 Zhi Zeng1,2†
Zhi Zeng1,2† Qiliang Lu1,2†
Qiliang Lu1,2† Yang Liu1,2†Junjun Zhao2,3†Qian Zhang1
Yang Liu1,2†Junjun Zhao2,3†Qian Zhang1 Linjun Hu1,2
Linjun Hu1,2 Zhan Shi4Yifeng Tu4
Zhan Shi4Yifeng Tu4 Zunqiang Xiao4
Zunqiang Xiao4 Qiuran Xu5*
Qiuran Xu5* Dongsheng Huang5*
Dongsheng Huang5*- 1The Medical College of Qingdao University, Qingdao, China
- 2Zhejiang Provincial People’s Hospital (People’s Hospital of Hangzhou Medical College), Hangzhou, China
- 3Graduate Department, Bengbu Medical College, Bengbu, China
- 4The Second Clinical Medical College of Zhejiang Chinese Medical University, Hangzhou, China
- 5The Key Laboratory of Tumor Molecular Diagnosis and Individualized Medicine of Zhejiang Province, Zhejiang Provincial People’s Hospital (People’s Hospital of Hangzhou Medical College), Hangzhou, China
Sorafenib a multi-target tyrosine kinase inhibitor, is the first-line drug for treating advanced hepatocellular carcinoma (HCC). Mechanistically, it suppresses tumor angiogenesis, cell proliferation and promotes apoptosis. Although sorafenib effectively prolongs median survival rates of patients with advanced HCC, its efficacy is limited by drug resistance in some patients. In HCC, this resistance is attributed to multiple complex mechanisms. Previous clinical data has shown that HIFs expression is a predictor of poor prognosis, with further evidence demonstrating that a combination of sorafenib and HIFs-targeted therapy or HIFs inhibitors can overcome HCC sorafenib resistance. Here, we describe the molecular mechanism underlying sorafenib resistance in HCC patients, and highlight the impact of hypoxia microenvironment on sorafenib resistance.
Introduction
The globally cancer statistics of 2018 show that liver cancer is the sixth most commonly diagnosed form of cancer, and a fourth cause of cancer-related deaths worldwide (1). Despite significant progress being made in development of therapies for early diagnosis and treatment therapies for HCC in recent years, over 50% of all HCC cases are still diagnosed at an advanced stage. Additionally, approximately 70% of all HCC patients relapse within five years of initial treatment (2). Current treatment options for HCC include radiotherapy, chemotherapy, local ablation and molecular targeted therapy (3). Several targeted inhibitors have also been developed and applied in clinical practice. For example, sorafenib, which acts as a multiple-target tyrosine kinase inhibitor (TKI), was the first systematic drug to be approved for advanced HCC patients based on results of two randomized clinical trials. Functionally, sorafenib inhibits proliferation and angiogenesis of tumor cells, thereby delaying HCC progression while effectively prolonging the survival time of patients (4). A previous Sorafenib Hepatocellular Carcinoma Assessment Randomized Protocol (SHARP) trial confirmed that the drug was safe and efficacious in patients with advanced HCC. In fact, these similar results were corroborated by findings in Asia-Pacific clinical trials (5, 6), in which sorafenib improved the clinical symptoms of about 30% of HCC patients. However, this group of patients reportedly develop resistance to sorafenib within 6 months of treatment (7). Previous studies indicate that sorafenib inhibits activity of various kinases, including Ras, Raf, MEK, and ERK, among others, and further targets VEGFR, c-KIT, PDGFR-β, and FLT-3, as well as other proteins that suppress tumor angiogenesis (8, 9). Moreover, sorafenib plays an anti-tumor role in HCC and other types of cancer, such as desmoid tumor, renal cell carcinoma, lung cancer and thyroid cancer (10–13). Although the drug effectively prolongs survival rates of HCC patients, its efficacy is significantly limited by development of drug-acquired resistance. The underlying mechanism of sorafenib resistance is complex. Previous studies have shown that the drug activates c-Jun, Akt pathway, epidermal growth factor receptor (EGFR), cancer stem cells enrichment, epithelial-mesenchymal transition (EMT) enhancement and reduces autophagy. Recently, other factors, such as dysregulation of miRNAs and lncRNAs in HCC have been implicated in sorafenib resistance (9, 14). Moreover, sorafenib reportedly induces hypoxia response in HCC, with dysregulation of hypoxia microenvironment and HIF expression shown to contribute to poor prognosis of HCC patients. In addition, sorafenib has also been implicated in effective inhibition of the HIF-1α/VEGFA signaling pathway (15). Weinberg et al. described six hallmarks of cancer, namely evasion of growth suppression, sustained proliferative signaling, induction of angiogenesis, resistance to cell death, replicative immortality, as well as activation of invasion and metastasis, and further demonstrated that these biological behaviors influence the degree of malignancy (16). Hypoxia, a common event that plays important roles in development and progression of malignant tumors, has been implicated in development of drug resistance and activation of tumor metastasis (17, 18). In the present study, we sought to clarify the underlying mechanism of sorafenib resistance, its relationship with the hypoxia microenvironment and the effect of targeting HIFs on sorafenib resistance in hepatocellular carcinoma.
Sorafenib Resistance In Hcc
Drug resistance is divided into the primary and acquired resistance, based on the time and sequence of exposure to the drug. Although both categories involve a complex chemical resistance mechanism, signaling pathways, characterized by up-/down-regulation and changes in molecular targets, represent the two most important factors (19). Elucidating the underlying mechanism of drug resistance is imperative to development of effective strategies to prevent or overcome its development (Figure 1).
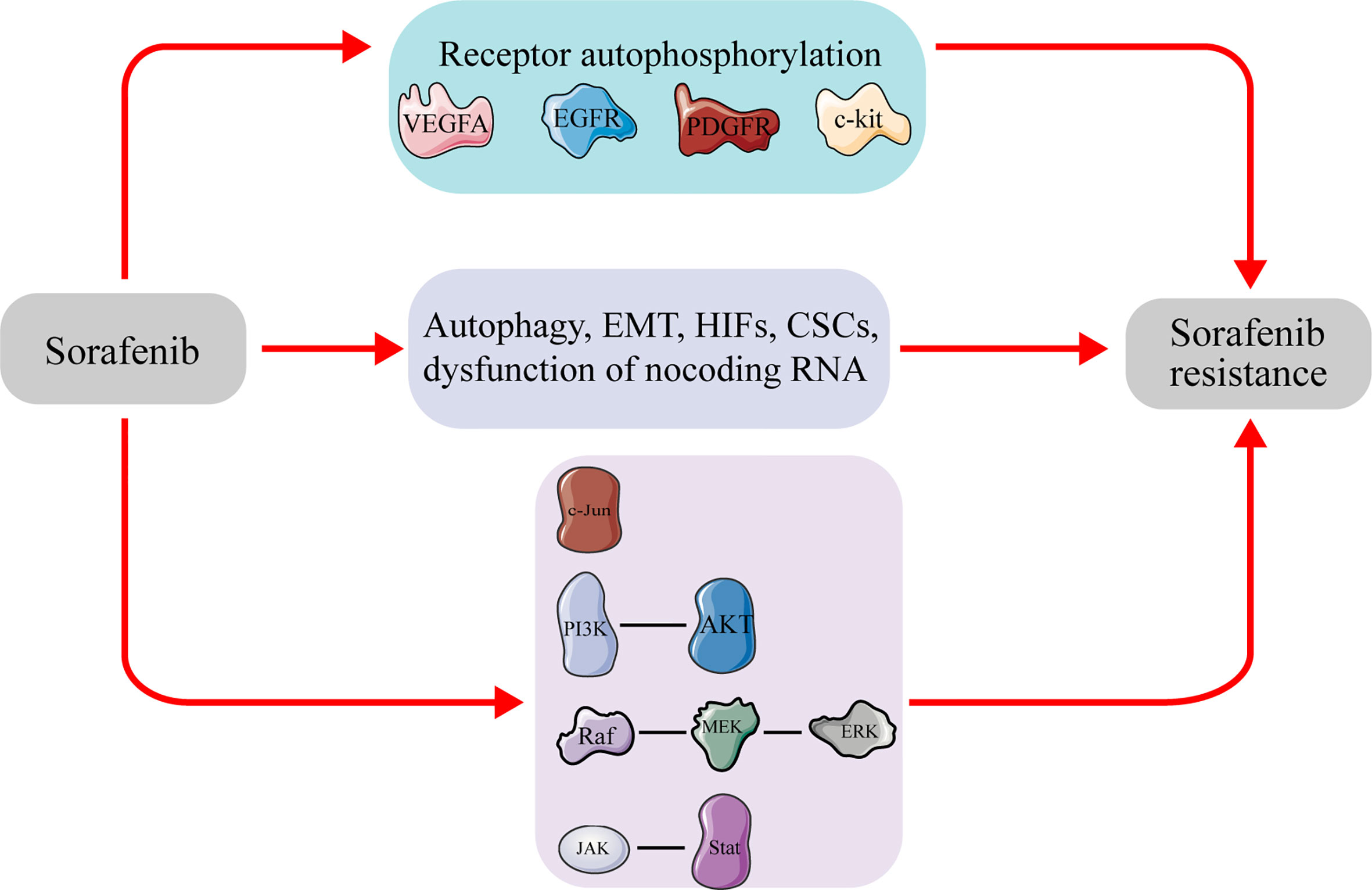
Figure 1 Molecules and signal pathways related to sorafenib resistance in hepatocellular carcinoma. Sustained sorafenib treatment will affect the expression of the molecules and activate pathways, leading to sorafenib resistance in hepatocellular carcinoma. VEGFA, vascular endothelial growth factor A; EGFR, epidermal growth factor receptor; PDGFR, Platelet-derived growth factor receptor; c-kit, tyrosine kinase receptors hepatocyte factor receptor; EMT, epithelial–mesenchymal transition; CSCs, cancer stem cells; PI3K, phosphatidylinositol-3-kinase; AKT, protein kinase B; ERK, extracellular signal-regulated kinase; JAK, janus tyrosine kinase; STAT, signal transducer and activator of transcription.
c-Jun also known as AP-1 transcription factor subunit. It’s located at 1p32-p31, Deletion and translocation of this chromosomal region has been associated with development of malignant tumors. A previous study reported that regulation of mitotic signals can activate AP-1 (20). while others have shown demonstrated its significance in hepatocyte activity and liver regeneration, as well as in development of hepatocellular carcinoma (21). Previous studies have also shown that c-Jun was remarkably activated in sorafenib-treated HCC cells, with its downregulation found to significantly elevate apoptosis of HCC cells induced by sorafenib (22). Another in vitro study found that sorafenib treatment could activate expression of c-Jun, while its inhibition significantly enhanced sorafenib-induced apoptosis in HCC cells (23). Results from a clinical trial revealed that HCC patients approached with sorafenib, the expression of Phosphorylation C-Jun was remarkably higher in the non-responder group than in the responder group (24). Therefore, c-Jun is probably one of the molecules that causes of HCC resistance to sorafenib.
The PI3K/AKT signaling pathway plays a role in regulation of apoptosis and chemotherapeutic resistance in malignant tumors. Previous studies have shown that sorafenib-mediated inhibition of AKT expression enhanced apoptosis induction in HCC cells (25). Moreover, Zhang et al. found that long term exposure of HCC cells to sorafenib could activate the PI3K/Akt signaling pathway, thereby whereas inhibiting PI3K using LY294002 could reverse sorafenib resistance (26). In another study, sorafenib effectively promoted AKT phosphorylation but did not significantly affect that of other proteins in the PI3K/AKT/mTOR signaling pathway (27). Therefore, activation of the PI3K/AKT signaling pathway is considered a compensatory mechanism for acquired sorafenib resistance. In fact, numerous studies have demonstrated that HCC cells with acquired sorafenib resistance exhibit significantly higher levels of phosphorylation AKT than parental cells, although suppression of AKT can reverse acquired sorafenib resistance (27, 28). The activation of AKT pathway has an important impact in sorafenib resistance in HCC.
Epidermal growth factor receptor (EGFR), which belongs to the protein kinase superfamily, can induce receptor dimerization and regulate autophosphorylation of tyrosine thereby causing cell proliferation. This phenomenon has been shown to be a potential indicator of sorafenib resistance in HCC cells. In HCC cells with higher EGFR expression, the efficacy of sorafenib is significantly weakened. A previous study demonstrated that sensitivity of cells to sorafenib could be increased by downregulating EGFR expression or inhibiting its kinase activity (29). In, Moreover, the EGFR pathway is overexpressed in HCC cells with acquired resistance to sorafenib, where it acts as the driving force for maintaining HCC cell proliferation under sorafenib (30).
Epithelial–mesenchymal transformation (EMT) contributes to migration and drug resistance, and is therefore an important cellular program (31). Previous studies have reported that EMT is associated with cancer chemotherapeutic resistance, with its inhibition found to reverse this drug-resistant outcome (32). For example, Fisher et al. established a genetically engineered mouse model and demonstrated the relationship between cancer drug resistance and EMT (33). Moreover, induction of EMT reportedly promotes tumor progression and sorafenib resistance in HCC. Although EMT development is inhibited by sorafenib, it has also been reported to promote chemotherapy resistance to sorafenib in HCC cells (34, 35). Previous studies have demonstrated that cancer stem cells (CSCs) are also involved in development of chemoresistance in HCC. In addition, there is crosstalk between EMT and CSCs, as evidenced by the fact that acquired EMT cells exhibit CSCs-like characteristics, while CSCs show mesenchymal phenotype (36), EMT activation is usually associated with enrichment of CSCs subsets in sorafenib resistance cells (37, 38). Thus CSCs markers have been used as predictors of sorafenib reaction. Notably, upregulation of CSC markers, CD90 and CD133, was shown to be a predictor for sorafenib resistance in HCC cells. Moreover, PTK2 activated CSC characteristics to promote tumor progression by inducing β-catenin nuclear accumulation in HCC cells thereby inducing sorafenib resistance (39). CD44 can be used as a marker for evaluating efficacy of sorafenib in HCC cells, as evidenced by its role in development of drug resistance (40). Other markers, such as CD13, EpCAM, and CD24 are also considered helpful for CSCs enrichment in HCC (41).
Autophagy, a self-degrading system that directs cells to eliminate abnormal proteins and dysfunctional organelles, plays an essential role in maintaining homeostasis in cells under stress, such as nutritional deficiency or hypoxia (42). In fact, autophagy plays a double-edged sword in different cancers by suppressing initiation of tumors but also supporting their progression. This mechanism further plays an important role in drug resistance, enabling tumor cells to maintain cell activity under metabolic and therapeutic stress. In fact, autophagy is often activated in radiotherapy, chemotherapy, and targeted therapy (43). In HCC, elevated autophagy reportedly regulated sorafenib resistance (44). Patients treated with sorafenib were found to overexpress Atg7 and had elevated autophagy activity, indicative of poor prognosis (45). However, another study demonstrated that sorafenib induced autophagy and further enhanced the drug’s effect on HCC, contrary to previous results (46). Furthermore, different HCC cell lines showed varied sensitivities to sorafenib, possibly due autophagy (47). The mechanism of autophagy has not been fully elucidated, and further research in this field is worthy of further study.
Numerous studies have reported that non-coding genes can also play an important role in development of chemotherapy resistance in cancers. For example, some miRNAs associated with sorafenib resistance have been identified, and can be used as biomarkers for predicting sorafenib treatment in HCC (Table 1).
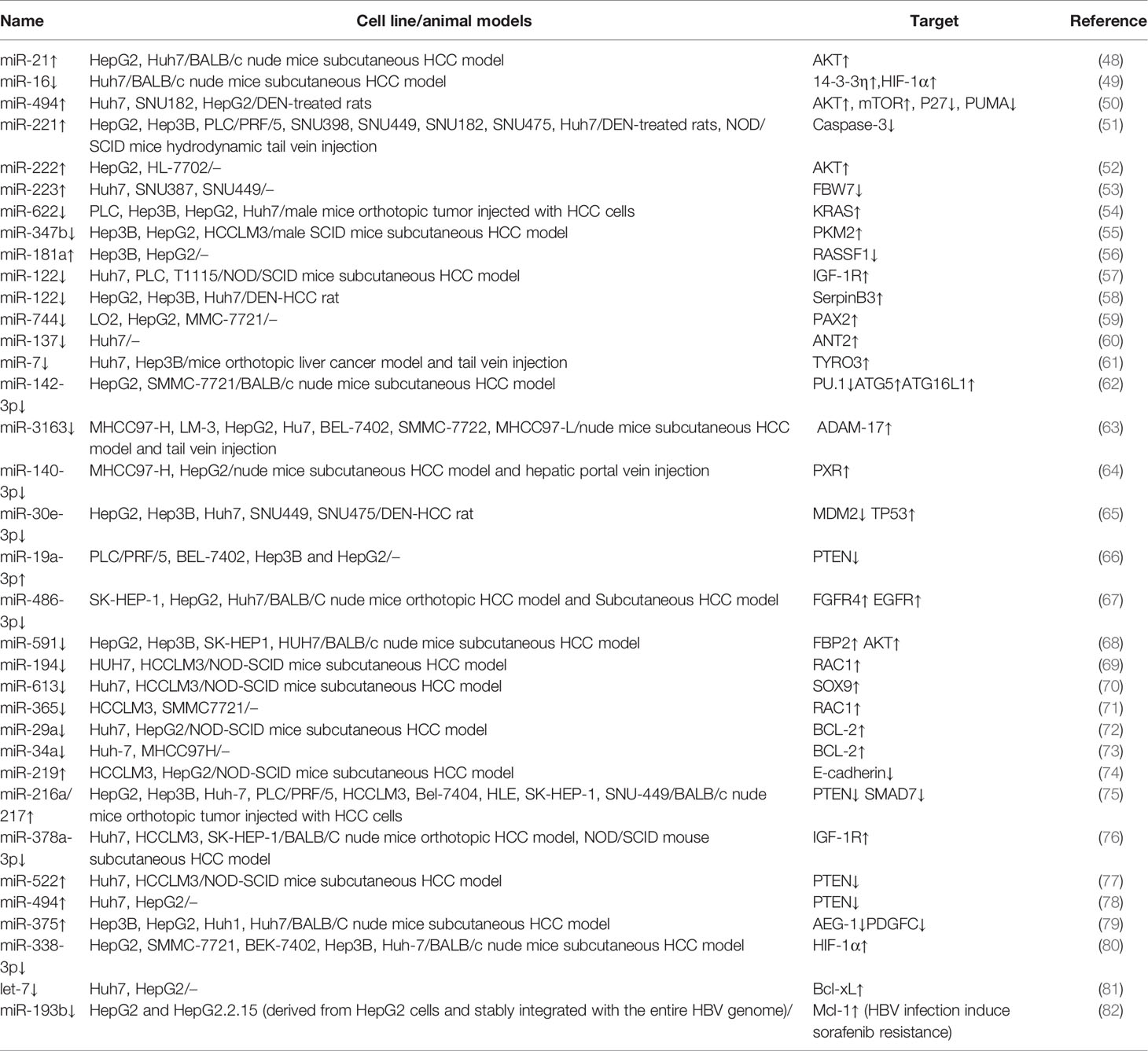
Table 1 miRNA and sorafenib resistance in HCC cells.
miRNAs play various functions like mediate proliferation, invasion and metastasis, angiogenesis, induction of hypoxia, and et al. For example, low expression of some miRNAs had been found to promote sorafenib-resistance of HCC cells, miRNAs although others may have an opposite effect. A previous study reported that miR-486-3p inhibits cell proliferation and induces apoptosis, however, it was downregulated in sorafenib-resistant HCC cell lines by up-regulating FGFR4 and EGFR activity (67). In contrast, miR-216a/217 cluster was significantly upregulated in HCC compared to normal cells. although this upregulation could activate the TGF-β and PI3K/AKT signaling pathways, thereby contributing to acquired sorafenib resistance in HCC cells (75). miRNAs functional mechanism is complicated and still controversial. Different miRNAs have different effects on HCC, moreover, the same miRNA could have different effects on different cancers. There is still plenty of room for research in this field. Apart from miRNAs, many lncRNAs have also been implicated in sorafenib resistance (Table 2).

Table 2 lncRNA and sorafenib resistance in HCC cells.
Hypoxic And Sorafenib-Resistance In Hcc
Hypoxia, which often occurs in many solid tumors, including HCC, is caused by faulty vascularization and vigorous metabolic activity, and has been associated with chemoresistance, increased invasiveness, and poor prognosis (100). Thus, suppressing hypoxia is considered a feasible approach for overcoming drug resistance. HIFs are transcription factors related to regulating angiogenesis, proliferation, glucose metabolism, tumor invasion and metastasis (100). Particularly, HIF-1α, -2α, -3α and -β are key factors that play a role in regulating a range of genes to control the hypoxia-induced signaling pathway. Since expression of the α-subunit is sensitive to oxygen, while the β-subunit is constitutively expressed, this review focuses on the α-subunit of HIFs (101). Among known α-subunits, HIF-1α and HIF-2α have been shown to regulate occurrence of hepatocellular carcinoma, while HIF-3α has generally been associated with inhibition of HIF-1α and HIF-2α activities (102, 103). Previous studies have shown that multiple factors are involved in hypoxia signal conversion from HIF-1α to HIF-2α (104). Moreover, adaptability of tumor cells to hypoxia changes with regulation of HIF-1α and HIF-2α. For example, hypoxia upregulates HIF-1α expression, and can cause it to bind to the hypoxia response element (HRE) of the target gene promoter, leading to transcription of related genes involved in hypoxia effects (105). HIF-1α is usually up-regulated in patients with liver cancer, with its overexpression associated with poor prognosis (106). Zhao et al. demonstrated that continuous sorafenib treatment could induce hypoxia and protect HCC cells to against the resulting apoptosis in HCC patients. During this process, HIF-1α was upregulated in untreated patients. Furthermore, HCC samples resistant to sorafenib exhibited HIF-1α levels above the sensitive groups (107).
Hypoxia not only activates HIF-1α in HCC, but also promotes production of VEGF and angiogenesis through HIF-1α activation (15). However, sorafenib reportedly suppresses HIF-1α synthesis, thereby causing a reduction in VEGF and tumor angiogenesis in HCC (108). Apart from that, suppression of sorafenib causes hypoxic response to be converted from HIF-1α to HIF-2α-dependent pathways, thereby promoting sorafenib resistance in hypoxic HCC cells. HIF-2α is also upregulated through a compensation mechanism, resulting in corresponding overexpression of VEGF and cyclin D1 (109).
The PI3K/AKT signaling pathway has also been widely associated with hypoxia-induced sorafenib resistance in HCC. Notably, several proteins have been shown to affect this pathway by reversing sorafenib induced hypoxia. Bort et al. reported downregulation of the AMPK/phosphorylated AMPK signaling pathway in sorafenib-resistant cells, with inhibition of this pathway via AMPK activation shown to affect sensitivities of HCC cells to sorafenib (110). Interestingly, down-regulation of AMPK also upregulates HIF-1α, and cooperates with c-myc to increase tumorigenesis, inducing and enhancing the CSCs of HCC cells, while its upregulation AMPK restores the sensitivity of HCC cells to sorafenib (111, 112). In addition, Yeh et al. showed that galectin-1 is elevated in sorafenib-resistant HCC cells, both in vitro and in vivo, promoting tumor metastasis and increasing tumor invasion, suggesting that galectin-1 plays a role in downstream regulation of the AKT/mTOR/HIF-1 signaling pathway (113). Hypoxia induces overexpression of AQP3 in hypoxic HCC cells, thereby altering sensitivity of these cells to sorafenib by activating the PI3K/Akt signaling pathway (114).
Other proteins have also been reported to alter resistance of HCC cells to sorafenib by acting on HIFs. For example, sorafenib was found to inhibit TIP30, thereby promoting EMT, which caused resistance to the drug (115, 116). Overexpression of HIF-2α was shown to downregulate TIP30 and promote EMT, although its down-regulation could reverse these effects (117). BNIP3 is also a hypoxic-regulated protein. Blanco et al. showed that HIFs stability and overexpression not only silenced the BNIP3 promoter, but also inhibited sorafenib-mediated apoptosis, thereby contributing to acquired drug resistance in HCC cells (118). On the other hand, RIT1, which belongs to the Ras superfamily, was shown to induce overexpression of RIT1 in HCC cells under HIF-1α-mediated hypoxia. Notably, sorafenib treatment could upregulate RIT1, while downregulating RIT1 in HCC cells could restore sensitivity of the cells to sorafenib (119). In addition, Long et al. found that PFKFB3 was upregulated in sorafenib-treated HCC cells. Notably, overexpressing PFKFB3 significantly enhanced sorafenib resistance in these cells by downregulating expression of apoptosis-related molecules, while blocking HIF-1α inhibited the enhancement of PFKFB3 (120). Apart from proteins, miRNAs have also been shown to play an important role in hypoxia. For example, 14-3-3η could stabilize HIF-1α and maintain resistance to sorafenib in HCC by inhibiting degradation of ubiquitin-proteasome-dependent protein, thereby maintaining CSCs. In addition, miR-16 was shown to reverse sorafenib resistance by inhibiting the 14-3-3η/HIF-1α/CSCs axis (49).
Taken together, the findings of these studies affirm the relationship between HIF expression disorder and sorafenib resistance, suggesting that hypoxia may significantly affect the therapeutic effect of sorafenib. Therefore, targeting these factors holds promise to future development of effective therapies to overcome drug resistance (Table 3).

Table 3 Hypoxia and sorafenib resistance in HCC cells.
Strategies To Overcome Sorafenib Resistance In Hcc By Target Hifs
Considering that HIFs participate in a variety of cancer-promoting pathways and regulate the biological behavior of HCC cells, targeting HIFs may be an effective treatment strategy. For example, since HIFs play a key role in development of HCC resistance to chemotherapy drugs, inhibiting them could be a feasible strategy to manage drug resistance in HCC cells. Sustained sorafenib treatment leads to increased hypoxia in the tumor, thus targeting HIFs can enhance efficacy of sorafenib. Previous studies have shown that several drugs can reverse sorafenib-resistance in HCC by targeting HIF (Figure 2).
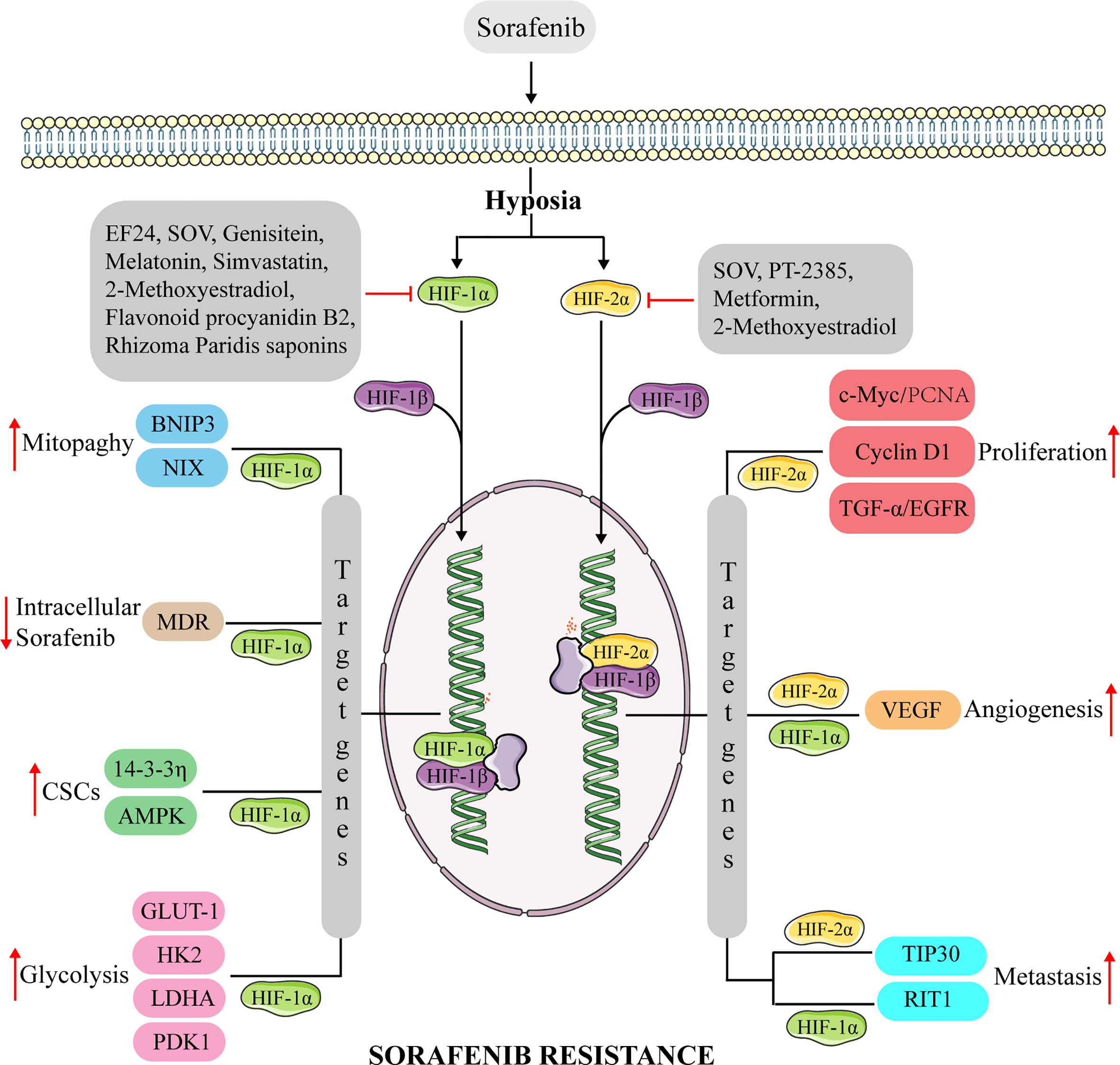
Figure 2 The mechanism of anti-HIFs overcoming sorafenib resistance in hepatocellular carcinoma. Continuous sorafenib treatment induce the dysregulation of HIF-1α and HIF-2α expression in hepatocellular carcinoma, promoting the transcription of multiple genes involved in proliferation, CSCs, metastasis, glycolysis, mitophagy, and angiogenesis. Causes hepatocellular carcinoma to develop resistance to sorafenib. Anti-HIFs could overcome this drug resistance. BNIP3, adenovirus E1B 19kDa-interacting protein 3; NIX, BNIP3-like protein X; MDR, multidrug resistance protein; AMPK, AMP-activated protein kinase; GULT-1, glucose transporter 1; HK2, hexokinase 2; LDHA, lactate dehydrogenase A; PDK1, pyruvate dehydrogenase kinase isoform 1; c-Myc, Myc proto-oncogene protein; TGF-α, transforming growth factor α; RIT1, Ras like without CAAX 1.
For example, sodium orthovanadate is a phosphate analogue that inhibits the cell cycle of sorafenib-resistant HCC cells by regulating cyclin B1 and CDK1 (arrest is in the G2/M phase). It also mediates a reduction in the mitochondrial membrane potential to induce apoptosis. A previous study demonstrated that sodium orthovanadate could down-regulate HIF-1α and HIF-2α, thereby causing a corresponding down-regulation of downstream molecules, such as VEGF, lactate dehydrogenase A and glucose transporter 1 (127). Similarly, melatonin was found to down-regulate HIF-1α protein synthesis by inhibiting the pathway of rapamycin complex-1/ribosomal protein S6 kinase β-1/ribosomal protein S6. In addition, co-administration of sorafenib and melatonin downregulated HIF-1α mitotic targets, NIX and BNIP3, thereby enhancing sensitivity of HCC cells to sorafenib (123). Notably, a combination of melatonin and sorafenib was shown to regulate the JNK/c-Jun signaling pathway, to synergistically suppress proliferation of HCC cells and induce apoptosis (130). Moreover, You et al. reported that a combination of metformin and sorafenib could synergistically inhibit HIF-2α expression, thereby increasing sensitivity of hypoxic HCC cells to sorafenib, and hindering EMT. This combination was also associated with inhibition of the growth of recurrent tumors and could significantly reduce the number of metastases in vivo (117). Feng et al. demonstrated that Simvastatin could suppress the HIF-1α/PPAR-γ/PKM2 signaling pathway by inhibiting PKM2-mediated glycolysis, thereby promoting and lowering apoptosis and proliferation of HCC cells, respectively. In addition, the drug also enhanced the effect of sorafenib in HCC cells (124). Another drug, 2-Methoxyestradiol, was shown to significantly downregulate HIF-1α and HIF-2α expression as well as that of downstream molecules such as VEGF, cyclin D1, and LDHA. Notably, its synergistic interaction with sorafenib reportedly inhibited proliferation of HCC cells and induced apoptosis both in vivo and in vitro, thereby inhibiting tumor angiogenesis (109).
Certain natural compounds have also shown efficacy in improving sorafenib-mediated treatment in drug-resistant liver cancer cells. For example, flavonoid procyanidin B2 was shown to downregulate PKM2 expression, thereby affecting the PKM2/HSP90/HIF-1α axis, inhibiting aerobic glycolysis, as well as proliferation and induction of apoptosis in HCC cells. Notably, co-treatment of procyanidin B2 and sorafenib could effectively improve the latter’s sensitivity in HCC cells (126). Genistein, a natural isoflavone that inhibits glycolysis, was shown to induce apoptosis and down-regulate GLUT-1 and HK2 by suppressing HIF-1α, thereby enhancing the effect of sorafenib on drug-resistant HCC cells both in vitro and in vivo (125). In addition, saponins derived from Rhizoma Paridis significantly downregulated mRNA expression and protein levels of HIF-1α, and further exhibited their anti-tumor activity by regulating glycolysis and lipid metabolism. Notably, a combination of these saponins with sorafenib could improve the anti-tumor effect in vivo. Previous studies have further shown that g sorafenib resistance in liver cancer cells can be overcome by preventing mitochondrial damage, inhibiting anaerobic glycolysis and suppressing lipid synthesis by targeting the PI3K/Akt/mTOR signaling pathway (121). For example, EF24 effectively reversed sorafenib resistance by degrading HIF-1α and inactivating NF-κB via a VHL tumor suppressor. A combination of EF24 with sorafenib was also found to generate a synergistic effect that enhanced the associated anti-tumor effect (129).
Previous studies have also reported that application of PT-2385 could specifically inhibit HIF-2α, to increase androgen receptors, suppress downstream factors such as STAT3, and activate the Akt and ERK signaling pathways, thereby improve sorafenib efficacy in HCC cells both in vivo and in vitro (128). In summary, inhibiting HIFs can effectively enhance the sensitivity of HCC cells to sorafenib and improve drug resistance.
Conclusion And Discussion
Although sorafenib is a safe and effective therapy for treating advanced HCC, development of drug resistance has been shown to reduce its benefits. The underlying mechanism of this resistance is complex and currently remains unclear. Primary drug resistance can be explained by the genetic heterogeneity of HCC. Elucidating the underlying mechanism of acquired drug resistance is important in guiding development of approaches to overcome or delay its development. Previous studies have demonstrated that sorafenib-acquired resistance involves multiple mechanisms, including crosstalk in the PI3K/Akt, MAPK, JAK-STAT, ERK and HIF signaling pathways, abnormal expression of proteins, such as PDGFR-β, c-KIT, FLT-3, VEGFR, EGFR, as well as EMT, cancer stem cells, and autophagy, among others. Abnormal regulation of miRNAs and lncRNAs, as well as development of hypoxia in HCC also play an important role in inducing acquired resistance to sorafenib.
For patients with advanced liver cancer, who have been exposed to sorafenib for a long time, the drug’s anti-angiogenic effect is expected to cause a decrease in microvessel density and enhance tumor hypoxia. Consequently, this induces the HIF-mediated cell adaptation mechanism to the hypoxic microenvironment. In other tumors, extensive researches have been done using gene therapy to target HIFs or adding HIFs inhibitors to current therapies, with a view to improve its effectiveness. Particularly, overexpression of HIFs in liver cancer has been reported, with sorafenib found to promote HIF activity. Notably, a combination of sorafenib with other drugs, to lower the level of or directly target HIFs, has been proven to improve efficacy of sorafenib, suppress the proliferation and promote apoptosis of HCC cells, as well as reduce the number of metastases and tumor volume both in vitro and in vivo. For patients with advanced HCC, who have failed sorafenib treatment, several drugs, such as lenvatinib, regorafenib, cabozantinib, and ramucirumab, have been approved for second-line treatment. However, sorafenib remains the mainstay for treating advanced HCC (131). The importance of overcoming sorafenib resistance in HCC cells cannot be overemphasized. For this, targeting HIFs and improving the tumor hypoxic microenvironment hold promise for future development of therapies to manage sorafenib resistance and improve prognosis of patients with advanced HCC.
Author Contributions
ZZ, QL, YL, and JZ conceived the project and wrote the manuscript. QZ, LH, ZX, YT, and ZS participated in data analysis. QX participated in discussion and language editing. DH reviewed the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was funded by the Co-construction of Provincial and Department Project (WKJ-ZJ-1919), the National Science and Technology Major Project for New Drug (No. 2017ZX09302003004), and the Key Research and Development Project of Zhejiang Science and Technology Department (2020C03008).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A, et al. Global Cancer Statistics 2018: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J Clin (2018) 68(6):394–424. doi: 10.3322/caac.21492
2. Llovet JM, Zucman–Rossi J, Pikarsky E, Sangro B, Schwartz M, Sherman M, et al. Hepatocellular Carcinoma. Nat Rev Dis Primers (2016) 2:16018. doi: 10.1038/nrdp.2016.18
3. Sia D, Villanueva A, Friedman SL, Llovet JM. Liver Cancer Cell of Origin, Molecular Class, and Effects on Patient Prognosis. Gastroenterology (2017) 152(4):745–61. doi: 10.1053/j.gastro.2016.11.048
4. Llovet JM, Ricci S, Mazzaferro V, Hilgard P, Gane E, Blanc J-F, et al. Sorafenib in Advanced Hepatocellular Carcinoma. N Engl J Med (2008) 359(4):378–90. doi: 10.1056/NEJMoa0708857
5. Vogel A, Saborowski A. Corrigendum to “Current Strategies for the Treatment of Intermediate and Advanced Hepatocellular Carcinoma” [Cancer Treatm. Rev. 82 (2019) 101946]. Cancer Treat Rev (2020) 83:101962. doi: 10.1016/j.ctrv.2019.101962
6. Cheng AL, Kang Y-K, Chen Z, Tsao C-J, Qin S, Kim JS, et al. Efficacy and Safety of Sorafenib in Patients in the Asia-Pacific Region With Advanced Hepatocellular Carcinoma: A Phase III Randomised, Double-Blind, Placebo-Controlled Trial. Lancet Oncol (2009) 10(1):25–34. doi: 10.1016/S1470-2045(08)70285-7
7. Ford R, Schwartz L, Dancey J, Dodd LE, Eisenhauer EA, Gwyther S, et al. Lessons Learned From Independent Central Review. Eur J Cancer (2009) 45(2):268–74. doi: 10.1016/j.ejca.2008.10.031
8. Liu L, Cao Y, Chen C, Zhang X, McNabola A, Wilkie D, et al. Sorafenib Blocks the RAF/MEK/ERK Pathway, Inhibits Tumor Angiogenesis, and Induces Tumor Cell Apoptosis in Hepatocellular Carcinoma Model PLC/PRF/5. Cancer Res (2006) 66(24):11851–8. doi: 10.1158/0008-5472.CAN-06-1377
9. Zhu YJ, Zheng B, Wang HY, Chen L. New Knowledge of the Mechanisms of Sorafenib Resistance in Liver Cancer. Acta Pharmacol Sin (2017) 38(5):614–22. doi: 10.1038/aps.2017.5
10. Gounder MM, Mahoney MR, Van Tine BA, Ravi V, Attia S, Deshpande HA, et al. Sorafenib for Advanced and Refractory Desmoid Tumors. N Engl J Med (2018) 379(25):2417–28. doi: 10.1056/NEJMoa1805052
11. Brose MS, Nutting CM, Jarzab B, Elisei R, Siena S, Bastholt L, et al. Sorafenib in Radioactive Iodine-Refractory, Locally Advanced or Metastatic Differentiated Thyroid Cancer: A Randomised, Double-Blind, Phase 3 Trial. Lancet (2014) 384(9940):319–28. doi: 10.1016/S0140-6736(14)60421-9
12. Escudier B, Eisen T, Stadler WM, Szczylik C, Oudard S, Staehler M, et al. Sorafenib for Treatment of Renal Cell Carcinoma: Final Efficacy and Safety Results of the Phase III Treatment Approaches in Renal Cancer Global Evaluation Trial. J Clin Oncol (2009) 27(20):3312–8. doi: 10.1200/JCO.2008.19.5511
13. Kim ES, Herbst RS, Wistuba II, Lee JJ, Blumenschein GR Jr, Tsao A, et al. The BATTLE Trial: Personalizing Therapy for Lung Cancer. Cancer Discov (2011) 1(1):44–53. doi: 10.1158/2159-8274.CD-10-0010
14. Niu L, Liu L, Yang S, Ren J, Lai PBS, Chen GG. New Insights Into Sorafenib Resistance in Hepatocellular Carcinoma: Responsible Mechanisms and Promising Strategies. Biochim Biophys Acta Rev Cancer (2017) 1868(2):564–70. doi: 10.1016/j.bbcan.2017.10.002
15. Liu LP, Ho RLK, Chen GG, Lai PBS. Sorafenib Inhibits Hypoxia-Inducible Factor-1alpha Synthesis: Implications for Antiangiogenic Activity in Hepatocellular Carcinoma. Clin Cancer Res (2012) 18(20):5662–71. doi: 10.1158/1078-0432.CCR-12-0552
16. Hanahan D, Weinberg RA. Hallmarks of Cancer: The Next Generation. Cell (2011) 144(5):646–74. doi: 10.1016/j.cell.2011.02.013
17. Gilkes DM, Semenza GL, Wirtz D. Hypoxia and the Extracellular Matrix: Drivers of Tumour Metastasis. Nat Rev Cancer (2014) 14(6):430–9. doi: 10.1038/nrc3726
18. Jing X, Yang F, Shao C, Wei K, Xie M, Shen H, et al. Role of Hypoxia in Cancer Therapy by Regulating the Tumor Microenvironment. Mol Cancer (2019) 18(1):157. doi: 10.1186/s12943-019-1089-9
19. Marin JJ, Romero MR, Briz O. Molecular Bases of Liver Cancer Refractoriness to Pharmacological Treatment. Curr Med Chem (2010) 17(8):709–40. doi: 10.2174/092986710790514462
20. Eferl R, Wagner EF. Ap-1: A Double-Edged Sword in Tumorigenesis. Nat Rev Cancer (2003) 3(11):859–68. doi: 10.1038/nrc1209
21. Eferl R, Ricci R, Kenner L, Zenz R, David J-P, Rath M, et al. Liver Tumor Development. c-Jun Antagonizes the Proapoptotic Activity of P53. Cell (2003) 112(2):181–92. doi: 10.1016/S0092-8674(03)00042-4
22. Xiang DM, Sun W, Zhou T, Zhang C, Cheng Z, Li S-C, et al. Oncofetal HLF Transactivates c-Jun to Promote Hepatocellular Carcinoma Development and Sorafenib Resistance. Gut (2019) 68(10):1858–71. doi: 10.1136/gutjnl-2018-317440
23. Chen W, Xiao W, Zhang K, Yin X, Lai J, Liang L, et al. Activation of c-Jun Predicts a Poor Response to Sorafenib in Hepatocellular Carcinoma: Preliminary Clinical Evidence. Sci Rep (2016) 6:22976. doi: 10.1038/srep22976
24. Hagiwara S, Kudo M, Nagai T, Inoue T, Ueshima K, Nishida N, et al. Activation of JNK and High Expression Level of CD133 Predict a Poor Response to Sorafenib in Hepatocellular Carcinoma. Br J Cancer (2012) 106(12):1997–2003. doi: 10.1038/bjc.2012.145
25. Morgensztern D, McLeod HL. Pi3k/Akt/mTOR Pathway as a Target for Cancer Therapy. Anticancer Drugs (2005) 16(8):797–803. doi: 10.1097/01.cad.0000173476.67239.3b
26. Zhang H, Wang Q, Liu J, Cao H. Inhibition of the PI3K/Akt Signaling Pathway Reverses Sorafenib-Derived Chemo-Resistance in Hepatocellular Carcinoma. Oncol Lett (2018) 15(6):9377–84. doi: 10.3892/ol.2018.8536
27. Li A, Zhang R, Zhang Y, Liu X, Wang R, Liu J, et al. BEZ235 Increases Sorafenib Inhibition of Hepatocellular Carcinoma Cells by Suppressing the PI3K/AKT/mTOR Pathway. Am J Transl Res (2019) 11(9):5573–85.
28. Wu CH, Wu X, Zhang HW. Inhibition of Acquired-Resistance Hepatocellular Carcinoma Cell Growth by Combining Sorafenib With Phosphoinositide 3-Kinase and Rat Sarcoma Inhibitor. J Surg Res (2016) 206(2):371–9. doi: 10.1016/j.jss.2016.08.014
29. Ezzoukhry Z, Louandre C, Trécherel E, Godin C, Chauffert B, Dupont S, et al. EGFR Activation Is a Potential Determinant of Primary Resistance of Hepatocellular Carcinoma Cells to Sorafenib. Int J Cancer (2012) 131(12):2961–9. doi: 10.1002/ijc.27604
30. Blivet-Van Eggelpoel MJ, Chettouh H, Fartoux L, Aoudjehane L, Barbu V, Rey C, et al. Epidermal Growth Factor Receptor and HER-3 Restrict Cell Response to Sorafenib in Hepatocellular Carcinoma Cells. J Hepatol (2012) 57(1):108–15. doi: 10.1016/j.jhep.2012.02.019
31. Du B, Shim JS. Targeting Epithelial-Mesenchymal Transition (EMT) to Overcome Drug Resistance in Cancer. Molecules (2016) 21(7):965. doi: 10.3390/molecules21070965
32. Shang Y, Cai X, Fan D. Roles of Epithelial-Mesenchymal Transition in Cancer Drug Resistance. Curr Cancer Drug Targets (2013) 13(9):915–29. doi: 10.2174/15680096113136660097
33. Fischer KR, Durrans A, Lee S, Sheng J, Li F, Wong STC, et al. Epithelial-to-Mesenchymal Transition Is Not Required for Lung Metastasis But Contributes to Chemoresistance. Nature (2015) 527(7579):472–6. doi: 10.1038/nature15748
34. Huang XY, Ke A-W, Shi G-M, Zhang X, Zhang C, Shi Y-H, et al. alphaB-Crystallin Complexes With 14-3-3zeta to Induce Epithelial-Mesenchymal Transition and Resistance to Sorafenib in Hepatocellular Carcinoma. Hepatology (2013) 57(6):2235–47. doi: 10.1002/hep.26255
35. van Malenstein H, Dekervel J, Verslype C, Van Cutsem E, Windmolders P, Nevens F, et al. Long-Term Exposure to Sorafenib of Liver Cancer Cells Induces Resistance With Epithelial-to-Mesenchymal Transition, Increased Invasion and Risk of Rebound Growth. Cancer Lett (2013) 329(1):74–83. doi: 10.1016/j.canlet.2012.10.021
36. Singh A, Settleman J. EMT, Cancer Stem Cells and Drug Resistance: An Emerging Axis of Evil in the War on Cancer. Oncogene (2010) 29(34):4741–51. doi: 10.1038/onc.2010.215
37. Chow AK-M, Ng L, Lam CS-C, Wong SK-M, Wan TM-H, Cheng NS-M, et al. The Enhanced Metastatic Potential of Hepatocellular Carcinoma (HCC) Cells With Sorafenib Resistance. PloS One (2013) 8(11):e78675. doi: 10.1371/journal.pone.0078675
38. Chen X, Lingala S, Khoobyari S, Nolta J, Zern MA, Wu J. Epithelial Mesenchymal Transition and Hedgehog Signaling Activation Are Associated With Chemoresistance and Invasion of Hepatoma Subpopulations. J Hepatol (2011) 55(4):838–45. doi: 10.1016/j.jhep.2010.12.043
39. Fan Z, Duan J, Wang L, Xiao S, Li L, Yan X, et al. PTK2 Promotes Cancer Stem Cell Traits in Hepatocellular Carcinoma by Activating Wnt/beta-Catenin Signaling. Cancer Lett (2019) 450:132–43. doi: 10.1016/j.canlet.2019.02.040
40. Fernando J, Malfettone A, Cepeda EB, Vilarrasa-Blasi R, Bertran E, Raimondi G, et al. A Mesenchymal-Like Phenotype and Expression of CD44 Predict Lack of Apoptotic Response to Sorafenib in Liver Tumor Cells. Int J Cancer (2015) 136(4):E161–72. doi: 10.1002/ijc.29097
41. Liu LL, Fu D, Ma Y, Shen X-Z. The Power and the Promise of Liver Cancer Stem Cell Markers. Stem Cells Dev (2011) 20(12):2023–30. doi: 10.1089/scd.2011.0012
42. Tian Y, Kuo C-F, Sir D, Wang L, Govindarajan S, Petrovic L, et al. Autophagy Inhibits Oxidative Stress and Tumor Suppressors to Exert Its Dual Effect on Hepatocarcinogenesis. Cell Death Differ (2015) 22(6):1025–34. doi: 10.1038/cdd.2014.201
43. Thorburn A, Thamm DH, Gustafson DL. Autophagy and Cancer Therapy. Mol Pharmacol (2014) 85(6):830–8. doi: 10.1124/mol.114.091850
44. Lu S, Yao Y, Xu G, Zhou C, Zhang Y, Sun J, et al. CD24 Regulates Sorafenib Resistance Via Activating Autophagy in Hepatocellular Carcinoma. Cell Death Dis (2018) 9(6):646. doi: 10.1038/s41419-018-0681-z
45. Luo T, Fu J, Xu A, Su B, Ren Y, Li N, et al. PSMD10/Gankyrin Induces Autophagy to Promote Tumor Progression Through Cytoplasmic Interaction With ATG7 and Nuclear Transactivation of ATG7 Expression. Autophagy (2016) 12(8):1355–71. doi: 10.1080/15548627.2015.1034405
46. Tai W-T, Shiau C-W, Chen H-L, Liu C-Y, Lin C-S, Cheng A-S, et al. Mcl-1-Dependent Activation of Beclin 1 Mediates Autophagic Cell Death Induced by Sorafenib and SC-59 in Hepatocellular Carcinoma Cells. Cell Death Dis (2013) 4:e485. doi: 10.1038/cddis.2013.18
47. Fischer TD, Wang J-H, Vlada A, Kim J-S, Behrns KE. Role of Autophagy in Differential Sensitivity of Hepatocarcinoma Cells to Sorafenib. World J Hepatol (2014) 6(10):752–8. doi: 10.4254/wjh.v6.i10.752
48. He C, Dong X, Zhai B, Jiang X, Dong D, Li B, et al. MiR-21 Mediates Sorafenib Resistance of Hepatocellular Carcinoma Cells by Inhibiting Autophagy Via the PTEN/Akt Pathway. Oncotarget (2015) 6(30):28867–81. doi: 10.18632/oncotarget.4814
49. Qiu Y, Shan W, Yang Y, Jin M, Dai Y, Yang R, et al. Reversal of Sorafenib Resistance in Hepatocellular Carcinoma: Epigenetically Regulated Disruption of 14-3-3eta/hypoxia-inducible Factor-1alpha. Cell Death Discovery (2019) 5:120. doi: 10.1038/s41420-019-0200-8
50. Pollutri D, Patrizi C, Marinelli S, Giovannini C, Trombetta E, Giannone FA, et al. The Epigenetically Regulated miR-494 Associates With Stem-Cell Phenotype and Induces Sorafenib Resistance in Hepatocellular Carcinoma. Cell Death Dis (2018) 9(1):4. doi: 10.1038/s41419-017-0076-6
51. Fornari F, Pollutri D, Patrizi C, La Bella TL, Marinelli S, Gardini C, et al. In Hepatocellular Carcinoma Mir-221 Modulates Sorafenib Resistance Through Inhibition of Caspase-3-Mediated Apoptosis. Clin Cancer Res (2017) 23(14):3953–65. doi: 10.1158/1078-0432.CCR-16-1464
52. Liu K, Liu S, Zhang W, Ji B, Wang Y, Liu Y. miR−222 Regulates Sorafenib Resistance and Enhance Tumorigenicity in Hepatocellular Carcinoma. Int J Oncol (2014) 45(4):1537–46. doi: 10.3892/ijo.2014.2577
53. Tang X, Yang W, Shu Z, Shen X, Zhang W, Cen C, et al. MicroRNA223 Promotes Hepatocellular Carcinoma Cell Resistance to Sorafenib by Targeting FBW7. Oncol Rep (2019) 41(2):1231–7. doi: 10.3892/or.2018.6908
54. Dietrich P, Koch A, Fritz V, Hartmann A, Bosserhoff AK, Hellerbrand C. Wild Type Kirsten Rat Sarcoma Is a Novel microRNA-622-Regulated Therapeutic Target for Hepatocellular Carcinoma and Contributes to Sorafenib Resistance. Gut (2018) 67(7):1328–41. doi: 10.1136/gutjnl-2017-315402
55. Zhang M, Zhang H, Hong H, Zhang Z. MiR-374b Re-Sensitizes Hepatocellular Carcinoma Cells to Sorafenib Therapy by Antagonizing PKM2-Mediated Glycolysis Pathway. Am J Cancer Res (2019) 9(4):765–78.
56. Azumi J, Tsubota T, Sakabe T, Shiota G. miR-181a Induces Sorafenib Resistance of Hepatocellular Carcinoma Cells Through Downregulation of RASSF1 Expression. Cancer Sci (2016) 107(9):1256–62. doi: 10.1111/cas.13006
57. Xu Y, Huang J, Ma L, Shan J, Shen J, Yang Z, et al. MicroRNA-122 Confers Sorafenib Resistance to Hepatocellular Carcinoma Cells by Targeting IGF-1R to Regulate RAS/RAF/ERK Signaling Pathways. Cancer Lett (2016) 371(2):171–81. doi: 10.1016/j.canlet.2015.11.034
58. Turato C, Fornari F, Pollutri D, Fassan M, Quarta S, Villano G, et al. Mir-122 Targets SerpinB3 and Is Involved in Sorafenib Resistance in Hepatocellular Carcinoma. J Clin Med (2019) 8(2). doi: 10.3390/jcm8020171
59. Wang G, Zhao W, Wang H, Qiu G, Jiang Z, Wei G, et al. Exosomal MiR-744 Inhibits Proliferation and Sorafenib Chemoresistance in Hepatocellular Carcinoma by Targeting Pax2. Med Sci Monit (2019) 25:7209–17. doi: 10.12659/MSM.919219
60. Lu AQ, Lv B, Qiu F, Wang X-Y, Cao X-H. Upregulation of miR-137 Reverses Sorafenib Resistance and Cancer-Initiating Cell Phenotypes by Degrading ANT2 in Hepatocellular Carcinoma. Oncol Rep (2017) 37(4):2071–8. doi: 10.3892/or.2017.5498
61. Kabir TD, Ganda C, Brown RM, Beveridge DJ, Richardson KL, Chaturvedi V, et al. A microRNA-7/growth Arrest Specific 6/TYRO3 Axis Regulates the Growth and Invasiveness of Sorafenib-Resistant Cells in Human Hepatocellular Carcinoma. Hepatology (2018) 67(1):216–31. doi: 10.1002/hep.29478
62. Zhang K, Chen J, Zhou H, Chen Y, Zhi Y, Zhang B, et al. Pu.1/microRNA-142-3p Targets ATG5/ATG16L1 to Inactivate Autophagy and Sensitize Hepatocellular Carcinoma Cells to Sorafenib. Cell Death Dis (2018) 9(3):312. doi: 10.1038/s41419-018-0344-0
63. Yang B, Wang C, Xie H, Wang Y, Huang J, Rong Y, et al. MicroRNA-3163 Targets ADAM-17 and Enhances the Sensitivity of Hepatocellular Carcinoma Cells to Molecular Targeted Agents. Cell Death Dis (2019) 10(10):784. doi: 10.1038/s41419-019-2023-1
64. Li J, Zhao J, Wang H, Li X, Liu A, Qin Q, et al. MicroRNA-140-3p Enhances the Sensitivity of Hepatocellular Carcinoma Cells to Sorafenib by Targeting Pregnenolone X Receptor. Onco Targets Ther (2018) 11:5885–94. doi: 10.2147/OTT.S179509
65. Gramantieri L, Pollutri D, Gagliardi M, Giovannini C, Quarta S, Ferracin M, et al. Mir-30e-3p Influences Tumor Phenotype Through MDM2/TP53 Axis and Predicts Sorafenib Resistance in Hepatocellular Carcinoma. Cancer Res (2020) 80(8):1720–34. doi: 10.1158/0008-5472.CAN-19-0472
66. Jiang XM, Yu XN, Liu TT, Zhu HR, Shi X, Bilegsaikhan E, et al. microRNA-19a-3p Promotes Tumor Metastasis and Chemoresistance Through the PTEN/Akt Pathway in Hepatocellular Carcinoma. BioMed Pharmacother (2018) 105:1147–54. doi: 10.1016/j.biopha.2018.06.097
67. Ji L, Lin Z, Wan Z, Xia S, Jiang S, Cen D, et al. miR-486-3p Mediates Hepatocellular Carcinoma Sorafenib Resistance by Targeting FGFR4 and EGFR. Cell Death Dis (2020) 11(4):250. doi: 10.1038/s41419-020-2413-4
68. Wang H, Tang Y, Yang D, Zheng L. MicroRNA-591 Functions as a Tumor Suppressor in Hepatocellular Carcinoma by Lowering Drug Resistance Through Inhibition of Far-Upstream Element-Binding Protein 2-Mediated Phosphoinositide 3-Kinase/Akt/Mammalian Target of Rapamycin Axis. Pharmacology (2019) 104(3-4):173–86. doi: 10.1159/000501162
69. Ran RZ, Chen J, Cui LJ, Lin XL, Fan MM, Cong ZZ, et al. miR-194 Inhibits Liver Cancer Stem Cell Expansion by Regulating RAC1 Pathway. Exp Cell Res (2019) 378(1):66–75. doi: 10.1016/j.yexcr.2019.03.007
70. Li B, Liu D, Yang P, Li HY, Wang D. miR-613 Inhibits Liver Cancer Stem Cell Expansion by Regulating SOX9 Pathway. Gene (2019) 707:78–85. doi: 10.1016/j.gene.2019.05.015
71. Jiang Z-B, Ma B-Q, Liu S-G, Li J, Yang G-M, Hou Y-B, et al. miR-365 Regulates Liver Cancer Stem Cells Via RAC1 Pathway. Mol Carcinog (2019) 58(1):55–65. doi: 10.1002/mc.22906
72. Song S, Sun K, Dong J, Zhao Y, Liu F, Liu H, et al. microRNA-29a Regulates Liver Tumor-Initiating Cells Expansion Via Bcl-2 Pathway. Exp Cell Res (2020) 387(2):111781. doi: 10.1016/j.yexcr.2019.111781
73. Yang F, Li Q-J, Gong Z-B, Zhou L, You N, Wang S, et al. MicroRNA-34a Targets Bcl-2 and Sensitizes Human Hepatocellular Carcinoma Cells to Sorafenib Treatment. Technol Cancer Res Treat (2014) 13(1):77–86. doi: 10.7785/tcrt.2012.500364
74. Si A, Wang L, Miao K, Zhang R, Ji H, Lei Z, et al. miR-219 Regulates Liver Cancer Stem Cell Expansion Via E-Cadherin Pathway. Cell Cycle (2019) 18(24):3550–61. doi: 10.1080/15384101.2019.1691762
75. Xia H, Ooi LL, Hui KM. MicroRNA-216a/217-induced Epithelial-Mesenchymal Transition Targets PTEN and SMAD7 to Promote Drug Resistance and Recurrence of Liver Cancer. Hepatology (2013) 58(2):629–41. doi: 10.1002/hep.26369
76. Lin Z, Xia S, Liang Y, Ji L, Pan Y, Jiang S, et al. LXR Activation Potentiates Sorafenib Sensitivity in HCC by Activating microRNA-378a Transcription. Theranostics (2020) 10(19):8834–50. doi: 10.7150/thno.45158
77. Han T, Zhang Y, Yang X, Han L, Li H, Chen T, et al. Mir-552 Regulates Liver Tumor-Initiating Cell Expansion and Sorafenib Resistance. Mol Ther Nucleic Acids (2020) 19:1073–85. doi: 10.1016/j.omtn.2019.12.043
78. Liu K, Liu S, Zhang W, Jia B, Tan L, Jin Z, et al. miR-494 Promotes Cell Proliferation, Migration and Invasion, and Increased Sorafenib Resistance in Hepatocellular Carcinoma by Targeting PTEN. Oncol Rep (2015) 34(2):1003–10. doi: 10.3892/or.2015.4030
79. Li D, Wang T, Sun F-F, Feng J-Q, Peng J-J, Li H, et al. MicroRNA-375 Represses Tumor Angiogenesis and Reverses Resistance to Sorafenib in Hepatocarcinoma. Cancer Gene Ther (2020) 8(1-2):126–40. doi: 10.1038/s41417-020-0191-x
80. Xu H, Zhao L, Fang Q, Sun J, Zhang S, Zhan C, et al. MiR-338-3p Inhibits Hepatocarcinoma Cells and Sensitizes These Cells to Sorafenib by Targeting Hypoxia-Induced Factor 1alpha. PloS One (2014) 9(12):e115565. doi: 10.1371/journal.pone.0115565
81. Shimizu S, Takehara T, Hikita H, Kodama T, Miyagi T, Hosui A, et al. The Let-7 Family of microRNAs Inhibits Bcl-xL Expression and Potentiates Sorafenib-Induced Apoptosis in Human Hepatocellular Carcinoma. J Hepatol (2010) 52(5):698–704. doi: 10.1016/j.jhep.2009.12.024
82. Mao K, Zhang J, He C, Xu K, Liu J, Sun J, et al. Restoration of miR-193b Sensitizes Hepatitis B Virus-Associated Hepatocellular Carcinoma to Sorafenib. Cancer Lett (2014) 352(2):245–52. doi: 10.1016/j.canlet.2014.07.004
83. Li W, Dong X, He C, Tan G, Li Z, Zhai B, et al. Lncrna SNHG1 Contributes to Sorafenib Resistance by Activating the Akt Pathway and Is Positively Regulated by miR-21 in Hepatocellular Carcinoma Cells. J Exp Clin Cancer Res (2019) 38(1):183. doi: 10.1186/s13046-019-1177-0
84. Zhang PF, Wang F, Wu J, Wu Y, Huang W, Liu D, et al. LncRNA SNHG3 Induces EMT and Sorafenib Resistance by Modulating the miR-128/CD151 Pathway in Hepatocellular Carcinoma. J Cell Physiol (2019) 234(3):2788–94. doi: 10.1002/jcp.27095
85. Ye J, Zhang R, Du X, Chai W, Zhou Q. Long Noncoding RNA SNHG16 Induces Sorafenib Resistance in Hepatocellular Carcinoma Cells Through Sponging Mir-140-5p. Onco Targets Ther (2019) 12:415–22. doi: 10.2147/OTT.S175176
86. Sui C, Dong Z, Yang Y, Zhang M, Dai B, Geng L, et al. Lncrna FOXD2-AS1 as a Competitive Endogenous RNA Against miR-150-5p Reverses Resistance to Sorafenib in Hepatocellular Carcinoma. J Cell Mol Med (2019) 23(9):6024–33. doi: 10.1111/jcmm.14465
87. Li X, Zhou Y, Yang L, Ma Y, Peng X, Yang S, et al. Lncrna NEAT1 Promotes Autophagy Via Regulating miR-204/ATG3 and Enhanced Cell Resistance to Sorafenib in Hepatocellular Carcinoma. J Cell Physiol (2020) 235(4):3402–13. doi: 10.1002/jcp.29230
88. Liu Y, Chen L, Yuan H, Guo S, Wu G. LncRNA DANCR Promotes Sorafenib Resistance Via Activation of IL-6/STAT3 Signaling in Hepatocellular Carcinoma Cells. Onco Targets Ther (2020) 13:1145–57. doi: 10.2147/OTT.S229957
89. Tang X, Zhang W, Ye Y, Li H, Cheng L, Zhang M, et al. Lncrna HOTAIR Contributes to Sorafenib Resistance Through Suppressing miR-217 in Hepatic Carcinoma. BioMed Res Int (2020) 2020:9515071. doi: 10.1155/2020/9515071
90. Shen Q, Jiang S, Wu M, Zhang L, Su X, Zhao D. LncRNA HEIH Confers Cell Sorafenib Resistance in Hepatocellular Carcinoma by Regulating miR-98-5p/PI3K/AKT Pathway. Cancer Manag Res (2020) 12:6585–95. doi: 10.2147/CMAR.S241383
91. Fan L, Huang X, Chen J, Zhang K, Gu Y-H, Sun J, et al. Long Noncoding Rna MALAT1 Contributes to Sorafenib Resistance by Targeting Mir-140-5p/Aurora-A Signaling in Hepatocellular Carcinoma. Mol Cancer Ther (2020) 19(5):1197–209. doi: 10.1158/1535-7163.MCT-19-0203
92. Zhi Y, Abudoureyimu M, Zhou H, Wang T, Feng B, Wang R, et al. Foxm1-Mediated LINC-ROR Regulates the Proliferation and Sensitivity to Sorafenib in Hepatocellular Carcinoma. Mol Ther Nucleic Acids (2019) 16:576–88. doi: 10.1016/j.omtn.2019.04.008
93. Cheng Z, Lei Z, Yang P, Si A, Xiang D, Zhou J, et al. Long Non-Coding RNA THOR Promotes Liver Cancer Stem Cells Expansion Via Beta-Catenin Pathway. Gene (2019) 684:95–103. doi: 10.1016/j.gene.2018.10.051
94. Tang S, Tan G, Jiang X, Han P, Zhai B, Dong X, et al. An Artificial lncRNA Targeting Multiple miRNAs Overcomes Sorafenib Resistance in Hepatocellular Carcinoma Cells. Oncotarget (2016) 7(45):73257–69. doi: 10.18632/oncotarget.12304
95. Quagliata L, Quintavalle C, Matter MS, Novello MS, di Tommaso C, Pressiani L, et al. High Expression of HOXA13 Correlates With Poorly Differentiated Hepatocellular Carcinomas and Modulates Sorafenib Response in In Vitro Models. Lab Invest (2018) 98(1):95–105. doi: 10.1038/labinvest.2017.107
96. Jin W, Chen L, Cai X, Zhang Y, Zhang J, Ma D, et al. Long non-Coding RNA TUC338 Is Functionally Involved in Sorafenib-Sensitized Hepatocarcinoma Cells by Targeting RASAL1. Oncol Rep (2017) 37(1):273–80. doi: 10.3892/or.2016.5248
97. Shi Y, Yang X, Xue X, Sun D, Cai P, Song Q, et al. Hanr Enhances Autophagy-Associated Sorafenib Resistance Through Mir-29b/ATG9A Axis in Hepatocellular Carcinoma. Onco Targets Ther (2020) 13:2127–37. doi: 10.2147/OTT.S229913
98. Xu Y, Liu Y, Li Z, Li H, Li X, Yan L, et al. Long Noncoding RNA H19 Is Involved in Sorafenib Resistance in Hepatocellular Carcinoma by Upregulating Mir675. Oncol Rep (2020) 44(1):165–73. doi: 10.3892/or.2020.7608
99. Schultheiss CS, Laggai S, Czepukojc B, Hussein UK, List M, Barghash A, et al. The Long Non-Coding RNA H19 Suppresses Carcinogenesis and Chemoresistance in Hepatocellular Carcinoma. Cell Stress (2017) 1(1):37–54. doi: 10.15698/cst2017.10.105
100. Wilson GK, Tennant DA, McKeating JA. Hypoxia Inducible Factors in Liver Disease and Hepatocellular Carcinoma: Current Understanding and Future Directions. J Hepatol (2014) 61(6):1397–406. doi: 10.1016/j.jhep.2014.08.025
101. Keith B, Johnson RS, Simon MC. HIF1alpha and HIF2alpha: Sibling Rivalry in Hypoxic Tumour Growth and Progression. Nat Rev Cancer (2011) 12(1):9–22. doi: 10.1038/nrc3183
102. Yang SL, Wu C, Xiong Z-F, Fang X. Progress on Hypoxia-Inducible Factor-3: Its Structure, Gene Regulation and Biological Function (Review). Mol Med Rep (2015) 12(2):2411–6. doi: 10.3892/mmr.2015.3689
103. Xiong XX, Qiu XY, Hu DX, Chen XQ. Advances in Hypoxia-Mediated Mechanisms in Hepatocellular Carcinoma. Mol Pharmacol (2017) 92(3):246–55. doi: 10.1124/mol.116.107706
104. Murugesan T, Rajajeyabalachandran G, Kumar S, Nagaraju S, Jegatheesan SK. Targeting HIF-2alpha as Therapy for Advanced Cancers. Drug Discov Today (2018) 23(7):1444–51. doi: 10.1016/j.drudis.2018.05.003
105. Al Hasawi N, Alkandari MF, Luqmani YA. Phosphofructokinase: A Mediator of Glycolytic Flux in Cancer Progression. Crit Rev Oncol Hematol (2014) 92(3):312–21. doi: 10.1016/j.critrevonc.2014.05.007
106. Semenza GL. Targeting HIF-1 for Cancer Therapy. Nat Rev Cancer (2003) 3(10):721–32. doi: 10.1038/nrc1187
107. Zhao D, Zhai B, He C, Tan G, Jiang X, Pan X, et al. Upregulation of HIF-2alpha Induced by Sorafenib Contributes to the Resistance by Activating the TGF-alpha/EGFR Pathway in Hepatocellular Carcinoma Cells. Cell Signal (2014) 26(5):1030–9. doi: 10.1016/j.cellsig.2014.01.026
108. Xu M, Zheng YL, Xie XY, Liang JY, Pan FS, Zheng SG, et al. Sorafenib Blocks the HIF-1alpha/VEGFA Pathway, Inhibits Tumor Invasion, and Induces Apoptosis in Hepatoma Cells. DNA Cell Biol (2014) 33(5):275–81. doi: 10.1089/dna.2013.2184
109. Ma L, Li G, Zhu H, Dong X, Zhao D, Jiang X, et al. 2-Methoxyestradiol Synergizes With Sorafenib to Suppress Hepatocellular Carcinoma by Simultaneously Dysregulating Hypoxia-Inducible Factor-1 and -2. Cancer Lett (2014) 355(1):96–105. doi: 10.1016/j.canlet.2014.09.011
110. Bort A, Spínola E, Rodríguez–Henche N, Dívaz–Laviada I. Capsaicin Exerts Synergistic Antitumor Effect With Sorafenib in Hepatocellular Carcinoma Cells Through AMPK Activation. Oncotarget (2017) 8(50):87684–98. doi: 10.18632/oncotarget.21196
111. Faubert B, Boily G, Izreig S, Griss T, Samborska B, Dong Z, et al. AMPK Is a Negative Regulator of the Warburg Effect and Suppresses Tumor Growth In Vivo. Cell Metab (2013) 17(1):113–24. doi: 10.1016/j.cmet.2012.12.001
112. Bort A, Sánchez BG, Mateos–Gómez A, Vara–Ciruelos D, Rodríguez–Henche N, Díaz–Laviada I. Targeting AMP-activated Kinase Impacts Hepatocellular Cancer Stem Cells Induced by Long-Term Treatment With Sorafenib. Mol Oncol (2019) 13(5):1311–31. doi: 10.1002/1878-0261.12488
113. Yeh CC, Hsu CH, Shao YY, Ho WC, Tsai MH, Feng WC, et al. Integrated Stable Isotope Labeling by Amino Acids in Cell Culture (SILAC) and Isobaric Tags for Relative and Absolute Quantitation (Itraq) Quantitative Proteomic Analysis Identifies Galectin-1 as a Potential Biomarker for Predicting Sorafenib Resistance in Liver Cancer. Mol Cell Proteomics (2015) 14(6):1527–45. doi: 10.1074/mcp.M114.046417
114. Malale K, Fu K, Qiu L, Zhan K, Gan X, Mei Z. Hypoxia-Induced Aquaporin-3 Changes Hepatocellular Carcinoma Cell Sensitivity to Sorafenib by Activating the PI3K/Akt Signaling Pathway. Cancer Manag Res (2020) 12:4321–33. doi: 10.2147/CMAR.S243918
115. Zhu M, Yin F, Fan X, Jing W, Chen R, Liu L, et al. Decreased TIP30 Promotes Snail-Mediated Epithelial-Mesenchymal Transition and Tumor-Initiating Properties in Hepatocellular Carcinoma. Oncogene (2015) 34(11):1420–31. doi: 10.1038/onc.2014.73
116. Zhang W, Sun HC, Wang WQ, Zhang QB, Zhuang PY, Xiong YQ, et al. Sorafenib Down-Regulates Expression of HTATIP2 to Promote Invasiveness and Metastasis of Orthotopic Hepatocellular Carcinoma Tumors in Mice. Gastroenterology (2012) 143(6):1641–1649 e5. doi: 10.1053/j.gastro.2012.08.032
117. You A, Cao M, Guo Z, Zuo B, Gao J, Zhou H, et al. Metformin Sensitizes Sorafenib to Inhibit Postoperative Recurrence and Metastasis of Hepatocellular Carcinoma in Orthotopic Mouse Models. J Hematol Oncol (2016) 9:20. doi: 10.1186/s13045-016-0253-6
118. Mendez-Blanco C, Fondevila F, Fernández-Palanca P, García–Palomo A, van Pelt J, Verslype C, et al. Stabilization of Hypoxia-Inducible Factors and BNIP3 Promoter Methylation Contribute to Acquired Sorafenib Resistance in Human Hepatocarcinoma Cells. Cancers (Basel) (2019) 11(12). doi: 10.3390/cancers11121984
119. Song Z, Liu T, Chen J, Ge C, Zhao F, Zhu M, et al. HIF-1alpha-Induced RIT1 Promotes Liver Cancer Growth and Metastasis and Its Deficiency Increases Sensitivity to Sorafenib. Cancer Lett (2019) 460:96–107. doi: 10.1016/j.canlet.2019.06.016
120. Long Q, Zou X, Song Y, Duan Z, Liu L. Pfkfb3/Hif-1alpha Feedback Loop Modulates Sorafenib Resistance in Hepatocellular Carcinoma Cells. Biochem Biophys Res Commun (2019) 513(3):642–50. doi: 10.1016/j.bbrc.2019.03.109
121. Yao J, Man S, Dong H, Yang L, Ma L, Gao W. Combinatorial Treatment of Rhizoma Paridis Saponins and Sorafenib Overcomes the Intolerance of Sorafenib. J Steroid Biochem Mol Biol (2018) 183:159–66. doi: 10.1016/j.jsbmb.2018.06.010
122. Liu F, Dong X, Lv H, Xiu P, Li T, Wang F, et al. Targeting Hypoxia-Inducible factor-2alpha Enhances Sorafenib Antitumor Activity Via Beta-Catenin/C-Myc-dependent Pathways in Hepatocellular Carcinoma. Oncol Lett (2015) 10(2):778–84. doi: 10.3892/ol.2015.3315
123. Prieto-Dominguez N, Méndez-Blanco S, Carbajo‐Pescador F, Fondevila A, García-Palomo J, González-Gallego J, et al. Melatonin Enhances Sorafenib Actions in Human Hepatocarcinoma Cells by Inhibiting mTORC1/p70S6K/HIF-1alpha and Hypoxia-Mediated Mitophagy. Oncotarget (2017) 8(53):91402–14. doi: 10.18632/oncotarget.20592
124. Feng J, Dai W, Mao Y, Wu L, Li J, Chen K, et al. Simvastatin Re-Sensitizes Hepatocellular Carcinoma Cells to Sorafenib by Inhibiting HIF-1alpha/PPAR-Gamma/PKM2-Mediated Glycolysis. J Exp Clin Cancer Res (2020) 39(1):24. doi: 10.1186/s13046-020-1528-x
125. Li S, Li J, Dai W, Zhang Q, Feng J, Wu L, et al. Genistein Suppresses Aerobic Glycolysis and Induces Hepatocellular Carcinoma Cell Death. Br J Cancer (2017) 117(10):1518–28. doi: 10.1038/bjc.2017.323
126. Feng J, Wu L, Ji J, Chen K, Yu Q, Zhang J, et al. PKM2 Is the Target of Proanthocyanidin B2 During the Inhibition of Hepatocellular Carcinoma. J Exp Clin Cancer Res (2019) 38(1):204. doi: 10.1186/s13046-019-1194-z
127. Jiang W, Li G, Li W, Wang P, Xiu P, Jiang X, et al. Sodium Orthovanadate Overcomes Sorafenib Resistance of Hepatocellular Carcinoma Cells by Inhibiting Na(+)/K(+)-ATPase Activity and Hypoxia-Inducible Pathways. Sci Rep (2018) 8(1):9706. doi: 10.1038/s41598-018-28010-y
128. Xu J, Zheng L, Chen J, Sun YH, Lin RA, Jin M, et al. Increasing AR by HIF-2alpha Inhibitor (PT-2385) Overcomes the Side-Effects of Sorafenib by Suppressing Hepatocellular Carcinoma Invasion Via Alteration of pSTAT3, pAKT and pERK Signals. Cell Death Dis (2017) 8(10):e3095. doi: 10.1038/cddis.2017.411
129. Liang Y, Zheng T, Song R, Wang J, Yin D, Wang L, et al. Hypoxia-Mediated Sorafenib Resistance can be Overcome by EF24 Through Von Hippel-Lindau Tumor Suppressor-Dependent HIF-1alpha Inhibition in Hepatocellular Carcinoma. Hepatology (2013) 57(5):1847–57. doi: 10.1002/hep.26224
130. Lin S, Hoffmann K, Gao C, Petrulionis M, Herr I, Schemmer P. Melatonin Promotes Sorafenib-Induced Apoptosis Through Synergistic Activation of JNK/c-jun Pathway in Human Hepatocellular Carcinoma. J Pineal Res (2017) 62(3). doi: 10.1111/jpi.12398
Keywords: sorafenib, hepatocellular carcinoma, HIF-1α, HIF-2α, sorafenib-resistance
Citation: Zeng Z, Lu Q, Liu Y, Zhao J, Zhang Q, Hu L, Shi Z, Tu Y, Xiao Z, Xu Q and Huang D (2021) Effect of the Hypoxia Inducible Factor on Sorafenib Resistance of Hepatocellular Carcinoma. Front. Oncol. 11:641522. doi: 10.3389/fonc.2021.641522
Received: 14 December 2020; Accepted: 16 June 2021;
Published: 07 July 2021.
Edited by:
Yongchuan Gu, The University of Auckland, New ZealandReviewed by:
Jiang Chen, Zhejiang University, ChinaYuelong Liang, Zhejiang University, China
Heping Kan, Southern Medical University, China
Copyright © 2021 Zeng, Lu, Liu, Zhao, Zhang, Hu, Shi, Tu, Xiao, Xu and Huang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Qiuran Xu, d2luZHdheTYyNkBzaW5hLmNvbQ==; Dongsheng Huang, ZHNodWFuZ0B6anUuZWR1LmNu
†These authors have contributed equally to this work and share first authorship