
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Oncol. , 09 March 2021
Sec. Gastrointestinal Cancers
Volume 11 - 2021 | https://doi.org/10.3389/fonc.2021.638171
 Yu-an Qiu1,2
Yu-an Qiu1,2 Jianping Xiong1Qin Fu3Yun Dong3Manran Liu4Meixi Peng4
Jianping Xiong1Qin Fu3Yun Dong3Manran Liu4Meixi Peng4 Wenjian Jin5Lixia Zhou6Xue Xu7Xianming Huang8Airong Fu8Guohui Xu9Gang Tu10Tenghua Yu3*
Wenjian Jin5Lixia Zhou6Xue Xu7Xianming Huang8Airong Fu8Guohui Xu9Gang Tu10Tenghua Yu3*Hepatocellular carcinoma (HCC) is an aggressive malignancy with a poor prognosis. Effective biomarkers and specific therapeutic targets for HCC are therefore urgently needed. G protein-coupled estrogen receptor (GPER) plays a crucial role in numerous cancer types; however, its functions in HCC require further exploration. In the present study, we found a remarkable difference in GPER staining between tumor tissue (100/141, 70.9%) and matched non-tumor tissue (27/30, 90.0%). Compared with the GPER-negative patients, the GPER-positive patients with HCC were closely associated with female sex, negative hepatitis B surface antigen, small tumor size, low serum alpha fetoprotein level, and longer overall survival. Treatment with GPER-specific agonist G1 led to the sustained and transient activation of the EGFR/ERK and EGFR/AKT signaling pathways, respectively, in the HCC cell lines HCCLM3 and SMMC-7721, which express high levels of GPER. Interestingly, G1-induced EGFR/ERK signaling, rather than EGFR/AKT signaling mediated by GPER, was involved in decreasing cell viability by blocking cell cycle progression, thereby promoting apoptosis and inhibiting cell growth. Clinical analysis indicated that simultaneous high expression of GPER and phosphorylated-ERK (p-ERK) predicted improved prognosis for HCC. Finally, the activation of GPER/ERK signaling remarkably suppressed tumor growth in an HCC xenograft model, and this result was consistent with the in vitro data. Our findings suggest that specific activation of the GPER/ERK axis may serve as a novel tumor-suppressive mechanism and that this axis could be a therapeutic target for HCC.
Hepatocellular carcinoma (HCC) represents approximately 90% of primary liver cancers and is the third leading cause of cancer-related deaths in China (approximately 422, 100 deaths in 2015) (1, 2). HCC is generally caused by hepatitis B virus infection, hepatitis C virus infection, and alcohol use (3). Surgery, local destructive therapies, and liver transplantation are the current therapeutic strategies for patients with early HCC (4). However, the recurrence rate in treated patients reaches an incidence of >70% at five years (5). Although some treatment processes, including genomic- and immune-based therapies, have been performed in clinical practice, HCC remains one of the deadliest cancers (6). The molecular mechanisms involved in HCC and its potential therapeutic targets must be explored to improve treatment efficacy.
The HCC rate in men is usually 2–4 times higher than that in women, suggesting that sex hormones may play vital roles in HCC pathogenesis (7, 8). Clinical data shows that higher HCC morbidity and mortality occur in male patients, and that estrogen replacement treatment reduces the risk and increases the survival time of female patients with HCC (9, 10).
Estrogen-stimulated actions are mediated by classical nuclear estrogen receptors (ERα and ERβ) (11). GPER, formerly known as G protein-coupled receptor 30, is a non-classical estrogen receptor that rapidly mediates the acute response of effector cells to estrogen and can be specifically activated by G1 (a specific agonist for GPER) (12–14). The activation of GPER can induce multiple downstream effectors, including adenylyl cyclase, epidermal growth factor receptor (EGFR), and phosphoinositide 3-kinase (PI3K), as well as Ca2+ mobilization (15–17). Many studies have demonstrated that GPER mediates biological effects in various malignant tumors, including reproductive organs (such as endometrial (18) and ovarian cancers (19)) and other hormonally responsive organs (such as lung (20) and prostate cancers (21)). The biological functions of GPER are inconsistent between different cells and organs. Our previous studies indicated that the activation of GPER enhances the viability of and confers multidrug resistance to breast cancer cells (22–24). Conversely, GPER activation inhibits cell proliferation and increases apoptosis in gastric and colorectal cancers (25, 26). Recent evidence has demonstrated that GPER reprograms the tumor microenvironment, mediates antiviral effects, and suppresses inflammation and fibrosis in HCC (27–29). However, the biological effects and mechanism of action of GPER on HCC cells have not yet been fully explored.
Therefore, the current study aimed to investigate the role of GPER and its potential molecular mechanisms in HCC. We elucidated that the interaction of specific agonist G1-triggered GPER activation and its downstream EGFR/ERK signaling plays a key role in decreasing the tumor viability of HCC. We compared GPER expression in HCC tissue with that in matched non-tumor counterparts and analyzed clinical HCC survival data. We treated two high-GPER-expressing HCC cell lines (i.e., HCCLM3 and SMMC-7721) and a low-GPER-expressing HepG2 cell line with the GPER-specific agonist G1, and measured GPER/EGFR signals and their downstream pathways. We confirmed our findings through immunohistochemical staining of GPER and p-ERK in HCC specimens and in vivo xenograft tumors. Our results provide novel insights into GPER-mediated protection against HCC and suggest that targeting the activation of GPER may represent a new therapeutic option for HCC.
HepG2, MHCC97-H, and HCCLM3 cells were maintained in Dulbecco’s modified Eagle’s medium (Gibco, MD, USA) supplemented with 10% fetal bovine serum (FBS) (Gibco). SMMC-7721 and HEP3B cells were maintained in RPMI 1640 (Gibco) and Eagle’s minimum essential medium (Gibco), respectively, supplemented with 10% FBS. Before the experiments, all cells were transferred to serum-free medium for 24 h.
GPER-specific agonist G1 and GPER-specific antagonist G15 were acquired from Tocris (Ellisville, MO, USA). EGFR inhibitor AG1478 (AG), MEK inhibitor U0126, and PI3K inhibitor wortmannin (WM) were obtained from Millipore (Temecula, CA, USA). The drugs were solubilized in dimethyl sulfoxide (Sigma–Aldrich). The following antibodies were purchased from Bioworld (St. Louis Park, MN, USA) and used for western blotting: ER (1:800), PR (1:500), EGFR (1:1,000), p-ERK (1:1,000), ERK (1:1,000), p-AKT (1:1,000), AKT (1:1,000), p-P38 (1:1,000), P38 (1:1,000), p-JNK (1:1,000), JNK (1:1,000), and p-PKA (1:1,000). GPER (1:250) was obtained from Abcam (Cambridge, MA, USA), and β-actin (1:1,000) was purchased from Zhongshan Golden Bridge (Beijing, China).
A total of 141 archival paraffin-embedded HCC specimens and 30 paired normal liver tissue samples were obtained from the Department of Pathology, Jiangxi Cancer Hospital (Nanchang, China). All patients who underwent surgery at the Jiangxi Cancer Hospital between 1999 and 2008 were diagnosed at the same center. The clinical information and overall survival (OS) data of 141 patients with HCC who underwent surgery were collected from the Department of Hepatobiliary Surgery of Jiangxi Cancer Hospital. The study was approved by the Ethics Committee of Jiangxi Cancer Hospital and complied with the Helsinki Declaration.
The cells were treated as follows: (1) cells treated with G1 (1 µM), or (2) before G1 treatment, cells were treated with G15 (1 µM, 30 min), AG (10 µM, 30 min), U0126 (10 µM, 30 min), and WM (10 µM, 30 min). Whole cell extracts were prepared in RIPA buffer with protease inhibitors (Beyotime, Shanghai, China). For the preparation of the membrane and cytosolic fractions, the treated cells were suspended in membrane and cytosolic protein extract buffer (Beyotime) according to the manufacturer’s protocol. Proteins were electrophoresed on a 10% sodium dodecyl sulphate–polyacrylamide gel electrophoresis (SDS–PAGE) gel and transferred onto PVDF membranes. Each primary antibody was incubated with the membranes overnight at 4°C. Membranes were then incubated with the appropriate horseradish peroxidase-conjugated secondary antibody for 1 h at room temperature. The membranes were washed and visualized using an enhanced chemiluminescence system (Amersham Pharmacia Biotech). The intensity of the immunoblot bands was quantified using Quantity One 4.62 software, and results were expressed as fold change relative to the control.
The GPER-specific siRNA (siGPER), non-specific control siRNA, pcDNA (Vector), and pcDNA/GPER (GPEROE) were purchased from Genechem (Shanghai, China). HCCLM3 and HepG2 cells were transiently transfected with Lipofectamine™ 2000 reagent according to the manufacturer’s instructions. Target sequences were obtained as previously described (22). The expression level of GPER protein after transfection was measured by western blotting.
Immunofluorescence assays were performed using a previously described method (30). Cells were grown on sterile coverslips for 24 h, fixed with 4% paraformaldehyde, treated with 0.1% Triton X-100, and blocked with 5% normal goat serum. After blocking, the cells were incubated with the primary antibody (1:200) against GPER at 4°C overnight, washed with phosphate-buffered saline (PBS), and incubated with FITC-labelled goat anti-rabbit secondary antibody (1:200; Zhongshan Golden Bridge) for 30 min at room temperature. Nuclei were stained with DAPI. Immunofluorescence images were collected using the Nikon Eclipse 80i microscope (Tokyo, Japan; 400× magnification).
Deparaffinized tissue sections (thickness = 4 μm) were heated at 95°C for 15 min in 10 mM citric acid buffer (pH 6.0) for antigen retrieval and treated with 3% H2O2 for 10 min to quench the endogenous peroxidase activity. The sections were incubated at 4°C for 16 h with primary antibodies against GPER and p-ERK at a 1:200 dilution. The sections treated with PBS served as the negative control. Sections were treated with horseradish peroxidase-labelled goat anti-rabbit IgG at 37°C for 30 min, followed by diaminobenzidine (Zhongshan Golden Bridge). Slides were counterstained with Mayer’s modified haematoxylin. The immunohistochemistry results were scored independently by two pathologists who were blinded to patient identity using semiquantitative scoring software (Image-Pro Plus 6.0). GPER and p-ERK expression levels were evaluated as previously described (31, 32).
Flow cytometry (FACSVantage SE; BD, Franklin Lakes, NJ, USA) was used to analyze the cell cycle and apoptosis, as described previously (22, 33). Cells were cultured in FBS-free conditioned medium for 24 h and treated with G1 (1 μM) alone or in combination with G15 (1 μM), AG (10 μM), U0126 (10 μM), and WM (10 μM) pre-treatment for 24 h to detect the DNA content of each phase in the cell cycle. For the cell apoptosis assay, cells were transferred into a conditioned medium without FBS and phenol for 24 h, treated with G1 (1 μM) for 48 h, collected, and stained with Annexin V/PI.
A total of 3 × 103 cells were seeded into a 96-well plate and assessed using the CCK-8 protocol (Dojindo Molecular Technologies, Rockville, MD) after treatment. All experiments were performed in duplicate. The absorbance of each well was measured at 450 nm using an enzyme-linked immunosorbent assay microplate reader.
Total RNA was extracted using TRIzol® (Invitrogen, Carlsbad, CA, USA) and reverse-transcribed using the PrimeScript RT kit (Takara, Dalian, China). Real-time qPCR was performed using SYBR Premix Ex TaqTM II (Takara). The cells were pre-treated to determine the mRNA expression of GPER. Gene expression was calculated using the ΔΔCT method. The primers used for each gene are listed in Supplementary Table 1.
Cells were seeded on 60 mm tissue culture plates at a density of 1 × 106 cells per well for 24 h, switched to a serum-starved medium for 5 h, and treated with G1 as described in the figure legends. Subsequently, the cells were washed twice with PBS, frozen, and thawed three times. The final cAMP levels were measured using the enzyme immunoassay kit (R&D System, Minneapolis, USA) according to the manufacturer’s instructions.
HCCLM3 and SMMC-7721 xenograft cell models were established in athymic nude mice aged 4–6 weeks, which were obtained from the Animal Experimental Center of Chongqing Medical University (Chongqing, China). Experiments were conducted in accordance with the guidelines on animal care and use established by the Chongqing Medical University Experimental Animal Management Committee. The animal study protocols were approved by the ethics committee of Chongqing Medical University.
A total of 5 × 106 cells were implanted into the left armpit. When tumors grew to 150–200 mm3 (5–6 weeks), the mice were randomly assigned to experimental groups (n = 5 per group). G1, G15, AG, and U0126 were dissolved and diluted in absolute ethanol. The compounds (10 µl) were added to an aqueous vehicle (90 µl, 0.9% NaCl with 0.1% albumin and 0.1% Tween-20). Ethanol (10 µl) was added to the aqueous vehicle (90 µl) and used as the control group. Mice were subcutaneously injected daily with 0.1 ml of G1 (1 μM) alone, or G1 (1 μM) in combination with the GPER-specific antagonist G15 (1 μM), AG (10 μM), or U0126 (10 μM). Body weights were monitored daily. At the end of the 56 day experiment, tumors were removed and measured using a Vernier calliper, and tumor volumes were calculated using the following equation: Tumor voulume = 1/2 × length × (width)2. The expression of specific proteins was analysed by immunoblotting to determine the effects of treatment.
A prognostic database was accessed online (http://kmplot.com/analysis//) and used to predict the prognostic value of GPER gene expression in patients with HCC under different parameters (23). Briefly, each percentile of gene expression between the lower and upper quartiles was calculated, and the best-performing threshold was used as the final cut-off in the univariate Cox regression analysis. The clinical data and gene expression data were integrated using the object-relational PostgreSQL database system (http://www.postgresql.org/).
Statistical analysis was performed using SPSS software, version 21.0. Data are expressed as the mean ± SD. A two-tailed Student’s t test was used to compare two groups, whereas one-way ANOVA followed by the Student–Newman–Keuls post hoc test was used to compare multiple groups. The association between GPER and p-ERK expression levels in patients with HCC was evaluated using the χ2 or Fisher’s exact test. OS analysis was performed for patients who underwent surgery until they died of HCC, and the OS was calculated using the Kaplan–Meier survival method. Differences between survival curves were tested using the log-rank test. P values < 0.05 were considered statistically significant.
A total of 141 archival paraffin-embedded HCC specimens and 30 paired non-tumor tissue specimens were eligible for analysis. GPER staining was located in the cytoplasm of both normal liver and tumor tissue (Figure 1A). GPER staining was observed in non-malignant tissue with a total positive rate of 90% (27/30), which was significantly higher than that in HCC (70.9%, 100/141; P = 0.0001; Figure 1B). Similarly, GPER protein levels in cancerous tissue from six patients with HCC was remarkably decreased compared to in paired non-cancerous tissue (Figure 1C).
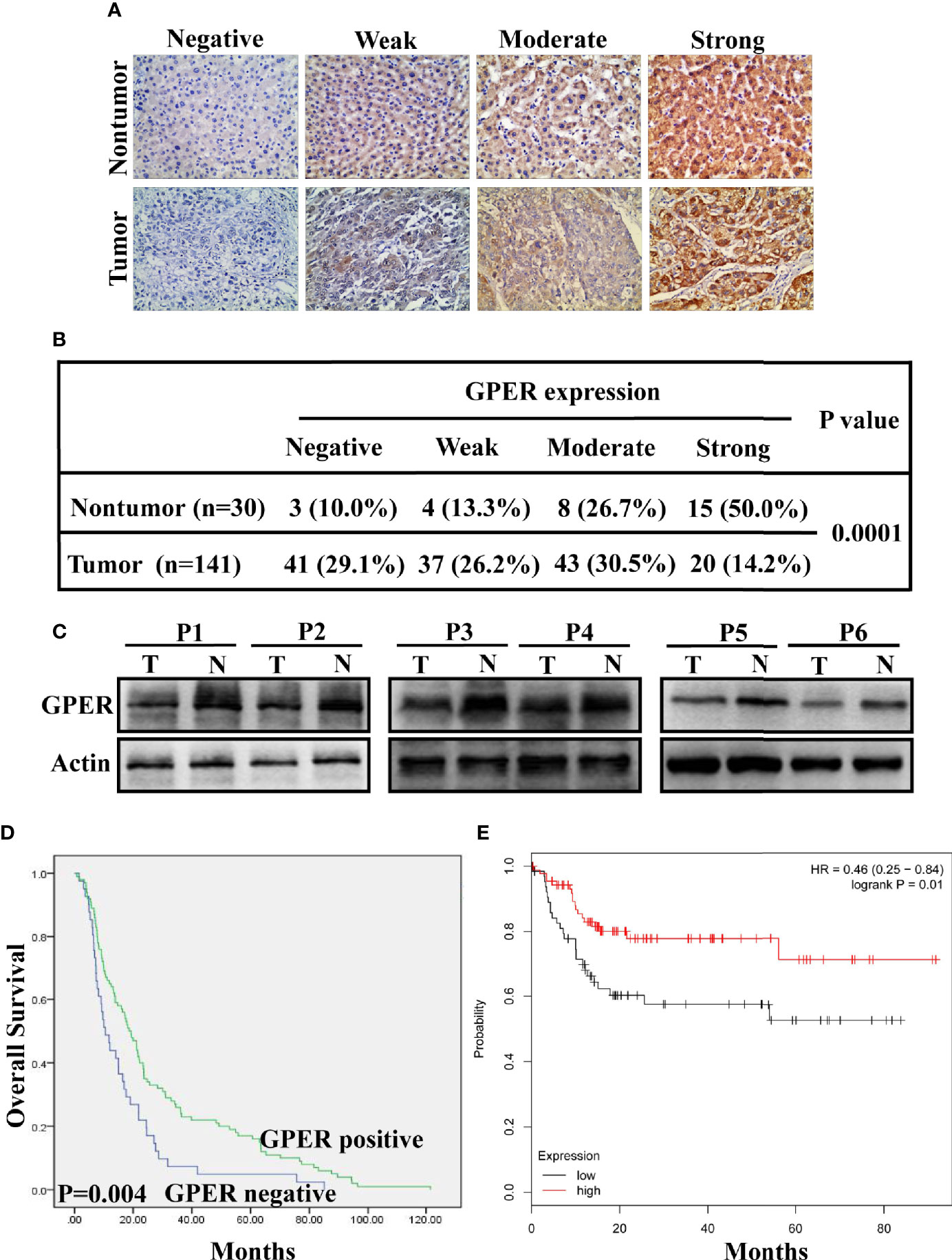
Figure 1 Clinical significance and prognostic value of GPER expression in hepatocellular carcinoma. (A) GPER expression (negative, weak, moderate, and strong positive staining) in representative non-tumor tissue (top) and HCC tissue (bottom) was analysed using immunohistochemistry (400× magnification, scale bar = 50 μm). (B) IHC analysis revealed significantly different GPER staining between HCC and matched non-tumor tissue (P = 0.0001). (C) Western blot of GPER protein in six pairs of frozen HCC tissue (T1–T6) and matched non-cancerous liver tissue (N1–N6). (D) High GPER level predicts a good overall survival (OS) in patients with HCC (P = 0.004). (E) GPER may be an important protective factor for HCC in Asia (P = 0.01). Data were extracted from a bioinformation database (http://kmplot.com/analysis/).
The correlation between GPER expression and clinicopathological characteristics was retrospectively analyzed to understand the role of GPER in patients with HCC. GPER immunostaining in tumor cells was significantly associated with sex (P = 0.043), negative HBsAg (P = 0.036), small tumor size (P = 0.002), and low serum AFP levels (P = 0.014, Table 1). GPER-positive patients showed better OS than GPER-negative patients (P = 0.004, Figure 1D). Similar data were also obtained from a bioinformatic database (http://kmplot.com/analysis/) by predicting the prognostic value of GPER in HCC patients from Asia (P = 0.01, Figure 1E). These data strongly indicate that GPER is downregulated during the cancer process and could serve as a prognostic marker that also plays a protective role in patients with HCC.
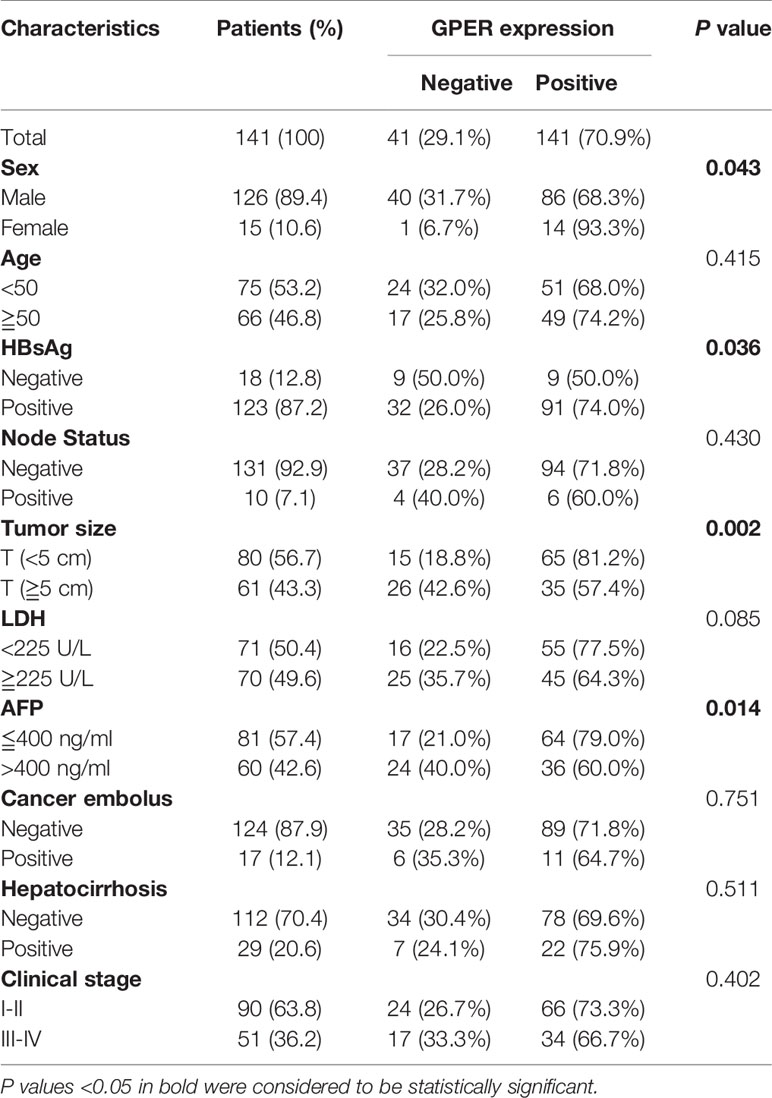
Table 1 Correlation between clinicopathological factors and GPER expression in patients with HCC.
The typical HCC SMMC-7721, HepG2, HEP3B, MHCC97-H, and HCCLM3 cell lines were used to investigate the expression of GPER in multiple HCC cell lines. GPER was expressed at low levels in HepG2, HEP3B, and MHCC97-H cells, but at high levels in HCCLM3 and SMMC-7721 cells (Figures 2A, B). The expression pattern of GPER (principally located in the cell cytoplasm) was also confirmed by immunofluorescence (Figure 2C) and western blotting (Supplementary Figure 1A), which was consistent with the IHC data. We selected high GPER-expressing HCCLM3 and SMMC-7721 cells, and low GPER-expressing HepG2 cells, to further study the role of GPER in HCC in vitro.
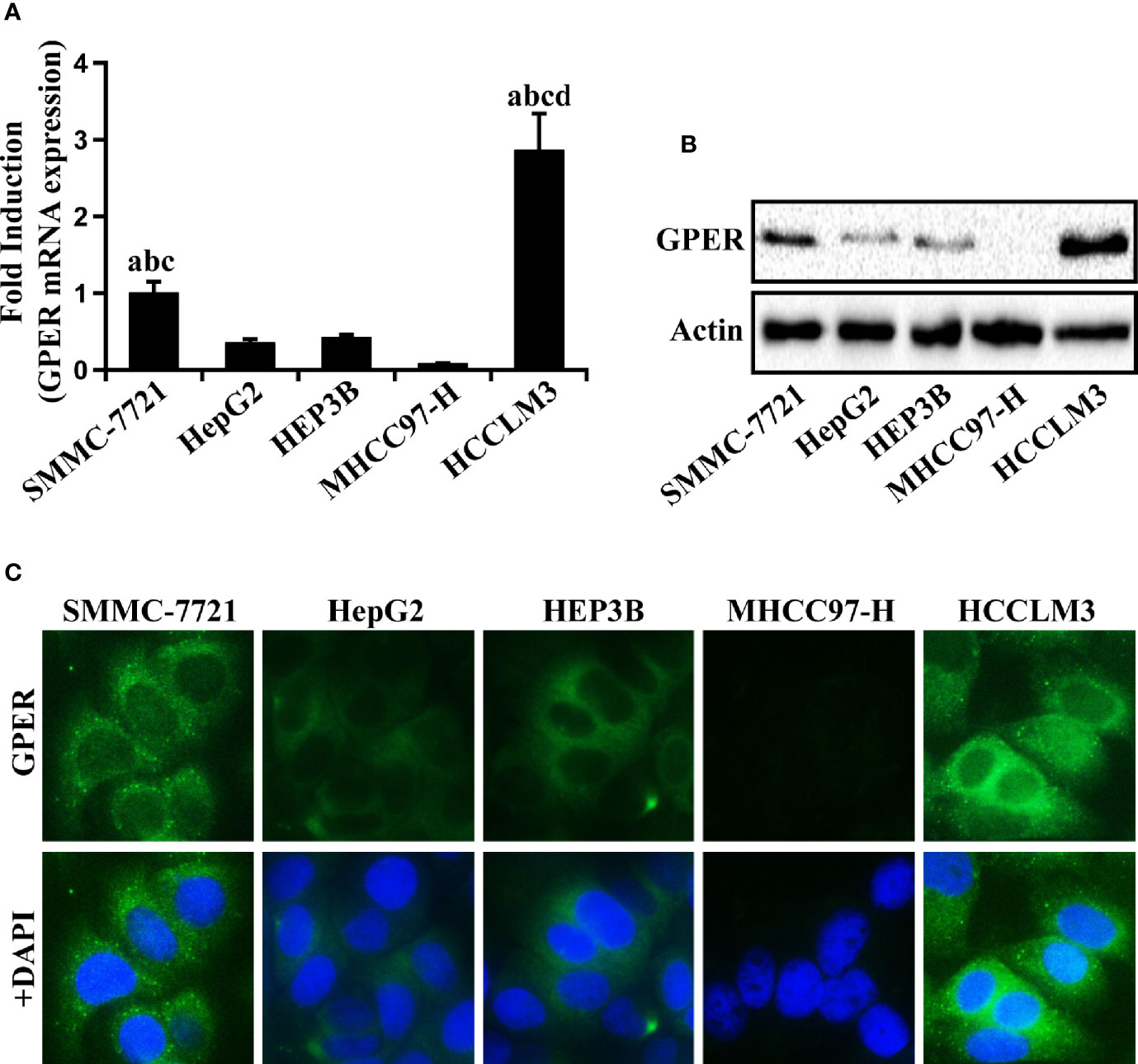
Figure 2 GPER expression in human HCC cells. (A, B) Real-time PCR and western blot of GPER in HCC SMMC-7721, HepG2, HEP3B, MHCC97-H, and HCCLM3 cell lines. The relative fold induction of mRNA in each cell line was calculated against the numerical value of GPER expression in SMMC-7721 cells. Data represents the mean ± SD from triplicate independent experiments (P < 0.05; a vs. HepG2; b vs. HEP3B; c vs. MHCC97-H; d vs. SMMC-7721). (C) The immunofluorescence localization of GPER (green) viewed in the above five HCC cell lines. The nucleus was stained blue with DAPI (scale bar = 100 μm, 400× magnification). Each experiment was repeated at least three times.
G1 can trigger GPER/EGFR signaling in malignant tumor cells (22–24, 34, 35). The effects of different doses of G1 on HCC cell growth at different time points were investigated. Since G1 concentrations >1 μM could not further inhibit the growth of HCC cells (Supplementary Figures 1B, C), 1 μM G1 was chosen for follow-up experiments. We hypothesized that GPER-mediated non-genomic signaling occurs in HCC cells, so the activation of GPER/EGFR signals and their downstream pathways was measured in HCC cells. After a short exposure to G1, significant rapid phosphorylation of EGFR, ERK, and AKT was triggered in HCCLM3 cells (Figure 3A). However, G1 had no effect on the activation of P38, JNK, and PKA signaling (Figure 3A). The intracellular cAMP levels in HCCLM3 cells were also not affected by G1 (Supplementary Figure 2A). Interestingly, the transient activation of AKT signaling was not observed after 3 h of G1 treatment, but a sustained increase in p-ERK expression was detected after 24 h of G1 treatment (Figure 3B). These data suggest that G1 enhances ERK and AKT signaling in HCC cells.
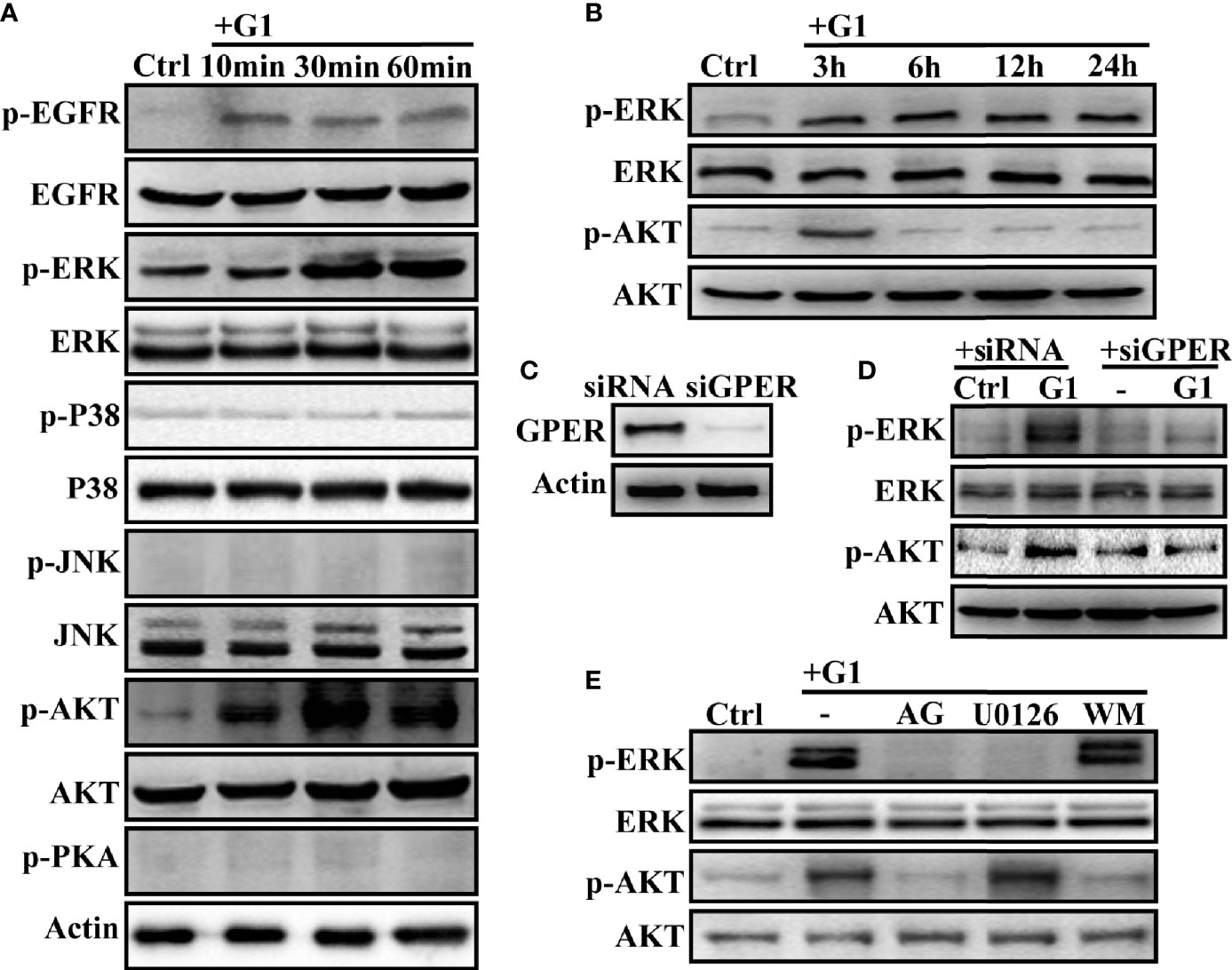
Figure 3 G1 induces the activation of the GPER/EGFR/ERK and GPER/EGFR/AKT signaling pathways in HCC cells (A) G1 induced the phosphorylation of EGFR, ERK, and AKT, but not P38, JNK, and PKA in HCCLM3 cells. The HCCLM3 cells were maintained in medium without FBS and phenol for 24 h and treated with 1 μM G1 for 10, 30, and 60 min. (B) G1-induced phosphorylation of ERK and AKT was detected for extended periods (3, 6, 12, and 24 h) in HCCLM3 cells. (C) GPER expression was knocked down by GPER-specific siRNA transfection of HCCLM3 cells. (D, E) G1-induced p-ERK and p-AKT expression levels were blocked by GPER interference or the specific inhibitors AG (1 μM), U0126 (10 μM), and WM (10 μM) in HCCLM3 cells. Each experiment was repeated at least three times.
Next, the siRNA-specific targeting of GPER (Figure 3C), GPER-specific antagonist G15 (1 μM), EGFR inhibitor AG (10 μM), MEK inhibitor U0126 (10 μM), and PI3K inhibitor WM (10 μM) were used to confirm GPER/EGFR/ERK and GPER/EGFR/AKT signaling in HCCLM3 and SMMC-7721 cells. After silencing the GPER gene with siRNA or pre-treatment with AG and U0126, the phosphorylation of ERK in HCCLM3 cells was blocked (Figures 3D, E). At the same time, p-AKT was significantly blocked by GPER interferents AG and WM (Figures 3D, E). Similar data were obtained for SMMC-7721 cells (Supplementary Figures 2B). However, WM failed to abolish the activation of p-ERK in the two high GPER-expressing cell lines (Figures 3D, E, Supplementary Figure 2B). Meanwhile, overexpression of GPER in HepG2 cells (Supplementary Figure 2C) had similar effects to that in HepG2/GPEROE cells (Supplementary Figures 2D, E). Taken together, our data demonstrated G1-induced GPER/EGFR/ERK and GPER/EGFR/AKT signaling in HCC cells.
The effect of GPER signaling on cell cycle and apoptosis was tested using flow cytometry, and cell growth was evaluated using the CCK-8 assay to further explore the role of GPER and its downstream EGFR/ERK and EGFR/AKT signaling pathways in HCC cells. Compared with control HCC cells, G1-treated cells had significantly decreased S phase ratio and increased apoptosis rate (P < 0.05) (Figures 4A, B and Supplementary Figures 3A, B). This phenomenon was reversed after GPER interference or pre-treatment with G15, AG, and U0126 (Figures 4C–F, Supplementary Figures 3C, 3D). Interestingly, these changes were not observed in WM-treated cells (Figures 4E, F, Supplementary Figures 3C, D). Furthermore, consistent with the results of flow cytometry assays, the inhibitory effect of G1 on cell viability was abolished by the GPER interferents G15, AG, and U0126 (Figures 4G, H, Supplementary Figure 3E). Similar data were also obtained in HepG2/GPEROE cells (Supplementary Figures 3F, G). Our results indicate that GPER-induced EGFR/ERK signaling, but not EGFR/AKT signaling, suppressed the viability of HCC cells.
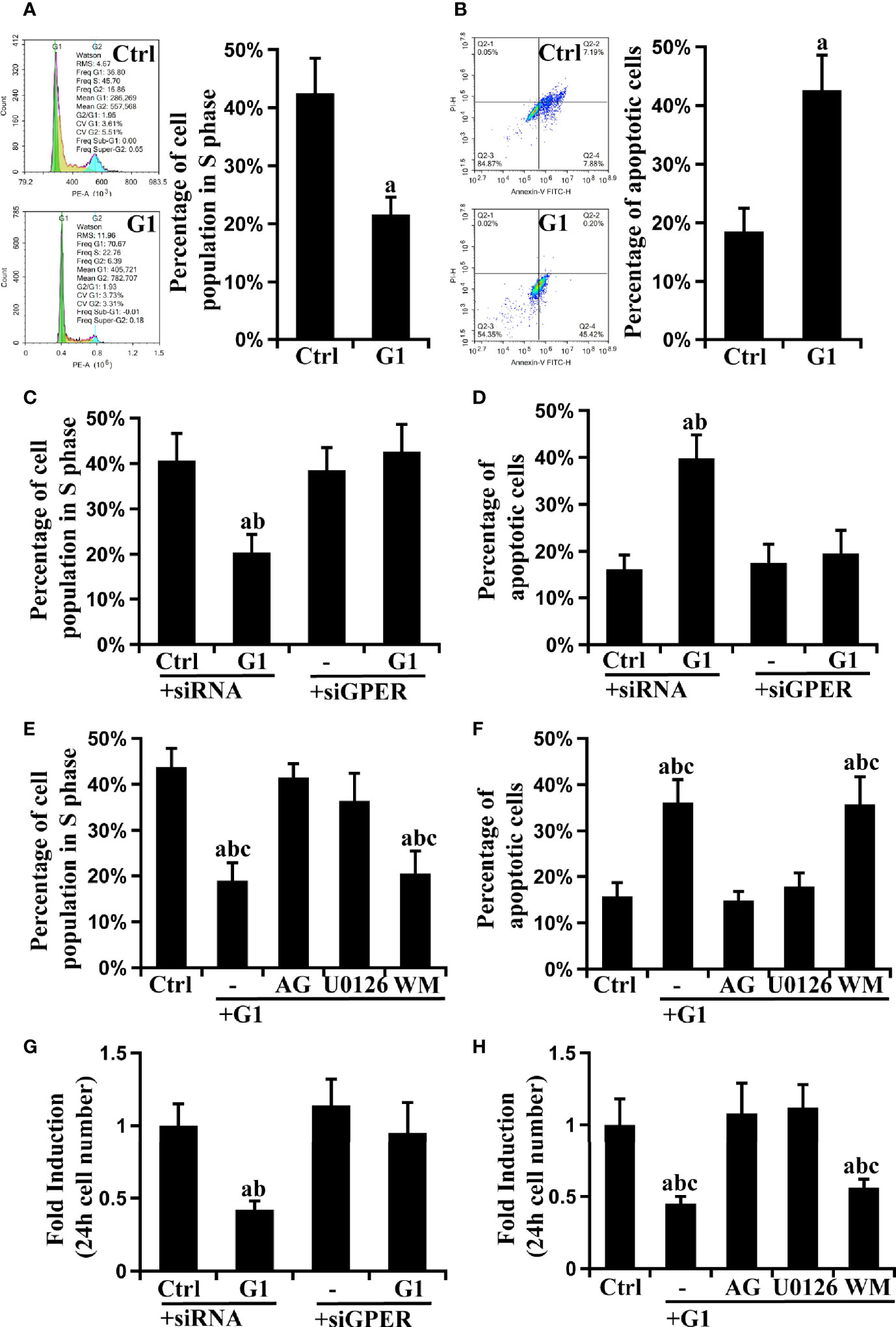
Figure 4 GPER-mediated EGFR/ERK signaling decreases cell viability in HCC cell lines. The GPER-specific agonist G1 blocked progression to the S phase of the cell cycle (A) and promoted apoptosis (B) in HCCLM3 cells (P < 0.05; a vs. ctrl). Synchronized cells were cultured with G1 and stained with propidium iodide or Annexin V-FITC/PI, and the cell cycle distribution and apoptosis were measured using flow cytometry. (C, D) The observed cell cycle and apoptosis blockade by G1 were reversed by GPER interference (P < 0.05; a vs. ctrl + siRNA, b vs. G1 + siGPER). (E, F) The observed cell cycle and apoptosis blockade by G1 were attenuated by specific inhibitors targeting EGFR (AG) or ERK (U0126), but not AKT (WM) (P < 0.05; a vs. ctrl, b vs. G1 + AG, c vs. G1 + U0126). The above data represents the mean ± SD from triplicate independent experiments. (G) The G1-inhibited cell growth was reversed by GPER interference (P < 0.05; a vs. ctrl + siRNA, b vs. G1 + siGPER). (H) The observed inhibition of cell growth by G1 was attenuated by AG and U0126 (P < 0.05; a vs. ctrl, b vs. G1 + AG, c vs. G1 + U0126). The above data represents the mean ± SD from five different experiments.
The IHC expression of p-ERK was examined in 141 HCC specimens to further validate the in vitro data. GPER was detected in the cytoplasm, whereas p-ERK mostly displayed nuclear staining (Figure 5A). GPER and p-ERK were expressed in 70.9% (100/141) and 59.6% (84/141) of tumor tissue, respectively. Furthermore, a significantly different IHC staining of p-ERK was observed in GPER-positive (72/100, 72.0%) and GPER-negative (12/41, 29.3%) HCC tissue (P < 0.0001, Figure 5B), indicating that the GPER/ERK pathway was strongly associated with GPER-positive patients. In addition, p-ERK expression was highly consistent with GPER protein levels in cancerous tissue from six patients with HCC (Figure 5C). Patients with elevated GPER/ERK signaling activation showed the longest OS time compared to those with other subtypes (P < 0.0001, Figure 5D). Thus, our data demonstrated that GPER-mediated ERK signaling might play a protective role in patients with HCC, which is similar to our in vitro observations.
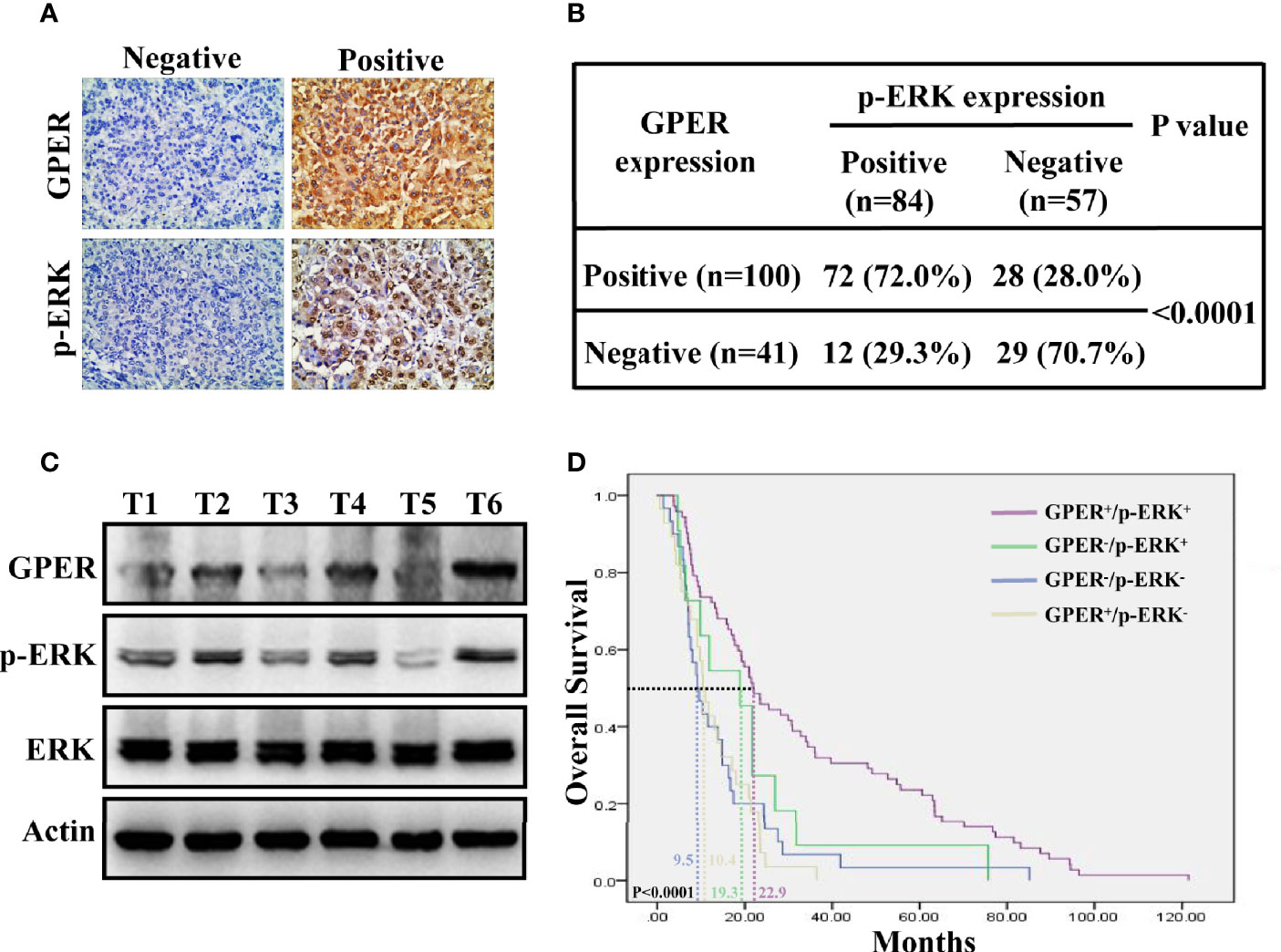
Figure 5 Co-expression of GPER and p-ERK predicts improved prognosis for HCC patients. (A) Representative GPER and p-ERK staining in 141 HCC specimens. GPER was detected in the cytoplasm, while p-ERK was mostly expressed in the nucleus (400× magnification, scale bar = 50 μm). (B) Significantly different IHC staining of p-ERK in GPER-positive tissue compared with that in GPER-negative tissue (P < 0.0001). (C) Western blot of GPER and p-ERK proteins in six frozen HCC tissue samples (T1–T6). (D) Kaplan–Meier plots for OS vs. GPER/p-ERK status. Patients with HCC and high GPER/ERK activation are predicted to have better outcomes than those with other subtypes. The median OS times of GPER-/p-ERK-, GPER+/p-ERK-, GPER-/p-ERK+ and GPER+/p-ERK+ patients were 9.5, 10.4, 19.3 and 22.9 months, respectively (P < 0.0001).
We confirmed that the G1/GPER pathway influenced the viability of HCC cells through EGFR/ERK signaling in vitro. HCC xenografts were then used to determine whether this signaling could affect tumorigenesis in vivo. Tumors were palpable for approximately 42 days in nude mice. Under treatment, the mean volume of G1 groups decreased by 0.20-fold compared with that of the control group over 56 days (Figures 6A, B, P < 0.05). However, the combined pre-treatment with G15, AG, or U0126 remarkably attenuated G1-inhibited growth in HCC xenografts during the intervention (Figures 6A, B). At the end of drug treatment, the combination groups (G1 + G15, G1 + AG, and G1 + U0126) had at least a fourfold increase in tumor volume compared to the G1 group (Figures 6A, B). Moreover, these drugs showed no evident toxicity because the body weights of the mice did not change significantly. Western blotting was performed to determine the protein expression levels of GPER, p-ERK, and Ki-67 in drug-treated HCC xenograft tumors. Similar to the in vitro data, G1 dramatically improved p-ERK expression, which could be blocked by G15, AG, and U0126, whereas xenograft tumors in all treatment groups possessed almost the same GPER protein levels (Figures 6C, D). G1 remarkably decreased Ki-67 expression, representing tumor proliferation ability (36), whereas the observed effects could be attributed to the indicated signaling pathway inhibitors (Figures 6C, D), and these results corresponded with previous tumor volume studies. These data imply that GPER is a tumor suppressor in HCC xenografts, and that the specific activation of GPER-mediated ERK signaling might present a potential therapeutic avenue for patients with HCC.
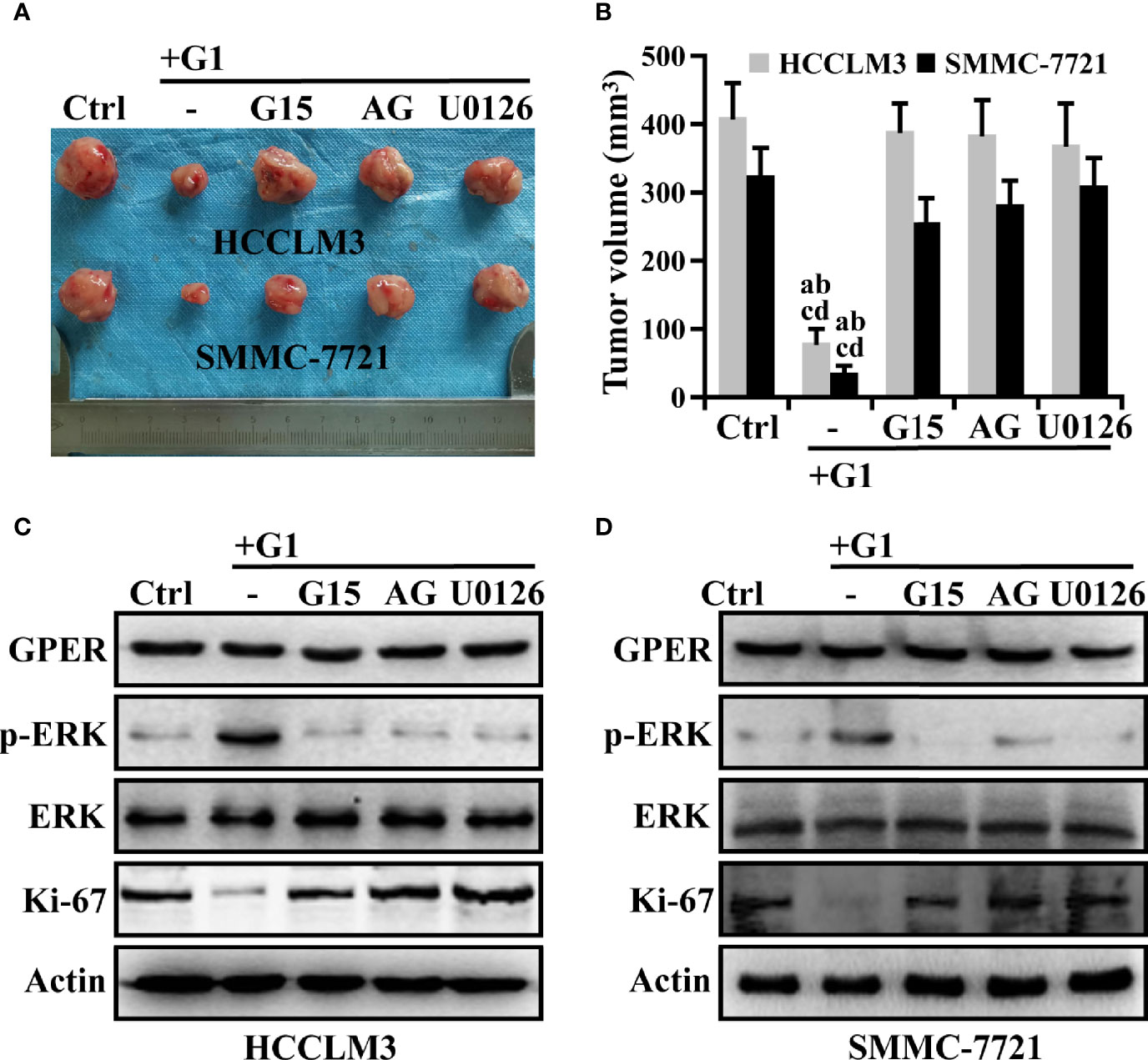
Figure 6 The inhibition of G1-induced GPER/EGFR/ERK signaling abrogates the repression of HCC xenograft growth. (A, B) Nude mice bearing HCC xenograft tumors were randomized to receive ethanol alone, G1 (1 μM) alone, or G1 (1 μM) in combination with the GPER-specific antagonist G15 (1 μM), AG (10 μM), or U0126 (10 μM). Images show HCC xenograft tumours in the monotherapy and combination therapy groups. (P < 0.05; a vs. ctrl, b vs. G1 + G15, c vs. G1 + AG, d vs. G1 + U0126). (C, D) Western blot detection of the protein expression levels of GPER, p-ERK, and Ki-67 in HCC xenograft tumors. Each experiment was repeated at least three times.
Estrogen receptors are thought to regulate HCC tumorigenesis and progression, but the role and mechanism of GPER in the development and progression of HCC have not been thoroughly studied. In the present study, GPER was downregulated in HCC tissue compared with that in matched non-tumor counterparts, and GPER-specific agonist G1-triggered GPER/EGFR/ERK signaling played a crucial role in decreasing the tumor viability of HCC, both in vitro and in vivo. GPER/ERK signaling is strongly associated with GPER-positive HCC tissue, and patients with simultaneous high expression of GPER and p-ERK showed improved clinical outcomes.
Using a small-scale cohort of 62 HCC samples, Wei et al. showed that GPER staining levels were significantly lower in HCC tissue than in matched non-tumor tissue (29). In the present study, we expanded upon the work by Wei et al. by confirming that GPER is remarkably downregulated in tumor tissue in a larger clinical cohort (141 cases). Moreover, a positive association between GPER expression and several indicators of improved clinical prognosis, such as female sex, negative HBsAg, small tumor size (<5 cm), and low serum level of AFP (≤400 ng/ml), was observed in patients with HCC, and GPER-positive patients exhibited longer OS than GPER-negative patients. These data indicate that GPER may act as a tumor suppressor in HCC. Conversely, Chaturantabut et al. found that human HCC samples (68 cases) have increased GPER expression levels compared with that in non-tumor tissue and that the activation of GPER promotes liver tumor development via the PI3K/mammalian target of rapamycin signaling in zebrafish (31). These inconsistent results may be attributed to differences in demographics and clinical sample size between these studies, highlighting the need for large, diverse clinical cohorts in future studies.
The GPER-specific agonist G1, which is wildly employed in the study of numerous cancer types (12, 13, 18–25, 27–31, 34–37), shows extremely high affinity for GPER, but not for classical ERs (14, 38, 39) and 25 other important G-protein coupled receptors (GPCRs) (40, 41), and G1 also had no activity in GPER-knockout mice (42). In the present study, we demonstrate that G1 can block cell cycle progression, promote apoptosis, and inhibit cell growth through GPER/EGFR/ERK signaling in HCCLM3 and SMMC-7721 cell lines. To our knowledge, our work is the first to prove that GPER is a critical factor in these two cell lines. The above data was corroborated by the overexpression of GPER in HepG2 cells. In agreement with our results, a previous study also showed that G1 antagonises the oncogenic actions of leptin in HCC cells by activating GPER/ERK signaling (37). Interestingly, the present study demonstrated that GPER-induced phosphorylation of EGFR, MAPK/ERK, and PI3K/AKT is remarkably upregulated in HCC cells. Despite this, other reported GPER downstream signals, including cAMP/PKA (16, 23), MAPK/JNK (43, 44), and MAPK/P38 (43, 45), were not detected in HCC cells, indicating that GPER may mediate different biological downstream pathways in diverse tumor types. Interestingly, GPER-mediated GPER/AKT signaling is transient, unlike GPER/ERK signaling, and does not contribute to tumor viability in HCC. The definite biological function of GPER-mediated GPER/AKT signaling in HCC should be investigated in detail in the future.
Activation of the ERK signaling pathway is generally associated with enhanced malignant cell survival, metastasis, and clinical drug resistance (46, 47). Targeted inhibition of ERK signaling can suppress hepatocarcinogenesis, and block the invasion and metastasis of HCC (48, 49). However, in the present study, G1 decreased HCC cell viability through the GPER/ERK pathway. Many studies have proposed that the sustained activation of ERK signaling increases cell death via cell cycle or apoptosis regulation in a variety of cancer types, including prostate, gastric, colon, and cervical cancers (21, 50–52), which supports our findings. While the overactivation of ERK signaling may be involved in cell death, the appropriate activation of ERK signaling may enhance tumor progression. The balance between the intensity and the duration of pro- or anti-cancer signals transmitted by ERK determines whether tumor cells proliferate or undergo apoptosis.
Our in vitro data are strongly supported by in vivo results, which show that G1 significantly inhibits tumor growth of HCC xenografts through the GPER/EGFR/ERK axis, and that the specific activation of the GPER/ERK pathway notably enhances tumor reduction. The GPER/ERK pathway is strongly associated with GPER-positive patients with HCC in clinics, and patients with high GPER/ERK activation have better clinical outcomes than those with other subtypes. Our findings further support the idea that the expression of GPER and its downstream signaling should be routinely evaluated in HCC tissue, and the targeted activation of GPER signaling is expected to be effective in improving the prognosis of patients with HCC.
In summary, in the present study we provide a novel insight into the role of GPER-mediated tumor suppression in patients with HCC, providing insight into the molecular basis for clinical observations. In brief, the GPER-specific agonist G1 promotes crosstalk between GPER and EGFR in HCC, and the downstream MAPK/ERK and PI3K/AKT signaling pathways are significantly activated. The GPER/EGFR/ERK axis is further responsible for blocking cell cycle progression, promoting apoptosis, and inhibiting cell growth in HCC cells, finally leading to reduced tumor viability both in vitro and in vivo (Figure 7). The activation of GPER/ERK signaling may be a potential treatment for patients with HCC. Further investigations into this signaling cascade, including preclinical and prospective studies, are therefore warranted.

Figure 7 Illustration depicting the specific role of GPER-mediated ERK signaling in decreasing the viability of HCC cells. The GPER-specific agonist G1 promoted crosstalk between GPER and EGFR in HCC cells, and the downstream MAPK/ERK and PI3K/AKT signaling pathways were significantly activated. Activation of the GPER/EGFR/ERK axis blocked cell cycle progression, promoted apoptosis, and inhibited cell growth in HCC cells, finally leading to reduced tumor viability, both in vitro and in vivo.
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding author.
The studies involving human participants were reviewed and approved by The Ethics Committee of Jiangxi Cancer Hospital. The patients/participants provided their written informed consent to participate in this study. The animal study was reviewed and approved by The Ethics Committee of Chongqing Medical University. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
All authors met the authorship requirements. Y-AQ participated in the design of the study and carried out most of the experiments. JX, QF, YD, ML, and MP participated in immunohistochemistry, RT-PCR, immunoblotting, and immunofluorescence studies. LZ, WJ, and XX participated in siRNA transfection, flow cytometry, CCK8, and in vivo assays. XH, AF, GX, and GT analyzed the data and performed the statistical analysis. TY designed the study and drafted the manuscript. All authors contributed to the article and approved the submitted version.
This work was supported by the Youths Program of the National Natural Science Foundation of China (81702641) (to TY), the Excellent Youths Program of the Natural Science Foundation of Jiangxi Province (2018ACB21042) (to TY), the Youths Program of the Natural Science Foundation of Jiangxi Province (20171BAB215046) (to TY), and the General Program of the Natural Science Foundation of Jiangxi Province (20192BAB205069) (to Y-AQ).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2021.638171/full#supplementary-material
AFP, alpha fetoprotein; AG, AG1478; AKT, protein-serine-threonine kinase; cAMP, cyclic adenosine monophosphate; CCK-8, Cell Counting Kit-8; Ctrl, control; DAB, daiminobenezidine; DAPI, 4,6-diamino-2-phenyl indole; DMEM, Dulbecco’s modified Eagle’s medium; EGFR, epidermal growth factor receptor; ERK, extracellular signal-regulated kinase; ERα, estrogen receptor alpha; ERβ, estrogen receptor beta; FBS, fetal bovine serum; FITC, fluorescein isothiocyanate; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; GPER, G-protein-coupled estrogen receptor; GPR30, G protein-coupled receptor 30; HBsAg, hepatitis B surface antigen; HCC, hepatocellular carcinoma; JNK, c-Jun N-terminal kinases; MAPK, mitogen-activated protein kinases; MEK, mitogen-activated protein kinase/extracellular signal-regulated kinase; mRNA, messenger RNA; OS, overall survival; P38, p38 mitogen-activated protein kinases; PBS, phosphate-buffered saline; PI, propidium iodide; PI3K, phosphatidylinositol 3-kinases; PKA, protein kinase A; RT-PCR, real-time PCR; SDS–PAGE, sodium dodecyl sulphate–polyacrylamide gel electrophoresis; siRNA, small interfering RNA; WM, Wortmannin.
1. Chen W, Zheng R, Baade PD, et al. Cancer statistics in China, 2015. CA: A Cancer J Clin (2016) 66(2):115–32. doi: 10.3322/caac.21338
2. Akinyemiju T, Abera S, Ahmed M, et al. The burden of primary liver cancer and underlying etiologies from 1990 to 2015 at the global, regional, and national level: results from the Global Burden of Disease Study 2015. JAMA Oncol (2017) 3(12):1683–91.
3. El–Serag HB, Rudolph KL. Hepatocellular carcinoma: epidemiology and molecular carcinogenesis. Gastroenterology (2007) 132(7):2557–76. doi: 10.1053/j.gastro.2007.04.061
4. Forner A, Reig M, Bruix J. Hepatocellular carcinoma. Lancet (2018) 391(10127):1301–14. doi: 10.1016/S0140-6736(18)30010-2
5. Lim KC, Chow PKH, Allen JC, Siddiqui FJ, Chan ES-Y, Tan S-B. Systematic review of outcomes of liver resection for early hepatocellular carcinoma within the Milan criteria. Br J Surg (2012) 99(12):1622–9. doi: 10.1002/bjs.8915
6. Khemlina G, Ikeda S, Kurzrock R. The biology of Hepatocellular carcinoma: implications for genomic and immune therapies. Mol Cancer (2017) 16(1):149. doi: 10.1186/s12943-017-0712-x
7. Zhang Y, Ren JS, Shi JF, et al. International trends in primary liver cancer incidence from 1973 to 2007. BMC Cancer (2015) 15(1):94. doi: 10.1186/s12885-015-1113-4
8. Kur P, Kolasa-Wołosiuk A, Misiakiewicz-Has K, Wiszniewska B. Sex Hormone-Dependent Physiology and Diseases of Liver. Int J Environ Res Public Health (2020) 17(8):2620. doi: 10.3390/ijerph17082620
9. Sukocheva OA. Estrogen, estrogen receptors, and hepatocellular carcinoma: Are we there yet? World J Gastroenterol (2018) 24(1):1–4. doi: 10.3748/wjg.v24.i1.1
10. Hassan MM, Botrus G, Abdel-Wahab R, et al. Estrogen replacement reduces risk and increases survival times of women with hepatocellular carcinoma. Clin Gastroenterol Hepatol (2017) 15(11):1791–9. doi: 10.1016/j.cgh.2017.05.036
11. Ikeda K, Inoue S. Estrogen receptors and their downstream targets in cancer. Arch Histol Cytol (2004) 67(5):435–42. doi: 10.1679/aohc.67.435
12. Yin H, Zhu Q, Liu M, et al. GPER promotes tamoxifen-resistance in ER+ breast cancer cells by reduced Bim proteins through MAPK/Erk-TRIM2 signaling axis. Int J Oncol (2017) 51(4):1191–8. doi: 10.3892/ijo.2017.4117
13. Yin J, Tu G, Peng M, et al. GPER-regulated lncRNA-Glu promotes glutamate secretion to enhance cellular invasion and metastasis in triple-negative breast cancer. FASEB J (2020) 34(3):4557–72. doi: 10.1096/fj.201901384RR
14. Bologa CG, Revankar CM, Young SM, et al. Virtual and biomolecular screening converge on a selective agonist for GPR30. Nat Chem Biol (2006) 2(4):207–12. doi: 10.1038/nchembio775
15. Prossnitz ER, Arterburn JB, Smith HO, Oprea TI, Sklar LA, Hathaway HJ. Estrogen signaling through the transmembrane G protein–coupled receptor GPR30. Annu Rev Physiol (2008) 70:165–90. doi: 10.1146/annurev.physiol.70.113006.100518
16. Zucchetti AE, Barosso IR, Boaglio AC, et al. G-protein-coupled receptor 30/adenylyl cyclase/protein kinase A pathway is involved in estradiol 17ß-d-glucuronide-induced cholestasis. Hepatology (2014) 59(3):1016–29. doi: 10.1002/hep.26752
17. Goswami C, Kuhn J, Dina OA, et al. Estrogen destabilizes microtubules through an ion-conductivity-independent TRPV1 pathway. J Neurochem (2011) 117(6):995–1008. doi: 10.1111/j.1471-4159.2011.07270.x
18. Li Y, Jia Y, Bian Y, et al. Autocrine motility factor promotes endometrial cancer progression by targeting GPER-1. Cell Commun Signaling (2019) 17(1):22. doi: 10.1186/s12964-019-0336-4
19. Yan Y, Jiang X, Zhao Y, Wen H, Liu G. Role of GPER on proliferation, migration and invasion in ligand-independent manner in human ovarian cancer cell line SKOV3. Cell Biochem Funct (2015) 33(8):552–9. doi: 10.1002/cbf.3154
20. Hsu LH, Chu NM, Kao SH. Estrogen, estrogen receptor and lung cancer. Int J Mol Sci (2017) 18(8):1713. doi: 10.3390/ijms18081713
21. Lau KM, Ma FMT, Xia JT, Chan QKY, Ng C-F, To K-F. Activation of GPR30 stimulates GTP-binding of Gαi1 protein to sustain activation of Erk1/2 in inhibition of prostate cancer cell growth and modulates metastatic properties. Exp Cell Res (2017) 350(1):199–209. doi: 10.1016/j.yexcr.2016.11.022
22. Yu T, Liu M, Luo H, et al. GPER mediates enhanced cell viability and motility via non-genomic signaling induced by 17β-estradiol in triple-negative breast cancer cells. J Steroid Biochem Mol Biol (2014) 143:392–403. doi: 10.1016/j.jsbmb.2014.05.003
23. Yu T, Yang G, Hou Y, et al. Cytoplasmic GPER translocation in cancer-associated fibroblasts mediates cAMP/PKA/CREB/glycolytic axis to confer tumor cells with multidrug resistance. Oncogene (2017) 36(15):2131–45. doi: 10.1038/onc.2016.370
24. Yu T, Cheng H, Ding Z, et al. GPER mediates decreased chemosensitivity via regulation of ABCG2 expression and localization in tamoxifen-resistant breast cancer cells. Mol Cell Endocrinol (2020) 506:110762. doi: 10.1016/j.mce.2020.110762
25. Lee SJ, Kim TW, Park GL, et al. G protein-coupled estrogen receptor-1 agonist induces chemotherapeutic effect via ER stress signaling in gastric cancer. BMB Rep (2019) 52(11):647–52. doi: 10.5483/BMBRep.2019.52.11.007
26. Liu Q, Chen Z, Jiang G, et al. Epigenetic down regulation of G protein-coupled estrogen receptor (GPER) functions as a tumor suppressor in colorectal cancer. Mol Cancer (2017) 16(1):87. doi: 10.1186/s12943-017-0654-3
27. Cortes E, Lachowski D, Rice A, et al. Tamoxifen mechanically deactivates hepatic stellate cells via the G protein-coupled estrogen receptor. Oncogene (2019) 38(16):2910–22. doi: 10.1038/s41388-018-0631-3
28. Ulitzky L, Lafer MM, KuKuruga MA, Silberstein E, Cehan N, Taylor DR. A new signaling pathway for HCV inhibition by estrogen: GPR30 activation leads to cleavage of occludin by MMP-9. PloS One (2016) 11(1):e0145212. doi: 10.1371/journal.pone.0145212
29. Wei T, Chen W, Wen L, et al. G protein-coupled estrogen receptor deficiency accelerates liver tumorigenesis by enhancing inflammation and fibrosis. Cancer Lett (2016) 382(2):195–202. doi: 10.1016/j.canlet.2016.08.012
30. Wen S, Hou Y, Fu L, et al. Cancer-associated fibroblast (CAF)-derived IL32 promotes breast cancer cell invasion and metastasis via integrin β3–p38 MAPK signalling. Cancer Lett (2019) 442:320–32. doi: 10.1016/j.canlet.2018.10.015
31. Chaturantabut S, Shwartz A, Evason KJ, et al. Estrogen Activation of G-Protein-Coupled Estrogen Receptor 1 Regulates Phosphoinositide 3-Kinase and mTOR Signaling to Promote Liver Growth in Zebrafish and Proliferation of Human Hepatocytes. Gastroenterology (2019) 156(6):1788–1804. e13. doi: 10.1053/j.gastro.2019.01.010
32. Shao YY, Chen CL, Ho MC, et al. Dissimilar immunohistochemical expression of ERK and AKT between paired biopsy and hepatectomy tissues of hepatocellular carcinoma. Anticancer Res (2012) 32(11):4865–70.
33. Tang X, Tu G, Yang G, et al. Autocrine TGF-β1/miR-200s/miR-221/DNMT3B regulatory loop maintains CAF status to fuel breast cancer cell proliferation. Cancer Lett (2019) 452:79–89. doi: 10.1016/j.canlet.2019.02.044
34. Mo Z, Liu M, Yang F, et al. GPR30 as an initiator of tamoxifen resistance in hormone-dependent breast cancer. Breast Cancer Res (2013) 15(6):1–15. doi: 10.1186/bcr3581
35. Yuan J, Liu M, Yang L, et al. Acquisition of epithelial-mesenchymal transition phenotype in the tamoxifen-resistant breast cancer cell: a new role for G protein-coupled estrogen receptor in mediating tamoxifen resistance through cancer-associated fibroblast-derived fibronectin and β1-integrin signaling pathway in tumor cells. Breast Cancer Res (2015) 17(1):69. doi: 10.1186/s13058-015-0579-y
36. Yang J, Li J, Liu B, et al. Long Noncoding RNA AK021443 Promotes Cell Proliferation and Migration by Regulating Epithelial–Mesenchymal Transition in Hepatocellular Carcinoma Cells. DNA Cell Biol (2018) 37(5):481–90. doi: 10.1089/dna.2017.4030
37. Shen M, Shi H. Estradiol and estrogen receptor agonists oppose oncogenic actions of leptin in HepG2 cells. PloS One (2016) 11(3):e0151455. doi: 10.1371/journal.pone.0151455
38. Dennis MK, Burai R, Ramesh C, et al. In vivo effects of a GPR30 antagonist. Nat Chem Biol (2009) 5(6):421–7. doi: 10.1038/nchembio.168
39. Dennis MK, Field AS, Burai R, et al. Identification of a GPER/GPR30 antagonist with improved estrogen receptor counterselectivity. J Steroid Biochem Mol Biol (2011) 127(3-5):358–66. doi: 10.1016/j.jsbmb.2011.07.002
40. Blasko E, Haskell CA, Leung S, et al. Beneficial role of the GPR30 agonist G-1 in an animal model of multiple sclerosis. J Neuroimmunol (2009) 214(1-2):67–77. doi: 10.1016/j.jneuroim.2009.06.023
41. Prossnitz ER, Barton M. The G-protein-coupled estrogen receptor GPER in health and disease. Nat Rev Endocrinol (2011) 7(12):715–26. doi: 10.1038/nrendo.2011.122
42. Haas E, Bhattacharya I, Brailoiu E, et al. Regulatory role of G protein-coupled estrogen receptor for vascular function and obesity. Circ Res (2009) 104(3):288–91. doi: 10.1161/CIRCRESAHA.108.190892
43. Jung J. Role of G Protein-Coupled Estrogen Receptor in Cancer Progression. Toxicol Res (2019) 35(3):209–14. doi: 10.5487/TR.2019.35.3.209
44. Davis RJ. Signal transduction by the JNK group of MAP kinases. Cell (2000) 103:239–52. doi: 10.1016/S0092-8674(00)00116-1
45. Sun Y, Liu WZ, Liu T, Feng X, Yang N, Zhou H-F. Signaling pathway of MAPK/ERK in cell proliferation, differentiation, migration, senescence and apoptosis. J Recept Signal Transduct (2015) 35(6):600–4. doi: 10.3109/10799893.2015.1030412
46. Wu XS, Wang XA, Wu WG, et al. MALAT1 promotes the proliferation and metastasis of gallbladder cancer cells by activating the ERK/MAPK pathway. Cancer Biol Ther (2014) 15(6):806–14. doi: 10.4161/cbt.28584
47. Shi M, He X, Wei W, Wang J, Zhang T, Shen X. Tenascin-C induces resistance to apoptosis in pancreatic cancer cell through activation of ERK/NF-κB pathway. Apoptosis (2015) 20(6):843–57. doi: 10.1007/s10495-015-1106-4
48. Shen Z, Zhang C, Qu L, et al. MKP-4 suppresses hepatocarcinogenesis by targeting ERK1/2 pathway. Cancer Cell Int (2019) 19(1):61. doi: 10.1186/s12935-019-0776-3
49. Li QT, Feng YM, Ke ZH, et al. KCNN4 promotes invasion and metastasis through the MAPK/ERK pathway in hepatocellular carcinoma. J Invest Med (2020) 68(1):68–74. doi: 10.1136/jim-2019-001073
50. Qu J, Zhao M, Teng Y, et al. Interferon-α sensitizes human gastric cancer cells to TRAIL-induced apoptosis via activation of the c-CBL-dependent MAPK/ERK pathway. Cancer Biol Ther (2011) 12(6):494–502. doi: 10.4161/cbt.12.6.15973
51. Dubey N, Peng BY, Lin CM, et al. NSC 95397 suppresses proliferation and induces apoptosis in colon cancer cells through MKP-1 and the ERK1/2 pathway. Int J Mol Sci (2018) 19(6):1625. doi: 10.3390/ijms19061625
Keywords: G protein-coupled estrogen receptor, cell viability, hepatocellular carcinoma, ERK signaling, therapeutic target
Citation: Qiu Y-a, Xiong J, Fu Q, Dong Y, Liu M, Peng M, Jin W, Zhou L, Xu X, Huang X, Fu A, Xu G, Tu G and Yu T (2021) GPER-Induced ERK Signaling Decreases Cell Viability of Hepatocellular Carcinoma. Front. Oncol. 11:638171. doi: 10.3389/fonc.2021.638171
Received: 05 December 2020; Accepted: 25 January 2021;
Published: 09 March 2021.
Edited by:
Kanjoormana Aryan Manu, Amala Cancer Research Centre, IndiaReviewed by:
Thomas Meigs, University of North Carolina at Asheville, United StatesCopyright © 2021 Qiu, Xiong, Fu, Dong, Liu, Peng, Jin, Zhou, Xu, Huang, Fu, Xu, Tu and Yu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tenghua Yu, eXV0ZW5naHVhMDEwN0BzaW5hLmNu
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.