 María Paz Saldías1,2†
María Paz Saldías1,2† Diego Maureira1,2†
Diego Maureira1,2† Octavio Orellana-Serradell1,2†
Octavio Orellana-Serradell1,2† Ian Silva1,2
Ian Silva1,2 Boris Lavanderos1,2
Boris Lavanderos1,2 Pablo Cruz1,2
Pablo Cruz1,2 Camila Torres1,2
Camila Torres1,2 Mónica Cáceres1,2,3
Mónica Cáceres1,2,3 Oscar Cerda1,2,3*
Oscar Cerda1,2,3*- 1Program of Cellular and Molecular Biology, Institute of Biomedical Sciences (ICBM), Faculty of Medicine, Universidad de Chile, Santiago, Chile
- 2Millennium Nucleus of Ion Channels-Associated Diseases (MiNICAD), Santiago, Chile
- 3The Wound Repair, Treatment, and Health (WoRTH) Initiative, Santiago, Chile
Breast cancer is one of the most frequent cancer types worldwide and the first cause of cancer-related deaths in women. Although significant therapeutic advances have been achieved with drugs such as tamoxifen and trastuzumab, breast cancer still caused 627,000 deaths in 2018. Since cancer is a multifactorial disease, it has become necessary to develop new molecular therapies that can target several relevant cellular processes at once. Ion channels are versatile regulators of several physiological- and pathophysiological-related mechanisms, including cancer-relevant processes such as tumor progression, apoptosis inhibition, proliferation, migration, invasion, and chemoresistance. Ion channels are the main regulators of cellular functions, conducting ions selectively through a pore-forming structure located in the plasma membrane, protein–protein interactions one of their main regulatory mechanisms. Among the different ion channel families, the Transient Receptor Potential (TRP) family stands out in the context of breast cancer since several members have been proposed as prognostic markers in this pathology. However, only a few approaches exist to block their specific activity during tumoral progress. In this article, we describe several TRP channels that have been involved in breast cancer progress with a particular focus on their binding partners that have also been described as drivers of breast cancer progression. Here, we propose disrupting these interactions as attractive and potential new therapeutic targets for treating this neoplastic disease.
Introduction
Breast Cancer Overview and Global Impact
Breast cancer is the second most frequent cancer type and the first cause of cancer-related deaths in women worldwide, with 627,000 deaths in 2018 (1). This neoplastic disease can be classified according to different parameters, providing further information about the tumor. In terms of diagnosis, breast tumors are histologically classified by their cellular origins in the mammary gland. If the tumor was originated by a ductal or lobular epithelial cell, it would be referred to as ductal or lobular carcinoma, respectively (2). For prognosis information, different molecular classifications have been developed. In this context, an important molecular classification is the "intrinsic subtype", which involves tumor classification according to the expression pattern of 50 genes related to cancer (PAM50). In PAM50, patients are sorted into six groups: Luminal A, Luminal B, HER2-enriched, Normal-like, Claudin-low, and Basal-like (3, 4). Another important molecular classification is the "surrogate intrinsic subtypes", which is the focus of this review. In this classification, tumors are grouped in five subtypes according to histologic features and expression of the Estrogen Receptor (ER), Progesterone Receptor (PR), and Human Epidermic Growth Factor Receptor 2 (HER2): Luminal A-like, Luminal B-like HER2−, Luminal B-like HER2+, HER2-enriched (non-luminal) and Triple-negative (TNBC). Luminal A-like, luminal B-like HER2- and luminal B-like HER2+ patients are the most common type, accounting for 75–80% of cases (4). Despite the above, these patients are usually treated with competitive inhibitors (also called hormonal therapy) that target the estrogen receptor, such as Tamoxifen or Fulvestrant. In cases where hormonal therapy fails, alternative treatments are employed, such as aromatase and mTOR inhibitors, which hinder and reduce estrogen synthesis. On the other hand, HER2-enriched tumors present increased levels of HER2, which is why the most efficient therapy consists of the use of Trastuzumab, a humanized monoclonal antibody against HER2. Even though Trastuzumab is the standard therapy for this group of patients, other HER2-related antibodies are available, such as Pertuzumab, an inhibitor of HER2 dimerization; Ado-trastuzumab (emtansine), an antibody–drug conjugate; and the tyrosine-kinase inhibitors lapatinib and neratinib, which inhibit both HER2 and the epithelial growth factor (EGFR) pathways (5). TNBC account for around 10–15% of breast cancer cases and are the hardest to treat (4). Since this group of patients is ER-, PR- and HER2-expression, there are no effective hormonal-based treatments or molecular therapies available. The treatment usually consists of surgery followed by chemotherapy and radiotherapy, whether individually or combined. However, its prognosis of TNBC remains poor (6).
Calcium Signaling in Breast Cancer
Calcium ions (Ca2+) regulate several cellular processes in different organisms (7–9). Spatio-temporal changes in the cytoplasmic Ca2+ concentration occur in response to different stimuli that will be interpreted by the cell, promoting specific cellular outputs (10). Different molecular entities regulate Ca2+ homeostasis (11) and allow the cell to establish large Ca2+ concentration gradients (15,000–20,000 times) maintaining extremely low intracellular Ca2+ levels (close to 100 nM) compared to the extracellular medium (1–1.5 mM) (9, 12).
Ca2+ signaling modulates various cellular processes such as gene expression, cell cycle progression, cell migration, autophagy apoptosis (13), fertilization (14), and synapsis (15). Accordingly, aberrant intracellular Ca2+ signaling has been implicated in the development of different pathologies, such as neurodegenerative (16), metabolic (17, 18), and autoimmune disorders (19). In the context of neoplastic diseases, intracellular Ca2+-dependent signaling is involved in various processes that promote tumorigenesis and cancer progression, such as proliferation, migration, angiogenesis, and evasion of apoptosis (reviewed by 20–23).
Recent studies have highlighted specific regulators of Ca2+ signaling due to their role in the progression of breast cancer (24). These Ca2+ signaling regulators have been involved in controlling of proliferation, metastasis, cell death, and drug resistance. Therefore, several molecular entities that participate in these events have been suggested as possible therapeutic targets for breast cancer treatment (25). Among these regulators, the Transient Receptor Potential (TRP) channels superfamily has emerged as a novel and interesting target for therapeutic intervention in the context of breast cancer.
The TRP channels superfamily is characterized by sharing a common structure of six transmembrane segments and acting as polymodal cellular sensors for a broad spectrum of physical and chemical stimuli (26). TRP channels have been divided into seven families according to their sequence similarity (27, 28): TRPA (ankyrin), TRPC (canonical), TRPM (melastatin), TRPML (mucolipin), TRPN (no mechanoreceptor potential C or nompC), TRPP (polycystin or polycystic kidney disease), and TRPV (vanilloid and a proposed sister family, TRPVL) and recently a new member has been identified, TRPS (soromelastatin) (29).
Recent reports suggest the important role of TRP channels in different diseases, including cardiovascular, neurological, metabolic and neoplastic disorders (30). In cancer, changes in the expression and function of TRP channels are directly related to cellular processes that contribute to the progression of cancer, such as cell proliferation, differentiation, angiogenesis, migration, invasion and chemoresistance of cancer cells to apoptotic-induced cell death, promoting resistance to chemotherapy treatments (31). Several studies have shown the effect of modulating the activity of TRP channels in various cancer models using agonists and antagonists with or without the use of chemotherapeutic drugs, proposing them as excellent therapeutic targets (31–33). However, most of these agonists or antagonists are not specific and affect several TRP channels, and can impact their activity in other tissues, causing adverse effects (34).
In this context, TRP channels can form homomeric complexes or co-assemble into heteromeric functional tetramers with subunits of the same family or between different families (35). Also, these channels interact with an extensive network of different regulatory and structural proteins (TRIP Database: (36, 37). Drug discovery has been characterized by searching for molecular targets to identify new and potentially beneficial agents to selectively target disease-specific mechanisms and pathways involved in these diseases (38). Protein–protein interactions (PPIs) mediate and regulate most cellular functions and processes. Also, several diseases are caused by aberrant interactions between proteins and their regulators or effectors. Thus, the searching and design of drugs to disrupt PPIs have become increasingly more relevant (39). In the context of cancer, one of the approaches to study the impact of certain genes or proteins on the tumor phenotype and differentiate between causal and driver genes is to analyze their network of interactions and their effect on specific functions and pathways (40). Recent studies demonstrate that ion channels can be crucial in the organization of large macromolecular complexes serving for the regulation of diverse cellular processes (41, 42), as well as transduction nodes on which several signaling pathways might converge (43). Indeed, ion channels’ expression is altered in several types of cancer. Also, their activity has been considered of great importance in the development and progression of the disease, including breast cancer (44, 45). Interestingly, not only ion channels are dysregulated in cancer, but also their regulators, effectors, and other interacting proteins. Here we discuss the role of TRP channel interactors involved in breast cancer and propose the modulation of these interactions as therapeutic targets (Figure 1 and Supplementary Table 1), opening a window for the design of more specific drugs against cancer.
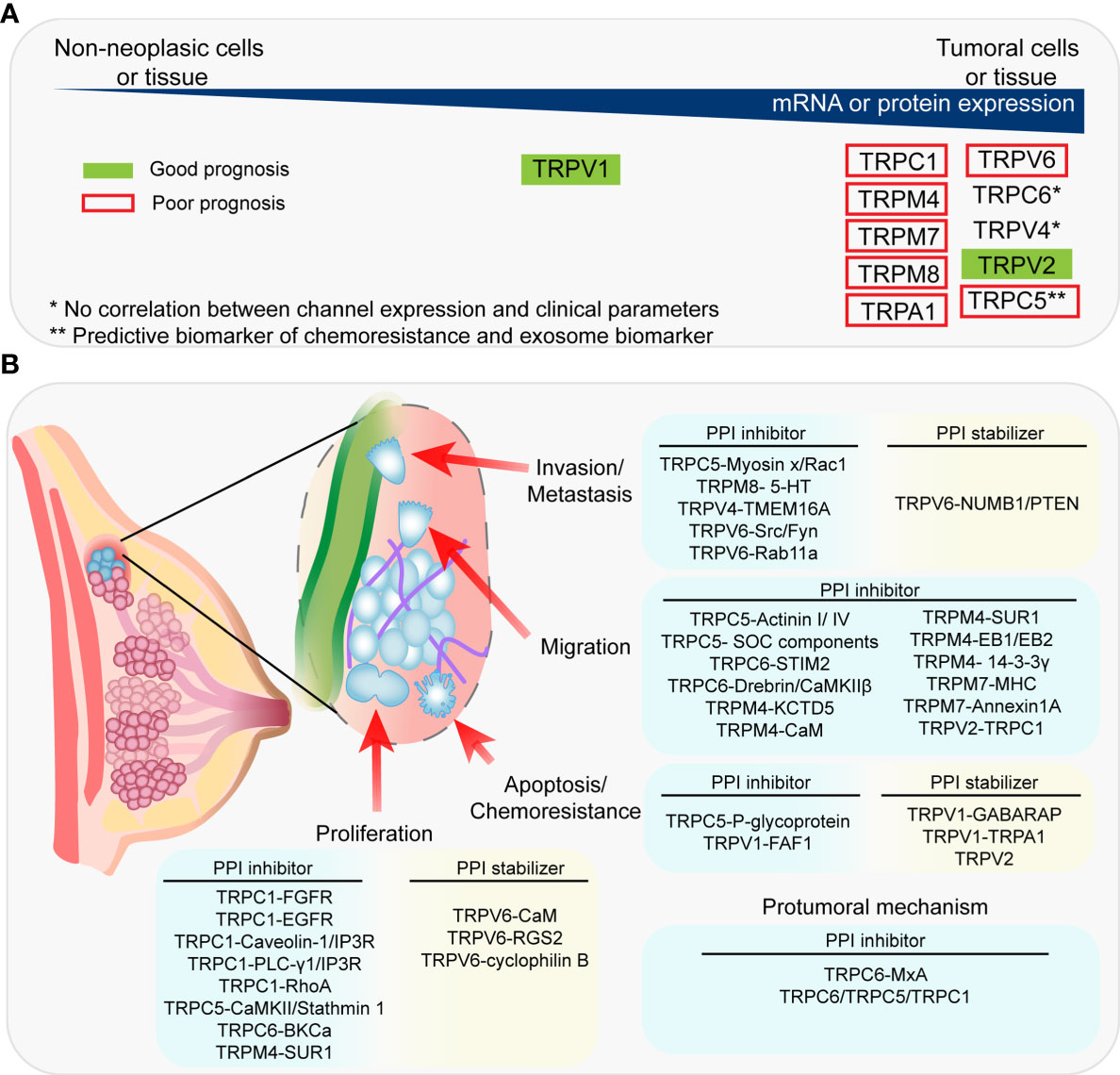
Figure 1 Association between the expression of TRP channels and clinical prognosis and PPIs proposed as therapeutic targets. (A) Summary of the expression of TRPC channels in tumoral vs non-tumoral tissue and the association between TRP channels and clinical prognosis. (B) Summary of the PPIs proposed as therapeutic targets and their associated-pro-tumoral processes.
TRP Channels and Their Interactors in Breast Cancer
TRPC1
TRPC1 is a non-selective cationic channel with a reported PCa/PNa ~1 in homotetrameric subunits (46–48). TRPC1 is expressed in diverse cell types such as smooth muscle, Central Nervous System (CNS) neurons, endothelial cells, platelets and salivary gland epithelial cells (46). Moreover, TRPC1 participates in endothelial permeability, vascular tone modulation (46), salivary fluid secretion (49), and synaptic plasticity (50). Mechanical stretch, receptor-dependent stimulation, diacylglycerol (DAG) (46, 51, 52) and ER-Ca2+ store depletion via STIM1 (53, 54) have been proposed as activation mechanisms.
The dysregulation in the activity of this channel has been reported in several type of cancer such as breast cancer, pancreatic cancer, lung cancer and others, where its overexpression has been related with poorly differentiated tumors and higher cell motility, proliferation and hypoxia-induced autophagy (55–58). Currently, it has been reported that TRPC1 promotes hypoxia-induced HIF1α-mediated EGFR signaling and expression of Epitelial–Mesenchymal Transition (EMT)-associated genes such as SNAIL in MDA-MB-468 cells (human TNBC cell line, Basal B). Consistently, TRPC1 expression is increased in Claudin-low human breast tumors and it has been associated with poor prognosis in patients with basal-like breast cancer (59). Importantly, TRPC1 is required for EGF-induced Ca2+ influx (48). Furthermore, TRPC1 interacts with FGFR1, and downregulation of TRPC1 inhibits proliferation in rat neural stem cells (60). Accordingly, TRPC1 silencing leads to decreased FGFR1 activation-induced Ca2+ signals. Both growth factor receptors, FGFR1 and EGFR, have crucial roles in cancer progression since their activation induces cell proliferation (61, 62). Moreover, FGFR1 expression is associated with resistance against endocrine therapy (63, 64), and EGFR is associated with poor prognosis in TNBC (61). These data suggest that TRPC1 might have important roles in FGFR1/EGFR-mediated breast cancer processes, which are still unaddressed. Since Ca2+ signaling is pivotal for these receptors’ downstream pathways, a novel therapeutic proposition might be to block TRPC1 activity or the functional interaction with either FGFR or EGFR.
TRPC1 interaction with Caveolin-1 is required for its proper plasma membrane targeting during Store-Operated Calcium Entry (SOCE) (65). Also, Caveolin-1 acts as a scaffold protein which mediates IP3R-TRPC1 interaction (66). Indeed, silencing Caveolin-1 decreases both SOCE and ER-Ca2+ depletion-induced association of TRPC1 to IP3Rs (67), indicating that Caveolin-1 plays an active role on Ca2+ homeostasis. The participation of Caveolin-1 in breast cancer has been extensively reviewed (68). Interestingly, Caveolin-1 participates in several breast cancer-progression events such as metastasis, cell cycle progression, and therapy resistance. Nevertheless, the current role of Caveolin-1 in breast cancer remains controversial due to different studies suggesting that this protein might act as a tumor suppressor. In contrast, others propose that it is instead an oncogene (68). Thus, the interplay between Caveolin-1 and TRPC1 in breast cancer is an open window for further research and future therapies. Similar to Caveolin-1, PLC-γ1 interacts with TRPC1 and IP3R during SOCE, suggesting a scaffolding role for this protein besides its IP3/DAG-production function (69).
PLC- γ1 overexpression has been proposed as a marker for metastasis development in early-stage luminal A and luminal B breast cancers (70). Interestingly, a synthetic peptide based on PLC-γ1 SH2 domains produces antitumoral effects in EGFR/HER-2 positive cells (71). Moreover, TRPC1 interacts with RhoA, a member of the Rho GTPases family. RhoA plays a crucial role in actin cytoskeleton dynamics, cell migration and contractility (72). Several authors have reported the opposite effects of RhoA in breast cancer models. Some studies state that RhoA prevents invasive processes (73, 74), while others propose that RhoA enhances cell invasiveness and proliferation of breast cancer cells (75, 76). This might be due to the fact that RhoA downstream effectors have opposing effects on pro-neoplastic processes (73). RhoA is suggested to promote TRPC1 association to IP3R during SOCE (77). Moreover, RhoA promotes the translocation of TRPC1 to the plasma membrane (77) and its activation (78), thus enhancing SOCE. Indeed, RhoA promotes cell migration of intestinal epithelial cells through the induction of SOCE via interaction with TRPC1 (78). Hence, TRPC1 might be an effector of RhoA activity, implying that this interaction’s specific disruption might impair TRPC1-dependent, breast cancer-associated pathways.
Interestingly, TRPC1 interaction with and activation by STIM1 confers a central role to this channel in the SOCE response contributing to the generation of the Store-Operated Currents (ISOC) (79). Moreover, TRPC1 interacts with several proteins that are implicated in the regulation of intracellular and ER Ca2+ concentrations, such as ORAI channels (80, 81), the SERCA pump (82, 83), PMCA (84), IP3R (82, 85), NCS-1 (86), and FKBP4 (87). Since Ca2+ signals are crucial for a myriad of cellular processes, it is not surprising that all these TRPC1-associated proteins have been proposed to play a role in breast cancer progression (88–93).
As the data suggests, the understanding of the dynamic cooperation between TRPC1-associated proteins mentioned above might shed light on the design and development of targeted therapeutics to selectively disrupt one or more of these interactions, depending on the differential features of the tumors.
TRPC5
TRPC5 is a nonselective cation channel permeable to Ca2+ with a reported PCa/PNa/PCs = 14.3/1.5/1, that can be activated following stimulation of receptors coupled to PLC in an intracellular Ca2+ dependent manner (94, 95). In cancer, this channel is mainly involved in angiogenesis and chemoresistance (96). Moreover, TRPC5 has been proposed as a predictive biomarker of chemoresistance in breast and colorectal cancer (97, 98). The expression of this ion channel is up-regulated together with P-glycoprotein, an efflux pump in Adriamycin-resistant breast cancer cells (MCF-7/ADM) (99, 100). In addition, the activation of this channel evokes Ca2+ currents, which induce P-glycoprotein over-expression, promoting chemoresistance (100). Moreover, TRPC5 was proposed as an exosome biomarker in breast cancer, which was demonstrated to have the capacity to transfer their chemoresistant phenotype to neighbor cells (101). Interestingly, this channel has been involved in Adriamycin resistance, mediating autophagy through the CaMKKβ/AMPKα/mTOR pathway (102).
TRPC5 interacts with several actin cytoskeleton regulators, such as Rac1 and several F-actin binding proteins such as Myosin X, α-actinin I, α -actinin IV and Neurabin II (103, 104). All these proteins have been involved in breast cancer progression. For instance, Myosin X is overexpressed in breast cancer tissue and breast cancer cell lines (MCF-7, BT-549 and MDA-MB-231) but not in non-cancerous breast cell lines (MCF-10A), promoting invadopodia formation, and metastasis in a Rac1-dependent manner (105). Rac1 is activated by TRPC5-mediated Ca2+ entry, promoting cell migration (103). Moreover, Rac1 is overexpressed and activated in the plasma membrane of tumor cells in aggressive breast cancer samples (106). These results suggest that TRPC5 interacts with Myosin X and Rac1, where the activation of Rac1 by TRPC5 or Myosin X might promote metastasis. Therefore, the disruption of these interactions could be beneficial for patients. On the other hand, α-actinin I and IV are actin binding proteins which localize in cell-cell adhesions, cell-matrix contact sites and in cellular protrusions, stabilizing these structures and linking membrane receptors with the cytoskeleton (107). The binding of α-actinin to Ca2+ reduces their affinity for actin (108). Moreover, the loss of α-actinin I in cell contact sites of TNBC cell lines (MDA-MB-231) promotes cell migration, whereas the same was reported for α-actinin IV in ER+/HER2- cell lines (MCF-7 cells) (109, 110). These results suggest that α-actinin I and IV act as tumor suppressor proteins, while their Ca2+-dependent delocalization promotes migration in breast cancer cells. Although we cannot attribute these proteins’ delocalization only to TRPC5-dependent Ca2+ entry, promoting the localization of α-actinin I and IV to cell contact structures or disrupt their binding to Ca2+ could be useful in breast cancer treatment.
TRPC5 interacts with other proteins that modulate Ca2+ signaling, such as TRPC1, TRPC6, IP3R3 and STIM1. Moreover, all of these TRPC channels interact with the IP3R3 and STIM1 (111, 112), which could be an intrinsic characteristic of Store Operated Channels (SOC). Interestingly, the interaction between TRPC1, STIM1 and IP3R3 is relevant in Ca2+ signaling for cancer progression (55). Although the role of TRPC5 in cellular signaling through Ca2+ is still unclear, it is accepted that TRPC5-mediated Ca2+ entry activates CaMKIIβ inducing its phosphorylation in T287 (113), modification that has been described as increased in breast cancer tissue and metastasis (114). In addition, in vitro studies in MDA-MB-231 cells reported that CaMKIIβ promotes cell invasion (114). Therefore, the activation of CaMKIIβ through TRPC5-mediated Ca2+ entry might promote breast cancer progression, making of this interaction an interesting therapeutic target. In summary, although the mechanism by which TRPC5-mediated Ca2+ entry could promote cancer progression requires further investigation, the data suggests that disrupting interactions between TRPC5 and others SOC components (including IP3R3) involved in Ca2+ signaling could become a therapeutic strategy in breast cancer.
Stathmin1 is a protein which active or non-phosphorylated form mediates depolymerization of microtubules in late mitosis (115). Interestingly, it has been reported that tumors with high levels of this protein show enhanced proliferation, angiogenesis and immune response evasion, mainly in basal like subtypes of breast cancer (116). Moreover, increased expression of Stathmin1 correlates with poor prognosis for patients with breast cancer (117). Moreover, Stahmin1 is inhibited through CaMKII-mediated phosphorylation in a Ca2+-dependent manner, which in turn is activated by TRPC5 (113, 118). These results suggest that TRPC5, CaMKII and Stathmin1 could be part of a complex and a novel pathway to study as a therapeutic target.
TRPC6
TRPC6 is a non-selective Ca2+ permeable channel with a PCa/PNa ~ 5, tightly receptor-operated and shows little basal activity (119). The main regulatory mechanism of TRPC6 is its activation by diacylglycerol (DAG) in response to the activation of G protein-coupled receptors. Additionally, other modulation mechanisms include regulation by Ca2+/Calmodulin (CaM) and binding of phosphoinositides (120). In humans, this channel is highly expressed in the lungs, placenta, ovary and spleen. It has important roles in regulating the heart and cardiopulmonary vasculature, podocyte functioning in the kidney, several neuronal processes, and several neoplastic diseases (121). In this context, TRPC6 was found upregulated in several types of cancer biopsies such as glioma, hepatocellular, gastric, esophageal and breast cancer, where it promotes migration invasion and proliferation (122–126). Specifically in breast cancer, studies in breast cancer cell lines showed high expression of TRPC6 in MCF-7 and MDA-MB-231 cells, whereas no expression was found in MCF-10A cells (122). Consistently, TRPC6 is the most overexpressed of all TRP channels in samples of human breast ductal adenocarcinomas compared to normal tissue. However, no correlation between TRPC6 expression and clinical parameters such as histological grade, tumor size, proliferation or invasiveness has been described (57). Although a precise role for TRPC6 in breast cancer has not been defined yet, there are studies relating to TRPC6 expression and activity with breast cancer progression in in vitro models. Moreover, TRPC6 promotes cell proliferation, migration and invasion in MCF-7 and MDA-MB-231 cell lines, an effect that is partially mediated by the modulation of surface expression of ORAI1 and ORAI3 Ca2+ channels (127). Recent studies showed that TRPC6 interacts and participates in conjunction with STIM2 to maintain cytosolic and endoplasmic reticulum Ca2+ concentrations in MCF-7 cells. Moreover, inhibition of TRPC6 activity leads to endoplasmic reticulum stress and caspase-3 activation (128). Conversely, TRPC6 associates with Large-Conductance Ca2+-Activated K+ (BKCa) channels, modulating their surface expression in podocytes (129). Interestingly, BKCa channels have been proposed as possible oncogenes in several types of cancer, including breast cancer, with augmented Ca2+ sensitivity, promoting shifts in the membrane potential that might promote tumor growth (130, 131). Thus, TRPC6-mediated BKCa modulation might contribute to tumor proliferation in breast cancer. TRPC6 also interacts with Fyn and Src tyrosine kinases, which positively regulate the activity of the channel through phosphorylation (132) and which have been reported as overexpressed in breast cancer cell lines and tissue (133, 134). Thus, Fyn and Src interaction with TRPC6 might be an important promoter of its activity and pro-tumoral effects in breast cancer. Moreover, TRPC6 activity is positively regulated by its interaction with the human myxovirus resistance protein 1 (MxA) (135), whose expression is higher in TNBC tumors than in other subtypes and is associated with a higher histologic grade (136). This data suggests that at least in TNBC, MxA-mediated TRPC6 increased activity might be a pro-tumoral mechanism promoting breast cancer progression. Another interesting interactor of TRPC6 is the actin-binding protein Drebrin, which, as we discuss previously, promotes the formation of cell protrusions in motile cells (104). Although a functional role for this interaction was not described, Drebrin interacts with and helps to localize and stabilize CaMKIIβ at actin filaments (137), which is a known positive regulator of TRPC6 activity (138), suggesting an interplay between Drebrin, CaMKIIβ and TRPC6, resulting in regulation of channel activity. Given the importance of both TRPC6 and Drebrin, this interaction and subsequent increase in TRPC6 activity might be one of the mechanisms by which Drebrin exerts its effects on breast cancer development, posing this interaction as an interesting and potential therapeutic target. Furthermore, TRPC6 interacts with several other TRPC family members of ion channels, such as TRPC1 and TRPC5 (139), downregulating the activity of the channel. As we mentioned above, these channels have also been reported as dysregulated in breast cancer. Thus, modulating the interplay between these TRPC channels might reduce Ca2+ influx and a subsequent reduction of tumor progression-associated processes.
TRPM4
TRPM4 channel, is a non-selective cation calcium-activated channel permeable to monovalent ions, mainly Na+ and K+, with a permeability ratio PNa/PCa = 11, which is activated by intracellular Ca2+ (140–142). TRPM4 is broadly expressed in several tissues (141) and modulates several cell functions, including insulin secretion (143), immune response (144), cell migration and contractility (145, 146). TRPM4 also plays an important role in disease and several evidences has emerged linking the expression of TRPM4 with the increase of migration, invasion and proliferation of certain cancers, such as lymphoma (147), cervical (148), colorectal channel, prostate cancer (149–151) and others (152). Moreover, upregulation of this channel is associated with poor prognosis in large B cell lymphoma, breast cancer and endometrial carcinoma, while its overexpression increases the recurrence risk in prostate cancer (153–156). Recently, independent groups showed that TRPM4 is overexpressed in breast cancer samples of ER+/PR+ and triple-negative subtypes compared with normal breast tissue (153, 157). Moreover, TRPM4 expression is increased in IIB, IIIA, and IV stages and higher lymph node spread (N1–N2), according to the TNM classification (153, 157). In addition, TRPM4 gene was significantly associated with the expression of estrogen response and epithelial–mesenchymal transition (EMT) gene sets (157). As such, the identification of TRPM4 interactions might allow us to understand the biological role of TRPM4 in breast cancer and to broaden the search for molecular targets for its treatment. KCTD5 (Potassium Channel Tetramerization Domain-containing protein 5) interacts with TRPM4, regulating cell migration and contractility, by acting as a positive regulator of TRPM4 activity. Interestingly, KCTD5 mRNA is significantly upregulated in breast cancer tumors versus normal breast tissue samples (153), consistent with transcriptomics data in public databases (158). These data suggest that the increased expression of KCTD5 could be a key determinant of the malignancy and aggressiveness of these types of tumors, by interacting with TRPM4, enhancing the activity of the channel and thus, promoting TRPM4-dependent cell migration. Along the same lines, another interactor that acts as a positive regulator of channel activity is CaM which modulates the NFAT and Akt pathway promoting survival, proliferation and migration in several breast cancer cell lines in a Ca2+ dependent manner (159, 160). TRPM4 contains five CaM-binding sites and that by eliminating any three sites at the C-terminal, it strongly alters the activation current by decreasing Ca2+ sensitivity and modifying its voltage-dependent activation (161). Therefore, in the context of breast cancer, increased activity, and channel sensitivity due to TRPM4–CaM interaction could promote cell migration and contractility of tumor cells.
Another interesting interactor is sulfonylurea receptor-1 (SUR1), which forms a functional channel with TRPM4, presenting biophysical properties of TRPM4 and pharmacological properties of SUR1. Woo et al. showed that this TRPM4-SUR1 co-assembly has double affinity and sensitivity for CaM and Ca2+, respectively (162). SUR1 is a subunit of the inward-rectifier potassium ion channels Kir6.X (Kir6.1 and Kir6.2), whose association forms the KATP channels (163). SUR1 has been reported as overexpressed in lung cancer (164) and breast cancer (165). Studies using glibenclamide, a drug that binds SUR1, have shown a cytostatic effect on MDA-MB-231 cells, inhibiting cell cycle progression (166). Interestingly, recent studies suggest that this drug acts as an antagonist of the TRPM4–SUR1 interaction (167). Therefore, the interruption of this interaction might decrease cell migration and cell cycle progression, possibly through phosphorylation of CaMKII and decreased nuclear translocation of NFAT (168), which have also been implicated in cancer progression (169).
In the context of trafficking mechanisms and binding partners, two interesting TRPM4 interactors have been involved in the progression of breast cancer. First, EB1 and EB2 proteins that regulate the trafficking, surface expression and activity of TRPM4 (170). Interestingly, EB1 promotes the proliferation of cancer cells and have implicated EB1 in the tumorigenesis of breast cancer (171–173). For instance, EB1 expression correlates with higher histological grade, and the incidence of lymph node metastasis (171). Since the TRPM4–EB1 interaction has been shown to decrease FA turnover, and cell migration and invasion in murine melanoma cell lines (B16-F10 cells) (170), the disruption of the TRPM4–EB1 interaction might reduce the aggressiveness and metastatic capacity of breast cancer cells. Conversely, 14-3-3γ increases the surface expression of TRPM4 in HEK293T cells (174). The 14-3-3 protein family binds to the phosphorylated Ser/Thr motifs of its target proteins, regulating them through different mechanisms (175). Recently, the role of this family in age related-diseases such as neurodegenerative diseases and cancer, has been demonstrated (176). In breast cancer 14-3-3γ localizes in pseudopodia of MDA-MB-231 cells, where 14-3-3γ knockdown diminishes pseudopodia formation and cell migration (177). In summary, the TRPM4-14-3-3γ interaction could promote the expression of the channel on the surface, increasing its activity, which, given the data, could have a synergistic effect on cell migration of breast cancer cells. Therefore, interrupting the TRPM4-EB and TRPM4-14-3-3γ interactions could be favorable for breast cancer patients, reducing cancer cells’ migration and potential metastasis.
TRPM7
TRPM7 is a member of the TRP family that permeates both Ca2+ and Mg2+. Moreover, free intracellular Mg2+ blocks this channel whereas ATP promotes its activation (178). In addition, TRPM7 possesses a unique C-terminal domain, which contains an active kinase domain (179). Interestingly, excision of the kinase domain enhances the TRPM7 ion channel activity (180). TRPM7 is broadly expressed in different tissues (181), and its ion channel function and kinase activity has been related to migration, cell proliferation and cell death in immune, hepatic, and different tumor cells (182). In the context of cancer, TRPM7 expression and activity has been associated with different types of cancer such as retinoblastoma (183), gastric cancer cells (184), head and neck carcinoma (185), and neuroblastoma (186, 187). In addition, TRPM7 expression is elevated in several histological subtypes, such as invasive ductal, invasive lobular (188) and in ER- adenocarcinoma (189). Moreover, TRPM7 expression correlates with breast cancer progression and diminished survival of patients (190). Interestingly, the TRPM7 kinase domain directly phosphorylates the myosin heavy chain (MHC) (191, 192), an effect that was corroborated in MDA-MB-231 cells (189). Moreover, overexpression of TRPM7 increases migration in MCF-7 and MDA-MB-213 cells, while a truncated version that lacks the kinase domain has no effect on cell migration (189). TRPM7 silencing in MDA-MB-231 cells induces a redistribution of the actin cytoskeleton, promoting cell contraction (190). These data suggest that TRPM7 might regulate cell migration in breast cancer through phosphorylation of the MHC, facilitating invasiveness and metastasis. Thus, disrupting the interaction of TRPM7 with MHC, preventing its phosphorylation, may be a novel target to reduce metastasis formation in breast cancer.
Another TRPM7 phosphorylation target is the lipid-binding in a Ca2+-dependent manner protein Annexin 1A (193). Phosphorylation of Annexin 1A by TRPM7 stabilizes its interaction with membrane lipids (194). Interestingly, Annexin 1A is related to the pathogenesis of several cancer types (195), including breast cancer, where it has been proposed as an indicator of poor prognosis (196, 197). Annexin 1A promotes migration and metastasis in the highly invasive cell lines MDA-MB-231 and 4T1 (murine TNBC cell line) cells (196, 198). Interestingly, Annexin 1A depletion in MCF-7 cells increases the formation of focal adhesions and stress fibers, promoting a contractile phenotype, which is related to the effects observed when TRPM7 was absent. All together, these results suggest that TRPM7 might modulate cell migration and invasion of breast cancer through the regulation of Annexin 1A activity, suggesting this interaction as a possible therapeutic target against breast cancer.
Smad2 is a transcriptional regulator related to TGF-ß -induced EMT in several cancer types (199). Interestingly, Smad2 phosphorylation was reduced upon TRPM7 silencing (200). High levels of Smad2 phosphorylation are proposed as a marker of poor prognosis in breast cancer (201). Thus, TRPM7 inhibitors could decrease the levels of Smad2 phosphorylation, ameliorating the prognosis of patients.
In summary, the TRPM7 kinase activity is a novel pharmacological target to modulate invasiveness and metastasis of breast cancer, through the indirect regulation of the activity of its phosphorylation targets such as MHC, Annexin 1A or even Smad2.
TRPM8
TRPM8 is a nonselective cationic ion channel with higher permeability for Ca2+ with a PCa/PNa ~1 (202). Notably, TRPM8 is a cold sensitive channel and is activated by cooling compounds such as menthol (203). Since TRPM8 is activated by cold temperatures, this channel is considered the molecular entity responsible for cold transduction in peripheral nerves (204). In the context of cancer, TRPM8 has been strongly involved in prostate cancer wherein it is related to tumor staging (205, 206). Moreover, this channel was found overexpressed in others types such as glioblastoma (207), pancreatic cancer (208), melanoma (209) and breast cancer (210). In breast cancer, TRPM8 expression is mainly upregulated in ER+ grade I adenocarcinomas (57, 211). Comparative analysis of TRPM8 mRNA levels shows higher expression in the highly invasive MDA-MB-231 cell line. Despite the precise analysis of TRPM8 expression in the different classifications of breast cancer, little is known about the participation of TRPM8 in the tumor pathology. For instance, TRPM8 silencing decreases migration and invasion in MDA-MB-231 cells, reducing the expression of EMT-related markers such as Akt, GSK-3ß phosphorylation and Snail expression. Conversely, TRPM8 overexpression in the low aggressive MCF-7 cell line increases invasion, migration and promotes EMT (212). The analysis of TRPM8 interacting proteins helps to hypothesize possible roles for this channel in the pathology of breast cancer. For instance, TRPM8 interacts with the Serotonin receptor 5-HT1B, enhancing the response of the channel to its agonists (213). In breast cancer, the 5-HT7 receptor is upregulated in triple negative cell lines such as the MDA-MB-231, HCC-1395, and Hs578T cells, increasing its invasion and proliferation through the PI3K/Akt pathway (214), an effect similar to that of TRPM8 (212), which suggests that 5-HT receptors might be a downstream effectors of TRPM8 activity.
TRPV1
TRPV1 is a ligand-gated non-selective channel whose activation allows the influx of Ca2 + and Na+ PCa/PNa = 9.6 (215, 216) and its selectivity depends on the nature and concentration of the agonist (217). TRPV1 can be activated by capsaicin and its analogues as well as by temperature, and pH changes (215) among others (216, 218–221). TRPV1 activity can be modulated by PKA (222), PKC (223) and CaMKII-dependent phosphorylation (224). Additionally, CaM and Inositol-4,5-biphosphate (PIP2) (225) binding have been reported to activate it. TRPV1 has been involved in processes such as thermoregulation and inflammatory nociception and is widely expressed in brain tissue, primary and secondary sensory neurons, arteriolar smooth muscle cells (226), skeletal muscle (227) and urothelium (228). TRPV1 has been associated with different diseases, including different types of cancer such as tongue squamous cell and prostate carcinoma (229, 230), hepatocellular carcinoma (231) and bladder carcinoma (232), skin cancer (233) and others (234–236). Although most studies report a tumor-suppressor role for TRPV1, its exact function in tumorigenesis is not clear. Increased expression of TRPV1 has been observed in all subtypes based on the molecular profile in breast cancer (237). In addition, TRPV1 expression pattern varies between cytoplasmic/membranous (165, 238) to a pattern in aggregates at the ER/Golgi, which was associated with a more aggressive tumoral stage (238).
Multiple studies have linked TRPV1 activity with an antitumoral role in breast cancer, promoting cell death (237, 239, 240) and decreased cell proliferation. Additionally, the activation of TRPV1 with agonists, modulators (MRS1477) and chemotherapeutic agents such as cisplatin induces cell death by apoptosis through depolarization of the mitochondrial membrane, ROS production and caspase activation (239, 240). Similarly, melatonin, in conjunction with the chemotherapeutic agent DOX, promotes apoptosis in MCF-7 cells through the activation of TRPV1 (241). Moreover, activation of TRPV1 using low doses of capsaicin may induce apoptosis in tumor cells, while higher doses of capsaicin activate necrosis (242). Wu et al. also determined that in MCF-7 cells, the expression levels of TRPV1 are decisive for capsaicin-induced cell death (243). Therefore, the use of TRPV1 agonists can be a complementary treatment to those currently used, promoting the sensitization of therapy-resistant tumors to cell death.
Fas-associated factor 1 (FAF1) is an adaptor protein for the Fas receptor and has been described to interact with TRPV1, reducing its response to capsaicin, heat and acid (244). A study performed in fibrosarcoma cells suggest that FAF1 acts as a negative regulator of cell death mediated by capsaicin-dependent activation of TRPV1. Moreover, FAF1 silencing increases fibrosarcoma cells susceptibility to apoptosis when cells were treated with capsaicin (245). However, in breast cancer, several studies have reported a tumor suppressor activity for FAF1, where this protein antagonizes Wnt signaling by stimulating β-catenin degradation (246). Additionally, FAF1 destabilizes TβRII on the cell surface limiting excessive TGF-β response, an effect that could supress metastasis formation (247). Furthermore, Xie et al. showed that there is a positive correlation between the survival of patients with metastasis-free breast cancer and the expression of FAF1. Therefore, the design of a drug to prevent the TRPV1/FAF1 interaction without affecting FAF1 tumor supressing activity could be a powerful tool in the treatment of breast cancer.
Another interesting interactor of TRPV1 is γ-aminobutyric acid type A (GABAA) receptor associated protein (GABARAP) where GABARAP increases TRPV1 expression, clustering at the cell surface and modulates TRPV1 gating and sensitivity (248). Moreover, GABARAP has been described as a tumor suppressor in breast cancer (249), where its mRNA and protein expression levels were significantly downregulated in invasive and ductal lobular carcinomas compared to normal breast tissue. Therefore, the design of a peptide that functionally emulates GABARAP that could promote its tumor suppressive role and increases the traffic of TRPV1 without altering its activity, could contribute to the treatment of breast cancer. Therefore, further studies into this interaction might represent an interesting new therapeutic approach.
TRPA1 is a nonselective cation channel permeable to Ca2+, Na+, and K+. TRPA1 detects a wide range of hazardous stimuli and is also involved in noxious cold and mechanical sensation (250). Different studies have demonstrated functional interaction between TRPA1 and TRPV1 (251, 252). Direct interaction of TRPV1 with TRPA1 exerts a Ca2+-dependent modulation on certain intrinsic properties of TRPA1 (253). Recently, it has been observed up-regulated expression of TRPA1 in breast cancer cells, promoting tolerance to oxidative stress in tumor cells. On the other hand, TRPA1 inhibition reduces tumor growth and improves sensitivity to chemotherapies (254). Interestingly, cell-permeable peptide that mimics the C-terminal of Tmem100-3Q, a modulator of the interaction between TRPA1 and TRPV1, selectively inhibit TRPA1-mediated activity in a TRPV1-dependent manner, generating a promising therapy for pain (255). Therefore, the design of peptides that promote the interaction of TRPV1–TRPA1 that might induce channel inhibition could be a potential treatment as has been observed.
TRPV2
TRPV2 is a non-selective cation channel showing Ca2+ permeability, with a PCa/PNa ~3, and can be activated by high temperatures (>52 °C) (256), hypoosmolality (257), cell stretching (258), cannabinoids (259), among others (260, 261). TRPV2 is mainly expressed in the central and peripheral nervous system (256, 262–264) and at the subcellular level it is present in intracellular membranes (265, 266) and plasma membrane (267). TRPV2 act as a mechanosensor, osmosensor (257) and contributes to nociception and thermoregulation. However, studies performed in TRPV2 knock out mice suggest that it might participate in other activities (262), such as neurogenesis and gliomagenesis (268). TRPV2 has been involved in pathological processes, such as the development of cardiomyopathies (269) and cancer (270). In the context of cancer, the overexpression of channel is associated with well differentiated tumours and an increase of the survival in hepatocellular carcinoma, glioma and glioblastoma, while the opposite effect was observed in oesophageal carcinoma, urothelial carcinoma and gastric cancer (271–276). Increased TRPV2 expression has been observed in breast cancer samples, especially in TNBC subtype patients, where TRPV2 activation improved DOX uptake by tumor cells and was associated with better prognosis (277). Different studies have shown the formation of heteromers between TRPV1 and TRPV2 (278–280). Even though there are no studies directly linking their co-expression in breast cancer as indicated in the previous section, TRPV1, as well as TRPV2 have been described as markers of good prognosis. Therefore, enhancing their interaction and activation might be an interesting therapeutic approach.
Another interactor of TRPV2 is Acyl-CoA binding domain containing 3 (ACBD3), which mediates the TRPV2–PKA interaction and subsequent PKA-mediated phosphorylation (281). ACBD3 is involved in the maintenance of Golgi structure and function through its interaction with the integral membrane protein. However, a recent study shows that ACBD3 overexpression correlates with poor prognosis in breast cancer, where this protein promotes tumorigenesis by activating the Wnt/β-catenin signaling pathway (282). Although more studies are required on how the TRPV2–ACBD3 interaction might contribute to the modulation of channel activity or downstream signaling pathways in breast cancer contexts, it represents a novel and interesting therapeutic target to prevent disease progression.
TRPV4
TRPV4 is a non-selective cationic channel with a reported relative permeability of PCa/PNa ~7 (283, 284). This channel is activated by multiple stimuli, such as osmotic changes, mechanical stretching, warm temperatures and arachidonic acid metabolites (285). It participates in several physiological processes such as vascular tone modulation, endothelial permeability, nociception and inflammatory responses in multiple organs (283, 285). Its expression has been reported in sensory and brain neurons, skeletal and smooth muscle as well as in epithelial tissue of several organs including the trachea, bladder, cornea, bile ducts and breast (283).
In the context of cancer, decreased TRPV4 expression has been reported in basal cell carcinoma, whereas an increase in its expression has been associated with poor survival in colorectal cancer (286, 287). Moreover, overexpression of this ion channel has been detected in breast cancer samples from human patients in comparison with normal tissue (288, 289). In this context, significantly higher expression was found in metastatic lesions compared with intraductal carcinomas (289) and in ER-negative compared with ER-positive samples (288). Moreover, TRPV4 expression has been associated with decreased survival of distant metastasis-free patients and enhanced EMT progression. This is consistent with its established role in non-metastatic murine breast cancer cell line (i.e. 4T07 cells), which involves the phosphorylation-dependent activation of Akt (289), pivotal kinase for EMT during breast carcinogenesis (290). Ca2+ signaling mediated by TRPV4 lead to phosphorylation-dependent Akt activation, causing a decrease of E-Cadherin expression (289). Interestingly, in keratinocytes it has been observed that TRPV4 forms a protein complex with β-catenin and E-cadherin in E-cadherin-dependent junctions, where TRPV4-mediated Ca2+ signals favours the maturation of these structures (291). This discrepancy might be explained by differences in the cellular model or due to alterations of regulatory mechanisms proper from neoplastic cells. Despite this, the presented data highly suggest that TRPV4 has an important role in breast cancer EMT progression via E-Cadherin regulation. Further studies are needed to unveil the possible functional association between these proteins in the context of breast cancer, which might grant valuable information for therapeutic approaches.
TRPV4 also interacts with Fyn and Src tyrosine kinase (292), protein associated with the progression and malignant features of diverse types of cancer (293). Similar to TRPV4, Fyn promotes EMT progression in breast cancer (294). Interestingly, Fyn-mediated phosphorylation of TRPV4 Tyr-253 residue is necessary for hypotonicity-induced Ca2+ entry, suggesting that Fyn is a positive regulator of TRPV4 (292). AQP5 associates with TRPV4 and is crucial for TRPV4-dependent Ca2+ influx promoted by hypotonic stimulus. AQP5 has been associated with breast cancer cells proliferation and migration (295) possibly by accelerating volume changes of the leading edge, contributing to actin cytoskeleton dynamics and cell shape changes during this process (296–298). Additionally, TRPV4 interaction with the Ca2+-activated Cl- channel TMEM16A is also involved in volume regulation. Different studies show that TRPV4-mediated Ca2+ influx promotes TMEM16A activation, leading to Cl− outward currents and subsequent water efflux (299, 300). It has been reported that TMEM16A is overexpressed in 78% of human breast cancer samples and that it modulates breast cancer progression through promotion of EGFR signaling in breast cancer cell lines (301). Thus, a possible interplay between Fyn, TRPV4, TMEM16A and AQP5 might occur during breast cancer progression and contribute to volume regulation during cell migration, leading to the development of a metastatic phenotype.
We previously mentioned the participation of IP3R3 and Caveolin-1 in breast cancer and their association with TRPC1. Interestingly, TRPV4 interaction with these proteins has been reported (302–304). Indeed, TRPV4 forms functional heterotetramers with TRPC1 (303) and is suggested to participate in SOCE (305). Moreover, the functional interaction between TRPC1 and TRPV4 participates in Ca2+ homeostasis of endothelial cells (305, 306). Thus, TRPV4 is crucial for the migration of breast cancer endothelial cells, an essential process for angiogenesis during tumor growth (307). In conclusion, the association of TRPV4 and TRPC1 could modulate tumor angiogenesis and involve Caveolin-1 and the IP3R3. Functional analysis of these interactions and their cellular outcomes remains to be explored to address this as a possible therapeutic target.
TRPV6
TRPV6 is a Ca2+ channel with a PCa/PNa ~130 (308), regulated by intracellular Ca2+ in a Ca2+ and Ca2+/CaM-dependent manner (309). TRPV6 channels are widely expressed in absorptions epitheliums, exocrine glands, placenta, epididymal epithelium and in vestibular and cochlear tissues, where it mediates transcellular transport and maintains physiological levels of Ca2+ (310–313). In neoplastic diseases, TRPV6 channel has been reported as overexpressed in breast, colon, ovary, prostate and thyroid carcinomas promoting proliferation (314). Moreover, increased levels of this channel have been associated with high survival rates and better prognosis in cervical and oesophageal carcinomas, whereas the opposite effect has been reported in pancreatic cancer (315–317). In breast cancer, TRPV6 mRNA increased levels have been reported in tumor tissue and its expression has been correlated with poor prognosis for patients (57, 318). In vitro analysis has shown that TRPV6 silencing reduces breast cancer cell proliferation and promotes apoptosis (318). Interestingly, TRPV6 expression is promoted by the activation of vitamin D3, estrogen and androgen receptors. This induced overexpression increases the basal intracellular levels of Ca2+ in breast cancer cells, positively regulating gene expression via the CaM/Calcineurin/NFAT pathway, which modulates proliferation and apoptosis (314). Interestingly, this channel interacts with several proteins with important roles in breast cancer.
CaM, is a Ca2+ -sensing protein that regulates intracellular Ca2+ levels and has the capability to activate or inactivate several target proteins in a Ca2+-dependent manner (319). CaM induced the slow inactivation of TRPV6 channel by direct interaction (320) and modulates positively the NFAT and Akt pathways, promoting survival, proliferation and migration in several breast cancer cell lines in a Ca2+-dependent manner (159, 160). These results suggest that CaM inactivates TRPV6, reducing Ca2+ entry. Interestingly, in breast cancer, rises in Ca2+ levels might activate the NFAT and Akt pathways. Thus, uncoupling the TRPV6–CaM interaction could promote Ca2+ influx and activate those pathways. Thus, promoting this interaction could be beneficial for patients.
TRPV6-dependent Ca2+ currents are regulated by phosphorylation of the channel in tyrosine residues. For example, Fyn and Src are tyrosine kinases, members of the Src family, which regulate several processes, such as cell proliferation, survival, differentiation, motility and angiogenesis (321). Although Fyn interaction with TRPV6 was reported, the specific residues phosphorylated by Fyn remain unknown, whereas Src phosphorylates TRPV6 specifically in residues Y161 and Y162, promoting Ca2+ entry (322). In breast cancer, Fyn has been described as a predictive biomarker of tamoxifen response, participating in tamoxifen resistance (323). Moreover, this protein is involved in maintaining a mesenchymal phenotype in MDA-MB-231 cells (294). Interestingly, Src promotes cell growth and survival in MDA-MB-468, while in the MCF-7 cells, this protein promotes spreading and motility. Moreover, a gain of function of this protein promotes bone metastasis in mice models (324). Thus, these results suggest that tyrosine phosphorylation in TRPV6 promotes its activity, which could enhance breast cancer malignancy. For this reason, disruption of the interaction between these kinases and TRPV6 channels could be a therapeutic alternative in breast cancer.
TRPV6 also forms a complex with tumor suppressor proteins with relevance in cancer development such as Numb1 and phosphatidylinositol 3,4,5-trisphosphate 3-phosphatase (PTEN). Numb1 is a cell fate determinant protein, which stabilizes p53, preventing its ubiquitination. This protein interacts with TRPV6, decreasing cell Ca2+ influx, although its mechanism in unclear. Moreover, it has been described that in MCF-7 cells, Numb1 knockdown increases the expression of this channel, whereas TRPV6 knockdown increases Numb1 expression (325). On the other hand, PTEN is a protein phosphatase with tumor suppressor activity which mediates the PI (3–5) P3 to PI (4, 5) P2 conversion, promoting inhibition of the PI3K/Akt pathway (326). Interestingly, this phosphatase forms a complex with TRPV6 and Numb1 in the prostate cancer cell lines (DU145). Moreover, knockdown of PTEN causes a positive regulation between Numb1 and TRPV6 expression in DU145 cells (327). In breast cancer, the absence of Numb correlates with reduced disease-free survival in patients (328). A meta-analysis of generated data about PTEN suggests that the loss of this protein could predict more aggressive forms of breast cancer and worse prognosis for patients (329). Thus, these results suggest that loss of Numb and PTEN correlate with worse prognosis in breast cancer. For this reason, promoting the interaction between TRPV6, NUMB1 and PTEN could be useful as a therapeutic approach in breast cancer.
TRPV6 also interacts with accessory proteins with reported activity in breast cancer. For example, cyclophilin B is a peptidyl-prolyl cis-trans isomerase which promotes changes in protein conformation. This chaperone was identified as a TRPV6 accessory protein, promoting TRPV6-dependent Ca2+ influx (330). Moreover, the knockdown of cyclophilin B in ER+/HER2− cell lines (T47D cells) downregulates the expression of several elements involved in cell proliferation, such as the progesterone and estrogen receptors (331). Another interesting protein is regulator of G-protein signaling 2 (RGS2) which corresponds to a negative regulator of G-protein coupled receptors (GPCR), increasing GTPase activity of Gα subunits and regulating several signaling pathways (332). This protein interacts with TRPV6 reducing its Na+ and Ca2+ currents, in a GPCR-independent manner (333). Interestingly, RGS2 is downregulated in MCF-7 cells but not in MCF-10A. Moreover, overexpression of this protein in MCF-7 cells reduces its proliferation (332). These results indicate that knockdown of cyclophilin B reduces TRPV6 currents and breast cancer cell proliferation. Moreover, overexpression of RGS2 reduces TRPV6 currents and breast cancer cell growth. Thus, we hypothesize that promoting these interactions could serve as a therapeutic approach in breast cancer.
An important component of TRPV6 trafficking machinery is Rab11. This protein regulates the budding, movement, and delivery of transport vesicles along different trafficking routes (334). Interestingly, Rab11 interacts with TRPV5 and TRPV6 mediating the trafficking of these proteins to the plasma membrane. Moreover, silencing of Rab11a promotes a decrease in membrane expression of both channels (309). In breast cancer cell lines, it has been described that Rab11a knockdown reduces proliferation, migration and invasion in an Akt-dependent manner (335, 336). These results reveal that downregulation of Rab11a reduces TRPV6-dependent Ca2+ currents and breast cancer cell malignancy. Thus, inhibiting the interaction between Rab11a and TRPV6 might constitute an interesting new target for breast cancer treatment.
TRP Channels and STIM/ORAI Complex
As we described above, TRP channels exert multiple effects on breast cancer cells, at least in part, through the modulation of Ca2+ signaling. The SOCE response is crucial for Ca2+ homeostasis, and represents the main path for its influx in non-excitable cells. This response is triggered after endoplasmic reticulum Ca2+ depletion through the establishment of the STIM1/ORAI1 protein complex. The coupling between these proteins promotes the gating mechanism of the highly selective Ca2+ channel ORAI1, which leads to the establishment of Ca2+ Release-Activated Ca2+ Currents (ICRAC) (337). Although this current was the first associated to SOCE, other non-selective currents were identified as part of this homeostatic response as well, broadening the concept to ISOC (338). This current has been associated with ORAI1 activity and with specific TRP channels, in particular with TRPC1 (80). Interestingly, SOCE has been widely associated with several types of cancer such as cervical, esophageal, breast, lung cancer and others (339). Moreover, a gain of function of this response has been related with a poor prognosis, bigger tumour size and poor differentiation (339). In addition, SOCE has been widely associated to breast cancer progression (340, 341). The expression of essential components of SOCE, such as ORAI1 and STIM1 are essential in mammary gland physiology during lactation, and also in breast cancer progression and invasion (342, 343). In addition to ORAI1, other ORAI isoforms have been linked to breast cancer. For instance, ORAI3 is a tumor promoting agent during cancer progression in ER+ malignant cells (344). Interestingly, increased levels of STIM1, TRPC1, ORAI1 and ORAI3 are correlated with poor prognosis of patients with aggressive basal breast cancer types, such as triple negative molecular subtypes (59, 345, 346). Although the exact mechanisms by which they could promote tumor progression are still not understood in detail, they might represent potential novel prognosis biomarkers in breast cancer.
In addition to TRPC1, other TRP channels have been related to SOCE and its signaling pathways. Several studies have described the physical and functional interaction between TRP channels and ORAI and STIM proteins (347). TRPC subfamily members are the most represented TRP channels forming complexes with SOCE components. In this context, TRPC3 (348) and TRPC6 (349) are proposed to interact with STIM1/ORAI1 complexes and promote Ca2+ entry by ISOC and the subsequent activation of signaling pathways related to cancer progression (350). TRPC1/STIM1/ORAI1 complex activity might participate in metabolic regulation through Ca2+ dependent control of mitochondrial function and autophagy (351, 352). Other TRP subfamilies are also involved in Ca2+ entry through SOCE. TRPA1 interacts with the STIM1/ORAI1 complex, interfering with complex assembly, and decreasing the SOCE response (353). On the other hand, TRPV1, although it does not physically interact with ORAI1, exerts a functional modulation on this channel, particularly by promoting the Ca2+ dependent inactivation of ORAI1, leading to a decreased Ca2+ entry (354). Since these TRP channels and SOCE components have been previously proposed as prognosis biomarkers (56), its physical and functional interactions could be a potential target for the modulation of breast cancer progression. Other SOCE components are also related physically and functionally with TRP channels. STIM2 has been reported to interact with TRPC1 and TRPC6 in MCF-7 cells, modulating the resting Ca2+ content through SOCE activation (81, 128). TRPC1 and TRPC4 are activated by a longer isoform of STIM1, STIM1L. These interactions have different effect to that observed in the activation of SOCE related channels (355). While ORAI1 is preferentially activated by STIM1, TRPC1 is preferentially activated by STIM1L (356). Additionally, SOCE-Associated Regulatory Factor (SARAF), which is the main negative modulator of SOCE, interacts with either STIM1/ORAI1 complex and/or TRPC1 (357) decreasing in Ca2+ influx. Since several TRP channels interact with STIM1 and ORAI1, these could be considered a signaling hub in which TRP and ORAI channels’ activity might converge. Furthermore, since these physical and functional interactions between TRP channels and several SOCE components are reported to modulate Ca2+ entry and subsequent activation of pro-oncogenic processes in breast cancer cells, interrupting these interactions could be an exciting and novel strategy to attenuate cancer progression. TRPC channels and STIM/ORAI expression and interaction have been proposed as potential breast cancer prognosis biomarkers as proposed (56, 339). Further studies to explore the role of TRP channels and STIM1/ORAI1 complexes and how these are related to prognosis and their potential utility as biomarkers are necessary.
Future Prospects for Other Possible Interactions of Ion Channels as Therapeutic Targets
As we previously discussed, the TRP channels interactome could be used as a novel therapeutic target in breast cancer by promoting or interrupting the discussed interactions, thus modulating channel activity and associated signaling pathways. As with others proteins, TRP channels can also be regulated by a plethora of post-translational modifications (PTM), which provide spatial–temporal control of several processes such as ion channel inactivation, activation, trafficking, endocytosis, and degradation (358–360). Interestingly, in several pathological conditions, including cancer, have been reported a dysregulation in the PTM balance. This dysregulation in PTM balance leads to several pathologies due to a gain or loss of function in crucial proteins (361–363). For example, tumor suppressors such as Rb, P53, and PTEN are targets of several PTM that mediated their inactivation or degradation, which promotes carcinogenesis (362). Although the relevance of PTM in cancer and TRP channels, further investigation is still required to identify new residues targets, the role of the modification in the channel, and the enzyme that mediates the PTM (359). Interestingly, only a few modifying enzymes have been reported as molecular partners of TRP channels such as Fyn, Src, PTEN, and KCTD5. However, all these presented relevant contributions in breast cancer and therefore could be exciting targets in molecular therapies (153, 294, 323, 324, 329). Further investigations are needed to identify more modifying enzymes and new putative targets in breast cancer and several others pathologies.
Modulation of PPIS as a Therapeutic Strategy for Breast Cancer
Almost all breast cancer patients will receive at some point chemotherapy as either first-line treatment or as adjuvant/neoadjuvant, which will bring with it serious side effects for the patient (364). Given the systemic nature of the treatment and its low cell specificity and toxicity, chemotherapy can affect almost every organ in the body, including the heart, lungs, brain and kidneys (365, 366). These side effects, along with the fact that most times chemotherapy does not have a curative effect on breast cancer, but only prolongs the patient’s life and alleviates the disease-associated symptoms (365, 367) makes the discovery of new therapies that will only affect tumor cells an important field of research. As was described in the previous sections, there is a myriad of TRP channels that have been associated with several clinical parameters and diverse aspects of breast cancer progression, making them interesting therapeutic targets. Moreover, several interactors of these channels have also been described as important regulators in the development of breast cancer. The fact that the expression of both the TRP channels and their interactors are highly dysregulated in breast cancer suggests that the modulation of these interactions might provide new specific and selective targets for therapeutic interventions in the field of breast cancer research. Since peptides to modulate PPIs are highly specific in regards to their targets, affecting only the cellular pathways associated with a specific interaction and in cells where both the channel and the interactor are expressed, they are attractive candidates for in vivo treatment due to their minimal off-target effects (368). Nevertheless, compared to the targeting of enzymes or receptors, the therapeutic intervention of PPIs has been more challenging, partly due to the large and less structured interface of these interactions (369–372). Despite that, some of the current methods for PPIs modulation have been used as therapeutic strategy in breast cancer (Figure 2).
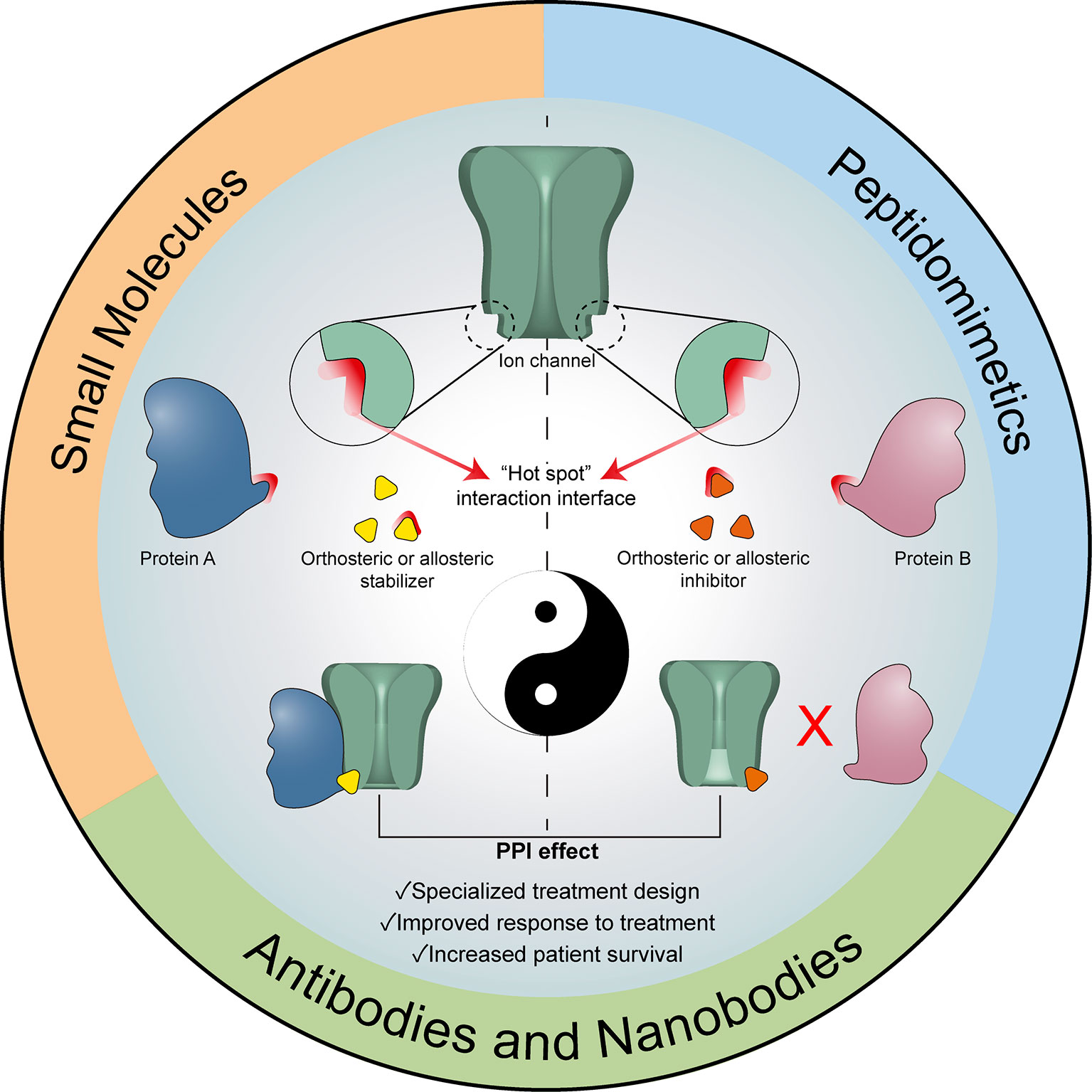
Figure 2 Modulation of ion channel-associated PPIs as therapeutic tool. Graphical representation of the modulation options for Ion channel-associated PPI, either through stabilizers or inhibitors of the interactions. All these strategies could constitute a useful complementary therapeutic tool contributing to increasing existing or personalized therapies.
One of the main challenges that PPI modulators have faced is that most interactions encompassed relatively large contact areas. Nevertheless, subsequent research lead to the description of "hotspots," which are a small subset of amino acid residues contributing most of the binding free energy in PPIs (373). These hotspots are rich in Tyr, Trp, Leu, Ile, Phe, and Arg, where the amino acids Trp, Arg, and Tyr promote hydrogen bonds forming between the interacting proteins, contributing to π-interactions and binding free energy (373). This definition of hotspots has led to the design of small molecules or peptides that target this small number of amino acids to modulate PPIs. An early example of this was the design of Tirofiban, a mimetic of the Arg-Gly-Asp tripeptide epitope of fibrinogen that binds to the αIIbβ3 integrin approved by the FDA (374).
Another therapeutic challenge that these molecules might have is their poor cell/tissue specificity and membrane penetration capacity. In this context, the use of cell-penetrating peptides (CPPs) has provided an important tool to improve these aspects. CPPs are short peptides (less than 30 residues) with low cytotoxicity that have the ability to cross biological membranes, which can be coupled to bioactive molecules to transport them inside cells (375, 376). Interestingly, it has been demonstrated that several of these peptides have tumor homing capacities or might be modified to discriminate between tumoral and non-tumoral cells, thus, when coupled to PPIs-modulators, they would reduce undesired off-target effects and improve their cellular uptake (377–380). Several methods have been developed to provide CPPs with tumoral specificity, such as the use of peptides that naturally bind to tumor-specific cell surface molecules (380, 381), peptides that are inactive until modified by a cancer-associated protease or by properties of the tumoral microenvironment (382, 383) and those that take advantage of cellular mechanisms dysregulated in tumoral cells (384). As we can see, the use of CPPs is a major tool to provide PPI-modulators with tumoral specificity and low side effects.
Several small molecules have been designed and are being used for the targeting of pro-tumoral PPIs in breast cancer. Eribulin and Ixabepilone both interfere with the tubulin-α–tubulin-β interaction inhibiting microtubule growth, ultimately causing G2-M phase cell cycle arrest and cell death through apoptosis, have been used for years in the treatment of metastatic breast cancer patients (385–388). LCL161 and Birinapant are two among several SMAC mimetics being used in breast cancer treatment, which are modeled after the N-terminal AVPI tetrapeptide of Smac, which binds to the BIR domains of the inhibitor of apoptosis (IAP) proteins, thus interfering with this interaction (389, 390). These are just examples of PPIs and drugs used to target them in breast cancer treatment. Further discovery of small molecules is guaranteed by using current techniques, such as High Throughput/virtual Screening and Fragment-based methodologies (391). These approaches will be relevant to the discovery and development of new drugs targeting ion channel-associated PPIs. In this context, although modulators of TRPC channels-associated PPIs have not been approved for human treatment yet, several studies using in vivo models have shown the efficacy of using systemically-administered peptides to disrupt ion channels-associated PPIs to treat diseases with no observed side effects, and high specificity for their targets (392–395).
Despite their positive characteristics, small peptides do have several limitations, such as a lack of stability, relatively low affinity, poor cell-penetrability and short plasma half-life (396, 397). Given this, the use of antibodies has become an essential tool to interrupt PPIs and the fastest-growing and most successful class of therapeutic biologics, given their intrinsic ability to interact potently with proteins along a broader surface spectrum and various epitopes and their inherent stability (398). In this context, daclizumab is an FDA-approved antibody that blocks the interaction between IL-2 and its receptor, which has shown clinical success in patients with metastatic breast cancer when applied in combination with immune therapy (399). Atezolizumab is a monoclonal antibody against programmed death ligand-1 (PD-L1), which blocks its interaction with Programmed cell death protein 1 (PD-1) (400). In a recent study, the treatment with Atezolizumab plus nab-paclitaxel prolonged progression-free survival among patients with metastatic TNBC (401). Moreover, in a pilot study in women with early-stage breast cancer, the use of ipilimumab and FDA approved (for melanoma treatment) monoclonal antibody against CTLA-4, which blocks the interaction with its ligand CD86, showed potentially favorable intratumoral and systemic immunologic effects in combination with cryoablation (402). Although the use of antibodies to modulate PPIs has become a new and exciting tool to modulate PPIs, we acknowledge the relative difficulty of using them in the context of ion channels and their interactors due to the inability of antibodies to cross the plasma membrane, which is where most of the interactions in ion channels occur. The use and generation of recombinant antibodies might improve those difficulties. Additionally, the development of nanobodies, small single-domain antigen-binding antibody fragments, has helped to improve many of the problems that normal monoclonal antibodies have. The smaller size of nanobodies helps them to better penetrate deeper into the tumor and have a more precise targeting than normal IgGs (403–405). Moreover, nanobodies can be conjugated to CPPs to overcome their inability to penetrate the plasma membrane and thus be able to modulate pro-tumoral PPIs occurring inside the cell with great specificity without affecting the functionality of the involved proteins in other signaling pathways (404, 406, 407).
The more broadly used (and FDA approved) approaches to modulate PPIs are the small molecules and antibodies. However, other methods are being proved and improved to make them more suitable for clinical use, such as peptidomimetics, long peptides with a structure derived from existing peptides or protein domains. These molecules tend to mimic natural interaction and conserve a protein-like structure (391, 408). Although no peptidomimetics have been approved for breast cancer treatment, the major advances in the field of cell-penetrating peptides (CPPs), will encourage the development of therapeutic peptides and help to overcome some of the shortcomings that these molecules have as possible therapeutics (375, 409). Moreover, in the field of antibodies, nanobodies’ discovery and use should promote more stable and simpler molecules (410, 411). Also, aptamers can also be used to modulate PPIs (412, 413). Finally, the combination of several of these methods, such as the conjugation of peptides with small molecules, oligoribonucleotides, or antibodies as a possible alternative to overcome each type’s disadvantages, has gained interest in the last years, especially in oncology (396).
Concluding Remarks
Cancer is a multifactorial disease, which progression depends on the dysregulation of several pathways involved in processes such as cell metabolism, immune evasion, avoiding cell death, inflammation, migration, and invasion. Interestingly, Ca2+ signaling is involved in many, if not all, of these processes. In this context, ion channels, especially TRP channel family members, are interesting proteins given their regulatory role on intracellular Ca2+ levels. Indeed, TRP channels expression and activity of several of them have been found commonly dysregulated in several types of cancer, including breast cancer. Hence, several TRP channels have been proposed as potential biomarkers of tumor progression and response to breast cancer treatment.
Interestingly, several proteins that interact with TRP channels have been reported as dysregulated in breast cancer, and thus, proposed as biomarkers for cancer progression. In this article, we have extensively reviewed the main TRP channels that might have a role in breast cancer progression, focusing on how their interaction with other reported biomarkers might constitute novel and exciting targets for intervention as possible therapies in this disease. Furthermore, we have presented several mechanisms by which these interactions might be regulating several aspects of breast cancer progression. However, although there are strong evidence and literature to support the interactions presented here, we are aware that most of them are based on reports in non-cancer models. Thus, further studies are needed to confirm these interactions and their possible pathological role in breast cancer. In conclusion, the identification and confirmation of new TRP-interactions, their role in breast cancer, and the development of drugs to modulate those interactions might provide new alternatives to help in the treatment of breast cancer patients, especially in those cases where there are no current treatments available or where treatments are non-selective and carry dangerous secondary effects with them.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding author.
Author Contributions
MS, DM, OO-S, IS, BL, PC, CT, MC, and OC wrote the article. MS designed and made the illustrations. All authors contributed to the article and approved the submitted version.
Funding
FONDECYT Grant 1200917 and Millennium Nucleus of Ion Channel-Associated Diseases (MiNICAD) to OC fund this research. MiNICAD is a Millennium Nucleus supported by the Iniciativa Científica Milenio of the National Agency of Research and Development (ANID). FONDECYT Grant 1181263 funds to MC. CONICYT Doctoral Fellowship Program funds to BL (#21181611), PC (#21180306), IS (#21180245), and MS (#21191141) and DM (#21201941). OO-S is funded by the MiNICAD Postdoctoral Fellowship Program.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2021.621614/full#supplementary-material
References
1. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global Cancer Statistics 2018: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J Clin (2018) 68(6):394–424. doi: 10.3322/caac.21492
2. Makki J. Diversity of Breast Carcinoma: Histological Subtypes and Clinical Relevance. Clin Med Insights Pathol (2015) 8:23–31. doi: 10.4137/CPath.S31563
3. Cheang MC, Martin M, Nielsen TO, Prat A, Voduc D, Rodriguez-Lescure A, et al. Defining Breast Cancer Intrinsic Subtypes by Quantitative Receptor Expression. Oncologist (2015) 20(5):474–82. doi: 10.1634/theoncologist.2014-0372
4. Harbeck N, Penault-Llorca F, Cortes J, Gnant M, Houssami N, Poortmans P, et al. Breast Cancer. Nat Rev Dis Primers (2019) 5(1):66. doi: 10.1038/s41572-019-0111-2
5. Godone RLN, Leitão GM, Araújo NB, Castelletti CHM, Lima-Filho JL, Martins DBG. Clinical and Molecular Aspects of Breast Cancer: Targets and Therapies. BioMed Pharmacother (2018) 106:14–34. doi: 10.1016/j.biopha.2018.06.066
6. Wahba HA, El-Hadaad HA. Current Approaches in Treatment of Triple-Negative Breast Cancer. Cancer Biol Med (2015) 12(2):106–16. doi: 10.7497/j.issn.2095-3941.2015.0030
7. Edel KH, Marchadier E, Brownlee C, Kudla J, Hetherington AM. The Evolution of Calcium-Based Signalling in Plants. Curr Biol (2017) 27(13):R667–R79. doi: 10.1016/j.cub.2017.05.020
8. Domínguez DC, Guragain M, Patrauchan M. Calcium Binding Proteins and Calcium Signaling in Prokaryotes. Cell Calcium (2015) 57(3):151–65. doi: 10.1016/j.ceca.2014.12.006
10. Parekh AB. Decoding Cytosolic Ca2+ Oscillations. Trends Biochem Sci (2011) 36(2):78–87. doi: 10.1016/j.tibs.2010.07.013
11. Bagur R, Hajnóczky G. Intracellular Ca. Mol Cell (2017) 66(6):780–8. doi: 10.1016/j.molcel.2017.05.028
12. Rizzuto R, Pozzan T. Microdomains of Intracellular Ca2+: Molecular Determinants and Functional Consequences. Physiol Rev (2006) 86(1):369–408. doi: 10.1152/physrev.00004.2005
13. Berridge MJ, Lipp P, Bootman MD. The Versatility and Universality of Calcium Signalling. Nat Rev Mol Cell Biol (2000) 1(1):11–21. doi: 10.1038/35036035
14. Wakai T, Vanderheyden V, Fissore RA. Ca2+ Signaling During Mammalian Fertilization: Requirements, Players, and Adaptations. Cold Spring Harb Perspect Biol (2011) 3(4). doi: 10.1101/cshperspect.a006767
15. Hagenston AM, Bading H. Calcium Signaling in Synapse-to-Nucleus Communication. Cold Spring Harb Perspect Biol (2011) 3(11):a004564. doi: 10.1101/cshperspect.a004564
16. Bezprozvanny I. Calcium Signaling and Neurodegenerative Diseases. Trends Mol Med (2009) 15(3):89–100. doi: 10.1016/j.molmed.2009.01.001
17. Rieusset J. Endoplasmic Reticulum-Mitochondria Calcium Signaling in Hepatic Metabolic Diseases. Biochim Biophys Acta Mol Cell Res (2017) 1864(6):865–76. doi: 10.1016/j.bbamcr.2017.01.001
18. Jayakumar S, Hasan G. Neuronal Calcium Signaling in Metabolic Regulation and Adaptation to Nutrient Stress. Front Neural Circuits (2018) 12:25. doi: 10.3389/fncir.2018.00025
19. Park YJ, Yoo SA, Kim M, Kim WU. The Role of Calcium-Calcineurin-NFAT Signaling Pathway in Health and Autoimmune Diseases. Front Immunol (2020) 11:195. doi: 10.3389/fimmu.2020.00195
20. Varghese E, Samuel SM, Sadiq Z, Kubatka P, Liskova A, Benacka J, et al. Anti-Cancer Agents in Proliferation and Cell Death: The Calcium Connection. Int J Mol Sci (2019) 20(12). doi: 10.3390/ijms20123017
21. Cui C, Merritt R, Fu L, Pan Z. Targeting Calcium Signaling in Cancer Therapy. Acta Pharm Sin B (2017) 7(1):3–17. doi: 10.1016/j.apsb.2016.11.001
22. Marchi S, Giorgi C, Galluzzi L, Pinton P. Ca2+ Fluxes and Cancer. Mol Cell (2020) 78(6):1055–69. doi: 10.1016/j.molcel.2020.04.017
23. Monteith GR, Prevarskaya N, Roberts-Thomson SJ. The Calcium-Cancer Signalling Nexus. Nat Rev Cancer (2017) 17(6):367–80. doi: 10.1038/nrc.2017.18
24. So CL, Saunus JM, Roberts-Thomson SJ, Monteith GR. Calcium Signalling and Breast Cancer. Semin Cell Dev Biol (2019) 94:74–83. doi: 10.1016/j.semcdb.2018.11.001
25. Roberts-Thomson SJ, Chalmers SB, Monteith GR. The Calcium-Signaling Toolkit in Cancer: Remodeling and Targeting. Cold Spring Harb Perspect Biol (2019) 11(8). doi: 10.1101/cshperspect.a035204
26. Ramsey IS, Delling M, Clapham DE. An Introduction to TRP Channels. Annu Rev Physiol (2006) 68:619–47. doi: 10.1146/annurev.physiol.68.040204.100431
27. Peng G, Shi X, Kadowaki T. Evolution of TRP Channels Inferred by Their Classification in Diverse Animal Species. Mol Phylogenet Evol (2015) 84:145–57. doi: 10.1016/j.ympev.2014.06.016
28. Venkatachalam K, Montell C. TRP Channels. Annu Rev Biochem (2007) 76:387–417. doi: 10.1146/annurev.biochem.75.103004.142819
29. Himmel NJ, Gray TR, Cox DN. Phylogenetics Identifies Two Eumetazoan Trpm Clades and An Eighth Trp Family, Trp Soromelastatin (Trps). Mol Biol Evol (2020) 37(7):2034–44. doi: 10.1093/molbev/msaa065
30. Smani T, Shapovalov G, Skryma R, Prevarskaya N, Rosado JA. Functional and Physiopathological Implications of TRP Channels. Biochim Biophys Acta (2015) 1853(8):1772–82. doi: 10.1016/j.bbamcr.2015.04.016
31. Santoni G, Maggi F, Morelli MB, Santoni M, Marinelli O. Transient Receptor Potential Cation Channels in Cancer Therapy. Med Sci (Basel) (2019) 7(12). doi: 10.3390/medsci7120108
32. Santoni G, Farfariello V. TRP Channels and Cancer: New Targets for Diagnosis and Chemotherapy. Endocr Metab Immune Disord Drug Targets (2011) 11(1):54–67. doi: 10.2174/187153011794982068
33. Prevarskaya N, Skryma R, Shuba Y. Ion Channels and the Hallmarks of Cancer. Trends Mol Med (2010) 16(3):107–21. doi: 10.1016/j.molmed.2010.01.005
34. Steinritz D, Stenger B, Dietrich A, Gudermann T, Popp T. Trps in Tox: Involvement of Transient Receptor Potential-Channels in Chemical-Induced Organ Toxicity-A Structured Review. Cells (2018) 7(8):235–56. doi: 10.3390/cells7080098
35. Zheng J. Molecular Mechanism of TRP Channels. Compr Physiol (2013) 3(1):221–42. doi: 10.1002/cphy.c120001
36. Chun JN, Lim JM, Kang Y, Kim EH, Shin YC, Kim HG, et al. A Network Perspective on Unraveling the Role of TRP Channels in Biology and Disease. Pflugers Arch (2014) 466(2):173–82. doi: 10.1007/s00424-013-1292-2
37. Shin YC, Shin SY, Chun JN, Cho HS, Lim JM, Kim HG, et al. TRIP Database 2.0: A Manually Curated Information Hub for Accessing TRP Channel Interaction Network. PLoS One (2012) 7(10):e47165. doi: 10.1371/journal.pone.0047165
38. Zinzalla G, Thurston DE. Targeting Protein-Protein Interactions for Therapeutic Intervention: A Challenge for the Future. Future Med Chem (2009) 1(1):65–93. doi: 10.4155/fmc.09.12
39. Petta I, Lievens S, Libert C, Tavernier J, De Bosscher K. Modulation of Protein-Protein Interactions for the Development of Novel Therapeutics. Mol Ther (2016) 24(4):707–18. doi: 10.1038/mt.2015.214
40. Cowen L, Ideker T, Raphael BJ, Sharan R. Network Propagation: A Universal Amplifier of Genetic Associations. Nat Rev Genet (2017) 18(9):551–62. doi: 10.1038/nrg.2017.38
41. Heijman J, Dobrev D. Ionenkanäle Als Teil Makromolekularer Multiproteinkomplexe: Klinische Signifikanz. Herzschrittmacherther und Elektrophysiol (2018) 29(1):30–5. doi: 10.1007/s00399-017-0542-y
42. Lee A, Fakler B, Kaczmarek LK, Isom LL. More Than a Pore: Ion Channel Signaling Complexes. J Neurosci (2014) 34(46):15159–69. doi: 10.1523/JNEUROSCI.3275-14.2014
43. Swayne LA, Altier C, Zamponi GW. The Truth in Complexes: Perspectives on Ion Channel Signaling Nexuses in the Nervous System. Front Cell Neurosci (2014) 8:406. doi: 10.3389/fncel.2014.00406
44. Litan A, Langhans SA. Cancer as a Channelopathy: Ion Channels and Pumps in Tumor Development and Progression. Front Cell Neurosci (2015) 9(March):1–11. doi: 10.3389/fncel.2015.00086
45. Prevarskaya N, Skryma R, Shuba Y. Ion Channels in Cancer: Are Cancer Hallmarks Oncochannelopathies? Physiol Rev (2018) 98(2):559–621. doi: 10.1152/physrev.00044.2016
46. Nesin V, Tsiokas L. Trpc1. Handb Exp Pharmacol (2014) 222:15–51. doi: 10.1007/978-3-642-54215-2_2
47. Kim J, Ko J, Myeong J, Kwak M, Hong C, So I. TRPC1 as a Negative Regulator for TRPC4 and TRPC5 Channels. Pflugers Archiv Eur J Physiol (2019) 471(8):1045–53. doi: 10.1007/s00424-019-02289-w
48. Tajeddine N, Gailly P. TRPC1 Protein Channel is Major Regulator of Epidermal Growth Factor Receptor Signaling. J Biol Chem (2012) 287(20):16146–57. doi: 10.1074/jbc.M112.340034
49. Ambudkar I. Calcium Signaling Defects Underlying Salivary Gland Dysfunction. Biochim Biophys Acta Mol Cell Res (2018) 1865(11 Pt B):1771–7. doi: 10.1016/j.bbamcr.2018.07.002
50. Lepannetier S, Gualdani R, Tempesta S, Schakman O, Seghers F, Kreis A, et al. Activation of TRPC1 Channel by Metabotropic Glutamate Receptor Mglur5 Modulates Synaptic Plasticity and Spatial Working Memory. Front Cell Neurosci (2018) 12:318. doi: 10.3389/fncel.2018.00318
51. Martín-Aragón Baudel MAS, Shi J, Large WA, Albert AP. Obligatory Role for Pkcδ in PIP(2) -Mediated Activation of Store-Operated TRPC1 Channels in Vascular Smooth Muscle Cells. J Physiol (2020) 598(18):3911–25. doi: 10.1113/JP279947
52. Lintschinger B, Balzer-Geldsetzer M, Baskaran T, Graier WF, Romanin C, Zhu MX, et al. Coassembly of Trp1 and Trp3 Proteins Generates Diacylglycerol- and Ca2+-sensitive Cation Channels. J Biol Chem (2000) 275(36):27799–805. doi: 10.1074/jbc.M002705200
53. Ambudkar IS, de Souza LB, Ong HL. Trpc1, Orai1, and STIM1 in SOCE: Friends in Tight Spaces. Cell Calcium (2017) 63:33–9. doi: 10.1016/j.ceca.2016.12.009
54. Baudel M, Shi J, Large WA, Albert AP. Insights Into Activation Mechanisms of Store-Operated Trpc1 Channels in Vascular Smooth Muscle. Cells (2020) 9(1). doi: 10.3390/cells9010179
55. Elzamzamy OM, Penner R, Hazlehurst LA. The Role of TRPC1 in Modulating Cancer Progression. Cells (2020) 9(2). doi: 10.3390/cells9020388
56. Ouadid-Ahidouch H, Dhennin-Duthille I, Gautier M, Sevestre H, Ahidouch A. TRP Channels: Diagnostic Markers and Therapeutic Targets for Breast Cancer? Trends Mol Med (2013) 19(2):117–24. doi: 10.1016/j.molmed.2012.11.004
57. Dhennin-Duthille I, Gautier M, Faouzi M, Guilbert A, Brevet M, Vaudry D, et al. High Expression of Transient Receptor Potential Channels in Human Breast Cancer Epithelial Cells and Tissues: Correlation With Pathological Parameters. Cell Physiol Biochem (2011) 28(5):813–22. doi: 10.1159/000335795
58. Jiang HN, Zeng B, Zhang Y, Daskoulidou N, Fan H, Qu JM, et al. Involvement of TRPC Channels in Lung Cancer Cell Differentiation and the Correlation Analysis in Human Non-Small Cell Lung Cancer. PLoS One (2013) 8(6):e67637. doi: 10.1371/journal.pone.0067637
59. Azimi I, Milevskiy MJG, Kaemmerer E, Turner D, Yapa K, Brown MA, et al. TRPC1 is a Differential Regulator of Hypoxia-Mediated Events and Akt Signalling in PTEN-deficient Breast Cancer Cells. J Cell Sci (2017) 130(14):2292–305. doi: 10.1242/jcs.196659
60. Fiorio Pla A, Maric D, Brazer SC, Giacobini P, Liu X, Chang YH, et al. Canonical Transient Receptor Potential 1 Plays a Role in Basic Fibroblast Growth Factor (bFGF)/FGF Receptor-1-Induced Ca2+ Entry and Embryonic Rat Neural Stem Cell Proliferation. J Neurosci (2005) 25(10):2687–701. doi: 10.1523/JNEUROSCI.0951-04.2005
61. Hsu JL, Hung MC. The Role of HER2, EGFR, and Other Receptor Tyrosine Kinases in Breast Cancer. Cancer Metastasis Rev (2016) 35(4):575–88. doi: 10.1007/s10555-016-9649-6
62. Wang S, Ding Z. Fibroblast Growth Factor Receptors in Breast Cancer. Tumour Biol (2017) 39(5):1010428317698370. doi: 10.1177/1010428317698370
63. Mao P, Cohen O, Kowalski KJ, Kusiel J, Buendia-Buendia JE, Cuoco MS, et al. Acquired FGFR and FGF Alterations Confer Resistance to Estrogen Receptor (ER) Targeted Therapy in ER+ Metastatic Breast Cancer. Clin Cancer Res (2020) 26(22):5974–89. doi: 10.1101/605436
64. Turner N, Pearson A, Sharpe R, Lambros M, Geyer F, Lopez-Garcia MA, et al. FGFR1 Amplification Drives Endocrine Therapy Resistance and is a Therapeutic Target in Breast Cancer. Cancer Res (2010) 70(5):2085–94. doi: 10.1158/0008-5472.CAN-09-3746
65. Brazer SC, Singh BB, Liu X, Swaim W, Ambudkar IS. Caveolin-1 Contributes to Assembly of Store-Operated Ca2+ Influx Channels by Regulating Plasma Membrane Localization of TRPC1. J Biol Chem (2003) 278(29):27208–15. doi: 10.1074/jbc.M301118200
66. Sundivakkam PC, Kwiatek AM, Sharma TT, Minshall RD, Malik AB, Tiruppathi C. Caveolin-1 Scaffold Domain Interacts With TRPC1 and IP3R3 to Regulate Ca2+ Store Release-Induced Ca2+ Entry in Endothelial Cells. Am J Physiol Cell Physiol (2009) 296(3):C403–13. doi: 10.1152/ajpcell.00470.2008
67. Hardin CD, Vallejo J. Dissecting the Functions of Protein-Protein Interactions: Caveolin as a Promiscuous Partner. Focus on “Caveolin-1 Scaffold Domain Interacts With TRPC1 and IP3R3 to Regulate Ca2+ Store Release-Induced Ca2+ Entry in Endothelial Cells”. Am J Physiol Cell Physiol (2009) 296(3):C387–9. doi: 10.1152/ajpcell.00663.2008
68. Qian XL, Pan YH, Huang QY, Shi YB, Huang QY, Hu ZZ, et al. Caveolin-1: A Multifaceted Driver of Breast Cancer Progression and its Application in Clinical Treatment. Onco Targets Ther (2019) 12:1539–52. doi: 10.2147/OTT.S191317
69. Tu CL, Chang W, Bikle DD. Phospholipase Cgamma1 is Required for Activation of Store-Operated Channels in Human Keratinocytes. J Invest Dermatol (2005) 124(1):187–97. doi: 10.1111/j.0022-202X.2004.23544.x
70. Lattanzio R, Iezzi M, Sala G, Tinari N, Falasca M, Alberti S, et al. PLC-Gamma-1 Phosphorylation Status is Prognostic of Metastatic Risk in Patients With Early-Stage Luminal-A and -B Breast Cancer Subtypes. BMC Cancer (2019) 19(1):747. doi: 10.1186/s12885-019-5949-x
71. Katterle Y, Brandt BH, Dowdy SF, Niggemann B, Zänker KS, Dittmar T. Antitumour Effects of PLC-gamma1-(SH2)2-TAT Fusion Proteins on EGFR/c-erbB-2-Positive Breast Cancer Cells. Br J Cancer (2004) 90(1):230–5. doi: 10.1038/sj.bjc.6601506
72. Jaffe AB, Hall A. Rho GTPASES: Biochemistry and Biology. Annu Rev Cell Dev Biol (2005) 21(1):247–69. doi: 10.1146/annurev.cellbio.21.020604.150721
73. Kalpana G, Figy C, Yeung M, Yeung KC. Reduced RhoA Expression Enhances Breast Cancer Metastasis With a Concomitant Increase in CCR5 and CXCR4 Chemokines Signaling. Sci Rep (2019) 9(1):16351. doi: 10.1038/s41598-019-52746-w
74. Humphries B, Wang Z, Li Y, Jhan JR, Jiang Y, Yang C. Arhgap18 Downregulation by Mir-200b Suppresses Metastasis of Triple-Negative Breast Cancer by Enhancing Activation of Rhoa. Cancer Res (2017) 77(15):4051–64. doi: 10.1158/0008-5472.CAN-16-3141
75. Pillé JY, Denoyelle C, Varet J, Bertrand JR, Soria J, Opolon P, et al. Anti-RhoA and anti-RhoC siRNAs Inhibit the Proliferation and Invasiveness of MDA-MB-231 Breast Cancer Cells In Vitro and In Vivo. Mol Ther (2005) 11(2):267–74. doi: 10.1016/j.ymthe.2004.08.029
76. Daubriac J, Han S, Grahovac J, Smith E, Hosein A, Buchanan M, et al. The Crosstalk Between Breast Carcinoma-Associated Fibroblasts and Cancer Cells Promotes RhoA-Dependent Invasion Via IGF-1 and PAI-1. Oncotarget (2018) 9(12):10375–87. doi: 10.18632/oncotarget.23735
77. Mehta D, Ahmmed GU, Paria BC, Holinstat M, Voyno-Yasenetskaya T, Tiruppathi C, et al. RhoA Interaction With Inositol 1,4,5-Trisphosphate Receptor and Transient Receptor Potential Channel-1 Regulates Ca2+ Entry: Role in Signaling Increased Endothelial Permeability. J Biol Chem (2003) 278(35):33492–500. doi: 10.1074/jbc.M302401200
78. Chung HK, Rathor N, Wang SR, Wang JY, Rao JN. Rhoa Enhances Store-Operated Ca2+ Entry and Intestinal Epithelial Restitution by Interacting With TRPC1 After Wounding. Am J Physiol Gastrointest Liver Physiol (2015) 309(9):G759–67. doi: 10.1152/ajpgi.00185.2015
79. Lopez JJ, Jardin I, Sanchez-Collado J, Salido GM, Smani T, Rosado JA. Trpc Channels in the SOCE Scenario. Cells (2020) 9(1). doi: 10.3390/cells9010126
80. Cheng KT, Liu X, Ong HL, Ambudkar IS. Functional Requirement for Orai1 in Store-Operated TRPC1-STIM1 Channels. J Biol Chem (2008) 283(19):12935–40. doi: 10.1074/jbc.C800008200
81. Berna-Erro A, Galan C, Dionisio N, Gomez LJ, Salido GM, Rosado JA. Capacitative and Non-Capacitative Signaling Complexes in Human Platelets. Biochim Biophys Acta (2012) 1823(8):1242–51. doi: 10.1016/j.bbamcr.2012.05.023
82. Redondo PC, Jardin I, Lopez JJ, Salido GM, Rosado JA. Intracellular Ca2+ Store Depletion Induces the Formation of Macromolecular Complexes Involving hTRPC1, hTRPC6, the Type II IP3 Receptor and SERCA3 in Human Platelets. Biochim Biophys Acta (2008) 1783(6):1163–76. doi: 10.1016/j.bbamcr.2007.12.008
83. Redondo PC, Salido GM, Pariente JA, Sage SO, Rosado JA. SERCA2b and 3 Play a Regulatory Role in Store-Operated Calcium Entry in Human Platelets. Cell Signal (2008) 20(2):337–46. doi: 10.1016/j.cellsig.2007.10.019
84. Singh BB, Liu X, Tang J, Zhu MX, Ambudkar IS. Calmodulin Regulates Ca2+-Dependent Feedback Inhibition of Store-Operated Ca2+ Influx by Interaction With a Site in the C Terminus of Trpc1. Mol Cell (2002) 9(4):739–50. doi: 10.1016/S1097-2765(02)00506-3
85. Rosado JA, Sage SO. Activation of Store-Mediated Calcium Entry by Secretion-Like Coupling Between the Inositol 1,4,5-Trisphosphate Receptor Type II and Human Transient Receptor Potential (hTrp1) Channels in Human Platelets. Biochem J (2001) 356(Pt 1):191–8. doi: 10.1042/bj3560191
86. Hannan MA, Kabbani N, Paspalas CD, Levenson R. Interaction With Dopamine D2 Receptor Enhances Expression of Transient Receptor Potential Channel 1 At the Cell Surface. Biochim Biophys Acta - Biomembranes (2008) 1778(4):974–82. doi: 10.1016/j.bbamem.2008.01.011
87. López E, Berna-Erro A, Salido GM, Rosado JA, Redondo PC. FKBP52 is Involved in the Regulation of SOCE Channels in the Human Platelets and MEG 01 Cells. Biochim Biophys Acta (2013) 1833(3):652–62. doi: 10.1016/j.bbamcr.2012.11.029
88. Mound A, Vautrin-Glabik A, Foulon A, Botia B, Hague F, Parys JB, et al. Downregulation of Type 3 Inositol (1,4,5)-Trisphosphate Receptor Decreases Breast Cancer Cell Migration Through an Oscillatory Ca. Oncotarget (2017) 8(42):72324–41. doi: 10.18632/oncotarget.20327
89. Peters AA, Milevskiy MJ, Lee WC, Curry MC, Smart CE, Saunus JM, et al. The Calcium Pump Plasma Membrane Ca(2+)-ATPase 2 (PMCA2) Regulates Breast Cancer Cell Proliferation and Sensitivity to Doxorubicin. Sci Rep (2016) 6:25505. doi: 10.1038/srep25505
90. Bong AHL, Robitaille M, Milevskiy MJG, Roberts-Thomson SJ, Monteith GR. NCS-1 Expression is Higher in Basal Breast Cancers and Regulates Calcium Influx and Cytotoxic Responses to Doxorubicin. Mol Oncol (2020) 14(1):87–104. doi: 10.1002/1878-0261.12589
91. Sehgal P, Szalai P, Olesen C, Praetorius HA, Nissen P, Christensen SB, et al. Inhibition of the Sarco/Endoplasmic Reticulum (ER) Ca(2+)-ATPase by Thapsigargin Analogs Induces Cell Death Via ER Ca(2+) Depletion and The Unfolded Protein Response. J Biol Chem (2017) 292(48):19656–73. doi: 10.1074/jbc.M117.796920
92. Xiong H, Chen Z, Zheng W, Sun J, Fu Q, Teng R, et al. FKBP4 is a Malignant Indicator in Luminal A Subtype of Breast Cancer. J Cancer (2020) 11(7):1727–36. doi: 10.7150/jca.40982
93. Tajbakhsh A, Pasdar A, Rezaee M, Fazeli M, Soleimanpour S, Hassanian SM, et al. The Current Status and Perspectives Regarding the Clinical Implication of Intracellular Calcium in Breast Cancer. J Cell Physiol (2018) 233(8):5623–41. doi: 10.1002/jcp.26277
94. Okada T, Shimizu S, Wakamori M, Maeda A, Kurosaki T, Takada N, et al. Molecular Cloning and Functional Characterization of a Novel Receptor-Activated TRP Ca2+ Channel From Mouse Brain. J Biol Chem (1998) 273(17):10279–87. doi: 10.1074/jbc.273.17.10279
95. Schaefer M, Plant TD, Obukhov AG, Hofmann T, Gudermann T, Schultz G. Receptor-Mediated Regulation of the Nonselective Cation Channels TRPC4 and TRPC5. J Biol Chem (2000) 275(23):17517–26. doi: 10.1074/jbc.275.23.17517
96. Gaunt HJ, Vasudev NS, Beech DJ. Transient Receptor Potential Canonical 4 and 5 Proteins as Targets in Cancer Therapeutics. Eur Biophys J (2016) 45(7):611–20. doi: 10.1007/s00249-016-1142-1
97. Wang T, Ning K, Lu TX, Hua D. Elevated Expression of TrpC5 and GLUT1 is Associated With Chemoresistance in Colorectal Cancer. Oncol Rep (2017) 37(2):1059–65. doi: 10.3892/or.2016.5322
98. Wang T, Ning K, Lu TX, Sun X, Jin L, Qi X, et al. Increasing Circulating Exosomes-Carrying TRPC5 Predicts Chemoresistance in Metastatic Breast Cancer Patients. Cancer Sci (2017) 108(3):448–54. doi: 10.1111/cas.13150
99. Binkhathlan Z, Lavasanifar A. P-Glycoprotein Inhibition as a Therapeutic Approach for Overcoming Multidrug Resistance in Cancer: Current Status and Future Perspectives. Curr Cancer Drug Targets (2013) 13(3):326–46. doi: 10.2174/15680096113139990076
100. Ma X, Cai Y, He D, Zou C, Zhang P, Lo CY, et al. Transient Receptor Potential Channel TRPC5 is Essential for P-glycoprotein Induction in Drug-Resistant Cancer Cells. Proc Natl Acad Sci USA (2012) 109(40):16282–7. doi: 10.1073/pnas.1202989109
101. Ma X, Chen Z, Hua D, He D, Wang L, Zhang P, et al. Essential Role for TrpC5-containing Extracellular Vesicles in Breast Cancer With Chemotherapeutic Resistance. Proc Natl Acad Sci USA (2014) 111(17):6389–94. doi: 10.1073/pnas.1400272111
102. Zhang P, Liu X, Li H, Chen Z, Yao X, Jin J, et al. TRPC5-Induced Autophagy Promotes Drug Resistance in Breast Carcinoma Via Camkkβ/Ampkα/mTOR Pathway. Sci Rep (2017) 7(1):3158. doi: 10.1038/s41598-017-03230-w
103. Tian D, Jacobo SM, Billing D, Rozkalne A, Gage SD, Anagnostou T, et al. Antagonistic Regulation of Actin Dynamics and Cell Motility by TRPC5 and TRPC6 Channels. Sci Signal (2010) 3(145):ra77. doi: 10.1126/scisignal.2001200
104. Goel M, Sinkins W, Keightley A, Kinter M, Schilling WP. Proteomic Analysis of TRPC5- and TRPC6-Binding Partners Reveals Interaction With the Plasmalemmal Na(+)/K(+)-Atpase. Pflugers Arch (2005) 451(1):87–98. doi: 10.1007/s00424-005-1454-y
105. Cao R, Chen J, Zhang X, Zhai Y, Qing X, Xing W, et al. Elevated Expression of Myosin X in Tumours Contributes to Breast Cancer Aggressiveness and Metastasis. Br J Cancer (2014) 111(3):539–50. doi: 10.1038/bjc.2014.298
106. Schnelzer A, Prechtel D, Knaus U, Dehne K, Gerhard M, Graeff H, et al. Rac1 in Human Breast Cancer: Overexpression, Mutation Analysis, and Characterization of a New Isoform, Rac1b. Oncogene (2000) 19(26):3013–20. doi: 10.1038/sj.onc.1203621
107. Otey CA, Carpen O. Alpha-Actinin Revisited: A Fresh Look At an Old Player. Cell Motil Cytoskelet (2004) 58(2):104–11. doi: 10.1002/cm.20007
108. Burridge K, Feramisco JR. Non-Muscle Alpha Actinins are Calcium-Sensitive Actin-Binding Proteins. Nature (1981) 294(5841):565–7. doi: 10.1038/294565a0
109. Kovac B, Mäkelä TP, Vallenius T. Increased α-Actinin-1 Destabilizes E-cadherin-based Adhesions and Associates With Poor Prognosis in Basal-Like Breast Cancer. PLoS One (2018) 13(5):e0196986. doi: 10.1371/journal.pone.0196986
110. Hsu KS, Kao HY. Alpha-Actinin 4 and Tumorigenesis of Breast Cancer. Vitam Horm (2013) 93:323–51. doi: 10.1016/B978-0-12-416673-8.00005-8
111. Tang J, Lin Y, Zhang Z, Tikunova S, Birnbaumer L, Zhu MX. Identification of Common Binding Sites for Calmodulin and Inositol 1,4,5-Trisphosphate Receptors on the Carboxyl Termini of Trp Channels. J Biol Chem (2001) 276(24):21303–10. doi: 10.1074/jbc.M102316200
112. Yuan JP, Zeng W, Huang GN, Worley PF, Muallem S. STIM1 Heteromultimerizes TRPC Channels to Determine Their Function as Store-Operated Channels. Nat Cell Biol (2007) 9(6):636–45. doi: 10.1038/ncb1590
113. Puram SV, Riccio A, Koirala S, Ikeuchi Y, Kim AH, Corfas G, et al. A TRPC5-Regulated Calcium Signaling Pathway Controls Dendrite Patterning in the Mammalian Brain. Genes Dev (2011) 25(24):2659–73. doi: 10.1101/gad.174060.111
114. Chi M, Evans H, Gilchrist J, Mayhew J, Hoffman A, Pearsall EA, et al. Phosphorylation of Calcium/Calmodulin-Stimulated Protein Kinase II At T286 Enhances Invasion and Migration of Human Breast Cancer Cells. Sci Rep (2016) 6:33132. doi: 10.1038/srep33132
115. Marklund U, Larsson N, Gradin HM, Brattsand G, Gullberg M. Oncoprotein 18 is a Phosphorylation-Responsive Regulator of Microtubule Dynamics. EMBO J (1996) 15(19):5290–8. doi: 10.1002/j.1460-2075.1996.tb00914.x
116. Askeland C, Wik E, Finne K, Birkeland E, Arnes JB, Collett K, et al. Stathmin Expression Associates With Vascular and Immune Responses in Aggressive Breast Cancer Subgroups. Sci Rep (2020) 10(1):2914. doi: 10.1038/s41598-020-59728-3
117. Obayashi S, Horiguchi J, Higuchi T, Katayama A, Handa T, Altan B, et al. Stathmin1 Expression is Associated With Aggressive Phenotypes and Cancer Stem Cell Marker Expression in Breast Cancer Patients. Int J Oncol (2017) 51(3):781–90. doi: 10.3892/ijo.2017.4085
118. Ohkawa N, Fujitani K, Tokunaga E, Furuya S, Inokuchi K. The Microtubule Destabilizer Stathmin Mediates the Development of Dendritic Arbors in Neuronal Cells. J Cell Sci (2007) 120(Pt 8):1447–56. doi: 10.1242/jcs.001461
119. Dryer SE, Kim EY. Permeation and Rectification in Canonical Transient Receptor Potential-6 (Trpc6) Channels. Front Physiol (2018) 9:1055. doi: 10.3389/fphys.2018.01055
120. Kwon Y, Hofmann T, Montell C. Integration of Phosphoinositide- and Calmodulin-Mediated Regulation of TRPC6. Mol Cell (2007) 25(4):491–503. doi: 10.1016/j.molcel.2007.01.021
121. Dietrich A, Gudermann T. TRPC6: Physiological Function and Pathophysiological Relevance. Handb Exp Pharmacol (2014) 222:157–88. doi: 10.1007/978-3-642-54215-2_7
122. Aydar E, Yeo S, Djamgoz M, Palmer C. Abnormal Expression, Localization and Interaction of Canonical Transient Receptor Potential Ion Channels in Human Breast Cancer Cell Lines and Tissues: A Potential Target for Breast Cancer Diagnosis and Therapy. Cancer Cell Int (2009) 9:23. doi: 10.1186/1475-2867-9-23
123. Xu J, Yang Y, Xie R, Liu J, Nie X, An J, et al. The NCX1/TRPC6 Complex Mediates Tgfβ-Driven Migration and Invasion of Human Hepatocellular Carcinoma Cells. Cancer Res (2018) 78(10):2564–76. doi: 10.1158/0008-5472.CAN-17-2061
124. Shi Y, Ding X, He ZH, Zhou KC, Wang Q, Wang YZ. Critical Role of TRPC6 Channels in G2 Phase Transition and the Development of Human Oesophageal Cancer. Gut (2009) 58(11):1443–50. doi: 10.1136/gut.2009.181735
125. Cai R, Ding X, Zhou K, Shi Y, Ge R, Ren G, et al. Blockade of TRPC6 Channels Induced G2/M Phase Arrest and Suppressed Growth in Human Gastric Cancer Cells. Int J Cancer (2009) 125(10):2281–7. doi: 10.1002/ijc.24551
126. Ding X, He Z, Zhou K, Cheng J, Yao H, Lu D, et al. Essential Role of TRPC6 Channels in G2/M Phase Transition and Development of Human Glioma. J Natl Cancer Inst (2010) 102(14):1052–68. doi: 10.1093/jnci/djq217
127. Jardin I, Diez-Bello R, Lopez JJ, Redondo PC, Salido GM, Smani T, et al. Trpc6 Channels are Required for Proliferation, Migration and Invasion of Breast Cancer Cell Lines by Modulation of Orai1 and Orai3 Surface Exposure. Cancers (Basel) (2018) 10(9). doi: 10.3390/cancers10090331
128. Sanchez-Collado J, Lopez JJ, Gonzalez-Gutierrez L, Cantonero C, Jardin I, Salido GM, et al. Functional Role of TRPC6 and STIM2 in Cytosolic and Endoplasmic Reticulum Ca2+ Content in Resting Estrogen Receptor-Positive Breast Cancer Cells. Biochem J (2020) 477(17):3183–97. doi: 10.1042/BCJ20200560
129. Kim EY, Alvarez-Baron CP, Dryer SE. Canonical Transient Receptor Potential Channel (TRPC)3 and TRPC6 Associate With Large-Conductance Ca2+-activated K+ (Bkca) Channels: Role in BKCa Trafficking to the Surface of Cultured Podocytes. Mol Pharmacol (2009) 75(3):466–77. doi: 10.1124/mol.108.051912
130. Mound A, Rodat-Despoix L, Bougarn S, Ouadid-Ahidouch H, Matifat F. Molecular Interaction and Functional Coupling Between Type 3 Inositol 1,4,5-Trisphosphate Receptor and BKCa Channel Stimulate Breast Cancer Cell Proliferation. Eur J Cancer (2013) 49(17):3738–51. doi: 10.1016/j.ejca.2013.07.013
131. Schickling BM, England SK, Aykin-Burns N, Norian LA, Leslie KK, Frieden-Korovkina VP. Bkca Channel Inhibitor Modulates the Tumorigenic Ability of Hormone-Independent Breast Cancer Cells Via the Wnt Pathway. Oncol Rep (2015) 33(2):533–8. doi: 10.3892/or.2014.3617
132. Hisatsune C, Kuroda Y, Nakamura K, Inoue T, Nakamura T, Michikawa T, et al. Regulation of TRPC6 Channel Activity by Tyrosine Phosphorylation. J Biol Chem (2004) 279(18):18887–94. doi: 10.1074/jbc.M311274200
133. Xie YG, Yu Y, Hou LK, Wang X, Zhang B, Cao XC. FYN Promotes Breast Cancer Progression Through Epithelial-Mesenchymal Transition. Oncol Rep (2016) 36(2):1000–6. doi: 10.3892/or.2016.4894
134. Mayer EL, Krop IE. Advances in Targeting SRC in the Treatment of Breast Cancer and Other Solid Malignancies. Clin Cancer Res (2010) 16(14):3526–32. doi: 10.1158/1078-0432.CCR-09-1834
135. Lussier MP, Cayouette S, Lepage PK, Bernier CL, Francoeur N, St-Hilaire M, et al. MxA, A Member of the Dynamin Superfamily, Interacts With the Ankyrin-Like Repeat Domain of TRPC. J Biol Chem (2005) 280(19):19393–400. doi: 10.1074/jbc.M500391200
136. Kim YA, Lee HJ, Heo SH, Park HS, Park SY, Bang W, et al. Mxa Expression is Associated With Tumor-Infiltrating Lymphocytes and is a Prognostic Factor in Triple-Negative Breast Cancer. Breast Cancer Res Treat (2016) 156(3):597–606. doi: 10.1007/s10549-016-3786-z
137. Yamazaki H, Sasagawa Y, Yamamoto H, Bito H, Shirao T. Camkiiβ is Localized in Dendritic Spines as Both Drebrin-Dependent and Drebrin-Independent Pools. J Neurochem (2018) 146(2):145–59. doi: 10.1111/jnc.14449
138. Shi J, Geshi N, Takahashi S, Kiyonaka S, Ichikawa J, Hu Y, et al. Molecular Determinants for Cardiovascular TRPC6 Channel Regulation by Ca2+/calmodulin-dependent Kinase II. J Physiol (2013) 591(11):2851–66. doi: 10.1113/jphysiol.2013.251249
139. Shi J, Ju M, Saleh SN, Albert AP, Large WA. TRPC6 Channels Stimulated by Angiotensin II are Inhibited by TRPC1/C5 Channel Activity Through a Ca2+- and PKC-dependent Mechanism in Native Vascular Myocytes. J Physiol (2010) 588(Pt 19):3671–82. doi: 10.1113/jphysiol.2010.194621
140. Launay P, Fleig A, Perraud AL, Scharenberg AM, Penner R, Kinet JP. TRPM4 is a Ca2+-activated Nonselective Cation Channel Mediating Cell Membrane Depolarization. Cell (2002) 109(3):397–407. doi: 10.1016/S0092-8674(02)00719-5
141. Mathar I, Jacobs G, Kecskes M, Menigoz A, Philippaert K, Vennekens R. Trpm4. Handb Exp Pharmacol (2014) 222:461–87. doi: 10.1007/978-3-642-54215-2_18
142. Demion M, Bois P, Launay P, Guinamard R. TRPM4, a Ca2+-activated Nonselective Cation Channel in Mouse Sino-Atrial Node Cells. Cardiovasc Res (2007) 73(3):531–8. doi: 10.1016/j.cardiores.2006.11.023
143. Cheng H, Beck A, Launay P, Gross SA, Stokes AJ, Kinet JP, et al. TRPM4 Controls Insulin Secretion in Pancreatic Beta-Cells. Cell Calcium (2007) 41(1):51–61. doi: 10.1016/j.ceca.2006.04.032
144. Launay P, Cheng H, Srivatsan S, Penner R, Fleig A, Kinet JP. TRPM4 Regulates Calcium Oscillations After T Cell Activation. Science (2004) 306(5700):1374–7. doi: 10.1126/science.1098845
145. Barbet G, Demion M, Moura IC, Serafini N, Léger T, Vrtovsnik F, et al. The Calcium-Activated Nonselective Cation Channel TRPM4 is Essential for the Migration But Not the Maturation of Dendritic Cells. Nat Immunol (2008) 9(10):1148–56. doi: 10.1038/ni.1648
146. Caceres M, Ortiz L, Recabarren T, Romero A, Colombo A, Leiva-Salcedo E, et al. Trpm4 Is a Novel Component of the Adhesome Required for Focal Adhesion Disassembly, Migration and Contractility. PLoS One (2015) 10(6):e0130540. doi: 10.1371/journal.pone.0130540
147. Loo SK, Ch’ng ES, Md Salleh MS, Banham AH, Pedersen LM, Moller MB, et al. TRPM4 Expression is Associated With Activated B Cell Subtype and Poor Survival in Diffuse Large B Cell Lymphoma. Histopathology (2017) 71(1):98–111. doi: 10.1111/his.13204
148. Narayan G, Bourdon V, Chaganti S, Arias-Pulido H, Nandula SV, Rao PH, et al. Gene Dosage Alterations Revealed by cDNA Microarray Analysis in Cervical Cancer: Identification of Candidate Amplified and Overexpressed Genes. Genes Chromosomes Cancer (2007) 46(4):373–84. doi: 10.1002/gcc.20418
149. Holzmann C, Kappel S, Kilch T, Jochum MM, Urban SK, Jung V, et al. Transient Receptor Potential Melastatin 4 Channel Contributes to Migration of Androgen-Insensitive Prostate Cancer Cells. Oncotarget (2015) 6(39):41783–93. doi: 10.18632/oncotarget.6157
150. Sagredo AI, Sagredo EA, Cappelli C, Báez P, Andaur RE, Blanco C, et al. TRPM4 Regulates Akt/GSK3-β Activity and Enhances β-Catenin Signaling and Cell Proliferation in Prostate Cancer Cells. Mol Oncol (2018) 12(2):151–65. doi: 10.1002/1878-0261.12100
151. Sagredo AI, Sagredo EA, Pola V, Echeverria C, Andaur R, Michea L, et al. TRPM4 Channel is Involved in Regulating Epithelial to Mesenchymal Transition, Migration, and Invasion of Prostate Cancer Cell Lines. J Cell Physiol (2019) 234(3):2037–50. doi: 10.1002/jcp.27371
152. Borgström A, Peinelt C, Stokłosa P. TRPM4 in Cancer-A New Potential Drug Target. Biomolecules (2021) 11(2). doi: 10.3390/biom11020229
153. Rivas J, Díaz N, Silva I, Morales D, Lavanderos B, Álvarez A, et al. KCTD5, a Novel TRPM4-regulatory Protein Required for Cell Migration as a New Predictor for Breast Cancer Prognosis. FASEB J (2020) 34(6):7847–65. doi: 10.1096/fj.201901195RRR
154. Loo SK, Ch’ng ES, Md Salleh MS, Banham AH, Pedersen LM, Møller MB, et al. TRPM4 Expression is Associated With Activated B Cell Subtype and Poor Survival in Diffuse Large B Cell Lymphoma. Histopathology (2017) 71(1):98–111. doi: 10.1111/his.13204
155. Berg KD, Soldini D, Jung M, Dietrich D, Stephan C, Jung K, et al. TRPM4 Protein Expression in Prostate Cancer: A Novel Tissue Biomarker Associated With Risk of Biochemical Recurrence Following Radical Prostatectomy. Virchows Arch (2016) 468(3):345–55. doi: 10.1007/s00428-015-1880-y
156. Li XC, Cheng Y, Yang X, Zhou JY, Dong YY, Shen BQ, et al. Decreased Expression of TRPM4 is Associated With Unfavorable Prognosis and Aggressive Progression of Endometrial Carcinoma. Am J Transl Res (2020) 12(7):3926–39.
157. Wong KK, Hussain FA. TRPM4 is Overexpressed in Breast Cancer Associated With Estrogen Response and Epithelial-Mesenchymal Transition Gene Sets. PLoS One (2020) 15(6):e0233884. doi: 10.1371/journal.pone.0233884
158. Mizuno H, Kitada K, Nakai K, Sarai A. PrognoScan: A New Database for Meta-Analysis of the Prognostic Value of Genes. BMC Med Genomics (2009) 2:18. doi: 10.1186/1755-8794-2-18
159. Coticchia CM, Revankar CM, Deb TB, Dickson RB, Johnson MD. Calmodulin Modulates Akt Activity in Human Breast Cancer Cell Lines. Breast Cancer Res Treat (2009) 115(3):545–60. doi: 10.1007/s10549-008-0097-z
160. Deb TB, Coticchia CM, Dickson RB. Calmodulin-Mediated Activation of Akt Regulates Survival of c-Myc-overexpressing Mouse Mammary Carcinoma Cells. J Biol Chem (2004) 279(37):38903–11. doi: 10.1074/jbc.M405314200
161. Nilius B, Prenen J, Tang J, Wang C, Owsianik G, Janssens A, et al. Regulation of the Ca2+ Sensitivity of the Nonselective Cation Channel TRPM4. J Biol Chem (2005) 280(8):6423–33. doi: 10.1074/jbc.M411089200
162. Woo SK, Kwon MS, Ivanov A, Gerzanich V, Simard JM. The Sulfonylurea Receptor 1 (Sur1)-Transient Receptor Potential Melastatin 4 (Trpm4) Channel. J Biol Chem (2013) 288(5):3655–67. doi: 10.1074/jbc.M112.428219
163. Lee KPK, Chen J, MacKinnon R. Molecular Structure of Human KATP in Complex With ATP and ADP. Elife (2017) 6. doi: 10.7554/eLife.32481
164. Xu K, Sun G, Li M, Chen H, Zhang Z, Qian X, et al. Glibenclamide Targets Sulfonylurea Receptor 1 to Inhibit P70s6k Activity and Upregulate Klf4 Expression to Suppress Non-Small Cell Lung Carcinoma. Mol Cancer Ther (2019) 18(11):2085–96. doi: 10.1158/1535-7163.MCT-18-1181
165. Uhlen M, Zhang C, Lee S, Sjöstedt E, Fagerberg L, Bidkhori G, et al. A Pathology Atlas of the Human Cancer Transcriptome. Science (2017) 357(6352). doi: 10.1126/science.aan2507
166. Núñez M, Medina V, Cricco G, Croci M, Cocca C, Rivera E, et al. Glibenclamide Inhibits Cell Growth by Inducing G0/G1 Arrest in the Human Breast Cancer Cell Line MDA-MB-231. BMC Pharmacol Toxicol (2013) 14:6. doi: 10.1186/2050-6511-14-6
167. Wilkinson CM, Brar PS, Balay CJ, Colbourne F. Glibenclamide, a Sur1-Trpm4 Antagonist, Does Not Improve Outcome After Collagenase-Induced Intracerebral Hemorrhage. PLoS One (2019) 14(5):e0215952. doi: 10.1371/journal.pone.0215952
168. Kurland DB, Gerzanich V, Karimy JK, Woo SK, Vennekens R, Freichel M, et al. The Sur1-Trpm4 Channel Regulates NOS2 Transcription in TLR4-activated Microglia. J Neuroinflamm (2016) 13(1):130. doi: 10.1186/s12974-016-0599-2
169. Mancini M, Toker A. NFAT Proteins: Emerging Roles in Cancer Progression. Nat Rev Cancer (2009) 9(11):810–20. doi: 10.1038/nrc2735
170. Blanco C, Morales D, Mogollones I, Vergara-Jaque A, Vargas C, Álvarez A, et al. EB1- and EB2-Dependent Anterograde Trafficking of TRPM4 Regulates Focal Adhesion Turnover and Cell Invasion. FASEB J (2019) 33(8):9434–52. doi: 10.1096/fj.201900136R
171. Dong X, Liu F, Sun L, Liu M, Li D, Su D, et al. Oncogenic Function of Microtubule End-Binding Protein 1 in Breast Cancer. J Pathol (2010) 220(3):361–9. doi: 10.1002/path.2662
172. Fujii K, Kondo T, Yokoo H, Yamada T, Iwatsuki K, Hirohashi S. Proteomic Study of Human Hepatocellular Carcinoma Using Two-Dimensional Difference Gel Electrophoresis With Saturation Cysteine Dye. Proteomics (2005) 5(5):1411–22. doi: 10.1002/pmic.200401004
173. Wang Y, Zhou X, Zhu H, Liu S, Zhou C, Zhang G, et al. Overexpression of EB1 in Human Esophageal Squamous Cell Carcinoma (ESCC) may Promote Cellular Growth by Activating Beta-Catenin/TCF Pathway. Oncogene (2005) 24(44):6637–45. doi: 10.1038/sj.onc.1208819
174. Cho CH, Kim E, Lee YS, Yarishkin O, Yoo JC, Park JY, et al. Depletion of 14-3-3γ Reduces the Surface Expression of Transient Receptor Potential Melastatin 4b (TRPM4b) Channels and Attenuates TRPM4b-mediated Glutamate-Induced Neuronal Cell Death. Mol Brain (2014) 7:52. doi: 10.1186/s13041-014-0052-3
175. Aitken A. 14-3-3 Proteins: A Historic Overview. Semin Cancer Biol (2006) 16(3):162–72. doi: 10.1016/j.semcancer.2006.03.005
176. Fan X, Cui L, Zeng Y, Song W, Gaur U, Yang M. 14-3-3 Proteins are on the Crossroads of Cancer, Aging, and Age-Related Neurodegenerative Disease. Int J Mol Sci (2019) 20(14). doi: 10.3390/ijms20143518
177. Hiraoka E, Mimae T, Ito M, Kadoya T, Miyata Y, Ito A, et al. Breast Cancer Cell Motility is Promoted by 14-3-3γ. Breast Cancer (2019) 26(5):581–93. doi: 10.1007/s12282-019-00957-4
178. Nadler MJ, Hermosura MC, Inabe K, Perraud AL, Zhu Q, Stokes AJ, et al. LTRPC7 is a Mg.ATP-regulated Divalent Cation Channel Required for Cell Viability. Nature (2001) 411(6837):590–5. doi: 10.1038/35079092
179. Ryazanova LV, Dorovkov MV, Ansari A, Ryazanov AG. Characterization of the Protein Kinase Activity of TRPM7/ChaK1, a Protein Kinase Fused to the Transient Receptor Potential Ion Channel. J Biol Chem (2004) 279(5):3708–16. doi: 10.1074/jbc.M308820200
180. Desai BN, Krapivinsky G, Navarro B, Krapivinsky L, Carter BC, Febvay S, et al. Cleavage of TRPM7 Releases the Kinase Domain From the Ion Channel and Regulates its Participation in Fas-induced Apoptosis. Dev Cell (2012) 22(6):1149–62. doi: 10.1016/j.devcel.2012.04.006
181. Runnels LW, Yue L, Clapham DE. Trp-PLIK, a Bifunctional Protein With Kinase and Ion Channel Activities. Science (2001) 291(5506):1043–7. doi: 10.1126/science.1058519
182. Runnels LW. TRPM6 and TRPM7: A Mul-TRP-PLIK-cation of Channel Functions. Curr Pharm Biotechnol (2011) 12(1):42–53. doi: 10.2174/138920111793937880
183. Hanano T, Hara Y, Shi J, Morita H, Umebayashi C, Mori E, et al. Involvement of TRPM7 in Cell Growth as a Spontaneously Activated Ca2+ Entry Pathway in Human Retinoblastoma Cells. J Pharmacol Sci (2004) 95(4):403–19. doi: 10.1254/jphs.FP0040273
184. Kim BJ, Park EJ, Lee JH, Jeon JH, Kim SJ, So I. Suppression of Transient Receptor Potential Melastatin 7 Channel Induces Cell Death in Gastric Cancer. Cancer Sci (2008) 99(12):2502–9. doi: 10.1111/j.1349-7006.2008.00982.x
185. Jiang J, Li MH, Inoue K, Chu XP, Seeds J, Xiong ZG. Transient Receptor Potential Melastatin 7-Like Current in Human Head and Neck Carcinoma Cells: Role in Cell Proliferation. Cancer Res (2007) 67(22):10929–38. doi: 10.1158/0008-5472.CAN-07-1121
186. Middelbeek J, Vrenken K, Visser D, Lasonder E, Koster J, Jalink K, et al. The TRPM7 Interactome Defines a Cytoskeletal Complex Linked to Neuroblastoma Progression. Eur J Cell Biol (2016) 95(11):465–74. doi: 10.1016/j.ejcb.2016.06.008
187. Visser D, Langeslag M, Kedziora KM, Klarenbeek J, Kamermans A, Horgen FD, et al. TRPM7 Triggers Ca2+ Sparks and Invadosome Formation in Neuroblastoma Cells. Cell Calcium (2013) 54(6):404–15. doi: 10.1016/j.ceca.2013.09.003
188. Meng X, Cai C, Wu J, Cai S, Ye C, Chen H, et al. TRPM7 Mediates Breast Cancer Cell Migration and Invasion Through the MAPK Pathway. Cancer Lett (2013) 333(1):96–102. doi: 10.1016/j.canlet.2013.01.031
189. Guilbert A, Gautier M, Dhennin-Duthille I, Rybarczyk P, Sahni J, Sevestre H, et al. Transient Receptor Potential Melastatin 7 is Involved in Oestrogen Receptor-Negative Metastatic Breast Cancer Cells Migration Through its Kinase Domain. Eur J Cancer (2013) 49(17):3694–707. doi: 10.1016/j.ejca.2013.07.008
190. Middelbeek J, Kuipers AJ, Henneman L, Visser D, Eidhof I, van Horssen R, et al. Trpm7 Is Required for Breast Tumor Cell Metastasis. Cancer Res (2012) 72(16):4250. doi: 10.1158/0008-5472.CAN-11-3863
191. Clark K, Langeslag M, van Leeuwen B, Ran L, Ryazanov AG, Figdor CG, et al. TRPM7, a Novel Regulator of Actomyosin Contractility and Cell Adhesion. EMBO J (2006) 25(2):290–301. doi: 10.1038/sj.emboj.7600931
192. Clark K, Middelbeek J, Dorovkov MV, Figdor CG, Ryazanov AG, Lasonder E, et al. The Alpha-Kinases TRPM6 and TRPM7, But Not eEF-2 Kinase, Phosphorylate the Assembly Domain of Myosin IIA, IIB and IIC. FEBS Lett (2008) 582(20):2993–7. doi: 10.1016/j.febslet.2008.07.043
193. Dorovkov MV, Ryazanov AG. Phosphorylation of Annexin I by TRPM7 Channel-Kinase. J Biol Chem (2004) 279(49):50643–6. doi: 10.1074/jbc.C400441200
194. Dorovkov MV, Kostyukova AS, Ryazanov AG. Phosphorylation of Annexin A1 by TRPM7 Kinase: A Switch Regulating the Induction of an α-Helix. Biochemistry (2011) 50(12):2187–93. doi: 10.1021/bi101963h
195. Tu Y, Johnstone CN, Stewart AG. Annexin A1 Influences in Breast Cancer: Controversies on Contributions to Tumour, Host and Immunoediting Processes. Pharmacol Res (2017) 119:278–88. doi: 10.1016/j.phrs.2017.02.011
196. de Graauw M, van Miltenburg MH, Schmidt MK, Pont C, Lalai R, Kartopawiro J, et al. Annexin A1 Regulates TGF-beta Signaling and Promotes Metastasis Formation of Basal-Like Breast Cancer Cells. Proc Natl Acad Sci USA (2010) 107(14):6340–5. doi: 10.1073/pnas.0913360107
197. Yom CK, Han W, Kim SW, Kim HS, Shin HC, Chang JN, et al. Clinical Significance of Annexin A1 Expression in Breast Cancer. J Breast Cancer (2011) 14(4):262–8. doi: 10.4048/jbc.2011.14.4.262
198. Bist P, Leow SC, Phua QH, Shu S, Zhuang Q, Loh WT, et al. Annexin-1 Interacts With NEMO and RIP1 to Constitutively Activate IKK Complex and NF-κb: Implication in Breast Cancer Metastasis. Oncogene (2011) 30(28):3174–85. doi: 10.1038/onc.2011.28
199. Kretzschmar M. Transforming Growth Factor-Beta and Breast Cancer: Transforming Growth Factor-Beta/SMAD Signaling Defects and Cancer. Breast Cancer Res (2000) 2(2):107–15. doi: 10.1186/bcr42
200. Fang L, Huang C, Meng X, Wu B, Ma T, Liu X, et al. Tgf-β1-Elevated TRPM7 Channel Regulates Collagen Expression in Hepatic Stellate Cells Via TGF-β1/Smad Pathway. Toxicol Appl Pharmacol (2014) 280(2):335–44. doi: 10.1016/j.taap.2014.08.006
201. de Kruijf EM, Dekker TJA, Hawinkels L, Putter H, Smit V, Kroep JR, et al. The Prognostic Role of TGF-β Signaling Pathway in Breast Cancer Patients. Ann Oncol (2013) 24(2):384–90. doi: 10.1093/annonc/mds333
202. Peier AM, Moqrich A, Hergarden AC, Reeve AJ, Andersson DA, Story GM, et al. A TRP Channel That Senses Cold Stimuli and Menthol. Cell (2002) 108(5):705–15. doi: 10.1016/S0092-8674(02)00652-9
203. McKemy DD, Neuhausser WM, Julius D. Identification of a Cold Receptor Reveals a General Role for TRP Channels in Thermosensation. Nature (2002) 416(6876):52–8. doi: 10.1038/nature719
204. Knowlton WM, Palkar R, Lippoldt EK, McCoy DD, Baluch F, Chen J, et al. A Sensory-Labeled Line for Cold: TRPM8-expressing Sensory Neurons Define the Cellular Basis for Cold, Cold Pain, and Cooling-Mediated Analgesia. J Neurosci (2013) 33(7):2837–48. doi: 10.1523/JNEUROSCI.1943-12.2013
205. Bai VU, Murthy S, Chinnakannu K, Muhletaler F, Tejwani S, Barrack ER, et al. Androgen Regulated TRPM8 Expression: A Potential mRNA Marker for Metastatic Prostate Cancer Detection in Body Fluids. Int J Oncol (2010) 36(2):443–50. doi: 10.3892/ijo_00000518
206. Du JD, Zheng X, Chen YL, Huang ZQ, Cai SW, Jiao HB, et al. Elevated Transient Receptor Potential Melastatin 8 (Trpm8) Expression is Correlated With Poor Prognosis in Pancreatic Cancer. Med Sci Monit (2018) 24:3720–5. doi: 10.12659/MSM.909968
207. Wondergem R, Bartley JW. Menthol Increases Human Glioblastoma Intracellular Ca2+, BK Channel Activity and Cell Migration. J BioMed Sci (2009) 16(1):90. doi: 10.1186/1423-0127-16-90
208. Liu J, Hu G, Gong Y, Yu Q, He B, Li W, et al. Silencing of TRPM8 Inhibits Aggressive Tumor Phenotypes and Enhances Gemcitabine Sensitivity in Pancreatic Cancer. Pancreatology (2018) 18(8):935–44. doi: 10.1016/j.pan.2018.08.011
209. Yamamura H, Ugawa S, Ueda T, Morita A, Shimada S. TRPM8 Activation Suppresses Cellular Viability in Human Melanoma. Am J Physiology-Cell Physiol (2008) 295(2):C296–301. doi: 10.1152/ajpcell.00499.2007
210. Tsavaler L, Shapero MH, Morkowski S, Laus R. Trp-p8, a Novel Prostate-Specific Gene, is Up-Regulated in Prostate Cancer and Other Malignancies and Shares High Homology With Transient Receptor Potential Calcium Channel Proteins. Cancer Res (2001) 61(9):3760–9.
211. Chodon D, Guilbert A, Dhennin-Duthille I, Gautier M, Telliez MS, Sevestre H, et al. Estrogen Regulation of TRPM8 Expression in Breast Cancer Cells. BMC Cancer (2010) 10:212. doi: 10.1186/1471-2407-10-212
212. Liu J, Chen Y, Shuai S, Ding D, Li R, Luo R. TRPM8 Promotes Aggressiveness of Breast Cancer Cells by Regulating EMT Via Activating AKT/GSK-3β Pathway. Tumour Biol (2014) 35(9):8969–77. doi: 10.1007/s13277-014-2077-8
213. Vinuela-Fernandez I, Sun L, Jerina H, Curtis J, Allchorne A, Gooding H, et al. The TRPM8 Channel Forms a Complex With the 5-HT(1B) Receptor and Phospholipase D That Amplifies its Reversal of Pain Hypersensitivity. Neuropharmacology (2014) 79:136–51. doi: 10.1016/j.neuropharm.2013.11.006
214. Gautam J, Banskota S, Regmi SC, Ahn S, Jeon YH, Jeong H, et al. Tryptophan Hydroxylase 1 and 5-HT(7) Receptor Preferentially Expressed in Triple-Negative Breast Cancer Promote Cancer Progression Through Autocrine Serotonin Signaling. Mol Cancer (2016) 15(1):75. doi: 10.1186/s12943-016-0559-6
215. Caterina MJ, Schumacher MA, Tominaga M, Rosen TA, Levine JD, Julius D. The Capsaicin Receptor: A Heat-Activated Ion Channel in the Pain Pathway. Nature (1997) 389(6653):816–24. doi: 10.1038/39807
216. Benítez-Angeles M, Morales-Lázaro SL, Juárez-González E, Rosenbaum T. Trpv1: Structure, Endogenous Agonists, and Mechanisms. Int J Mol Sci (2020) 21(10). doi: 10.3390/ijms21103421
217. Bautista D, Julius D. Fire in the Hole: Pore Dilation of the Capsaicin Receptor TRPV1. Nature Neuroscience (2008) 11:528–9. doi: 10.1038/nn0508-528
218. Gunthorpe MJ, Harries MH, Prinjha RK, Davis JB, Randall A. Voltage- and Time-Dependent Properties of the Recombinant Rat Vanilloid Receptor (Rvr1). J Physiol (2000) 525(Pt 3):747–59. doi: 10.1111/j.1469-7793.2000.t01-1-00747.x
219. Ho KW, Lambert WS, Calkins DJ. Activation of the TRPV1 Cation Channel Contributes to Stress-Induced Astrocyte Migration. Glia (2014) 62(9):1435–51. doi: 10.1002/glia.22691
220. Aneiros E, Cao L, Papakosta M, Stevens E, Phillips S, Grimm C. The Biophysical and Molecular Basis of TRPV1 Proton Gating. EMBO J (2011) 30(6):994–1002. doi: 10.1038/emboj.2011.19
221. Boonen B, Alpizar YA, Sanchez A, Lopez-Requena A, Voets T, Talavera K. Differential Effects of Lipopolysaccharide on Mouse Sensory TRP Channels. Cell Calcium (2018) 73:72–81. doi: 10.1016/j.ceca.2018.04.004
222. Bhave G, Zhu W, Wang H, Brasier DJ, Oxford GS, Gereau RW. cAMP-dependent Protein Kinase Regulates Desensitization of the Capsaicin Receptor (VR1) by Direct Phosphorylation. Neuron (2002) 35(4):721–31. doi: 10.1016/S0896-6273(02)00802-4
223. Bhave G, Hu HJ, Glauner KS, Zhu W, Wang H, Brasier DJ, et al. Protein Kinase C Phosphorylation Sensitizes But Does Not Activate the Capsaicin Receptor Transient Receptor Potential Vanilloid 1 (TRPV1). Proc Natl Acad Sci USA (2003) 100(21):12480–5. doi: 10.1073/pnas.2032100100
224. Jung J, Shin JS, Lee SY, Hwang SW, Koo J, Cho H, et al. Phosphorylation of Vanilloid Receptor 1 by Ca2+/calmodulin-dependent Kinase II Regulates its Vanilloid Binding. J Biol Chem (2004) 279(8):7048–54. doi: 10.1074/jbc.M311448200
225. Prescott ED, Julius D. A Modular PIP2 Binding Site as a Determinant of Capsaicin Receptor Sensitivity. Science (2003) 300(5623):1284–8. doi: 10.1126/science.1083646
226. Cavanaugh DJ, Chesler AT, Jackson AC, Sigal YM, Yamanaka H, Grant R, et al. Trpv1 Reporter Mice Reveal Highly Restricted Brain Distribution and Functional Expression in Arteriolar Smooth Muscle Cells. J Neurosci (2011) 31(13):5067–77. doi: 10.1523/JNEUROSCI.6451-10.2011
227. Cristino L, de Petrocellis L, Pryce G, Baker D, Guglielmotti V, Di Marzo V. Immunohistochemical Localization of Cannabinoid Type 1 and Vanilloid Transient Receptor Potential Vanilloid Type 1 Receptors in the Mouse Brain. Neuroscience (2006) 139(4):1405–15. doi: 10.1016/j.neuroscience.2006.02.074
228. Birder LA, Nakamura Y, Kiss S, Nealen ML, Barrick S, Kanai AJ, et al. Altered Urinary Bladder Function in Mice Lacking the Vanilloid Receptor TRPV1. Nat Neurosci (2002) 5(9):856–60. doi: 10.1038/nn902
229. Czifra G, Varga A, Nyeste K, Marincsák R, Tóth BI, Kovács I, et al. Increased Expressions of Cannabinoid Receptor-1 and Transient Receptor Potential Vanilloid-1 in Human Prostate Carcinoma. J Cancer Res Clin Oncol (2009) 135(4):507–14. doi: 10.1007/s00432-008-0482-3
230. Sánchez MG, Sánchez AM, Collado B, Malagarie-Cazenave S, Olea N, Carmena MJ, et al. Expression of the Transient Receptor Potential Vanilloid 1 (TRPV1) in LNCaP and PC-3 Prostate Cancer Cells and in Human Prostate Tissue. Eur J Pharmacol (2005) 515(1-3):20–7. doi: 10.1016/j.ejphar.2005.04.010
231. Miao X, Liu G, Xu X, Xie C, Sun F, Yang Y, et al. High Expression of Vanilloid Receptor-1 is Associated With Better Prognosis of Patients With Hepatocellular Carcinoma. Cancer Genet Cytogenet (2008) 186(1):25–32. doi: 10.1016/j.cancergencyto.2008.05.011
232. Lazzeri M, Vannucchi MG, Spinelli M, Bizzoco E, Beneforti P, Turini D, et al. Transient Receptor Potential Vanilloid Type 1 (TRPV1) Expression Changes From Normal Urothelium to Transitional Cell Carcinoma of Human Bladder. Eur Urol (2005) 48(4):691–8. doi: 10.1016/j.eururo.2005.05.018
233. Bode AM, Cho YY, Zheng D, Zhu F, Ericson ME, Ma WY, et al. Transient Receptor Potential Type Vanilloid 1 Suppresses Skin Carcinogenesis. Cancer Res (2009) 69(3):905–13. doi: 10.1158/0008-5472.CAN-08-3263
234. Amantini C, Mosca M, Nabissi M, Lucciarini R, Caprodossi S, Arcella A, et al. Capsaicin-Induced Apoptosis of Glioma Cells is Mediated by TRPV1 Vanilloid Receptor and Requires P38 MAPK Activation. J Neurochem (2007) 102(3):977–90. doi: 10.1111/j.1471-4159.2007.04582.x
235. Amantini C, Ballarini P, Caprodossi S, Nabissi M, Morelli M, Lucciarini R, et al. Triggering of Transient Receptor Potential Vanilloid Type 1 (TRPV1) by Capsaicin Induces Fas/CD95-mediated Apoptosis of Urothelial Cancer Cells in an ATM-dependent Manner. Carcinogenesis (2009) 30(8):1320–9. doi: 10.1093/carcin/bgp138
236. Xu S, Zhang L, Cheng X, Yu H, Bao J, Lu R. Capsaicin Inhibits the Metastasis of Human Papillary Thyroid Carcinoma BCPAP Cells Through the Modulation of the TRPV1 Channel. Food Funct (2018) 9(1):344–54. doi: 10.1039/C7FO01295K
237. Weber LV, Al-Refae K, Wölk G, Bonatz G, Altmüller J, Becker C, et al. Expression and Functionality of TRPV1 in Breast Cancer Cells. Breast Cancer (Dove Med Press) (2016) 8:243–52. doi: 10.2147/BCTT.S121610
238. Lozano C, Córdova C, Marchant I, Zúñiga R, Ochova P, Ramírez-Barrantes R, et al. Intracellular Aggregated TRPV1 is Associated With Lower Survival in Breast Cancer Patients. Breast Cancer (Dove Med Press) (2018) 10:161–8. doi: 10.2147/BCTT.S170208
239. Nur G, Nazıroğlu M, Deveci HA. Synergic Prooxidant, Apoptotic and TRPV1 Channel Activator Effects of Alpha-Lipoic Acid and Cisplatin in MCF-7 Breast Cancer Cells. J Recept Signal Transduct Res (2017) 37(6):569–77. doi: 10.1080/10799893.2017.1369121
240. Nazıroğlu M, Çiğ B, Blum W, Vizler C, Buhala A, Marton A, et al. Targeting Breast Cancer Cells by MRS1477, a Positive Allosteric Modulator of TRPV1 Channels. PLoS One (2017) 12(6):e0179950. doi: 10.1371/journal.pone.0179950
241. Koşar PA, Nazıroğlu M, Övey İ, Çiğ B. Synergic Effects of Doxorubicin and Melatonin on Apoptosis and Mitochondrial Oxidative Stress in MCF-7 Breast Cancer Cells: Involvement of TRPV1 Channels. J Membr Biol (2016) 249(1-2):129–40. doi: 10.1007/s00232-015-9855-0
242. Ramírez-Barrantes R, Córdova C, Gatica S, Rodriguez B, Lozano C, Marchant I, et al. Transient Receptor Potential Vanilloid 1 Expression Mediates Capsaicin-Induced Cell Death. Front Physiol (2018) 9:682. doi: 10.3389/fphys.2018.00682
243. Wu TTL, Peters AA, Tan PT, Roberts-Thomson SJ, Monteith GR. Consequences of Activating the Calcium-Permeable Ion Channel TRPV1 in Breast Cancer Cells With Regulated TRPV1 Expression. Cell Calcium (2014) 56(2):59–67. doi: 10.1016/j.ceca.2014.04.006
244. Kim S, Kang C, Shin CY, Hwang SW, Yang YD, Shim WS, et al. TRPV1 Recapitulates Native Capsaicin Receptor in Sensory Neurons in Association With Fas-associated Factor 1. J Neurosci (2006) 26(9):2403–12. doi: 10.1523/JNEUROSCI.4691-05.2006
245. Ghosh AK, Basu S. Fas-Associated Factor 1 is a Negative Regulator in Capsaicin Induced Cancer Cell Apoptosis. Cancer Lett (2010) 287(2):142–9. doi: 10.1016/j.canlet.2009.06.007
246. Zhang L, Zhou F, Li Y, Drabsch Y, Zhang J, van Dam H, et al. Fas-Associated Factor 1 is a Scaffold Protein That Promotes β-Transducin Repeat-Containing Protein (β-TrCP)-Mediated β-Catenin Ubiquitination and Degradation. J Biol Chem (2012) 287(36):30701–10. doi: 10.1074/jbc.M112.353524
247. Xie F, Jin K, Shao L, Fan Y, Tu Y, Li Y, et al. FAF1 Phosphorylation by AKT Accumulates TGF-β Type II Receptor and Drives Breast Cancer Metastasis. Nat Commun (2017) 8:15021. doi: 10.1038/ncomms15021
248. Laínez S, Valente P, Ontoria-Oviedo I, Estévez-Herrera J, Camprubí-Robles M, Ferrer-Montiel A, et al. GABAA Receptor Associated Protein (GABARAP) Modulates TRPV1 Expression and Channel Function and Desensitization. FASEB J (2010) 24(6):1958–70. doi: 10.1096/fj.09-151472
249. Klebig C, Seitz S, Arnold W, Deutschmann N, Pacyna-Gengelbach M, Scherneck S, et al. Characterization of {Gamma}-Aminobutyric Acid Type A Receptor-Associated Protein, A Novel Tumor Suppressor, Showing Reduced Expression in Breast Cancer. Cancer Res (2005) 65(2):394–400.
250. Zygmunt PM, Högestätt ED. Trpa1. Handb Exp Pharmacol (2014) 222:583–630. doi: 10.1007/978-3-642-54215-2_23
251. Salas MM, Hargreaves KM, Akopian AN. TRPA1-Mediated Responses in Trigeminal Sensory Neurons: Interaction Between TRPA1 and TRPV1. Eur J Neurosci (2009) 29(8):1568–78. doi: 10.1111/j.1460-9568.2009.06702.x
252. Akopian AN, Ruparel NB, Jeske NA, Hargreaves KM. Transient Receptor Potential TRPA1 Channel Desensitization in Sensory Neurons is Agonist Dependent and Regulated by TRPV1-directed Internalization. J Physiol (2007) 583(Pt 1):175–93. doi: 10.1113/jphysiol.2007.133231
253. Staruschenko A, Jeske NA, Akopian AN. Contribution of TRPV1-TRPA1 Interaction to the Single Channel Properties of the TRPA1 Channel. J Biol Chem (2010) 285(20):15167–77. doi: 10.1074/jbc.M110.106153
254. Takahashi N, Chen HY, Harris IS, Stover DG, Selfors LM, Bronson RT, et al. Cancer Cells Co-Opt the Neuronal Redox-Sensing Channel TRPA1 to Promote Oxidative-Stress Tolerance. Cancer Cell (2018) 33(6):985–1003.e7. doi: 10.1016/j.ccell.2018.05.001
255. Weng HJ, Patel KN, Jeske NA, Bierbower SM, Zou W, Tiwari V, et al. Tmem100 Is a Regulator of TRPA1-TRPV1 Complex and Contributes to Persistent Pain. Neuron (2015) 85(4):833–46. doi: 10.1016/j.neuron.2014.12.065
256. Caterina MJ, Rosen TA, Tominaga M, Brake AJ, Julius D. A Capsaicin-Receptor Homologue With a High Threshold for Noxious Heat. Nature (1999) 398(6726):436–41. doi: 10.1038/18906
257. Muraki K, Iwata Y, Katanosaka Y, Ito T, Ohya S, Shigekawa M, et al. Trpv2 Is a Component of Osmotically Sensitive Cation Channels in Murine Aortic Myocytes. Circ Res (2003) 93(9):829–38. doi: 10.1161/01.RES.0000097263.10220.0C
258. Sugio S, Nagasawa M, Kojima I, Ishizaki Y, Shibasaki K. Transient Receptor Potential Vanilloid 2 Activation by Focal Mechanical Stimulation Requires Interaction With the Actin Cytoskeleton and Enhances Growth Cone Motility. FASEB J (2017) 31(4):1368–81. doi: 10.1096/fj.201600686RR
259. Qin N, Neeper MP, Liu Y, Hutchinson TL, Lubin ML, Flores CM. TRPV2 is Activated by Cannabidiol and Mediates CGRP Release in Cultured Rat Dorsal Root Ganglion Neurons. J Neurosci (2008) 28(24):6231–8. doi: 10.1523/JNEUROSCI.0504-08.2008
260. Hu HZ, Gu Q, Wang C, Colton CK, Tang J, Kinoshita-Kawada M, et al. 2-Aminoethoxydiphenyl Borate is a Common Activator of TRPV1, TRPV2, and TRPV3. J Biol Chem (2004) 279(34):35741–8. doi: 10.1074/jbc.M404164200
261. Bang S, Kim KY, Yoo S, Lee SH, Hwang SW. Transient Receptor Potential V2 Expressed in Sensory Neurons is Activated by Probenecid. Neurosci Lett (2007) 425(2):120–5. doi: 10.1016/j.neulet.2007.08.035
262. Park U, Vastani N, Guan Y, Raja SN, Koltzenburg M, Caterina MJ. TRP Vanilloid 2 Knock-Out Mice are Susceptible to Perinatal Lethality But Display Normal Thermal and Mechanical Nociception. J Neurosci (2011) 31(32):11425–36. doi: 10.1523/JNEUROSCI.1384-09.2011
263. Nedungadi TP, Dutta M, Bathina CS, Caterina MJ, Cunningham JT. Expression and Distribution of TRPV2 in Rat Brain. Exp Neurol (2012) 237(1):223–37. doi: 10.1016/j.expneurol.2012.06.017
264. Wainwright A, Rutter AR, Seabrook GR, Reilly K, Oliver KR. Discrete Expression of TRPV2 Within the Hypothalamo-Neurohypophysial System: Implications for Regulatory Activity Within the Hypothalamic-Pituitary-Adrenal Axis. J Comp Neurol (2004) 474(1):24–42. doi: 10.1002/cne.20100
265. Saito M, Hanson PI, Schlesinger P. Luminal Chloride-Dependent Activation of Endosome Calcium Channels: Patch Clamp Study of Enlarged Endosomes. J Biol Chem (2007) 282(37):27327–33. doi: 10.1074/jbc.M702557200
266. Abe K, Puertollano R. Role of TRP Channels in the Regulation of the Endosomal Pathway. Physiology (2011) 26(1):14–22. doi: 10.1152/physiol.00048.2010
267. Shibasaki K, Ishizaki Y, Mandadi S. Astrocytes Express Functional TRPV2 Ion Channels. Biochem Biophys Res Commun (2013) 441(2):327–32. doi: 10.1016/j.bbrc.2013.10.046
268. Santoni G, Amantini C. The Transient Receptor Potential Vanilloid Type-2 (Trpv2) Ion Channels in Neurogenesis and Gliomagenesis: Cross-Talk Between Transcription Factors and Signaling Molecules. Cancers (2019) 11(3):322. doi: 10.3390/cancers11030322
269. Katanosaka Y, Iwasaki K, Ujihara Y, Takatsu S, Nishitsuji K, Kanagawa M, et al. TRPV2 is Critical for the Maintenance of Cardiac Structure and Function in Mice. Nat Commun (2014) 5(1):1–14. doi: 10.1038/ncomms4932
270. Santoni G, Amantini C, Maggi F, Marinelli O, Santoni M, Nabissi M, et al. The TRPV2 Cation Channels: From Urothelial Cancer Invasiveness to Glioblastoma Multiforme Interactome Signature. Lab Invest (2020) 100(2):186–98. doi: 10.1038/s41374-019-0333-7
271. Alptekin M, Eroglu S, Tutar E, Sencan S, Geyik MA, Ulasli M, et al. Gene Expressions of TRP Channels in Glioblastoma Multiforme and Relation With Survival. Tumour Biol (2015) 36(12):9209–13. doi: 10.1007/s13277-015-3577-x
272. Liu G, Xie C, Sun F, Xu X, Yang Y, Zhang T, et al. Clinical Significance of Transient Receptor Potential Vanilloid 2 Expression in Human Hepatocellular Carcinoma. Cancer Genet Cytogenet (2010) 197(1):54–9. doi: 10.1016/j.cancergencyto.2009.08.007
273. Nabissi M, Morelli MB, Amantini C, Farfariello V, Ricci-Vitiani L, Caprodossi S, et al. TRPV2 Channel Negatively Controls Glioma Cell Proliferation and Resistance to Fas-Induced Apoptosis in ERK-dependent Manner. Carcinogenesis (2010) 31(5):794–803. doi: 10.1093/carcin/bgq019
274. Zoppoli P, Calice G, Laurino S, Ruggieri V, La Rocca F, La Torre G, et al. Trpv2 Calcium Channel Gene Expression and Outcomes in Gastric Cancer Patients: A Clinically Relevant Association. J Clin Med (2019) 8(5). doi: 10.3390/jcm8050662
275. Caprodossi S, Lucciarini R, Amantini C, Nabissi M, Canesin G, Ballarini P. Transient Receptor Potential Vanilloid Type 2 (TRPV2) Expression in Normal Urothelium and in Urothelial Carcinoma of Human Bladder: Correlation With the Pathologic Stage Eur Urol (2008) 54(3):612–30. doi: 10.1016/j.eururo.2007.10.016
276. Zhou K, Zhang SS, Yan Y, Zhao S. Overexpression of Transient Receptor Potential Vanilloid 2 is Associated With Poor Prognosis in Patients With Esophageal Squamous Cell Carcinoma. Med Oncol (2014) 31(7):17. doi: 10.1007/s12032-014-0017-5
277. Elbaz M, Ahirwar D, Xiaoli Z, Zhou X, Lustberg M, Nasser MW, et al. TRPV2 is a Novel Biomarker and Therapeutic Target in Triple Negative Breast Cancer. Oncotarget (2018) 9(71):33459–70. doi: 10.18632/oncotarget.9663
278. Liapi A, Wood JN. Extensive Co-Localization and Heteromultimer Formation of the Vanilloid Receptor-Like Protein TRPV2 and the Capsaicin Receptor TRPV1 in the Adult Rat Cerebral Cortex. Eur J Neurosci (2005) 22(4):825–34. doi: 10.1111/j.1460-9568.2005.04270.x
279. Rutter AR, Ma QP, Leveridge M, Bonnert TP. Heteromerization and Colocalization of TrpV1 and TrpV2 in Mammalian Cell Lines and Rat Dorsal Root Ganglia. Neuroreport (2005) 16(16):1735–9. doi: 10.1097/01.wnr.0000185958.03841.0f
280. Hellwig N, Albrecht N, Harteneck C, Schultz G, Schaefer M. Homo- and Heteromeric Assembly of TRPV Channel Subunits. J Cell Sci (2005) 118(Pt 5):917–28. doi: 10.1242/jcs.01675
281. Stokes AJ, Shimoda LM, Koblan-Huberson M, Adra CN, Turner H. A TRPV2-PKA Signaling Module for Transduction of Physical Stimuli in Mast Cells. J Exp Med (2004) 200(2):137–47. doi: 10.1084/jem.20032082
282. Huang Y, Yang L, Pei YY, Wang J, Wu H, Yuan J, et al. Overexpressed ACBD3 has Prognostic Value in Human Breast Cancer and Promotes the Self-Renewal Potential of Breast Cancer Cells by Activating the Wnt/beta-catenin Signaling Pathway. Exp Cell Res (2018) 363(1):39–47. doi: 10.1016/j.yexcr.2018.01.003
283. Garcia-Elias A, Mrkonjić S, Jung C, Pardo-Pastor C, Vicente R, Valverde MA. The TRPV4 Channel. Handb Exp Pharmacol (2014) 222:293–319. doi: 10.1007/978-3-642-54215-2_12
284. Voets T, Prenen J, Vriens J, Watanabe H, Janssens A, Wissenbach U, et al. Molecular Determinants of Permeation Through the Cation Channel TRPV4. J Biol Chem (2002) 277(37):33704–10. doi: 10.1074/jbc.M204828200
285. White JPM, Cibelli M, Urban L, Nilius B, McGeown JG, Nagy I. Trpv4: Molecular Conductor of a Diverse Orchestra. Physiol Rev (2016) 96(3):911–73. doi: 10.1152/physrev.00016.2015
286. Fusi C, Materazzi S, Minocci D, Maio V, Oranges T, Massi D, et al. Transient Receptor Potential Vanilloid 4 (TRPV4) is Downregulated in Keratinocytes in Human non-Melanoma Skin Cancer. J Invest Dermatol (2014) 134(9):2408–17. doi: 10.1038/jid.2014.145
287. Liu X, Zhang P, Xie C, Sham KWY, Ng SSM, Chen Y, et al. Activation of PTEN by Inhibition of TRPV4 Suppresses Colon Cancer Development. Cell Death Dis (2019) 10(6):460. doi: 10.1038/s41419-019-1700-4
288. Lee WH, Choong LY, Mon NN, Lu S, Lin Q, Pang B, et al. Trpv4 Regulates Breast Cancer Cell Extravasation, Stiffness and Actin Cortex. Sci Rep (2016) 6:27903. doi: 10.1038/srep27903
289. Lee WH, Choong LY, Jin TH, Mon NN, Chong S, Liew CS, et al. TRPV4 Plays a Role in Breast Cancer Cell Migration Via Ca(2+)-dependent Activation of AKT and Downregulation of E-cadherin Cell Cortex Protein. Oncogenesis (2017) 6(5):e338. doi: 10.1038/oncsis.2017.39
290. Li CJ, Chu PY, Yiang GT, Wu MY. The Molecular Mechanism of Epithelial-Mesenchymal Transition for Breast Carcinogenesis. Biomolecules (2019) 9(9). doi: 10.3390/biom9090476
291. Sokabe T, Fukumi-Tominaga T, Yonemura S, Mizuno A, Tominaga M. The TRPV4 Channel Contributes to Intercellular Junction Formation in Keratinocytes. J Biol Chem (2010) 285(24):18749–58. doi: 10.1074/jbc.M110.103606
292. Xu H, Zhao H, Tian W, Yoshida K, Roullet JB, Cohen DM. Regulation of a Transient Receptor Potential (TRP) Channel by Tyrosine Phosphorylation. SRC Family Kinase-Dependent Tyrosine Phosphorylation of TRPV4 on TYR-253 Mediates its Response to Hypotonic Stress. J Biol Chem (2003) 278(13):11520–7. doi: 10.1074/jbc.M211061200
293. Elias D, Ditzel HJ. Fyn is an Important Molecule in Cancer Pathogenesis and Drug Resistance. Pharmacol Res (2015) 100:250–4. doi: 10.1016/j.phrs.2015.08.010
294. Lee GH, Yoo KC, An Y, Lee HJ, Lee M, Uddin N, et al. FYN Promotes Mesenchymal Phenotypes of Basal Type Breast Cancer Cells Through STAT5/NOTCH2 Signaling Node. Oncogene (2018) 37(14):1857–68. doi: 10.1038/s41388-017-0114-y
295. Jung HJ, Park JY, Jeon HS, Kwon TH. Aquaporin-5: A Marker Protein for Proliferation and Migration of Human Breast Cancer Cells. PLoS One (2011) 6(12):e28492. doi: 10.1371/journal.pone.0028492
296. Jensen HH, Login FH, Koffman JS, Kwon TH, Nejsum LN. The Role of Aquaporin-5 in Cancer Cell Migration: A Potential Active Participant. Int J Biochem Cell Biol (2016) 79:271–6. doi: 10.1016/j.biocel.2016.09.005
297. Papadopoulos MC, Saadoun S, Verkman AS. Aquaporins and Cell Migration. Pflugers Arch (2008) 456(4):693–700. doi: 10.1007/s00424-007-0357-5
298. Wang J, Feng L, Zhu Z, Zheng M, Wang D, Chen Z, et al. Aquaporins as Diagnostic and Therapeutic Targets in Cancer: How Far We are? J Transl Med (2015) 13:96. doi: 10.1186/s12967-015-0439-7
299. Takayama Y, Shibasaki K, Suzuki Y, Yamanaka A, Tominaga M. Modulation of Water Efflux Through Functional Interaction Between TRPV4 and TMEM16A/anoctamin 1. FASEB J (2014) 28(5):2238–48. doi: 10.1096/fj.13-243436
300. Derouiche S, Takayama Y, Murakami M, Tominaga M. TRPV4 Heats Up ANO1-dependent Exocrine Gland Fluid Secretion. FASEB J (2018) 32(4):1841–54. doi: 10.1096/fj.201700954R
301. Wang H, Zou L, Ma K, Yu J, Wu H, Wei M, et al. Cell-Specific Mechanisms of TMEM16A Ca(2+)-Activated Chloride Channel in Cancer. Mol Cancer (2017) 16(1):152. doi: 10.1186/s12943-017-0720-x
302. Fernandes J, Lorenzo IM, Andrade YN, Garcia-Elias A, Serra SA, Fernández-Fernández JM, et al. IP3 Sensitizes TRPV4 Channel to the Mechano- and Osmotransducing Messenger 5’-6’-Epoxyeicosatrienoic Acid. J Cell Biol (2008) 181(1):143–55. doi: 10.1083/jcb.200712058
303. Ma X, Nilius B, Wong JW, Huang Y, Yao X. Electrophysiological Properties of Heteromeric TRPV4-C1 Channels. Biochim Biophys Acta (2011) 1808(12):2789–97. doi: 10.1016/j.bbamem.2011.07.049
304. Ma X, Cao J, Luo J, Nilius B, Huang Y, Ambudkar IS, et al. Depletion of Intracellular Ca2+ Stores Stimulates the Translocation of Vanilloid Transient Receptor Potential 4-C1 Heteromeric Channels to the Plasma Membrane. Arterioscler Thromb Vasc Biol (2010) 30(11):2249–55. doi: 10.1161/ATVBAHA.110.212084
305. Ma X, Cheng KT, Wong CO, O’Neil RG, Birnbaumer L, Ambudkar IS, et al. Heteromeric TRPV4-C1 Channels Contribute to Store-Operated Ca2+ Entry in Vascular Endothelial Cells. Cell Calcium (2011) 50(6):502–9. doi: 10.1016/j.ceca.2011.08.006
306. Ma X, Qiu S, Luo J, Ma Y, Ngai CY, Shen B, et al. Functional Role of Vanilloid Transient Receptor Potential 4-Canonical Transient Receptor Potential 1 Complex in Flow-Induced Ca2+ Influx. Arterioscler Thromb Vasc Biol (2010) 30(4):851–8. doi: 10.1161/ATVBAHA.109.196584
307. Fiorio Pla A, Ong HL, Cheng KT, Brossa A, Bussolati B, Lockwich T, et al. TRPV4 Mediates Tumor-Derived Endothelial Cell Migration Via Arachidonic Acid-Activated Actin Remodeling. Oncogene (2012) 31(2):200–12. doi: 10.1038/onc.2011.231
308. Yue L, Peng JB, Hediger MA, Clapham DE. CaT1 Manifests the Pore Properties of the Calcium-Release-Activated Calcium Channel. Nature (2001) 410(6829):705–9. doi: 10.1038/35070596
309. van de Graaf SF, Hoenderop JG, Bindels RJ. Regulation of TRPV5 and TRPV6 by Associated Proteins. Am J Physiol Renal Physiol (2006) 290(6):F1295–302. doi: 10.1152/ajprenal.00443.2005
310. Hoenderop JG, Nilius B, Bindels RJ. Epithelial Calcium Channels: From Identification to Function and Regulation. Pflugers Arch (2003) 446(3):304–8. doi: 10.1007/s00424-003-1045-8
311. Suzuki Y, Kovacs CS, Takanaga H, Peng JB, Landowski CP, Hediger MA. Calcium Channel TRPV6 is Involved in Murine Maternal-Fetal Calcium Transport. J Bone Miner Res (2008) 23(8):1249–56. doi: 10.1359/jbmr.080314
312. Weissgerber P, Kriebs U, Tsvilovskyy V, Olausson J, Kretz O, Stoerger C, et al. Excision of Trpv6 Gene Leads to Severe Defects in Epididymal Ca2+ Absorption and Male Fertility Much Like Single D541A Pore Mutation. J Biol Chem (2012) 287(22):17930–41. doi: 10.1074/jbc.M111.328286
313. Yamauchi D, Nakaya K, Raveendran NN, Harbidge DG, Singh R, Wangemann P, et al. Expression of Epithelial Calcium Transport System in Rat Cochlea and Vestibular Labyrinth. BMC Physiol (2010) 10:1. doi: 10.1186/1472-6793-10-1
314. Stewart JM. TRPV6 as A Target for Cancer Therapy. J Cancer (2020) 11(2):374–87. doi: 10.7150/jca.31640
315. Sun F, Xiao L, Jang XX, Xiong Y, Li Q, Yue XJ, et al. TRPV6 is a Prognostic Marker in Early-Stage Cervical Squamous Cell Carcinoma. Tumour Biol (2016) 37:15743–51. doi: 10.1007/s13277-016-5368-4
316. Song H, Dong M, Zhou J, Sheng W, Li X, Gao W. Expression and Prognostic Significance of TRPV6 in the Development and Progression of Pancreatic Cancer. Oncol Rep (2018) 39(3):1432–40. doi: 10.3892/or.2018.6216
317. Zhang SS, Xie X, Wen J, Luo KJ, Liu QW, Yang H, et al. TRPV6 Plays a New Role in Predicting Survival of Patients With Esophageal Squamous Cell Carcinoma. Diagn Pathol (2016) 11:14. doi: 10.1186/s13000-016-0457-7
318. Bolanz KA, Hediger MA, Landowski CP. The Role of TRPV6 in Breast Carcinogenesis. Mol Cancer Ther (2008) 7(2):271–9. doi: 10.1158/1535-7163.MCT-07-0478
319. Chin D, Means AR. Calmodulin: A Prototypical Calcium Sensor. Trends Cell Biol (2000) 10(8):322–8. doi: 10.1016/S0962-8924(00)01800-6
320. Niemeyer BA, Bergs C, Wissenbach U, Flockerzi V, Trost C. Competitive Regulation of CaT-like-mediated Ca2+ Entry by Protein Kinase C and Calmodulin. Proc Natl Acad Sci U S A (2001) 98(6):3600–5. doi: 10.1073/pnas.051511398
321. Thomas SM, Brugge JS. Cellular Functions Regulated by Src Family Kinases. Annu Rev Cell Dev Biol (1997) 13:513–609. doi: 10.1146/annurev.cellbio.13.1.513
322. Sternfeld L, Anderie I, Schmid A, Al-Shaldi H, Krause E, Magg T, et al. Identification of Tyrosines in the Putative Regulatory Site of the Ca2+ Channel TRPV6. Cell Calcium (2007) 42(1):91–102. doi: 10.1016/j.ceca.2006.11.008
323. Elias D, Vever H, Lænkholm AV, Gjerstorff MF, Yde CW, Lykkesfeldt AE, et al. Gene Expression Profiling Identifies FYN as An Important Molecule in Tamoxifen Resistance and a Predictor of Early Recurrence in Patients Treated With Endocrine Therapy. Oncogene (2015) 34(15):1919–27. doi: 10.1038/onc.2014.138
324. Finn RS. Targeting Src in Breast Cancer. Ann Oncol (2008) 19(8):1379–86. doi: 10.1093/annonc/mdn291
325. Kim SY, Yang D, Myeong J, Ha K, Kim SH, Park EJ, et al. Regulation of Calcium Influx and Signaling Pathway in Cancer Cells Via TRPV6-Numb1 Interaction. Cell Calcium (2013) 53(2):102–11. doi: 10.1016/j.ceca.2012.10.005
326. Maehama T, Dixon JE. The Tumor Suppressor, PTEN/MMAC1, Dephosphorylates the Lipid Second Messenger, Phosphatidylinositol 3,4,5-Trisphosphate. J Biol Chem (1998) 273(22):13375–8. doi: 10.1074/jbc.273.22.13375
327. Kim SY, Hong C, Wie J, Kim E, Kim BJ, Ha K, et al. Reciprocal Positive Regulation Between TRPV6 and NUMB in PTEN-Deficient Prostate Cancer Cells. Biochem Biophys Res Commun (2014) 447(1):192–6. doi: 10.1016/j.bbrc.2014.03.123
328. Rennstam K, McMichael N, Berglund P, Honeth G, Hegardt C, Rydén L, et al. Numb Protein Expression Correlates With a Basal-Like Phenotype and Cancer Stem Cell Markers in Primary Breast Cancer. Breast Cancer Res Treat (2010) 122(2):315–24. doi: 10.1007/s10549-009-0568-x
329. Li S, Shen Y, Wang M, Yang J, Lv M, Li P, et al. Loss of PTEN Expression in Breast Cancer: Association With Clinicopathological Characteristics and Prognosis. Oncotarget (2017) 8(19):32043–54. doi: 10.18632/oncotarget.16761
330. Stumpf T, Zhang Q, Hirnet D, Lewandrowski U, Sickmann A, Wissenbach U, et al. The Human TRPV6 Channel Protein is Associated With Cyclophilin B in Human Placenta. J Biol Chem (2008) 283(26):18086–98. doi: 10.1074/jbc.M801821200
331. Fang F, Flegler AJ, Du P, Lin S, Clevenger CV. Expression of Cyclophilin B is Associated With Malignant Progression and Regulation of Genes Implicated in the Pathogenesis of Breast Cancer. Am J Pathol (2009) 174(1):297–308. doi: 10.2353/ajpath.2009.080753
332. Lyu JH, Park DW, Huang B, Kang SH, Lee SJ, Lee C, et al. RGS2 Suppresses Breast Cancer Cell Growth Via a MCPIP1-dependent Pathway. J Cell Biochem (2015) 116(2):260–7. doi: 10.1002/jcb.24964
333. Schoeber JP, Topala CN, Wang X, Diepens RJ, Lambers TT, Hoenderop JG, et al. RGS2 Inhibits the Epithelial Ca2+ Channel TRPV6. J Biol Chem (2006) 281(40):29669–74. doi: 10.1074/jbc.M606233200
334. Bhuin T, Roy JK. Rab Proteins: The Key Regulators of Intracellular Vesicle Transport. Exp Cell Res (2014) 328(1):1–19. doi: 10.1016/j.yexcr.2014.07.027
335. Li W, Li G, Fan Z, Liu T. Tumor-Suppressive microRNA-452 Inhibits Migration and Invasion of Breast Cancer Cells by Directly Targeting RAB11A. Oncol Lett (2017) 14(2):2559–65. doi: 10.3892/ol.2017.6426
336. Wang B, Yang Z, Wang H, Cao Z, Zhao Y, Gong C, et al. MicroRNA-320a Inhibits Proliferation and Invasion of Breast Cancer Cells by Targeting RAB11A. Am J Cancer Res (2015) 5(9):2719–29.
337. Prakriya M, Lewis RS. Store-Operated Calcium Channels. Physiol Rev (2015) 95(4):1383–436. doi: 10.1152/physrev.00020.2014
338. Parekh AB, Putney JW. Store-Operated Calcium Channels. Physiol Rev (2005) 85(2):757–810. doi: 10.1152/physrev.00057.2003
339. Chen YF, Lin PC, Yeh YM, Chen LH, Shen MR. Store-Operated Ca2+ Entry in Tumor Progression: From Molecular Mechanisms to Clinical Implications. Cancers (Basel) (2019) 11(7). doi: 10.3390/cancers11070899
340. Jardin I, Lopez JJ, Salido GM, Rosado JA. Store-Operated Ca 2+ Entry in Breast Cancer Cells: Remodeling and Functional Role. Int J Mol Sci (2018) 19(12). doi: 10.3390/ijms19124053
341. Mo P, Yang S. The Store-Operated Calcium Channels in Cancer Metastasis: From Cell Migration, Invasion to Metastatic Colonization. Front Biosci (Landmark Ed) (2018) 23:1241–56. doi: 10.2741/4641
342. Yang S, Zhang JJ, Huang XY. Orai1 and STIM1 are Critical for Breast Tumor Cell Migration and Metastasis. Cancer Cell (2009) 15(2):124–34. doi: 10.1016/j.ccr.2008.12.019
343. McAndrew D, Grice DM, Peters AA, Davis FM, Stewart T, Rice M, et al. ORAI1-Mediated Calcium Influx in Lactation and in Breast Cancer. Mol Cancer Ther (2011) 10(3):448–60. doi: 10.1158/1535-7163.MCT-10-0923
344. Motiani RK, Zhang X, Harmon KE, Keller RS, Matrougui K, Bennett JA, et al. Orai3 is an Estrogen Receptor α-Regulated Ca²⁺ Channel That Promotes Tumorigenesis. FASEB J (2013) 27(1):63–75. doi: 10.1096/fj.12-213801
345. Yang Y, Jiang Z, Wang B, Chang L, Liu J, Zhang L, et al. Expression of STIM1 is Associated With Tumor Aggressiveness and Poor Prognosis in Breast Cancer. Pathol Res Pract (2017) 213(9):1043–7. doi: 10.1016/j.prp.2017.08.006
346. Azimi I, Milevskiy MJG, Chalmers SB, Yapa KTDS, Robitaille M, Henry C, et al. ORAI1 and ORAI3 in Breast Cancer Molecular Subtypes and the Identification of ORAI3 as a Hypoxia Sensitive Gene and a Regulator of Hypoxia Responses. Cancers (Basel) (2019) 11(2). doi: 10.3390/cancers11020208
347. Jardin I, Rosado JA. STIM and Calcium Channel Complexes in Cancer. Biochim Biophys Acta (2016) 1863(6 Pt B):1418–26. doi: 10.1016/j.bbamcr.2015.10.003
348. Woodard GE, López JJ, Jardín I, Salido GM, Rosado JA. TRPC3 Regulates Agonist-Stimulated Ca2+ Mobilization by Mediating the Interaction Between Type I Inositol 1,4,5-Trisphosphate Receptor, RACK1, and Orai1. J Biol Chem (2010) 285(11):8045–53. doi: 10.1074/jbc.M109.033605
349. Liao Y, Erxleben C, Yildirim E, Abramowitz J, Armstrong DL, Birnbaumer L. Orai Proteins Interact With TRPC Channels and Confer Responsiveness to Store Depletion. Proc Natl Acad Sci U S A (2007) 104(11):4682–7. doi: 10.1073/pnas.0611692104
350. Liao Y, Erxleben C, Abramowitz J, Flockerzi V, Zhu MX, Armstrong DL, et al. Functional Interactions Among Orai1, Trpcs, and STIM1 Suggest a STIM-regulated Heteromeric Orai/TRPC Model for SOCE/Icrac Channels. Proc Natl Acad Sci USA (2008) 105(8):2895–900. doi: 10.1073/pnas.0712288105
351. Sukumaran P, Sun Y, Vyas M, Singh BB. TRPC1-Mediated Ca²⁺ Entry is Essential for the Regulation of Hypoxia and Nutrient Depletion-Dependent Autophagy. Cell Death Dis (2015) 6:e1674. doi: 10.1038/cddis.2015.7
352. Villalobos C, Hernández-Morales M, Gutiérrez LG, Núñez L. TRPC1 and ORAI1 Channels in Colon Cancer. Cell Calcium (2019) 81:59–66. doi: 10.1016/j.ceca.2019.06.003
353. Albarrán L, Lopez JJ, Dionisio N, Smani T, Salido GM, Rosado JA. Transient Receptor Potential Ankyrin-1 (TRPA1) Modulates Store-Operated Ca(2+) Entry by Regulation of STIM1-Orai1 Association. Biochim Biophys Acta (2013) 1833(12):3025–34. doi: 10.1016/j.bbamcr.2013.08.014
354. Bastián-Eugenio CE, Bohórquez-Hernández A, Pacheco J, Sampieri A, Asanov A, Ocelotl-Oviedo JP, et al. Heterologous Calcium-Dependent Inactivation of Orai1 by Neighboring TRPV1 Channels Modulates Cell Migration and Wound Healing. Commun Biol (2019) 2:88. doi: 10.1038/s42003-019-0338-1
355. Antigny F, Sabourin J, Saüc S, Bernheim L, Koenig S, Frieden M. TRPC1 and TRPC4 Channels Functionally Interact With STIM1L to Promote Myogenesis and Maintain Fast Repetitive Ca. Biochim Biophys Acta Mol Cell Res (2017) 1864(5):806–13. doi: 10.1016/j.bbamcr.2017.02.003
356. Dyrda A, Koenig S, Frieden M. STIM1 Long and STIM1 Gate Differently TRPC1 During Store-Operated Calcium Entry. Cell Calcium (2020) 86:102134. doi: 10.1016/j.ceca.2019.102134
357. Albarrán L, López JJ, Gómez LJ, Salido GM, Rosado JA. SARAF Modulates TRPC1, But Not TRPC6, Channel Function in a STIM1-independent Manner. Biochem J (2016) 473(20):3581–95. doi: 10.1042/BCJ20160348
358. Liu X, Yao X, Tsang SY. Post-Translational Modification and Natural Mutation of TRPC Channels. Cells (2020) 9(1). doi: 10.3390/cells9010135
359. Voolstra O, Huber A. Post-Translational Modifications of TRP Channels. Cells (2014) 3(2):258–87. doi: 10.3390/cells3020258
360. Srikanth S, Ribalet B, Gwack Y. Regulation of CRAC Channels by Protein Interactions and Post-Translational Modification. Channels (Austin) (2013) 7(5):354–63. doi: 10.4161/chan.23801
361. Han ZJ, Feng YH, Gu BH, Li YM, Chen H. The Post-Translational Modification, SUMOylation, and Cancer (Review). Int J Oncol (2018) 52(4):1081–94. doi: 10.3892/ijo.2018.4280
362. Chen L, Liu S, Tao Y. Regulating Tumor Suppressor Genes: Post-Translational Modifications. Signal Transduct Target Ther (2020) 5(1):90. doi: 10.1038/s41392-020-0196-9
363. Heo KS. Regulation of Post-Translational Modification in Breast Cancer Treatment. BMB Rep (2019) 52(2):113–8. doi: 10.5483/BMBRep.2019.52.2.017
364. Waks AG, Winer EP. Breast Cancer Treatment: A Review. JAMA (2019) 321(3):288–300. doi: 10.1001/jama.2018.19323
365. Schirrmacher V. From Chemotherapy to Biological Therapy: A Review of Novel Concepts to Reduce the Side Effects of Systemic Cancer Treatment (Review). Int J Oncol (2019) 54(2):407–19. doi: 10.3892/ijo.2018.4661
366. Nurgali K, Jagoe RT, Abalo R. Editorial: Adverse Effects of Cancer Chemotherapy: Anything New to Improve Tolerance and Reduce Sequelae? Front Pharmacol (2018) 9:245. doi: 10.3389/fphar.2018.00245
367. Bianchini G, Balko JM, Mayer IA, Sanders ME, Gianni L. Triple-Negative Breast Cancer: Challenges and Opportunities of a Heterogeneous Disease. Nat Rev Clin Oncol (2016) 13(11):674–90. doi: 10.1038/nrclinonc.2016.66
368. Calejo AI, Taskén K. Targeting Protein-Protein Interactions in Complexes Organized by A Kinase Anchoring Proteins. Front Pharmacol (2015) 6:192. doi: 10.3389/fphar.2015.00192
369. Petta I, Lievens S, Libert C, Tavernier J, De Bosscher K. Modulation of Protein-Protein Interactions for the Development of Novel Therapeutics. Mol Ther (2016) 24(4):707–18. doi: 10.1038/mt.2015.214
370. Scott DE, Bayly AR, Abell C, Skidmore J. Small Molecules, Big Targets: Drug Discovery Faces the Protein-Protein Interaction Challenge. Nat Rev Drug Discovery (2016) 15(8):533–50. doi: 10.1038/nrd.2016.29
371. Ruffalo M, Bar-Joseph Z. Protein Interaction Disruption in Cancer. BMC Cancer (2019) 19(1):1–10. doi: 10.1186/s12885-019-5532-5
372. Cheng SS, Yang GJ, Wang W, Leung CH, Ma DL. The Design and Development of Covalent Protein-Protein Interaction Inhibitors for Cancer Treatment. J Hematol Oncol (2020) 13(1):26. doi: 10.1186/s13045-020-00850-0
373. Mabonga L, Kappo AP. Protein-Protein Interaction Modulators: Advances, Successes and Remaining Challenges. Biophys Rev (2019) 11(4):559–81. doi: 10.1007/s12551-019-00570-x
374. Fuller JC, Burgoyne NJ, Jackson RM. Predicting Druggable Binding Sites At the Protein-Protein Interface. Drug Discovery Today (2009) 14(3-4):155–61. doi: 10.1016/j.drudis.2008.10.009
375. Xie J, Bi Y, Zhang H, Dong S, Teng L, Lee RJ, et al. Cell-Penetrating Peptides in Diagnosis and Treatment of Human Diseases: From Preclinical Research to Clinical Application. Front Pharmacol (2020) 11(May):1–23. doi: 10.3389/fphar.2020.00697
376. Tripathi PP, Arami H, Banga I, Gupta J, Gandhi S. Cell Penetrating Peptides in Preclinical and Clinical Cancer Diagnosis and Therapy. Oncotarget (2018) 9(98):37252–67. doi: 10.18632/oncotarget.26442
377. Habault J, Poyet JL. Recent Advances in Cell Penetrating Peptide-Based Anticancer Therapies. Molecules (2019) 24(5). doi: 10.3390/molecules24050927
378. Regberg J, Srimanee A, Langel U. Applications of Cell-Penetrating Peptides for Tumor Targeting and Future Cancer Therapies. Pharmaceuticals (Basel) (2012) 5(9):991–1007. doi: 10.3390/ph5090991
379. Jagot-Lacoussiere L, Kotula E, Villoutreix BO, Bruzzoni-Giovanelli H. Poyet Jl. A Cell-Penetrating Peptide Targeting AAC-11 Specifically Induces Cancer Cells Death. Cancer Res (2016) 76(18):5479–90. doi: 10.1158/0008-5472.CAN-16-0302
380. Kang RH, Jang JE, Huh E, Kang SJ, Ahn DR, Kang JS, et al. A Brain Tumor-Homing Tetra-Peptide Delivers a Nano-Therapeutic for More Effective Treatment of a Mouse Model of Glioblastoma. Nanoscale Horiz (2020) 5(8):1213–25. doi: 10.1039/D0NH00077A
381. Zhou N, Wu J, Qin YY, Zhao XL, Ding Y, Sun LS, et al. Novel Peptide MT23 for Potent Penetrating and Selective Targeting in Mouse Melanoma Cancer Cells. Eur J Pharm Biopharm (2017) 120:80–8. doi: 10.1016/j.ejpb.2017.08.011
382. Cheng CJ, Bahal R, Babar IA, Pincus Z, Barrera F, Liu C, et al. MicroRNA Silencing for Cancer Therapy Targeted to the Tumour Microenvironment. Nature (2015) 518(7537):107–10. doi: 10.1038/nature13905
383. Olson ES, Aguilera TA, Jiang T, Ellies LG, Nguyen QT, Wong EH, et al. In Vivo Characterization of Activatable Cell Penetrating Peptides for Targeting Protease Activity in Cancer. Integr Biol (Camb) (2009) 1(5-6):382–93. doi: 10.1039/b904890a
384. Karagiota A, Kourti M, Simos G, Mylonis I. Hif-1α-Derived Cell-Penetrating Peptides Inhibit ERK-dependent Activation of HIF-1 and Trigger Apoptosis of Cancer Cells Under Hypoxia. Cell Mol Life Sci (2019) 76(4):809–25. doi: 10.1007/s00018-018-2985-7
385. Cortes J, O’Shaughnessy J, Loesch D, Blum JL, Vahdat LT, Petrakova K, et al. Eribulin Monotherapy Versus Treatment of Physician’s Choice in Patients With Metastatic Breast Cancer (EMBRACE): A Phase 3 Open-Label Randomised Study. Lancet (2011) 377(9769):914–23. doi: 10.1016/S0140-6736(11)60070-6
386. Doherty MK, Morris PG. Eribulin for the Treatment of Metastatic Breast Cancer: An Update on its Safety and Efficacy. Int J Women’s Health (2015) 7:47–58. doi: 10.2147/IJWH.S74462
387. Kingston DGI. Tubulin-Interactive Natural Products as Anticancer Agents. J Natural Products (2009) 72(3):507–15. doi: 10.1021/np800568j
388. Li J, Ren J, Sun W. Systematic Review of Ixabepilone for Treating Metastatic Breast Cancer. Breast Cancer (2017) 24(2):171–9. doi: 10.1007/s12282-016-0717-0
389. Bardia A, Parton M, Kummel S, Estevez LG, Huang CS, Cortes J, et al. Paclitaxel With Inhibitor of Apoptosis Antagonist, LCL161, for Localized Triple-Negative Breast Cancer, Prospectively Stratified by Gene Signature in a Biomarker-Driven Neoadjuvant Trial. J Clin Oncol (2018) 36(31):3126–33. doi: 10.1200/JCO.2017.74.8392
390. Lalaoui N, Merino D, Giner G, Vaillant F, Chau D, Liu L, et al. Targeting Triple-Negative Breast Cancers With the Smac-mimetic Birinapant. Cell Death Differentiation (2020) 27:2768–80. doi: 10.1038/s41418-020-0541-0
391. Corbi-Verge C, Kim PM. Motif Mediated Protein-Protein Interactions as Drug Targets. Cell Commun Signaling (2016) 14(1):1–12. doi: 10.1186/s12964-016-0131-4
392. Schulien AJ, Yeh CY, Orange BN, Pav OJ, Hopkins MP, Moutal A, et al. Targeted Disruption of Kv2.1-VAPA Association Provides Neuroprotection Against Ischemic Stroke in Mice by Declustering Kv2.1 Channels. Sci Adv (2020) 6(27). doi: 10.1126/sciadv.aaz8110
393. Brittain JM, Duarte DB, Wilson SM, Zhu W, Ballard C, Johnson PL, et al. Suppression of Inflammatory and Neuropathic Pain by Uncoupling CRMP-2 From the Presynaptic Ca²⁺ Channel Complex. Nat Med (2011) 17(7):822–9. doi: 10.1038/nm.2345
394. Fischer A, Rosen AC, Ensslin CJ, Wu S, Lacouture ME. Pruritus to Anticancer Agents Targeting the EGFR, BRAF, and CTLA-4. Dermatol Ther (2013) 26(2):135–48. doi: 10.1111/dth.12027
395. Fischer MJ, Btesh J, McNaughton PA. Disrupting Sensitization of Transient Receptor Potential Vanilloid Subtype 1 Inhibits Inflammatory Hyperalgesia. J Neurosci (2013) 33(17):7407–14. doi: 10.1523/JNEUROSCI.3721-12.2013
396. Fosgerau K, Hoffman T. Peptide Therapeutics: Current Status and Future Directions. Drug Discovery Today (2015) 20(1):122–8. doi: 10.1016/j.drudis.2014.10.003
397. Lau JL, Dunn MK. Therapeutic Peptides: Historical Perspectives, Current Development Trends, and Future Directions. Bioorg Med Chem (2018) 26(10):2700–7. doi: 10.1016/j.bmc.2017.06.052
398. Nero TL, Morton CJ, Holien JK, Wielens J, Parker MW. Oncogenic Protein Interfaces: Small Molecules, Big Challenges. Nat Rev Cancer (2014) 14(4):248–62. doi: 10.1038/nrc3690
399. Rech AJ, Mick R, Martin S, Recio A, Aqui NA, Powell DJ Jr, et al. CD25 Blockade Depletes and Selectively Reprograms Regulatory T Cells in Concert With Immunotherapy in Cancer Patients. Sci Trans Med (2012) 4(134). doi: 10.1126/scitranslmed.3003330
400. Yang J, Hu L. Immunomodulators Targeting the PD-1/PD-L1 Protein-Protein Interaction: From Antibodies to Small Molecules. Med Res Rev (2019) 39(1):265–301. doi: 10.1002/med.21530
401. Schmid P, Adams S, Rugo HS, Schneeweiss A, Barrios CH, Iwata H, et al. Atezolizumab and Nab-Paclitaxel in Advanced Triple-Negative Breast Cancer. New Engl J Med (2018) 379(22):2108–21. doi: 10.1056/NEJMoa1809615
402. McArthur HL, Diab A, Page DB, Yuan J, Solomon SB, Sacchini V, et al. A Pilot Study of Preoperative Single-Dose Ipilimumab and/or Cryoablation in Women With Early-Stage Breast Cancer With Comprehensive Immune Profiling. Clin Cancer Res (2016) 22(23):5729–37. doi: 10.1158/1078-0432.CCR-16-0190
403. Jovčevska I, Muyldermans S. The Therapeutic Potential of Nanobodies. BioDrugs (2020) 34(1):11–26. doi: 10.1007/s40259-019-00392-z
404. Yang EY, Shah K. Nanobodies: Next Generation of Cancer Diagnostics and Therapeutics. Front Oncol (2020) 10:1182. doi: 10.3389/fonc.2020.01182
405. Arezumand R, Alibakhshi A, Ranjbari J, Ramazani A, Muyldermans S. Nanobodies As Novel Agents for Targeting Angiogenesis in Solid Cancers. Front Immunol (2017) 8:1746. doi: 10.3389/fimmu.2017.01746
406. Schumacher D, Helma J, Schneider AFL, Leonhardt H, Hackenberger CPR. Nanobodies: Chemical Functionalization Strategies and Intracellular Applications. Angew Chem Int Ed Engl (2018) 57(9):2314–33. doi: 10.1002/anie.201708459
407. Herce HD, Schumacher D, Schneider AFL, Ludwig AK, Mann FA, Fillies M, et al. Cell-Permeable Nanobodies for Targeted Immunolabelling and Antigen Manipulation in Living Cells. Nat Chem (2017) 9(8):762–71. doi: 10.1038/nchem.2811
408. Mabonga L, Kappo AP. Peptidomimetics: A Synthetic Tool for Inhibiting Protein–Protein Interactions in Cancer. Int J Pept Res Ther (2020) 26(1):225–41. doi: 10.1007/s10989-019-09831-5
409. Borrelli A, Tornesello AL, Tornesello ML, Buonaguro FM. Cell Penetrating Peptides as Molecular Carriers for Anti-Cancer Agents. Molecules (2018) 23(2). doi: 10.3390/molecules23020295
410. Jamnani FR, Rahbarizadeh F, Shokrgozar MA, Ahmadvand D, Mahboudi F, Sharifzadeh Z. Targeting High Affinity and Epitope-Distinct Oligoclonal Nanobodies to HER2 Over-Expressing Tumor Cells. Exp Cell Res (2012) 318(10):1112–24. doi: 10.1016/j.yexcr.2012.03.004
411. Kijanka M, Dorresteijn B, Oliveira S, van Bergen en Henegouwen PM. Nanobody-Based Cancer Therapy of Solid Tumors. Nanomedicine (2015) 10(1):161–74. doi: 10.2217/nnm.14.178
412. Bojadzic D, Buchwald P. Toward Small-Molecule Inhibition of Protein–Protein Interactions: General Aspects and Recent Progress in Targeting Costimulatory and Coinhibitory (Immune Checkpoint) Interactions. Curr Topics Med Chem (2018) 18(8):674–99. doi: 10.2174/1568026618666180531092503
Keywords: TRP channels, protein–protein interactions, calcium signaling, breast cancer, interactomics
Citation: Saldías MP, Maureira D, Orellana-Serradell O, Silva I, Lavanderos B, Cruz P, Torres C, Cáceres M and Cerda O (2021) TRP Channels Interactome as a Novel Therapeutic Target in Breast Cancer. Front. Oncol. 11:621614. doi: 10.3389/fonc.2021.621614
Received: 26 October 2020; Accepted: 31 March 2021;
Published: 10 June 2021.
Edited by:
Zhijie Jason Liu, The University of Texas Health Science Center at San Antonio, United StatesReviewed by:
Jorge Morales-Montor, National Autonomous University of Mexico, MexicoRenzo Luciano Boldorini, Università degli Studi del Piemonte Orientale, Italy
Copyright © 2021 Saldías, Maureira, Orellana-Serradell, Silva, Lavanderos, Cruz, Torres, Cáceres and Cerda. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Oscar Cerda, b3NjYXJjZXJkYUB1Y2hpbGUuY2w=
†These authors have contributed equally to this work