
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Oncol. , 20 May 2021
Sec. Pharmacology of Anti-Cancer Drugs
Volume 11 - 2021 | https://doi.org/10.3389/fonc.2021.651693
This article is part of the Research Topic Clinical Therapeutic Development Against Cancers Resistant to Targeted Therapies View all 31 articles
 Xuewei Zhu1Li Liu2Yang Wang3Jianan Cong4Zhang Lin5Yongsen Wang6Qi Liu7Leiming Wang8
Xuewei Zhu1Li Liu2Yang Wang3Jianan Cong4Zhang Lin5Yongsen Wang6Qi Liu7Leiming Wang8 Ben Yang5*Tao Li9*
Ben Yang5*Tao Li9*Cisplatin-based chemotherapy and radiotherapy are the main first-line treatment strategies for nasopharyngeal carcinoma (NPC) patients. Unfortunately, resistance is a major obstacle in the clinical management of NPC patients. We prove that the expression level of high-mobility group box 1 (HMGB1) is dramatically increased in resistant NPC cells than that in sensitive cells. HMGB1 induces the expression and secretion of IL6, which leads to constitutive autocrine activation of the JAK2/STAT3 pathway and eventually contributes to chemoresistance in NPC cells. Long non-coding RNAs (lncRNAs) have been identified as key regulators involved in drug resistance. In this study, using GO analysis of the biological process and differential expression analysis, we find 12 significantly altered IncRNAs in NPC cell lines, which may be involved in regulating gene expression. Furthermore, we determine that elevated lncRNA MIAT level upregulates HMGB1 expression, contributing to cisplatin resistance in NPC cells. We find that the deficiency of the lncRNA MIAT/HMGB1 axis, inhibition of JAK2/STAT3, or neutralization of IL6 by antibodies significantly re-sensitizes resistant NPC cells to cisplatin in resistant NPC cells. Moreover, we provide the in vivo evidence that the deficiency of HMGB1 reduces cisplatin-resistant tumor growth. Most importantly, we provide clinical evidence showing that the expression level of the lncRNA MIAT/HMGB1/IL6 axis is elevated in resistant NPC tumors, which is highly correlated with poor clinical outcome. Our findings identify a novel chemoresistance mechanism regulated by the lncRNA MIAT/HMGB1/IL6 axis, which indicates the possibilities for lncRNA MIAT, HMGB1, and IL6 as biomarkers for chemoresistance and targets for developing novel strategies to overcome resistance in NPC patients.
Nasopharyngeal carcinoma (NPC) is a type of squamous cell carcinoma that arises from the nasopharynx’s epithelial lining (1). The prevalence of NPC in southern China, Southeast Asia, Northern Africa, Greenland, and Inuits of Alaska endangers human health (1, 2). Clinical evidence and experimental studies indicated that Epstein-Barr virus (EBV) infection, genetic susceptibility, and environmental factors are associated with pathogenic factors of NPC (3, 4). Currently, chemotherapy and radiotherapy are the front-line treatment of NPC. Even though these therapeutic options increased the survival of patients with locally advanced NPC and distant metastasis (DM), resistance to radiotherapy and chemotherapy remains a major clinical obstacle for the treatment of NPC (5–7). Therefore, there is an urgent need to explore the mechanisms underlying radio or chemoresistance for NPC and develop more effective therapeutic strategies.
Recent evidence has revealed that many cytokines/chemokines/growth factors and their receptors are dysregulated in cancers, building up a network involved in regulating tumor progressions, invasion, and especially resistance to radiotherapy and chemotherapy (8, 9). Notably, the interleukin-6 (IL6) cytokine family has been reported to be involved in cancer invasion and proliferation (10). Interestingly, IL6, as a member of the IL6 family and a pleiotropic cytokine, has been shown to accumulate in solid tumors of human colorectal cancer, breast cancer, gastric cancer, prostate cancer, and osteosarcoma (11–14). IL6 is a key player in multiple pathophysiological properties associated with cancer progression and drug resistance (9, 15, 16). The interleukin-6 receptor (IL6R) engages through gp130 ligands and activates the JAK2/STAT3 pathway. IL6R has been shown to be involved in tumor proliferation, survival, invasion, and angiogenesis (17–19). IL6 binding to the IL6R/gp130 complex activates the JAK2/STAT3 pathway and induces its own production in an autocrine manner (13, 20).
High-mobility group box 1(HMGB1) is a nonhistone and highly mobile DNA-binding protein, which functions as a nuclear factor that enhances gene transcription and regulates DNA repair and chromatin remodeling (21, 22). HMGB1 is also recognized as a damage-associated molecular pattern molecule (DAMP) released by necrotic cells and monocytes in response to cell death, inflammation stimuli, and various environmental stressors (21–23). Particularly, dysfunction of intracellular and extracellular HMGB1 has been implicated in the proliferation and metastasis of various cancers, including hepatocellular carcinoma, colorectal cancer, melanoma, and breast cancer (24–27). In many cases, extracellular HMGB1 acts as a pro-tumor protein due to its cytokine, chemokine, and growth factor activity, whereas intracellular HMGB1 promotes drug resistance owing to the pro-autophagy activity and regulation of gene transcription (28–30). However, the function of HMGB1 in chemoresistance remains unknown.
Long non-coding RNAs (lncRNAs), known as non-coding single-strand RNAs, play an essential role as carcinogenic genes or tumor suppressors in the development of human cancer (31–34). Myocardial infarction-associated transcript (MIAT) was first identified as an lncRNA in 2006 and is considered as a regulator in the nucleus interacting with nuclear factors, while as a competitive endogenous RNA (ceRNA) in the cytoplasm (31, 35, 36).
In this study, we demonstrated that the lncRNA MIAT/HMGB1 axis is highly upregulated in cisplatin-resistant nasopharyngeal carcinoma cell lines. The lncRNA MIAT/HMGB1 axis contributes to cisplatin resistance by regulating IL6 expression and secretion. Autocrine IL6 promotes resistance by activating the JAK2/STAT3 pathway in NPC. HMGB1 deficiency overcomes cisplatin resistance in vivo. Clinical evidence shows that the expression level of the lncRNA MIAT/HMGB1/IL6 axis is elevated in resistant NPC tumors, which is highly correlated with the poor survival of patients. Thus, the lncRNA MIAT/HMGB1/IL6 axis could be considered as a promising candidate for therapy in chemoresistant NPC.
NPC cell lines CNE-2 and HONE-1 were cultured in Roswell Park Memorial Institute 1640 medium with 10% fetal bovine serum (FBS) and 100 IU/ml of penicillin/streptomycin. Cells were maintained at 37°C and 5% CO2 in a humidified incubator. Resistant cells were generated according to previous studies (37). Briefly, CNE-2 resistant cells (CNE-2 CR) and HONE-1 resistant cells (HONE-1 CR) were generated by exposing CNE-2 wild-type cells (CNE-2 WT) or HONE-1 wild-type cells (HONE-1 WT) to increasing concentrations of cisplatin once per week for at least three months.
A cell viability assay was performed as previously described (37). Briefly, cells were seeded in a 96-well plate to a final concentration of 5,000 cells per well and cultured to allow adherence. Then cells were exposed to cisplatin at varying concentrations and incubated for 72 h. Cell viability was detected using a Sulforhodamine B (SRB) assay. Cells were fixed by 10% trichloroacetic acid (TCA) then followed by staining using 0.02% SRB, finally the SRB was extracted with Tris buffer (10 mM of tris-HCl, pH 10.5) and the absorbance was measured at 510 nm by a microplate reader (MTX Lab Systems).
Transfections of siRNA were performed with Lipofectamine™ RNAi MAX (Invitrogen) according to the manufacturer’s instructions. Gl2 (luciferase) siRNA was used as a negative control. The HMGB1 plasmid (Addgene #31609) was transfected using Lipofectamine 3000 (Invitrogen) according to the manufacturer’s instructions.
HMGB1 KO cell line CNE-2 was generated using CRISPR/Cas9 as previously described (37).
RNA isolation and qRT-PCR assays were performed as described previously (37). Total RNA from cells was extracted with the miRNeasy Mini Kit (Qiagen), and complementary DNA was synthesized using moloney murine leukemia virus (M-MLV) reverse transcriptase with random primers. cDNA was generated with the BioTeke super RT kit (Bioteke) according to the manufacture’s protocol. qRT-PCR was performed using SYBR Premix Ex TaqTM (TaKaRa). The relative expressions of genes were calculated by normalization to the internal control Actin. Primers are listed in Supplementary Table 1.
Western blot was performed as described previously (37). The cells were collected in ice-cold PBS, and lysed in RIPA buffer containing protease inhibitor and phosphatase inhibitor cocktail (Santa Cruz Biotechnology). The protein concentrations were determined by use of Bradford methods and boiled in SDS sample buffer (50 mM of Tris [pH 6.8]; 100 mM of DTT; 2% SDS; 0.1% bromophenol blue; 10% glycerol). The proteins were separated on sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and transferred onto nitrocellulose membranes. Membranes were then incubated in blocking solution (5% non-fat milk in 20 mM of TRIS-HCl, 150 mM of NaCl, 0.1% Tween-20) (TBST), followed by incubation with the indicated primary antibodies at 4°C overnight. Then secondary antibodies were incubated and detected with an enhanced chemiluminescence reaction.
The whole data of NPC cell lines CNE-2 and HONE-1 underwent GO analysis of the biological process and differential expression analysis, which were implemented in the R programing language.
EdU staining was conducted using the Click-iT™ Plus EdU Alexa Fluor™ 594 Flow Cytometry Assay Kit (Thermo Fisher) as per the manufacturer’s instructions. EdU-positive cells were analyzed by a flow cytometer.
Eight-week-old nude mice (female, athymic Foxn1nu; Vital River, Beijing, China) were inoculated subcutaneously by injecting CNE-2 CR or CNE-2 CR-KO cells (5 x 106) into the dorsal flank of each mouse. Cisplatin was injected twice per week, for a total of two weeks (5 mg/kg, intraperitoneally) when the tumor volume reached 100 mm3. Tumor volume was calculated from caliper measurements by the formula 1/2(L × W2). Tumor volume and body weight were measured twice per week.
Immunohistochemistry (IHC) staining was performed as previously described. An antibody against Ki67 was used for specific recognition of corresponding proteins in the xenograft tumor samples. The quantification of cells with positive staining per field was analyzed.
The tumor tissues from sensitive and resistant patients were from China Japan Union Hospital of Jilin University. The present study was approved by the institutional ethics committee. Survival rate of patients was analyzed using a Kaplan-Meier plotter (https://kmplot.com/analysis/).
Data were presented as mean ± SD of at least three independent experiments. Statistical analysis of data was performed by Student’s t test and one-way ANOVA using Graph Pad Prism 7 software for two groups and multiple group comparison. P<0.05 represents the statistically significant difference.
To discover new mechanisms of resistance, pathways, and targets for novel drug combinations that can overcome cisplatin resistance, we established resistant NPC cells using two parent NPC cell lines CNE-2 and HONE-1, according to methods described previously (37). We obtained sub-cell lines, CNE-2 CR and HONE-1 CR, which could grow in a high concentration of cisplatin (Figures 1A, B). The SRB cell survival assay revealed that the IC50 of cisplatin in CNE-2 CR and HONE-1 CR increased 3-fold (Figures 1A, B). We further used these two pairs of cell lines to screen for factors that may contribute to chemoresistance.
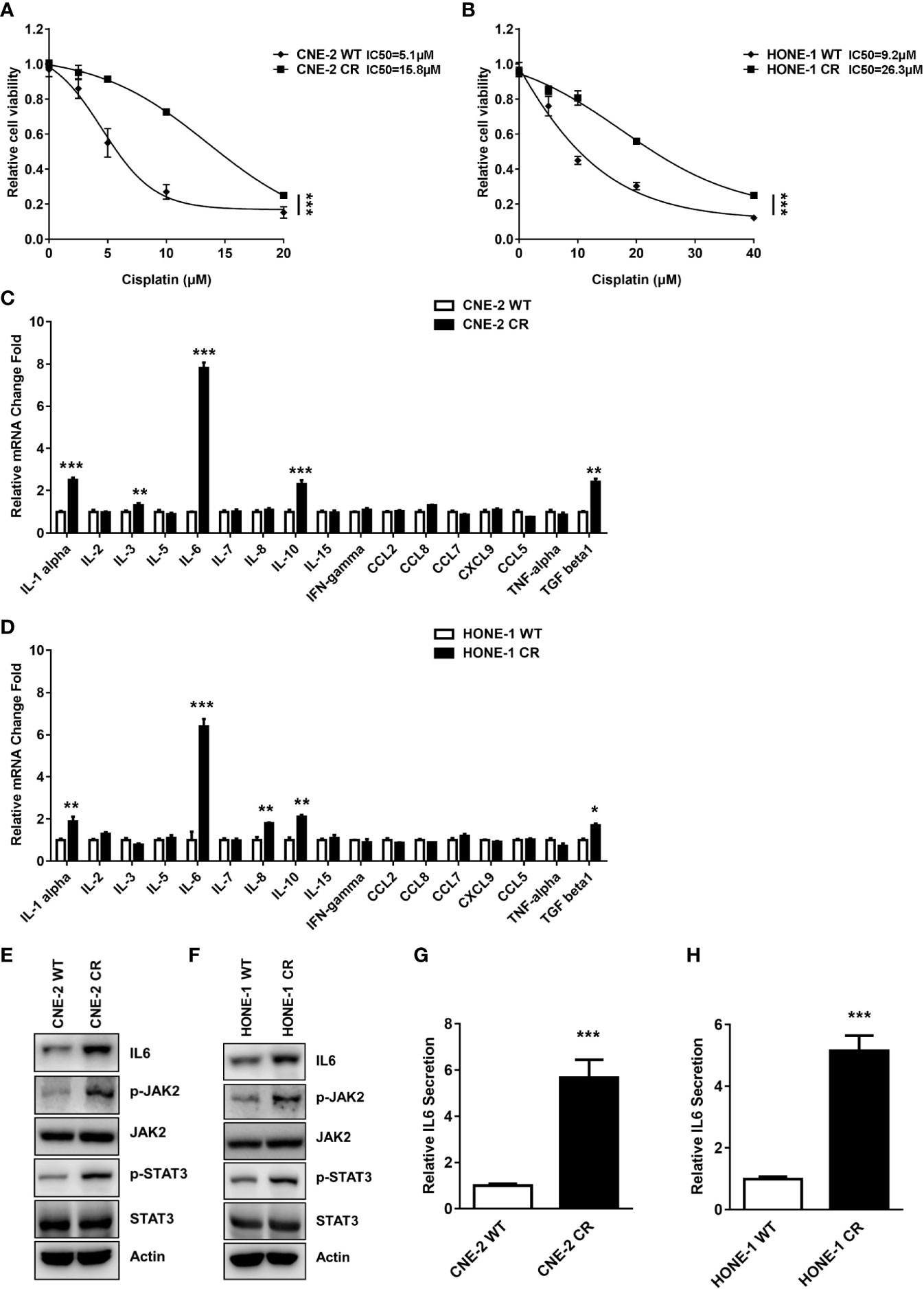
Figure 1 The expression and secretion levels of IL6 were elevated in resistant NPC cells. (A, B) Establishment of cisplatin-resistant NPC cells. Cell proliferation was assessed by SRB assay, ***P < 0.001. (C, D) The mRNA levels of some cytokines and chemokines in parent wild-type NPC cell lines (CNE-2 WT and HONE-1 WT) and resistant cell lines (CNE-2 CR and HONE-1 CR) were detected by qPCR, *P < 0.05, **P < 0.01, ***P < 0.001. (E, F) The protein levels of IL6, p-JAK2, JAK2, p-STAT3, STAT3, and Actin were detected by western blotting. (G, H) IL6 secretion levels were detected by an ELISA kit, ***P < 0.001.
Cytokines and chemokines have been reported to participate in the progression of tumor growth, invasion, metastasis, and drug resistance. The expressions of cytokines and chemokines including IL6, IL-11, TGF-b, IL-5, IL-10, and IL-1b have significant differences after ionizing radiation (IR) or cisplatin treatment in various tumors (8, 11, 38, 39). Thus, cytokines or chemokines are designated as potential biomarkers for diagnosis and treatment in cancers. We hypothesized that cytokines or chemokines and their activated receptors might be involved in resistance to cisplatin. To further test this possibility, we detected the mRNA expression levels of some cytokines and chemokines in parent wild-type sensitive NPC cell lines (CNE-2 WT and HONE-1 WT) and resistant cell lines (CNE-2 CR and HONE-1 CR). Interestingly, the IL6 mRNA level was significantly increased in both resistant cell lines compared to the sensitive cell lines (Figures 1C, D), indicating that an elevated level of IL6 may cause resistance.
To verify if IL6 expression and its downstream pathway are upregulated in cisplatin resistant cells, we detected the IL6 protein level and its downstream pathway in cisplatin sensitive and resistant cells. Consistently, we found the protein level of IL6 was significantly increased in both resistant cell lines (Figures 1E, F). Furthermore, we tested the activation of IL6 downstream signaling via the JAK2/STAT3 pathway. The phosphorylation levels of JAK2 (Y1007/1008) and STAT3 (Y694) were both upregulated in resistant cells, while the total protein level of JAK2 and STAT3 were not altered when compared to the sensitive cells (Figures 1E, F). Our data indicated that IL6 is upregulated, which activates the JAK2/STAT3 pathway in resistant cells.
Given that IL6 mRNA and protein levels are increased in both resistant cells, we hypothesized that IL6 in the medium of resistant cells may cause, in an autocrine fashion, the activation of a downstream pathway, which eventually results in resistance to cisplatin. We tested the level of IL6 secreted in resistant cells. Consistently, the secretion level of IL6 in the medium of both resistant cells was significantly elevated compared to their sensitive counterparts (Figures 1G, H).
Given the above findings of the elevated expression and secretion levels of IL6, we hypothesized that IL6-mediated autocrine activation of the JAK2/STAT3 pathway may contribute to the induction of resistance in NPC. We showed that the stimulation of recombinant IL6 treatment turned the sensitive cells toward resistance to cisplatin (Figures 2A, B). To confirm whether the elevated IL6 secretion level in the medium of resistant cells causes resistance to cisplatin, the conditioned medium from sensitive cells and resistant cells were collected to culture sensitive cells. Indeed, both sensitive cells CNE-2 WT and HONE-1 WT cultured with the conditioned medium from resistant cells displayed an elevated resistance to cisplatin, compared to the cells cultured in the conditioned medium from sensitive cells (Figures 2C, D). However, this elevated resistance was abolished by an IL6 neutralization antibody (Figures 2C, D). Meanwhile, the activation level of the JAK2/STAT3 pathway was also dramatically decreased by an IL6 neutralization antibody (Figures 2E, F). These results indicate that the secreted IL6 acts as an autocrine factor to induce cisplatin resistance in NPC cells via the JAK2/STAT3 pathway. Furthermore, we found that IL6 deficiency by specific siRNA resulted in a decreased IL6 secretion level (Figures 2G, H). More importantly, IL6 deficiency suppressed the cell survival of both resistant cell lines by downregulating the activation of the JAK2/STAT3 pathway, indicating that IL6 deficiency re-sensitizes resistant cells to cisplatin (Figures 2I–L).
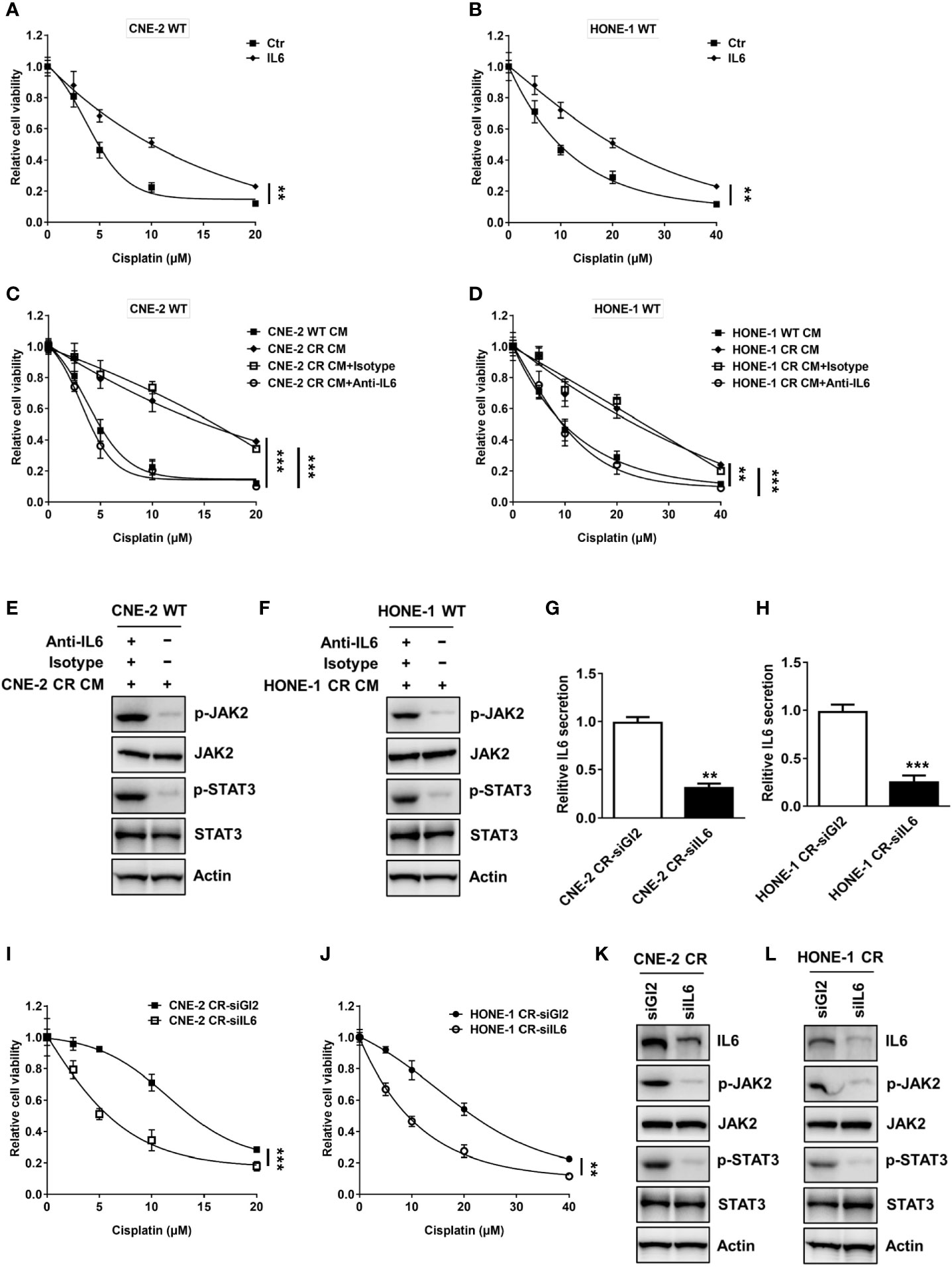
Figure 2 Activation of IL6 promotes cisplatin resistance. (A, B) Cell proliferation was assessed by SRB assay after recombinant IL6 treatment, **P < 0.01. (C, D) Cell proliferation was assessed by SRB assay after incubation with conditional medium from wild-type cells or resistant cells, and isotype control or anti-IL6, **P < 0.01, ***P < 0.001. (E, F) The protein levels of p-JAK2, JAK2, p-STAT3, STAT3, and Actin were detected by western blotting in CNE-2 WT and HONE-1 WT cells treated with conditional medium from resistant cells, and isotype control or anti-IL6. (G, H) IL6 secretion levels were detected by an ELISA kit after IL6 siRNA transfection, ***P < 0.001. (I, J) Cell proliferation was assessed by SRB assay after IL6 siRNA transfection, ***P < 0.001. (K, L) The protein levels of IL6, p-JAK2, JAK2, p-STAT3, STAT3, and Actin were detected by western blotting after IL6 siRNA transfection.
Previous studies have reported that HMGB1 is associated with cytokines or chemokines production (21, 23, 28, 40–42). We tested whether HMGB1 is involved in the upregulation of IL6. Interestingly, we observed that the basal levels of HMGB1 in both resistant cells CNE-2 CR and HONE-1 CR were significantly elevated (Figures 3A, B). To determine whether HMGB1 regulates IL6 expression, we knocked down HMGB1 with siRNA and detected the expression of IL6 and the activation of the downstream JAK2/STAT3 pathway. HMGB1 deficiency significantly reduced IL6 expression, as well as the phosphorylation level of JAK2 and STAT3 (Figures 3C, D). Importantly, HMGB1 deficiency re-sensitized resistant cells to cisplatin (Figures 3E, F). Similar results were observed from the EdU cell proliferation assay. The resistant cells with HMGB1 deficiency showed less proliferation when exposed to cisplatin compared to the cells in the control group (Figures 3G, H). To confirm whether HMGB1 contributes to cisplatin resistance in NPC cells via targeting IL6 directly, we established HMGB1 knock-out cells in resistant NPC cells (CNE-2 CR KO) for rescue experiments (Figures 4A, B). The results showed that HMGB1 KO cells in resistant cells re-sensitized to cisplatin, which was polished by recombinant IL6 treatment. Consistently, the elevated resistance caused by ectopic overexpression of HMGB1 in sensitive CNE-2 WT cells was polished by the treatment of the IL6 neutralization antibody (Figures 4C, D). To further validate the HMGB1 function, we rescued HMGB1 by overexpressing it in HMGB1 KO resistant cells. Results showed that overexpressed HMGB1 impaired the sensitivity to cisplatin in HMGB1 KO resistant cells (Figures 4E, F). Taken together, these results suggest that HMGB1 contributes to cisplatin resistance by directly upregulating IL6 expression in NPC cells.
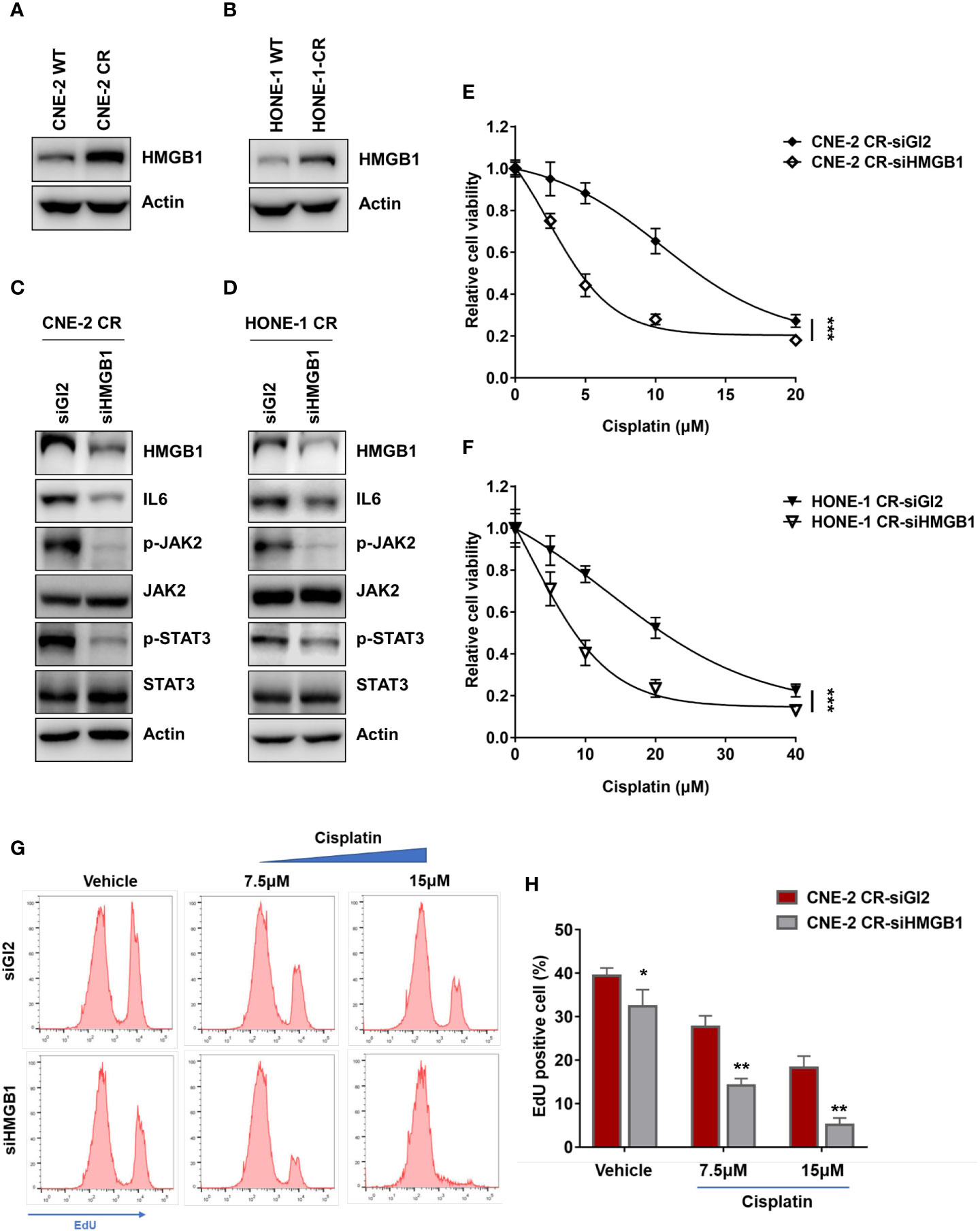
Figure 3 HMGB1 is upregulated in NPC cisplatin-resistant cells. (A, B) The protein levels of HMGB1 and Actin were detected by western blotting. (C, D) The protein levels of HMGB1, IL6, p-JAK2, JAK2, p-STAT3, STAT3, and Actin were detected by western blotting after HMGB1 siRNA transfection. (E, F) Cell proliferation was assessed by SRB assay, after HMGB1 siRNA transfection ***P < 0.001. (G, H) A cell proliferation assay was performed using an EdU assay and analyzed by flow cytometry after siRNA transfection. *P < 0.05, **P < 0.01.
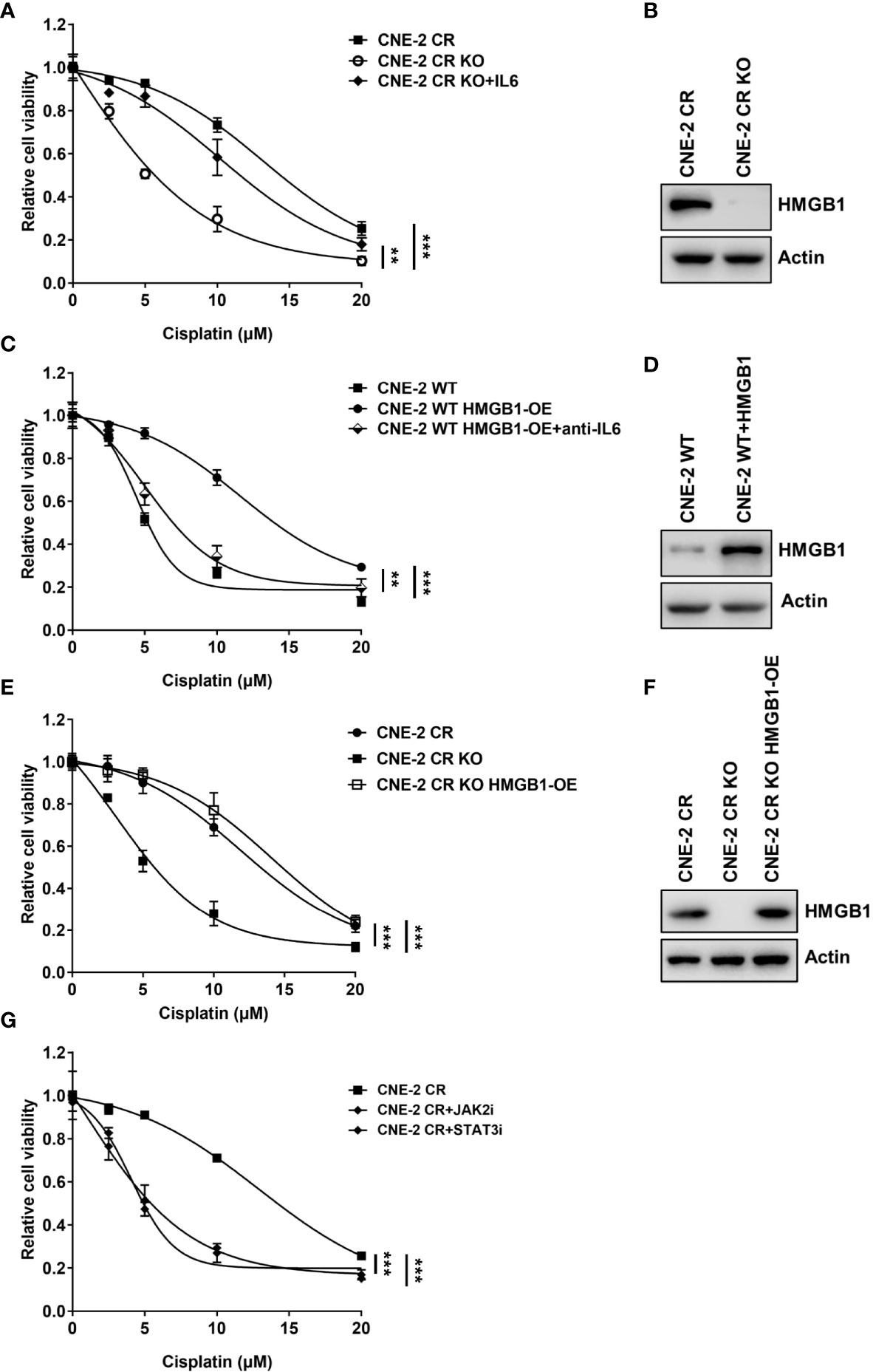
Figure 4 HMGB1 upregulates expression of IL6 in resistance. (A) Cell proliferation was assessed by SRB assay after recombinant IL6 treatment in CNE-2 CR and CNE-2 CR-KO cells, **P < 0.01, ***P < 0.001. (B) The protein levels of HMGB1 and Actin were detected by western blotting in CNE-2 CR and CNE-2 CR-KO cells. (C) Cell proliferation was assessed by SRB assay after anti-IL6 treatment in CNE-2 WT and CNE-2 HMGB1 OE cells, **P < 0.01, ***P < 0.001. (D) The protein levels of HMGB1 and Actin were detected by western blotting after HMGB1 was overexpressed in CNE-2 WT cells. (E) Cell proliferation was assessed by SRB assay after HMGB1 was overexpressed in CNE-2 CR OK cells. (F) The protein levels of HMGB1 and Actin were detected by western blotting in CNE-2 CR and CNE-2 CR KO and CNE-2 CR KO with HMGB1 overexpressed cells. (G) Cell proliferation was assessed by SRB assay after treatment with JAK2 inhibitor LY2784544 or STAT3 inhibitor stattic, ***P < 0.001.
Given that HMGB1 elevated IL6 level, which induces the activation of the JAK2/STAT3 pathway, we explored the approach to overcome cisplatin resistance via treating resistant cells with JAK2 inhibitor LY2784544 or STAT3 inhibitor stattic. Similarly, both the JAK2 inhibitor and STAT3 inhibitor exhibited a synergistic effect with cisplatin on resistant cells (Figure 4G) which suggests possibilities to develop novel drug combinations with chemotherapy to overcome resistance in NPC.
To evaluate whether HMGB1 deficiency could re-sensitize cisplatin in vivo, we established xenograft tumors by injecting CNE-2 CR or CNE-2 CR-KO cells, followed by cisplatin treatment (Figure 5A). We found that HMGB1 deficiency without cisplatin treatment did not affect tumor growth. Strikingly, HMGB1 deficiency showed a dramatically synergistic effect with cisplatin in resistant xenograft tumors with no significant difference in body weight (Figures 5B–E). Similarly, the Ki67 immunohistochemistry (IHC) results showed that cell proliferation was impaired in CNE-2-KO xenograft tumors with cisplatin treatment (Figures 5F, G).
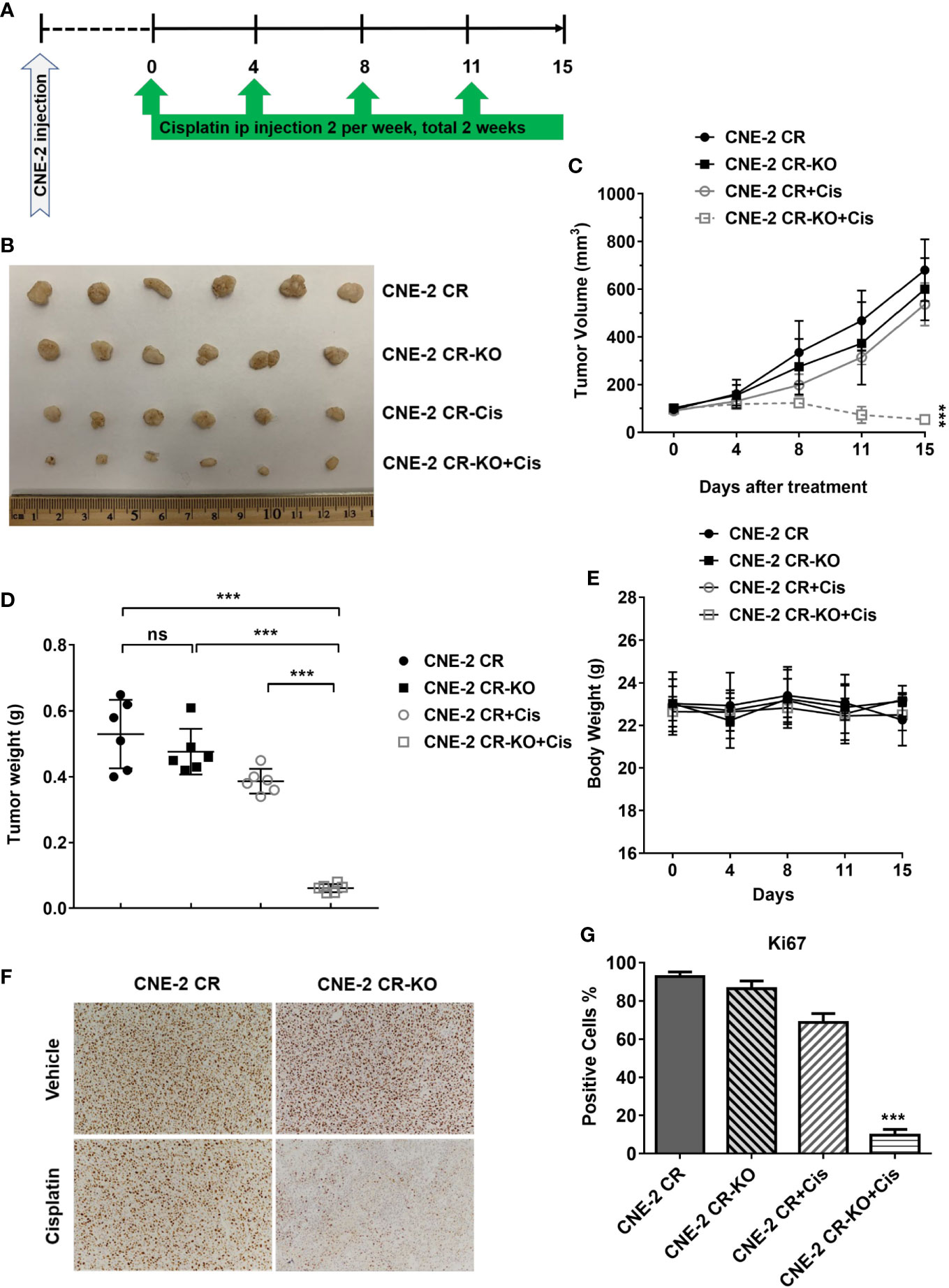
Figure 5 HMGB1 deficiency overcomes cisplatin resistance in vivo. (A) The scheme of xenograft tumors and treatment. (B) The photograph of the representative tumors from mice in each treatment group. Ruler scale is shown in cm. (C) Growth curves of CNE-2 CR xenograft tumors treated with vehicle and cisplatin (5 mg/kg, intraperitoneally) for two weeks, n = 6 mice/group, ***P < 0.001. (D) The tumor weight in each treatment group, ***P < 0.001. (E) The body weight from mice in each treatment group. (F–G) Immunohistochemistry staining of Ki67 in xenograft tumor samples. Ki67 positive cells were quantified. NS, Non-significant.
lncRNAs have been reported to regulate the growth, proliferation, invasion, metastasis, and drug resistance of various cancers (32, 33, 34). lncRNAs participate in transcriptional modulation, splicing regulation, post-transcriptional process, chromatin remodeling, or protein-protein, protein-DNA, and protein-RNA interactions (43, 44, 45). We, therefore, compared the nasopharyngeal carcinoma cell lines CNE-2 and HONE1 to a normal nasopharynx cell line for the detection of differentially expressed lncRNAs (Figure 6A and Supplementary Figure 1). The GO analysis results showed differentially expressed genes enrichment in the pathway of negative regulation of posttranscriptional gene silencing and negative regulation of gene silencing by RNA, which had been reported to be associated with chemotherapy resistance in various cancers (Figure 6B). This result indicates that these lncRNAs may be involved in the regulation of HMGB1 expression. We overlapped the differentially expressed lncRNAs in CNE-2 and HONE-1 cell lines. Twelve lncRNAs (LINC01238, TCL6, LINC01006, TNRC6C-AS1, MMP25-AS1, LINC00954, A1BG-AS1, lnc MIAT, LINC00173, LINC00342, CD27-AS1, and TSPOAP1-AS1) were found dramatically differentially expressed compared with the normal nasopharynx cell line, which may be involved in the regulation of HMGB1 expression (Figure 6C).
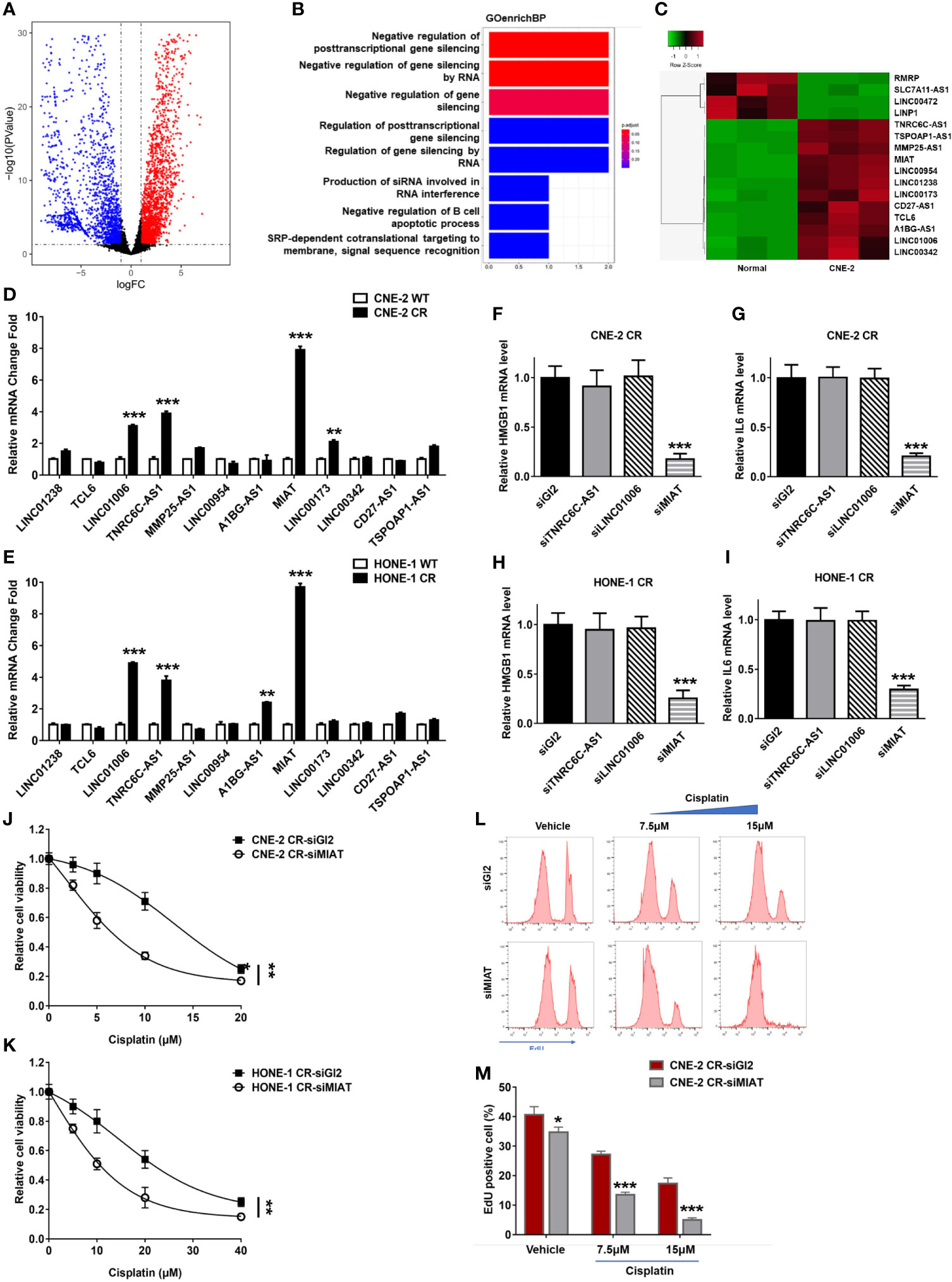
Figure 6 lnc MIAT contributes to cisplatin resistance via regulating HMGB1 in NPC cells. (A) Volcano plot showing differentially expressed lncRNAs in CNE-2 compared with normal nasopharynx cells. A change is considered significant if the change is >2-fold with a p-value of <0.05. (B) GO analysis of the biological process in CNE-2 cells. (C) Heatmap showing the selected differentially expressed lncRNAs in CNE-2 after overlap with those in HONE-1. (D, E) lncRNA levels were detected by qPCR, **P < 0.01, ***P < 0.001. (F–I) The expression levels of HMGB1 and IL6 were detected by qPCR after siRNA transfection of lncRNAs, **P < 0.01, ***P < 0.001. (J, K) Cell proliferation was assessed by SRB assay, after MIAT siRNA transfection ***P < 0.001. (L, M) Cell proliferation assay was performed using an EdU assay and analyzed by flow cytometry after siRNA transfection. *P < 0.05.
To determine the roles of lncRNAs involved in cisplatin resistance in NPC cells, we tested the expression levels of these twelve lncRNAs in resistant NPC cells. We found that TNRC6C-AS1, LINC01006, and lncRNA MIAT were upregulated in both resistant NPC cells CNE-2 CR and HONE-1 CR (Figures 6D, E). To determine which lncRNA may be involved in regulating HMGB1 in resistant cells, we detected their effect on HMGB1 expression via defecting these lncRNAs by siRNA individually. Notably, lncRNA MIAT deficiency decreased HMGB1 expression as well as IL6 expression, while TNRC6C-AS1 and LINC01006 did not show an effect on HMGB1 and IL6 (Figures 6F–I). This result indicates that lncRNA MIAT participates in HMGB1 expression in resistance. To further confirm whether lncRNA MIAT deficiency overcomes resistance, we tested synergy with cisplatin in NPC resistant cells and results showed that lncRNA MIAT deficiency re-sensitized cells to cisplatin (Figures 6J, K). Similar results were obtained from the EdU cell proliferation assay. The resistant cells with lncRNA MIAT deficiency showed less proliferation when exposed to cisplatin (Figures 6L, M). Taken together, we found that lncRNA MIAT participated in cisplatin resistance via regulating the HMGB1/IL6 axis in NPC cells.
To directly evaluate the correlation between the lncRNA MIAT/HMGB1/IL6 axis and tumor resistance in NPC patients, we collected samples from cisplatin-sensitive patients and the patients developed resistance. We obtained four sensitive tumor samples and four resistant tumor samples and analyzed the expression levels of lncRNA MIAT, HMGB1, and IL6 (Figures 7A–D). We found that all the resistant patients had elevated lncRNA MIAT, HMGB1, and IL6 expression levels. Inspired by this finding of the correlation between the lncRNA MIAT/HMGB1/IL6 axis and resistance in NPC patients, we further evaluated the correlation between the expression of the lncRNA MIAT/HMGB1/IL6 axis and survival rate. NPC is the most common head neck squamous cell carcinoma (HNSCC) derived from the epithelial cells at the nasopharynx. Given that NPC samples are relatively rare to obtain, we analyzed the data from HNSCC patients in the datasets and correlated the expression levels of lncRNA MIAT, HMGB1, and IL6 with the survival rate of these patients using a Kaplan-Meier plotter (https://kmplot.com/analysis/). Indeed, patients exhibited a worse 5-year PPS rate when the expression level of lncRNA MIAT, HMGB1, or IL6 was higher (Figures 7E–G). Consistently, the high expression level of the lncRNA MIAT/HMGB1/IL6 axis significantly correlated with poor patient survival (Figure 7H). Thus, this clinical evidence strongly indicates that the expression level of the lncRNA MIAT/HMGB1/IL6 axis is elevated in resistant NPC tumors, which is highly correlated with poor survival.
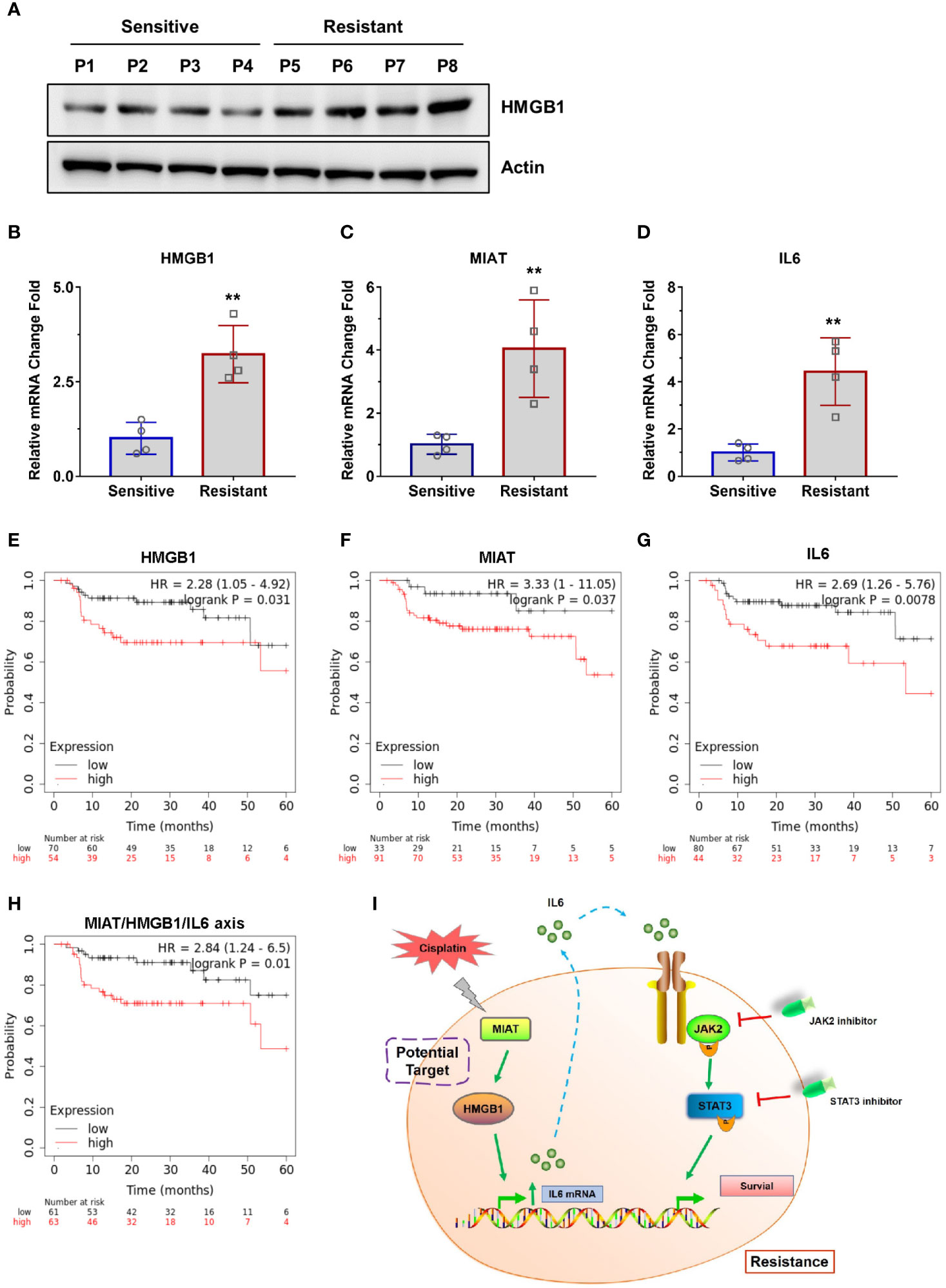
Figure 7 Clinical evidence of the lncRNA MIAT/HMGB1/IL6 axis. (A) The protein levels of HMGB1 and Actin were detected by western blotting in patient samples. (B–D) The mRNA levels of IL6, HMGB1, and lncRNA MIAT were detected by qPCR in patient samples, **P < 0.01. (E–H) Survival rate of patients was analyzed using a Kaplan-Meier plotter (https://kmplot.com/analysis/). (I) Working model of the lncRNA MIAT/HMGB1/IL6 axis in regulating cisplatin resistance in NPC cells.
In this study, we showed the elevated lncRNA MIAT/HMGB1 axis is responsible for IL6-mediated activation of the JAK2/STAT3 pathway to endow NPC cells with cisplatin resistance (Figure 7I). We have provided the evidence that targeting lncRNA MIAT, HMGB1, or IL6 is capable of re-sensitizing cells to cisplatin. In recent years, there has been more and more evidence that lncRNAs can be considered as new and valuable molecules that are involved in the regulation of the growth, proliferation, invasion, metastasis, and drug resistance of cancer cells (32, 33, 34). lncRNAs participate in transcriptional modulation, splicing regulation, post-transcriptional process, chromatin remodeling, or protein-protein, protein-DNA, and protein-RNA interactions (43, 44, 45). Myocardial infarction-associated transcript (MIAT) known as retina non-coding RNA2, locates on the human chromosome 12q12.1 (31, 46). It has been reported that lncRNA MIAT acts as an oncogene upregulated in various cancers such as gastric cancer, liver cancer, colorectal cancer, and lung cancer (31). lncRNA MIAT is involved in regulating proliferation, migration, invasion, drug resistance, and cell cycle by targeting some proteins, including MMP9, SF1, TDP43, ZEB1, MYO1B, SGK1, WNT9A, DDX5, and HDAC4 (31). However, the role of lncRNA MIAT in cisplatin-resistant NPC cells is barely known. For the first time, our study determined that MIAT, as well as HMGB1, is upregulated in cisplatin-resistant NPC cells. More importantly, we have revealed that MIAT contributes to cisplatin resistance via upregulating HMGB1 expression and then elevating IL6 expression and secretion levels. However, the mechanism by which MIAT regulates HMGB1 expression remains to be defined in a future study. Given that MIAT has been revealed to play a regulatory role in the cytoplasm via the mechanism of competitive endogenous RNA (ceRNA) to target various microRNAs including miR-141, miR-29A-3p, miR-133a-5p, miR-184, miR-34a, miR-1246, and miR-150 (31), we assume that MIAT regulates HMGB1 expression via targeting some microRNAs.
HMGB1 was first discovered as one of a group of chromatin-associated proteins. Structurally, HMGB1 protein contains two homologous DNA-binding domains with a negatively charged C-terminal region (47). Nuclear HMGB1 binds to DNA to stabilize the nucleosome and regulate chromatin remodeling, DNA repair, and gene transcription (22). HMGB1 has been shown to be a transcriptional co-factor of multiple genes, including p53, p73, NF-κB, and estrogen receptor (22, 30). Therefore, it is reasonable that HMGB1 participates in upregulating IL6 expression. Most importantly, for the first time, we demonstrated that the expression level of HMGB1 is much higher in resistant NPC cells than in sensitive cells, suggesting that HMGB1 may be associated with resistance. Indeed, we have found that HMGB1 deficiency re-sensitizes resistant cells to cisplatin. Extracellular HMGB1 also acts as a pro-tumor protein due to its cytokine, chemokine, and growth factor activity (28–30). It has been previously reported that HMGB1 binds to lipoteichoic acid and enhances TNF-α and IL6 production in an inflammatory response (40). Consistent with this report, our findings have revealed that HMGB1 upregulates IL6 expression to contribute to resistance, because HMGB1 has dual nucleus and extracellular functions. This also opens up new possibilities that the secreted HMGB1 may also improve tumor cell survival and invasion. Moreover, we found that HMGB1 deficiency overcomes cisplatin resistance in vivo, which provides evidence that HMGB1 could be a potential target for the following therapy of NPC patients.
We have also provided evidence that IL6 acts as an autocrine factor to activate the downstream JAK2/STAT3 pathway, which eventually transforms sensitive cells into resistant cells. Therefore, it provides the possibility that IL6 can be potentially used as a biomarker for diagnosing, preventing, and treating cisplatin resistance in clinical application.
Most importantly, we have provided clinical evidence showing that the expression level of the lncRNA MIAT/HMGB1/IL6 axis is elevated in resistant NPC tumors, which is highly correlated with the poor survival of patients. Therefore, lncRNA MIAT, HMGB1, and IL6 are promising targets in drug development to overcome resistance in NPC patients. Additionally, it is well known that the JAK inhibitor (LY2784544) we used in this study is currently in a Phase 2 clinical trial for its potential use in treating myeloproliferative neoplasms (NCT01594723). During the Phase 1 trials, patients showed good tolerance to LY2784544 and clinical improvement at a dose of 120 mg/day (48). Our studies have indicated that LY2784544 can efficiently re-sensitize cisplatin-resistant NPC cells to cisplatin via inhibition of JAK2, which eventually blocks IL6 autocrine activity. Therefore, the application of specific JAK2 inhibitors, including LY2784544 is likely to lead to a promising and efficient combination approach with chemotherapy as the first-line treatment or the treatment strategy for relapsed patients after radiotherapy (Figure 7I). We have also demonstrated that the STAT3 inhibitor (stattic) overcomes resistance, which indicates that the STAT3 inhibitor is promising for drug innovation (Figure 7I). Taken together, our study provides the impetus to discover new strategies, for instance, the development of efficient agents, including JAK2/STAT3 inhibitors and humanized monoclonal antibodies against IL6 to re-sensitize resistant NPC cells or other cancers that were resistant to chemotherapy.
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding authors.
The animal study was reviewed and approved by Laboratory Animal Welfare Ethics Committee of Jilin University. The patients study was approved by the institutional ethics committee of Jilin University.
BY and TL designed the project. XZ, LL, YW, JC, ZL, YSW, QL, and LW performed the experiments and analyzed the data. XZ and LL prepared the manuscript. XZ and YSW performed the experiments in manuscript revision process. BY and TL contributed to manuscript revision. All authors contributed to the article and approved the submitted version.
Author YSW was employed by the company Harbin Boshixuan Technology Co.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest
We would like to acknowledge funding from the NSFC (Natural Science Foundation of China) (81600787) and the Jilin Province Science and Technology Institute (20170520028JH) received by XZ.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2021.651693/full#supplementary-material
1. Chen YP, Chan ATC, Le QT, Blanchard P, Sun Y, Ma J. Nasopharyngeal Carcinoma. Lancet (London England) (2019) 394:64–80. doi: 10.1016/S0140-6736(19)30956-0
2. Lo KW, Chung GT, To KF. Deciphering the Molecular Genetic Basis of NPC Through Molecular, Cytogenetic, and Epigenetic Approaches. Semin Cancer Biol (2012) 22:79–86. doi: 10.1016/j.semcancer.2011.12.011
3. Chua MLK, Wee JTS, Hui EP, Chan ATC. Nasopharyngeal Carcinoma. Lancet (London England) (2016) 387:1012–24. doi: 10.1016/S0140-6736(15)00055-0
4. Lee HM, Okuda KS, González FE, Patel V. Current Perspectives on Nasopharyngeal Carcinoma. Adv Exp Med Biol (2019) 1164:11–34. doi: 10.1007/978-3-030-22254-3_2
5. Hui EP, Ma BB, Leung SF, King AD, Mo F, Kam MK, et al. Randomized Phase II Trial of Concurrent Cisplatin-Radiotherapy With or Without Neoadjuvant Docetaxel and Cisplatin in Advanced Nasopharyngeal Carcinoma. J Clin Oncol (2009) 27:242–9. doi: 10.1200/JCO.2008.18.1545
6. Xu T, Tang J, Gu M, Liu L, Wei W, Yang H. Recurrent Nasopharyngeal Carcinoma: A Clinical Dilemma and Challenge. Curr Oncol (Toronto Ont) (2013) 20:e406–419. doi: 10.3747/co.20.1456
7. Lee AW, Ma BB, Ng WT, Chan AT. Management of Nasopharyngeal Carcinoma: Current Practice and Future Perspective. J Clin Oncol (2015) 33:3356–64. doi: 10.1200/JCO.2015.60.9347
8. Jin YB, Zhang GY, Lin KR, Chen XP, Cui JH, Wang YJ, et al. Changes of Plasma Cytokines and Chemokines Expression Level in Nasopharyngeal Carcinoma Patients After Treatment With Definitive Intensity-Modulated Radiotherapy (IMRT). PloS One (2017) 12:e0172264. doi: 10.1371/journal.pone.0172264
9. Salazar N, Castellan M, Shirodkar SS, Lokeshwar BL. Chemokines and Chemokine Receptors as Promoters of Prostate Cancer Growth and Progression Crit Rev Eukaryot Gene Expr (2013) 23:77–91. doi: 10.1615/CritRevEukaryotGeneExpr.2013006905
10. Unver N, McAllister F. IL-6 Family Cytokines: Key Inflammatory Mediators as Biomarkers and Potential Therapeutic Targets. Cytokine Growth Factor Rev (2018) 41:10–7. doi: 10.1016/j.cytogfr.2018.04.004
11. Hara M, Nagasaki T, Shiga K, Takahashi H, Takeyama H. High Serum Levels of Interleukin-6 in Patients With Advanced or Metastatic Colorectal Cancer: The Effect on the Outcome and the Response to Chemotherapy Plus Bevacizumab. Surg Today (2017) 47:483–9. doi: 10.1007/s00595-016-1404-7
12. Sansone P, Bromberg J. Targeting the interleukin-6/Jak/stat Pathway in Human Malignancies. J Clin Oncol (2012) 30:1005–14. doi: 10.1200/JCO.2010.31.8907
13. Li J, He K, Liu P, Xu LX. Iron Participated in Breast Cancer Chemoresistance by Reinforcing IL-6 Paracrine Loop. Biochem Biophys Res Commun (2016a) 475:154–60. doi: 10.1016/j.bbrc.2016.05.064
14. Ara T, Declerck YA. Interleukin-6 in Bone Metastasis and Cancer Progression. Eur J Cancer (Oxford Engl 1990) (2010) 46:1223–31. doi: 10.1016/j.ejca.2010.02.026
15. Yang J, Wang Y, Gao Y, Shao J, Zhang XJ, Yao Z. Reciprocal Regulation of 17beta-Estradiol, Interleukin-6 and Interleukin-8 During Growth and Progression of Epithelial Ovarian Cancer. Cytokine (2009) 46:382–91. doi: 10.1016/j.cyto.2009.03.013
16. Bondurant KL, Lundgreen A, Herrick JS, Kadlubar S, Wolff RK, Slattery ML. Interleukin Genes and Associations With Colon and Rectal Cancer Risk and Overall Survival. Int J Cancer (2013) 132:905–15. doi: 10.1002/ijc.27660
17. Huang C, Yang G, Jiang T, Huang K, Cao J, Qiu Z. Effects of IL-6 and AG490 on Regulation of Stat3 Signaling Pathway and Invasion of Human Pancreatic Cancer Cells In Vitro. J Exp Clin Cancer Res (2010) 29:51. doi: 10.1186/1756-9966-29-51
18. Groner B, Lucks P, Borghouts C. The Function of Stat3 in Tumor Cells and Their Microenvironment. Semin Cell Dev Biol (2008) 19:341–50. doi: 10.1016/j.semcdb.2008.06.005
19. Corcoran RB, Contino G, Deshpande V, Tzatsos A, Conrad C, Benes CH, et al. STAT3 Plays a Critical Role in KRAS-induced Pancreatic Tumorigenesis. Cancer Res (2011) 71:5020–9. doi: 10.1158/0008-5472.CAN-11-0908
20. Keller P, Keller C, Carey AL, Jauffred S, Fischer CP, Steensberg A, et al. Interleukin-6 Production by Contracting Human Skeletal Muscle: Autocrine Regulation by IL-6. Biochem Biophys Res Commun (2003) 310:550–4. doi: 10.1016/j.bbrc.2003.09.048
21. Lotze MT, Tracey KJ. High-Mobility Group Box 1 Protein (HMGB1): Nuclear Weapon in the Immune Arsenal. Nat Rev Immunol (2005) 5:331–42. doi: 10.1038/nri1594
22. Müller S, Scaffidi P, Degryse B, Bonaldi T, Ronfani L, Agresti A, et al. New EMBO Members’ Review: The Double Life of HMGB1 Chromatin Protein: Architectural Factor and Extracellular Signal. EMBO J (2001) 20:4337–40. doi: 10.1093/emboj/20.16.4337
23. Hou CH, Fong YC, Tang CH. HMGB-1 Induces IL-6 Production in Human Synovial Fibroblasts Through C-Src, Akt and NF-κB Pathways. J Cell Physiol (2011) 226:2006–15. doi: 10.1002/jcp.22541
24. Rapoport BL, Steel HC, Theron AJ, Heyman L, Smit T, Ramdas Y, et al. High Mobility Group Box 1 in Human Cancer. Cells (2020) 9(7):1664. doi: 10.3390/cells9071664
25. Vijayakumar EC, Bhatt LK, Prabhavalkar KS. High Mobility Group Box-1 (HMGB1): A Potential Target in Therapeutics. Curr Drug Targets (2019) 20:1474–85. doi: 10.2174/1389450120666190618125100
26. Chen G, Shi Y, Zhang Y, Sun J. CircRNA_100782 Regulates Pancreatic Carcinoma Proliferation Through the IL6-STAT3 Pathway. Onco Targets Ther (2017) 10:5783–94. doi: 10.2147/OTT.S150678
27. Kang R, Chen R, Zhang Q, Hou W, Wu S, Cao L, et al. HMGB1 in Health and Disease. Mol Aspects Med (2014) 40:1–116. doi: 10.1016/j.mam.2014.05.001
28. Yang J, Shah R, Robling AG, Templeton E, Yang H, Tracey KJ, et al. HMGB1 is a Bone-Active Cytokine. J Cell Physiol (2008) 214:730–9. doi: 10.1002/jcp.21268
29. Bald T, Quast T, Landsberg J, Rogava M, Glodde N, Lopez-Ramos D, et al. Ultraviolet-Radiation-Induced Inflammation Promotes Angiotropism and Metastasis in Melanoma. Nature (2014) 507:109–13. doi: 10.1038/nature13111
30. Sims GP, Rowe DC, Rietdijk ST, Herbst R, Coyle AJ. HMGB1 and RAGE in Inflammation and Cancer. Annu Rev Immunol (2010) 28:367–88. doi: 10.1146/annurev.immunol.021908.132603
31. Da CM, Gong CY, Nan W, Zhou KS, Wu ZL, Zhang HH. The Role of Long non-Coding RNA MIAT in Cancers. Biomed Pharmacother (2020) 129:110359. doi: 10.1016/j.biopha.2020.110359
32. Renganathan A, Felley-Bosco E. Long non-coding Rnas in Cancer and Therapeutic Potential. Adv Exp Med Biol (2017) 1008:199–222. doi: 10.1007/978-981-10-5203-3_7
33. Li J, Tian H, Yang J, Gong Z. Long non-coding RNAs Regulate Cell Growth, Proliferation, and Apoptosis. DNA Cell Biol (2016b) 35:459–70. doi: 10.1089/dna.2015.3187
34. Weidle UH, Birzele F, Kollmorgen G, Rüger R. Long non-coding RNAs and Their Role in Metastasis. Cancer Genomics Proteomics (2017) 14:143–60. doi: 10.21873/cgp.20027
35. Ishii N, Ozaki K, Sato H, Mizuno H, Susumu S, Takahashi A, et al. Identification of a Novel non-Coding RNA, MIAT, That Confers Risk of Myocardial Infarction. J Hum Genet (2006) 51:1087–99. doi: 10.1007/s10038-006-0070-9
36. Venables JP, Klinck R, Koh C, Gervais-Bird J, Bramard A, Inkel L, et al. Cancer-Associated Regulation of Alternative Splicing. Nat Struct Mol Biol (2009) 16:670–6. doi: 10.1038/nsmb.1608
37. Zhu X, Cong J, Lin Z, Sun J, Yang B, Li A. Inhibition of HMGB1 Overcomes Resistance to Radiation and Chemotherapy in Nasopharyngeal Carcinoma. Onco Targets Ther (2020) 13:4189–99. doi: 10.2147/OTT.S239243
38. Pan S, Wang J, Wu A, Guo Z, Wang Z, Zheng L, et al. Radiation Exposure-Induced Changes in the Immune Cells and Immune Factors of Mice With or Without Primary Lung Tumor. Dose Response (2020) 18:1559325820926744. doi: 10.1177/1559325820926744
39. Bhat AA, Nisar S, Maacha S, Carneiro-Lobo TC, Akhtar S, Siveen KS, et al. Cytokine-Chemokine Network Driven Metastasis in Esophageal Cancer; Promising Avenue for Targeted Therapy. Mol Cancer (2021) 20:2. doi: 10.1186/s12943-020-01294-3
40. Kwak MS, Lim M, Lee YJ, Lee HS, Kim YH, Youn JH, et al. HMGB1 Binds to Lipoteichoic Acid and Enhances TNF-α and IL-6 Production Through HMGB1-Mediated Transfer of Lipoteichoic Acid to CD14 and TLR2. J Innate Immun (2015) 7:405–16. doi: 10.1159/000369972
41. Hirano H, Yoshioka T, Yunoue S, Fujio S, Yonezawa H, Niiro T, et al. TLR4, IL-6, IL-18, MyD88 and HMGB1 are Highly Expressed in Intracranial Inflammatory Lesions and the IgG4/IgG Ratio Correlates With TLR4 and IL-6. Neuropathology (2012) 32:628–37. doi: 10.1111/j.1440-1789.2012.01310.x
42. Li J, Wang FP, She WM, Yang CQ, Li L, Tu CT, et al. Enhanced High-Mobility Group Box 1 (HMGB1) Modulates Regulatory T Cells (Treg)/T Helper 17 (Th17) Balance Via Toll-Like Receptor (TLR)-4-interleukin (IL)-6 Pathway in Patients With Chronic Hepatitis B. J Viral Hepat (2014) 21:129–40. doi: 10.1111/jvh.12152
43. Sanchez Calle A, Kawamura Y, Yamamoto Y, Takeshita F, Ochiya T. Emerging Roles of Long non-Coding RNA in Cancer. Cancer Sci (2018) 109:2093–100. doi: 10.1111/cas.13642
44. Noh JH, Kim KM, McClusky WG, Abdelmohsen K, Gorospe M. Cytoplasmic Functions of Long non-Coding RNAs. Wiley Interdiscip Rev RNA (2018) 9:e1471. doi: 10.1002/wrna.1471
45. Mercer TR, Mattick JS. Structure and Function of Long Non-Coding RNAs in Epigenetic Regulation. Nat Struct Mol Biol (2013) 20:300–7. doi: 10.1038/nsmb.2480
46. Rapicavoli NA, Poth EM, Blackshaw S. The Long non-Coding RNA RNCR2 Directs Mouse Retinal Cell Specification. BMC Dev Biol (2010) 10:49. doi: 10.1186/1471-213X-10-49
47. Goodwin GH, Johns EW. The Isolation and Purification of the High Mobility Group (HMG) Nonhistone Chromosomal Proteins. Methods Cell Biol (1977) 16:257–67. doi: 10.1016/S0091-679X(08)60104-1
Keywords: cisplatin resistance, HMGB1, IL6, NPC, lncRNA MIAT
Citation: Zhu X, Liu L, Wang Y, Cong J, Lin Z, Wang Y, Liu Q, Wang L, Yang B and Li T (2021) lncRNA MIAT/HMGB1 Axis Is Involved in Cisplatin Resistance via Regulating IL6-Mediated Activation of the JAK2/STAT3 Pathway in Nasopharyngeal Carcinoma. Front. Oncol. 11:651693. doi: 10.3389/fonc.2021.651693
Received: 26 January 2021; Accepted: 06 April 2021;
Published: 20 May 2021.
Edited by:
Wen Zhou, Central South University, ChinaReviewed by:
Qianjin Liao, Central South University, ChinaCopyright © 2021 Zhu, Liu, Wang, Cong, Lin, Wang, Liu, Wang, Yang and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ben Yang, eWFuZ2JlbkBqbHUuZWR1LmNu
Tao Li, bHQ2MDY0MDNAamx1LmVkdS5jbg==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.