 Liang Wu1†
Liang Wu1† Minghao Jiang1†
Minghao Jiang1† Ping Yu1
Ping Yu1 Jianfeng Li1,2
Jianfeng Li1,2 Wen Ouyang1
Wen Ouyang1 Chong Feng1,2
Chong Feng1,2 Wei Li Zhao1*
Wei Li Zhao1* Yuting Dai1,2*Jinyan Huang1*
Yuting Dai1,2*Jinyan Huang1*- 1Shanghai Institute of Hematology, State Key Laboratory of Medical Genomics, National Research Center for Translational Medicine, Shanghai Rui Jin Hospital, Shanghai Jiao Tong University School of Medicine, Shanghai, China
- 2School of Life Sciences and Biotechnology, Shanghai Jiao Tong University, Shanghai, China
B cell precursor acute lymphoblastic leukemia (BCP-ALL) is a blood cancer that originates from the abnormal proliferation of B-lymphoid progenitors. Cell population components and cell–cell interaction in the bone marrow microenvironment are significant factors for progression, relapse, and therapy resistance of BCP-ALL. In this study, we identified specifically expressed genes in B cells and myeloid cells by analyzing single-cell RNA sequencing data for seven BCP-ALL samples and four healthy samples obtained from a public database. Integrating 1356 bulk RNA sequencing samples from a public database and our previous study, we found a total of 57 significant ligand–receptor pairs (24 upregulated and 33 downregulated) in the autocrine crosstalk network of B cells. Via assessment of the communication between B cells and myeloid cells, another 29 ligand–receptor pairs were discovered, some of which notably affected survival outcomes. A score-based model was constructed with least absolute shrinkage and selection operator (LASSO) using these ligand–receptor pairs. Patients with higher scores had poorer prognoses. This model can be applied to create predictions for both pediatric and adult BCP-ALL patients.
Introduction
B cell precursor acute lymphoblastic leukemia (BCP-ALL) is a hematological malignant neoplasm caused by the abnormal proliferation and accumulation of B-lymphoblastic progenitor cells in the bone marrow (1). Although the 5-year survival rate of pediatric BCP-ALL has surpassed 90% in some developed countries, it remains a main factor in cancer-related death in children and has high morbidity (2, 3). Chemotherapy and targeted therapy are effective treatments for the majority of incipient BCP-ALL patients. However, about 15–20% of such patients will relapse within 5 years, become drug resistant, and eventually die (4). This is in part due to the high heterogeneity of BCP-ALL and to extensive remodeling of the immune microenvironment (5).
Bulk RNA sequencing (RNA-seq) is widely used to analyze the transcriptomic landscape of BCP-ALL. It can reflect the average expression level of various cell types in bone marrow or peripheral blood as a whole. However, our knowledge of the microenvironment of leukemia cells is limited to only bulk RNA-seq data. As single-cell RNA sequencing (scRNA-seq) technology in cancer research becomes increasingly promoted and applied, it has come to provide insights into the analysis of the complexity of cellular composition as well as the heterogeneity of the tumor microenvironment (TME) (6, 7). The use of scRNA-seq can help us gain a deep understanding of the pathogenesis of BCP-ALL (5).
TME plays a crucial role in tumorigenesis and tumor progression, drug tolerance, and immune infiltration (8). The process of tumor development is inhibited by immune cells, and conversely, tumor cells secrete immunoregulatory factors and constantly reshape the microenvironment, leading to a change in the microenvironment in favor of tumor growth and invasions (9–12).
The communication among various cells in TME is mainly mediated through ligand–receptor interactions either in soluble or membrane bound form (13). Checkpoint inhibitors that operate based on the ligand–receptor interaction have become powerful tools for clinical therapy (14). In recent years, several studies have been conducted on the cell–cell crosstalk of TME based on scRNA-seq. For example, Kumar et al. characterized cell–cell communication across all cell types in the microenvironment of mouse tumor models, including melanoma, breast cancer, and lung cancer, and found that the expression of individual ligand–receptor pairs was closely linked to tumor growth rate (15). By analyzing single-cell data in glioma, Shi et al. found that cellular interactions between glioma stem cells and tumor-associated macrophages could affect the prognosis of glioma patients (16). These works provide the references and analytical workflow for cell–cell communications.
However, current research on cell–cell communication focuses on solid tumors. Our understanding of intercellular interactions in leukemia, such as BCP-ALL, remains limited. Previous research has found the extensive remodeling of the TME in BCP-ALL, and a non-classic mononuclear subpopulation is enriched within the myeloid compartment. This subpopulation has prognostic implications for BCP-ALL (5). How myeloid cells affect tumorigenesis and the communication between myeloid and neoplastic B cells in the BCP-ALL TME has not been fully explored. To investigate cell–cell communication in BCP-ALL in depth, we analyzed scRNA-seq data of seven BCP-ALL samples and four healthy samples. Among the seven BCP-ALL samples, five of them are ETV6-RUNX1 fusion. They belong to low-risk subtype and occurs mostly in children. Two of them are BCR-ABL1 fusion (also called Ph+), which belong to high-risk subtype (17, 18). Totally 57 ligand–receptor pairs were found in the autocrine crosstalk network of tumor-related B cells, and 29 were detected in the paracrine crosstalk network between B cells and myeloid cells. A robust least absolute shrinkage and selection operator (LASSO) regression model was constructed using ligand–receptor pairs to predict prognoses for both pediatric and adult BCP-ALL patients.
Materials and Methods
Datasets
The scRNA-seq data related to BCP-ALL in recent five years was searched from Gene Expression Omnibus (GEO, https://www.ncbi.nlm.nih.gov/geo/) and only the dataset GSE134759 was found. Bulk RNA-seq and clinical data of BCP-ALL used for survival analysis and prognostic model construction was downloaded from the Therapeutically Applicable Research to Generate Effective Treatments (TARGET, https://ocg.cancer.gov/programs/target). The TARGET ALL P2 cohort with 532 samples was obtained by R package TGCAbiolinks (v2.16.3). And 133 primary diagnosis BCP-ALL samples whose definition was primary blood derived cancer (bone marrow) were used in the downstream analysis. Another bulk RNA-seq and the clinical dataset was collected from five significant patient cohorts (19–26), including 1,223 BCP-ALL cases available from our previous study (17). This dataset was used for Spearman’s correlation calculation and prognostic model validation. The 36 tumor cohorts of The Cancer Genome Atlas (TCGA) used for validating the model were downloaded via R package TGCAbiolinks (v2.16.3). Ligand–receptor pairs were collected from several public databases (13, 27).
scRNA-seq Data Analysis
All steps for scRNA-seq data processing and cell–cell communication analysis as well as for the machine learning model development described below were performed with R (v4.0.1). For the seven BCP-ALL and four healthy samples, cells for which less than 500 genes or over 10% genes derived from the mitochondrial genome were first filtered out. To remove doublets, cells with more than 5,000 genes were also filtered. All of the 11 samples were preprocessed and normalized using SCTransform, with default parameters implemented in Seurat (v3.5.1) package individually (28, 29). Seurat anchor-based integration method was used to correct the batch and merge multiple samples (30). Cell-type annotation was performed by R package cellassign (v0.99.21) in conjunction with manual comparison of the expression of marker genes among different clusters (31). The pheatmap (v1.0.12) was used to plot heatmap for cell-type annotation using 5,000 randomly selected cells. This was only done to plot the heatmap. The inferCNV (v1.4.0) was used to calculate the copy number variation (CNV) levels of tumor samples.
Cell–Cell Communication Analysis
The differential expression of genes between the BCP-ALL samples and healthy samples separately for B cells and myeloid cells was compared using MAST (v1.14.0) (32). Significant genes with adjusted P-value < 0.05 were mapped to ligand–receptor pair databases. To further investigate the correlations in the ligand–receptor pairs, Spearman’s correlation coefficient was calculated to check the co-expression level of individual pairs. Any pair with an adjusted P-value < 0.05 and coefficient > 0.3 was considered to be significant. Gene set enrichment analysis (GSEA) was performed using fgsea (v1.14.0). Pathway enrichment analysis was performed using clusterProfiler (v3.16.1) (33).
Survival Analysis
Kaplan-Meier and log-rank tests were performed using the survival (v3.2-3) and survminer (v0.4.8) packages to construct and compare survival curves for the LASSO prediction model or specific genes. For specific genes, the patients were divided into high- or low-expression groups according to the mean expression of this gene, and P-value < 0.05 was considered to denote significance.
Machine Learning Model Development
The LASSO regression model implemented in the glmnet (v4.0-2) package was fitted to predict the patient prognosis based on ligand–receptor pairs between B cells and myeloid cells. LASSO regression penalizes the data-fitting standard by eliminating predictive variables with less information to generate simpler and more interpretable models. To evaluate the variability and reproducibility of the estimates produced by the LASSO Cox regression model, we repeated the regression fitting process for each of the 1,000 leave-10%-out cross-validation evaluations. Genes with non-zero coefficient estimates were retained across all 1,000 evaluations. For each of these genes, the final model coefficient was taken as the average of the coefficient estimates obtained for the set of cross-validation evaluations. The recursive partitioning survival model available in the rpart (v4.1-15) package was used to dichotomize patients into low- and high-score groups. Multivariable Cox-proportional hazard model was used to check the independent prognostic effect. The risk group was defined by our previous study (17). In pediatric BCP-ALL, patients with TCF3-PBX1, ETV6-RUNX1/-like, DUX4 fusions, ZNF384/ZNF362 fusions, and high hyperdiploidy (51–65/67 chromosomes) were defined as low-risk. Patients with hyperdiploidy (≤50 chromosomes), PAX5 and CRLF2 fusions were defined as intermediate-risk. While patients with MEF2D fusions, BCR-ABL1/Ph-like, and KMT2A fusions were defines as high-risk. And in adult BCP-ALL, patients with DUX4 fusions, ZNF384/ZNF362 fusions, and hyperdiploidy were defined as intermediate-risk, and patients with MEF2D fusions, TCF3-PBX1, BCR-ABL1/Ph-like, and KMT2A fusions were defined as high-risk (17).
Results
Cellular Heterogeneity Within the Immune Microenvironment of BCP-ALL
To delineate the cellular diversity of the BCP-ALL microenvironment, we analyzed the scRNA-seq data for seven newly diagnosed BCP-ALL samples (five with ETV6-RUNX1 and two with BCR-ABL1, Ph+) and four healthy samples. After initial quality control was conducted (see methods), and all samples were merged using anchor-based integration, 58,518 cells (Figure 1A) were enrolled for downstream analyses (38,860 from BCP-ALL, 19,658 from healthy samples). Little difference was seen in the cell distribution of tumor and normal samples (Figure 1B). This may be due to the special sample preparation method for BCP-ALL, where 20% CD19+ B cells was mixed with 80% CD19-CD45+ non-B cells (5). The profiles separated by subtype of BCP-ALL were also very similar (Figure S1A). Cell-type annotation was performed using cellassign (31), and then the top genes upregulated in each cluster were examined and visualized (Figure S1B).All 58,518 cells were assigned to six distinct cell types: B cells (25.2%), erythrocytic cells (0.7%), hematopoietic stem and progenitor cells (HSPC 3.1%), myeloid cells (11.1%), natural killer (NK) cells (6.3%), and T cells (53.5%, Figures 1C, D). All 11 samples contained each of the six cell types (Figure 1E). After assessing the differences of non-tumor cell subsets between BCP-ALL and healthy samples, only the proportion of myeloid cells was significantly different (Figure S1C), which could imply a special role for myeloid in the bone marrow of BCP-ALL. According to the expression level of MME (an important cell surface marker in the diagnosis of human ALL), the vast majority of B cells present in neoplastic samples were leukemic cells of a pre-B phenotype (Figures 1F, S1B). The malignity of these B cells was also confirmed by inferred CNV level. Among the different cell types in the seven BCP-ALL samples, B cells had the highest CNV level (Figure S1D). A comparison of the CNV level of B cells between BCP-ALL and healthy samples found a significant difference (Figure 1G).
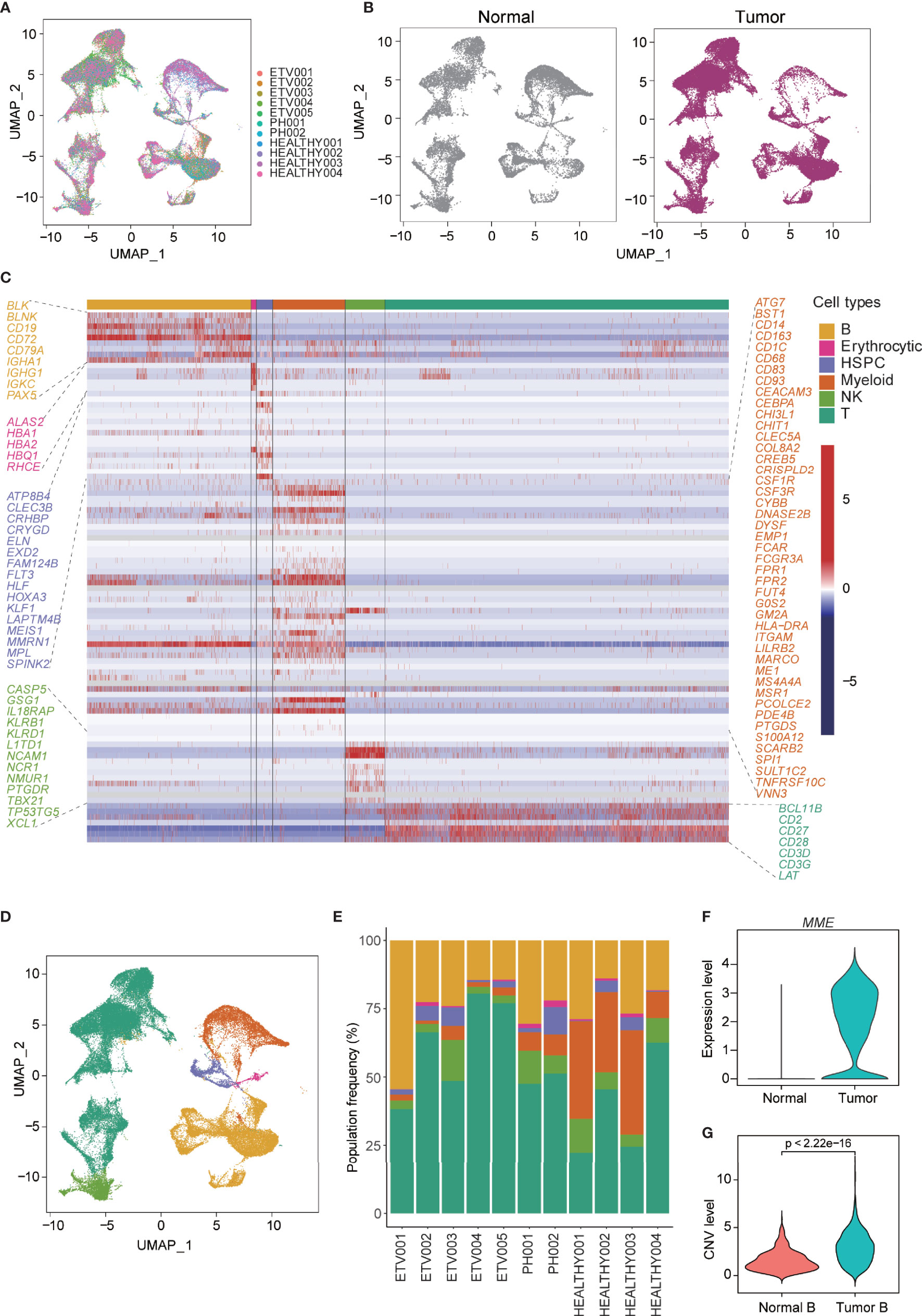
Figure 1 Single-cell profiling and cell-type identification in both healthy and BCP-ALL samples. (A) Distribution of 58,518 cells from 11 samples shown by uniform manifold approximation and projection (UMAP). (B) UMAP plot showing similar cell distributions in normal and tumor samples. (C) Gene expression heatmap of marker genes for the identification of six cell types. (D) UMAP visualization of six marker-based cell types. Cell types are colored as in (C). (E) Stacked barplots showing the frequencies of six cell types in all of the 11 samples. Cell types are colored as in (C). (F) Expression level of MME of B cells from normal and tumor samples. (G) Inferred CNV level of B cells from normal and tumor samples.
Specific Ligand–Receptor Pairs Reveal an Autocrine Crosstalk Network in BCP-ALL
The cell–cell communication level can be reflected in the expression of ligands and their special receptors. For this reason, first, we detected the intracellular communication network of B cells. Only those ligand–receptor pairs in B cells of BCP-ALL samples that had significantly high or low expression passed the filtration. We supposed that these pairs were more closely associated with leukemogenesis. As shown in Figure S2A, we performed differential expression testing between tumor B cells and non-tumor B cells. Then, these genes were mapped to public ligand–receptor databases (see Materials and Methods) (13, 27). And 152 upregulated and 206 downregulated genes were identified. Finally, the expression correlation between the individual ligands and their corresponding receptors was examined using bulk RNA-seq data obtained from our previous study (17). Only the 296 samples with ETV6-RUNX1 and BCR-ABL1 subtypes were used. After these strict criteria were applied, 24 upregulated and 33 downregulated ligand–receptor pairs were detected in total (see Materials and Methods, Figures 2A, B, S3A, B, Tables S1, S2).
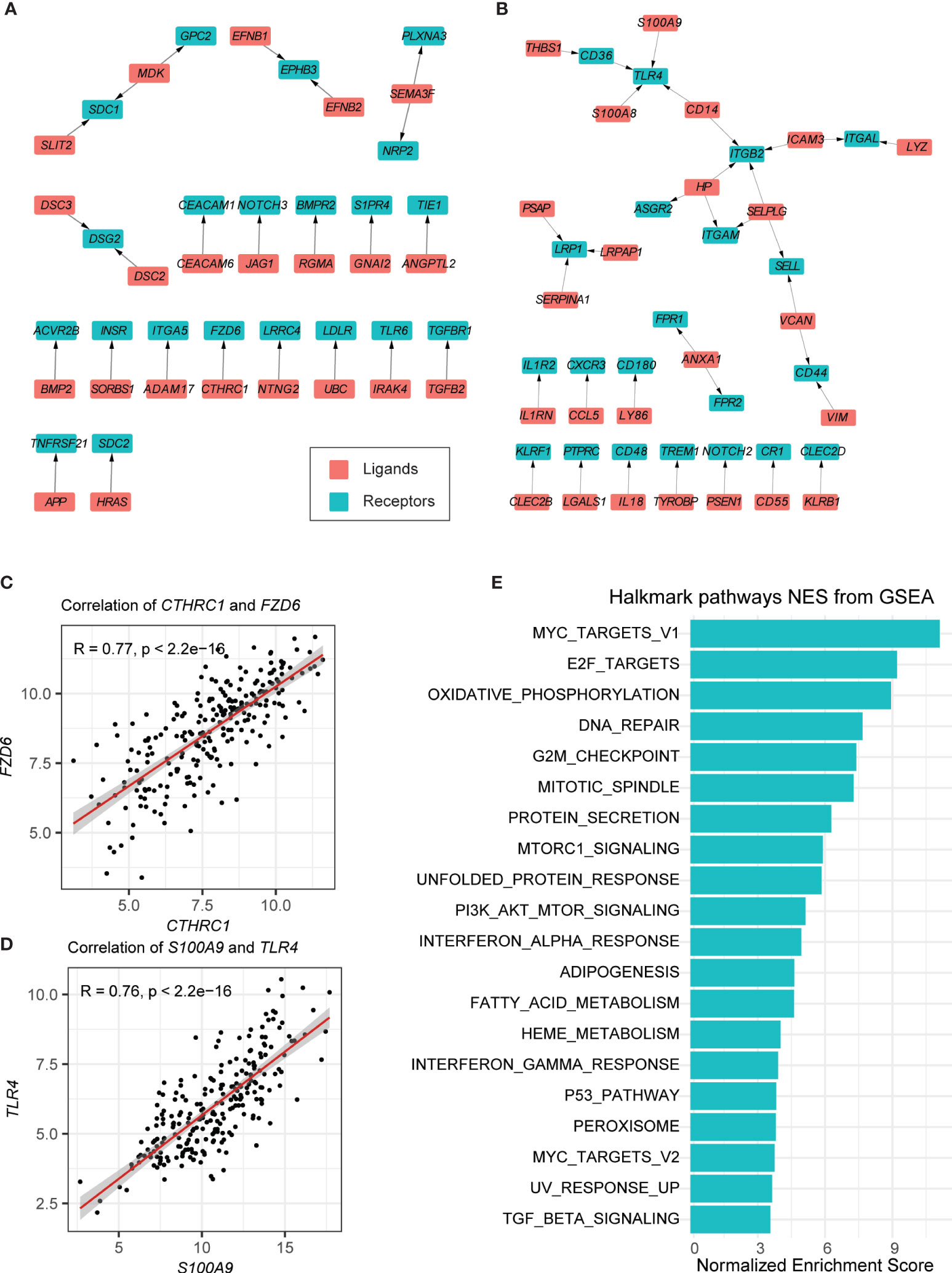
Figure 2 Autocrine ligand–receptor pairs network in tumor-related B cells. (A, B) Ligand–receptor pairs that were upregulated (A) and downregulated (B) in B cells. Red and green squares represent ligands and receptors, respectively, and arrows point from ligands to receptors. (C, D) Spearman correlation coefficients of two ligand–receptor pairs (CTHRC-FZD6 and S100A9-TLR4). (E) GSEA of the hallmark pathways in tumor-related B cells.
In the upregulated pairs, the B-cell leukemogenesis gene FZD6 and its ligand CTHRC1 were upregulated in several solid tumors, associated with increased cell migration and tumor invasion (34, 35). The analytical results showed that FZD6 and CTHRC1 were both highly expressed in the B cells of tumor samples (Figure 2C). It should be noted that APP is highly expressed in acute myeloid leukemia (AML), which may promote cancer cell proliferation and metastasis (36). In our results, we found that APP and its binding partner TNFRSF21 were also highly expressed in tumor-related B-cells (Figure S2B). MDK (a cytokine and growth factor with complex biological functions involved in cancer development and progression) (37), together with its two receptors (SDC1 and GPC2) were highly expressed in the B-cells of BCP-ALL samples (Figures S2C, D).
Among the downregulated pairs, the receptor genes TLR4, ITGB2, and LRP1, located in the center of the ligand–receptor network, with three to four ligands connected respectively (Figure 2B). This may imply that they play an important role in anti-tumorigenesis. Previous studies on these genes suggested that TLR4 is required for protective immune response and to kill cancer cells (Figure 2D) (38). ITGB2 has been found to participate in cell adhesion and cell-surface mediated signaling (39). Lower expression of LRP1 is associated with the aggressive phenotypes and inferior clinical outcomes in some cancers (40, 41). It should be noted that ITGAM also has low expression in the B cells of tumor samples (Figure S2E). This has been reported as negative regulator of immune suppression and a target for cancer immune therapy (42).
We also conducted GSEA on B cells in BCP-ALL and healthy samples (Figure 2E). The enriched pathways in the HALLMARK database of neoplastic B cells were correlated with cell cycle progressions, such as the E2F targets and the G2M checkpoint, suggesting that most B cells in neoplastic samples are immature B cell progenitors. Other canonical tumor-related pathways, such as the MYC targets and the p53 pathway, were also enriched in neoplastic B cells (Figure 2E).
Cell–Cell Communication From B Cells to Myeloid Cells
Previous studies have reported that myeloid cells might play a central role in the immune microenvironment of BCP-ALL (5, 43). Investigation of the crosstalk of B cells with myeloid cells is important for understanding the BCP-ALL TME. Thus, we performed differential expression testing between the myeloid cells of tumor samples and healthy samples. Ligands that were highly expressed in B cells and the receptors that were highly expressed in myeloid cells were selected. After calculating the Spearman’s correlation coefficient, 11 ligand–receptor pairs were identified (Figures 3A, S5A, B, Table S3). Interestingly, we found that some of these 11 ligand–receptor pairs were the same as those found in the autocrine crosstalk of B cells, such as UBC-LDLR and MDK-GPC2 (Figures S4A, B). This partly indicates the consistency in the process of leukemogenesis within the bone marrow environment. Of note, patients with higher expression level for UBC tend to have worse clinical outcomes (Figure 3B). MDK has similar survival trends (Figure 3C). The other ligand–receptor pairs that were specifically present in the crosstalk of B cells to myeloid cells, also have a crucial influence on tumorigenesis. For example, ABCA1 is an auspicious therapy target in prostate cancer (Figure S4C) (44). A previous study has shown that high expression of ADRB2 is significantly linked to early treatment failure in ALL (45) (Figure S4D). In the survival analyses of these specially expressed ligand–receptor pairs, patients with higher expression of LIN7C or NRTN are prone to poor prognosis (Figures 3D, E). Gene Ontology (GO) analysis indicated that these 11 ligand–receptor pairs are mainly associated with the biological processes of cell migration and cell development (Figure 3F).
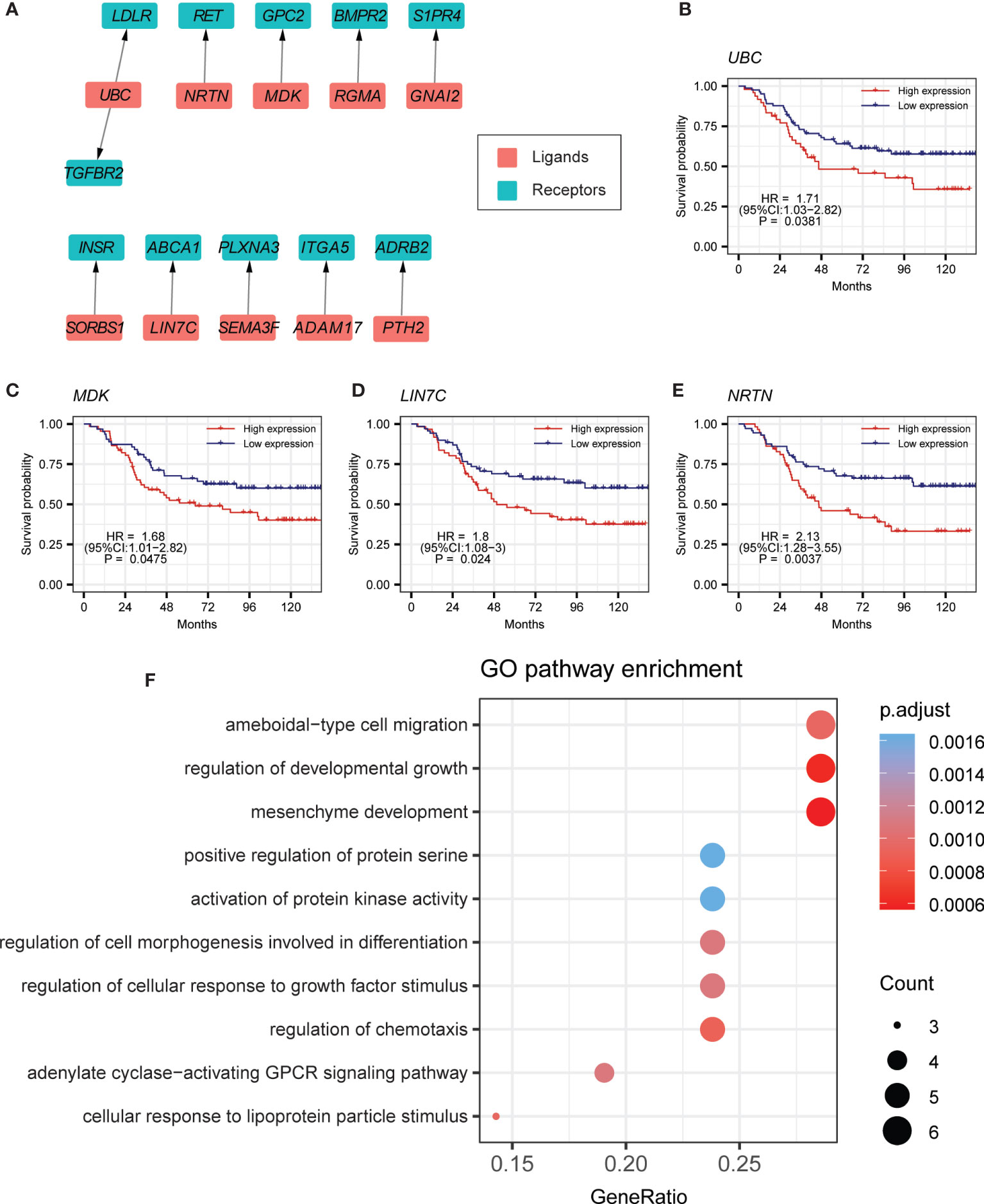
Figure 3 Cell–cell-communication from B cells to myeloid cells. (A) Ligand–receptor pairs of the signaling network from B cells to myeloid cells. Red and green squares represent ligands and receptors, respectively, and arrows point from ligands to receptors. (B–E) Kaplan-Meier survival for UBC, MDK, LIN7C, and NRTN in curated TARGET BCP-ALL P2 cohort. (F) GO pathway enrichment analysis for ligand–receptor pairs in the crosstalk from B cells to myeloid cells.
Cell–Cell Communication From Myeloid Cells to B Cells
We also further identified cell–cell communication from myeloid cells to B cells, built on the expression of differentially expressed ligand–receptor pairs. Ligands and receptors that were separately highly expressed in myeloid and B cells were tested. In all, 18 ligand–receptor pairs passed the strict criteria (Figures 4A, S6A, B, Table S4), and about half of them match autocrine pairs of tumor-related B cells. This suggested that many interactions could be simultaneously activated by malignant or normal cells in the process of leukemogenesis. Intriguingly, the ligand B2M had three receptors, indicating its important role in crosstalk from myeloid cells to B cells (Figures S4E–G). And patients with higher expression level for B2M tended to have worse OS (Figure 4B). We also found that LAMB1 and its receptor ITGB4 were overexpressed in myeloid cells and B cells, respectively (Figure S4H). Patients with higher expression of LAMB1 have a superior prognosis (Figure 4C). ITGB4 is also a significant prognostic indicator tested by the TARGET cohort (Figure 4D). Besides, patients with higher expression of HRAS and VEGFB have worse prognoses (Figures 4E, F, S4I, J). Both of them are closely related to tumorigenesis and progression. GO analysis indicated that these ligand–receptor pairs in the crosstalk from myeloid cells to B cells were mainly related to leukocyte migration, cell proliferation, and cell activation (Figure 4G).

Figure 4 Cell–cell-communication from myeloid cells to B cells. (A) Ligand–receptor pairs of the signaling network from myeloid cells to B cells. Red and green squares represent ligands and receptors, respectively, and arrows point from ligands to receptors. (B–F) Kaplan-Meier survival for B2M, LAMB1, ITGB4, HRAS, and VEGFB in the curated TARGET BCP-ALL P2 cohort. (G) GO pathway enrichment analysis for ligand–receptor pairs in the crosstalk from myeloid cells to B cells.
LASSO Model Based on Ligand–Receptor Pairs Precisely Predicted BCP-ALL Patient Prognosis
The results of cell–cell communication in BCP-ALL revealed that significantly expressed ligand–receptor pairs might play a key role in leukemogenesis and progression. A machine learning model was built to predict the prognosis for BCP-ALL patients based on these pairs identified above. The principal component analysis was performed, with the expression level of ligand–receptor pairs in 14 different BCP-ALL subtypes which were classified in our previous study (17). The result showed little difference in the expression level of these ligand–receptor pairs across all the 14 BCP-ALL subgroups (Figure S7A).
To develop the prognostic model, a curated TARGET cohort with 133 BCP-ALL samples was used as training cohort and samples from our previous BCP-ALL cohort were used as validation cohort (see methods). The overall process is shown in Figure 5A (46, 47). First, we fitted a LASSO regression model using the expression levels of ligand–receptor pairs. After performing 1,000 leave-10%-out cross-validation replications, the coefficients of 18 genes were found to be non-zero in at least one of these 1,000 evaluations (Table S5). And the coefficients of 11 genes were presented in at least 950 of 1,000 analyses (Figure S7B). Then we calculated an LR (ligand–receptor) score for each patient using the expression of these 15 genes, weighted by the regression coefficients, as defined in the LASSO model. The equation is LR score = (ITGB4 × −0.263) + (SDC1 × 0.177) + (GPC2 × −0.13) + (TLR6 × −0.0838) + (CEACAM1 × −0.0607) + (JAG1 × 0.058) + (NOTCH3 × 0.0501) + (LDLR × −0.0469) + (ACVR2B × −0.0511) + (SLIT2 × −0.0191) + (TIE1 × −0.00592). We further used a recursive partitioning Cox regression model to dichotomize patients. After pruning the regression tree, patients in the curated TARGET cohorts with different LR scores were divided into a low-LR score group (n = 65, 50%), and a high-LR score group (n= 65, 50%). The overall survival (OS) of these two groups is remarkably different. Higher LR scores were predictive of inferior OS in TARGET cohort (HR = 8.27, 95% CI = 4.27–16.04, p < 0.0001) (Figure 5B).
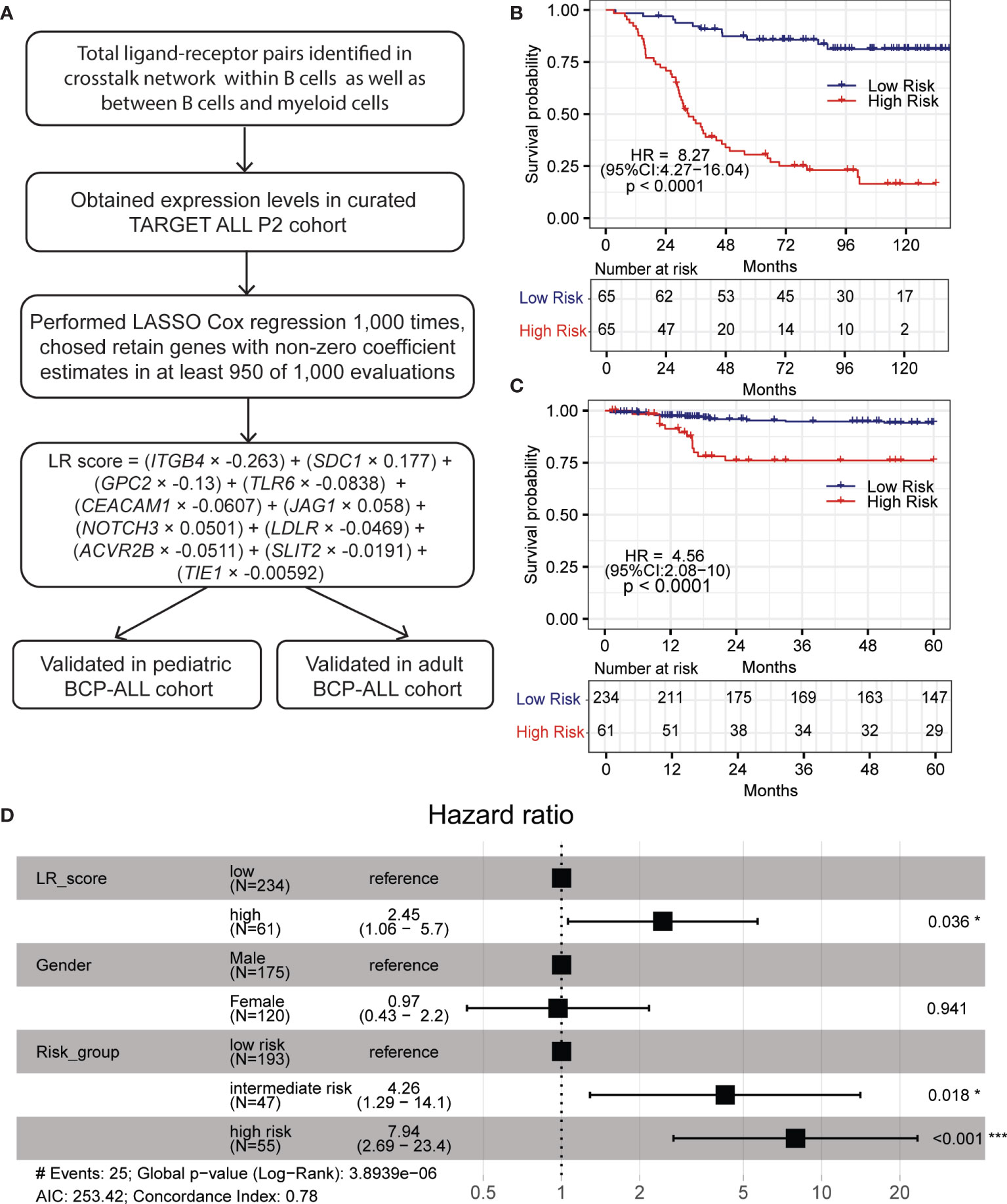
Figure 5 LR score based on LASSO regression model predicts inferior OS in pediatric BCP-ALL patients. (A) Overall scheme for constructing LASSO prognostic model. (B, C) High LR scores predict poor OS in the TARGET and pediatric validation cohort, respectively. (D) Forest plot of multivariable Cox-proportional hazard model showing LR score as an independent prognostic factor for OS in the pediatric validation cohort. Within forest plot, * indicates P-value < 0.05, *** indicates P-value < 0.001.
To further confirm the robustness of the LASSO model, an independent cohort with 295 pediatric and 85 adult BCP-ALL patients was used as a validation cohort (19, 24, 25, 48). The LR score was computed with the equation defined above. A similar result was observed in pediatric patients. Based on the recursive portioning cutoff, the high-LR score group (n=61, 21%) demonstrated worse OS than the low-LR score group (n = 234, 79%), and the range of HR was 4.56 (95% CI = 2.08–10, p < 0.0001, Figure 5C). Although these ligand–receptor pairs were identified using scRNA-seq data of pediatric BCP-ALL patients, the prognostic power of the LR score in the 97 adult BCP-ALL patients (17) was also significant (HR = 2.99, 95% CI = 1.26–7.14, p = 0.009, Figure S7C). Multivariate analysis was performed with the Cox-proportional hazard model to check the individual risk factor. In the pediatric validation cohort, after adjusting for gender and risk group, the LR score remained an independent predictor of worse OS (HR = 2.45, 95% CI = 1.06–5.7, p = 0.036 Figure 5D). The same was true for the adult validation cohort (HR = 2.8, 95% CI = 1.18–6.8, p = 0.019, Figure S7D). All of these results demonstrate that the robust machine learning model built with ligand–receptor pairs has promise for identifying high-risk BCP-ALL patients and may have a role as a primary consideration for developing different treatment strategies.
Discussion
Cellular composition and cell–cell communication are two important aspects of TME. In hematological malignant neoplasms, such as BCP-ALL, a deep understanding of cell–cell interactions in the bone marrow can help us to investigate the leukemogenesis and progression and support the development of new drugs and therapies. In the current omics studies of BCP-ALL, which mainly focus on bulk RNA-seq, the promotion of scRNA-seq reveals the landscape of TME at cellular level resolution and make it possible to investigate the cell–cell communications. In this work, we analyzed a large scale of scRNA-seq profile in seven BCP-ALL pediatric samples and four healthy samples. By classifying and identifying each cell cluster (Figures 1D, E), we found B cells from both BCP-ALL and healthy samples were mixed. This may indicate that the biological characteristics of proliferating tumor B cells were presented in a way that was similar to normal B cells. However, compared to both B cells from healthy samples and other cell types from BCP-ALL samples, tumor B cells had higher CNV level (Figure 1G), revealing that the accumulation of genetic abnormalities was mainly focused on B cells during leukemic progression.
T cells appeared in the largest proportion in the TME of BCP-ALL. Although previous studies of solid tumors have explored the interaction between T cells and tumor cells (49, 50), other work revealed that myeloid cells also play an important role in the TME of BCP-ALL (5). However, our understanding of the interactions involved in myeloid cells in TME remains limited. In this study, we focused on the two ways to explore the cell–cell communication: the autocrine way for B cells of tumor samples, and the paracrine way between myeloid cells and malignant B cells. Interestingly, this revealed that a considerable number of ligand–receptor pairs were closely associated with tumorigenesis and progression. For example, the CTHRC1-FZD6 pair and the APP-TNFRSF21 pair may significantly promote tumorigenesis and the proliferation of cancer cell (34–36). And the LAMB1-ITGB4 pair has been hypothesized to be involved in tumor invasion and EMT (51). LAMB1 has also been shown to be a potential biomarker for some cancers, such as colorectal cancer and multiple myeloma (52, 53). Pairs of UBC-LDLR and MDK-GPC2 are widely overexpressed in various cell types of the BCP-ALL bone marrow microenvironment, participating in many processes of tumor development (37, 54). The ligand gene B2M takes center stage in the crosstalk from myeloid cells to B cells. It has been demonstrated in several studies that the elevated expression level of B2M is historically associated with poor outcome in several lymphoproliferative disorders, such as AML, myelodysplastic syndrome, and ALL (55). Similar results were found in our study (Figure 4B). Several genes in ligand–receptor pairs showed significant correlations with the clinical outcomes of pediatric BCP-ALL patients. To better predict prognosis, a machine learning model based on LASSO regression was built based on the determined ligand–receptor pairs. In the pediatric validation cohort, the prognosis for the high-LR score group was significantly worse than for the low-LR score group. Although these ligand–receptor pairs were assessed with pediatric BCP-ALL samples, our prognostic model achieved good performance in the adult validation cohort. This suggests that the prognostic model could help support the clinical decisions for both adult and pediatric BCP-ALL patients. And to further test the predictive efficiency of LR score, we applied our model in 36 tumor cohorts of TCGA. The results showed that LR score had good predictive power in a considerable number of tumors, such as acute myeloid leukemia (AML), skin cutaneous melanoma (SKCM) and uveal melanoma (UVM), of which AML was the most significant. It may indicate that LR score has strong predictive potential for prognosis in hematological malignant neoplasms (Figure S8).
In conclusion, via integrated analyses of scRNA-seq and bulk RNA-seq data for BCP-ALL, we presented a comprehensive landscape of the autocrine crosstalk network of neoplastic B cells and the paracrine communication network between B cells and myeloid cells. Based on the significant ligand–receptor pairs, a LASSO regression model was built to predict the prognoses for both pediatric and adult patients. These identifications shed light on BCP-ALL pathogenesis and have the potential to improve the clinical diagnosis for BCP-ALL patients.
Data Availability Statement
Publicly available datasets were analyzed in this study. This data can be found here: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE134759.
Author Contributions
JH conceived, designed, and supervised the study with WZ, LW, and YD. LW collected and analyzed data, wrote the draft of the manuscript. MJ, PY, JL, WO, WZ, and CF analyzed the data and reviewed the manuscript. JH and YD oversaw the bioinformatics data analyses and modified and improved the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the National Natural Science Foundation of China (No. 82070147, 81570122, 81770205), the National Key Research and Development Program (No. SQ2019YFE010340), and the Shanghai Municipal Education Commission-Gaofeng Clinical Medicine Grant Support (20161303).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2021.639013/full#supplementary-material
References
1. Franquiz MJ, Short NJ. Blinatumomab for the treatment of adult B-cell acute lymphoblastic leukemia: Toward a new era of targeted immunotherapy. Biol Targets Ther (2020) 14:23–34. doi: 10.2147/BTT.S202746
2. Conter V, Valsecchi MG, Parasole R, Putti MC, Locatelli F, Barisone E, et al. Childhood high-risk acute lymphoblastic leukemia in first remission: Results after chemotherapy or transplant from the AIEOP ALL 2000 study. Blood (2014) 123:1470–8. doi: 10.1182/blood-2013-10-532598
3. Hunger SP, Mullighan CG. Acute lymphoblastic leukemia in children. N Engl J Med (2015) 373:1541–52. doi: 10.1056/NEJMra1400972
4. Locatelli F, Schrappe M, Bernardo ME, Rutella S. How i treat relapsed childhood acute lymphoblastic leukemia. Blood (2012) 120:2807–16. doi: 10.1182/blood-2012-02-265884
5. Witkowski MT, Dolgalev I, Evensen NA, Ma C, Chambers T, Roberts KG, et al. Extensive Remodeling of the Immune Microenvironment in B Cell Acute Lymphoblastic Leukemia. Cancer Cell (2020) 37:867–82.e12. doi: 10.1016/j.ccell.2020.04.015
6. Kolodziejczyk AA, Kim JK, Svensson V, Marioni JC, Teichmann SA. The Technology and Biology of Single-Cell RNA Sequencing. Mol Cell (2015) 58:610–20. doi: 10.1016/j.molcel.2015.04.005
7. Luecken MD, Theis FJ. Current best practices in single-cell RNA-seq analysis: a tutorial. Mol Syst Biol (2019) 15:e8746. doi: 10.15252/msb.20188746
8. Wu T, Dai Y. Tumor microenvironment and therapeutic response. Cancer Lett (2017) 387:61–8. doi: 10.1016/j.canlet.2016.01.043
9. Smyth MJ, Dunn GP, Schreiber RD. Cancer Immunosurveillance and Immunoediting: The Roles of Immunity in Suppressing Tumor Development and Shaping Tumor Immunogenicity. Adv Immunol (2006) 90:1–50. doi: 10.1016/S0065-2776(06)90001-7
10. Swann JB, Smyth MJ. Immune surveillance of tumors. J Clin Invest (2007) 117:1137–46. doi: 10.1172/JCI31405
11. Mittal D, Gubin MM, Schreiber RD, Smyth MJ. New insights into cancer immunoediting and its three component phases-elimination, equilibrium and escape. Curr Opin Immunol (2014) 27:16–25. doi: 10.1016/j.coi.2014.01.004
12. Austin R, Smyth MJ, Lane SW. Harnessing the immune system in acute myeloid leukaemia. Crit Rev Oncol Hematol (2016) 103:62–77. doi: 10.1016/j.critrevonc.2016.04.020
13. Ramilowski JA, Goldberg T, Harshbarger J, Kloppman E, Lizio M, Satagopam VP, et al. A draft network of ligand-receptor-mediated multicellular signalling in human. Nat Commun (2015) 6:1–11. doi: 10.1038/ncomms8866
14. Dempke WCM, Fenchel K, Uciechowski P, Dale SP. Second- and third-generation drugs for immuno-oncology treatment—The more the better? Eur J Cancer (2017) 74:55–72. doi: 10.1016/j.ejca.2017.01.001
15. Kumar MP, Du J, Lagoudas G, Jiao Y, Sawyer A, Drummond DC, et al. Analysis of Single-Cell RNA-Seq Identifies Cell-Cell Communication Associated with Tumor Characteristics. Cell Rep (2018) 25:1458–68.e4. doi: 10.1016/j.celrep.2018.10.047
16. Yuan D, Tao Y, Chen G, Shi T. Systematic expression analysis of ligand-receptor pairs reveals important cell-to-cell interactions inside glioma. Cell Commun Signal (2019) 17:1–10. doi: 10.1186/s12964-019-0363-1
17. Li JF, Dai YT, Lilljebjörn H, Shen SH, Cui BW, Bai L, et al. Transcriptional landscape of B cell precursor acute lymphoblastic leukemia based on an international study of 1,223 cases. Proc Natl Acad Sci USA (2018) 115:E11711–20. doi: 10.1073/pnas.1814397115
18. Gu Z, Churchman ML, Roberts KG, Moore I, Zhou X, Nakitandwe J, et al. PAX5-driven subtypes of B-progenitor acute lymphoblastic leukemia. Nat Genet (2019) 51:296–307. doi: 10.1038/s41588-018-0315-5
19. Roberts KG, Morin RD, Zhang J, Hirst M, Zhao Y, Su X, et al. Genetic Alterations Activating Kinase and Cytokine Receptor Signaling in High-Risk Acute Lymphoblastic Leukemia. Cancer Cell (2012) 22:153–66. doi: 10.1016/j.ccr.2012.06.005
20. Lilljebjörn H, Henningsson R, Hyrenius-Wittsten A, Olsson L, Orsmark-Pietras C, Von Palffy S, et al. Identification of ETV6-RUNX1-like and DUX4-rearranged subtypes in paediatric B-cell precursor acute lymphoblastic leukaemia. Nat Commun (2016) 7:11790. doi: 10.1038/ncomms11790
21. Qian M, Zhang H, Kham SKY, Liu S, Jiang C, Zhao X, et al. Whole-transcriptome sequencing identifies a distinct subtype of acute lymphoblastic leukemia with predominant genomic abnormalities of EP300 and CREBBP. Genome Res (2017) 27:185–95. doi: 10.1101/gr.209163.116
22. Yasuda T, Tsuzuki S, Kawazu M, Hayakawa F, Kojima S, Ueno T, et al. Recurrent DUX4 fusions in B cell acute lymphoblastic leukemia of adolescents and young adults. Nat Genet (2016) 48:569–74. doi: 10.1038/ng.3535
23. Liu YF, Wang BY, Zhang WN, Huang JY, Li BS, Zhang M, et al. Genomic Profiling of Adult and Pediatric B-cell Acute Lymphoblastic Leukemia. EBioMedicine (2016) 8:173–83. doi: 10.1016/j.ebiom.2016.04.038
24. Gu Z, Churchman M, Roberts K, Li Y, Liu Y, Harvey RC, et al. Genomic analyses identify recurrent MEF2D fusions in acute lymphoblastic leukaemia. Nat Commun (2016) 7:1–10. doi: 10.1038/ncomms13331
25. Roberts KG, Li Y, Payne-Turner D, Harvey RC, Yang Y-L, Pei D, et al. Targetable Kinase-Activating Lesions in Ph-like Acute Lymphoblastic Leukemia. N Engl J Med (2014) 371:1005–15. doi: 10.1056/nejmoa1403088
26. Churchman ML, Low J, Qu C, Paietta EM, Kasper LH, Chang Y, et al. Efficacy of Retinoids in IKZF1-Mutated BCR-ABL1 Acute Lymphoblastic Leukemia. Cancer Cell (2015) 28:343–56. doi: 10.1016/j.ccell.2015.07.016
27. Cabello-Aguilar S, Alame M, Kon-Sun-Tack F, Fau C, Lacroix M, Colinge J. SingleCellSignalR: inference of intercellular networks from single-cell transcriptomics. Nucleic Acids Res (2020) 48:e55. doi: 10.1093/nar/gkaa183
28. Macosko EZ, Basu A, Satija R, Nemesh J, Shekhar K, Goldman M, et al. Highly parallel genome-wide expression profiling of individual cells using nanoliter droplets. Cell (2015) 161:1202–14. doi: 10.1016/j.cell.2015.05.002
29. Hafemeister C, Satija R. Normalization and variance stabilization of single-cell RNA-seq data using regularized negative binomial regression. Genome Biol (2019) 20:1–15. doi: 10.1186/s13059-019-1874-1
30. Stuart T, Butler A, Hoffman P, Hafemeister C, Papalexi E, Mauck WM, et al. Comprehensive Integration of Single-Cell Data. Cell (2019) 177:1888–902.e21. doi: 10.1016/j.cell.2019.05.031
31. Zhang AW, O’Flanagan C, Chavez EA, Lim JLP, Ceglia N, McPherson A, et al. Probabilistic cell-type assignment of single-cell RNA-seq for tumor microenvironment profiling. Nat Methods (2019) 16:1007–15. doi: 10.1038/s41592-019-0529-1
32. Finak G, McDavid A, Yajima M, Deng J, Gersuk V, Shalek AK, et al. MAST: A flexible statistical framework for assessing transcriptional changes and characterizing heterogeneity in single-cell RNA sequencing data. Genome Biol (2015) 16:1–13. doi: 10.1186/s13059-015-0844-5
33. Yu G, Wang LG, Han Y, He QY. ClusterProfiler: An R package for comparing biological themes among gene clusters. Omi A J Integr Biol (2012) 16:284–7. doi: 10.1089/omi.2011.0118
34. Wu QL, Zierold C, Ranheim EA. Dysregulation of frizzled 6 is a critical component of B-cell leukemogenesis in a mouse model of chronic lymphocytic leukemia. Blood (2009) 113:3031–9. doi: 10.1182/blood-2008-06-163303
35. Mei D, Zhu Y, Zhang L, Wei W. The Role of CTHRC1 in Regulation of Multiple Signaling and Tumor Progression and Metastasis. Mediators Inflammation (2020) 2020:9578701. doi: 10.1155/2020/9578701
36. Yu G, Yin C, Jiang L, Xu D, Zheng Z, Wang Z, et al. Amyloid precursor protein has clinical and prognostic significance in AML1-ETO-positive acute myeloid leukemia. Oncol Lett (2018) 15:917–25. doi: 10.3892/ol.2017.7396
37. Filippou PS, Karagiannis GS, Constantinidou A. Midkine (MDK) growth factor: a key player in cancer progression and a promising therapeutic target. Oncogene (2020) 39:2040–54. doi: 10.1038/s41388-019-1124-8
38. Awasthi S. Toll-like receptor-4 modulation for cancer immunotherapy. Front Immunol (2014) 5:328. doi: 10.3389/fimmu.2014.00328
39. Moore SW, Sidler D, Zaahl MG. The ITGB2 immunomodulatory gene (CD18), enterocolitis, and Hirschsprung’s disease. J Pediatr Surg (2008) 43:1439–44. doi: 10.1016/j.jpedsurg.2007.12.057
40. Meng H, Chen G, Zhang X, Wang Z, Thomas DG, Giordano TJ, et al. Stromal LRP1 in lung adenocarcinoma predicts clinical outcome. Clin Cancer Res (2011) 17:2426–33. doi: 10.1158/1078-0432.CCR-10-2385
41. Huang XY, Shi GM, Devbhandari RP, Ke AW, Wang Y, Wang XY, et al. Low level of Low-density lipoprotein receptor-related protein 1 predicts an unfavorable prognosis of hepatocellular carcinoma after curative resection. PloS One (2012) 7:e32775. doi: 10.1371/journal.pone.0032775
42. Schmid MC, Khan SQ, Kaneda MM, Pathria P, Shepard R, Louis TL, et al. Integrin CD11b activation drives anti-tumor innate immunity. Nat Commun (2018) 9:1–14. doi: 10.1038/s41467-018-07387-4
43. Yang J, Zhang L, Yu C, Yang XF, Wang H. Monocyte and macrophage differentiation: Circulation inflammatory monocyte as biomarker for inflammatory diseases. Biomark Res (2014) 2:1. doi: 10.1186/2050-7771-2-1
44. Xiong T, Xu G, Huang X, Lu K, Xie W, Yin K, et al. ATP−binding cassette transporter A1: A promising therapy target for prostate cancer (Review). Mol Clin Oncol (2017) 8:9. doi: 10.3892/mco.2017.1506
45. Pottier N, Paugh SW, Ding C, Pei D, Yang W, Das S, et al. Promoter polymorphisms in the β-2 adrenergic receptor are associated with drug-induced gene expression changes and response in acute lymphoblastic leukemia. Clin Pharmacol Ther (2010) 88:854–61. doi: 10.1038/clpt.2010.212
46. Ng SWK, Mitchell A, Kennedy JA, Chen WC, McLeod J, Ibrahimova N, et al. A 17-gene stemness score for rapid determination of risk in acute leukaemia. Nature (2016) 540:433–7. doi: 10.1038/nature20598
47. Elsayed AH, Rafiee R, Cao X, Raimondi S, Downing JR, Ribeiro R, et al. A six-gene leukemic stem cell score identifies high risk pediatric acute myeloid leukemia. Leukemia (2020) 34:735–45. doi: 10.1038/s41375-019-0604-8
48. Pui CH, Yang JJ, Hunger SP, Pieters R, Schrappe M, Biondi A, et al. Childhood acute lymphoblastic leukemia: Progress through collaboration. J Clin Oncol (2015) 33:2938–48. doi: 10.1200/JCO.2014.59.1636
49. Zhou JX, Taramelli R, Pedrini E, Knijnenburg T, Huang S. Extracting Intercellular Signaling Network of Cancer Tissues using Ligand-Receptor Expression Patterns from Whole-tumor and Single-cell Transcriptomes. Sci Rep (2017) 7:1–15. doi: 10.1038/s41598-017-09307-w
50. Chen Z, Yang X, Bi G, Liang J, Hu Z, Zhao M, et al. Ligand-receptor interaction atlas within and between tumor cells and t cells in lung adenocarcinoma. Int J Biol Sci (2020) 16:2205–19. doi: 10.7150/ijbs.42080
51. Li XL, Liu L, Li DD, He YP, Guo LH, Sun LP, et al. Integrin β4 promotes cell invasion and epithelial-mesenchymal transition through the modulation of Slug expression in hepatocellular carcinoma. Sci Rep (2017) 7:40464. doi: 10.1038/srep40464
52. Lin Q, Lim HSR, Lin HL, Tan HT, Lim TK, Cheong WK, et al. Analysis of colorectal cancer glyco-secretome identifies laminin β-1 (LAMB1) as a potential serological biomarker for colorectal cancer. Proteomics (2015) 15:3905–20. doi: 10.1002/pmic.201500236
53. Peng Y, Wu D, Li F, Zhang P, Feng Y, He A. Identification of key biomarkers associated with cell adhesion in multiple myeloma by integrated bioinformatics analysis. Cancer Cell Int (2020) 20:1–16. doi: 10.1186/s12935-020-01355-z
54. Sahasrabuddhe AA, Elenitoba-Johnson KSJ. Role of the ubiquitin proteasome system in hematologic malignancies. Immunol Rev (2015) 263:224–39. doi: 10.1111/imr.12236
Keywords: BCP-ALL, scRNA-seq, ligand–receptor pairs, machine learning, prognosis
Citation: Wu L, Jiang M, Yu P, Li J, Ouyang W, Feng C, Zhao WL, Dai Y and Huang J (2021) Single-Cell Transcriptome Analysis Identifies Ligand–Receptor Pairs Associated With BCP-ALL Prognosis. Front. Oncol. 11:639013. doi: 10.3389/fonc.2021.639013
Received: 08 December 2020; Accepted: 25 January 2021;
Published: 10 March 2021.
Edited by:
Martina Seiffert, German Cancer Research Center (DKFZ), GermanyReviewed by:
Laura Llaó Cid, German Cancer Research Center (DKFZ), GermanyDeepshi Thakral, All India Institute of Medical Sciences, India
Copyright © 2021 Wu, Jiang, Yu, Li, Ouyang, Feng, Zhao, Dai and Huang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Wei-Li Zhao, emhhby53ZWlsaUB5YWhvby5jb20=; Yuting Dai, Zm9ybHlubmFAc2p0dS5lZHUuY24=; Jinyan Huang, aHVhbmdqeUBzanR1LmVkdS5jbg==
†These authors have contributed equally to this work