secretion system (T4SS), leading to the promotion of genetic instability, epithelial–mesenchymal transformation and eventual carcinogenesis (145). In addition, CagA bound to ASPP2, thereby inhibiting the binding of ASPP2 to p53, leading to decreased apoptosis and promoting the formation of GC (146, 147).
H. pylori could activate the PI3K/AKT pathway (148, 149) or the MAPK/ERK pathway (150) to activate the ubiquitin ligases murine double minute (MDM2, also known as HDM2), which promoted ubiquitination and proteasomal degradation of p53. On the other hand, p53 can activate MDM2 to form a negative feedback loop that ensures low levels of p53 in unstressed cells (151). Related proteins of the p53-MDM2 feedback loop were distinctly expressed at different stages of GC development (152). In the case of H. pylori infection, MDM2 expression was found to be significantly elevated in the progression from chronic gastritis to GC (152). Interestingly, MDM2 was bound to RB through a central acidic domain in U20S, C33A, SAOS-2 cells (153), and could promote proteasomal degradation of RB in cells of osteosarcoma, cervical cancer, non-small cell lung cancer and temperature-sensitive murine ts20 cells (153, 154).
H. pylori could also induce a subtype-specific damage response mechanism of p53 in a T4SS-dependent manner (155). Specifically, H. pylori induced Δ133p53 and Δ160p53, which encode for N-terminally truncated isoforms of p53 protein, thereby inhibiting the activity of p53. Δ133p53 also activated the NF-κB pathway and caused up-regulation of its downstream target genes, leading to the inhibition of apoptosis indirectly (155). There is also cross talk between p53 and NF-κB pathway that results in reduced apoptosis and the occurrence of tumor (156). Besides, some H. pylori products were associated with the RB-E2F pathway in vitro (157, 158). By isolating and cloning genes encoding for the two secretory H. pylori proteins CagA and HspB, and transfecting them into AGS cell line, researchers found that CagA and HspB directly promoted the growth of GC cells by facilitating G1-S transition of the cell cycle through the upregulation of cyclin D3 and subsequent RB phosphorylation (158). Interestingly, unknown soluble factor(s) released by H. pylori in cell culture medium might inhibit RB phosphorylation by increasing the level of p27, leading to inhibition of cell cycle progression in gastric epithelial cells (157). The primary interactions between H. pylori and the RB and p53 tumor suppressor pathways are summarized in Figure 3.
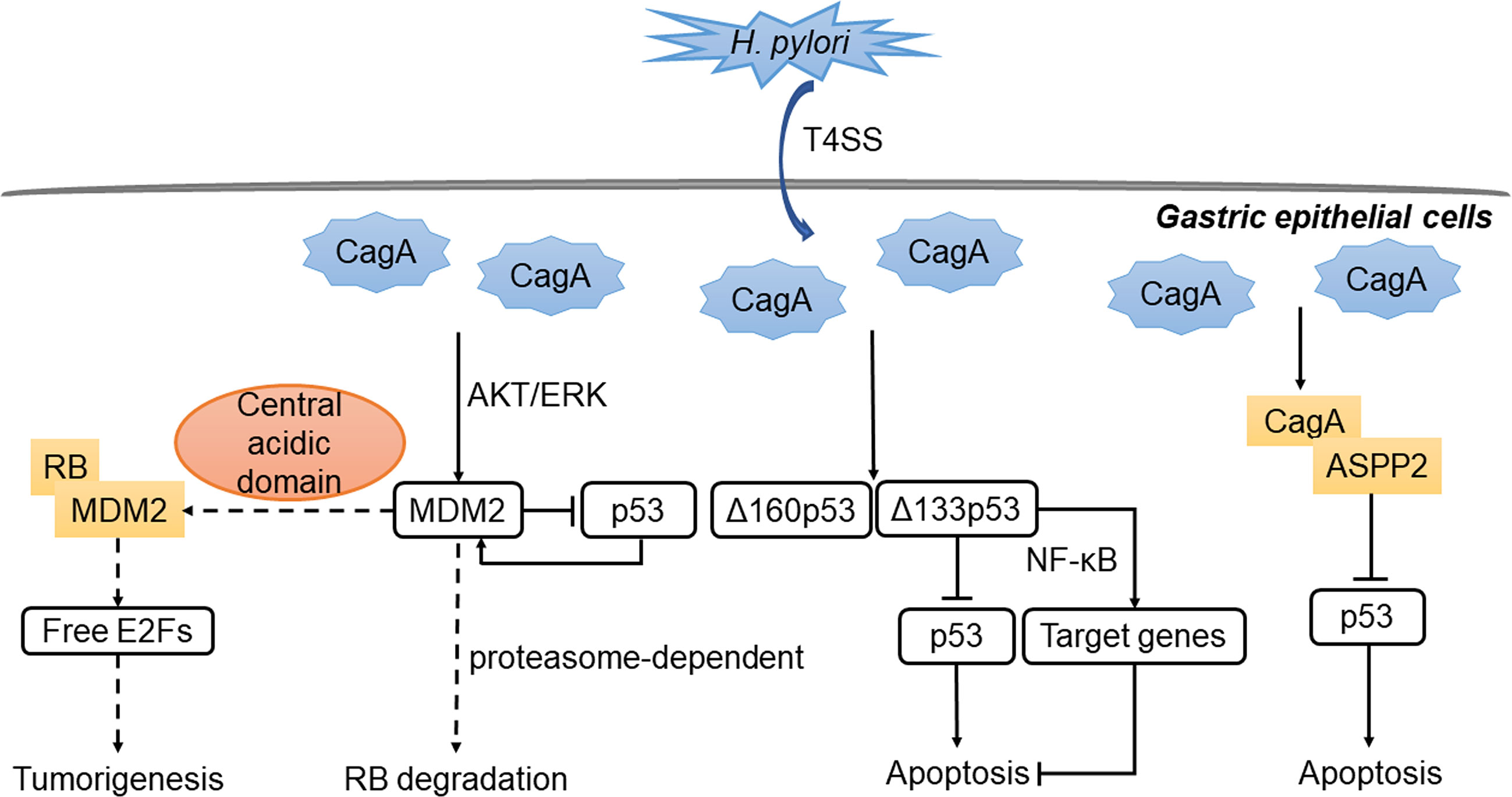
Figure 3 Regulation of p53 and RB by H. pylori in stomach. The virulence factor CagA is introduced into gastric epithelial cells by T4SS. CagA binds to ASPP2, thereby inhibiting the binding of ASPP2 to p53, leading to decreased apoptosis. Besides, MDM2 is activated through the AKT/ERK pathway and form a negative feedback loop with p53. MDM2 can bind to RB and inhibits the function of RB-E2F repressor. Meanwhile, the expression of the isoforms of p53 inhibits the activity of p53 or activates NF-κB target genes. The outcome is the down-regulation of apoptosis and the occurrence of gastric cancer.
Studies based on clinical practice have shown that treatment of H. pylori reduced the risk of precancerous lesions converting to GC, but the degree of risk reduction depended on the population and the extent of damage already present at the time of eradication (159–161). The close association between H. pylori and the RB and p53 suppressor pathways provides us with the possibility of combinational therapy, such as H. pylori eradication combined with targeted intervention of MDM2 or other related molecules, which may greatly improve the therapeutic effect. Interestingly, the positive index of E2F nuclear staining was higher in H. pylori-infected gastric mucosa than in non-infected gastritis samples, and E2F1 was co-localized with proliferating cell nuclear antigen (PCNA) (162). The positive index of E2F1 decreased after H. pylori successful eradication (162). This study suggests that enhanced expression of E2F may be involved in the occurrence and development of H. pylori-infected GC by promoting cell cycle progression (162). Therefore, we speculate that E2F-targeted therapy may be more effective in patients with H. pylori-infected gastritis and H. pylori-infected GC, and has the potential to be applied in the prevention and treatment of GC.
Potential Therapeutic Targets of the Rb-E2f Pathway in Gastric Cancer
Because GC is often asymptomatic in the early to middle stage of the disease progression, it is often diagnosed at an advanced stage with limited treatment options. Currently the primary treatment strategy for GC is still surgeries, complemented with chemotherapy and radiotherapy (163). Most patients still have low survival rates and high recurrence rates (163). Therefore, it is particularly important to find more effective treatment strategies and preventive measures for GC. Through studies on the function of tumor suppressor genes and mechanisms of related pathways such as the RB-E2F pathway, we may be able to find novel therapeutic targets and develop more effective treatment strategies for GC. Antagonists of CDK can block the action of the cyclin D1-CDK4 complex to target the RB-E2F pathway for cancer therapy (164). Flavopiridol is a broad-spectrum CKI commonly used in clinical practice of solid tumors (165). A phase II clinical trial showed that flavopiridol alone had no significant antitumor effect on advanced GC (166), pending changes in regimen and combination with other agents. Selective CDK4/6 inhibitors palbociclib, ribociclib and abemaciclib have been developed and are undergoing clinical trials in a variety of cancers (167). Palbociclib is in phase II trials in patients with advanced GC with limited single-agent activity (168). Some of the genetic characteristics of GC help us stratify patients for the most effective drug therapies. Studies showed that high levels of cyclin E protein in GC correspond to increased resistance to palbociclib (169). The methylation of p16 increased the sensitivity of GC cells to abemaciclib, suggesting that abemaciclib is more effective in patients with hypermethylated p16 (170). Targeted therapies with a single drug are likely to develop drug resistance, but combinations of drugs are more effective in controlling the disease. For example, palbociclib had a synergistic effect with 5-FU in the treatment of GC cells (169). Combination of human epidermal growth factor receptor 2 (HER2) inhibitor pyrotinib and CDK4/6 inhibitor SHR6390 was thought to be a more effective treatment strategy for HER2-positive metastatic GC (171). In Table 2, we summarize the CKIs currently used in clinical trials and preclinical studies for GC. Notably, CDK4/6 inhibitor relies on RB to induce cell growth arrest (172). In order to improve therapeutic efficacy and precision, we need to develop new therapeutic strategies, such as the use of multiple CDK4/6 inhibitors to enhance cell cycle arrest and selective targeting of RB-deficient tumors (172). Immunotherapy based on immune checkpoint block is being applied in the clinical treatment of advanced GC, such as anti-PD-1 therapy (163). However, most GC cases are not sensitive to immune checkpoint inhibitor monotherapy, so patients may need combinational therapies to improve response to the PD-1 therapy or other immune checkpoint inhibitors (163). If CDK4/6 inhibitors can be combined with immunotherapy in the treatment of GC in the future, perhaps a better therapeutic effect will be achieved.
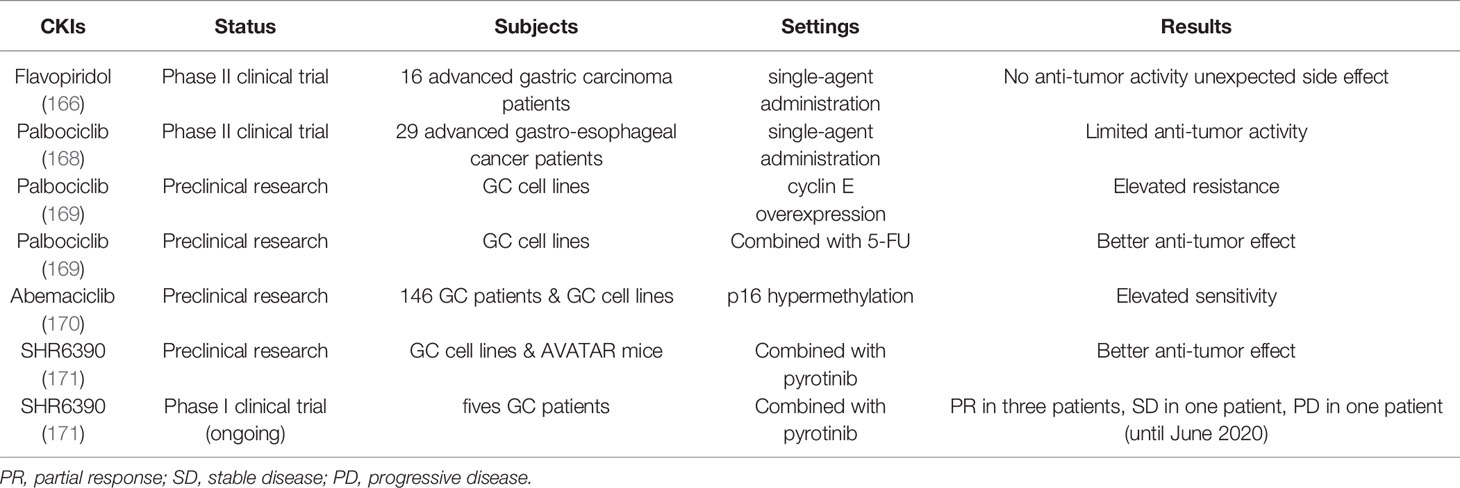
Table 2 CKIs for clinical trials and preclinical studies in GC.
Using an E2F promoter-regulated adenovirus carrying the p16 gene could combine the apoptosis induced by p16 gene and oncolysis induced by virus replication to have antitumor effect on GC (173). This kind of replication-competent adenovirus (RCAd) provided a new view of cancer therapies (173). At present, oncolytic virus has become an active research field on cancer targeted therapies (174). Advances in genetic engineering can help scientists create oncolytic viruses that target cancer cells with different types of mutations to achieve better therapeutic effects (174). If oncolytic virus and immunotherapy are properly combined in GC, it is possible to achieve a synergistic anti-cancer effect. However, since there are significant uncertainties on potential side effects and viral penetration efficiencies in solid tumors like GC, such a therapy option still has a long way to go before it can be used in clinical practice of GC.
H. pylori eradication therapy has been widely used in clinical practice and significantly reduced the risk of GC (160, 175). An experiment using H. pylori-infected p27-deficient mice showed that H. pylori eradication through an antibiotic combinational therapy could reduce gastric inflammation and hinder precancerous lesions such as gastric ulcer and dysplasia, thus preventing GC (176). Interestingly, H. pylori induced cytoplasmic localization of p27, resulting in loss of tumor suppressor function of p27 and correspondingly poor prognosis of patients (177). CDK4/6 inhibitors play the same role as p27, so patients with p27 cytoplasmic localization after H. pylori infection may respond better to CDK4/6 inhibitors. In addition, H. pylori activates many cell cycle-related genes, such as E2F1 and cyclin D1 (178), suggesting that current CDK4/6 inhibitors and potential RB-E2F targeting agents may be more effective in H. pylori-infected patients. H. pylori infection can also change the epigenetics of cells, such as increased p16 methylation (179). In this regard, H. pylori-infected GC patients may be more sensitive to abemaciclib (170).
Intervention of RB-E2F pathway has not been commonly used in GC as virtually all such options remain in preclinical stages or in clinical trials. Nevertheless, targeted therapies based on key components of the RB-E2F pathway, combined with immunotherapy, oncolytic viral therapy, and/or H. pylori eradication are likely a viable option for developing more effective treatment strategies for GC.
Conclusions and Perspective
In summary, RB-E2F pathway plays important roles in the occurrence and development of GC. Current understanding on the role and mechanism of the three pocket proteins in GC are insufficient, especially for p107. The functional consequences of epigenetic regulation of RB and cytoplasmic localization of p130, as well as the cooperative functions of these three pocket proteins in GC have great potentials to be explored. Most studies involving E2Fs in GC have focused on E2F1 and E2F4. We need a better understanding of the roles of other E2F family members in GC. In addition, whether E2F1 can be used as a viable therapeutic target remain to be determined. Since current data on the dual role of E2F1 in GC came from GC cell lines or xenograft mouse models, using transgenic mouse models will likely provide more significant insights on how E2F1 is involved in the various process of GC, including initiation, progression, and drug resistance. In view of the importance of H. pylori in GC and its complex interaction with the RB-E2F pathway, H. pylori eradication therapy combined with targeted therapy may achieve more effective therapeutic outcomes. It is worth noting that the molecular typing of GC is an important advance as we are now able to tailor our studies to different genetic alterations for each molecular subtype, thereby facilitating precision medicine. In the clinical application of RB-E2F pathway for GC, essentially all targeted therapy options remain in preclinical stages or in clinical trials. Therefore, there is an urgent need to facilitate our research efforts on translating research data into clinical practice. Nevertheless, targeting key components of the RB-E2F pathway for the development of more effective therapies of GC offers both significant promises and challenges to the scientific community.
Author Contributions
Both authors (TW and LW) prepared, revised and edited this manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the start-up funds from the China Medical University.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to acknowledge all members in Dr. Wu’s laboratory for their critical comments on this manuscript.
References
1. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global Cancer Statistics 2018: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J Clin (2018) 68:394–424. doi: 10.3322/caac.21492
2. Tan P, Yeoh KG. Genetics and Molecular Pathogenesis of Gastric Adenocarcinoma. Gastroenterology (2015) 149:1153–62.e3. doi: 10.1053/j.gastro.2015.05.059
3. Tsugane S, Sasazuki S. Diet and the Risk of Gastric Cancer: Review of Epidemiological Evidence. Gastric Cancer (2007) 10(2):75–83. doi: 10.1007/s10120-007-0420-0
4. Noto JM, Gaddy JA, Lee JY, Piazuelo MB, Friedman DB, Colvin DC, et al. Iron Deficiency Accelerates Helicobacter Pylori-Induced Carcinogenesis in Rodents and Humans. J Clin Invest (2013) 123(1):479–92. doi: 10.1172/JCI64373
5. Lauren P. The Two Histological Main Types of Gastric Carcinoma: Diffuse and So-Called Intestinal-Type Carcinoma. An Attempt at a Histo-Clinical Classification. Acta Pathol Microbiol Scand (1965) 64:31–49. doi: 10.1111/apm.1965.64.1.31
6. Lee WH, Bookstein R, Hong F, Young LJ, Shew JY, Lee EY. Human Retinoblastoma Susceptibility Gene: Cloning, Identification, and Sequence. Science (1987) 235:1394–9. doi: 10.1126/science.3823889
7. Kovesdi I, Reichel R, Nevins JR. Identification of a Cellular Transcription Factor Involved in E1A Trans-Activation. Cell (1986) 45:219–28. doi: 10.1016/0092-8674(86)90386-7
8. Yee AS, Reichel R, Kovesdi I, Nevins JR. Promoter Interaction of the E1A-inducible Factor E2F and its Potential Role in the Formation of a Multi-Component Complex. EMBO J (1987) 6:2061–8. doi: 10.1002/j.1460-2075.1987.tb02471.x
9. Kent LN, Leone G. The Broken Cycle: E2F Dysfunction In Cancer. Nat Rev Cancer (2019) 19:326–38. doi: 10.1038/s41568-019-0143-7
10. Fischer M, Müller GA. Cell Cycle Transcription Control: DREAM/MuvB and RB-E2F Complexes. Crit Rev Biochem Mol Biol (2017) 52:638–62. doi: 10.1080/10409238.2017.1360836
11. Hanahan D, Weinberg RA. Hallmarks of Cancer: The Next Generation. Cell (2011) 144(5):646–74. doi: 10.1016/j.cell.2011.02.013
12. Cito L, Pentimalli F, Forte I, Mattioli E, Giordano A. Rb Family Proteins in Gastric Cancer (Review). Oncol Rep (2010) 24:1411–8. doi: 10.3892/or_00001000
13. Xanthoulis A, Tiniakos DG. E2F Transcription Factors and Digestive System Malignancies: How Much do We Know. World J Gastroenterol (2013) 19:3189–98. doi: 10.3748/wjg.v19.i21.3189
14. Muñoz N, Correa P, Cuello C, Duque E. Histologic Types of Gastric Carcinoma in High- and Low-Risk Areas. Int J Cancer (1968) 3:809–18. doi: 10.1002/ijc.2910030614
15. Correa P, Sasano N, Stemmermann GN, Haenszel W. Pathology of Gastric Carcinoma in Japanese Populations: Comparisons Between Miyagi Prefecture, Japan, and Hawaii. J Natl Cancer Inst (1973) 51:1449–59. doi: 10.1093/jnci/51.5.1449
16. Liu GY, Liu KH, Zhang Y, Wang YZ, Wu XH, Lu YZ, et al. Alterations of Tumor-Related Genes do Not Exactly Match the Histopathological Grade in Gastric Adenocarcinomas. World J Gastroenterol (2010) 16:1129–37. doi: 10.3748/wjg.v16.i9.1129
17. Correa P. Human Gastric Carcinogenesis: A Multistep and Multifactorial Process–First American Cancer Society Award Lecture on Cancer Epidemiology and Prevention. Cancer Res (1992) 52:6735–40.
18. Yuasa Y. Control of Gut Differentiation and Intestinal-Type Gastric Carcinogenesis. Nat Rev Cancer (2003) 3:592–600. doi: 10.1038/nrc1141
19. de Vries AC, van Grieken NC, Looman CW, Casparie MK, de Vries E, Meijer GA, et al. Gastric Cancer Risk in Patients With Premalignant Gastric Lesions: A Nationwide Cohort Study in the Netherlands. Gastroenterology (2008) 134:945–52. doi: 10.1053/j.gastro.2008.01.071
20. Huang KK, Ramnarayanan K, Zhu F, Srivastava S, Xu C, Tan A, et al. Genomic and Epigenomic Profiling of High-Risk Intestinal Metaplasia Reveals Molecular Determinants of Progression to Gastric Cancer. Cancer Cell (2018) 33:137–50.e5. doi: 10.1016/j.ccell.2017.11.018
21. Becker KF, Atkinson MJ, Reich U, Becker I, Nekarda H, Siewert JR, et al. E-Cadherin Gene Mutations Provide Clues to Diffuse Type Gastric Carcinomas. Cancer Res (1994) 54:3845–52.
22. Kalamohan K, Periasamy J, Bhaskar Rao D, Barnabas GD, Ponnaiyan S, Ganesan K. Transcriptional Coexpression Network Reveals the Involvement of Varying Stem Cell Features With Different Dysregulations in Different Gastric Cancer Subtypes. Mol Oncol (2014) 8:1306–25. doi: 10.1016/j.molonc.2014.04.005
23. Cancer Genome Atlas Research Network. Comprehensive Molecular Characterization of Gastric Adenocarcinoma. Nature (2014) 513(7517):202–9. doi: 10.1038/nature13480
24. Fukayama M, Hino R, Uozaki H. Epstein-Barr Virus and Gastric Carcinoma: Virus-Host Interactions Leading to Carcinoma. Cancer Sci (2008) 99(9):1726–33. doi: 10.1111/j.1349-7006.2008.00888.x
25. Burke AP, Yen TS, Shekitka KM, Sobin LH. Lymphoepithelial Carcinoma of the Stomach With Epstein-Barr Virus Demonstrated by Polymerase Chain Reaction. Mod Pathol (1990) 3:377–80.
26. Ohtani N, Brennan P, Gaubatz S, Sanij E, Hertzog P, Wolvetang E, et al. Epstein-Barr Virus LMP1 Blocks P16ink4a-RB Pathway by Promoting Nuclear Export of E2F4/5. J Cell Biol (2003) 162:173–83. doi: 10.1083/jcb.200302085
27. Chung YJ, Kim KM, Choi JR, Choi SW, Rhyu MG. Relationship Between Intratumor Histological Heterogeneity and Genetic Abnormalities in Gastric Carcinoma With Microsatellite Instability. Int J Cancer (1999) 82:782–8. doi: 10.1002/(SICI)1097-0215(19990909)82:6<782::AID-IJC2>3.0.CO;2-#
28. Leung SY, Yuen ST, Chung LP, Chu KM, Wong MP, Branicki FJ, et al. Microsatellite Instability, Epstein-Barr Virus, Mutation of Type II Transforming Growth Factor Beta Receptor and BAX in Gastric Carcinomas in Hong Kong Chinese. Br J Cancer (1999) 79:582–8. doi: 10.1038/sj.bjc.6690092
29. Pizzi S, Azzoni C, Bassi D, Bottarelli L, Milione M, Bordi C. Genetic Alterations in Poorly Differentiated Endocrine Carcinomas of the Gastrointestinal Tract. Cancer (2003) 98:1273–82. doi: 10.1002/cncr.11621
30. Lei Z, Tan IB, Das K, Deng N, Zouridis H, Pattison S, et al. Identification of Molecular Subtypes of Gastric Cancer With Different Responses to PI3-kinase Inhibitors and 5-Fluorouracil. Gastroenterology (2013) 145:554–65. doi: 10.1053/j.gastro.2013.05.010
31. Suzuki T, Yasui W, Yokozaki H, Naka K, Ishikawa T, Tahara E. Expression of the E2F Family in Human Gastrointestinal Carcinomas. Int J Cancer (1999) 81:535–8. doi: 10.1002/(SICI)1097-0215(19990517)81:4<535::AID-IJC5>3.0.CO;2-4
32. Lee J, Park CK, Park JO, Lim T, Park YS, Lim HY, et al. Impact of E2F-1 Expression on Clinical Outcome of Gastric Adenocarcinoma Patients With Adjuvant Chemoradiation Therapy. Clin Cancer Res (2008) 14:82–8. doi: 10.1158/1078-0432.CCR-07-0612
33. Souza RF, Yin J, Smolinski KN, Zou TT, Wang S, Shi YQ, et al. Frequent Mutation of the E2F-4 Cell Cycle Gene in Primary Human Gastrointestinal Tumors. Cancer Res (1997) 57:2350–3.
34. Ogata S, Tamura G, Endoh Y, Sakata K, Ohmura K, Motoyama T. Microsatellite Alterations and Target Gene Mutations in the Early Stages of Multiple Gastric Cancer. J Pathol (2001) 194:334–40. doi: 10.1002/path.895
35. Korourian A, Roudi R, Shariftabrizi A, Kalantari E, Sotoodeh K, Madjd Z. Differential Role of Wnt Signaling and Base Excision Repair Pathways in Gastric Adenocarcinoma Aggressiveness. Clin Exp Med (2017) 17(4):505–17. doi: 10.1007/s10238-016-0443-0
36. Feakins RM, Nickols CD, Bidd H, Walton SJ. Abnormal Expression of pRb, p16, and Cyclin D1 in Gastric Adenocarcinoma and its Lymph Node Metastases: Relationship With Pathological Features and Survival. Hum Pathol (2003) 34(12):1276–82. doi: 10.1016/j.humpath.2003.07.005
37. Kishimoto I, Mitomi H, Ohkura Y, Kanazawa H, Fukui N, Watanabe M. Abnormal Expression of p16(INK4a), Cyclin D1, Cyclin-Dependent Kinase 4 and Retinoblastoma Protein in Gastric Carcinomas. J Surg Oncol (2008) 98:60–6. doi: 10.1002/jso.21087
38. Arici DS, Tuncer E, Ozer H, Simek G, Koyuncu A. Expression of Retinoblastoma and Cyclin D1 in Gastric Carcinoma. Neoplasma (2009) 56:63–7. doi: 10.4149/neo_2009_01_63
39. Songun I, van de Velde CJ, Hermans J, Pals ST, Verspaget HW, Vis AN, et al. Expression of Oncoproteins and the Amount of Eosinophilic and Lymphocytic Infiltrates can be Used as Prognostic Factors in Gastric Cancer. Dutch Gastric Cancer Group (DGCG). Br J Cancer (1996) 74:1783–8. doi: 10.1038/bjc.1996.630
40. Xue L, Ouyang Q, Li J, Meng X, Li Y, Xing L, et al. Different Roles for p16(INK) (4a) -Rb Pathway and INK4a/ARF Methylation Between Adenocarcinomas of Gastric Cardia and Distal Stomach. J Gastroenterol Hepatol (2014) 29:1418–26. doi: 10.1111/jgh.12547
41. Zhou Q, Zou JX, Chen YL, Yu HZ, Wang LD, Li YX, et al. Altered Expression of Tumor Suppressors p16 and Rb in Gastric Carcinogenesis. World J Gastroenterol (1997) 3:262. doi: 10.3748/wjg.v3.i4.262
42. Lan J, Xiong YY, Lin YX, Wang BC, Gong LL, Xu HS, et al. Helicobacter Pylori Infection Generated Gastric Cancer Through P53-Rb Tumor-Suppressor System Mutation and Telomerase Reactivation. World J Gastroenterol (2003) 9(1):54–8. doi: 10.3748/wjg.v9.i1.54
43. Guo L, Huang C, Ji QJ. Aberrant Promoter Hypermethylation of p16, Survivin, and Retinoblastoma in Gastric Cancer. Bratisl Lek Listy (2017) 118:164–8. doi: 10.4149/BLL_2017_033
44. Cito L, Indovina P, Forte IM, Pentimalli F, Di Marzo D, Somma P, et al. pRb2/p130 Localizes to the Cytoplasm in Diffuse Gastric Cancer. J Cell Physiol (2015) 230:802–5. doi: 10.1002/jcp.24805
45. Gao P, Zhou GY, Liu Y, Li JS, Zhen JH, Yuan YP. Alteration of Cyclin D1 in Gastric Carcinoma and Its Clinicopathologic Significance. World J Gastroenterol (2004) 10:2936–9. doi: 10.3748/wjg.v10.i20.2936
46. Arber N, Gammon MD, Hibshoosh H, Britton JA, Zhang Y, Schonberg JB, et al. Overexpression of Cyclin D1 Occurs in Both Squamous Carcinomas and Adenocarcinomas of the Esophagus and in Adenocarcinomas of the Stomach. Hum Pathol (1999) 30:1087–92. doi: 10.1016/S0046-8177(99)90227-7
47. Song SH, Jong HS, Choi HH, Kang SH, Ryu MH, Kim NK, et al. Methylation of Specific CpG Sites in the Promoter Region Could Significantly Down-Regulate p16(INK4a) Expression in Gastric Adenocarcinoma. Int J Cancer (2000) 87:236–40. doi: 10.1002/1097-0215(20000715)87:2<236::AID-IJC14>3.0.CO;2-M
48. He XS, Rong YH, Su Q, Luo Q, He DM, Li YL, et al. Expression of p16 Gene and Rb Protein in Gastric Carcinoma and Their Clinicopathological Significance. World J Gastroenterol (2005) 11:2218–23. doi: 10.3748/wjg.v11.i15.2218
49. Mattioli E, Vogiatzi P, Sun A, Abbadessa G, Angeloni G, D’Ugo D, et al. Immunohistochemical Analysis of Prb2/P130, VEGF, Ezh2, p53, p16(INK4A), p27(KIP1), P21(WAF1), Ki-67 Expression Patterns in Gastric Cancer. J Cell Physiol (2007) 210:183–91. doi: 10.1002/jcp.20833
50. Tsujimoto H, Hagiwara A, Sugihara H, Hattori T, Yamagishi H. Promoter Methylations of p16INK4a and p14ARF Genes in Early and Advanced Gastric Cancer. Correlations of the Modes of Their Occurrence With Histologic Type. Pathol Res Pract (2002) 198(12):785–94. doi: 10.1078/0344-0338-00337
51. Hanazono K, Natsugoe S, Stein HJ, Aikou T, Hoefler H, Siewert JR. Distribution of p53 Mutations in Esophageal and Gastric Carcinomas and the Relationship With p53 Expression. Oncol Rep (2006) 15:821–4. doi: 10.3892/or.15.4.821
52. Henley SA, Dick FA. The Retinoblastoma Family of Proteins and Their Regulatory Functions in the Mammalian Cell Division Cycle. Cell Div (2012) 7:10. doi: 10.1186/1747-1028-7-10
53. Indovina P, Marcelli E, Casini N, Rizzo V, Giordano A. Emerging Roles of RB Family: New Defense Mechanisms Against Tumor Progression. J Cell Physiol (2013) 228:525–35. doi: 10.1002/jcp.24170
54. Morris EJ, Dyson NJ. Retinoblastoma Protein Partners. Adv Cancer Res (2001) 82:1–54. doi: 10.1016/S0065-230X(01)82001-7
55. Friend SH, Bernards R, Rogelj S, Weinberg RA, Rapaport JM, Albert DM, et al. A Human DNA Segment With Properties of the Gene That Predisposes to Retinoblastoma and Osteosarcoma. Nature (1986) 323(6089):643–6. doi: 10.1038/323643a0
56. Macaluso M, Montanari M, Giordano A. Rb Family Proteins as Modulators of Gene Expression and New Aspects Regarding the Interaction With Chromatin Remodeling Enzymes. Oncogene (2006) 25:5263–7. doi: 10.1038/sj.onc.1209680
57. Haas-Kogan DA, Kogan SC, Levi D, Dazin P, T’Ang A, Fung YK, et al. Inhibition of Apoptosis by the Retinoblastoma Gene Product. EMBO J (1995) 14(3):461–72. doi: 10.1002/j.1460-2075.1995.tb07022.x
58. Bookstein R, Rio P, Madreperla SA, Hong F, Allred C, Grizzle WE, et al. Promoter Deletion and Loss of Retinoblastoma Gene Expression in Human Prostate Carcinoma. Proc Natl Acad Sci USA (1990) 87(19):7762–6. doi: 10.1073/pnas.87.19.7762
59. Hui AM, Li X, Makuuchi M, Takayama T, Kubota K. Over-Expression and Lack of Retinoblastoma Protein are Associated With Tumor Progression and Metastasis in Hepatocellular Carcinoma. Int J Cancer (1999) 84(6):604–8. doi: 10.1002/(SICI)1097-0215(19991222)84:6<604::AID-IJC11>3.0.CO;2-Y
60. zur Hausen A, Sarbia M, Heep H, Willers R, Gabbert HE. Retinoblastoma-Protein (prb) Expression and Prognosis in Squamous-Cell Carcinomas of the Esophagus. Int J Cancer (1999) 84(6):618–22. doi: 10.1002/(SICI)1097-0215(19991222)84:6<618::AID-IJC14>3.0.CO;2-I
61. Varley JM, Armour J, Swallow JE, Jeffreys AJ, Ponder BA, T’Ang A, et al. The Retinoblastoma Gene is Frequently Altered Leading to Loss of Expression in Primary Breast Tumours. Oncogene (1989) 4(6):725–9.
62. Reissmann PT, Simon MA, Lee WH, Slamon DJ. Studies of the Retinoblastoma Gene in Human Sarcomas. Oncogene (1989) 4(7):839–43.
63. Furukawa Y, DeCaprio JA, Belvin M, Griffin JD. Heterogeneous Expression of the Product of the Retinoblastoma Susceptibility Gene in Primary Human Leukemia Cells. Oncogene (1991) 6(8):1343–6.
64. Cairns P, Proctor AJ, Knowles MA. Loss of Heterozygosity at the RB Locus is Frequent and Correlates With Muscle Invasion in Bladder Carcinoma. Oncogene (1991) 6(12):2305–9.
65. Mayol X, Graña X, Baldi A, Sang N, Hu Q, Giordano A. Cloning of a New Member of the Retinoblastoma Gene Family (pRb2) Which Binds to the E1A Transforming Domain. Oncogene (1993) 8(9):2561–6.
66. Yeung RS, Bell DW, Testa JR, Mayol X, Baldi A, Graña X, et al. The Retinoblastoma-Related Gene, RB2, Maps to Human Chromosome 16q12 and Rat Chromosome 19. Oncogene (1993) 8(12):3465–8.
67. Ewen ME, Xing YG, Lawrence JB, Livingston DM. Molecular Cloning, Chromosomal Mapping, and Expression of the cDNA for p107, A Retinoblastoma Gene Product-Related Protein. Cell (1991) 66(6):1155–64. doi: 10.1016/0092-8674(91)90038-Z
68. Classon M, Dyson N. p107 and p130: Versatile Proteins With Interesting Pockets. Exp Cell Res (2001) 264(1):135–47. doi: 10.1006/excr.2000.5135
69. Schade AE, Fischer M, DeCaprio JARB. p130 and p107 Differentially Repress G1/S and G2/M Genes After p53 Activation. Nucleic Acids Res (2019) 47:11197–208. doi: 10.1093/nar/gkz961
70. Schade AE, Oser MG, Nicholson HE, DeCaprio JA. Cyclin D-CDK4 Relieves Cooperative Repression of Proliferation and Cell Cycle Gene Expression by DREAM and RB. Oncogene (2019) 38:4962–76. doi: 10.1038/s41388-019-0767-9
71. Robanus-Maandag E, Dekker M, van der Valk M, Carrozza ML, Jeanny JC, Dannenberg JH, et al. p107 is a Suppressor of Retinoblastoma Development in pRb-deficient Mice. Genes Dev (1998) 12:1599–609. doi: 10.1101/gad.12.11.1599
72. MacPherson D, Conkrite K, Tam M, Mukai S, Mu D, Jacks T. Murine Bilateral Retinoblastoma Exhibiting Rapid-Onset, Metastatic Progression and N-myc Gene Amplification. EMBO J (2007) 26:784–94. doi: 10.1038/sj.emboj.7601515
73. Shin MK, Pitot HC, Lambert PF. Pocket Proteins Suppress Head and Neck Cancer. Cancer Res (2012) 72:1280–9. doi: 10.1158/0008-5472.CAN-11-2833
74. Santos M, Ruiz S, Lara MF, Segrelles C, Moral M, Martínez-Cruz AB, et al. Susceptibility of pRb-Deficient Epidermis to Chemical Skin Carcinogenesis is Dependent on the p107 Allele Dosage. Mol Carcinog (2008) 47:815–21. doi: 10.1002/mc.20426
75. MacPherson D, Sage J, Kim T, Ho D, McLaughlin ME, Jacks T. Cell Type-Specific Effects of Rb Deletion in the Murine Retina. Genes Dev (2004) 18:1681–94. doi: 10.1101/gad.1203304
76. Dannenberg JH, Schuijff L, Dekker M, van der Valk M, te Riele H. Tissue-Specific Tumor Suppressor Activity of Retinoblastoma Gene Homologs p107 and P130. Genes Dev (2004) 18:2952–62. doi: 10.1101/gad.322004
77. Leone G, Nuckolls F, Ishida S, Adams M, Sears R, Jakoi L, et al. Identification of a Novel E2F3 Product Suggests a Mechanism for Determining Specificity of Repression by Rb Proteins. Mol Cell Biol (2000) 20:3626–32. doi: 10.1128/MCB.20.10.3626-3632.2000
78. DeGregori J, Johnson DG. Distinct and Overlapping Roles for E2F Family Members in Transcription, Proliferation and Apoptosis. Curr Mol Med (2006) 6:739–48. doi: 10.2174/1566524010606070739
79. Attwooll C, Lazzerini DE, Helin K. The E2F Family: Specific Functions and Overlapping Interests. EMBO J (2004) 23(24):4709–16. doi: 10.1038/sj.emboj.7600481
80. Girling R, Partridge JF, Bandara LR, Burden N, Totty NF, Hsuan JJ, et al. A New Component of the Transcription Factor DRTF1/E2F. Nature (1993) 365:468. doi: 10.1038/365468d0
81. Danielian PS, Friesenhahn LB, Faust AM, West JC, Caron AM, Bronson RT, et al. E2f3a and E2f3b Make Overlapping But Different Contributions to Total E2f3 Activity. Oncogene (2008) 27:6561–70. doi: 10.1038/onc.2008.253
82. Chong JL, Tsai SY, Sharma N, Opavsky R, Price R, Wu L, et al. E2f3a and E2f3b Contribute to the Control of Cell Proliferation and Mouse Development. Mol Cell Biol (2009) 29:414–24. doi: 10.1128/MCB.01161-08
83. Liban TJ, Thwaites MJ, Dick FA, Rubin SM. Structural Conservation and E2F Binding Specificity Within the Retinoblastoma Pocket Protein Family. J Mol Biol (2016) 428:3960–71. doi: 10.1016/j.jmb.2016.08.017
84. Takahashi Y, Rayman JB, Dynlacht BD. Analysis of Promoter Binding by the E2F and pRB Families In Vivo: Distinct E2F Proteins Mediate Activation and Repression. Genes Dev (2000) 14:804–16.
85. Chou NH, Chen HC, Chou NS, Hsu PI, Tseng HH. Expression of Altered Retinoblastoma Protein Inversely Correlates With Tumor Invasion in Gastric Carcinoma. World J Gastroenterol (2006) 12:7188–91. doi: 10.3748/wjg.v12.i44.7188
86. Shats I, Deng M, Davidovich A, Zhang C, Kwon JS, Manandhar D, et al. Expression Level Is a Key Determinant of E2F1-Mediated Cell Fate. Cell Death Differ (2017) 24:626–37. doi: 10.1038/cdd.2017.12
87. Pierce AM, Schneider-Broussard R, Gimenez-Conti IB, Russell JL, Conti CJ, Johnson DG. E2F1 Has Both Oncogenic and Tumor-Suppressive Properties in a Transgenic Model. Mol Cell Biol (1999) 19:6408–14. doi: 10.1128/MCB.19.9.6408
88. Johnson DG, Cress WD, Jakoi L, Nevins JR. Oncogenic Capacity of the E2F1 Gene. Proc Natl Acad Sci USA (1994) 91:12823–7. doi: 10.1073/pnas.91.26.12823
89. Yamasaki L, Jacks T, Bronson R, Goillot E, Harlow E, Dyson NJ. Tumor Induction and Tissue Atrophy in Mice Lacking E2F-1. Cell (1996) 85:537–48. doi: 10.1016/S0092-8674(00)81254-4
90. DeGregori J, Leone G, Miron A, Jakoi L, Nevins JR. Distinct Roles for E2F Proteins in Cell Growth Control and Apoptosis. Proc Natl Acad Sci USA (1997) 94:7245–50. doi: 10.1073/pnas.94.14.7245
91. Field SJ, Tsai FY, Kuo F, Zubiaga AM, Kaelin WG Jr, Livingston DM, et al. E2F-1 Functions in Mice to Promote Apoptosis and Suppress Proliferation. Cell (1996) 85:549–61. doi: 10.1016/S0092-8674(00)81255-6
92. Zhang X, Ni Z, Duan Z, Xin Z, Wang H, Tan J, et al. Overexpression of E2F mRNAs Associated With Gastric Cancer Progression Identified by the Transcription Factor and miRNA Co-Regulatory Network Analysis. PloS One (2015) 10:e0116979. doi: 10.1371/journal.pone.0116979
93. Liu X, Hu C. Novel Potential Therapeutic Target for E2F1 and Prognostic Factors of E2F1/2/3/5/7/8 in Human Gastric Cancer. Mol Ther Methods Clin Dev (2020) 18:824–38. doi: 10.1016/j.omtm.2020.07.017
94. Xie Y, Wang C, Li L, Ma Y, Yin Y, Xiao Q. Overexpression of E2F-1 Inhibits Progression of Gastric Cancer In Vitro. Cell Biol Int (2009) 33(6):640–9. doi: 10.1016/j.cellbi.2009.02.015
95. Wei WY, Yan LH, Wang XT, Li L, Cao WL, Zhang XS, et al. E2F-1 Overexpression Inhibits Human Gastric Cancer MGC-803 Cell Growth In Vivo. World J Gastroenterol (2015) 21(2):491–501. doi: 10.3748/wjg.v21.i2.491
96. Atienza C, Elliott MJ, Dong YB, Yang HL, Stilwell A, Liu TJ, et al. Adenovirus-Mediated E2F-1 Gene Transfer Induces an Apoptotic Response in Human Gastric Carcinoma Cells That Is Enhanced by Cyclin Dependent Kinase Inhibitors. Int J Mol Med (2000) 6(1):55–63. doi: 10.3892/ijmm.6.1.55
97. Wang XT, Xie YB, Xiao Q. Lentivirus-Mediated RNA Interference Targeting E2F-1 Inhibits Human Gastric Cancer MGC-803 Cell Growth In Vivo. Exp Mol Med (2011) 43(11):638–45. doi: 10.3858/emm.2011.43.11.072
98. Yan LH, Wang XT, Yang J, Kong FB, Lian C, Wei WY, et al. Reversal of Multidrug Resistance in Gastric Cancer Cells by E2F-1 Downregulation In Vitro and In Vivo. J Cell Biochem (2014) 115(1):34–41. doi: 10.1002/jcb.24652
99. DeGregori J, Kowalik T, Nevins JR. Cellular Targets for Activation by the E2F1 Transcription Factor Include DNA Synthesis- and G1/S-Regulatory Genes. Mol Cell Biol (1995) 15:4215–24. doi: 10.1128/mcb.15.8.4215
100. Kowalik TF, DeGregori J, Schwarz JK, Nevins JR. E2F1 Overexpression in Quiescent Fibroblasts Leads to Induction of Cellular DNA Synthesis and Apoptosis. J Virol (1995) 69:2491–500. doi: 10.1128/JVI.69.4.2491-2500.1995
101. Whitfield ML, George LK, Grant GD, Perou CM. Common Markers of Proliferation. Nat Rev Cancer (2006) 6:99–106. doi: 10.1038/nrc1802
102. Holmberg C, Helin K, Sehested M, Karlström O. E2f-1-Induced p53-Independent Apoptosis in Transgenic Mice. Oncogene (1998) 17:143–55. doi: 10.1038/sj.onc.1201915
103. Lindström MS, Wiman KG. Myc and E2F1 Induce p53 Through p14ARF-Independent Mechanisms in Human Fibroblasts. Oncogene (2003) 22:4993–5005. doi: 10.1038/sj.onc.1206659
104. Elliott MJ, Dong YB, Yang H, McMasters KM. E2F-1 Up-Regulates c-Myc and p14(ARF) and Induces Apoptosis in Colon Cancer Cells. Clin Cancer Res (2001) 7:3590–7.
105. Bates S, Phillips AC, Clark PA, Stott F, Peters G, Ludwig RL, et al. p14ARF Links the Tumour Suppressors RB and P53. Nature (1998) 395:124–5. doi: 10.1038/25867
106. Sherr CJ, McCormick F. The RB and p53 Pathways in Cancer. Cancer Cell (2002) 2:103–12. doi: 10.1016/S1535-6108(02)00102-2
107. Wu Q, Li G, Xu F. Resected Gastric Cancer With D2 Dissection: Advances in Adjuvant Chemoradiotherapy and Radiotherapy Techniques. Expert Rev Anticancer Ther (2015) 15:703–13. doi: 10.1586/14737140.2015.1042863
108. Qiao L, Wong BC. Targeting Apoptosis as an Approach for Gastrointestinal Cancer Therapy. Drug Resist Updat (2009) 12:55–64. doi: 10.1016/j.drup.2009.02.002
109. Engelmann D, Pützer BM. Translating DNA Damage Into Cancer Cell Death-a Roadmap for E2F1 Apoptotic Signalling and Opportunities for New Drug Combinations to Overcome Chemoresistance. Drug Resist Updat (2010) 13:119–31. doi: 10.1016/j.drup.2010.06.001
110. Schwemmle S, Pfeifer GP. Genomic Structure and Mutation Screening of the E2F4 Gene in Human Tumors. Int J Cancer (2000) 86(5):672–7. doi: 10.1002/(SICI)1097-0215(20000601)86:5<672::AID-IJC11>3.0.CO;2-X
111. Woo DK, Lee WA, Kim YI, Kim WH. Microsatellite Instability and Alteration of E2F-4 Gene in Adenosquamous and Squamous Cell Carcinomas of the Stomach. Pathol Int (2000) 50(9):690–5. doi: 10.1046/j.1440-1827.2000.01105.x
112. Fischer M, Quaas M, Steiner L, Engeland K. The P53-P21-DREAM-CDE/CHR Pathway Regulates G2/M Cell Cycle Genes. Nucleic Acids Res (2016) 44:164–74. doi: 10.1093/nar/gkv927
113. Wang D, Russell JL, Johnson DG. E2F4 and E2F1 Have Similar Proliferative Properties But Different Apoptotic and Oncogenic Properties. Vivo Mol Cell Biol (2000) 20:3417–24. doi: 10.1128/mcb.20.10.3417-3424.2000
114. Dingar D, Konecny F, Zou J, Sun X, von Harsdorf R. Anti-Apoptotic Function of the E2F Transcription Factor 4 (E2F4)/p130, A Member of Retinoblastoma Gene Family in Cardiac Myocytes. J Mol Cell Cardiol (2012) 53:820–8. doi: 10.1016/j.yjmcc.2012.09.004
115. Poppy Roworth A, Ghari F, La Thangue NB. To Live or Let Die - Complexity Within the E2F1 Pathway. Mol Cell Oncol (2015) 2:e970480. doi: 10.4161/23723548.2014.970480
116. Arellano M, Moreno S. Regulation of CDK/cyclin Complexes During the Cell Cycle. Int J Biochem Cell Biol (1997) 29:559–73. doi: 10.1016/S1357-2725(96)00178-1
117. Hydbring P, Malumbres M, Sicinski P. Non-Canonical Functions of Cell Cycle Cyclins and Cyclin-Dependent Kinases. Nat Rev Mol Cell Biol (2016) 17:280–92. doi: 10.1038/nrm.2016.27
118. Serrano M, Hannon GJ, Beach D. A New Regulatory Motif in Cell-Cycle Control Causing Specific Inhibition of Cyclin D/CDK4. Nature (1993) 366:704–7. doi: 10.1038/366704a0
119. Nobori T, Miura K, Wu DJ, Lois A, Takabayashi K, Carson DA. Deletions of the Cyclin-Dependent Kinase-4 Inhibitor Gene in Multiple Human Cancers. Nature (1994) 368:753–6. doi: 10.1038/368753a0
120. Kamb A, Gruis NA, Weaver-Feldhaus J, Liu Q, Harshman K, Tavtigian SV, et al. A Cell Cycle Regulator Potentially Involved in Genesis of Many Tumor Types. Science (1994) 264:436–40. doi: 10.1126/science.8153634
121. Kim H, Kim YH, Kim SE, Kim NG, Noh SH, Kim H. Concerted Promoter Hypermethylation of hMLH1, p16INK4A, and E-Cadherin in Gastric Carcinomas With Microsatellite Instability. J Pathol (2003) 200(1):23–31. doi: 10.1002/path.1325
122. Sakuma K, Chong JM, Sudo M, Ushiku T, Inoue Y, Shibahara J, et al. High-Density Methylation of p14ARF and p16INK4A in Epstein-Barr Virus-Associated Gastric Carcinoma. Int J Cancer (2004) 112:273–8. doi: 10.1002/ijc.20420
123. Ojima H, Saito K, Yamauchi H, Yamaki E, Idetu A, Hosouchi Y, et al. P16 Protein Abnormality in Epstein-Barr Virus-Associated Gastric Carcinomas. Anticancer Res (2006) 26:933–7.
124. Myung N, Kim MR, Chung IP, Kim H, Jang JJ. Loss of p16 and p27 is Associated With Progression of Human Gastric Cancer. Cancer Lett (2000) 153(1-2):129–36. doi: 10.1016/S0304-3835(00)00359-1
125. Lee WA, Woo DK, Kim YI, Kim WH. p53, p16 and RB Expression in Adenosquamous and Squamous Cell Carcinomas of the Stomach. Pathol Res Pract (1999) 195(11):747–52. doi: 10.1016/S0344-0338(99)80116-2
126. Yoo YD, Choi JY, Lee SJ, Kim JS, Min BR, Lee YI, et al. TGF-Beta-Induced Cell-Cycle Arrest Through the P21(WAF1/CIP1)-G1 Cyclin/Cdks-p130 Pathway in Gastric-Carcinoma Cells. Int J Cancer (1999) 83(4):512–7. doi: 10.1002/(SICI)1097-0215(19991112)83:4<512::AID-IJC13>3.0.CO;2-Z
127. Lv H, Liu R, Fu J, Yang Q, Shi J, Chen P, et al. Epithelial Cell-Derived Periostin Functions as a Tumor Suppressor in Gastric Cancer Through Stabilizing p53 and E-Cadherin Proteins Via the Rb/E2F1/p14ARF/Mdm2 Signaling Pathway. Cell Cycle (2014) 13(18):2962–74. doi: 10.4161/15384101.2014.947203
128. Lv BB, Ma RR, Chen X, Zhang GH, Song L, Wang SX, et al. E2F1-Activated SPIN1 Promotes Tumor Growth Via a MDM2-p21-E2F1 Feedback Loop in Gastric Cancer. Mol Oncol (2020) 10:2629–45. doi: 10.1002/1878-0261.12778
129. Zhou X, Ji H, Ye D, Li H, Liu F, Li H, et al. Knockdown of ATAD2 Inhibits Proliferation and Tumorigenicity Through the Rb-E2F1 Pathway and Serves as a Novel Prognostic Indicator in Gastric Cancer. Cancer Manag Res (2020) 12:337–51. doi: 10.2147/CMAR.S228629
130. Shiozaki A, Otsuji E, Marunaka Y. Intracellular Chloride Regulates the G(1)/S Cell Cycle Progression in Gastric Cancer Cells. World J Gastrointest Oncol (2011) 3(8):119–22. doi: 10.4251/wjgo.v3.i8.119
131. Linzer DI, Levine AJ. Characterization of a 54K Dalton Cellular SV40 Tumor Antigen Present in SV40-Transformed Cells and Uninfected Embryonal Carcinoma Cells. Cell (1979) 17(1):43–52. doi: 10.1016/0092-8674(79)90293-9
132. Levine AJ. p53, The Cellular Gatekeeper for Growth and Division. Cell (1997) 88:323–31. doi: 10.1016/S0092-8674(00)81871-1
133. Samuels-Lev Y, O’Connor DJ, Bergamaschi D, Trigiante G, Hsieh JK, Zhong S, et al. ASPP Proteins Specifically Stimulate the Apoptotic Function of P53. Mol Cell (2001) 8:781–94. doi: 10.1016/S1097-2765(01)00367-7
134. Patel S, George R, Autore F, Fraternali F, Ladbury JE, Nikolova PV. Molecular Interactions of ASPP1 and ASPP2 With the p53 Protein Family and the Apoptotic Promoters PUMA and Bax. Nucleic Acids Res (2008) 36:5139–51. doi: 10.1093/nar/gkn490
135. Engeland K. Cell Cycle Arrest Through Indirect Transcriptional Repression by P53: I Have a DREAM. Cell Death Differ (2018) 25:114–32. doi: 10.1038/cdd.2017.172
136. Anwar W, Armstrong BK, Correa P, Forman D, Gentile JM, Haswell-Elkins M, et al. Schistosomes, Liver Flukes and Helicobacter Pylori. In: Iarc Working Group on the Evaluation of Carcinogenic Risks to Humans, vol. 61. Lyon. p. 1–241. IARC Monogr Eval Carcinog Risks Hum.
137. Hooi J, Lai WY, Ng WK, Suen M, Underwood FE, Tanyingoh D, et al. Global Prevalence of Helicobacter Pylori Infection: Systematic Review and Meta-Analysis. Gastroenterology (2017) 153:420–9. doi: 10.1053/j.gastro.2017.04.022
138. Niwa T, Tsukamoto T, Toyoda T, Mori A, Tanaka H, Maekita T, et al. Inflammatory Processes Triggered by Helicobacter Pylori Infection Cause Aberrant DNA Methylation in Gastric Epithelial Cells. Cancer Res (2010) 70:1430–40. doi: 10.1158/0008-5472.CAN-09-2755
139. Kouraklis G, Katsoulis IE, Theocharis S, Tsourouflis G, Xipolitas N, Glinavou A, et al. Does the Expression of Cyclin E, pRb, and p21 Correlate With Prognosis in Gastric Adenocarcinoma. Dig Dis Sci (2009) 54(5):1015–20. doi: 10.1007/s10620-008-0464-y
140. Boyacioglu SO, Kasap E, Yuceyar H, Korkmaz M. Alteration in Methylation Pattern of Retinoblastoma 1 Gene Promotor Region in Intestinal Metaplasia With or Without Helicobacter Pylori and Gastric Cancer Patients. Adv Clin Exp Med (2016) 25(3):465–70. doi: 10.17219/acem/38842
141. Yamaoka Y. Mechanisms of Disease: Helicobacter Pylori Virulence Factors. Nat Rev Gastroenterol Hepatol (2010) 7:629–41. doi: 10.1038/nrgastro.2010.154
142. Gu H. Role of Flagella in the Pathogenesis of Helicobacter Pylori. Curr Microbiol (2017) 74:863–9. doi: 10.1007/s00284-017-1256-4
143. Moran AP. Lipopolysaccharide in Bacterial Chronic Infection: Insights From Helicobacter Pylori Lipopolysaccharide and Lipid A. Int J Med Microbiol (2007) 297:307–19. doi: 10.1016/j.ijmm.2007.03.008
144. Cho SJ, Kang NS, Park SY, Kim BO, Rhee DK, Pyo S. Induction of Apoptosis and Expression of Apoptosis Related Genes in Human Epithelial Carcinoma Cells by Helicobacter Pylori VacA Toxin. Toxicon (2003) 42(6):601–11. doi: 10.1016/j.toxicon.2003.08.003
145. Denic M, Touati E, De Reuse H. Review: Pathogenesis of Helicobacter Pylori Infection. Helicobacter (2020) 25 Suppl 1:e12736. doi: 10.1111/hel.12736
146. Buti L, Spooner E, Van der Veen AG, Rappuoli R, Covacci A, Ploegh HL. Helicobacter Pylori Cytotoxin-Associated Gene A (CagA) Subverts the Apoptosis-Stimulating Protein of p53 (ASPP2) Tumor Suppressor Pathway of the Host. Proc Natl Acad Sci USA (2011) 108:9238–43. doi: 10.1073/pnas.1106200108
147. Nešić D, Buti L, Lu X, Stebbins CE. Structure of the Helicobacter Pylori CagA Oncoprotein Bound to the Human Tumor Suppressor ASPP2. Proc Natl Acad Sci USA (2014) 111:1562–7. doi: 10.1073/pnas.1320631111
148. Wei J, Nagy TA, Vilgelm A, Zaika E, Ogden SR, Romero-Gallo J, et al. Regulation of p53 Tumor Suppressor by Helicobacter Pylori in Gastric Epithelial Cells. Gastroenterology (2010) 139:1333–43. doi: 10.1053/j.gastro.2010.06.018
149. Shu X, Yang Z, Li ZH, Chen L, Zhou XD, Xie Y, et al. Helicobacter Pylori Infection Activates the Akt-Mdm2-p53 Signaling Pathway in Gastric Epithelial Cells. Dig Dis Sci (2015) 60:876–86. doi: 10.1007/s10620-014-3470-2
150. Bhardwaj V, Noto JM, Wei J, Andl C, El-Rifai W, Peek RM, et al. Helicobacter Pylori Bacteria Alter the p53 Stress Response Via ERK-HDM2 Pathway. Oncotarget (2015) 6:1531–43. doi: 10.18632/oncotarget.2828
151. Michael D, Oren M. The P53-Mdm2 Module and the Ubiquitin System. Semin Cancer Biol (2003) 13:49–58. doi: 10.1016/S1044-579X(02)00099-8
152. Yang Z, Shu X, Chen L, Chen J, Xie Y, Lu NH. Expression of P53-MDM2 Feedback Loop Related Proteins in Different Gastric Pathologies in Relation to Helicobacter Pylori Infection: Implications in Gastric Carcinogenesis. Clin Res Hepatol Gastroenterol (2012) 36:235–43. doi: 10.1016/j.clinre.2011.11.009
153. Sdek P, Ying H, Zheng H, Margulis A, Tang X, Tian K, et al. The Central Acidic Domain of MDM2 Is Critical in Inhibition of Retinoblastoma-Mediated Suppression of E2F and Cell Growth. J Biol Chem (2004) 279:53317–22. doi: 10.1074/jbc.M406062200
154. Sdek P, Ying H, Chang DL, Qiu W, Zheng H, Touitou R, et al. MDM2 Promotes Proteasome-Dependent Ubiquitin-Independent Degradation of Retinoblastoma Protein. Mol Cell (2005) 20:699–708. doi: 10.1016/j.molcel.2005.10.017
155. Wei J, Noto J, Zaika E, Romero-Gallo J, Correa P, El-Rifai W, et al. Pathogenic Bacterium Helicobacter Pylori Alters the Expression Profile of p53 Protein Isoforms and p53 Response to Cellular Stresses. Proc Natl Acad Sci USA (2012) 109(38):E2543–50. doi: 10.1073/pnas.1205664109
156. Ak P, Levine AJ. p53 and NF-κb: Different Strategies for Responding to Stress Lead to a Functional Antagonism. FASEB J (2010) 24(10):3643–52. doi: 10.1096/fj.10-160549
157. Sommi P, Savio M, Stivala LA, Scotti C, Mignosi P, Prosperi E, et al. Helicobacter Pylori Releases a Factor(s) Inhibiting Cell Cycle Progression of Human Gastric Cell Lines by Affecting Cyclin E/cdk2 Kinase Activity and Rb Protein Phosphorylation Through Enhanced p27(KIP1) Protein Expression. Exp Cell Res (2002) 281(1):128–39. doi: 10.1006/excr.2002.5629
158. De Luca A, Baldi A, Russo P, Todisco A, Altucci L, Giardullo N, et al. Coexpression of Helicobacter Pylori’s Proteins CagA and HspB Induces Cell Proliferation in AGS Gastric Epithelial Cells, Independently From the Bacterial Infection. Cancer Res (2003) 63(19):6350–6.
159. Choi IJ, Kook MC, Kim YI, Cho SJ, Lee JY, Kim CG, et al. Helicobacter Pylori Therapy for the Prevention of Metachronous Gastric Cancer. N Engl J Med (2018) 378:1085–95. doi: 10.1056/NEJMoa1708423
160. Kumar S, Metz DC, Ellenberg S, Kaplan DE, Goldberg DS. Risk Factors and Incidence of Gastric Cancer After Detection of Helicobacter Pylori Infection: A Large Cohort Study. Gastroenterology (2020) 158:527–36.e7. doi: 10.1053/j.gastro.2019.10.019
161. Choi IJ, Kim CG, Lee JY, Kim YI, Kook MC, Park B, et al. Family History of Gastric Cancer and Helicobacter Pylori Treatment. N Engl J Med (2020) 382:427–36. doi: 10.1056/NEJMoa1909666
162. Isomoto H, Furusu H, Shin M, Ohnita K, Miyazaki M, Omagari K, et al. Enhanced Expression of Transcription Factor E2F in Helicobacter Pylori-Infected Gastric Mucosa. Helicobacter (2002) 7:152–62. doi: 10.1046/j.1523-5378.2002.00075.x
163. Smyth EC, Nilsson M, Grabsch HI, van Grieken NC, Lordick F. Gastric Cancer. Lancet (2020) 396:635–48. doi: 10.1016/S0140-6736(20)31288-5
164. Dickson MA. Molecular Pathways: CDK4 Inhibitors for Cancer Therapy. Clin Cancer Res (2014) 20:3379–83. doi: 10.1158/1078-0432.CCR-13-1551
165. Roskoski R. Cyclin-Dependent Protein Serine/Threonine Kinase Inhibitors as Anticancer Drugs. Pharmacol Res (2019) 139:471–88. doi: 10.1016/j.phrs.2018.11.035
166. Schwartz GK, Ilson D, Saltz L, O’Reilly E, Tong W, Maslak P, et al. Phase II Study of the Cyclin-Dependent Kinase Inhibitor Flavopiridol Administered to Patients With Advanced Gastric Carcinoma. J Clin Oncol (2001) 19:1985–92. doi: 10.1200/JCO.2001.19.7.1985
167. Schettini F, De Santo I, Rea CG, De Placido P, Formisano L, Giuliano M, et al. Cdk 4/6 Inhibitors as Single Agent in Advanced Solid Tumors. Front Oncol (2018) 8:608. doi: 10.3389/fonc.2018.00608
168. Karasic TB, O’Hara MH, Teitelbaum UR, Damjanov N, Giantonio BJ, d’Entremont TS, et al. Phase II Trial of Palbociclib in Patients With Advanced Esophageal or Gastric Cancer. Oncologist (2020) 25:e1864–1864e1868. doi: 10.1634/theoncologist.2020-0681
169. Min A, Kim JE, Kim YJ, Lim JM, Kim S, Kim JW, et al. Cyclin E Overexpression Confers Resistance to the CDK4/6 Specific Inhibitor Palbociclib in Gastric Cancer Cells. Cancer Lett (2018) 430:123–32. doi: 10.1016/j.canlet.2018.04.037
170. Bae HJ, Kang SK, Kwon WS, Jeong I, Park S, Kim TS, et al. p16 Methylation is a Potential Predictive Marker for Abemaciclib Sensitivity in Gastric Cancer. Biochem Pharmacol (2021) 183:114320. doi: 10.1016/j.bcp.2020.114320
171. Chen Z, Xu Y, Gong J, Kou F, Zhang M, Tian T, et al. Pyrotinib Combined With CDK4/6 Inhibitor in HER2-Positive Metastatic Gastric Cancer: A Promising Strategy From AVATAR Mouse to Patients. Clin Transl Med (2020) 10:e148. doi: 10.1002/ctm2.148
172. Knudsen ES, Pruitt SC, Hershberger PA, Witkiewicz AK, Goodrich DW. Cell Cycle and Beyond: Exploiting New Rb1 Controlled Mechanisms for Cancer Therapy. Trends Cancer (2019) 5:308–24. doi: 10.1016/j.trecan.2019.03.005
173. Ma J, Ma J, He X, He X, Wang W, Wang W, et al. E2F Promoter-Regulated Oncolytic Adenovirus With p16 Gene Induces Cell Apoptosis and Exerts Antitumor Effect on Gastric Cancer. Dig Dis Sci (2009) 54:1425–31. doi: 10.1007/s10620-008-0543-0
174. Bronisz A, Chiocca EA. A Cross-Talk Network That Facilitates Tumor Virotherapy. Nat Med (2015) 21:426–7. doi: 10.1038/nm.3857
175. Leung WK, Wong I, Cheung KS, Yeung KF, Chan EW, Wong A, et al. Effects of Helicobacter Pylori Treatment on Incidence of Gastric Cancer in Older Individuals. Gastroenterology (2018) 155:67–75. doi: 10.1053/j.gastro.2018.03.028
176. Zhang S, Lee DS, Morrissey R, Aponte-Pieras JR, Rogers AB, Moss SF. Early or Late Antibiotic Intervention Prevents Helicobacter Pylori-Induced Gastric Cancer in a Mouse Model. Cancer Lett (2015) 359:345–51. doi: 10.1016/j.canlet.2015.01.028
177. Wen S, So Y, Singh K, Slingerland JM, Resnick MB, Zhang S, et al. Promotion of Cytoplasmic Mislocalization of p27 by Helicobacter Pylori in Gastric Cancer. Oncogene (2012) 31:1771–80. doi: 10.1038/onc.2011.362
178. De Luca A, De Falco M, Iaquinto S, Iaquinto G. Effects of Helicobacter Pylori Infection on Cell Cycle Progression and the Expression of Cell Cycle Regulatory Proteins. J Cell Physiol (2004) 200:334–42. doi: 10.1002/jcp.20022
Keywords: gastric cancer, RB-E2F pathway, pocket protein, E2F family, Helicobacter pylori, p53
Citation: Wu T and Wu L (2021) The Role and Clinical Implications of the Retinoblastoma (RB)-E2F Pathway in Gastric Cancer. Front. Oncol. 11:655630. doi: 10.3389/fonc.2021.655630
Received: 19 January 2021; Accepted: 07 May 2021;
Published: 31 May 2021.
Edited by:
Kanjoormana Aryan Manu, Amala Cancer Research Centre, IndiaReviewed by:
Martin Fischer, Fritz Lipmann Institute (FLI), GermanyDavid N. Arnosti, Michigan State University, United States
Copyright © 2021 Wu and Wu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lizhao Wu, bHp3dUBjbXUuZWR1LmNu