 Jie Qiu
Jie Qiu
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oncol. , 25 February 2021
Sec. Women's Cancer
Volume 11 - 2021 | https://doi.org/10.3389/fonc.2021.628359
This article is part of the Research Topic Metabolic Abnormalities and Breast Cancer: Challenges from Bench to Bedside View all 19 articles
Female breast cancer is a complex, multifactorial disease. Studies have shown that hyperglycemia is one of the most important contributing factors to increasing the risk of breast cancer that also has a major impact on the efficacy of chemotherapy. At the cellular level, hyperglycemia can promote the proliferation, invasion, and migration of breast cancer cells and can also induce anti-apoptotic responses to enhance the chemoresistance of tumors via abnormal glucose metabolism. In this article, we focus on the latest progress in defining the mechanisms of chemotherapy resistance in hyperglycemic patients including the abnormal behaviors of cancer cells in the hyperglycemic microenvironment and the impact of abnormal glucose metabolism on key signaling pathways. To better understand the advantages and challenges of breast cancer treatments, we explore the causes of drug resistance in hyperglycemic patients that may help to better inform the development of effective treatments.
Breast cancer (BC) is the most common malignancy in women all over the world (1) and has several known risk factors including age, sex, obesity, estrogen levels, and family history (2). Recent studies have shown that hyperglycemia is an important risk factor in the development of BC (3). In BC, hyperglycemia is associated with an increased prevalence and mortality but also has major impacts on the efficacy of chemotherapy efficacy and can lead to chemoresistance. The development and progression of BC involves the dysfunction of several molecular processes including abnormal glucose metabolism, abnormal insulin levels, insulin resistance, distorted signal pathways, oxidative stress, and enhanced inflammatory processes (4, 5).
The “Warburg” effect is considered to be the most important feature of glucose metabolism in tumors (6). Under hypoxic conditions, aerobic glycolysis in tumor cells significantly changes compared to aerobic oxidation and so cancer cells upregulate the processes of glycolysis and the catabolism of glucose to form lactate. This process is accompanied by ATP production. However, ATP produced by glycolysis is not sufficient to support the survival of cancer cells and so the rate of glucose uptake and the fermentation of glucose to lactate are both increased (7). Sufficient energy supply activates cellular signaling pathways, promotes the abnormal activity of tumor cells, and induces an anti-apoptotic response and chemotherapy resistance (8). The reprogramming of glucose metabolism accelerates the conversion of glycolysis and changes the acidity of the microenvironment which acts to promote the expression of angiogenic factors and enhance tumor metastasis (9).
Hyperglycemia in patients is mostly accompanied by dyslipidemia. It has been reported that glycolipid metabolism may have a synergistic influence on chemotherapy resistance (10). Obesity leads to an accumulation of lipids and increases the circulating levels of fatty acids that enhance insulin resistance and hyperinsulinemia eventually leading to hyperglycemia and diabetes (1). However, glucose also can act as a substrate for fatty acid synthase which is a key enzyme responsible for the de novo synthesis of fatty acids (11, 12). The prognosis for women with BC is adversely affected by the comorbidities of hyperlipidemia and hyperglycemia. In this review, we primarily focus on the effects of crucial glucose metabolic pathways to explore how abnormal blood glucose levels influence the pathology of BC cells and reduce the efficacy of chemotherapy to drive chemoresistance.
Hyperglycemia can provide nutrients for the rapid proliferation of breast cancer cells. Studies have reported that high concentrations of glucose significantly increase the proliferation of BC cells (such as MDA-MB-231 and MCF-7) (13, 14) through a mechanism involving activation of epidermal growth factor receptors (EGFRs) (15). EGFR is frequently overexpressed in triple-negative BC and is associated with poor prognosis. It has been reported that the GTPase activating protein (GAP) USP6NL that is involved in endocytosis and signal transduction is also overexpressed in BC, particularly the basal-like subtype (16). BC cells with high levels of USP6NL show delayed inactivation of EGFR leading to chronic activation of AKT which maintains the stability of the glucose transporter 1 (GLUT1) on the plasma membrane leading to increased glucose metabolism (17). GLUT1 transports and absorbs glucose through the plasma membrane to provide energy for BC cells and to promote cellular proliferation and invasion (18, 19). When BC cells lack sensitivity to respond to changes in glucose concentration, elevated USP6NL can compensate for this deficiency and stabilize GLUT1 by activated AKT, shows that its glycolysis ability depends on the protein.
Long-term hyperglycemia results in insulin resistance, hyperinsulinemia, and dysfunctional signaling through the insulin-like growth factor-1 pathway (20). It has been reported that IGF-1 participates in estrogen receptor signal transduction through IGF-1 receptor/ER interactions. This process is bidirectional as these interactions can regulate the proliferation, apoptosis, and differentiation of breast epithelial cells, resulting in increased risk of endocrine-related cancers particularly in post-menopausal women (21).
It has also been reported that hyperglycemia promotes the proliferation of malignant BC epithelial cells by increasing activity of the leptin/insulin-like growth factor-1 receptor signaling pathway and causing activation of the Protein Kinase B/mechanistic target of rapamycin (AKT/mTOR) pathway (22). Luey et al. found that the type I IGF receptor is expressed widely in BC cells and mediates the effects of IGFs on cell proliferation and migration. IGF-1 activates both the PI3K/Akt and Grb2/Ras/MAP-kinase pathways to increasing the invasive capacity of BC cells (23). The abnormal activation of the classical PI3K/AKT/mTOR signaling pathway leads to a poor prognosis due to increased tumor cell proliferation, metastasis, and drug resistance. Based on these data, targeting the PI3K/AKT/mTOR signaling pathway may be a potential therapeutic strategy in the treatment of BC (24).
The regulation of key transcription factors and inflammatory mediators in the glycolytic phenotype of BC remains an area of intense investigation (25). Hypoxia is a common phenomenon within the tumor environment. The cellular hypoxic response is mediated by hypoxia-inducible factor-1 (HIF-1) which is a crucial regulatory factor of glycolysis in cancer cells. HIF-1 is an oxygen-sensing transcription factor that regulates the consumption of glucose through oxidation or glycolysis (26). Using short interfering RNA (siRNA) to silence the expression of HIF-1, Chen et al. showed that HIF-1 KO significantly inhibited the extracellular acidification rate (ECAR), glucose consumption rate, and production of lactic acid in BC cells. HIF-1 silencing has also been shown to reduce the expression of metabolic enzymes and transporters in tumor cells and reversing resistance to apoptosis in BC cells following chemotherapy treatment (27).
Hypoxia-inducible factor 1α-induced glycolysis is essential for the activation of inflammatory macrophages. For example, the high infiltration of M2 tumor-associated macrophages is an extremely important feature of inflammatory BC (28). HIF-1 regulates metabolism during hypoxia and its transcriptional activity is also induced by T cell activation in response to hypoxia (29). These changes promote metabolic reprogramming and lead to the upregulation of genes encoding glycolytic promoting enzymes such as pyruvate kinase (PKM1), hexokinase 2 (HK2), and GLUT1 (30–32). Upregulated expression of these genes can also produce a false hypoxic state in tumors to promote angiogenesis, migration, and metastasis (33) that are closely related to the occurrence and development of BC tumors.
Epithelial-mesenchymal transformation (EMT) is an important mechanism that promotes the migration, invasion, and metastasis of cancer cells (29, 34). Zielinska et al. showed that hyperglycemia can induce matrix-specific EMT to promote the Warburg effect by upregulating glucose uptake, lactate release, and the expression of specific glycolytic enzymes and transporters. They also found that silencing fatty acid synthase (FASN) reversed the effects of hyperglycemia on the levels of EMT markers leading to increased expression of E-cadherin and decreased the expression of vimentin and fibronectin. Upregulation of these proteins is indicative of EMT and associated with metastatic progression (34). Taken together, these studies highlight the importance of hyperglycemia in the development and progression of BC (Figure 1).
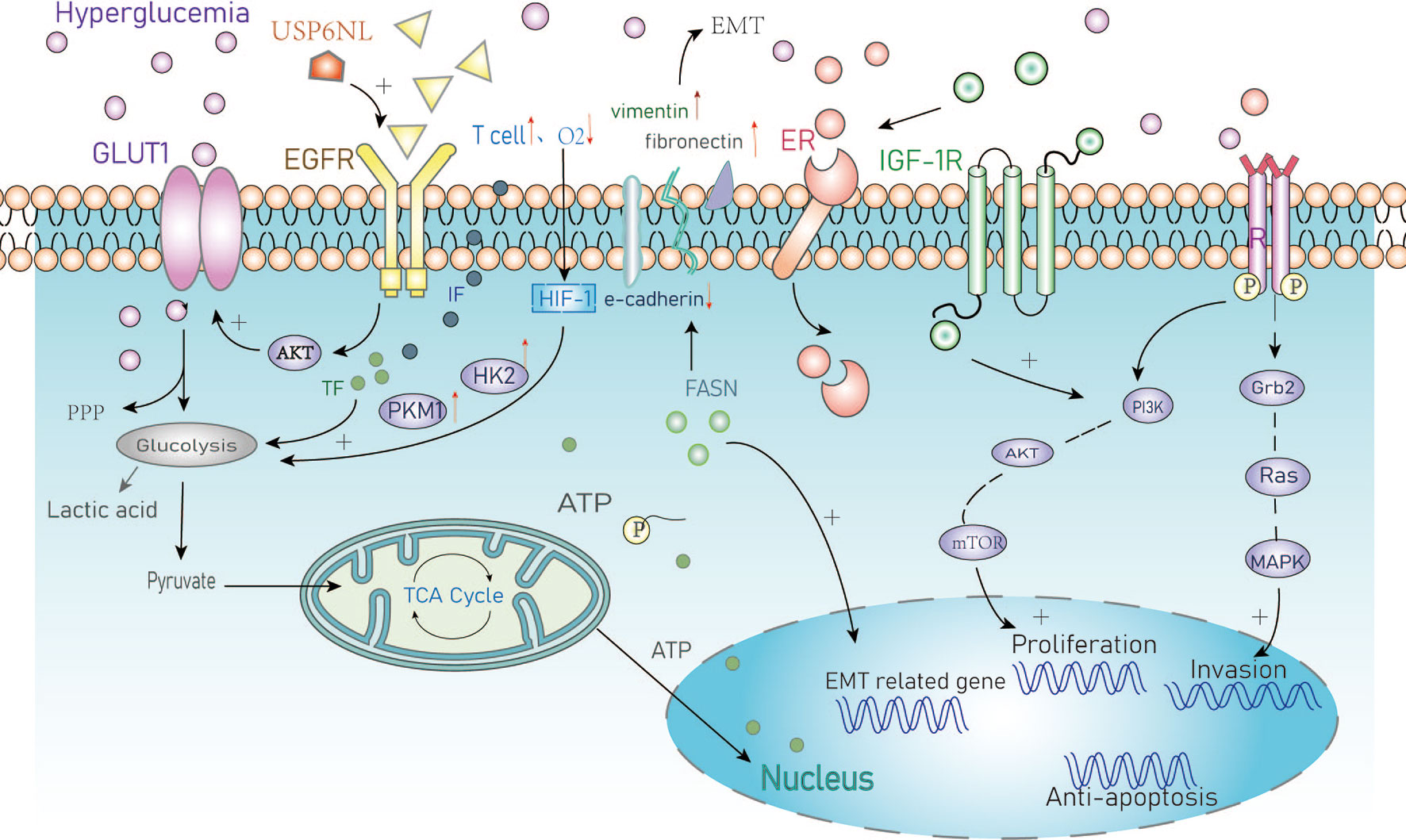
Figure 1 Summary of the cellular metabolic effects of hyperglycemia in cancer. GLUT1, Glucose transporter 1; PPP, Pentose phosphate pathway; EGFR, Epidermal growth factor receptor; IF, Inflammatory factors; TF, Transcription factors; HIF, Hypoxia-inducible factor-1; HK2, Hexokinase2; PKM1, Pyruvate kinase M1; EMT, Epithelial-mesenchymal transition; FASN/FAS, Fatty acid synthase; ER, Estrogen receptor; IGF, Insulin growth factor; PI3K, Intracellular phosphatidylinositol kinase; AKT, Protein kinase B; mTOR, Mammalian target of rapamycin; Grb2, Growth factor receptor-bound protein2; MAPK, Mitogen-activated protein kinase.
P53 inhibits cellular transformation and activates tumor cell responses to chemotherapy drugs. Homeodomain-interaction protein kinase 2 (HIPK2) is a nuclear serine/threonine kinase that mediates p53-dependent apoptotic pathways in tumor cells (8). Hyperglycemic environments can trigger the degradation of the HIPK2 protein and upregulate the expression of mutant p53 to inhibit p53-induced apoptosis (35). Overexpression of mutant p53 is positively correlated with high expression of the P-glycoprotein which is a known protein biomarker of chemotherapeutic resistance. Upregulation of P-glycoprotein means can facilitate drug resistance and promote tumor progression, however, this mechanism has not been frequently reported in BC.
Rac1 is a small GTP binding protein. Li et al. found it is overexpressed and associated with multidrug resistance to neoadjuvant chemotherapy (NAC) (36). Rac1 activates aldosterone A and ERK signals and upregulates glycolysis, particularly the pentose phosphate pathway (PPP). This leads to an increase in nucleotide metabolism that can protect BC cells from DNA damage caused by chemotherapy (37). The PPP metabolite pentose phosphate is critical for nucleic acid biosynthesis and NADPH is essential for fatty acid synthesis as well as the mitigation of cellular oxidative stress (38) (Figure 2). The overexpression of Rac1 and the abnormal activation of the PPP pathway in BC patients with hyperglycemia can result in resistance to DNA damage as well as reduced oxidative stress. This can lead to therapeutic resistance that contributes to poor outcomes in BC patients.
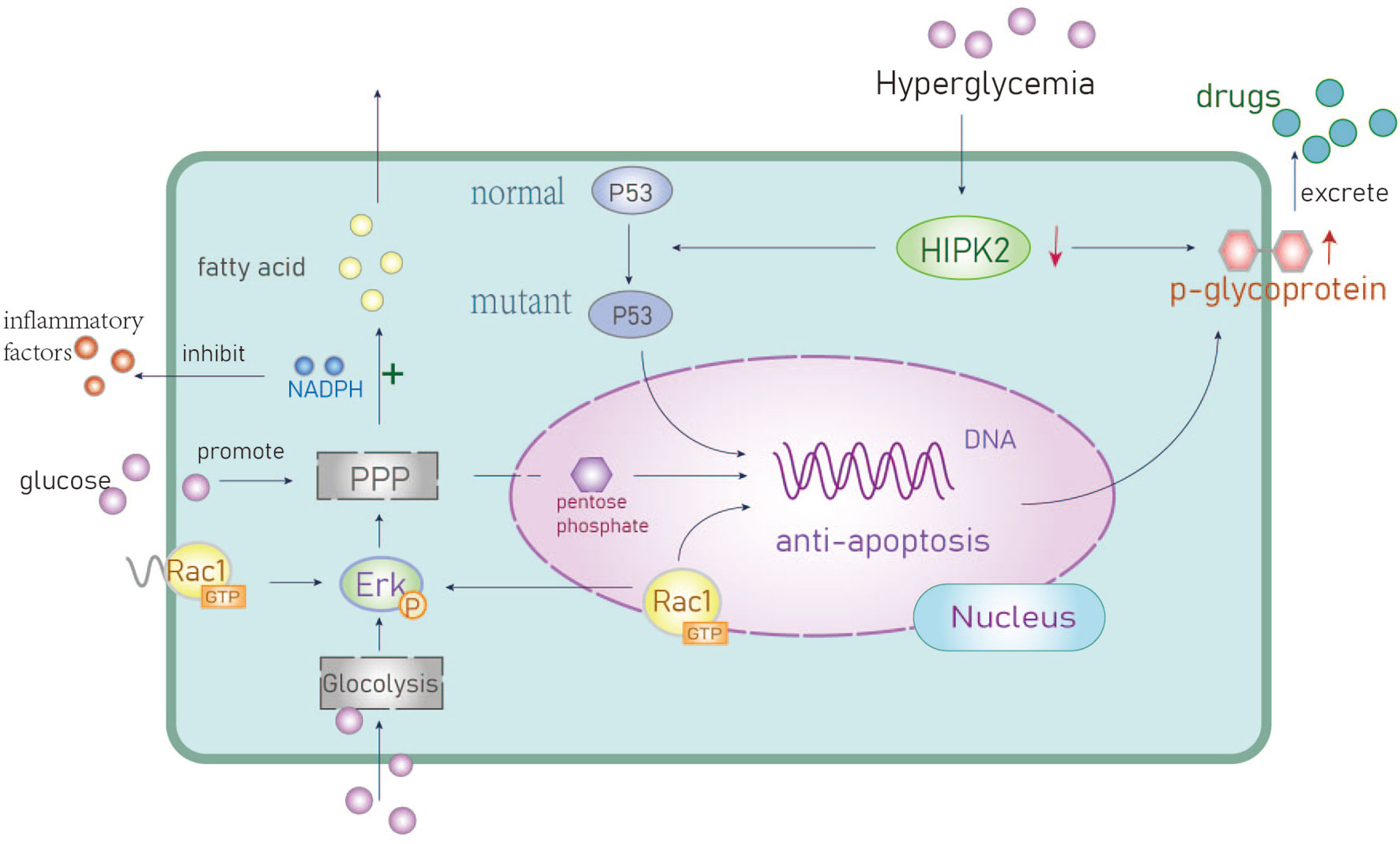
Figure 2 The anti-apoptotic mechanisms of hyperglycemia. Activation of anti-apoptotic genes promotes chemotherapeutic drug resistance. PPP, Pentose phosphate pathway; HIPK2, Homologous domain interacting protein kinase1; Erk, extracellular regulated protein kinase; P53, a tumor suppressor gene.
Adriamycin (ADR) resistant BC cells have increased glucose metabolism (39). It has been reported that ADR containing chemotherapy plans can effectively induce insulin resistance in cells (40). The fibroblast growth factor (FGFs)-FGFR signaling pathway is involved in many biological processes such as embryonic development, wound healing, and angiogenesis. Xu et al. postulated that high expression of FGFR4 is a key regulator in ADR resistant cells that is associated with poor survival (41). Activation of FGFR4 signaling leads to phosphorylation of FGF receptor substrate 2 (FRS2) and further activation of downstream MAPK/ERK signaling. Pharmacological inhibition of the FGFR4-FRS2-ERK signaling pathway has been shown to decrease chemoresistance and the glycolytic phenotypes of ADR-resistant cells (42). MQA et al. hypothesized that glucose metabolism has a major impact on the expression of insulin-like growth factor binding protein2 (IGFBP-2) which is an essential regulator of the IGF signaling axis. The continuous secretion of IGFBP-2 promotes chemotherapy resistance in BC and can be abrogated by silencing of IGFBP-2 expression. This results in reversing chemotherapy resistance induced by high glucose levels and resensitizes BC cells to ADR (5). The combination of 3-BRPY (a glycolysis inhibitor) and ADR has been shown to reduce total ATP and lactic acid levels (43) yet these specific mechanisms remain to be fully understood.
Paclitaxel is a commonly used chemotherapy drug in the treatment of TNBC to which patients commonly develop resistance (44). Studies have reported the relationship between lactate dehydrogenase A (LDHA) and paclitaxel resistance. The downregulation of LDHA combined with paclitaxel with oxamate (an analogue of pyruvate) has been shown to result in a two-fold increase in the sensitivity to paclitaxel suggesting that targeted glycolytic enzymes may resensitize drug-resistant cells to paclitaxel. These data indicate that LDHA is a potential therapeutic target for overcoming paclitaxel resistance and in BC (45). Moreover, the most common mechanism of paclitaxel resistance is through drug efflux from the ATP binding cassette transporter. The energy required for drug efflux mainly comes from the glycolytic pathway and the P-glycoprotein is an important factor that mediates drug outflow causing drug resistance (46).
Cisplatin is a common chemotherapy drug to which BC patients often develop resistance mediated by the altered expression of glycolytic enzymes and glucose transporters (47). Considering the role of GLUT1 as an important glucose transporter (17, 18), GLUT1 may be involved in cisplatin resistance in BC cells under hyperglycemia. He et al. found that the overexpression of the oncogene TRIM59 in non-small cell lung cancer cells was related to cisplatin resistance and was mediated by the glycolysis-related gene HK2. Based on these data, we hypothesize that TRIM59 is involved in the metabolic reprogramming of cisplatin-resistant cancer cells by regulating the expression of HK2 (48), however, this has not yet been reported in the field of BC.
Tamoxifen is the most commonly used endocrine therapy in BC and patients often develop drug resistance due to enhanced glycolysis in ER-positive BC. He et al. demonstrated that activation of the EGFR signaling pathway and its downstream glycolytic genes play an important role in tamoxifen-resistant BC cells (49). Tamoxifen acts by inhibiting the mitochondrial respiratory complex I to reduce ATP levels and activate AMPK. These alterations induce apoptosis by suppressing mTOR (50). Hyperglycemia can activate the AKT/mTOR/AMPK signaling pathway which is involved in tamoxifen resistance (51). Also, Huang et al. recently reported that tamoxifen inhibits the proliferation of gallbladder cancer cells by impairing glucose metabolism (52). It is known that the occurrence of gall bladder cancer may be related to estrogen receptors suggesting a potential link with the effects of tamoxifen. This deserves in the field of breast cancer deeply.
Trastuzumab (Herceptin) is an antibody targeted against HER2. Aerobic glycolysis can be inhibited by the ErbB2 (HER2)-heat shock factor1 (HSF1)-LDHA pathway. The sensitivity to trastuzumab has been associated with LDHA activity (53). PKM2 is another key glycolytic enzyme that has been implicated in response to trastuzumab in BC (54). PKM2 is a rate-limiting enzyme in glycolysis that has been proposed as an early marker of the treatment response to trastuzumab in BC patients with metastasis (55). The PI3K/AKT signaling pathway has multiple control points in the glucose metabolism pathway including glucose transporters and enzymes that regulate glycolysis. Studies have proposed that trastuzumab combined with PI3K/AKT inhibitors may be used to improve responses to treatment in cancer (56). In particular, one study showed differences in glucose metabolism between ER+/HER2−/+ subtypes of BC cell lines exposed to palbociclib. In ER+/HER2-palbociclib sensitive cells, aerobic glycolysis and glucose catabolism were enhanced in ER+/HER2+ Palbociclib resistance (57) (Figure 3). However, the mechanism through which targeted drugs promote aerobic glycolysis in BC remains to be fully determined.
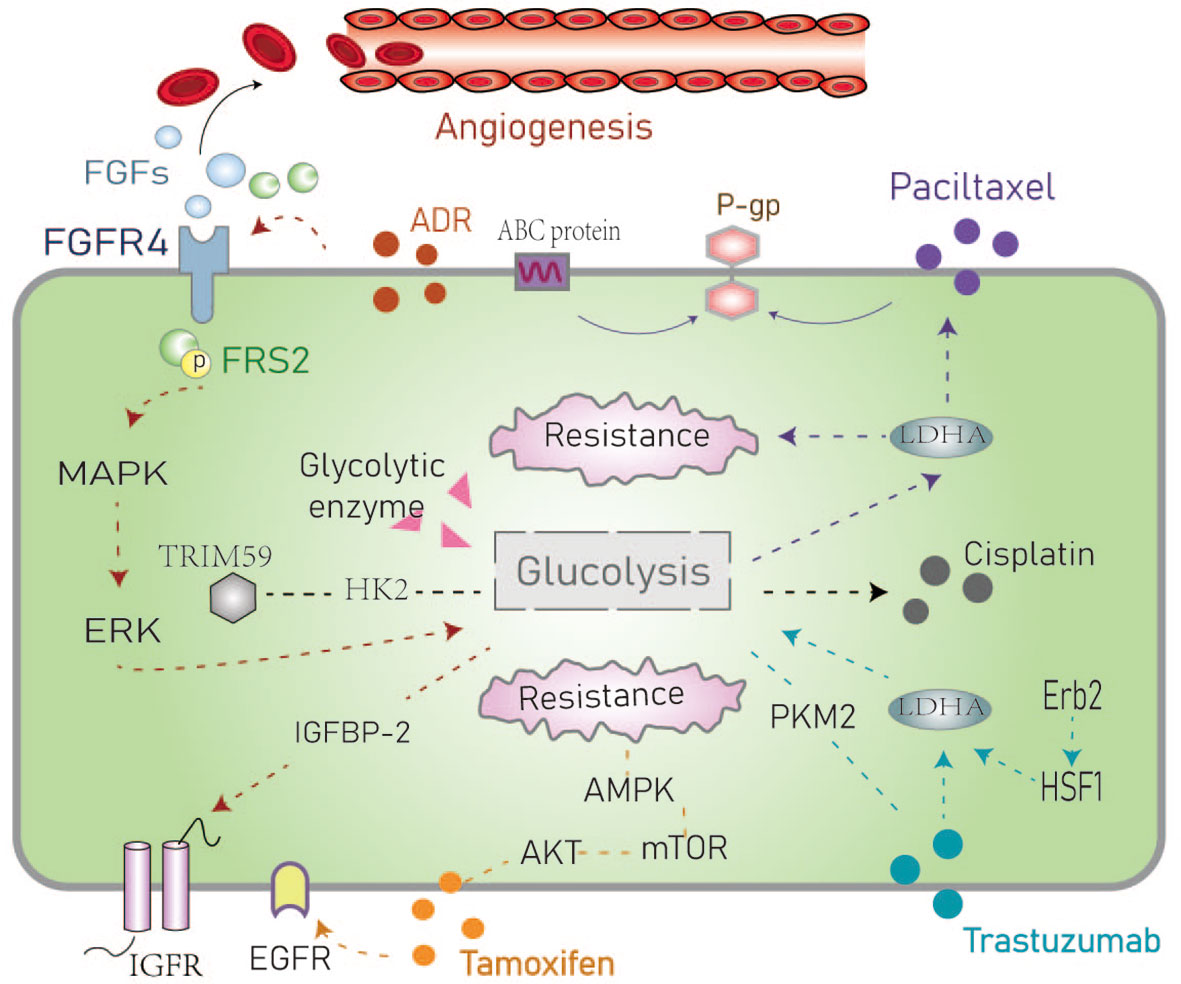
Figure 3 Schematic representation of the resistance mechanisms of common chemotherapeutic drugs in the hyperglycemic environment. The dotted line represents the route in the text. ADR, Adriamycin; FGF/FGFR, Fibroblast growth factor and its receptor; FRS, Fibroblast growth factor receptor substrate; IGFBP-1, Insulin-like growth factor-binding protein-1; ABC protein, ATP binding cassette transporter; P-glycoprotein; AMPK, AMP-activated protein kinase; PKM2, Pyruvate kinase M2; HSF1, Heat shock factor1; LDHA, Lactate dehydrogenase.
BC cells undergo metabolic reprogramming that usually includes enhanced glycolysis and increased activity of the tricarboxylic acid cycle (58). Scholars put forward the blood glucose changes three pathways in TME: VEGF and its receptors, cell to cell, and cell to extracellular matrix (ECM) adhesion proteins. What cause BC cells (MDA-MB-231) to metastatic mutations to bone and brain (59). It can be seen that the blood glucose load in the microenvironment is very important in tumor growth. We talked about the changes caused by hyperglycemia in the microenvironment, such as the increase in PH, the high concentration of lactic acid, the production of inflammatory factors, the imbalance of ROS, and the impact on immune cells.
The TME is mainly composed of tumor cells, surrounding immune and inflammatory cells, fibroblasts, interstitial tissue, capillaries, various cytokines, and chemokines. The TME is characterized by high levels of H2O2 and glutathione (GSH), low pH, and hypoxia that have important implications for responses to treatment (60).
The TME plays an important role in the metabolic adaptation and survival of tumor cells. The inflammatory microenvironment can directly induce aerobic glycolysis (61). Studies have found that hyperglycemia and insulin can induce mesenchymal phenotypes through the generation of reactive oxygen species (ROS) (62). Under hyperglycemic or pathological microenvironments, imbalances between the production and removal of ROS and the production of chronic inflammatory markers (such as IL6, TNF-α, and COX-2) under hyperglycemic conditions can induce anti-apoptotic activities and EMT in cells (63).
The Warburg effect causes tumor cells to continuously export and accumulate lactic acid in the TME (5, 64). Chen and colleagues reported that high levels of lactic acid are related to the incidence of distant metastasis. LIN28B promotes the secretion of lactate and enhances the stem cell properties of cancer cells. Overexpression LIN28B increases the rate of extracellular acidification, glucose uptake, and lactic acid secretion both in vivo and in vitro (65). The impact of the acidic TME on cancer stem cells (CSCs) is thought to result in tumor relapse, therapeutic resistance, and metastasis (66). Moreover, lactic acid is also a key factor involved in angiogenesis and immune evasion. Lactic acid leads to extracellular pH acidification within the microenvironment that is also related to clinical prognosis (67). The production of lactic acid depends on the levels of key enzymes in the glycolytic pathway. LDH also plays an important role in regulating the nutritional exchange between tumor cells and stroma cells. Studies have found LDHA targeting tumor cells and LDHB targeting stromal cells influence tumor proliferation. These data suggest that it may be beneficial to block lactate exchange between tumor and stroma as a potential therapeutic strategy (68).
Uridine diphosphate glucose (UDP)-sugars are generated as intermediate products of glucose metabolism. The levels of these sugars are significantly increased in BC and have been shown to promote the accumulation of hyaluronic acid which is a known promoter of the disease. The metabolism of hyaluronic acid within the TME a key factor that drives invasive growth and metastasis (69, 70). In ductal and lobular BC, the levels of UDP-sugars are significantly increased. These observations suggest that blocking the excessive supply of UDP-sugars and reducing the content of hyaluronic acid may be potential therapeutic strategies in BC patients with glucose metabolism disorders (71, 72) (Figure 4).
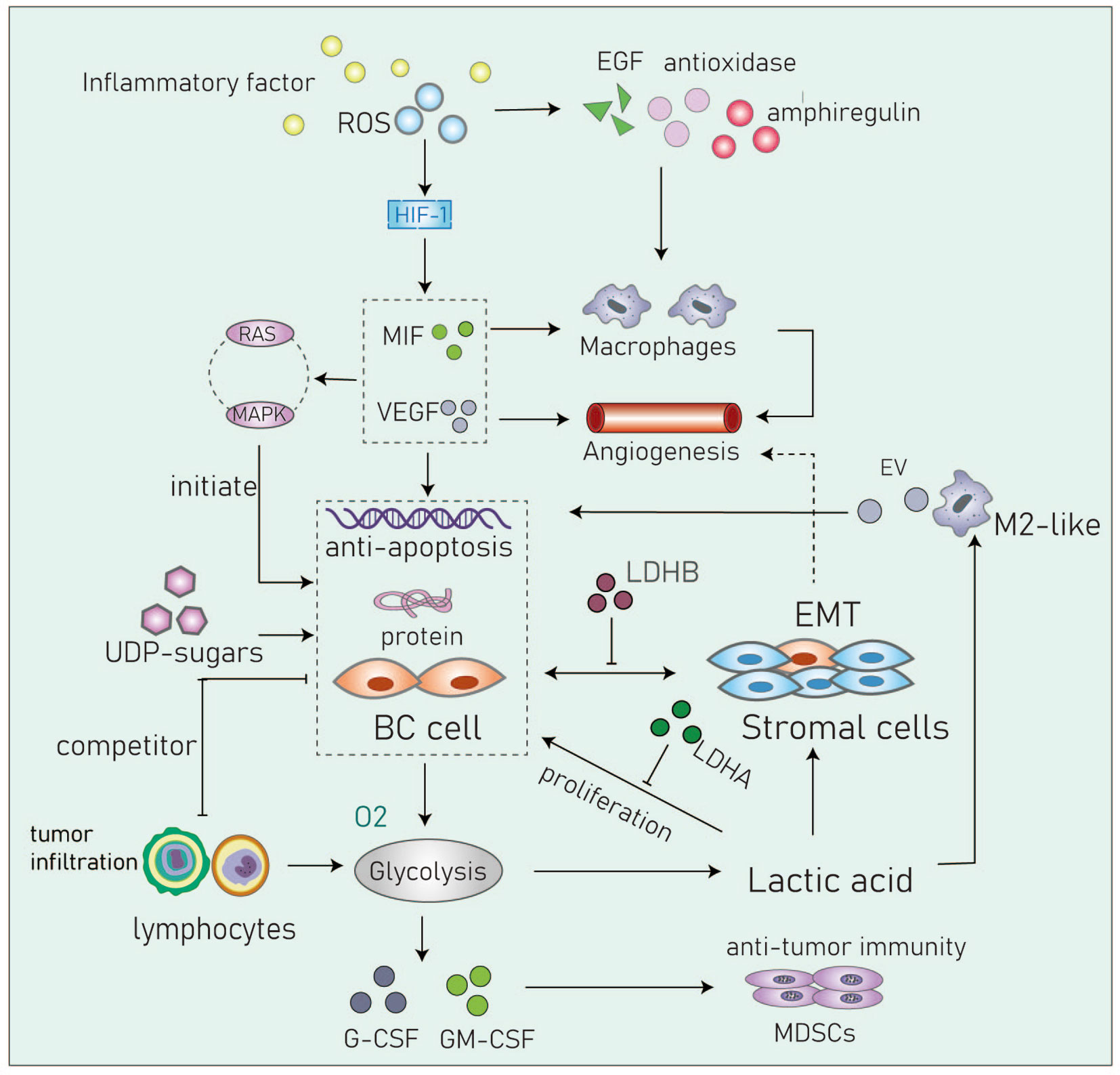
Figure 4 Cancer cells in the acidic microenvironment can escape the immune system and eventually lead to tumor progression. Inflammatory factor, IL6, TNF-α, COX-2 and so on; RDS, Reactive oxygen species; MIF, Migration inhibitory factor; VEGF, Vascular endothelial growth factor; UDP-sugar, Uridine diphosphate glucose; G-CSF, Granulocyte colony stimulating factor; GM-CSF, Granulocyte macrophage colony stimulating factor; MDSCs, Myeloid-derived suppressor cells; EV, Extracellular vesicle, transmit myeloid-specific lncRNA and HIF-1α-stabilizing lncRNA.
Cancer cells within the TME have higher levels of reactive oxygen species (ROS) compared to normal cells. In basal-like and BRCA1-associated BC, it has been shown that ROS levels are associated with the expression and activity of the transcription factor aryl hydrocarbon receptor (AHR). These changes promote the transcription of antioxidant enzymes, epidermal growth factor receptor (EGFR) ligands, and the bidirectional regulatory factor AREG. AHR can attract monocytes into the TME and activate macrophages to promote angiogenesis (73, 74). Parekh et al. captured multinucleated cells in chemotherapy-resistance triple-negative BC cells. They showed that these cells are non-proliferate but can significantly regulate the TME by elevating levels of ROS and by stabilizing HIF-1. These processes contribute to increased levels of vascular endothelial growth factor (VEGF) and macrophage migration inhibitory factor (MIF) and can induce chemotherapeutic resistance by upregulating anti-apoptotic proteins through the RAS/MAPK pathway (75).
The highly acidic TME formed by glycolysis may affect the infiltration of immune cells eventually leading to immune escape and cancer progression (76). The immune infiltrates of the TME consist of lymphocytes and bone marrow cells. In this section, we briefly review the changes in immune cells in the hyperglycemia TME.
Macrophages are the most abundant type of immune cell found in the TME. M2 macrophages are closely related to the occurrence and development of tumors. Lactic acid can promote macrophages towards an M2-like phenotype that is associated with adverse clinical characteristics such as large tumor volumes, higher histological grades, ER negativity, higher recurrence rates, and lower rates of survival (76, 77). It has been demonstrated that tumor-associated macrophages (TAMs) enhance aerobic glycolysis and apoptotic resistance in BC cells via the transmission of extracellular vesicles (EV) that contain a myeloid-specific HIF-1α-stabilizing long noncoding RNA (HISLA). Lactic acid released by glycolytic tumor cells upregulates macrophage HISLA and forms a feed-forward loop between TAMs and tumor cells in the TME (27).
Tumor-infiltrating lymphocytes (TIL) comprise B and T cells. Cytotoxic CD8+T lymphocytes (CTL) are the most abundant TILs in the TME of BC but helper CD4+T cells and NK cells are also present (78). Tumor cells compete with these cells for glucose. Abundant lactate production by tumor cells has been shown to inhibit MCT1-mediated lactate export by TILs leading to decreased cell proliferation, cytokine production, and/or cytolysis. This phenomenon will inhibit the glycolysis of T cells and inhibit their tumor-killing function along with functional damage to NK cells (79).
Myeloid-derived suppressor cells (MDSCs) inhibit anti-tumor immunity. In TNBC mouse models, studies have found glycolysis inhibits the expression of granulocyte colony-stimulating factor (G-CSF) and granulocyte macrophage colony-stimulating factor (GM-CSF) resulting in the decreased expression of MDSCs and promotion of tumor immunosuppression (80). Although other immune cells such as dendritic cells, mast cells and granulocytes are also present in the BC TME, studies are required to better define their metabolic interactions with tumor cells (Figure 4).
In BC patients, hyperglycemia during chemotherapy will increase the resistance to treatment (81) and so reducing blood glucose levels is particularly important. Drugs that alter glucose metabolism can be beneficial in improving the effects of conventional chemotherapy drugs commonly used in cancer treatment. The choice of chemotherapy plan and cycle time should be carefully considered in hyperglycemic patients.
Metformin was developed in the late 1970s as an anti-hyperglycemia drug (82). It has now been widely tested for an anticancer agent along with other hyperglycemic drugs (3). Metformin can alter cancer metabolism and mitochondrial function. It can also regulate key signaling pathways such as the Ras/Raf MEK/ERK PI3K/Akt and mTOR pathways to increase cell death and inhibit many cellular processes including proliferation, migration, EMT, invasion, and metastasis (83). Metformin also changes signal transmission of the Warburg effect during tumor development and can inhibit glucose uptake by cancer cells (84). It reduces circulating hormone levels, particularly estrogens, that are associated with the development of postmenopausal BC (85).
Additionally, studies have shown that the thiazolidinediones are insulin sensitizers that can also inhibit the growth of BC cells. Pioglitazone acts as an agonist of the tumor suppressor PPAR. When PPAR is activated, the levels of free fatty acid (FFA) and eicosanoid are reduced, and VEGF-induced angiogenesis is inhibited. Also, the proliferation and migration of cancer cells are inhibited through the JAK2/STAT3 pathway (86).
Insulin and insulin analogs such as sulfonylureas and glitinides have powerful hypoglycemic effects. Studies have reported that insulin downregulates IGF-BPs and sex hormone-binding proteins (SHBGs) leading to IGF and hormone-dependent BC (21). Type 2 diabetes and insulin therapy may be independently associated with a poorer prognosis in BC. Premenopausal women with diabetes tend to develop breast tumors that do not express hormone receptors making their treatment extremely challenging (87, 88).
The antidiabetic drugs described above may also have potential roles as anti-cancer drugs. A total of 46 studies of metformin in BC patients have been registered in ClinicalTrials.gov. 17 studies have completed, 14 studies are recruiting, 4 studies were terminated due to inapplicable factors such as long rest intervals, changes in treatment methods, slow accumulation of patients’ number, and data loss. And we also have listed completed in Table 1 and recruiting trials in Table 2. Hope to be helpful to interested readers.

Table 1 Completed researches on ClinicalTrials.gov. We excluded some studies that have too few participants, or inapplicable conclusions.
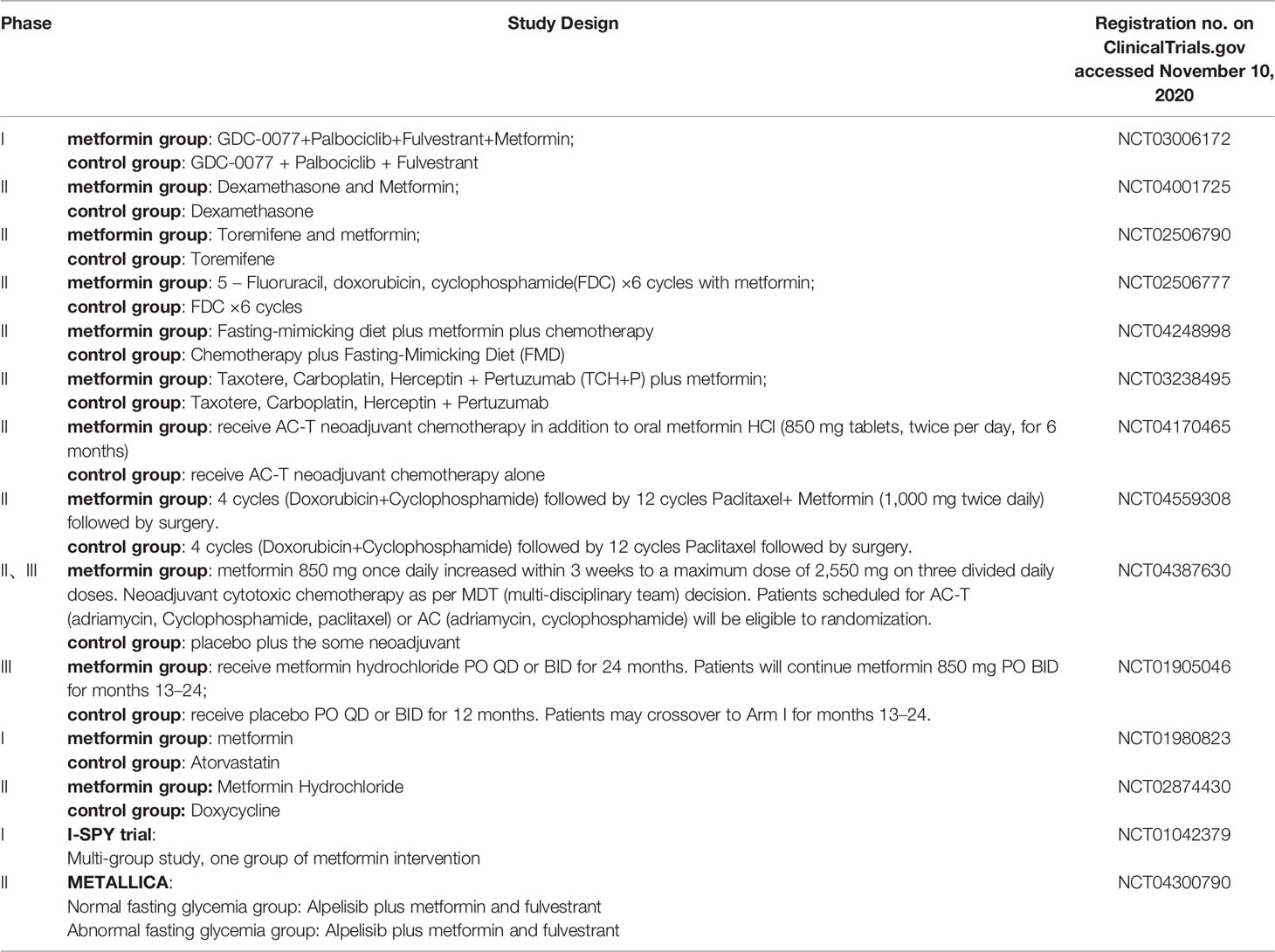
Table 2 Recruiting researches on ClinicalTrials.gov.
Combining the above table, we can see that the role of metformin on its anti-tumor effects are researched continuously. In addition, there are reported retrospective studies in the past have shown that the use of metformin combined with neoadjuvant chemotherapy can enable BC patients to obtain a higher pCR rate (89). The METTEN study shows that the addition of metformin plus trastuzumab to neoadjuvant chemotherapy can effectively increase the pCR rate of early her2-positive BC patients (90). The METEOR study also provides evidence of the neoadjuvant metformin plus letrozole for anti-tumor effects in non-diabetic postmenopausal ER-positive patients (91).
Although metformin has shown good antitumor activity in BC, there remain many challenges concerning how best to further optimize treatment. The efficacy of metformin as an anticancer drug depends largely on the glucose concentration in the TME and so treating diabetes in cancer patients may be necessary. There are relatively few studies concerning glitazones and other antidiabetic drugs. The heterogeneity and differences in diseases mean that it is necessary to adopt personalized treatments and using precision medicine approaches to tailor treatments at the individual patient level.
Patients with hyperglycemia are resistant to chemotherapy drugs. Metformin resistance is no exception (92). Scherbakov et al. demonstrated for the first time that structural activation of Akt/Snail1/E-Cadherin signaling leads to cross-resistance of BC cells to metformin and tamoxifen (93).
Reports have shown that glycolysis inhibitors such as 2-deoxy-d-glucose (2DG) combined with metformin can also significantly reduced the survival of BC cells (94). It has been reported in the literature that the combined treatments using glycolysis inhibitors and anti-glycemic drugs are feasible in humans, however, these approaches have not yet translated to clinical evaluation.
PI3K/AKT/mTOR and CDK4/6 inhibitors are emerging drugs for the treatment of ER-positive and human epidermal growth factor receptor-2 (HER2) negative metastatic BC (95). The PI3K/AKT/mTOR pathway is an important oncogenic signaling pathway in BC that can be activated by hyperglycemia to promote the proliferation of malignant BC epithelial cells (56, 95). Everolimus is an mTOR inhibitor that can target the rapamycin pathway. However, because of its toxic effect and its efficacy it has limited applications in the clinic (96). Gerke et al. have found that everolimus combined with metformin had a combined anti-cancer effect and a common inhibitory effect on glucose metabolism, tumor cell growth, and colony formation (97). However, hyperglycemia is also the most common adverse reaction of everolimus (98). Metformin may be used to prevent and/or treat everolimus-induced hyperglycemia and may enhance its anticancer effects yet there are relatively few clinical research studies in this area.
CDK4/6 inhibitors have rapidly translated from preclinical studies to clinical evaluation (99). Palbociclib can downregulate glucose uptake by GLUT-1 through the RB/E2F/C-MYC signaling pathway to reprogram glucose metabolism. It also inhibits the expression of HIF-1, a key regulator of tumor progression (100). In vitro experiments have shown that ER+/HER2- BC cells are sensitive to palbociclib under conditions of enhanced aerobic glycolysis whilst ER+/HER2+ cells show enhanced glycolytic catabolism with the development of palbociclib resistance. These metabolic phenotypes may have potential prognostic value (101).
Abnormal glucose metabolism is an important clinical problem in many types of BC. Recent studies have shown that somatic and BC cells from patients with hyperglycemia or metabolic abnormalities have elevated acidity in the TME accompanied by increased ROS and other changes in energy homeostasis. Hypoxia within the TME leads to the abnormal activation of cancer cell behaviors including enhanced proliferation, invasion, and metastasis. The reprogramming of metabolic changes in the glycolysis pathway increases the supply of substrates to cancer cells. However, tumor cells need to adapt to new conditions within the TME which can affect the survival and prognosis of BC patients.
The treatment of BCs with abnormal metabolism remains a major clinical challenge. For example, metformin can be used as an anticancer drug and can also control hyperglycemia or diabetes in patients. However, the efficacy of metformin depends on the glucose concentration within the TME and the sensitivity of patients to metformin. Other anti-sugar drugs also have anti-cancer effects but have not been studied in detail. Consequently, individualized treatment plans are required to optimize BC treatments in patients with hyperglycemia.
JQ and QZ contributed equally to this paper. JQ wrote the manuscript and composed the figures for this article. QZ reviewed the literature and contributed to the writing of the article. XM contributed to the editing and composition of the final version. All authors contributed to the article and approved the submitted version.
This work was supported by a special program from the Chinese National Natural Science Funds (81973861 to XM), the Key Construction Project Co-sponsored by Province and Ministry (WKJ-ZJ-2116 to XM), the Key Project for Medical and Health in Zhejiang Province (2016ZDA004 to XM), Zhejiang Medicine and Health Technology Plan Project(WKJ-ZJ-1803-01-XM), the Medical and Health Projects in Zhejiang Province (2020KY495 to QZ) and Zhejiang Traditional Chinese Medicine Project(2020 ZQ 009-QZ).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Kang C, LeRoith D, Gallagher EJ. Diabetes, Obesity, and Breast Cancer. Endocrinology (2018) 159(11):3801–12. doi: 10.1210/en.2018-00574
2. Mohamed HT, El-Shinawi M, Nouh MA, Bashtar AR, Elsayed ET, Schneider RJ, et al. Inflammatory breast cancer: high incidence of detection of mixed human cytomegalovirus genotypes associated with disease pathogenesis. Front Oncol (2014) 4:246. doi: 10.3389/fonc.2014.00246
3. Samuel SM, Varghese E, Varghese S, Büsselberg D. Challenges and perspectives in the treatment of diabetes associated breast cancer. Cancer Treat Rev (2018) 70:98–111. doi: 10.1016/j.ctrv.2018.08.004
4. Belardi V, Gallagher EJ, Novosyadlyy R, LeRoith D. Insulin and IGFs in obesity-related breast cancer. J Mammary Gland Biol Neoplasia (2013) 18(3-4):277–89. doi: 10.1007/s10911-013-9303-7
5. Al Qahtani A, Holly J, Perks C. Hypoxia negates hyperglycemia-induced chemo-resistance in breast cancer cells: the role of insulin-like growth factor binding protein 2. Oncotarget (2017) 8(43):74635–48. doi: 10.18632/oncotarget.20287
6. Warburg O. On the origin of cancer cells. Science (1956) 123:309–14. doi: 10.1126/science.123.3191.309
7. Pavlides S, Whitaker-Menezes D, Castello-Cros R, Flomenberg N, Witkiewicz AK, Frank PG, et al. The reverse Warburg effect: aerobic glycolysis in cancer associated fibroblasts and the tumor stroma. Cell Cycle (2009) 8(23):3984–4001. doi: 10.4161/cc.8.23.10238
8. Li W, Zhang X, Sang H, Zhou Y, Shang C, Wang Y, et al. Effects of hyperglycemia on the progression of tumor diseases. J Exp Clin Cancer Res (2019) 38(1):327. doi: 10.1186/s13046-019-1309-6
9. Donzelli S, Milano E, Pruszko M, Sacconi A, Masciarelli S, Iosue I, et al. Expression of ID4 protein in breast cancer cells induces reprogramming of tumour-associated macrophages. Breast Cancer Res (2018) 20(1):59. doi: 10.1186/s13058-018-0990-2
10. Yang L, He Z, Yao J, Tan R, Zhu Y, Li Z, et al. Regulation of AMPK-related glycolipid metabolism imbalances redox homeostasis and inhibits anchorage independent growth in human breast cancer cells. Redox Biol (2018) 17:180–91. doi: 10.1016/j.redox.2018.04.016
11. Wulaningsih W, Vahdaninia M, Rowley M, Holmberg L, Garmo H, Malmstrom H, et al. Prediagnostic serum glucose and lipids in relation to survival in breast cancer patients: a competing risk analysis. BMC Cancer (2015) 15:913. doi: 10.1186/s12885-015-1928-z
12. Balaban S, Lee LS, Varney B, Aishah A, Gao Q, Shearer RF, et al. Heterogeneity of fatty acid metabolism in breast cancer cells underlies differential sensitivity to palmitate-induced apoptosis. Mol Oncol (2018) 12(9):1623–38. doi: 10.1002/1878-0261.12368
13. Hou Y, Zhou M, Xie J, Chao P, Feng Q, Wu J. High glucose levels promote the proliferation of breast cancer cells through GTPases. Breast Cancer (Dove Med Press) (2017) 9:429–36. doi: 10.2147/BCTT.S135665
14. Zhao J, Zeng D, Liu Y, Luo Y, Ji S, Li X, et al. Selenadiazole derivatives antagonize hyperglycemia-induced drug resistance in breast cancer cells by activation of AMPK pathways. Metallomics (2017) 9:535–45. doi: 10.1039/C7MT00001D
15. Steelman LS, Fitzgerald T, Lertpiriyapong K, Cocco L, Follo MY, Martelli AM, et al. Critical Roles of EGFR Family Members in Breast Cancer and Breast Cancer Stem Cells: Targets for Therapy. Curr Pharm Des (2016) 22:2358–88. doi: 10.2174/1381612822666160304151011
16. Huang F, Shi Q, Li Y, Xu L, Xu C, Chen F, et al. HER2/EGFR-AKT Signaling Switches TGFβ from Inhibiting Cell Proliferation to Promoting Cell Migration in Breast Cancer. Cancer Res (2018) 78:6073–85. doi: 10.1158/0008-5472.CAN-18-0136
17. Avanzato D, Pupo E, Ducano N, Isella C, Bertalot G, Luise C, et al. High USP6NL Levels in Breast Cancer Sustain Chronic AKT Phosphorylation and GLUT1 Stability Fueling Aerobic Glycolysis. Cancer Res (2018) 78:3432–44. doi: 10.1158/0008-5472.CAN-17-3018
18. Barron CC, Bilan PJ, Tsakiridis T, Tsiani E. Facilitative glucose transporters: Implications for cancer detection, prognosis and treatment. Metabolism (2016) 65:124–39. doi: 10.1016/j.metabol.2015.10.007
19. Oh S, Kim H, Nam K, Shin I. Glut1 promotes cell proliferation, migration and invasion by regulating epidermal growth factor receptor and integrin signaling in triple-negative breast cancer cells. BMB Rep (2017) 50:132–7. doi: 10.5483/bmbrep.2017.50.3.189
20. Dabrowski M, Szymanska-Garbacz E, Miszczyszyn Z, Derezinski T, Czupryniak L. Risk factors for cancer development in type 2 diabetes: a retrospective case-control study. BMC Cancer (2016) 16:785. doi: 10.1186/s12885-016-2836-6
21. Ferroni P, Riondino S, Buonomo O, Palmirotta R, Guadagni F, Roselli M. Type 2 Diabetes and Breast Cancer: The Interplay between Impaired Glucose Metabolism and Oxidant Stress. Oxid Med Cell Longev (2015) 2015:183928. doi: 10.1155/2015/183928
22. Rostoker R, Abelson S, Bitton-Worms K, Genkin I, Ben-Shmuel S, Dakwar M, et al. Highly specific role of the insulin receptor in breast cancer progression. Endocr Relat Cancer (2015) 22:145–57. doi: 10.1530/ERC-14-0490
23. Luey BC, May FEB. Insulin-like growth factors are essential to prevent anoikis in oestrogen-responsive breast cancer cells: importance of the type I IGF receptor and PI3-kinase/Akt pathway. Mol Cancer (2016) 15:8. doi: 10.1186/s12943-015-0482-2
24. Jia L, Huang S, Yin X, Zan Y, Guo Y, Han L. Quercetin suppresses the mobility of breast cancer by suppressing glycolysis through Akt-mTOR pathway mediated autophagy induction. Life Sci (2018) 208:123–30. doi: 10.1016/j.lfs.2018.07.027
25. Lee S, Hallis SP, Jung K-A, Ryu D, Kwak MK. Impairment of HIF-1α-mediated metabolic adaption by NRF2-silencing in breast cancer cells. Redox Biol (2019) 24:101210. doi: 10.1016/j.redox.2019.101210
26. Shrivastava R, Singh V, Asif M, Negi MPS, Bhadauria S. Oncostatin M upregulates HIF-1α in breast tumor associated macrophages independent of intracellular oxygen concentration. Life Sci (2018) 194:59–66. doi: 10.1016/j.lfs.2017.12.017
27. Chen F, Chen J, Yang L, Liu J, Zhang X, Zhang Y, et al. Extracellular vesicle-packaged HIF-1α-stabilizing lncRNA from tumour-associated macrophages regulates aerobic glycolysis of breast cancer cells. Nat Cell Biol (2019) 21:498–510. doi: 10.1038/s41556-019-0299-0
28. Wei R, Mao L, Xu P, Zheng X, Hackman RM, Mackenzie GG, et al. Suppressing glucose metabolism with epigallocatechin-3-gallate (EGCG) reduces breast cancer cell growth in preclinical models. Food Funct (2018) 9:5682–96. doi: 10.1039/C8FO01397G
29. Valeta-Magara A, Gadi A, Volta V, Walters B, Arju R, Giashuddin S, et al. Inflammatory Breast Cancer Promotes Development of M2 Tumor-Associated Macrophages and Cancer Mesenchymal Cells through a Complex Chemokine Network. Cancer Res (2019) 79:3360–71. doi: 10.1158/0008-5472.CAN-17-2158
30. Patel CH, Leone RD, Horton MR, Powell JD. Targeting metabolism to regulate immune responses in autoimmunity and cancer. Nat Rev Drug Discovery (2019) 18:669–88. doi: 10.1038/s41573-019-0032-5
31. Pollizzi KN, Powell JD. Integrating canonical and metabolic signalling programmes in the regulation of T cell responses. Nat Rev Immunol (2014) 14:435–46. doi: 10.1038/nri3701
32. Tiwari P, Blank A, Cui C, Schoenfelt KQ, Zhou G, Xu Y, et al. Metabolically activated adipose tissue macrophages link obesity to triple-negative breast cancer. J Exp Med (2019) 216:1345–58. doi: 10.1084/jem.20181616
33. Varghese E, Samuel SM, Líšková A, Samec M, Kubatka P, Büsselberg D. Targeting Glucose Metabolism to Overcome Resistance to Anticancer Chemotherapy in Breast Cancer. Cancers (Basel) (2020) 12(8):2252. doi: 10.3390/cancers12082252
34. Zielinska HA, Holly JMP, Bahl A, Perks CM. Inhibition of FASN and ERα signalling during hyperglycaemia-induced matrix-specific EMT promotes breast cancer cell invasion via a caveolin-1-dependent mechanism. Cancer Lett (2018) 419:187–202. doi: 10.1016/j.canlet.2018.01.028
35. Wang X-N, Wang K-Y, Zhang X-S, Yang C, Li XY. 4-Hydroxybenzoic acid (4-HBA) enhances the sensitivity of human breast cancer cells to adriamycin as a specific HDAC6 inhibitor by promoting HIPK2/p53 pathway. Biochem Biophys Res Commun (2018) 504:812–9. doi: 10.1016/j.bbrc.2018.08.043
36. Li Q, Qin T, Bi Z, Hong H, Ding L, Chen J, et al. Rac1 activates non-oxidative pentose phosphate pathway to induce chemoresistance of breast cancer. Nat Commun (2020) 11(1):1456. doi: 10.1038/s41467-020-15308-7
37. Kazanietz MG, Caloca MJ. The Rac GTPase in cancer: from old concepts to new paradigms. Cancer Res (2017) 77:5445–51. doi: 10.1158/0008-5472.CAN-17-1456
38. Ganapathy-Kanniappan S. Rac1 repression reverses chemoresistance by targeting tumor metabolism [published online ahead of print, 2020 Aug 31]. Cancer Biol Ther (2020) 21(10):888–90. doi: 10.1080/15384047.2020.1809923
39. Chen Q, Meng YQ, Xu XF, Gu J. Blockade of GLUT1 by WZB117 resensitizes breast cancer cells to adriamycin. Anticancer Drugs (2017) 28(8):880–7. doi: 10.1097/CAD.0000000000000529
40. Arunachalam S, Tirupathi Pichiah PB, Achiraman S. Doxorubicin treatment inhibits PPARy and may induce lipotoxicity by mimicking a type 2 diabetes-like condition in rodent models. FEBS Lett (2013) 587:105–10. doi: 10.1016/j.febslet.2012.11.019
41. Xu M, Chen Sz, Yang Wb, Cheng X, Ye Y, Mao J, et al. FGFR4 Links Glucose Metabolism and Chemotherapy Resistance in Breast Cancer. Cell Physiol Bio Chem (2018) 47:151–60. doi: 10.1159/000489759
42. Tiong KH, Tan BS, Choo HL, Chung FF, Hii LW, Tan SH, et al. Fibroblast growth factor receptor 4 (FGFR4) and fibroblast growth factor 19 (FGF19) autocrine enhance breast cancer cells survival. Oncotarget (2016) 7(36):57633–50. doi: 10.18632/oncotarget.9328
43. Bean JF, Qiu Y-Y, Yu S, Clark S, Chu F, Madonna MB. Glycolysis inhibition and its effect in doxorubicin resistance in neuroblastoma. J Pediatr Surg (2014) 49:981–4. doi: 10.1016/j.jpedsurg.2014.01.037
44. Mustacchi G, De Laurentiis M. The role of taxanes in triple-negative breast cancer: Literature review. Drug Des Dev Ther (2015) 9:4303–18. doi: 10.2147/DDDT.S86105
45. Zhou M, Zhao Y, Ding Y, Liu H, Liu Z, Fodstad O, et al. Warburg effect in chemosensitivity: targeting lactate dehydrogenase-A re-sensitizes taxol-resistant cancer cells to taxol. Mol Cancer (2010) 9:33. doi: 10.1186/1476-4598-9-33
46. Chen Z, Shi T, Zhang L, Zhu P, Deng M, Huang C, et al. Mammalian drug efflux transporters of the ATP binding cassette (ABC) family in multidrug resistance: A review of the past decade. Cancer Lett (2016) 370:153–64. doi: 10.1016/j.canlet.2015.10.010
47. Zaal EA, Berkers CR. The Influence of Metabolism on Drug Response in Cancer. Front Oncol (2018) 8:500. doi: 10.3389/fonc.2018.00500
48. He R, Hongxu L. TRIM59 knockdown blocks cisplatin resistance in A549/DDP cells through regulating PTEN/AKT/HK2. Gene (2020) 747:144553. doi: 10.1016/j.gene.2020.144553
49. He M, Jin Q, Chen C, Liu Y, Ye X, Jiang Y, et al. The miR-186-3p/EREG axis orchestrates tamoxifen resistance and aerobic glycolysis in breast cancer cells. Oncogene (2019) 38(28):5551–65. doi: 10.1038/s41388-019-0817-3
50. Woo SH, Seo SK, Park Y, Kim EK, Seong MK, Kim HA, et al. Dichloroacetate potentiates tamoxifen-induced cell death in breast cancer cells via downregulation of the epidermal growth factor receptor. Oncotarget (2016) 7:59809–19. doi: 10.18632/oncotarget.10999
51. Woo YM, Shin Y, Lee EJ, Lee S, Jeong SH, Kong HK, et al. Inhibition of Aerobic Glycolysis Represses Akt/mTOR/HIF-1α Axis and Restores Tamoxifen Sensitivity in Antiestrogen-Resistant Breast Cancer Cells. PloS One (2015) 10(7):e0132285. doi: 10.1371/journal
52. Huang S, Wang H, Chen W, Zhan M, Xu S, Huang X, et al. Tamoxifen inhibits cell proliferation by impaired glucose metabolism in gallbladder cancer. J Cell Mol Med (2020) 24(2):1599–613. doi: 10.1111/jcmm.14851
53. Castagnoli L, Iorio E, Dugo M, Koschorke A, Faraci S, Canese R, et al. Intratumor lactate levels reflect HER2 addiction status in HER2-positive breast cancer. J Cell Physiol (2019) 234:1768–79. doi: 10.1002/jcp.27049
54. Ma C, Zu X, Liu K, Bode AM, Dong Z, Liu Z, et al. Knockdown of Pyruvate Kinase M Inhibits Cell Growth and Migration by Reducing NF-kB Activity in Triple-Negative Breast Cancer Cells. Mol Cells (2019) 42(9):628–36. doi: 10.14348/molcells.2019.0038
55. Hoopmann M, Warm M, Mallmann P, Thomas A, Göhring UJ, Schöndorf T. Tumor M2 pyruvate kinase–determination in breast cancer patients receiving trastuzumab therapy. Cancer Lett (2002) 187:223–8. doi: 10.1016/S0304-3835(02)00404-4
56. Fleming IN, Andriu A, Smith TA. Early changes in [18F] FDG incorporation by breast cancer cells treated with trastuzumab in normoxic conditions: role of the Akt-pathway, glucose transport and HIF-1α. Breast Cancer Res Treat (2014) 144(2):241–8. doi: 10.1007/s10549-014-2858-1
57. Lorito N, Bacci M, Smiriglia A, Mannelli M, Parri M, Comito G, et al. Glucose Metabolic Reprogramming of ER Breast Cancer in Acquired Resistance to the CDK4/6 Inhibitor Palbociclib+. Cells (2020) 9:668. doi: 10.3390/cells9030668
58. Dias AS, Almeida CR, Helguero LA, Duarte IF. Metabolic crosstalk in the breast cancer microenvironment. Eur J Cancer (2019) 121:154–71. doi: 10.1016/j.ejca.2019.09.002
59. Adham SA, Al Rawahi H, Habib S, Al Moundhri MS, Viloria-Petit A, Coomber BL. Modeling of hypo/hyperglycemia and their impact on breast cancer progression related molecules. PloS One (2014) 9(11):e113103. doi: 10.1371/journal.pone.0113103
60. Soysal SD, Tzankov A, Muenst SE. Role of the Tumor Microenvironment in Breast Cancer. Pathobiology (2015) 82(3-4):142–52. doi: 10.1159/000430499
61. Mittal S, Brown NJ, Holen I. The breast tumor microenvironment: role in cancer development, progression and response to therapy. Expert Rev Mol Diagn (2018) 18(3):227–43. doi: 10.1080/14737159.2018.1439382
62. Flores-López LA, Martínez-Hernández MG, Viedma-Rodríguez R, Díaz-Flores M, Baiza-Gutman LA. High glucose and insulin enhance uPA expression, ROS formation and invasiveness in breast cancer-derived cells. Cell Oncol (Dordr) (2016) 39(4):365–78. doi: 10.1007/s13402-016-0282-8
63. Jiang GM, Xie WY, Wang HS, Du J, Wu BP, Xu W, et al. Curcumin combined with FAPalphac vaccine elicits effective antitumor response by targeting indolamine-2,3-dioxygenase and inhibiting EMT induced by TNF-αlpha in melanoma. Oncotarget (2015) 6:25932–42. doi: 10.18632/oncotarget.4577
64. Kim J, DeBerardinis RJ. Mechanisms and Implications of Metabolic Heterogeneity in Cancer. Cell Metab (2019) 30:434–46. doi: 10.1016/j.cmet.2019.08.013
65. Chen C, Bai L, Cao F, Wang S, He H, Song M, et al. Targeting LIN28B reprograms tumor glucose metabolism and acidic microenvironment to suppress cancer stemness and metastasis. Oncogene (2019) 38(23):4527–39. doi: 10.1038/s41388-019-0735-4
66. de la Cruz-López KG, Castro-Muñoz LJ, Reyes-Hernández DO, García-Carrancá A, Manzo-Merino J. Lactate in the Regulation of Tumor Microenvironment and Therapeutic Approaches. Front Oncol (2019) 9:1143. doi: 10.3389/fonc.2019.01143
67. Ding J, Karp JE, Emadi A. Elevated lactate dehydrogenase (LDH) can be a marker of immune suppression in cancer: Interplay between hematologic and solid neoplastic clones and their microenvironments. Cancer Biomark (2017) 19(4):353–63. doi: 10.3233/CBM-160336
68. Mishra D, Banerjee D. Lactate Dehydrogenases as Metabolic Links between Tumor and Stroma in the Tumor Microenvironment [published correction appears in Cancers (Basel). 2020 Apr 09;12(4):]. Cancers (Basel) (2019) 11(6):750. doi: 10.3390/cancers11060750
69. Oikari S, Kettunen T, Tiainen S, Häyrinen J, Masarwah A, Sudah M, et al. UDP-sugar accumulation drives hyaluronan synthesis in breast cancer. Matrix Biol (2018) 67:63–74. doi: 10.1016/j
70. Gao R, Liu Y, Li D, Xun J, Zhou W, Wang P, et al. PFKFB4 Promotes Breast Cancer Metastasis via Induction of Hyaluronan Production in a p38-Dependent Manner. Cell Physiol Biochem (2018) 50(6):2108–23. doi: 10.1159/000495055
71. Arnold JM, Gu F, Ambati CR, Rasaily U, Ramirez-Pena E, Joseph R, et al. UDP-glucose 6-dehydrogenase regulates hyaluronic acid production and promotes breast cancer progression [published correction appears in Oncogene. 2020 Mar 25;]. Oncogene (2020) 39(15):3089–101. doi: 10.1038/s41388-019-0885-4
72. Li L, Liu X, Sanders KL, Edwards JL, Ye J, Si F, et al. TLR8-Mediated Metabolic Control of Human Treg Function: A Mechanistic Target for Cancer Immunotherapy. Cell Metab (2019) 29(1):103–23.e5. doi: 10.1016/j.cmet.2018.09.020
73. Zhang L, Li S. Lactic acid promotes macrophage polarization through MCT-HIF1α signaling in gastric cancer. Exp Cell Res (2020) 388(2):111846. doi: 10.1016/j.yexcr.2020.111846
74. Kubli SP, Bassi C, Roux C, Wakeham A, Göbl C, Zhou W, et al. AhR controls redox homeostasis and shapes the tumor microenvironment in BRCA1-associated breast cancer. Proc Natl Acad Sci USA (2019) 116(9):3604–13. doi: 10.1073/pnas.1815126116
75. Parekh A, Das S, Parida S, Das CK, Dutta D, Mallick SK, et al. Multi-nucleated cells use ROS to induce breast cancer chemo-resistance in vitro and in vivo. Oncogene (2018) 37(33):4546–61. doi: 10.1038/s41388-018-0272-6
76. Gill KS, Fernandes P, O’Donovan TR, McKenna SL, Doddakula KK, Power DG, et al. Glycolysis inhibition as a cancer treatment and its role in an antitumor immune response. Biochim Biophys Acta (2016) 1866(1):87–105. doi: 10.1016/j.bbcan.2016.06.005
77. Yang M, Li Z, Ren M, Li S, Zhang L, Zhang X, et al. Stromal infiltration of tumor-associated macrophages conferring poor prognosis of patients with basal-like breast carcinoma. J Cancer (2018) 9:2308e16. doi: 10.7150/jca.25155
78. Stanton SE, Disis ML. Clinical significance of tumor-infiltrating lymphocytes in breast cancer. J Immunother Cancer (2016) 4:59. doi: 10.1186/s40425-016-0165-6MLA
79. Brand A, Singer K, Koehl GE, Kolitzus M, Schoenhammer G, Thiel A, et al. LDHA-associated lactic acid production blunts tumor immunosurveillance by T and NK cells. Cell Metabol (2016) 24:657e71. doi: 10.1016/j.cmet.2016.08.011
80. Li W, Tanikawa T, Kryczek I, Xia H, Li G, Wu K, et al. Aerobic Glycolysis Controls Myeloid-Derived Suppressor Cells and Tumor Immunity via a Specific CEBPB Isoform in Triple-Negative Breast Cancer. Cell Metab (2018) 28(1):87–103.e6. doi: 10.1016/j.cmet.2018.04.022
81. Zeng L, Zielinska HA, Arshad A, Shield JP, Bahl A, Holly JM, et al. Hyperglycemia-induced chemoresistance in breast cancer cells: role of the estrogen receptor. Endocr Relat Cancer (2016) 23:125–34. doi: 10.1530/ERC-15-0507
82. Bailey CJ. Metformin: historical overview. Diabetologia (2017) 60(9):1566–76. doi: 10.1007/s00125-017-4318-z
83. Daugan M, Dufaÿ Wojcicki A, d’Hayer B, Boudy V. Metformin: An anti-diabetic drug to fight cancer. Pharmacol Res (2016) 113(Pt A):675–85. doi: 10.1016/j.phrs.2016.10.006
84. Bradley Conor A. Diabetes: Metformin in breast cancer. Nat Rev Endocrinol (2017) 13:251. doi: 10.1038/nrendo.2017.37
85. Pimentel I, Chen BE, Lohmann AE, Ennis M, Ligibel J, Shepherd L, et al. The effect of metformin vs placebo on sex hormones in CCTG MA.32. J Natl Cancer Inst (2020). doi: 10.1093/jnci/djaa082
86. Jiao XX, Lin SY, Lian SX, Qiu YR, Li ZH, Chen ZH, et al. The inhibition of the breast cancer by PPARγ agonist pioglitazone through JAK2/STAT3 pathway. Neoplasma (2020) 67(4):834–42. doi: 10.4149/neo_2020_190805N716
87. Mu L, Zhu N, Zhang J, Xing F, Li D, Wang X. Type 2 diabetes, insulin treatment and prognosis of breast cancer. Diabetes Metab Res Rev (2017) 33(1). doi: 10.1002/dmrr.2823 10.1002/dmrr.2823.
88. Luque RM, López-Sánchez LM, Villa-Osaba A, Luque IM, Santos-Romero AL, Yubero-Serrano EM, et al. Breast cancer is associated to impaired glucose/insulin homeostasis in premenopausal obese/overweight patients. Oncotarget (2017) 8(46):81462–74. doi: 10.18632/oncotarget.20399
89. Jiralerspong S, Palla SL, Giordano SH, Meric-Bernstam F, Liedtke C, Barnett CM, et al. Metformin and pathologic complete responses to neoadjuvant chemotherapy in diabetic patients with breast cancer. J Clin Oncol (2009) 27:3297–302. doi: 10.1200/JCO.2009.19.6410
90. Martin-Castillo B, Pernas S, Dorca J, Álvarez I, Martínez S, Pérez-Garcia JM, et al. A phase 2 trial of neoadjuvant metformin in combination with trastuzumab and chemotherapy in women with early HER2-positive breast cancer: the METTEN study. Oncotarget (2018) 9(86):35687–704. doi: 10.18632/oncotarget.26286
91. Kim J, Lim W, Kim EK, Kim MK, Paik NS, Jeong SS, et al. Phase II randomized trial of neoadjuvant metformin plus letrozole versus placebo plus letrozole for estrogen receptor positive postmenopausal breast cancer (METEOR). BMC Cancer (2014) 14:170. doi: 10.1186/1471-2407-14-170
92. De A, Kuppusamy G. Metformin in breast cancer: preclinical and clinical evidence. Curr Probl Cancer (2020) 44:100488. doi: 10.1016/j.currproblcancer.2019.06.003
93. Scherbakov AM, Sorokin DV, Tatarskiy VV Jr, Prokhorov NS, Semina SE, Berstein LM, et al. The phenomenon of acquired resistance to metformin in breast cancer cells: The interaction of growth pathways and estrogen receptor signaling. IUBMB Life (2016) 68(4):281–92. doi: 10.1002/iub.1481
94. Vella V, Nicolosi ML, Giuliano M, Morrione A, Malaguarnera R, Belfiore A. Insulin Receptor Isoform A Modulates Metabolic Reprogramming of Breast Cancer Cells in Response to IGF2 and Insulin Stimulation. Cells (2019) 8(9):1017. doi: 10.3390/cells8091017
95. Kalinsky K, Sparano JA, Zhong X, Andreopoulou E, Taback B, Wiechmann L, et al. Pre-surgical trial of the AKT inhibitor MK-2206 in patients with operable invasive breast cancer: a New York Cancer Consortium trial. Clin Transl Oncol (2018) 20(11):1474–83. doi: 10.1007/s12094-018-1888-2
96. André F, O’Regan R, Ozguroglu M, Toi M, Xu B, Jerusalem G, et al. Everolimus for women with trastuzumab-resistant, HER2-positive, advanced breast cancer (BOLERO-3): a randomised, double-blind, placebo-controlled phase 3 trial. Lancet Oncol (2014) 15:580–91. doi: 10.1016/S1470-2045(14)70138-X
97. Ariaans G, Jalving M, Vries EG, Jong S. Anti-tumor effects of everolimus and metformin are complementary and glucose-dependent in breast cancer cells. BMC Cancer (2017) 17(1):232. doi: 10.1186/s12885-017-3230-8
98. Morviducci L, Rota F, Rizza L, Di Giacinto P, Ramponi S, Nardone MR, et al. Everolimus is a new anti-cancer molecule: Metabolic side effects as lipid disorders and hyperglycemia. Diabetes Res Clin Pract (2018) 143:428–31. doi: 10.1016/j.diabres.2018.04.001
99. Pernas S, Tolaney SM, Winer EP, Goel S. CDK4/6 inhibition in breast cancer: current practice and future directions. Ther Adv Med Oncol (2018) 10:1758835918786451. doi: 10.1177/1758835918786451
100. Cretella D, Fumarola C, Bonelli M, Alfieri R, La Monica S, Digiacomo G, et al. Pre-treatment with the CDK4/6 inhibitor palbociclib improves the efficacy of paclitaxel in TNBC cells. Sci Rep (2019) 9(1):13014. doi: 10.1038/s41598-019-49484-4
Keywords: hyperglycemia, chemotherapy resistance, chemoresistance, breast cancer, glucose metabolism
Citation: Qiu J, Zheng Q and Meng X (2021) Hyperglycemia and Chemoresistance in Breast Cancer: From Cellular Mechanisms to Treatment Response. Front. Oncol. 11:628359. doi: 10.3389/fonc.2021.628359
Received: 11 November 2020; Accepted: 05 January 2021;
Published: 25 February 2021.
Edited by:
Xiaosong Chen, Shanghai Jiao Tong University, ChinaReviewed by:
Chuangui Song, Fujian Medical University, ChinaCopyright © 2021 Qiu, Zheng and Meng. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xuli Meng, mxlmail@126.com
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.