 Ilenia Sana
Ilenia Sana Maria Elena Mantione
Maria Elena Mantione Piera Angelillo1,2
Piera Angelillo1,2 Marta Muzio
Marta Muzio
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oncol. , 01 April 2021
Sec. Hematologic Malignancies
Volume 11 - 2021 | https://doi.org/10.3389/fonc.2021.651057
This article is part of the Research Topic Role of the Antigen Receptor in the Pathogenesis of B-Cell Lymphoid Malignancies View all 10 articles
In recent years significant progress has been made in the clinical management of chronic lymphocytic leukemia (CLL) as well as other B-cell malignancies; targeting proximal B-cell receptor signaling molecules such as Bruton Tyrosine Kinase (BTK) and Phosphoinositide 3-kinase (PI3Kδ) has emerged as a successful treatment strategy. Unfortunately, a proportion of patients are still not cured with available therapeutic options, thus efforts devoted to studying and identifying new potential druggable targets are warranted. B-cell receptor stimulation triggers a complex cascade of signaling events that eventually drives the activation of downstream transcription factors including Nuclear Factor of Activated T cells (NFAT). In this review, we summarize the literature on the expression and function of NFAT family members in CLL where NFAT is not only overexpressed but also constitutively activated; NFAT controls B-cell anergy and targeting this molecule using specific inhibitors impacts on CLL cell viability. Next, we extend our analysis on other mature B-cell lymphomas where a distinct pattern of expression and activation of NFAT is reported. We discuss the therapeutic potential of strategies aimed at targeting NFAT in B-cell malignancies not overlooking the fact that NFAT may play additional roles regulating the inflammatory microenvironment.
In recent years there have been significant improvements in the field of chronic lymphocytic leukemia (CLL) from both bench and bedside perspectives. CLL cells are addicted to different microenvironmental stimuli with a key role being played by the B-cell receptor (BCR) stimulation and/or constitutive cell autonomous BCR activation (1) leading to cell survival and proliferation. On this scientific basis, several lines of research led to the development of small molecule inhibitors of the kinases that transmit the signals from the proximal BCR signaling complex to the downstream Transcription Factors (TFs) [i.e. Bruton Tyrosine Kinase (BTK) and Phosphoinositide 3-kinases delta (PI3Kδ)] (2, 3). CLL is a disease of the elderly, but it can also affect younger patients, who often are the most difficult to treat given the long disease history and the frequent need for multiple lines of treatment. To date, the only curative approach for CLL patients, unfortunately, is allogeneic bone marrow transplantation, a procedure burdened by a high rate of morbidity and mortality especially in older patients. Nowadays, one of the most powerful available tools for the treatment of CLL, is a novel small molecule that inhibits BTK, Ibrutinib. The drug was approved by the US Food and Drug Administration in 2014 for the treatment of relapsed refractory CLL and for CLL patients with the 17p deletion/mutation, known to frequently be chemo refractory. Ibrutinib was granted approval for first-line treatment of CLL in March 2016; since then, data from real world practice consistently shows a significant improvement in survival curves, in keeping with what investigators previously observed in clinical trials (4, 5). BTK is not the only target of novel non chemotherapeutic agents, as both BTK and PI3Kδ inhibitors are highly effective even for the treatment of refractory or relapsed disease. These drugs are designed to be administered until relapse/progression or unacceptable toxicity. Patients are therefore kept under follow-up during the administration for early detection of signs of clinical progression or toxicity. Several factors have been proposed as predictive markers for the emergence of resistance (i.e. prolonged lymphocytosis) (6, 7), which is currently an unmet clinical need. Novel therapeutic strategies are needed to cure refractory patients and to perhaps achieve deeper response with the intent of fully eradicating the disease.
In this context, we hypothesize that exploring other downstream signaling mediators including transcription factors may reveal novel vulnerabilities of malignant B cells, which could be of aid in treating CLL and other B cell malignancies. Given that resistance to targeted agents often occurs by mutation of the target kinase (8, 9), it is reasonable to hypothesize that blocking downstream signaling molecules could be a strategy to block the transmission of the survival signal to the nucleus. One of the transcription factors involved in B-cell antigen receptor signaling is NFAT, Nuclear factor of activated T-lymphocytes [others being Nuclear Factor kappa-light-chain-enhancer of activated B cells (NFκB), cMyc and activator protein 1 (AP1)]. Here, we briefly describe the biology of NFAT followed by a discussion on the expression pattern and functional role of NFAT transcription factors in CLL and other lymphoid malignancies.
The NFAT family of TFs includes five members grouped by the presence of the REL homology region (RHR), a highly conserved DNA binding domain that confers a unique DNA binding specificity to these proteins (see Figure 1 for a schematic representation). Several alternative names exist for each NFAT member, and they are all reported in Table 1 together with essential information on each gene. Herein, we refer to the NFAT family members with their official names; NFAT1, NFAT2, NFAT3, NFAT4, and NFAT5.
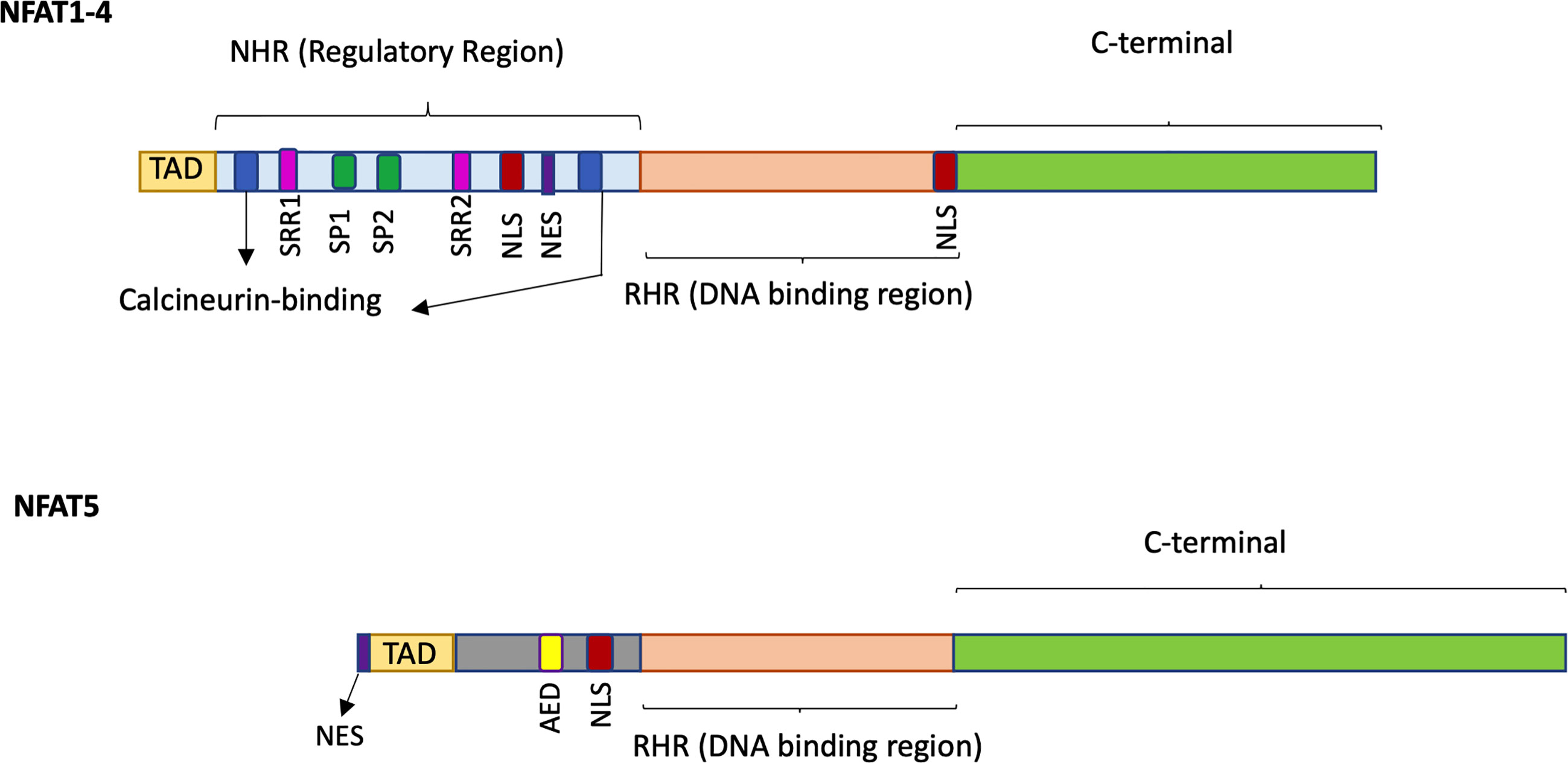
Figure 1 Schematic representation of the structure of NFAT1-4 and NFAT5. NFAT proteins contain a REL homology region (RHR), the most conserved domain, which binds to DNA and is common to all five NFAT family members. The NFAT homology region (NHR) is the regulatory region conserved in NFAT1-4 members but not in NFAT5 and contains two calcineurin-binding sites, serine residues (SSR, SP), a Nuclear Localization Sequence (NLS) and a Nuclear Export Signal (NES). NFAT5 does not display the calcineurin binding site but owns an auxiliary export domain (AED) and a NLS and possess a nuclear export sequence (NES) located at the first 19 amino acids. All NFAT family members have a N-terminal Transactivation Domain (TAD) and a C-terminal domain which are the less conserved regions.
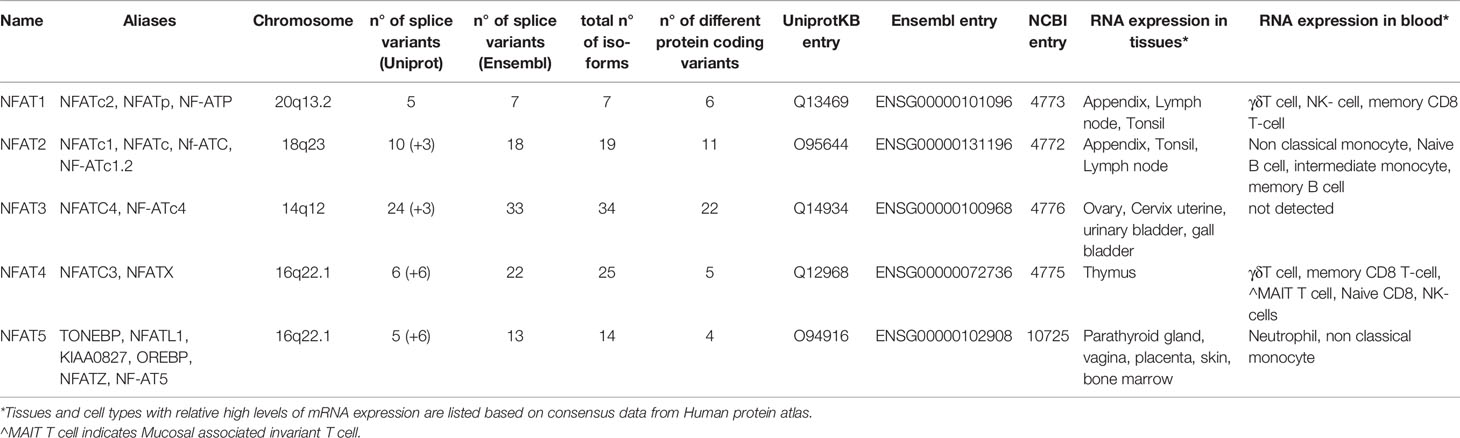
Table 1 Summary of key features of human NFAT genes, mRNAs and proteins.
Four NFAT proteins share sequence homology in the N-term regulatory region (NHR) responsible for the modulation by Calcium signaling, while NFAT5 is induced by osmotic stress (10). Briefly, calcium-dependent NFATs are normally retained in an inactive state into the cytoplasm of the cells by different kinases that phosphorylate the NHR domain (11). The stimulation of receptors such as the BCR in B-cells and the T-cell receptor (TCR) in T-cells (12) generates a cascade that induces calcium mobilization through the activation of phospholipase C (PLCγ) that hydrolyzes phosphatidylinositol-3,4-bisphosphate (PIP2) leading to the release of diacylglycerol (DAG) and inositol-1,4,5-trisphosphate (IP3). IP3 binds to the IP3 receptor and causes the release of Ca2+ from the endoplasmic reticulum and the consequent extracellular influx of Ca2+ in the cytosol by specific calcium channels (13, 14) (see Figure 2 for a schematic representation). The presence of Ca2+ ions cause the binding of calmodulin (CaM) to the calcineurin phosphatase, leading to the dephosphorylation and activation of NFAT serine residues in the regulatory domain. Nuclear importing factors then mediate NFAT translocation into the nucleus where it binds the DNA alone or with other factors such as AP1, Stat3, GATA, c-Fos, c-Jun, and NFκB, to regulate gene expression either activating or silencing target genes; most of which are immune-related (15, 16). NFAT inactivation and the following relocation into the cytoplasm is operated by several kinases including glycogen-synthase kinase 3β [please refer to the following recent reviews for molecular details on NFAT proteins (16–19)].
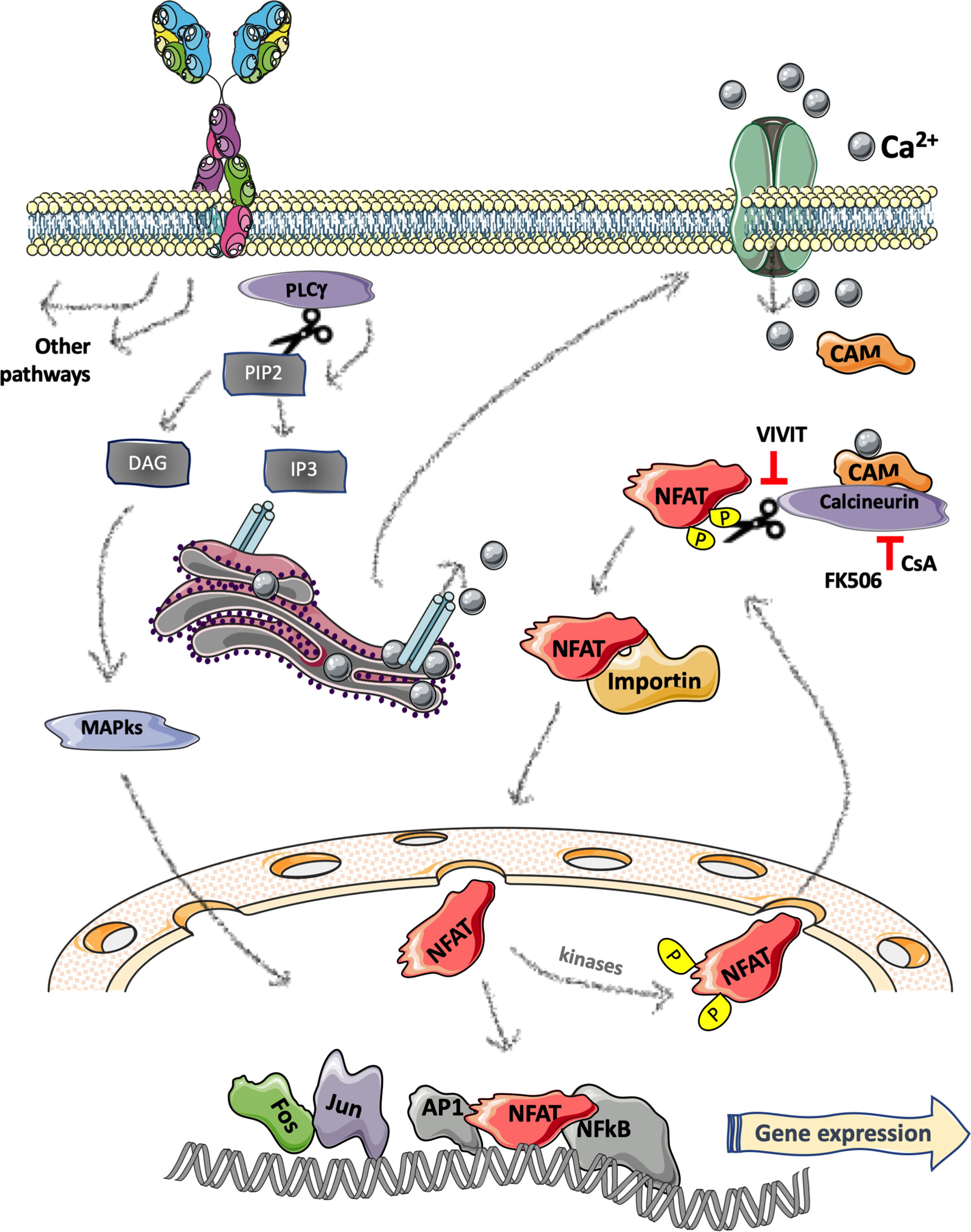
Figure 2 Schematic representation of B-cell receptor induced NFAT pathway. Stimulation of the B-cell receptor (BCR) activates a cascade resulting in phospholipase C (PLCγ) activation and hydrolyzation of phosphatidylinositol-3,4-bisphosphate (PIP2) generating the release of two second messengers: diacylglycerol (DAG) and inositol-1,4,5-trisphosphate (IP3). IP3 binds to the IP3 receptor located on the endoplasmic reticulum leading to release of Ca2+ from the ER and an extracellular influx of Ca2+ in the cytosol that causes the binding of calmodulin (CaM) to the calcineurin phosphatase and the consequent dephosphorylation and activation of NFAT. The nuclear translocation of NFAT is mediated by nuclear importing factors (e.g., importin). In the nucleus, NFAT binds to the DNA either alone or with other factors regulating gene expression. Figure 2 was created using Servier Medical Art (https://smart.servier.com).
The molecular activation of NFAT5 is more complex and still partially defined. Under isotonic conditions, there is a continuous shuttling of NFAT5 between the cytoplasm and the nucleus that can be regulated by tonicity stress. While hypotonic stress promotes nuclear export of the protein, hypertonic conditions induce transcription, translation and nuclear import of NFAT5 (20, 21). With its well-known tonicity-related regulatory role, it is emerging that several isotonic stimuli can promote NFAT5 activity; for instance, triggering of innate immunity receptors such as Toll-like receptors and consequent activation of reactive oxygen species (ROS) and mitogen activated protein kinases (MAPK) results in NFAT5 activation that, interestingly, shows distinctive features with respect to the osmotic activated response (22). However, the interplay between these two mechanisms of activation and the exact molecular pathways involved are still elusive and yet to be fully uncovered.
To dissect the role of these TFs, several studies analyzed the phenotype of specific NFAT-deficient mice as well as combinations of two or more genetic deletions. For a comprehensive description of the murine lines please refer to online resources (e.g., the International Mouse Phenotyping Consortium: https://www.mousephenotype.org; MGI-Mouse Genome Informatics-: http://www.informatics.jax.org/). Herein, we focus on the results related to lymphocytes and the B-cell lineage as this may help in understanding and interpreting the specific role of individual NFAT proteins in B-cell malignancies.
With the notable exception of NFAT3, for which no specific studies analyzed the B-cell compartment, deletion of individual NFAT or double inactivation of NFAT genes in vivo revealed the importance of this TF family in the regulation of lymphocyte differentiation, proliferation, apoptosis, cytokine production, and inflammation. The vast majority of the immunological studies, as well as the B cell-centered ones, focused on NFAT1 and NFAT2 that regulate different B-cell populations in diverse ways (23). Between the two, NFAT2 deficiency has a more severe impact; indeed, the global loss of NFAT2 determines prenatal lethality around day 14/15 upon gestation (24, 25). However, chimeric mice with lymphocyte-restrained NFAT2 loss showed defects in BCR-mediated proliferation of B cells (26), and a peculiar deficiency in peritoneal CD5+ B1a B-cells (27). Specifically, the defect in BCR-induced proliferation is determined by NFAT2-dependent expression of CD22, Rcan1, Tnfsf14, FasL and other key proteins of the BCR signaling pathway. In addition, the abrogation of NFAT2-mediated calcium flux response facilitates activation-induced cell death (AICD), which leads to the loss of CD5+ peritoneal B1a cells. Moreover, the lack of NFAT2-mediated repression of IL-10 production, impacts on IFN-γ production by CD4+ T-cells, impairing the capacity of B-cells to stimulate T cell proliferation (28). Mice lacking NFAT2 in pro-B cells have deficient expression of an essential TF determining B-cell lineage fate, EBF1, and a similar phenotype of EBF1-deficient mice (29) with defective Immunoglobulin (Ig) gene rearrangement, and pre-BCR formation which impairs B cell development and leads to severe B-cell lymphopenia (30). The early developmental role of NFAT2 in peripheral B-cells was endorsed by a recent study that characterized lymphocyte dissemination in mice bearing deletion of NFAT2 in CD19-positive cells. The authors confirmed NFAT2-dependent deficiency in peritoneal CD5+ B1a B-cells that was accompanied by increased immature and mature follicular B cell populations (31). At later stages, NFAT2 loss of function causes functional defects only of mature B cells that promote mild clinical course of experimental autoimmune encephalomyelitis (28).
NFAT1-null mice have a normal development and a less severe phenotype. Nevertheless, after 6 months a proportion of the litters showed alterations of the immune system such as lymph node hyperplasia and splenomegaly accompanied by enlarged germinal centers and pronounced retardation in the involution of the thymus. NFAT1 deficient mice also displayed a hyperproliferative syndrome, higher B and T cell counts, dysregulated production of IL-4, and higher primary and secondary immune responses (32–34). Notably, NFAT1 plays a pivotal role in regulating the response of B-cells to self-antigen, balancing the processes of anergy and self-tolerance (35). Moreover, NFAT1 in B cells, controls and represses the expression of Cyclin E1 and E2, taking control of cell cycle progression and proliferation rates (36).
NFAT4 shows a more restricted role in regulating T cell activity; however, double deficient mice for NFAT1 and NFAT4 experience lymphadenopathy, splenomegaly, and a strongly activated phenotype with a substantial increase in serum IgE and IgG1 levels, similar to single knock-out mice (37). One of the origins of the lymphadenopathy was attributed to the observed resistance to apoptosis, due to decreased FasL expression and defective AICD induction (38). Moreover, the absence of both NFAT1 and NFAT4 drives naive CD4 T cells into Th2 cell differentiation even in the absence of endogenous IL-4, and boosts their responsiveness to TCR-mediated activation and secretion of Th2-type lymphokines (39). The elevated Th2 cytokine production also leads to hyperactivation of mature follicular B cells but not of marginal zone (MZ) B cells. This evidence indirectly links the loss of both NFAT1 and NFAT4 to the altered B cell phenotype of these mice, which have a lower representation of MZ B cells and a higher number of mature follicular B cells (40).
Focusing on NFAT5, the tonicity-responsive member of the family, its complete loss of function results in gestational lethality. Heterozygous animals show a phenotype marked by lymphoid hypocellularity, with thymus and spleen hypoplasia, defective antigen-specific antibody responses (in particular IgG secretion) and less mature CD4 and CD8 cells in the spleen and lymph nodes (41). These indications highlight the role for NFAT5-mediated adaptation to physiologic osmotic stress for lymphocyte-mediated immunity, with a putative B-cell centered role on T-cell dependent Ig response and proliferation, specifically under hypertonic conditions (42). Interestingly, NFAT5 showed a tonicity-independent role in the development and activation of macrophages where NFAT5 accumulation and the following increased expression of target genes such as TNF and IL6 can be mediated by Toll-like receptors and NFκB pathway activation (43). NFκB-mediated expression of NFAT5 also has a crucial role in pre–T-cell receptor thymocytes where it regulates the expression of the prosurvival factors A1 and Bcl2, and attenuates the proapoptotic p53/Noxa axis (44).
From all these observations it emerges that not only are NFAT1, NFAT2, and NFAT4 involved in the regulation and homeostasis of B-cells and BCR signaling, but NFAT5 also plays a crucial role which has yet to be fully characterized. On the contrary, no functional data are available for NFAT3. Nonetheless, both NFAT1 and NFAT2 are expressed by distinct B-cell malignancies as described in detail below, while less information is available on NFAT3, NFAT4, and NFAT5.
The category of non-Hodgkin lymphomas (NHL) comprises a large spectrum of entities ranging from indolent to highly aggressive diseases (45). Pathogenesis of most NHLs is unknown. For some subtypes, a chronic immune stimulation role has been suggested, thus, gaining insight on the multistep mechanism that leads to malignant transformation is key for the development of new treatments.
NFAT has neither been highlighted as a prominent B-cell lymphoma-associated molecule, nor a frequently mutated gene; however, distinct studies reported specific molecular and functional features of NFAT family members that may open up interesting novel therapeutic perspectives.
First, NFAT2 could be detected by IHC in lymphoid cells in routine biopsies of several hematologic malignancies, while nuclear NFAT2 was observed in a proportion of Burkitt and diffuse large B cell lymphoma (DLBCL) samples, suggesting an ongoing activation of the pathway in this type of lymphoma (46). In contrast, NFAT2, but not NFAT1, is downregulated by promoter methylation in Hodgkin’s lymphoma cells (47). To note, the pattern of expression of NFAT does not fully reflect its role, as the activity of this TF is regulated by different mechanisms that eventually control the shuttling between the nucleus and the cytoplasm.
By using patients’ derived cells and cell lines, Pham et al. not only reported constitutive activation of NFAT2 in large B-cell lymphoma, but also demonstrated that NFAT2 and NFκB cooperate to drive CD40L expression, which in turn triggers pro-survival signaling. NFAT siRNA inhibitors as well as drugs targeting NFAT activation blocked CD40L expression and induced apoptosis (48). The same group also demonstrated that both large B-cell lymphoma and mantle cell lymphoma constitutively express NFAT1 and NFAT2 that control BLyS expression and survival signaling again in cooperation with NFκB (49). Another interesting target of NFAT2 is c-Myc that can be transcriptionally upregulated by NFAT2 through an epigenetic chromatin remodeling in DLBCL (50).
On the contrary, a tumor suppressive role of NFAT1 was proposed in DLBCL where its downregulation correlated with increased cyclin E expression; specifically, NFAT1 directly controlled cyclin E induction by binding to its promoter in lymphoma cell lines (36). Moreover, NFAT1 was implicated in mediating BCR-induced cell death in Hodgkin lymphoma cells, a phenomenon observed in selected cell lines; both Cyclosporin and FK506 inhibited apoptotic signaling, further supporting the functional involvement of NFAT (51).
Regarding the mechanisms responsible for NFAT overexpression and activation in lymphoma patients, while most of the studies suggest transcriptional and/or post-transcriptional mechanisms of regulation of NFAT in B-cell lymphoma, there is also evidence of NFAT gene amplification in a proportion of DLBCL of the ABC type (52). An increase of hexosamine biosynthetic pathway and O-GlcNAc metabolism plays a critical role in DLBCL cell proliferation, and is responsible for the observed NFκB and NFAT activation (53). Constitutive ongoing BCR signaling may also explain constitutive NFAT2 activation in DLBCL (54). However, a recent report suggested a BCR-independent, Calcium-dependent pathway towards NFAT2 activation in DLBCL (55, 56).
Finally, in addition to protein phosphorylation and splicing, distinct post-translational modifications modulate its activity including acetylation, SUMOylation and cleavage (57). However, no specific data on these mechanisms operating in B-cell malignancies are available.
CLL involves mature clonal B-cells that accumulate in the peripheral blood and lymphoid organs where they receive supportive signals form the microenvironment. The BCR, the key protein for every B-lymphocyte, not surprisingly modulates CLL cells biology as well; several signaling molecules downstream of the BCR such as kinases and TFs are involved, and they have been recently targeted for therapeutic purposes (58, 59). Among the most relevant BCR-mediated TFs, we focus on NFAT, not overlooking the fact that NFκB as well as other TFs play a relevant role and may complement the activity of NFAT itself.
In 1996, Schuh et al. demonstrated for the first time that in contrast to normal B-cell, malignant cells isolated from the peripheral blood of patients with CLL show nuclear/active NFAT1 even in the absence of in vitro stimulation; in parallel, NFκB and AP1 activation were also observed (60). CLL cells show higher mRNA levels of expression of both NFAT1 and NFAT2 as compared to normal lymphocytes (61); hypomethylation of the NFAT2 promoter region as well the first intron region may explain higher levels of both mRNA and protein in CLL as compared to normal B-cells types (62). A comprehensive study on the epigenome and regulatory chromatic landscape of CLL highlighted that active chromatin regions were enriched for binding motifs of NFAT (as well as FOX and TCF/LEF transcription families) (63). When different groups of CLL patients with different clinic-biological characteristics were analyzed, NFAT promoter hypomethylation correlated with clinical staging (64). More recently, looking for markers of a specific subset of CLL, bearing trisomy of chromosome 12, Abruzzo at el. discovered that NFAT1, NFAT2, and NFAT4 mRNAs are significantly overexpressed (65).
Based on all these somewhat descriptive analyses, several authors suggested that BCR-mediated NFAT2 overexpression may be implicated in CLL pathobiology and may potentially be targeted for therapeutic purposes. Along this line of reasoning, several questions emerged: Which are the target genes of NFAT transcription factors in leukemic cells? Which are the functional consequences of NFAT hyperactivation in CLL? Which are the molecular mechanisms regulating NFAT activation in malignant cells?
No specific NFAT ChIP-seq analysis was performed in CLL cells, thus hampering a broad view of all its target genes; nevertheless, independent studies demonstrated that distinct genes that are typically expressed by leukemic cells are directly regulated by NFAT family members in different cell types.
CD23 is a receptor for FcE, and a distinctive molecule expressed on the surface of CLL cells as well as released in the serum (66). Two different isoforms regulated by two different promoters exist, namely CD23a and CD23b. The CD23b promoter is specifically regulated by NFAT1 and NFAT2 in concert with STAT6 (67); in contrast, CD23a expression is regulated by Notch2 (68). CD23 expression can also be upregulated by BCR stimulation in CLL cells where blocking NFAT prevents CD23 induction (61).
CD5 expression is regulated by NFAT in normal B-cell populations (27, 69, 70); however, it is not known if the same occurs in CLL where it is distinctively expressed on the cell surface.
LCK was recently identified as a direct NFAT2 target gene in both human and mouse CLL samples (71). CD5 mediated IL10 production is regulated by NFAT in CLL cells; by cooperating with STAT3, NFAT2 binds to IL5 and IL13 promoters and the IL10 enhancer to upregulate their expression (72).
NFAT2 is constitutively active in approximately half of CLL cases, the same than that characterized by concomitant MAPK phosphorylation and anergy in terms of response to the BCR. Despite constitutive basal levels of NFAT2 activation, it can be further induced after BCR stimulation, at least in a group of responding cases that are characterized by an adverse clinical outcome (61). Not only is NFAT overexpressed and activated in CLL, but NFAT binding sites are hypomethylated in leukemic samples suggesting the overactivation of target genes that may be related to autoreactive BCR (73).
To assess the functional role of NFAT during disease development and/progression, different approaches were used including ablation of NFAT2 in a mouse model of leukemia, and the use of drugs targeting NFAT activation. The two approaches address different questions whilst also being complementary; while genetic inactivation results in complete inhibition of a single molecule in vivo over time, drug treatment suddenly interrupts different NFAT family members as well as any additional off targets. Here, we focus on genetic approaches while describing potential drug targeting in a dedicated paragraph below for both CLL and lymphoma.
Overexpression of the TCL1 oncogene in the B-cell lineage results in the development of a malignant disease resembling human CLL, as characterized by the accumulation of CD5-positive clonal B-cells in the peripheral blood and lymphoid organs (74); this is a widely used and accepted mouse model of leukemia. Leukemic cells of this model show constitutive activation of NFAT2 and somewhat anergic features (71), as previously shown for a group of CLL cases (75). Genetic inactivation of NFAT2 in the B-cell types of these mice led to rapid acceleration of leukemia development and progression toward and aggressive disease resembling Richter transformation, occurring in a small proportion of CLL patients (71). The authors suggest that NFAT2 is a key regulator of anergy in CLL. To better understand if NFAT deletion impacted directly on different BCR downstream signaling or rather on clonal selection of different BCR recombination, Muller et al. analyzed clonal evolution in leukemic mice and found that NFAT2 signaling in CLL cells precipitates the oligoclonal selection of preferentially unmutated BCRs (76).
Overall, these data suggest that NFAT2, being implicated in the maintenance of anergy, may restrict leukemia development; however, at the same time, anergic signaling provides a survival advantage to the cells. It is important to evaluate the effect of NFAT2 inhibition after leukemia development, as data from primary patient samples suggest that it may have therapeutic activity by interrupting anergy (77).
Given the central role of NFAT in regulating the adaptive immune response, it has been thoroughly scrutinized as a “specific” drug target to achieve immunosuppression in the context of organ transplantation, or to dampen excessive autoimmune manifestations. The most widely used drugs targeting the NFAT pathways are Cyclosporine A (CsA) and Tacrolimus, both originally isolated from fungi, acting with slightly different mechanisms of action. Briefly, as schematically reported in Figure 2, they inhibit the activity of the phosphatase Calcineurin, thus augmenting the phosphorylation state of all its substrates including NFAT1-4 family members; to do so, CsA binds to Cyclophilin while FK-506 (an alternative name of Tacrolimus) binds to FKBP12 (78). Both drugs are widely used to prevent graft-rejection or to treat autoimmune diseases; however, several years ago, anecdotical reports suggested its potential positive effect in the context of CLL (79, 80).
Nowadays, accumulating preclinical data from many different labs suggest that targeting NFAT may represent a novel therapeutic approach to treat at least a subset of NFAT-positive B-cell malignancies. In particular, CsA, FK-506 as well as a short cell-permeable NFAT-specific inhibitory peptide (called VIVIT as according to its core aminoacidic sequence) have been tested in vitro and in vivo in mouse models of leukemia and lymphoma (81, 82). To note, VIVIT peptide directly binds to the NFAT-docking portion of Calcineurin, thus being more selective towards these transcription factors as compared to CsA.
Targeting CLL cells with the VIVIT peptide blocks BCR-mediated NFAT2 activation and target genes in responsive cells (61, 71); at the same time, VIVIT blocks constitutive NFAT activation in CLL, thus rescuing leukemic cells from anergy (77). More importantly, targeting spontaneous NFAT activation with VIVIT not only blocked related biochemical pathways but also induced cell death in vitro and delayed leukemia progression in vivo in mouse models (77). These observations suggest that NFAT controls survival signals in anergic cells, though, at the same time keeps the cells in an indolent state. In fact, as mentioned above, deleting the whole NFAT2 gene from cells before leukemia development, triggers faster accumulation of the disease and progression (71).
Cyclosporin and FK506 inhibited NFAT signaling leading to the abrogation of pro-survival effects present in CLL cells, even in the presence of supportive signaling given by stroma cells (64). Interestingly, the use of the BTK signaling inhibitor, a clinically approved drug targeting upstream BCR signaling, abrogated NFAT activity in leukemic cells suggesting that downstream NFAT activation may represent a novel therapeutic target in the cases where resistance to Ibrutinib arise due to mutations in upstream molecules of the signaling cascade (64).
Targeting the upstream BTK kinase with different inhibitors, including Ibrutinib, blocks NFAT2 together with its targets including IL10; to note, IL10 can control PDL1 expression preferentially in the ABC subset of DLBCL thus suggesting that targeting NFAT may impact on the expression of this molecule, therefore being relevant for anti-tumor immunity (54). Along this line, a recent paper confirmed chronic NFAT activation in ABC-DLBCL controlling IL10 production and demonstrated the efficacy of calcineurin inhibitors in blocking NFAT signaling and reducing proliferation. However, the authors proposed a BCR-independent mechanism of NFAT activation, yet a BCR-dependent expression of NFAT protein that is dependent on NFκB signaling. Interestingly, blocking calcineurin synergized with BCL2 and MCL1 inhibitors to kill lymphoma cells (55).
Based on all these observations, using CsA, FK-506, or novel formulations of VIVT appear to be a promising therapeutic perspective. However, the concomitant immunosuppressive activity of these drugs may potentially counterbalance the direct anti-tumor activity, and it should be carefully considered for any putative future clinical approach (83). Along this line, it will be crucial to design and engineer novel drugs targeting NFAT in the malignant cells only. Finally, several additional drugs targeting NFAT are emerging, and it will be important to test them either alone or in combination with signaling inhibitors using advanced preclinical models and primary tumor samples.
The B-cell receptor is the key molecule regulating the pathobiology of both normal and malignant B-lymphocytes; accordingly, targeting the kinases proximal to the BCR (such as BTK and PI3Kδ), has emerged as a successful treatment strategy. Nevertheless, BCR targeting is in some cases not sufficient to achieve disease control and, eventually, tumor cells find alternative mechanisms to survive and proliferate. For this reason, efforts devoted to studying and identifying additional new potential druggable targets along the BCR signaling cascade are warranted. In this review, we explored the biology of a key family of transcription factors that are activated after BCR stimulation, namely NFATs.
First, to obtain insight into the intrinsic role of different NFATs in the B-cell context, we reported an overview of genetically modified mice where a dual role of NFAT1 and NFAT2 emerged, regulating both cell proliferation and cell death after BCR stimulation. Next, we detailed the expression, activation status, and functional role of different NFAT family members in CLL and other B-cell malignancies. NFAT1 and NFAT2 were described to be not only overexpressed but also functionally implicated in the regulation of malignant B-cell biology. In contrast, no information was available on NFAT3 and NFAT4 in this context, while a recent paper demonstrated that NFAT5 is overexpressed in CLL where it facilitates malignant cells survival and activation (84); yet, NFAT5 activation is not dependent on BCR but it is regulated by osmotic stress and inflammatory stimuli. Overall, we described a cell autonomous function of BCR-related NFAT activation in leukemia and lymphoma cells. Targeting this molecule using a specific inhibitor was shown to be beneficial in treating CLL and lymphoma in preclinical models; however, NFAT deletion in mouse models broke anergy with a paradoxical induction of leukemia progression in vivo (85).
It has been known for years now that the NFAT family of transcription factors are involved in several processes which are central for immune system function, inflammation, and the development of both autoimmune and neoplastic diseases (86). This is particularly important in light of the role that the tumor microenvironment has been recognized to have for tumor survival and progression. NFAT may exert additional effects in different cell types, including stroma cells, by regulating inflammation and inflammation-associated cancer, as previously reported by other reviews on this topic (86, 87). To note, inflammation is a hallmark of CLL as well as other lymphoid malignancies where infiltrating immune cells, stroma, and vessels contribute to shape a complex tumor microenvironment (88–90). With this in mind it is reasonable to hypothesize that inhibition of the NFAT pathway could be effective for the treatment of lymphoproliferative disease since it affects cell function and survival both on and off the tumor. Recently Bucher et al. have reported strong evidence showing that NFAT signaling is chronically activated in DLBCL regulating cell survival and inflammatory cytokines (55, 56). In particular ABC DLBCL cells seem to be particularly dependent on the activation of the NFAT pathway. Moreover, data shows that blockade of signals generated from BCR activation is not able to affect NFAT1/2 phosphorylation or translocation to the nucleus. These findings suggest that other mechanisms could be responsible for the pathway activation. In summary, evidence has accumulated showing that the NFAT family controls biological processes on and off the tumor, which should be carefully analyzed in the context of targeting for any future treatment of lymphoproliferative diseases.
All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
This work was supported by Ministero della Salute under Ricerca Finalizzata 2018–ID. RF-2018-12367072 project—P.I. MM and co-P.I. PA; AIRC under IG 2019–ID. 23088 project—P.I. MM.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
BCR, B cell receptor; BTK, Bruton Tyrosine Kinase; CLL, Chronic Lymphocytic Leukemia; DLBCL, Diffuse large B-cell lymphoma; NFAT, Nuclear Factor of Activated T cells; NFκB, Nuclear Factor kappa-light-chain-enhancer of activated B cells; PI3K, Phosphoinositide 3-kinases; TF, Transcription Factor.
1. ten Hacken E, Burger JA. Microenvironment interactions and B-cell receptor signaling in Chronic Lymphocytic Leukemia: Implications for disease pathogenesis and treatment. Biochim Biophys Acta - Mol Cell Res (2016) 1863:401–13. doi: 10.1016/j.bbamcr.2015.07.009
2. Bosch F, Dalla-Favera R. Chronic lymphocytic leukaemia: from genetics to treatment. Nat Rev Clin Oncol (2019) 16:684–701. doi: 10.1038/s41571-019-0239-8
3. Schiattone L, Ghia P, Scarfò L. The evolving treatment landscape of chronic lymphocytic leukemia. Curr Opin Oncol (2019) 31:568–73. doi: 10.1097/CCO.0000000000000585
4. Shanafelt TD, Wang XV, Kay NE, Hanson CA, O’Brien S, Barrientos J, et al. Ibrutinib–Rituximab or Chemoimmunotherapy for Chronic Lymphocytic Leukemia. N Engl J Med (2019) 381:432–43. doi: 10.1056/nejmoa1817073
5. Moreno C, Greil R, Demirkan F, Tedeschi A, Anz B, Larratt L, et al. Ibrutinib plus obinutuzumab versus chlorambucil plus obinutuzumab in first-line treatment of chronic lymphocytic leukaemia (iLLUMINATE): a multicentre, randomised, open-label, phase 3 trial. Lancet Oncol (2019) 20:43–56. doi: 10.1016/S1470-2045(18)30788-5
6. Rossi D, Gaidano G. Lymphocytosis and ibrutinib treatment of CLL. Blood (2014) 123(12):1772–4. doi: 10.1182/blood-2014-01-549493
7. Woyach JA, Furman RR, Liu T-M, Ozer HG, Zapatka M, Ruppert AS, et al. Resistance Mechanisms for the Bruton’s Tyrosine Kinase Inhibitor Ibrutinib. N Engl J Med (2014) 370:2286–94. doi: 10.1056/nejmoa1400029
8. Kittai AS, Woyach JA. Resistance Mechanisms to Targeted Agents in Chronic Lymphocytic Leukemia. Cancer J (United States) (2019) 25:428–35. doi: 10.1097/PPO.0000000000000406
9. Sedlarikova L, Petrackova A, Papajik T, Turcsanyi P, Kriegova E. Resistance-associated mutations in chronic lymphocytic leukemia patients treated with novel agents. Front Oncol (2020) 10:894. doi: 10.3389/fonc.2020.00894
10. Müller MR, Rao A. NFAT, immunity and cancer: A transcription factor comes of age. Nat Rev Immunol (2010) 10:645–56. doi: 10.1038/nri2818
11. Okamura H, Aramburu J, García-Rodríguez C, Viola JPB, Raghavan A, Tahiliani M, et al. Concerted dephosphorylation of the transcription factor NFAT1 induces a conformational switch that regulates transcriptional activity. Mol Cell (2000) 6:539–50. doi: 10.1016/S1097-2765(00)00053-8
12. Gwack Y, Feske S, Srikanth S, Hogan PG, Rao A. Signalling to transcription: Store-operated Ca2+ entry and NFAT activation in lymphocytes. Cell Calcium (2007) 42:145–56. doi: 10.1016/j.ceca.2007.03.007
13. Kar P, Nelson C, Parekh AB. Selective activation of the transcription factor NFAT1 by calcium microdomains near Ca2+ release-activated Ca2+ (CRAC) channels. J Biol Chem (2011) 286:14795–803. doi: 10.1074/jbc.M111.220582
14. Medyouf H, Ghysdael J. The calcineurin/NFAT signaling pathway: A novel therapeutic target in leukemia and solid tumors. Cell Cycle (2008) 7:297–303. doi: 10.4161/cc.7.3.5357
15. Hogan PG, Chen L, Nardone J, Rao A. Transcriptional regulation by calcium, calcineurin, and NFAT. Genes Dev (2003) 17:2205–32. doi: 10.1101/gad.1102703
16. Qin J-JJ, Nag S, Wang W, Zhou J, Zhang W-DD, Wang H, et al. NFAT as cancer target: Mission possible? Elsevier (2014) 1846(2):297–331. doi: 10.1016/j.bbcan.2014.07.009
17. Gwack Y, Sharma S, Nardone J, Tanasa B, Iuga A, Srikanth S, et al. A genome-wide Drosophila RNAi screen identifies DYRK-family kinases as regulators of NFAT. Nature (2006) 441:646–50. doi: 10.1038/nature04631
18. Okamura H, Garcia-Rodriguez C, Martinson H, Qin J, Virshup DM, Rao A. A Conserved Docking Motif for CK1 Binding Controls the Nuclear Localization of NFAT1. Mol Cell Biol (2004) 24:4184–95. doi: 10.1128/mcb.24.10.4184-4195.2004
19. Beals CR. Nuclear Export of NF-ATc Enhanced by Glycogen Synthase Kinase-3. Science (1997) 275:1930–3. doi: 10.1126/science.275.5308.1930
20. Miyakawa H, Woo SK, Dahl SC, Handler JS, Kwon HM. Tonicity-responsive enhancer binding protein, a Rel-like protein that stimulates transcription in response to hypertonicity. Proc Natl Acad Sci USA (1999) 96(5):2538–42. doi: 10.1073/pnas.96.5.2538
21. Woo SK, Lee S, Kwon HM. TonEBP transcriptional activator in the cellular response to increased osmolality. Pflugers Arch Eur J Physiol (2002) 444:579–85. doi: 10.1007/s00424-002-0849-2
22. Kim NH, Hong BK, Choi SY, Kwon HM, Cho CS, Yi EC, et al. Reactive oxygen species regulate context-dependent inhibition of NFAT5 target genes. Exp Mol Med (2013) 45:e32. doi: 10.1038/emm.2013.61
23. Peng SL, Gerth AJ, Ranger AM, Glimcher LH. NFATc1 and NFATc2 together control both T and B cell activation and differentiation. Immunity (2001) 14:13–20. doi: 10.1016/S1074-7613(01)00085-1
24. De La Pompa JL, Timmerman LA, Takimoto H, Yoshida H, Elia AJ, Samper E, et al. Role of the NF-ATc transcription factor in morphogenesis of cardiac valves and septum. Nature (1998) 392:182–6. doi: 10.1038/32419
25. Ranger AM, Grusby MJ, Hodge MR, Gravallese EM, De La Brousse FC, Hoey T, et al. The transcription factor NF-ATc is essential for cardiac valve formation. Nature (1998) 392:186–90. doi: 10.1038/32426
26. Yoshida H, Nishina H, Takimoto H, Marengère LEM, Wakeham AC, Bouchard D, et al. The transcription factor NF-ATc1 regulates lymphocyte proliferation and Th2 cytokine production. Immunity (1998) 8:115–24. doi: 10.1016/S1074-7613(00)80464-1
27. Berland R, Wortis HH. Normal B-1a cell development requires B cell-intrinsic NFATc1 activity. Proc Natl Acad Sci USA (2003) 100:13459–64. doi: 10.1073/pnas.2233620100
28. Bhattacharyya S, Deb J, Patra AK, Pham DAT, Chen W, Vaeth M, et al. NFATc1 affects mouse splenic B cell function by controlling the calcineurin-NFAT signaling network. J Exp Med (2011) 208:823–39. doi: 10.1084/jem.20100945
29. Lin H, Grosschedl R. Failure of B–cell differentiation in mice lacking the transcription factor EBF. Nature (1995) 376:263–7. doi: 10.1038/376263a0
30. Giampaolo S, Wójcik G, Klein-Hessling S, Serfling E, Patra AK. B cell development is critically dependent on NFATc1 activity. Cell Mol Immunol (2019) 16:508–20. doi: 10.1038/s41423-018-0052-9
31. Märklin M, Heitmann JS, Kauer J, Wirths S, Müller MR. Genetic loss of NFAT2 (NFATc1) impairs B cell development of B1 and B2 B cells. Cell Immunol (2020) 349:104048. doi: 10.1016/j.cellimm.2020.104048
32. Xanthoudakis S, Viola JPB, Shaw KTY, Luo C, Wallace JD, Bozza PT, et al. An enhanced imune respones in mice lackinh the transcription factor NFAT1. Science (1996) 272:892–5. doi: 10.1126/science.272.5263.892
33. Ranger AM, Hodge MR, Gravallese EM, Oukka M, Davidson L, Alt FW, et al. Delayed lymphoid repopulation with defects in IL-4-driven responses produced by inactivation of NF-ATc. Immunity (1998) 8:125–34. doi: 10.1016/S1074-7613(00)80465-3
34. Hodge MR, Ranger AM, De La Brousse FC, Hoey T, Grusby MJ, Glimcher LH. Hyperproliferation and dysregulation of IL-4 expression in NF-ATp-deficient mice. Immunity (1996) 4(4):397–405. doi: 10.1016/S1074-7613(00)80253-8
35. Barrington RA, Borde M, Rao A, Carroll MC. Involvement of NFAT1 in B cell self-tolerance. J Immunol (2006) 177:1510–5. doi: 10.4049/jimmunol.177.3.1510
36. Teixeira LK, Carrossini N, Sécca C, Kroll JE, DaCunha DC, Faget DV, et al. NFAT1 transcription factor regulates cell cycle progression and cyclin E expression in B lymphocytes. Cell Cycle (2016) 15:2346–59. doi: 10.1080/15384101.2016.1203485
37. Ranger AM, Oukka M, Rengarajan J, Glimcher LH. Inhibitory function of two NFAT family members in lymphoid homeostasis and Th2 development. Immunity (1998) 9:627–35. doi: 10.1016/S1074-7613(00)80660-3
38. Rengarajan J, Mittelstadt PR, Mages HW, Gerth AJ, Kroczek RA, Ashwell JD, et al. Sequential Involvement of NFAT and Egr Transcription Factors in FasL Regulation. Immunity (2000) 12:293–300. doi: 10.1016/S1074-7613(00)80182-X
39. Rengarajan J, Tang B, Glimcher LH. Nfatc2 and nfatc3 regulate th2 differentiation and modulate tcr-responsiveness of naïve th cells. Nat Immunol (2002) 3:48–54. doi: 10.1038/ni744
40. Samanta DN, Palmetshofer A, Marinkovic D, Wirth T, Serfling E, Nitschke L. B Cell Hyperresponsiveness and Expansion of Mature Follicular B Cells but Not of Marginal Zone B Cells in NFATc2/c3 Double-Deficient Mice. J Immunol (2005) 174:4797–802. doi: 10.4049/jimmunol.174.8.4797
41. Berga-Bolanos R, Alberdi M, Buxade M, Aramburu J, Lopez-Rodriguez C. NFAT5 induction by the pre-T-cell receptor serves as a selective survival signal in T-lymphocyte development. Proc Natl Acad Sci (2013) 110:16091–6. doi: 10.1073/pnas.1215934110
42. Go WY, Liu X, Roti MA, Liu F, Ho SN. NFATS/TonEBP mutant mice define osmotic stress as a critical feature of the lymphoid microenvironment. Proc Natl Acad Sci USA (2004) 101:10673–8. doi: 10.1073/pnas.0403139101
43. Buxadé M, Lunazzi G, Minguillón J, Iborra S, Berga-Bolaños R, del Val M, et al. Gene expression induced by Toll-like receptors in macrophages requires the transcription factor NFAT5. J Exp Med (2012) 209:379–93. doi: 10.1084/jem.20111569
44. Berga-Bolaños R, Drews-Elger K, Aramburu J, López-Rodríguez C. NFAT5 Regulates T Lymphocyte Homeostasis and CD24-Dependent T Cell Expansion under Pathologic Hypernatremia. J Immunol (2010) 185:6624–35. doi: 10.4049/jimmunol.1001232
45. Swerdlow SH, Campo E, Pileri SA, Lee Harris N, Stein H, Siebert R, et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood (2016) 127:2375–90. doi: 10.1182/blood-2016-01-643569
46. Marafiot T, Pozzobon M, Hansmann M-L, Ventura R, Pileri SA, Roberton H, et al. The NFATc1 transcription factor is widely expressed in white cells and translocates from the cytoplasm to the nucleus in a subset of human lymphomas. Br J Haematol (2005) 128:333–42. doi: 10.1111/j.1365-2141.2004.05313.x
47. Akimzhanov A, Krenacs L, Schlegel T, Klein-Hessling S, Bagdi E, Stelkovics E, et al. Epigenetic changes and suppression of the nuclear factor of activated T cell 1 (NFATC1) promoter in human lymphomas with defects in immunoreceptor signaling. Am J Pathol (2008) 172:215–24. doi: 10.2353/ajpath.2008.070294
48. Pham LV, Tamayo AT, Yoshimura LC, Lin-Lee YC, Ford RJ. Constitutive NF-κB and NFAT activation in aggressive B-cell lymphomas synergistically activates the CD154 gene and maintains lymphoma cell survival. Blood (2005) 106:3940–7. doi: 10.1182/blood-2005-03-1167
49. Fu L, Lin-Lee YC, Pham LV, Tamayo A, Yoshimura L, Ford RJ. Constitutive NF-κB and NFAT activation leads to stimulation of the BLyS survival pathway in aggressive B-cell lymphomas. Blood (2006) 107:4540–8. doi: 10.1182/blood-2005-10-4042
50. Pham LV, Tamayo AT, Li C, Bueso-Ramos C, Ford RJ. An epigenetic chromatin remodeling role for NFATc1 in transcriptional regulation of growth and survival genes in diffuse large B-cell lymphomas. Blood (2010) 116:3899–906. doi: 10.1182/blood-2009-12-257378
51. Kondo E, Harashima A, Takabatake T, Takahashi H, Matsuo Y, Yoshino T, et al. NF-ATc2 induces apoptosis in Burkitt’s lymphoma cells through signaling via the B cell antigen receptor. Eur J Immunol (2003) 33:1–11. doi: 10.1002/immu.200390000
52. Lenz G, Wright GW, Emre NCT, Kohlhammer H, Dave SS, Davis RE, et al. Molecular subtypes of diffuse large B-cell lymphoma arise by distinct genetic pathways. Proc Natl Acad Sci USA (2008) 105:13520–5. doi: 10.1073/pnas.0804295105
53. Pham LV, Bryant JL, Mendez R, Chen J, Tamayo AT, Xu-Monette ZY, et al. Targeting the hexosamine biosynthetic pathway and O-linked N-acetylglucosamine cycling for therapeutic and imaging capabilities in diffuse large B-cell lymphoma. Oncotarget (2016) 7:80599–611. doi: 10.18632/oncotarget.12413
54. Li L, Zhang J, Chen J, Xu-Monette ZY, Miao Y, Xiao M, et al. B-cell receptor–mediated NFATc1 activation induces IL-10/STAT3/PD-L1 signaling in diffuse large B-cell lymphoma. Blood (2018) 132:1805–17. doi: 10.1182/blood-2018-03-841015
55. Bucher P, Erdmann T, Grondona P, Xu W, Schmitt A, Schürch C, et al. Targeting chronic NFAT activation with calcineurin inhibitors in diffuse large B-cell lymphoma. Blood (2020) 135:121–32. doi: 10.1182/blood.2019001866
56. Muppidi JR. A role for NFAT signaling in ABC-DLBCL. Blood (2020) 135:81. doi: 10.1182/blood.2019004199
57. Lee N, Kim D, Kim W-U. Role of NFAT5 in the Immune System and Pathogenesis of Autoimmune Diseases. Front Immunol (2019) 10:270. doi: 10.3389/fimmu.2019.00270
58. Fabbri G, Dalla-Favera R. The molecular pathogenesis of chronic lymphocytic leukaemia. Nat Rev Cancer (2016) 16:145–62. doi: 10.1038/nrc.2016.8
59. Arnason JE, Brown JR. Targeting B Cell Signaling in Chronic Lymphocytic Leukemia. Curr Oncol Rep (2017) 19:1–13. doi: 10.1007/s11912-017-0620-7
60. Schuh K, Avots A, Tony HP, Serfling E, Kneitz C. Nuclear NF-ATp is a hallmark of unstimulated B cells from B-CLL patients. Leuk Lymphoma (1996) 23:583–92. doi: 10.3109/10428199609054868
61. Le Roy C, Deglesne PA, Chevallier N, Beitar T, Eclache V, Quettier M, et al. The degree of BCR and NFAT activation predicts clinical outcomes in chronic lymphocytic leukemia. Blood (2012) 120:356–65. doi: 10.1182/blood-2011-12-397158
62. Pei L, Choi JH, Liu J, Lee EJ, McCarthy B, Wilson JM, et al. Genome-wide DNA methylation analysis reveals novel epigenetic changes in chronic lymphocytic leukemia. Epigenetics (2012) 7:567–78. doi: 10.4161/epi.20237
63. Beekman R, Chapaprieta V, Russiñol N, Vilarrasa-Blasi R, Verdaguer-Dot N, Martens JHA, et al. The reference epigenome and regulatory chromatin landscape of chronic lymphocytic leukemia. Nat Med (2018) 24:868–80. doi: 10.1038/s41591-018-0028-4
64. Wolf C, Garding A, Filarsky K, Bahlo J, Robrecht S, Becker N, et al. NFATC1 activation by DNA hypomethylation in chronic lymphocytic leukemia correlates with clinical staging and can be inhibited by ibrutinib. Int J Cancer (2018) 142:322–33. doi: 10.1002/ijc.31057
65. Abruzzo LV, Herling CD, Calin GA, Oakes C, Barron LL, Banks HE, et al. Trisomy 12 chronic lymphocytic leukemia expresses a unique set of activated and targetable pathways. Haematologica (2018) 103:2069–78. doi: 10.3324/haematol.2018.190132
66. Rawstron AC, Kreuzer KA, Soosapilla A, Spacek M, Stehlikova O, Gambell P, et al. Reproducible diagnosis of chronic lymphocytic leukemia by flow cytometry: An European Research Initiative on CLL (ERIC) & European Society for Clinical Cell Analysis (ESCCA) Harmonisation project. Cytom Part B - Clin Cytom (2018) 94:121–8. doi: 10.1002/cyto.b.21595
67. Kneitz C, Goller M, Tony HP, Simon A, Stibbe C, König T, et al. The CD23b promoter is a target for NF-AT transcription factors in B-CLL cells. Biochim Biophys Acta - Mol Basis Dis (2002) 1588:41–7. doi: 10.1016/S0925-4439(02)00114-X
68. Hubmann R, Schwarzmeier JD, Shehata M, Hilgarth M, Duechler M, Dettke M, et al. Notch2 is involved in the overexpression of CD23 in B-cell chronic lymphocytic leukemia. Blood (2002) 99:3742–7. doi: 10.1182/blood.v99.10.3742
69. Teutsch M, Higer M, Wang D, Wortis HW. Induction of CD5 on B and T cells is suppressed by cyclosporin A, FK-520 and rapamycin. Int Immunol (1995) 7:381–92. doi: 10.1093/intimm/7.3.381
70. Berland R, Wortis HH. An NFAT-dependent enhancer is necessary for anti-IgM-mediated induction of murine CD5 expression in primary splenic B cells. J Immunol (1998) 161:277–85.
71. Märklin M, Heitmann JS, Fuchs AR, Truckenmüller FM, Gutknecht M, Bugl S, et al. NFAT2 is a critical regulator of the anergic phenotype in chronic lymphocytic leukaemia. Nat Commun (2017) 8:1–14. doi: 10.1038/s41467-017-00830-y
72. Garaud S, Morva A, Lemoine S, Hillion S, Bordron A, Pers J-O, et al. CD5 Promotes IL-10 Production in Chronic Lymphocytic Leukemia B Cells through STAT3 and NFAT2 Activation. J Immunol (2011) 186:4835–44. doi: 10.4049/jimmunol.1003050
73. Oakes CC, Seifert M, Assenov Y, Gu L, Przekopowitz M, Ruppert AS, et al. DNA methylation dynamics during B cell maturation underlie a continuum of disease phenotypes in chronic lymphocytic leukemia. Nat Genet (2016) 48:253–64. doi: 10.1038/ng.3488
74. Bichi R, Shinton SA, Martin ES, Koval A, Calin GA, Cesari R, et al. Human chronic lymphocytic leukemia modeled in mouse by targeted TCL1 expression. Proc Natl Acad Sci USA (2002) 99:6955–60. doi: 10.1073/pnas.102181599
75. Muzio M, Apollonio B, Scielzo C, Frenquelli M, Vandoni I, Boussiotis V, et al. Constitutive activation of distinct BCR-signaling pathways in a subset of CLL patients: a molecular signature of anergy. Blood (2008) 112:188–95. doi: 10.1182/blood-2007-09-111344
76. Müller DJ, Wirths S, Fuchs AR, Märklin M, Heitmann JS, Sturm M, et al. Loss of NFAT2 expression results in the acceleration of clonal evolution in chronic lymphocytic leukemia. J Leukoc Biol (2019) 105:531–8. doi: 10.1002/JLB.2AB0218-076RR
77. Apollonio B, Scielzo C, Bertilaccio MTS, Ten Hacken E, Scarfò L, Ranghetti P, et al. Targeting B-cell anergy in chronic lymphocytic leukemia. Blood (2013) 121:3879–88. doi: 10.1182/blood-2012-12-474718
78. Lee JU, Kim LK, Choi JM. Revisiting the concept of targeting NFAT to control T cell immunity and autoimmune diseases. Front Immunol (2018) 9:2747. doi: 10.3389/fimmu.2018.02747
79. Wright SJ, Keating MJ. Cyclosporine a in chronic lymphocytic leukemia: Dual anti-leukemic and immunosuppressive role? Leuk Lymphoma (1995) 20:131–6. doi: 10.3109/10428199509054764
80. Kyasa MJ, Parrish RS, Schichman SA, Zent CS. Autoimmune cytopenia does not predict poor prognosis in chronic lymphocytic eukemia/small lymphocytic lymphoma. Am J Hematol (2003) 74:1–8. doi: 10.1002/ajh.10369
81. Yu H, Van Berkel TJC, Biessen EAL. Therapeutic potential of VIVIT, a selective peptide inhibitor of nuclear factor of activated T cells, in cardiovascular disorders. Cardiovasc Drug Rev (2007) 25:175–87. doi: 10.1111/j.1527-3466.2007.00011.x
82. Aramburu J, Yaffe MB, López-Rodríguez C, Cantley LC, Hogan PG, Rao A. Affinity-driven peptide selection of an NFAT inhibitor more selective than cyclosporin A. Science (1999) 285:2129–33. doi: 10.1126/science.285.5436.2129
83. Nishii K, Katayama N, Mitani H, Matsumoto T, Miwa H, Kita K, et al. Effects of cyclosporin A on refractory B-cell chronic lymphocytic leukemia. Int J Hematol (2000) 71:59–65.
84. Chen BL, Li Y, Xu S, Nie Y, Zhang J. NFAT5 Regulated by STUB1, Facilitates Malignant Cell Survival and p38 MAPK Activation by Upregulating AQP5 in Chronic Lymphocytic Leukemia. Biochem Genet (2021), 123456789. doi: 10.1007/s10528-021-10040-3
85. Bachireddy P, Wu CJ. Arresting the Inflammatory Drive of Chronic Lymphocytic Leukemia with Ibrutinib. Clin Cancer Res (2016) 22:1547–9. doi: 10.1158/1078-0432.CCR-15-3106
86. Pan M-G, Xiong Y, Chen F. NFAT gene family in inflammation and cancer. Curr Mol Med (2013) 13:543–54. doi: 10.2174/1566524011313040007
87. Zanoni I, Granucci F. Regulation and dysregulation of innate immunity by NFAT signaling downstream of pattern recognition receptors (PRRs). Eur J Immunol (2012) 42:1924–31. doi: 10.1002/eji.201242580
88. Caligaris-Cappio F. Inflammation, the microenvironment and chronic lymphocytic leukemia. Haematologica (2011) 96:353–5. doi: 10.3324/haematol.2010.039446
89. Schulz A, Toedt G, Zenz T, Stilgenbauer S, Lichter P, Seiffert M. Inflammatory cytokines and signaling pathways are associated with survival of primary chronic lymphocytic leukemia cells in vitro: a dominant role of CCL2. Haematologica (2011) 96:408–16. doi: 10.3324/haematol.2010.031377
Keywords: nuclear factor of activated T cells, B-cell receptor, chronic lymphocytic leukemia, lymphoma, lymphoid malignancies
Citation: Sana I, Mantione ME, Angelillo P and Muzio M (2021) Role of NFAT in Chronic Lymphocytic Leukemia and Other B-Cell Malignancies. Front. Oncol. 11:651057. doi: 10.3389/fonc.2021.651057
Received: 08 January 2021; Accepted: 15 March 2021;
Published: 01 April 2021.
Edited by:
Franco Fais, University of Genoa, ItalyReviewed by:
Magdalena Winiarska, Medical University of Warsaw, PolandCopyright © 2021 Sana, Mantione, Angelillo and Muzio. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Marta Muzio, bXV6aW8ubWFydGFAaHNyLml0
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.