 Kevin Roarty1,2
Kevin Roarty1,2 Gloria V. Echeverria1,2,3,4*
Gloria V. Echeverria1,2,3,4*- 1Department of Molecular and Cellular Biology, Baylor College of Medicine, Houston, TX, United States
- 2Dan L. Duncan Cancer Center, Baylor College of Medicine, Houston, TX, United States
- 3Lester and Sue Smith Breast Center, Baylor College of Medicine, Houston, TX, United States
- 4Department of Medicine, Baylor College of Medicine, Houston, TX, United States
While numerous therapies are highly efficacious in early-stage breast cancers and in particular subsets of breast cancers, therapeutic resistance and metastasis unfortunately arise in many patients. In many cases, tumors that are resistant to standard of care therapies, as well as tumors that have metastasized, are treatable but incurable with existing clinical strategies. Both therapy resistance and metastasis are multi-step processes during which tumor cells must overcome diverse environmental and selective hurdles. Mechanisms by which tumor cells achieve this are numerous and include acquisition of invasive and migratory capabilities, cell-intrinsic genetic and/or epigenetic adaptations, clonal selection, immune evasion, interactions with stromal cells, entering a state of dormancy or senescence, and maintaining self-renewal capacity. To overcome therapy resistance and metastasis in breast cancer, the ability to effectively model each of these mechanisms in the laboratory is essential. Herein we review historic and the current state-of-the-art laboratory model systems and experimental approaches used to investigate breast cancer metastasis and resistance to standard of care therapeutics. While each model system has inherent limitations, they have provided invaluable insights, many of which have translated into regimens undergoing clinical evaluation. We will discuss the limitations and advantages of a variety of model systems that have been used to investigate breast cancer metastasis and therapy resistance and outline potential strategies to improve experimental modeling to further our knowledge of these processes, which will be crucial for the continued development of effective breast cancer treatments.
Introduction
Breast Cancer Metastasis and Therapy Resistance
Breast cancer is the most commonly diagnosed cancer in women and results in 40,000 deaths in the United States annually. The presence of hormone receptors (HR), specifically estrogen receptor (ER) and progesterone receptor (PR), together with expression and amplification of the human epidermal growth factor receptor type 2 (HER2), help to broadly classify breast cancer into three main clinical subtypes: ER/PR+, HER2+, or triple negative (TNBC). The histological classification of breast cancer by HR status largely dictates treatment decisions today. However, the molecular stratification of breast cancer, discovered almost 20 years ago by cDNA microarray of breast tumors, further unmasked intrinsic subtypes of breast cancer. These molecular portraits of breast cancer, now based on a 50-gene classifier (PAM50) (1, 2), help depict the molecular heterogeneity both within and across these intrinsic subtypes and offer a molecular complement to histological classifications. Although standard of care (SOC) therapeutic regimens vary amongst the major breast cancer subtypes, therapy resistance and metastasis remain shared clinical issues for all types of breast cancer.
Metastatic breast cancer accounts for the vast majority of breast cancer related deaths. However, our understanding of this process is still largely evolving. Further complicating this multistep process is the inherent heterogeneity present within a patient’s tumor (intra-tumor heterogeneity) and the fact that the intrinsic classification of breast cancer shapes both the timing and location of metastatic relapse. For instance, while HR+ breast cancers tend to home to the bone and lymph nodes, TNBCs exhibit a preference for visceral organs like the lungs, liver, and brain. HER2+ breast cancers tend to metastasize to the brain after averting HER2-targeted therapies. Additionally, the timing of metastatic presentation also differs by breast cancer subtype, with HR+ tumors typically recurring later than TNBCs after initial presentation and treatment of the primary disease. The predilection of subtypes to home to specific locations in the body, the subtype-dependent variation in recurrence windows, as well as how particular subpopulations of tumor cells within these cancers accomplish metastatic steps remain imperative questions to answer in order to mitigate breast cancer mortality.
Despite considerable appreciation for the subtype-specific differences in timing and location of metastatic disease, knowledge is lacking on how to accurately predict a tumor’s metastatic fitness as well as the specific biological mechanisms instructing the stage-specific steps of the metastatic cascade. The development of in vitro and in vivo models over several decades has helped illuminate the metastatic process. Considerable work remains to improve such models in order to gain molecular insights into metastasis and therapeutic resistance, the primary culprits of cancer-related deaths.
Laboratory Models of Breast Cancer
Metastasis is a multistep process that requires the successful dissemination of tumor cells from the primary site, vascular entry (intravasation) and transit to a distant site, exit (extravasation) from the vasculature into the secondary site, and finally seeding and colonization in the secondary organ site. Importantly, the accomplishment of only one phase of the metastatic cascade by the tumor cell does not necessarily predict successful fulfillment of metastasis as a whole. Thus, experimental models and interpretation of the mechanisms derived from these models is imperative in order to differentiate successful from unsuccessful metastasis and the consequential events dictating a tumor cell’s fitness to evade, spread, and thrive a distant site from the breast. The multistep nature of metastasis and the heterogeneity exhibited within breast cancer warrants the continued use and development of laboratory models to accurately reflect this complicated process in order to discover therapeutic interventions. To date, a compilation of experimental models has shed light on mechanisms surrounding invasion and dissemination, tumor cell dormancy, organ tropism, and microenvironment interactions (Figure 1). How these biological events are shaped by therapeutic interventions adds another level of complexity surrounding metastasis and disease recurrence.
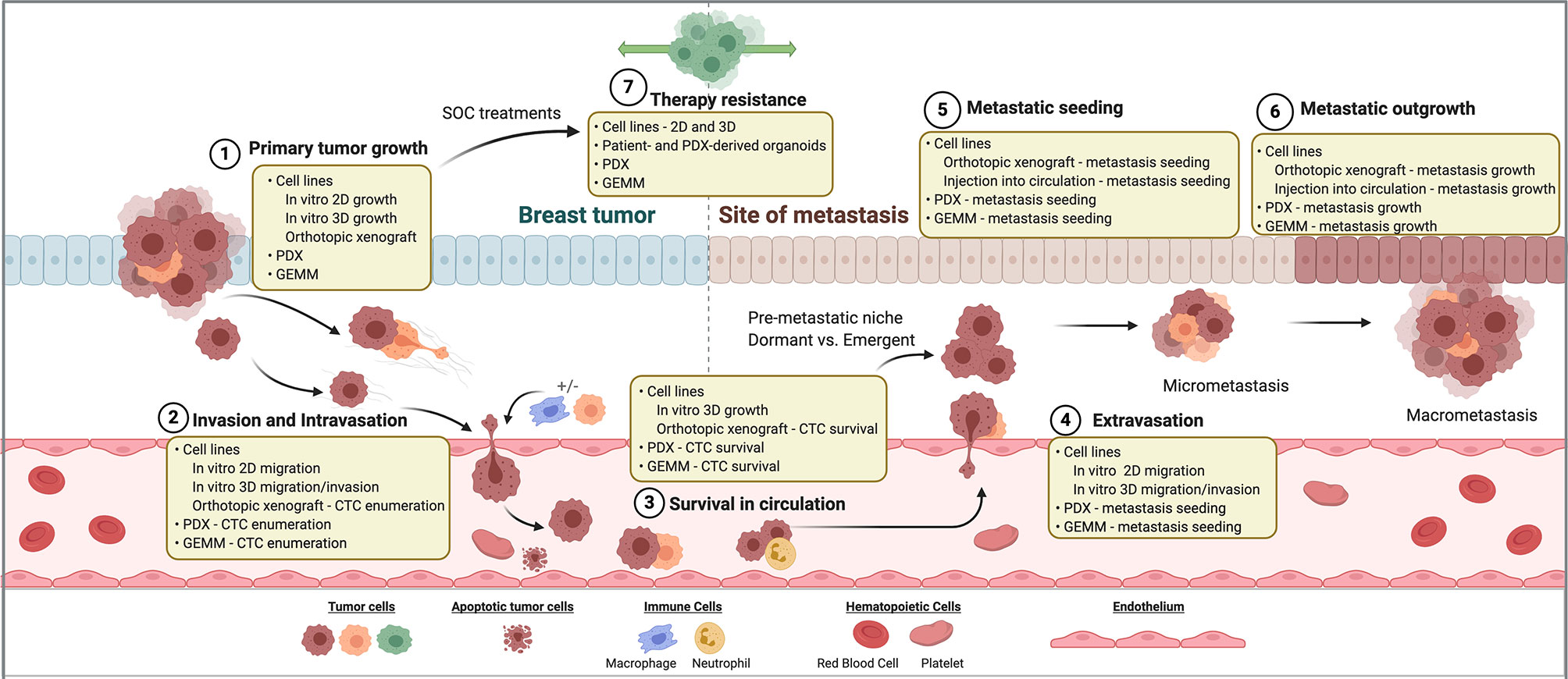
Figure 1 Breast cancer models for investigating therapy resistance and metastasis. Steps of the metastatic cascade and SOC therapy resistance are diagrammed. For each step, classes of laboratory models that may be used to investigate its biology are listed. SOC, standard of care. PDX, patient-derived xenograft. GEMM, genetically engineered mouse model. CTC, circulating tumor cell.
Mechanisms of therapy resistance in breast cancer are diverse amongst breast cancer subtypes and mechanism of action of each therapy. Mechanisms of therapy resistance have been found to be particularly different in the cases of molecularly targeted versus cytotoxic chemotherapies. Therapeutic resistance can be intrinsic, or pre-existing in tumors prior to drug exposure, or acquired following drug treatment. Both intrinsic and acquired resistance can be achieved through clonal evolution (de novo acquisition of mutations or genomic structural changes), clonal dynamics (enrichment and/or depletion of genomic subclones through Darwinian selection), epigenetic adaptations (chromatin modification, transcriptional and post-transcriptional cellular plasticity, microenvironmental crosstalk, metabolic regulation), and acquisition or maintenance of cancer stem-like cell (CSC) features. While some genomic mechanisms of therapy resistance have been appreciated for decades, models to study epigenetic-mediated mechanisms of resistance have been developed more recently. As an added layer of complexity, many non-genomic resistance mechanisms have been found to be reversible, such as drug tolerant or persister cell states. Thus, elucidating the temporal nature of resistance mechanisms is of utmost importance to effectively identify appropriate therapeutic windows. Laboratory models to investigate these complex mechanisms will be discussed below (Figure 1).
Models of Metastasis
The establishment of distant metastasis necessitates the cancer cells to overcome several key hurdles along the journey from the primary tumor to a distant organ. Numerous in vitro and in vivo models have enabled the exploration of mechanisms surrounding the various steps of metastasis, yet the accurate recapitulation of the multi-step process of the metastatic cascade varies drastically from model to model. Though metastasis is traditionally viewed as a linear series of events, often accomplished by the fittest of cancer cells (3), numerous questions remain surrounding not only the mechanisms governing these discrete steps, but also concepts surrounding dormancy and the emergence of metastatic lesions after months to years. The metastatic cascade can also be impacted by somatic mutation-driven mechanisms. For example, numerous ESR1 mutations and gene fusions have been identified in metastatic or liquid biopsies from ER+ breast cancer patients. Introduction of many of these mutations into in vitro and in vivo laboratory models (some even naturally occur in patient-derived xenografts, PDXs) has enabled demonstration that they functionally drive metastasis through aberrant ESR1 signaling (4–7). Further description of these mutations can be found in our discussion of therapy resistance in ER+ breast cancer. On the other hand, the metastatic cascade can also be driven by non-genetic (i.e. epigenetic) mechanisms that can be modeled in the laboratory, such as tumor cell-microenvironmental interactions. The continued mystery surrounding multiple facets of breast cancer metastasis and the need to develop therapies around this advanced stage of disease requires a renewed approach by investigators to develop and use models with increased physiological relevance, whether in vitro or in vivo. Specifically, how experimental models accurately reflect early versus late recurrences, distinguish metastatic risk among patients, and provide an accurate approximation of the metastatic process that can be extrapolated to patients remain imperative questions to answer. The model platforms, as well as the advantages and disadvantages of various systems, will be summarized in the current section (Table 1).
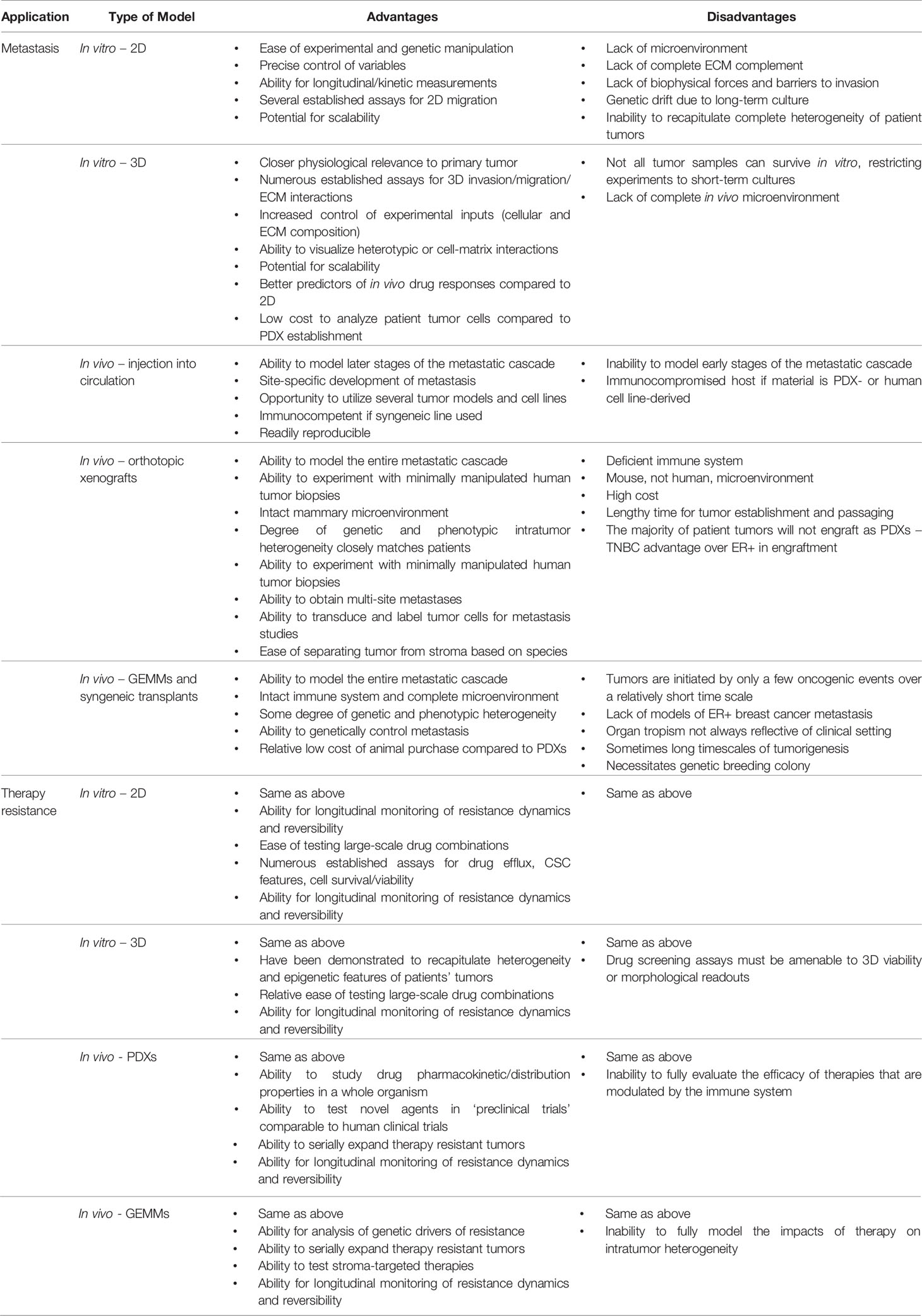
Table 1 Benefits and drawbacks of laboratory models to study breast cancer therapy resistance and metastasis.
In Vitro Models of Metastasis—2D
In vitro models encompass a variety of assays of different structural, microenvironmental, and cellular composition that provide controlled experimental systems to extrapolate cellular processes implicated in the metastatic cascade. Given the elusive biology of metastasis in vivo, in vitro models offer a surrogate approach to interrogate mechanisms responsible for fulfilling discrete steps in the metastatic cascade. Typically, these approaches have been instrumental to examine the functional implications of a particular gene or pathway in metastasis and provide a defined platform to quantitatively assess cell function associated with cell proliferation, survival, invasion, adhesion, and cell–cell and -microenvironment interactions. Additionally, the adoption of more heterogenous cell models through genetically engineered mouse models (GEMMs), PDXs, or primary cells directly from patients for in vitro studies has the potential to significantly enhance our understanding of metastasis.
The initial steps of metastasis require that tumor cells disseminate or invade from the breast. This initial step of metastasis requires that cells gain migration capacity. The scratch or wound healing assay is one such in vitro assay in a two-dimensional (2D) space that measures the ability of a monolayer of tumor cells to fill a “wounded” area created experimentally by introducing a scratch through the cell sheet. Often these assays are applied to studies that query the function of a particular gene in the regulation of migration properties. The application of live-cell microscopy can provide a level of quantitation that enables the establishment of cell migration kinetics over time. Despite the relative ease of the 2D invasion assay, the scalable nature of the method, and the relative flexibility of the system with multiple cell inputs, these 2D cell models differ considerably from in vivo models. Namely, their spatial organization, cell interactions, and intercellular signaling can differ substantially from the physiologically complex three-dimensional (3D) space of a tumor. Indeed, drug screening outcomes in 2D systems often fail to accurately recapitulate the in vivo setting (8, 9).
Cancer cell invasion and dissemination often involve chemotaxis, the directed movement of cells by an extracellular gradient. Boyden chamber assays enable the experimental evaluation of these phenomena by the seeding of cells on an upper chamber and monitoring the migration of cells through a defined porous membrane toward a chemoattractant in the bottom well. Given the separation of migrating vs. refractory cells on the bottom and upper chambers, respectively, migration-competent cells can be recovered and evaluated in response to a particular chemical or physical gradient in an effort to identify subpopulations with potentially distinct invasive potentials. These approaches helped establish the bone-tropic mouse mammary 4T1 carcinoma cells from repeated chemotactic selection in vitro (10). Adaptations of the Boyden chamber have evolved to include additional matrices and cell types to enable the evaluation of other metastatic steps, such as intravasation and extravasation. The modified Boyden chamber assay, for example, includes Matrigel, fibronectin, or collagen I to the trans-well porous membrane in order to model the extracellular matrix (ECM), a critical component in cellular migration. The addition of macrophages and endothelial cells to such a modified trans-well system, termed the subluminal to luminal trans-endothelial migration assay (iTEM), identified the presence of macrophages as an important niche factor for invasive tumor cells highly expressing an actin regulatory protein, MenaINV, to traverse the endothelium during intravasation (11–13). The plating of endothelial cells within this system provided an additional component that enabled the evaluation of invasion through cell-cell junctions of the endothelium and the ECM.
In Vitro Models of Metastasis—3D
3D models have gained considerable attention lately to better recapitulate the multicellular interactions of tumor cells within a defined ECM. These models can be generated from GEMMs, breast cancer cell lines, PDX tumors, or tumors obtained directly from breast cancer patients. In contrast to 2D in vitro systems, 3D approaches provide a platform to study cellular heterogeneity, cellular plasticity, cell-cell, and cell-ECM interactions and have evolved to provide a more physiologically relevant in vitro platform to interrogate the metastatic program (Table 1). Since the advent of organoid cultures for the investigation of cell organization and polarity in 3D basement membrane contexts, molecular insights into the heterogeneity of the primary tumor now demand the adaptation of the 3D system to accurately reflect the level of complexity in vivo. 3D organoid biobanks have emerged as a comprehensive representation of the phenotypic and molecular heterogeneity from patient tumors (14–16). In addition to GEMM and cell line models, they represent an extremely powerful resource for ongoing development of engineered 3D systems as models for metastasis and therapeutic resistance.
3D systems rely on the ECM, known to be intricately involved in breast cancer metastasis. The ECM of both primary tumor and distant metastatic sites are composed of insoluble proteins (e.g., collagen, laminin, fibronectin, and elastin), glycosaminoglycans, and proteogIycans. In particular, the deposition, remodeling, and crosslinking of ECM within the primary tumor regulates both mechanical and biochemical cues for the cancer cells, and “stiffer” tumors often exhibit poorer prognosis (17). Multiple 3D organoid models have implicated matrix composition as a critical regulator of tumor cell transit. For instance, the mode of migration by carcinoma cells, specifically single or collective in nature, is impacted by the presence of Type 1 collagen, independent of the genetic state of the tumor cell (18). Similarly, the conserved cytokeratin 14 (K14+) basal epithelial program orchestrates collective leader-follower cell behaviors during tumor cell invasion in 3D Type 1 Collagen (19). Friedl and colleagues demonstrated that leader cell function depends on a gap junction Cx43-dependent/ADORA1 axis in mediating collective cancer cell invasion (20). Interestingly, cadherins and ECM confinement further cooperate to determine unjamming transitions, coordinated vs. uncoordinated collective cell movements, and fluidization of tumor cells, impacting states of cell transit at matrix bottlenecks (21). Introduction of microfluidic systems by soft lithography techniques to such organoid models further revealed the importance of a chemotactic SDF1/CXCR4 gradient necessary for positioning K14+ leader cells within invasive cellular collectives (22).
While the above 3D organoid models largely focus on mechanisms of tumor cell invasion within the ECM, organotypic cultures have recently evolved in their level of sophistication to address biological questions related to additional stages within the metastatic cascade. For instance, immune cell introduction into 3D organoid models of invasion addresses the immunosurveillance bottleneck encountered by tumor cells, revealing important functions for natural killer cell and tumor cell crosstalk on the invasion of K14+ cells (23). Reconstitution of 3D cultures of established breast cancer cell lines with immune cells offers additional models to interrogate immune- and tumor-cell interactions in vitro (24). Organotypic models have more recently been developed to model the metastatic niche, where questions of tumor cell dormancy and colonization can be addressed. For instance, in vitro co-cultures of organotypic microvascular niches and disseminated tumor cells (DTCs) identified the importance of the microvascular niche in distinguishing states of tumor cell dormancy versus emergence based on thrombospondin-1 and TGF-β availability (25). Additional complex organotypic cultures, such as the Bone-In-Culture-Array (BICA) have been developed to determine mechanisms of early-stage bone colonization (26, 27).
Organotypic cultures of the metastatic niche provide an important platform for drug screening. For instance, BICA revealed the utility of danusertib, an Aurora kinase family inhibitor, as a potential therapeutic inhibiting early-stage bone colonization (26). Moreover, DTCs were protected from chemotherapy by an α5β3 and α4β1 integrin-mediated interaction with the perivascular niche (28). Using organotypic cultures, integrin inhibitors disrupted this protection and rendered DTCs susceptible to chemotherapy. Thus, tailored drug screening using organotypic cultures of breast cancer cells and cells of the microenvironment offer more high-throughput and less costly alternatives to therapeutic testing in vivo.
In Vivo Experimental Models of Metastasis
Experimental metastasis refers to the introduction of tumors cells directly into the vascular system, circumventing the early stages of the metastatic cascade. This approach has been useful to explore the functional roles of distinct genes in metastatic colonization and to test therapeutic agents in late-stage metastasis. Importantly, experimental models of metastasis simulate extravasation and colonization in the secondary site, reflecting later stages of disease, as opposed to spontaneous models (described below), which model the full extent of the metastatic cascade. In the case of PDXs and human cell lines, the majority of these injection studies are conducted in immunodeficient mice, precluding analysis of the immune system. Despite this limited snapshot of the metastatic process, the application of such an approach by Fidler and colleagues sparked the landmark discovery that only subpopulations of cells possess metastatic abilities, and these could be clonally selected to derive lines with enhanced metastatic seeding to a particular organ (29).
Importantly, experimental models of metastasis are largely dictated by the site of injection and inherent tropism of the tumor cells. Although these studies rely heavily on the lodging of tumor cells into the first capillary bed encountered downstream of the location of vascular delivery, mechanisms of Paget’s seed-and-soil hypothesis have been pursued to identify factors involved in organ-specific metastasis (3). For instance, lateral tail vein injections largely result in pulmonary metastases (30), intracardiac injections prompt metastasis in the bone and brain (31), intracarotid injection similarly route to the brain, and intra-iliac artery injections selectively seed bone metastasis (32). Using such approaches, studies were performed to identify genes that orchestrate breast cancer metastasis to specific organs. One widely used model, the lung-tropic MDA-MB-231 LM2 cells, was derived by selection of a subline from the parental MDA-MB-231 TNBC cells with greater metastatic proclivity to the lungs (30). Similar bone-tropic (33) and brain-tropic (31) sublines of MDA-MB-231 cells were also derived using similar methodologies. Experimental metastasis models have been instrumental to establish metastatic derivatives of other human and mouse breast cancer cell lines, such as MCF7 (34), 4T1 (35), and T47D (36). Thus, collective efforts over the years have leveraged the experimental metastasis model and the utility of such a model to dissect mechanisms of extravasation and tumor cell colonization. While noteworthy, these studies exclude earlier stages of metastasis, limiting the full physiological comparison to appropriately model aspects of the selective pressures encountered by tumor cells within the earlier stages of the metastatic cascade, the potential interclonal tumor cell interactions required throughout the metastatic process, additional tumor-host cell interactions during transit, and the elusive biology surrounding tumor cell dormancy. Despite these limitations, experimental models of metastasis have provided a reproducible approach to interrogate aspects of metastatic fitness. Recently, a sophisticated strategy involving lentiviral barcoding and scaling across several human basal-like cell lines as proof-of-principle used pan-cancer PRISM cell line pools for high-throughput metastatic potential mapping (37). Using this approach, an altered lipid metabolism state was associated with brain metastasis in basal-like breast cancer. Though this pan-cancer “MetMap” lacked the context of an intact immune system, such a study provides a valuable resource to probe metastatic potential across tumor types.
In Vivo Orthotopic Xenograft Models of Metastasis
A major advantage of orthotopic models of breast cancer metastasis, in which breast cancer cells are engrafted into the mammary glands of mice, is that they capture all steps of the metastatic cascade. These models enable direct comparison of primary tumors, circulating tumor cells (CTCs), and metastases matched within the same animal. Importantly, some models metastasize to multiple secondary sites, enabling comparisons of tumor cells growing in distinct secondary organ microenvironments. Numerous breast cancer cell lines have been orthotopically xenografted into mice for CTC and metastasis studies (33, 38, 39). PDX models, in which never-cultured biopsies are obtained from patients and directly engrafted into mice, have been found to capture molecular features and heterogeneity of originating patients’ tumors and serve as a renewable resource of minimally manipulated human tumor cells (16, 40–42). The primary disadvantages of these models are: 1) the requirement of using immune-compromised mice, thus precluding assessment of the impact of a fully intact immune system on metastasis, 2) the often-lengthy duration of experiments, regularly up to 12 months, and 3) the costly nature of immune-compromised animal purchase and long-term housing. A major need in the field is the broad implementation of xenograft models in mice with ‘humanized’ immune systems.
Ideally, PDXs should reflect the full range of cellular heterogeneity and disease progression across breast cancers. The PDX consortium, a shared effort comprised of several academic institutions, has amassed 537 PDX lines representing 500 patients (40). An open question remains regarding how accurately these PDXs reproduce the metastatic behavior of the patient’s tumor, as well as more general metastatic characteristics associated with breast cancer subtype. Although considerable evidence exists that these PDXs can produce CTCs and generate micro- and macroscopic metastatic lesions within several distant sits in the mouse (41, 43–45), a full credentialization of the metastatic propensity of this vast tissue resource remains an evolving collective task. Given that ER+ cancers typically exhibit longer latency and a proclivity to metastasize to bone, the development of humanized mouse models in which breast cancer PDXs metastasize to human bone implants has created a highly reliable system to interrogate late-stage metastasis to the bone (46). Specifically, bone discs from femoral heads of patients undergoing hip replacement surgery were implanted subcutaneously into NOD/SCID mice. This model system resembles a prior human-in-mouse bone system where breast cancer cell lines, instead of PDXs, were used (47). Nonetheless, human bone was the preferred site of metastasis for ER+ PDXs over mouse bone, while TNBC PDXs metastasized at a lower rate to bone, but with an increased frequency of visceral metastasis. Thus, PDX models can accurately recapitulate site-specific preferences of metastasis for breast cancer subtypes.
Spontaneously arising metastases in PDX models, sometimes even to distinct secondary organs, enable powerful comparisons that are usually impossible in the clinical setting due to limited availability of metastatic specimens. PDX models have been found to faithfully recapitulate secondary organ tropisms of their originating patient tumor (40, 41, 43). A major benefit of PDX models is the difference in species between the tumor and stromal compartments, enabling relative ease of separating these in the laboratory and informatically. While markers universally recognizing human tumor cells are uncommon, human CD298 has been used with success to isolate viable human tumor cells from early- and late-stage PDX mammary tumors and lung metastases (48, 49). Obtaining macroscopic metastatic lesions from PDX models, especially in secondary organ sites aside from the lung, is extremely uncommon. Incorporating survival surgery, in which mammary tumors are grown nearing ethical tumor burden endpoints, then resected, enables monitoring of mice for longer periods to allow detectable metastatic lesions to arise. This approach has been used with success in several PDX models, some of which metastasize robustly to multiple secondary organs. This methodology is majorly bolstered by incorporation of in vivo imaging constructs (e.g. bioluminescent markers), allowing in vivo and ex vivo detection of metastatic lesions from multiple secondary organs of the mouse (50, 51). These models have also enabled comparison of tumor cell subpopulations growing as primary tumors, CTCs, and metastatic lesions. In particular, in vivo modeling of CTC tumor cell biology to capture vascular transit has been demonstrated directly from patient blood specimens together with in vivo validation in cell line xenografts. The differential labeling of the MDA-MB-231 LM2 cell line with eGFP and mCherry fluorescence enabled the detection of multicolor CTC clusters in circulation, which were later shown to be oligoclonal precursors of metastasis to the lung requiring plakoglobin for collective tumor cell transit (52). Interestingly, such CTC collectives preferentially arose in hypoxic areas of the tumor, as demonstrated in patient and cell line specimens (53). Similar studies using MDA-MB-231 or murine 4T1 cell lines further demonstrated the requirement of neutrophils to facilitate CTC cluster cell cycle entry, heightening metastatic conditioning in the circulation (54). While powerful, extrapolation of such approaches to cell line or PDX models necessitates prior introduction of lentiviral or alternative cell labels for accurate tracking and identification of rare cell populations in vivo.
Enrichment and Screening of Metastasis With In Vivo Xenograft Models
To identify genes suppressing colonization of the lung, a high-throughput RNAi screen of ~1,000 genes was conducted by intravenously injecting pools of mouse mammary tumor 4T1 cells expressing siRNA constructs into Balb/c mice (55). Bioluminescence imaging was used to quantify lung colonization for each of 48 pools, and next-generation sequencing was used to identify siRNAs enriched in lung lesions. This screen identified alpha-N-acetylgalactosaminide alpha-2,6-sialyltransferase 2 (St6GalNAc2) as a novel metastasis suppressor that acts through its O-glycanation of the surface of tumor cells. A major advantage of this approach is use of immune-competent Balb/c mice. While this screen focused on the final steps of the metastatic cascade (colonization and outgrowth in the secondary organ site), additional screens encompassing the entire metastatic cascade from the orthotopic site are warranted in order to piece together mediators of specific phases of metastasis. Genetic screens focused on the regulation of CTCs have shed light on important regulators of CTC composition and function during vascular transit. One such screen entailed a CRISPR-Cas9 loss-of-function mini-pool screen in vivo to evaluate guide RNA dropouts, with Vcam1 identified as a necessary factor for CTC-neutrophil interactions (54). Additionally, an in vivo genome-wide CRISPR activation screen was performed on CTCs to screen for pro-metastatic genes. Together with single cell RNA sequencing from patient CTC specimens, Rpl15-dependent ribosomal protein upregulation was implicated in proliferative and survival cues for CTCs in vivo (56).
Orthotopic xenograft models have been a rich model system with which to conduct in vivo functional genomics screens for genes driving or suppressing metastasis. A recent study employed TNBC PDX tumor cells transduced with an ORF library orthotopically injected into mice, then utilized bioluminescence imaging to obtain lung metastases. Genes decreasing lung metastasis latency were then identified by next generation sequencing of lung lesions (57). This custom ORF library was constructed to over-express genes identified from differential expression analysis of human genes identified by RNA sequencing of lung metastases and matched mammary tumors from PDX models and successfully identified a validated driver of breast cancer metastasis, CEACAM5, that is currently under clinical investigation. While in vivo metastasis screens are arguably one of the most powerful approaches available to identify genes with a bona fide function in the metastatic cascade, these screens are costly and, especially in the case of orthotopic xenografts, can require long periods of time. Thus, focusing such screens on a prioritized subset of genes is critical to minimize the cost and scale of this undertaking.
Genetically Engineered Mouse Models and Syngeneic In Vivo Transplant Models of Metastasis
A considerable number of GEMMs exist that utilize constitutive or inducible transgenic approaches to model tumor progression and metastasis. By far the most widely used system is the Mouse Mammary Tumor Virus (MMTV) LTR promoter, among several other promoters (WAP, BLG, and C- (3)1) (58), that has been used to readily drive the expression of transgenes specifically in the mammary epithelium. Key oncogenes explored within the mammary epithelium include ErbB2/Neu (59), polyoma middle T antigen (PyMT) (60), Simian virus 40 (SV40) (61), Wnt1 (62), TGF-α (63), c-Myc (64), and H-Ras (58). MMTV-Neu and MMTV-PyMT represent two of the most well-characterized transgenic mouse models of mammary tumorigenesis, which readily metastasize to the lung, albeit at different rates (58).
By far, the most widely utilized models over the past 20 years include the MMTV-Neu and MMTV-PyMT models. MMTV-neu transgenic mice develop multifocal mammary tumors at a median age of 7.5 months and metastasize to the lungs (65–67). MMTV-PyMT mice, on the other hand, metastasize with higher frequency and shorter latency (60). Recent integrative genomic analyses of both models identified critical parallels with human breast cancers, particularly copy number alterations in key ECM and other proteins that drive metastasis in these models (68). Over the years, both models were instrumental in establishing the biological functions for the TGF-β (69–72), Wnt (73), and EGF (72) pathways in breast cancer progression and metastasis. Importantly, these models incorporated the thorough examination of endogenous tumor–stroma interactions associated with metastatic progression (74). As genetic and technological advances developed, higher resolution cell biology and live microscopy approaches unveiled previously furtive cellular interactions occurring along the metastatic cascade. Findings from such studies unveiled important tumor cell-macrophage interactions critical for vascular leakage and intravasation (75–77). MMTV-PyMT transgenic mice were also utilized to uncover collective tumor cell interactions during invasion, ultimately responsible for oligoclonal metastasis (78). Follow-up studies further implicated nanolumenal signaling between tumor cell clusters via the molecule epigen during oligoclonal metastasis (79). To more accurately depict breast cancer subtype, the TP53-null syngeneic transplant model of mammary tumorigenesis comprises a biobank of tumors that reflect heterogeneity of human breast cancers at the molecular and histological levels (80–82). Importantly, the TP53-null syngeneic transplantable GEMM harbors an intact immune system, which has been an instrumental modulator of metastatic propensity to the lung (83). Given the molecular and histological representation of cellular heterogeneity, this transplant model has enabled the study of various aspects of the metastasis and therapeutic resistance (83, 84) Establishment of organ-tropic models from this heterogeneous GEMM will provide an invaluable resource to study the contributions of inter- and intra-tumor heterogeneity (Roarty, unpublished). The foremost advantage of these GEMMs is the ability to experimentally probe the entirety of the metastatic cascade in the context of an intact immune system.
Spontaneous models of metastasis also hold great promise to unravel mechanisms of tumor cell dormancy in the metastatic niche. A persistent mystery in cancer biology is the “lag” or emergence of metastasis several months, years, or decades following removal of the patient’s primary tumor. Although it is appreciated that the time-to-relapse and cancer cell tropism exhibited in breast cancer are dictated largely by the intrinsic subtype of the tumor (85), the exact timing of dissemination during cancer progression and how such fleeing cells later emerge as metastatic lesions remains unknown. Several lines of evidence demonstrate a lack of linearity in the metastatic process. In patients, disseminated tumor cells in the bone marrow were found to harbor fewer genetic alterations than the primary lesion, suggesting that these precursors arose earlier rather than later in advanced stages of disease progression (86). Mouse models have molecularly exposed this lack of linearity seen in humans (87), where non-invasive mammary intraepithelial neoplasia (MIN), arising in both MMTV-neu and MMTV-PyMT transgenic models, were capable of releasing disseminated cells into the circulation of mice, leading to micrometastasis within the bone marrow and lungs (88). Such early disseminated cancer cells can fulfill all steps of metastasis, as has been demonstrated in the MMTV-neu model, where Wnt signaling and a hybrid EMT-dependent program enable metastasis after a period of dormancy (89). The switch from dormant to active metastatic states is an ongoing area of investigation, but one that is yielding interesting findings of the constant interplay between cancer cells and their extracellular and immune microenvironment in this process (25, 90–94). Thus, the utility of mouse models to interrogate the molecular regulation of dormant versus active metastatic states will be an imperative endeavor to provide important therapeutic insights.
The recent success of immune checkpoint inhibitors (ICIs) in improving patient outcomes has only amplified a growing interest the application of such therapies to breast cancer (95). Syngeneic models of metastasis offer a unique opportunity to interrogate the immune landscape and immune cell responses in the tumor microenvironment. Early work in the MMTV-PyMT transgenic model, harboring a homozygous null mutation for the gene encoding the macrophage growth factor, colony-stimulating factor-1 (CSF-1), demonstrated that macrophages were necessary for metastatic progression in vivo (96). As mentioned above, tumor-associated macrophages play multiple roles in promoting cancer metastasis by secreting epidermal growth factor (EGF) to promote motility, invasion, and ECM degradation by cancer cells (97). Such models have additionally implicated adaptive immune cells, IL-4 expressing CD4+ T lymphocytes, in indirectly promoting invasion and metastasis by regulating the phenotype and effector function of CD11b+Gr1-F4/80+ macrophages, ultimately modulating EGF signaling within the cancer cells (98). Other murine models like the K14cre;Cdh1f/f;Trp53f/f (KEP) model further highlighted the importance of immune cell function in tumor progression by demonstrating a role for neutrophil expansion during tumor progression by a γδT cell/IL-17/neutrophil axis (99). Separately, in the syngeneic TP53 null transplant model of mammary tumorigenesis, the dichotomous distribution of macrophages and neutrophils in murine tumor models was identified, further emphasizing the need for improved characterization of inter-patient heterogeneity of the myeloid compartment (100). At present, TNBC represents the most promising candidate for ICIs given the presence of immune cell infiltrates in subsets of these patients and a higher somatic mutation burden relative to non-TNBC. More recently, the utilization of “mutagenized” tumors by overexpression of the APOBEC3B enzyme in credentialed GEMMs further demonstrated the utility of mouse models in the identification of mechanisms of response to ICI therapy involving B cells and CD4+ T follicular helper cells (101). Future studies using relevant mouse models will be imperative to uncover the spatiotemporal exchanges between cancer and immune cells across both the primary metastatic cellular landscape in an effort to effectively develop novel immunotherapeutic approaches for advanced-stage breast cancers.
Standard of Care Therapy Resistance in Breast Cancer
Although SOC regimens vary amongst the major breast cancer subtypes, therapeutic resistance is a major clinical issue in each. The foremost classes of targeted therapy used in ER+ breast cancer are selective estrogen receptor modulators (SERMs; e.g. tamoxifen), selective estrogen degraders (SERDs; e.g. fulvestrant) or aromatase inhibitors (102). SOC for HER2+ breast cancers include anti-HER agents such as small molecule inhibitors, HER2 blocking antibodies, or HER2 antibody drug conjugates (ADCs). As TNBC lacks these cell surface proteins, SOC agents in this setting are currently limited to cytotoxic chemotherapies. In the case of BRCA1/2 deleterious mutant carriers, patients who are often triple negative, PARP inhibitors are currently approved for use in the metastatic setting and are under investigation for use in the neoadjuvant setting.
Mechanisms of therapy resistance can be categorized as: 1) acquired either permanently or reversibly, and either clonally or sub-clonally, following treatment, or 2) pre-existing clonally or sub-clonally prior to treatment. Acquired or pre-existing resistance can be mediated by genomic events (mutations, copy number alterations, genomic structural variants), transcriptional programs, epigenetic modification of chromatin, post-transcriptional regulation of RNA and/or protein levels, and metabolic rewiring. Reversible resistance is often referred to as drug-tolerance or adaptation of “persister” cell phenotypes. These molecular changes can ultimately mediate resistance by enhancing efflux or breakdown of drugs, blocking drug uptake, inhibiting drug-mediated apoptosis, adaptive programs of repair and survival, or protection of CSC features. Tumor cell extrinsic mechanisms driving resistance such as immune system escape, have also been identified. Furthermore, tumor cell dormancy has been found to contribute to therapy resistance, especially in the ER+ subtype with characteristically late-arising metastatic/therapy resistant relapses. In contrast, TNBCs typically exhibit relapses on the scale of only a few years after diagnosis (103). Here we discuss the variety of experimental models that have been used to gain insights into breast cancer therapy resistance.
In Vitro Models of Therapy Resistance—2D
Established breast cancer cell lines provide a tractable platform with which to functionally dissect the roles of putative drivers of resistance discovered by profiling patients’ biopsies. Although these models lack the often important microenvironmental cues of in vivo systems, they have provided valuable insights about the biology of breast cancer resistance. While systematic analyses of SOC therapy resistance mechanisms across a multitude of models within each major breast cancer subtype are yet incomplete, some studies have provided snapshots of these mechanisms in defined contexts as described below (Figure 1, Table 1).
Drug Tolerant States, Epigenetic Phenotypes, and Metabolic Rewiring
Breast cancer cell lines offer the opportunity to study intra-tumoral heterogeneity and cellular plasticity as they pertain to therapeutic resistance. In an effort to investigate targeted therapies not yet approved as SOC for breast cancers, modeling of the “drug tolerant persister” (DTP) cell subpopulation in basal-like breast cancer cell lines after acute treatment with therapies such as MEK or BRAF inhibitors revealed that epigenetic plasticity, rather than Darwinian selection, was associated with resistance. This study demonstrated that targeting this epigenetic plasticity with a BET inhibitor abrogated the DTP state and cell survival (104). Acute treatment of a broad panel of cancer cell lines, including HER2-positive breast cancer cell lines, with tyrosine kinase targeted inhibitors revealed chromatin modification-mediated adaptation of the DTP state is a common feature of cancer cells (105). A study of ER+ breast cancer cell lines revealed the histone demethylase KDM5 contributed to fulvestrant resistance. KDM5 was found to drive transcriptomic intra-tumor heterogeneity as evidenced by single cell RNA sequencing of cell lines. Single cell analyses and cellular barcode-mediated lineage tracing revealed that the fulvestrant-resistant phenotype pre-existed in a low-abundance genomic subclone prior to treatment of cell lines (106). Furthermore, cell line-based studies of resistance to experimental epigenetic-targeted therapies such as BET bromodomain inhibitors have revealed potential synergistic drug combinations that may prove useful clinically in the future (107, 108).
Treatment of TNBC cell lines with SOC chemotherapy was found to result in adaptation of a polyploid “giant cell” phenotype, a morphological feature that has been observed in chemotherapy-treated human breast tumors (109). These resistant cell lines were characterized by metabolic reprogramming that may provide novel therapeutic opportunities for treating chemoresistant TNBCs. Other studies of acute chemotherapy treatment of MCF7 cells revealed increased expression of proteins related to apoptosis signaling and redox homeostasis (110). Serial analyses of pre- and post-chemotherapy TNBC biopsies has nominated putative drivers and suppressors of adaptive survival programs in post-chemotherapy residual disease. Functionalization of the putative resistance drivers MYC and MCL1 in TNBC cell lines revealed they mediated CSC features through rewiring of mitochondrial oxidative phosphorylation (111). Conversely, the putative resistance suppressor DUSP4 was found to be silenced in post-chemotherapy TNBCs, thus removing its inhibition of ERK signaling (112). Taken together, these studies revealed that breast cancer cell lines can model dynamic, reversible mechanisms of SOC therapy resistance. It is possible these epigenetic mechanisms of therapy resistance are prominent in the context of TNBC due to the lack of a unifying oncogenic driver in this subtype.
ER and HER2 Pathway Resistance Mechanisms
Numerous ESR1 mutations and gene fusions, reviewed recently (113), have been identified in patient tumor sequencing data associated with resistance and relapse in HR+ positive breast cancers. Many of these mutations have been introduced into breast cancer cell lines for mechanistic studies. For example, the K303R ESR1 mutation, identified in patient tumor sequencing data and ectopically expressed in the MCF7 ER+ cell line, was demonstrated to confer aromatase inhibitor resistance through increased downstream PI3K and IGF1R pathway activation (114, 115). Recurrent ESR1 activating mutations, such as Y537S, Y537N, and D538G, and gene fusions such as ESR1-YAP1 and ESR1-PCDH11X, frequently identified in metastatic ER+ breast cancers, have been introduced into breast cancer cell lines to reveal their role in driving SERM and SERD resistance and to identify collateral lethalities associated with these frequently observed mutations (4–7). ESR1 mutations associated with resistance in breast cancer patients have also been found to naturally occur in ER+ breast cancer cell lines grown under long-term estrogen deprivation (LTED). These LTED cell lines eventually resume proliferation in the absence of estrogen supplementation and were found to harbor a subclonal Y537C mutation (116). Thus, cell lines naturally evolving estrogen-independent growth mechanisms provide an additional system with which to study ESR1 biology. Recently, loss of neurofibromin (NF1), identified in breast cancer patient sequencing data as associated with poor outcomes, was demonstrated in ER+ breast cancer cell lines to function as a transcriptional co-repressor of ER. These findings were then translated in vivo using cell line xenografts and PDXs, enabling preclinical trials demonstrating novel therapeutic combinations to treat NF1-low ER+ breast tumors (117).
Anti-HER2 therapy resistance mechanisms include genetic alteration of HER2 itself, reactivation of downstream HER2 signaling, or activation of compensatory pathways (118). These mechanisms have been investigated in a multitude of HER2-positive breast cancer cell lines. For example, long-term exposure of HER2+ cell lines to anti-HER2 drugs revealed that resistance could be conferred through upregulation of ER signaling (119). Xenograftment of HER2-amplified cell lines or ER+ cell lines genetically engineered to over-express HER2 has provided a platform with which to compare the efficacies of anti-HER2 agents in combination with anti-estrogen and targeted therapies (120, 121). Recent studies of HER2+ cell lines and genetically engineered mouse models (MMTV-rtTA/HER2) revealed HER2 therapy resistance can be mediated by cyclin D1/CDK4 and EGFR signaling, providing promising therapeutic targets to overcome resistance that are currently in clinical testing (122).
Cancer Stem-Like Cells and Drug Efflux
Numerous studies have demonstrated a critical role for CSCs or tumor-initiating cells (TICs) in driving breast tumorigenesis, resistance, and metastasis. These cells can be distinguished from the non-TIC population based on cell surface marker expression (123) and have been identified in human tumors, breast cancer cell lines, GEMMs, and PDX models. Studies in breast cancer cell lines have demonstrated that following exposure to SOC chemotherapies, CSC, TIC, and EMT features and functions can be elevated in cells of the various major subtypes of breast cancer (124–127). These models have provided a robust platform with which to characterize and target transcriptional and signaling regulators of CSC features. Furthermore, breast cancer cell lines with mesenchymal properties were found to exhibit more chemoresistance than were epithelial-like or “hybrid EMT” breast cancer cells (128). As opposed to administering chemotherapies to breast cancer cells grown on plastic, HER2+ breast cancer cells have been xenografted into immune-compromised mice which were then treated with chemotherapy. Ex vivo analyses of cells derived from those tumors revealed that chemotherapy exposure in vivo had enriched for CSC/TIC features that were maintained in cultures derived from those tumors (129).
Subsets of breast CSCs, termed the “side population”, have been identified that have high expression of drug efflux proteins and are resistant to chemotherapeutics due to their ability to expel drugs from within the cells. This population has been observed in breast cancer cell lines (130). Breast cancer cell lines were used to determine that ROR1, an upstream regulator of the drug efflux pump ABCB1, contributes to chemotherapy resistance and is correlated with CSC features and poor therapeutic responses (131). Importantly, the CSC and drug efflux features of breast cancer in vitro models have also been observed in biopsies obtained directly from patients. Development of anti-CSC therapies is a major topic of current investigation in the field and is expected to perturb both therapy resistance and metastasis.
In Vitro Models of Therapy Resistance—3D
Recent advances in 3D organoid culturing methodologies have revolutionized the ability to test SOC and investigational agents in patient- and PDX-derived cells. A major advantage of these organoid models is the relatively low cost and high efficiency when compared with mouse PDX establishment. A biobank of 95 patient-derived primary and metastatic breast cancer organoids was recently described that preserves many of the histologic and genomic features of donor patient’s tumors. These organoids were leveraged for high-throughput drug screening. Interestingly, direct comparison of tamoxifen response in patients with their matched organoid cultures revealed congruent responses (14). Similarly, organoids have been derived from orthotopic PDX models, enabling high-throughput drug screening with panels of SOC and experimental compounds, providing novel avenues for preclinical drug testing (132) and synergistic combinations (133). Direct genomic and pharmacologic comparisons of organoids in vitro and tumors derived from orthotopic xenotransplantation into mice has revealed a high degree of concordance (16). Together, these studies reveal that patient- and PDX-derived organoid cultures are promising platform with which to efficiently and speedily test the efficacies of SOC and investigational therapies for clinical translation. There is a great deal of excitement that the relative speed and ease of investigational drug testing in patient-derived organoid cultures, when compared with establishment of PDX mice, will finally enable rapid, real-time, implementation of personalized therapies tailored for patients exhibiting resistance to SOC therapies.
In Vivo Cell Line Xenograft PDX Models of Therapy Resistance
PDX models enable experimentation with minimally manipulated human tumor cells in an organismal microenvironment, one that albeit lacks a fully functional immune system. Several studies have utilized these models to study SOC therapy resistance, revealing novel biological insights and trends matching those observed in patients’ tumors. These models also afford the ability to study the conjoined phenotypes of metastasis and therapy resistance, which often co-occur in models and in patients. Two main approaches have been used with these models: 1) discovery-based approaches in which SOC agents are administered to PDXs, then tumors are sampled longitudinally to identify mechanisms of resistance, and 2) preclinical testing approaches monitoring the efficacy of experimental agents or combinations with SOC.
As an example of a discovery approach, treatment of TNBC PDX models with standard front-line chemotherapies revealed diverse responses across models derived from distinct patients. A subset of models harbored resistance accompanied by a reversible drug-tolerant phenotypic state in the absence of clonal selection. Lentiviral barcode-mediated clonal tracking in these models enabled monitoring of clonal architecture throughout treatment in vivo and, combined with transcriptomic profiling, revealed targeted therapy options that were translated into preclinical trials in PDXs (134). Studies such as these have revealed novel therapeutic avenues such as oxidative phosphorylation inhibition in the case of TNBC (134, 135). A longitudinal profiling study of long-term single-agent taxane treatment of TNBC PDX models delineated dynamic maintenance of TIC populations as resistance arose (136). A study of BRCA1-deficient PDX models was conducted to longitudinally characterize resistance to SOC chemotherapies and PARP inhibitors. This study identified previously known, as well as novel, mechanisms of BRCA1 reactivation, including de novo gene fusion events (137). In each of these studies, aspects of these resistance mechanisms were validated in unmanipulated patients’ biopsies, revealing that PDX models are effective tools with which to discover bona fide resistance drivers with clinical relevance.
In the second type of approach, PDX models have also proven a robust platform with which to test the efficacy of experimental and repurposed anticancer drugs, such as BET bromodomain inhibitors in TNBC (138). In the HER2+ breast cancer setting, PDXs were instrumental in demonstrating the efficacy of CDK4/6 inhibition in overcoming anti-HER2 therapy resistance (122). As discussed above, ESR1 mutations contribute to therapy resistance and metastasis in ER+ breast cancers. PDX models bearing naturally occurring ESR1 mutations have been valuable tools with which to test endocrine therapies (5) and targeted inhibitors against oncogenic kinases such as RON to overcome endocrine therapy resistance (139). Furthermore, use of PDX models affords the capacity to test the efficacy of stroma-targeted therapies such as anti-angiogenesis agents (140) and endothelium-targeted chimeric antigen receptor T cells (141). As these models lack an intact immune system, most PDX studies to date have focused on tumor cell-intrinsic mechanisms of resistance. It will be of vital importance to expand these studies to PDX models with ‘humanized’ immune system components as those technologies evolve in the future.
In Vivo GEMMs of Therapy Resistance
Preclinical GEMMs, in addition to their ability to model several aspects of tumor progression, can be leveraged to provide insights into the mechanisms of therapy response and resistance. One such model recapitulated BRCA1-mutated breast cancer by means of K14Cre;Brca1fl/fl;Trp53fl/fl (KBIP) genetics. In particular, these tumors exhibited a hypersensitivity to platinum drugs and PARP inhibitors, yet like patients, GEMMs succumbed to acquired resistance (142, 143). These tumors up-regulated drug efflux transporters and homologous recombination. GEMMs have also enabled the identification of several other mechanisms of therapeutic resistance, involving a stroma-related gene signature as a predictor of resistance to neoadjuvant chemotherapy (144, 145), stromal-derived exosome uptake as a determinant of radiation- and chemotherapy-resistance (146), and tumor-associated fibroblast promotion of Her2-targeted resistance through FGFR2 (147). Given the accurate reflection of breast cancer subtypes by GEMMs, the testing of new drugs, combinations, and schedules can be evaluated in such models to provide predictive value for patients (148). Much like the isolation and selection of metastatic derivatives, GEMMs can be used to serially expand therapeutically resistant tumors, propagate them, and then test and screen for therapeutic vulnerabilities in the resistant setting (149). Relative to PDX models, lower cost is a significant advantage to the use of GEMMs; however, they only represent surrogates to their patient counterparts and do not always reflect the complex genomic intra-tumor heterogeneity observed in breast cancer patients’ tumors.
Tumor Dormancy and Microenvironmental Impacts on Therapy Resistance
As described above, tumor cell dormancy in the context of DTCs that have seeded at metastatic sites but not yet outgrown, is a major issue due to their ability to evade therapeutic treatment and their long-term survivability (27). DTCs have been found to persist at metastatic sites, often undetected by standard clinical means, for many years and are thought to lead to the often-late relapses observed in ER+ cancers. Available models to study metastatic dormancy were recently reviewed (150). DTCs were identified in the bone marrow of Balb/c immune-competent mice following orthotopic implantation of mouse mammary tumor 4T1 cells and surgical resection of primary tumors. These DTCs were shielded from killing by standard cytotoxic chemotherapies by the bone marrow microenvironment (specifically, the vascular endothelium). Therapeutic inhibition of the interaction between DTCs and the endothelium prevented eventual bone metastasis in these models (28). Numerous studies describing the role of tumor cell dormancy in therapy resistance have been reviewed recently (151). For example, in ER+ breast cancer cells made resistant to endocrine therapy, dormancy gene expression signatures were identified by single cell RNA sequencing (152). Furthermore, in vitro dormancy models have been used to demonstrate bone marrow secreted factors are able to induce ‘re-awakening’ (i.e. growth) of dormant ER+ breast cancer cells (153).
The contribution of stroma to therapy resistance is also an active area of investigation, especially leveraging in vivo models comprising stromal compartments. For example, analysis of BRCA mutant TNBCs unexpectedly revealed extensive macrophage infiltration in this subtype. Use of ex vivo macrophage cultures, PARP-deficient GEMMs, and BRCA-deficient xenografts revealed that PARP1 aides in macrophage development and that combination of a PARP inhibitor with a CSF1 receptor-blocking antibody enhanced tumor responses in the BRCA-mutant setting (154). Numerous studies using in vitro and xenograft models have also revealed a functional role for cancer-associated fibroblasts in SOC therapy resistance in breast cancers (155), as recently reviewed (156). Studies such as these have clearly demonstrated that the roles of dormancy, therapy resistance, microenvironmental crosstalk, and metastasis are closely intertwined.
Functional Genomics Screens for Mediators of Breast Cancer Resistance
Genome-wide shRNA screening in breast cancer cell lines has enabled high-throughput identification of genes required for cell viability in the context of various oncogenic drivers and have informed synergistic drug combinations (157, 158). Leveraging shRNA screens in defined genetic backgrounds of well characterized cell lines, such as in the context of PTEN-null lines, has enabled identification of vulnerabilities relevant to genetic driver events recurrent in breast cancer patient populations (159). Knock-down screens in the context of SOC therapeutic treatment are only beginning to be adopted and can provide insights into functional mediators of therapy resistance. A barcoded RNAi screen in a HER2 positive cell line revealed trastuzumab resistance could be conferred only by PTEN loss out of a library targeting approximately 8,000 genes. The importance of this pathway was corroborated by the finding that PIK3CA oncogenic mutations similarly conferred resistance to trastuzumab (160). A study conducting genome-wide shRNA screens in 77 breast cancer cell lines revealed functional vulnerabilities of breast cancer cells en masse. When compared with high-throughput drug screening data generated in these lines, cross-referencing gene essentiality with drug resistance data in cell lines yielded valuable insights into putative mediators of drug resistance (161). Future expanded application of screening methodologies in the context of therapeutic treatments in breast cancer cell lines and organoids is expected to reveal valuable biological insights and potential therapeutic combinations.
In vivo functional genomics screens hold further promise to yield clinically relevant insights into mediators of therapy resistance. Several groups have leveraged high-throughput shRNA or CRISPR/Cas9 libraries subsequently xenografted into immune-compromised mice in other cancer contexts (162, 163). These technologies are only beginning to be leveraged in breast cancer models and have not been applied to the issue of SOC therapy resistance as of yet. One recent study revealed genes required for in vivo tumorigenic capacity in subcutaneously xenografted TNBC cell lines, revealing genes involved in CSC feature maintenance (164). A unique screening strategy was used to identify tumor cell genes involved in immune-microenvironment communication. A murine TNBC cell line was transduced with a genome-wide shRNA library, then subcutaneously transplanted into immune-competent and immune-compromised mice. This novel screening approach revealed several genes that were functionally validated to mediate in vivo sensitivity to immune recognition, providing potential targets for future immune therapies (165). In vivo screening is limited by library complexity achievable in tumor models, as well as cost of animal acquisition and maintenance. However, application of shRNA and CRISPR/Cas9 libraries in orthotopically xenografted breast cancer cell lines and PDX models, as well as genetically engineered mouse models, upon treatment with SOC therapies is expected to provide invaluable insights into clinically relevant functional drivers of resistance in breast cancer.
Concluding Remarks
Therapy resistance and metastasis continue to be the two major causes of breast cancer mortality. The research works reviewed herein have provided valuable insights into mechanisms driving metastatic recurrence and treatment resistance. Continued advancements in the field are needed to push scientific boundaries to provide comprehensive insights into clinically relevant mechanisms of cancer relapse. Additionally, as therapies generate alterations in the tumor biology, modeling appropriate disease outcomes will be imperative in order to accurately predict metastatic behaviors. Acquisition, expansion, and ease-of-use of PDX models with ‘humanized’ microenvironmental components is expected to revolutionize the field. Use of these humanized PDX models for gene and protein expression profiling, lineage tracing, clonal tracking, comparison of multi-site metastases, longitudinal profiling throughout therapeutic treatment, and high-throughput ORF and CRISPR/Cas9 screening are expected to provide unprecedented biological insights. By including a more physiologically relevant immune system, results from these studies may be more readily translatable to the clinic. Moreover, in vitro 3D organoid applications composed of multi-component platforms that recapitulate an appropriate tumor microenvironment will provide the ability to experimentally interrogate meaningful cell and biological interactions driving disease progression and could theoretically provide real-time personalized therapeutic information for patients. As laboratory and clinical research progress, the next generation of therapies will become the new “standard of care”. As these develop, novel mechanisms of resistance to those agents should be anticipated and deeply investigated in the laboratory. With useful models, the mysteries of metastasis and recurrence will gradually be unraveled with time.
Author Contributions
KR and GE conceived of and wrote the manuscript. All authors contributed to the article and approved the submitted version.
Funding
The authors are supported by a CPRIT faculty recruitment award RR200009 to GE, National Institutes of Health (NIH) 1K22CA241113-01 to GE and 5K22CA207463 to KR, and Susan G. Komen CCR18548284 to KR. The content is solely the responsibility of the authors and does not necessarily represent the official views of NIH, Susan G. Komen, or CPRIT.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
GE is a Cancer Prevention Research Institute of Texas (CPRIT) Scholar in Cancer Research. KR is a Dan L. Duncan Cancer Center (DLDCCC) Faculty Scholar at Baylor College of Medicine. Figure 1 was made using BioRender.
References
1. Parker JS, Mullins M, Cheang MC, Leung S, Voduc D, Vickery T, et al. Supervised risk predictor of breast cancer based on intrinsic subtypes. J Clin Oncol (2009) 27:1160–7. doi: 10.1200/JCO.2008.18.1370
2. Perou CM, Sorlie T, Eisen MB, van de Rijn M, Jeffrey SS, Rees CA, et al. Molecular portraits of human breast tumours. Nature (2000) 406:747–52. doi: 10.1038/35021093
3. Fidler IJ. The pathogenesis of cancer metastasis: the ‘seed and soil’ hypothesis revisited. Nat Rev Cancer (2003) 3:453–8. doi: 10.1038/nrc1098
4. Gelsomino L, Gu G, Rechoum Y, Beyer AR, Pejerrey SM, Tsimelzon A, et al. ESR1 mutations affect anti-proliferative responses to tamoxifen through enhanced cross-talk with IGF signaling. Breast Cancer Res Treat (2016) 157:253–65. doi: 10.1007/s10549-016-3829-5
5. Li S, Shen D, Shao J, Crowder R, Liu W, Prat A, et al. Endocrine-therapy-resistant ESR1 variants revealed by genomic characterization of breast-cancer-derived xenografts. Cell Rep (2013) 4:1116–30. doi: 10.1016/j.celrep.2013.08.022
6. Toy W, Shen Y, Won H, Green B, Sakr RA, Will M, et al. ESR1 ligand-binding domain mutations in hormone-resistant breast cancer. Nat Genet (2013) 45:1439–45. doi: 10.1038/ng.2822
7. Lei JT, Shao J, Zhang J, Iglesia M, Chan DW, Cao J, et al. Functional Annotation of ESR1 Gene Fusions in Estrogen Receptor-Positive Breast Cancer. Cell Rep (2018) 24:1434–44.e7. doi: 10.1016/j.celrep.2018.07.009
8. Wang F, Weaver VM, Petersen OW, Larabell CA, Dedhar S, Briand P, et al. Reciprocal interactions between beta1-integrin and epidermal growth factor receptor in three-dimensional basement membrane breast cultures: a different perspective in epithelial biology. Proc Natl Acad Sci USA (1998) 95:14821–6. doi: 10.1073/pnas.95.25.14821
9. Weaver VM, Lelievre S, Lakins JN, Chrenek MA, Jones JC, Giancotti F, et al. beta4 integrin-dependent formation of polarized three-dimensional architecture confers resistance to apoptosis in normal and malignant mammary epithelium. Cancer Cell (2002) 2:205–16. doi: 10.1016/S1535-6108(02)00125-3
10. Chia J, Kusuma N, Anderson R, Parker B, Bidwell B, Zamurs L, et al. Evidence for a role of tumor-derived laminin-511 in the metastatic progression of breast cancer. Am J Pathol (2007) 170:2135–48. doi: 10.2353/ajpath.2007.060709
11. Roh-Johnson M, Bravo-Cordero JJ, Patsialou A, Sharma VP, Guo P, Liu H, et al. Macrophage contact induces RhoA GTPase signaling to trigger tumor cell intravasation. Oncogene (2014) 33:4203–12. doi: 10.1038/onc.2013.377
12. Roussos ET, Balsamo M, Alford SK, Wyckoff JB, Gligorijevic B, Wang Y, et al. Mena invasive (MenaINV) promotes multicellular streaming motility and transendothelial migration in a mouse model of breast cancer. J Cell Sci (2011) 124:2120–31. doi: 10.1242/jcs.086231
13. Pignatelli J, Goswami S, Jones JG, Rohan TE, Pieri E, Chen X, et al. Invasive breast carcinoma cells from patients exhibit MenaINV- and macrophage-dependent transendothelial migration. Sci Signal (2014) 7:ra112. doi: 10.1126/scisignal.2005329
14. Sachs N, de Ligt J, Kopper O, Gogola E, Bounova G, Weeber F, et al. A Living Biobank of Breast Cancer Organoids Captures Disease Heterogeneity. Cell (2018) 172:373–86.e10. doi: 10.1016/j.cell.2017.11.010
15. Rosenbluth JM, Schackmann RCJ, Gray GK, Selfors LM, Li CM, Boedicker M, et al. Organoid cultures from normal and cancer-prone human breast tissues preserve complex epithelial lineages. Nat Commun (2020) 11:1711. doi: 10.1038/s41467-020-15548-7
16. Bruna A, Rueda OM, Greenwood W, Batra AS, Callari M, Batra RN, et al. A Biobank of Breast Cancer Explants with Preserved Intra-tumor Heterogeneity to Screen Anticancer Compounds. Cell (2016) 167:260–74.e22. doi: 10.1016/j.cell.2016.08.041
17. Esbona K, Inman D, Saha S, Jeffery J, Schedin P, Wilke L, et al. COX-2 modulates mammary tumor progression in response to collagen density. Breast Cancer Res (2016) 18:35. doi: 10.1186/s13058-016-0695-3
18. Nguyen-Ngoc KV, Cheung KJ, Brenot A, Shamir ER, Gray RS, Hines WC, et al. ECM microenvironment regulates collective migration and local dissemination in normal and malignant mammary epithelium. Proc Natl Acad Sci USA (2012) 109:E2595–604. doi: 10.1073/pnas.1212834109
19. Cheung KJ, Gabrielson E, Werb Z, Ewald AJ. Collective invasion in breast cancer requires a conserved basal epithelial program. Cell (2013) 155:1639–51. doi: 10.1016/j.cell.2013.11.029
20. Khalil AA, Ilina O, Vasaturo A, Venhuizen JH, Vullings M, Venhuizen V, et al. Collective invasion induced by an autocrine purinergic loop through connexin-43 hemichannels. J Cell Biol (2020) 219. doi: 10.1083/jcb.201911120
21. Ilina O, Gritsenko PG, Syga S, Lippoldt J, La Porta CAM, Chepizhko O, et al. Cell-cell adhesion and 3D matrix confinement determine jamming transitions in breast cancer invasion. Nat Cell Biol (2020) 22:1103–15. doi: 10.1038/s41556-020-0552-6
22. Hwang PY, Brenot A, King AC, Longmore GD, George SC. Randomly Distributed K14(+) Breast Tumor Cells Polarize to the Leading Edge and Guide Collective Migration in Response to Chemical and Mechanical Environmental Cues. Cancer Res (2019) 79:1899–912. doi: 10.1158/0008-5472.CAN-18-2828
23. Chan IS, Knutsdottir H, Ramakrishnan G, Padmanaban V, Warrier M, Ramirez JC, et al. Cancer cells educate natural killer cells to a metastasis-promoting cell state. J Cell Biol (2020) 219. doi: 10.1083/jcb.202001134
24. Saraiva DP, Matias AT, Braga S, Jacinto A, Cabral MG. Establishment of a 3D Co-culture With MDA-MB-231 Breast Cancer Cell Line and Patient-Derived Immune Cells for Application in the Development of Immunotherapies. Front Oncol (2020) 10:1543. doi: 10.3389/fonc.2020.01543
25. Ghajar CM, Peinado H, Mori H, Matei IR, Evason KJ, Brazier H, et al. The perivascular niche regulates breast tumour dormancy. Nat Cell Biol (2013) 15:807–17. doi: 10.1038/ncb2767
26. Wang H, Tian L, Goldstein A, Liu J, Lo HC, Sheng K, et al. Bone-in-culture array as a platform to model early-stage bone metastases and discover anti-metastasis therapies. Nat Commun (2017) 8:15045. doi: 10.1038/ncomms15045
27. Wang H, Yu C, Gao X, Welte T, Muscarella AM, Tian L, et al. The osteogenic niche promotes early-stage bone colonization of disseminated breast cancer cells. Cancer Cell (2015) 27:193–210. doi: 10.1016/j.ccell.2014.11.017
28. Carlson P, Dasgupta A, Grzelak CA, Kim J, Barrett A, Coleman IM, et al. Targeting the perivascular niche sensitizes disseminated tumour cells to chemotherapy. Nat Cell Biol (2019) 21:238–50. doi: 10.1038/s41556-018-0267-0
29. Fidler IJ, Nicolson GL. Organ selectivity for implantation survival and growth of B16 melanoma variant tumor lines. J Natl Cancer Inst (1976) 57:1199–202. doi: 10.1093/jnci/57.5.1199
30. Minn AJ, Gupta GP, Siegel PM, Bos PD, Shu W, Giri DD, et al. Genes that mediate breast cancer metastasis to lung. Nature (2005) 436:518–24. doi: 10.1038/nature03799
31. Bos PD, Zhang XH, Nadal C, Shu W, Gomis RR, Nguyen DX, et al. Genes that mediate breast cancer metastasis to the brain. Nature (2009) 459:1005–9. doi: 10.1038/nature08021
32. Yu C, Wang H, Muscarella A, Goldstein A, Zeng HC, Bae Y, et al. Intra-iliac Artery Injection for Efficient and Selective Modeling of Microscopic Bone Metastasis. J Vis Exp (2016) 2016(115):53982. doi: 10.3791/53982
33. Kang Y, Siegel PM, Shu W, Drobnjak M, Kakonen SM, Cordon-Cardo C, et al. A multigenic program mediating breast cancer metastasis to bone. Cancer Cell (2003) 3:537–49. doi: 10.1016/S1535-6108(03)00132-6
34. Pavlovic M, Arnal-Estape A, Rojo F, Bellmunt A, Tarragona M, Guiu M, et al. Enhanced MAF Oncogene Expression and Breast Cancer Bone Metastasis. J Natl Cancer Inst (2015) 107:djv256. doi: 10.1093/jnci/djv256
35. Aslakson CJ, Miller FR. Selective events in the metastatic process defined by analysis of the sequential dissemination of subpopulations of a mouse mammary tumor. Cancer Res (1992) 52:1399–405.
36. Gawrzak S, Rinaldi L, Gregorio S, Arenas EJ, Salvador F, Urosevic J, et al. MSK1 regulates luminal cell differentiation and metastatic dormancy in ER(+) breast cancer. Nat Cell Biol (2018) 20:211–21. doi: 10.1038/s41556-017-0021-z
37. Jin X, Demere Z, Nair K, Ali A, Ferraro GB, Natoli T, et al. A metastasis map of human cancer cell lines. Nature (2020) 588:331–6. doi: 10.1038/s41586-020-2969-2
38. Cleris L, Daidone MG, Fina E, Cappelletti V. The Detection and Morphological Analysis of Circulating Tumor and Host Cells in Breast Cancer Xenograft Models. Cells (2019) 8. doi: 10.3390/cells8070683
39. Minn AJ, Kang Y, Serganova I, Gupta GP, Giri DD, Doubrovin M, et al. Distinct organ-specific metastatic potential of individual breast cancer cells and primary tumors. J Clin Invest (2005) 115:44–55. doi: 10.1172/JCI22320
40. Dobrolecki LE, Airhart SD, Alferez DG, Aparicio S, Behbod F, Bentires-Alj M, et al. Patient-derived xenograft (PDX) models in basic and translational breast cancer research. Cancer Metastasis Rev (2016) 35:547–73. doi: 10.1007/s10555-016-9653-x
41. Zhang X, Claerhout S, Prat A, Dobrolecki LE, Petrovic I, Lai Q, et al. A renewable tissue resource of phenotypically stable, biologically and ethnically diverse, patient-derived human breast cancer xenograft models. Cancer Res (2013) 73:4885–97. doi: 10.1158/0008-5472.CAN-12-4081
42. Eirew P, Steif A, Khattra J, Ha G, Yap D, Farahani H, et al. Dynamics of genomic clones in breast cancer patient xenografts at single-cell resolution. Nature (2015) 518:422–6. doi: 10.1038/nature13952
43. DeRose YS, Wang G, Lin YC, Bernard PS, Buys SS, Ebbert MT, et al. Tumor grafts derived from women with breast cancer authentically reflect tumor pathology, growth, metastasis and disease outcomes. Nat Med (2011) 17:1514–20. doi: 10.1038/nm.2454
44. Powell E, Shao J, Yuan Y, Chen HC, Cai S, Echeverria GV, et al. p53 deficiency linked to B cell translocation gene 2 (BTG2) loss enhances metastatic potential by promoting tumor growth in primary and metastatic sites in patient-derived xenograft (PDX) models of triple-negative breast cancer. Breast Cancer Res (2016) 18:13. doi: 10.1186/s13058-016-0673-9
45. Gomez-Miragaya J, Moran S, Calleja-Cervantes ME, Collado-Sole A, Pare L, Gomez A, et al. The Altered Transcriptome and DNA Methylation Profiles of Docetaxel Resistance in Breast Cancer PDX Models. Mol Cancer Res (2019) 17:2063–76. doi: 10.1158/1541-7786.MCR-19-0040
46. Lefley D, Howard F, Arshad F, Bradbury S, Brown H, Tulotta C, et al. Development of clinically relevant in vivo metastasis models using human bone discs and breast cancer patient-derived xenografts. Breast Cancer Res (2019) 21:130. doi: 10.1186/s13058-019-1220-2
47. Kuperwasser C, Dessain S, Bierbaum BE, Garnet D, Sperandio K, Gauvin GP, et al. A mouse model of human breast cancer metastasis to human bone. Cancer Res (2005) 65:6130–8. doi: 10.1158/0008-5472.CAN-04-1408
48. Lawson DA, Bhakta NR, Kessenbrock K, Prummel KD, Yu Y, Takai K, et al. Single-cell analysis reveals a stem-cell program in human metastatic breast cancer cells. Nature (2015) 526:131–5. doi: 10.1038/nature15260
49. Davis RT, Blake K, Ma D, Gabra MBI, Hernandez GA, Phung AT, et al. Transcriptional diversity and bioenergetic shift in human breast cancer metastasis revealed by single-cell RNA sequencing. Nat Cell Biol (2020) 22:310–20. doi: 10.1038/s41556-020-0477-0
50. Echeverria GV, Powell E, Seth S, Ge Z, Carugo A, Bristow C, et al. High-resolution clonal mapping of multi-organ metastasis in triple negative breast cancer. Nat Commun (2018) 9:5079. doi: 10.1038/s41467-018-07406-4
51. Powell E, Shao J, Yuan Y, Chen H-C, Cai S, Echeverria GV, et al. p53 deficiency linked to B cell translocation gene 2 (BTG2) loss enhances metastatic potential by promoting tumor growth in primary and metastatic sites in patient-derived xenograft (PDX) models of triple-negative breast cancer. Breast Cancer Res BCR (2016) 18:13. doi: 10.1186/s13058-016-0673-9
52. Aceto N, Bardia A, Miyamoto DT, Donaldson MC, Wittner BS, Spencer JA, et al. Circulating tumor cell clusters are oligoclonal precursors of breast cancer metastasis. Cell (2014) 158:1110–22. doi: 10.1016/j.cell.2014.07.013
53. Donato C, Kunz L, Castro-Giner F, Paasinen-Sohns A, Strittmatter K, Szczerba BM, et al. Hypoxia Triggers the Intravasation of Clustered Circulating Tumor Cells. Cell Rep (2020) 32:108105. doi: 10.1016/j.celrep.2020.108105
54. Szczerba BM, Castro-Giner F, Vetter M, Krol I, Gkountela S, Landin J, et al. Neutrophils escort circulating tumour cells to enable cell cycle progression. Nature (2019) 566:553–7. doi: 10.1038/s41586-019-0915-y
55. Murugaesu N, Iravani M, van Weverwijk A, Ivetic A, Johnson DA, Antonopoulos A, et al. An in vivo functional screen identifies ST6GalNAc2 sialyltransferase as a breast cancer metastasis suppressor. Cancer Discov (2014) 4:304–17. doi: 10.1158/2159-8290.CD-13-0287
56. Ebright RY, Lee S, Wittner BS, Niederhoffer KL, Nicholson BT, Bardia A, et al. Deregulation of ribosomal protein expression and translation promotes breast cancer metastasis. Science (2020) 367:1468–73. doi: 10.1126/science.aay0939
57. Powell E, Shao J, Picon HM, Bristow C, Ge Z, Peoples M, et al. A functional genomic screen in vivo identifies CEACAM5 as a clinically relevant driver of breast cancer metastasis. NPJ Breast Cancer (2018) 4:9. doi: 10.1038/s41523-018-0062-x
58. Taneja P, Frazier DP, Kendig RD, Maglic D, Sugiyama T, Kai F, et al. MMTV mouse models and the diagnostic values of MMTV-like sequences in human breast cancer. Expert Rev Mol Diagn (2009) 9:423–40. doi: 10.1586/erm.09.31
59. Siegel PM, Dankort DL, Hardy WR, Muller WJ. Novel activating mutations in the neu proto-oncogene involved in induction of mammary tumors. Mol Cell Biol (1994) 14:7068–77. doi: 10.1128/MCB.14.11.7068
60. Rao T, Ranger JJ, Smith HW, Lam SH, Chodosh L, Muller WJ. Inducible and coupled expression of the polyomavirus middle T antigen and Cre recombinase in transgenic mice: an in vivo model for synthetic viability in mammary tumour progression. Breast Cancer Res (2014) 16:R11. doi: 10.1186/bcr3603
61. Green JE, Shibata MA, Yoshidome K, Liu ML, Jorcyk C, Anver MR, et al. The C3(1)/SV40 T-antigen transgenic mouse model of mammary cancer: ductal epithelial cell targeting with multistage progression to carcinoma. Oncogene (2000) 19:1020–7. doi: 10.1038/sj.onc.1203280
62. Li Y, Hively WP, Varmus HE. Use of MMTV-Wnt-1 transgenic mice for studying the genetic basis of breast cancer. Oncogene (2000) 19:1002–9. doi: 10.1038/sj.onc.1203273
63. Matsui Y, Halter SA, Holt JT, Hogan BL, Coffey RJ. Development of mammary hyperplasia and neoplasia in MMTV-TGF alpha transgenic mice. Cell (1990) 61:1147–55. doi: 10.1016/0092-8674(90)90077-R
64. Andrechek ER, Cardiff RD, Chang JT, Gatza ML, Acharya CR, Potti A, et al. Genetic heterogeneity of Myc-induced mammary tumors reflecting diverse phenotypes including metastatic potential. Proc Natl Acad Sci USA (2009) 106:16387–92. doi: 10.1073/pnas.0901250106
65. Muller WJ, Sinn E, Pattengale PK, Wallace R, Leder P. Single-step induction of mammary adenocarcinoma in transgenic mice bearing the activated c-neu oncogene. Cell (1988) 54:105–15. doi: 10.1016/0092-8674(88)90184-5
66. Guy CT, Cardiff RD, Muller WJ. Activated neu induces rapid tumor progression. J Biol Chem (1996) 271:7673–8. doi: 10.1074/jbc.271.13.7673
67. Guy CT, Webster MA, Schaller M, Parsons TJ, Cardiff RD, Muller WJ. Expression of the neu protooncogene in the mammary epithelium of transgenic mice induces metastatic disease. Proc Natl Acad Sci USA (1992) 89:10578–82. doi: 10.1073/pnas.89.22.10578
68. Rennhack JP, To B, Swiatnicki M, Dulak C, Ogrodzinski MP, Zhang Y, et al. Integrated analyses of murine breast cancer models reveal critical parallels with human disease. Nat Commun (2019) 10:3261. doi: 10.1038/s41467-019-11236-3
69. Muraoka RS, Koh Y, Roebuck LR, Sanders ME, Brantley-Sieders D, Gorska AE, et al. Increased malignancy of Neu-induced mammary tumors overexpressing active transforming growth factor beta1. Mol Cell Biol (2003) 23:8691–703. doi: 10.1128/MCB.23.23.8691-8703.2003
70. Muraoka-Cook RS, Kurokawa H, Koh Y, Forbes JT, Roebuck LR, Barcellos-Hoff MH, et al. Conditional overexpression of active transforming growth factor beta1 in vivo accelerates metastases of transgenic mammary tumors. Cancer Res (2004) 64:9002–11. doi: 10.1158/0008-5472.CAN-04-2111
71. Siegel PM, Shu W, Cardiff RD, Muller WJ, Massague J. Transforming growth factor beta signaling impairs Neu-induced mammary tumorigenesis while promoting pulmonary metastasis. Proc Natl Acad Sci USA (2003) 100:8430–5. doi: 10.1073/pnas.0932636100
72. Pierce DF Jr, Gorska AE, Chytil A, Meise KS, Page DL, Coffey RJ Jr, et al. Mammary tumor suppression by transforming growth factor beta 1 transgene expression. Proc Natl Acad Sci USA (1995) 92:4254–8. doi: 10.1073/pnas.92.10.4254
73. Kwan H, Pecenka V, Tsukamoto A, Parslow TG, Guzman R, Lin TP, et al. Transgenes expressing the Wnt-1 and int-2 proto-oncogenes cooperate during mammary carcinogenesis in doubly transgenic mice. Mol Cell Biol (1992) 12:147–54. doi: 10.1128/MCB.12.1.147
74. Pickup M, Novitskiy S, Moses HL. The roles of TGFbeta in the tumour microenvironment. Nat Rev Cancer (2013) 13:788–99. doi: 10.1038/nrc3603
75. Wyckoff JB, Wang Y, Lin EY, Li JF, Goswami S, Stanley ER, et al. Direct visualization of macrophage-assisted tumor cell intravasation in mammary tumors. Cancer Res (2007) 67:2649–56. doi: 10.1158/0008-5472.CAN-06-1823
76. Harney AS, Arwert EN, Entenberg D, Wang Y, Guo P, Qian BZ, et al. Real-Time Imaging Reveals Local, Transient Vascular Permeability, and Tumor Cell Intravasation Stimulated by TIE2hi Macrophage-Derived VEGFA. Cancer Discov (2015) 5:932–43. doi: 10.1158/2159-8290.CD-15-0012
77. Arwert EN, Harney AS, Entenberg D, Wang Y, Sahai E, Pollard JW, et al. A Unidirectional Transition from Migratory to Perivascular Macrophage Is Required for Tumor Cell Intravasation. Cell Rep (2018) 23:1239–48. doi: 10.1016/j.celrep.2018.04.007
78. Cheung KJ, Padmanaban V, Silvestri V, Schipper K, Cohen JD, Fairchild AN, et al. Polyclonal breast cancer metastases arise from collective dissemination of keratin 14-expressing tumor cell clusters. Proc Natl Acad Sci USA (2016) 113:E854–63. doi: 10.1073/pnas.1508541113
79. Wrenn ED, Yamamoto A, Moore BM, Huang Y, McBirney M, Thomas AJ, et al. Regulation of Collective Metastasis by Nanolumenal Signaling. Cell (2020) 183:395–410.e19. doi: 10.1016/j.cell.2020.08.045
80. Zhang M, Behbod F, Atkinson RL, Landis MD, Kittrell F, Edwards D, et al. Identification of tumor-initiating cells in a p53-null mouse model of breast cancer. Cancer Res (2008) 68:4674–82. doi: 10.1158/0008-5472.CAN-07-6353
81. Pfefferle AD, Herschkowitz JI, Usary J, Harrell JC, Spike BT, Adams JR, et al. Transcriptomic classification of genetically engineered mouse models of breast cancer identifies human subtype counterparts. Genome Biol (2013) 14:R125. doi: 10.1186/gb-2013-14-11-r125
82. Herschkowitz JI, Zhao W, Zhang M, Usary J, Murrow G, Edwards D, et al. Comparative oncogenomics identifies breast tumors enriched in functional tumor-initiating cells. Proc Natl Acad Sci USA (2012) 109:2778–83. doi: 10.1073/pnas.1018862108
83. Welte T, Kim IS, Tian L, Gao X, Wang H, Li J, et al. Oncogenic mTOR signalling recruits myeloid-derived suppressor cells to promote tumour initiation. Nat Cell Biol (2016) 18:632–44. doi: 10.1038/ncb3355
84. Pfefferle AD, Agrawal YN, Koboldt DC, Kanchi KL, Herschkowitz JI, Mardis ER, et al. Genomic profiling of murine mammary tumors identifies potential personalized drug targets for p53-deficient mammary cancers. Dis Model Mech (2016) 9:749–57. doi: 10.1242/dmm.025239
85. Kennecke H, Yerushalmi R, Woods R, Cheang MC, Voduc D, Speers CH, et al. Metastatic behavior of breast cancer subtypes. J Clin Oncol (2010) 28:3271–7. doi: 10.1200/JCO.2009.25.9820
86. Schmidt-Kittler O, Ragg T, Daskalakis A, Granzow M, Ahr A, Blankenstein TJ, et al. From latent disseminated cells to overt metastasis: genetic analysis of systemic breast cancer progression. Proc Natl Acad Sci USA (2003) 100:7737–42. doi: 10.1073/pnas.1331931100
87. Pantel K, Brakenhoff RH, Brandt B. Detection, clinical relevance and specific biological properties of disseminating tumour cells. Nat Rev Cancer (2008) 8:329–40. doi: 10.1038/nrc2375
88. Husemann Y, Geigl JB, Schubert F, Musiani P, Meyer M, Burghart E, et al. Systemic spread is an early step in breast cancer. Cancer Cell (2008) 13:58–68. doi: 10.1016/j.ccr.2007.12.003
89. Harper KL, Sosa MS, Entenberg D, Hosseini H, Cheung JF, Nobre R, et al. Mechanism of early dissemination and metastasis in Her2(+) mammary cancer. Nature (2016) 540:588–92. doi: 10.1038/nature20609
90. Gao H, Chakraborty G, Lee-Lim AP, Mo Q, Decker M, Vonica A, et al. The BMP inhibitor Coco reactivates breast cancer cells at lung metastatic sites. Cell (2012) 150:764–79. doi: 10.1016/j.cell.2012.06.035
91. Barkan D, El Touny LH, Michalowski AM, Smith JA, Chu I, Davis AS, et al. Metastatic growth from dormant cells induced by a col-I-enriched fibrotic environment. Cancer Res (2010) 70:5706–16. doi: 10.1158/0008-5472.CAN-09-2356
92. Aguirre Ghiso JA, Kovalski K, Ossowski L. Tumor dormancy induced by downregulation of urokinase receptor in human carcinoma involves integrin and MAPK signaling. J Cell Biol (1999) 147:89–104. doi: 10.1083/jcb.147.1.89
93. Barkan D, Kleinman H, Simmons JL, Asmussen H, Kamaraju AK, Hoenorhoff MJ, et al. Inhibition of metastatic outgrowth from single dormant tumor cells by targeting the cytoskeleton. Cancer Res (2008) 68:6241–50. doi: 10.1158/0008-5472.CAN-07-6849
94. Albrengues J, Shields MA, Ng D, Park CG, Ambrico A, Poindexter ME, et al. Neutrophil extracellular traps produced during inflammation awaken dormant cancer cells in mice. Science (2018) 361. doi: 10.1126/science.aao4227
95. Sharma P, Allison JP. The future of immune checkpoint therapy. Science (2015) 348:56–61. doi: 10.1126/science.aaa8172
96. Lin EY, Nguyen AV, Russell RG, Pollard JW. Colony-stimulating factor 1 promotes progression of mammary tumors to malignancy. J Exp Med (2001) 193:727–40. doi: 10.1084/jem.193.6.727
97. Wyckoff J, Wang W, Lin EY, Wang Y, Pixley F, Stanley ER, et al. A paracrine loop between tumor cells and macrophages is required for tumor cell migration in mammary tumors. Cancer Res (2004) 64:7022–9. doi: 10.1158/0008-5472.CAN-04-1449
98. DeNardo DG, Barreto JB, Andreu P, Vasquez L, Tawfik D, Kolhatkar N, et al. CD4(+) T cells regulate pulmonary metastasis of mammary carcinomas by enhancing protumor properties of macrophages. Cancer Cell (2009) 16:91–102. doi: 10.1016/j.ccr.2009.06.018
99. Coffelt SB, Kersten K, Doornebal CW, Weiden J, Vrijland K, Hau CS, et al. IL-17-producing gammadelta T cells and neutrophils conspire to promote breast cancer metastasis. Nature (2015) 522:345–8. doi: 10.1038/nature14282
100. Kim IS, Gao Y, Welte T, Wang H, Liu J, Janghorban M, et al. Immuno-subtyping of breast cancer reveals distinct myeloid cell profiles and immunotherapy resistance mechanisms. Nat Cell Biol (2019) 21:1113–26. doi: 10.1038/s41556-019-0373-7
101. Hollern DP, Xu N, Thennavan A, Glodowski C, Garcia-Recio S, Mott KR, et al. B Cells and T Follicular Helper Cells Mediate Response to Checkpoint Inhibitors in High Mutation Burden Mouse Models of Breast Cancer. Cell (2019) 179:1191–206.e21. doi: 10.1016/j.cell.2019.10.028
102. Strasser-Weippl K, Goss PE. Advances in adjuvant hormonal therapy for postmenopausal women. J Clin Oncol (2005) 23:1751–9. doi: 10.1200/JCO.2005.11.038
103. Symmans WF, Wei C, Gould R, Yu X, Zhang Y, Liu M, et al. Long-Term Prognostic Risk After Neoadjuvant Chemotherapy Associated With Residual Cancer Burden and Breast Cancer Subtype. J Clin Oncol (2017) 35:1049–60. doi: 10.1200/JCO.2015.63.1010
104. Risom T, Langer EM, Chapman MP, Rantala J, Fields AJ, Boniface C, et al. Differentiation-state plasticity is a targetable resistance mechanism in basal-like breast cancer. Nat Commun (2018) 9:3815. doi: 10.1038/s41467-018-05729-w
105. Sharma SV, Lee DY, Li B, Quinlan MP, Takahashi F, Maheswaran S, et al. A chromatin-mediated reversible drug-tolerant state in cancer cell subpopulations. Cell (2010) 141:69–80. doi: 10.1016/j.cell.2010.02.027
106. Hinohara K, Wu HJ, Sebastien V, McDonald TO, Igarashi KJ, Yamamoto KN, et al. KDM5 Histone Demethylase Activity Links Cellular Transcriptomic Heterogeneity to Therapeutic Resistance. Cancer Cell (2019) 35:330–2. doi: 10.1016/j.ccell.2019.01.012
107. Ge JY, Shu S, Kwon M, Jovanovic B, Murphy K, Gulvady A, et al. Acquired resistance to combined BET and CDK4/6 inhibition in triple-negative breast cancer. Nat Commun (2020) 11:2350. doi: 10.1038/s41467-020-16170-3
108. Shu S, Wu HJ, Ge JY, Zeid R, Harris IS, Jovanovic B, et al. Synthetic Lethal and Resistance Interactions with BET Bromodomain Inhibitors in Triple-Negative Breast Cancer. Mol Cell (2020) 78:1096–113.e8. doi: 10.1016/j.molcel.2020.04.027
109. Sirois I, Aguilar-Mahecha A, Lafleur J, Fowler E, Vu V, Scriver M, et al. A Unique Morphological Phenotype in Chemoresistant Triple-Negative Breast Cancer Reveals Metabolic Reprogramming and PLIN4 Expression as a Molecular Vulnerability. Mol Cancer Res (2019) 17:2492–507. doi: 10.1158/1541-7786.MCR-19-0264
110. Sommer AK, Hermawan A, Ljepoja B, Frohlich T, Arnold GJ, Wagner E, et al. A proteomic analysis of chemoresistance development via sequential treatment with doxorubicin reveals novel players in MCF7 breast cancer cells. Int J Mol Med (2018) 42:1987–97. doi: 10.3892/ijmm.2018.3781
111. Lee KM, Giltnane JM, Balko JM, Schwarz LJ, Guerrero-Zotano AL, Hutchinson KE, et al. MYC and MCL1 Cooperatively Promote Chemotherapy-Resistant Breast Cancer Stem Cells via Regulation of Mitochondrial Oxidative Phosphorylation. Cell Metab (2017) 26:633–47.e7. doi: 10.1158/1538-7445.AM2016-3328
112. Balko JM, Cook RS, Vaught DB, Kuba MG, Miller TW, Bhola NE, et al. Profiling of residual breast cancers after neoadjuvant chemotherapy identifies DUSP4 deficiency as a mechanism of drug resistance. Nat Med (2012) 18:1052–9. doi: 10.1038/nm.2795
113. Lei JT, Gou X, Seker S, Ellis MJ. ESR1 alterations and metastasis in estrogen receptor positive breast cancer. J Cancer Metastasis Treat (2019) 5. doi: 10.20517/2394-4722.2019.12
114. Barone I, Cui Y, Herynk MH, Corona-Rodriguez A, Giordano C, Selever J, et al. Expression of the K303R estrogen receptor-alpha breast cancer mutation induces resistance to an aromatase inhibitor via addiction to the PI3K/Akt kinase pathway. Cancer Res (2009) 69:4724–32. doi: 10.1158/0008-5472.CAN-08-4194
115. Barone I, Iacopetta D, Covington KR, Cui Y, Tsimelzon A, Beyer A, et al. Phosphorylation of the mutant K303R estrogen receptor alpha at serine 305 affects aromatase inhibitor sensitivity. Oncogene (2010) 29:2404–14. doi: 10.1038/onc.2009.520
116. Martin LA, Ribas R, Simigdala N, Schuster E, Pancholi S, Tenev T, et al. Discovery of naturally occurring ESR1 mutations in breast cancer cell lines modelling endocrine resistance. Nat Commun (2017) 8:1865. doi: 10.1038/s41467-017-01864-y
117. Zheng ZY, Anurag M, Lei JT, Cao J, Singh P, Peng J, et al. Neurofibromin Is an Estrogen Receptor-alpha Transcriptional Co-repressor in Breast Cancer. Cancer Cell (2020) 37:387–402.e7. doi: 10.1016/j.ccell.2020.02.003
118. Rimawi MF, De Angelis C, Schiff R. Resistance to Anti-HER2 Therapies in Breast Cancer. Am Soc Clin Oncol Educ Book (2015), e157–64. doi: 10.14694/EdBook_AM.2015.35.e157
119. Wang YC, Morrison G, Gillihan R, Guo J, Ward RM, Fu X, et al. Different mechanisms for resistance to trastuzumab versus lapatinib in HER2-positive breast cancers–role of estrogen receptor and HER2 reactivation. Breast Cancer Res (2011) 13:R121. doi: 10.1186/bcr3067
120. Arpino G, Gutierrez C, Weiss H, Rimawi M, Massarweh S, Bharwani L, et al. Treatment of human epidermal growth factor receptor 2-overexpressing breast cancer xenografts with multiagent HER-targeted therapy. J Natl Cancer Inst (2007) 99:694–705. doi: 10.1093/jnci/djk151
121. Rimawi MF, Wiechmann LS, Wang YC, Huang C, Migliaccio I, Wu MF, et al. Reduced dose and intermittent treatment with lapatinib and trastuzumab for potent blockade of the HER pathway in HER2/neu-overexpressing breast tumor xenografts. Clin Cancer Res (2011) 17:1351–61. doi: 10.1158/1078-0432.CCR-10-1905
122. Goel S, Wang Q, Watt AC, Tolaney SM, Dillon DA, Li W, et al. Overcoming Therapeutic Resistance in HER2-Positive Breast Cancers with CDK4/6 Inhibitors. Cancer Cell (2016) 29:255–69. doi: 10.1016/j.ccell.2016.02.006
123. Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci USA (2003) 100:3983–8. doi: 10.1073/pnas.0530291100
124. Cheng GZ, Chan J, Wang Q, Zhang W, Sun CD, Wang LH. Twist transcriptionally up-regulates AKT2 in breast cancer cells leading to increased migration, invasion, and resistance to paclitaxel. Cancer Res (2007) 67:1979–87. doi: 10.1158/0008-5472.CAN-06-1479
125. Zhang S, Zhang H, Ghia EM, Huang J, Wu L, Zhang J, et al. Inhibition of chemotherapy resistant breast cancer stem cells by a ROR1 specific antibody. Proc Natl Acad Sci USA (2019) 116:1370–7. doi: 10.1073/pnas.1816262116
126. Samanta D, Gilkes DM, Chaturvedi P, Xiang L, Semenza GL. Hypoxia-inducible factors are required for chemotherapy resistance of breast cancer stem cells. Proc Natl Acad Sci USA (2014) 111:E5429–38. doi: 10.1073/pnas.1421438111
127. Asiedu MK, Ingle JN, Behrens MD, Radisky DC, Knutson KL. TGFbeta/TNF(alpha)-mediated epithelial-mesenchymal transition generates breast cancer stem cells with a claudin-low phenotype. Cancer Res (2011) 71:4707–19. doi: 10.1158/0008-5472.CAN-10-4554
128. Liu X, Li J, Cadilha BL, Markota A, Voigt C, Huang Z, et al. Epithelial-type systemic breast carcinoma cells with a restricted mesenchymal transition are a major source of metastasis. Sci Adv (2019) 5:eaav4275. doi: 10.1126/sciadv.aav4275
129. Yu F, Yao H, Zhu P, Zhang X, Pan Q, Gong C, et al. let-7 regulates self renewal and tumorigenicity of breast cancer cells. Cell (2007) 131:1109–23. doi: 10.1016/j.cell.2007.10.054
130. Hirschmann-Jax C, Foster AE, Wulf GG, Nuchtern JG, Jax TW, Gobel U, et al. A distinct “side population” of cells with high drug efflux capacity in human tumor cells. Proc Natl Acad Sci USA (2004) 101:14228–33. doi: 10.1073/pnas.0400067101
131. Fultang N, Illendula A, Lin J, Pandey MK, Klase Z, Peethambaran B. ROR1 regulates chemoresistance in Breast Cancer via modulation of drug efflux pump ABCB1. Sci Rep (2020) 10:1821. doi: 10.1038/s41598-020-58864-0
132. Powell RT, Redwood A, Liu X, Guo L, Cai S, Zhou X, et al. Pharmacologic profiling of patient-derived xenograft models of primary treatment-naive triple-negative breast cancer. Sci Rep (2020) 10:17899. doi: 10.1038/s41598-020-74882-4
133. Turner TH, Alzubi MA, Harrell JC. Identification of synergistic drug combinations using breast cancer patient-derived xenografts. Sci Rep (2020) 10:1493. doi: 10.1038/s41598-020-58438-0
134. Echeverria GV, Ge Z, Seth S, Zhang X, Jeter-Jones S, Zhou X, et al. Resistance to neoadjuvant chemotherapy in triple-negative breast cancer mediated by a reversible drug-tolerant state. Sci Transl Med (2019) 11. doi: 10.1158/1538-7445.SABCS18-GS5-05
135. Molina JR, Sun Y, Protopopova M, Gera S, Bandi M, Bristow C, et al. An inhibitor of oxidative phosphorylation exploits cancer vulnerability. Nat Med (2018) 24:1036–46. doi: 10.1038/s41591-018-0052-4
136. Gomez-Miragaya J, Palafox M, Pare L, Yoldi G, Ferrer I, Vila S, et al. Resistance to Taxanes in Triple-Negative Breast Cancer Associates with the Dynamics of a CD49f+ Tumor-Initiating Population. Stem Cell Rep (2017) 8:1392–407. doi: 10.1016/j.stemcr.2017.03.026
137. Ter Brugge P, Kristel P, van der Burg E, Boon U, de Maaker M, Lips E, et al. Mechanisms of Therapy Resistance in Patient-Derived Xenograft Models of BRCA1-Deficient Breast Cancer. J Natl Cancer Inst (2016) 108. doi: 10.1093/jnci/djw148
138. Shu S, Lin CY, He HH, Witwicki RM, Tabassum DP, Roberts JM, et al. Response and resistance to BET bromodomain inhibitors in triple-negative breast cancer. Nature (2016) 529:413–7. doi: 10.1158/1557-3125.ADVBC15-B16
139. Dustin D, Gu G, Beyer AR, Herzog SK, Edwards DG, Lin H, et al. RON signalling promotes therapeutic resistance in ESR1 mutant breast cancer. Br J Cancer (2020) 124(1):191–206. doi: 10.1038/s41416-020-01174-z
140. Hoey T, Yen WC, Axelrod F, Basi J, Donigian L, Dylla S, et al. DLL4 blockade inhibits tumor growth and reduces tumor-initiating cell frequency. Cell Stem Cell (2009) 5:168–77. doi: 10.1016/j.stem.2009.05.019
141. Byrd TT, Fousek K, Pignata A, Szot C, Samaha H, Seaman S, et al. TEM8/ANTXR1-Specific CAR T Cells as a Targeted Therapy for Triple-Negative Breast Cancer. Cancer Res (2018) 78:489–500. doi: 10.1158/0008-5472.CAN-16-1911
142. Rottenberg S, Nygren AO, Pajic M, van Leeuwen FW, van der Heijden I, van de Wetering K, et al. Selective induction of chemotherapy resistance of mammary tumors in a conditional mouse model for hereditary breast cancer. Proc Natl Acad Sci USA (2007) 104:12117–22. doi: 10.1073/pnas.0702955104
143. Rottenberg S, Jaspers JE, Kersbergen A, van der Burg E, Nygren AO, Zander SA, et al. High sensitivity of BRCA1-deficient mammary tumors to the PARP inhibitor AZD2281 alone and in combination with platinum drugs. Proc Natl Acad Sci USA (2008) 105:17079–84. doi: 10.1073/pnas.0806092105
144. Farmer P, Bonnefoi H, Anderle P, Cameron D, Wirapati P, Becette V, et al. A stroma-related gene signature predicts resistance to neoadjuvant chemotherapy in breast cancer. Nat Med (2009) 15:68–74. doi: 10.1038/nm.1908
145. Jaspers JE, Kersbergen A, Boon U, Sol W, van Deemter L, Zander SA, et al. Loss of 53BP1 causes PARP inhibitor resistance in Brca1-mutated mouse mammary tumors. Cancer Discov (2013) 3:68–81. doi: 10.1158/2159-8290.CD-12-0049
146. Boelens MC, Wu TJ, Nabet BY, Xu B, Qiu Y, Yoon T, et al. Exosome transfer from stromal to breast cancer cells regulates therapy resistance pathways. Cell (2014) 159:499–513. doi: 10.1016/j.cell.2014.09.051
147. Fernandez-Nogueira P, Mancino M, Fuster G, Lopez-Plana A, Jauregui P, Almendro V, et al. Tumor-Associated Fibroblasts Promote HER2-Targeted Therapy Resistance through FGFR2 Activation. Clin Cancer Res (2020) 26:1432–48. doi: 10.1158/1078-0432.CCR-19-0353
148. Usary J, Zhao W, Darr D, Roberts PJ, Liu M, Balletta L, et al. Predicting drug responsiveness in human cancers using genetically engineered mice. Clin Cancer Res (2013) 19:4889–99. doi: 10.1158/1078-0432.CCR-13-0522
149. Usary J, Darr DB, Pfefferle AD, Perou CM. Overview of Genetically Engineered Mouse Models of Distinct Breast Cancer Subtypes. Curr Protoc Pharmacol (2016) 72:14 38 1–14 38 11. doi: 10.1002/0471141755.ph1438s72
150. Montagner M, Sahai E. In vitro Models of Breast Cancer Metastatic Dormancy. Front Cell Dev Biol (2020) 8:37. doi: 10.3389/fcell.2020.00037
151. De Angelis ML, Francescangeli F, La Torre F, Zeuner A. Stem Cell Plasticity and Dormancy in the Development of Cancer Therapy Resistance. Front Oncol (2019) 9:626. doi: 10.3389/fonc.2019.00626
152. Hong SP, Chan TE, Lombardo Y, Corleone G, Rotmensz N, Bravaccini S, et al. Single-cell transcriptomics reveals multi-step adaptations to endocrine therapy. Nat Commun (2019) 10:3840. doi: 10.1038/s41467-019-11721-9
153. Tivari S, Lu H, Dasgupta T, De Lorenzo MS, Wieder R. Reawakening of dormant estrogen-dependent human breast cancer cells by bone marrow stroma secretory senescence. Cell Commun Signal (2018) 16:48. doi: 10.1186/s12964-018-0259-5
154. Mehta AK, Cheney EM, Hartl CA, Pantelidou C, Oliwa M, Castrillon JA, et al. Targeting immunosuppressive macrophages overcomes PARP inhibitor resistance in BRCA1-associated triple-negative breast cancer. Nat Cancer (2020) 2:66–82. doi: 10.1038/s43018-020-00148-7
155. Su S, Chen J, Yao H, Liu J, Yu S, Lao L, et al. CD10(+)GPR77(+) Cancer-Associated Fibroblasts Promote Cancer Formation and Chemoresistance by Sustaining Cancer Stemness. Cell (2018) 172:841–56.e16. doi: 10.1016/j.cell.2018.01.009
156. Fiori ME, Di Franco S, Villanova L, Bianca P, Stassi G, De Maria R. Cancer-associated fibroblasts as abettors of tumor progression at the crossroads of EMT and therapy resistance. Mol Cancer (2019) 18:70. doi: 10.1186/s12943-019-0994-2
157. Guest ST, Kratche ZR, Irish JC, Wilson RC, Haddad R, Gray JW, et al. Functional oncogene signatures guide rationally designed combination therapies to synergistically induce breast cancer cell death. Oncotarget (2016) 7:36138–53. doi: 10.18632/oncotarget.9147
158. Turner-Ivey B, Guest ST, Irish JC, Kappler CS, Garrett-Mayer E, Wilson RC, et al. KAT6A, a chromatin modifier from the 8p11-p12 amplicon is a candidate oncogene in luminal breast cancer. Neoplasia (2014) 16:644–55. doi: 10.1016/j.neo.2014.07.007
159. Tang YC, Ho SC, Tan E, Ng AWT, McPherson JR, Goh GYL, et al. Functional genomics identifies specific vulnerabilities in PTEN-deficient breast cancer. Breast Cancer Res (2018) 20:22. doi: 10.1186/s13058-018-0949-3
160. Berns K, Horlings HM, Hennessy BT, Madiredjo M, Hijmans EM, Beelen K, et al. A functional genetic approach identifies the PI3K pathway as a major determinant of trastuzumab resistance in breast cancer. Cancer Cell (2007) 12:395–402. doi: 10.1016/j.ccr.2007.08.030
161. Marcotte R, Sayad A, Brown KR, Sanchez-Garcia F, Reimand J, Haider M, et al. Functional Genomic Landscape of Human Breast Cancer Drivers, Vulnerabilities, and Resistance. Cell (2016) 164:293–309. doi: 10.1016/j.cell.2015.11.062
162. Chen S, Sanjana NE, Zheng K, Shalem O, Lee K, Shi X, et al. Genome-wide CRISPR screen in a mouse model of tumor growth and metastasis. Cell (2015) 160:1246–60. doi: 10.1016/j.cell.2015.02.038
163. Zender L, Xue W, Zuber J, Semighini CP, Krasnitz A, Ma B, et al. An oncogenomics-based in vivo RNAi screen identifies tumor suppressors in liver cancer. Cell (2008) 135:852–64. doi: 10.1016/j.cell.2008.09.061
164. Wolf J, Muller-Decker K, Flechtenmacher C, Zhang F, Shahmoradgoli M, Mills GB, et al. An in vivo RNAi screen identifies SALL1 as a tumor suppressor in human breast cancer with a role in CDH1 regulation. Oncogene (2014) 33:4273–8. doi: 10.1038/onc.2013.515
Keywords: breast cancer, metastasis, chemoresistance, genetically engineered mouse models, patient derived xenograft (PDX) model, cancer cell lines
Citation: Roarty K and Echeverria GV (2021) Laboratory Models for Investigating Breast Cancer Therapy Resistance and Metastasis. Front. Oncol. 11:645698. doi: 10.3389/fonc.2021.645698
Received: 23 December 2020; Accepted: 28 January 2021;
Published: 10 March 2021.
Edited by:
Ariella Hanker, University of Texas Southwestern Medical Center, United StatesReviewed by:
David A. Gewirtz, Virginia Commonwealth University, United StatesRosalin Mishra, University of Cincinnati, United States
Isaac Chan, University of Texas Southwestern Medical Center, United States
Brian Lehmann, Vanderbilt University Medical Center, United States
Copyright © 2021 Roarty and Echeverria. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Gloria V. Echeverria, Z2xvcmlhLmVjaGV2ZXJyaWFAYmNtLmVkdQ==