
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Oncol., 26 April 2021
Sec. Thoracic Oncology
Volume 11 - 2021 | https://doi.org/10.3389/fonc.2021.642190
This article is part of the Research TopicTherapeutic Strategies in EGFR Mutant Lung CancerView all 26 articles
 Alessandro Leonetti1
Alessandro Leonetti1 Roberta Minari1*
Roberta Minari1* Giulia Mazzaschi1Letizia Gnetti2
Giulia Mazzaschi1Letizia Gnetti2 Silvia La Monica3
Silvia La Monica3 Roberta Alfieri3Nicoletta Campanini2Michela Verzè1Andrea Olivani4Luigi Ventura5
Roberta Alfieri3Nicoletta Campanini2Michela Verzè1Andrea Olivani4Luigi Ventura5 Marcello Tiseo1,3
Marcello Tiseo1,3Introduction: Small cell lung cancer (SCLC) transformation represents a mechanism of resistance to osimertinib in EGFR-mutated lung adenocarcinoma, which dramatically impacts patients' prognosis due to high refractoriness to conventional treatments.
Case Description: We present the case of a patient who developed a SCLC phenotypic transformation as resistance mechanism to second-line osimertinib for T790M-positive EGFR-mutated NSCLC. Our patient received platinum–etoposide doublet following SCLC switch and achieved a modest clinical benefit which lasted 4 months. NGS and IHC analyses for p53 and Rb were performed on subsequent liver biopsies, revealing baseline TP53 mutation and complete absence of p53 and Rb expression. Primary cell cultures were established following a liver biopsy at the time of SCLC transformation, and drug sensitivity assays showed meaningful cell growth inhibition when osimertinib was added to platinum–etoposide compared with control (p < 0.05). A review of the current literature regarding SCLC transformation after failure of osimertinib was performed.
Conclusions: Based on retrospective data available to date, platinum–etoposide chemotherapy is the preferred treatment choice in the occurrence of SCLC transformation after osimertinib failure. The extension of osimertinib in combination with chemotherapy in the occurrence of SCLC transformation as resistance mechanism to osimertinib is a matter of debate. The combination of osimertinib and platinum–etoposide was effective in inhibiting cell growth in our primary cell cultures. Clinical studies are needed to further explore this combination in the occurrence of SCLC transformation as a resistance mechanism to osimertinib.
The epidermal growth factor receptor (EGFR)-tyrosine kinase inhibitor (TKI) osimertinib constitutes a milestone for the treatment of advanced EGFR-mutated non-small cell lung cancer (NSCLC), both in the second line after failure of the previous generation of EGFR-TKIs due to the onset of T790M mutation and in the first line, regardless of T790M status (1). Despite remarkable activity exerted by osimertinib in this clinical setting, several resistance mechanisms have been described (2). Among these, small cell lung cancer (SCLC) phenotypic transformation represents a critical issue for clinicians, since effective therapeutic strategies to apply in this circumstance are lacking to date.
Herein, we report the case of a patient who developed a SCLC switch as resistance mechanism to second-line osimertinib for T790M-positive EGFR-mutated NSCLC, whose pre-clinical studies revealed a promising activity of prolonged osimertinib in combination with chemotherapy. Moreover, we performed a literature review to summarize the underlying mechanism and clinical features of SCLC transformation following osimertinib treatment, including current and future therapeutic opportunities.
In September 2017, due to persistence of dry cough, a never-smoker 63-year-old woman underwent computed tomography (CT) scan which showed a lesion to the upper left pulmonary lobe associated with ipsilateral hilar lymph nodes. The subsequent positron emission tomography (PET) showed increased glucose uptake at both lesions. Left upper lobectomy and mediastinal lymphadenectomy were performed in November 2017, and the pathologic examination revealed an EGFR-mutated (exon 19 deletion) adenocarcinoma of the lung with stage pT3N2, R1 for microscopic residual disease at the bronchial margin. At the post-operative CT scan performed in January 2018, a recurrence of disease was documented, involving bilateral pulmonary metastases and left pleural effusion (Figure 1). Due to the presence of sensitizing EGFR mutation, the patient started gefitinib treatment, achieving partial response of the disease, with almost a complete disappearance of bilateral pulmonary nodules and a decrease of left pleural effusion. The benefit was maintained until October 2018, when the onset of multiple liver metastases and bone lesions was documented. A liver biopsy (liver biopsy 1, LB1) was subsequently performed in order to explore putative resistance mechanisms to gefitinib, revealing the presence of secondary T790M EGFR mutation in the context of exon 19 deletion. The presence of EGFR activating and T790M mutations was also confirmed on liquid biopsy (Figure 2). The patient promptly started osimertinib, which led to complete metabolic response on the liver and osteoblastic reaction of pre-existing bone lesions. Osimertinib therapy was continued until December 2019, when liver lesions increased. The patient underwent a liver biopsy on a new-onset lesion (LB2), which showed a phenotypic switch to SCLC.
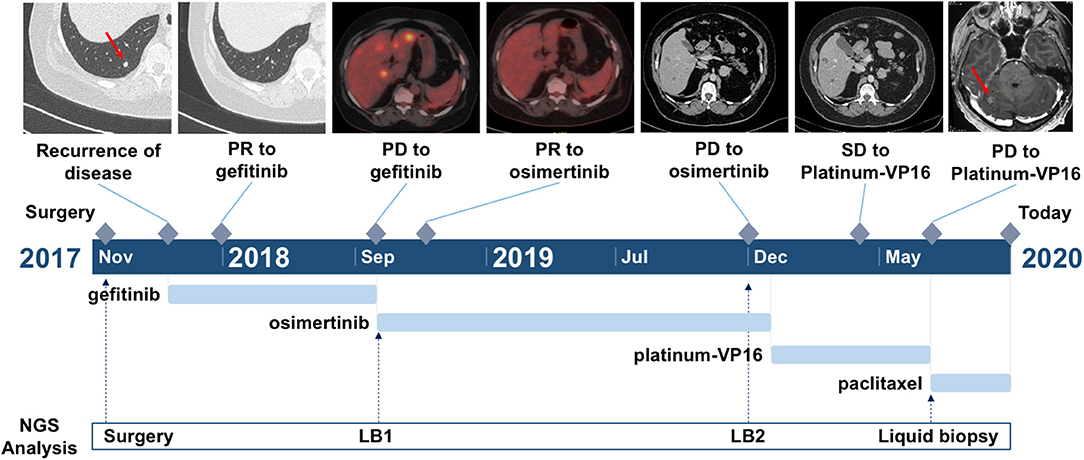
Figure 1. Timeline of the clinical course of the patient. Arrows indicate the link between the clinical event and the date of liquid biopsy time point.
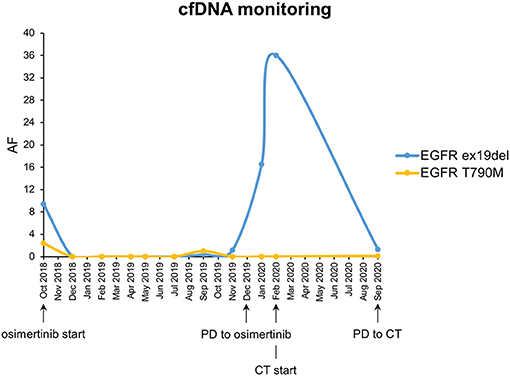
Figure 2. CfDNA monitoring of EGFR ex19del and EGFR T790M mutations. Each dot corresponds to a different liquid biopsy time point. AF, allele frequency; CT, chemotherapy with platinum–etoposide.
Following LB2, primary cell line establishment was attempted. Tissue from liver metastasis was enzymatically digested using the Tumor Dissociation Kit (Miltenyi Biotec, Bergisch Gladbach, Germany), and the gentleMACS™ Dissociator (Miltenyi Biotec) was used for the mechanical dissociation in a closed and sterile system. The single-cell suspension was cultured in a 1:1 ratio of Ham's nutrient mixture F-12:DMEM, 10% FBS, 2 mM glutamine, 1× mammary epithelial growth supplement (MEGS, Life Technologies Corp., CA), and a Rho-associated protein kinase (ROCK) inhibitor.
A drug screening was performed in the primary cell culture testing osimertinib alone, cisplatin plus etoposide, and osimertinib combined with cisplatin plus etoposide. As shown in Figure 3, tumor cells were sensitive to the combination of osimertinib with chemotherapeutic agents (p < 0.05 vs. control) even if the results did not reach statistical significance vs. single drug treatments. Unfortunately, after a few weeks, the cells stopped their proliferation and it was not possible to perform additional experiments and to establish a stable cell line.
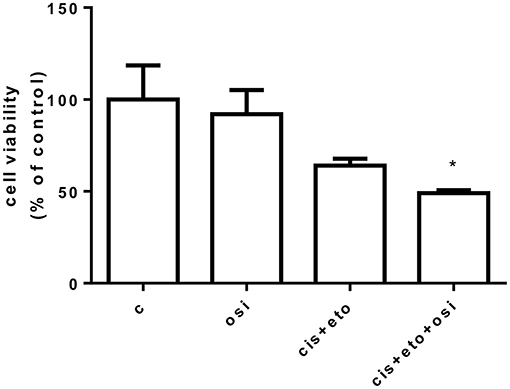
Figure 3. Drug screening in a primary cell line following osimertinib resistance. Primary cells were treated with 250 nM osimertinib, 0.150 μg/ml etoposide, and 0.05 μg/ml cisplatin. After 72 h, cell proliferation was assessed by CellTiter 96® AQueous One Solution Cell Proliferation Assay (MTS). Data are expressed as percent of cell viability vs. control cells and are means ± SD of three measurements. *p < 0.05.
The patient underwent platinum–etoposide doublet in February 2020, and chemotherapy granted stability of the disease, as documented at the CT scan after three cycles. Unfortunately, following further three cycles, the patient experienced liver progression and central nervous system (CNS) progression due to the onset of multiple brain metastases (Figure 1). A further liver biopsy (LB3) was conducted on a new-onset liver lesion, with diagnosis of pure adenocarcinoma. At the time of writing the manuscript, the patient has been receiving weekly paclitaxel and whole-brain radiotherapy was performed. A liquid biopsy was carried out at the time of chemotherapy switch.
SCLC transformation clearly emerged as a mechanism of resistance to osimertinib. Nonetheless, next-generation sequencing (NGS) and immunohistochemistry (IHC) of Rb1 and p53 were performed in order to characterize the proficiency of the initial tumor to evolve in a neuroendocrine differentiated subtype. DNA was extracted from the liver biopsy undertaken before osimertinib treatment (LB1) and on SCLC-transformed liver lesion at osimertinib progression (LB2). Molecular analyses on LB3 were not conducted due to insufficient material. NGS study was performed with Solid Tumor Solution panel, Sophia Genetics, on MiSeq Platform, Illumina. No other putative resistance mechanisms to osimertinib were underlined (Table 1) and variants on TP53 were found on LB1. The presence of those variants was retrospectively confirmed by NGS also in the lobectomy samples.
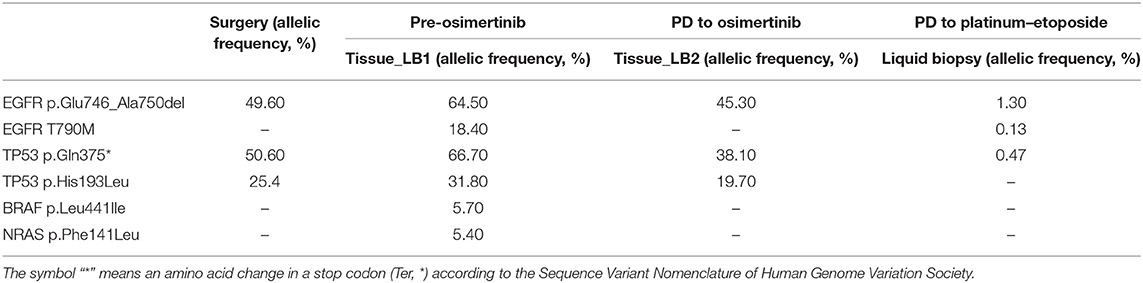
Table 1. NGS analyses on available samples.
Expression of p53 and Rb1 was evaluated with IHC on lobectomy tissue, LB1 and LB2, as previously described (3). IHC analysis was not performed on LB3 due to insufficient material. The evaluation of Rb1 showed the complete absence of expression in all analyzed samples, while p53 presented an abnormal pattern of expression consistent with inactivation. In fact, p53 was negative in the surgery tissue, overexpressed in LB1, and scattered positive in LB2 (Figure 4).
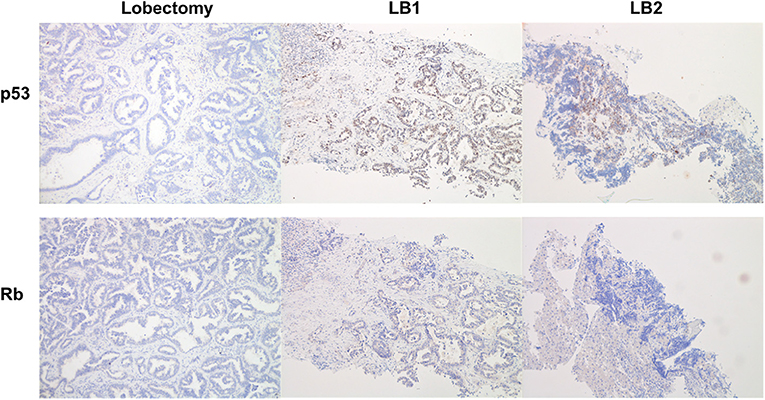
Figure 4. Immunohistochemistry analysis for p53 (upper line) and Rb (lower line). The resected tumor (lobectomy) showed a negativity for both p53 and Rb. In the first liver biopsy (LB1), p53 was overexpressed, whereas Rb was negative; the histotype of LB1 is that of a NSCLC/adenocarcinoma with morphology similar to lobectomy. In the second liver biopsy (LB2), we detected only scattered cells positive for p53 and negative for Rb; the histotype is that of a SCLC also documented by positivity for neuroendocrine markers.
Moreover, NGS analysis was carried out on liquid biopsy collected after disease progression to third-line platinum–etoposide with AVENIO ctDNA Expanded Panel, Roche, on NextSeq Platform, Illumina. Interestingly, EGFR T790M showed up again with the known activating mutation and with two TP53 non-sense variants (Table 1). The presence of EGFR mutations on liquid biopsy were confirmed also with ddPCR (Figure 2).
Dissecting the mechanisms of acquired resistance to osimertinib and other third-generation EGFR-TKIs represents an area of active investigation (2, 4, 5). Nonetheless, the role of histologic transformations, and more specifically SCLC switch, remains partly uncovered. This could be ascribed to the lack of analyses conducted on tissue samples; in fact, in both registrational trials, AURA and FLAURA, the delineation of genomic profiles of osimertinib-resistant NSCLC has been performed by NGS on plasma samples (6–8). Tissue biopsy at the time of progression to osimertinib plays a critical role in order to unravel SCLC transformation (9). Recent data from the six largest series of osimertinib-resistant cases to date reported an overall incidence of SCLC transformation ranging from 2 to 15% (10). As documented by Oxnard et al., among 28 patients who developed disease progression to second-line osimertinib and lost T790M, SCLC transformation resulted the most frequent mechanism of resistance accounting for 21% of all the recognized causes (11). Along the same line, Lee et al. reported small cell transformation as EGFR-independent resistance process in 5 over 36 tested cases (12). A lower proportion of SCLC-switched cases (4–6%) was present in “first-line” and “latter-line” cohorts included in the study by Schoenfeld et al. (13) and in osimertinib-resistant population described by Piotrowska et al. (14) and Michels et al. (15). In the real-world study conducted by Le et al., although potentially affected by the pure retrospective nature of the analysis, the incidence significantly decreased to 2% (16).
Although histologic transformation is a well-known phenomenon, the comprehension of how it occurs and leads to EGFR-TKI resistance is still incomplete. SCLC transformation was first described in 2006 in a 45-year-old never-smoker woman with advanced EGFR-mutant adenocarcinoma after erlotinib failure (17). Since this initial observation, several additional cases have been reported (18), all confirmed by positive immunohistochemical staining for synaptophysin, chromogranin, or CD56/NCAM.
Different hypotheses have been proposed concerning the origin of SCLC as a mechanism of resistance to EGFR-TKIs. Initial studies on SCLC-transformed cancers have revealed relevant similarities to de novo SCLCs, most remarkably the inactivation of tumor suppression via RB1 (19) and TP53 (20). SCLC and NSCLC histologies might coexist within the same initial tumor, with the SCLC subtype becoming dominant after an initial response to EGFR-TKIs (18). Conversely, other lines of evidence support the assumption of a trans-differentiation process of the original EGFR-mutated adenocarcinoma under the pressure of TKIs (18, 21). Of interest, most of the SCLC-switched tumors retained the same EGFR mutation after transformation (22).
The identification of biomolecular mediators of treatment-dependent SCLC transformation represents a fundamental goal to subsequently develop therapeutic interventions. Triple mutant adenocarcinomas (EGFR/RB1/TP53) are considered at higher risk for transformation to SCLC (23). Moreover, a rapid increase in the serum levels of neurone specific enolase (NSE) together with a poor response to EGFR-TKIs usually indicates a transformation from adenocarcinoma to SCLC (24). Along the same line, the assessment of the pro-gastrin-releasing peptide (pro-GRP) has also been recommended for the early prediction of disease transformation (25).
Since most of the SCLC-transformed cases harbor typical neuroendocrine differentiation, platinum–etoposide chemotherapy remains the current standard treatment at the time of SCLC switch (26). Even though SCLC-transformed cases achieve similar objective response rate from chemotherapy than primary SCLC (around 80%), the prognosis of the former is usually worse than the latter even after a favorable response (27). Ferrer et al. recently performed a retrospective study on 61 SCLC-transformed cases, either with EGFR mutation or not (22). In this study, overall survival (OS) from the initial diagnosis was lower in the EGFR-mutated group compared with the non-EGFR-mutant group; however, OS from the time of transformation into SCLC was comparable between the two groups (22).
The early introduction of platinum–etoposide chemotherapy along with osimertinib may act as an effective therapeutic strategy to eradicate emerging SCLC subclones and prevent the phenotypic transformation in EGFR-mutated patients with a high risk of SCLC switch (ongoing clinical trial, Clinicaltrials.gov: NCT03567642). Other approaches that might be pursued involve targeting cell cycle vulnerabilities generated upon RB1 loss through the use of Aurora kinase (AURKA or AURKB) inhibitors (28, 29) or applying epigenetic therapy, mainly directly against reprogramming factors such as EZH2 (30).
In the present study, we reported a case of osimertinib resistance driven by SCLC switch in an EGFR-mutated NSCLC patient. At the time of progression to osimertinib, due to phenotypic transformation, our patient received standard platinum–etoposide chemotherapy, achieving only modest clinical benefit.
Overall, it could be difficult to determine whether SCLC arises by transformation from NSCLC, rather than being a new tumor or being present simultaneously with the NSCLC from the beginning. Since SCLC is characterized by rapid growth and is not controlled by EGFR-TKIs, a simultaneous SCLC–NSCLC mixed tumor is expected to recur quickly. Our patient benefited from ~2 years of EGFR-TKIs; hence, it is unlikely that SCLC was part of the initial presentation.
A fundamental issue is represented by the identification of biomarkers able to predict SCLC transformation. Current evidence supports TP53 and RB1 mutations as potential predictors of phenotypic switch in EGFR-mutated NSCLC (23). In our case, histological examination at diagnosis showed the complete absence of p53 and Rb at IHC, likely underlying TP53 and RB1 baseline alterations. In addition, TP53 mutations were detected by tissue NGS analysis before starting osimertinib, suggesting that the patient had a high risk of developing SCLC as a resistance mechanism.
To date, platinum–etoposide chemotherapy is the only viable treatment approach with a confirmed clinical efficacy in counteracting SCLC after failure of EGFR-TKIs. Given the positive results of the exploratory analysis of the IMpower150 trial in EGFR-mutated patients (31), a combination strategy of carboplatin–paclitaxel plus atezolizumab and bevacizumab after failure of previous EGFR-TKIs could be envisaged in this peculiar subset of patients, considering the proven efficacy of chemotherapy plus atezolizumab for the frontline treatment of extensive stage SCLC (32). Whether continued EGFR-TKI might gain additional clinical benefit is still a matter of debate, considering that SCLC is generally resistant to EGFR inhibition. Against this notion, a recent study reported a successful treatment with osimertinib in a synchronous SCLC and adenocarcinoma histology (33).
The continuation of osimertinib and its potential association with chemotherapy is still under investigation (34), also in view of the results obtained from the phase III IMPRESS study that did not demonstrate any PFS or OS improvement by continuing gefitinib vs. placebo in combination with second-line, platinum-based chemotherapy in EGFR-mutated NSCLC (35). In our report, drug screening assay on primary cell cultures from post-osimertinib biopsy showed increased sensitivity to the combination of osimertinib with chemotherapeutic drugs compared with control (p < 0.5), suggesting a potential effective therapeutic option. Moreover, it is likely that the progression to platinum–etoposide was driven by the EGFR-positive component in our case. To support this, the liver biopsy performed after chemotherapy (LB3) showed pure adenocarcinoma histology, and the liquid biopsy revealed the restoration of EGFR T90M. In this view, we assume that the interruption of EGFR pressure might have unleashed EGFR-positive clones resulting in inexorable treatment failure. Considering that our patient experienced CNS disease progression to platinum–etoposide, the excellent CNS penetration of osimertinib and its effectiveness on brain metastases might lead to continue osimertinib along with platinum-based chemotherapy in this occurrence.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found here: NCBI BioProject, Accession No: PRJNA698448.
Ethical review and approval was not required for the study on human participants in accordance with the local legislation and institutional requirements. The patients/participants provided their written informed consent to participate in this study. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
AL, RM, and MT designed the manuscript. AL and GM collected the clinical data and performed the literature review. RM conducted the molecular analyses. LG and NC conducted the IHC analyses. SL and RA isolated tumor cells from biopsy tissue and performed the drug screening assay. AO performed the liver biopsies. LV performed the thoracic surgery. All the authors contributed to the writing of the manuscript and approved the submitted version.
This work was supported by Associazione Italiana per la Ricerca sul Cancro (AIRC), Milan Grant IG2017-20074 (MT).
MT has been on advisory boards and received speakers' fees for AstraZeneca, Pfizer, Eli-Lilly, BMS, Novartis, Roche, MSD, Boehringer Ingelheim, Otsuka, Takeda, and Pierre Fabre. MT has received research grants from AstraZeneca and Boehringer Ingelheim. AL received speakers' fees for AstraZeneca.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. NCCN Clinical Practice Guidelines in Oncology (NCCN Guidelines®) - Non-Small Cell Lung Cancer - Version 4,.2021 (2021). Available online at: https://www.nccn.org/ (accessed April 9, 2021).
2. Leonetti A, Sharma S, Minari R, Perego P, Giovannetti E, Tiseo M. Resistance mechanisms to osimertinib in EGFR-mutated non-small cell lung cancer. Br J Cancer. (2019) 121:725–37. doi: 10.1038/s41416-019-0573-8
3. Minari R, Bordi P, Del Re M, Facchinetti F, Mazzoni F, Barbieri F, et al. Primary resistance to osimertinib due to SCLC transformation: issue of T790M determination on liquid re-biopsy. Lung Cancer. (2018) 115:21–7. doi: 10.1016/j.lungcan.2017.11.011
4. Schmid S, Li JJN, Leighl NB. Mechanisms of osimertinib resistance and emerging treatment options. Lung Cancer. (2020) 147:123–9. doi: 10.1016/j.lungcan.2020.07.014
5. Schoenfeld AJ, Yu HA. The evolving landscape of resistance to osimertinib. J Thorac Oncol. (2020) 15:18–21. doi: 10.1016/j.jtho.2019.11.005
6. Mok TS, Wu Y-L, Ahn M-J, Garassino MC, Kim HR, Ramalingam SS, et al. Osimertinib or platinum-pemetrexed in EGFR T790M-positive lung cancer. N Engl J Med. (2017) 376:629–40. doi: 10.1056/NEJMoa1612674
7. Papadimitrakopoulou VA, Wu Y-L, Han J-Y, Ahn M-J, Ramalingam SS, John T, et al. Analysis of resistance mechanisms to osimertinib in patients with EGFR T790M advanced NSCLC from the AURA3 study. Ann Oncol. (2018) 29:viii741. doi: 10.1093/annonc/mdy424.064
8. Soria J-C, Ohe Y, Vansteenkiste J, Reungwetwattana T, Chewaskulyong B, Lee KH, et al. Osimertinib in untreated EGFR-mutated advanced non-small-cell lung cancer. N Engl J Med. (2018) 378:113–25. doi: 10.1056/NEJMoa1713137
9. Piotrowska Z, Hata AN. Resistance to first-line osimertinib in EGFR-mutant NSCLC: tissue is the issue. Clin Cancer Res. (2020) 26:2441–3. doi: 10.1158/1078-0432.CCR-20-0097
10. Piper-Vallillo AJ, Sequist LV, Piotrowska Z. Emerging treatment paradigms for EGFR-mutant lung cancers progressing on osimertinib: a review. J Clin Oncol. (2020) 38:2926–36. doi: 10.1200/JCO.19.03123
11. Oxnard GR, Hu Y, Mileham KF, Husain H, Costa DB, Tracy P, et al. Assessment of resistance mechanisms and clinical implications in patients with EGFR T790M-positive lung cancer and acquired resistance to osimertinib. JAMA Oncol. (2018) 4:1527–34. doi: 10.1001/jamaoncol.2018.2969
12. Lee J, Kim HS, Lee B, Kim HK, Sun JM, Ahn JS, et al. Genomic landscape of acquired resistance to third-generation EGFR tyrosine kinase inhibitors in EGFR T790M-mutant non-small cell lung cancer. Cancer. (2020) 126:2704–12. doi: 10.1002/cncr.32809
13. Schoenfeld AJ, Chan JM, Kubota D, Sato H, Rizvi H, Daneshbod Y, et al. Tumor analyses reveal squamous transformation and off-target alterations as early resistance mechanisms to first-line osimertinib in EGFR-mutant lung cancer. Clin Cancer Res. (2020) 26:2654–63. doi: 10.1158/1078-0432.CCR-19-3563
14. Piotrowska Z, Isozaki H, Lennerz JK, Gainor JF, Lennes IT, Zhu VW. Landscape of acquired resistance to osimertinib in EGFR-mutant NSCLC and clinical validation of combined EGFR and RET inhibition with osimertinib and BLU-667 for acquired RET fusion. Cancer Discov. (2018) 8:1529–39. doi: 10.1158/2159-8290.CD-18-1022
15. Michels S, Heydt C, van Veggel B, Deschler-Baier B, Pardo N, Monkhorst K, et al. Genomic profiling identifies outcome-relevant mechanisms of innate and acquired resistance to third-generation epidermal growth factor receptor tyrosine kinase inhibitor therapy in lung cancer. JCO Precis Oncol. (2019) 3:PO.18.00210. doi: 10.1200/PO.18.00210
16. Le X, Puri S, Negrao MV, Nilsson MB, Robichaux J, Boyle T, et al. Landscape of EGFR-dependent and -independent resistance mechanisms to osimertinib and continuation therapy beyond progression in EGFR-mutant NSCLC. Clin Cancer Res. (2018) 24:6195–203. doi: 10.1158/1078-0432.CCR-18-1542
17. Zakowski MF, Ladanyi M, Kris MG. EGFR mutations in small-cell lung cancers in patients who have never smoked [18]. N Engl J Med. (2006) 355:213–5. doi: 10.1056/NEJMc053610
18. Oser MG, Niederst MJ, Sequist LV, Engelman JA. Transformation from non-small-cell lung cancer to small-cell lung cancer: molecular drivers and cells of origin. Lancet Oncol. (2015) 16:e165–72. doi: 10.1016/S1470-2045(14)71180-5
19. Niederst MJ, Sequist LV, Poirier JT, Mermel CH, Lockerman EL, Garcia AR, et al. RB loss in resistant EGFR mutant lung adenocarcinomas that transform to small-cell lung cancer. Nat Commun. (2015) 6:6377. doi: 10.1038/ncomms7377
20. Lee J-K, Lee J, Kim S, Kim S, Youk J, Park S, et al. Clonal history and genetic predictors of transformation into small-cell carcinomas from lung adenocarcinomas. J Clin Oncol. (2017) 35:3065–74. doi: 10.1200/JCO.2016.71.9096
21. Shaurova T, Zhang L, Goodrich DW, Hershberger PA. Understanding lineage plasticity as a path to targeted therapy failure in EGFR-mutant non-small cell lung cancer. Front Genet. (2020) 11:1–15. doi: 10.3389/fgene.2020.00281
22. Ferrer L, Giaj Levra M, Brevet M, Antoine M, Mazieres J, Rossi G, et al. A brief report of transformation from NSCLC to SCLC: molecular and therapeutic characteristics. J Thorac Oncol. (2019) 14:130–4. doi: 10.1016/j.jtho.2018.08.2028
23. Offin M, Chan JM, Tenet M, Rizvi HA, Shen R, Riely GJ, et al. Concurrent RB1 and TP53 alterations define a subset of EGFR-mutant lung cancers at risk for histologic transformation and inferior clinical outcomes. J Thorac Oncol. (2019) 14:1784–93. doi: 10.1016/j.jtho.2019.06.002
24. Liu Y. Small cell lung cancer transformation from EGFR-mutated lung adenocarcinoma: a case report and literatures review. Cancer Biol Ther. (2018) 19:445–9. doi: 10.1080/15384047.2018.1435222
25. Norkowski E, Ghigna MR, Lacroix L, Le Chevalier T, Fadel É, Dartevelle P, et al. Small-cell carcinoma in the setting of pulmonary adenocarcinoma: new insights in the era of molecular pathology. J Thorac Oncol. (2013) 8:1265–71. doi: 10.1097/JTO.0b013e3182a407fa
26. van Meerbeeck JP, Fennell DA, De Ruysscher DKM. Small-cell lung cancer. Lancet. (2011) 378:1741–55. doi: 10.1016/S0140-6736(11)60165-7
27. Jiang S-Y, Zhao J, Wang M-Z, Huo Z, Zhang J, Zhong W, et al. Small-cell lung cancer transformation in patients with pulmonary adenocarcinoma. Medicine (Baltimore). (2016) 95:e2752. doi: 10.1097/MD.0000000000002752
28. Gong X, Du J, Parsons SH, Merzoug FF, Webster Y, Iversen PW, et al. Aurora a kinase inhibition is synthetic lethal with loss of the RB1 tumor suppressor gene. Cancer Discov. (2019) 9:248–63. doi: 10.1158/2159-8290.CD-18-0469
29. Oser MG, Fonseca R, Chakraborty AA, Brough R, Spektor A, Jennings RB, et al. Cells lacking the RB1 tumor suppressor gene are hyperdependent on aurora B kinase for survival. Cancer Discov. (2019) 9:230–47. doi: 10.1158/2159-8290.CD-18-0389
30. Ishak CA, Marshall AE, Passos DT, White CR, Kim SJ, Cecchini MJ, et al. An RB-EZH2 complex mediates silencing of repetitive DNA sequences. Mol Cell. (2016) 64:1074–87. doi: 10.1016/j.molcel.2016.10.021
31. Mok TSK, Socinski MA, Reck M, Jotte RM, Lim DW-T, Cappuzzo F, et al. IMpower150: an exploratory analysis of efficacy outcomes in patients with EGFR mutations. Ann Oncol. (2018) 29:ix177–8. doi: 10.1093/annonc/mdy483.008
32. Horn L, Mansfield AS, Szczesna A, Havel L, Krzakowski M, Hochmair MJ, et al. First-line atezolizumab plus chemotherapy in extensive-stage small-cell lung cancer. N Engl J Med. (2018) 379:2220–9. doi: 10.1056/NEJMoa1809064
33. Batra U, Nathany S, Sharma M, Mehta A, Jain P, Bansal A. Successful treatment of EGFR-mutant synchronous SCLC and lung adenocarcinoma with osimertinib. JTO Clin Res Rep. (2021) 2:100098. doi: 10.1016/j.jtocrr.2020.100098
34. Jänne P, Planchard D, Howarth P, Todd A, Kobayashi K. OA07.01 osimertinib plus platinum/pemetrexed in newly-diagnosed advanced EGFRm-positive NSCLC; the phase 3 FLAURA2 study. J Thorac Oncol. (2019) 14:S222–3. doi: 10.1016/j.jtho.2019.08.441
35. Soria JC, Wu YL, Nakagawa K, Kim SW, Yang JJ, Ahn MJ, et al. Gefitinib plus chemotherapy versus placebo plus chemotherapy in EGFR-mutation-positive non-small-cell lung cancer after progression on first-line gefitinib (IMPRESS): a phase 3 randomised trial. Lancet Oncol. (2015) 16:990–8. doi: 10.1016/S1470-2045(15)00121-7
Keywords: NSCLC, EGFR, osimertinib resistance, SCLC transformation, phenotype switch
Citation: Leonetti A, Minari R, Mazzaschi G, Gnetti L, La Monica S, Alfieri R, Campanini N, Verzè M, Olivani A, Ventura L and Tiseo M (2021) Small Cell Lung Cancer Transformation as a Resistance Mechanism to Osimertinib in Epidermal Growth Factor Receptor-Mutated Lung Adenocarcinoma: Case Report and Literature Review. Front. Oncol. 11:642190. doi: 10.3389/fonc.2021.642190
Received: 15 December 2020; Accepted: 12 February 2021;
Published: 26 April 2021.
Edited by:
Yaxiong Zhang, Sun Yat-sen University Cancer Center (SYSUCC), ChinaReviewed by:
Alessandro Morabito, Istituto Nazionale Tumori Fondazione G. Pascale (IRCCS), ItalyCopyright © 2021 Leonetti, Minari, Mazzaschi, Gnetti, La Monica, Alfieri, Campanini, Verzè, Olivani, Ventura and Tiseo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Roberta Minari, cm9taW5hcmlAYW8ucHIuaXQ=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.