 Jacob Handlos Grauslund1†Morten Orebo Holmström1†Nicolai Grønne Jørgensen1Uffe Klausen1Stine Emilie Weis-Banke1Daniel El Fassi2,3Claudia Schöllkopf2Mette Borg Clausen2Lise Mette Rahbek Gjerdrum4Marie Fredslund Breinholt5Julie Westerlin Kjeldsen1Morten Hansen1Steffen Koschmieder6Nicolas Chatain6Guy Wayne Novotny2Jesper Petersen2Lasse Kjær7Vibe Skov7Özcan Met1,8Inge Marie Svane1Hans Carl Hasselbalch7‡Mads Hald Andersen1,8*‡
Jacob Handlos Grauslund1†Morten Orebo Holmström1†Nicolai Grønne Jørgensen1Uffe Klausen1Stine Emilie Weis-Banke1Daniel El Fassi2,3Claudia Schöllkopf2Mette Borg Clausen2Lise Mette Rahbek Gjerdrum4Marie Fredslund Breinholt5Julie Westerlin Kjeldsen1Morten Hansen1Steffen Koschmieder6Nicolas Chatain6Guy Wayne Novotny2Jesper Petersen2Lasse Kjær7Vibe Skov7Özcan Met1,8Inge Marie Svane1Hans Carl Hasselbalch7‡Mads Hald Andersen1,8*‡- 1National Center for Cancer Immune Therapy (CCIT-DK), Department of Oncology, Copenhagen University Hospital, Herlev, Denmark
- 2Department of Hematology, Copenhagen University Hospital, Herlev, Denmark
- 3Department of Medicine, Copenhagen University, Copenhagen, Denmark
- 4Department of Pathology, Zealand University Hospital, Roskilde, Denmark
- 5Department of Pathology, Copenhagen University Hospital, Herlev, Denmark
- 6Department of Hematology, Oncology, Hemostaseology and Stem Cell Transplantation, Faculty of Medicine, RWTH Aachen University, Aachen, Germany
- 7Department of Hematology, Zealand University Hospital, Roskilde, Denmark
- 8Institute for Immunology and Microbiology, Copenhagen University, Copenhagen, Denmark
Background: The calreticulin (CALR) exon 9 mutations that are identified in 20% of patients with Philadelphia chromosome negative chronic myeloproliferative neoplasms (MPN) generate immunogenic antigens. Thus, therapeutic cancer vaccination against mutant CALR could be a new treatment modality in CALR-mutant MPN.
Methods: The safety and efficacy of vaccination with the peptide CALRLong36 derived from the CALR exon 9 mutations was tested in a phase I clinical vaccination trial with montanide as adjuvant. Ten patients with CALRmut MPN were included in the trial and received 15 vaccines over the course of one year. The primary end point was evaluation of safety and toxicity of the vaccine. Secondary endpoint was assessment of the immune response to the vaccination epitope (www.clinicaltrials.gov identifier NCT03566446).
Results: Patients had a median age of 59.5 years and a median disease duration of 6.5 years. All patients received the intended 15 vaccines, and the vaccines were deemed safe and tolerable as only two grade three AE were detected, and none of these were considered to be related to the vaccine. A decline in platelet counts relative to the platelets counts at baseline was detected during the first 100 days, however this did not translate into neither a clinical nor a molecular response in any of the patients. Immunomonitoring revealed that four of 10 patients had an in vitro interferon (IFN)-γ ELISPOT response to the CALRLong36 peptide at baseline, and four additional patients displayed a response in ELISPOT upon receiving three or more vaccines. The amplitude of the immune response increased during the entire vaccination schedule for patients with essential thrombocythemia. In contrast, the immune response in patients with primary myelofibrosis did not increase after three vaccines.
Conclusion: Therapeutic cancer vaccination with peptide vaccines derived from mutant CALR with montanide as an adjuvant, is safe and tolerable. The vaccines did not induce any clinical responses. However, the majority of patients displayed a marked T-cell response to the vaccine upon completion of the trial. This suggests that vaccines directed against mutant CALR may be used with other cancer therapeutic modalities to enhance the anti-tumor immune response.
Introduction
The Philadelphia chromosome-negative chronic myeloproliferative neoplasms (MPN) are chronic blood cancers originating in the hematopoietic stem cells in the bone marrow (1, 2). MPN have overlapping signs and symptoms and thus may be difficult to distinguish from one another. Essential thrombocythemia (ET) and polycythemia vera (PV) are the non-advanced MPN, with patients presenting elevated levels of peripheral blood cells, whereas primary myelofibrosis is the advanced disease characterized by bone marrow fibrosis, and patients often present with cytopenia. Prefibrotic or early myelofibrosis (PreMF) was recently acknowledged as a distinct MPN with patients presenting with early signs of myelofibrosis (3). Allogeneic hematopoietic stem cell transplantation (alloHSCT) remains the only curative treatment for MPN, and as patients with ET and PV may live for several decades (4, 5) alloHSCT, which has a high treatment-related mortality, is not used for the treatment of these diseases. AlloHSCT is used for younger and fit patients with PMF (6), but as the majority of patients with PMF are of older age, alloHSCT is not used routinely for these patients. Due to the elevated blood cell counts and the presence of the MPN-driver mutations per se, patients with ET and PV have a dramatically increased risk of thromboembolic episodes and hemorrhage. Hence, the treatment for ET and PV aims to lower this risk with cytoreductive therapy (7) such as hydroxyurea (HU), anagrelide (ANA), and pegylated interferon alpha (IFN-α). HU is a weak chemotherapeutic agent that reduces peripheral blood cell counts, whereas ANA is solely used to reduce the platelet count. IFN-α is a cytokine that in the past has been used to treat several cancers, such as multiple myeloma, chronic myelogenous leukemia (CML), malignant melanoma and renal cancer (8) but in general with a low-rate of success except partly for CML (9). In MPN, IFN-α can induce major molecular remissions, complete hematological responses (10, 11), and even normalization of bone marrow architecture in some patients (11). As IFN-α is an immunostimulatory cytokine, it has been speculated that one of its mechanisms of action is to normalize the immune environment in patients with MPN (12, 13).
Several lines of evidence show that the immune system is severely dysregulated in MPN (13–15). Cytokine levels and the immune phenotype in patients differ from those in healthy donors. This immune derangement is likely partially driven by the driver mutations identified in MPN, as they all affect the JAK-STAT signaling pathway which is of paramount importance in the regulation of the immune-system (16). The mutational landscape in MPN is highly homogeneous, as approximately 90% of patients harbor a mutation in the Janus kinase-2 (JAK2), myeloproliferative leukemia virus (MPL), or calreticulin (CALR) gene. The JAK2V617F mutation has been identified in 98% of patients with PV and in 50% of patients with ET and PMF (17, 18). The MPL mutations are identified in 5–10% of patients with ET and PMF (19), and the CALR exon 9 mutations are found in 20–25% of patients with ET and PMF (20, 21). Whereas the JAK2 and MPL mutations generate single-amino-acid substitutions in their respective proteins, the CALR deletion or insertion mutations result in frameshift mutations that generate a novel mutant C-terminus different from the wild-type (wt) CALR C-terminus (20, 21). Interestingly, peptides derived from the mutant C-terminus are recognized by peripheral blood mononuclear cells (PBMCs) isolated both from patients with CALR-mutant (CALRmut) MPN and from healthy donors (22, 23), and T cells isolated from patients with CALRmut MPN recognize and kill autologous CALRmut cells in a CALRmut-dependent manner (22). These data have established that the CALR mutations encode tumor-specific antigens (TSAs) that are recognized by patient T cells.
Studies on other TSAs have shown that they may be targeted by the immune system (24), and clinical trials have used this in the setting of therapeutic cancer vaccination, where vaccination with peptides derived from the TSA is aimed at inducing/enhancing the tumor-specific immune response (25). The first TSAs targeted by therapeutic cancer vaccines arose from RAS mutations, which are the most common somatic mutations in human cancer (26). Initial studies showed that patient T cells respond to stimulation in vitro with epitopes derived from mutant RAS (27, 28), and that these T cells can kill HLA-matched RAS-mutant cancer cells (29). These results spurred the first therapeutic cancer vaccines using TSA-derived peptides, where vaccination against mutant KRAS induced or enhanced an immune response specific to the KRAS mutation-derived TSA (30). Several clinical vaccination trials testing vaccination against mutant KRAS have shown a survival benefit to patients who develop an immune response to the vaccination antigen (31–33), thus establishing the potential of TSA-specific therapeutic cancer vaccines.
Given the high immunogenicity of the CALRmut-derived TSA, we initiated a phase I clinical vaccination trial with a CALRmut-derived peptide in 10 patients with CALRmut MPN. Safety and tolerability were the primary end points, attainment of immune response to the CALRmut TSA was the secondary end point, and clinical response was the tertiary end point. The vaccine was safe and tolerable, and although the vaccines did not induce any clinical responses, they induced marked alterations in the immune phenotype and immune response to the vaccination antigen.
Subject, Materials and Methods
Study Design
This was a phase I clinical vaccination trial testing the safety and immunogenicity of vaccination with the CALRLong36 peptide with montanide as an adjuvant. Patients from the Departments of Hematology at Herlev and Gentofte Hospital, Copenhagen, Denmark, as well as Zealand University Hospital, Roskilde, Denmark were enrolled in the trial between August 30th, 2018 and February 17th 2019 with the last vaccination administered January 14th 2020. The study was conducted in accordance with the Helsinki Declaration, and Good Clinical Practice recommendations. All participants provided written informed consent before enrollment in the trial. The protocol was approved by the Ethics Committee of the Zealand Region, the National Board of Health, and the Danish Data Protection Agency, and registered at www.clinicaltrials.gov (NCT03566446; date of registration June 25th, 2018). One patient from whom we wished to isolate PBMC after the end of the study provided informed consent for this in accordance with an approval from the Ethics Committee of Zealand Region (approval number SJ-456).
As therapeutic cancer vaccines have not been tested earlier in patients with MPN, we had no basis for any power calculations. Based on our previous experience with phase I clinical vaccination trials, we chose to include 10 patients in the trial. Main inclusion criteria were a diagnosis of ET, PreMF, or PMF according to the WHO criteria, verified mutation in exon 9 of CALR, and Eastern Cooperative Oncology Group performance status of ≤2. A full list of the inclusion and exclusion criteria can be found in Supplementary Figure 1. Patients were allowed concurrent treatment with IFN-α, HU, or ANA in any combination, but no other anti-neoplastic or anti-MPN treatments were allowed.
Vaccine Composition and Treatment Schedule
Each vaccine was composed of 200 µg CALRLong36, a 36-amino acid peptide (RMRRMRRTRRKMRRKMSPARPRTSCREACLQGWTEA) spanning the entire mutant C-terminus created by the most frequent CALR mutations. The peptide was provided by Polypeptide (Strasbourg, France). The peptide was dissolved in 500 µL sterile water and mixed with 500 µL montanide ISA-51 just prior to administration. The vaccine was administered subcutaneously, and patients were vaccinated with a total of 15 doses, with the first six doses administered every second week and the final nine doses every fourth week. A Gant chart of the vaccination schedule is provided in Supplementary Figure 2.
Evaluation of Adverse Events and Clinical Response
Adverse events (AE) were assessed according to the Common Terminology Criteria for Adverse Events (CTCAE) version 4.03. Before receiving the first vaccination, each patient underwent a full medical examination and had an electrocardiogram recorded. This was repeated before each of the first three vaccinations, and then before the 7th and after the 15th vaccination. Biochemical tests on peripheral blood were performed before each vaccination. The patients underwent a full bone marrow examination and analysis by next-generation sequencing (NGS) performed on peripheral blood prior to the first vaccination and at end of study. The clinical response was evaluated according to the response criteria described by the European LeukemiaNet (34, 35).
Evaluation of the CALRmut Allelic Burden and Next-Generation Sequencing
PCR detection of CALR exon 9 frameshift mutations was performed as described by Klampfl et al. (20). Primers used are: CALR Forward: 5′-Fam-GGCAAGGCCCTGAGGTGT-3′, CALR Reverse: 5′-GGCCTCAGTCCAGCCCTG-3′. The PCR product was run on a ABI3500 Genetic Analyzer (Thermo Fisher Scientific) and mutated CALR allele burden calculated as 100*AUC (area under the curve) for mutated CALR/(AUC for mutated + AUC wildtype).
Next generation sequencing was performed on a custom made panel of 42 genes designed to capture mutations implicated in myeloid malignancies (ASXL1, BCOR, BRAF, CALR, CEBPA, CBL, CSF3R, CUX1, DNMT3A, ETV6, EZH2, FLT3, GATA2, GNAS, IDH1, IDH2, JAK2, KIT, KRAS, MLL3, MPL, MYD88, NF1, NFE2, NOTCH1, NPM1, NRAS, PHF6, PPM1D, PTPN11, RB1, RIT, RUNX1, SETBP1, SF3B1, SH2B3, SRSF2, STAG2, TET2, TP53, U2AF1, ZRSR2). Genomic DNA was purified from peripheral blood at baseline and 12 months after the first vaccination of 10 CALR mutated MPN patients. Libraries were prepared using the Nextera Flex for Enrichment protocol (Illumina® Inc, San Diego, CA, USA), and 2 × 150 bp paired-end sequencing was done on the NextSeq 500 platform (Illumina® Inc, San Diego, CA, USA). Quality control of all sequencing runs was assessed using the Illumina Sequencing Analysis Viewer (SAV) software. CLC Genomics Workbench v.12.0.3 software was applied to align sequencing data to the human reference genome (GRCh37/hg19) and to call variants from the mapped reads. Annotation and filtering of variants were performed using the VarSeq™ v.2.2.1 software (Golden Helix, Inc., Bozeman, MT, USA). Variants with coverage <100x, variant allele frequency (VAF) <1%, introns, germline, and SNPs with minor allele frequency >1% (ExAC variant frequencies, Broad Institute, MA, USA) were excluded. A mutation with VAF <1% in either a baseline or treatment sample was retained if the mutation had a VAF >1% in the paired sample. In addition, only variants classified as either pathogenic or likely pathogenic were retained for further analysis.
Isolation of Bone Marrow and Peripheral Blood Mononuclear Cells
Blood samples for isolation of PBMCs were obtained at baseline, after three vaccinations, after six vaccinations, and after 15 vaccinations. Samples were kept at room temperature (RT) for ≤5 h until handling. PBMCs were isolated by gradient centrifugation of heparinized blood on Lymphoprep (STEMCELL Technologies) in LeucoSep tubes (Greiner Bio-One). Isolated PBMCs were cryopreserved in 90% human serum (Sigma-Aldrich) with 10% DMSO (Sigma-Aldrich), using controlled-rate freezing (Cool-Cell, Biocision) in a −80°C freezer. The next day, the ampoules were transferred to −140°C.
Heparinized bone marrow samples (10 mL in a heparinized tube) were obtained at baseline and after 15 vaccinations. Ortho-Lysing Buffer diluted 10× in H20 was added to half of the sample, followed by centrifugation and incubation for 15 min in the dark. The other half of the sample was handled and cryopreserved following the same procedure as for PBMCs.
Delayed-Type Hypersensitivity and Skin-Infiltrating Lymphocytes
Presence of tumor-specific T cells in biopsies from delayed-type hypersensitivity (DTH) testing post-vaccination is correlated with clinical outcome (36). We assessed the presence of vaccine-reactive cells at DTH sites after six vaccinations. On the lower back, we performed three intradermal injections of CALRLong36 without adjuvant and one control injection of sterile water, without peptide. At 48 h post-DTH injection, skin reaction was measured and punch biopsies were taken from the sites of CALRLong36-containing injections and cut into fragments. Fragments were cultured in 24-well plates for 3–5 weeks in RPMI-1640 with 10% human serum and 100 U/mL or 6,000 U/mL interleukin-2 (IL-2) with penicillin, streptomycin, and fungizone. Three times weekly, half the medium was replaced with fresh medium containing IL-2. Skin-infiltrating lymphocytes (SKILs) emigrated from the biopsies. After 3–5 weeks, SKILs were harvested and tested in ELISPOT assays (see below). The remaining SKILs were cryopreserved, as described for PBMCs.
IFN-γ ELISPOT Assay
In order to assess the immune response against CALRLong36, we used IFN-γ ELISPOT assays performed on PBMCs as described in detail earlier (37). In short, PBMCs were thawed and stimulated with CALRLong36 and then stimulated with IL-2 (120 U/mL) the next day. The cells were incubated for 14 days in x-vivo (Lonza, Belgium) supplemented with 5% human serum before restimulation with CALRLong36 in the ELISPOT wells with unstimulated wells as negative controls. The ELISPOT plates were analyzed using the ImmunoSpot Series 2.0 Analyzer (CTL, Shaker Heights, Ohio). ELISPOT assays were performed in triplicate experiments with a concentration of 2.5–3.5 × 105 cells/well. For graphical representation the spot counts were normalized to 3 × 105 cells/well. The mean spot count per experiment was calculated by subtracting the mean spot count of the negative control wells from the mean spot count in the peptide-stimulated wells. ELISPOT assays on SKILs were run in triplicates or quadruplicates with 3 × 105 cells/well.
Intracellular Cytokine Staining
In order to identify the phenotype of cells releasing IFN-γ and TNF-α upon stimulation with CALRLong36 we used intracellular cytokine staining on in vitro-stimulated PBMC. The in vitro stimulation followed the same steps as for ELISPOT analysis and the stimulation, staining, and flow cytometric analysis of PBMCs followed our previously described method (37).
Phenotyping of Peripheral Blood Mononuclear Cells Using Fluorescence-Activated Cell Sorting (FACS)
Cryopreserved PBMCs from different timepoints were thawed and washed in preheated PBS. The Fc-receptors were blocked by incubation with human IgG (human IgG, 50 µg/ml), and dead cells were stained using Fixable Near-IR Dead cell stain kit (Thermo-Fisher) and incubated at 4°C for 10 min. Next, cells were stained by fluorochrome labelled antibodies and stained in the dark at 4°C for 20 min. After staining, the cells were washed and acquired using a NovoCyte Quanteon Flow Cytometer (Agilent, Santa Clara, CA). Gating strategy is provided in Supplementary Figure 3 and a list of antibodies used in Supplementary Figure 4. Data were analyzed using the NovoExpress 1.4.1 software. All gates at baseline were applied to all timepoints. Illustration of data were performed using Graphpad Prism v 8.0 (GraphPad Software. Inc.).
Statistical Analysis
ELISPOT responses were analyzed using the distribution-free resampling (DFR) method (38). Differences in immune subset populations between different time points were analyzed using the Wilcoxon matched-pairs signed-rank test. As this exploratory analysis was descriptive and done post hoc, no formal multiple testing corrections were performed. p values ≤ 0.05 were considered significant. All analyses were performed in Graphpad Prism v 8.0 (GraphPad Software. Inc.), apart from the DFR analyses which were performed using the statistical analysis program R.
Results
Patient Characteristics
Eleven patients were included and evaluated by examination of the bone marrow biopsies and blood samples prior to initiation of vaccinations. Due to compliance issues, one patient was excluded, and thus 10 patients continued in the trial. All patients received the planned 15 vaccine doses over the course of one year. The male:female ratio was 5:5, the median age at inclusion was 59.5 years (range 41-73 years), and the median duration of disease was 6.5 years (range 2–26 years) (Tables 1 and 2). At study entry, four patients had ET, three patients PMF, two patients post-essential thrombocythemia MF (PET-MF) and one patient prefibrotic/early myelofibrosis (PreMF). All but one patient had received MPN-directed therapy including HU, ANA, or INF-α prior to enrollment. Of the nine treated patients, eight were treated with INF-α and one with ANA at the start of vaccination. This one patient treated with ANA had received IFN-α earlier, however IFN-α had been withdrawn due to side effects. Median platelet count was 283 × 109/l (range 145 × 109/l–728 × 109/l), median hemoglobin count was 7.95 mmol/l (range 6.5 mmol/l–9.2 mmol/l), and median leucocyte count was 4.9 × 109/l (range 3.3 × 109/l–10.5 × 109/l). Four patients presented with splenomegaly on clinical examination. Four patients had the type 1 CALR-mutation, one patient a type-1-like CALR-mutation and five had the type 2 CALR-mutation. The median CALRmut VAF was 40.5% (range, 1–49%).

Table 1 Patient characteristics.
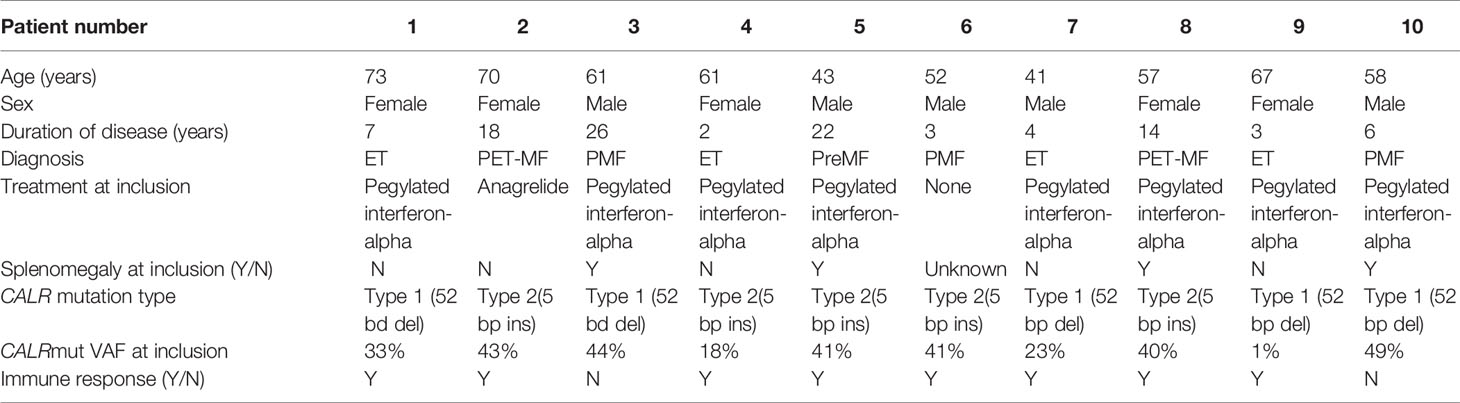
Table 2 Individual patient data for all patients in the trial.
Adverse Events and Safety Profile
All patients experienced AE and these were both local and systemic (Table 3). No vaccination related AE ≥ grade 3 were observed. The most common reactions were injection site specific with grade 2 in nine of ten patients at any time. Most common systemic reaction was flu like symptoms in five patients. Two grade 3 AE were registered: One vasovagal reaction during bone marrow biopsy at the end of study, and one patient experienced hypertension before the first vaccine was administered. One autoimmune reaction was registered (see below) but this reaction was not deemed to be triggered by the vaccines.
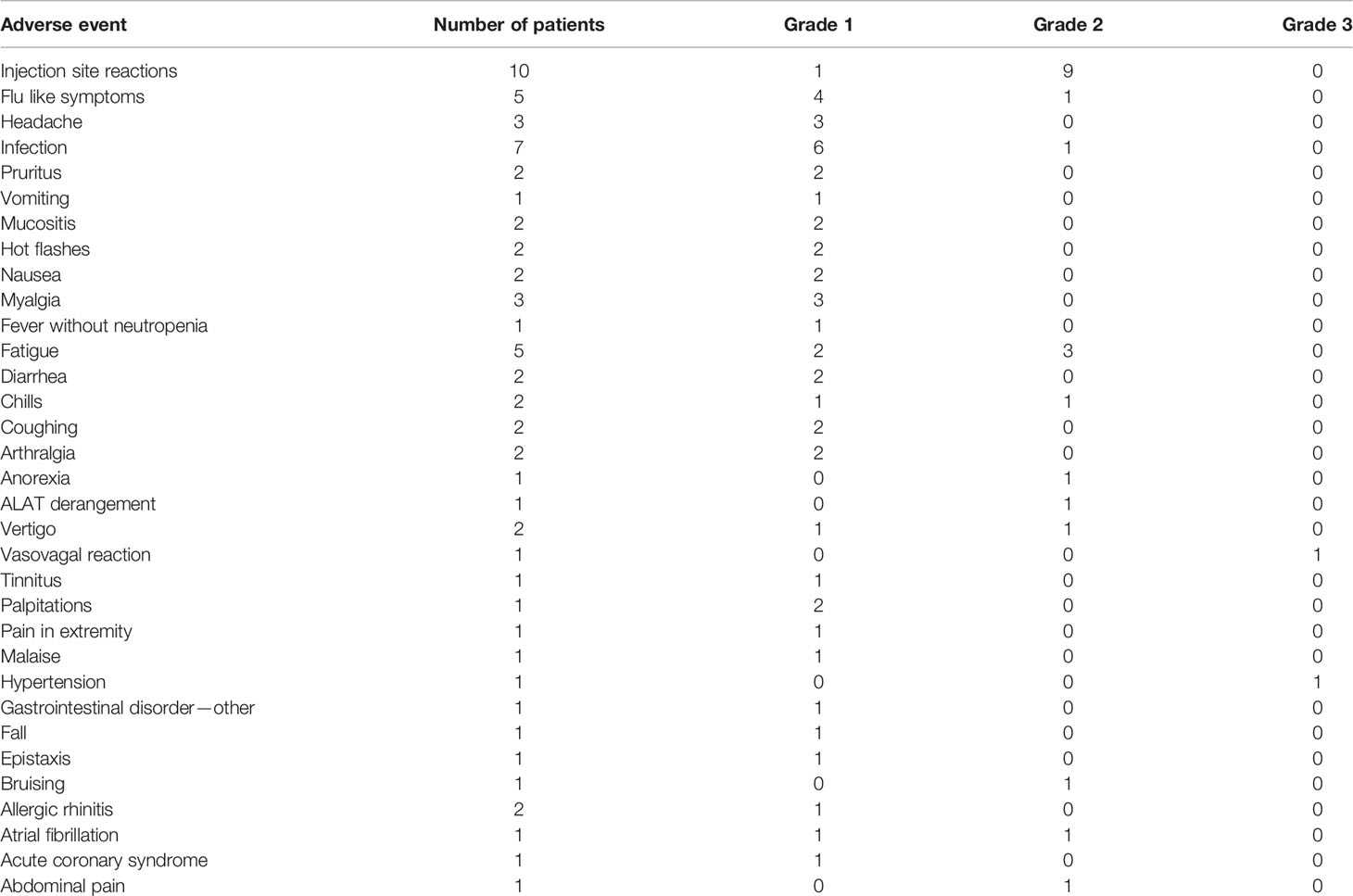
Table 3 Adverse events registered during the trial.
Clinical Response and Variations in CALRmut Variant Allele Frequency
As the CALR mutations mainly affect the megakaryocytic lineage, and patients present with marked thrombocythemia, we expected that the vaccinations would induce a decrease in platelet counts. Looking at each patient individually, we did not demonstrate any decrease in platelet counts (Figure 1A). Of note, one patient (patient 6), who did not receive any therapy at inclusion in the trial, was started on IFN-α after 7th vaccination due to high platelet counts. Another patient (patient 7) had to stop IFN-α after 8 vaccinations due to elevated liver enzymes and concomitant development of smooth muscle antibodies (SMA). Before and during the first vaccines, the patient developed increasing levels of alanine aminotransferase (ALAT) below upper normal limit. After 5 vaccinations the level of ALAT reached a maximum of 241 U/l (upper normal limit 70 U/L). SMA were detected in the blood, and examination of archived samples taken at study inclusion prior to vaccine initiation showed low levels of SMA. After IFN-α was stopped the patient continued his vaccinations as planned and normalized his liver function.
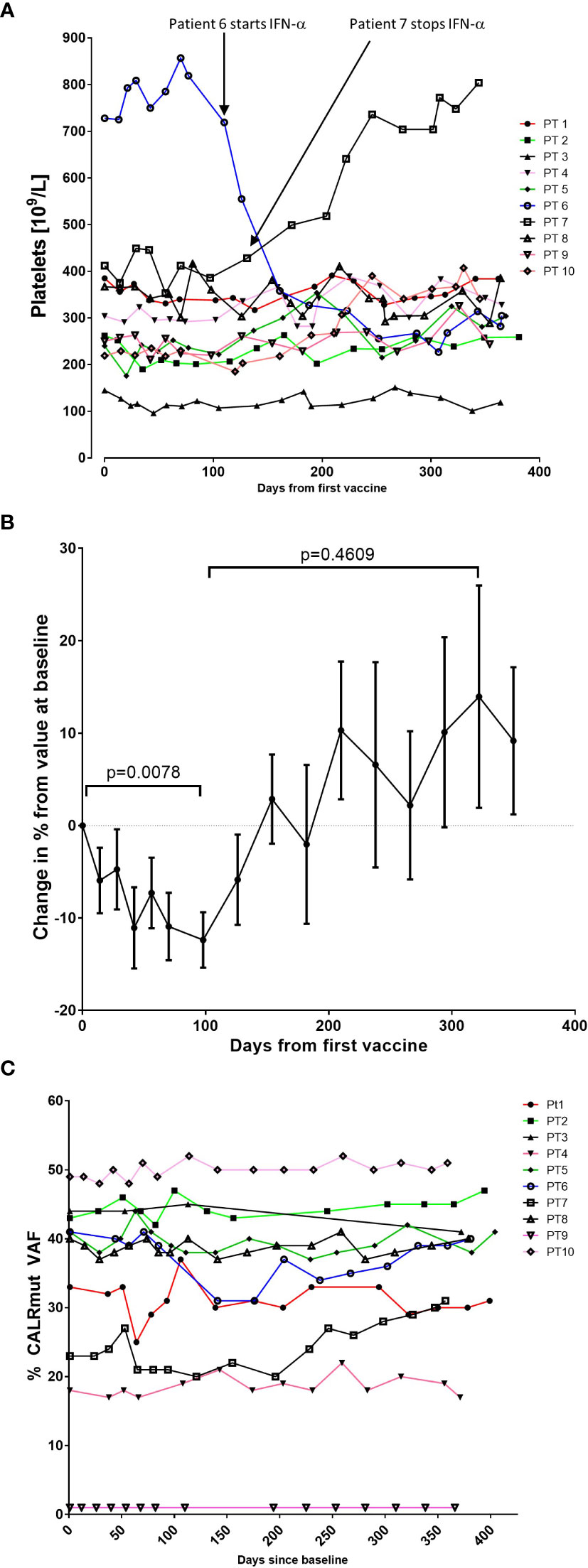
Figure 1 Variation in platelet counts and CALRmut variant allele frequency (VAF) during the trial. (A) Platelet counts (109/L) for each patient during the trial. (B) To get a better impression of the cumulative change in platelet counts, the relative change in platelet counts from baseline was calculated for each patient. Each dot represents the mean change in platelet count in percent relative to the platelet count at baseline. Error bars depict the standard error of the mean. Statistical testing was performed with the Wilcoxon signed-rank test. (C) Changes in the CALRmut VAF over time.
We detected a relative decrease in platelet count during the first 100–120 days after the first vaccination that was statistically significant (Figure 1B). Platelet levels increased thereafter to a level above baseline levels, but this increase was not statistically significant (Figure 1B). We expected to identify a decrease in the CALRmut variant allele frequency (VAF) in peripheral blood. The CALRmut VAF showed some fluctuations over time, which is also our experience in our normal clinical setting. However, the CALRmut VAF displayed no substantial decrease nor increase in any of the patients (Figure 1C) even in the patient that displayed a CALRmut VAF of only 1% at baseline. As described, patients underwent a full bone marrow examination at baseline and at end of study. No morphological or cytological changes were detected in patients during the vaccination schedule. Analysis of additional mutations quantified by NGS showed that seven of 10 patients harbored additional mutations at baseline and three patients only had a CALR mutation with no additional mutations (Table 4). However, at end of study one of these three patients had developed an additional mutation. Of note, among the additional mutations identified by NGS, several genes of importance in myeloid cancers such as ASXL1, TET2, EZH2, SF3B1, and DNMT3A were detected. Three patients displayed a mutation in TP53. The majority of patients did not experience marked alterations in mutant VAF as measured by NGS. However, patient 6 showed a decrease in DNMT3A (20% VAF to 12% VAF), whereas ASXL1 increased from 0.66% to 2.1%. Patient 8 was the only patient who demonstrated a marked expansion of a subclone as GNAS mutant VAF increased from 0.99% to 10% during the vaccination schedule. Additionally, patient 4 developed a new STAG2-mutant subclone and patient 10 developed a BCOR-mutant subclone during the vaccination schedule.
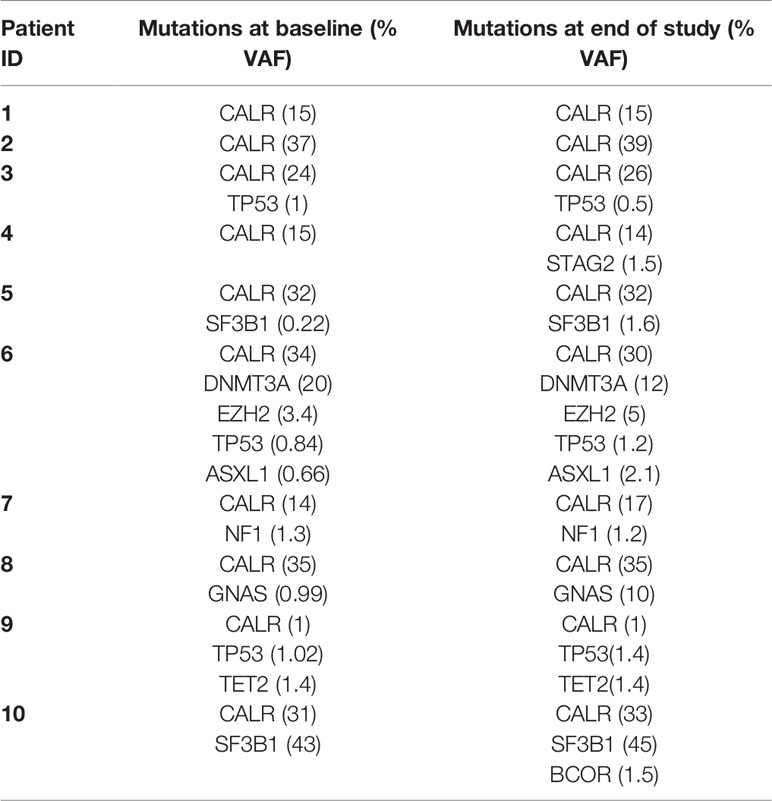
Table 4 Mutant variant allele frequency (VAF) of CALR and additional mutations identified by next generation sequencing.
Immune Response to the Vaccination Antigen in Peripheral Blood Mononuclear Cells
Patient PBMC isolated at baseline and after three, six, and 15 vaccinations were analyzed for immune responses against the vaccination antigen using IFN-γ ELISPOT. We detected a DFR2x-defined immune response in four of 10 patients at baseline (Figure 2A). Interestingly, the vaccinations induced immune responses in several patients that did not show an immune response at baseline: Seven of 10 patients displayed at least a DFR-defined response after three vaccinations, six of 10 had at least a DFR-defined response after six vaccinations, and seven of 10 had at least a DFR-defined response at end of study (Figure 2A). The amplitude of the responses increased over time (Figure 2B), but interestingly the response in patients with MF did not increase after the third vaccination, whereas the amplitude of the response in patients with ET increased during the entire vaccination schedule (Figure 2C). The difference in response amplitude between patients with ET and PMF were statistically significant at 6th vaccination and at the end of the study (Figure 2C). Two patients (patients 3 and 10), both of whom had PMF, did not attain an immune response at all during the vaccination schedule.
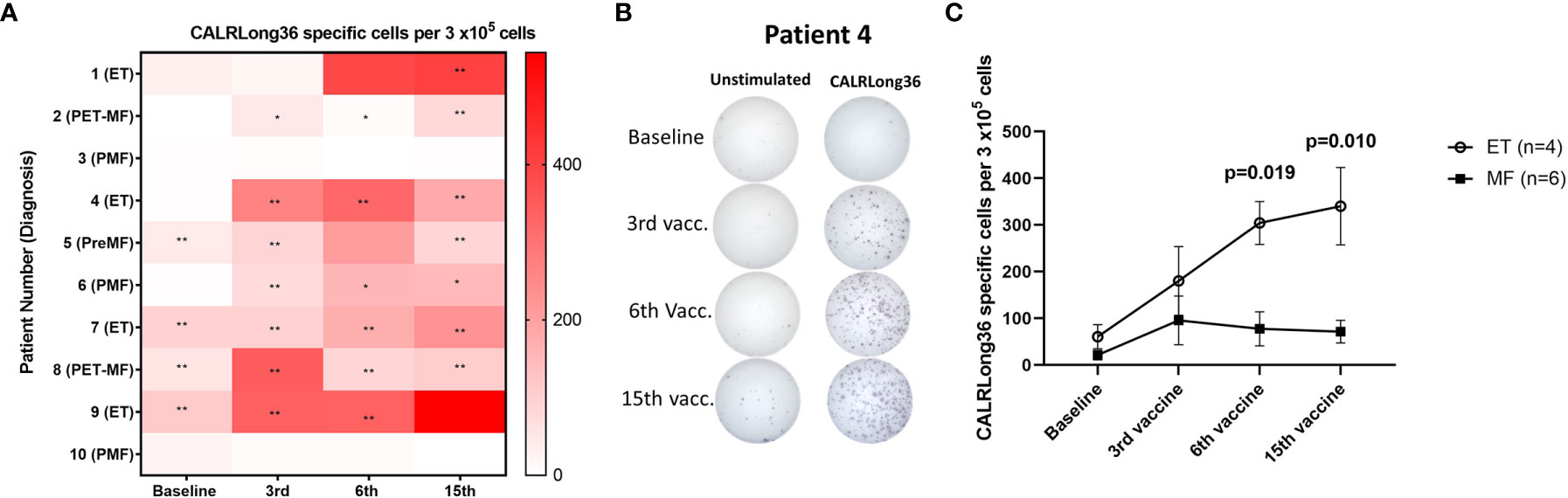
Figure 2 Immune responses to CALRLong36 in IFN-γ ELISPOT. (A) Heat map depicting the responses to CALRLong36 in patient peripheral blood mononuclear cells at each time point for all patients. The number of CALRLong36-specific cells was calculated by subtracting the mean spots in the control wells from the mean spots in the peptide-stimulated wells. The analysis was only performed in duplicates for patient 1 and 5 at the 6th vaccination and for patient 9 at the 15th vaccination, which prevented us from performing statistical analysis of the results. All other experiments were performed in triplicates. * Indicates a statistically significant response according to the DFR-rule. ** Indicates a statistically significant response according to the DFR(2x)-rule (38). (B) Representative image from (A, C) ELISPOT responses over time in patients with essential thrombocythemia (ET) and patients with myelofibrosis (MF) with each dot representing the mean of normalized spots. Error bars depict the standard error of the mean. Statistical testing was performed using the Mann-Whitney test.
The phenotype of the cytokine-producing cells was evaluated using intracellular cytokine staining (ICS). From the eight patients who showed an immune response to CALRLong36 in ELISPOT, we analyzed PBMC isolated at the time point where the response in ELISPOT was deemed to be highest. For patients 3 and 10, who did not display an immune response in ELISPOT, we chose to analyze PBMC isolated after 15 vaccinations. Not surprisingly, we did not identify a response in patients who did not show a response in ELISPOT. Additionally, patient 5 had a very low fraction of CD3+ T cells, making us unable to gate on a satisfactory number of T cells for our analysis. However, of the remaining seven patients, six displayed a CD4+ T-cell response to stimulation with CALRLong36 (Figures 3A, C), and two patients displayed a CD8+ T-cell response (Figures 3B, D). Patient 7 demonstrated both a CD4+ and a CD8+ T-cell response, whereas patient 2 demonstrated only a CD8+ T-cell response. Patient 2 provided informed consent for us to isolate PBMC at several time points after the end of study and showed a sustained CD8+ T-cell response to CALRLong36 even at 37 and 49 weeks after the last vaccination (Supplementary Figure 5).
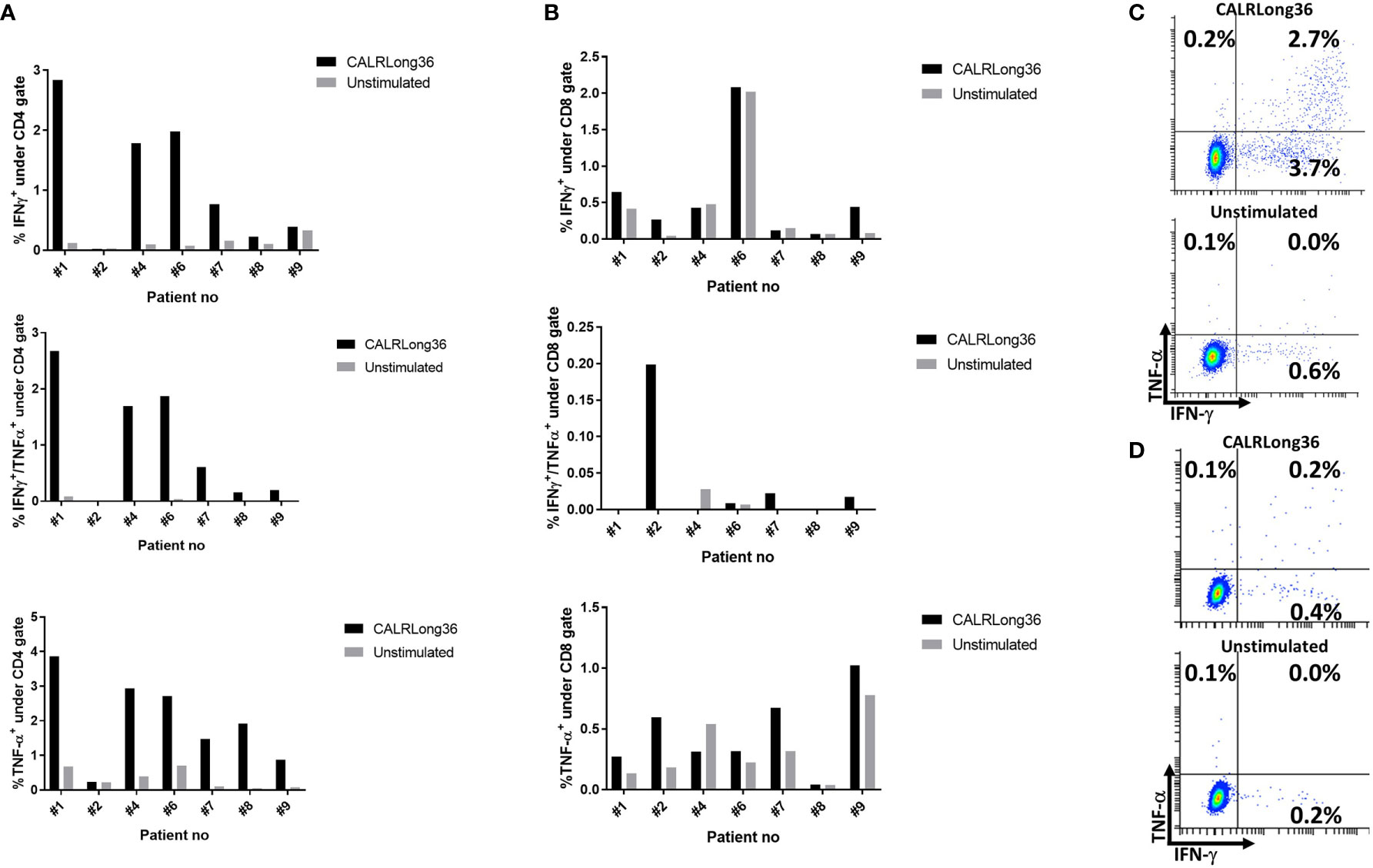
Figure 3 Responses against CALRLong36 identified by intracellular cytokine staining in patients with a response in ELISPOT. (A) Responses in CD4+-gated T cells with IFN-γ secreting cells (top), IFN-γ/TNF-α secreting cells (middle). and TNF-α secreting cells (bottom). (B) Responses in CD8+-gated T cells with IFN-γ secreting cells (top), IFN-γ/TNF-α secreting cells (middle), and TNF-α secreting cells (bottom). (C) An example of a CD4+ T-cell response. (D) An example of a CD8+ T-cell response.
Delayed-Type Hypersensitivity and Response in Skin-Infiltrating Lymphocytes
Ten patients received intradermal injections with CALRLong36 as described in Materials and Methods. Of note, none of the patients demonstrated an induration at the site of intradermal injections. In order to investigate if the injections could have induced a migration of CALRLong36-specific T cells to the injection site, 10 patients were subjected to a punch biopsy at the injection site. Punch biopsies were cultured as described previously (39) to expand specific T cells in the biopsy. Surprisingly, even though none of the patients displayed an induration at the injection sites, we were able to expand skin-infiltrating lymphocytes (SKILs) from five patients by culturing with low dose IL-2 (100 U/mL) and SKILs from seven patients by culturing with high dose IL-2 (6,000 U/mL). The SKILs were tested in IFN-γ ELISPOT for response to CALRLong36. Only patient 6 displayed a response (Figure 4); the responses in the remaining patients were either absent or the background was too high (Supplementary Figure 6).
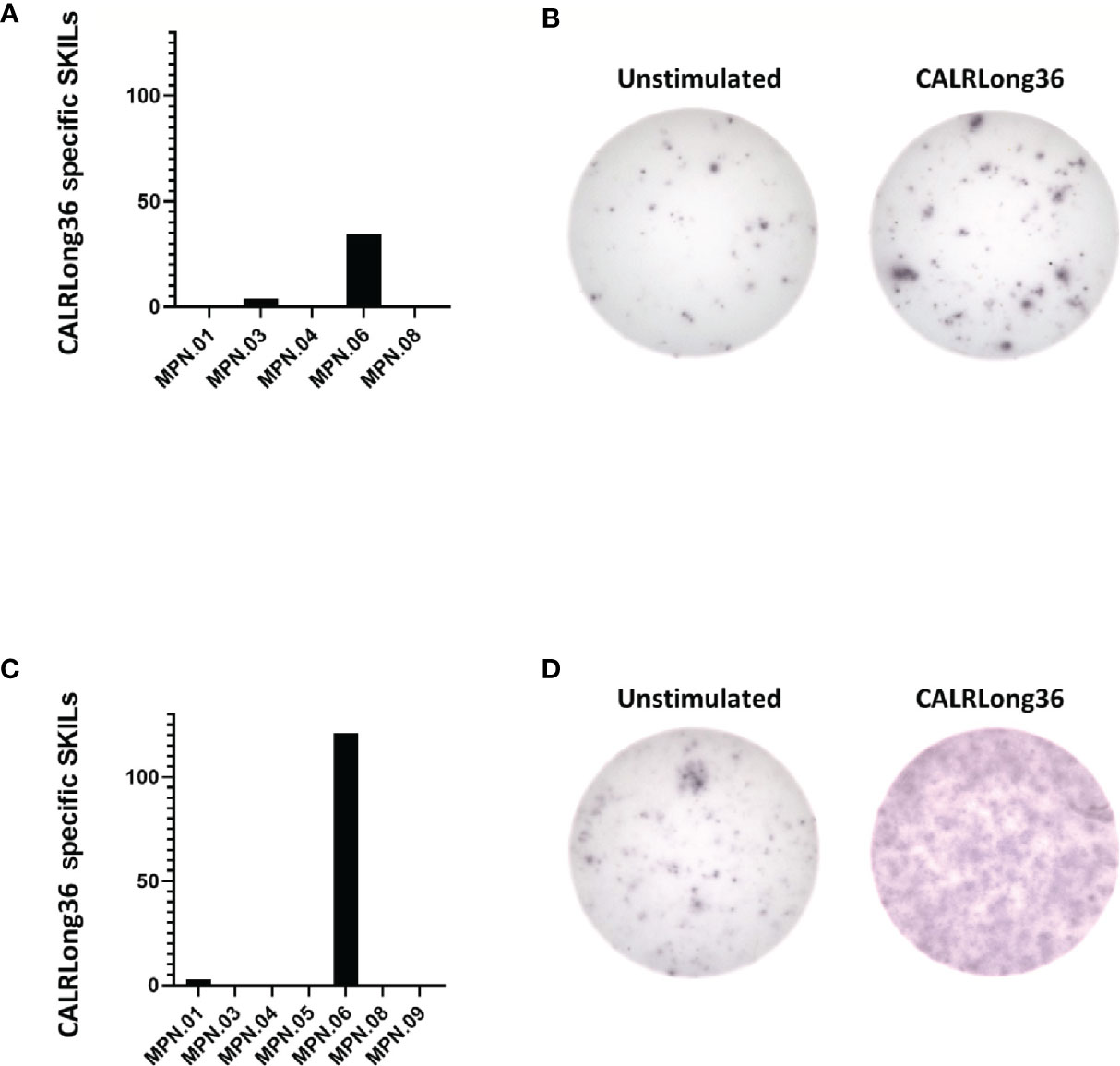
Figure 4 Responses in skin-infiltrating lymphocytes (SKILs) against CALRLong36. SKILs were harvested as described in the Materials and Methods section and were then expanded in either high-dose IL-2 (6,000 U/mL) culture medium or low-dose IL-2 (100 U/mL) culture medium. Cells were harvested and analyzed by IFN-γ ELISPOT for a response against CALRLong36. (A) Normalized numbers of cells specific to CALRLong36 in SKILs from patient 1, 3, 4, 6 and 8 cultured in low dose IL-2. (B) Response in SKILs from patients 6 expanded in low dose IL-2. (C) Normalized numbers of cells specific to CALRLong36 in SKILs from patient 1, 3, 4, 5, 6, 8 and 9 cultured in high dose IL-2. (D) Response in SKILs from patients 6 expanded in high dose IL-2.
Phenotype of Peripheral Blood Mononuclear Cells During the Vaccination Schedule
Alterations in the composition of immune cells in peripheral blood were analyzed during the vaccination schedule. No alterations were identified in the number of T cells in general, nor in the numbers of CD4+ and CD8+ T cells. Neither did we detect any changes in the levels of CD14+ monocytes. B cells increased slightly during the study, but returned to baseline levels at the end of the trial. NK cells decreased initially, but towards the end of the study they reached their level at baseline (Figure 5A). Interestingly, we identified a statistically significant decrease in the number of circulating CD4+ central memory T cells (TCM) in combination with a statistically significant increase in the number of CD4+ effector memory T cells (TEM) (Figure 5B). Changes in the CD8+ T-cell fraction were not dramatic, but we detected a somewhat paradoxical increase in the number of CD8+ naïve T cells (Tnaïve) during the vaccination schedule, and levels of Tnaïve only returned to baseline levels at the end of the study period (Figure 5C). Conversely, the number of CD8+ TEMRA decreased after the first three vaccinations, after which it increased to reach the levels at baseline (Figure 5C). Levels of PD-1 on both T cells in total and in CD4+ and CD8+ T cells remained constant, as did the levels of regulatory T cells (Treg) (Figure 5D). As PD-1 expression is increased on T cells in patients with MPN (40, 41) we sought to analyze differences in PD-1 expression between ET- and PMF-patients. Most interestingly, we showed that at the third and sixth vaccination, CD4+ T cells from patients with MF expressed significantly higher levels of PD-1 compared with patients with ET (Figure 6A). A difference in expression levels was also detected at baseline and at the 15th vaccination, but the difference was not statistically significant (Figure 6A). On the other hand, expression of PD-1 by CD8+ T cells did not differ between patients with ET and PMF (Figure 6B). No significant changes were identified in the different NK cell subsets (Figure 7A). Of note, levels of monocytic myeloid-derived suppressor cells (mMDSC) increased initially but decreased to a level lower than that at baseline (Figure 7B), and the CD4+/CD8+ T-cell ratio remained constant throughout the study (Figure 7C).
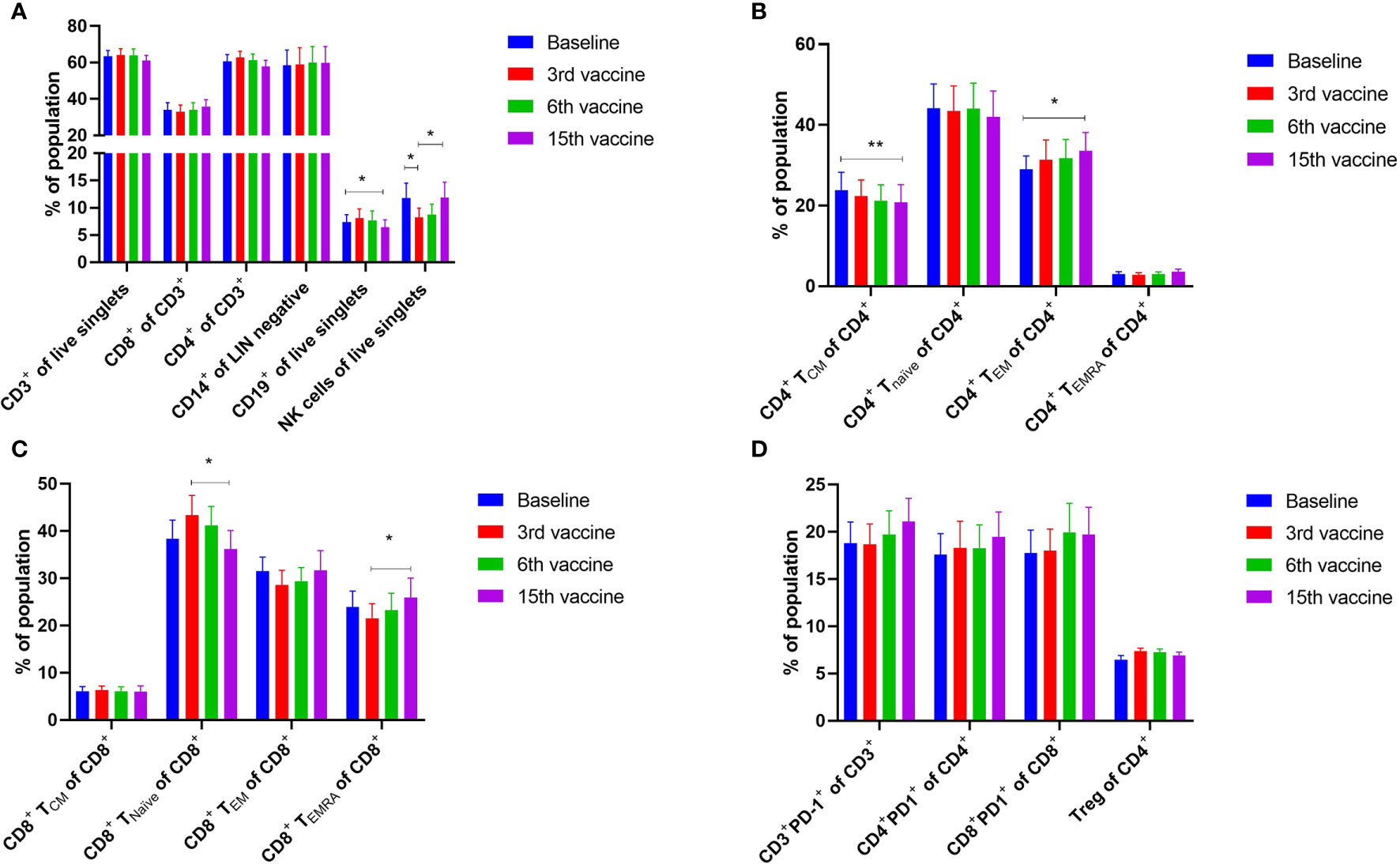
Figure 5 Phenotyping of peripheral blood mononuclear cells (PBMC) and T cells by fluorescence-activated cell sorting. (A) Quantification of the mean PBMC subsets in peripheral blood of vaccinated patients. (B) Quantification of CD4+ central memory T cells (TCM), CD4+ naïve T cells (Tnaïve), CD4+ effector memory T cells (TEM), and CD4+ effector memory T cells re-expressing CD45RA (TEMRA). (C) Quantification of CD8+ central memory T cells (TCM), CD8+ naïve T cells (Tnaïve), CD8+ effector memory T cells (TEM), and CD8+ effector memory T cells re-expressing CD45RA (TEMRA). (D) Expression levels of PD-1 on T cells and CD4+ and CD8+ T cells, and number of regulatory T cells (Treg). Statistical testing was performed using the Wilcoxon signed-rank test. Bars represent standard error of the mean. * denotes p ≤ 0.05, ** denotes p ≤ 0.01.
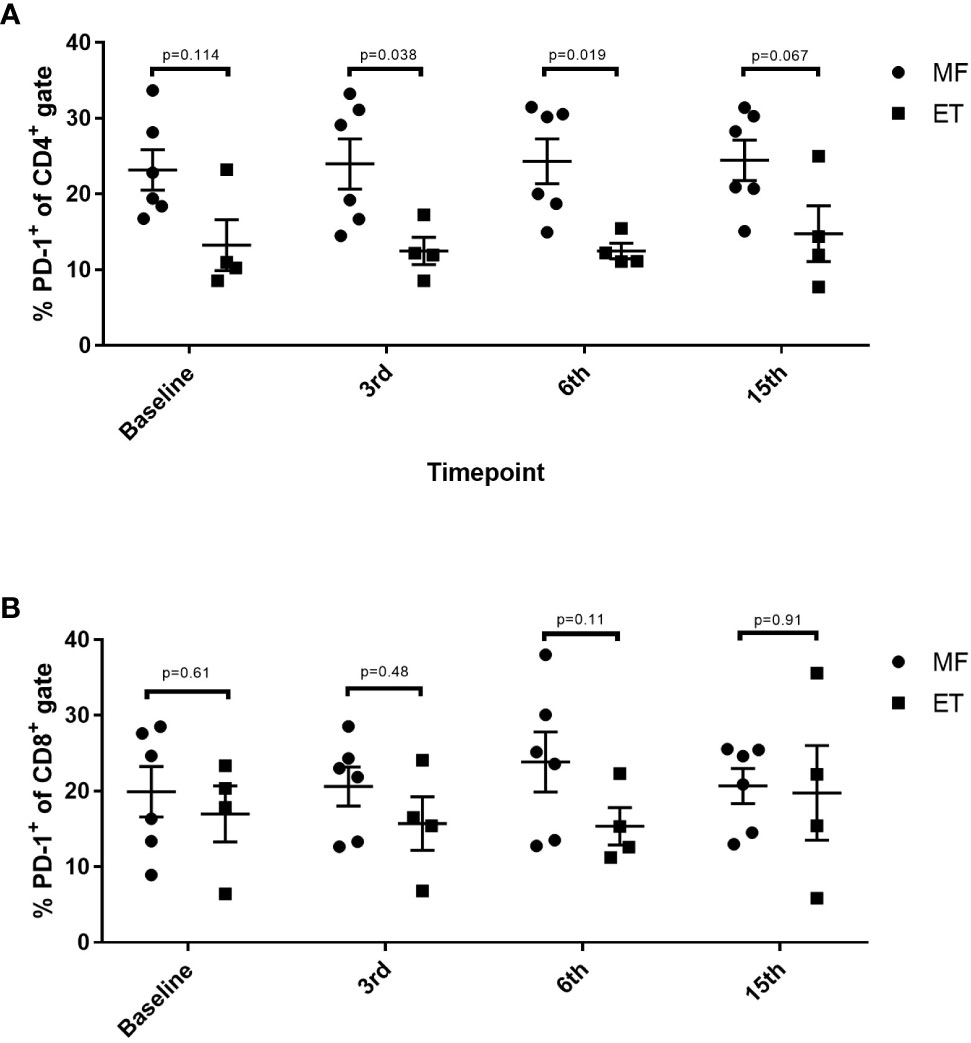
Figure 6 Changes in levels of PD-1 expression in CD4+ and CD8+ T cells in patients with essential thrombocythemia (ET) and primary myelofibrosis (PMF) during the vaccination schedule. (A) PD-1 expression on CD4+ T cells. (B) PD-1 expression on CD8+ T cells. Statistical testing was performed using the Mann-Whitney test. Bars represent standard error of the mean.
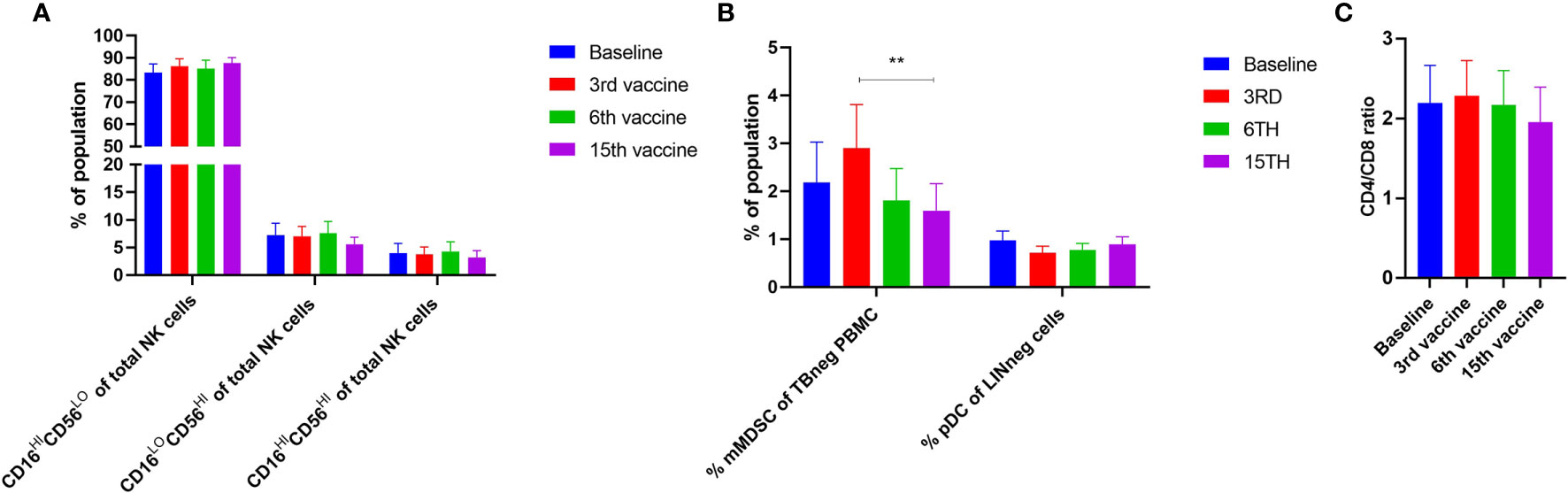
Figure 7 Phenotyping of NK cells and other cells fractions in PBMC by fluorescence-activated cell sorting. (A) Levels of different NK cell subsets. (B) Alterations in the levels of monocytic MDSC (mMDSC) and plasmacytoid dendritic cells (pDC). (C) Quantification of the CD4+/CD8+ ratio during the trial. Statistical testing was performed using the Wilcoxon signed-rank test. Bars represent standard error of the mean. ** denotes p ≤ 0.01.
Discussion
In order to attain a clinical response using cancer immune therapy, it is of utmost importance that the tumor cells express immunogenic antigens that may be targeted by specific T cells (42). The RAS mutations generate several immunogenic neo-antigens that are targets of specific T cells, and therapeutic cancer vaccines targeting these neo-antigens have shown a survival benefit in patients with an immune response to the vaccine epitope (33, 43). Vaccination of AML patients with immunogenic tumor-associated antigens such as Wilms tumor antigen-1 (WT1) and NY-ESO have also demonstrated a survival benefit for patients with an immune response (44, 45). The NPM1 mutations identified in a substantial proportion of patients with AML generate highly immunogenic antigens (46), and it has been shown that patients with an intact immune response to NPM1-derived neo-antigens have significantly longer overall survival compared with patients without a response (47). As such, the existence of a neo-antigen-specific immune response is believed to be highly important in attaining a therapeutic effect. In recent years, research has mainly focused on the potentially large number of patient-specific, mutated neoantigens. They comprise the predicted products of the numerous mutations revealed by exome sequencing of primary tumors, and it was demonstrated that it is not only the number of mutations that predict outcome but also the immunogenicity of the antigens generated by the somatic mutations (48).
We have shown earlier that patients with CALRmut MPN harbor T cells specific to neo-antigens derived from the mutant CALR C-terminus. However, patient T-cell responses are less frequent and weaker than the responses identified in healthy donors. This could indicate that patients with CALRmut MPN harbor an exhausted CALRmut-specific immune response, and we speculated that therapeutic cancer vaccination with a CALRmut-derived peptide could enhance the tumor-specific immune response in patients and lead to a clinical response. As the CALR mutations have been shown to be early mutations residing in the highly undifferentiated HSC (21), a strong CALRmut-specific immune response in patients may be of concern, as a strong immune response could potentially eradicate all CALRmut HSC in the patients, and thus, result in bone marrow failure. Only a few side effects were identified in our study, with only two grade 3 AE. One patient developed hypertension during the trial, but this occurred before the administration of the first vaccine, thus it is impossible that the vaccines elicited this AE. Another patient experienced a vasovagal episode just prior to the end-of-study bone marrow biopsy. The episode could be explained simply by the fact that bone marrow biopsies are highly uncomfortable and somewhat painful for the patients. Apart from these, no severe AE (SAE) were observed. The most frequent AE registered in this trial, infection, fatigue and myalgia, are complaints that are common in patients with MPN. The injection-site reactions experienced by all patients and the flu-like symptoms experienced by 50% of the patients are likely reactions to the montanide adjuvant in the vaccine. The occurrence of these AE could actually be regarded as a beneficial sign, as they indicate that a proper inflammatory response to the vaccine was induced. In conclusion, this vaccination trial adds weight to the notion that cancer vaccines only very rarely induce SAE (49). The patient that experienced a transient increase in ALAT showed evidence of increased SMA even before study entry, thus this AE is not believed to be induced by the vaccines.
Given the immunogenicity of the CALR mutations, we expected the vaccines to induce a clinical response in patients. As the CALR mutations mainly affect the megakaryocytic lineage (50–53) and patients with CALRmut MPN display higher platelet counts than their JAK2V617F+ counterparts (54, 55), we expected that a clinical response would be identified by a decline in platelet counts. Indeed, we detected a decline in platelet counts during the first 100 days, after which the numbers increased. Importantly, apart from patient 6 and 7, no patients had any adjustments in their MPN-directed therapy during the trial. Thus, this cannot explain the changes detected in platelet counts. A potential cause of the decline in platelets is a tumor-specific immune response that targets the megakaryocytes and thus reduces platelet production. Importantly, the subsequent increase in platelet counts coincided with the conversion of biweekly vaccinations into monthly vaccinations. To our belief, the decline in platelet counts during the first period of time could be explained by a tumor specific T-cell response and certainly needs further investigation in future trials.
The decline in platelet counts did not translate into a decline in the CALRmut VAF. In daily practice the VAF oscillates over time but largely remains constant, which was also recorded in our cohort. Theoretically, some patients were more likely to display a response to the vaccines than others. Thus, patient number 6 was not receiving any treatment at inclusion, but after seven vaccinations it was decided to start treatment with IFN-α (45 µg weekly) due to a high platelet count. As IFN-α is a highly immunostimulatory cytokine, we believed that the addition of IFN-α to the vaccines would induce a molecular response. Patient number 9 displayed a very low CALRmut VAF of only 1%. Earlier reports on therapeutic cancer vaccines suggest that patients with a low tumor burden are more likely to benefit from therapy (56, 57). Since patient 9 had a relatively low tumor burden we believed that the low number of tumor cells would not impede the tumor-specific immune response. Despite the low tumor burden in this patient, the vaccines did not induce a clinical nor molecular response. However, it is intriguing to consider, whether the lack of responses might be attributed to the additional mutations in both TP53 and TET2, rendering the CALR-mutant clone resistant to immune mediated clearing.
Our results clearly demonstrate that a lack of clinico-hematological and molecular response to the CALRmut-vaccine is not explicitly due to incompetent CALRLong36-specific immune responses. Thus, eight patients displayed strong immune responses to CALRLong36. Of these eight patients, four showed an immune response at baseline that was later enhanced by the vaccines. In the remaining four patients who displayed an immune response to CALRLong36, the response was absent at baseline but induced during the course of the trial. Two patients, both of whom have PMF, did not display an immune response against the vaccine. Interestingly, we found that the immune response in patients with ET increased during the vaccination course, in contrast to patients with PMF in whom the amplitude of the immune response remained stable after the first three vaccinations. We believe that this could be due to the more severe immune dysfunction in patients with PMF, and the results are on par with our earlier results showing that patients with ET have more frequent responses than patients with PMF (23, 58, 59). Recently it was demonstrated that monocyte-derived dendritic cells (moDC) from patients with PMF display lower levels of costimulatory molecules compared with healthy-donor moDC, and that these moDC have inferior priming potential (60). This could explain the weaker responses identified in patients with PMF. In contrast, T cells from patients with CALRmut MPN are more prone to activation, and Treg from CALRmut patients display a lower inhibitory potential than Treg from CALRwt patients (60), which should counteract the deficiency in moDC. We hypothesized that the differences in immune responses might be partially explained by the occurrence of additional mutations, however we did not identify any association between lack of immune response and additional mutations identified by NGS.
Earlier reports have shown that the majority of CALRmut-specific immune responses identified are CD4+ T-cell responses (22, 23, 41, 61). In this trial, six of seven patients showed a CD4+ T-cell response, and one of these displayed a CD8+ T-cell response as well. One patient displayed only a CD8+ T-cell response, and this response was sustained even after cessation of vaccination. Earlier trials have shown that a sustained tumor-specific CD8+ T-cell response is important in attaining a clinical response (62), and the apparent lack of CD8+ T-cell responses could explain the lack of clinical results. However, the low frequency of CD8+ T-cell responses could also be due to our experimental setup during the ICS, as the cells are only allowed to incubate with the peptide for 5 h, thus leaving only a little time for antigen-presenting cells to process and present possible CD8+ epitopes. Arshad et al. showed that the CALR mutations inhibit the presentation of peptides by human leukocyte antigen (HLA)-I molecules (63), as the CALR protein is an important chaperone in the assembly of the peptide:HLA-I complex. The low number of CD8+ T-cell responses identified could thus also be explained by the inability of patient antigen-presenting cells to process and present high-quality HLA-I-restricted epitopes. We do therefore find it noteworthy that two of the seven patients analyzed showed a CALRmut-specific CD8+ T-cell response.
Earlier peptide vaccination trials have evaluated the immune response not only by measuring response in PBMC but also by measuring the immune response in SKILs and the occurrence of a DTH induration from the vaccination antigen after intradermal injection. Collectively, patients with tumor-specific T cells at the DTH site have a greater chance of a clinical response to vaccination (64, 65). Strikingly, no patients displayed a DTH induration in this trial. We performed punch biopsy at the site of the intradermal injection in all patients and were able to grow SKILs from eight patients. These SKILs were tested in IFN-γ ELISPOT assays and only one patient displayed a response to CALRLong36. The apparent lack of DTH in our patient cohort is striking. First, because the antigen is highly immunogenic, and second because the majority of patients receiving intradermal injections with antigen in other trials show a DTH. As no patients demonstrated a DTH, we speculate that this was due to a general feature, either of CALRLong36 that prevents it from being processed and presented by dermal dendritic cells, or of MPN patients in general, who might harbor a deficiency in the dermal immune system that renders the dermal immune cells hyporesponsive to stimuli. Our current trial testing vaccinations with epitopes derived from programmed death ligand (PD-L)-1 and arginase (ARG)-1 (NCT04051307) will shed further light on this question. Of note, patients with psoriasis, an autoimmune skin disease primarily driven by T cells (66), are at an elevated risk of developing PMF (67), which could indicate that the dermal immune system in at least patients with PMF is dysregulated.
As therapeutic cancer vaccines generally do not induce any systemic effects, a marked alteration in the immune phenotype of patients was not expected. However, as patients with mutated CALR are heterozygous for the mutations, and the mutant VAF reaches almost 50% in all patients, essentially all cells of myeloid origin are believed to carry the mutation. Additionally, we have shown that T and B cells may also harbor the mutation (68). As such, T-cell mediated targeting of the CALR mutations has the potential to have a noticeable effect on the phenotype of circulating immune cells. Alterations in the phenotype of effector or suppressor immune cells could also be expected. We identified an increase in the number of CD4+ TEM, whereas CD4+ TCM levels declined. We believe that these data are highly interesting, as they could imply that the immune system reaches a more effector-like phenotype as TCM differentiate into TEM, which are more active in cytotoxic killing of tumor cells (69, 70). CD8+ Tnaïve increased initially, and then declined. The decline might be explained by the priming of Tnaïve by the vaccine, while the subsequent increase in CD8+ TEMRA might be explained by the sustained presentation of antigen to T cells that subsequently terminally differentiate and become hyporesponsive. Riley et al. monitored the phenotype of circulating immune cells in patients with JAK2V617F+ PV treated with IFN-α extensively and identified an increase in Treg numbers in these patients (71). Another study demonstrated an expansion of CD56hi NK cells in patients receiving IFN-α for more than a year (72). We did not identify any changes in the levels of Tregs nor in the numbers of NK cell subsets, but the total number of NK cells declined after the first vaccines, after which the levels increased to baseline levels after 15 vaccinations. As noted above, the main CALRmut cells in the PBMC fraction are monocytes, and a clinical response to therapy would likely be reflected by decreasing numbers of CD14+ monocytes in the PBMC fraction, but such changes were not identified. Monocytic myeloid-derived suppressor cells (mMDSC) are cells of myeloid origin that are highly immunosuppressive (73). Patients with MPN have elevated levels of mMDSC in their peripheral blood (74), which could impede the tumor-specific immune response. Patients experienced a small increase in peripheral blood mMDSC followed by a decrease, a phenomenon also identified in CD19+ B cells. Neither of these were highly significant and to our thinking are not alterations reflecting the effect of the vaccinations. Earlier studies of the expression of PD-1 on T cell in patients with MPN have shown that PD-1 expression is increased on patient T cells compared to healthy donor T cells (40, 41). We did not have a healthy donor cohort for comparison in this study, but compared our subjects’ levels with the normal levels identified in earlier reports (40, 41). Looking at PBMC from CALRmut patients only, Cimen-Bozkus et al. found that 19.2% of CD3+ T cells were PD-1+, which is comparable to our results (41). The PD-1 expression on CD4+ and CD8+ T-cells are also comparable to our results, and in another report analyzing PBMC from both JAK2V617F+ and CALRmut patients, the numbers of PD-1+ CD4+ and CD8+ T-cells were even higher (40). Of note, both reports show that patient T cells have significantly higher expression of PD-1 compared to healthy-donor T cells, and several additional exhaustion markers such as CTLA-4 have also been shown to be enhanced in T cells from CALRmut patients (41).
As the majority of responses to mutant CALR in both patients and healthy donors are CD4+ T cell responses (23, 41, 61), and mutant CALR is expressed on the surface of mutant cells (53), one would expect the occurrence of CALRmut specific antibodies. However, the occurrence of such antibodies has not been reported. Of note, CALRmut cells secrete mutant CALR to the extracellular compartment (75, 76), and patients with PMF exhibit elevated levels of circulating CALR compared to healthy donors (77). This could explain the apparent lack of antibodies specific to mutant CALR, as circulating mutant CALR could neutralize the formed antibodies making these undetectable and additionally impede on the anti-tumor effect of the antibodies. Other possible effects of circulating mutant CALR could be that the mutant protein induces tolerance in specific T cells due to chronic antigen stimulation.
One of the main conclusions from this trial is that even though therapeutic cancer vaccination against an antigen derived from the CALR exon 9 mutations induces T-cell responses specific to the vaccination antigen, this does not translate into a molecular response. Thus, future trials employing therapeutic cancer vaccinations against mutant CALR should aim to combine the vaccines with other treatments. This should be highly feasible as the vaccination is well tolerated. One obvious treatment to combine with a vaccine would be IFN-α. IFN-α enhances the Th1 immune response (8) and, specifically in MPN, the expression of genes related to antigen processing and presentation (78). However, the majority of patients in our trial were receiving IFN-α, and we cannot rule out that the dose administered is too low to enhance the effect of the vaccine. Immune-checkpoint inhibitors (ICPI) have demonstrated strong clinical potential in several solid cancers and in the treatment of Hodgkin lymphoma (79), and the potential of using ICPI to treat MPN is highly intriguing. A recent report showed that treatment of CALRmut MPN patients with ICPI induces an immune response in patient PBMC stimulated in vitro with both the peptide and ICPI (41), adding impetus to the notion that ICPI can be used to enhance a neo-antigen-specific immune response in patients with CALRmut MPN. However, data on the clinical effect of ICPI are yet to be reported. Considering the immense number of adverse events observed following treatment with ICPI, caution should be used when treating patients with non-advanced MPN, as several less toxic treatment modalities are at hand for these patients. It has been shown that patients with CALRmut PMF have better overall survival after alloHSCT compared with patients with CALRwt PMF (80). This could be explained by recognition and clearing of residual CALRmut cells by donor T cells. In the setting of alloHSCT, donor lymphocyte infusion (DLI) can be used for patients who do not attain complete remission after alloHSCT. DLI acts by enhancing the graft-versus-leukemia effect and has shown its potency in the treatment of PMF (81). Interestingly, a patient with NPM1-mutant AML was brought into remission by DLI, and after engraftment the authors detected NPM1mut-specific T-cell responses by ELISPOT (82). One probable mechanism beyond the therapeutic effect of the DLI may be that NPM1mut-specific T cells in the infusion product have recognized and killed residual AML blasts. Vaccination of HSCT donors before harvest of the DLI product could potentially enhance the number of CALRmut-specific T cells in the infusion product, thus increasing the likelihood of DLI-mediated clearing of residual CALRmut cells. Another highly active compound in MPN is ruxolitinib which is a very potent anti-inflammatory drug that inhibits JAK1-2 signaling. Ruxolitinib decreases constitutional symptoms and reduces splenomegaly in a majority of patients (83). However, combination of ruxolitinib and vaccines is not believed to induce clinico-hematological responses in patients since ruxolitinib, due to its JAK1-2 inhibiting properties, attenuates the functionality of dendritic cells, NK cells and T cells (84–86). Hence, the combination of ruxolitinib with cancer immune therapeutic modalities is not believed to result in clinical responses.
As noted above, several studies have shown that patients with MPN harbor an increased number of MDSC in peripheral blood, and patient T cells express elevated amounts of PD-1 in conjunction with elevated amounts of PD-L1 (40). Together, these factors render the immune environment in MPN highly immunosuppressive, which could also explain the lack of response to the vaccines. As mentioned, one way to circumvent this immune suppression would be ICPI. However, it has recently been reported that the immune system itself harbors autoreactive anti-regulatory T cells that are able to kill regulatory immune cells through the recognition of regulatory proteins and enzymes (87). T cells specific to important immunoregulatory molecules such as PD-L1, PD-L2, indoleamine 2,3-dioxygenase (IDO), and ARG-1/2 have been described (88–92), and vaccination with IDO- and PD-L1-derived epitopes have already shown clinical benefit to patients with stage IV non-small-cell lung cancer and metastatic melanoma (93, 94). As patients with MPN have increased levels of several of these regulatory proteins, we believe that induction of an anti-regulatory immune response through vaccination with one or several anti-regulatory T cell epitopes will enhance the anti-regulatory immune responses which ultimately will lower the immune suppression in patients (95). In combination with CALRmut-specific therapeutic cancer vaccination, these anti-regulatory vaccines have the potential to induce a clinical response, especially as patients with MPN harbor T cells specific to both PD-L1- and ARG-1-derived epitopes (58, 59). This finding spurred us to initiate a phase I/II vaccination trial testing the combination of PD-L1- and ARG-1-derived epitopes in patients with ET and PV (NCT04051307).
Immune tolerance might be another barrier to the vaccines having an effect. Patients with MPN may live for decades with their disease. As chronic antigen stimulation has been shown to induce tolerance in antigen-specific T cells, we speculate that specific T cells in patients with CALRmut MPN are exhausted and the effect of vaccines is simply not enough to revert this tolerance. Recently, a Danish population-based study showed that healthy donors may harbor CALR exon 9 mutations with a low VAF without showing any signs of MPN (96). Thus, we believe that CALRmut MPN may evolve due to immune escape after prolonged exposure of CALRmut antigens to T cells, and speculate that therapeutic cancer vaccination at a very early stage, if possible the preclinical stage, will induce a clinical effect. Of note, CALRmut healthy donors identified in the study by Cordua et al. harbor CALRmut-specific immune responses, supporting the notion that these individuals hold the CALRmut HSC at bay through T-cell-mediated elimination of mutant cells (97). Thus, we suggest early up-front vaccination therapy in order to minimize the risk of T-cell exhaustion and concurrently induce a T-cell response when the tumor burden is still relatively low. Although therapeutic cancer vaccination in healthy individuals is controversial, it is a feasible option given the low frequency of serious AE identified in this trial.
In conclusion, this clinical phase I vaccination trial with an epitope derived from the CALR exon 9 mutations showed that the vaccine is safe and tolerable. The vaccines either induced or enhanced an existing CALRmut-specific immune response in the majority of patients. However, the immune responses did not translate into a clinical response as no patients experienced any improvement in their disease status. Since the vaccines have a good safety profile, we suggest combining the vaccine with other immune therapeutic modalities in order to induce a clinical response.
Data Availability Statement
The data sets presented in this article are not readily available because Danish Law and the General Data Protection Rules prohibits this. Requests to access the data sets should be directed to molecular biologist VS at vihs@regionsjaelland.dk.
Ethics Statement
The studies involving human participants were reviewed and approved by Zealand Region Ethics Committee. The patients/participants provided their written informed consent to participate in this study.
Author Contributions
JHG conducted the trial, analyzed the data, and wrote the manuscript. MOH concieved the project, performed experiments, analyzed data, and wrote the manuscript. NJ analyzed data. UK analyzed data. SW-B performed experiments and analyzed the data. CS and MC recruited, treated patients, and analyzed data. LG and MB performed experiments and analyzed data. JK analyzed data. MH analyzed data and provided vital reagent. SK and NC analyzed data. GN, JP, LK, and VS performed experiments, analyzed data, and wrote the manuscript. ÖM analyzed data and provided vital reagents. IS analyzed data and provided vital reagent. HH and MHA conceived the project, analyzed data, and wrote the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This study was supported in part by Kræftens Bekæmpelse grant number R149-A10159-16-S47 and a pre-seed grant from the Novo Nordisk Foundation.
Conflict of Interest
MOH, HH, and MA have filed a patent regarding the CALR exon 9 mutations as a target for cancer immune therapy. The patent has been transferred to University Hospital Zealand, Zealand Region and Copenhagen University Hospital at Herlev, Capital Region according to Danish Law concerning inventions made at public research institutions.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank laboratory technician Merete Jonassen for her excellent assistance in performing the immunomonitoring for this study. We thank laboratory technicians Sandra Ullitz Færch, Barbara Thaysen and Bettina Johansen for isolation of peripheral blood mononuclear cells from the patients and for preparation of the vaccines in the study.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2021.637420/full#supplementary-material
Supplementary Figure 1 | Detailed in- and exclusion criteria.
Supplementary Figure 2 | Gant chart showing the vaccination schedule and additional examination/analyses. Abbreviations: V=vaccine, HB=health blood sample, SB=study blood sample, PE=physical examination, ECG=electrocardiogram, BM=bone marrow examination, DTH=delayed type hypersensitivity, NGS=next generation sequencing
Supplementary Figure 3 | Gating strategy for identification of immune cell subsets in vaccinated patients.
Supplementary Figure 4 | List of antibodies used for identification of immune cell substest in vaccinated patients.
Supplementary Figure 5 | Sustained CD8+ T-cell response in patient 2 even after cessation of vaccination, identified by intracellular cytokine staining. (A) CD8+ T-cell response in patient PBMC at 37 weeks after cessation of therapy. (B) CD8+ T-cell response in patient PBMC at 49 weeks after cessation of therapy.
Supplementary Figure 6 | Response in skin-infiltrating lymphocytes showing response in both the CALRLong36-stimulated wells and the negative control wells.
References
1. Campbell PPJ, Green AAR. The Myeloproliferative Disorders. N Engl J Med (2006) 355:2452–66. doi: 10.1056/NEJMra063728
2. Spivak JL. Myeloproliferative Neoplasms. N Engl J Med (2017) 376:2168–81. doi: 10.1056/NEJMra1406186
3. Arber DA, Orazi A, Hasserjian R, Borowitz MJ, Le Beau MM, Bloomfield CD, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood (2016) 127:2391–406. doi: 10.1182/blood-2016-03-643544.The
4. Wolanskyj AP, Schwager SM, Mcclure RF, Larson DR, Tefferi A. Essential Thrombocythemia Beyond the First Decade : Life Expectancy, Long-term Complication Rates, and Prognostic Factors. Mayo Clin Proc (2006) 81:159–66. doi: 10.1016/S0025-6196(11)61664-9
5. Mesa RA, Silverstein MN, Jacobsen SJ, Wollan PC, Tefferi A. Population-based incidence and survival figures in essential thrombocythemia and agnogenic myeloid metaplasia: An olmsted county study, 1976-1995. Am J Hematol (1999) 61:10–5. doi: 10.1002/(SICI)1096-8652(199905)61:1<10::AID-AJH3>3.0.CO;2-I
6. Deeg HJ, Gooley TA, Flowers MED, Sale GE, Slattery JT, Anasetti C, et al. Allogeneic hematopoietic stem cell transplantation for myelofibrosis. Transplantation (2003) 102:3912–8. doi: 10.1182/blood-2003-06-1856.Supported
7. Barbui T, Barosi G, Birgegard G, Cervantes F, Finazzi G, Griesshammer M, et al. Philadelphia-negative classical myeloproliferative neoplasms: critical concepts and management recommendations from European leukemiaNet. J Clin Oncol (2011) 29:761–70. doi: 10.1200/JCO.2010.31.8436
8. Belardelli F, Ferrantini M, Proietti E, Kirkwood JM. Interferon-alpha in tumor immunity and immunotherapy. Cytokine Growth Factor Rev (2002) 13:119–34. doi: 10.1016/s1359-6101(01)00022-3
9. Talpaz M, Hehlmann R, Quintás-Cardama A, Mercer J, Cortes J. Re-emergence of interferon-α in the treatment of chronic myeloid leukemia. Leukemia (2013) 27:803–12. doi: 10.1038/leu.2012.313
10. Kiladjian J, Cassinat B, Chevret S, Turlure P, Cambier N, Roussel M, et al. Pegylated interferon-alfa-2a induces complete hematologic and molecular responses with low toxicity in polycythemia vera. Blood (2008) 112:3065–72. doi: 10.1182/blood-2008-03-143537
11. Rank CU, Bjerrum OW, Larsen TS, Kjær L, de Stricker K, Riley CH, et al. Minimal residual disease after long-term interferon-alpha2 treatment: a report on hematological, molecular and histomorphological response patterns in 10 patients with essential thrombocythemia and polycythemia vera. Leuk Lymphoma (2016) 57:348–54. doi: 10.3109/10428194.2015.1049171
12. Hasselbalch HC, Holmström MO. Perspectives on interferon-alpha in the treatment of polycythemia vera and related myeloproliferative neoplasms: minimal residual disease and cure? Semin Immunopathol (2019) 41:5–19. doi: 10.1007/s00281-018-0700-2
13. Kiladjian J-J, Giraudier S, Cassinat B. Interferon-alpha for the therapy of myeloproliferative neoplasms: Targeting the malignant clone. Leukemia (2015) 30:1–6. doi: 10.1038/leu.2015.326
14. Barosi G. An Immune Dysregulation in MPN. Curr Hematol Malig Rep (2014) 9:331–9. doi: 10.1007/s11899-014-0227-0
15. Holmström MO, Hasselbalch HC, Andersen MH. Cancer immune therapy for Philadelphia chromosome-negative chronic myeloproliferative neoplasms. Cancers (Basel) (2020) 12:1–21. doi: 10.3390/cancers12071763
16. Villarino AV, Kanno Y, Ferdinand JR, O’Shea JJ. Mechanisms of Jak/STAT Signaling in Immunity and Disease. J Immunol (2015) 194:21–7. doi: 10.4049/jimmunol.1401867
17. Levine RL, Wadleigh M, Cools J, Ebert BL, Wernig G, Huntly BJP, et al. Activating mutation in the tyrosine kinase JAK2 in polycythemia vera, essential thrombocythemia, and myeloid metaplasia with myelofibrosis. Cancer Cell (2005) 7:387–97. doi: 10.1016/j.ccr.2005.03.023
18. Kralovics R, Passamonti F, Buser AAS, Teo S-SS-S, Tiedt R, Passweg JRJ, et al. A Gain-of-Function Mutation of JAK2 in Myeloproliferative Disorders. N Engl J Med (2005) 352:1779–90. doi: 10.1056/NEJMoa051113
19. Pardanani AD, Levine RL, Lasho T, Pikman Y, Mesa RA, Wadleigh M, et al. MPL515 mutations in myeloproliferative and other myeloid disorders: A study of 1182 patients. Blood (2006) 108:3472–6. doi: 10.1182/blood-2006-04-018879
20. Klampfl T, Gisslinger H, Harutyunyan AS, Nivarthi H, Rumi E, Milosevic JD, et al. Somatic mutations of calreticulin in myeloproliferative neoplasms. N Engl J Med (2013) 369:2379–90. doi: 10.1056/NEJMoa1311347
21. Nangalia J, Massie CE, Baxter EJ, Nice FL, Gundem G, Wedge DC, et al. Somatic CALR mutations in myeloproliferative neoplasms with nonmutated JAK2. N Engl J Med (2013) 369:2391–405. doi: 10.1056/NEJMoa1312542
22. Holmstrom MO, Martinenaite E, Ahmad SM, Met O, Friese C, Kjaer L, et al. The calreticulin (CALR) exon 9 mutations are promising targets for cancer immune therapy. Leukemia (2018) 32:429–37. doi: 10.1038/leu.2017.214
23. Holmström MO, Riley CH, Svane IM, Hasselbalch HC, Andersen MH, Holmstrom MO, et al. The CALR exon 9 mutations are shared neoantigens in patients with CALR mutant chronic myeloproliferative neoplasms. Leukemia (2016) 30:2413–6. doi: 10.1038/leu.2016.233
24. Coulie PG, Van den Eynde BJ, van der Bruggen P, Boon T. Tumour antigens recognized by T lymphocytes: at the core of cancer immunotherapy. Nat Rev Cancer (2014) 14:135–46. doi: 10.1038/nrc3670
25. Melief CJM, van Hall T, Arens R, Ossendorp F, van der Burg SH. Therapeutic cancer vaccines. J Clin Invest (2015) 125:1–12. doi: 10.1172/JCI80009
26. Prior IA, Lewis PD, Mattos C. A comprehensive survey of ras mutations in cancer. Cancer Res (2012) 72:2457–67. doi: 10.1158/0008-5472.CAN-11-2612
27. Qin H, Chen W, Takahashi M, Disis ML, Byrd DR, McCahill L, et al. CD4+ T-Cell Immunity to Mutated ras Protein in Pancreatic and Colon Cancer Patients. Cancer Res (1995) 55:2984–7.
28. Gedde-Dahl T, Eriksen JA, Thorsby E, Gaudernack G. T-cell responses against products of oncogenes: Generation and characterization of human T-cell clones specific for p21 ras-derived synthetic peptides. Hum Immunol (1992) 33:266–74. doi: 10.1016/0198-8859(92)90334-J
29. Fossum B, Olsen AC, Thorsby E, Gaudernack G. CD8+ T cells from a patient with colon carcinoma, specific for a mutant p21-Ras-derived peptide (Gly13–>Asp), are cytotoxic towards a carcinoma cell line harbouring the same mutation. Cancer Immunol Immunother (1995) 40:165–72. doi: 10.1007/BF01517348
30. Gjertsen KM, Bakka A, Breivik J, Saeterdal I. Vaccination with mutant ras peptides and induction of T-cell responsiveness in pancreatic carcinoma patients carrying the corresponding RAS mutation. Lancet (1995) 346:1399–400. doi: 10.1016/s0140-6736(95)92408-6
31. Rahma OE, Hamilton JM, Wojtowicz M, Dakheel O, Bernstein S, Liewehr DJ, et al. The immunological and clinical effects of mutated ras peptide vaccine in combination with IL-2, GM-CSF, or both in patients with solid tumors. J Transl Med (2014) 12:1–12. doi: 10.1186/1479-5876-12-55
32. Toubaji A, Achtar M, Provenzano M, Herrin VE, Behrens R, Hamilton M, et al. Pilot study of mutant ras peptide-based vaccine as an adjuvant treatment in pancreatic and colorectal cancers. Cancer Immunol Immunother (2008) 57:1413–20. doi: 10.1007/s00262-008-0477-6
33. Wedén S, Klemp M, Gladhaug IP, Mãller M, Eriksen JA, Gaudernack G, et al. Long-term follow-up of patients with resected pancreatic cancer following vaccination against mutant K-ras. Int J Cancer (2011) 128:1120–8. doi: 10.1002/ijc.25449
34. Tefferi A, Cervantes F, Mesa R, Passamonti F, Verstovsek S, Vannucchi AM, et al. Revised response criteria for myelofibrosis: International Working Group-Myeloproliferative Neoplasms Research and Treatment (IWG-MRT) and European LeukemiaNet (ELN) consensus report. Blood (2013) 122:1395–8. doi: 10.1182/blood-2013-03-488098
35. Barosi G, Mesa R, Finazzi G, Harrison C, Kiladjian JJ, Lengfelder E, et al. Revised response criteria for polycythemia vera and essential thrombocythemia: An ELN and IWG-MRT consensus project. Blood (2013) 121:4778–81. doi: 10.1182/blood-2013-01-478891
36. de Vries IJM, Bernsen MR, Lesterhuis WJ, Scharenborg NM, Strijk SP, Gerritsen M-JP, et al. Immunomonitoring tumor-specific T cells in delayed-type hypersensitivity skin biopsies after dendritic cell vaccination correlates with clinical outcome. J Clin Oncol (2005) 23:5779–87. doi: 10.1200/JCO.2005.06.478
37. Holmström MO, Andersen MH. Healthy donors harbor memory T cell responses to RAS neo-antigens. Cancers (Basel) (2020) 12:3045. doi: 10.3390/cancers12103045
38. Moodie Z, Price L, Gouttefangeas C, Mander A, Janetzki S, Löwer M, et al. Response definition criteria for ELISPOT assays revisited. Cancer Immunol Immunother (2010) 59:1489–501. doi: 10.1007/s00262-010-0875-4
39. Jørgensen NG, Klausen U, Grauslund JH, Helleberg C, Aagaard TG, Do TH, et al. Peptide Vaccination Against PD-L1 With IO103 a Novel Immune Modulatory Vaccine in Multiple Myeloma : A Phase I First-in-Human Trial. Front Immunol (2020) 11:1–11. doi: 10.3389/fimmu.2020.595035
40. Wang JC, Chen C, Kundra A, Kodali S, Pandey A, Wong C, et al. Programmed Cell Death Receptor (PD-1) Ligand (PD-L1) expression in Philadelphia chromosome-negative myeloproliferative neoplasms. Leuk Res (2019) 79:52–9. doi: 10.1016/j.leukres.2019.02.010
41. Bozkus CC, Roudko V, Finnigan JP, Mascarenhas J, Hoffman R, Iancu-Rubin C, et al. Immune checkpoint blockade enhances shared neoantigen-induced T-cell immunity directed against mutated calreticulin in myeloproliferative neoplasms. Cancer Discov (2019) 9:1192–207. doi: 10.1158/2159-8290.CD-18-1356
42. Schumacher TN, Schreiber RD. Neoantigens in cancer immunotherapy. Science (2015) 348:69–74. doi: 10.1126/science.aaa4971
43. Khleif SN, Abrams SI, Hamilton JM, Bergmann-Leitner E, Chen A, Bastian A, et al. A phase I vaccine trial with peptides reflecting ras oncogene mutations of solid tumors. J Immunother (1999) 22:155–65. doi: 10.1097/00002371-199903000-00007
44. Griffiths EA, Srivastava P, Matsuzaki J, Brumberger Z, Wang ES, Kocent J, et al. NY-ESO-1 Vaccination in Combination with Decitabine Induces Antigen-Specific T-Lymphocyte Responses in Patients with Myelodysplastic Syndrome. Clin Cancer Res (2017) 24:1019–29. doi: 10.1158/1078-0432.CCR-17-1792
45. Maslak PG, Dao T, Bernal Y, Chanel SM, Zhang R, Frattini M, et al. Phase 2 trial of a multivalent WT1 peptide vaccine (galinpepimut-S) in acute myeloid leukemia. Blood Adv (2018) 2:224–34. doi: 10.1182/bloodadvances.2017014175
46. Greiner J, Ono Y, Hofmann S, Schmitt A, Mehring E, Götz M, et al. Mutated regions of nucleophosmin 1 elicit both CD4+ and CD8+ T-cell responses in patients with acute myeloid leukemia. Blood (2016) 97:1282–90. doi: 10.1182/blood-2011-11-394395.There
47. Greiner J, Schneider V, Schmitt M, Götz M, Döhner K, Wiesneth M, et al. Immune responses against the mutated region of cytoplasmatic NPM1 might contribute to the favorable clinical outcome of AML patients with NPM1 mutations (NPM1mut). Blood (2013) 122:1087–8. doi: 10.1182/blood-2013-04-496844
48. Balachandran VP, Luksza M, Zhao JN, Makarov V, Moral JA, Remark R, et al. Identification of unique neoantigen qualities in long-term survivors of pancreatic cancer. Nature (2017) 551:S12–6. doi: 10.1038/nature24462
49. Rahma OE, Gammoh E, Simon RM, Khleif SN. Is the “3+3” dose-escalation Phase i Clinical trial design suitable for therapeutic cancer vaccine development? A recommendation for alternative design. Clin Cancer Res (2014) 20:4758–67. doi: 10.1158/1078-0432.CCR-13-2671
50. Elf S, Abdelfattah NS, Chen E, Perales-Pat??n J, Rosen EA, Ko A, et al. Mutant calreticulin requires both its mutant C-terminus and the thrombopoietin receptor for oncogenic transformation. Cancer Discov (2016) 6:368–81. doi: 10.1158/2159-8290.CD-15-1434
51. Nivarthi H, Chen D, Cleary C, Kubesova B, Jäger R, Bogner E, et al. Thrombopoietin receptor is required for the oncogenic function of CALR mutants. Leukemia (2016) 30:1759–63. doi: 10.1038/leu.2016.32
52. Chachoua I, Pecquet C, El-Khoury M, Nivarthi H, Albu RI, Marty C, et al. Thrombopoietin receptor activation by myeloproliferative neoplasm associated calreticulin mutants. Blood (2016) 127:1325–35. doi: 10.1182/blood-2015-11-681932
53. Araki M, Yang Y, Masubuchi N, Hironaka Y, Takei H, Morishita S, et al. Activation of the thrombopoietin receptor by mutant calreticulin in CALR-mutant myeloproliferative neoplasms. Blood (2016) 127:1307–16. doi: 10.1182/blood-2015-09-671172
54. Tefferi A, Guglielmelli P, Larson DR, Finke C, Wassie EA, Pieri L, et al. Long-term survival and blast transformation in molecularly annotated essential thrombocythemia, polycythemia vera, and myelofibrosis. Blood (2014) 124:2507–13. doi: 10.1182/blood-2014-05-579136
55. Rumi E, Pietra D, Pascutto C, Guglielmelli P, Martínez-Trillos A, Casetti I, et al. Clinical effect of driver mutations of JAK2, CALR, or MPL in primary myelofibrosis. Blood (2014) 124:1062–9. doi: 10.1182/blood-2014-05-578435
56. Qazilbash MH, Wieder E, Thall PF, Wang X, Rios R, Lu S, et al. PR1 peptide vaccine induces specific immunity with clinical responses in myeloid malignancies. Leukemia (2017) 31:697–704. doi: 10.1038/leu.2016.254
57. Kenter GG, Welters MJP, Valentijn a RPM, Lowik MJG, Berends-van der Meer D, Vloon APG, et al. Vaccination against HPV-16 oncoproteins for vulvar intraepithelial neoplasia. N Engl J Med (2009) 361:1838–47. doi: 10.1056/NEJMoa0810097
58. Holmström MO, Riley CH, Skov V, Svane IM, Hasselbalch HC, Andersen MH. Spontaneous T-cell responses against the immune check point programmed-death-ligand 1 (PD-L1) in patients with chronic myeloproliferative neoplasms correlate with disease stage and clinical response. Oncoimmunology (2018) 7:e1433521. doi: 10.1080/2162402X.2018.1433521
59. Aaboe-Jørgensen M, Holmström MO, Martinenaite E, Riley CH, Hasselbalch HC, Hald Andersen M. Spontaneous T-cell responses against Arginase-1 in chronic myeloproliferative neoplasms relative to disease stage and type of driver mutation. Oncoimmunology (2018) 7(9):e1468957. doi: 10.1080/2162402X.2018.1468957
60. Romano M, Sollazzo D, Trabanelli S, Barone M, Polverelli N, Perricone M, et al. Mutations in JAK2 and Calreticulin genes are associated with specific alterations of the immune system in myelofibrosis. Oncoimmunology (2017) 6:e1345402. doi: 10.1080/2162402X.2017.1345402
61. Holmström MO, Ahmad SM, Klausen U, Bendtsen SK, Martinenaite E, Riley CH, et al. High frequencies of circulating memory T cells specific for calreticulin exon 9 mutations in healthy individuals. Blood Cancer J (2019) 9:8. doi: 10.1038/s41408-018-0166-4
62. Blankenstein T, Coulie PG, Gilboa E, Jaffee EM. The determinants of tumour immunogenicity. Nat Rev Cancer (2012) 12:307–13. doi: 10.1038/nrc3246
63. Arshad N, Cresswell P. Tumor-associated calreticulin variants functionally compromise the peptide loading complex and impair its recruitment of MHC-I. J Biol Chem (2018) 293:9555–69. doi: 10.1074/jbc.RA118.002836
64. Aarntzen EHJG, Bol K, Schreibelt G, Jacobs JFM, Lesterhuis WJ, Van Rossum MM, et al. Skin-test infiltrating lymphocytes early predict clinical outcome of dendritic cell-based vaccination in metastatic melanoma. Cancer Res (2012) 72:6102–10. doi: 10.1158/0008-5472.CAN-12-2479
65. Westdorp H, Creemers JHA, Van Oort IM, Schreibelt G, Gorris MAJ, Mehra N, et al. Blood-derived dendritic cell vaccinations induce immune responses that correlate with clinical outcome in patients with chemo-naive castration-resistant prostate cancer. J Immunother Cancer (2019) 7:1–15. doi: 10.1186/s40425-019-0787-6
66. Cai Y, Fleming C, Yan J. New insights of T cells in the pathogenesis of psoriasis. Cell Mol Immunol (2012) 9:302–9. doi: 10.1038/cmi.2012.15
67. Kristinsson SY, Landgren O, Samuelsson J, Björkholm M, Goldin LR. Autoimmunity and the risk of myeloproliferative neoplasms. Haematologica (2010) 95:1216–20. doi: 10.3324/haematol.2009.020412
68. Kjaer L, Holmström MO, Cordua S, Andersen MH, Svane IM, Thomassen M, et al. Sorted Peripheral Blood Cells Identify CALR Mutations in B- and T-lymphocytes. Leuk Lymphoma (2017) 59:973–7. doi: 10.1080/10428194.2017.1359743
69. Sallusto F, Geginat J, Lanzavecchia A. Central memory and effector memory T cell subsets: Function, generation, and maintenance. Annu Rev Immunol (2004) 22:745–63. doi: 10.1146/annurev.immunol.22.012703.104702
70. Sallusto F, Lenig D, Förster R, Lipp M, Lanzavecchia A. Two subsets of memory T lymphocytes with distinct homing potentials and effector functions. Nature (1999) 401:708–12. doi: 10.1038/44385
71. Riley CH, Jensen MK, Brimnes MK, Hasselbalch HC, Bjerrum OW, Straten PT, et al. Increase in circulating CD4+CD25+Foxp3+ T cells in patients with Philadelphia-negative chronic myeloproliferative neoplasms during treatment with IFN-α. Blood (2011) 118:2170–3. doi: 10.1182/blood-2011-03-340992
72. Riley CH, Hansen M, Brimnes MK, Hasselbalch HC, Bjerrum OW, Straten PT, et al. Expansion of circulating CD56(bright) natural killer cells in patients with JAK2-positive chronic myeloproliferative neoplasms during treatment with interferon-α. Eur J Haematol (2015) 94:227–34. doi: 10.1111/ejh.12420
73. Talmadge JE, Gabrilovich DI. History of myeloid-derived suppressor cells. Nat Rev Cancer (2013) 13:739–52. doi: 10.1038/nrc3581
74. Wang JC, Kundra A, Andrei M, Baptiste S, Chen C, Wong C. Myeloid-derived suppressor cells in patients with myeloproliferative neoplasm. Leuk Res (2016) 43:39–43. doi: 10.1016/j.leukres.2016.02.004
75. Han L, Schubert C, Köhler J, Schemionek M, Isfort S, Brümmendorf TH, et al. Calreticulin-mutant proteins induce megakaryocytic signaling to transform hematopoietic cells and undergo accelerated degradation and Golgi-mediated secretion. J Hematol Oncol (2016) 9:45. doi: 10.1186/s13045-016-0275-0
76. Liu P, Zhao L, Loos F, Marty C, Xie W, Martins I, et al. Immunosuppression by Mutated Calreticulin Released from Malignant Cells. Mol Cell (2020) 77:1–13. doi: 10.1016/j.molcel.2019.11.004
77. Sollazzo D, Forte D, Polverelli N, Perricone M, Romano M, Luatti S, et al. Circulating Calreticulin Is Increased in Myelofibrosis: Correlation with Interleukin-6 Plasma Levels, Bone Marrow Fibrosis, and Splenomegaly. Mediators Inflamm (2016) 2016:5860657. doi: 10.1155/2016/5860657
78. Skov V, Riley CH, Thomassen M, Kjær L, Stauffer Larsen T, Bjerrum OW, et al. The impact of interferon-alpha2 on HLA genes in patients with polycythemia vera and related neoplasms. Leuk Lymphoma (2017) 58:1914–21. doi: 10.1080/10428194.2016.1262032
79. Lesokhin AM, Ansell SM, Armand P, Scott EC, Halwani A, Gutierrez M, et al. Nivolumab in Patients With Relapsed or Refractory Hematologic Malignancy: Preliminary Results of a Phase Ib Study. J Clin Oncol (2016) 34:2698–704. doi: 10.1200/JCO.2015.65.9789
80. Panagiota V, Thol F, Markus B, Fehse B, Alchalby H, Badbaran A, et al. Prognostic effect of calreticulin mutations in patients with myelofibrosis after allogeneic hematopoietic stem cell transplantation. Leukemia (2014) 28:1552–5. doi: 10.1038/leu.2014.66
81. Klyuchnikov E, Holler E, Bornhäuser M, Kobbe G, Nagler A, Shimoni A, et al. Donor lymphocyte infusions and second transplantation as salvage treatment for relapsed myelofibrosis after reduced-intensity allografting. Br J Haematol (2012) 159:172–81. doi: 10.1111/bjh.12013
82. Hofmann S, Götz M, Scheider V, Guillaume P, Bunjes D, Döhner H, et al. Donor Lymphocyte Infusion Induces Polyspecific CD8+ T-Cell Responses With Concurrent Molecular Remission in Acute Myeloid Leukemia With NPM1 Mutation. J Clin Oncol (2013) 31:2013–5. doi: 10.1200/JCO.2012.48.6183
83. Verstovsek S, Mesa Ra, Gotlib J, Levy RS, Gupta V, DiPersio JF, et al. A double-blind, placebo-controlled trial of ruxolitinib for myelofibrosis. N Engl J Med (2012) 366:799–807. doi: 10.1056/NEJMoa1110557
84. Heine A, Andrea S, Held E, Daecke SN, Wallner S, Yajnanarayana SP, et al. The JAK-inhibitor ruxolitinib impairs dendritic cell function in vitro and in vivo. Blood (2013) 122:1192–202. doi: 10.1182/blood-2013-03-484642
85. Schönberg K, Rudolph J, Vonnahme M, Yajnanarayana SP, Cornez I, Hejazi M, et al. JAK inhibition impairs NK cell function in myeloproliferative neoplasms. Cancer Res (2015) 75:2187–99. doi: 10.1158/0008-5472.CAN-14-3198
86. Parampalli Yajnanarayana S, Stübig T, Cornez I, Alchalby H, Schönberg K, Rudolph J, et al. JAK1/2 inhibition impairs T cell function in vitro and in patients with myeloproliferative neoplasms. Br J Haematol (2015) 169:824–33. doi: 10.1021/ac901991x
87. Andersen MH. Anti-regulatory T cells. Semin Immunopathol (2017) 39:317–26. doi: 10.1007/s00281-016-0593-x
88. Munir S, Andersen GH, Met Ö, Donia M, Frøsig TM, Larsen SK, et al. HLA-restricted CTL that are specific for the immune checkpoint ligand PD-L1 occur with high frequency in cancer patients. Cancer Res (2013) 73:1764–76. doi: 10.1158/0008-5472.CAN-12-3507
89. Ahmad SM, Martinenaite E, Holmström M, Jørgensen MA, Met Ö, Nastasi C, et al. The inhibitory checkpoint, PD-L2, is a target for effector T cells: Novel possibilities for immune therapy. Oncoimmunology (2017) 7(2):e1390641. doi: 10.1080/2162402X.2017.1390641
90. Martinenaite E, Mortensen REJ, Hansen M, Orebo Holmström M, Munir Ahmad S, Grønne Dahlager Jørgensen N, et al. Frequent adaptive immune responses against arginase-1. Oncoimmunology (2017) 7:e1404215. doi: 10.1080/2162402X.2017.1404215
91. Weis-Banke SE, Hübbe ML, Holmström MO, Jørgensen MA, Bendtsen SK, Martinenaite E, et al. The metabolic enzyme arginase-2 is a potential target for novel immune modulatory vaccines. Oncoimmunology (2020) 9. doi: 10.1080/2162402X.2020.1771142
92. Munir S, Larsen SK, Iversen TZ, Donia M, Klausen TW, Svane IM, et al. Natural CD4 + T-cell responses against indoleamine 2,3-dioxygenase. PLoS One (2012) 7:e34568. doi: 10.1371/journal.pone.0034568
93. Iversen TZ, Engell-Noerregaard L, Ellebaek E, Andersen R, Larsen SK, Bjoern J, et al. Long-lasting disease stabilization in the absence of toxicity in metastatic lung cancer patients vaccinated with an epitope derived from indoleamine 2,3 dioxygenase. Clin Cancer Res (2014) 20:221–32. doi: 10.1158/1078-0432.CCR-13-1560
94. Svane I-M, Kjeldsen JW, Lorentzen CL, Martinenaite E, Andersen MH. LBA48 Clinical efficacy and immunity of combination therapy with nivolumab and IDO/PD-L1 peptide vaccine in patients with metastatic melanoma: A phase I/II trial. Ann Oncol (2020) 31:S1176. doi: 10.1016/j.annonc.2020.08.2278
95. Andersen MH. The T-win® technology: immune-modulating vaccines. Semin Immunopathol (2019) 41:87–95. doi: 10.1007/s00281-018-0695-8
96. Cordua S, Kjaer L, Skov V, Pallisgaard N, Hasselbalch HC, Ellervik C. Prevalence and phenotypes of JAK2 V617F and Calreticulin mutations in a Danish general population. Blood (2019) 134:469–79. doi: 10.1182/blood.2019001113
Keywords: myeloproliferative neoplasms, cancer immune therapy, cancer vaccines, neo-antigen, calreticulin
Citation: Handlos Grauslund J, Holmström MO, Jørgensen NG, Klausen U, Weis-Banke SE, El Fassi D, Schöllkopf C, Clausen MB, Gjerdrum LMR, Breinholt MF, Kjeldsen JW, Hansen M, Koschmieder S, Chatain N, Novotny GW, Petersen J, Kjær L, Skov V, Met Ö, Svane IM, Hasselbalch HC and Andersen MH (2021) Therapeutic Cancer Vaccination With a Peptide Derived From the Calreticulin Exon 9 Mutations Induces Strong Cellular Immune Responses in Patients With CALR-Mutant Chronic Myeloproliferative Neoplasms. Front. Oncol. 11:637420. doi: 10.3389/fonc.2021.637420
Received: 03 December 2020; Accepted: 18 January 2021;
Published: 26 February 2021.
Edited by:
Bing Xu, Xiamen University, ChinaReviewed by:
Giuseppe Alberto Palumbo, University of Catania, ItalyGarima Pandey, Moffitt Cancer Center, United States
Copyright © 2021 Handlos Grauslund, Holmström, Jørgensen, Klausen, Weis-Banke, El Fassi, Schöllkopf, Clausen, Gjerdrum, Breinholt, Kjeldsen, Hansen, Koschmieder, Chatain, Novotny, Petersen, Kjær, Skov, Met, Svane, Hasselbalch and Andersen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mads Hald Andersen, mads.hald.andersen@regionh.dk
†These authors have contributed equally to this work and share first authorship
‡These authors have contributed equally to this work and share senior authorship