 Chryssoula Kordella
Chryssoula Kordella Eleftheria Lamprianidou
Eleftheria Lamprianidou- Department of Hematology, University Hospital of Alexandroupolis, Democritus University of Thrace, Alexandroupolis, Greece
Abnormal DNA methylation patterns are thought to drive the pathobiology of high-risk myelodysplastic syndromes (HR-MDS) and acute myeloid leukemia (AML). Sixteen years after their initial approval, the hypomethylating agents (HMAs), 5-azacytidine (AZA) and 5-aza-2′-deoxycytidine, remain the mainstay of treatment for HR-MDS and AML. However, a connection of the hypomethylating or additional effects of HMAs with clinical responses remains yet to be shown, and the mode of action of HMAs remains obscure. Given the relatively short-lived responses and the inevitable development of resistance in HMAs, a thorough understanding of the antineoplastic mechanisms employed by HMAs holds critical importance. Recent data in cancer cell lines demonstrate that reactivation of endogenous retroelements (EREs) and induction of a cell-intrinsic antiviral response triggered by RNA neotranscripts may underlie the antitumor activity of HMAs. However, data on primary CD34+ cells derived from patients with HR-MDS failed to confirm a link between HMA-mediated ERE modulation and clinical response. Though difficult to reconcile the apparent discrepancy, it is possible that HMAs mediate their effects in more advanced levels of differentiation where cells become responsive to interferon, whereas, inter-individual variations in the process of RNA editing and, in particular, in the ADAR1/OAS/RNase L pathway may also confound the associations of clinical response with the induction of viral mimicry. Further ex vivo studies along with clinical correlations in well-annotated patient cohorts are warranted to decipher the role of ERE derepression in the antineoplastic mechanisms of HMAs.
Introduction
In an attempt to model embryonic development, C.H. Waddington coined the term “epigenetics,” as “the causal interactions between genes and their products, which bring the phenotype into being.” (1). However, the current notion of epigenetics refers to the processes that mediate heritable changes in gene expression without changing the primary DNA sequence (2). DNA methylation, post-translational modifications of histone proteins, and post-transcriptional gene regulation by long non-coding RNA are key epigenetic mechanisms that are collectively referred to as the epigenome. The role of epigenetic alterations in promoting and maintaining cancerous growth is now well-established (3, 4). Malignant cells harbor mutations in almost all gene encoding epigenetic regulators, such as chromatin-modifying enzymes (5). In addition, the cancer epigenome exhibits aberrancies in virtually all the epigenetic control characteristics, particularly in DNA methylation, where cancer-specific hypermethylation at CpG-rich sites or CpG islands in the promoter regions theoretically lead to repression in the expression of critical tumor suppressor genes (6, 7).
Long before deciphering the complexity of DNA methylation, inhibitors of DNA methyltransferase 1 (DNMT1), the enzyme which catalyzes the addition of the methyl group to the cytosine residues, were developed (8, 9) and approved for the treatment of myelodysplastic syndromes (MDS) and acute myeloid leukemia (AML). 5-azacytidine (AZA) and decitabine (DAC) are chemical nucleoside analogs of cytidine with identical ring structure, which is attached to the ribose sugar of AZA and deoxyribose of DAC (10). Both agents induce hypomethylation after incorporation into DNA and/or RNA of highly proliferating cells and depletion of DNMT1 (11). Nevertheless, despite the wide use of the two hypomethylating agents (HMAs), the exact mechanism of action and the genetic and cellular level where HMAs exert their effects remain largely unidentified (12).
HMAs in Myeloid Malignancies
Myelodysplastic syndromes and AML comprise two heterogeneous groups of clonal hematopoietic disorders sharing several common molecular defects (13, 14). In early 1970, AZA was first administered to patients with AML using a higher than the current dosing, which resulted in limited efficacy and severe cytotoxicity (8). Several decades later, AZA was approved for the treatment of MDS, on the basis of a less intensive regimen and of the administration via a subcutaneous route (15, 16). Two formulations of AZA are also approved for AML, parenteral formulation as an induction therapy for unfit patients (17), and oral formulation as maintenance treatment for patients who achieved complete remission and are ineligible for hematopoietic cell transplantation (18). Treatment with DAC was initially investigated in pediatric acute lymphoblastic leukemia (ALL) and then in MDS and AML, showing promising antitumor effects but with dose-limiting toxicities (19). Currently, DAC is approved for AML in Europe and for AML and high risk myelodysplastic syndromes (HR-MDS) in the USA (20, 21). The effectiveness, ease of use, and the favorable toxicity profile have rendered HMAs as the backbone for combination regimens for clinical trials in AML and MDS. However, the median overall survival with HMA monotherapy is ~13–16 months for patients with MDS (22) and less than a year for the ones with AML (17). In addition, both primary and secondary, i.e., after an initial response, failure to HMAs confer a grave (23–25) outcome, and there is currently no approach to overcome the inevitable development of resistance to HMAs (26).
Immune Mechanisms of Action of HMAs
Hypomethylating agents induce global hypomethylation and purportedly have pleiotropic effects. Beyond the presumed derepression of tumor suppressor genes, HMA-mediated hypomethylation potentially affects cell cycle control, DNA repair, apoptosis, cell signaling, angiogenesis and control of cancer cell invasion, and metastasis (27, 28). In addition, the dissociation between the degree of HMA-induced demethylation and clinical response (29) points to alternative, DNMT-independent mechanisms, such as direct cytotoxicity via inhibition of protein synthesis and activation of DNA damage pathways (30) and immunomodulation. Epigenetic silencing of immune-response genes by DNA methylation characterizes the cancer genome (31), and HMAs can restore numerous pathways of cancer immune evasion (32). Consequently, the antileukemic activity of HMA could be, at least partially, immune-mediated, but literature reports are often contradictory. HMA can promote the antitumor response by several mechanisms encompassing increased tumor immunogenicity and enhanced the cellular- and cytokine-mediated effector T-cell tumor lysis (32, 33). Conversely, HMA may inhibit T-cell proliferation and pro-inflammatory cytokine secretion with simultaneous induction of regulatory T cells (Tregs) (34, 35), while the HMA-induced increase in the expression of immune checkpoint molecules in clonal CD34+ progenitors is associated with refractory disease in patients with high-risk MDS (36). Partially reconciling these antithetic effects, another study demonstrated that HMAs are probably effective only in tumors with an “immune evasion” gene expression signature (37). HMAs can also directly affect leukemic and immune signaling pathways, and perturbed signaling networks in malignancies are not only detected in cancer cells but also in the cellular components of tumor immunity (38, 39). We have shown that the AZA-mediated restoration of the pathological signal transducer and activator of transcription (STAT) biosignature in both CD34+ and CD4+ T cells is strongly linked with a favorable clinical outcome in patients with high-risk MDS (40, 41). However, in accordance with the selective immunological activity of HMAs, no effect on the STAT networks was observed in patients who were refractory to AZA. Though not readily interpretable, the highly diverse clinical course of patients treated with HMA (42) and the equally heterogeneous, sample-specific responses to HMAs observed in multiomics studies (43, 44) argue for a multifaceted mechanism of the action of HMAs, potentially influenced by the molecular background of individual patients.
Another candidate antineoplastic mechanism of HMAs is the reactivation of human endogenous retroelements (EREs) (45) and the induction of viral mimicry (46). Recently, several intriguing reports rekindled interest on the ERE-induced antitumor immune response by demonstrating the upregulation of endogenous double-stranded RNAs (dsRNAs) and the induction of type I and III interferon (IFN) responses- in cancer cells treated with epigenetic agents (47–53).
Endogenous Retroelements
Endogenous retroelements comprise a significant part of the human genome, composing about 43% of the genome (54). EREs are distinguished into non-long terminal repeats (non-LTRs), which include the long and short interspersed nuclear elements (LINEs and SINEs), and LTR elements. With the exception of about 100 young elements, the rest of the LINEs reside as inactive fragments (55). The most common SINEs and the more abundant mobile elements in the human genome are Alu elements which comprise up to 11% of the genome. Alus have a variable impact on gene functions, and up to 0.3% of all human genetic disorders are associated with Alu-mediated recombination (56). Human endogenous retroviruses (HERVs) and mammalian apparent LTR retrotransposons (MaLRs) are part of the LTR elements (57–59). HERVs are remnants of germ-line integrations of exogenous retroviruses which in the past have infected the host (57, 60) and occupied ~8% of our genomes (54). ERVs are the only EREs that could be transmitted from one cell to another (60, 61). However, most of the HERVs have lost this ability due to mutations and recombination events that have been accumulated for years during the evolution. Recently acquired HERVs have preserved their copies intact and retain the ability to produce infectious particles, while evolutionarily old LTRs are more likely inactivated and not replication-competent (62).
EREs and Leukemogenesis
Since the first discovery of transposable elements (63), hypothetical links between repetitive elements and tumorigenesis have started to emerge, and a multitude of ERE-mediated mechanisms ultimately leading to the disruption of genomic integrity have been reported (57, 64, 65). The term “onco-exaptation” has been coined to describe the exploitation of the epigenetic and transcriptional dysregulation in cancer by EREs (66). Reactivation of EREs may promote carcinogenesis via the activation of cryptic promoters and the formation of chimeric transcripts with gene regulatory properties (64, 67). Specifically, the data for leukemogenesis are scarce, and a direct demonstration of leukemogenic potential of EREs in the primary AML cells is still lacking. Rearrangements of the MLL gene in AML have been associated with Alu-mediated recombination events (68–70), while the induction of AML in a xenograft mouse model for primary myelofibrosis (PMF) was attributed to unrestricted replication and subsequent viremia of murine leukemia virus (MuLV), potentially due to a paracrine mechanism in peptide mass fingerprinting (PMF) (71). A recent study demonstrated that specific HERVs can also act as oncogenic enhancers in AML. Using genome and epigenome editing approaches in AML cell lines, the authors detected six ERV families which bore chromatin signatures of enhancers and could regulate the host gene expression. It is to be noted that the deletion of the AML-specific LTR2B elements decreased proliferation and induced the apoptosis of AML cell lines by reducing the expression of the apolipoprotein C1 (APOC1), an oncogene (72).
EREs, Tumor Immunity, and HMAs
In contrast to their putative role in oncogenesis and leukemogenesis, EREs can potentially promote antitumor immunity. Epigenetic deregulation in tumors results in the expression of several ERE antigens not expressed in healthy tissues which can, at least theoretically, trigger immune sensing and induce potent adaptive antitumor responses (58). Cytotoxic CD8+ T-cell responses against epitopes of certain HERV proteins have been reported in several tumors (73–76), and the in silico analysis of local cytolytic activity in 18 untreated tumors identified a set of three tumor-specific ERVs (TSERVs), untraceable in the corresponding normal tissues (77). Importantly, the immune pathways were enriched in tumors with the highest expression of TSERVs, whereas, the cytolytic activity in several tumor types correlated with the expression of other HERVs. Upregulation of EREs in AML was linked to specific mutations, but, rather unexpectedly, not with DNMT3A and TET2 mutations (51, 78). IDH1 and TP53 mutations were associated with suppression in the expression of ERE (78), consistent with the low immunogenicity of IDH1-mutated tumors (79) and the role of TP53 dysfunction in tumor-immune evasion (80). Activating mutations of the SET-binding protein 1 gene (SETBP1) and the overexpression of wild-type SETDB1 are associated with aggressive diseases and poor outcomes in myeloid neoplasms (81). The disruption of SETDB1 in AML cell lines triggers a type-I IFN antiviral response by desilencing both the LTR and non-LTR elements, indicating that the evasion of innate immune sensing of EREs possibly underlies the poor prognostic impact of SETDB1 alterations in AML and MDS (51).
Regarding the therapeutic derepression of EREs, Jaenisch et al. (45) first showed that AZA can reactivate silent retroviral genomes in mice, whereas, in 1999, Karpf et al. (82) reported the induction of IFN responsive genes by AZA in HT29 colon adenocarcinoma cells. Two recent articles revisited these phenomena and addressed their role in the antineoplastic mechanisms employed by synthetic azanucleosides. Utilizing the in vitro assays in colorectal cancer cells (CRCs), Roulois et al. (47) demonstrated that a low dose of DAC can induce the formation of dsRNAs into cancer-initiating cells (CICs). These dsRNAs were mainly derived from endogenous retroviral elements and activated the melanoma differentiation-associated protein 5 (MDA5), a cytosolic pattern recognition receptor, the mitochondrial antiviral-signaling protein (MAVS), its downstream signaling modules, and IFN regulatory factor 7 (IRF7). Triggering the MDA5/MAVS/IRF7 axis culminates in the induction of an IFN type-III response, upregulation of IFN-stimulated genes (ISGs), and setting the CICs into a “virus-infected” state. Analogous findings were reported by Chiapinelli et al. (48), who observed a HMA-mediated induction of IFN type-I response in ovarian cancer cell lines via ERV demethylation and dsRNA formation. Also, AZA-induced viral defense gene levels discriminated epithelial ovarian cancer tumors into good (high levels) and poor (low levels) prognosis, whereas, an intense viral defense signature was associated with a better outcome in patients with melanoma treated with the anti-CTLA-4 therapy. Given the pleiotropic function of IFNs in immune response (83), the authors further used murine models to show a synergistic effect of the combination of AZA with anti-CTLA-4 antibody (48).
In line with the above reports, the antitumor activity of other agents targeting the epigenetic machinery was also based on the induction of ERE-mediated viral mimicry. Experiments in breast cancer cell lines and patient-derived xenograft models revealed that CDK4/6 inhibitors upregulate the ERV3-1 gene by reducing the activity of DNMT1, leading to a type-III IFN viral mimicry response (49). Ablation of the histone demethylase LSD1 in cancer cell lines also induced the reexpression of HERVs, LINE1, and the AluYa5 sub-family of Alu elements, which in turn mediated a dsRNA-diven type-I IFN antitumor response, without affecting either global methylation or the DNMT1 levels (50). Moreover, the derepression of LINE1 by histone deacetylase (HDAC) inhibitors led to the increased death of drug-resistant cancer cell lines by permitting a chemotherapy-induced antiviral defense response (52), whereas, the combination of epigenetic therapies enhanced the viral defense response and increased the antitumor efficacy (84, 85). However, the global patterns of the reexpression of ERE and the heterogeneous investigational approaches of the above studies hampered the identification of the exact origin of drug-induced immunogenic EREs. By using an MDA5-protection assay and RNA-sequencing (RNA-seq) in CRCs, Mehdipour et al. (53) identified inverted-repeat Alus (IRAlus) as the major source of DAC-induced immunogenic dsRNAs. Almost 90% of the MDA-protected RNA was IRAlus, whereas, the LTR elements were represented only by 1.37% and the ERV-derived dsRNAs had no role in the induction of viral mimicry (53).
In contrast to the majority of the abovementioned studies that used cancer cells lines or murine models, we investigated the effect of AZA on the pattern of ERE expression on CD34+ primary hematopoietic stem cells (HSCs) derived from patients with MDS undergoing treatment with AZA (86). By using RNA-seq, sophisticated bioinformatics tools, and the de novo assembly, we charted a complete transcriptional profile of EREs in the bone marrow of HSCs from healthy donors and patients with AML, MDS, and chronic myelomonocytic leukemia (CMML), before and after AZA administration. Even though the transcription of EREs increased after six cycles of AZA, this effect was equally observed in both patients with complete remission (CR) and failure to AZA. An analysis of an independent dataset (accession number: SRP067631) in a comparable cohort (87) revealed identical results, clearly suggesting that the response to AZA cannot be predicted by the global upregulation of EREs. We further analyzed the AZA-mediated modulation of specific ERE groups of loci, but, again, we were unable to track significant differences based on the treatment response. To address the possibility of an ERE-induced antiviral response only in patients with CR, we also assessed the alterations of IFN-inducible LTR elements or IFN-signature genes (ISGs) in our patients and the aforementioned cohort (87). No induction after AZA therapy or correlation with its outcome was observed either after six cycles of AZA or as early as day 15 after the first cycle of AZA, thus pre-cluding a sustained IFN signature in HSCs as a mechanism of the AZA activity. In keeping with our results, no correlation between an IFN response in the bone marrow of CD34+ cells and the response to AZA and DAC was shown in a cohort of 55 patients with MDS and CMML, whereas, the inflammatory signaling was reduced instead of enhanced after HMA administration (88). Also, in a heterogeneous cohort of patients with myeloid and lymphoid malignancies, evolutionarily young EREs were pre-ferentially upregulated in responders to AZA. However, the same EREs were also upregulated in few non-responders, whereas, the analyses were performed in diverse cell subpopulations, thus adding another layer of complexity to the interpretation of the findings (89). Of note both in vitro (48) and in patients treated with HMAs (88), no differential regulation of EREs or induction of an IFN response was noted between AZA and DAC, whereas, guadecitabine also appears to induce an ERE-mediated immune response, but this has not been formally shown (90).
The discrepancy of findings in the pre-clinical data of patients with MDS might be in part due to the intrinsically high constitutive expression of ISGs and the resistance of HSCs to IFN stimulation (91). In contrast, the differentiated cells downregulated the expression of ISGs and became responsive to IFN (91), indicating that the level of cellular differentiation might be an important factor in determining the mode of action of HMAs. Another possibility is the antagonizing role of the ADAR1 enzyme in the cytotoxic effect of AZA. ADAR1 edits dsRNA through the conversion of adenosine to inosine, resulting in the destabilization of RNA duplexes (92), repression of the 2′,5′-oligoadenylate synthetase (OAS)-RNase L pathway (93), and blocking the activation of the MDA5 receptor (94). Inactivating mutations of ADAR1 are present in Aicardi–Goutières syndrome (AGS), an autoimmune disorder, and are accompanied by an intense type-I IFN signature and upregulation of ISGs (95). Knockout of the IFN-inducible ADAR1 p150 isoform in a lung adenocarcinoma cell line enhanced the cytotoxic potential of AZA via reactivation of the single-stranded RNA-specific endoribonuclease RNAase L, an antiviral enzyme (93). Mehdipour and colleagues (53) also elegantly showed that the efficacy of HMAs is interdependent on the expression of ADAR1 and activity. DAC induced the expression of ADAR1 in CRCs and depleted immunogenic dsRNAs, while ADAR1-knockdown of both the constitutively expressed (p110) and the interferon-inducible (p150) forms resulted in persistent upregulation of ISGs and an enhanced activation of MAVS. Further confirming the bidirectional interaction between HMAs and ADAR1, a low dose DAC in CRC xenograft models was efficacious only in mice injected with ADAR1-knockdown CRCs. Collectively, the above data suggest that the differential expression of ADAR1 enzyme and/or regulators of the ADAR1/OAS/RNase L pathway may account for the disconnection between the clinical response and the induction of viral mimicry observed in our study. Several tumors express high levels of ADAR1 (96) and display dysregulated RNA editing (97), but, in others, the depletion of the ADAR1 may instead promote tumor progression and metastasis (98, 99). In AML, ADAR1 is variably expressed (100) and a JAK2-dependent upregulation of ADAR1 p150 takes place during blast crisis in chronic myeloid leukemia (CML) (101), but there is a lack of correlations between ADAR1 levels and activity and clinical outcomes. Importantly, the expression and activity of ADAR1 increases during the differentiation of myeloid cell lines (100); therefore, measurements on more differentiated myeloid forms rather than the leukemic blasts will probably be more relevant for clinical associations. Based on the above considerations, an updated model of the viral mimicry hypothesis is provided in Figure 1.
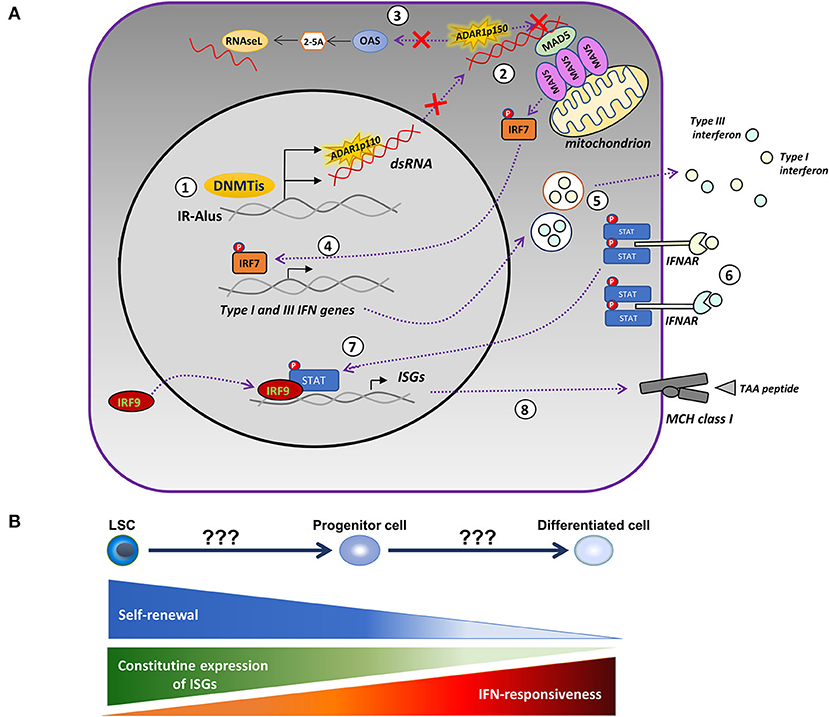
Figure 1. Modified model of the HMA-induced viral mimicry in leukemic cells. (A) Treatment with DNA-methyltransferase inhibitors (DNMTis), such as azacytidine (AZA) and decitabine (DAC), reactivate repressed inverted-repeat Alus (IRAlus), leading to the formation of double-stranded RNAs (dsRNAs). The same agents upregulate the expression of the RNA-editing enzyme, ADAR1 (1). The pattern recognition receptor, MDA5, senses the dsRNAs located in the cytoplasm, leading to the aggregation of the mitochondrial antiviral-signaling protein (MAVS) and to the phosphorylation of interferon (IFN) regulatory factor 7 (IRF7). However, increased ADAR1 activity depletes dsRNAs and prevents the activation of MDA5 (2), whereas, it further inhibits the OAS/RNAse L apoptotic pathway (3). In the absence of ADAR1 upregulation, activated IRF7 translocates into the nucleus inducing the transcription of type-I and III IFN (4), which are then secreted into the tumor microenvironment (5) and bind to their receptors, causing the phosphorylation of signal transducer and activator of transcription (STAT) proteins (6). Activated STATs associate with the IRF9 and move into the nucleus (7), where they induce the expression of IFN-stimulated genes (ISGs) and the major histocompatibility complex (MHC) molecules (8) by increasing the ability of tumor cells to present tumor-associated antigens (TAAs). (B) The competence of HMAs to induce a viral mimicry state potentially depends on the level of cellular differentiation. The latter defines the permissiveness to IFN-mediated induction of ISGs and the degree of upregulation of ADAR1.
Conclusion
More than 50 years after their discovery, HMAs remain as the only approved compound for the treatment of HR-MDS and the main therapeutic option for unfit patients with AML. Paradoxically, after 16 years of clinical experience, the mechanism of action of HMAs is still under investigation, a fact that poses obvious obstacles in bypassing the resistance and developing rational combinations with other agents. Endogenous retroelements, once viewed as parasitic elements, are currently enjoying a resurgence of interest regarding their role in the mechanism of action of HMAs. Aside from its potential use as a predictor of response to immunotherapy, the HMA-mediated induction of immunogenic EREs appears to sensitize immune-refractory tumors to checkpoint inhibition. However, a robust clinical proof confirming a cause–effect relationship of the induction of viral mimicry with the efficacy of HMAs is currently lacking. In addition, the combination of HMAs with immune checkpoint inhibitors demonstrates the modest efficacy in clinical trials for patients with MDS (102). While a multitude of issues pertaining to treatment schedule, dosing, and pharmacological attributes may account for the discordance between research findings and clinical efficacy, the interpatient-diversity of the tumor-immune system interactions is an obvious obstacle that has to be thoroughly interrogated before assigning a mechanistic role of ERE reactivation in the clinical activity of HMAs. A deeper understanding of the regulation of HMA-mediated reactivation of EREs at the single cell level and large-scale correlations of the experimental findings with clinical information is required to circumvent the limitations of both HMA and immune therapy in myeloid neoplasms.
Author Contributions
CK and IK wrote the manuscript and created the figure. CK, EL, and IK revised the manuscript. All authors contributed to the article and approved the submitted version.
Funding
CK is supported by the Greece and the European Union (European Social Fund—ESF), through the operational program, Human Resources Development, Education and Lifelong Learning, in the context of the project, Strengthening Human Resources Research Potential via Doctorate Research (MIS-5000432), implemented by the State Scholarships Foundation (IKY).
Conflict of Interest
IK have received research funding from Celgene Corporation and received honoraria from Genesis Pharma Hellas.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We are grateful to George Kassiotis, Prof. Emmanouil Spanoudakis, and Prof. Katerina Chlichlia for providing important intellectual assistance.
References
2. Berger SL, Kouzarides T, Shiekhattar R, Shilatifard A. An operational definition of epigenetics. Genes Dev. (2009) 23:781–3. doi: 10.1101/gad.1787609
3. Jones PA, Baylin SB. The epigenomics of cancer. Cell. (2007) 128:683–92. doi: 10.1016/j.cell.2007.01.029
4. Suva ML, Riggi N, Bernstein BE. Epigenetic reprogramming in cancer. Science. (2013) 339:1567–70. doi: 10.1126/science.1230184
5. Plass C, Pfister SM, Lindroth AM, Bogatyrova O, Claus R, Lichter P. Mutations in regulators of the epigenome and their connections to global chromatin patterns in cancer. Nature Rev Genet. (2013) 14:765–80. doi: 10.1038/nrg3554
6. Baylin SB, Jones PA. A decade of exploring the cancer epigenome-biological and translational implications. Nat Rev Cancer. (2011) 11:726–34. doi: 10.1038/nrc3130
7. Dawson MA, Kouzarides T. Cancer epigenetics: from mechanism to therapy. Cell. (2012) 150:12–27. doi: 10.1016/j.cell.2012.06.013
8. Sorm F, Piskala A, Cihak A, Vesely J. 5-Azacytidine, a new, highly effective cancerostatic. Experientia. (1964) 20:202–3. doi: 10.1007/BF02135399
9. Sorm F, Vesely J. The activity of a new antimetabolite, 5-Azacytidine, against lymphoid leukaemia in Ak mice. Neoplasma. (1964) 11:123–30.
10. Derissen EJ, Beijnen JH, Schellens JH. Concise drug review: azacitidine and decitabine. Oncologist. (2013) 18:619–24. doi: 10.1634/theoncologist.2012-0465
11. Stresemann C, Lyko F. Modes of action of the DNA methyltransferase inhibitors azacytidine and decitabine. Int J Cancer. (2008) 123:8–13. doi: 10.1002/ijc.23607
12. Odenike O. Incorporating novel approaches in the management of MDS beyond conventional hypomethylating agents. Hematology Am Soc Hematol Educ Program. (2017) 2017:460–9. doi: 10.1182/asheducation-2017.1.460
13. Hemmati S, Haque T, Gritsman K. Inflammatory signaling pathways in preleukemic and leukemic stem cells. Front Oncol. (2017) 7:265. doi: 10.3389/fonc.2017.00265
15. Silverman LR, Demakos EP, Peterson BL, Kornblith AB, Holland JC, Odchimar-Reissig R, et al. Randomized controlled trial of azacitidine in patients with the myelodysplastic syndrome: a study of the cancer and leukemia group B. J Clin Oncol. (2002) 20:2429–40. doi: 10.1200/JCO.2002.04.117
16. Fenaux P, Mufti GJ, Hellstrom-Lindberg E, Santini V, Finelli C, Giagounidis A, et al. Efficacy of azacitidine compared with that of conventional care regimens in the treatment of higher-risk myelodysplastic syndromes: a randomised, open-label, phase III study. Lancet Oncol. (2009) 10:223–32. doi: 10.1016/S1470-2045(09)70003-8
17. Dombret H, Seymour JF, Butrym A, Wierzbowska A, Selleslag D, Jang JH, et al. International phase 3 study of azacitidine vs conventional care regimens in older patients with newly diagnosed AML with >30% blasts. Blood. (2015) 126:291–9. doi: 10.1182/blood-2015-01-621664
18. Wei AH, Döhner H, Pocock C, Montesinos P, Afanasyev B, Dombret H, et al. The QUAZAR AML-342 001 maintenance trial: results of a phase III international, randomized, double-blind, Placebo-343 controlled study of CC-486 (oral formulation of azacitidine) in patients with acute myeloid leukemia 344 (AML) in first remission. Blood. (2019) 134((Supplement.2):LBA-3):105–19. doi: 10.1182/blood-2019-132405
19. de Vos D, van Overveld W. Decitabine: a historical review of the development of an epigenetic drug. Ann Hematol. (2005) 84(Suppl. 1):3–8. doi: 10.1007/s00277-005-0008-x
20. Kantarjian HM, Thomas XG, Dmoszynska A, Wierzbowska A, Mazur G, Mayer J, et al. Multicenter, randomized, open-label, phase III trial of decitabine versus patient choice, with physician advice, of either supportive care or low-dose cytarabine for the treatment of older patients with newly diagnosed acute myeloid leukemia. J Clin Oncol. (2012) 30:2670–7. doi: 10.1200/JCO.2011.38.9429
21. Kantarjian H, Oki Y, Garcia-Manero G, Huang X, O'Brien S, Cortes J, et al. Results of a randomized study of 3 schedules of low-dose decitabine in higher-risk myelodysplastic syndrome and chronic myelomonocytic leukemia. Blood. (2007) 109:52–7. doi: 10.1182/blood-2006-05-021162
22. Zeidan AM, Stahl M, DeVeaux M, Giri S, Huntington S, Podoltsev N, et al. Counseling patients with higher-risk MDS regarding survival with azacitidine therapy: are we using realistic estimates? Blood Cancer J. (2018) 8:55. doi: 10.1038/s41408-018-0081-8
23. Prebet T, Gore SD, Esterni B, Gardin C, Itzykson R, Thepot S, et al. Outcome of high-risk myelodysplastic syndrome after azacitidine treatment failure. J Clin Oncol. (2011) 29:3322–7. doi: 10.1200/JCO.2011.35.8135
24. Kadia TM, Jabbour E, Kantarjian H. Failure of hypomethylating agent-based therapy in myelodysplastic syndromes. Semin Oncol. (2011) 38:682–92. doi: 10.1053/j.seminoncol.2011.04.011
25. Kotsianidis I, Papageorgiou SG, Pappa V, Galanopoulos AG, Viniou NA, Vassilakopoulos TP, et al. Azacytidine failure revisited: an appraisal based on real-life data from the MDS registry of the hellenic myelodysplastic syndrome study group (HMDS). Mediterr J Hematol Infect Dis. (2019) 11:e2019045. doi: 10.4084/mjhid.2019.045
26. Santini V. How I treat MDS after hypomethylating agent failure. Blood. (2019) 133:521–9. doi: 10.1182/blood-2018-03-785915
27. Agrawal K, Das V, Vyas P, Hajduch M. Nucleosidic DNA demethylating epigenetic drugs-A comprehensive review from discovery to clinic. Pharmacol Ther. (2018) 188:45–79. doi: 10.1016/j.pharmthera.2018.02.006
28. Sigalotti L, Fratta E, Coral S, Cortini E, Covre A, Nicolay HJ, et al. Epigenetic drugs as pleiotropic agents in cancer treatment: biomolecular aspects and clinical applications. J Cell Physiol. (2007) 212:330–44. doi: 10.1002/jcp.21066
29. Voso MT, Santini V, Fabiani E, Fianchi L, Criscuolo M, Falconi G, et al. Why methylation is not a marker predictive of response to hypomethylating agents. Haematologica. (2014) 99:613–9. doi: 10.3324/haematol.2013.099549
30. Stresemann C, Bokelmann I, Mahlknecht U, Lyko F. Azacytidine causes complex DNA methylation responses in myeloid leukemia. Mol Cancer Ther. (2008) 7:2998–3005. doi: 10.1158/1535-7163.MCT-08-0411
31. Heninger E, Krueger TE, Lang JM. Augmenting antitumor immune responses with epigenetic modifying agents. Front Immunol. (2015) 6:29. doi: 10.3389/fimmu.2015.00029
32. Topper MJ, Vaz M, Marrone KA, Brahmer JR, Baylin SB. The emerging role of epigenetic therapeutics in immuno-oncology. Nat Rev Clin Oncol. (2020) 17:75–90. doi: 10.1038/s41571-019-0266-5
33. Lindblad KE, Goswami M, Hourigan CS, Oetjen KA. Immunological effects of hypomethylating agents. Expert Rev Hematol. (2017) 10:745–52. doi: 10.1080/17474086.2017.1346470
34. Sanchez-Abarca LI, Gutierrez-Cosio S, Santamaria C, Caballero-Velazquez T, Blanco B, Herrero-Sanchez C, et al. Immunomodulatory effect of 5-azacytidine (5-azaC): potential role in the transplantation setting. Blood. (2009) 115:107–21doi: 10.1182/blood-2009-03-210393
35. Lal G, Bromberg JS. Epigenetic mechanisms of regulation of Foxp3 expression. Blood. (2009) 114:3727–35. doi: 10.1182/blood-2009-05-219584
36. Yang H, Bueso-Ramos C, DiNardo C, Estecio MR, Davanlou M, Geng QR, et al. Expression of PD-L1, PD-L2, PD-1 and CTLA4 in myelodysplastic syndromes is enhanced by treatment with hypomethylating agents. Leukemia. (2014) 28:1280–8. doi: 10.1038/leu.2013.355
37. Li H, Chiappinelli KB, Guzzetta AA, Easwaran H, Yen RW, Vatapalli R, et al. Immune regulation by low doses of the DNA methyltransferase inhibitor 5-azacitidine in common human epithelial cancers. Oncotarget. (2014) 5:587–98. doi: 10.18632/oncotarget.1782
38. Yu H, Pardoll D, Jove R. STATs in cancer inflammation and immunity: a leading role for STAT3. Nat Rev Cancer. (2009) 9:798–809. doi: 10.1038/nrc2734
39. Scholl C, Gilliland DG, Frohling S. Deregulation of signaling pathways in acute myeloid leukemia. Semin Oncol. (2008) 35:336–45. doi: 10.1053/j.seminoncol.2008.04.004
40. Miltiades P, Lamprianidou E, Vassilakopoulos TP, Papageorgiou SG, Galanopoulos AG, Kontos CK, et al. The Stat3/5 signaling biosignature in hematopoietic stem/progenitor cells predicts response and outcome in myelodysplastic syndrome patients treated with azacitidine. Clin Cancer Res. (2016) 22:1958–68. doi: 10.1158/1078-0432.CCR-15-1288
41. Lamprianidou EKC, Kazachenka A, Zoulia E, Bernard E, Filia A, Laidou S, et al. Modulation of IL-6/STAT3 signaling axis in CD4+FOXP3- T cells represents a potential antitumor mechanism of azacitidine. Blood Adv. (2020) 5:129–42. doi: 10.1182/bloodadvances.2020002351
42. Lamprianidou E, Zoulia E, Bernard E, Kordella C, Papoutselis M, Bezirgiannidou Z, et al. Multifaceted modes of action of azacytidine: a riddle wrapped up in an enigma. Leuk Lymphoma. (2019) 60:3277–81. doi: 10.1080/10428194.2019.1627542
43. Klco JM, Spencer DH, Lamprecht TL, Sarkaria SM, Wylie T, Magrini V, et al. Genomic impact of transient low-dose decitabine treatment on primary AML cells. Blood. (2013) 121:1633–43. doi: 10.1182/blood-2012-09-459313
44. Leung KK, Nguyen A, Shi T, Tang L, Ni X, Escoubet L, et al. Multiomics of azacitidine-treated AML cells reveals variable and convergent targets that remodel the cell-surface proteome. Proc Natl Acad Sci U S A. (2019) 116:695–700. doi: 10.1073/pnas.1813666116
45. Jaenisch R, Schnieke A, Harbers K. Treatment of mice with 5-azacytidine efficiently activates silent retroviral genomes in different tissues. Proc Natll Acad Sci U S A. (1985) 82:1451–5. doi: 10.1073/pnas.82.5.1451
46. Jones PA, Ohtani H, Chakravarthy A, De Carvalho DD. Epigenetic therapy in immune-oncology. Nat Rev Cancer. (2019) 19:151–61. doi: 10.1038/s41568-019-0109-9
47. Roulois D, Loo Yau H, Singhania R, Wang Y, Danesh A, Shen SY, et al. DNA-demethylating agents target colorectal cancer cells by inducing viral mimicry by endogenous transcripts. Cell. (2015) 162:961–73. doi: 10.1016/j.cell.2015.07.056
48. Chiappinelli KB, Strissel PL, Desrichard A, Li H, Henke C, Akman B, et al. Inhibiting DNA methylation causes an interferon response in cancer via dsRNA including endogenous retroviruses. Cell. (2015) 162:974–86. doi: 10.1016/j.cell.2015.07.011
49. Goel S, DeCristo MJ, Watt AC, BrinJones H, Sceneay J, Li BB, et al. CDK4/6 inhibition triggers anti-tumour immunity. Nature. (2017) 548:471–5. doi: 10.1038/nature23465
50. Sheng W, LaFleur MW, Nguyen TH, Chen S, Chakravarthy A, Conway JR, et al. LSD1 Ablation stimulates anti-tumor immunity and enables checkpoint blockade. Cell. (2018) 174:549–63 e19. doi: 10.1016/j.cell.2018.05.052
51. Cuellar TL, Herzner AM, Zhang X, Goyal Y, Watanabe C, Friedman BA, et al. Silencing of retrotransposons by SETDB1 inhibits the interferon response in acute myeloid leukemia. J Cell Biol. (2017) 216:3535–49. doi: 10.1083/jcb.201612160
52. Guler GD, Tindell CA, Pitti R, Wilson C, Nichols K, KaiWai Cheung T, et al. Repression of stress-induced LINE-1 expression protects cancer cell subpopulations from lethal drug exposure. Cancer cell. (2017) 32:221–37 e13. doi: 10.1016/j.ccell.2017.07.002
53. Mehdipour P, Marhon SA, Ettayebi I, Chakravarthy A, Hosseini A, Wang Y, et al. Epigenetic therapy induces transcription of inverted SINEs and ADAR1 dependency. Nature. (2020) 588:169–73. doi: 10.1038/s41586-020-2844-1
54. Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, Baldwin J, et al. Initial sequencing and analysis of the human genome. Nature. (2001) 409:860–921. doi: 10.1038/35057062
55. Beck CR, Garcia-Perez JL, Badge RM, Moran JV. LINE-1 elements in structural variation and disease. AnnualRreview Genomics Hum Genet. (2011) 12:187–215. doi: 10.1146/annurev-genom-082509-141802
56. Batzer MA, Deininger PL. Alu repeats and human genomic diversity. Nature Rev Genet. (2002) 3:370–9. doi: 10.1038/nrg798
57. Kassiotis G. Endogenous retroviruses and the development of cancer. J Immunol. (2014) 192:1343–9. doi: 10.4049/jimmunol.1302972
58. Kassiotis G, Stoye JP. Immune responses to endogenous retroelements: taking the bad with the good. Nat Rev Immunol. (2016) 16:207–19. doi: 10.1038/nri.2016.27
59. Burns KH, Boeke JD. Human transposon tectonics. Cell. (2012) 149:740–52. doi: 10.1016/j.cell.2012.04.019
60. Dewannieux M, Heidmann T. Endogenous retroviruses: acquisition, amplification and taming of genome invaders. Curr Opin Virol. (2013) 3:646–56. doi: 10.1016/j.coviro.2013.08.005
61. van der Laan LJ, Lockey C, Griffeth BC, Frasier FS, Wilson CA, Onions DE, et al. Infection by porcine endogenous retrovirus after islet xenotransplantation in SCID mice. Nature. (2000) 407:90–4. doi: 10.1038/35024089
62. Kassiotis G, Stoye JP. Making a virtue of necessity: the pleiotropic role of human endogenous retroviruses in cancer. Philos Trans R Soc Lond B Biol Sci. (2017) 372. doi: 10.1098/rstb.2016.0277
63. Mc CB. The origin and behavior of mutable loci in maize. Proc Natll Acad Sci U S A. (1950) 36:344–55. doi: 10.1073/pnas.36.6.344
64. Burns KH. Transposable elements in cancer. Nat Rev Cancer. (2017) 17:415–24. doi: 10.1038/nrc.2017.35
65. Ishak CA, Classon M, De Carvalho DD. Deregulation of retroelements as an emerging therapeutic opportunity in cancer. Trends in cancer. (2018) 4:583–97. doi: 10.1016/j.trecan.2018.05.008
66. Babaian A, Romanish MT, Gagnier L, Kuo LY, Karimi MM, Steidl C, et al. Onco-exaptation of an endogenous retroviral LTR drives IRF5 expression in Hodgkin lymphoma. Oncogene. (2016) 35:2542–6. doi: 10.1038/onc.2015.308
67. Jang HS, Shah NM, Du AY, Dailey ZZ, Pehrsson EC, Godoy PM, et al. Transposable elements drive widespread expression of oncogenes in human cancers. Nat Genet. (2019) 51:611–7. doi: 10.1038/s41588-019-0373-3
68. Strout MP, Marcucci G, Bloomfield CD, Caligiuri MA. The partial tandem duplication of ALL1 (MLL) is consistently generated by Alu-mediated homologous recombination in acute myeloid leukemia. Proc Natll Acad Sci U S A. (1998) 95:2390–5. doi: 10.1073/pnas.95.5.2390
69. So CW, Ma ZG, Price CM, Dong S, Chen SJ, Gu LJ, et al. MLL self fusion mediated by Alu repeat homologous recombination and prognosis of AML-M4/M5 subtypes. Cancer Res. (1997) 57:117–22.
70. Schichman SA, Caligiuri MA, Strout MP, Carter SL, Gu Y, Canaani E, et al. ALL-1 tandem duplication in acute myeloid leukemia with a normal karyotype involves homologous recombination between Alu elements. Cancer Res. (1994) 54:4277–80.
71. Triviai I, Ziegler M, Bergholz U, Oler AJ, Stubig T, Prassolov V, et al. Endogenous retrovirus induces leukemia in a xenograft mouse model for primary myelofibrosis. Proc Natll Acad Sci U S A. (2014) 111:8595–600. doi: 10.1073/pnas.1401215111
72. Deniz O, Ahmed M, Todd CD, Rio-Machin A, Dawson MA, Branco MR. Endogenous retroviruses are a source of enhancers with oncogenic potential in acute myeloid leukaemia. Nat Commun. (2020) 11:3506. doi: 10.1038/s41467-020-17206-4
73. Schiavetti F, Thonnard J, Colau D, Boon T, Coulie PG. A human endogenous retroviral sequence encoding an antigen recognized on melanoma by cytolytic T lymphocytes. Cancer Res. (2002) 62:5510–6.
74. Takahashi Y, Harashima N, Kajigaya S, Yokoyama H, Cherkasova E, McCoy JP, et al. Regression of human kidney cancer following allogeneic stem cell transplantation is associated with recognition of an HERV-E antigen by T cells. J Clin Invest. (2008) 118:1099–109. doi: 10.1172/JCI34409
75. Mullins CS, Linnebacher M. Endogenous retrovirus sequences as a novel class of tumor-specific antigens: an example of HERV-H env encoding strong CTL epitopes. Cancer Immunol Immunother. (2012) 61:1093–100. doi: 10.1007/s00262-011-1183-3
76. Wang-Johanning F, Radvanyi L, Rycaj K, Plummer JB, Yan P, Sastry KJ, et al. Human endogenous retrovirus K triggers an antigen-specific immune response in breast cancer patients. Cancer Res. (2008) 68:5869–77. doi: 10.1158/0008-5472.CAN-07-6838
77. Rooney MS, Shukla SA, Wu CJ, Getz G, Hacohen N. Molecular and genetic properties of tumors associated with local immune cytolytic activity. Cell. (2015) 160:48–61. doi: 10.1016/j.cell.2014.12.033
78. Colombo AR, Triche T Jr, Ramsingh G. Transposable element expression in acute myeloid leukemia transcriptome and prognosis. Sci Rep. (2018) 8:16449. doi: 10.1038/s41598-018-34189-x
79. Thorsson V, Gibbs DL, Brown SD, Wolf D, Bortone DS, Ou Yang TH, et al. The immune landscape of cancer. Immunity. (2018) 48:812–30 e14. doi: 10.1016/j.immuni.2018.03.023
80. Cui Y, Guo G. Immunomodulatory function of the tumor suppressor p53 in host immune response and the tumor microenvironment. Int J Mol Sci. (2016) 17:1942. doi: 10.3390/ijms17111942
81. Makishima H. Somatic SETBP1 mutations in myeloid neoplasms. Int J Hematol. (2017) 105:732–42. doi: 10.1007/s12185-017-2241-1
82. Karpf AR, Peterson PW, Rawlins JT, Dalley BK, Yang Q, Albertsen H, et al. Inhibition of DNA methyltransferase stimulates the expression of signal transducer and activator of transcription 1, 2, and 3 genes in colon tumor cells. Proc Natll Acad Sci U S A. (1999) 96:14007–12. doi: 10.1073/pnas.96.24.14007
83. Borden EC, Sen GC, Uze G, Silverman RH, Ransohoff RM, Foster GR, et al. Interferons at age 50: past, current and future impact on biomedicine. Nat Rev Drug Discov. (2007) 6:975–90. doi: 10.1038/nrd2422
84. Liu M, Ohtani H, Zhou W, Orskov AD, Charlet J, Zhang YW, et al. Vitamin C increases viral mimicry induced by 5-aza-2'-deoxycytidine. Proc Natll Acad Sci U S A. (2016) 113:10238–44. doi: 10.1073/pnas.1612262113
85. Liu M, Thomas SL, DeWitt AK, Zhou W, Madaj ZB, Ohtani H, et al. Dual inhibition of DNA and histone methyltransferases increases viral mimicry in ovarian cancer cells. Cancer Res. (2018) 78:5754–66. doi: 10.1158/0008-5472.CAN-17-3953
86. Kazachenka A, Young GR, Attig J, Kordella C, Lamprianidou E, Zoulia E, et al. Epigenetic therapy of myelodysplastic syndromes connects to cellular differentiation independently of endogenous retroelement derepression. Genome Med. (2019) 11:86. doi: 10.1186/s13073-019-0707-x
87. Unnikrishnan A, Papaemmanuil E, Beck D, Deshpande NP, Verma A, Kumari A, et al. Integrative genomics identifies the molecular basis of resistance to azacitidine therapy in myelodysplastic syndromes. Cell Rep. (2017) 20:572–85. doi: 10.1016/j.celrep.2017.06.067
88. Montalban-Bravo G, Class CA, Ganan-Gomez I, Kanagal-Shamanna R, Sasaki K, Richard-Carpentier G, et al. Transcriptomic analysis implicates necroptosis in disease progression and prognosis in myelodysplastic syndromes. Leukemia. (2020) 34:872–81. doi: 10.1038/s41375-019-0623-5
89. Ohtani H, Orskov AD, Helbo AS, Gillberg L, Liu M, Zhou W, et al. Activation of a subset of evolutionarily young transposable elements and innate immunity are linked to clinical responses to 5-azacytidine. Can Res. (2020) 80:2441–50. doi: 10.1158/0008-5472.CAN-19-1696
90. Nahas MR, Stroopinsky D, Rosenblatt J, Cole L, Pyzer AR, Anastasiadou E, et al. Hypomethylating agent alters the immune microenvironment in acute myeloid leukaemia (AML) and enhances the immunogenicity of a dendritic cell/AML vaccine. Br J Haematol. (2019) 185:679–90. doi: 10.1111/bjh.15818
91. Wu X, Dao Thi VL, Huang Y, Billerbeck E, Saha D, Hoffmann HH, et al. Intrinsic immunity shapes viral resistance of stem cells. Cell. (2018) 172:423–38 e25. doi: 10.1016/j.cell.2017.11.018
92. Levanon EY, Eisenberg E, Yelin R, Nemzer S, Hallegger M, Shemesh R, et al. Systematic identification of abundant A-to-I editing sites in the human transcriptome. Nat Biotechnol. (2004) 22:1001–5. doi: 10.1038/nbt996
93. Banerjee S, Gusho E, Gaughan C, Dong B, Gu X, Holvey-Bates E, et al. OAS-RNase L innate immune pathway mediates the cytotoxicity of a DNA-demethylating drug. Proc Natll Acad Sci U S A. (2019) 116:5071–6. doi: 10.1073/pnas.1815071116
94. Liddicoat BJ, Piskol R, Chalk AM, Ramaswami G, Higuchi M, Hartner JC, et al. RNA editing by ADAR1 prevents MDA5 sensing of endogenous dsRNA as nonself. Science. (2015) 349:1115–20. doi: 10.1126/science.aac7049
95. Rice GI, Kasher PR, Forte GM, Mannion NM, Greenwood SM, Szynkiewicz M, et al. Mutations in ADAR1 cause Aicardi-Goutieres syndrome associated with a type I interferon signature. Nat Genet. (2012) 44:1243–8. doi: 10.1038/ng.2414
96. Xu LD, Ohman M. ADAR1 editing and its role in cancer. Genes (Basel). (2018) 10:12. doi: 10.3390/genes10010012
97. Han L, Diao L, Yu S, Xu X, Li J, Zhang R, et al. The genomic landscape and clinical relevance of A-to-I RNA editing in human cancers. Cancer Cell. (2015) 28:515–28. doi: 10.1016/j.ccell.2015.08.013
98. Shoshan E, Mobley AK, Braeuer RR, Kamiya T, Huang L, Vasquez ME, et al. Reduced adenosine-to-inosine miR-455-5p editing promotes melanoma growth and metastasis. Nat Cell Biol. (2015) 17:311–21. doi: 10.1038/ncb3110
99. Gumireddy K, Li A, Kossenkov AV, Sakurai M, Yan J, Li Y, et al. The mRNA-edited form of GABRA3 suppresses GABRA3-mediated Akt activation and breast cancer metastasis. Nat Commun. (2016) 7:10715. doi: 10.1038/ncomms10715
100. Rossetti C, Picardi E, Ye M, Camilli G, D'Erchia AM, Cucina L, et al. RNA editing signature during myeloid leukemia cell differentiation. Leukemia. (2017) 31:2824–32. doi: 10.1038/leu.2017.134
101. Zipeto MA, Court AC, Sadarangani A, Delos Santos NP, Balaian L, Chun HJ, et al. ADAR1 activation drives leukemia stem cell self-renewal by impairing Let-7 biogenesis. Cell Stem Cell. (2016) 19:177–91. doi: 10.1016/j.stem.2016.05.004
Keywords: endogenous retroelements, hypomethylating agents, 5-azacytidine, decitabine, myelodysplastic syndromes, acute myeloid leukemia
Citation: Kordella C, Lamprianidou E and Kotsianidis I (2021) Mechanisms of Action of Hypomethylating Agents: Endogenous Retroelements at the Epicenter. Front. Oncol. 11:650473. doi: 10.3389/fonc.2021.650473
Received: 07 January 2021; Accepted: 04 February 2021;
Published: 09 March 2021.
Edited by:
Nicholas Short, University of Texas MD Anderson Cancer Center, United StatesReviewed by:
Yue Wei, University of Texas MD Anderson Cancer Center, United StatesThomas Cluzeau, Centre Hospitalier Universitaire de Nice, France
Copyright © 2021 Kordella, Lamprianidou and Kotsianidis. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ioannis Kotsianidis, aWtvdHNpYW5AbWVkLmR1dGguZ3I=