 Benedetta Apollonio
Benedetta Apollonio Nikolaos Ioannou
Nikolaos Ioannou Despoina Papazoglou
Despoina Papazoglou Alan G. Ramsay
Alan G. Ramsay
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oncol. , 25 March 2021
Sec. Hematologic Malignancies
Volume 11 - 2021 | https://doi.org/10.3389/fonc.2021.626818
This article is part of the Research Topic New Insights into the Complexity of Tumor Immunology in B-cell Malignancies: Tumor Immunology and Immunotherapy View all 13 articles
Cancers, including lymphomas, develop in complex tissue environments where malignant cells actively promote the creation of a pro-tumoral niche that suppresses effective anti-tumor effector T cell responses. Research is revealing that the tumor microenvironment (TME) differs between different types of lymphoma, covering inflamed environments, as exemplified by Hodgkin lymphoma, to non-inflamed TMEs as seen in chronic lymphocytic leukemia (CLL) or diffuse-large B-cell lymphoma (DLBCL). In this review we consider how T cells and interferon-driven inflammatory signaling contribute to the regulation of anti-tumor immune responses, as well as sensitivity to anti-PD-1 immune checkpoint blockade immunotherapy. We discuss tumor intrinsic and extrinsic mechanisms critical to anti-tumor immune responses, as well as sensitivity to immunotherapies, before adding an additional layer of complexity within the TME: the immunoregulatory role of non-hematopoietic stromal cells that co-evolve with tumors. Studying the intricate interactions between the immune-stroma lymphoma TME should help to design next-generation immunotherapies and combination treatment strategies to overcome complex TME-driven immune suppression.
There is a clinical need to identify novel treatments for lymphoid malignancies (1). Effective immunotherapy promotes the killing of cancer cells by cytotoxic T cells. Immune checkpoint blockade has demonstrated that reinvigorating anti-tumor immune activity can induce durable responses across multiple cancer types and serves as an illustrative example of therapeutically targeting the tumor microenvironment (TME) (2). The most promising clinical responses to PD-1 blockade have been seen in classical Hodgkin lymphoma (cHL) and primary mediastinal B cell lymphoma (PMBL) with up to 87% overall response rate (ORR) detected in relapsed/refractory (R/R) cHL (3). However, the efficacy of anti-PD-1 immunotherapy in non-Hodgkin lymphomas (NHLs) including diffuse large B-cell lymphoma (DLBCL) and follicular lymphoma (FL) has been more modest (4). Unexpectedly, no activity was seen in a trial of anti-PD-1 therapy for R/R chronic lymphocytic leukemia (CLL) although PD-L1-PD-1-mediated T cell dysfunction has been described (5–7). This clinical experience suggests that profound immunosuppressive barriers operate within the TME.
The current immuno-oncology era is directing attention towards the relevance of studying the composition and function of the immune TME, together with genomic analysis. In this review, we discuss the current understanding concerning the role of T cells and inflammatory signaling (“inflamed” versus “non-inflamed” immune TMEs) in generating both endogenous anti-tumor immune responses, as well as sensitivity to immunotherapy. We describe how cancer immunology has been the subject of intense research in the solid tumor field but has been comparatively ill-defined in the lymphomas. We highlight tumor intrinsic and extrinsic mechanisms of resistance in generating an effective anti-tumor immune response, before introducing an understudied area of lymphoma research—the role of non-hematopoietic stromal cell types in regulating anti-tumor immunity, that could also represent a targetable obstacle that immune cells face in the cancer-immunity cycle.
It is known that the immune system can paradoxically both inhibit and promote tumor development through a dynamic process where complex interactions between malignant cells and the immune system determine the fate of tumors, termed cancer immunoediting, which progresses through three phases: elimination, equilibrium, and escape (8, 9). During the elimination phase, the immune system recognizes and eradicates transformed cells; however, some tumor clones can avoid elimination leading to the equilibrium phase during which tumor growth is kept in check (10). However, chronic inflammation as well as evolutionary pressure from immune cells, can allow tumor sub-clones to escape immune surveillance, leading to tumor outgrowth and disease (10). Although cancer immunoediting has been defined using murine models, next generation technology is now in a position to shed light on the relevance of these concepts for human patients and clinical observations (11).
The immune composition of the TME is a major determinant of tumor progression through these phases and includes innate (natural killer cells, macrophages, neutrophils, mast cells and dendritic cells), and adaptive (T and B cells) immune cells. The composition of the TME in B cell lymphomas that arise in secondary lymphoid tissues, can vary from resembling normal reactive lymph nodes with germinal centers as seen in FL, to tumor effacement as exemplified by CLL and DLBCL (12). In common with solid cancers, malignant cells actively influence the composition of the TME and educate surrounding immune and stromal cells that can acquire pro-tumorigenic and immunomodulatory activity.
Particular cell types of the innate and the adaptive immune system can function in a tumor-promoting or inhibitory way with neutrophils, M2-polarized tumor-associated macrophages (TAMs), TH2 CD4+ T cells and TRegs generally considered as pro-tumor cells, whereas, M1-macrophages/TAMs, TH1 CD4+ T cells and cytotoxic CD8+ T cells are associated with anti-tumor functions (13). Importantly, in a significant proportion of cancer patients, including the lymphomas, there is evidence of an active anti-tumor immune response directed against tumor-specific (neoantigens) (14, 15) or tumor-associated antigens (16). However, although acute inflammatory signals can stimulate adaptive immunity, chronic inflammation can be antagonistic and promote immune suppression. Numerous studies in solid cancer have shown that an activated adaptive immune response involving effective antigen presentation and interferon (IFN) signaling, as well as sufficient numbers of T cells in the TME, is associated with a favorable prognosis (13, 17, 18).
The crucial role of T cell lymphocytes as the main regulators and effectors in anti-tumor immunity has been well established. Seminal studies have revealed the role of immune surveillance and the vital contribution made by adaptive immune cells in suppressing the formation of tumors (19–21). Murine models have shown that tumor killing is mainly mediated by cytotoxic CD8+ T cells in both solid (13) and B cell lymphomas (22). Elegant studies using the transgenic Eµ-TCL1 mouse model of CLL have shown that CD8+ T cells play an important role in controlling disease development in an IFNγ-dependent manner (23). CD8+ T cells, following successful priming, recognize antigens presented by tumor cells on their surface in complexes with HLA class I molecules and kill their targets, primarily via the release of cytotoxic molecules such as perforin and granzymes (24, 25). While the anti-tumor role of CD8+ is generally accepted, the role of CD4+ T cells in cancer immunity is controversial. TH1 CD4+ T cells, via the secretion of pro-inflammatory cytokines such as IFNγ and IL-2, can activate antigen presentation and costimulatory function on antigen-presenting cells (APCs), promote the effector differentiation of CD8+ T cells and enhance their migration (26). However, as mentioned above, some subsets of CD4+ cells can exhibit pro-tumor functions. TH2-polarized CD4+ T cells secrete cytokines that can limit CTL differentiation and proliferation such as IL-10 and IL-4, while CD4+ TRegs (FOXP3+) have significant immunosuppressive functions through various mechanisms and their pro-tumor role has been described in both solid tumors and lymphomas (13, 27–29). In CLL, in vitro and xenograft murine models have demonstrated a pro-tumor effect of the CD4+ T cell compartment in patients, with correlations of CD4+ subset counts and clinical outcome supporting this role (28, 30). However, research using Eµ-TCL1-based murine models has suggested a more complex role of CD4+ T cells with some studies showing an anti-tumor function (31), while others demonstrating a dispensable role (23). Such differences likely reflect differences in the assay systems used, and the degree to which they model complex immune TMEs, as well as highlighting the divergent role of TH1 versus TH2-polarized CD4+ T cell subsets and their plasticity. Interestingly, CD4+ T cells specific for immunoglobulin-derived neoantigens in mantle cell lymphoma (MCL) were shown to express granzyme B and possess cytolytic function against autologous tumor cells following their engagement and expansion (14, 15), as previously described for granzyme B+ tumor-reactive CD4+ T cells in solid cancer (32). However, despite their crucial roles in controlling tumor growth, T cells become dysfunctional due to exhaustion in the chronically inflamed TME. T cell exhaustion is characterized by low proliferative capacity and limited effector function, due to chronic tumor antigen exposure (33, 34). This, in combination with a multitude of additional mechanisms covered later in this review, allow some tumors to escape both endogenous and therapy-mediated anti-tumor responses.
Numerous studies have established the important role of the type II IFN, IFNγ as a key player in host anti-tumor immunity through direct anti-tumor and indirect immunoregulatory actions (19, 35–39). IFNs can suppress tumors directly by triggering pro-apoptotic signaling or inhibiting their proliferation, while increasing MHC expression (40), antigen presentation (41) and promoting tumor immunogenicity, a critical process for effective tumor recognition and elimination by the immune system (19). IFNγ strongly promotes inflammatory responses and has been shown to augment the function of tumor-infiltrating immune cells including CD4+ TH1 cells, CD8+ T cells, dendritic cells and macrophages, while suppressing TH2 cells and Tregs (20, 39). Despite its pleiotropic effects, it has been demonstrated that IFNγ-expressing T cells play a dominant role in mediating effective anti-tumor activity (42).
Although the majority of studies have used solid tumor models, the importance of IFNγ in anti-tumor immunity has also been shown in B cell malignancy models. Effective lymphoma immune surveillance using murine models was shown to be mediated by tumor-specific CD4+ T cells and associated with proinflammatory cytokines, particularly those that promote a TH1 phenotype including IFNγ, IL-2, and IL-12 (43). As discussed above, a non-redundant role of IFNγ-expressing CD8+ T cells in suppressing CLL progression was demonstrated using a murine transgenic model (23). Recent work has revealed that a subset of DLBCL harboring a T cell-inflamed TME (discussed later in this review) expressed a number of inflammatory and effector cytokine pathways including IFNγ and TNFα (44). Moreover, a TH1 cytokine profile including type II IFN T cell responses has been associated with a favorable prognosis and response to traditional chemotherapy, the immunomodulatory drug lenalidomide and anti-PD-1 immunotherapy in NHL patients (44–48).
Type I interferons (including IFNα and IFNβ) can be produced by all nucleated cells and act on both tumor and immune cells (38, 49). Even though some decades-old studies had suggested an anti-tumor activity for IFNα/β, only recently, their role in the elimination of tumor cells and immunoediting has started to be elucidated (50, 51). Several studies have established their direct anti-tumor effect which is mediated through growth inhibition and/or induction of apoptosis by the regulation of the cell cycle (G1 phase arrest) and activation of both intrinsic and extrinsic apoptotic pathways (TRAIL, FAS and FASL among others) (52, 53). Moreover, type I IFNs have been shown to increase both HLA class I and tumor antigen expression and consequently, increase immune recognition and successful generation of an anti-tumor response (54, 55).
In addition to direct effects on tumor cells, there is substantial evidence showing that type I IFNs mainly function through stimulating anti-tumor immune responses that is relevant for both natural and therapy-induced immunity. The autocrine and paracrine circuits within the immune TME that are triggered by type I IFN show similarity with IFNγ signaling but they do not overlap completely (50, 56). Type I IFN deficient dendritic cells (DCs) in murine TME models have been correlated with ineffective T cell priming (57, 58). In common with IFNγ, the ability of type I IFN to promote immune-mediated anti-tumor activity is also through their effects on T cells. More specifically, type I IFNs have been reported to play an important role in TH1 CD4+ T cell polarization as well as promoting the survival and effector function of CD8+ cytotoxic T cells by increasing granzyme expression (51, 59, 60). In addition, type I IFNs have been shown to negatively regulate the numbers and activity of TRegs (61) and myeloid-derived suppressor cells (MDSCs) (62).
Remarkably, in the context of hematological malignancies, one of the earliest studies indicating a role of type I IFNs in promoting immune-mediated anti-tumor responses was reported in a mouse lymphocytic leukemia model (63). Recent work has demonstrated that avadomide, a cereblon E3 ligase modulator (CELMoD), can induce IFN signaling in DLBCL B cells that triggered tumor apoptosis (64). In addition, the immunomodulatory drug lenalidomide has been shown to induce IFNβ signaling in DLBCL that promotes tumor cell death (65). In contrast, lenalidomide and avadomide have anti-proliferative effects on CLL cells that are likely a direct effect of IFN signaling induction but does not induce direct tumor B cell apoptosis (66, 67). Interestingly, the fusion of IFNα to anti-CD20 antibody induced a superior anti-lymphoma effect than anti-CD20 alone by direct and potent killing of type I IFNα receptor-positive lymphoma cells (68). It was further demonstrated that an important additional mechanism of action of tumor-directed antibodies, in addition to direct anti-tumor effects, was the induction of type I IFN in the TME that activated DC cross-presentation and T cell activation (69). This work highlights the potential of next-generation Ab-based immunotherapy (Ab–IFN fusion) (70) to induce direct anti-tumor effects and reconnect suppressed innate and adaptive immune responses in the TME (69). However, it is worth noting that earlier work in CLL has described conflicting data regarding a pro-survival effect when treating CLL cells with recombinant type I (71–73) or type II IFNs (74), that may be associated with studies using peripheral blood-derived CLL cells, rather than tissue culture or in vivo models that mimic activated CLL TME biology (75). Recently, impaired IFN signaling in CLL B cells (loss of IFN regulatory factor 4, IRF4 using a murine model or its reduced expression in human CLL cells) has been linked to downregulated antigen presentation and co-stimulatory molecules that prevented the generation of activated, exhausted T cell responses and was associated with accelerated disease progression. In addition to this tumor immune evasion mechanism, T cells from treatment naïve CLL patients have been shown to express deregulated IFN type I and II signaling genes compared to healthy age-matched control T cells (67), in keeping with impaired IFN signaling in T cells representing a common immune defect in cancer (76). Together these recent findings provide evidence that reduced IFN signaling in the CLL TME could contribute the development of an immunosuppressive/non-inflamed TME.
Analysis of solid cancer tumors including colorectal, melanoma, and ovarian cancer among others has established the prognostic significance of the type, density, and localization (immune contexture) of immune cells that reside within the TME for predicting a patient’s overall survival (77–80). The prognostic power of these variables is so powerful that they led to the development of a new scoring system termed the ‘immunoscore’, which has been internationally validated for colorectal carcinomas (CRC) (81). The immunoscore is based on the quantification and localization of CD3+ and CD8+ lymphocytes within tumors and their invasive margin and evidence so far suggests that this alternative scoring system is a more robust classification system for CRC for predicting survival than the classical tumor-node-metastasis staging system (81, 82). This was the first attempt of cancer classification/stratification through a system which was not tumor-based but rather immune-based, leading to a basic distinction of TMEs as T cell inflamed (or hot) that contain high numbers of infiltrated T cells, versus non-inflamed (or cold) phenotypes that are non-infiltrated (3, 18).
Transcriptome profiling studies have revealed additional molecular characteristics of T cell inflamed TMEs including the number of tumor-infiltrating T cells and the expression of IFN-inducible activated T cell biology gene signatures including granzymes, chemokines, PD-L1, as well as tumor mutational burden and neoantigen load (83, 84). In contrast, non-inflamed TMEs are generally characterized by low or absent infiltration of CD8+ cytotoxic T cells, low frequency of neoantigen expression, and a paucity of IFN signaling (18, 85). It should be noted that solid cancer and B cell lymphoma subtype TMEs fall into a spectrum of T cell inflamed to non-inflamed phenotypes reflecting expected heterogeneity, and can include “excluded” phenotypes where understudied stromal cells may present a barrier to infiltrating T cells (18). Applying the concept of inflamed versus non-inflamed TME phenotypes to lymphoma is challenging due to two major characteristics of these cancers. First, lymphomas are cancers of immune cells and as a consequence, tumor cells can regulate immunological functions themselves, making the tumor-immune cell interaction significantly more complex than solid tumors. Secondly, the origin and residence of malignant cells in most B cell lymphomas are secondary lymphoid organs that serve as sites of immune cell surveillance. Therefore, the presence of T cell subsets or inflammatory signatures in the TME may also represent active non-tumor immune responses. For example, cytomegalovirus causes a marked expansion of virus-specific T cells in CLL patients (86). However, the role of Epstein–Barr virus (EBV) in the pathogenesis of some lymphomas may also be a source of antigens for tumor T cell recognition (87). Regardless, a classification of lymphomas based on inflamed versus non-inflamed immune landscapes (‘immune contexture’) and responsiveness to immune checkpoint blockade has been described (3).
In the current era of immunotherapy, the characterization of tumors according to their immune landscape is more relevant than ever (88), as IFNγ-expressing T cell inflamed tumors have shown increased sensitivity to anti-PD-1 therapy with both predictive and prognostic significance (83, 89–91). PD-1 has been shown to be a negative regulator of pre-existing immune responses in tumors, thus blocking the interaction of PD-1 with its ligands (PD-L1 or PD-L2) prevents this inhibitory signaling and allows tumor-specific T cells to remain activated and kill tumor cells. Pre-clinical studies in solid cancer have suggested that anti-PD-1 therapy cannot function in the absence of primed tumor antigen-specific CD8+ cytotoxic T cells which express high levels of PD-1 (exhausted phenotype) (2, 92, 93). However, high-dimensional correlative analysis of circulating immune cells from melanoma patients receiving anti-PD-1 immunotherapy has revealed a more complex role of the T cell compartment during therapy. In particular, higher numbers of PD-1+, CTLA-4+, IL-17A+, IFNγ+ activated memory CD4+ and CD8+ T cells including granzyme B+ CD4+ T cells were detected after therapy and in responding patients (94). Interestingly, the frequency of classical CD14+ HLA-DR+ monocytes prior to therapy was found to be a strong predictor of response, thought to reflect myeloid expansion triggered by IFNγ produced by activated tumor-specific and infiltrated T cells in patients who are more likely to become responders.
Inflamed lymphomas such as cHL tumors have shown increased sensitivity to anti-PD-1 blockade therapy and are characterized by an unusually high immune cell infiltrate, an inflammatory PD-L1+ TME, a high mutational burden (95) and genetic alterations that facilitate cancer immunoediting escape including aberrant MHC class I molecule expression (96). Intriguingly, the majority of T cells that infiltrate HL tumors are IFNγ-expressing TH1-polarized CD4+ T cells (96, 97), which is again at odds with the concept that PD-1 inhibition works solely through the reactivation of MHC class I-restricted CD8+ T cells (98), given that cHL is sensitive to this immunotherapy. Indeed, recent immune monitoring studies have shown that circulating cytotoxic granzyme B+, PD-1+ CD4+ T cells, as well as PD-1+ differentiated effector CD8+ T cells are detected in HL patients. Interestingly, only CD4+ T cell receptor (TCR) diversity at baseline and during therapy correlated with responses to anti-PD-1 therapy (99). Additionally, the study identified an IFN-experienced circulating CD68+ CD4+ Granzyme B+ innate effector subset in patients who responded to therapy. This correlative data highlights the potential cytotoxic anti-tumor capability of specific CD4+ T cell subsets that is relevant for both solid cancers (94) and lymphomas (99–102). It is also interesting to note that the presence of a baseline T cell inflamed TME has correlated with improved responses to other therapies including adoptive cell therapy in solid cancer models (103), that may have relevance for CAR T therapy in lymphoma (104), and the CELMoD avadomide which induced higher responses in immune-rich DLBCL TMEs (105). However, although a proportion of DLBCL (44) and FL tumors (106, 107) exhibit features of T cell inflamed TMEs, the immune landscape of the NHLs is more heterogenous with most harboring a cold or non-inflamed environment, and are typically resistant to anti-PD-1 therapy. A recent phase 2 study of anti-PD-1 in R/R DLBCL following autologous transplant reported an ORR of 10%, although some patients did show encouraging disease stabilization and durable responses (108). Non-inflamed lymphomas are thought to contain fewer infiltrating immune cells, particularly T cells, that can predict poor survival in DLBCL (109). Sparse infiltration of immune cells is linked to lymphoma intrinsic oncogenic pathways that prevent the recruitment or retention of immune cells in TME lymphoid tissues or downregulate APC capability (110). Interestingly, non-inflamed lymphomas typically lack genetic aberrations that facilitate immune escape (111). We have recently demonstrated that CLL lymph nodes show salient features of non-inflamed tumors including sparse numbers of CD8+ T cells relative to reactive non-malignant tissues and low PD-L1 expression in the TME, as well as profound T cell functional exhaustion (67).
It is now clear that distinct mechanisms may confer sensitivity and resistance to different types of immunotherapy in tumors that show a large degree of heterogeneity in their immune TMEs, falling within the extremes of the inflamed versus non-inflamed classification. The key factors that shape the TME include tumor immunogenicity, oncogenic pathways, and genetic alterations that regulate T cell infiltration and function (3, 112). The sculpting of the immune TME is inherently interconnected with the cancer immunoediting process. Available evidence from studies of patients treated with immune checkpoint blockade drugs suggests that immunoediting takes place not only during tumor progression but, at least in some form, also in response to therapy (9). Several factors of immune escape and resistance to immunotherapy (innate or acquired) that have been characterized to date, can be broadly divided in tumor-intrinsic and tumor-extrinsic mechanisms (113, 114). While the bulk of evidence regarding the role of these processes in regulating anti-tumor immunity and response to immunotherapy comes from solid tumor research, recent studies are revealing similar mechanisms operative in the lymphomas.
Tumor intrinsic mechanisms generally include genetic aberrations that can affect antigen recognition and influence immune function and immune contexture in TMEs including neoantigen load (Figure 1). HLA class I and II are required for the display and presentation of tumor-associated antigens and consequently an effective adaptive anti-tumor response. Several studies have reported HLA class I and II molecule loss or downregulation in lymphomas through genetic, epigenetic or transcriptional mechanisms. Intriguingly, acquisition of recurrent genetic alterations in genes encoding antigen presentation machinery appears to be a shared characteristic of T cell inflamed lymphomas such as cHL (115) and a subset of DLBCLs (44). This likely represents a critical cancer immunoediting escape mechanism used by lymphoma cells to evade the effector activity of lymphoma-specific CD8+ and CD4+ T cells. Impaired or loss of expression of HLA class I molecules is frequently detected in HL and DLBCL but has not been reported or is extremely rare in indolent lymphomas such as MCL, marginal zone lymphoma and CLL (16, 115–118). The most common mechanism leading to this altered HLA expression is caused by mutations and deletions in the β2-microglobulin gene, although direct genetic alterations of the HLA I genes have also been reported (115, 117, 119). HLA class II molecule downregulation is also observed in HL and DLBCL and mediated mainly at a transcriptional level through inactivating mutations CIITA, as well as homozygous deletions of chromosome 6p21.3 (116, 136–139).
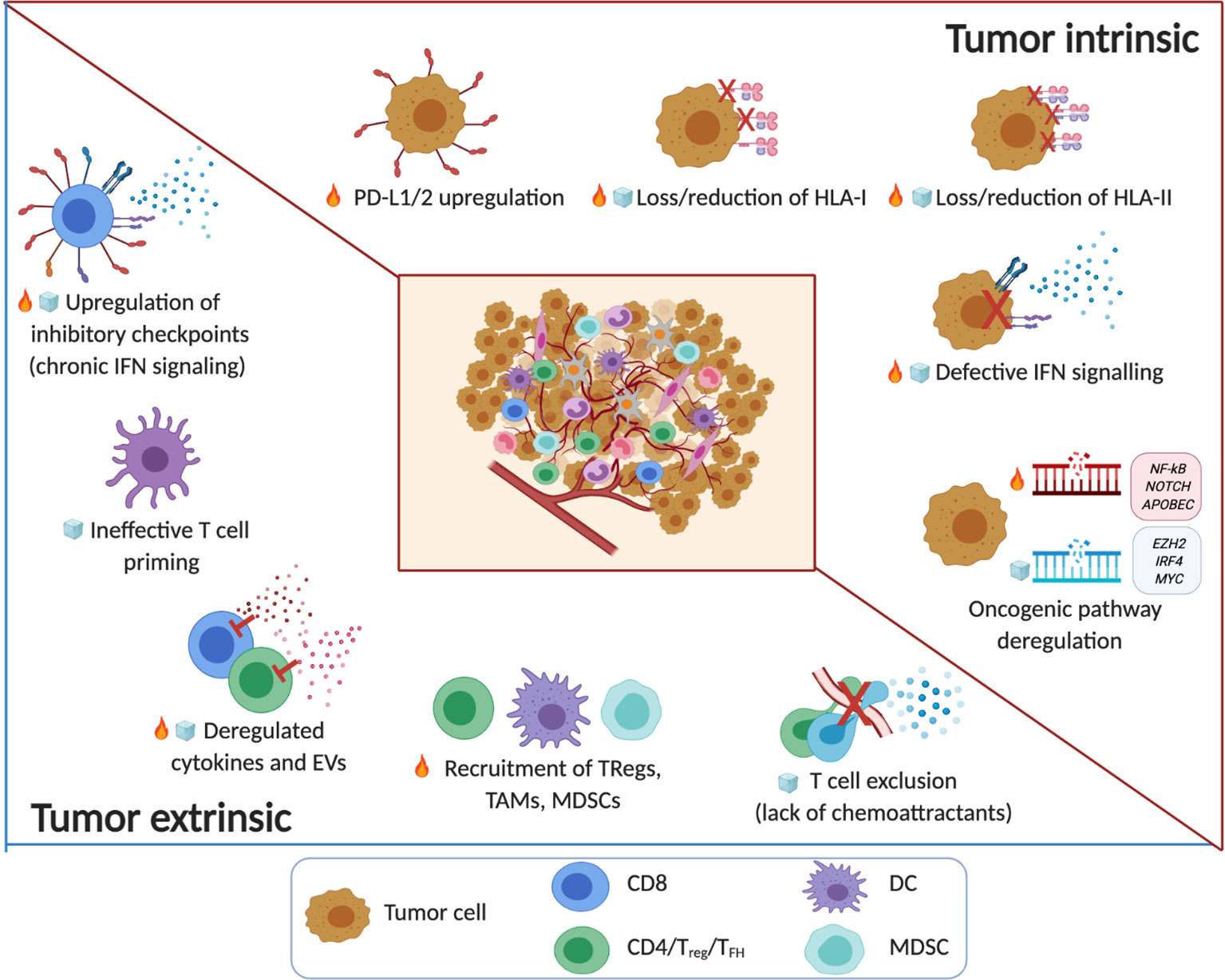
Figure 1 The immune TME in B cell malignancies. Tumor intrinsic and tumor extrinsic mechanisms associated with noninflamed/cold ( ) or inflamed/hot (
) or inflamed/hot ( ) TMEs in B cell malignancy. The key factors that shape the TME include tumor immunogenicity, oncogenic pathways and genetic alterations that regulate T cell infiltration and function. Note: the timeline or sequence of events during the evolution of the altered pro-tumor, yet immunoprivileged TME is a current topic in the field. Emerging evidence from patients treated with immune checkpoint blockade drugs suggests that cancer immunoediting takes place not only during tumor progression but also in response to therapy (e.g. the acquisition of mutations that contribute to ‘defective IFN signaling’) (8–11). Several factors of immune escape and resistance to immunotherapy (innate or acquired) that have been characterized to date, can be broadly divided in tumor-intrinsic and tumor-extrinsic mechanisms. Tumor intrinsic mechanisms generally include genetic aberrations that can affect antigen recognition (‘loss/reduction of HLA-I/-II molecules’) (16, 115–119) and influence immune function (‘PD-L1/2 upregulation’) (120–122) and immune contexture in TMEs (‘oncogenic pathway deregulation’) including neoantigen load (83, 84). Tumor cell extrinsic factors that regulate anti-tumor immunity, immune evasion or resistance to immunotherapy involve non-tumor cellular and molecular components within the immune TME including ‘upregulation of inhibitory immune checkpoints’ (linked to chronic IFN signaling) (123–125), the ‘recruitment of TAMs, MDSCs, TRegs’ and stromal cells, as well as ‘deregulated cytokines and EVs’ (126–130), ‘ineffective T cell priming’ and ‘T cell exclusion’ (131–135). (Created with Biorender.com).
) TMEs in B cell malignancy. The key factors that shape the TME include tumor immunogenicity, oncogenic pathways and genetic alterations that regulate T cell infiltration and function. Note: the timeline or sequence of events during the evolution of the altered pro-tumor, yet immunoprivileged TME is a current topic in the field. Emerging evidence from patients treated with immune checkpoint blockade drugs suggests that cancer immunoediting takes place not only during tumor progression but also in response to therapy (e.g. the acquisition of mutations that contribute to ‘defective IFN signaling’) (8–11). Several factors of immune escape and resistance to immunotherapy (innate or acquired) that have been characterized to date, can be broadly divided in tumor-intrinsic and tumor-extrinsic mechanisms. Tumor intrinsic mechanisms generally include genetic aberrations that can affect antigen recognition (‘loss/reduction of HLA-I/-II molecules’) (16, 115–119) and influence immune function (‘PD-L1/2 upregulation’) (120–122) and immune contexture in TMEs (‘oncogenic pathway deregulation’) including neoantigen load (83, 84). Tumor cell extrinsic factors that regulate anti-tumor immunity, immune evasion or resistance to immunotherapy involve non-tumor cellular and molecular components within the immune TME including ‘upregulation of inhibitory immune checkpoints’ (linked to chronic IFN signaling) (123–125), the ‘recruitment of TAMs, MDSCs, TRegs’ and stromal cells, as well as ‘deregulated cytokines and EVs’ (126–130), ‘ineffective T cell priming’ and ‘T cell exclusion’ (131–135). (Created with Biorender.com).
Another mechanism contributing to immune evasion in lymphoma is the genetic overexpression of PD-L1, that is also a common feature of checkpoint blockade-sensitive T cell inflamed TMEs including cHL and PMBL (3). Upregulated expression of PD-1 ligands has been found to be driven by genetic alterations driving amplification of structural variations (SVs) of the chromosome region 9p24.1 which contains the loci for PD-L1, PD-L2 and JAK2 (termed the PDJ amplicon) (120–122). PD-L1 expression is less prevalent in DLBCL, but PD-L1 gene alterations have also been detected in a subset of DLBCLs, particularly the non-germinal center B cell (GCB) subtypes, harboring T cell inflamed phenotypes with high numbers of infiltrating T cells, downregulated HLA expression and upregulation of inflammatory NF-kB, TNFα and IFNγ gene pathways (44, 120, 140). Recurrent SVs in the 3′ UTR of the PD-L1 gene, which are thought to stabilize PD-L1 transcripts leading to increased protein expression, have also been uniquely described in DLBCL (141).
As mentioned earlier, type I and II IFNs are important mediators of both innate and adaptive anti-tumor immune function and are key players in cancer immunoediting. Importantly, defective IFN signaling in tumor cells has emerged as a major tumor-intrinsic resistance mechanism during immune checkpoint blockade therapy (114, 142, 143). Loss-of-function mutations in IFNγ receptor signaling pathway genes JAK1 and JAK2 have been seen in melanoma patients who developed late relapses after initial successful anti-PD-1 therapy (144), as well as patients with primary resistance to this immunotherapy (145). Mutations in IFNγ-related genes were also observed in non-responsive patients following anti-CTLA-4 checkpoint therapy (146). This loss of IFNγ receptor signaling allows the tumor to evade the effects of IFNγ produced by anti-tumor T cells that would reduce tumor antigen presentation capability, decrease the expression of IFN-associated chemoattractants and promote insensitivity to the anti-proliferative and pro-apoptotic effects of IFNγ in tumor cells. Several studies have found evidence of deregulation of tumor-intrinsic IFN signaling in lymphoma but this has not yet been directly linked to immunotherapy resistance. Suppression of type I interferon by STAT3 has been described in non-GCB DLBCL, with inhibition of STAT3 activity using ruxolitinib inducing a synergistic growth inhibition effect when combined with the IFN-inducing immunomodulatory drug lenalidomide (147). Evidence for a CLL cell-intrinsic IFN signaling defect was described earlier in this review with low expression of IRF4 associated with inferior prognosis and associated studies supporting a novel tumor immune evasion mechanism (148). This IFN signaling defect reduced expression of tumor genes required to activate T cells including antigen processing and presentation, that could contribute to resistance to immunotherapies in this disease.
In addition to altering intrinsic tumor cell properties, oncogenic signaling has been found to contribute to the immune contexture of inflamed and non-inflamed TMEs. Genetic aberrations affecting MYC, p53 and NF-kB among other genetic events can dictate the immune landscape (84, 149). Importantly, pathways including WNT-β-catenin and MAPK signaling, as well as those associated with loss of PTEN, have all been implicated in driving intrinsic resistance to immune checkpoint blockade in solid tumors (114). Recent genomic studies in DLBCL have revealed associations between alterations in oncogenes or tumor suppressors including PTEN, EZH2 and TP53 and reduced expression of genes linked to immune cell activation (150). However, mechanistic data on how oncogenic alterations promote a non-inflamed immune TME or immunotherapy resistance in lymphomas is currently lacking. Double hit GCB DLBCLs with MYC, BCL2, and/or BCL6 gene rearrangements have been shown to contain low number of infiltrating T cells (111). These lymphomas are enriched for EZH2 activating mutations that have recently been shown to underlie acquired deficiency in MHC I and II expression and low T cell infiltrates in murine lymphoid TMEs (110). Interestingly, MYC has been shown to regulate the anti-tumor immune response in murine models of T cell acute lymphoblastic leukemia (ALL) and liver cancer by inducing the expression of CD47 and/or PD-L1 immune checkpoint molecules and recruiting TAMs, while excluding T cells among other mechanisms (151–153). However, it should be noted that the ability of oncogenes like MYC to control the recruitment and function of immune cells in tumors can be counteracted by other genetic aberrations as has been seen using the Eµ-MYC lymphoma model (149, 154), that highlights the complexity of defining the roles of oncogenic alterations in modulating immune cell landscapes both within and between complex molecular lymphoma subtypes. Conversely, oncogenes and tumor suppression genes can equally foster an inflammatory immune TME. One notable example in solid cancer is NF-kB that controls cell survival and proliferation, but also production of inflammatory cytokines (149). In common, recurrent genetic modifications that lead to NF-kB activation have been described in inflamed lymphomas including cHL, PMBL and a subset of T cell-rich DLBCLs that contain PD-L1 SVs and downregulated HLA expression (44, 155, 156). In addition, oncogenic NOTCH signaling has been implicated in the regulation of inflammatory DLBCLs that harbor genetic immune escape mechanisms including inactivating CD70 and FAS mutations (157–159). Overall, further studies involving pre-clinical murine models will be required to define the how specific oncogenic signaling pathways contribute to tumor-immune cell interactions and T cell recruitment in lymphoma TMEs.
Several studies have shown that tumor neoantigens can function as targets for anti-tumor T cells and there is a positive correlation between tumor mutational burden and response to immune checkpoint blockade across cancers (160–162). In particular, deficiency in DNA repair mechanisms has been found to be the main driver of genomic instability and has been associated with response to immunotherapy (163). Interestingly, mutations that lead to DNA mismatch repair defects and microsatellite instability, as well as APOBEC mutational signatures have been identified in PD-1 blockade sensitive ‘‘inflamed’’ lymphomas such as cHL and PMBL; however, these seem more rare in other B cell malignancies (164–166). On the other hand, low tumor immunogenicity with low frequency of neoantigen generation as is seen in CLL, is linked to reduced intrinsic sensitivity to immune checkpoint blockade (5, 167). However, recent antigen-presentation profiling work is revealing that B cell lymphomas including CLL can present immunoglobulin neoantigens (15), with preferential MHC-II presentation that has implications for promoting the cytolytic TH1 differentiation of neoantigen-specific CD4+ T cells with immunotherapy, as has been demonstrated in solid cancer (168).
Recent studies have demonstrated that metabolically active tumor cells chronically deprive the TME of essential nutrients that affect T cell effector function and promote the creation of a tolerogenic TME [metabolic reprogramming in the lymphomas has been reviewed elsewhere (169–171)].
Tumor cell extrinsic factors that regulate anti-tumor immunity, immune evasion or resistance to immunotherapy involve non-tumor cellular and molecular components within the immune TME including inhibitory immune checkpoints, TAMs, MDSCs, TRegs and stromal cells (Figure 1). Although tumor extrinsic components have been linked to cold or non-inflamed TMEs and response to therapy, it should be noted that their roles in cancer immunology are highly dynamic and context dependent, including the nature and duration of the driving forces implicated. For example, cytokines within immune TMEs including IL-10 (131), IL-6 (132) and TGFβ (133) or extracellular vesicles (EVs) (134, 135) are known to have complex, context-dependent effects on both immune and cancer cells. Although there is evidence that these secreted factors have relevant immunomodulatory activity in B cell malignancies including CLL (172–176) and FL (177), our understanding of the hierarchy and cooperation required between cytokines and chemokines or EVs and their cellular sources for the licensing of immune evasion or the promotion of anti-tumor immune responses is currently ill-defined.
Type I and II IFNs can act as double-edged swords in cancer, promoting both feedforward immune activation responses described earlier, as well as feedback inhibitory mechanisms. Importantly, persistent IFN signaling in cancer, in common with chronic virus infection, can be immunosuppressive by inducing PD-L1, IDO and LAG-3 in the immune TME (123–125). Indeed, prolonged type I and II IFN-driven expression of multiple immune checkpoint ligands, receptors and inhibitory pathways linked to exhaustion including PD-1, LAG-3 and TIM-3, have been shown to be upregulated on T cell subsets during adaptive or acquired resistance to immune checkpoint therapy (125, 178, 179). It is plausible that combination checkpoint inhibition may be able to bypass negative feedback and multiple inhibitory receptors as has been demonstrated using a CLL murine model with dual anti-PD-1 and LAG-3 blockade (180). Interestingly, CD8+ CAR T cells co-expressing PD-1 with either LAG-3 or TIM-3 were associated with poor responses in CLL, whereas patients who had complete and durable remissions were infused with CAR T cells containing lower frequencies of these exhausted phenotypes. Moreover, the clinical effectiveness of CAR T cells in CLL was increased when co-stimulatory receptor CD27+ was expressed on CAR T cells that may reflect a less exhausted and functionally competent T cell phenotype (‘intrinsic T cell fitness’) (181). This data supports the relevance of multiple inhibitory pathways induced by IFN signaling that could hinder CAR T therapy and promote therapy resistance.
TAMs are an important subset of terminally differentiated myeloid cells that regulate anti-tumor immunity and response to therapy in many cancers including lymphoma (126, 127). Early work demonstrated that TAMs promoted chemotherapy resistance in cHL and in DLBCL in the pre-rituximab treatment era (182, 183). However, TAMs have been shown to mediate antibody-dependent cellular phagocytosis of rituximab engaged malignant B cells, highlighting therapeutic context. Studies have demonstrated the capability of TAMs to directly suppress T cells through PD-L1 expression, as well as the production of cytokines such as IL-10 and TGF-β or enzymes that can limit effector activity (126). Interestingly, inflamed cHLs harbor high numbers of PD-L1+ TAMs, that presumably represent an active immune evasive strategy elicited within the TME to suppress lymphoma-specific PD-1+ CD4+ T cells (184). In addition, it has been shown that lymphoma cells can upregulate expression of the membrane anti-phagocytic protein CD47 that allows them to escape elimination by TAMs. Combining anti-CD47 blocking antibody with rituximab has elicited synergistic anti-tumor activity in pre-clinical models and early clinical results have been promising (185, 186). These studies highlight the potential to harness anti-lymphoma TAM activity that could be combined with immune checkpoint blockade therapy to re-educate their pro-tumor, immunosuppressive activity and optimize immunotherapy. In addition, MDSCs have also emerged as potentially important regulators of immune responses, through the production of several immunosuppressive factors (ARG1, NO, PGE2) (187), and their presence in solid cancer TMEs correlates with decreased efficacy of immunotherapies including checkpoint blockade and adoptive T cell therapy (128, 129). However, a role in contributing to immunotherapy resistance in B cell malignancy has not been defined to date (188). Notably, in CLL it has been demonstrated that MDSCs, characterized by a CD14+HLA-DRlo phenotype, can be induced by malignant B cells in vitro to suppress T cell effector function and promote TReg-differentiation mediated by upregulation of indoleamine 2,3-dioxygenase (IDO) (189).
TRegs comprise a subtype of CD4+ cells which are mainly defined by the expression of FOXP3 and play an important role in maintaining self-tolerance (190). TRegs suppress effector T cells by secretion of inhibitory cytokines including IL-10 and TGFβ, as well as direct cell contact. Indeed, many murine studies of solid cancer have shown that the depletion of TRegs cells from the TME can enhance or restore anti-tumor immunity (130). Studies in CLL using the TCL1 transgenic model indicated that partial depletion of TRegs numbers did not impact on CLL disease progression but did result in enhanced CD8+ T cell functional capacity (191). Increased numbers of circulating Tregs have been found in CLL patients and correlated with disease progression (192, 193), and lymph node tissue was shown to harbor twice the number of these suppressive cells than the peripheral blood compartment (194). Most immune checkpoint molecules including PD-1 and CTLA-4 are expressed on TRegs but the effects of checkpoint inhibitors on this T cell subset and treatment response remain unclear. Intriguingly, studies have suggested that anti-PD-1 immunotherapy might enhance the immunosuppressive function of TRegs (130, 195), whereas anti-CTLA-4 inhibitors might deplete these cells (196). It is possible that TRegs cells may coincide with lymphoma-specific T cells, indicating a potentially immune-responsive tumor. Correlative studies to determine the impact of TRegs on clinical outcomes for lymphoma patients receiving immunotherapies should be informative, as well as the development of targeted treatment approaches to deplete these cells in order to activate anti-tumor immunity (130).
Emerging evidence has now demonstrated that the creation of a “cold” TME requires the coordinated intervention of several other non-immune cell types that co-evolve with the tumor. Cancer-reprogrammed stromal cells [endothelial cells (ECs) and fibroblasts] acquire altered features that promote tumor progression and contribute to immunosuppression (197). This is achieved by a plethora of tightly orchestrated mechanisms influencing both spatial organization and function of immune subsets within the TME.
Extensive studies of tumor-associated endothelial cells (TECs) in solid cancer are highlighting not only the significance of angiogenesis in disease progression but also the role of tumor-reprogrammed ECs in recruiting and polarizing immune cells in the TME (198). EC-mediated T cell trafficking, a crucial and highly dynamic process, is initiated by the release of chemokines, to attract circulating leukocytes, followed by selectin-dependent leukocyte rolling on the endothelium and integrin-induced trans-endothelial migration (199). Multiple steps in this tightly controlled process are disrupted in tumors.
The tumor vasculature has been found to be less functional with abnormal ‘leaky’ vessels (200), contributing to the hypoxic state that characterizes the majority of solid tumors. In this hypoxic environment, ECs release nitric oxide (NO) that suppresses the expression of leukocyte adhesion molecules (VCAM-1, ICAM-1) (201). Decreased ICAM-1 expression on TECs has been shown to impair T cell extravasation and has been associated with decreased numbers of tumor-infiltrating lymphocytes (TILs) (202).
Together with ECs, cancer-associated fibroblasts (CAFs), the resident fibroblasts activated in a chronic inflamed TME, have been shown to actively shape the immune infiltrate. The CAF secretome promotes recruitment and polarization of regulatory cells from both the innate and adaptive immune arms (203). Recently, a subpopulation of immunoregulatory CAFs (CAF-S1) has been functionally involved in the attraction, retention and activation of TRegs in the breast cancer TME through the secretion of CXCL12 (204). Moreover, other CAF-derived factors like Chi3L1, MCP-1 and SDF-1 drive the recruitment of monocytes and their polarization into pro-tumoral, immunosuppressive M2-macrophages in breast and prostate cancers (205–207), while CAF-derived IL-6 has been shown to promote immunosuppressive neutrophils in hepatocellular carcinoma (208). In addition, CAF-driven abnormal extracellular matrix (ECM) deposition and remodeling have been shown to induce the physical trapping of anti-tumor T cells to prevent effective tumor access (209).
Besides their role in mapping the spatial organization of immune cells in the TME, stromal cells can also directly influence or suppress endogenous anti-tumor immune responses through additional mechanisms, including antigen presentation. ECs upregulate both MHC-I and MHC-II in response to inflammatory cytokines (such as INFγ) acting as semi-professional non-hematopoietic APCs (210). As a consequence, ECs can mediate Ag-specific stimulation of effector memory CD4+ and CD8+ T cells (211–215). Interestingly, Motz and colleagues found that FasL expression on the vasculature of human and mouse solid tumors induced specific killing of CD8+ TILs, but not Tregs, thus skewing the lymphocyte infiltrate towards a more regulatory phenotype (216). Moreover, ECs can upregulate PD-L1 and PD-L2 molecules inhibiting T cell activation and cytotoxic capacity (217–220).
Along the same lines, CAFs also directly impact the cytolytic activity of anti-tumor effector T cells through a number of different mechanisms. Prostaglandin E2 (PGE2) and NO produced by tumor-associated fibroblasts dampen CD8+ T cells proliferation in pancreatic and breast cancer respectively (221, 222) and CAFs expressing tumor antigens promote CD8+ apoptosis via PD-L2 and FasL expression (223). Overall, these intricate studies have shown that stromal cells shape the TME by affecting the recruitment and function of different adaptive and innate immune cells in solid tumors, but accumulating evidence is now revealing that similar mechanisms are operative in B cell malignancies.
In lymphoid organs [lymph nodes (LNs), spleen and bone marrow (BM)] resident stromal cells engage in bidirectional interactions with lymphocytes that directly contribute to the shaping and function of the immune system (224, 225). Lymphocytes enter the LN via both afferent lymphatics formed by lymphatic endothelial cells (LECs) and peripheral circulation through high endothelial venues (HEVs). Lymphocyte homing to secondary lymphoid organs is coordinated by the expression of integrin and adhesion molecules, as well as chemokine–chemokine receptors on ECs lining HEVs (199). Emerging evidence also shows that LN ECs also acquire distinct immunomodulatory roles such as priming T cells (226), while BM ECs critically influence bone marrow remodeling and hematopoiesis, also contributing to the differentiation of immature B cells (225). Besides ECs, lymphocytes also closely interact with stromal cells of mesenchymal origin: mesenchymal stromal cells (MSCs) in the BM and both fibroblastic reticular cells (FRCs) and follicular dendritic cells (FDCs) in the LNs. MSCs primarily function to create a reticular network in the BM and to produce factors essential to lymphoid lineage development and differentiation. MSCs are also a key component of the regenerative system and they possess the ability to migrate to a damaged tissue, promote its repair and suppress the associated inflammation through a number of immunomodulatory factors (227). In the LN, FRCs and FDCs maintain immune system homeostasis by guiding the correct compartmentalization of immune cells through the display and secretion of chemokines and cytokines. They also actively participate in the control of immune responses by regulating the recruitment and the activation status of myeloid cells and lymphocytes using similar mechanisms to the ones described for MSCs (228). Thus, it is reasonable to hypothesize that stromal cells could similarly participate in shaping the immune TME, with potential contribution to the immunosuppressive state in B cell malignancies.
Several studies have shown that stromal cell architecture is altered in both the BM and LN tissue compartments of patients with different hematological malignancies (229, 230). Increased proliferation of VEGFR-1+ neovasculature has been observed in aggressive lymphomas (DLBCL and Burkitt’s lymphoma), and an elevated numbers of αSMA+ mesenchymal cells described in indolent NHLs including CLL and small lymphocytic lymphoma (SLL) (231). Despite this general distinction, the stromal landscape of hematological malignancies is far more complex, with evidence that both ECs and fibroblasts cooperate in shaping disease biology and promoting tumor progression. Angiogenesis contributes to CLL development and correlates with disease progression (232–234). In line with this observation, overexpression of vascular endothelial growth factor (VEGF) in tissue biopsies has been shown to be a promising prognostic factor for NHL (235). In aggressive DLBCL, Lenz and colleagues described two different stromal gene signatures (stromal 1 and stromal 2) associated with patient survival following CHOP and R-CHOP treatment. The ‘stromal-2’ signature comprised of angiogenesis-associated genes (including CD31, VEGF), increased tumor blood-vessel density and correlated with poor prognosis following R-CHOP. On the other hand, the prognostically favorable stromal-1 signature showed enrichment for fibroblasts and ECM-associated genes (236). The importance of fibroblasts in aggressive lymphoma was functionally demonstrated in the Eµ-Myc mouse model, where expanded FRCs created a pro-tumor niche directing the homing and survival of lymphoma cells in the spleen (237). It has also been demonstrated in CLL using the Em-TCL1 murine model, that splenic stromal cells co-evolve with disease, in particular an expansion and reprogramming of splenic fibroblasts that produce the pro-B cell cytokine CXCL13 (238). To complicate the picture further, endothelial and mesenchymal cells are intimately linked to each other as exemplified by PDGF-activated MSCs in CLL that were shown to up-regulate VEGF production and promote the ‘angiogenic switch’ associated with disease progression (239). The importance of crosstalk in CLL between tumor cells and stroma cells has been recently reviewed by Dubois et al. (240).
Altered vascular patterns and angiogenesis characterize aggressive and indolent lymphomas (241, 242). Tumor B cells secrete pro-angiogenic factors such as VEGF (243, 244), that promote the activation of endothelial cells and neoangiogenesis. In turn, as highlighted by numerous studies in CLL (245–248) and other hematological malignancies (249, 250), activated ECs critically contribute to malignant B cell survival through factors such as B-cell activating factor (BAFF) (245, 251, 252) (Figure 2A).
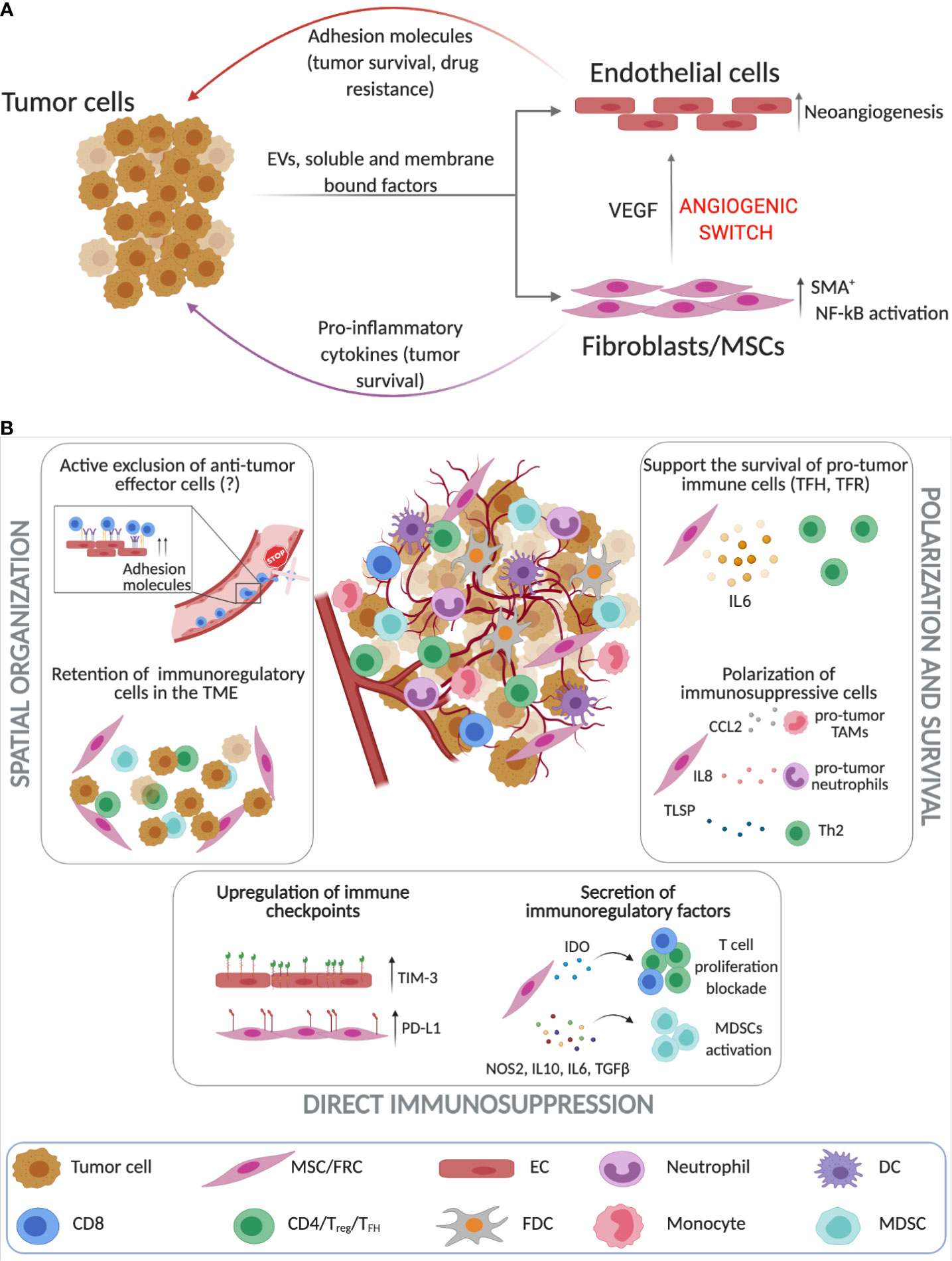
Figure 2 Stroma cells as key players in regulating immune responses in the TME of B cell malignancies. (A) Tumor cells and tissue-resident stroma (endothelial cells (ECs) and FRCs [fibroblastic reticular cells)/MSCs (mesenchymal stromal cells)] engage into complex bidirectional interactions that promote cancer progression while simultaneously altering the stroma cell phenotype which can then further contribute to resistance to therapy. Tumor B cells induce neoangiogensis and the upregulation of adhesion molecules on ECs (243–245, 248, 252). Similarly, lymphoma cells through cell-to-cell contact interactions, secretion of soluble factors and extracellular vesicles (EVs) promote the activation of FRCs and MSCs that contribute to increased tumor survival and neoangiogensis (135, 253–255). (B) Unlike solid tumors the investigation of the immunosuppressive roles of stroma cells in the lymphoma TME is still in its infancy. Stromal cells play a crucial role in spatial organization of the TME as they can retain immunoregulatory cells and possibly actively exclude anti-tumor effector cell populations (237, 253, 256–261). Additionally, lymphoma ECs and FRCs upregulate immune checkpoints such as TIM-3 and PD-L1 (261, 262) and secrete immunoregulatory factors such as IDO and IL-10 that block T cell proliferation, while activating immunosuppressive cells (263–266). FRCs have been also found to drive the survival of pro-tumoral immune subsets such as TFH, TH2 and TAMs (257–260). (Created with Biorender.com).
Moreover, CLL and HL cells secrete EVs that promote the acquisition of a CAF-like phenotype in previously healthy ECs and fibroblasts from different origins (267, 268). Importantly, tumor-derived EVs can also modulate other cellular components of the TME (269), and their role in B-cell malignancies has been recently reviewed (135).
In multiple myeloma (MM), the combination of both soluble (SDF-1α) and membrane-bound factors (integrins) induce stroma activation (ECs and BM-MSCs) (263), while in FL and CLL, tumor-derived TNFα and lymphotoxin (LT) are involved in the remodeling of BM-MSCs and LN-FDCs respectively (253, 254). These tumor-specific factors ultimately converge in the activation of NF-kB-dependent transcriptional programs (253, 263) that promote the secretion of pro-inflammatory soluble mediators involved in the enhancement of cancer survival (255) and in the modulation of TME immune infiltration (Figure 2A).
As described above, tumor-activated stroma has a direct impact on the retention of tumor B cells and could therefore impact the recruitment or exclusion of other lymphocytes via similar mechanisms. The upregulation of adhesion molecules on EC surfaces (250) may negatively impact T cell trans-endothelial migration, which is a critical step of T cell homing into the LNs (270). In CLL, CD4+ and CD8+ T cells exhibit impaired integrin lymphocyte function-associated antigen-1 (LFA-1)–driven migration (271). Altered expression of adhesion molecules on TECs, together with T cell motility defects, could therefore block effective T cell trafficking into LN tissues.
Although studies showing a direct impact of activated endothelium on T cell recruitment in hematological malignancies are lacking, recently de Weerdt et al. revealed that CLL LNs contained twice the amount of TRegs and a lower frequency of cytotoxic lymphocytes compared to the peripheral blood compartment (194). Under inflammatory conditions, activated ECs induce the expansion of TRegs (215, 216) and the proliferation of memory CD4+ T cells (272), and it could be speculated that similar mechanisms occur in the CLL TME.
Despite the lack of evidence for ECs, several studies have demonstrated the contribution of mesenchymal cells in the regulation of immune infiltration in B cell malignancies. In a mouse model of aggressive lymphoma, where tumor cells rely less on the TME for survival, the co-injection of MSCs with tumor cells induced a marked increase of immune cells including CD4+ T cells, CD11b+ cells, CD4+Foxp3+ TRegs, and CD11b+Ly6C+Ly6G− MDSCs (256), demonstrating that the CAF secretome can modulate the recruitment and/or polarization of different immune subpopulations. In another model of aggressive lymphoma (Eµ-Myc), T cell zone resident stromal cells were shown to selectively retain CD4+ T cells in the TME to support tumor progression via CD40L signaling (237).
In FL, where tumor cells strongly depend on TME interactions, stromal-derived IL-6 supports the survival of T-follicular helper (TFH) and T-follicular regulatory (TFR) cells (257). TFH cells, that sustain lymphoma through a number of mechanisms (258), were also shown to establish a feedback loop with stromal cells. IL-4, the predominant cytokine produced by TFH, was shown to trigger production of CXCL12 by FRC-like stromal cells, thus supporting FL tumor cell activation and survival (259). Additionally, FL-associated stroma (BM-MSCs) derived CCL2 and IL-8 were shown to promote the recruitment of monocytes/TAMs and neutrophils respectively, using in vitro assays (253, 260). Once in contact with FL-MSCs, monocytes/TAMs have been demonstrated to acquire an immunosuppressive phenotype with a reduced capacity to respond to pro-inflammatory stimuli such as LPS, while neutrophils supported the inflammatory stromal phenotype. Also in MM, malignant B cells have been shown to induce MSCs to secrete pro-tumor cytokines, as well thymic stromal lymphopoietin (TSLP) that activated TH2-type inflammation in the BM TME resulting in tumor progression (261) (Figure 2B).
Another important mechanism of how stromal cells contribute to an immunosuppressive TME is through the expression of immune checkpoint molecules. Lymphoma-derived ECs preferentially express TIM-3. Expression levels of TIM-3 in the B-cell lymphoma endothelium has been correlated with poor prognosis and TIM-3+ ECs suppressed the activation of CD4+ T cells inhibiting TH1 polarization in vitro and in vivo (273). In DLBCL, besides its expression on tumor and immune cells described earlier in this review, PD-L1 has also been associated to the non-malignant cellular compartment (“microenvironmental PD-L1”or mPD-L1) (262). Our own work has reported that PD-L1 upregulation on lymphoma-educated stromal cells can dampen TIL cytolytic killing activity against DLBCL tumor cells, highlighting that stromal cells expressing inhibitory ligands can directly modulate anti-tumor immune responses (274).
In addition to expression of immunomodulatory immune checkpoints, both LN and BM-derived stromal cells can adopt a number of other immunosuppressive mechanisms to modulate immune responses (Figure 2B). In MM, MSCs exhibit a distinctive gene expression profile compared to healthy MSCs, characterized by the expression of a number of immunoregulatory factors and cytokines including NOS2, IL-10, IL-6 and TGF-β, involved in the generation and activation of MDSCs (263, 264). In FL, MSCs also produce prostaglandin E2 (PGE2) that not only promotes neutrophil survival, but can have additional effect on other immune subpopulations (260). In physiological conditions, MSCs and LN-fibroblasts produce IDO to keep activated immune responses in check by inhibiting T cell proliferation, a mechanism also described in solid tumors (265). In FL, MSCs-derived IDO was also shown to repress not only T cell proliferation, but also the pro-lymphoma activity of stromal cells, highlighting the complex effects of cytokines in the TME (266). Clearly, our understanding of the immunomodulatory function of the different stromal cell populations that reside within lymphoma TMEs is still in its infancy. However, it is becoming clear that different non-hemopoietic cells within lymphoid TMEs can alter the recruitment, polarization and function of immune cells, suggesting that the tumor stroma may also influence immune responses elicited by current immunotherapy.
A number of studies have shown that the TME can interfere with clinical response to tumor-targeting therapy via different mechanisms. Reduced macrophage infiltration in the BM of ALL mice limits the response to anti-CD20 antibodies (alemtuzumab) (275), while adhesion to BM-derived stromal cells provides protection to CLL cells from rituximab-induced apoptosis (276). On the other side, tumor-targeting drugs can have indirect effects on the TME that may boost clinical responses. In CLL, there is evidence that ibrutinib treatment alleviates T cell exhaustion (277), while chemotherapy-induced cancer cell death promotes the exposure of tumor neoantigens and could re-invigorate anti-tumor immune responses (278). Understanding how the TME is shaped by currently available treatments can help to understand resistance mechanisms and to design combination therapies to boost clinical responses.
The immuno-oncology era has introduced a vast array of drugs designed to engage and promote innate and adaptive immune responses within the TME. Thus, lymphoma therapy is changing dramatically with the introduction of several new therapeutic approaches, including the use of checkpoint inhibitors (3). However, as described here, only a subset of patients achieve long-lasting responses to anti-PD-1 monotherapy even if they harbor inflamed TMEs. This clinical experience, together with the lack of clinical activity of anti-PD-1 in NHL (108) and CLL (5), has highlighted the need to incorporate checkpoint blockade therapies into more powerful combinations to unleash the power of anti-tumor immune cells, with potential therapeutic partners including CELMoDs and immunomodulatory drugs, CAR T cells and bispecific antibodies (Figure 3).
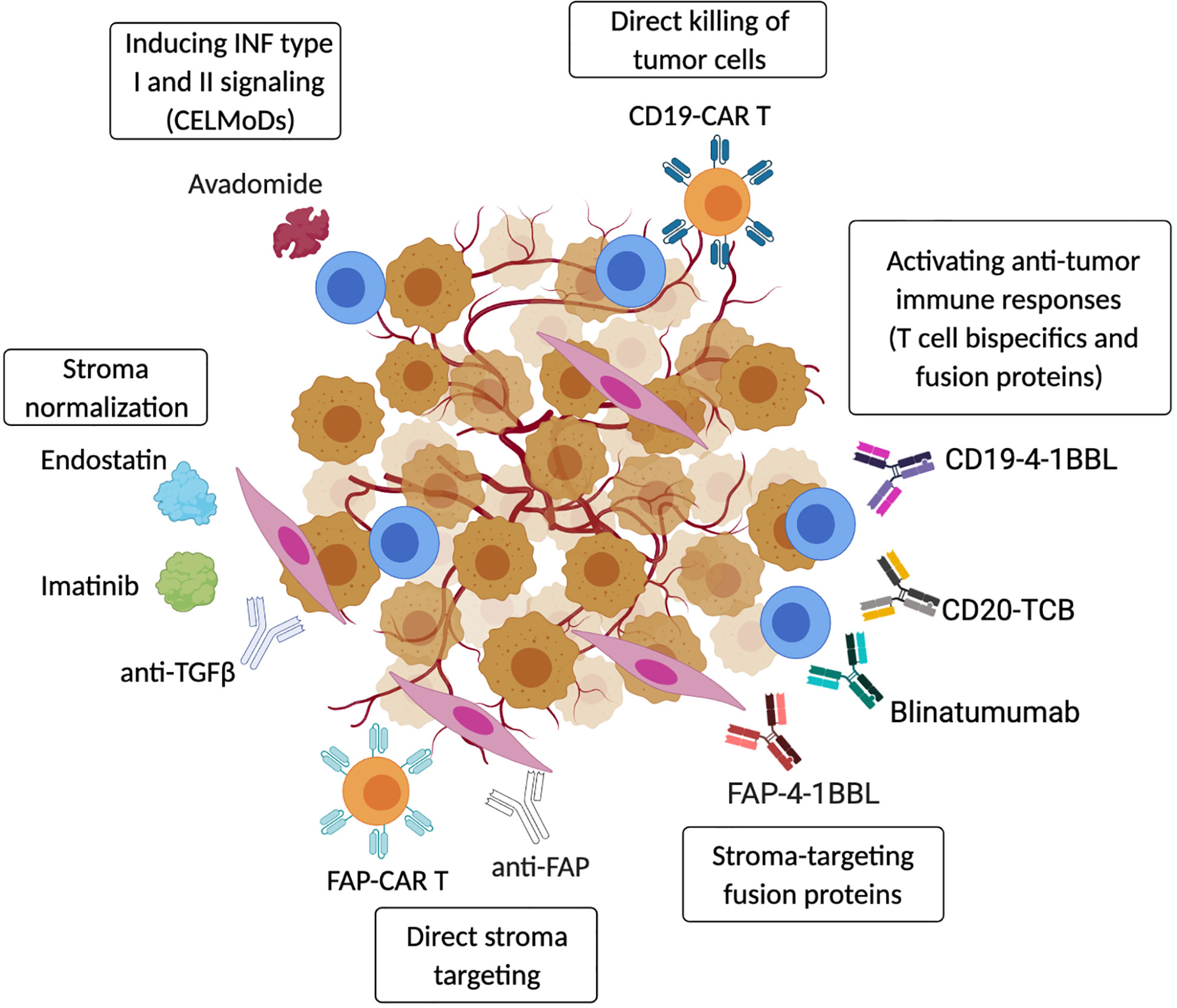
Figure 3 Targeting strategies to activate anti-tumor immunity in the TME. Therapeutic strategies include re-activation of autologous anti-tumor immune responses using CELMoDs (e.g. avadomide) (67) or T-cell bispecific antibodies and fusion proteins (CD20-TCB, CD19-4-1BBL, Blinatumumab) (279–282). Anti-tumor immunity can also be induced by transferring CAR-T/NK effector cells that directly target cancer cells (CD19-CAR-T) (283). In addition, cancer-associated stroma can be efficiently targeted to boost anti-tumor immunity, and fibroblasts-specific pharmacological approaches (anti-FAP antibodies or CAR-T) have been shown to induce tumor regression (284). Tumor stroma can also be ‘normalized’ by blocking or neutralizing cancer-secreted factors that promote stromal cell activation (Endostatin, Imatinib, anti-TGFβ) (285–287). Moreover, the presence of cancer-stroma-specific proteins can be used to activate tissue resident anti-tumor T cells using stroma-specific/T cells co-stimulatory fusion proteins (FAP-4-1BBL) (282). (Created with Biorender.com).
As discussed earlier, there is substantial evidence demonstrating that type I and II IFN signaling is required within TMEs to prevent development of an immunosuppressive state (37, 49). Recent work by our group has demonstrated that the CELMoD avadomide can induce type I and II IFN signaling in the T cell compartment that sensitizes CLL to anti-PD-1 or anti-PD-L1 checkpoint blockers (67). Avadomide was shown to trigger a feedforward cascade of reinvigorated T cell responses, as well as IFN-inducible feedback inhibition through upregulation of PD-L1. Patient-derived xenograft tumor models revealed that inducing IFN-driven T cell responses with avadomide could convert non-inflamed CLL tumors into CD8+ T cell-inflamed TMEs that responded to anti-PD-L1/PD-1-based combination therapy. This pre-clinical study provides encouraging proof of concept that inducing inflammatory IFN type I and II signaling in patient T cells can successfully re-shape anti-tumor T cell responses and sensitize CLL to immunotherapy (67).
The re-activation of autologous anti-tumor immune responses has been also demonstrated in a number of different hematological malignancies by using “off the shelf” bispecific T cell engager (BiTE) antibodies. This dual binding of both neoplastic cells and tumor-infiltrating lymphocytes is providing an attractive therapeutic approach for B cell malignancies (279). Blinatumumab, a BiTE with dual specificity for CD3 and CD19, has shown activity in ALL and in different NHL, and has been approved by FDA in 2014 (280). However, due to its short half-life, Blinatumumab requires continuous infusion for weeks, causing patients discomfort. Moreover, the occurrence of different resistance mechanisms described for ALL patients (effector T cell exhaustion/dysfunction and expansion of TRegs), prompted the optimization and testing of alternative BiTE drugs in recent years [reviewed in (288)]. Among them, the T cell bispecific (TCB) antibody drug CD20-TCB (RG6026), with its 2:1 CD20-CD3 binding format, has shown superior potency in T cell activation and increased half-life in pre-clinical settings (281). CD20-TCB can be efficiently combined with the bispecific antibody fusion protein CD19-4-1BBL, that provides co-stimulatory signals to tumor-engaged immune cells (T cells or NK cells) and this combination immunotherapy has been shown to promote intratumoral TILs accumulation and increased anti-tumor efficacy in preclinical models of NHL (282). These pre-clinical results suggest that combination immunotherapy may achieve better clinical responses and overcome immune suppression in the TME. Currently, BiTEs and CD20 TCBs, including RG6026, are being evaluated in B cell malignancy patients with activity detected.
CD19-directed CAR-T cells have shown remarkable efficiency in the treatment of ALL and aggressive NHL (283, 289, 290) and, along the same line, new CD19-CAR-NK cells are showing effective anti-tumor activity with minor side effects (291). Indeed, CAR-T cell therapy has been the first FDA-approved cellular therapy in lymphoma [covered in a recent review (4)]. Despite this clinical success, emerging data is showing that durable responses induced by CAR Ts are seen in only a subset of patients and their anti-tumor activity depends on the persistence of CARs (181). While peripheral blood analysis of treated patients has shown that CAR-T concentrations peak at 14 days post-infusion (292), recent data has demonstrated that only a minimal amount of CAR-T cells infiltrate TME tissue at 5 days post-infusion, and that virtually no CAR-Ts were present in the TME 10 days following infusion (104). These results support the concept that the stroma-rich TME may contain ill-defined immunosuppressive mechanisms that interfere with the effective trafficking and optimal activation of anti-tumor immune cells using current immunotherapies.
Combining immunotherapy with drugs that target stromal cells is an attractive, next-generation treatment strategy. Different approaches can be adopted to target stromal cells in cancer including the direct targeting of fibroblasts/ECs or their associated proteins, activated signaling pathways or secreted factors. The majority of studies to date targeting tumor stroma have used solid cancer models, establishing a strong rationale for developing therapies for hematological malignancies. A strong case in point is fibroblast activating protein (FAP), a cell-surface serine protease which is expressed at high levels on tumor stroma and has been considered a suitable therapeutic candidate on CAFs for some time. Several FAP-specific pharmacological approaches (vaccines, CAR-T cells) designed to selectively target CAFs, were shown to induce tumor regression in different murine models (284). However, depletion of FAP+ fibroblasts has been shown to cause profound systemic immune-mediated toxicity, indicating their importance not only in cancer but also in physiological tissue functions (293). This example suggests that caution should be taken in considering direct elimination of stromal cells forming the TME given their critical functions in tissue architecture and immune homeostasis. Therefore, therapies designed to ‘normalize’ or ‘re-educate’ aberrant stromal cells by targeting or overcoming their pro-tumor, immune evasive pathways could be more clinically relevant (Figure 3).
To this direction, stromal cell normalization can be achieved by directly blocking or neutralizing cancer-secreted factors that promote stromal cell activation. To date, in lymphoma this approach has been exploited through the use of endostatin, an endogenous inhibitor of angiogenesis which inhibits matrix metalloproteinases (MMP) activity and blocks VEGF binding to VEGFR-2. Administration of endostatin in lymphoma-bearing mice was shown to delay tumor growth (285). Imatinib, a PDGFRβ inhibitor, has also been shown to disrupt lymphoma angiogenesis by targeting vascular pericytes (286). Moreover, as PDGFR ligation also has a role in promoting fibroblast activation, the use of specific inhibitors can also affect fibroblast differentiation and function in the TME. As described above, TGFβ, TNFα and LT are all master regulators of immunosuppressive fibroblast function in hematological malignancies, and the use of neutralizing antibodies against these molecules could provide an interesting approach for stroma normalization, as already demonstrated in solid tumors (294). Combined blockade of TGFβ and anti-PD-1 in MM has been shown to promote anti-tumor T cell activation and proliferation, indicating that targeting the different immunosuppressive pathways occurring in the TME can reactivate endogenous anti-tumor immune responses and favor tumor clearance (287).
In conclusion, understanding the functional role of stroma and its specific features will help to design new combination immunotherapies to improve clinical responses. An example of effective immune-stroma dual targeting therapy comes from recent work from Claus and colleagues in solid tumors (282), where the combination of tumor antigen-TCB (CEA-TCB) with a stroma-specific/T cell co-stimulatory fusion protein (FAP-4-1BBL) promoted tumor remission and accumulation of activated CD8+ in the TME (Figure 3).
Immunotherapy has revolutionized cancer treatment, achieving significant responses in patients with B cell malignancies, although sensitivity and resistance remain major challenges. Despite the extreme inflamed vs non-inflamed classification of immune TMEs, most B cell malignancies and their subtypes show a high degree of heterogeneity that likely influences responses to immunotherapy. For this reason, it is crucial for further research to fully unravel TME complexity including the major tumor intrinsic and extrinsic drivers of immune composition and spatial organization of immune and stromal cell subsets. Defining how malignant B cells alter both immune and stromal cells, as well as how these reprogrammed cells contribute to the creation of a pro-tumor and immunosuppressive TME will be essential to design next-generation immunotherapies and combination treatment strategies to overcome TME-driven immune suppression and optimize therapy for patients.
All authors contributed to the article and approved the submitted version.
AGR has received research funding to Institution from Bristol-Myers Squibb Roche Glycart AG and AstraZeneca.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
CLL, chronic lymphocytic leukemia; SLL, small lymphocytic lymphoma; DLBCL, diffuse large B-cell lymphoma; cHL, classical non-Hodgkin lymphoma; NHL, non-Hodgkin lymphoma; MM, multiple myeloma; PMBL, primary mediastinal B-cell lymphoma; FL, follicular lymphoma; MCL, mantle-cell lymphoma; ALL, acute lymphoblastic leukemia; CRC, colorectal carcinomas; EBV, Epstein–Barr virus; GCB, non-germinal center B-cell subtype; R/R relapsed/refractory; ORR, overall response rate; TME, tumor microenvironment; LN(s), lymph nodes; BM, bone marrow; TILs, tumor-infiltrating lymphocytes; TAMs, tumor-associated macrophages; APCs, antigen-presenting cells; DCs, dendritic cells; NK(s), natural killer cells; FDCs, follicular dendritic cells; MDSCs, myeloid-derived suppressor cells; ECs, endothelial cells; TECs, tumor-associated endothelial cells; LECs, lymphatic endothelial cells; BECs, blood endothelial cells; HEVs, high endothelial venules; FRCs, fibroblastic reticular cells; CAFs, cancer-associated fibroblasts; MSCs, mesenchymal stromal cells; PD-1, programmed cell death protein 1; IFN, interferon; HLA, human leukocyte antigen; IL-, Interleukin-; MHC, major histocompatibility complex; TRAIL, TNF-related apoptosis-inducing ligand; CELMoD, cereblon E3 ligase modulator; IRF4, IFN regulatory factor 4; CTLA-4, cytotoxic T-lymphocyte-associated protein 4; CAR T cells, chimeric antigen receptor T cells; JAK2, Janus kinase 2; NF-kB, nuclear factor kappa-light-chain-enhancer of activated B cells; STAT3, signal transducer and activator of transcription 3; MAPK, mitogen-activated protein kinase; PTEN, phosphatase and tensin homolog; EZH2, enhancer of zeste homolog 2; TP53, tumor protein P53; BCL2, B-cell lymphoma 2; TGF-β, transforming growth factor beta; EVs, extracellular vesicles; ARG1, arginase 1; IDO, indoleamine-pyrrole 2,3-dioxygenase; LAG-3, lymphocyte activation gene-3; TIM-3, T-cell immunoglobulin and mucin-domain containing-3; FOXP3, forkhead box p3; NO, nitric oxide; ICAM-1, intercellular adhesion molecule 1; VCAM-1, vascular cell adhesion protein 1; CXCL, C-X-C motif chemokine ligand; CXCR, C-X-C motif chemokine receptor; CCR, (C-C motif) chemokine receptor; SDF-1, stromal cell-derived factor 1; MCP-1, monocyte chemoattractant protein-1; Chi3L1, chitinase 3 like 1; ECM, extracellular matrix; PGE2, prostaglandin E2; VEGF, vascular endothelial growth factor; αSMA, alpha smooth muscle actin; (R-)CHOP, (rituximab-), cyclophosphamide, doxorubicin hydrochloride (hydroxydaunorubicin), vincristine sulfate (Oncovin), and prednisone; PDGF, platelet-derived growth factor; BAFF, B-cell activating factor; LT, lymphotoxin; TSLP, thymic stromal lymphopoietin; NOS2, nitric oxide synthase 2; FAP, fibroblast activating protein.
CLL: chronic lymphocytic leukemia
SLL: small lymphocytic lymphoma
DLBCL: diffuse large B-cell lymphoma
cHL: classical non-Hodgkin lymphoma
NHL: non-Hodgkin lymphoma
MM: multiple myeloma
PMBL: primary mediastinal B-cell lymphoma
FL: follicular lymphoma
MCL: mantle-cell lymphoma
ALL: acute lymphoblastic leukemia
CRC: colorectal carcinomas
EBV: Epstein–Barr virus
GCB: non-germinal center B-cell subtype
R/R: relapsed/refractory
ORR: overall response rate
TME: tumor microenvironment
LN(s): lymph nodes
BM: bone marrow
TILs: tumor-infiltrating lymphocytes
TAMs: tumor-associated macrophages
APCs: antigen-presenting cells
DCs: dendritic cells
NK(s): natural killer cells
FDCs: follicular dendritic cells
MDSCs: myeloid-derived suppressor cells
ECs: endothelial cells
TECs: tumor-associated endothelial cells
LECs: lymphatic endothelial cells
BECs: blood endothelial cells
HEVs: high endothelial venules
FRCs: fibroblastic reticular cells
CAFs: cancer-associated fibroblasts
MSCs: mesenchymal stromal cells
PD-1: programmed cell death protein 1
IFN: interferon
HLA: human leukocyte antigen
IL-: Interleukin-
MHC: major histocompatibility complex
TRAIL: TNF-related apoptosis-inducing ligand
CELMoD: cereblon E3 ligase modulator
IRF4: IFN regulatory factor 4
CTLA-4: cytotoxic T-lymphocyte-associated protein 4
CAR T cells: chimeric antigen receptor T cells
JAK2: Janus kinase 2
NF-kB: nuclear factor kappa-light-chain-enhancer of activated B cells
STAT3: signal transducer and activator of transcription 3
MAPK: mitogen-activated protein kinase
PTEN: phosphatase and tensin homolog
EZH2: enhancer of zeste homolog 2
TP53: tumor protein P53
BCL2: B-cell lymphoma 2
TGF-β: transforming growth factor beta
EVs: extracellular vesicles
ARG1: arginase 1
IDO: indoleamine-pyrrole 2,3-dioxygenase
LAG-3: lymphocyte activation gene-3
TIM-3: T-cell immunoglobulin and mucin-domain containing-3
FOXP3: forkhead box p3
NO: nitric oxide
ICAM-1: intercellular adhesion molecule 1
VCAM-1: vascular cell adhesion protein 1
CXCL: C-X-C motif chemokine ligand
CXCR: C-X-C motif chemokine receptor
CCR: (C-C motif) chemokine receptor
SDF-1: stromal cell-derived factor 1
MCP-1: monocyte chemoattractant protein-1
Chi3L1: chitinase 3 like 1
ECM: extracellular matrix
PGE2: prostaglandin E2
VEGF: vascular endothelial growth factor
αSMA: alpha smooth muscle actin
R-)CHOP: (rituximab-) cyclophosphamide doxorubicin hydrochloride (hydroxydaunorubicin) vincristine sulfate (Oncovin) and prednisone
PDGF: platelet-derived growth factor
BAFF: B-cell activating factor
LT: lymphotoxin
TSLP: thymic stromal lymphopoietin
NOS2: nitric oxide synthase 2
FAP: fibroblast activating protein
1. Weinstock DM, Dalla-Favera R, Gascoyne RD, Leonard JP, Levy R, Lossos IS, et al. A roadmap for discovery and translation in lymphoma. Blood (2015) 125(13):2175–7. doi: 10.1182/blood-2015-01-623777
2. Ribas A, Wolchok JD. Cancer immunotherapy using checkpoint blockade. Science (2018) 359(6382):1350–5. doi: 10.1126/science.aar4060
3. Kline J, Godfrey J, Ansell SM. The immune landscape and response to immune checkpoint blockade therapy in lymphoma. Blood (2020) 135(8):523–33. doi: 10.1182/blood.2019000847
4. Ansell SM, Lin Y. Immunotherapy of lymphomas. J Clin Invest (2020) 130(4):1576–85. doi: 10.1172/JCI129206
5. Ding W, LaPlant BR, Call TG, Parikh SA, Leis JF, He R, et al. Pembrolizumab in patients with CLL and Richter transformation or with relapsed CLL. Blood (2017) 129(26):3419–27. doi: 10.1182/blood-2017-02-765685
6. Ramsay AG, Clear AJ, Fatah R, Gribben JG. Multiple inhibitory ligands induce impaired T-cell immunologic synapse function in chronic lymphocytic leukemia that can be blocked with lenalidomide: establishing a reversible immune evasion mechanism in human cancer. Blood (2012) 120(7):1412–21. doi: 10.1182/blood-2012-02-411678
7. Brusa D, Serra S, Coscia M, Rossi D, D’Arena G, Laurenti L, et al. The PD-1/PD-L1 axis contributes to T-cell dysfunction in chronic lymphocytic leukemia. Haematologica (2013) 98(6):953–63. doi: 10.3324/haematol.2012.077537
8. de Visser KE, Eichten A, Coussens LM. Paradoxical roles of the immune system during cancer development. Nat Rev Cancer (2006) 6(1):24–37. doi: 10.1038/nrc1782
9. O’Donnell JS, Teng MWL, Smyth MJ. Cancer immunoediting and resistance to T cell-based immunotherapy. Nat Rev Clin Oncol (2019) 16(3):151–67. doi: 10.1038/s41571-018-0142-8
10. Mittal D, Gubin MM, Schreiber RD, Smyth MJ. New insights into cancer immunoediting and its three component phases–elimination, equilibrium and escape. Curr Opin Immunol (2014) 27:16–25. doi: 10.1016/j.coi.2014.01.004
11. Teng MW, Galon J, Fridman WH, Smyth MJ. From mice to humans: developments in cancer immunoediting. J Clin Invest (2015) 125(9):3338–46. doi: 10.1172/JCI80004
12. Scott DW, Gascoyne RD. The tumour microenvironment in B cell lymphomas. Nat Rev Cancer (2014) 14(8):517–34. doi: 10.1038/nrc3774
13. Disis ML. Immune regulation of cancer. J Clin Oncol (2010) 28(29):4531–8. doi: 10.1200/JCO.2009.27.2146
14. Khodadoust MS, Olsson N, Wagar LE, Haabeth OA, Chen B, Swaminathan K, et al. Antigen presentation profiling reveals recognition of lymphoma immunoglobulin neoantigens. Nature (2017) 543(7647):723–7. doi: 10.1038/nature21433
15. Khodadoust MS, Olsson N, Chen B, Sworder B, Shree T, Liu CL, et al. B-cell lymphomas present immunoglobulin neoantigens. Blood (2019) 133(8):878–81. doi: 10.1182/blood-2018-06-845156
16. Kowalewski DJ, Schuster H, Backert L, Berlin C, Kahn S, Kanz L, et al. HLA ligandome analysis identifies the underlying specificities of spontaneous antileukemia immune responses in chronic lymphocytic leukemia (CLL). Proc Natl Acad Sci U S A (2015) 112(2):E166–75. doi: 10.1073/pnas.1416389112
17. Steer HJ, Lake RA, Nowak AK, Robinson BWS. Harnessing the immune response to treat cancer. Oncogene (2010) 29(48):6301–13. doi: 10.1038/onc.2010.437
18. Galon J, Bruni D. Approaches to treat immune hot, altered and cold tumours with combination immunotherapies. Nat Rev Drug Discov (2019) 18(3):197–218. doi: 10.1038/s41573-018-0007-y
19. Shankaran V, Ikeda H, Bruce AT, White JM, Swanson PE, Old LJ, et al. IFNgamma and lymphocytes prevent primary tumour development and shape tumour immunogenicity. Nature (2001) 410(6832):1107–11. doi: 10.1038/35074122
20. Alspach E, Lussier DM, Schreiber RD. Interferon gamma and Its Important Roles in Promoting and Inhibiting Spontaneous and Therapeutic Cancer Immunity. Cold Spring Harb Perspect Biol (2019) 11(3). doi: 10.1101/cshperspect.a028480
21. Albert ML, Darnell RB. Paraneoplastic neurological degenerations: keys to tumour immunity. Nat Rev Cancer (2004) 4(1):36–44. doi: 10.1038/nrc1255
22. Smyth MJ, Thia KY, Street SE, MacGregor D, Godfrey DI, Trapani JA. Perforin-mediated cytotoxicity is critical for surveillance of spontaneous lymphoma. J Exp Med (2000) 192(5):755–60. doi: 10.1084/jem.192.5.755
23. Hanna BS, Roessner PM, Yazdanparast H, Colomer D, Campo E, Kugler S, et al. Control of chronic lymphocytic leukemia development by clonally-expanded CD8(+) T-cells that undergo functional exhaustion in secondary lymphoid tissues. Leukemia (2019) 33(3):625–37. doi: 10.1038/s41375-018-0250-6
24. Groscurth P, Filgueira L. Killing Mechanisms of Cytotoxic T Lymphocytes. Physiology (1998) 13(1):17–21. doi: 10.1152/physiologyonline.1998.13.1.17
25. Farhood B, Najafi M, Mortezaee K. CD8(+) cytotoxic T lymphocytes in cancer immunotherapy: A review. J Cell Physiol (2019) 234(6):8509–21. doi: 10.1002/jcp.27782
26. Ahrends T, Borst J. The opposing roles of CD4+ T cells in anti-tumour immunity. Immunology (2018) 154(4):582–92. doi: 10.1111/imm.12941
27. Wang J, Ke X-Y. The Four types of Tregs in malignant lymphomas. J Hematol Oncol (2011) 4(1):50. doi: 10.1186/1756-8722-4-50
28. Bagnara D, Kaufman MS, Calissano C, Marsilio S, Patten PEM, Simone R, et al. A novel adoptive transfer model of chronic lymphocytic leukemia suggests a key role for T lymphocytes in the disease. Blood (2011) 117(20):5463–72. doi: 10.1182/blood-2010-12-324210
29. Lad DP, Varma S, Varma N, Sachdeva MU, Bose P, Malhotra P. Regulatory T-cells in B-cell chronic lymphocytic leukemia: their role in disease progression and autoimmune cytopenias. Leuk Lymphoma (2013) 54(5):1012–9. doi: 10.3109/10428194.2012.728287
30. Elston L, Fegan C, Hills R, Hashimdeen SS, Walsby E, Henley P, et al. Increased frequency of CD4(+) PD-1(+) HLA-DR(+) T cells is associated with disease progression in CLL. Br J Haematol (2020) 188(6):872–80. doi: 10.1111/bjh.16260
31. Kocher T, Asslaber D, Zaborsky N, Flenady S, Denk U, Reinthaler P, et al. CD4+ T cells, but not non-classical monocytes, are dispensable for the development of chronic lymphocytic leukemia in the TCL1-tg murine model. Leukemia (2016) 30(6):1409–13. doi: 10.1038/leu.2015.307
32. Quezada SA, Simpson TR, Peggs KS, Merghoub T, Vider J, Fan X, et al. Tumor-reactive CD4(+) T cells develop cytotoxic activity and eradicate large established melanoma after transfer into lymphopenic hosts. J Exp Med (2010) 207(3):637–50. doi: 10.1084/jem.20091918
33. Xia A, Zhang Y, Xu J, Yin T, Lu X-J. T Cell Dysfunction in Cancer Immunity and Immunotherapy. Front Immunol (2019) 10:1719–. doi: 10.3389/fimmu.2019.01719
34. Wherry EJ, Kurachi M. Molecular and cellular insights into T cell exhaustion. Nat Rev Immunol (2015) 15(8):486–99. doi: 10.1038/nri3862
35. Kaplan DH, Shankaran V, Dighe AS, Stockert E, Aguet M, Old LJ, et al. Demonstration of an interferon gamma-dependent tumor surveillance system in immunocompetent mice. Proc Natl Acad Sci U S A (1998) 95(13):7556–61. doi: 10.1073/pnas.95.13.7556
36. Street SE, Cretney E, Smyth MJ. Perforin and interferon-gamma activities independently control tumor initiation, growth, and metastasis. Blood (2001) 97(1):192–7. doi: 10.1182/blood.V97.1.192
37. Dunn GP, Koebel CM, Schreiber RD. Interferons, immunity and cancer immunoediting. Nat Rev Immunol (2006) 6(11):836–48. doi: 10.1038/nri1961
38. Parker BS, Rautela J, Hertzog PJ. Antitumour actions of interferons: implications for cancer therapy. Nat Rev Cancer (2016) 16(3):131–44. doi: 10.1038/nrc.2016.14
39. Ivashkiv LB. IFNgamma: signalling, epigenetics and roles in immunity, metabolism, disease and cancer immunotherapy. Nat Rev Immunol (2018) 18(9):545–58. doi: 10.1038/s41577-018-0029-z
40. Propper DJ, Chao D, Braybrooke JP, Bahl P, Thavasu P, Balkwill F, et al. Low-dose IFN-gamma induces tumor MHC expression in metastatic malignant melanoma. Clin Cancer Res (2003) 9(1):84–92.
41. Boyer CM, Dawson DV, Neal SE, Winchell LF, Leslie DS, Ring D, et al. Differential induction by interferons of major histocompatibility complex-encoded and non-major histocompatibility complex-encoded antigens in human breast and ovarian carcinoma cell lines. Cancer Res (1989) 49(11):2928–34.
42. Koebel CM, Vermi W, Swann JB, Zerafa N, Rodig SJ, Old LJ, et al. Adaptive immunity maintains occult cancer in an equilibrium state. Nature (2007) 450(7171):903–7. doi: 10.1038/nature06309
43. Haabeth OA, Lorvik KB, Hammarstrom C, Donaldson IM, Haraldsen G, Bogen B, et al. Inflammation driven by tumour-specific Th1 cells protects against B-cell cancer. Nat Commun (2011) 2:240. doi: 10.1038/ncomms1239
44. Godfrey J, Tumuluru S, Bao R, Leukam M, Venkataraman G, Phillip J, et al. PD-L1 gene alterations identify a subset of diffuse large B-cell lymphoma harboring a T-cell-inflamed phenotype. Blood (2019) 133(21):2279–90. doi: 10.1182/blood-2018-10-879015
45. Jones EA, Pringle JH, Angel CA, Rees RC. Th1/Th2 cytokine expression and its relationship with tumor growth in B cell non-Hodgkin’s lymphoma (NHL). Leuk Lymphoma (2002) 43(6):1313–21. doi: 10.1080/10428190290026385
46. Monti S, Savage KJ, Kutok JL, Feuerhake F, Kurtin P, Mihm M, et al. Molecular profiling of diffuse large B-cell lymphoma identifies robust subtypes including one characterized by host inflammatory response. Blood (2005) 105(5):1851–61. doi: 10.1182/blood-2004-07-2947
47. Mori T, Takada R, Watanabe R, Okamoto S, Ikeda Y. T-helper (Th)1/Th2 imbalance in patients with previously untreated B-cell diffuse large cell lymphoma. Cancer Immunol Immunother (2001) 50(10):566–8. doi: 10.1007/s00262-001-0232-8
48. Aue G, Sun C, Liu D, Park JH, Pittaluga S, Tian X, et al. Activation of Th1 Immunity within the Tumor Microenvironment Is Associated with Clinical Response to Lenalidomide in Chronic Lymphocytic Leukemia. J Immunol (2018) 201(7):1967–74. doi: 10.4049/jimmunol.1800570
49. Zitvogel L, Galluzzi L, Kepp O, Smyth MJ, Kroemer G. Type I interferons in anticancer immunity. Nat Rev Immunol (2015) 15(7):405–14. doi: 10.1038/nri3845
50. Kim R, Emi M, Tanabe K. Cancer immunoediting from immune surveillance to immune escape. Immunology (2007) 121(1):1–14. doi: 10.1111/j.1365-2567.2007.02587.x
51. Musella M, Manic G, De Maria R, Vitale I, Sistigu A. Type-I-interferons in infection and cancer: Unanticipated dynamics with therapeutic implications. Oncoimmunology (2017) 6(5):e1314424–e. doi: 10.1080/2162402X.2017.1314424
52. Sangfelt, Strander H. Apoptosis and cell growth inhibition as antitumor effector functions of interferons. Med Oncol (2001) 18(1):3–14. doi: 10.1385/MO:18:1:3
53. Thyrell L, Erickson S, Zhivotovsky B, Pokrovskaja K, Sangfelt O, Castro J, et al. Mechanisms of Interferon-alpha induced apoptosis in malignant cells. Oncogene (2002) 21(8):1251–62. doi: 10.1038/sj.onc.1205179
54. Dunn IS, Haggerty TJ, Kono M, Durda PJ, Butera D, Macdonald DB, et al. Enhancement of human melanoma antigen expression by IFN-beta. J Immunol (2007) 179(4):2134–42. doi: 10.4049/jimmunol.179.4.2134
55. Lanza L, Peirano L, Bosco O, Contini P, Filaci G, Setti M, et al. Interferons up-regulate with different potency HLA class I antigen expression in M14 human melanoma cell line. Possible interaction with glucocorticoid hormones. Cancer Immunol Immunother (1995) 41(1):23–8. doi: 10.1007/BF01788956
56. Dunn GP, Bruce AT, Sheehan KC, Shankaran V, Uppaluri R, Bui JD, et al. A critical function for type I interferons in cancer immunoediting. Nat Immunol (2005) 6(7):722–9. doi: 10.1038/ni1213
57. Fuertes MB, Kacha AK, Kline J, Woo SR, Kranz DM, Murphy KM, et al. Host type I IFN signals are required for antitumor CD8+ T cell responses through CD8{alpha}+ dendritic cells. J Exp Med (2011) 208(10):2005–16. doi: 10.1084/jem.20101159
58. Diamond MS, Kinder M, Matsushita H, Mashayekhi M, Dunn GP, Archambault JM, et al. Type I interferon is selectively required by dendritic cells for immune rejection of tumors. J Exp Med (2011) 208(10):1989–2003. doi: 10.1084/jem.20101158
59. Guillot B, Portales P, Thanh AD, Merlet S, Dereure O, Clot J, et al. The expression of cytotoxic mediators is altered in mononuclear cells of patients with melanoma and increased by interferon-alpha treatment. Br J Dermatol (2005) 152(4):690–6. doi: 10.1111/j.1365-2133.2005.06512.x
60. Katlinski KV, Gui J, Katlinskaya YV, Ortiz A, Chakraborty R, Bhattacharya S, et al. Inactivation of Interferon Receptor Promotes the Establishment of Immune Privileged Tumor Microenvironment. Cancer Cell (2017) 31(2):194–207. doi: 10.1016/j.ccell.2017.01.004
61. Bacher N, Raker V, Hofmann C, Graulich E, Schwenk M, Baumgrass R, et al. Interferon-alpha suppresses cAMP to disarm human regulatory T cells. Cancer Res (2013) 73(18):5647–56. doi: 10.1158/0008-5472.CAN-12-3788
62. Zoglmeier C, Bauer H, Noerenberg D, Wedekind G, Bittner P, Sandholzer N, et al. CpG blocks immunosuppression by myeloid-derived suppressor cells in tumor-bearing mice. Clin Cancer Res (2011) 17(7):1765–75. doi: 10.1158/1078-0432.CCR-10-2672
63. Gresser I, Maury C, Brouty-Boyé D. Mechanism of the antitumour effect of interferon in mice. Nature (1972) 239(5368):167–8. doi: 10.1038/239167a0
64. Hagner PR, Man HW, Fontanillo C, Wang M, Couto S, Breider M, et al. CC-122, a pleiotropic pathway modifier, mimics an interferon response and has antitumor activity in DLBCL. Blood (2015) 126(6):779–89. doi: 10.1182/blood-2015-02-628669
65. Yang Y, Shaffer AL,3, Emre NC, Ceribelli M, Zhang M, Wright G, et al. Exploiting synthetic lethality for the therapy of ABC diffuse large B cell lymphoma. Cancer Cell (2012) 21(6):723–37. doi: 10.1016/j.ccr.2012.05.024
66. Kater AP, Tonino SH, Egle A, Ramsay AG. How does lenalidomide target the chronic lymphocytic leukemia microenvironment? Blood (2014) 124(14):2184–9. doi: 10.1182/blood-2014-05-578286
67. Ioannou N, Hagner PR, Stokes M, Gandhi AK, Apollonio B, Fanous M, et al. Triggering interferon signaling in T cells with avadomide sensitizes CLL to anti-PD-L1/PD-1 immunotherapy. Blood (2020). doi: 10.1182/blood.2020006073
68. Xuan C, Steward KK, Timmerman JM, Morrison SL. Targeted delivery of interferon-alpha via fusion to anti-CD20 results in potent antitumor activity against B-cell lymphoma. Blood (2010) 115(14):2864–71. doi: 10.1182/blood-2009-10-250555
69. Yang X, Zhang X, Fu ML, Weichselbaum RR, Gajewski TF, Guo Y, et al. Targeting the Tumor Microenvironment with Interferon-β Bridges Innate and Adaptive Immune Responses. Cancer Cell (2014) 25(1):37–48. doi: 10.1016/j.ccr.2013.12.004
70. Zhang L, Tai Y-T, Ho MZG, Qiu L, Anderson KC. Interferon-alpha-based immunotherapies in the treatment of B cell-derived hematologic neoplasms in today’s treat-to-target era. Exp Hematol Oncol (2017) 6(1):20. doi: 10.1186/s40164-017-0081-6
71. Jewell AP, Worman CP, Lydyard PM, Yong KL, Giles FJ, Goldstone AH. Interferon-alpha up-regulates bcl-2 expression and protects B-CLL cells from apoptosis in vitro and in vivo. Br J Haematol (1994) 88(2):268–74. doi: 10.1111/j.1365-2141.1994.tb05017.x
72. Tomic J, Lichty B, Spaner DE. Aberrant interferon-signaling is associated with aggressive chronic lymphocytic leukemia. Blood (2011) 117(9):2668–80. doi: 10.1182/blood-2010-05-285999
73. Ngo VN, Young RM, Schmitz R, Jhavar S, Xiao W, Lim K-H, et al. Oncogenically active MYD88 mutations in human lymphoma. Nature (2011) 470(7332):115–9. doi: 10.1038/nature09671
74. Buschle M, Campana D, Carding SR, Richard C, Hoffbrand AV, Brenner MK. Interferon gamma inhibits apoptotic cell death in B cell chronic lymphocytic leukemia. J Exp Med (1993) 177(1):213–8. doi: 10.1084/jem.177.1.213
75. Herishanu Y, Perez-Galan P, Liu D, Biancotto A, Pittaluga S, Vire B, et al. The lymph node microenvironment promotes B-cell receptor signaling, NF-kappaB activation, and tumor proliferation in chronic lymphocytic leukemia. Blood (2011) 117(2):563–74. doi: 10.1182/blood-2010-05-284984
76. Critchley-Thorne RJ, Simons DL, Yan N, Miyahira AK, Dirbas FM, Johnson DL, et al. Impaired interferon signaling is a common immune defect in human cancer. Proc Natl Acad Sci U S A (2009) 106(22):9010–5. doi: 10.1073/pnas.0901329106
77. Mlecnik B, Tosolini M, Kirilovsky A, Berger A, Bindea G, Meatchi T, et al. Histopathologic-based prognostic factors of colorectal cancers are associated with the state of the local immune reaction. J Clin Oncol (2011) 29(6):610–8. doi: 10.1200/JCO.2010.30.5425
78. Galon J, Costes A, Sanchez-Cabo F, Kirilovsky A, Mlecnik B, Lagorce-Pagès C, et al. Type, Density, and Location of Immune Cells Within Human Colorectal Tumors Predict Clinical Outcome. Science (2006) 313(5795):1960–4. doi: 10.1126/science.1129139
79. Jacquelot N, Roberti MP, Enot DP, Rusakiewicz S, Semeraro M, Jégou S, et al. Immunophenotyping of Stage III Melanoma Reveals Parameters Associated with Patient Prognosis. J Invest Dermatol (2016) 136(5):994–1001. doi: 10.1016/j.jid.2015.12.042
80. Rådestad E, Klynning C, Stikvoort A, Mogensen O, Nava S, Magalhaes I, et al. Immune profiling and identification of prognostic immune-related risk factors in human ovarian cancer. OncoImmunology (2019) 8(2):e1535730. doi: 10.1080/2162402X.2018.1535730
81. Pages F, Mlecnik B, Marliot F, Bindea G, Ou FS, Bifulco C, et al. International validation of the consensus Immunoscore for the classification of colon cancer: a prognostic and accuracy study. Lancet (2018) 391(10135):2128–39. doi: 10.1016/S0140-6736(18)30789-X
82. Galon J, Mlecnik B, Bindea G, Angell HK, Berger A, Lagorce C, et al. Towards the introduction of the ‘Immunoscore’ in the classification of malignant tumours. J Pathol (2014) 232(2):199–209. doi: 10.1002/path.4287
83. Ayers M, Lunceford J, Nebozhyn M, Murphy E, Loboda A, Kaufman DR, et al. IFN-gamma-related mRNA profile predicts clinical response to PD-1 blockade. J Clin Invest (2017) 127(8):2930–40. doi: 10.1172/JCI91190
84. Trujillo JA, Sweis RF, Bao R, Luke JJ. T Cell-Inflamed versus Non-T Cell-Inflamed Tumors: A Conceptual Framework for Cancer Immunotherapy Drug Development and Combination Therapy Selection. Cancer Immunol Res (2018) 6(9):990–1000. doi: 10.1158/2326-6066.CIR-18-0277
85. Gajewski TF. The Next Hurdle in Cancer Immunotherapy: Overcoming the Non-T-Cell-Inflamed Tumor Microenvironment. Semin Oncol (2015) 42(4):663–71. doi: 10.1053/j.seminoncol.2015.05.011
86. Pourgheysari B, Bruton R, Parry H, Billingham L, Fegan C, Murray J, et al. The number of cytomegalovirus-specific CD4+ T cells is markedly expanded in patients with B-cell chronic lymphocytic leukemia and determines the total CD4+ T-cell repertoire. Blood (2010) 116(16):2968–74. doi: 10.1182/blood-2009-12-257147
87. Keane C, Gould C, Jones K, Hamm D, Talaulikar D, Ellis J, et al. The T-cell Receptor Repertoire Influences the Tumor Microenvironment and Is Associated with Survival in Aggressive B-cell Lymphoma. Clin Cancer Res (2017) 23(7):1820–8. doi: 10.1158/1078-0432.CCR-16-1576
88. Bruni D, Angell HK, Galon J. The immune contexture and Immunoscore in cancer prognosis and therapeutic efficacy. Nat Rev Cancer (2020). doi: 10.1038/s41568-020-0285-7
89. Fehrenbacher L, Spira A, Ballinger M, Kowanetz M, Vansteenkiste J, Mazieres J, et al. Atezolizumab versus docetaxel for patients with previously treated non-small-cell lung cancer (POPLAR): a multicentre, open-label, phase 2 randomised controlled trial. Lancet (2016) 387(10030):1837–46. doi: 10.1016/S0140-6736(16)00587-0
90. Cristescu R, Mogg R, Ayers M, Albright A, Murphy E, Yearley J, et al. Pan-tumor genomic biomarkers for PD-1 checkpoint blockade-based immunotherapy. Science (2018) 362(6411). doi: 10.1126/science.aar3593
91. Grasso CS, Tsoi J, Onyshchenko M, Abril-Rodriguez G, Ross-Macdonald P, Wind-Rotolo M, et al. Conserved Interferon-γ Signaling Drives Clinical Response to Immune Checkpoint Blockade Therapy in Melanoma. Cancer Cell (2020) 38(4):500–15.e3. doi: 10.1016/j.ccell.2020.08.005
92. Fife BT, Guleria I, Gubbels Bupp M, Eagar TN, Tang Q, Bour-Jordan H, et al. Insulin-induced remission in new-onset NOD mice is maintained by the PD-1-PD-L1 pathway. J Exp Med (2006) 203(12):2737–47. doi: 10.1084/jem.20061577
93. Wei SC, Levine JH, Cogdill AP, Zhao Y, Anang N-AAS, Andrews MC, et al. Distinct Cellular Mechanisms Underlie Anti-CTLA-4 and Anti-PD-1 Checkpoint Blockade. Cell (2017) 170(6):1120–33.e17. doi: 10.1016/j.cell.2017.07.024
94. Krieg C, Nowicka M, Guglietta S, Schindler S, Hartmann FJ, Weber LM, et al. High-dimensional single-cell analysis predicts response to anti-PD-1 immunotherapy. Nat Med (2018) 24(2):144–53. doi: 10.1038/nm.4466
95. Wienand K, Chapuy B, Stewart C, Dunford AJ, Wu D, Kim J, et al. Genomic analyses of flow-sorted Hodgkin Reed-Sternberg cells reveal complementary mechanisms of immune evasion. Blood Adv (2019) 3(23):4065–80. doi: 10.1182/bloodadvances.2019001012
96. Cader FZ, Schackmann RCJ, Hu X, Wienand K, Redd R, Chapuy B, et al. Mass cytometry of Hodgkin lymphoma reveals a CD4(+) regulatory T-cell-rich and exhausted T-effector microenvironment. Blood (2018) 132(8):825–36. doi: 10.1182/blood-2018-04-843714
97. Greaves P, Clear A, Owen A, Iqbal S, Lee A, Matthews J, et al. Defining characteristics of classical Hodgkin lymphoma microenvironment T-helper cells. Blood (2013) 122(16):2856–63. doi: 10.1182/blood-2013-06-508044
98. Ribas A, Wolchok JD. Combining cancer immunotherapy and targeted therapy. Curr Opin Immunol (2013) 25(2):291–6. doi: 10.1016/j.coi.2013.02.011
99. Cader FZ, Hu X, Goh WL, Wienand K, Ouyang J, Mandato E, et al. A peripheral immune signature of responsiveness to PD-1 blockade in patients with classical Hodgkin lymphoma. Nat Med (2020) 26(9):1468–79. doi: 10.1038/s41591-020-1006-1
100. Lindqvist CA, Christiansson LH, Thorn I, Mangsbo S, Paul-Wetterberg G, Sundstrom C, et al. Both CD4+ FoxP3+ and CD4+ FoxP3- T cells from patients with B-cell malignancy express cytolytic markers and kill autologous leukaemic B cells in vitro. Immunology (2011) 133(3):296–306. doi: 10.1111/j.1365-2567.2011.03439.x
101. Porakishvili N, Roschupkina T, Kalber T, Jewell AP, Patterson K, Yong K, et al. Expansion of CD4+ T cells with a cytotoxic phenotype in patients with B-chronic lymphocytic leukaemia (B-CLL). Clin Exp Immunol (2001) 126(1):29–36. doi: 10.1046/j.1365-2249.2001.01639.x
102. Yang ZZ, Kim HJ, Villasboas JC, Chen YP, Price-Troska T, Jalali S, et al. Expression of LAG-3 defines exhaustion of intratumoral PD-1(+) T cells and correlates with poor outcome in follicular lymphoma. Oncotarget (2017) 8(37):61425–39. doi: 10.18632/oncotarget.18251
103. Spranger S, Dai D, Horton B, Gajewski TF. Tumor-Residing Batf3 Dendritic Cells Are Required for Effector T Cell Trafficking and Adoptive T Cell Therapy. Cancer Cell (2017) 31(5):711–23 e4. doi: 10.1016/j.ccell.2017.04.003
104. Chen PH, Lipschitz M, Weirather JL, Jacobson C, Armand P, Wright K, et al. Activation of CAR and non-CAR T cells within the tumor microenvironment following CAR T cell therapy. JCI Insight (2020) 5(12). doi: 10.1172/jci.insight.134612
105. Carpio C, Bouabdallah R, Ysebaert L, Sancho JM, Salles G, Cordoba R, et al. Avadomide monotherapy in relapsed/refractory DLBCL: safety, efficacy, and a predictive gene classifier. Blood (2020) 135(13):996–1007. doi: 10.1182/blood.2019002395
106. Dave SS, Wright G, Tan B, Rosenwald A, Gascoyne RD, Chan WC, et al. Prediction of survival in follicular lymphoma based on molecular features of tumor-infiltrating immune cells. N Engl J Med (2004) 351(21):2159–69. doi: 10.1056/NEJMoa041869
107. Byers RJ, Sakhinia E, Joseph P, Glennie C, Hoyland JA, Menasce LP, et al. Clinical quantitation of immune signature in follicular lymphoma by RT-PCR-based gene expression profiling. Blood (2008) 111(9):4764–70. doi: 10.1182/blood-2007-10-115915
108. Ansell SM, Minnema MC, Johnson P, Timmerman JM, Armand P, Shipp MA, et al. Nivolumab for Relapsed/Refractory Diffuse Large B-Cell Lymphoma in Patients Ineligible for or Having Failed Autologous Transplantation: A Single-Arm, Phase II Study. J Clin Oncol (2019) 37(6):481–9. doi: 10.1200/JCO.18.00766
109. Xu-Monette ZY, Xiao M, Au Q, Padmanabhan R, Xu B, Hoe N, et al. Immune Profiling and Quantitative Analysis Decipher the Clinical Role of Immune-Checkpoint Expression in the Tumor Immune Microenvironment of DLBCL. Cancer Immunol Res (2019) 7(4):644–57. doi: 10.1158/2326-6066.CIR-18-0439
110. Ennishi D, Takata K, Béguelin W, Duns G, Mottok A, Farinha P, et al. Molecular and Genetic Characterization of MHC Deficiency Identifies EZH2 as Therapeutic Target for Enhancing Immune Recognition. Cancer Discov (2019) 9(4):546–63. doi: 10.1158/2159-8290.CD-18-1090
111. Ennishi D, Jiang A, Boyle M, Collinge B, Grande BM, Ben-Neriah S, et al. Double-Hit Gene Expression Signature Defines a Distinct Subgroup of Germinal Center B-Cell-Like Diffuse Large B-Cell Lymphoma. J Clin Oncol (2019) 37(3):190–201. doi: 10.1200/JCO.18.01583
112. Zappasodi R, Merghoub T, Wolchok JD. Emerging Concepts for Immune Checkpoint Blockade-Based Combination Therapies. Cancer Cell (2018) 33(4):581–98. doi: 10.1016/j.ccell.2018.03.005
113. Pitt JM, Vetizou M, Daillere R, Roberti MP, Yamazaki T, Routy B, et al. Resistance Mechanisms to Immune-Checkpoint Blockade in Cancer: Tumor-Intrinsic and -Extrinsic Factors. Immunity (2016) 44(6):1255–69. doi: 10.1016/j.immuni.2016.06.001
114. Kalbasi A, Ribas A. Tumour-intrinsic resistance to immune checkpoint blockade. Nat Rev Immunol (2020) 20(1):25–39. doi: 10.1038/s41577-019-0218-4
115. Reichel J, Chadburn A, Rubinstein PG, Giulino-Roth L, Tam W, Liu Y, et al. Flow sorting and exome sequencing reveal the oncogenome of primary Hodgkin and Reed-Sternberg cells. Blood (2015) 125(7):1061–72. doi: 10.1182/blood-2014-11-610436
116. Riemersma SA, Jordanova ES, Schop RF, Philippo K, Looijenga LH, Schuuring E, et al. Extensive genetic alterations of the HLA region, including homozygous deletions of HLA class II genes in B-cell lymphomas arising in immune-privileged sites. Blood (2000) 96(10):3569–77. doi: 10.1182/blood.V96.10.3569.h8003569_3569_3577
117. Challa-Malladi M, Lieu YK, Califano O, Holmes AB, Bhagat G, Murty VV, et al. Combined genetic inactivation of beta2-Microglobulin and CD58 reveals frequent escape from immune recognition in diffuse large B cell lymphoma. Cancer Cell (2011) 20(6):728–40. doi: 10.1016/j.ccr.2011.11.006
118. Roemer MG, Advani RH, Redd RA, Pinkus GS, Natkunam Y, Ligon AH, et al. Classical Hodgkin Lymphoma with Reduced beta2M/MHC Class I Expression Is Associated with Inferior Outcome Independent of 9p24.1 Status. Cancer Immunol Res (2016) 4(11):910–6. doi: 10.1158/2326-6066.CIR-16-0201
119. Drenou B, Tilanus M, Semana G, Alizadeh M, Birebent B, Grosset JM, et al. Loss of heterozygosity, a frequent but a non-exclusive mechanism responsible for HLA dysregulation in non-Hodgkin’s lymphomas. Br J Haematol (2004) 127(1):40–9. doi: 10.1111/j.1365-2141.2004.05151.x
120. Green MR, Monti S, Rodig SJ, Juszczynski P, Currie T, O’Donnell E, et al. Integrative analysis reveals selective 9p24.1 amplification, increased PD-1 ligand expression, and further induction via JAK2 in nodular sclerosing Hodgkin lymphoma and primary mediastinal large B-cell lymphoma. Blood (2010) 116(17):3268–77. doi: 10.1182/blood-2010-05-282780
121. Roemer MG, Advani RH, Ligon AH, Natkunam Y, Redd RA, Homer H, et al. PD-L1 and PD-L2 Genetic Alterations Define Classical Hodgkin Lymphoma and Predict Outcome. J Clin Oncol (2016) 34(23):2690–7. doi: 10.1200/JCO.2016.66.4482
122. Twa DD, Chan FC, Ben-Neriah S, Woolcock BW, Mottok A, Tan KL, et al. Genomic rearrangements involving programmed death ligands are recurrent in primary mediastinal large B-cell lymphoma. Blood (2014) 123(13):2062–5. doi: 10.1182/blood-2013-10-535443
123. Snell LM, McGaha TL, Brooks DG. Type I Interferon in Chronic Virus Infection and Cancer. Trends Immunol (2017) 38(8):542–57. doi: 10.1016/j.it.2017.05.005
124. Castro F, Cardoso AP, Gonçalves RM, Serre K, Oliveira MJ. Interferon-Gamma at the Crossroads of Tumor Immune Surveillance or Evasion. Front Immunol (2018) 9(847). doi: 10.3389/fimmu.2018.00847
125. Benci JL, Xu B, Qiu Y, Wu TJ, Dada H, Twyman-Saint Victor C, et al. Tumor Interferon Signaling Regulates a Multigenic Resistance Program to Immune Checkpoint Blockade. Cell (2016) 167(6):1540–54.e12. doi: 10.1016/j.cell.2016.11.022
126. Noy R, Pollard JW. Tumor-associated macrophages: from mechanisms to therapy. Immunity (2014) 41(1):49–61. doi: 10.1016/j.immuni.2014.06.010
127. Cassetta L, Pollard JW. Targeting macrophages: therapeutic approaches in cancer. Nat Rev Drug Discov (2018) 17(12):887–904. doi: 10.1038/nrd.2018.169
128. Kodumudi KN, Weber A, Sarnaik AA, Pilon-Thomas S. Blockade of myeloid-derived suppressor cells after induction of lymphopenia improves adoptive T cell therapy in a murine model of melanoma. J Immunol (2012) 189(11):5147–54. doi: 10.4049/jimmunol.1200274
129. Meyer C, Cagnon L, Costa-Nunes CM, Baumgaertner P, Montandon N, Leyvraz L, et al. Frequencies of circulating MDSC correlate with clinical outcome of melanoma patients treated with ipilimumab. Cancer Immunol Immunother (2014) 63(3):247–57. doi: 10.1007/s00262-013-1508-5
130. Togashi Y, Shitara K, Nishikawa H. Regulatory T cells in cancer immunosuppression - implications for anticancer therapy. Nat Rev Clin Oncol (2019) 16(6):356–71. doi: 10.1038/s41571-019-0175-7
131. Zhang H, Wang Y, Hwang ES, He YW. Interleukin-10: An Immune-Activating Cytokine in Cancer Immunotherapy. J Clin Oncol (2016) 34(29):3576–8. doi: 10.1200/JCO.2016.69.6435
132. Fisher DT, Appenheimer MM, Evans SS. The two faces of IL-6 in the tumor microenvironment. Semin Immunol (2014) 26(1):38–47. doi: 10.1016/j.smim.2014.01.008
133. Yang L. TGFbeta, a potent regulator of tumor microenvironment and host immune response, implication for therapy. Curr Mol Med (2010) 10(4):374–80. doi: 10.2174/156652410791317039
134. Möller A, Lobb RJ. The evolving translational potential of small extracellular vesicles in cancer. Nat Rev Cancer (2020). doi: 10.1038/s41568-020-00299-w
135. Gargiulo E, Morande PE, Largeot A, Moussay E, Paggetti J. Diagnostic and Therapeutic Potential of Extracellular Vesicles in B-Cell Malignancies. Front Oncol (2020) 10:580874. doi: 10.3389/fonc.2020.580874
136. Steidl C, Shah SP, Woolcock BW, Rui L, Kawahara M, Farinha P, et al. MHC class II transactivator CIITA is a recurrent gene fusion partner in lymphoid cancers. Nature (2011) 471(7338):377–81. doi: 10.1038/nature09754
137. Tada K, Maeshima AM, Hiraoka N, Yamauchi N, Maruyama D, Kim S-W, et al. Prognostic significance of HLA class I and II expression in patients with diffuse large B cell lymphoma treated with standard chemoimmunotherapy. Cancer Immunol Immunother (2016) 65(10):1213–22. doi: 10.1007/s00262-016-1883-9
138. Rimsza LM, Roberts RA, Miller TP, Unger JM, LeBlanc M, Braziel RM, et al. Loss of MHC class II gene and protein expression in diffuse large B-cell lymphoma is related to decreased tumor immunosurveillance and poor patient survival regardless of other prognostic factors: a follow-up study from the Leukemia and Lymphoma Molecular Profiling Project. Blood (2004) 103(11):4251–8. doi: 10.1182/blood-2003-07-2365
139. Cycon KA, Rimsza LM, Murphy SP. Alterations in CIITA constitute a common mechanism accounting for downregulation of MHC class II expression in diffuse large B-cell lymphoma (DLBCL). Exp Hematol (2009) 37(2):184–94. doi: 10.1016/j.exphem.2008.10.001
140. Tanaka Y, Maeshima AM, Nomoto J, Makita S, Fukuhara S, Munakata W, et al. Expression pattern of PD-L1 and PD-L2 in classical Hodgkin lymphoma, primary mediastinal large B-cell lymphoma, and gray zone lymphoma. Eur J Haematol (2018) 100(5):511–7. doi: 10.1111/ejh.13033
141. Kataoka K, Shiraishi Y, Takeda Y, Sakata S, Matsumoto M, Nagano S, et al. Aberrant PD-L1 expression through 3’-UTR disruption in multiple cancers. Nature (2016) 534(7607):402–6. doi: 10.1038/nature18294
142. Budhwani M, Mazzieri R, Dolcetti R. Plasticity of Type I Interferon-Mediated Responses in Cancer Therapy: From Anti-tumor Immunity to Resistance. Front Oncol (2018) 8:322–. doi: 10.3389/fonc.2018.00322
143. Sucker A, Zhao F, Pieper N, Heeke C, Maltaner R, Stadtler N, et al. Acquired IFNγ resistance impairs anti-tumor immunity and gives rise to T-cell-resistant melanoma lesions. Nat Commun (2017) 8:15440. doi: 10.1038/ncomms15440
144. Zaretsky JM, Garcia-Diaz A, Shin DS, Escuin-Ordinas H, Hugo W, Hu-Lieskovan S, et al. Mutations Associated with Acquired Resistance to PD-1 Blockade in Melanoma. N Engl J Med (2016) 375(9):819–29. doi: 10.1056/NEJMoa1604958
145. Shin DS, Zaretsky JM, Escuin-Ordinas H, Garcia-Diaz A, Hu-Lieskovan S, Kalbasi A, et al. Primary Resistance to PD-1 Blockade Mediated by JAK1/2 Mutations. Cancer Discov (2017) 7(2):188–201. doi: 10.1158/2159-8290.CD-16-1223
146. Gao J, Shi LZ, Zhao H, Chen J, Xiong L, He Q, et al. Loss of IFN-gamma Pathway Genes in Tumor Cells as a Mechanism of Resistance to Anti-CTLA-4 Therapy. Cell (2016) 167(2):397–404.e9. doi: 10.1016/j.cell.2016.08.069
147. Lu L, Zhu F, Zhang M, Li Y, Drennan AC, Kimpara S, et al. Gene regulation and suppression of type I interferon signaling by STAT3 in diffuse large B cell lymphoma. Proc Natl Acad Sci (2018) 115(3):E498–505. doi: 10.1073/pnas.1715118115
148. Asslaber D, Qi Y, Maeding N, Steiner M, Denk U, Hopner JP, et al. B-cell-specific IRF4 deletion accelerates chronic lymphocytic leukemia development by enhanced tumor immune evasion. Blood (2019) 134(20):1717–29. doi: 10.1182/blood.2019000973
149. Wellenstein MD, de Visser KE. Cancer-Cell-Intrinsic Mechanisms Shaping the Tumor Immune Landscape. Immunity (2018) 48(3):399–416. doi: 10.1016/j.immuni.2018.03.004
150. Reddy A, Zhang J, Davis NS, Moffitt AB, Love CL, Waldrop A, et al. Genetic and Functional Drivers of Diffuse Large B Cell Lymphoma. Cell (2017) 171(2):481–94.e15. doi: 10.1016/j.cell.2017.09.027
151. Casey SC, Tong L, Li Y, Do R, Walz S, Fitzgerald KN, et al. MYC regulates the antitumor immune response through CD47 and PD-L1. Science (2016) 352(6282):227–31. doi: 10.1126/science.aac9935
152. Xu Y, Poggio M, Jin HY, Shi Z, Forester CM, Wang Y, et al. Translation control of the immune checkpoint in cancer and its therapeutic targeting. Nat Med (2019) 25(2):301–11. doi: 10.1038/s41591-018-0321-2
153. Filip D, Mraz M. The role of MYC in the transformation and aggressiveness of ‘indolent’ B-cell malignancies. Leuk Lymphoma (2020) 61(3):510–24. doi: 10.1080/10428194.2019.1675877
154. Schuster C, Berger A, Hoelzl MA, Putz EM, Frenzel A, Simma O, et al. The cooperating mutation or “second hit” determines the immunologic visibility toward MYC-induced murine lymphomas. Blood (2011) 118(17):4635–45. doi: 10.1182/blood-2010-10-313098
155. Liu WR, Shipp MA. Signaling pathways and immune evasion mechanisms in classical Hodgkin lymphoma. Blood (2017) 130(21):2265–70. doi: 10.1182/blood-2017-06-781989
156. Feuerhake F, Kutok JL, Monti S, Chen W, LaCasce AS, Cattoretti G, et al. NFkappaB activity, function, and target-gene signatures in primary mediastinal large B-cell lymphoma and diffuse large B-cell lymphoma subtypes. Blood (2005) 106(4):1392–9. doi: 10.1182/blood-2004-12-4901
157. Compagno M, Lim WK, Grunn A, Nandula SV, Brahmachary M, Shen Q, et al. Mutations of multiple genes cause deregulation of NF-kappaB in diffuse large B-cell lymphoma. Nature (2009) 459(7247):717–21. doi: 10.1038/nature07968
158. Chapuy B, Stewart C, Dunford AJ, Kim J, Kamburov A, Redd RA, et al. Molecular subtypes of diffuse large B cell lymphoma are associated with distinct pathogenic mechanisms and outcomes. Nat Med (2018) 24(5):679–90. doi: 10.1038/s41591-018-0016-8
159. Schmitz R, Wright GW, Huang DW, Johnson CA, Phelan JD, Wang JQ, et al. Genetics and Pathogenesis of Diffuse Large B-Cell Lymphoma. N Engl J Med (2018) 378(15):1396–407. doi: 10.1056/NEJMoa1801445
160. Kim JY, Kronbichler A, Eisenhut M, Hong SH, van der Vliet HJ, Kang J, et al. Tumor Mutational Burden and Efficacy of Immune Checkpoint Inhibitors: A Systematic Review and Meta-Analysis. Cancers (Basel) (2019) 11(11). doi: 10.3390/cancers11111798
161. Goodman AM, Kato S, Bazhenova L, Patel SP, Frampton GM, Miller V, et al. Tumor Mutational Burden as an Independent Predictor of Response to Immunotherapy in Diverse Cancers. Mol Cancer Ther (2017) 16(11):2598–608. doi: 10.1158/1535-7163.MCT-17-0386
162. Rooney MS, Shukla SA, Wu CJ, Getz G, Hacohen N. Molecular and genetic properties of tumors associated with local immune cytolytic activity. Cell (2015) 160(1-2):48–61. doi: 10.1016/j.cell.2014.12.033
163. Bever KM, Le DT. DNA repair defects and implications for immunotherapy. J Clin Invest (2018) 128(10):4236–42. doi: 10.1172/JCI122010
164. Chapuy B, Stewart C, Dunford AJ, Kim J, Wienand K, Kamburov A, et al. Genomic analyses of PMBL reveal new drivers and mechanisms of sensitivity to PD-1 blockade. Blood (2019) 134(26):2369–82. doi: 10.1182/blood.2019002067
165. de Miranda NFCC, Peng R, Georgiou K, Wu C, Falk Sörqvist E, Berglund M, et al. DNA repair genes are selectively mutated in diffuse large B cell lymphomas. J Exp Med (2013) 210(9):1729–42. doi: 10.1084/jem.20122842
166. Volpe G, Gamberi B, Pastore C, Roetto A, Pautasso M, Parvis G, et al. Analysis of microsatellite instability in chronic lymphoproliferative disorders. Ann Hematol (1996) 72(2):67–71. doi: 10.1007/BF00641310
167. Schumacher TN, Schreiber RD. Neoantigens in cancer immunotherapy. Science (2015) 348(6230):69–74. doi: 10.1126/science.aaa4971
168. Tran E, Turcotte S, Gros A, Robbins PF, Lu YC, Dudley ME, et al. Cancer immunotherapy based on mutation-specific CD4+ T cells in a patient with epithelial cancer. Science (2014) 344(6184):641–5. doi: 10.1126/science.1251102
169. Franchina DG, He F, Brenner D. Survival of the fittest: Cancer challenges T cell metabolism. Cancer Lett (2018) 412:216–23. doi: 10.1016/j.canlet.2017.10.014
170. Arruga F, Gyau BB, Iannello A, Vitale N, Vaisitti T, Deaglio S. Immune Response Dysfunction in Chronic Lymphocytic Leukemia: Dissecting Molecular Mechanisms and Microenvironmental Conditions. Int J Mol Sci (2020) 21(5). doi: 10.3390/ijms21051825
171. Bottcher M, Baur R, Stoll A, Mackensen A, Mougiakakos D. Linking Immunoevasion and Metabolic Reprogramming in B-Cell-Derived Lymphomas. Front Oncol (2020) 10:594782. doi: 10.3389/fonc.2020.594782
172. Alhakeem SS, McKenna MK, Oben KZ, Noothi SK, Rivas JR, Hildebrandt GC, et al. Chronic Lymphocytic Leukemia-Derived IL-10 Suppresses Antitumor Immunity. J Immunol (2018) 200(12):4180–9. doi: 10.4049/jimmunol.1800241
173. Buggins AG, Patten PE, Richards J, Thomas NS, Mufti GJ, Devereux S. Tumor-derived IL-6 may contribute to the immunological defect in CLL. Leukemia (2008) 22(5):1084–7. doi: 10.1038/sj.leu.2405015
174. Smallwood DT, Apollonio B, Willimott S, Lezina L, Alharthi A, Ambrose AR, et al. Extracellular vesicles released by CD40/IL-4-stimulated CLL cells confer altered functional properties to CD4+ T cells. Blood (2016) 128(4):542–52. doi: 10.1182/blood-2015-11-682377
175. Lotz M, Ranheim E, Kipps TJ. Transforming growth factor beta as endogenous growth inhibitor of chronic lymphocytic leukemia B cells. J Exp Med (1994) 179(3):999–1004. doi: 10.1084/jem.179.3.999
176. Manna A, Kellett T, Aulakh S, Lewis-Tuffin LJ, Dutta N, Knutson K, et al. Targeting CD38 is lethal to Breg-like chronic lymphocytic leukemia cells and Tregs, but restores CD8+ T-cell responses. Blood Adv (2020) 4(10):2143–57. doi: 10.1182/bloodadvances.2019001091
177. Yang ZZ, Grote DM, Xiu B, Ziesmer SC, Price-Troska TL, Hodge LS, et al. TGF-β upregulates CD70 expression and induces exhaustion of effector memory T cells in B-cell non-Hodgkin’s lymphoma. Leukemia (2014) 28(9):1872–84. doi: 10.1038/leu.2014.84
178. Koyama S, Akbay EA, Li YY, Herter-Sprie GS, Buczkowski KA, Richards WG, et al. Adaptive resistance to therapeutic PD-1 blockade is associated with upregulation of alternative immune checkpoints. Nat Commun (2016) 7:10501. doi: 10.1038/ncomms10501
179. Jacquelot N, Yamazaki T, Roberti MP, Duong CPM, Andrews MC, Verlingue L, et al. Sustained Type I interferon signaling as a mechanism of resistance to PD-1 blockade. Cell Res (2019) 29(10):846–61. doi: 10.1038/s41422-019-0224-x
180. Wierz M, Pierson S, Guyonnet L, Viry E, Lequeux A, Oudin A, et al. Dual PD1/LAG3 immune checkpoint blockade limits tumor development in a murine model of chronic lymphocytic leukemia. Blood (2018) 131(14):1617–21. doi: 10.1182/blood-2017-06-792267
181. Fraietta JA, Lacey SF, Orlando EJ, Pruteanu-Malinici I, Gohil M, Lundh S, et al. Determinants of response and resistance to CD19 chimeric antigen receptor (CAR) T cell therapy of chronic lymphocytic leukemia. Nat Med (2018) 24(5):563–71. doi: 10.1038/s41591-018-0010-1
182. Steidl C, Lee T, Shah SP, Farinha P, Han G, Nayar T, et al. Tumor-associated macrophages and survival in classic Hodgkin’s lymphoma. N Engl J Med (2010) 362(10):875–85. doi: 10.1056/NEJMoa0905680
183. Cai QC, Liao H, Lin SX, Xia Y, Wang XX, Gao Y, et al. High expression of tumor-infiltrating macrophages correlates with poor prognosis in patients with diffuse large B-cell lymphoma. Med Oncol (2012) 29(4):2317–22. doi: 10.1007/s12032-011-0123-6
184. Carey CD, Gusenleitner D, Lipschitz M, Roemer MGM, Stack EC, Gjini E, et al. Topological analysis reveals a PD-L1-associated microenvironmental niche for Reed-Sternberg cells in Hodgkin lymphoma. Blood (2017) 130(22):2420–30. doi: 10.1182/blood-2017-03-770719
185. Advani R, Flinn I, Popplewell L, Forero A, Bartlett NL, Ghosh N, et al. CD47 Blockade by Hu5F9-G4 and Rituximab in Non-Hodgkin’s Lymphoma. N Engl J Med (2018) 379(18):1711–21. doi: 10.1056/NEJMoa1807315
186. Jaiswal S, Chao MP, Majeti R, Weissman IL. Macrophages as mediators of tumor immunosurveillance. Trends Immunol (2010) 31(6):212–9. doi: 10.1016/j.it.2010.04.001
187. Veglia F, Perego M, Gabrilovich D. Myeloid-derived suppressor cells coming of age. Nat Immunol (2018) 19(2):108–19. doi: 10.1038/s41590-017-0022-x
188. Betsch A, Rutgeerts O, Fevery S, Sprangers B, Verhoef G, Dierickx D, et al. Myeloid-derived suppressor cells in lymphoma: The good, the bad and the ugly. Blood Rev (2018) 32(6):490–8. doi: 10.1016/j.blre.2018.04.006
189. Jitschin R, Braun M, Buttner M, Dettmer-Wilde K, Bricks J, Berger J, et al. CLL-cells induce IDOhi CD14+HLA-DRlo myeloid-derived suppressor cells that inhibit T-cell responses and promote TRegs. Blood (2014) 124(5):750–60. doi: 10.1182/blood-2013-12-546416
190. Plitas G, Rudensky AY. Regulatory T Cells: Differentiation and Function. Cancer Immunol Res (2016) 4(9):721–5. doi: 10.1158/2326-6066.CIR-16-0193
191. Hanna BS, Roessner PM, Scheffold A, Jebaraj BMC, Demerdash Y, Öztürk S, et al. PI3Kδ inhibition modulates regulatory and effector T-cell differentiation and function in chronic lymphocytic leukemia. Leukemia (2019) 33(6):1427–38. doi: 10.1038/s41375-018-0318-3
192. Palma M, Gentilcore G, Heimersson K, Mozaffari F, Nasman-Glaser B, Young E, et al. T cells in chronic lymphocytic leukemia display dysregulated expression of immune checkpoints and activation markers. Haematologica (2017) 102(3):562–72. doi: 10.3324/haematol.2016.151100
193. D’Arena G, Laurenti L, Minervini MM, Deaglio S, Bonello L, De Martino L, et al. Regulatory T-cell number is increased in chronic lymphocytic leukemia patients and correlates with progressive disease. Leuk Res (2011) 35(3):363–8. doi: 10.1016/j.leukres.2010.08.010
194. de Weerdt I, Hofland T, de Boer R, Dobber JA, Dubois J, van Nieuwenhuize D, et al. Distinct immune composition in lymph node and peripheral blood of CLL patients is reshaped during venetoclax treatment. Blood Adv (2019) 3(17):2642–52. doi: 10.1182/bloodadvances.2019000360
195. Kamada T, Togashi Y, Tay C, Ha D, Sasaki A, Nakamura Y, et al. PD-1+ regulatory T cells amplified by PD-1 blockade promote hyperprogression of cancer. Proc Natl Acad Sci (2019) 116(20):9999–10008. doi: 10.1073/pnas.1822001116
196. Simpson TR, Li F, Montalvo-Ortiz W, Sepulveda MA, Bergerhoff K, Arce F, et al. Fc-dependent depletion of tumor-infiltrating regulatory T cells co-defines the efficacy of anti-CTLA-4 therapy against melanoma. J Exp Med (2013) 210(9):1695–710. doi: 10.1084/jem.20130579
197. Blonska M, Agarwal NK, Vega F. Shaping of the tumor microenvironment: Stromal cells and vessels. Semin Cancer Biol (2015) 34:3–13. doi: 10.1016/j.semcancer.2015.03.002
198. Lanitis E, Irving M, Coukos G. Targeting the tumor vasculature to enhance T cell activity. Curr Opin Immunol (2015) 33:55–63. doi: 10.1016/j.coi.2015.01.011
199. von Andrian UH, Mempel TR. Homing and cellular traffic in lymph nodes. Nat Rev Immunol (2003) 3(11):867–78. doi: 10.1038/nri1222
200. Carmeliet P, Jain RK. Principles and mechanisms of vessel normalization for cancer and other angiogenic diseases. Nat Rev Drug Discov (2011) 10(6):417–27. doi: 10.1038/nrd3455
201. Takahashi M, Ikeda U, Masuyama J, Funayama H, Kano S, Shimada K. Nitric oxide attenuates adhesion molecule expression in human endothelial cells. Cytokine (1996) 8(11):817–21. doi: 10.1006/cyto.1996.0109
202. Buckanovich RJ, Facciabene A, Kim S, Benencia F, Sasaroli D, Balint K, et al. Endothelin B receptor mediates the endothelial barrier to T cell homing to tumors and disables immune therapy. Nat Med (2008) 14(1):28–36. doi: 10.1038/nm1699
203. Erez N, Truitt M, Olson P, Arron ST, Hanahan D. Cancer-Associated Fibroblasts Are Activated in Incipient Neoplasia to Orchestrate Tumor-Promoting Inflammation in an NF-kappaB-Dependent Manner. Cancer Cell (2010) 17(2):135–47. doi: 10.1016/j.ccr.2009.12.041
204. Costa A, Kieffer Y, Scholer-Dahirel A, Pelon F, Bourachot B, Cardon M, et al. Fibroblast Heterogeneity and Immunosuppressive Environment in Human Breast Cancer. Cancer Cell (2018) 33(3):463–79.e10. doi: 10.1016/j.ccell.2018.01.011
205. Gok Yavuz B, Gunaydin G, Gedik ME, Kosemehmetoglu K, Karakoc D, Ozgur F, et al. Cancer associated fibroblasts sculpt tumour microenvironment by recruiting monocytes and inducing immunosuppressive PD-1(+) TAMs. Sci Rep (2019) 9(1):3172. doi: 10.1038/s41598-019-39553-z
206. Cohen N, Shani O, Raz Y, Sharon Y, Hoffman D, Abramovitz L, et al. Fibroblasts drive an immunosuppressive and growth-promoting microenvironment in breast cancer via secretion of Chitinase 3-like 1. Oncogene (2017) 36(31):4457–68. doi: 10.1038/onc.2017.65
207. Comito G, Giannoni E, Segura CP, Barcellos-de-Souza P, Raspollini MR, Baroni G, et al. Cancer-associated fibroblasts and M2-polarized macrophages synergize during prostate carcinoma progression. Oncogene (2014) 33(19):2423–31. doi: 10.1038/onc.2013.191
208. Cheng Y, Li H, Deng Y, Tai Y, Zeng K, Zhang Y, et al. Cancer-associated fibroblasts induce PDL1+ neutrophils through the IL6-STAT3 pathway that foster immune suppression in hepatocellular carcinoma. Cell Death Dis (2018) 9(4):422. doi: 10.1038/s41419-018-0458-4
209. Joyce JA, Fearon DT. T cell exclusion, immune privilege, and the tumor microenvironment. Science (2015) 348(6230):74–80. doi: 10.1126/science.aaa6204
210. Carman CV, Martinelli R. T Lymphocyte-Endothelial Interactions: Emerging Understanding of Trafficking and Antigen-Specific Immunity. Front Immunol (2015) 6:603. doi: 10.3389/fimmu.2015.00603
211. Berg M, Wingender G, Djandji D, Hegenbarth S, Momburg F, Hammerling G, et al. Cross-presentation of antigens from apoptotic tumor cells by liver sinusoidal endothelial cells leads to tumor-specific CD8+ T cell tolerance. Eur J Immunol (2006) 36(11):2960–70. doi: 10.1002/eji.200636033
212. Bordry N, Broggi MAS, de Jonge K, Schaeuble K, Gannon PO, Foukas PG, et al. Lymphatic vessel density is associated with CD8(+) T cell infiltration and immunosuppressive factors in human melanoma. Oncoimmunology (2018) 7(8):e1462878. doi: 10.1080/2162402X.2018.1462878
213. Dengler TJ, Pober JS. Human vascular endothelial cells stimulate memory but not naive CD8+ T cells to differentiate into CTL retaining an early activation phenotype. J Immunol (2000) 164(10):5146–55. doi: 10.4049/jimmunol.164.10.5146
214. Khayyamian S, Hutloff A, Buchner K, Grafe M, Henn V, Kroczek RA, et al. ICOS-ligand, expressed on human endothelial cells, costimulates Th1 and Th2 cytokine secretion by memory CD4+ T cells. Proc Natl Acad Sci U S A (2002) 99(9):6198–203. doi: 10.1073/pnas.092576699
215. Lim WC, Olding M, Healy E, Millar TM. Human Endothelial Cells Modulate CD4(+) T Cell Populations and Enhance Regulatory T Cell Suppressive Capacity. Front Immunol (2018) 9:565. doi: 10.3389/fimmu.2018.00565
216. Motz GT, Santoro SP, Wang LP, Garrabrant T, Lastra RR, Hagemann IS, et al. Tumor endothelium FasL establishes a selective immune barrier promoting tolerance in tumors. Nat Med (2014) 20(6):607–15. doi: 10.1038/nm.3541
217. Dieterich LC, Ikenberg K, Cetintas T, Kapaklikaya K, Hutmacher C, Detmar M. Tumor-Associated Lymphatic Vessels Upregulate PDL1 to Inhibit T-Cell Activation. Front Immunol (2017) 8:66. doi: 10.3389/fimmu.2017.00066
218. Lane RS, Femel J, Breazeale AP, Loo CP, Thibault G, Kaempf A, et al. IFNgamma-activated dermal lymphatic vessels inhibit cytotoxic T cells in melanoma and inflamed skin. J Exp Med (2018) 215(12):3057–74. doi: 10.1084/jem.20180654
219. Nambiar DK, Aguilera T, Cao H, Kwok S, Kong C, Bloomstein J, et al. Galectin-1-driven T cell exclusion in the tumor endothelium promotes immunotherapy resistance. J Clin Invest (2019) 129(12):5553–67. doi: 10.1172/JCI129025
220. Rodig N, Ryan T, Allen JA, Pang H, Grabie N, Chernova T, et al. Endothelial expression of PD-L1 and PD-L2 down-regulates CD8+ T cell activation and cytolysis. Eur J Immunol (2003) 33(11):3117–26. doi: 10.1002/eji.200324270
221. Gorchs L, Fernandez Moro C, Bankhead P, Kern KP, Sadeak I, Meng Q, et al. Human Pancreatic Carcinoma-Associated Fibroblasts Promote Expression of Co-inhibitory Markers on CD4(+) and CD8(+) T-Cells. Front Immunol (2019) 10:847. doi: 10.3389/fimmu.2019.00847
222. Cremasco V, Astarita JL, Grauel AL, Keerthivasan S, MacIsaac K, Woodruff MC, et al. FAP Delineates Heterogeneous and Functionally Divergent Stromal Cells in Immune-Excluded Breast Tumors. Cancer Immunol Res (2018) 6(12):1472–85. doi: 10.1158/2326-6066.CIR-18-0098
223. Lakins MA, Ghorani E, Munir H, Martins CP, Shields JD. Cancer-associated fibroblasts induce antigen-specific deletion of CD8 (+) T Cells to protect tumour cells. Nat Commun (2018) 9(1):948. doi: 10.1038/s41467-018-03347-0
224. Krishnamurty AT, Turley SJ. Lymph node stromal cells: cartographers of the immune system. Nat Immunol (2020) 21(4):369–80. doi: 10.1038/s41590-020-0635-3
225. Nombela-Arrieta C, Isringhausen S. The Role of the Bone Marrow Stromal Compartment in the Hematopoietic Response to Microbial Infections. Front Immunol (2016) 7:689. doi: 10.3389/fimmu.2016.00689
226. Vokali E, Yu SS, Hirosue S, Rincon-Restrepo M, VD F, Scherer S, et al. Lymphatic endothelial cells prime naive CD8(+) T cells into memory cells under steady-state conditions. Nat Commun (2020) 11(1):538. doi: 10.1038/s41467-019-14127-9
227. Poggi A, Varesano S, Zocchi MR. How to Hit Mesenchymal Stromal Cells and Make the Tumor Microenvironment Immunostimulant Rather Than Immunosuppressive. Front Immunol (2018) 9:262. doi: 10.3389/fimmu.2018.01342
228. Fletcher AL, Acton SE, Knoblich K. Lymph node fibroblastic reticular cells in health and disease. Nat Rev Immunol (2015) 15(6):350–61. doi: 10.1038/nri3846
229. Kadowaki I, Ichinohasama R, Harigae H, Ishizawa K, Okitsu Y, Kameoka J, et al. Accelerated lymphangiogenesis in malignant lymphoma: possible role of VEGF-A and VEGF-C. Br J Haematol (2005) 130(6):869–77. doi: 10.1111/j.1365-2141.2005.05695.x
230. Vega F, Medeiros LJ, Lang WH, Mansoor A, Bueso-Ramos C, Jones D. The stromal composition of malignant lymphoid aggregates in bone marrow: variations in architecture and phenotype in different B-cell tumours. Br J Haematol (2002) 117(3):569–76. doi: 10.1046/j.1365-2141.2002.03497.x
231. Ruan J, Hyjek E, Kermani P, Christos PJ, Hooper AT, Coleman M, et al. Magnitude of stromal hemangiogenesis correlates with histologic subtype of non-Hodgkin’s lymphoma. Clin Cancer Res (2006) 12(19):5622–31. doi: 10.1158/1078-0432.CCR-06-1204
232. Ibrahim S, Jilani I, O’Brien S, Rogers A, Manshouri T, Giles F, et al. Clinical relevance of the expression of the CD31 ligand for CD38 in patients with B-cell chronic lymphocytic leukemia. Cancer (2003) 97(8):1914–9. doi: 10.1002/cncr.11264
233. McCabe D, Bacon L, O’Regan K, Condron C, O’Donnell JR, Murphy PT. CD38 expression on B-cell chronic lymphocytic leukemic cells is strongly correlated with vascular endothelial growth factor expression. Leukemia (2004) 18(3):649–50. doi: 10.1038/sj.leu.2403282
234. Molica S, Vacca A, Ribatti D, Cuneo A, Cavazzini F, Levato D, et al. Prognostic value of enhanced bone marrow angiogenesis in early B-cell chronic lymphocytic leukemia. Blood (2002) 100(9):3344–51. doi: 10.1182/blood-2002-01-0084
235. Yang J, Li W, He X, Zhang G, Yue L, Chai Y. VEGF overexpression is a valuable prognostic factor for non-Hodgkin’s lymphoma evidence from a systemic meta-analysis. Dis Markers (2015) 2015:786790. doi: 10.1155/2015/786790
236. Lenz G, Wright G, Dave SS, Xiao W, Powell J, Zhao H, et al. Stromal gene signatures in large-B-cell lymphomas. N Engl J Med (2008) 359(22):2313–23. doi: 10.1056/NEJMoa0802885
237. Rehm A, Mensen A, Schradi K, Gerlach K, Wittstock S, Winter S, et al. Cooperative function of CCR7 and lymphotoxin in the formation of a lymphoma-permissive niche within murine secondary lymphoid organs. Blood (2011) 118(4):1020–33. doi: 10.1182/blood-2010-11-321265
238. Farinello D, Wozinska M, Lenti E, Genovese L, Bianchessi S, Migliori E, et al. A retinoic acid-dependent stroma-leukemia crosstalk promotes chronic lymphocytic leukemia progression. Nat Commun (2018) 9(1):1787. doi: 10.1038/s41467-018-04150-7
239. Ding W, Knox TR, Tschumper RC, Wu W, Schwager SM, Boysen JC, et al. Platelet-derived growth factor (PDGF)-PDGF receptor interaction activates bone marrow-derived mesenchymal stromal cells derived from chronic lymphocytic leukemia: implications for an angiogenic switch. Blood (2010) 116(16):2984–93. doi: 10.1182/blood-2010-02-269894
240. Dubois N, Crompot E, Meuleman N, Bron D, Lagneaux L, Stamatopoulos B. Importance of Crosstalk Between Chronic Lymphocytic Leukemia Cells and the Stromal Microenvironment: Direct Contact, Soluble Factors, and Extracellular Vesicles. Front Oncol (2020) 10:1422. doi: 10.3389/fonc.2020.01422
241. Passalidou E, Stewart M, Trivella M, Steers G, Pillai G, Dogan A, et al. Vascular patterns in reactive lymphoid tissue and in non-Hodgkin’s lymphoma. Br J Cancer (2003) 88(4):553–9. doi: 10.1038/sj.bjc.6600742
242. Song G, Li Y, Jiang G. Role of VEGF/VEGFR in the pathogenesis of leukemias and as treatment targets (Review). Oncol Rep (2012) 28(6):1935–44. doi: 10.3892/or.2012.2045
243. Badoux X, Bueso-Ramos C, Harris D, Li P, Liu Z, Burger J, et al. Cross-talk between chronic lymphocytic leukemia cells and bone marrow endothelial cells: role of signal transducer and activator of transcription 3. Hum Pathol (2011) 42(12):1989–2000. doi: 10.1016/j.humpath.2011.02.027
244. Kay NE, Bone ND, Tschumper RC, Howell KH, Geyer SM, Dewald GW, et al. B-CLL cells are capable of synthesis and secretion of both pro- and anti-angiogenic molecules. Leukemia (2002) 16(5):911–9. doi: 10.1038/sj.leu.2402467
245. Buggins AG, Pepper C, Patten PE, Hewamana S, Gohil S, Moorhead J, et al. Interaction with vascular endothelium enhances survival in primary chronic lymphocytic leukemia cells via NF-kappaB activation and de novo gene transcription. Cancer Res (2010) 70(19):7523–33. doi: 10.1158/0008-5472.CAN-10-1634
246. Cols M, Barra CM, He B, Puga I, Xu W, Chiu A, et al. Stromal endothelial cells establish a bidirectional crosstalk with chronic lymphocytic leukemia cells through the TNF-related factors BAFF, APRIL, and CD40L. J Immunol (2012) 188(12):6071–83. doi: 10.4049/jimmunol.1102066
247. Enzler T, Kater AP, Zhang W, Widhopf GF,2, HY C, Lee J, et al. Chronic lymphocytic leukemia of Emu-TCL1 transgenic mice undergoes rapid cell turnover that can be offset by extrinsic CD257 to accelerate disease progression. Blood (2009) 114(20):4469–76. doi: 10.1182/blood-2009-06-230169
248. Maffei R, Fiorcari S, Bulgarelli J, Martinelli S, Castelli I, Deaglio S, et al. Physical contact with endothelial cells through beta1- and beta2- integrins rescues chronic lymphocytic leukemia cells from spontaneous and drug-induced apoptosis and induces a peculiar gene expression profile in leukemic cells. Haematologica (2012) 97(6):952–60. doi: 10.3324/haematol.2011.054924
249. Drusbosky L, Gars E, Trujillo A, McGee C, Meacham A, Wise E, et al. Endothelial cell derived angiocrine support of acute myeloid leukemia targeted by receptor tyrosine kinase inhibition. Leuk Res (2015) 39(9):984–9. doi: 10.1016/j.leukres.2015.05.015
250. Pezeshkian B, Donnelly C, Tamburo K, Geddes T, Madlambayan GJ. Leukemia Mediated Endothelial Cell Activation Modulates Leukemia Cell Susceptibility to Chemotherapy through a Positive Feedback Loop Mechanism. PloS One (2013) 8(4):e60823. doi: 10.1371/journal.pone.0060823
251. Parameswaran R, Lim M, Fei F, Abdel-Azim H, Arutyunyan A, Schiffer I, et al. Effector-mediated eradication of precursor B acute lymphoblastic leukemia with a novel Fc-engineered monoclonal antibody targeting the BAFF-R. Mol Cancer Ther (2014) 13(6):1567–77. doi: 10.1158/1535-7163.MCT-13-1023
252. Qin H, Wei G, Sakamaki I, Dong Z, Cheng WA, Smith DL, et al. Novel BAFF-Receptor Antibody to Natively Folded Recombinant Protein Eliminates Drug-Resistant Human B-cell Malignancies In Vivo. Clin Cancer Res (2018) 24(5):1114–23. doi: 10.1158/1078-0432.CCR-17-1193
253. Guilloton F, Caron G, Menard C, Pangault C, Ame-Thomas P, Dulong J, et al. Mesenchymal stromal cells orchestrate follicular lymphoma cell niche through the CCL2-dependent recruitment and polarization of monocytes. Blood (2012) 119(11):2556–67. doi: 10.1182/blood-2011-08-370908
254. Heinig K, Gatjen M, Grau M, Stache V, Anagnostopoulos I, Gerlach K, et al. Access to follicular dendritic cells is a pivotal step in murine chronic lymphocytic leukemia B-cell activation and proliferation. Cancer Discov (2014) 4(12):1448–65. doi: 10.1158/2159-8290.CD-14-0096
255. Lutzny G, Kocher T, Schmidt-Supprian M, Rudelius M, Klein-Hitpass L, Finch AJ, et al. Protein kinase c-beta-dependent activation of NF-kappaB in stromal cells is indispensable for the survival of chronic lymphocytic leukemia B cells in vivo. Cancer Cell (2013) 23(1):77–92. doi: 10.1016/j.ccr.2012.12.003
256. Lee MJ, Park SY, Ko JH, Lee HJ, Ryu JS, Park JW, et al. Mesenchymal stromal cells promote B-cell lymphoma in lacrimal glands by inducing immunosuppressive microenvironment. Oncotarget (2017) 8(39):66281–92. doi: 10.18632/oncotarget.19971
257. Brady MT, Hilchey SP, Hyrien O, Spence SA, Bernstein SH. Mesenchymal stromal cells support the viability and differentiation of follicular lymphoma-infiltrating follicular helper T-cells. PloS One (2014) 9(5):e97597. doi: 10.1371/journal.pone.0097597
258. Ame-Thomas P, Le Priol J, Yssel H, Caron G, Pangault C, Jean R, et al. Characterization of intratumoral follicular helper T cells in follicular lymphoma: role in the survival of malignant B cells. Leukemia (2012) 26(5):1053–63. doi: 10.1038/leu.2011.301
259. Pandey S, Mourcin F, Marchand T, Nayar S, Guirriec M, Pangault C, et al. IL-4/CXCL12 loop is a key regulator of lymphoid stroma function in follicular lymphoma. Blood (2017) 129(18):2507–18. doi: 10.1182/blood-2016-08-737239
260. Gregoire M, Guilloton F, Pangault C, Mourcin F, Sok P, Latour M, et al. Neutrophils trigger a NF-kappaB dependent polarization of tumor-supportive stromal cells in germinal center B-cell lymphomas. Oncotarget (2015) 6(18):16471–87. doi: 10.18632/oncotarget.4106
261. Nakajima S, Fujiwara T, Ohguchi H, Onishi Y, Kamata M, Okitsu Y, et al. Induction of thymic stromal lymphopoietin in mesenchymal stem cells by interaction with myeloma cells. Leuk Lymphoma (2014) 55(11):2605–13. doi: 10.3109/10428194.2014.881478
262. Kiyasu J, Miyoshi H, Hirata A, Arakawa F, Ichikawa A, Niino D, et al. Expression of programmed cell death ligand 1 is associated with poor overall survival in patients with diffuse large B-cell lymphoma. Blood (2015) 126(19):2193–201. doi: 10.1182/blood-2015-02-629600
263. Frassanito MA, Rao L, Moschetta M, Ria R, Di Marzo L, De Luisi A, et al. Bone marrow fibroblasts parallel multiple myeloma progression in patients and mice: in vitro and in vivo studies. Leukemia (2014) 28(4):904–16. doi: 10.1038/leu.2013.254
264. Giallongo C, Tibullo D, Parrinello NL, La Cava P, Di Rosa M, Bramanti V, et al. Granulocyte-like myeloid derived suppressor cells (G-MDSC) are increased in multiple myeloma and are driven by dysfunctional mesenchymal stem cells (MSC). Oncotarget (2016) 7(52):85764–75. doi: 10.18632/oncotarget.7969
265. Gieniec KA, Butler LM, Worthley DL, Woods SL. Cancer-associated fibroblasts-heroes or villains? Br J Cancer (2019) 121(4):293–302. doi: 10.1038/s41416-019-0509-3
266. Maby-El Hajjami H, Ame-Thomas P, Pangault C, Tribut O, DeVos J, Jean R, et al. Functional alteration of the lymphoma stromal cell niche by the cytokine context: role of indoleamine-2,3 dioxygenase. Cancer Res (2009) 69(7):3228–37. doi: 10.1158/0008-5472.CAN-08-3000
267. Dorsam B, Bosl T, Reiners KS, Barnert S, Schubert R, Shatnyeva O, et al. Hodgkin Lymphoma-Derived Extracellular Vesicles Change the Secretome of Fibroblasts Toward a CAF Phenotype. Front Immunol (2018) 9:1358. doi: 10.3389/fimmu.2018.01358
268. Paggetti J, Haderk F, Seiffert M, Janji B, Distler U, Ammerlaan W, et al. Exosomes released by chronic lymphocytic leukemia cells induce the transition of stromal cells into cancer-associated fibroblasts. Blood (2015) 126(9):1106–17. doi: 10.1182/blood-2014-12-618025
269. Haderk F, Schulz R, Iskar M, Cid LL, Worst T, Willmund KV, et al. Tumor-derived exosomes modulate PD-L1 expression in monocytes. Sci Immunol (2017) 2(13). doi: 10.1126/sciimmunol.aah5509
270. Denucci CC, Mitchell JS, Shimizu Y. Integrin function in T-cell homing to lymphoid and nonlymphoid sites: getting there and staying there. Crit Rev Immunol (2009) 29(2):87–109. doi: 10.1615/CritRevImmunol.v29.i2.10
271. Ramsay AG, Evans R, Kiaii S, Svensson L, Hogg N, Gribben JG. Chronic lymphocytic leukemia cells induce defective LFA-1-directed T-cell motility by altering Rho GTPase signaling that is reversible with lenalidomide. Blood (2013) 121(14):2704–14. doi: 10.1182/blood-2012-08-448332
272. Taflin C, Favier B, Baudhuin J, Savenay A, Hemon P, Bensussan A, et al. Human endothelial cells generate Th17 and regulatory T cells under inflammatory conditions. Proc Natl Acad Sci U S A (2011) 108(7):2891–6. doi: 10.1073/pnas.1011811108
273. Huang X, Bai X, Cao Y, Wu J, Huang M, Tang D, et al. Lymphoma endothelium preferentially expresses Tim-3 and facilitates the progression of lymphoma by mediating immune evasion. J Exp Med (2010) 207(3):505–20. doi: 10.1084/jem.20090397
274. Apollonio B JP, Phillips B, Kuhnl A, Salisbury J, Zacharioudakis G, Sutto LA, et al. Diffuse Large B-Cell Lymphoma Remodels the Fibroblastic Reticular Network That Acquires Aberrant Immunosuppressive Capabilities; Implications for the Regulation of Anti-Tumor Immunity in the Immuno-Oncology Era. Blood (2018) 132. doi: 10.1182/blood-2018-99-116409
275. Pallasch CP, Leskov I, Braun CJ, Vorholt D, Drake A, Soto-Feliciano YM, et al. Sensitizing protective tumor microenvironments to antibody-mediated therapy. Cell (2014) 156(3):590–602. doi: 10.1016/j.cell.2013.12.041
276. Mraz M, Zent CS, Church AK, Jelinek DF, Wu X, Pospisilova S, et al. Bone marrow stromal cells protect lymphoma B-cells from rituximab-induced apoptosis and targeting integrin alpha-4-beta-1 (VLA-4) with natalizumab can overcome this resistance. Br J Haematol (2011) 155(1):53–64. doi: 10.1111/j.1365-2141.2011.08794.x
277. Long M, Beckwith K, Do P, Mundy BL, Gordon A, Lehman AM, et al. Ibrutinib treatment improves T cell number and function in CLL patients. J Clin Invest (2017) 127(8):3052–64. doi: 10.1172/JCI89756
278. Cook KW, Durrant LG, Brentville VA. Current Strategies to Enhance Anti-Tumour Immunity. Biomedicines (2018) 6(2). doi: 10.3390/biomedicines6020037
279. Labrijn AF, Janmaat ML, Reichert JM, Parren P. Bispecific antibodies: a mechanistic review of the pipeline. Nat Rev Drug Discov (2019) 18(8):585–608. doi: 10.1038/s41573-019-0028-1
280. Przepiorka D, Ko CW, Deisseroth A, Yancey CL, Candau-Chacon R, Chiu HJ, et al. FDA Approval: Blinatumomab. Clin Cancer Res (2015) 21(18):4035–9. doi: 10.1158/1078-0432.CCR-15-0612
281. Bacac M, Colombetti S, Herter S, Sam J, Perro M, Chen S, et al. CD20-TCB with Obinutuzumab Pretreatment as Next-Generation Treatment of Hematologic Malignancies. Clin Cancer Res (2018) 24(19):4785–97. doi: 10.1158/1078-0432.CCR-18-0455
282. Claus C, Ferrara C, Xu W, Sam J, Lang S, Uhlenbrock F, et al. Tumor-targeted 4-1BB agonists for combination with T cell bispecific antibodies as off-the-shelf therapy. Sci Transl Med (2019) 11(496). doi: 10.1126/scitranslmed.aav5989
283. Locke FL, Ghobadi A, Jacobson CA, Miklos DB, Lekakis LJ, Oluwole OO, et al. Long-term safety and activity of axicabtagene ciloleucel in refractory large B-cell lymphoma (ZUMA-1): a single-arm, multicentre, phase 1-2 trial. Lancet Oncol (2019) 20(1):31–42. doi: 10.1016/S1470-2045(18)30864-7
284. Jiang GM, Xu W, Du J, Zhang KS, Zhang QG, Wang XW, et al. The application of the fibroblast activation protein alpha-targeted immunotherapy strategy. Oncotarget (2016) 7(22):33472–82. doi: 10.18632/oncotarget.8098
285. Bertolini F, Fusetti L, Mancuso P, Gobbi A, Corsini C, Ferrucci PF, et al. Endostatin, an antiangiogenic drug, induces tumor stabilization after chemotherapy or anti-CD20 therapy in a NOD/SCID mouse model of human high-grade non-Hodgkin lymphoma. Blood (2000) 96(1):282–7. doi: 10.1182/blood.V96.1.282.013k09_282_287
286. Ruan J, Luo M, Wang C, Fan L, Yang SN, Cardenas M, et al. Imatinib disrupts lymphoma angiogenesis by targeting vascular pericytes. Blood (2013) 121(26):5192–202. doi: 10.1182/blood-2013-03-490763
287. Kwon M, Kim CG, Lee H, Cho H, Kim Y, Lee EC, et al. PD-1 Blockade Reinvigorates Bone Marrow CD8(+) T Cells from Patients with Multiple Myeloma in the Presence of TGFbeta Inhibitors. Clin Cancer Res (2020) 26(7):1644–55. doi: 10.1158/1078-0432.CCR-19-0267
288. Velasquez MP, Bonifant CL, Gottschalk S. Redirecting T cells to hematological malignancies with bispecific antibodies. Blood (2018) 131(1):30–8. doi: 10.1182/blood-2017-06-741058
289. Schuster SJ, Bishop MR, Tam CS, Waller EK, Borchmann P, McGuirk JP, et al. Tisagenlecleucel in Adult Relapsed or Refractory Diffuse Large B-Cell Lymphoma. N Engl J Med (2019) 380(1):45–56. doi: 10.1056/NEJMoa1804980
290. Maude SL, Laetsch TW, Buechner J, Rives S, Boyer M, Bittencourt H, et al. Tisagenlecleucel in Children and Young Adults with B-Cell Lymphoblastic Leukemia. N Engl J Med (2018) 378(5):439–48. doi: 10.1056/NEJMoa1709866
291. Liu E, Marin D, Banerjee P, Macapinlac HA, Thompson P, Basar R, et al. Use of CAR-Transduced Natural Killer Cells in CD19-Positive Lymphoid Tumors. N Engl J Med (2020) 382(6):545–53. doi: 10.1056/NEJMoa1910607
292. Neelapu SS, Locke FL, Bartlett NL, Lekakis LJ, Miklos DB, Jacobson CA, et al. Axicabtagene Ciloleucel CAR T-Cell Therapy in Refractory Large B-Cell Lymphoma. N Engl J Med (2017) 377(26):2531–44. doi: 10.1056/NEJMoa1707447
293. Tran E, Chinnasamy D, Yu Z, Morgan RA, Lee CC, Restifo NP, et al. Immune targeting of fibroblast activation protein triggers recognition of multipotent bone marrow stromal cells and cachexia. J Exp Med (2013) 210(6):1125–35. doi: 10.1084/jem.20130110
Keywords: immunotherapy, tumor microenviroment, stroma, anti-PD1, lymphoma, interferon, T cells, CAR T
Citation: Apollonio B, Ioannou N, Papazoglou D and Ramsay AG (2021) Understanding the Immune-Stroma Microenvironment in B Cell Malignancies for Effective Immunotherapy. Front. Oncol. 11:626818. doi: 10.3389/fonc.2021.626818
Received: 06 November 2020; Accepted: 04 January 2021;
Published: 25 March 2021.
Edited by:
Jérôme Paggetti, Luxembourg Institute of Health, LuxembourgReviewed by:
Marek Mraz, Central European Institute of Technology (CEITEC), CzechiaCopyright © 2021 Apollonio, Ioannou, Papazoglou and Ramsay. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Alan G. Ramsay, YWxhbi5yYW1zYXlAa2NsLmFjLnVr
†These authors share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.