 Yunus Akkoc
Yunus Akkoc Nesibe Peker
Nesibe Peker Arzu Akcay2
Arzu Akcay2 Devrim Gozuacik
Devrim Gozuacik- 1Koç University Research Center for Translational Medicine (KUTTAM), Istanbul, Turkey
- 2Yeni Yüzyıl University, School of Medicine, Private Gaziosmanpaşa Hospital, Department of Pathology, Istanbul, Turkey
- 3Koç University School of Medicine, Istanbul, Turkey
- 4Sabancı University Nanotechnology Research and Application Center (SUNUM), Istanbul, Turkey
Metastasis and relapse account for the great majority of cancer-related deaths. Most metastatic lesions are micro metastases that have the capacity to remain in a non-dividing state called “dormancy” for months or even years. Commonly used anticancer drugs generally target actively dividing cancer cells. Therefore, cancer cells that remain in a dormant state evade conventional therapies and contribute to cancer recurrence. Cellular and molecular mechanisms of cancer dormancy are not fully understood. Recent studies indicate that a major cellular stress response mechanism, autophagy, plays an important role in the adaptation, survival and reactivation of dormant cells. In this review article, we will summarize accumulating knowledge about cellular and molecular mechanisms of cancer dormancy, and discuss the role and importance of autophagy in this context.
Introduction
Cancer is the cause of death for millions of people every year, hence it’s one of the most devastating disease. Detection and diagnosis at early stages of cancer remarkably improve the chance of cure. However, the incidence of cancer continues to rise due various factors, including tobacco use, air pollution, obesity, increased life expectancy and cancer-causing infections. First approach in the treatment of cancer is usually surgical resection of the primary tumor, often followed by chemotherapy and/or radiotherapy. Besides, recent advances in targeted therapies and immunotherapies help to reduce the tumor burden. Thanks to high resolution diagnostic tools, advances in tumor ablation techniques, drug combinations, and targeted therapeutics, 5-year survival rates are improved for some cancer types, yet overall cancer survival rates for patients suffering from advanced disease are still low. A major reason for such discrepancy is the spread of cancer cells to organs other than the primary site and formation of the metastatic lesions. In other words, metastasis is among the leading causes of cancer-related deaths.
Metastasis of cancer to distant organs requires a sequential and complex chain of events. Cancer cells need to undergo several mutations and adaptations in order to gain motility and invasiveness, intravasate (migration into vessels), survive in the blood circulation and the lymphatics, extravasate, nestle and grow at secondary sites. Metastasis and survival of cancer cells at secondary sites are also affected by “the soil” in which tumor cells are seeded, namely the tumor microenvironment or stroma (1).
Mutations promoting epithelial-to-mesenchymal transition (EMT) greatly contribute to metastasis of cancer cells. Cells of normal tissues are tightly regulated by cell-to-cell and cell-to-matrix interactions. During cancerous transformation, epithelial cells may acquire mesenchymal cell-like properties, including loss of critical epithelial markers (e.g., E-cadherin, α-catenin), and expression of mesenchymal markers (e.g., N-cadherin and vimentin) (2). A transcriptional program orchestrates this transformation (e.g., ZEB1/2, Snail etc.) (3–5). Remodeling of epithelial junctions and cytoskeleton promotes motility and invasiveness of cancer cells (6). Cancer cells that are now motile and invasive, penetrate through the tissue extracellular matrix (ECM) and spread to lymph nodes and secondary sites through blood and lymph vessels. Seeding to metastatic sites and metastatic growth require reversal of this process, namely mesenchymal-to-epithelial transition (MET).
Advances in the last decade showed that in many tumor types, a small population of progenitor cancer cells, namely cancer stem cells (CSC), are responsible for the evolution and progression of the disease and metastasis (7, 8). Cancer cells and CSC might spread from primary tumors at various stages of tumor progression. These disseminated cells or clusters of cells (disseminated tumor cells, DTC) continue their evolution in their new tumor niches and they generally acquire genetic and epigenetic signatures that are different from the tumor of origin (9–13). Although aggressive proliferation of DTC might result in overt metastasis, latency periods lasting for months or even years were observed. During the latency period that spans the time between tumor formation and recurrence (also known as relapse), some cancer cells stay in a “dormant” state, a state of balanced proliferation or no proliferation at all (14, 15). At least some of these dormant cells have capacity to reactivate and form new metastatic lesions. Recurrent tumors were associated with drug resistance and aggressive behavior. So, most patients with recurrent disease show a very poor prognosis (16–18). For this reason, as an important mechanism contributing to tumor recurrence, cancer dormancy became a focus of attention in recent years.
There are two major mechanisms of cancer dormancy, namely, tumor mass dormancy and tumor cell dormancy (or cellular dormancy) (Figure 1). In tumor mass dormancy, proliferation of tumor cells counterbalanced by cellular demise and the tumor mass is preserved to a certain extent. A reason for limited tumor growth is hypoxia and inefficient nutrient supply due vascularization defects (angiogenic dormancy). Trimming of tumor cells by the cells of the immune system is another mechanism limiting tumor growth and expansion (immunological dormancy). On the other hand, cellular dormancy involves transition to a quiescent, cell cycle-arrest state, while cells retain the capacity to perpetuate neoplastic behavior when reactivated. In this review, we will mainly focus on the role of autophagy in cellular dormancy.
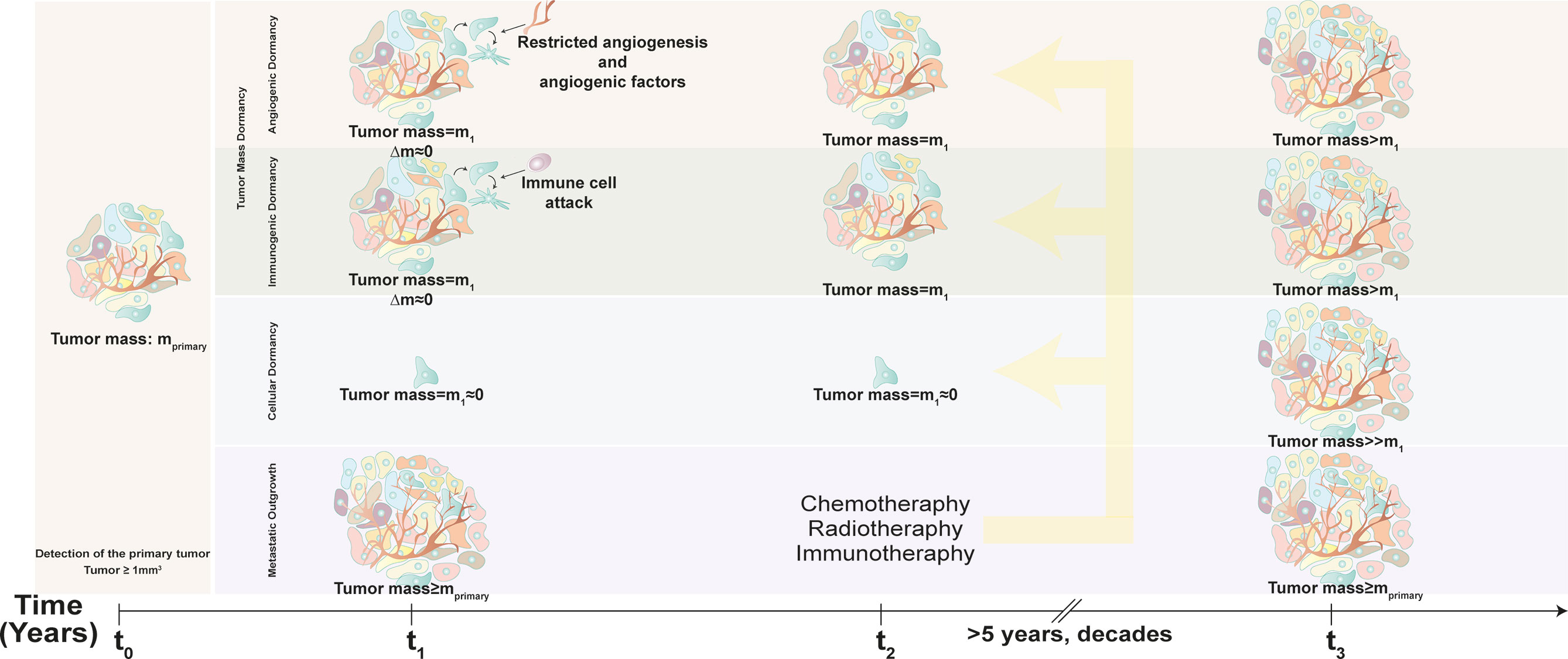
Figure 1 Time-dependent progression of metastasis and dormancy. Conventional diagnostic tumor scans are able to detect tumors bigger than 1 mm3 (tumor mass = mprimary). After diagnosis with cancer (time = t0), patient may undergo chemotherapy, radiotherapy or adjuvant therapy, yet dormant cells escape and become resistant to these treatments (time = t2), and awaken after years or even decades (time = t3). In tumor dormancy, tumor mass (m1) stagnates due to limited neovascularization and constant immune cell attack that balance tumor cell demise and proliferation. After the latency period, dormant tumor cells awaken and lead to tumor outgrowth (tumor mass>m1). In cellular dormancy, cancer cells hibernate as single cells or small clusters (tumor mass = m1≈0) and lead to massive tumor growth (tumor mass≥mprimary) following exit from dormancy.
Autophagy and Cancer Dormancy
Mechanisms of Mammalian Autophagy
Autophagy activation was reported as a novel characteristic of dormant cells in different tumor types (19). Three major types of autophagy were described: Macroautophagy, chaperone-mediated autophagy (CMA) and microautophagy. Although a clear connection between cancer and CMA was established (20), according to our knowledge, so far no study directly connecting CMA to cancer dormancy was published. Similarly, microautophagy was not studied in this context either. On the other hand, the number of studies implicating a role for macroautophagy in cancer dormancy continues to increase. Macroautophagy (autophagy herein) is an evolutionarily conserved catabolic process and an important stress response in all eukaryotic cells. Activation of autophagy leads to the clearance of various cellular components, including damaged organelles (e.g., mitochondria) as well as unfolded proteins and abnormal protein aggregates. As such, autophagy helps cells to combat stress, thereby contributes to survival. Mechanisms orchestrating autophagy activation, autophagic vesicle (autophagosome) formation and autophagic degradation were studied in detail.
Autophagic machinery primarily relies on the activity of ATG (autophagy-related) proteins (Figure 2). Following exposure to stress, activation of a core pathway involving ATG proteins leads to formation of double-membrane structures (phagophores) around target molecules and organelles. Phagophores eventually elongate and seal, forming closed vesicular structures called autophagosomes or autophagic vesicles. Autophagosomes fuse with late endosomes or lysosomes, to form autolysosomes. Lytic enzymes in the lumen of autolysosomes are responsible for the degradation of cargos carried by autophagosomes.
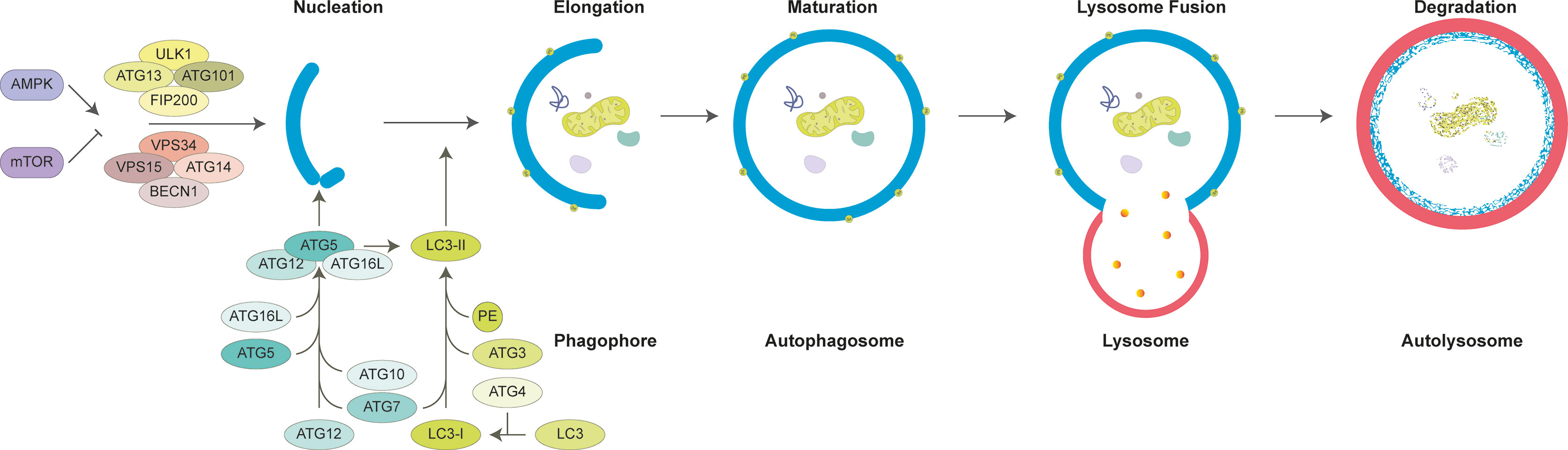
Figure 2 General mechanism of mammalian autophagy. Autophagy is tightly controlled by the activity of AMPK and mTOR. Under nutrient deprivation, AMPK activates autophagy, yet mTOR inhibition is relieved. Subsequent activation of ULK1 and BECN1 complexes promotes formation of phagophore. ATG5-12-16L complex and ATG8 family protein LC3 are required for elongation and closure of phagophore. Fully mature double-layered autophagosome containing cargo molecules fuses with late endosomes and lysosomes. Autophagosomes and their cargo are degraded through lysosomal enzymes and recycled into cytosol for reuse.
Autophagic activity is tightly controlled by protein complexes containing the mTOR kinase: mTORC1 and mTORC2 (21). These protein complexes are highly responsive to cellular cues, such as nutrient and growth factor availability, and in the active state, they work to inhibit autophagy (22). PKB/AKT pathway provides input from growth-related signals in order to regulate the mTOR complexes and autophagy. AMPK pathway, an energy sensor of the cell that monitors AMP/ATP ratios, comes into play when energy levels are low (23–25). While the mTORC1 has been documented to regulate autophagy directly, mTORC2 complex provides regulatory and feedback signals from insulin receptor phosphoinositide 3-kinase signaling (26–28). Under nutrient-rich conditions, mTORC1 keeps ULK1 and ATG13 in an inactive state. Nutrient deprivation leads to dephosphorylation of mTORC1 sites on ULK1 and ATG13 (24). ULK1 then autophosphorylates and phosphorylates its partner proteins ATG13 and FIP200 (29, 30). By this way, ULK1 activation initiates a cascade of events that promotes autophagosome formation.
Phagophore nucleation results from phosphorylation of lipids by the VPS34 lipid kinase complex (the class III PI3K, PI3KC3), BECN1 (Beclin-1), AMBRA1 and ATG14 (31, 32). Phosphorylation of inositol lipids on cellular membranes, such as ER membranes, leads to accumulation of phosphatidylinositol 3-phosphates (PI3P) (33). PI3P formation at membrane sites called omegasomes (or cradles), through recruitment of proteins with PI3P-binding domains, such as WIPI1/2 proteins and DFCP1 (34–36).
Proteins from the ubiquitin-like ATG8 family control elongation of phagophores through the activity of two key protein complexes. ATG12-ATG5–ATG16L1 complex facilitates coupling of ATG8 proteins, including MAP1LC3 (LC3) and GABARAPs, to phosphatidylethanolamine (PE) molecules on elongating membranes (37). Lipidated ATG8 proteins on autophagic membranes allow growth and closure of phagophore membranes (38, 39). Autophagy can be selective or non-selective. In the latter case, autophagy receptors, such as SQSTM1/p62, bridge between ATG8 proteins and ubiquitylated targets and direct them to autophagosomes. Hence, assessment of lipidation of ATG8s, especially LC3 lipidation, is a widely accepted as a powerful approach for monitoring autophagic activity (40, 41).
Mature autophagosomes are then transported along microtubules toward late endosomes and lysosomes. SNARE proteins facilitate fusion of autophagosomal outer membrane with endosomal or lysosomal membranes (42). Lysosomal acidic hydrolases degrade autophagic cargos into their building blocks (e.g. proteins into amino acids), which in turn are recycled to cytosol and used in the synthesis of new cellular components. As such, autophagy functions as a cellular degradation and recycling mechanism that allows cells to survive under stressful conditions.
Dysregulation of autophagy pathway is associated with various diseases, including cancer (43–45). In fact, autophagy plays an important yet context-dependent role at various stages of cancer progression and metastasis (45, 46). In early stages of cancer, control of ROS accumulation, prevention of DNA damage and genome instability require functional autophagic activity, eliminating damaged mitochondria and misfolded/aggregated proteins (47). Conversely, in established tumors and especially those tumors that grow relatively faster (e.g., tumors with K-RAS activation), tumor supporting role of autophagy is prominent. In this context, autophagy compensates for increased metabolic demands, originating from nutrient and energy deficiency, hypoxia and acidosis (48). Tumor stage-dependent dual role of autophagy might be explained in some models by hypoxia-triggered switches involving proteins, such as RAC3 (49). Moreover, autophagy was involved in various tumor progression- and metastasis-associated phenomena, including cell cycle regulation, stem cell behavior, extracellular matrix interactions, EMT, anoikis, tumor cell-stroma interactions, angiogenesis, immune responses and treatment resistance (50–54). In line with these observations, a number of autophagy genes and proteins show tumor suppressor or oncogenic activities (45, 55, 56).
In spite of the importance of autophagy in cancer formation and progression, contribution and molecular mechanisms of autophagy to cancer dormancy was not explored in detail in different cancer types and models. As summarized below, an increasing number of recent studies begins to provide evidence about a direct involvement of autophagy in cancer dormancy.
Mechanisms of Cellular Dormancy
Cellular dormancy is characterized by a halt in cancer cell proliferation and entrance to a quiescence-like state. This non-proliferative state of existence may last for months or years. Moreover, no matter how long the dormant state is, some cells retain the capacity to reactivate and re-enter to a proliferative state (57–59). So, cellular dormancy is defined as a reversible mechanism.
Dormant cells usually reside and survive in the G0-G1 phase of the cell cycle. Hence, they lack proliferation markers (e.g., Ki67) as well as markers of apoptosis (e.g., active-caspases) and senescence (e.g., beta-galactosidase) (60–62). Not surprisingly, several changes in cell cycle regulatory molecules were observed. For instance, cyclin-dependent kinase (CDK) inhibitors p27Kip1 and p21Cip1/WAF1 controlled the non-proliferative state during hematopoietic stem cell dormancy (63). In another example, adhesion of lymphoma cells to bone marrow stromal cells resulted in cell cycle arrest, involving post-transcriptional regulation of Skp2, a component of p27Kip1 and p21Cip1/WAF1 containing SCF complex (64).
Another regulator of dormancy-associated cell cycle arrest was identified as the DREAM complex. The complex consists of p130 or p107 (Retinoblastoma-like pocket proteins), MuvB and E2F protein. MuvB was defined as a core component in the transcriptional regulation of cell cycle genes by the DREAM complex (65). In dormant cells, elevated p130 levels were shown to facilitate DREAM complex formation and regulate its transcriptional effects (65, 66). On the other hand, high levels of p107 were detected only in proliferating cells. Regulatory kinases DYRK1A and DYRK1B phosphorylated a subunit of MuvB, namely LIN52, and activated DREAM complex assembly during entry to the non-proliferative state (67). Additionally, these kinases stabilized p27Kip1 and induced cyclin D turnover, further contributing to the non-proliferative state (68, 69).
Mitogen-activated kinase (MAPK) pathway plays a central role in the regulation of dormancy. A number of dormancy-related factors and their cognate receptors were associated with a shift in the balance between proliferative ERK1/2 versus non-proliferative p38 MAPKs. Independent studies conducted in different cancer cell types, including breast, prostate, melanoma cells, supported the involvement of p38 pathway in cancer dormancy (70), and activation of this pathway contributed to the proliferation arrest in this context (71, 72). For instance, p38 kinases were stimulated by the activity of TGF-β2/TGFβRIII, which in turn supported dormancy of head and neck squamous cell carcinoma cells in the bone marrow (73). In addition, as a paracrine factor, secreted TGF-β2 from osteoblasts in the bone microenvironment contributed to prostate cancer dormancy through activation of p38 (74). Dormant cells secreted high levels of TGF-β2, creating an autocrine loop in the regulation of dormancy (75). In line with this, results revealed that proliferating cells have low TGF-β2 levels (75).
Urokinase plasminogen activator receptor (uPAR) pathway was described as another dormancy-associated pathway. In HNSCC, status of the uPAR was directly related with the dormancy capacity of cells in vivo. In this tumor type, interaction of uPAR with α5β1 integrin dictated formation of insoluble fibronectin fibrils and blocked the activation of p38 (76). Conversely, decreased uPAR levels were detected correlated with ERK1/2 pathway attenuation (76). Moreover, downregulation of uPAR inhibited focal adhesion kinase (FAK) phosphorylation and downstream Src activity, facilitating the dormant state in vivo (77, 78).
In addition to the HNSCC model, FAK and Src-related mechanisms were also studied in breast cancer dormancy. Activation of Src by CXCL2/CXCR4 signaling correlated with prolonged survival of DTC in the bone marrow niche via phosphoinositide 3-kinase (PI3K)/AKT pathway (79, 80). Interestingly, Src-assisted dormancy was secondary organ-dependent and its downregulation had no effect on lung metastasis of breast cancer cells (79, 80).
Bone morphogenetic proteins (BMP) were mainly involved in dormancy regulating tumor stroma interactions. Investigations on prostate cancer revealed that, BMP7 (bone morphogenetic protein 7) regulated dormancy of prostate cancer cells through affecting cancer stem cell population. This effect required BMPR2 (BMP receptor 2) expression and activation of dormancy-associated downstream signaling components, such as p38, p21 and the metastasis suppressor NDRG1 (81). In another study, bi-directional communication between tumor cells and stroma was revealed. Dormant prostate cancer cells, but not proliferative cells, secreted SPARC, a factor which stimulated BMP7 expression from bone marrow stromal cells, contributing to the maintenance of dormant phenotype (82). Another BMP protein, BMP4, was studied in the context of breast cancer. High levels of BMP4 expression correlated with entrance of cancer cells to a dormant state in the lung. In this organ, dormancy activation was associated with ALK2/3 and SMAD1/5 signaling (83). In this system, extracellular BMP antagonist DAND5 counteracted BMP4-assisted dormancy and promoted the proliferative state (84).
Dormancy-related signaling pathways lead to the activation of a specific gene regulatory program. For instance, BHLHE41 transcription factor was documented among the downstream effectors of p38-regulated dormancy in HNSCC (72). In another study, Kim et al. identified BHLHE41 and NR2F1 as key factors promoting ER positive breast cancer dormancy in an in vivo xenograft mice model (85). Importance of BHLHE41 to breast cancer dormancy was further confirmed in a 3D endosteal bone niche model containing bone marrow-derived cells and endothelial cells (86).
NR2F1 belongs to the NR2F family of cancer-related transcription factors (77). Dormant cancer cells were found to express high levels of NR2F1 in comparison to their proliferative counterparts (79, 87). Moreover, high NR2F1 and TGF-β2 expression were characterized as a dormancy signature in prostate cancer DTC (87). Moreover, transcription of another p38-regulated gene, SOX9, was directly controlled by NR2F1 binding elements in its promoter (87). NR2F1-SOX9 axis was also regulated by microenvironment-derived retinoic acid (RA) signaling and RARβ (87). In addition to other targets, NR2F1 promoted expression of the CXCL12 and its receptor CXCR4 and induced-cell cycle arrest in salivary adenoid cystic carcinoma cells (79).
Receptor tyrosine kinases, including TYRO3, AXL and MER, were critically involved in the dormancy phenotype of certain cancer types. For example, activation of AXL or TYRO3 receptor kinases by GAS6 secreted from osteoblast cells, contributed to the establishment of metastatic dormancy of prostate cancer cells in the bone marrow (75, 88, 89). In another example, dormant state was triggered in lymphoblastic leukemia cells by GAS6 to MER binding (90). On the other hand, AXL was found to be an important regulator of myeloid lineage-related gene expression and dormancy in myeloma cells (91).
Although generally considered as a pathway involved in cancer dissemination and metastasis (92), Wnt pathway was implicated in dormancy control in a context- and stimulus-dependent manner (93, 94). For instance, DKK1-dependent inhibition of Wnt3a signaling induced growth arrest and entry to dormancy (95). On the other hand, activation of Wnt5a pathway was responsible for the entrance of prostate cancer cells to a non-proliferative dormant state (94).
Overall, several cytokines, growth factors and signaling pathways involving kinases as well as transcription factors were identified as regulators of dormancy. Although we are far from having a complete picture, pathways regulating dormancy are being better defined. A summary of known proteins and pathways studied in vitro and in vivo were shown in Tables 1 and 2 respectively, and the reader is referred to recent review articles about dormancy for further details (58, 191, 192).
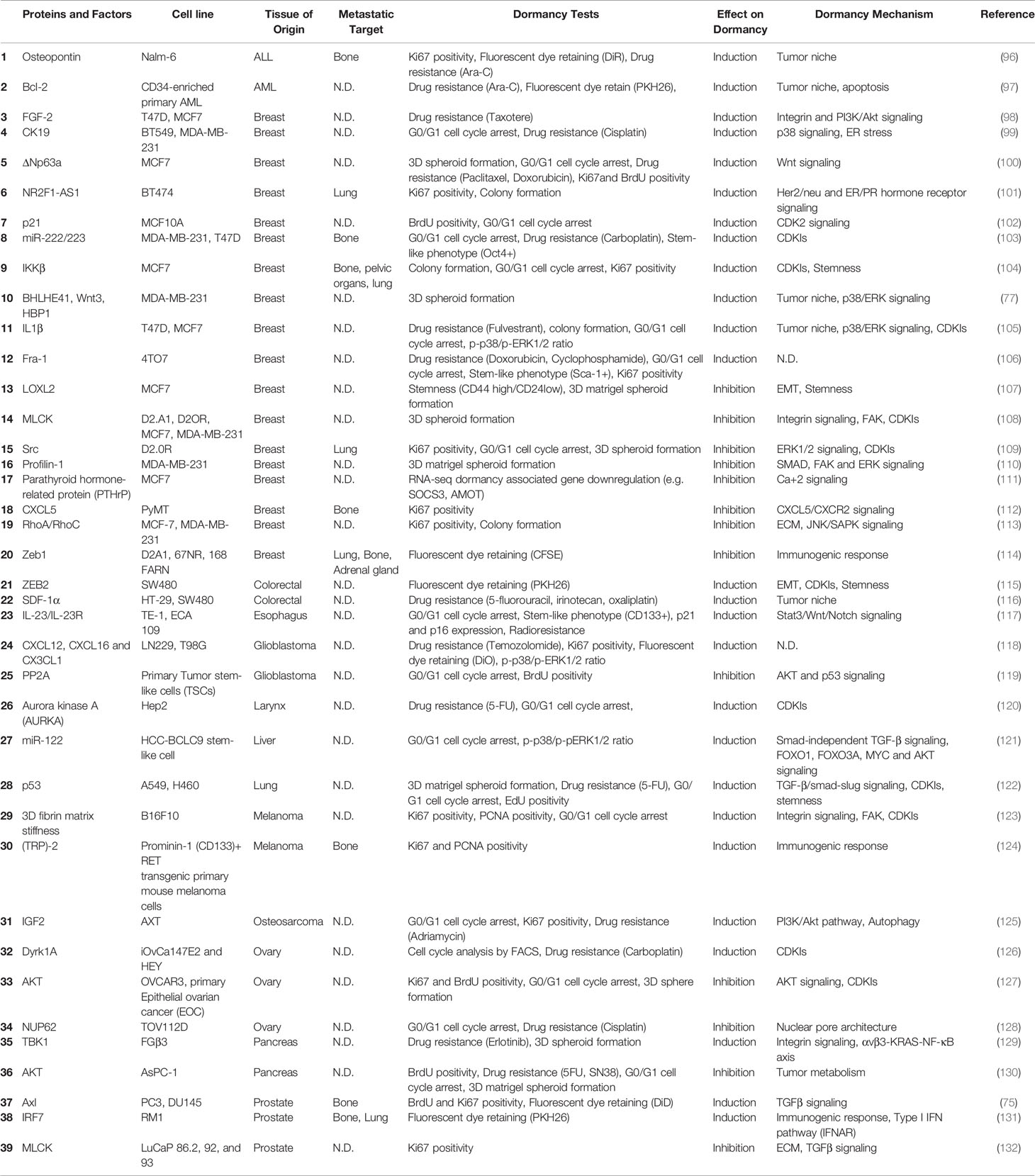
Table 1 Summary of in vitro dormancy models and mechanisms.
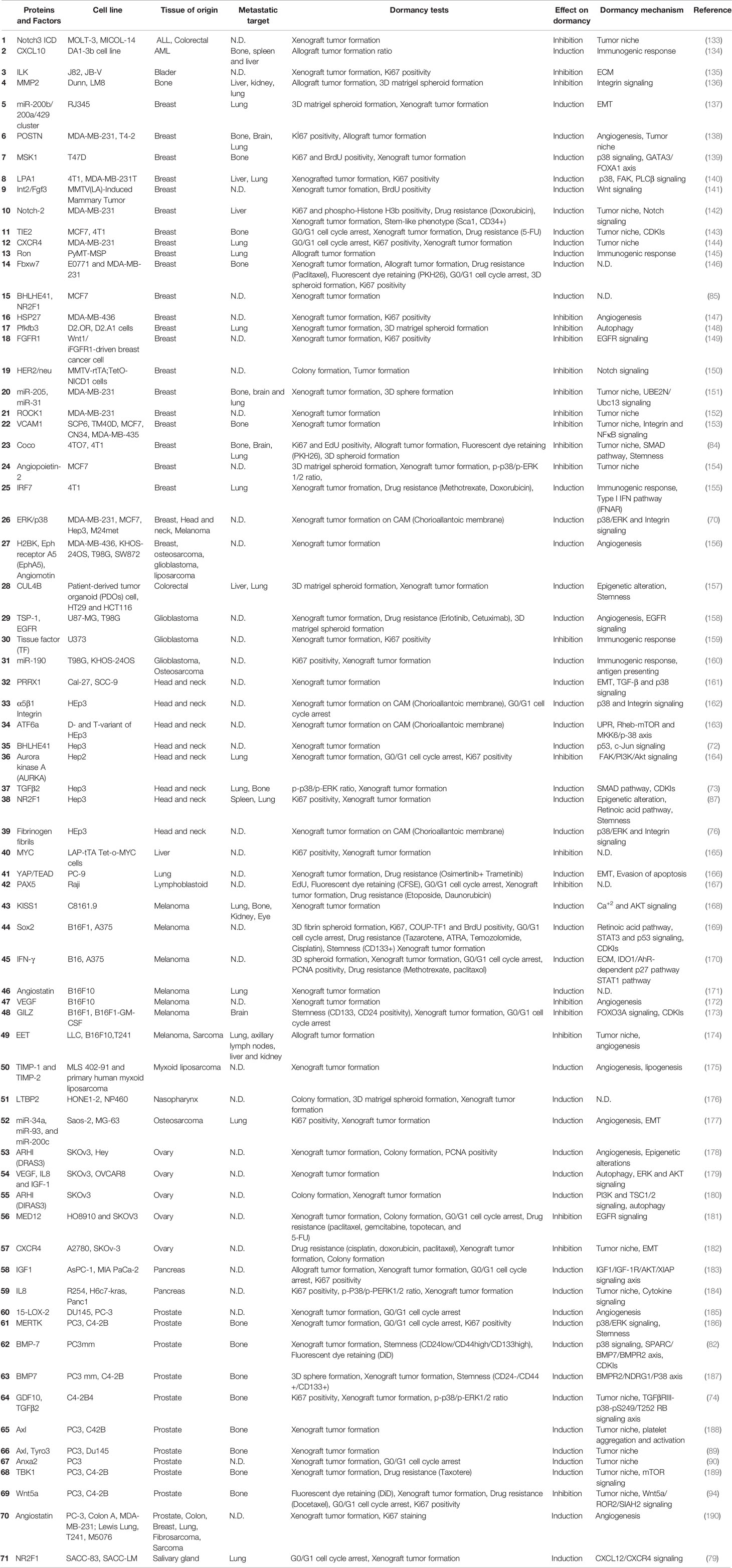
Table 2 Summary of in vivo dormancy models and mechanisms.
Role of Autophagy in the Context of Cancer Dormancy
In addition to recycling long-lived proteins, autophagy plays a key role in the management of energy crisis, control of reactive oxygen accumulation through destruction of damaged mitochondria, and in the elimination of unfolded and misfolded proteins. Studies in the last decade indicated that autophagy is involved in various stages of cancer formation and progression (193–197). As mentioned previously, autophagy plays a role in various events leading to tumor cell survival, resistance to treatment and metastasis. Hence, autophagy emerges as one of the critical determinants of the dormant state. In fact, several independent studies using cancer cells-derived from a wide variety cancer types, including breast, ovary, gastrointestinal tract, pancreas and bone cancers and their respective mice tumor or xenograft models showed that, autophagy is highly active in dormant cancer cells (125, 148, 180, 198–200). Some of these observations were even supported by the analysis of patient-derived tissue samples (201), yet molecular details of how and why autophagy contributes to the dormant phenotype are not well known. In this section, we will overview the current literature on autophagy-dormancy connection.
Autophagy-Dormancy Connection: Experimental Evidence
Studies using different experimental set-ups, different cancer cell types and models revealed that, malignant cells entering a non-proliferative, dormancy-like but reversible cycle arrest state showed increased autophagic activity (Table 3) (148, 180, 199). In this context, dormant cancer cells were more sensitive to autophagy inhibition compared to their proliferating counterparts and inhibition of autophagy was lethal in most cases. Moreover, inhibition of autophagy in dormant cancer cells changed their metastatic behavior in vivo in mice (148, 199).
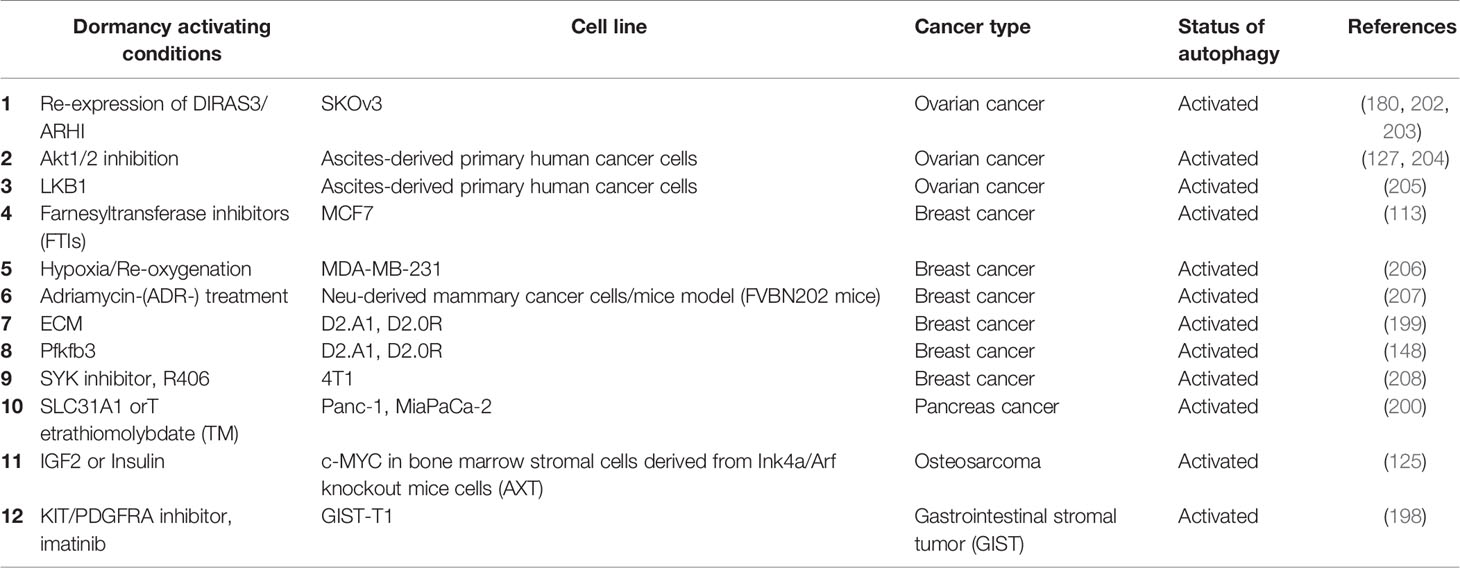
Table 3 Dormancy models with documented autophagic activity.
For instance, in gastrointestinal stromal tumor (GIST) cells, treatment with a KIT/PDGFRA inhibitor, imatinib, induced a dormancy-like quiescent state during which cells entered cell cycle arrest through accumulation of the cell cycle inhibitor p27 (198). Autophagy activation was observed under these conditions, and inhibition of autophagy using a genetic or chemical (chloroquine or quinacrine treatment) approach resulted in the loss of cell viability, and increased the anti-tumor efficacy of imatinib in in vitro and in vivo tests.
Contribution of autophagy to ovarian cancer dormancy was studied in detail. DIRAS3 (or ARHI) is a maternally imprinted tumor suppressor that is frequently downregulated in breast and ovarian cancers (209, 210). Re-expression of DIRAS3 in cancer cells robustly induced autophagy (180, 202, 211). Interestingly, although DIRAS3 expression resulted in the apoptotic death of cancer cells in culture (203), it promoted a dormancy-like state in vivo (180, 202). Re-expression of DIRAS3 in a Tet-inducible manner, stimulated autophagy in ovarian cancer xenografts, and led to a reversible inhibition of tumor growth and entry to a dormant state. Downregulation of the tumor suppressor was sufficient for the establishment of overt metastatic tumors. In this model, inhibition of DIRAS3-induced autophagy by chloroquine (a lysosomal autophagy inhibitor) reduced tumor growth, further underscoring the importance of autophagic activity to DIRAS3-related dormancy (202).
In other ovarian cancer studies, ascites-derived primary cancer cells from patients with high-grade serous ovarian cancer and ovarian cancer cell lines were used. Treatment of these cells with an allosteric AKT inhibitor Akti-1/2 induced a dormancy-like cytostatic response, and under these conditions, autophagic activity was significantly increased (204).
The role of autophagy in dormancy was studied in detail in breast cancer cell culture and animal models. Autophagy activation was observed during a dormancy-like arrest state of MCF7 breast cancer cells that were cultured with farnesyl transferase inhibitors (FTIs) (212). In MDA-MB-231 breast cancer cells, repetitive long-term hypoxia/reoxygenation cycles resulted in a low proliferation state and dormancy-like reversible cell cycle arrest (206). In another study, a dormancy-like state was induced by an adriamycin- (ADR-) treatment in vitro regimen using breast cancer cells from a Neu-driven cancer mice model (FVBN202 mice). Autophagy activation was also observed in this model of dormancy (207).
Other groups used two breast cancer cell lines derived from murine mammary hyperplastic alveolar nodules for modeling dormant versus proliferation states of this cancer type. D2.A1 cells were metastatic and D2.0R cells were dormancy-prone under certain growth conditions. In this system, autophagic activity of dormant D2.0R cells was found to be significantly higher than that of D2.A1 metastatic cells (148, 199). Both autophagosome and autolysosome numbers were increased, autophagy receptor (e.g. SQSTM1/p62) degradation was observed, indicating that autophagy in dormant cells was fully functional (148). In line with studies in other cancer types, dormant breast cancer cells were sensitive to autophagy inhibition whereas proliferative cells were resistant (199). Following tail vein injection to mice, most D2.0R cells stayed dormant in the lungs. Autophagy-related gene expression and autophagic activity in micrometastatic dormant lesions of D2.OR cells were observed higher as compared to the metastatic lesions of D2.A1 cells (148, 199).
Dormancy was also investigated in pancreatic duct adenocarcinoma (PDAC). In this cancer type, elevated levels of copper were associated with the degree of cancer progression. Interestingly, blockage of copper absorption by targeting the copper transporter 1 (SLC31A1) or usage of copper chelator tetrathiomolybdate (TM) inhibited proliferation of cancer cells and induced a dormancy-like arrest state (200). Under these conditions, autophagy was activated, and it was responsible for PDAC cell survival both in vitro and in vivo tests. Indeed, inhibition of autophagy caused an increase in in vitro cell death and decreased in vivo tumor burden. These results further provided evidence about the role of autophagy in the survival of dormant cells.
Dormancy in osteosarcoma following chemotherapy, has been associated with increased levels of IGF2 (125). Chronic exposure of osteosarcoma cells to IGF2 or insulin in combination with serum deprivation, successfully established an in vitro dormancy and drug-resistance model in osteosarcoma (125). Under these conditions, autophagy was activated.
Analysis of patient-derived samples provided further evidence about the importance of autophagy in cancer dormancy (201). In primary ovarian tumor tissue sections, LC3 localization in punctate structures was observed in only 21–23% of cases. In contrast, LC3 puncta, hence an upregulation of the autophagic activity was observed in more than 80% of tumor nodules found on the peritoneal surface of patients at second-look operations following primary chemotherapy. These results point out to a significant increase in autophagy in dormant ovarian cancer cells seeded in the peritoneum compared to primary tumor samples. These results underline the relevance and importance of experimental observations about dormancy-autophagy connection.
Role of Autophagy in Dormancy Establishment, Dormant Cell Survival, and Reactivation
Autophagy controls dormant cell survival and behavior in many ways. In DIRAS3-induced ovarian cancer dormancy model, ARHI re-expression enabled SKOv3 ovarian cancer cells to remain dormant when they were grown in mice as xenografts (180). Reduction of ARHI levels in dormant cells caused xenografts to grow faster, and inhibition of ARHI-induced autophagy with chloroquine dramatically blocked regrowth of tumors.
In the D2.0R dormant and D2.A1 metastatic breast cancer cell models, autophagy was critical for the maintenance of the dormant phenotype in cancer cells and their survival. In 3D cultures, dormant D2.0R cells lost viability following treatment with autophagy inhibitors hydroxychloroquine, bafilomycin or 3-methyladenine, while non-dormant counterparts were not affected (199). Knockdown of autophagy genes Atg3, Atg7, p62 or FIP200, resulted in the outgrowth of dormant cells in 3D cell cultures. Moreover, Atg3-deficient D2.0R cells showed an increased capacity to create pulmonary tumors in mice (148). Similarly, in the ADR-induced dormancy model of Neu-driven breast cancer, mice that were i.v. injected with ADR-treated Atg5 knockdown cancer cells developed lung metastasis significantly sooner than those that were injected with wild-type dormant cells. As expected, a higher frequency of Ki67 positive, polyploid-like cells was observed in ADR-treated Atg5 knockdown mammary tumors (207). In line with these results, autophagy was downregulated in proliferating metastatic cells, but it was found to be necessary for a dormant-to-proliferative switch before the establishment of overt metastatic lesions (199). Consequently, treatment with autophagy inhibitors after the development of proliferative lesions (i.e. lesions that moved beyond the dormant-to-proliferative switch) had lesser impact on the metastatic burden (199).
These observations indicate that autophagy plays an active role in the initiation and maintenance of the dormant state, as well as during the switch from dormancy to a proliferative state.
Role of Autophagy in the Clearance of Mitochondria and Regulation of Metabolism in Dormant Cancer Cells
Mitochondria are at the center of cellular energy metabolism control. A side product of oxidative phosphorylation is reactive oxygen species (ROS), and dysfunctional or damaged mitochondria are more prone to produce ROS. A life-threatening outcome of ROS accumulation at a cellular level is oxidation of building blocks such as proteins and lipids, as well as damage to DNA. A selective form of autophagy, mitophagy is a major mechanism that eliminates dysfunctional and damaged mitochondria and that ensures control of the mitochondrial mass in cells.
Cancer cells are able to stay in a dormant state for months or even years. Hence in dormant cells, in addition to elimination of unfolded/misfolded proteins and other building blocks, regulation of the mitochondrial mass and prevention of ROS accumulation should be of utmost importance for long-term survival and the preservation of reactivation capacity after transition to the proliferative state. Additionally, control of mitochondrial mass and function should be critical for metabolic reprogramming of dormant cells. Indeed, increased autophagic activity was associated with mitophagy in several models of cancer dormancy.
For instance, mitophagy was activated during DIRAS3-induced dormancy of ovarian cancer cells. Following DIRAS3 induction by the Tet-on system in ovarian cancer stable cell lines, TMRM uptake by mitochondria was decreased, indicating accumulation of depolarized mitochondria in these cells. Reduced TOM20 mitochondrial protein levels and mitochondrial mass as assessed through mitotracker staining were reported in dormant cells. As such, dormancy state was associated with a higher rate of mitochondrial depolarization, and mitophagy was increased as a mechanism to eliminate depolarized mitochondria and to limit ROS accumulation (213).
In the D2.0R breast cancer model of dormancy, autophagy protein LC3 was found to colocalize with mitochondria in cells growing in a matrix supporting dormancy. During mitophagy, PINK-assisted ubiquitylation of mitochondrial proteins by E3 ligases such as PARKIN prime mitochondria for mitophagic degradation. Indeed, accumulation of mitophagy-associated full length PINK and degradation of mitochondrial protein TOM20 was reported under these experimental conditions. Additionally, autophagy inhibition using HQ caused an accumulation of damaged mitochondria as well as ROS. Following suppression of the autophagic activity, dormant cells suffered from DNA damage, caspase-3 activation was prominent, and cells eventually died. Mitochondrial ROS scavengers prevented cell death, indicating that an important function of autophagy in dormant cells is the maintenance of healthy mitochondrial mass, hence limitation of ROS-induced damage (199). Similarly, in the TM-treated PDAC cell model of dormancy, inhibition of autophagy by CQ increased ROS accumulation and resulted in cell death, further showing that ROS limiting activity of autophagy is central to dormant cancer cell survival in different cancer models (200).
Autophagic and mitophagy activation during dormancy was associated with metabolic changes in cells. In the ovarian cancer dormancy model, induction of DIRAS3 resulted in a higher glycolytic rate and mitochondrial respiration rate was decreased (213). Indeed, ATP levels of were found to be attenuated in different models of dormancy (200, 205, 213). Moreover, in dormant cells, an increase in glucose and glutamate uptake was accompanied by extracellular lactate accumulation (213). Under these conditions, increased glucose uptake was coupled to an upregulation of glycolysis and glutaminolysis, and all these changes were autophagy dependent. In this context, blockage of these metabolic pathways resulted in decreased cell viability (213). In autophagic tumors in vivo, free valine, glycine, and alanine concentrations were increased at statistically significant levels, indicating that bulk degradation of proteins by autophagy was also accelerated and it further supported metabolic activities of dormant cancer cells (213).
Molecular Mechanisms of Autophagy in Dormant Cells
To date, molecular mechanisms governing autophagy activation during dormancy and autophagy signaling pathways that are involved are largely unknown. Yet, studies on autophagy-dormancy connection provided hints about the involvement of certain autophagy-related proteins and pathways in the process.
Among the upstream signaling pathways regulating autophagy, inhibition of the PI3K/AKT pathway emerges as a common observation. In many reports, mTOR pathway that is downstream to AKT was shown to be inhibited in dormant cells. As mentioned above, mTOR is a central regulator of autophagy, and its inhibition correlates with autophagy activation in various systems (214–217).
In ovarian cancer dormancy model, DIRAS3 expression resulted in the inhibition of signaling through PI3K/AKT and Ras/MAP through enhancing internalization and degradation of the epidermal growth factor receptor. As a result, mTOR signaling downstream to the AKT pathway was also inhibited (180, 201). Indeed, downregulation the pathway by DIRAS3 resulted in a decrease in the activation of mTOR downstream pathway proteins, such as p70S6K and pS6, proteins that are involved in the regulation of cell size and protein synthesis (180). Autophagic activity was strongly stimulated under these conditions.
In line with the DIRAS3 model, ascites-derived ovarian cancer spheroids were in a dormant state that was associated with AKT downregulation and autophagy activation (127, 205). In fact, inhibition of the AKT pathway in ovarian cancer cells using specific inhibitors of the AKT kinase, namely Akti-1/2, was sufficient to direct the entry of cells to a dormant-like state (127). Downregulation of AKT and mTOR pathway was also observed in the osteosarcoma dormancy model (125) and breast cancer cells entering dormancy following exposure to long-term hypoxia/reoxygenation cycles (206). Similarly, mTOR and its downstream pathways were reported to be inhibited in the imatinib-induced GIST cell dormancy model (198).
Another key protein regulating autophagy activation is the energy sensor kinase AMPK. Increased AMP/ATP ratio correlates with problems in energy status of cells, and leads to the activation of AMPK (218). Hypoxia is another signal that can activate AMPK (219). Following activation, AMPK was shown to phosphorylate TSC2 and interfere with the activity of the GTPase RHEB, an activator of mTORC1 signaling; the net result is autophagy activation (220, 221). Another mechanism through which the kinase contributes to autophagy activation direct phosphorylation of the autophagy protein ULK1 (23, 24).
In dormant cells, intracellular ATP levels are decreased (74, 205, 213). Consequently, AMPK activation was reported in different experimental models of dormancy. For instance, in ovarian tumor cells, LKB1 and AMPK expression and activity were increased during spheroid formation and dormancy (205). The study showed that LKB1 (and possibly AMPK) was required for the survival of ovarian cancer cells in a dormant state. Moreover, AMPK activation in proliferating ovarian cancer cells caused them to enter cell cycle arrest (205). Similarly, an increase in AMPK activity was observed during DIRAS-3-induced dormancy of ovarian cancer cells (180) and chronic hypoxia-induced dormancy of breast cancer cells (206).
Transcriptional upregulation of autophagy-related genes was observed in dormancy models. Independent groups reported the upregulation of key autophagy genes, including LC3, ATG4, ATG5, ATG7 and BECN1, in dormant cells (148, 199). A mechanistic explanation on the transcriptional regulation of autophagy gene expression in the dormant state came from studies using ovarian cancer cells. During DIRAS3-induced dormancy, mTOR inhibition promoted translocation of transcription factors FOXO3a and TFEB to the nucleus (222). The end result was a FOXO3a-dependent upregulation of autophagy proteins ATG4 and LC3 and Rab7, a mediator of autophagosome-autolysosome fusion (222). Similar to FOXO3a, TFEB translocation to the nucleus activated transcription of various autophagy-related genes (223, 224).
Interestingly DIRAS3 itself was subject to transcriptional regulation downstream to the mTOR pathway (225). Under conditions of nutrient deprivation, mTOR inhibition resulted in the dissociation of E2F1 and E2F4 from the DIRAS3 promoter, leading to the proteasomal degradation of these transcription factors. Dissociation of E2F1 and E2F4 from DIRAS3 promoter allowed transcriptional upregulation of the gene and activated autophagy. On the other hand, another transcription factor, CEBPα, positively regulated DIRAS3 expression and autophagy. Hence, transcriptional loops involving DIRAS3 might contribute to further activate and sustain autophagy during nutrient deprivation and possibly during dormancy (225).
DIRAS3 was directly participating to autophagy regulating protein complexes. In fact, DIRAS3 was shown to stabilize the autophagy initiation complex consisting of VPS34 (PIK3C3), BECN1 and ATG14 (201). DIRAS3 binding to BECN1 destabilized BECN1-BCL2 inhibitory complexes, displaced BCL2 and allowed recruitment of BECN1 protein by autophagy-related VPS34 lipid kinase complex. Binding of DIRAS3 to BECN1 facilitated association of BECN1 with VPS34 and ATG14. DIRAS3 was also shown to directly bind VPS34. Altogether, DIRAS3 enhanced VPS34 lipid kinase activity that is required for autophagosome formation and autophagy activation (201). Moreover, DIRAS3-mediated stabilization of the initiation complex and subsequent autophagy activation was necessary for dormant cell survival after chemotherapy (201).
On the other hand in mice, knockdown of Atg7 but not Becn1 decreased numbers of tumors formed by dormancy-prone cells in a TGFβ-induced inflammatory background, indicating that requirement for Becn1 gene in dormancy-related autophagy and tumor cell survival might be tumor and cell type-dependent (199).
Role of Autophagic Degradation in Dormancy
Data that was presented above show that autophagic activity is prominently higher in dormant cancer cells compared to their actively proliferating counterparts. Several studies provided clues about the nature of autophagy in this setting. In addition to an increase in autophagosome numbers, autolysosome formation and autolysosomal degradation was reported to be upregulated during dormancy. Autolysosomal degradation of the selective autophagy receptor p62/SQSTM1 as well as the LC3 protein itself was reported in many studies (148, 199). Inhibition of the autolysosomal activity by chemicals, such as chloroquine and its derivatives, changed the behavior of dormant cells, influenced cell survival, dormant cell reactivation and metastatic capacity. As explained above, mitochondria are among the targets of selective autophagic degradation during dormancy. Therefore, metabolic outcomes of autophagic activity in dormant cells might be attributed to mitophagy and non-selective autophagic degradation of cellular components, including long-lived proteins. So far, the role of selective autophagy in cancer dormancy is not well studied, and there are only a few reported examples.
A dormancy-related direct target of autophagy was identified using the D2.0R model of breast cancer dormancy. In this study, Pfkfb3 (6-phosphofructo-2-kinase/fructose 2,6-biphosphatase 3) was identified as a gene that was highly expressed in metastatic cells but downregulated in dormant cancer cells (148). Pfkfb3 is a key regulator of glycolysis rate in cells, and its expression was shown to promote metastatic tumor growth. An inverse correlation between dormancy-related autophagic activity and Pfkfb3 levels was observed. So, the role of autophagy in the degradation of Pfkfb3 protein was studied in dormant cancer cells. Indeed, Pfkfb3 protein accumulated when autophagic degradation was inhibited using CQ or autophagy gene knockdown. The protein was polyubiquitylated, and in this state, it directly interacted with the ubiquitin-associated domain (UBA) of the p62 protein. Strikingly, Pfkfb3 downregulation in metastatic D2.A1 breast cancer cells prevented their growth and delayed establishment of metastatic lesions. Conversely, autophagy inhibition and Pfkfb3 upregulation correlated with reactivation of dormant of D2.0R cells. The study showed that, although proteasomal degradation also contributes to the determination of protein’s half-life, selective degradation by autophagy is important in the control of Pfkfb3 protein levels in dormant cells. Hence, the study provides an example where tumor dormancy and recurrence rely on autophagic clearance of metabolic regulators (148).
Another autophagy target was identified among factors regulating EMT-MET (epithelial-mesenchymal and mesenchymal- epithelial transitions) during metastasis. SYK is a non-receptor tyrosine kinase mediating signaling events downstream to several transmembrane receptors, including the B-cell receptor (BCR). Decreased expression of SYK mRNA correlated with decreased survival in breast cancer patients (226). P-bodies are cytoplasmic foci containing mRNA, miRNA and mRNA-binding proteins, and they are involved in the regulation of mRNA half-life and translation control. During TGFβ-induced EMT, accumulation of P-bodies was observed. SYK concentrated in P-bodies, and SYK activity and autophagy was necessary for controlled clearance of P-bodies during MET and metastasis (208, 227). Hence in this system, SYK promoted removal of P-bodies through autophagy and supported activation dormant cancer cells, allowing initiation of cancer metastatic outgrowth (208).
There are possibly other direct or indirect targets of autophagy that are involved in dormancy maintenance and a dormant-to-proliferative switch. Further studies will reveal the identity of these key factors that are degraded by dormancy-associated autophagic activity.
Treatment Responses, Dormancy, and Autophagy
Current cancer treatment approaches are usually unable to result in the total elimination of disseminated cancer cells and micrometastases. They even seem to create a selective pressure on cancer cells promoting their escape from cell death by entering to a dormant state. Since dormant cells are not actively proliferating, they are in general resistant to chemotherapy and radiotherapy approaches that mainly result in DNA damage and block cell division. A body of literature provide evidence about the role of autophagy in the treatment resistance of growing primary tumors and overt metastases (228–231). Since autophagic activity is increased in dormant cells, autophagy might be a contributing factor in the observed robustness of dormant cells when faced with anticancer insults.
A number of studies tested the contribution of autophagy to treatment resistance that is observed in dormant cells. First observation is some drugs that were utilized in order to create models of dormancy, they themselves induced autophagy upregulation in cells. For example, treatment of cancer cells with imatinib (198), farnesyltransferase inhibitors (212), AKT inhibitor (204), and adriamycin (207), resulted in the upregulation of autophagic activity. Usage of antimalarial lysomorphic inhibitors of autolysosomal activity, such as Chloroquine, Hydroxychloroquine or quinacrine, as a combination treatment along with chemotherapy agents blocked autophagy under these conditions and generally resulted in the death of dormant cells and even elimination of tumors (125, 180, 202, 207). Combination of chemotherapy with genetic approaches gave similar results as well (125, 202, 207). So, capacity to activate sustained autophagy in response to cancer therapies might be one of the critical factors favoring the selection of dormant cells. This “autophagy addiction” might be exploited for the elimination of disseminated dormant cells in patients. On the other hand, considering indications about the role of autophagy in the dormant-to-proliferative switch, inhibition of autophagy might promote reactivation of dormant cancer cells, leading them to reenter an active proliferative state that renders them again susceptible to antiproliferative cancer treatments. On the other hand, crizotinib, an ALK inhibitor was shown to further activate autophagy and trigger apoptosis of dormant ovarian cancer cells (232). Either way, all these studies underline the therapeutic potential of autophagy manipulation in the context of dormancy.
Conclusion
Drug resistance and cancer dormancy are the two important causes of incurable metastatic disease that results in the loss of millions of lives from cancer-related deaths every year. Autophagy emerges as an integral part of the dormancy phenomenon. Autophagy activation was observed in dormant cells originating from different types of cancers, in cancer cellular models and animal models, as well as in patient-derived cells and tissues.
Autophagic activity was shown to confer survival advantage, treatment resistance and resilience to dormant cells. An important contribution of autophagy to dormant cell survival was related to the limitation of ROS accumulation. Autophagy is an important mechanism for the elimination of depolarized mitochondria, damaged peroxisomes and other organelles, as well as cytosolic long-lived proteins that are prone to aggregate and accumulate in the cytosol when exposed to excessive oxidative damage. Protection and preservation of the genetic material from ROS damage during long-lasting non-proliferative periods that may last for months or years, such as those observed during dormancy, is also an important challenge. For cells to preserve the reactivation capacity, dormant cells should be able to limit the number and extent of mutations they accumulate during periods of cell cycle arrest. Potency and efficacy of DNA repair pathways in dormant cells is not clear and further studies are required (233–235). At this point, studies in normal stem cell quiescence might give indications about the faith of DNA in cells that reside in long-term dormant periods. These studies indicated that in quiescent cells, DNA damage burden may even be higher in older cells than younger ones, and that repair process only begins following entry to cell cycle (236). Hence, these data underline the fact that, limitation of ROS accumulation by the autophagic activity contributes significantly to the survival of dormant cells.
Autophagy seems to play a critical role in the maintenance of dormant phenotype. Autophagy-deficient cells were not able to enter or stay in a dormant state compared to controls. Mechanisms through which autophagy controls the dormant phenotype are not clear. So far, only a few dormancy-related targets of autophagy were described. Pfkfb3 protein was identified as a target of selective autophagy in dormant cells. In fact, in addition to being a regulator of glycolysis, Pfkfb3 was shown to translocate to nuclei and its product fructose 2,6-biphosphate was shown to inhibit cell cycle inhibitor p27Kip and activate cyclin D3, resulting in progression from the G1 phase to the S phase (237). Moreover, Pfkfb3 was also involved in the upregulation of CDK1 and Cdc25 expression promoting entry to mitosis (237). Therefore, selective targeting of key proteins involved in cell cycle by autophagy, such as Pfkfb3, may be an important function of autophagy in the entry to and maintenance of the dormant phenotype.
P-bodies were also reported as selective targets of autophagy in a dormancy context. Whether autophagic degradation control a general downregulation of P-bodies or whether it is selectively and deliberately targeting a cell cycle- and dormancy-relevant subset of mRNAs, miRNAs and/or proteins is not clear.
Characterization of a full list of selective autophagy targets during dormancy will allow a better understanding of the role and contribution of autophagic degradation to the dormant phenotype.
Mechanistic aspects of autophagy signaling during dormancy are being better understood. In fact, AKT and related growth factor pathways seem to emerge as an important regulators of autophagy activation in dormant cells. Downstream to AKT, mTOR pathway components are inhibited, resulting in the activation of autophagy proteins, including ULK1. A decline in ATP levels in dormant cells may activate the energy sensor systems LKB1 and AMPK and further inhibit the mTOR pathway through TSC2 phosphorylation and activate autophagy by phosphorylating ULK1. Deficiency of nutrients, such as amino acids, possibly contribute to inhibition of mTOR on the lysosomes (238). mTOR inhibition allows activation of factors FoxO3a and TFEB, that transcriptionally upregulate autophagy-related genes.
Although most components of the canonical autophagy pathways were reported to be involved in dormancy-related autophagy, some studies questioned the contribution of key proteins, such as BECN1. Others placed BECN1 containing complexes at the center of autophagy regulation in dormant cells. ATG5 and ATG7 were reported to be important as well. p62/SQSTM1 was degraded in several independent models of dormancy, indicating that it may be an important mediator of selective autophagy under these conditions. Whether other autophagy receptors contribute to the autophagy pathway in dormant cells need further investigation.
Inhibition of autophagy by drugs or genetic methods was reported to impede dormancy, affect cell survival, or lead cells to enter into a proliferative phase, in which cancer cells are more susceptible to be eliminated by common cancer therapeutic strategies. Hence, approaches involving promotion of dormancy or reactivation of dormant cells should both necessitate study and manipulation of autophagy. A detailed understanding of mechanisms, regulatory pathways and specific targets of autophagy in the context of dormancy will certainly contribute to a better management of metastatic and recurrent disease, and maybe allow one day total elimination of disseminated cells and micrometastases in all cancer patients.
Author Contributions
DG designed the structure of the review and wrote and edited the review article. YA and NP wrote the review article and prepared the tables and figures. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by Scientific and Technological Research Council of Turkey (TUBITAK) grant number 216S489.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Liu Q, Zhang H, Jiang X, Qian C, Liu Z, Luo D. Factors involved in cancer metastasis: A better understanding to “seed and soil” hypothesis. Mol Cancer (2017) 16. doi: 10.1186/s12943-017-0742-4
2. Dongre A, Weinberg RA. New insights into the mechanisms of epithelial–mesenchymal transition and implications for cancer. Nat Rev Mol Cell Biol (2019) 20:69–84. doi: 10.1038/s41580-018-0080-4
3. Korpal M, Lee ES, Hu G, Kang Y. The miR-200 family inhibits epithelial-mesenchymal transition and cancer cell migration by direct targeting of E-cadherin transcriptional repressors ZEB1 and ZEB2. J Biol Chem (2008) 283:14910–4. doi: 10.1074/jbc.C800074200
4. Nieto MA. The snail superfamily of zinc-finger transcription factors. Nat Rev Mol Cell Biol (2002). doi: 10.1038/nrm757
5. Carver EA, Jiang R, Lan Y, Oram KF, Gridley T. The Mouse Snail Gene Encodes a Key Regulator of the Epithelial-Mesenchymal Transition. Mol Cell Biol (2001) 21:8184–8. doi: 10.1128/mcb.21.23.8184-8188.2001
6. Chaffer CL, San Juan BP, Lim E, Weinberg RA. EMT, cell plasticity and metastasis. Cancer Metastasis Rev (2016) 35:645–54. doi: 10.1007/s10555-016-9648-7
7. Bennett DC, Peachey LA, Durbin H, Rudland PS. A possible mammary stem cell line. Cell (1978) 15:283–98. doi: 10.1016/0092-8674(78)90104-6
8. Hager JC, Heppner GH, Stanley W, Richardson AM, Fligiel S. Characterization of a Variant-Producing Tumor Cell Line from a Heterogeneous Strain BALB/cfC3H Mouse Mammary Tumor. Cancer Res (1981) 41:1293–300.
9. Schardt JA, Meyer M, Hartmann CH, Schubert F, Schmidt-Kittler O, Fuhrmann C, et al. Genomic analysis of single cytokeratin-positive cells from bone marrow reveals early mutational events in breast cancer. Cancer Cell (2005) 8:227–39. doi: 10.1016/j.ccr.2005.08.003
10. Hüsemann Y, Geigl JB, Schubert F, Musiani P, Meyer M, Burghart E, et al. Systemic Spread Is an Early Step in Breast Cancer. Cancer Cell (2008) 13:58–68. doi: 10.1016/j.ccr.2007.12.003
11. Magbanua MJM, Sosa EV, Roy R, Eisenbud LE, Scott JH, Olshen A, et al. Genomic profiling of isolated circulating tumor cells from metastatic breast cancer patients. Cancer Res (2013) 73:30–40. doi: 10.1158/0008-5472.CAN-11-3017
12. Pixberg CF, Raba K, Müller F, Behrens B, Honisch E, Niederacher D, et al. Analysis of DNA methylation in single circulating tumor cells. Oncogene (2017) 36:3223–31. doi: 10.1038/onc.2016.480
13. Lu Z, Zou J, Li S, Topper MJ, Tao Y, Zhang H, et al. Epigenetic therapy inhibits metastases by disrupting premetastatic niches. Nature (2020) 579:284–90. doi: 10.1038/s41586-020-2054-x
14. Holmgren L, O’reilly MS, Folkman J. Dormancy of micrometastases: Balanced proliferation and apoptosis in the presence of angiogenesis suppression. Nat Med (1995) 1:149–53. doi: 10.1038/nm0295-149
15. Karrison TG, Ferguson DJ, Meier P. Dormancy of mammary carcinoma after mastectomy. J Natl Cancer Inst (1999) 91:80–5. doi: 10.1093/jnci/91.1.80
16. Kobayashi H, Mochizuki H, Sugihara K, Morita T, Kotake K, Teramoto T, et al. Characteristics of recurrence and surveillance tools after curative resection for colorectal cancer: A multicenter study. Surgery (2007) 141:67–75. doi: 10.1016/j.surg.2006.07.020
17. Brookman-May SD, May M, Shariat SF, Novara G, Zigeuner R, Cindolo L, et al. Time to recurrence is a significant predictor of cancer-specific survival after recurrence in patients with recurrent renal cell carcinoma - Results from a comprehensive multi-centre database (CORONA/SATURN-Project). BJU Int (2013) 112:909–16. doi: 10.1111/bju.12246
18. Lee J, Kim SH, Kang BJ. Prognostic Factors of Disease Recurrence in Breast Cancer Using Quantitative and Qualitative Magnetic Resonance Imaging (MRI) Parameters. Sci Rep (2020) 10. doi: 10.1038/s41598-020-64564-6
19. Sosa MS, Bragado P, Debnath J, Aguirre-Ghiso JA. Regulation of tumor cell dormancy by tissue microenvironments and autophagy. Adv Exp Med Biol (2013) 734:73–89. doi: 10.1007/978-1-4614-1445-2_5
20. Arias E, Cuervo AM. Pros and Cons of Chaperone-Mediated Autophagy in Cancer Biology. Trends Endocrinol Metab (2020) 31:53–66. doi: 10.1016/j.tem.2019.09.007
21. Jung CH, Ro SH, Cao J, Otto NM, Kim DH. MTOR regulation of autophagy. FEBS Lett (2010) 584:1287–95. doi: 10.1016/j.febslet.2010.01.017
22. Zoncu R, Efeyan A, Sabatini DM. MTOR: From growth signal integration to cancer, diabetes and ageing. Nat Rev Mol Cell Biol (2011). doi: 10.1038/nrm3025
23. Egan DF, Shackelford DB, Mihaylova MM, Gelino S, Kohnz RA, Mair W, et al. Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science (80- ) (2011) 331:456–61. doi: 10.1126/science.1196371
24. Kim J, Kundu M, Viollet B, Guan KL. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol (2011) 13:132–41. doi: 10.1038/ncb2152
25. Tripathi DN, Chowdhury R, Trudel LJ, Tee AR, Slack RS, Walker CL, et al. Reactive nitrogen species regulate autophagy through ATM-AMPK-TSC2-mediated suppression of mTORC1. Proc Natl Acad Sci USA (2013) 110. doi: 10.1073/pnas.1307736110
26. Bar-Peled L, Sabatini DM. Regulation of mTORC1 by amino acids. Trends Cell Biol (2014) 24:400–6. doi: 10.1016/j.tcb.2014.03.003
27. Dikic I, Elazar Z. Mechanism and medical implications of mammalian autophagy. Nat Rev Mol Cell Biol (2018) 19:349–64. doi: 10.1038/s41580-018-0003-4
28. Ozturk DG, Kocak M, Akcay A, Kinoglu K, Kara E, Buyuk Y, et al. MITF-MIR211 axis is a novel autophagy amplifier system during cellular stress. Autophagy (2019) 15:375–90. doi: 10.1080/15548627.2018.1531197
29. Hosokawa N, Hara T, Kaizuka T, Kishi C, Takamura A, Miura Y, et al. Nutrient-dependent mTORCl association with the ULK1-Atg13-FIP200 complex required for autophagy. Mol Biol Cell (2009) 20:1981–91. doi: 10.1091/mbc.E08-12-1248
30. Jung CH, Jun CB, Ro SH, Kim YM, Otto NM, Cao J, et al. ULK-Atg13-FIP200 complexes mediate mTOR signaling to the autophagy machinery. Mol Biol Cell (2009) 20:1992–2003. doi: 10.1091/mbc.E08-12-1249
31. Di Bartolomeo S, Corazzari M, Nazio F, Oliverio S, Lisi G, Antonioli M, et al. The dynamic interaction of AMBRA1 with the dynein motor complex regulates mammalian autophagy. J Cell Biol (2010) 191:155–68. doi: 10.1083/jcb.201002100
32. Karanasios E, Stapleton E, Manifava M, Kaizuka T, Mizushima N, Walker SA, et al. Dynamic association of the ULK1 complex with omegasomes during autophagy induction. J Cell Sci (2013) 126:5224–38. doi: 10.1242/jcs.132415
33. Hamasaki M, Furuta N, Matsuda A, Nezu A, Yamamoto A, Fujita N, et al. Autophagosomes form at ER-mitochondria contact sites. Nature (2013) 495:389–93. doi: 10.1038/nature11910
34. Polson HEJ, De Lartigue J, Rigden DJ, Reedijk M, Urbé S, Clague MJ, et al. Mammalian Atg18 (WIPI2) localizes to omegasome-anchored phagophores and positively regulates LC3 lipidation. Autophagy (2010) 6:506–22. doi: 10.4161/auto.6.4.11863
35. Dooley HC, Razi M, Polson HEJ, Girardin SE, Wilson MI, Tooze SA. WIPI2 Links LC3 Conjugation with PI3P, Autophagosome Formation, and Pathogen Clearance by Recruiting Atg12-5-16L1. Mol Cell (2014) 55:238–52. doi: 10.1016/j.molcel.2014.05.021
36. Axe EL, Walker SA, Manifava M, Chandra P, Roderick HL, Habermann A, et al. Autophagosome formation from membrane compartments enriched in phosphatidylinositol 3-phosphate and dynamically connected to the endoplasmic reticulum. J Cell Biol (2008) 182:685–701. doi: 10.1083/jcb.200803137
37. Rawet Slobodkin M, Elazar Z. The Atg8 family: Multifunctional ubiquitin-like key regulators of autophagy. Essays Biochem (2013) 55:51–64. doi: 10.1042/BSE0550051
38. Noda NN, Ohsumi Y, Inagaki F. Atg8-family interacting motif crucial for selective autophagy. FEBS Lett (2010) 584:1379–85. doi: 10.1016/j.febslet.2010.01.018
39. Ichimura Y, Kumanomidou T, Sou YS, Mizushima T, Ezaki J, Ueno T, et al. Structural basis for sorting mechanism of p62 in selective autophagy. J Biol Chem (2008) 283:22847–57. doi: 10.1074/jbc.M802182200
40. Yoshii SR, Mizushima N. Monitoring and measuring autophagy. Int J Mol Sci (2017) 18. doi: 10.3390/ijms18091865
41. Mizushima N, Yoshimori T, Levine B. Methods in Mammalian Autophagy Research. Cell (2010) 140:313–26. doi: 10.1016/j.cell.2010.01.028
42. Liu R, Zhi X, Zhong Q. ATG14 controls SNARE-mediated autophagosome fusion with a lysosome. Autophagy (2015) 11:847–9. doi: 10.1080/15548627.2015.1037549
43. Peker N, Gozuacik D. Autophagy as a Cellular Stress Response Mechanism in the Nervous System. J Mol Biol (2020) 432:2560–88. doi: 10.1016/j.jmb.2020.01.017
44. Bayraktar O, Oral O, Kocaturk NM, Akkoc Y, Eberhart K, Kosar A, et al. IBMPFD disease-causing mutant VCP/p97 proteins are targets of autophagic-lysosomal degradation. PloS One (2016) 11. doi: 10.1371/journal.pone.0164864
45. Gozuacik D, Akkoc Y, Gulfem Ozturk D, Kocak M. Autophagy-Regulating microRNAs and Cancer. Front Oncol (2017) 7. doi: 10.3389/fonc.2017.00065
46. Karakaş HE, Gözüaçik D. Autophagy and cancer. Turkish J Biol (2014) 38:720–39. doi: 10.3906/biy-1408-16
47. Mathew R, Karp CM, Beaudoin B, Vuong N, Chen G, Chen HY, et al. Autophagy Suppresses Tumorigenesis through Elimination of p62. Cell (2009) 137:1062–75. doi: 10.1016/j.cell.2009.03.048
49. Fernandez Larrosa PN, Alvarado CV, Rubio MF, Ruiz Grecco M, Micenmacher S, Martinez-Noel GA, et al. Nuclear receptor coactivator RAC3 inhibits autophagy. Cancer Sci (2012) 103:2064–71. doi: 10.1111/cas.12019
50. Kocaturk NM, Akkoc Y, Kig C, Bayraktar O, Gozuacik D, Kutlu O. Autophagy as a molecular target for cancer treatment. Eur J Pharm Sci (2019) 134:116–37. doi: 10.1016/j.ejps.2019.04.011
51. Unal O, Akkoc Y, Kocak M, Nalbat E, Dogan-Ekici AI, Yagci Acar H, et al. Treatment of breast cancer with autophagy inhibitory microRNAs carried by AGO2-conjugated nanoparticles. J Nanobiotechnol (2020) 18. doi: 10.1186/s12951-020-00615-4
52. Mathiassen SG, De Zio D, Cecconi F. Autophagy and the cell cycle: A complex landscape. Front Oncol (2017) 7. doi: 10.3389/fonc.2017.00051
53. Vlahakis A, Debnath J. The Interconnections between Autophagy and Integrin-Mediated Cell Adhesion. J Mol Biol (2017) 429:515–30. doi: 10.1016/j.jmb.2016.11.027
54. Gerada C, Ryan KM. Autophagy, the innate immune response and cancer. Mol Oncol (2020) 14:1913–29. doi: 10.1002/1878-0261.12774
55. Gozuacik D, Kimchi A. Autophagy as a cell death and tumor suppressor mechanism. Oncogene (2004) 23:2891–906. doi: 10.1038/sj.onc.1207521
56. Morselli E, Galluzzi L, Kepp O, Vicencio JM, Criollo A, Maiuri MC, et al. Anti- and pro-tumor functions of autophagy. Biochim Biophys Acta - Mol Cell Res (2009) 1793:1524–32. doi: 10.1016/j.bbamcr.2009.01.006
57. Hadfield G. The dormant cancer cell. Obstet Gynecol Surv (1955) 10:916–9. doi: 10.1097/00006254-195512000-00045
58. Sosa MS, Bragado P, Aguirre-Ghiso JA. Mechanisms of disseminated cancer cell dormancy: An awakening field. Nat Rev Cancer (2014) 14:611–22. doi: 10.1038/nrc3793
59. Yeh AC, Ramaswamy S. Mechanisms of cancer cell dormancy-another hallmark of cancer? Cancer Res (2015) 75:5014–22. doi: 10.1158/0008-5472.CAN-15-1370
60. Spiliotaki M, Mavroudis D, Kapranou K, Markomanolaki H, Kallergi G, Koinis F, et al. Evaluation of proliferation and apoptosis markers in circulating tumor cells of women with early breast cancer who are candidates for tumor dormancy. Breast Cancer Res (2014) 16. doi: 10.1186/s13058-014-0485-8
61. Marlow R, Honeth G, Lombardi S, Cariati M, Hessey S, Pipili A, et al. A novel model of dormancy for bone metastatic breast cancer cells. Cancer Res (2013) 73:6886–99. doi: 10.1158/0008-5472.CAN-13-0991
62. Darzynkiewicz Z, Juan G, Srour EF. Differential staining of DNA and RNA. Curr Protoc Cytom (2004). doi: 10.1002/0471142956.cy0703s30
63. Zou P, Yoshihara H, Hosokawa K, Tai I, Shinmyozu K, Tsukahara F, et al. P57 Kip2 and p27 Kip1 cooperate to maintain hematopoietic stem cell quiescence through interactions with Hsc70. Cell Stem Cell (2011) 9:247–61. doi: 10.1016/j.stem.2011.07.003
64. Lwin T, Hazlehurst LA, Dessureault S, Lai R, Bai W, Sotomayor E, et al. Cell adhesion induces p27Kip1-associated cell-cycle arrest through down-regulation of the SCFSkp2 ubiquitin ligase pathway in mantle-cell and other non-Hodgkin B-cell lymphomas. Blood (2007) 110:1631–8. doi: 10.1182/blood-2006-11-060350
65. Sadasivam S, DeCaprio JA. The DREAM complex: Master coordinator of cell cycle-dependent gene expression. Nat Rev Cancer (2013) 13:585–95. doi: 10.1038/nrc3556
66. Schmit F, Korenjak M, Mannefeld M, Schmitt K, Franke C, Von Eyss B, et al. LINC, a human complex that is related to pRB-containing complexes in invertebrates regulates the expression of G2/M genes. Cell Cycle (2007) 6:1903–13. doi: 10.4161/cc.6.15.4512
67. Litovchick L, Florens LA, Swanson SK, Washburn MP, Decaprio JA. DYRK1A protein kinase promotes quiescence and senescence through DREAM complex assembly. Genes Dev (2011) 25:801–13. doi: 10.1101/gad.2034211
68. Besson A, Gurian-West M, Chen X, Kelly-Spratt KS, Kemp CJ, Roberts JM. A pathway in quiescent cells that controls p27Kip1 stability, subcellular localization, and tumor suppression. Genes Dev (2006) 20:47–64. doi: 10.1101/gad.1384406
69. Deng X, Ewton DZ, Friedman E. Mirk dyrk1B maintains the viability of quiescent pancreatic cancer cells by reducing levels of reactive oxygen species. Cancer Res (2009) 69:3317–24. doi: 10.1158/0008-5472.CAN-08-2903
70. Aguirre-Ghiso JA, Estrada Y, Liu D, Ossowski L. ERKMAPK activity as a determinant of tumor growth and dormancy; regulation by p38SAPK. Cancer Res (2003) 63:1684–95. doi: 10.1016/j.urolonc.2003.12.012
71. Khaled AR, Bulavin DV, Kittipatarin C, Li WQ, Alvarez M, Kim K, et al. Cytokine-driven cell cycling is mediated through Cdc25A. J Cell Biol (2005) 169:755–63. doi: 10.1083/jcb.200409099
72. Adam AP, George A, Schewe D, Bragado P, Iglesias BV, Ranganathan AC, et al. Computational identification of a p38 SAPK-regulated transcription factor network required for tumor cell quiescence. Cancer Res (2009) 69:5664–72. doi: 10.1158/0008-5472.CAN-08-3820
73. Bragado P, Estrada Y, Parikh F, Krause S, Capobianco C, Farina HG, et al. TGF-β2 dictates disseminated tumour cell fate in target organs through TGF-β-RIII and p38α/β signalling. Nat Cell Biol (2013) 15:1351–61. doi: 10.1038/ncb2861
74. Yu-Lee LY, Yu G, Lee YC, Lin SC, Pan J, Pan T, et al. Osteoblast-secreted factors mediate dormancy of metastatic prostate cancer in the bone via activation of the TGFbRIII–p38MAPK–pS249/ T252RB pathway. Cancer Res (2018) 78:2911–24. doi: 10.1158/0008-5472.CAN-17-1051
75. Yumoto K, Eber MR, Wang J, Cackowski FC, Decker AM, Lee E, et al. Axl is required for TGF-β2-induced dormancy of prostate cancer cells in the bone marrow. Sci Rep (2016) 6. doi: 10.1038/srep36520
76. Aguirre-Ghiso JA, Liu D, Mignatti A, Kovalski K, Ossowski L. Urokinase receptor and fibronectin regulate the ERKMAPK to p38MAPK activity ratios that determine carcinoma cell proliferation or dormancy in vivo. Mol Biol Cell (2001) 12:863–79. doi: 10.1091/mbc.12.4.863
77. Litchfield LM, Riggs KA, Hockenberry AM, Oliver LD, Barnhart KG, Cai J, et al. Identification and characterization of nucleolin as a COUP-TFII coactivator of retinoic acid receptor β transcription in breast cancer cells. PloS One (2012) 7. doi: 10.1371/journal.pone.0038278
78. Aguirre Ghiso JA. Inhibition of FAK signaling activated by urokinase receptor induces dormancy in human carcinoma cells in vivo. Oncogene (2002) 21:2513–24. doi: 10.1038/sj.onc.1205342
79. Gao XL, Zheng M, Wang HF, Dai LL, Yu XH, Yang X, et al. NR2F1 contributes to cancer cell dormancy, invasion and metastasis of salivary adenoid cystic carcinoma by activating CXCL12/CXCR4 pathway. BMC Cancer (2019) 19. doi: 10.1186/s12885-019-5925-5
80. Zhang XHF, Wang Q, Gerald W, Hudis CA, Norton L, Smid M, et al. Latent Bone Metastasis in Breast Cancer Tied to Src-Dependent Survival Signals. Cancer Cell (2009) 16:67–78. doi: 10.1016/j.ccr.2009.05.017
81. Kobayashi A, Okuda H, Xing F, Pandey PR, Watabe M, Hirota S, et al. Bone morphogenetic protein 7 in dormancy and metastasis of prostate cancer stem-like cells in bone. J Exp Med (2011) 208:2641–55. doi: 10.1084/jem.20110840
82. Sharma S, Xing F, Liu Y, Wu K, Said N, Pochampally R, et al. Secreted protein acidic and rich in cysteine (sparc) mediates metastatic dormancy of prostate cancer in bone. J Biol Chem (2016) 291:19351–63. doi: 10.1074/jbc.M116.737379
83. Holtzhausen A, Golzio C, How T, Lee YH, Schiemann WP, Katsanis N, et al. Novel bone morphogenetic protein signaling through Smad2 and Smad3 to regulate cancer progression and development. FASEB J (2014) 28:1248–67. doi: 10.1096/fj.13-239178
84. Gao H, Chakraborty G, Lee-Lim AP, Mo Q, Decker M, Vonica A, et al. The BMP inhibitor Coco reactivates breast cancer cells at lung metastatic sites. Cell (2012) 150:764–79. doi: 10.1016/j.cell.2012.06.035
85. Kim RS, Avivar-Valderas A, Estrada Y, Bragado P, Sosa MS, Aguirre-Ghiso JA, et al. Dormancy signatures and metastasis in estrogen receptor positive and negative breast cancer. PloS One (2012) 7. doi: 10.1371/journal.pone.0035569
86. McGrath J, Panzica L, Ransom R, Withers HG, Gelman IH. Identification of genes regulating breast cancer dormancy in 3D bone endosteal niche cultures. Mol Cancer Res (2019) 17:860–9. doi: 10.1158/1541-7786.MCR-18-0956
87. Sosa MS, Parikh F, Maia AG, Estrada Y, Bosch A, Bragado P, et al. NR2F1 controls tumour cell dormancy via SOX9- and RARβ-driven quiescence programmes. Nat Commun (2015) 7. doi: 10.1038/ncomms7170
88. Shiozawa Y, Pedersen EA, Patel LR, Ziegler AM, Havens AM, Jung Y, et al. GAS6/AXL axis regulates prostate cancer invasion, proliferation, and survival in the bone marrow niche. Neoplasia (2010) 12:116–27. doi: 10.1593/neo.91384
89. Taichman RS, Patel LR, Bedenis R, Wang J, Weidner S, Schumann T, et al. GAS6 Receptor Status Is Associated with Dormancy and Bone Metastatic Tumor Formation. PloS One (2013) 8. doi: 10.1371/journal.pone.0061873
90. Shiozawa Y, Pedersen EA, Taichman RS. GAS6/Mer axis regulates the homing and survival of the E2A/PBX1-positive B-cell precursor acute lymphoblastic leukemia in the bone marrow niche. Exp Hematol (2010)38:132–40. doi: 10.1016/j.exphem.2009.11.002
91. Khoo WH, Ledergor G, Weiner A, Roden DL, Terry RL, McDonald MM, et al. A niche-dependent myeloid transcriptome signature defines dormant myeloma cells. Blood (2019) 134:30–43. doi: 10.1182/blood.2018880930
92. Harper KL, Sosa MS, Entenberg D, Hosseini H, Cheung JF, Nobre R, et al. Mechanism of early dissemination and metastasis in Her2+ mammary cancer. Nature (2016) 540:588–92. doi: 10.1038/nature20609
93. Buczacki SJA, Popova S, Biggs E, Koukorava C, Buzzelli J, Vermeulen L, et al. Itraconazole targets cell cycle heterogeneity in colorectal cancer. J Exp Med (2018) 215:1891–912. doi: 10.1084/jem.20171385
94. Ren D, Dai Y, Yang Q, Zhang X, Guo W, Ye L, et al. Wnt5a induces and maintains prostate cancer cells dormancy in bone. J Exp Med (2019) 216:428–49. doi: 10.1084/jem.20180661
95. Malladi S, MacAlinao DG, Jin X, He L, Basnet H, Zou Y, et al. Metastatic Latency and Immune Evasion through Autocrine Inhibition of WNT. Cell (2016) 165:45–60. doi: 10.1016/j.cell.2016.02.025
96. Boyerinas B, Zafrir M, Yesilkanal AE, Price TT, Hyjek EM, Sipkins DA. Adhesion to osteopontin in the bone marrow niche regulates lymphoblastic leukemia cell dormancy. Blood (2013) 121:4821–31. doi: 10.1182/blood-2012-12-475483
97. Yu N, Seedhouse C, Russell N, Pallis M. Quantitative assessment of the sensitivity of dormant AML cells to the BAD mimetics ABT-199 and ABT-737. Leuk Lymphoma (2018) 59:2447–53. doi: 10.1080/10428194.2018.1434884
98. Najmi S, Korah R, Chandra R, Abdellatif M, Wieder R. Flavopiridol blocks integrin-mediated survival in dormant breast cancer cells. Clin Cancer Res (2005) 11:2038–46. doi: 10.1158/1078-0432.CCR-04-1083
99. Bambang IF, Lu D, Li H, Chiu LL, Lau QC, Koay E, et al. Cytokeratin 19 regulates endoplasmic reticulum stress and inhibits ERp29 expression via p38 MAPK/XBP-1 signaling in breast cancer cells. Exp Cell Res (2009) 315:1964–74. doi: 10.1016/j.yexcr.2009.02.017
100. Amin R, Morita-Fujimura Y, Tawarayama H, Semba K, Chiba N, Fukumoto M, et al. δNp63α induces quiescence and downregulates the BRCA1 pathway in estrogen receptor-positive luminal breast cancer cell line MCF7 but not in other breast cancer cell lines. Mol Oncol (2016) 10:575–93. doi: 10.1016/j.molonc.2015.11.009
101. Sanchez Calle A, Yamamoto T, Kawamura Y, Hironaka-Mitsuhashi A, Ono M, Tsuda H, et al. Long non-coding NR2F1-AS1 is associated with tumor recurrence in estrogen receptor-positive breast cancers. Mol Oncol (2020) 14:2271–87. doi: 10.1002/1878-0261.12704
102. Wesley Overton K, Spencer SL, Noderer WL, Meyer T, Wang CL. Basal p21 controls population heterogeneity in cycling and quiescent cell cycle states. Proc Natl Acad Sci USA (2014) 111:E4386–93. doi: 10.1073/pnas.1409797111
103. Bliss SA, Sinha G, Sandiford OA, Williams LM, Engelberth DJ, Guiro K, et al. Mesenchymal stem cell-derived exosomes stimulate cycling quiescence and early breast cancer dormancy in bone marrow. Cancer Res (2016) 76:5832–44. doi: 10.1158/0008-5472.CAN-16-1092
104. El-Shennawy L, Dubrovskyi O, Kastrati I, Danes JM, Zhang Y, Whiteley HE, et al. Coactivation of estrogen receptor and IKKb induces a dormant metastatic phenotype in ER-Positive breast cancer. Cancer Res (2018) 78:974–84. doi: 10.1158/0008-5472.CAN-17-1686
105. Huang J, Woods P, Normolle D, Goff JP, Benos PV, Stehle CJ, et al. Downregulation of estrogen receptor and modulation of growth of breast cancer cell lines mediated by paracrine stromal cell signals. Breast Cancer Res Treat (2017) 161:229–43. doi: 10.1007/s10549-016-4052-0
106. Lu D, Chen S, Tan X, Li N, Liu C, Li Z, et al. Fra-1 promotes breast cancer chemosensitivity by driving cancer stem cells from dormancy. Cancer Res (2012) 72:3451–6. doi: 10.1158/0008-5472.CAN-11-2536
107. Weidenfeld K, Schif-Zuck S, Abu-Tayeh H, Kang K, Kessler O, Weissmann M, et al. Dormant tumor cells expressing LOXL2 acquire a stem-like phenotype mediating their transition to proliferative growth. Oncotarget (2016) 7:71362–77. doi: 10.18632/oncotarget.12109
108. Barkan D, Kleinman H, Simmons JL, Asmussen H, Kamaraju AK, Hoenorhoff MJ, et al. Inhibition of metastatic outgrowth from single dormant tumor cells by targeting the cytoskeleton. Cancer Res (2008) 68:6241–50. doi: 10.1158/0008-5472.CAN-07-6849
109. Touny LHE, Vieira A, Mendoza A, Khanna C, Hoenerhoff MJ, Green JE. Combined SFK/MEK inhibition prevents metastatic outgrowth of dormant tumor cells. J Clin Invest (2014) 124:156–68. doi: 10.1172/JCI70259
110. Chakraborty S, Jiang C, Gau D, Oddo M, Ding Z, Vollmer L, et al. Profilin-1 deficiency leads to SMAD3 upregulation and impaired 3D outgrowth of breast cancer cells. Br J Cancer (2018) 119:1106–17. doi: 10.1038/s41416-018-0284-6
111. Johnson RW, Sun Y, Ho PWM, Chan ASM, Johnson JA, Pavlos NJ, et al. Parathyroid hormone-related protein negatively regulates tumor cell dormancy genes in a PTHR1/cyclic AMP-independent manner. Front Endocrinol (Lausanne) (2018) 9:241. doi: 10.3389/fendo.2018.00241
112. Romero-Moreno R, Curtis KJ, Coughlin TR, Cristina Miranda-Vergara M, Dutta S, Natarajan A, et al. The CXCL5/CXCR2 axis is sufficient to promote breast cancer colonization during bone metastasis. Nat Commun (2019) 10. doi: 10.1038/s41467-019-12108-6
113. Chatterjee M, Van Golen KL. Farnesyl transferase inhibitor treatment of breast cancer cells leads to altered RhoA and RhoC GTPase activity and induces a dormant phenotype. Int J Cancer (2011) 129:61–9. doi: 10.1002/ijc.25655
114. De Cock JM, Shibue T, Dongre A, Keckesova Z, Reinhardt F, Weinberg RA. Inflammation triggers Zeb1-dependent escape from tumor latency. Cancer Res (2016) 76:6778–84. doi: 10.1158/0008-5472.CAN-16-0608
115. Francescangeli F, Contavalli P, De Angelis ML, Careccia S, Signore M, Haas TL, et al. A pre-existing population of ZEB2+ quiescent cells with stemness and mesenchymal features dictate chemoresistance in colorectal cancer. J Exp Clin Cancer Res (2020) 39:1–17. doi: 10.1186/s13046-019-1505-4
116. Heckmann D, Maier P, Laufs S, Wenz F, Zeller WJ, Fruehauf S, et al. CXCR4 expression and treatment with SDF-1α or plerixafor modulate proliferation and chemosensitivity of colon cancer cells. Transl Oncol (2013) 6:124–32. doi: 10.1593/tlo.12268
117. Zhou Y, Su Y, Zhu H, Wang X, Li X, Dai C, et al. Interleukin-23 receptor signaling mediates cancer dormancy and radioresistance in human esophageal squamous carcinoma cells via the Wnt/Notch pathway. J Mol Med (2019) 97:177–88. doi: 10.1007/s00109-018-1724-8
118. Adamski V, Hattermann K, Kubelt C, Cohrs G, Lucius R, Synowitz M, et al. Entry and exit of chemotherapeutically-promoted cellular dormancy in glioblastoma cells is differentially affected by the chemokines CXCL12, CXCL16, and CX3CL1. Oncogene (2020) 39:4421–35. doi: 10.1038/s41388-020-1302-8
119. Hofstetter CP, Burkhardt JK, Shin BJ, Gürsel DB, Mubita L, Gorrepati R, et al. Protein phosphatase 2A mediates dormancy of glioblastoma multiforme-derived tumor stem-like cells during hypoxia. PloS One (2012) 7. doi: 10.1371/journal.pone.0030059
120. Yang LY, Shan YM, Zhang Y, Zhou EH, Chen XP, Zhang H. Aurora kinase A induces chemotherapy resistance through revival of dormant cells in laryngeal squamous cell carcinoma. Head Neck (2019) 41:2239–48. doi: 10.1002/hed.25689
121. Boix L, López-Oliva JM, Rhodes AC, Bruix J. Restoring miR122 in human stem-like hepatocarcinoma cells, prompts tumor dormancy through Smad-independent TGF-ß pathway. Oncotarget (2016) 7:71309–29. doi: 10.18632/oncotarget.11885
122. Dai Y, Wang L, Tang J, Cao P, Luo Z, Sun J, et al. Activation of anaphase-promoting complex by p53 induces a state of dormancy in cancer cells against chemotherapeutic stress. Oncotarget (2016) 7:25478–92. doi: 10.18632/oncotarget.8172
123. Liu Y, Lv J, Liang X, Yin X, Zhang L, Chen D, et al. Fibrin stiffness mediates dormancy of tumor-repopulating cells via a Cdc42-driven Tet2 epigenetic program. Cancer Res (2018) 78:3926–37. doi: 10.1158/0008-5472.CAN-17-3719
124. Flores-Guzmán F, Utikal J, Umansky V. Dormant tumor cells interact with memory CD8+ T cells in RET transgenic mouse melanoma model. Cancer Lett (2020) 474:74–81. doi: 10.1016/j.canlet.2020.01.016
125. Shimizu T, Sugihara E, Yamaguchi-Iwai S, Tamaki S, Koyama Y, Kamel W, et al. IGF2 preserves osteosarcoma cell survival by creating an autophagic state of dormancy that protects cells against chemotherapeutic stress. Cancer Res (2014) 74:6531–41. doi: 10.1158/0008-5472.CAN-14-0914
126. MacDonald J, Ramos-Valdes Y, Perampalam P, Litovchick L, DiMattia GE, Dick FA. A systematic analysis of negative growth control implicates the DREAM complex in cancer cell dormancy. Mol Cancer Res (2017) 15:371–81. doi: 10.1158/1541-7786.MCR-16-0323-T
127. Correa RJM, Peart T, Valdes YR, Dimattia GE, Shepherd TG. Modulation of AKT activity is associated with reversible dormancy in ascites-derived epithelial ovarian cancer spheroids. Carcinogenesis (2012) 33:49–58. doi: 10.1093/carcin/bgr241
128. Kinoshita Y, Kalir T, Rahaman J, Dottino P, Kohtz DS. Alterations in nuclear pore architecture allow cancer cell entry into or exit from drug-resistant dormancy. Am J Pathol (2012) 180:375–89. doi: 10.1016/j.ajpath.2011.09.024
129. Cheng J, Cashman JR. PAWI-2 overcomes tumor stemness and drug resistance via cell cycle arrest in integrin β3-KRAS-dependent pancreatic cancer stem cells. Sci Rep (2020) 10:1–11. doi: 10.1038/s41598-020-65804-5
130. Endo H, Okuyama H, Ohue M, Inoue M. Dormancy of cancer cells with suppression of AKT activity contributes to survival in chronic hypoxia. PloS One (2014) 9:1–12. doi: 10.1371/journal.pone.0098858
131. Owen KL, Gearing LJ, Zanker DJ, Brockwell NK, Khoo WH, Roden DL, et al. Prostate cancer cell-intrinsic interferon signaling regulates dormancy and metastatic outgrowth in bone. EMBO Rep (2020) 21. doi: 10.15252/embr.202050162
132. Ruppender N, Larson S, Lakely B, Kollath L, Brown L, Coleman I, et al. Cellular adhesion promotes prostate cancer cells escape from dormancy. PloS One (2015) 10. doi: 10.1371/journal.pone.0130565
133. Indraccolo S, Minuzzo S, Masiero M, Pusceddu I, Persano L, Moserle L, et al. Cross-talk between tumor and endothelial cells involving the Notch3-DII4 interaction marks escape from tumor dormancy. Cancer Res (2009) 69:1314–23. doi: 10.1158/0008-5472.CAN-08-2791
134. Saudemont A, Jouy N, Hetuin D, Quesnel B. NK cells that are activated by CXCL10 can kill dormant tumor cells that resist CTL-mediated lysis and can express B7-H1 that stimulates T cells. Blood (2005) 105:2428–35. doi: 10.1182/blood-2004-09-3458
135. Hurst RE, Hauser PJ, Kyker KD, Heinlen JE, Hodde JP, Hiles MC, et al. Suppression and Activation of the Malignant Phenotype by Extracellular Matrix in Xenograft Models of Bladder Cancer: A Model for Tumor Cell “Dormancy.” PloS One (2013) 8. doi: 10.1371/journal.pone.0064181
136. Arlt MJE, Banke IJ, Bertz J, Ram Kumar RM, Muff R, Born W, et al. Reduced latency in the metastatic niche contributes to the more aggressive phenotype of LM8 compared to dunn osteosarcoma cells. Sarcoma (2013) 2013. doi: 10.1155/2013/404962
137. Watson KL, Jones RA, Bruce A, Moorehead RA. The miR-200b/200a/429 cluster prevents metastasis and induces dormancy in a murine claudin-low mammary tumor cell line. Exp Cell Res (2018) 369:17–26. doi: 10.1016/j.yexcr.2018.04.024
138. Ghajar CM, Peinado H, Mori H, Matei IR, Evason KJ, Brazier H, et al. The perivascular niche regulates breast tumour dormancy. Nat Cell Biol (2013) 15:807–17. doi: 10.1038/ncb2767
139. Gawrzak S, Rinaldi L, Gregorio S, Arenas EJ, Salvador F, Urosevic J, et al. MSK1 regulates luminal cell differentiation and metastatic dormancy in ER + breast cancer. Nat Cell Biol (2018) 20:211–21. doi: 10.1038/s41556-017-0021-z
140. Marshall JCA, Collins JW, Nakayama J, Horak CE, Liewehr DJ, Steinberg SM, et al. Effect of inhibition of the lysophosphatidic acid receptor 1 on metastasis and metastatic dormancy in breast cancer. J Natl Cancer Inst (2012) 104:1306–19. doi: 10.1093/jnci/djs319
141. Gattelli A, Cirio MC, Quaglino A, Schere-Levy C, Martinez N, Binaghi M, et al. Progression of pregnancy-dependent mouse mammary tumors after long dormancy periods. Involvement of Wnt pathway activation. Cancer Res (2004) 64:5193–9. doi: 10.1158/0008-5472.CAN-03-3992
142. Capulli M, Hristova D, Valbret Z, Carys K, Arjan R, Maurizi A, et al. Notch2 pathway mediates breast cancer cellular dormancy and mobilisation in bone and contributes to haematopoietic stem cell mimicry. Br J Cancer (2019) 121:157–71. doi: 10.1038/s41416-019-0501-y
143. Drescher F, Juárez P, Arellano DL, Serafín-Higuera N, Olvera-Rodriguez F, Jiménez S, et al. TIE2 induces breast cancer cell dormancy and inhibits the development of osteolytic bone metastases. Cancers (Basel) (2020) 12. doi: 10.3390/cancers12040868
144. Nobutani K, Shimono Y, Mizutani K, Ueda Y, Suzuki T, Kitayama M, et al. Downregulation of CXCR4 in metastasized breast cancer cells and implication in their dormancy. PloS One (2015) 10. doi: 10.1371/journal.pone.0130032
145. Eyob H, Ekiz HA, DeRose YS, Waltz SE, Williams MA, Welm AL. Inhibition of Ron kinase blocks conversion of micrometastases to overt metastases by boosting antitumor immunity. Cancer Discovery (2013) 3:751–60. doi: 10.1158/2159-8290.CD-12-0480
146. Shimizu H, Takeishi S, Nakatsumi H, Nakayama KI. Prevention of cancer dormancy by Fbxw7 ablation eradicates disseminated tumor cells. JCI Insight (2019) 4. doi: 10.1172/jci.insight.125138
147. Straume O, Shimamura T, Lampa MJG, Carretero J, AM Ø, Jia D, et al. Suppression of heat shock protein 27 induces long-term dormancy in human breast cancer. Proc Natl Acad Sci USA (2012) 109:8699–704. doi: 10.1073/pnas.1017909109
148. La Belle Flynn A, Calhoun BC, Sharma A, Chang JC, Almasan A, Schiemann WP. Autophagy inhibition elicits emergence from metastatic dormancy by inducing and stabilizing Pfkfb3 expression. Nat Commun (2019) 10. doi: 10.1038/s41467-019-11640-9
149. Holdman XB, Welte T, Rajapakshe K, Pond A, Coarfa C, Mo Q, et al. Upregulation of EGFR signaling is correlated with tumor stroma remodeling and tumor recurrence in FGFR1-driven breast cancer. Breast Cancer Res (2015) 17:1–17. doi: 10.1186/s13058-015-0649-1
150. Abravanel DL, Belka GK, Pan TC, Pant DK, Collins MA, Sterner CJ, et al. Notch promotes recurrence of dormant tumor cells following HER2/neu-targeted therapy. J Clin Invest (2015) 125:2484–96. doi: 10.1172/JCI74883
151. Vallabhaneni KC, Penfornis P, Xing F, Hassler Y, Adams KV, Mo YY, et al. Stromal cell extracellular Vesicular cargo mediated regulation of Breast Cancer cell Metastasis Via Ubiquitin conjugating Enzyme E2 N pathway. Oncotarget (2017) 8:109861–76. doi: 10.18632/oncotarget.22371
152. Bartosh TJ, Ullah M, Zeitouni S, Beaver J, Prockop DJ. Cancer cells enter dormancy after cannibalizing mesenchymal stem/stromal cells (MSCs). Proc Natl Acad Sci USA (2016) 113:E6447–56. doi: 10.1073/pnas.1612290113
153. Lu X, Mu E, Wei Y, Riethdorf S, Yang Q, Yuan M, et al. VCAM-1 Promotes Osteolytic Expansion of Indolent Bone Micrometastasis of Breast Cancer by Engaging α4β1-Positive Osteoclast Progenitors. Cancer Cell (2011) 20:701–14. doi: 10.1016/j.ccr.2011.11.002
154. Han HH, Kim BG, Lee JH, Kang S, Kim JE, Cho NH. Angiopoietin-2 promotes ER+ breast cancer cell survival in bone marrow niche. Endocr Relat Cancer (2016) 23:609–23. doi: 10.1530/ERC-16-0086
155. Lan Q, Peyvandi S, Duffey N, Huang YT, Barras D, Held W, et al. Type I interferon/IRF7 axis instigates chemotherapy-induced immunological dormancy in breast cancer. Oncogene (2019) 38:2814–29. doi: 10.1038/s41388-018-0624-2
156. Almog N, Ma L, Raychowdhury R, Schwager C, Erber R, Short S, et al. Transcriptional switch of dormant tumors to fast-growing angiogenic phenotype. Cancer Res (2009) 69:836–44. doi: 10.1158/0008-5472.CAN-08-2590
157. Li Y, Hu H, Wang Y, Fan Y, Yang Y, Guo B, et al. CUL4B contributes to cancer stemness by repressing tumor suppressor miR34a in colorectal cancer. Oncogenesis (2020) 9. doi: 10.1038/s41389-020-0206-3
158. Tiram G, Ferber S, Ofek P, Eldar-Boock A, Ben-Shushan D, Yeini E, et al. Reverting the molecular fingerprint of tumor dormancy as a therapeutic strategy for glioblastoma. FASEB J (2018) 32:5835–50. doi: 10.1096/fj.201701568R
159. Magnus N, Garnier D, Meehan B, McGraw S, Lee TH, Caron M, et al. Tissue factor expression provokes escape from tumor dormancy and leads to genomic alterations. Proc Natl Acad Sci USA (2014) 111:3544–9. doi: 10.1073/pnas.1314118111
160. Almog N, Briggs C, Beheshti A, Ma L, Wilkie KP, Rietman E, et al. Transcriptional changes induced by the tumor dormancy-associated microRNA-190. Transcription (2013) 4:177–91. doi: 10.4161/trns.25558
161. Jiang J, Zheng M, Zhang M, Yang X, Li L, Wang SS, et al. PRRX1 Regulates Cellular Phenotype Plasticity and Dormancy of Head and Neck Squamous Cell Carcinoma Through miR-642b-3p. Neoplasia (United States) (2019) 21:216–29. doi: 10.1016/j.neo.2018.12.001
162. Aguirre Ghiso JA, Kovalski K, Ossowski L. Tumor dormancy induced by downregulation of urokinase receptor in human carcinoma involves integrin and MAPK signaling. J Cell Biol (1999) 147:89–103. doi: 10.1083/jcb.147.1.89
163. Schewe DM, Aguirre-Ghiso JA. ATF6α-Rheb-mTOR signaling promotes survival of dormant tumor cells in vivo. Proc Natl Acad Sci USA (2008) 105:10519–24. doi: 10.1073/pnas.0800939105
164. Yang LY, He CY, Chen XH, Su LP, Liu BY, Zhang H. Aurora kinase A revives dormant laryngeal squamous cell carcinoma cells via FAK/PI3K/Akt pathway activation. Oncotarget (2016) 7:48346–59. doi: 10.18632/oncotarget.10233
165. Shachaf CM, Kopelman AM, Arvanitis C, Karlsson Å, Beer S, Mandl S, et al. MYC inactivation uncovers pluripotent differentiation and tumour dormancy in hepatocellular cancer. Nature (2004) 431:1112–7. doi: 10.1038/nature03043
166. Kurppa KJ, Liu Y, To C, Zhang T, Fan M, Vajdi A, et al. Treatment-Induced Tumor Dormancy through YAP-Mediated Transcriptional Reprogramming of the Apoptotic Pathway. Cancer Cell (2020) 37:104–22.e12. doi: 10.1016/j.ccell.2019.12.006
167. Liang X, Gu J, Li TJ, Zhao L, Fu X, Zhang W, et al. PAX5 haploinsufficiency induce cancer cell dormancy in Raji cells. Exp Cell Res (2018) 367:30–6. doi: 10.1016/j.yexcr.2018.02.010
168. Nash KT, Phadke PA, Navenot JM, Hurst DR, Accavitti-Loper MA, Sztul E, et al. Requirement of KISS1 secretion for multiple organ metastasis suppression and maintenance of tumor dormancy. J Natl Cancer Inst (2007) 99:309–21. doi: 10.1093/jnci/djk053
169. Jia Q, Yang F, Huang W, Zhang Y, Bao B, Li K, et al. Low levels of sox2 are required for melanoma tumor-repopulating cell dormancy. Theranostics (2019) 9:424–35. doi: 10.7150/thno.29698
170. Liu Y, Liang X, Yin X, Lv J, Tang K, Ma J, et al. Blockade of IDO-kynurenine-AhR metabolic circuitry abrogates IFN-γ-induced immunologic dormancy of tumor-repopulating cells. Nat Commun (2017) 8. doi: 10.1038/ncomms15207
171. Ambs S, Dennis S, Fairman J, Wright M, Papkoff J. Inhibition of tumor growth correlates with the expression level of a human angiostatin transgene in transfected B16f10 melanoma cells. Cancer Res (1999) 59:5773–7.
172. Malafa MP, Fokum FD, Smith LK, Louis A. Inhibition of angiogenesis and promotion of melanoma dormancy by vitamin E succinate. Ann Surg Oncol (2002) 9:1023–32. doi: 10.1245/ASO.2002.03.580
173. Touil Y, Segard P, Ostyn P, Begard S, Aspord C, El MacHhour R, et al. Melanoma dormancy in a mouse model is linked to GILZ/FOXO3A-dependent quiescence of disseminated stem-like cells. Sci Rep (2016) 6. doi: 10.1038/srep30405
174. Panigrahy D, Edin ML, Lee CR, Huang S, Bielenberg DR, Butterfield CE, et al. Epoxyeicosanoids stimulate multiorgan metastasis and tumor dormancy escape in mice. J Clin Invest (2012) 122:178–91. doi: 10.1172/JCI58128
175. Dossi R, Frapolli R, Di Giandomenico S, Paracchini L, Bozzi F, Brich S, et al. Antiangiogenic activity of trabectedin in myxoid liposarcoma: Involvement of host TIMP-1 and TIMP-2 and tumor thrombospondin-1. Int J Cancer (2015) 136:721–9. doi: 10.1002/ijc.29023
176. Chen H, Ko JMY, Wong VCL, Hyytiainen M, Keski-Oja J, Chua D, et al. LTBP-2 confers pleiotropic suppression and promotes dormancy in a growth factor permissive microenvironment in nasopharyngeal carcinoma. Cancer Lett (2012) 325:89–98. doi: 10.1016/j.canlet.2012.06.005
177. Tiram G, Segal E, Krivitsky A, Shreberk-Hassidim R, Ferber S, Ofek P, et al. Identification of Dormancy-Associated MicroRNAs for the Design of Osteosarcoma-Targeted Dendritic Polyglycerol Nanopolyplexes. ACS Nano (2016) 10:2028–45. doi: 10.1021/acsnano.5b06189
178. Lyu T, Jia N, Wang J, Yan X, Yu Y, Lu Z, et al. Expression and epigenetic regulation of angiogenesis-related factors during dormancy and recurrent growth of ovarian carcinoma. Epigenetics (2013) 8:1330–46. doi: 10.4161/epi.26675
179. Mao W, Peters HL, Sutton MN, Orozco AF, Pang L, Yang H, et al. The role of vascular endothelial growth factor, interleukin 8, and insulinlike growth factor in sustaining autophagic DIRAS3-induced dormant ovarian cancer xenografts. Cancer (2019) 125:1267–80. doi: 10.1002/cncr.31935
180. Lu Z, Luo RZ, Lu Y, Zhang X, Yu Q, Khare S, et al. The tumor suppressor gene ARHI regulates autophagy and tumor dormancy in human ovarian cancer cells. J Clin Invest (2008) 118:3917–29. doi: 10.1172/JCI35512
181. Luo XL, Deng CC, Su XD, Wang F, Chen Z, Wu XP, et al. Loss of MED12 induces tumor dormancy in human epithelial ovarian cancer via downregulation of EGFR. Cancer Res (2018) 78:3532–43. doi: 10.1158/0008-5472.CAN-18-0134
182. Lee HH, Bellat V, Law B. Chemotherapy induces adaptive drug resistance and metastatic potentials via phenotypic CXCR4-expressing cell state transition in ovarian cancer. PloS One (2017) 12. doi: 10.1371/journal.pone.0171044
183. Rajbhandari N, Lin Wc, Wehde BL, Triplett AA, Wagner KU. Autocrine IGF1 Signaling Mediates Pancreatic Tumor Cell Dormancy in the Absence of Oncogenic Drivers. Cell Rep (2017) 18:2243–55. doi: 10.1016/j.celrep.2017.02.013
184. Lenk L, Pein M, Will O, Gomez B, Viol F, Hauser C, et al. The hepatic microenvironment essentially determines tumor cell dormancy and metastatic outgrowth of pancreatic ductal adenocarcinoma. Oncoimmunology (2018) 7:1–14. doi: 10.1080/2162402X.2017.1368603
185. Tang Y, Wang MT, Chen Y, Yang D, Che M, Honn KV, et al. Downregulation of vascular endothelial growth factor and induction of tumor dormancy by 15-lipoxygenase-2 in prostate cancer. Int J Cancer (2009) 124:1545–51. doi: 10.1002/ijc.24118
186. Cackowski F, Eber MR, Rhee J, Decker A, Yumoto K, Berry JE, et al. Mer Tyrosine Kinase Regulates Disseminated Prostate Cancer Cellular Dormancy Graphical Abstract HHS Public Access. J Cell Biochem (2017) 118:891–902. doi: 10.1002/jcb.25768
187. Komatsu M, Kurokawa H, Waguri S, Taguchi K, Kobayashi A, Ichimura Y, et al. The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat Cell Biol (2010) 12:213–23. doi: 10.1038/ncb2021
188. Axelrod HD, Valkenburg KC, Amend SR, Hicks JL, Parsana P, Torga G, et al. AXL is a putative tumor suppressor and dormancy regulator in prostate cancer. Mol Cancer Res (2019) 17:356–69. doi: 10.1158/1541-7786.MCR-18-0718
189. Kim JK, Jung Y, Wang J, Joseph J, Mishra A, Hill EE, et al. TBK1 regulates prostate cancer dormancy through mTOR inhibition. Neoplasia (United States) (2013) 15:1064–74. doi: 10.1593/neo.13402
190. O’Reilly MS, Holmgren L, Chen C, Folkman J. Angiostatin induces and sustains dormancy of human primary tumors in mice. Nat Med (1996) 2:689–92. doi: 10.1038/nm0696-689
191. Phan TG, Croucher PI. The dormant cancer cell life cycle. Nat Rev Cancer (2020) 20:398–411. doi: 10.1038/s41568-020-0263-0
192. Aguirre-Ghiso JA, Sosa MS. Emerging Topics on Disseminated Cancer Cell Dormancy and the Paradigm of Metastasis. Annu Rev Cancer Biol (2018) 2:377–93. doi: 10.1146/annurev-cancerbio-030617-050446
193. Mortensen M, Soilleux EJ, Djordjevic G, Tripp R, Lutteropp M, Sadighi-Akha E, et al. The autophagy protein Atg7 is essential for hematopoietic stem cell maintenance. J Exp Med (2011) 208:455–67. doi: 10.1084/jem.20101145
194. Lazova R, Camp RL, Klump V, Siddiqui SF, Amaravadi RK, Pawelek JM. Punctate LC3B expression is a common feature of solid tumors and associated with proliferation, metastasis, and poor outcome. Clin Cancer Res (2012) 18:370–9. doi: 10.1158/1078-0432.CCR-11-1282
195. Yang A, Rajeshkumar NV, Wang X, Yabuuchi S, Alexander BM, Chu GC, et al. Autophagy is critical for pancreatic tumor growth and progression in tumors with p53 alterations. Cancer Discovery (2014) 10:1683–4. doi: 10.1158/2159-8290.CD-14-0362
196. Cianfanelli V, Fuoco C, Lorente M, Salazar M, Quondamatteo F, Gherardini PF, et al. AMBRA1 links autophagy to cell proliferation and tumorigenesis by promoting c-Myc dephosphorylation and degradation. Nat Cell Biol (2015) 17:20–30. doi: 10.1038/ncb3072
197. Mowers EE, Sharifi MN, Macleod KF. Autophagy in cancer metastasis. Oncogene (2017) 36:1619–30. doi: 10.1038/onc.2016.333
198. Gupta A, Roy S, Lazar AJF, Wang WL, McAuliffe JC, Reynoso D, et al. Autophagy inhibition and antimalarials promote cell death in gastrointestinal stromal tumor (GIST). Proc Natl Acad Sci USA (2010) 107:14333–8. doi: 10.1073/pnas.1000248107
199. Vera-Ramirez L, Vodnala SK, Nini R, Hunter KW, Green JE. Autophagy promotes the survival of dormant breast cancer cells and metastatic tumour recurrence. Nat Commun (2018) 9. doi: 10.1038/s41467-018-04070-6
200. Yu Z, Zhou R, Zhao Y, Pan Y, Liang H, Zhang JS, et al. Blockage of SLC31A1-dependent copper absorption increases pancreatic cancer cell autophagy to resist cell death. Cell Prolif (2019) 52. doi: 10.1111/cpr.12568
201. Lu Z, Baquero MT, Yang H, Yang M, Reger AS, Kim C, et al. DIRAS3 regulates the autophagosome initiation complex in dormant ovarian cancer cells. Autophagy (2014) 10:1071–92. doi: 10.4161/auto.28577
202. Washington MN, Suh G, Orozco AF, Sutton MN, Yang H, Wang Y, et al. ARHI (DIRAS3)-mediated autophagy-associated cell death enhances chemosensitivity to cisplatin in ovarian cancer cell lines and xenografts. Cell Death Dis (2015) 6. doi: 10.1038/cddis.2015.208
203. Bao JJ, Le XF, Wang RY, Yuan J, Wang L, Atkinson EN, et al. Reexpression of the tumor suppressor gene ARHI induces apoptosis in ovarian and breast cancer cells through a caspase-independent calpain-dependent pathway. Cancer Res (2002) 62:7264–72.
204. Correa RJM, Valdes YR, Peart TM, Fazio EN, Bertrand M, McGee J, et al. Combination of AKT inhibition with autophagy blockade effectively reduces ascites-derived ovarian cancer cell viability. Carcinogenesis (2014) 35:1951–61. doi: 10.1093/carcin/bgu049
205. Peart T, Valdes YR, Correa RJM, Fazio E, Bertrand M, McGee J, et al. Intact LKB1 activity is required for survival of dormant ovarian cancer spheroids. Oncotarget (2015) 6:22424–38. doi: 10.18632/oncotarget.4211
206. Carcereri de Prati A, Butturini E, Rigo A, Oppici E, Rossin M, Boriero D, et al. Metastatic Breast Cancer Cells Enter Into Dormant State and Express Cancer Stem Cells Phenotype Under Chronic Hypoxia.s. J Cell Biochem (2017) 118:3237–48. doi: 10.1002/jcb.25972
207. Aqbi HF, Tyutyunyk-Massey L, Keim RC, Butler SE, Thekkudan T, Joshi S, et al. Autophagy-deficient breast cancer shows early tumor recurrence and escape from dormancy. Oncotarget (2018) 9:22128–37. doi: 10.18632/oncotarget.25197
208. Shinde A, Hardy SD, Kim D, Akhand SS, Jolly MK, Wang WH, et al. Spleen tyrosine kinase–mediated autophagy is required for epithelial–mesenchymal plasticity and metastasis in breast cancer. Cancer Res (2019) 79:1831–43. doi: 10.1158/0008-5472.CAN-18-2636
209. Wang L, Hoque A, Luo RZ, Yuan J, Lu Z, Nishimoto A, et al. Loss of the expression of the tumor suppressor gene ARHI is associated with progression of breast cancer. Clin Cancer Res (2003) 9:3660–6.
210. Lu Z, Luo RZ, Peng H, Rosen DG, Atkinson EN, Warneke C, et al. Transcriptional and posttranscriptional down-regulation of the imprinted tumor suppressor gene ARHI (DRAS3) in ovarian cancer. Clin Cancer Res (2006) 12:2404–13. doi: 10.1158/1078-0432.CCR-05-1036
211. Zou CF, Jia L, Jin H, Yao M, Zhao N, Huan J, et al. Re-expression of ARHI (DIRAS3) induces autophagy in breast cancer cells and enhances the inhibitory effect of paclitaxel. BMC Cancer (2011) 11. doi: 10.1186/1471-2407-11-22
212. Chaterjee M, van Golen KL. Breast Cancer Stem Cells Survive Periods of Farnesyl-Transferase Inhibitor-Induced Dormancy by Undergoing Autophagy. Bone Marrow Res (2011) 2011:1–7. doi: 10.1155/2011/362938
213. Ornelas A, McCullough CR, Lu Z, Zacharias NM, Kelderhouse LE, Gray J, et al. Induction of autophagy by ARHI (DIRAS3) alters fundamental metabolic pathways in ovarian cancer models. BMC Cancer (2016) 16. doi: 10.1186/s12885-016-2850-8
214. Kamada Y, Funakoshi T, Shintani T, Nagano K, Ohsumi M, Ohsumi Y. Tor-mediated induction of autophagy via an Apg1 protein kinase complex. J Cell Biol (2000) 150:1507–13. doi: 10.1083/jcb.150.6.1507
215. Ravikumar B, Vacher C, Berger Z, Davies JE, Luo S, Oroz LG, et al. Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat Genet (2004) 36:585–95. doi: 10.1038/ng1362
216. Takeuchi H, Kondo Y, Fujiwara K, Kanzawa T, Aoki H, Mills GB, et al. Synergistic augmentation of rapamycin-induced autophagy in malignant glioma cells by phosphatidylinositol 3-kinase/protein kinase B inhibitors. Cancer Res (2005) 65:3336–46. doi: 10.1158/0008-5472.CAN-04-3640
217. Tanemura M, Ohmura Y, Deguchi T, MacHida T, Tsukamoto R, Wada H, et al. Rapamycin causes upregulation of autophagy and impairs islets function both in vitro and in vivo. Am J Transplant (2012) 12:102–14. doi: 10.1111/j.1600-6143.2011.03771.x
218. Corradetti MN, Inoki K, Bardeesy N, DePinho RA, Guan KL. Regulation of the TSC pathway by LKB1: Evidence of a molecular link between tuberous sclerosis complex and Peutz-Jeghers syndrome. Genes Dev (2004) 18:1533–8. doi: 10.1101/gad.1199104
219. Mungai PT, Waypa GB, Jairaman A, Prakriya M, Dokic D, Ball MK, et al. Hypoxia Triggers AMPK Activation through Reactive Oxygen Species-Mediated Activation of Calcium Release-Activated Calcium Channels. Mol Cell Biol (2011) 31:3531–45. doi: 10.1128/mcb.05124-11
220. Meijer AJ, Codogno P. Regulation and role of autophagy in mammalian cells. Int J Biochem Cell Biol (2004) 36:2445–62. doi: 10.1016/j.biocel.2004.02.002
221. Inoki K, Li Y, Xu T, Guan KL. Rheb GTpase is a direct target of TSC2 GAP activity and regulates mTOR signaling. Genes Dev (2003) 17:1829–34. doi: 10.1101/gad.1110003
222. Lu Z, Yang H, Sutton MN, Yang M, Clarke CH, Liao WSL, et al. ARHI (DIRAS3) induces autophagy in ovarian cancer cells by downregulating the epidermal growth factor receptor, inhibiting PI3K and Ras/MAP signaling and activating the FOXo3a-mediated induction of Rab7. Cell Death Differ (2014) 21:1275–89. doi: 10.1038/cdd.2014.48
223. Palmieri M, Impey S, Kang H, di Ronza A, Pelz C, Sardiello M, et al. Characterization of the CLEAR network reveals an integrated control of cellular clearance pathways. Hum Mol Genet (2011) 20:3852–66. doi: 10.1093/hmg/ddr306
224. Settembre C, Di Malta C, Polito VA, Arencibia MG, Vetrini F, Erdin S, et al. TFEB links autophagy to lysosomal biogenesis. Science (80- ) (2011) 332:1429–33. doi: 10.1126/science.1204592
225. Sutton MN, Huang GY, Zhou J, Mao W, Langley R, Lu Z, et al. Amino acid deprivation-induced autophagy requires upregulation of DIRAS3 through reduction of E2F1 and E2F4 transcriptional repression. Cancers (Basel) (2019) 11. doi: 10.3390/cancers11050603
226. Blancato J, Graves A, Rashidi B, Moroni M, Tchobe L, Ozdemirli M, et al. SYK allelic loss and the role of Syk-regulated genes in breast cancer survival. PloS One (2014) 9. doi: 10.1371/journal.pone.0087610
227. Krisenko MO, Higgins RL, Ghosh S, Zhou Q, Trybula JS, Wang WH, et al. Syk is recruited to stress granules and promotes their clearance through autophagy. J Biol Chem (2015) 290:27803–15. doi: 10.1074/jbc.M115.642900
228. Du H, Yang W, Chen L, Shi M, Seewoo V, Wang J, et al. Role of autophagy in resistance to oxaliplatin in hepatocellular carcinoma cells. Oncol Rep (2012) 27:143–50. doi: 10.3892/or.2011.1464
229. Shimizu S, Takehara T, Hikita H, Kodama T, Tsunematsu H, Miyagi T, et al. Inhibition of autophagy potentiates the antitumor effect of the multikinase inhibitor sorafenib in hepatocellular carcinoma. Int J Cancer (2012) 131:548–57. doi: 10.1002/ijc.26374
230. Qu X, Sheng J, Shen L, Su J, Xu Y, Xie Q, et al. Autophagy inhibitor chloroquine increases sensitivity to cisplatin in QBC939 cholangiocarcinoma cells by mitochondrial ROS. PloS One (2017) 12. doi: 10.1371/journal.pone.0173712
231. Tong M, Che N, Zhou L, Luk ST, Kau PW, Chai S, et al. Efficacy of annexin A3 blockade in sensitizing hepatocellular carcinoma to sorafenib and regorafenib. J Hepatol (2018) 69:826–39. doi: 10.1016/j.jhep.2018.05.034
232. Blessing AM, Santiago-O’Farrill JM, Mao W, Pang L, Ning J, Pak D, et al. Elimination of dormant, autophagic ovarian cancer cells and xenografts through enhanced sensitivity to anaplastic lymphoma kinase inhibition. Cancer (2020). doi: 10.1002/cncr.32985
233. Nishikawa S, Ishii H, Haraguchi N, Kano Y, Fukusumi T, Ohta K, et al. Genotoxic therapy stimulates error-prone DNA repair in dormant hepatocellular cancer stem cells. Exp Ther Med (2012) 3:959–62. doi: 10.3892/etm.2012.522
234. Evans EB, Lin SY. New insights into tumor dormancy: Targeting DNA repair pathways. World J Clin Oncol (2015) 6:80–8. doi: 10.5306/wjco.v6.i5.80
235. Skvortsova I, Debbage P, Kumar V, Skvortsov S. Radiation resistance: Cancer stem cells (CSCs) and their enigmatic pro-survival signaling. Semin Cancer Biol (2015) 35:39–44. doi: 10.1016/j.semcancer.2015.09.009
236. Beerman I, Seita J, Inlay MA, Weissman IL, Rossi DJ. Quiescent hematopoietic stem cells accumulate DNA damage during aging that is repaired upon entry into cell cycle. Cell Stem Cell (2014) 15:37–50. doi: 10.1016/j.stem.2014.04.016
237. Yalcin A, Clem BF, Simmons A, Lane A, Nelson K, Clem AL, et al. Nuclear targeting of 6-phosphofructo-2-kinase (PFKFB3) increases proliferation via cyclin-dependent kinases. J Biol Chem (2009) 284:24223–32. doi: 10.1074/jbc.M109.016816
Keywords: autophagy, dormancy, recurrence, relapse, cancer, metastasis
Citation: Akkoc Y, Peker N, Akcay A and Gozuacik D (2021) Autophagy and Cancer Dormancy. Front. Oncol. 11:627023. doi: 10.3389/fonc.2021.627023
Received: 07 November 2020; Accepted: 22 January 2021;
Published: 19 March 2021.
Edited by:
Daniel Hector Grasso, Consejo Nacional de Investigaciones Científicas y Técnicas (CONICET), ArgentinaReviewed by:
Gabriele Multhoff, Technical University of Munich, GermanyPablo Nicolas Fernandez Larrosa, Consejo Nacional de Investigaciones Científicas y Técnicas (CONICET), Argentina
Copyright © 2021 Akkoc, Peker, Akcay and Gozuacik. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Devrim Gozuacik, ZGdvenVhY2lrQGt1LmVkdS50cg==
†These authors have contributed equally to this work and share first authorship