 Xiuchao Geng
Xiuchao Geng Yuhao Zhang
Yuhao Zhang Zhaomu Zeng
Zhaomu Zeng Zhongrui Zhu
Zhongrui Zhu Hong Wang
Hong Wang Wentao Yu
Wentao Yu Qiang Li
Qiang Li
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Oncol., 18 March 2021
Sec. Head and Neck Cancer
Volume 11 - 2021 | https://doi.org/10.3389/fonc.2021.629718
This article is part of the Research TopicRNA modifiers in Head and Neck CancerView all 8 articles
N6-methyladenosine (m6A) plays crucial roles in a diverse range of physiological and pathological processes, and it is believed that it tremendously promotes neoplasia and progression. However, knowledge of the molecular characteristics of m6A modification, its prognostic value, and the infiltration of immune cell populations in head and neck squamous cell carcinoma (HNSCC) is still insufficient. Therefore, a pan-cancer genomic analysis was systematically performed here by examining m6A regulators at the molecular level within 33 multiple cancer types, and the correlations between the expression of m6A molecules were researched using datasets from The Cancer Genome Atlas (TCGA). Based on the above analysis, insulin-like growth factor 2 mRNA-binding protein 2 (IGF2BP2) is upregulated in HNSCC and may serve as an independent prognostic factor of overall survival, thus showing potential as a prognostic biomarker in HNSCC. Genetic alteration analyses elucidated the reasons for the abnormal upregulation of IGF2BP2 in HNSCC. As a result, IGF2BP2 was selected for further univariate and multivariate analyses. The functions of the related genes were annotated through gene set enrichment analysis, and the activation states of multiple biological pathways were shown by gene set variation analysis. We found that LRRC59 and STIP1 may act as IGF2BP2-associated genes to have a regulatory function in the m6A modification. In addition, we found that the status of immune cell infiltration was correlated with the level of IGF2BP2 gene expression. Our results provide supplementation at the molecular level for epigenetic regulation in HNSCC and insight into effective immunotherapy targets and strategies.
The classic epigenetic modifications are DNA methylation, histone modification, and chromatin remodeling, which take part in many basic biological activities of carcinogenesis, including malignant development, and are associated with the prognosis of various kinds of cancer, including head and neck squamous cell carcinoma (HNSC, HNSCC) (1). In the past, reports on tumor methylation modifications have concentrated mostly on DNA methylation and offered potential biomarkers for cancer diagnosis (2). In addition to these traditional epigenetic modifications, mRNA modification is known as another epigenetic regulation of gene expression. Its dynamic regulation and disorder are closely related to translation control, RNA splicing defects, and the occurrence, maintenance, and progression of various tumors (3, 4). The degree of RNA modification is highly sensitive to changes in the cell microenvironment or the transformation of physiological states, and RNA modification changes will in turn influence cell regulation and adaptation (5). Among the types of RNA modifications, N6-methyladenosine (m6A) is recognized as the first and most common modification in eukaryotic mRNA (6).
m6A modification has been studied for 40 years. As a posttranscriptional modification with dynamic and reversible characteristics, m6A methylation has a profound impact on gene regulation (7), and its biological importance has also been emphasized by recent research progress. m6A modification participates in numerous basic biological processes, such as mRNA stability, mRNA translation, RNA splicing, and the phase separation potential of mRNA (8–10). Many physiological processes of the human body are closely related to m6A, such as spermatogenesis (11) and spermatogonial differentiation (12), hematopoietic stem cell development (13), and antiviral innate immunity (14). Moreover, a disturbance in m6A modification is associated with the pathogenesis of diverse cellular processes, including reduced cell proliferation, impaired self-renewal ability, developmental defects, and cell death (15). m6A modification has crucial functions in many human diseases, including infertility (16), nervous system diseases (17), early embryonic retardation (18), immune diseases, and multiple cancers (19). Currently, m6A modification, especially the role of its regulatory enzymes (writers, erasers, and readers), in cancer biology has been recognized as a prominent hot spot in terms of tumorigenesis, malignant progression, and potential biological target screening. Writers form a multisubunit methyltransferase complex to upregulate the level of m6A, while erasers are m6A demethylases, making this event reversible. Readers can decode m6A methylation information and convert it into effectors of functional signals. Recently, increasing evidence has shown that changes in m6A regulators can promote the development of several cancers (20), including glioblastoma, breast and cervical cancers, liver and gallbladder cancers, and bladder and prostate cancers (21–23). However, despite some progress in revealing their role in multiple cancers, the characteristics of m6A modification in HNSCC are still very inadequate.
HNSCC is a highly invasive malignant tumor that originates from the mucous membrane of the hypopharynx, larynx, oral cavity, and oropharynx, accounting for 90% of all head and neck cancers (24). HNSCC has more than 650000 confirmed cases worldwide, causing more than 300000 deaths every year (25) and ranking as the sixth most common and fatal cancer throughout the world (26). Although much progress has been made in the treatment methods (surgery, chemotherapy, and radiotherapy), the clinical prognosis of HNSCC patients is still poor. The 5-year survival rate of patients is still approximately 50%, which remains to be improved, and suggests that approximately 30-50% of HNSCC patients will experience local recurrence and distant metastasis (24). Therefore, the search for specific molecular markers of HNSCC is of great significance for better understanding of its progress and for developing new therapeutic methods. At present, an increasing number of studies have concentrated on the mechanism of pathogenesis, especially genetic and epigenetic events, at the molecular level (27). Currently, m6A modification has been reported to play a vital function in cell infiltration in the tumor microenvironment, and the m6A modification pattern can be used to help identify different immune phenotypes in gastric cancer (28). In addition, HNSCC was one of the first diseases for which immunotherapy was developed, which has greatly changed the therapeutic prospects of cancer patients. However, until now, there have been few reports about the relationship between m6A methyltransferase and clinicopathological features, its prognostic value, and the infiltration of immune cells in HNSCC. Accordingly, the function of m6A modification in tumor diagnosis, prognosis, and immune cell infiltration in HNSCC remains to be explored and could provide treatment targets for HNSCC.
As a result, we investigated the expression modes of 24 m6A regulatory genes in 33 cancers and explored the differential expression of IGF2BP2 in HNSCC and its clinical significance. Based on the clinical and sequencing data of The Cancer Genome Atlas (TCGA)-HNSCC cohort, the relationships among differential expression, genetic alterations, and clinicopathological features, including survival, were analyzed. Gene set enrichment analysis (GSEA) and gene set variation analysis (GSVA) were used to analyze the molecular mechanism of IGF2BP2 abnormalities in HNSCC. On the other hand, the characteristics of m6A genes, including IGF2BP2, were analyzed to examine their relationship with immune cell infiltration in HNSCC. The purpose of this study was to provide supplementation at the molecular level for epigenetic regulation in HNSCC and ideas for effective immunotherapy targets and strategies.
For datasets in the TCGA, the R package TCGAbiolinks (29) was used to download RNA sequencing data and full clinical annotation information from the Genomic Data Commons (GDC, https://portal.gdc.cancer.gov/) and was specialized for an integrated analysis of GDC data (29). Patients for whom survival information was lacking were excluded from further evaluation. Information on somatic mutation data for HNSCC patients was obtained from the TCGA database as well. The expression of m6A regulatory genes in 33 cancers was analyzed with the TIMER2.0 database (http://timer.comp-genomics.org/) (30), in which the Wilcoxon test (also named the Mann–Whitney test) was applied to evaluate the P value. Sequencing and copy number variation (CNV) data provided by cBioPortal (http://www.cbioportal.org/) for cancer genomics were used to detect the genetic alterations in m6A regulatory proteins within 504 HNSCC patients/samples. IGF2BP2 mRNA expression, somatic mutations, CNVs, and the level of methylation in HNSCC were analyzed with UCSC Xena (https://xenabrowser.net/heatmap/). Twenty-four interactions between m6A regulators were elucidated with the help of the STRING database (http://www.string-db.org/). Immune cell infiltration was analyzed using the TIMER2.0 database.
Univariate and multivariate Cox regression analyses were used to assess the prognostic value of m6A regulators through their expression pattern in the TCGA. Both the “survival” and “survminer” (https://CRAN.R-project.org/package=survminer) R packages were used to investigate every gene. Four hundred and twenty-nine HNSCC patients were divided into different subgroups (high and low groups) by their characteristics and median gene expression values.
The TCGAbiolinks R package was used to download TCGA-HNSC read count data. Patients with HNSCC were assigned to high and low IGF2BP2 groups according to the median cutoff value. A total of 11,297 long noncoding RNAs (lncRNAs) were ultimately screened for differential expression analyses. The DEseq2 R package (version 1.26.0) in R software (https://www.r-project.org/) was used to perform data standardization and calculate differences in the expression data. An absolute log2-fold change (FC) > 1 and p-value < 0.05 were considered the criteria for the differential expression of lncRNAs. The ggplot2 R package was used to draw a volcano plot.
The differential expression profiles and survival outcomes of 24 m6A regulator genes among 33 cancer types were assessed using the GSCALite web server (31). Only 14 cancer types (THCA, KIRP, BLCA, LIHC, HNSC, BRCA, LUAD, PRAD, ESCA, KICH, LUSC, KIRC, STAD, and COAD) had paired samples (paired tumor-normal tissue) with available expression data. GSCALite was also used to study the effect (activation or inhibition) of m6A molecules on pathways related to cancer. In GSCALite, the scores of 7876 samples, 10 cancer-related pathways, and 32 cancer types (acute myeloid leukemia (AML) was excluded) were obtained from reversed-phase protein array (RPPA) data from The Cancer Proteome Atlas (TCPA). A total of ten cancer-related pathways, including apoptosis, PI3K/AKT, and TSC/mTOR, were identified, and m6A regulators that impacts (activate or inhibit) at least five different cancers were revealed by GSCALite.
GSEA was performed with the JAVA program of the MSigDB v6.1 acquired from the website of the Broad Institute (http://software.broadinstitute.org/gsea/) (32). Then, we applied GSEA with standard settings as implemented in the R package clusterProfiler to assign pathway activity estimates to each sample. Finally, 18419 genes were included in the GSEA program, where “c2.cp.kegg.v7.1.symbols.gmt” was regarded as the hallmark gene set (33). A normalized p-value < 0.05 and a false discovery rate < 0.25 of the hallmark gene set were considered significantly enriched.
GSVA was performed with the “GSVA” R package to determine the differences in physiological processes based on m6A modification patterns. GSVA is widely employed as a method to estimate variation in signaling pathways and physiological processes within a nonparametric and unsupervised method for samples of an expression dataset (34). Thus, the gene set “c2.cp.kegg.v7.1.symbols.gmt” was accessed through the MSigDB for GSVA. An adjusted p-value < 0.05 was considered statistically significant.
The Human Protein Atlas (https://www.proteinatlas.org/) provides pathological specimens for the immunohistochemical analysis of prognostic RNAs, such as m6A methyltransferases. A survival analysis of associated genes in HNSCC was also performed. Information on the patients, staining intensity, and quantity can be obtained online.
SPSS 20.0 (SPSS, Chicago, IL), R software V3.6.2 and GraphPad Prism 8 (GraphPad Software, Inc., La Jolla, CA, USA) were applied for statistical analysis. Continuous variables are presented as the mean ± standard deviation. Univariate and multivariate Cox regression analyses were used to illustrate their prognostic value and associations with clinical characteristics at the molecular/pathological level. Kaplan–Meier curves with a two-sided log-rank test were used to compare the overall survival (OS) of the patients in the high and low expression groups. p-values < 0.05 were considered statistically significant.
The genome-wide omics data, consisting of more than 10,000 human samples of 33 cancer types, were acquired from the TCGA for analyses (Supplementary Material: Table S1). The literature was reviewed, and a catalog of 24 genes that function mainly as regulators of m6A modifications was curated here: 8 writers (WTAP, METTL3, METTL14, ZC3H13, RBM15, RBM15B, KIAA1429 (VIRMA), and CBLL1), 2 erasers (ALKBH5, FTO) and 14 readers (HNRNPA2B1, HNRNPC, YTHDC1, YTHDC2, YTHDF1, YTHDF2, YTHDF3, IGF2BP1, IGF2BP2, IGF2BP3, RBMX, FMR1, ELAVL1, and LRPPRC). The proportions of readers, writers, and erasers among m6A regulators are listed in Figure 1A. The correlations between the expression of 24 m6A regulators in the TCGA-HNSC cohort were determined by Pearson correlation analysis, and the results are displayed in Figure 1B. The TIMER database was used for analysis, and we found that m6A regulators were abnormally expressed in cancers in a cancer-specific pattern. Compared with their paired normal controls, RBM15, METTL3, and IGF2BP1/2/3 were obviously upregulated in the TCGA-HNSC dataset, while WTAP, METTL14, ZC3H13, YTHDF2, YTHDF3, ALKBH5, FTO, CBLL1, and LRPPRC were not upregulated (Figure 1C). To better visualize the interactions between the 24 m6A regulators, their relationships were determined with the STRING database (Figure 1D). The GSCALite web server was used, and we found twenty-four m6A regulators to be differentially expressed in 14 cancer types (Figure 1E). We found that IGF2BP1/2/3 were upregulated in HNSCC, consistent with the data from TIMER.
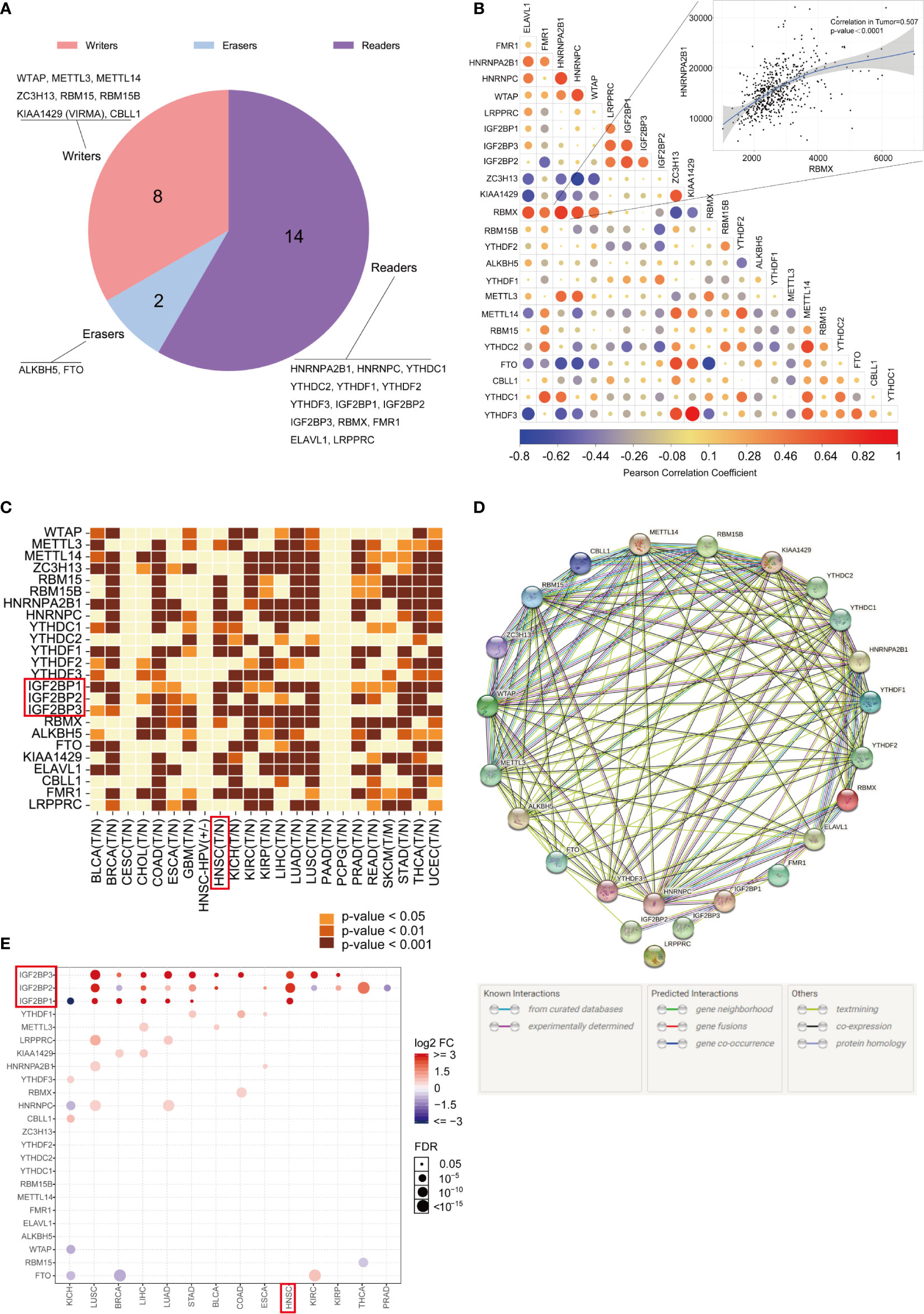
Figure 1 Gene expression landscape of m6A regulators in pan-cancer. (A) The proportions of readers, writers, and erasers among m6A regulators. (B) Correlations between the expression of 24 m6A regulators. The correlation between RBMX and HNRNPA2B1 is illustrated as a scatter plot (Pearson correlation). A positive correlation is indicated in red, and a negative correlation is indicated in blue. The color intensity and size of the circle are proportional to the correlation coefficient. (C) The expression profiles of the 24 m6A regulatory genes in the TCGA pan-cancer dataset originating from the TIMER database. (D) The interactions among 24 m6A regulatory genes (STRING). (E) GSCALite was used to analyze the gene expression profiles of 24 m6A regulatory genes in 14 types of cancer.
Among the different types of cancer, the prevalent genetic and expression alterations in m6A regulators could largely give rise to the breakthrough of translational medicine, as m6A regulatory genes were closely related to patient OS across 33 cancer types in our study. It was also implied by our results that 24 m6A regulatory genes were related to the OS of patients with at least one cancer type, where some of the genes, including IGF2BP1/2/3, showed oncogenic features, and their increased expression was related to worse survival in patients with various cancers. In particular, upregulation of the IGF2BP2 gene was associated with poor patient survival in seven cancer types, including HNSCC (Supplementary Material: Figure S1A).
To further explore the underlying mechanisms by which m6A regulatory genes participate in multiple cancers at the molecular level, the effects (activation or inhibition) of m6A regulatory genes on pathways accompanying cancers were analyzed with GSCALite. It was found that m6A regulatory genes are functionally expressed and are related to oncogenic activation or inhibitory pathways (Supplementary Material: Figure S1B). A large number of activated pathways, namely, the cell cycle, epithelial-mesenchymal transition (EMT), DNA damage response, PI3K-AKT, RTK, and RAS/MAPK, were found to be associated with the expression of IGF2BP2, RBMX, and HNRNPC. These results confirmed that m6A regulators are functionally related to multiple cancers.
We found that single nucleotide polymorphism (SNP) occupied the first place in variant type (Supplementary Material: Figure S2A). SNP analysis showed that missense mutations were the most frequent variants in the m6A-regulatory genes (Supplementary Material: Figure S2B). The most frequent SNV type was C > T (Supplementary Material: Figure S2C).
In addition, we summarized the incidence of single nucleotide variants (SNVs) in 24 m6A regulators in 33 pan-cancer samples with GSCALite. Among 1443 samples, 1002 harbored mutations in m6A regulators, with a frequency of 69.44%, among which ZC3H13 showed the highest mutation frequency, followed by KIAA1429 (Supplementary Material: Figure S2D).
Considering the relationship between m6A methyltransferases and the malignant progression of HNSCC, univariate and multivariate survival analyses were performed to further determine the prognostic value of m6A methyltransferases in HNSCC using Cox proportional hazards models of their expression levels in the TCGA dataset. A total of 429 HNSCC patients were assigned to two groups (high and low) based on the median expression value of each gene. Stage IV disease and high LRPPRC and IGF2BP2 expression levels were shown to be independent prognostic factors for OS. Multivariate analysis showed that patients aged ≥60 years, with stage IV disease, of African American race, and high ELVAL1 and IGF2BP2 expression were risk factors for poor OS (hazard ratio (HR)> 1) (Figure 2). As shown in Supplementary Material: Table S2, IGF2BP2 was correlated with T-stage based on the median cutoff of IGF2BP2 (P = 0.023). However, IGF2BP2 did not correlate with clinical characteristics such as age, race, stage, or sex. ELVAL1 was correlated with the clinical characteristic race (P = 0.014). LRPPRC was correlated with the clinical characteristics sex (P = 0.016), T-stage (P = 0.044), and race (P = 0.040) (Supplementary Material: Table S3).
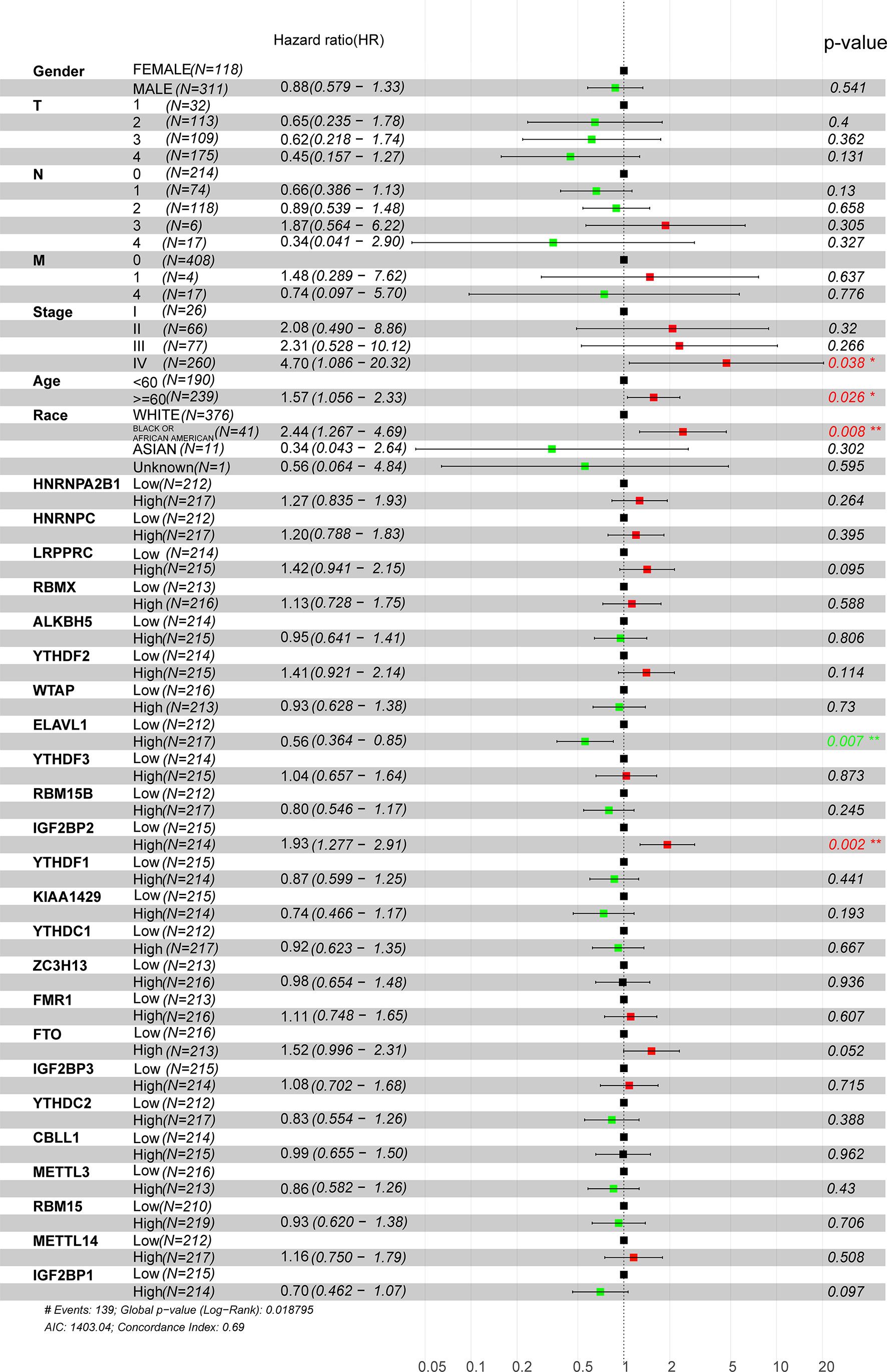
Figure 2 Univariate Cox regression analysis of m6A regulators in HNSCC patients in the TCGA dataset. Tumor node metastasis classification: T (tumor), N (lymph node), and M (metastasis).
Based on the above analysis, we next selected IGF2BP2 for the multivariate analysis of 24 m6A regulatory genes. The expression of the IGF2BP2 gene in pan-cancer was examined with TIMER (Figure 3A), and we verified the expression and prognostic value of the IGF2BP2 gene in HNSCC using GEPIA2 (Figures 3B, C). Kaplan–Meier curve analysis also indicated that patients with higher IGF2BP2 expression experienced significantly worse OS than their counterparts (Figure 3D).
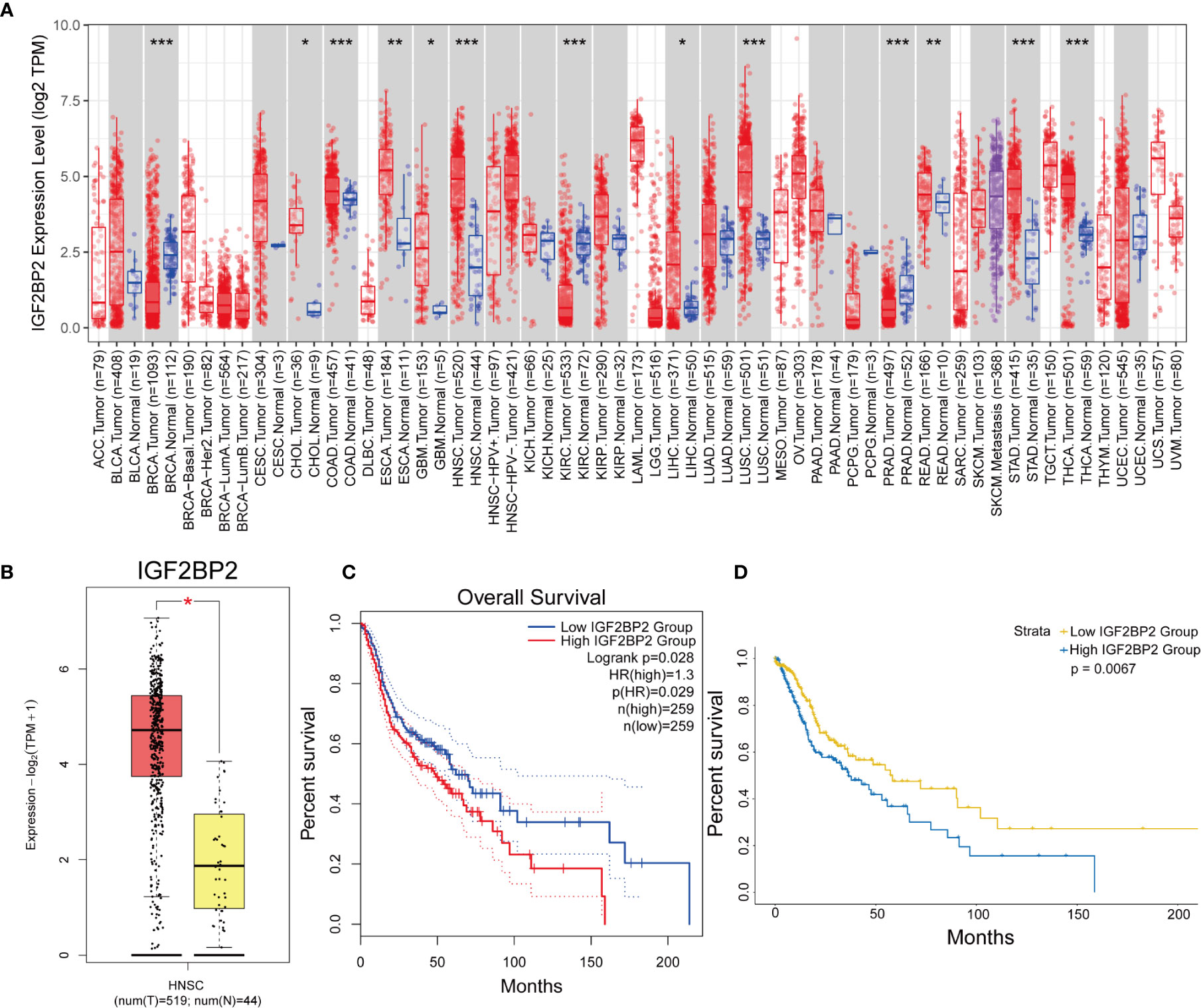
Figure 3 The expression and prognostic value of IGF2BP2 in HNSCC. (A) The expression of the IGF2BP2 gene in pan-cancer was analyzed with TIMER. (*p < 0.05, **p < 0.01, ***p < 0.001). (B, C) The expression and prognostic value of the IGF2BP2 gene in HNSCC was analyzed with GEPIA2. (HR, hazard ratio) (D) Kaplan–Meier survival curves illustrate the difference in OS based on the expression level of IGF2BP2.
The incidence of genetic alterations in the 24 m6A regulators in HNSCC (TCGA, Firehose Legacy) was analyzed by using the cBioPortal tool. Twenty-four m6A methylation regulators showed genetic alterations in 227 (45%) queried patients/samples (504 patients/samples). The most common genetic alterations were amplification and deep deletion mutations. In our analysis, IGF2BP2 exhibited the highest mutation frequency (an alteration rate of 20%), followed by KIAA1429 (VIRMA) (an alteration rate of 8%), while both METTL14 and YTHDF2 showed few mutations in HNSCC samples (Supplementary Material: Figure S3A). Furthermore, the potential mechanism of dysregulated IGF2BP2 was studied because it was identified as an independent prognostic factor for OS. In the UCSC Xena database, 604 samples from the TCGA-HNSC dataset were analyzed, which revealed that IGF2BP2 expression was associated with CNV and partial DNA methylation sites but not with somatic mutations (Supplementary Material: Figure S3B). In conclusion, CNV and DNA methylation may give rise to the abnormal upregulation of IGF2BP2 in HNSCC.
To identify the mechanisms controlling the tumorigenesis and poor prognosis of HNSCC at the transcriptional level, PROMO and Cistrome Data Browser were used to predict the transcription factor binding sites for IGF2BP2 (Supplementary Material: Table S4). The transcription factor CCAAT/enhancer-binding protein beta (CEBPB; C/EBP-β) was obtained after taking the intersections. IGF2BP2 (chr3:185643129-185821141) was found to be a putative target (score=2.843) of the transcription factor CEBPB by searching the ChIPseq results (GSM1010802) in Cistrome DB: 46089 (35) (Figure 4A). The same result was validated in the hTFtarget database (http://bioinfo.life.hust.edu.cn/hTFtarget/) (36). The motif of the transcription factor CEBPB is shown in Figure 4B (ID: MC00121, hits: 3187, cutoff: 6.012, z score: -107.2, -10log(pval): 690.776). A statistical plot showed the top 20 factors that regulate IGF2BP2 queried from Cistrome (Figure 4C), and CEBPB ranked first. In summary, the transcription factor CEBPB is most likely the transcription factor that regulates IGF2BP2.
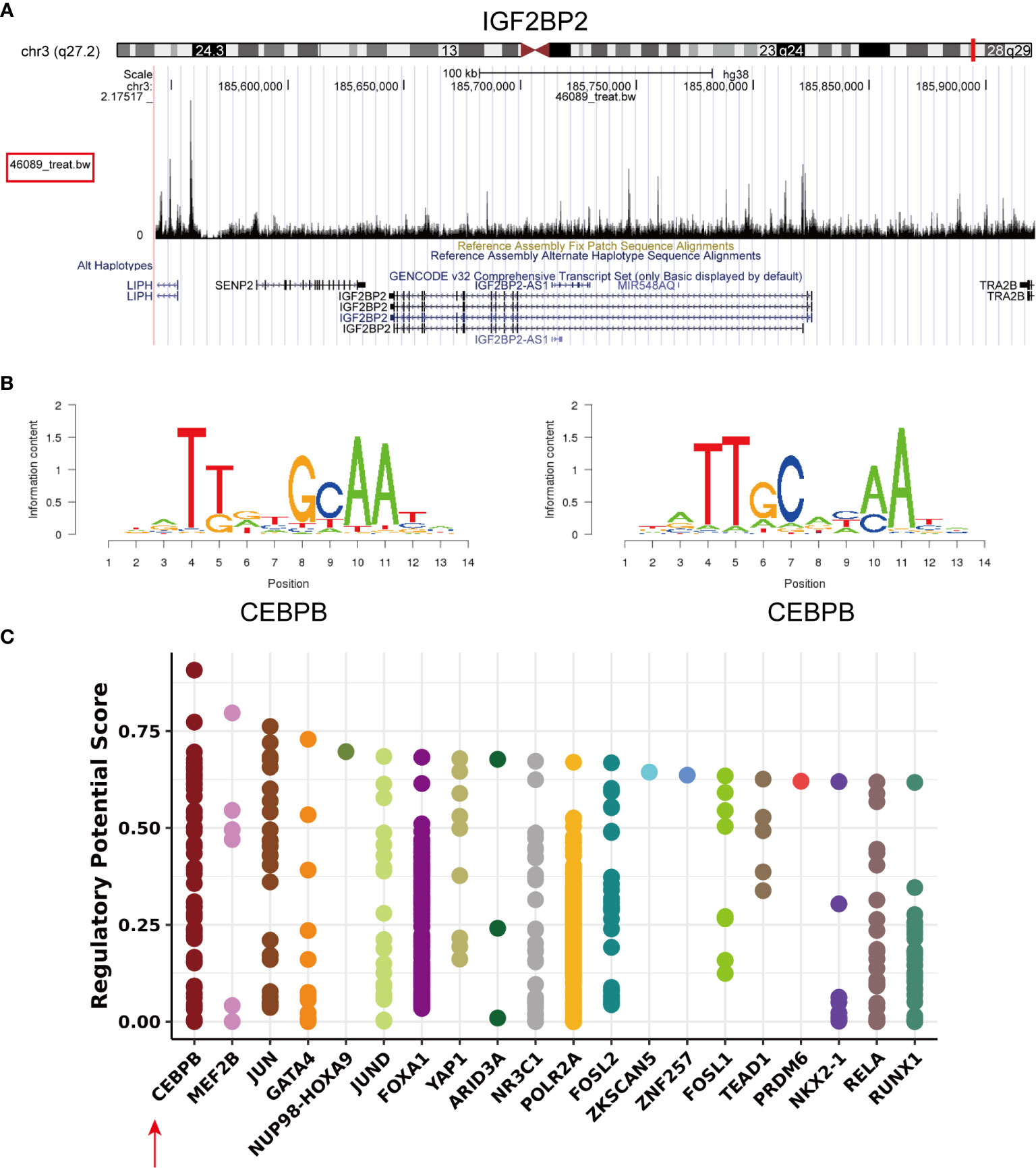
Figure 4 Transcription factor prediction of IGF2BP2. (A) Visual presentation of the high binding peak of the transcription factor CEBPB in the promoter region of IGF2BP2 (B) Representative sequence logo of the binding specificity of the transcription factor CEBPB queried from Cistrome. (C) Static plot of the top 20 factors that regulate IGF2BP2. Y axis shows the RP score. X axis shows different factors. Dots on the x axis line indicate the same factor.
Initially, we first collated an “A” list of dysregulated genes in HNSCC (n=5481) (log2FC cutoff = 0.58, p-value cutoff = 0.01), a “B” list of the top 1000 similar gene correlations of IGF2BP2 (n=1000), and then a “C” list of genes related to the survival of HNSCC patients (n=500). The number of genes in each list is displayed as a Venn diagram (Figure 5A). Twelve genes were common to all three gene sets. Therefore, they may act as IGF2BP2-associated genes and play a regulatory role in m6A modification. We analyzed their relationships with IGF2BP2 again. For example, we found that leucine-rich repeat-containing protein 59 (LRRC59) and stress-induced phosphoprotein 1 (STIP1) were correlated with IGF2BP2 (R=0.53 and R=0.53, respectively) (Figure 5B). LRRC59 and STIP1 were upregulated in HNSCC according to GEPIA2 (log2FC cutoff = 1, p-value cutoff = 0.01) (Figure 5C). High LRRC59 and STIP1 expression was correlated with poor OS in HNSCC patients (Figure 5D). It was predicted that IGF2BP2 facilitates the recruitment of RBPs to m6A-modified target mRNAs. By analyzing m6A Var, we analyzed the RBPs of potential associated genes, and their relationships were visualized with Cytoscape (37) (Figure 5E).
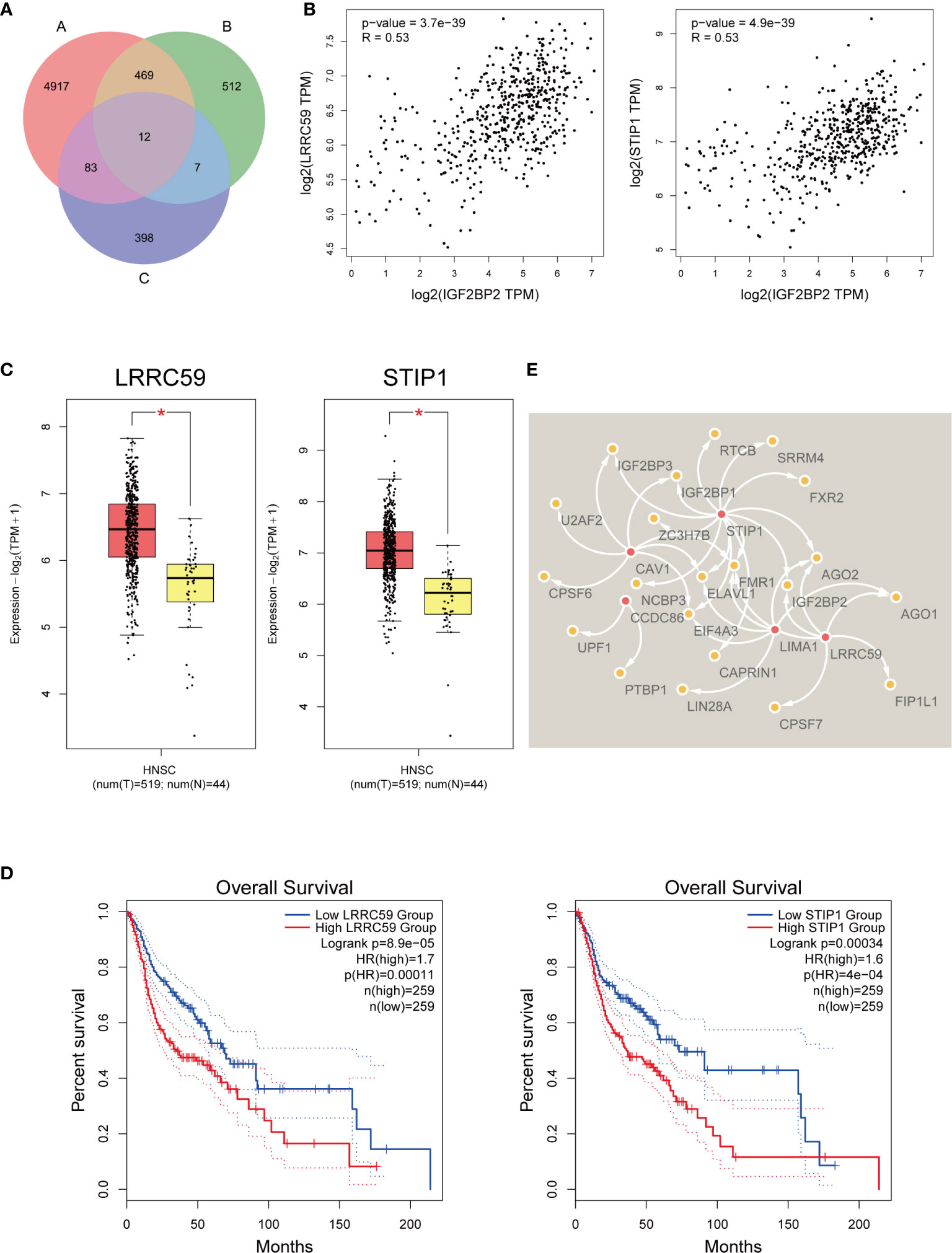
Figure 5 IGF2BP2-associated genes that mediate m6A modification. (A) Venn chart of IGF2BP2-related genes in HNSCC. (B) The correlation of LRRC59, STIP1, and IGF2BP2. (C) The expression of LRRC59 and STIP1 was confirmed with GEPIA2. T, tumor tissue. N, normal tissue. * means p-value < 0.01. (D) Kaplan-Meier survival curves illustrate the difference in OS according to the expression of LRRC59 and STIP1. (E) The relationships of RBPs and IGF2BP2-associated genes with the most potential.
Based on the above analysis, a total of 301 upregulated and 149 downregulated lncRNAs are significantly related to IGF2BP2 expression (Figure 6A). Among them, some lncRNAs are associated with HNSCC. The lncRNA SOX2-OT was reported to inhibit PTEN expression to facilitate laryngeal squamous cell carcinoma (LSCC) development through EZH2-mediated H3K27me3 (38). The potential target miRNAs (at least four of seven platforms predicted) and corresponding lncRNAs were predicted with starBase v2.0 (http://starbase.sysu.edu.cn/) (39). Finally, a Sankey plot consisting of lncRNAs, microRNAs (miRNAs), and two mRNAs (LRRC59 and STIP1) was constructed to illustrate a competing endogenous RNA (ceRNA) regulatory network (Figure 6B).
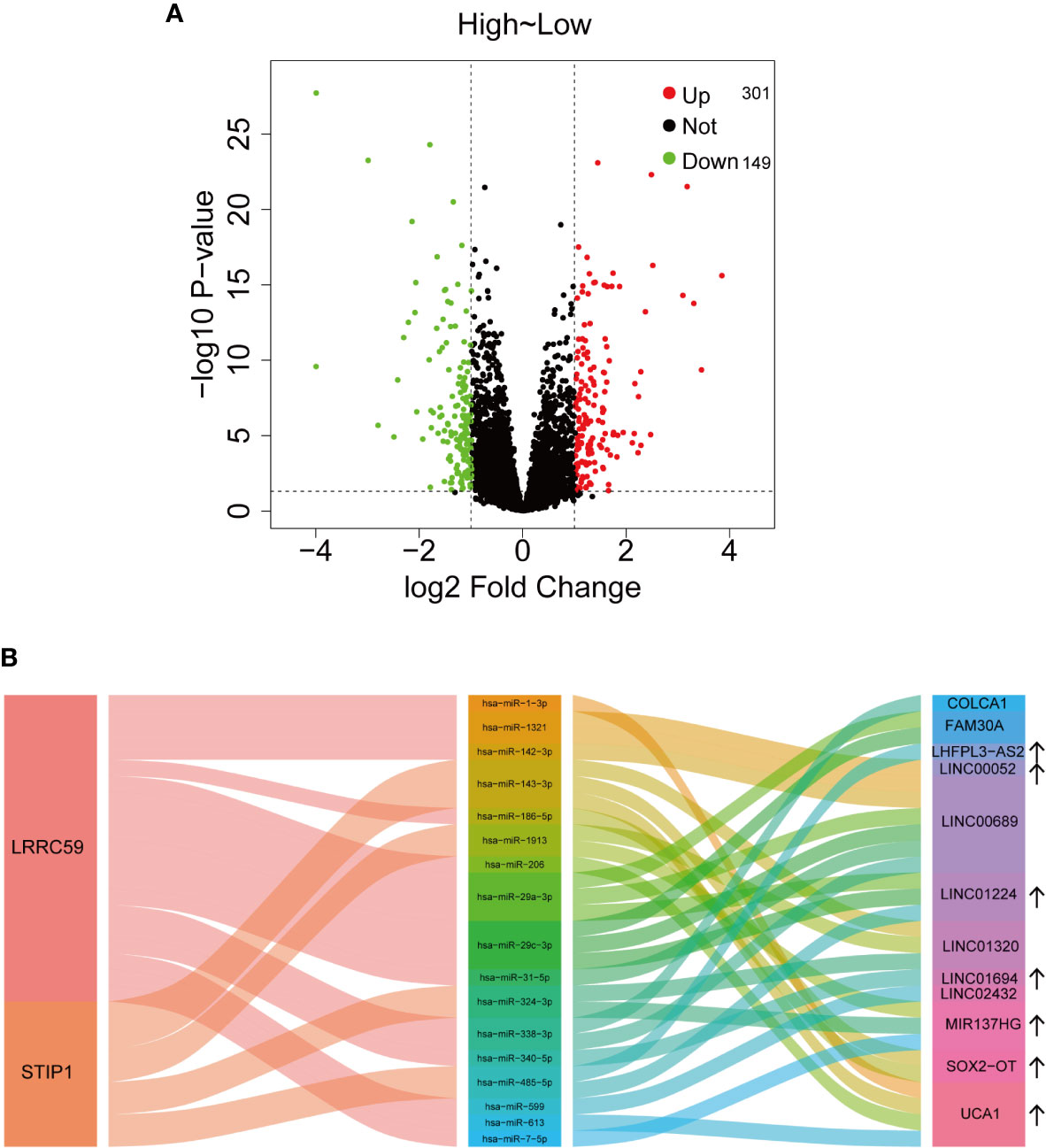
Figure 6 Potential regulatory mechanisms of IGF2BP2. (A) The dysregulated lncRNAs associated with IGF2BP2 expression. (B) Sankey plot illustrating the ceRNA regulatory network.
With the help of GSEA, the malignant hallmarks of tumors were investigated. The results showed that the tumor hallmarks primary immunodeficiency (NES=-2.21, normalized P < 0.0001), intestinal immune network for IgA production (NES=-1.95, normalized P < 0.0001), PPAR signaling pathway (NES=-1.80, normalized P< 0.001), basal cell carcinoma (NES=1.53, normalized P =0.013), Wnt signaling pathway (NES=1. 30, normalized P < 0.001), and Hedgehog signaling pathway (NES=1. 26, normalized P =0.080) were enriched in the IGF2BP2 group (Figure 7A).
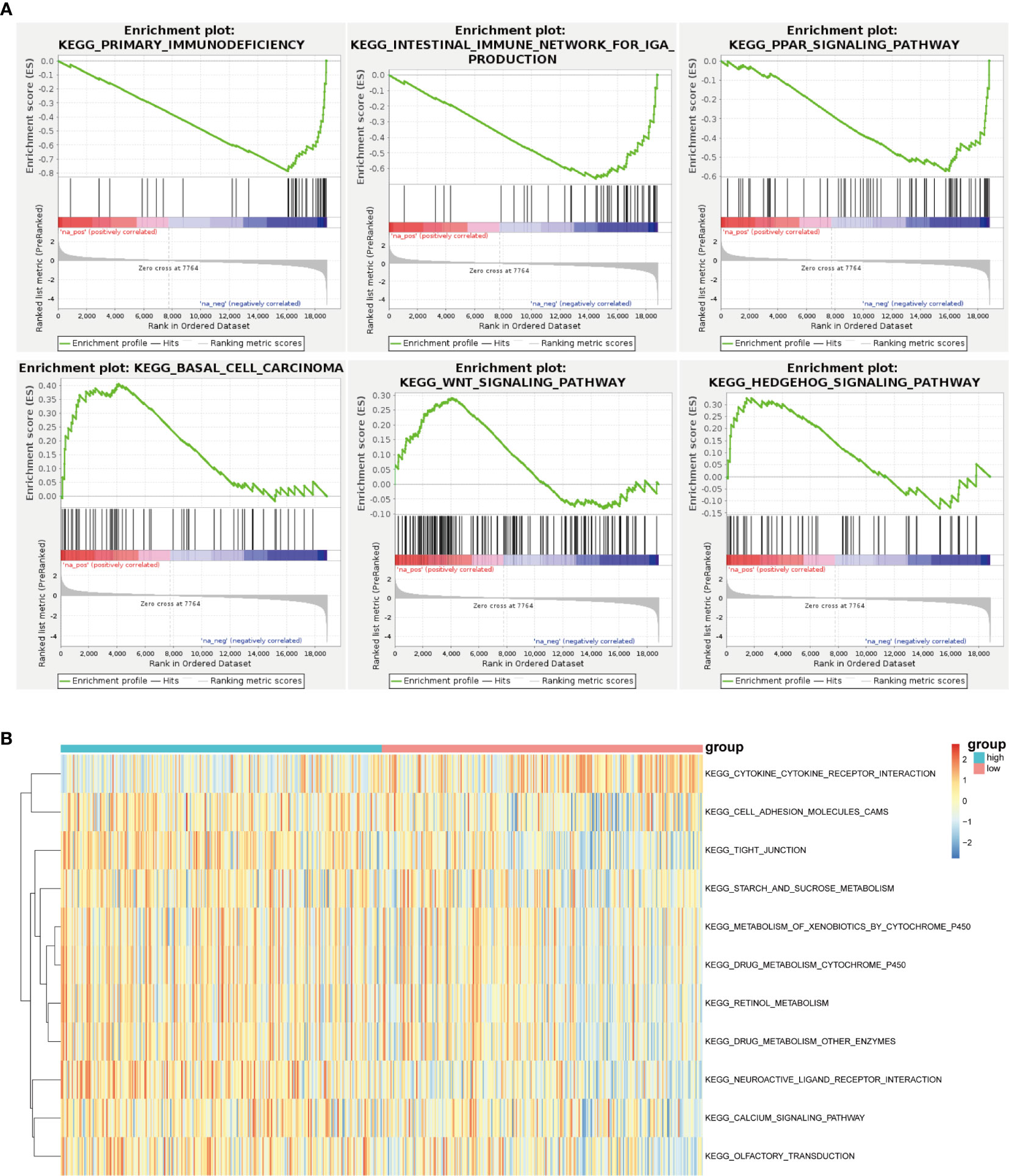
Figure 7 Unsupervised consensus analysis of m6A regulatory genes. (A) GSEA revealed that the IGF2BP2 group was enriched in the hallmarks malignant progression and tumor immunology. (B) GSVA revealed the state of biological pathways in the high and low IGF2BP2 groups. The column represents the TCGA-HNSCC samples.
Furthermore, GSVA was applied to illustrate the biological behaviors among distinct m6A modification patterns. As shown in Figure 7B, the high IGF2BP2 group was markedly enriched in cell adhesion molecules, neuroactive ligand receptor interaction pathways, and calcium signaling pathways. In our study, it was concluded that low IGF2BP2 expression was enriched in the cytokine receptor interaction pathway.
Based on the above analyses, we speculate that the IGF2BP2-mediated m6A methylation modification has significant associations with the malignant progression and tumor immunology of HNSCC.
To evaluate the expression differences in target genes at the protein level, immunohistochemistry data were obtained from pathological specimens from The Human Protein Atlas. We found that LRRC59 and STIP1 were dysregulated between HNSCC tumor tissues and normal tissues (Figure 8).
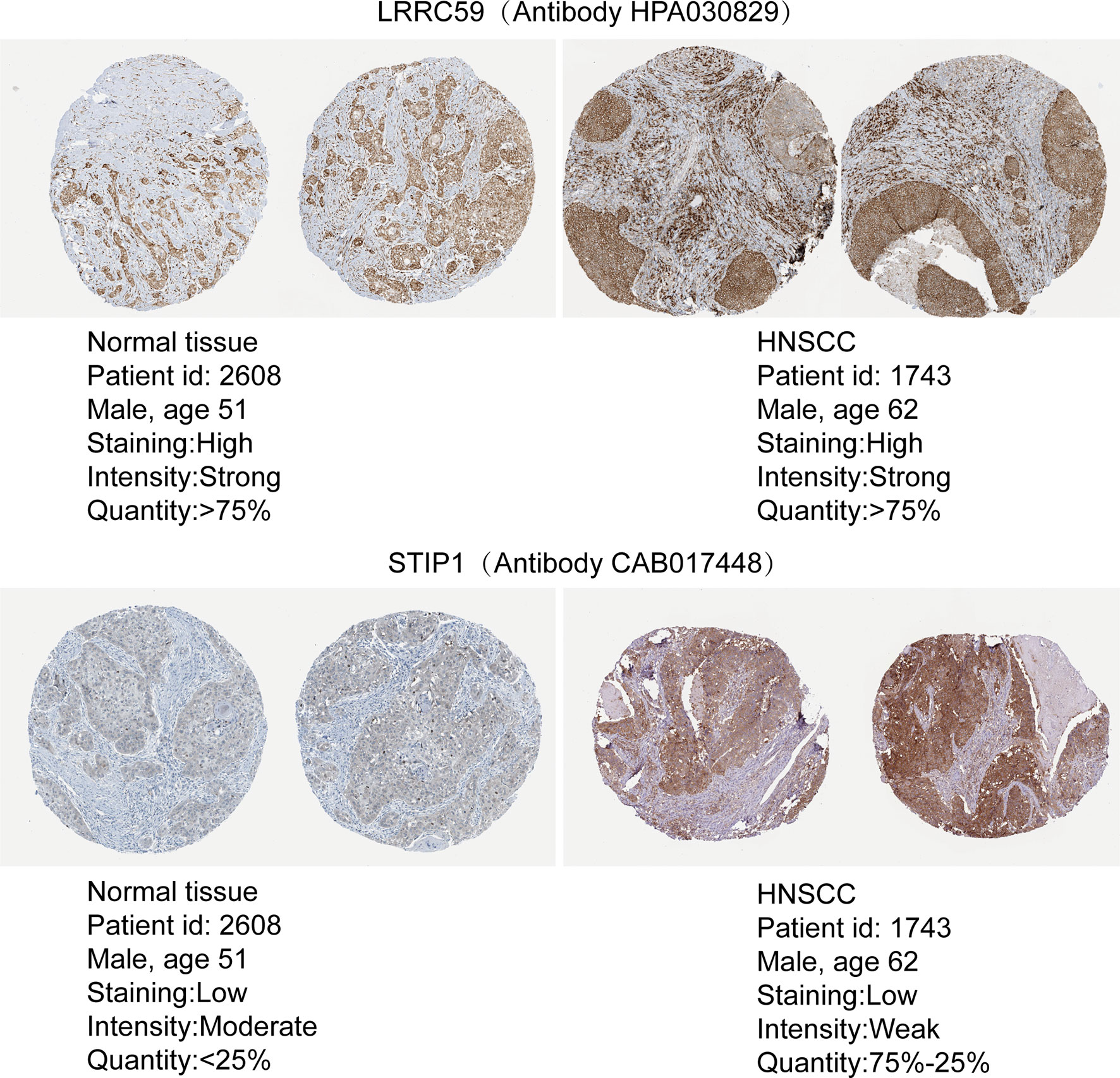
Figure 8 The protein expression of m6A target genes. A Comparison of the protein expression of LRRC59 and STIP1 in HNSCC tumor tissues and normal tissues.
After determining the malignant hallmarks of IGF2BP2, the relationship between IGF2BP2 expression and tumor immune cell infiltration in cancers was explored. First, the correlation of IGF2BP2 expression and immune infiltration levels was evaluated in diverse cancer types. The relationship between IGF2BP2 and 21 kinds of immune cells indicated a negative correlation between IGF2BP2 and B cells, CD4+ T cells, follicular helper T cells, regulatory T cells (Tregs), myeloid-derived suppressor cells, and cancer-associated fibroblast cells in HNSCC (Figure 9A). For example, the expression level of IGF2BP2 had a negative correlation with the immune infiltration level of B cells (Spearman r = -0.624, p=6.39e-11). The mutation frequency of the IGF2BP2 gene in each TCGA cancer type is shown as a bar plot (Figure 9B). We found that HNSCC has the lowest IGF2BP2 mutation frequency among multiple cancer types. Therefore, IGF2BP2 gene mutations may have a weak relationship with immune cell infiltration across 22 multiple cancer types and immune cell types. The relative proportions of IGF2BP2 in different sCNA states in all tumor types are shown as a stacked bar chart (Figure 9C). We found that high amplification, arm-level gain, and diploidy were the most common states. It was also found that the distribution of immune cell infiltration in different sCNA states was different. For example, the differential T cell CD8+ (TIMER) and monocyte (XCELL) infiltration levels were observed in these sCNA states in HNSCC (Figures 9D, E).
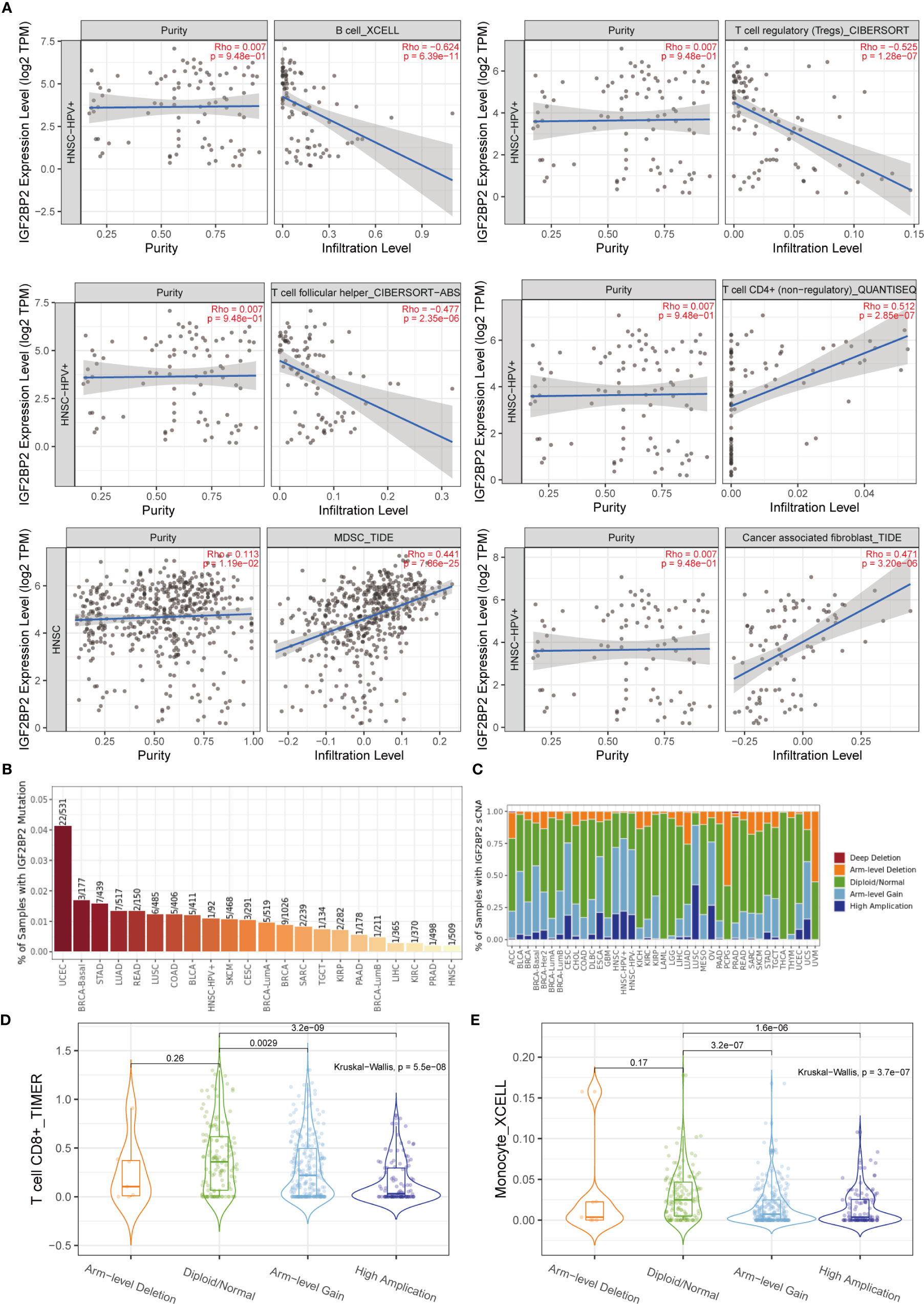
Figure 9 Relationship between the tumor-immune microenvironment and IGF2BP2 expression. (A) The relationship between IGF2BP2 and immune cell infiltration in HNSCC. (B) Bar plot of IGF2BP2 mutation frequency in each TCGA cancer type. (C) The stacked bar chart shows the relative proportions of IGF2BP2 in different sCNA states in all tumor types. (D) Differential T cell CD8+ infiltration level in HNSCC (TIMER). (E) Differential monocyte infiltration level in HNSCC (XCELL).
The abnormal epigenetic modification of RNA is important to tumorigenesis and cancer patient prognosis. As the most common modification type in eukaryotic mRNA, m6A is characterized by the N6 methylation of adenosine. Its role in cancer has gradually been recognized (6). It was recently found that m6A methylation has an indispensable function in innate immunity and the tumor microenvironment. This study focused on the abnormal gene expression of m6A regulators in pan-cancer and the molecular characteristics, prognostic value, and immune characteristics of m6A regulators in HNSCC.
In this study, we generated a catalog of 24 genes that act as regulators of m6A (8 writers, 2 erasers and 14 readers). After exploring their expression landscape in pan-cancer, it was noted that most m6A regulatory genes were abnormally expressed in tumors in a cancer-specific manner, and all the m6A regulatory genes were correlated with patient OS in at least one cancer type. These results were consistent with the findings of previous studies. For instance, METTL3 was found to be upregulated in colorectal cancer (CRC), and high METTL3 expression was associated with poor OS in CRC patients (40). Similarly, IGF2BP2 was upregulated and found to be related to a poor prognosis in pancreatic cancer (41) and esophagogastric junction adenocarcinoma (42). Moreover, it was found that combined levels of METTL3 and YTHDF1 could reflect the malignancy state and prognosis of hepatocellular carcinoma (HCC) (43). Our findings showed that most m6A regulatory genes were upregulated or downregulated in HNSCC, suggesting that these genes might be related to the carcinogenic activity of tumor cells and/or the prognosis of HNSCC patients. Among these m6A regulatory genes, IGF2BP2 was upregulated in HNSCC tumor tissues, and high IGF2BP2 expression was associated with poor patient OS in seven cancer types, including HNSCC. Similar studies in other cancers, such as colorectal, pancreatic, and breast cancers, have reported similar results (44–46). We found that the expression of IGF2BP2, RBMX, and HNRNPC was correlated with various activated cancer-related pathways, providing insight into the molecular mechanisms related to tumor progression. Furthermore, m6A regulators are useful as biomarkers for prognostic stratification (47). Our results showed that IGF2BP2 expression was negatively correlated with the prognosis of HNSCC, so IGF2BP2 is very likely an oncogene in HNSCC. In addition, CNV and DNA methylation, but not somatic mutations, give rise to the abnormal upregulation of IGF2BP2 in HNSCC based on data from TCGA-HNSCC patients. This result further shows that epigenetic modifications are also pivotal to the aberrant expression of IGF2BP2, an epigenetic modifier (41). This research suggests that the abnormal expression of m6A regulatory genes such as IGF2BP2 is common in HNSCC, but relatively little information on the function of IGF2BP2 in tumorigenesis is currently available, and its precise expression and mechanisms require further investigation.
Regarding the mechanisms controlling the tumorigenesis and poor prognosis of HNSCC at the transcriptional level, we found that the transcription factor CEBPB was the most likely to bind to the promoter region of IGF2BP2. Dysregulated IGF2BP2 could also affect the associated downstream genes via the m6A modification to accelerate cancer progression (41). Our study showed that 12 genes, including LRRC59 and STIP1, may act as IGF2BP2-associated genes to play a regulatory role in the m6A modification. LRRC59 and STIP1 have been reported to be closely associated with the progression of human malignancies. For instance, high LRRC59 expression is closely related to poor survival in LUAD patients and an aggressive phenotype of breast cancer (48). STIP1 is also upregulated in LUAD and may be associated with the enhanced proliferation, adhesion, and migration and inhibition of apoptosis in LUAD cells via the JAK2/STAT3 signaling pathway and EMT (49). Similar findings have been reported in HCC and ovarian, pancreatic, renal, and gastric cancers (50, 51). Noncoding RNAs are associated with the regulation of the m6A modification and can further affect the malignant progression of cancer (52, 53). Interestingly, a series of abnormally expressed m6A-modified miRNAs or lncRNAs could perform their function by acting as ceRNAs (54, 55), suggesting potential mechanisms in the dysregulation of IGF2BP2-associated genes in HNSCC. For example, IGF2BP2 serves as a reader of m6A-modified lncRNA-DANCR and can stabilize DANCR, which reversely contributes to tumor stemness-like properties and the pathogenesis of pancreatic cancer (41). So, in order to predict the key factors that exist in this regulatory relationship, we derived a regulatory network of IGF2BP2-associated genes, miRNAs, and lncRNAs.
In recent years, there has been increasing interest in m6A modification in multiple tumor biological processes and signaling pathways not limited to the growth of tumor stem cells, neoplasia (56–58), RNA metabolism (59), and the DNA damage response after radiotherapy and chemotherapy (60). By performing GSEA, the malignant hallmarks of IGF2BP2 in HNSCC were analyzed and may include basal cell carcinoma, the Wnt signaling pathway, the Hedgehog signaling pathway, and some signaling pathways related to tumor immunology, such as primary immunodeficiency and the intestinal immune network for IgA production. GSVA showed that the high IGF2BP2 group was markedly enriched in the cell adhesion molecule CAMS, neuroactive ligand receptor interaction pathways, and the calcium signaling pathway. This study uncovered that IGF2BP2-mediated m6A methylation was strongly correlated with the malignant progression of HNSCC and tumor immunology signaling pathways. The subsequent assays revealed a negative correlation between IGF2BP2 and multiple tumor-infiltrating immune cells, such as B cells, CD4+ T cells, follicular helper T cells, and Tregs. Our findings may provide novel insight into immunotherapy targets in HNSCC.
However, this study also has some limitations. For example, the potential mechanisms by which IGF2BP2 is upregulated are only descriptive. In the future, more basic functional mechanistic studies will be needed to validate our findings, and the clinical application of m6A methyltransferase in HNSCC needs further exploration.
In conclusion, by systematically analyzing the gene expression landscape of 24 m6A regulators in pan-cancer, IGF2BP2 was found to be highly expressed in HNSCC, is correlated with tumor progression and a low survival rate, and is involved in tumor immune invasion. More importantly, IGF2BP2 might be a promising candidate as a molecular biomarker for monitoring HNSCC development and provide ideal guidance for selecting therapeutic strategies. This study provides a supplement at the molecular level for epigenetic regulation in HNSCC and ideas for effective immunotherapy targets and strategies.
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding authors.
XCG and YHZ were the major contributors to the writing and revision of the manuscript. ZMZ and ZRZ collected the related references and participated in discussions. XCG and QL made substantial contributions to the conception or design of the work. QL, WTY, and HW approved the final version of the manuscript. All authors contributed to the article and approved the submitted version.
This work was financially supported by grants from the Natural Science Foundation of Hebei Province (no. H2020201050), the National Innovation and Entrepreneurship Training Plan in 2020 (no. 202014432031), Science and Technology Capacity Improvement Projects of Hebei University of Chinese Medicine in 2019 (no. KTZ2019019), Outstanding Student Scientific Research Ability Improvement Projects of Hebei University of Chinese Medicine in 2019 (no. YXZ2019002), and Graduate Innovative Ability Training Projects of Hebei Province in 2020 (no. XCXZZBS2020002) (no. hbu2020bs003).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We would like to give special thanks to Shaoguang Sun, Professor of the Department of Biochemistry and Molecular Biology of Hebei Medical University, for his guidance and assistance in data analysis.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2021.629718/full#supplementary-material
Supplementary Figure 1 | Relationships between m6A regulatory genes and pan-cancer. (A) summary of the relevance between clinical patient survival and expression of m6A regulators. Red dots illustrate high expression of m6A regulators related to poor survival, and blue dots illustrate prolonged survival. Only genes with a Kaplan-Meier log-rank test p-value < 0.05 are shown. (B) Heatmap represents the percentage of cancers in which the pathway may be activated or inhibited by 24 m6A regulators analyzed with GSCALite. Red represents the percentage of cancers whose pathways are likely activated, and blue represents the percentage of cancers whose pathways are likely inhibited.
Supplementary Figure 2 | SNP and SNV mutations in m6A regulatory genes. (A–C) SNP types in 24 m6A regulatory genes (D) The mutation frequencies of 24 m6A regulators in 1443 patients from the TCGA pan-cancer cohort. Individual patients are represented in the column. The right bar plot shows the proportion of each variant type.
Supplementary Figure 3 | Genetic alterations in m6A regulatory genes. (A) Genetic alteration profiles of 24 m6A regulatory genes in the TCGA dataset. The data were obtained from the cBioPortal for Cancer Genomics. (B) Heatmap of the correlations between IGF2BP2 and somatic mutations (MC3 gene-level nonsilent mutation), CNV, and methylation in HNSCC via UCSC Xena.
Supplementary Table 1 | Information on thirty-three cancer types from the TCGA.
Supplementary Table 2 | Correlation of IGF2BP2 expression and clinicopathologic characteristics in HNSCC.
Supplementary Table 3 | Correlation of ELVAL1 and LRPPRC expression and clinicopathologic characteristics.
Supplementary Table 4 | Prediction of IGF2BP2 transcription factors (based on binding to its promoter) with PROMO (dissimilarity rate =0%).
1. Michalak EM, Burr ML, Bannister AJ, Dawson MA. The roles of DNA, RNA and histone methylation in ageing and cancer. Nat Rev Mol Cell Biol (2019) 20:573–89. doi: 10.1038/s41580-019-0143-1
2. Koch A, Joosten SC, Feng Z, de Ruijter TC, Draht MX, Melotte V, et al. Analysis of DNA methylation in cancer: location revisited. Nat Rev Clin Oncol (2018) 15:459–66. doi: 10.1038/s41571-018-0004-4
3. Lan Q, Liu PY, Haase J, Bell JL, Hüttelmaier S, Liu T. The Critical Role of RNA m(6)A Methylation in Cancer. Cancer Res (2019) 79:1285–92. doi: 10.1158/0008-5472.Can-18-2965
4. Zhang Y, Geng X, Li Q, Xu J, Tan Y, Xiao M, et al. m6A modification in RNA: biogenesis, functions and roles in gliomas. J Exp Clin Cancer Res (2020) 39:192. doi: 10.1186/s13046-020-01706-8
5. Delaunay S, Frye M. RNA modifications regulating cell fate in cancer. Nat Cell Biol (2019) 21:552–9. doi: 10.1038/s41556-019-0319-0
6. Deng X, Su R, Weng H, Huang H, Li Z, Chen J. RNA N(6)-methyladenosine modification in cancers: current status and perspectives. Cell Res (2018) 28:507–17. doi: 10.1038/s41422-018-0034-6
7. Frye M, Harada BT, Behm M, He C. RNA modifications modulate gene expression during development. Science (2018) 361:1346–9. doi: 10.1126/science.aau1646
8. Li A, Chen YS, Ping XL, Yang X, Xiao W, Yang Y, et al. Cytoplasmic m(6)A reader YTHDF3 promotes mRNA translation. Cell Res (2017) 27:444–7. doi: 10.1038/cr.2017.10
9. Tang C, Klukovich R, Peng H, Wang Z, Yu T, Zhang Y, et al. ALKBH5-dependent m6A demethylation controls splicing and stability of long 3’-UTR mRNAs in male germ cells. Proc Natl Acad Sci U S A (2018) 115:E325–33. doi: 10.1073/pnas.1717794115
10. Ries RJ, Zaccara S, Klein P, Olarerin-George A, Namkoong S, Pickering BF, et al. m(6)A enhances the phase separation potential of mRNA. Nature (2019) 571:424–8. doi: 10.1038/s41586-019-1374-1
11. Tang C, Xie Y, Yu T, Liu N, Wang Z, Woolsey RJ, et al. m(6)A-dependent biogenesis of circular RNAs in male germ cells. Cell Res (2020) 30:211–28. doi: 10.1038/s41422-020-0279-8
12. Xu K, Yang Y, Feng GH, Sun BF, Chen JQ, Li YF, et al. Mettl3-mediated m(6)A regulates spermatogonial differentiation and meiosis initiation. Cell Res (2017) 27:1100–14. doi: 10.1038/cr.2017.100
13. Zhang C, Chen Y, Sun B, Wang L, Yang Y, Ma D, et al. m(6)A modulates haematopoietic stem and progenitor cell specification. Nature (2017) 549:273–6. doi: 10.1038/nature23883
14. Zheng Q, Hou J, Zhou Y, Li Z, Cao X. The RNA helicase DDX46 inhibits innate immunity by entrapping m(6)A-demethylated antiviral transcripts in the nucleus. Nat Immunol (2017) 18:1094–103. doi: 10.1038/ni.3830
15. Liu N, Pan T. N6-methyladenosine–encoded epitranscriptomics. Nat Struct Mol Biol (2016) 23:98–102. doi: 10.1038/nsmb.3162
16. Ding C, Zou Q, Ding J, Ling M, Wang W, Li H, et al. Increased N6-methyladenosine causes infertility is associated with FTO expression. J Cell Physiol (2018) 233:7055–66. doi: 10.1002/jcp.26507
17. Chen X, Yu C, Guo M, Zheng X, Ali S, Huang H, et al. Down-Regulation of m6A mRNA Methylation Is Involved in Dopaminergic Neuronal Death. ACS Chem Neurosci (2019) 10:2355–63. doi: 10.1021/acschemneuro.8b00657
18. Geula S, Moshitch-Moshkovitz S, Dominissini D, Mansour AA, Kol N, Salmon-Divon M, et al. Stem cells. m6A mRNA methylation facilitates resolution of naïve pluripotency toward differentiation. Science (2015) 347:1002–6. doi: 10.1126/science.1261417
19. Li Y, Xiao J, Bai J, Tian Y, Qu Y, Chen X, et al. Molecular characterization and clinical relevance of m(6)A regulators across 33 cancer types. Mol Cancer (2019) 18:137. doi: 10.1186/s12943-019-1066-3
20. He L, Li H, Wu A, Peng Y, Shu G, Yin G. Functions of N6-methyladenosine and its role in cancer. Mol Cancer (2019) 18:176. doi: 10.1186/s12943-019-1109-9
21. Wang S, Sun C, Li J, Zhang E, Ma Z, Xu W, et al. Roles of RNA methylation by means of N(6)-methyladenosine (m(6)A) in human cancers. Cancer Lett (2017) 408:112–20. doi: 10.1016/j.canlet.2017.08.030
22. Chen M, Wong CM. The emerging roles of N6-methyladenosine (m6A) deregulation in liver carcinogenesis. Mol Cancer (2020) 19:44. doi: 10.1186/s12943-020-01172-y
23. Dixit D, Prager BC, Gimple RC, Poh HX, Wang Y, Wu Q, et al. The RNA m6A reader YTHDF2 maintains oncogene expression and is a targetable dependency in glioblastoma stem cells. Cancer Discovery (2020) 11:480–99. doi: 10.1158/2159-8290.Cd-20-0331
24. Rai V, Aggarwal SK, Verma SS, Awasthee N, Dhasmana A, Aggarwal S, et al. Epoxyazadiradione exhibit activities in head and neck squamous cell carcinoma by targeting multiple pathways. Apoptosis (2020) 25:763–82. doi: 10.1007/s10495-020-01633-1
25. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin (2018) 68:394–424. doi: 10.3322/caac.21492
26. Carlisle JW, Steuer CE, Owonikoko TK, Saba NF. An update on the immune landscape in lung and head and neck cancers. CA Cancer J Clin (2020) 70:505–17. doi: 10.3322/caac.21630
27. Zhao Q, Zhao Y, Hu W, Zhang Y, Wu X, Lu J, et al. m(6)A RNA modification modulates PI3K/Akt/mTOR signal pathway in Gastrointestinal Cancer. Theranostics (2020) 10:9528–43. doi: 10.7150/thno.42971
28. Zhang B, Wu Q, Li B, Wang D, Wang L, Zhou YL. m(6)A regulator-mediated methylation modification patterns and tumor microenvironment infiltration characterization in gastric cancer. Mol Cancer (2020) 19:53. doi: 10.1186/s12943-020-01170-0
29. Colaprico A, Silva TC, Olsen C, Garofano L, Cava C, Garolini D, et al. TCGAbiolinks: an R/Bioconductor package for integrative analysis of TCGA data. Nucleic Acids Res (2016) 44:e71. doi: 10.1093/nar/gkv1507
30. Li T, Fu J, Zeng Z, Cohen D, Li J, Chen Q, et al. TIMER2.0 for analysis of tumor-infiltrating immune cells. Nucleic Acids Res (2020) 48:W509–w514. doi: 10.1093/nar/gkaa407
31. Liu CJ, Hu FF, Xia MX, Han L, Zhang Q, Guo AY. GSCALite: a web server for gene set cancer analysis. Bioinformatics (2018) 34:3771–2. doi: 10.1093/bioinformatics/bty411
32. Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A (2005) 102:15545–50. doi: 10.1073/pnas.0506580102
33. Liberzon A, Birger C, Thorvaldsdóttir H, Ghandi M, Mesirov JP, Tamayo P. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst (2015) 1:417–25. doi: 10.1016/j.cels.2015.12.004
34. Hänzelmann S, Castelo R, Guinney J. GSVA: gene set variation analysis for microarray and RNA-seq data. BMC Bioinf (2013) 14:7. doi: 10.1186/1471-2105-14-7
35. Gertz J, Savic D, Varley KE, Partridge EC, Safi A, Jain P, et al. Distinct properties of cell-type-specific and shared transcription factor binding sites. Mol Cell (2013) 52:25–36. doi: 10.1016/j.molcel.2013.08.037
36. Zhang Q, Liu W, Zhang HM, Xie GY, Miao YR, Xia M, et al. hTFtarget: A Comprehensive Database for Regulations of Human Transcription Factors and Their Targets. Genomics Proteomics Bioinf (2020) 18:120–8. doi: 10.1016/j.gpb.2019.09.006
37. Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res (2003) 13:2498–504. doi: 10.1101/gr.1239303
38. Tai Y, Ji Y, Liu F, Zang Y, Xu D, Ma S, et al. Long noncoding RNA SOX2-OT facilitates laryngeal squamous cell carcinoma development by epigenetically inhibiting PTEN via methyltransferase EZH2. IUBMB Life (2019) 71:1230–9. doi: 10.1002/iub.2026
39. Li JH, Liu S, Zhou H, Qu LH, Yang JH. starBase v2.0: decoding miRNA-ceRNA, miRNA-ncRNA and protein-RNA interaction networks from large-scale CLIP-Seq data. Nucleic Acids Res (2014) 42:D92–97. doi: 10.1093/nar/gkt1248
40. Peng W, Li J, Chen R, Gu Q, Yang P, Qian W, et al. Upregulated METTL3 promotes metastasis of colorectal Cancer via miR-1246/SPRED2/MAPK signaling pathway. J Exp Clin Cancer Res (2019) 38:393. doi: 10.1186/s13046-019-1408-4
41. Hu X, Peng WX, Zhou H, Jiang J, Zhou X, Huang D, et al. IGF2BP2 regulates DANCR by serving as an N6-methyladenosine reader. Cell Death Differ (2020) 27:1782–94. doi: 10.1038/s41418-019-0461-z
42. Tang W, Chen S, Liu J, Liu C, Wang Y, Kang M. Investigation of IGF1, IGF2BP2, and IGFBP3 variants with lymph node status and esophagogastric junction adenocarcinoma risk. J Cell Biochem (2019) 120:5510–8. doi: 10.1002/jcb.27834
43. Zhou Y, Yin Z, Hou B, Yu M, Chen R, Jin H, et al. Expression profiles and prognostic significance of RNA N6-methyladenosine-related genes in patients with hepatocellular carcinoma: evidence from independent datasets. Cancer Manag Res (2019) 11:3921–31. doi: 10.2147/cmar.S191565
44. Chen S, Qiu H, Liu C, Wang Y, Tang W, Kang M. Relationship between IGF2BP2 and IGFBP3 polymorphisms and susceptibility to non-small-cell lung cancer: a case-control study in Eastern Chinese Han population. Cancer Manag Res (2018) 10:2965–75. doi: 10.2147/cmar.S169222
45. McMullen ER, Gonzalez ME, Skala SL, Tran M, Thomas D, Djomehri SI, et al. CCN6 regulates IGF2BP2 and HMGA2 signaling in metaplastic carcinomas of the breast. Breast Cancer Res Treat (2018) 172:577–86. doi: 10.1007/s10549-018-4960-2
46. Xu X, Yu Y, Zong K, Lv P, Gu Y. Up-regulation of IGF2BP2 by multiple mechanisms in pancreatic cancer promotes cancer proliferation by activating the PI3K/Akt signaling pathway. J Exp Clin Cancer Res (2019) 38:497. doi: 10.1186/s13046-019-1470-y
47. Du J, Hou K, Mi S, Ji H, Ma S, Ba Y, et al. Malignant Evaluation and Clinical Prognostic Values of m6A RNA Methylation Regulators in Glioblastoma. Front Oncol (2020) 10:208. doi: 10.3389/fonc.2020.00208
48. Li D, Xing Y, Tian T, Guo Y, Qian J. Overexpression of LRRC59 Is Associated with Poor Prognosis and Promotes Cell Proliferation and Invasion in Lung Adenocarcinoma. Onco Targets Ther (2020) 13:6453–63. doi: 10.2147/ott.S245336
49. Guo X, Yan Z, Zhang G, Wang X, Pan Y, Huang M. STIP1 Regulates Proliferation and Migration of Lung Adenocarcinoma Through JAK2/STAT3 Signaling Pathway. Cancer Manag Res (2019) 11:10061–72. doi: 10.2147/cmar.S233758
50. Luo X, Liu Y, Ma S, Liu L, Xie R, Li M, et al. STIP1 is over-expressed in hepatocellular carcinoma and promotes the growth and migration of cancer cells. Gene (2018) 662:110–7. doi: 10.1016/j.gene.2018.03.076
51. Jing Y, Liang W, Liu J, Zhang L, Wei J, Zhu Y, et al. Stress-induced phosphoprotein 1 promotes pancreatic cancer progression through activation of the FAK/AKT/MMP signaling axis. Pathol Res Pract (2019) 215:152564. doi: 10.1016/j.prp.2019.152564
52. Dai F, Wu Y, Lu Y, An C, Zheng X, Dai L, et al. Crosstalk between RNA m(6)A Modification and Non-coding RNA Contributes to Cancer Growth and Progression. Mol Ther Nucleic Acids (2020) 22:62–71. doi: 10.1016/j.omtn.2020.08.004
53. Geng X, Zhang Y, Li Q, Xi W, Yu W, Shi L, et al. Screening and functional prediction of differentially expressed circular RNAs in human glioma of different grades. Aging (Albany NY) (2020) 13:1989–2014. doi: 10.18632/aging.202192
54. Geng X, Jia Y, Zhang Y, Shi L, Li Q, Zang A, et al. Circular RNA: biogenesis, degradation, functions and potential roles in mediating resistance to anticarcinogens. Epigenomics (2020) 12:267–83. doi: 10.2217/epi-2019-0295
55. Geng X, Lin X, Zhang Y, Li Q, Guo Y, Fang C, et al. Exosomal circular RNA sorting mechanisms and their function in promoting or inhibiting cancer. Oncol Lett (2020) 19:3369–80. doi: 10.3892/ol.2020.11449
56. Cui Q, Shi H, Ye P, Li L, Qu Q, Sun G, et al. m(6)A RNA Methylation Regulates the Self-Renewal and Tumorigenesis of Glioblastoma Stem Cells. Cell Rep (2017) 18:2622–34. doi: 10.1016/j.celrep.2017.02.059
57. Pan Y, Ma P, Liu Y, Li W, Shu Y. Multiple functions of m(6)A RNA methylation in cancer. J Hematol Oncol (2018) 11:48. doi: 10.1186/s13045-018-0590-8
58. Chai RC, Wu F, Wang QX, Zhang S, Zhang KN, Liu YQ, et al. m(6)A RNA methylation regulators contribute to malignant progression and have clinical prognostic impact in gliomas. Aging (Albany NY) (2019) 11:1204–25. doi: 10.18632/aging.101829
59. Dai D, Wang H, Zhu L, Jin H, Wang X. N6-methyladenosine links RNA metabolism to cancer progression. Cell Death Dis (2018) 9:124. doi: 10.1038/s41419-017-0129-x
Keywords: m6A regulators, IGF2BP2, head and neck squamous cell carcinoma (HNSCC), pan-cancer, prognosis, immune cell infiltration
Citation: Geng X, Zhang Y, Zeng Z, Zhu Z, Wang H, Yu W and Li Q (2021) Molecular Characteristics, Prognostic Value, and Immune Characteristics of m6A Regulators Identified in Head and Neck Squamous Cell Carcinoma. Front. Oncol. 11:629718. doi: 10.3389/fonc.2021.629718
Received: 15 November 2020; Accepted: 26 February 2021;
Published: 18 March 2021.
Edited by:
Baofa Sun, Beijing Institute of Genomics (CAS), ChinaReviewed by:
Shunmin He, Institute of Biophysics (CAS), ChinaCopyright © 2021 Geng, Zhang, Zeng, Zhu, Wang, Yu and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Qiang Li, cWlhbmdsaTIwMjBAMTYzLmNvbQ==; Wentao Yu, eXd0YXdlbkAxNjMuY29t; Hong Wang, Ym9zc3dAdmlwLnNpbmEuY29t
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.