 Francianne G. Andrade1
Francianne G. Andrade1 Suellen V. M. Feliciano1
Suellen V. M. Feliciano1 Ingrid Sardou-Cezar1Gisele D. Brisson1
Ingrid Sardou-Cezar1Gisele D. Brisson1 Filipe V. dos Santos-Bueno1Danielle T. Vianna2
Filipe V. dos Santos-Bueno1Danielle T. Vianna2 Luísa V. C. Marques1
Luísa V. C. Marques1 Eugênia Terra-Granado1Ilana Zalcberg2
Eugênia Terra-Granado1Ilana Zalcberg2 Marceli de O. Santos3Juliana T. Costa4
Marceli de O. Santos3Juliana T. Costa4 Elda P. Noronha1
Elda P. Noronha1 Luiz C. S. Thuler5
Luiz C. S. Thuler5 Joseph L. Wiemels6
Joseph L. Wiemels6 Maria S. Pombo-de-Oliveira1* and the Brazilian Collaborative Study Group of Acute Leukemia
Maria S. Pombo-de-Oliveira1* and the Brazilian Collaborative Study Group of Acute Leukemia- 1Pediatric Hematology-Oncology Program, Research Center, Instituto Nacional de Câncer, Rio de Janeiro, Brazil
- 2Laboratory of Molecular Biology, Bone Marrow Transplantation Center, Instituto Nacional de Câncer, Rio de Janeiro, Brazil
- 3Surveillance and Prevention, Instituto Nacional de Cancer, Rio de Janeiro, Brazil
- 4Department of Pediatric Hematology-Oncology, Hospital Martagão Gesteira, Salvador, Brazil
- 5Clinical Research Department, Research Center, Instituto Nacional de Câncer, Rio de Janeiro, Brazil
- 6Center for Genetic Epidemiology, Keck School of Medicine, University of Southern California, Los Angeles, CA, United States
Previous studies have suggested a variation in the incidence of acute promyelocytic leukemia (APL) among the geographic regions with relatively higher percentages in the Latin American population. We aimed to explore the population burden of pediatric APL, gathering information from the population-based cancer registry (PBCR) and the diagnosis of APL obtained through incident cases from a hospital-based cohort. The homozygous deletion in glutathione S-transferases (GSTs) leads to a loss of enzyme detoxification activity, possibly affecting the treatment response. Mutations in the RAS pathway genes are also considered to be a key component of the disease both in the pathogenesis and in the outcomes. We have assessed mutations in a RAS–MAP kinase pathway (FLT3, PTPN11, and K-/NRAS) and GST variant predisposition risk in the outcome. Out of the 805 children and adolescents with acute myeloid leukemia (AML) who are registered in the PBCR, 35 (4.3%) were APL cases. The age-adjusted incidence rate (AAIR) was 0.03 per 100,000 person-years. One-hundred and sixty-three patients with APL were studied out of 931 AML cases (17.5%) from a hospital-based cohort. Mutations in FLT3, KRAS, and NRAS accounted for 52.1% of the cases. Patients with APL presented a 5-year probability of the overall survival (OS) of 67.3 ± 5.8%. A GST-theta 1 (GSTT1) null genotype conferred adverse prognosis, with an estimated hazard ratio of 2.8, 95% confidence interval (CI) 1.2–6.9. We speculate that the GSTT1 polymorphism is associated with therapeutics and would allow better OS of patients with APL with a GSTT1 null genotype.
Introduction
Acute promyelocytic leukemia (APL) is a relatively uniform subtype of acute myeloid leukemia (AML) characterized by the accumulation of abnormal promyelocytes in the bone marrow (BM). In about >95% of the APL cases, a chimeric fusion gene that drives the pathogenesis originates in the chromosome translocation 15q24 and PML-RARα is a successful target for therapy using all-trans retinoic acid (ATRA) and arsenic trioxide (ATO) (1). Epidemiological studies reported the APL incidence rate in the adult population of 0.32 per 100,000 persons in the USA (2), and a high frequency in the Latin American, Southern European, and African childhood population (3). A wide variation in the relative frequency of pediatric APL among the worldwide population suggests that genetic predisposition and/or environmental exposures may influence susceptibility to breakage at the site involved in translocation and that ancestry and exposures via occupations or other factors should be studied for APL risk.
Inherited polymorphisms in the enzymes that activate or detoxify chemotherapeutic agents and the enzymes that protect cells from a reactive oxygen species generated by drugs may also influence the effectiveness of environmental exposures on health, treatment efficacy, and long-term outcomes. Glutathione S-transferases (GSTs) are a family of phase II detoxification enzymes, which act either by the direct conjugation of a drug with glutathione or by neutralizing drug-induced reactive compounds. The homozygous deletion of GSTs (named as null genotype) leads to a complete loss of enzyme activity, possibly affecting the treatment response (4). A profound polymorphic variance of these enzymes may be significant within promyelocytes, which are metabolically active cells containing high myeloperoxidase levels necessary for their primary functions, within primary azurophilic granules (5). In the context of somatic aberrations, APL is almost universally defined by the PML–RARα fusion gene, while the RAS pathway mutations are additionally a key component of the disease both in the pathogenesis and outcome. Mutations in the RAS–MAP kinase pathway genes are recurrent in leukemia, acting in leukemogenesis and the outcome (6).
To strengthen the ties of observational and molecular epidemiological studies of childhood APL in the Latin population, we explored the population burden of pediatric APL in Brazil, gathering the information from a population-based cancer registry (PBCR) and the APL diagnosis obtained through the incident cases from a hospital-based cohort. We have assessed both the mutations in the RAS–MAP kinase pathway (FLT3, PTPN11, and K-/NRAS) and the predisposition risk of a GST variant in the outcome. We built this data with our unique and complementary network study group of childhood leukemia in Brazil to assess the impact of biomarkers in the APL cases in the overall survival (OS) (7).
Results
Acute Promyelocytic Leukemia Incidence Rate
The coverage area of PBCR is estimated to be about 20% of the Brazilian population. Table 1 shows the number of APL cases, sex ratio, age-specific incidence rates (ASIRs), crude rates, and age-adjusted incidence rates (AAIRs) for the Brazilian regions. Out of 805 AML cases in children and adolescents who were registered in the PBCR (2000–2009), 35 cases were APL (4.3%) (data not shown). About <3% of the patients were identified with the death certificate. The male:female ratio was 1.2:1.0. The AAIR of APL was 0.03 per 100,000 person-years [95% confidence interval (CI) 0.03–0.03]. AAIR varied according to the macro-geographical region, with a higher incidence in the South region of Brazil in comparison with other areas. The incidence trends in AAIR (0–19 years) for the APL cases in the whole country remain stable throughout the period. The annual percent change (APC) is 14.1% (95% CI −2.9–34.0), although we observed fluctuations in all the Brazilian regions (Figure 1; Supplementary Table 1).
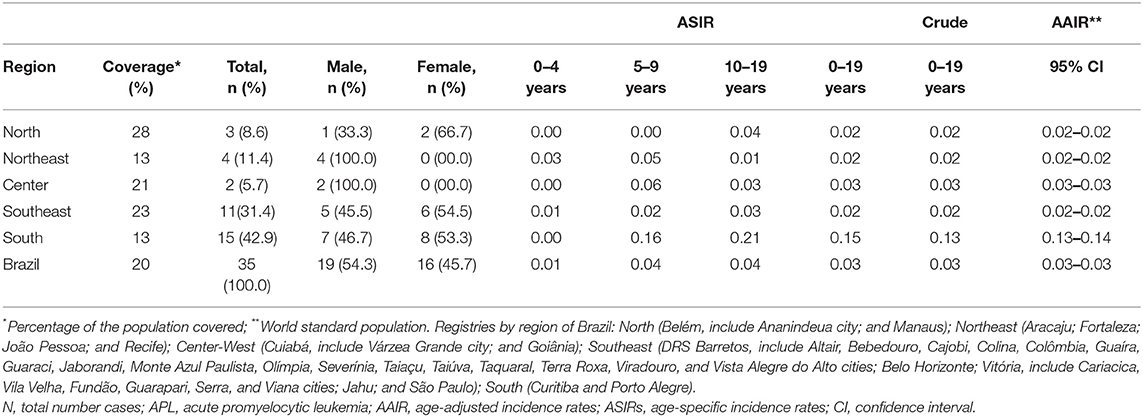
Table 1. Data on the incidence rate of APL cases from 15 Brazilian population-based cancer registries, 2000–2009.
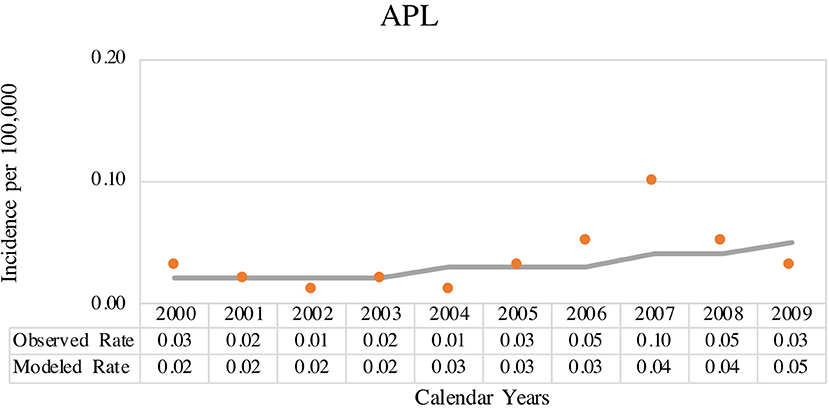
Figure 1. Joinpoint analysis for age-adjusted incidence rate (AAIR) (0–19 years, standard world population) from acute promyelocytic leukemia (APL) in Brazilian population-based cancer registry (PBCR), 2000–2009. Solid circles indicate the data for observed age-adjusted rate and solid line indicates the data for modeled age-adjusted rate.
Acute Promyelocytic Leukemia in a Hospital-Based Cohort
We have characterized 163 APL (17.5%) of 931 AML cases in a time frame of 17 years in the macro-geographical Brazilian regions, as shown in Table 2. Patients presented a median age at the diagnosis of 10.1 (0.75–19) years. The male:female ratio was 1.2:1.0 with a prevalence of non-blacks (89.4%). The prevalence of APL cases from the Northeast and Southeast regions was 41.1 and 28.2%, respectively; the South region presented the lower frequency (2.5%). Cases were sent from 33 medical centers, including seven hospitals that fed the PBCRs information data. We observed an increased frequency of APL cases from 2010 to 2018.
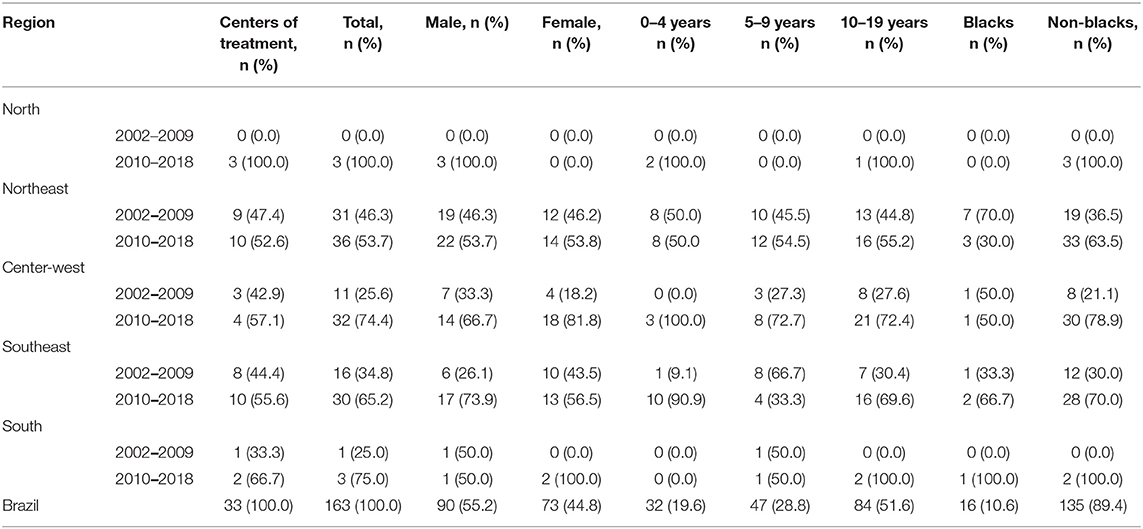
Table 2. The distribution frequency of APL diagnosed according to microregion, age, sex, and ethnicities, Brazil, 2002–2018.
The APL cases were clinical-morphological characterized by hyper granular subtype (86.5%) median of white blood cells (WBCs) count of 13.1 × 109/L (min,1 × 109/L, max 800 × 109/L). Twenty-two patients died in the first 10 days after the clinical diagnosis. Early death occurrence was observed in 10 patients who died before starting any treatment. The main cause of death was hemorrhage (60.8%) next to septicemia complications (29.4%). About 130 patients (130/151, 86.1%) presented the PML–RARα fusion, with breaks distributed in intron 3 [breakpoint cluster regions (bcr) 3] of the PML gene (30/53, 56.6%), intron 6 (bcr 1; 18/53, 34.0%), and exon 6 (bcr 2; 5/53, 9.4%). Overall, the mutations in FLT3, KRAS, and NRAS accounted for 52.1% of the cases.
FLT3 Mutations in Pediatric Acute Promyelocytic Leukemia
As shown in Table 3, 52 patients (52/120, 43.3%) presented FLT3 mutations, including 6 (5.0%) mutations in the tyrosine kinase domain (TKD) in codon 835 and 46 (38.0%) internal tandem duplications (ITDs) in exons 11/12. Patients with FLT3 mutations had a median age at the diagnosis of 11.2 years, similar to the observation done for the patients with FLT3 wild-type (9.8 years; p = 0.264); cases at an early age (≤ 2 years of age) presented no mutations. Most patients with FLT3 mutations presented high WBC count (>10 × 109/L) and low platelet count (≤ 40 × 109/L) (Table 3; Supplementary Figure 1). We found an association of FLT3 mutations with a variant promyelocytic subtype (M3v or microgranular) in 21.2% of the cases and a high frequency of PML-RARα bcr 3; most patients without the mutations presented PML-RARα bcr 1 (Table 3). The median of the allelic frequency ratio of FLT3 ITD mutations was 0.35. We used the median as a cutoff to compare low (≤ 0.35) and high (>0.35) allelic ratios according to the demographic and laboratory features. As described in Supplementary Table 2, we observed an association of high allelic ratio of FLT3 ITD and patients who were aged from 2 to 10 years old, males, as well as having a high WBC count (>10 × 109/L).
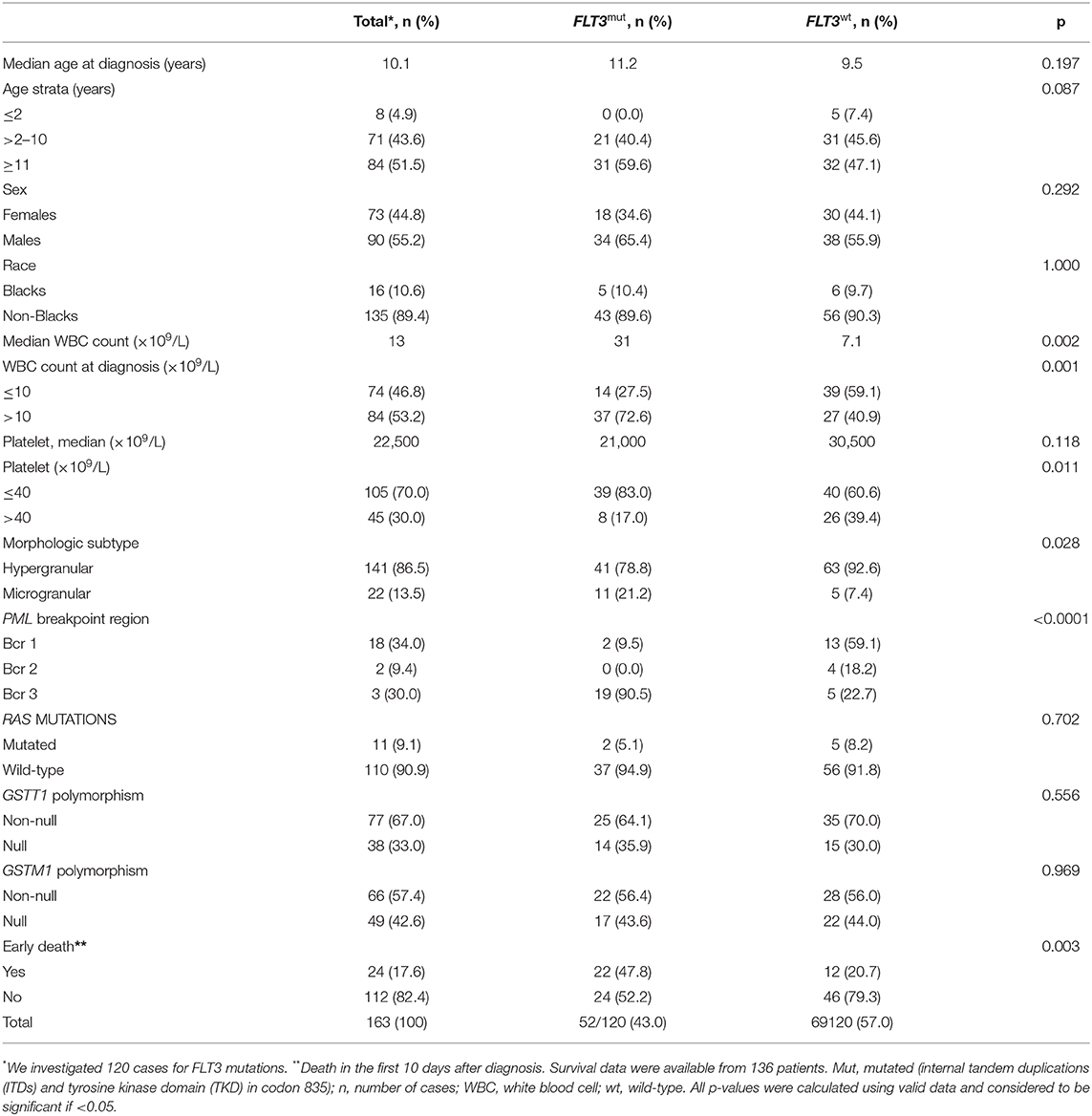
Table 3. Characteristics of APL according to FLT3 mutations (ITD/DTK).
Genetic Susceptibility
A GSTM1 null genotype occurred in 42.6% (49/115) of the cases and 40.3% (160/397) of the controls (p = 0.658) while a GST-theta 1 (GSTT1) null genotype was present in 33.0% (38/115) of the cases and 22.7% (90/397) of the controls (p = 0.024). We estimated the risk associations between genetic polymorphisms and APL as odds ratio (OR) 1.10 (95% CI 0.72–1.68) for the GSTM1 null genotype and OR 1.68 (95% CI 1.07–2.65) for the GSTT1 null genotype, respectively (Supplementary Table 3). We have tested risk associations between genetic alterations in GSTM1/GSTT1 and patients with APL harboring FLT3 mutations. There was no association between GSTM1/GSTT1 deletions and FLT3 mutations (crude OR = 0.98, 95% CI 0.42–2.29 and 1.31, 95% CI 0.54–3.19, respectively).
Survival
The data from 136 patients (83.4%) were available for the survival analysis. In the first 10 days after the diagnosis (early death), death affected 17.6% (24/136) of the patients, and we excluded these cases from the analysis. Patients with APL presented the probability of OS for 5 years of 67.3 ± 5.8% and a mean of 45.3 months (95% CI 40.2–50.4 months). Because of a small number of cases in each Brazilian geographic region category, we grouped patients in three localities (North/Northeast, Center-West, and South/Southeast). We observed no statistically significant differences in the outcome according to the locality of treatment, as well as to age, WBC and platelet counts, and the presence of RAS or FLT3 mutations. Although the patients with an undetected PML-RARα presented a slightly lower probability of OS, the difference was not statistically significant (Supplementary Table 4). A GSTT1 null genotype conferred adverse prognosis compared to wild-type genotype (probability of OS for 5 years 48.2 ± 13.4% and 80.5 ± 6.1%, respectively; p = 0.015) shown in Figure 2. The Cox regression model estimated the crude hazard ratio for GSTT1 deletions of 2.8, 95% CI 1.2–6.9, p = 0.021. No adjusting variable was retained in a multivariate model (Supplementary Table 5).
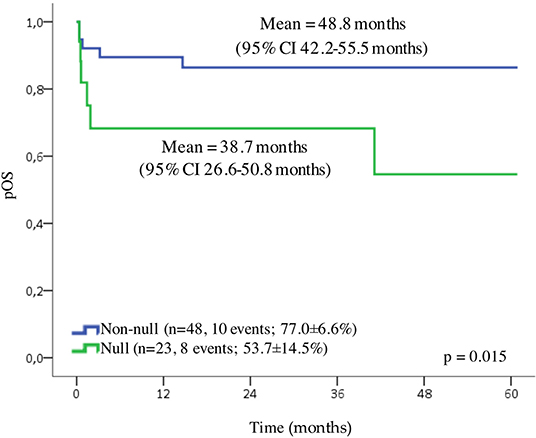
Figure 2. Overall survival (OS) of pediatric cases of APL. Probability of the OS, according to GSTT1 polymorphisms.
Discussion
Here, we report a comprehensive overview of the diagnostic characteristics and survival of APL pediatric cases in Brazil for the first time. Using two large data sources, we described the incidence of APL in children and adolescents from a population with Latino genetic background. We tested the hypothesis that polymorphisms in the genes of metabolic enzymes, i.e., GST, might affect the detoxification of chemotherapeutic compounds and found that a complete loss of the GSTT1 is associated with poor outcome.
Acute promyelocytic leukemia is a rare disease with an incidence rate in the US population of 0.27 per 100,000 in 2000–2008, increasing over time (0.11 per 100,000 person-years in 1975–1990). In young patients (<20 years of age), the disease is far rarer, with 0.06 per 100,000 compared to 0.36 per 100,000 among the patients diagnosed at ≥60 years of age, respectively (2). From PBCR with a coverage of 20% of the Brazilian population, we found an AAIR among the children of 0.03 per 100,000 persons in 2000–2009. An incidence of childhood cancer has been previously reported for Brazil; however, this is the first time APL was stratified from AMLs and compared to a robust hospital-based population (8–10). Other population-based studies have reported APL age-adjusted incidence of 0.08 cases per 100,000 person-years in Canada (11) and, in children, 2.5 per million person-years in Italy (12), 1.39 per million people in Spain, 0.54 per million person-years in Great Britain, 0.4 per million person-years in France, and an overall rate of 0.0–0.1 per 100,000 in the European population (13, 14). Environmental exposures and genetic predisposition, which make chromosomic sites to involve in the PML-RARα origin more prone to break in specific settings, may explain the epidemiological variation giving a hint into the pathogenesis of APL (15). As previously reported, region-specific environmental exposures might be associated with the underlying causal factors. The high rate in the South of the country might result from the main local economic activities such as agriculture and oil and gas industries (8). We observed a contrast in the incidence rate in the South region of Brazil in comparison to the other areas. Historically, the ancestry in the North has a considerable influence on Amerindian roots, the Northeast presents a strong African influence from slavery, and the European immigrants mostly settled in the South. European ancestry proportions range from 60.6% in the Northeast to 77.7% in the South (16). The macro-regions are primarily for statistical purposes and intriguingly mirror environmental exposures, and genetic background, which might result in the high incidence of APL observed in the South. A population-based study in Brazil has previously demonstrated regional differences in the incidence of AML (including APL) (9). The bias might be in the better quality of PBCR from the South than the other regions, measured by the percentage of the cases confirmed microscopically and <20% of the cases collected through death certificates. As noted by Feliciano et al. (8), the rates of unspecified leukemia were inversely proportional to the APL rates in the different Brazilian geographic regions, which led to the conjecture that the cases of APL could be included in the group of unspecified leukemia. However, we cannot rule out some case capture differences in the different regions of Brazil included in this study as another potential explanation of varied rates among the areas.
With an awareness of the potential challenges of the PBCR data representation of the Brazilian population, we also analyzed a hospital-based cancer registry covering >90% of pediatric leukemia in Brazil. PBCR mostly covers the people living in capital cities, and the hospital-based cancer registry does not exclude the patients according to the place of residence. We have built up a large population of leukemia patients over the past two decades by using an innovative clinical path resource for pediatric oncologists in Brazil, enhancing clinical treatments, and providing patient participation opportunities in epidemiologic studies (7). Our diagnostic resource offers a quality support mechanism and may yield a complete case capture. The rarity of an APL diagnosis directed our incidence estimates by calculating the proportion of APL among the AML cases. In combination with the immunophenotypic and molecular characterization, the well-defined morphological features of blast cells have reliably allowed the identification of APL.
We might explain the differences in the absolute number of APL cases between PBCR and a hospital-based cancer registry by using the different coverages of the registries and the period of analysis, and the inherent classification problems in the data sources. Despite the overlapping cases from the hospital based and the PBCR, several PBCR still lack this criteria and APL cases might be included in the AML group and the “no otherwise specified” entity (ICD-O-3 9861) (17–19).
We reported a proportion of 17.5% of APL among the AML cases referred to 52 Brazilian institutions. Jacomo et al. (20) have reported that the patients with APL represented 28.2% of the AML cases from 12 Brazilian institutions. This information could be biased by using the different patient referral patterns to specific hospitals and by including only the patients who were 15 years or older (15). The highest proportions (15–58%) of childhood APL were reported in the South and Central America studies, including Costa Rica, Chile, Bolivia, Guatemala, Honduras, and El Salvador. In African countries, APL represented around 15–20% of pediatric AML cases (Nigeria, South Africa, Tunisia, and Sudan), 10–15% of the AML cases in Egypt, and 5–10% of the AML cases in Malawi. Proportions of APL were similarly variable in the Middle East (3.4% in Saudi Arabia and 34.5% in Iraq) and Asia where China and Pakistan describe a proportion of 36.2 and 43.5%, respectively (3).
The translocation (14, 16) (q24;q21) that results in the chimeric gene/protein PML-RARα is a defining abnormality of APL. Using the techniques of fluorescence in situ hybridization (FISH) and reverse transcriptase PCR (RT-PCR), we could not detect PML-RARα in 13.9% of the cases. Zhao et al. (21) described the absence of PML-RARα in 18.8% of pediatric APL cases and the FISH and RT-PCR sensitivities as 91.30 and 88.64%, respectively. In the conflicting results, a next-generation sequencing performed in seven FISH/RT-PCR PML-RARα negative samples revealed six PML-RARα samples and one NPM-RARα (21). Atypical PML-RARα results in negative RT-PCR and/or FISH are a consequence of complex rearrangements, alternative splicing within PML or RARα, as well as through insertions/deletions beyond the limit of resolution (22–25). The effects of storage temperature and duration of whole blood on RNA quality might also affect the assay results, especially in a multicentric study in which the samples undergo environmental variations (26). When a next-generation sequencing is not available, clues to the diagnosis are the highly characteristic morphological, immunophenotypic, and clinical features (27).
The breakpoint in the RARa gene falls into intron 2 while the breakpoints in PML are distributed in intron 6 (bcr 1), exon 6 (bcr 2), and intron 3 (bcr 3), in 55, 8, and 40% of cases, respectively (28). We observed frequencies of bcr 1 in 34.0%, bcr 2 in 9.4%, and bcr 3 in 56.6% of the cases. Previous studies reported bcr 1 in 42.9–75% of the patients, with the highest proportions in Hispanic/Latino patients (15, 29–33). Our concordant findings fall into FLT3 wild-type patients (bcr 1 = 59.1%), meaning that secondary mutational events might give a growth advantage for pre-leukemic clones rather than a genetic background of a specific population. The bcr clusters of PML and RARa in therapy-related APL in genomic regions that are particularly susceptible to cleavage by using topoisomerase II inhibitors corroborates this hypothesis (3).
While all patients with APL will have a private or low frequency of recurrent mutations, we expect all APLs to harbor the mutations in a limited number of pathways, which reflect the homogeneous nature of the disease. Our findings of recurrent mutations in KRAS/NRAS genes in 8.8% of the APL cases were slightly lower than the observation by others, and the mutations in FLT3 (43.3%) were similar to the previous studies (20–53%) (21, 33–35). A higher frequency of FLT3 mutations in older patients is believed to result from a multistep process of the accumulation of secondary mutations (36). Other authors also reported the association of FLT3 mutations with high WBC count, bcr 3 in PML gene, and microgranular morphology (29, 37–39). For FLT3 ITD mutations, the allelic ratio refers to the number of ITD-mutated alleles compared to the wild-type allele number. This ratio is not only influenced by the amount of malignant vs. non-malignant cells in the tested sample but also by the percentage of cells with 0, 1, or 2 mutated alleles. The allelic ratio reflects the clonal burden of the FLT3 ITD-mutated cells within the leukemia cell population (40). Like secondary events, FLT3 mutations occur in a subset of the leukemic cell population only, resulting in both the upregulation of genes associated with cell division and proliferative signals in vitro. Thus, FLT3 mutations could readily account for the in vivo association with higher WBC count and are at very high risk for death (38, 41). On the contrary to the previously described ones, we did not find an association of FLT3 mutations with the OS (33).
Despite laboratory and molecular support for the diagnosis, our early death rate was higher compared to clinical trials. Rego et al. (42) described that 15% of the deaths occurred between a diagnosis and the first morphologic evaluation of BM (30 days), with 5% (9 cases) of the deaths within the first 7 days of treatment from the International Consortium on APL gathering patients >15 years of age from Brazil, Mexico, Chile, and Uruguay. Adès et al. (43) reported 2.8 and 6.8% of the deaths in the first 30 days of treatment in the LPA 99 trial (Spain, Argentina, The Netherlands, and the Czech Republic) and APL 2000 trial (France, Switzerland, and Belgium), respectively. Avvisati et al. (44) reported 5.5% (44 patients) of the deaths during the induction, and three patients died before starting the treatment in the AIDA 0493 trial (Italy, The Netherlands, and Germany). The causes of early death include a delay in referral to specialized centers and in recognizing APL due to its rarity, the availability of ATRA in the centers, and supportive care management. Access to such supportive care is more complicated outside the major cities in Brazil. The international effort to recognize and treat APL reduced early mortality and increased OS in developing countries. Action has been made to ensure prompt availability of ATRA in all centers, the definition of pre-existing transfusion medicine services, and hematology laboratories able to perform basic fluorescence microscopy, data report, and the establishment of a reference laboratory for molecular diagnosis (42). Further educational training of healthcare providers to recognize APL as a medical emergency will impact early death and improve survival rates mainly because most of the patients are first admitted in emergency rooms or intensive care units (45).
In the present study, OS was lower compared to collaborative studies and trials from developed countries in which a 5-year OS for APL reaches ~95% (46, 47). Our patients treated according to the BFM-based protocol received more intensive treatment than those treated according to the GIMEMA-based protocol. Nowadays, both GIMEMA and BFM study groups recommend low or no dose chemotherapy for low-risk patients and ATRA combined with ATO for APL treatment (48, 49). In Brazil, physicians use ATO as a first-line therapy for the last 3 years, and a uniform protocol has been developed in Latin America for pediatric APL (50).
After overcoming the complications in the first month of treatment, some patients will face failure in the treatment and risk relapse attributable to genetic susceptibility. Davies et al. (51) showed for the first time an association of a GSTT1 null genotype and an increase of deaths in remission patients with AML after intensive chemotherapy, including a high-dose anthracycline and cytarabine. GSTs are phase II detoxification enzymes that metabolize cancer chemotherapeutic agents, insecticides, herbicides, carcinogens, and oxidative stress products, catalyzing the conjugation of glutathione to these substances (4). Anthracyclines used in treating leukemia, such as daunorubicin, generate high levels of reactive oxygen species and activate GSTs activity, which protects cells (52). Polymorphic gene deletions in GSTT1 or GSTM1 result in the absence of protein and may thus play a role in both leukemogenesis and in response to the treatment (53, 54). We found evidence of an association here between the GSTT1 polymorphisms and the outcome of APL. However, current and previous studies are still somewhat preliminary to prove an association of GSTT1 status and the outcome of acute leukemia due to small sample sizes, the differences in the chemotherapy regimens, and the age of the population included in the analyses (54–56). The strengths of the study are the clinical and laboratory characterization of a large cohort of children with APL with the relevance of GST polymorphisms associated with the outcome. The original results support partially our hypothesis. We described here an overview of the epidemiological, molecular, and clinical features of pediatric APL in Brazil. We confirmed a high proportion of APL among the AML cases in the Brazilian population, suggesting the influence of genetic background in leukemogenesis although the multicenter and retrospective data collection limits our work. This approach might be affected by slight differences in treatment strategies, making us unable to evaluate the therapeutic protocol efficacy and causes of early death. However, we found new evidence of chemotherapy metabolism effects on survival. A GSTT1 null genotype, which results in a loss of enzyme activity, was associated with a poorer response to therapy. Since only the individuals who had a homozygous-variant genotype (i.e., completely deficient) were classified and the heterozygous individuals were not distinguished from the wild-type patients, we believe there is an underestimation of the impact of GSTT1 on the outcome. A limitation of this study should be addressed to the results obtained in the PBCR inconsistencies which are evident when compared with the data obtained in hospital-based data (HBD) and with other PBCR worldwide. The differences between the PBCR and the HBD result from a variation in the reported diagnosis, for instance, in the PBCR the majority of APL either reported in AML or in not-otherwise specified categories, while in the HBD the diagnosis is based on cytogenetic-molecular tests.
Materials and Methods
Study Subjects
Our approach was to bring together the data on PBCR, APL diagnosis, biomarkers characterization, and genetic predisposition. Figure 3 shows a brief study design and how the data were built.
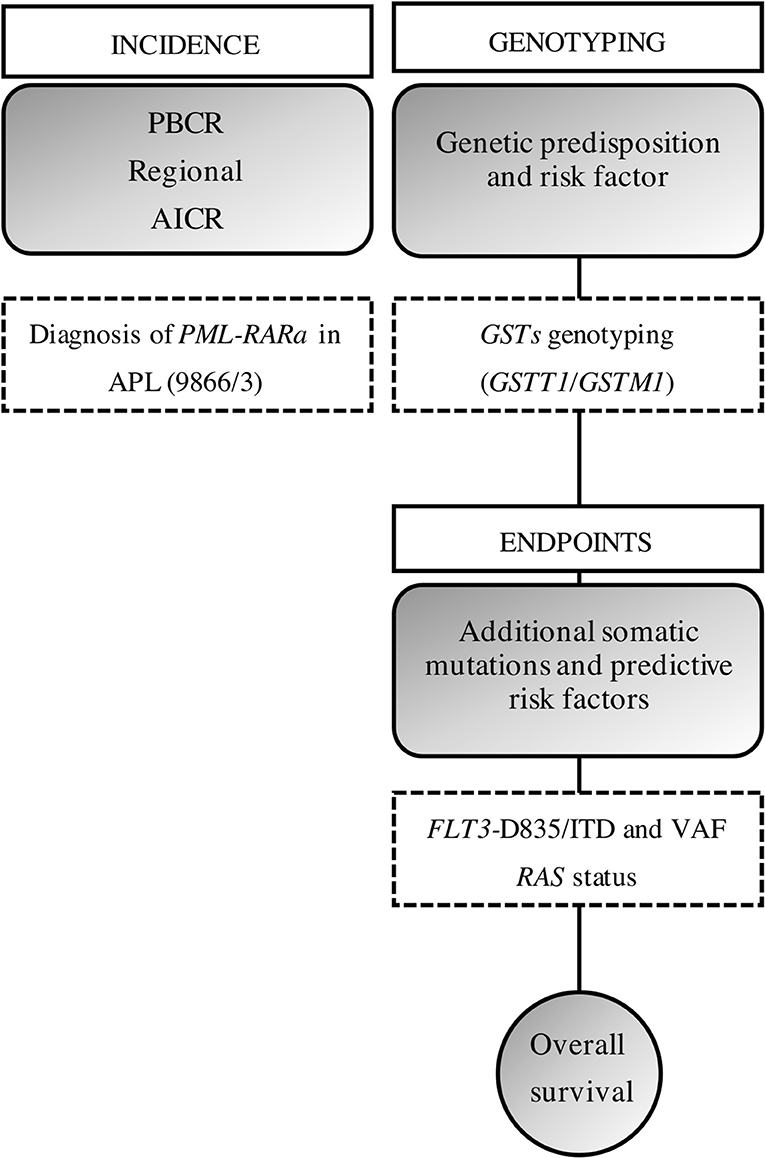
Figure 3. Study design. We gathered the information from a PBCR and the diagnosis of obtained through the incident cases from a hospital-based cohort. AAIR, age-adjusted incidence rate; ASIR, age-specific incident rate; ITD, internal tandem duplications; VAF, variant allelic frequency.
Population-Based Cancer Registries
The incidence data of APL (patients aged 0–19 years at the diagnosis) were obtained from databases of 15 PBCR across the Brazilian region from the period of 2000–2009 (8). Brazilian cancer registries correspond to local records by State, located in Capital and metropolitan surroundings, which gather the information on incidence cancers in the contributing registration areas. The PBCR distribution represents the Brazilian macro-geographical regions (shown in Supplementary Figure 2). The data were collected according to the International Agency for Research on Cancer (IARC) requirements for Cancer Registries according to quality data parameters, such as percentage of the AML cases diagnosed and confirmed microscopically, and <20% of those identified from the death certificate only (57, 58). Cases were classified according to the Third Edition of the International Classification of Diseases for Oncology (ICD-O-3/9866). Only data sets with high-quality data were eligible for inclusion in the analyses. Leukemia data were extracted from the Instituto National de Câncer and population demography from the Instituto Brasileiro de Geografia e Estatística website (https://www.inca.gov.br/BasePopIncidencias/Home.action; https://datasus.saude.gov.br).
Hospital-Based Cohort
Acute promyelocytic leukemia in children and adolescents following the PBCR age strata was diagnosed (2002–2018) in 33 Brazilian Oncological Centers and in the Pediatric Hematology-Oncology Program, Instituto Nacional de Câncer, Brazil, that is a reference laboratory for molecular diagnosis and pathology of childhood leukemia (59). Inclusion criteria were the cases with a diagnosis of APL according to morphology, immunophenotyping, or the presence of PML-RARα in BM and peripheral blood (PB) samples from the patients aged 0–19 years old. Molecular analysis for the detection of PML-RARα was based on the results of the good quality of a diagnostic material. The data set with demographic and clinical information were obtained from the clinicians at the time of diagnosis and complementary data during the follow-up of patients, such as the occurrence of relapse (if any, with data and local) and the status of the patient (death/alive).
Biological specimens (BM and PB samples) were collected previously for any chemotherapy treatment and to test minimal residual diseases. A predefined set of variables was collected, including sex, age, ethnicity, place of residence, significant clinical symptoms, WBC count, platelets count, and blast cells percentage. The institution staff of the patients provided these data via an online system (https://imunomolecular.inca.gov.br/).
The diagnostic algorithm tests included BM morphology, immunophenotyping, and molecular cytogenetics as described in Supplementary Figure 3. Briefly, samples were sent with the clinical report and suspicious of APL. Samples were immediately prepared for the identification of a PML–RARα fusion gene by using FISH. Immunophenotyping was performed and the APL diagnosis was released to clinicians in about12 h after the samples were received.
Molecular Diagnosis
Total RNA was purified from BM cells using TRIzol™ reagent (Invitrogen, Carlsbad, CA, USA) and directed for a complementary DNA (cDNA) synthesis following a transcriptase reverse enzyme addition. cDNA integrity was evaluated throughout the GAPDH amplification. Genomic DNA was purified from the BM by using a TRIzol™ reagent (Invitrogen, CA, USA), the QIAmp DNA Blood Mini Kit (Qiagen, CA, USA), or the HiYield Genomic DNA Mini Kit (Real Biotech Corporation, Taiwan).
PML-RARα was identified by using FISH with commercial probes (PML/RARa Translocation Probe Dual Fusion; Cytocell, Cambridge, UK). A RT-PCR was performed for the identification of PML-RARα according to a previous study (28). The bcr is localized within a 15 kb of the RARα intron 2 and three regions of the PML locus in intron 6, exon 6, and intron 3, respectively, named as bcr1, bcr2, and bcr3. A combination of four primer pairs was used to amplify RT-PCR products with different sizes allowing to identify transcripts with breaks in intron 6, exon 6, and intron 3 of PML and the variable breakpoint positions. Primer sequences were described by Van Dongen et al. (28). A nested amplification was included to improve the sensitivity of the reaction. Both reactions followed RT-PCR temperatures and cycle times: initial melting: 95°C for 30 s, 35 cycles of 94°C for 30 s (melting), 65°C for 60 s (annealing), and 72°C for 60 s (extension). The evaluation of the PCR products was performed with 10 ml of the products in a 1.5% agarose gel electrophoresis. The PML-RARα screening has been included in the APL diagnosis algorithm since 2009. The used positive control was the sample from APL with PML-RARα positive using FISH methods and confirmed by direct sanger sequencing.
Mutation Detection
The mutations in hotspot regions of RAS–MAP kinase pathway signaling genes (FLT3, NRAS, KRAS, and PTPN11) were analyzed by direct sequencing according to a previous study (59). The analysis included the cases with available biological materials (valid data; Supplementary Table 6).
Briefly, FLT3 mutations were examined at the TKD in codon 835 and the juxtamembrane domain in exons 11/12 as ITD. The variant allele frequency ratio of FLT3 ITD was assessed by a 6-carboxyfluorescein and PCR forward primer 5-end-labeled fragment analysis. About 1 μl of the product was analyzed in ABI3130XL (Applied Biosystems, CA, USA). The area of mutant peaks on the electropherogram was analyzed using the ChimerMarker® software (SoftGenetics, PA, USA). The FLT3 ITD mutant allelic burden was calculated as the ratio of the area under the curve of mutant and wild-type alleles (mutant/total). NRAS/KRAS status was determined by searching the mutations in exon 1 (codons 12/13), and PTPN11 mutations were screened in exons 3 and 13 (60). Primers and PCR conditions are described in Supplementary Table 7.
Glutathione S-transferase Genotyping
GSTM1 and GSTT1 homozygous deletions were detected by using a multiplex PCR as described in a study (61). The null genotype definition consisted of both copies of the genes GSTM1 or GSTT1 to be deleted while the non-null genotype was defined as at least one copy of the gene presented. The attributable risk conferred by genetic susceptibility was tested in a case-control study. An unmatched control group of healthy samples was used to estimate the susceptibility risk. It consisted of 416 umbilical cord blood samples randomly selected from the National Umbilical Cord and Placental Blood Bank. The samples were collected from healthy newborns at birth and are described in a study (62).
Treatment
Patients were not enrolled in a supervised clinical trial. APL management initially followed the same recommendations in the International Consortium on APL study of adult patients (50). Overall, the pediatric Brazilian institutions have established therapeutic protocols guided by two international clinical protocols: (i) the GIMEMA-based protocol that consisted of induction phase with ATRA and anthracycline, followed by consolidation phase with ATRA/Anthracycline and Cytarabine (three cycles), and maintenance with ATRA/Mercaptopurine and methotrexate (2 years) (48) and (ii) the BFM-AML-based protocol that consisted of the management of the patients with APL as low-risk AMLs with the addition of ATRA. Briefly, the induction-phase included ATRA, anthracycline, cytarabine, etoposide, followed by consolidation with ATRA, alternating anthracycline, cytarabine, and etoposide (three cycles), and 1-year maintenance of continuous thioguanine and one low dose of cytarabine monthly (49, 50).
Ethical Research
All collaborators have reviewed the ethical board and approved the APL treatment care according to the local laws and guidelines, following the Declaration of Helsinki. Informed consent was obtained from the parents of the children for the research assessments. The Ethical and Research Committee approved this study of the Instituto National de Câncer (#186.688).
Statistical Analysis
Population-Based Cancer Registries
Incidence rates were calculated as the annual number of cases per 100,000 persons per year, according to the geographical regions (North, Northeast, Center-West, Southeast, and South), based on a 10-year period of available information for all PBCR (2000–2009). AAIR per 100,000 person-year was calculated using the World Standard Population as described in a previous study (8). The 95% CI AAIR was calculated using the Poisson approximation. Incidence trends were performed using the Joinpoint regression model to identify the changes in APL incidence rates over time. To minimize the variability inherent in a small number of observations, the logarithm of the rates was used in the total data set. APC and average annual percent ranges (AAPC) were estimated and reported with corresponding 95% CI for each of the linear segments identified between two Joinpoints. The software was provided by the Surveillance Research Program of the US National Cancer Institute, version 4.6.0.0 (https://surveillance.cancer.gov/joinpoint).
Hospital-Based Cohort
Descriptive analyses were performed through continuous variables to measure central tendency and dispersion; medians were compared using the non-parametric Mann–Whitney U-test. Categorical variables such as demographic variables (sex and ethnicity) were compared using χ2 or the Fisher's exact test. Crude OR and 95% CI were assessed using an unconditional logistic regression to estimate the magnitude of risk associations between GSTM1/GSTT1 deletions and FLT3 mutations. Ethnicity was categorized into “blacks” and “non-blacks” as determined by the parents or guardians of the children, as a means of creating only two heterogeneity groups, since racial categorization in Brazil is a poor predictor of genomic ancestry (16). Age was also considered a categorical variable with three groups strata: (i) ≤ 2 years old; (ii) 2–10 years old; and (iii) >11 years old.
The OS was measured from the date of diagnosis to the last follow-up or death due to any cause. Patients who did not experience an event were censored at the time of the last known contact or at the date of the end of follow-up (December 31, 2017). The Kaplan–Meier survival analysis method was used to calculate the 5-year probabilities of OS, and estimated survival values were compared using the log-rank test to verify the association of one genetic alteration in the outcome of patients. An association between independent variables and the outcome was performed using the Cox proportional-hazard regression model with an estimated hazard ratio, and 95% CI were presented. Variables with p <0.25 in the univariate analysis (OS) or clinically relevant were included in the multivariate model following a stepwise forward method to adjust for confounders. All p-values were calculated using valid data and considered to be significant if <0.05. All analyses were performed using SPSS 20.0 (SPSS, Chicago, IL, USA, 2004).
Conclusions
We speculate that the GSTT1 polymorphism associated with prompt therapeutic protocols would permit survival improvements in the OS of patients with APL presenting a GSTT1 null genotype.
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation, but following the General Data Protection Regulation (GDPR, current version OJL 127,23.5.2018, adapted in Brazil under name Lei Geral de Proteção de Dados Pessoais, LGPD 13.709).
Ethics Statement
The studies involving human participants were reviewed and approved by The Ethical and Research Committee of the Instituto National de Câncer, Rio de Janeiro, Brazil (CEP#186.688). Written informed consent to participate in this study was provided by the participants' legal guardian/next of kin.
Author Contributions
MSPO: conceptualization. FA, IS-C, GB, FS-B, DV, LM, ET-G, and EN: gene mutation tests. FA, SF, and LT: formal statistical analysis. MSPO and IR: resources. FA, SF, and MS: data curation. FA: writing—original draft preparation. MSPO, JW, and JC: writing—review and editing. MSPO and JW: supervision. MP and FA: project administration. All authors critically reviewed and approved the final draft of the manuscript.
Funding
This research was funded by SwissBridge Fundation- Fundação do Câncer, Rio de Janeiro,Brazil (# L0J SWB 2014 SUB-PROJECT 1.A). MP has been a scholar of CNPq # 310877/2019-9; FA has been supported by Fundação Carlos Chagas Filho de Amparo à Pesquisa do Estado do Rio de Janeiro E-26/200.216/2017 and Ministry of Health of Brazil/INCA.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank the Brazilian Collaborative Study Group of Childhood Leukemia for providing samples and clinical data. We thank Alython Araujo Chung Filho, Vitória Carvalho, Bruno de Almeida Lopes, and Luciana Mayumi Gutiyama for technical support.
Brazilian Collaborative Study Group of Acute Leukemia (EMiLI)
Renata Sarkis Alves1, Renata Pereira de Souza Barros1, Rosania Maria Basegio2, Patrícia Carneiro de Brito3, José Carlos Córdoba4, Imaruí Costa5, Eloisa Cartaxo Eloy Fialho6, Teresa Cristina Cardoso Fonseca7, Isis Maria Quezado Magalhães4, Glaceanne Torres da Luz Mamede6, Eda Manzo8, Rebeca Ferreira Marques9, Gustavo Ribeiro Neves10, Andrea Gadelha Nobrega6, Claudia Teresa Oliveira11, Renato de Paula Guedes Oliveira12, Sidnei Epelman12, Ana Maria Marinho da Silva13, Luciana Nunes Silva13, Marcelo dos Santos Souza2, Regiana Quinto de Souza7, Adriano Nori Rodrigues Taniguchi9, Luciana Garcia Trujillo14, and Alayde Vieira Wanderley15.
1Hospital Estadual dos Servidores do Estado, RJ; 2Hospital Regional de Mato Grosso do Sul Rosa Pedrossian, Campo Grande, MS; 3Hospital Araújo Jorge, Goiânia, GO; 4Hematology-Oncology of Hospital da Criança de Brasília, DF; 5Hospital Infantil Joana de Gusmão, Florianópilis, SC; 6Hospital Napoleão Laureano, João pessoa, PB; 7Santa Casa, Hospital Manoel Novaes, Itabuna, BA; 8Hospital Amaral Carvalho, Jaú, SP; 9Hospital de Clínicas de Porto Alegre, Porto Alegre, RS; 10Gpaci Hospital Sarina Rolim Caracante, Sorocaba, SP; 11Hospital Estadual de Bauru, SP; 12Hospital Santa Marcelina, São Paulo, SP; 13Hospital Martagão Gesteira, Salvador, BA; 14Hospital Estadual Mario Covas, Santo André, SP; 15hospital Oncológico Infantil Octávio Lobo, Belém, PA.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2021.642744/full#supplementary-material
References
1. Thé H de, Pandolfi PP, Chen Z. Acute promyelocytic leukemia: a paradigm for oncoprotein-targeted cure. Cancer Cell. (2017) 32:552–60. doi: 10.1016/j.ccell.2017.10.002
2. Chen Y, Kantarjian H, Wang H, Cortes J, Ravandi F. Acute promyelocytic leukemia: a population-based study on incidence and survival in the United States, 1975-2008. Cancer. (2012) 118:5811–8. doi: 10.1002/cncr.27623
3. Zhang L, Samad A, Pombo-de-Oliveira MS, Scelo G, Smith MT, Feusner J, et al. Global characteristics of childhood acute promyelocytic leukemia. Blood Rev. (2015) 29:101–25. doi: 10.1016/j.blre.2014.09.013
4. Hayes JD, Flanagan JU, Jowsey IR. Glutathione transferases. Annu Rev Pharmacol Toxicol. (2003) 45:51–88. doi: 10.1146/annurev.pharmtox.45.120403.095857
5. Guibal FC, Alberich-Jorda M, Hirai H, Ebralidze A, Levantini E, Ruscio AD, et al. Identification of a myeloid committed progenitor as the cancer-initiating cell in acute promyelocytic leukemia. Blood. (2009) 114:5415–25. doi: 10.1182/blood-2008-10-182071
6. de Rooij J, Zwaan C, van den Heuvel-Eibrink M. Pediatric AML: from biology to clinical management. J Clin Med. (2015) 4:127–49. doi: 10.3390/jcm4010127
7. Pombo de Oliveira MS, Koifman S, Vasconcelos GM, Emerenciano M, de Oliveira Novaes C. Brazilian collaborative study group of infant acute leukemia. Development and perspective of current Brazilian studies on the epidemiology of childhood leukemia. Blood Cells Mol Dis. (2009) 42:121–5. doi: 10.1016/j.bcmd.2008.10.012
8. Feliciano SVM, Santos M de O, Pombo-de-Oliveira MS, Aquino JÂP de, Aquino TA de, Arregi MMU, et al. Incidence and mortality of myeloid malignancies in children, adolescents and Young adults in Brazil: a population-based study. Cancer Epidemiol. (2019) 62:101583. doi: 10.1016/j.canep.2019.101583
9. Reis R de S, Camargo B de, Santos M de O, Oliveira JM de, Silva FA, Pombo-de-Oliveira MS. Childhood leukemia incidence in Brazil according to different geographical regions. Pediatr Blood Cancer. (2010) 56:58–64. doi: 10.1002/pbc.22736
10. Reis R de S, Santos M de O, de Camargo B, Oliveira JFP, Thuler LCS, Pombo-de-Oliveira MS. Early childhood leukemia incidence trends in Brazil. Pediatr Hematol Oncol. (2016) 33:83–93. doi: 10.3109/08880018.2015.1130763
11. Paulson K, Serebrin A, Lambert P, Bergeron J, Everett J, Kew A, et al. Acute promyelocytic leukaemia is characterized by stable incidence and improved survival that is restricted to patients managed in leukaemia referral centres: a pan-Canadian epidemiological study. Br J Haematol. (2014) 166:660–6. doi: 10.1111/bjh.12931
12. Maule MM, Dama E, Mosso ML, Magnani C, Pastore G, Merletti F. Childhood cancer registry of piedmont. High incidence of acute promyelocytic leukemia in children in northwest Italy, 1980-2003: a report from the Childhood Cancer Registry of Piedmont. Leukemia. (2008) 22:439–41. doi: 10.1038/sj.leu.2404916
13. Marcos-Gragera R, Cervantes-Amat M, Vicente ML, de Sanjosé S, Guallar E, Godoy C, et al. Population-based incidence of childhood leukaemias and lymphomas in Spain (1993-2002). Eur J Cancer Prev Off J Eur Cancer Prev Organ. (2010) 19:247–55. doi: 10.1097/CEJ.0b013e328339e2f3
14. Visser O, Trama A, Maynadié M, Stiller C, Marcos-Gragera R, De Angelis R, et al. Incidence, survival and prevalence of myeloid malignancies in Europe. Eur J Cancer. (2012) 48:3257–66. doi: 10.1016/j.ejca.2012.05.024
15. Douer D, Santillana S, Ramezani L, Samanez C, Slovak ML, Lee MS, et al. Acute promyelocytic leukaemia in patients originating in Latin America is associated with an increased frequency of the bcr1 subtype of the PML/RARalpha fusion gene. Br J Haematol. (2003) 122:563–70. doi: 10.1046/j.1365-2141.2003.04480.x
16. Pena SDJ, Pietro GD, Fuchshuber-Moraes M, Genro JP, Hutz MH, Kehdy F de SG, et al. The genomic ancestry of individuals from different geographical regions of Brazil Is More Uniform Than Expected. PLoS ONE. (2011) 6:17063. doi: 10.1371/journal.pone.0017063
17. Bennett JM, Catovsky D, Daniel MT, Flandrin G, Galton DA, Gralnick HR, et al. Proposals for the classification of the acute leukaemias. French-American-British (FAB) co-operative group. Br J Haematol. (1976) 33:451–8. doi: 10.1111/j.1365-2141.1976.tb03563.x
18. Bennett JM, Catovsky D, Daniel MT, Flandrin G, Galton DA, Gralnick HR, et al. A variant form of hypergranular promyelocytic leukaemia (M3). Br J Haematol. (1980) 44:169–70. doi: 10.1111/j.1365-2141.1980.tb01195.x
19. Arber DA, Orazi A, Hasserjian R, Thiele J, Borowitz MJ, Beau MML, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. (2016) 127:2391–405. doi: 10.1182/blood-2016-03-643544
20. Jacomo RH, Melo RAM, Souto FR, Mattos ER de, Oliveira CT de, Fagundes EM, et al. Clinical features and outcomes of 134 Brazilians with acute promyelocytic leukemia who received ATRA and anthracyclines. Haematologica. (2007) 92:1431–2. doi: 10.3324/haematol.10874
21. Zhao J, Liang J-W, Xue H-L, Shen S-H, Chen J, Tang Y-J, et al. The genetics and clinical characteristics of children morphologically diagnosed as acute promyelocytic leukemia. Leukemia. (2019) 33:1387–99. doi: 10.1038/s41375-018-0338-z
22. Walz C, Grimwade D, Saussele S, Lengfelder E, Haferlach C, Schnittger S, et al. Atypical mRNA fusions in PML-RARA positive, RARA-PML negative acute promyelocytic leukemia. Genes Chromosom Cancer. (2010) 49:471–9. doi: 10.1002/gcc.20757
23. Welch JS, Westervelt P, Ding L, Larson DE, Klco JM, Kulkarni S, et al. Use of whole-genome sequencing to diagnose a cryptic fusion oncogene. JAMA. (2011) 305:1577–84. doi: 10.1001/jama.2011.497
24. Blanco EM, Curry CV, Lu XY, Sarabia SF, Redell MS, Lopez-Terrada DH, et al. Cytogenetically cryptic and FISH-negative PML/RARA rearrangement in acute promyelocytic leukemia detected only by PCR: an exceedingly rare phenomenon. Cancer Genet. (2014) 207:48–9. doi: 10.1016/j.cancergen.2014.01.001
25. Gruver AM, Rogers HJ, Cook JR, Ballif BC, Schultz RA, Batanian JR, et al. Modified array-based comparative genomic hybridization detects cryptic and variant PML-RARA rearrangements in acute promyelocytic leukemia lacking classic translocations. Diagn Mol Pathol Am J Surg Pathol Part B. (2013) 22:10–21. doi: 10.1097/PDM.0b013e31825b8326
26. Huang L-H, Lin P-H, Tsai K-W, Wang L-J, Huang Y-H, Kuo H-C, et al. The effects of storage temperature and duration of blood samples on DNA and RNA qualities. PloS ONE. (2017) 12:e0184692. doi: 10.1371/journal.pone.0184692
27. Rashidi A, Fisher SI. FISH-negative, cytogenetically cryptic acute promyelocytic leukemia. Blood Cancer J. (2015) 5:e320. doi: 10.1038/bcj.2015.47
28. van Dongen JJ, Macintyre EA, Gabert JA, Delabesse E, Rossi V, Saglio G, et al. Standardized RT-PCR analysis of fusion gene transcripts from chromosome aberrations in acute leukemia for detection of minimal residual disease. Report of the BIOMED-1 Concerted Action: investigation of minimal residual disease in acute leukemia. Leukemia. (1999) 13:1901–28. doi: 10.1038/sj.leu.2401592
29. González M, Barragán E, Bolufer P, Chillón C, Colomer D, Borstein R, et al. Pretreatment characteristics and clinical outcome of acute promyelocytic leukaemia patients according to the PML-RAR alpha isoforms: a study of the PETHEMA group. Br J Haematol. (2001) 114:99–103. doi: 10.1046/j.1365-2141.2001.02915.x
30. Burnett AK, Grimwade D, Solomon E, Wheatley K, Goldstone AH. Presenting white blood cell count and kinetics of molecular remission predict prognosis in acute promyelocytic leukemia treated with all-trans retinoic acid: result of the Randomized MRC Trial. Blood. (1999) 93:4131–43. doi: 10.1182/blood.V93.12.4131
31. Ruiz-Argüelles GJ, Garcés-Eisele J, Reyes-Núñez V, Gómez-Rangel JD, Ruiz-Delgado GJ. More on geographic hematology: the breakpoint cluster regions of the PML/RARalpha fusion gene in Mexican Mestizo patients with promyelocytic leukemia are different from those in Caucasians. Leuk Lymphoma. (2004) 45:1365–8. doi: 10.1080/10428190310001657344
32. Chatterjee T, Gupta S, Sharma S, Ganguli P. Distribution of different PML/RARα bcr Isoforms in indian acute promyelocytic leukemia (APL) patients and clinicohematological correlation. Mediterr J Hematol Infect Dis. (2014) 6:e2014004. doi: 10.4084/mjhid.2014.004
33. Lucena-Araujo AR, Kim HT, Jacomo RH, Melo RA, Bittencourt R, Pasquini R, et al. Internal tandem duplication of the FLT3 gene confers poor overall survival in patients with acute promyelocytic leukemia treated with all-trans retinoic acid and anthracycline-based chemotherapy: an International Consortium on Acute Promyelocytic Leukemia study. Ann Hematol. (2014) 93:2001–10. doi: 10.1007/s00277-014-2142-9
34. Madan V, Shyamsunder P, Han L, Mayakonda A, Nagata Y, Sundaresan J, et al. Comprehensive mutational analysis of primary and relapse acute promyelocytic leukemia. Leukemia. (2016) 30:1672–81. doi: 10.1038/leu.2016.69
35. Fasan A, Haferlach C, Perglerovà K, Kern W, Haferlach T. Molecular landscape of acute promyelocytic leukemia at diagnosis and relapse. Haematologica. (2017) 102:e222–24. doi: 10.3324/haematol.2016.162206
36. Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA, Kinzler KW. Cancer genome landscapes. Science. (2013) 339:1546–58. doi: 10.1126/science.1235122
37. Callens C, Chevret S, Cayuela J-M, Cassinat B, Raffoux E, de Botton S, et al. Prognostic implication of FLT3 and Ras gene mutations in patients with acute promyelocytic leukemia (APL): a retrospective study from the European APL Group. Leukemia. (2005) 19:1153–60. doi: 10.1038/sj.leu.2403790
38. Gale RE, Hills R, Pizzey AR, Kottaridis PD, Swirsky D, Gilkes AF, et al. Relationship between FLT3 mutation status, biologic characteristics, and response to targeted therapy in acute promyelocytic leukemia. Blood. (2005) 106:3768–76. doi: 10.1182/blood-2005-04-1746
39. Kuchenbauer F, Schoch C, Kern W, Hiddemann W, Haferlach T, Schnittger S. Impact of FLT3 mutations and promyelocytic leukaemia-breakpoint on clinical characteristics and prognosis in acute promyelocytic leukaemia. Br J Haematol. (2005) 130:196–202. doi: 10.1111/j.1365-2141.2005.05595.x
40. Levis M. FLT3 mutations in acute myeloid leukemia: what is the best approach in 2013? Hematol Am Soc Hematol Educ Program. (2011) 2013:220–6. doi: 10.1182/asheducation-2013.1.220
41. Kutny MA, Moser BK, Laumann K, Feusner JH, Gamis A, Gregory J, et al. FLT3 mutation status is a predictor of early death in pediatric acute promyelocytic leukemia: a report from the Children's Oncology Group. Pediatr Blood Cancer. (2012) 59:662–7. doi: 10.1002/pbc.24122
42. Rego EM, Kim HT, Ruiz-Argüelles GJ, Undurraga MS, Uriarte M del R, Jacomo RH, et al. Improving acute promyelocytic leukemia (APL) outcome in developing countries through networking, results of the International Consortium on APL. Blood. (2013) 121:1935–43. doi: 10.1182/blood-2012-08-449918
43. Adès L, Sanz MA, Chevret S, Montesinos P, Chevallier P, Raffoux E, et al. Treatment of newly diagnosed acute promyelocytic leukemia (APL): a comparison of French-Belgian-Swiss and PETHEMA results. Blood. (2008) 111:1078–84. doi: 10.1182/blood-2007-07-099978
44. Avvisati G, Lo-Coco F, Paoloni FP, Petti MC, Diverio D, Vignetti M, et al. AIDA 0493 protocol for newly diagnosed acute promyelocytic leukemia: very long-term results and role of maintenance. Blood. (2011) 117:4716–25. doi: 10.1182/blood-2010-08-302950
45. Rego EM. Impact of the ICAL on the treatment of acute leukemia. Blood Adv. (2017) 1:516. doi: 10.1182/bloodadvances.2017002147
46. Kutny MA, Geyer S, Laumann KM, Gregory J, Willman CL, Stock W, et al. Outcome for pediatric acute promyelocytic leukemia patients at Children's Oncology Group sites on the Leukemia Intergroup Study CALGB 9710 (Alliance). Pediatr Blood Cancer. (2019) 66:e27542. doi: 10.1002/pbc.27542
47. Testi AM, Pession A, Diverio D, Grimwade D, Gibson B, de Azevedo AC, et al. Risk-adapted treatment of acute promyelocytic leukemia: results from the International Consortium for Childhood APL. Blood. (2018) 132:405–12. doi: 10.1182/blood-2018-03-836528
48. Testi AM, Biondi A, Lo Coco F, Moleti ML, Giona F, Vignetti M, et al. GIMEMA-AIEOPAIDA protocol for the treatment of newly diagnosed acute promyelocytic leukemia (APL) in children. Blood. (2005) 106:447–53. doi: 10.1182/blood-2004-05-1971
49. Creutzig U, Zimmermann M, Bourquin J-P, Dworzak MN, Fleischhack G, Graf N, et al. Randomized trial comparing liposomal daunorubicin with idarubicin as induction for pediatric acute myeloid leukemia: results from Study AML-BFM 2004. Blood. (2013) 122:37–43. doi: 10.1182/blood-2013-02-484097
50. Pagnano KBB, Rego EM, Rohr S, Chauffaille M de L, Jacomo RH, Bittencourt R. Guidelines on the diagnosis and treatment for acute promyelocytic leukemia: Associação Brasileira de Hematologia, Hemoterapia e Terapia Celular Guidelines Project: Associação Médica Brasileira - 2013. Rev Bras de Hematol Hemoter. (2014) 36:71–89. doi: 10.5581/1516-8484.20140018
51. Davies SM, Robison LL, Buckley JD, Tjoa T, Woods WG, Radloff GA, et al. Glutathione S-transferase polymorphisms and outcome of chemotherapy in childhood acute myeloid leukemia. J Clin Oncol Off J Am Soc Clin Oncol. (2001) 19:1279–87. doi: 10.1200/JCO.2001.19.5.1279
52. L'Ecuyer T, Allebban Z, Thomas R, Vander Heide R. Glutathione S-transferase overexpression protects against anthracycline-induced H9C2 cell death. Am J Physiol Heart Circ Physiol. (2004) 286:H2057–64. doi: 10.1152/ajpheart.00778.2003
53. Ye Z, Song H. Glutathione s-transferase polymorphisms (GSTM1, GSTP1 and GSTT1) and the risk of acute leukaemia: a systematic review and meta-analysis. Eur J Cancer. (2005) 41:980–9. doi: 10.1016/j.ejca.2005.01.014
54. Weiss JR, Kopecky KJ, Godwin J, Anderson J, Willman CL, Moysich KB, et al. Glutathione S-transferase (GSTM1, GSTT1 and GSTA1) polymorphisms and outcomes after treatment for acute myeloid leukemia: pharmacogenetics in Southwest Oncology Group (SWOG) clinical trials. Leukemia. (2006) 20:2169–71. doi: 10.1038/sj.leu.2404421
55. Yee SW, Mefford JA, Singh N, Percival M-E, Stecula A, Yang K, et al. Impact of polymorphisms in drug pathway genes on disease-free survival in adults with acute myeloid leukemia. J Hum Genet. (2013) 58:353–61. doi: 10.1038/jhg.2013.38
56. Xiao Q, Deng D, Li H, Ye F, Huang L, Zhang B, et al. GSTT1 and GSTM1 polymorphisms predict treatment outcome for acute myeloid leukemia: a systematic review and meta-analysis. Ann Hematol. (2014) 93:1381–90. doi: 10.1007/s00277-014-2050-z
57. Muir CS, Percy C. Cancer registration: principles and methods. Classification and coding of neoplasms. IARC Sci Publ. (1988) 1991:64–81.
58. Boyle P, Parkin DM. Cancer registration: principles and methods. Statistical methods for registries. IARC Sci Publ. (1989) 1991:126–158.
59. Andrade FG Noronha EP Brisson GD Bueno F dos SV Cezar IS Terra-Granado E . Molecular characterization of pediatric acute myeloid leukemia: results of a multicentric study in Brazil. Arch Med Res. (2016) 47:656–67. doi: 10.1016/j.arcmed.2016.11.015
60. Santos-Bueno FV, Andrade FG, Sardou-Cezar I, Mendes-de-Almeida DP, Chung-Filho AA, Brisson GD, et al. Childhood myeloid neoplasms with PTPN11 mutations in Brazil. Clin Lymphoma Myeloma Leuk. (2020) 20:e496–505. doi: 10.1016/j.clml.2020.04.009
61. Abdel-Rahman SZ, el-Zein RA, Anwar WA, Au WW. A multiplex PCR procedure for polymorphic analysis of GSTM1 and GSTT1 genes in population studies. Cancer Lett. (1996) 107:229–33. doi: 10.1016/0304-3835(96)04832-X
62. Brisson GD, de Almeida Lopes B, Andrade FG, dos Santos Bueno FV, Sardou-Cezar I, de Aguiar Gonçalves BA, et al. EPHX1 rs1051740 T>C (Tyr113His) is strongly associated with acute myeloid leukemia and KMT2A rearrangements in early age. Arch Toxicol. (2018) 92:2001–12. doi: 10.1007/s00204-018-2198-8
Keywords: acute pediatric leukemia, childhood Incidence, prognosis, PML-RARA fusion gene, glutathione S-transferase
Citation: Andrade FG, Feliciano SVM, Sardou-Cezar I, Brisson GD, Santos-Bueno FV, Vianna DT, Marques LVC, Terra-Granado E, Zalcberg I, Santos MO, Costa JT, Noronha EP, Thuler LCS, Wiemels JL, Pombo-de-Oliveira MS and the Brazilian Collaborative Study Group of Acute Leukemia (2021) Pediatric Acute Promyelocytic Leukemia: Epidemiology, Molecular Features, and Importance of GST-Theta 1 in Chemotherapy Response and Outcome. Front. Oncol. 11:642744. doi: 10.3389/fonc.2021.642744
Received: 16 December 2020; Accepted: 08 February 2021;
Published: 19 March 2021.
Edited by:
Marcos De Lima, Case Western Reserve University, United StatesReviewed by:
Eduardo Rego, São Paulo State University, BrazilJignesh D. Dalal, Case Western Reserve University, United States
Copyright © 2021 Andrade, Feliciano, Sardou-Cezar, Brisson, Santos-Bueno, Vianna, Marques, Terra-Granado, Zalcberg, Santos, Costa, Noronha, Thuler, Wiemels, Pombo-de-Oliveira and the Brazilian Collaborative Study Group of Acute Leukemia. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Maria S. Pombo-de-Oliveira, bXBvbWJvQGluY2EuZ292LmJy