 Zhiqin LiSigrid A. Langhans*
Zhiqin LiSigrid A. Langhans*- Nemours Biomedical Research, Alfred I. duPont Hospital for Children, Wilmington, DE, United States
After leukemia, tumors of the brain and spine are the second most common form of cancer in children. Despite advances in treatment, brain tumors remain a leading cause of death in pediatric cancer patients and survivors often suffer from life-long consequences of side effects of therapy. The 5-year survival rates, however, vary widely by tumor type, ranging from over 90% in more benign tumors to as low as 20% in the most aggressive forms such as glioblastoma. Even within historically defined tumor types such as medulloblastoma, molecular analysis identified biologically heterogeneous subgroups each with different genetic alterations, age of onset and prognosis. Besides molecularly driven patient stratification to tailor disease risk to therapy intensity, such a diversity demonstrates the need for more precise and disease-relevant pediatric brain cancer models for research and drug development. Here we give an overview of currently available in vitro and in vivo pediatric brain tumor models and discuss the opportunities that new technologies such as 3D cultures and organoids that can bridge limitations posed by the simplicity of monolayer cultures and the complexity of in vivo models, bring to accommodate better precision in drug development for pediatric brain tumors.
Introduction
Brain tumors are the most common solid tumors and the leading cause of cancer-related death in children. The incidence and mortality rate of primary brain and other central nervous system tumors have not changed significantly in recent years, with an average incidence rate of 5.65 per 100,000 population and an average mortality rate of 0.72 per 100,000 population for the 0 to 14 years age group from 2011 to 2015 in the United States (1). In the past, the diagnosis and classification of brain tumors had largely relied on histological characteristics derived from hematoxylin and eosin-staining, and immunohistochemical detection of lineage-associated proteins. However, more and more evidence shows that histologically similar brain tumors sometimes have distinct molecular features; they respond differently to the treatment and have various prognosis as well. In addition, some histologically ambiguous tumors may largely rely on their molecular characterization for their diagnosis and treatment plan. In 2016, the World Health Organization (WHO) updated classification of central nervous system tumors by incorporating molecular features into traditional histological characteristics for more accurate diagnosis, prognosis predictions, and treatments (2–6). With the overall success rate of new anticancer drugs remaining low (7, 8), we will need to switch from “one size fits all” treatments to more specific individualized strategies, to increase treatment efficacy, to reduce complications due to treatment, and to improve the translation rate of anti-cancer drugs. Brain tumor models that can mimic tumor initiation and progression, and predict a tumor’s response to treatments in vivo are fundamental to achieve this goal. In this review, we provide an overview of the most common pediatric brain tumors and currently available well-annotated in vitro and in vivo tumor models. We also discuss the advantages and limitations of each model, which need to be considered when choosing an appropriate tumor model that best suits the experimental purpose.
Common Pediatric Brain Tumors and Molecular Subgrouping
Gliomas
Glioma is the most common pediatric primary brain tumor, representing approximately 47% of brain tumor cases in the age group of 0-19 years. Glioma can originate from all glia cell types and 75% of these glial tumors are astrocytoma (1). Glioma are highly heterogeneous tumors, ranging from low-grade glioma (LGG) to high-grade glioma (HGG) depending on the tumor malignant status.
LGG is the most common glioma, which is typically nonmalignant and slow growing. Histologically, LGGs include pilocytic astrocytoma (PA), pilomyxoid astrocytoma (PMA), oligoastrocytoma, subependymal giant cell astrocytoma (SEGA), pleomorphic xanthoastrocytomas (PXA), oligodendroglioma, ganglioglioma, dysembryoplastic neuroepithelial tumors, etc., among which pilocytic astrocytoma is the most common form. The aberrant Ras-mitogen activated protein kinase (MAPK) signaling pathway is mainly reported in LGG. The mutations usually occur at BRAF in this pathway, including the KIAA1549-BRAF fusion and BRAF V600E mutant, which lead to constitutive activation of the MAPK pathway. Furthermore, kRAS, FGFR1, MYB/MYBL1, NTRK2, NF1, TSC1/2 and other genetic alteration have also been identified in pediatric LGG. Unlike adult LGG, IDH mutations are almost absent in children (3–6, 9–11). In some cases, the molecular alteration is associated with a specific tumor type. For example, KIAA1549- BRAF fusion is mostly found in pilocytic astrocytoma (PA), while BRAF V600E is frequently detected in pleomorphic xanthoastrocytoma (PMA) and gangliogliomas (12).
HGG is relatively uncommon in pediatric glioma, accounting for around 20% of cases. However, HGGs are diffusely infiltrating malignant tumors and they are usually aggressive with an overall very poor prognosis; some patients succumb to the tumor within one year after diagnosis. Based on distinct histological and radiological features, HGG is subclassified into anaplastic astrocytoma, diffuse intrinsic pontine glioma (DIPG) and glioblastoma multiforme (GBM). Mutations in histone genes were first discovered in pediatric HGGs, and now serve as a hallmark of this glioma type. Histone mutations often vary according to HGG locations. In tumors arising from the midline and pons, K27M mutations in H3F3A (encoding histone H3.3) or HIST1H3B/C (encoding histone H3.1) are very common, which lead to a global decrease of H3 K27 trimethylation by inhibiting polycomb repressive complex 2 (PRC2) activity through sequestration of its catalytic subunit EZH2; while G34R (or rarely G34V) mutations in H3F3A (encoding histone H3.3) are mostly reported in hemispheric HGGs. In addition, the RTK/RAS/PI3K pathway (e.g., PDGFRA, PIK3CA, PIK3R1, or PTEN) and the p53/Rb pathway (e.g., TP53, CDKN2A, CDK4/6, CCND1-3) are also dysregulated in pediatric HGG (3, 4, 9–11, 13–15). Recent studies discovered some overlap in molecular profiling between LGG and HGG. BRAF V600E and FGFR1 mutations are found both in LGG and HGG (9, 10), which suggests that LGG and HGG might share a similar biological mechanism of tumor pathogenesis.
Ependymal Tumors
Ependymomas represent 5.5% of all pediatric primary brain tumor cases in the age group of 0 to 14 (1). Ependymomas are thought to originate from radial glia cells of the ependymal lining of the ventricles and the central canal. Histologically, ependymomas are classified into 4 groups: subependymoma, myxopapillary ependymoma, classic ependymoma, and anaplastic ependymoma, of which classic and anaplastic ependymoma are the most common subtypes in children. Classic ependymoma is further subclassified into 3 subtypes: papillary, clear cell, and tanycytic ependymoma based on their histological features (4, 5).
The molecular characteristics of ependymoma is usually associated with its location. Over 90% of pediatric ependymomas arise in the infratentorial and supratentorial regions. The infratentorial posterior fossa (PF) ependymomas are generally subclassified into Group A (PF-EPN-A) and Group B (PF-EPN-B) based on their DNA methylation profiling. PF-EPN-A tumors are hypermethylated, and mostly found in infants and young children, who have a poorer outcome compared to those with PF-EPN-B tumors, which are typically seen in adolescents and adults. Supratentorial (ST) ependymomas in children have two major subgroups: RELA fusion-positive (ST-EPN-RELA) ependymoma and YAP1 fusion-positive (ST-EPN-YAP1) ependymoma. ST-EPN-RELA usually harbors the fusion protein of C11orf95 and RELA, which constitutively activates the NF-κB pathway by enriching a RELA-encoded transcription factor p65. In ST-EPN-YAP1 ependymoma, transcriptional coactivator YAP1 fuses with other genes such as MAMLD1 and FAM118B and can upregulate Notch signaling. Compared to ST-EPN-YAP1, ST-EPN-RELA is more frequently observed in children and has worse prognosis. The major treatment plan for ependymoma is surgical resection plus adjuvant radiological therapies. Benefits of chemotherapy have not been reported, yet (3–6, 16–18).
Medulloblastoma
Medulloblastoma is the most common pediatric embryonal tumor originating from precursor cells in the cerebellum or dorsal brainstem. Like other embryonal tumors, medulloblastoma is highly proliferative and predisposed to metastasis. Histologically, medulloblastoma is classified into four different types: classic, desmoplastic/nodular, extensive nodularity and large cell/anaplastic. Medulloblastoma is one of the most heterogeneous brain tumors and currently has the best characterized molecular features. There are four distinct subgroups: wingless/integrated (WNT), sonic hedgehog (SHH), Group 3, and Group 4, which have different genetic alterations, phenotypes and prognostics.
The WNT subgroup accounts for around 10% of medulloblastoma; it mostly occurs in older children. The most common mutation in this subgroup is in the CTNNB1 gene, which encodes β-catenin, a major player in cell cycle control and embryogenesis. The overexpression of nuclear β-catenin is often used as a diagnostic indicator in this subgroup. Monosomy chromosome 6 is another hallmark of the WNT subgroup, occurring in ~80-85% of patients, usually in conjunction with CTNNB1 mutations. DDX3X, SMARCA4, and TP53 mutations also have been reported in WNT-activated medulloblastomas (3–6, 19).
The SHH subgroup represents approximately 30% of medulloblastoma. SHH-activated medulloblastoma is highly heterogeneous and many key molecules in the SHH signaling pathway such as SUFU, smoothened (SMO), PTCH1, GLI1 and GLI2 have been dysregulated in this subgroup. Besides those, other genetic aberrations, like MYCN amplification or TP53 mutation, are also involved in SHH-activated medulloblastoma formation (19, 20). The outcome varies in SHH-activated medulloblastoma and although metastasis is not common, if a patient has a metastatic tumor, the outcome is usually worse. Moreover, patients with TP53 mutations or MYCN amplification usually have a poorer prognosis. The ongoing therapies using small molecule inhibitors target almost all affected molecules in the SHH pathway (3–6, 13, 19).
Group 3 composes around 25% of medulloblastoma. It is the most aggressive form and metastasis is very common in this subgroup. Unlike WNT- and SHH-activated medulloblastoma, the Group 3 tumors are less defined; some studies showed MYC amplification leading to tumor formation in this subgroup. Other possible pathways, such as TNFβ, have been found in around 20% of Group 3 medulloblastoma. The prognosis is overall poor for this subgroup, especially for patients with MYC amplification (4–6).
Group 4 is the most prevalent subgroup, comprising approximately 35% of medulloblastoma. Like Group 3, it has not been biologically characterized. The loss of chromosome 8, 11 and 17p or gain of chromosome 7 and 17q have been identified in this subgroup. In addition, amplification of CDK6, MYCN and SNCAP1 as well as aberrant ERBB4-SRC signaling and nuclear factor kappa B (NF-κB) have also been observed in Group 4 medulloblastoma (3–6, 19, 21, 22).
In Vivo Brain Tumor Models
Most animals rarely develop spontaneous brain tumors (23) and they can be used to generate experimental models for brain tumor studies (24). A good animal model should have high incidence rate, can recapitulate original tumor’s histopathological and molecular features, and can manifest the human response to drug treatment. Numerous animal brain tumor models have been developed so far. These models can be used to investigate biological mechanisms of brain tumors and their microenvironment and for preclinical testing of novel, promising therapeutic regimens. To date, most animal models are generated with rodents, and in this review we will focus on rat and mouse models of brain tumors and discuss zebrafish brain tumor models. There are three major methods to generate animal models in brain tumor research: carcinogen induced animal models, xenograft animal models and genetically engineered animal models (23–26).
Carcinogen-Induced Brain Tumor Models
Rats are widely used when generating a carcinogen-induced brain tumor model, since the tumor induction in rat strains is much more effective than in mice (23). The most common carcinogens used to generate animal brain tumors are chemical carcinogens and viruses. N-nitrosourea and its derivatives have been reported to induce most common gliomas in rats, including astrocytoma, oligodendroglioma, and ependymal tumors. The embryos are much more susceptible to the chemical carcinogens, and transplacental injection is often used to administer the chemical compounds to pregnant animals (24). Injection of ethylnitrosourea to pregnant rats at gestational day 20 induced brain tumors in all of 25 pups born (27). Chemical carcinogens can also be applied to rodents through oral, intravenous or local exposure after they are born, but repeated administration might be necessary to increase induction efficacy, especially when working with older animals (23–26). The cell lines established from these chemical induced glioma models include C6, 9L, T9, F98, RG2, BT4C and CNS-1 and have been widely used in brain tumor studies (28–31). In addition to chemical carcinogens, oncogenic viruses may also be used to induce brain tumors. Both RNA viruses, such as Rous sarcoma virus-1 (RSV-1) and DNA viruses, such as adenovirus can induce brain tumors. Intracerebral injection of RSV caused malignant brain tumors in newborn pups (32). Different injection sites caused distinct tumor types (33). It has also been reported that injection of human adenovirus 12 virus (AD12) into mouse brain induced medulloblastoma or glioblastoma (34). These chemical carcinogens and oncogenic viruses are prevalent in the human environment; thus, this model can imitate natural tumorigenesis especially when animals are exposed in early development. The induced tumors can be continuously passed in animals and retain relatively stable biological characteristics. However, carcinogen induced tumor models lack consistency in tumor types, locations and biological characteristics. Moreover, the induced brain tumors are histologically and biologically different from human tumors.
Xenograft Models of Human Brain Tumors
Xenograft models are usually made by transplanting established cancer cell lines or brain tumor tissues derived from patients (patient-derived xenograft, PDX) or animal models into host animals. The established cancer cell lines grow very fast in vitro with well-defined biological characteristics, which makes them applicable to generate xenograft models. The cell lines generated from carcinogen-induced rodent tumors or from transgenic mice can be cultured and transplanted into syngeneic hosts with competent immune system (35). However, the cancer cell lines are a homogenous population lacking tumor heterogeneity and the induced tumors are rarely infiltrative. Moreover, cancer cell lines will gradually lose the original tumor phenotypes and genetic features during in vitro culture. Patient tissues can also be dissociated and cultured in neurobasal serum-free medium. This selects highly tumorigenic subpopulations with stem cell-like characteristics that can be grown as neurospheres before implantation into host mice (23, 24, 35, 36). In addition, the tumor tissues derived from patients can be directly transplanted into recipient animals without in vitro culture. Engraftments grown in these animals include tumor tissues as well as their surrounding stroma in early passage. They retain histological and molecular characteristic of original tumors, interaction between tumor and host, and a tumor’s responses to drug treatment. With this they are a more representative and reliable in vivo brain tumor model than those generated from cultured cells (23, 25).
In most cases to generate PDX, host animals are immunodeficient mice. The early xenografts were transplanted into nude mice, which are the first generation of immunodeficient mice. Nude mice not only lack body fur but also have no thymus. Thus, these mice have a defective adaptive immune response as they do not have T lymphocytes. Nevertheless, they still have functional B and NK cells, and an intact innate immune response causes a low engraftment rate in these mice. Later, the severe combined immunodeficient (SCID) mice that lack both functional T and B lymphocytes were generated. The engraftment efficacy has improved on SCID mice, but these mice still have remnant NK cells, hindering the engraftment rate. To eliminate the effect of NK cells, SCID mice were crossbred with Beige mice to establish SCID/Beige mice that have severely impaired NK cells and macrophages, and no mature T and B lymphocytes. SCID/Beige mice display a better engraftment rate, leading to more feasible PDX models. Since then, more immunodeficient mice strains have been established to improve engraftment and increase the success rate of PDX, such as non-obese diabetic (NOD)/SCID mice and its derivative mice (NOG, NSG and NOJ), and BALB/c background immunocompromised mice (BRG and BRJ) (37, 38). Different immunocompromised mouse strains have various sensitivity to chemotherapy or radiation, which needs to be considered when choosing an appropriate animal model. For example, BALB/c mice are very sensitive to radiation and SCID mice are sensitive to γ-irradiation and thus are not useful for radiotherapy related studies (39). Immunodeficient mice can also be modified by receiving human bone marrow to reconstitute a human immune response. These humanized mice provide an opportunity to even more closely recapitulate human brain tumors, to study the effect of the immune system on brain tumor pathogenesis, and to evaluate immunotherapies (24, 38, 39).
Xenografts can be administrated in two different ways: heterotopic xenograft and orthotopic xenograft. Heterotopic xenografts, which most typically are achieved through subcutaneous injection, are a popular method in cancer research. They are simple and convenient to observe and monitor tumors and to evaluate drug efficacies by measuring the tumor volume. However, the microenvironment of tumors induced in this way is different from the original tumor and it cannot faithfully recapitulate the original tumor initiation and progression. In addition, there is no blood brain barrier around these subcutaneous tumors, so this model cannot accurately reflect the anti-cancer drug efficacies. Orthotopic xenografts usually apply tumor cells/or tissues to the location where the original tumor is found in patients. Orthotopic xenografts can better mimic the original tumor pathogenesis, retain histological and molecular characteristics of original tumors as well as tumor host interactions (23, 25, 38, 39). However, even orthotopic xenografts may not completely maintain the histological characteristics of human tumors. Some intracranial glioblastoma xenograft models lack necrotic features and fail to show endothelial proliferation (40). To date, most available pediatric brain tumor PDX models represent glioblastoma, diffuse midline glioma, ependymoma, and medulloblastoma. The establishment of PDX models for less aggressive brain tumors, such as pilocytic astrocytoma, has been less successful due to a very low tumor engraftment rate (39).
The development of pediatric brain tumor PDX models emerged over thirty years ago (24, 38). In recent years, with the raised interest in some pediatric brain tumor types and increased availability of tumor tissues, more and more PDX models have been generated. In 2018, a brain tumor biology study sponsored by the Children’s Oncology Group led to generation of 30 orthotopic pediatric brain tumor PDX models, including medulloblastoma, high grade glioma and ependymoma. These PDX models are valuable tools to investigate subtype specific pediatric brain tumors, since they preserve the original tumors’ histological and molecular features and remain relatively stable when being passaged in mice (41). The scientists from St. Jude Children’s Research Hospital also successfully generated 37 novel orthotopic PDX models derived from pediatric brain tumor patients including 22 medulloblastomas and 5 ependymomas, which also maintain original tumors’ histological features and are genetically faithful to corresponding patient tumors (42). The Mayo Clinic Brain Tumor Patient-Derived Xenograft (PDX) National Resource has also established a repository of glioblastoma PDX models with highly characterized molecular subtype and phenotype. Another study collected DIPG samples from patient autopsies and biopsies at 8 different international institutions and generated 22 in vivo xenograft models, covering the main molecular subtypes including H3.3 K27M and H3.1 K27M mutations (43, 44).
With the advance in gene editing technology, neural stem cells (NSC) can be genetically engineered to acquire tumorigenic capability and used to generate xenograft models (35). Transplantation of NSCs that overexpressed myc alone, or with oncogene gfi1 or gfi1b into the cerebella of immunocompromised mice induced Group 3 medulloblastoma (45–48). Funato et al. successfully transformed neural progenitor cells derived human embryonic stem cells with a constitutively active form of the PDGFRA, a small hairpin RNA (shRNA) against p53 and H3.3K27M to model pediatric DIPG with H3.3K27M mutation (49). In addition, NSCs co-expressing PDGFRB and H3.3K27M were injected into the pons of SCID mice and induced tumors similar to human H3K27M DIPGs (50). The first mouse model of ependymoma was generated by implanting embryonic cerebral Ink4a/Arf−/− NSCs overexpressing Ephb2 into the cerebrum of immunocompromised mice (51). More recently, induced pluripotent stem (iPS) cells are also being used to generate brain tumor xenograft models. iPS-derived neural stem cells generated from Gorlin syndrome patients, who are carrying a germline mutation in PTCH1 and are predisposed to medulloblastoma, were transplanted into mouse cerebellum. These cells formed tumors that mimic SHH-driven medulloblastoma (52, 53).
Xenograft models are a valuable tool in cancer research and drug screening. The National Cancer Institute recently decided to use PDX models to replace a panel of 60 human cancer cell lines (NCI-60) as a model for drug screening (54). However, there are some limitations of xenograft models. First, the generation of some xenograft models is challenging, but the success rate is increased with more aggressive and highly malignant tumors. Second, xenografts usually require many cells at a time, which is not naturally occurring in patients. Third, the transplantation procedure can disrupt the blood brain barrier, which is a key factor when evaluating drug efficacy. Fourth, the host animal for xenografts are usually immunodeficient mice, which cannot be used to discover the contribution of immune system in tumor initiation and development. Fifth, the engrafted human tumor stroma structure will be lost over time, replaced by the host mice’s own microenvironment. Sixth, genetic and phenotypic drifts gradually occur as the xenograft tumors are propagated through mice. Last, maintenance of PDX models is costly and labor intensive (22–25, 35, 38, 39, 55).
Mouse Models With Genetic Engineering of Brain Tumors
In recent years, with rapid advances in gene editing techniques, genetically engineered mouse models (GEMMs) have gained popularity in brain tumor research. Unlike PDXs, GEMMs can recapitulate tumor initiation and development in animals with native immune system and intact blood brain barrier and undisrupted microenvironment. This makes GEMMs more attractive as models for tumor mechanism and drug discovery studies (23, 24, 26, 56). Moreover, other genetically engineered animal models, such as mice expressing enhanced green fluorescent protein (EGFP) or humanized mice carrying human functional biological system are valuable tools in brain tumor research, especially in studies about tumor host interactions and human-specific pathogenesis and therapies (57, 58).
GEMMs for cancer research can be generated by introduction of oncogenes or disruption of tumor suppressor genes in embryonic stem (ES) cells or zygotes (25) and include both transgenic mice and knockout mice. In addition to oncogenes and tumor suppressor genes, key molecules in tumor signaling pathways can also be utilized to develop GEMMs (Table 1). The conventional knockout models alter target gene expression in all tissues throughout the whole mouse. These constitutive changes often lead to more severe phenotypes with contribution by the brain tumor itself as well as other conditions. This makes data analysis and interpretation more difficult and less accurate. In some cases, this global knockout can even be lethal in animals. To overcome this issue, conditional or inducible conditional knockout has been developed, in which the target gene can be edited in a tissue specific and/or time-dependent way. The most common tool to make conditional knockout mice is the Cre-loxP system. Cre is a recombinase and its expression can be driven under the control of a tissue-specific promoter. When Cre is induced, it can recognize the loxP sites and catalyze the recombination, so the target gene flanked with two loxP sites in the same orientation will be excised. To achieve precise temporal specificity in the Cre-loxP system, Cre can be fused with a hormone responsive element, and induced by the exogenous inducers tamoxifen or tetracycline (35, 106).

Table 1 The common GEMMs of pediatric brain tumors.
The generation of germline GEMMs usually needs an extensive breeding scheme, which is time-consuming and expensive. Thus, virus mediated gene transfer is introduced to deliver Cre recombinase to somatic cells to establish non-germline GEMMs, which retains the ability of spatial and temporal gene regulation, at the same time also reduces the cost and time by bypassing complicated breeding (25). Replication-competent avian sarcoma-leukosis virus long terminal repeat with splice acceptor/tumor virus A (RCAS/TVA) is a commonly used system. RCAS is a retrovirus that enters specific cells via binding to its specific cell surface receptor TVA. TVA is only expressed in avian cells, but mammalian cells can gain the expression through genetic engineering (107). RACS/TVA based GEMMs have some advantages over Cre-loxP based models. The virus transduction rate is quite low, so only a small fraction of cells can acquire the expression of target genes. This makes the model close to natural tumorigenesis, since studies have shown that only small amounts of cancer stem cells are key players in tumor initiation (35). Moreover, genetically-engineered mammalian cells can get multiple RACS infection simultaneously or sequentially, which makes this model suitable to study the effect of multiple genes on tumorigenesis (107).
Recently, short palindromic clustered regularly interspaced repeats/CRISPR associated protein 9 (CRISPR/Cas9) technology has become a powerful tool to generate GEMMs. The CRISPR/Cas9 system is a groundbreaking gene editing technique; it consists of two necessary components: single strand guide RNA, which can recognize the target genomic DNA sequence, and endonuclease cas9, which can break the double-stranded DNA at the target sequence site. Then random or targeted gene editing can be achieved by DNA repair through error-prone non-homologous end joining (NHEJ) and high-fidelity homology directed repair (HDR) pathways. CRISPR/Cas9 can efficiently introduce gene modification on virtually any genetic background, both in germline and somatic cells. It turns conventionally tedious and expensive genetic engineering into a simple, fast and affordable procedure and dramatically broadens the application of GEMMs in tumor research (35, 108).
In 2019, MADR (mosaic analysis by dual recombinase-mediated cassette exchange) was introduced as a simpler, higher-throughput method to generate stable, defined copy number somatic transgenic animals. MADR was designed to overcome limitations of some of the previously described methods to generate mouse models (98). This includes the limited payloads and possible immune reactions when using viruses, the unpredictable genomic integration patterns, epigenetic transgenic silencing, transgene copy number variability, and overexpression artifacts such as cytotoxicity and transcriptional squelching when using viruses or transposons, or the variability and unintended off-target genomic alterations of CRISPR/Cas9 systems (98).
To date, the majority of pediatric brain tumor GEMMs are SHH-activated medulloblastoma (Table 1), which are generated by modifying SHH signaling genes, such as PTCH, SMO, or SUFU (22, 24). The first mouse model of medulloblastoma was established by disrupting ptch1 (61). Thereafter, the combination of a ptch mutation with inactivation of tumor suppressors, including TP53 or cyclin D-dependent kinase inhibitor p18Ink4c, was used to generate different SHH driven medulloblastoma models with shorter latency and higher penetrance (22, 55, 109). SHH medulloblastoma models were also developed by overexpressing Shh, alone or in combination with mycn or bcl2 with the RACS-TVA system (76, 77). Most WNT activated medulloblastoma models were generated by targeting the gene ctnnb1 in progenitor cells of the dorsal brainstem. However, ctnnb1 aberration alone was not sufficient to form medulloblastoma and the combination with TP53 mutation was needed to drive tumor initiation (59). Co-occurrence of pik3ca mutation significantly accelerated the formation of WNT medulloblastoma and dramatically increased tumor penetrance in mice (60). Most GEMMs of Group 3 medulloblastoma were developed by targeting myc. Newborn mice with myc overexpression in the cerebellum through the RCAS/TVA system induced Group 3 medulloblastoma, but tumor formation required tp53 loss or bcl-2 overexpression (81). Conditional enforced co-expression of myc and a dominant-negative form of Trp53 (Trp53DN) in embryonic cerebellar progenitor cells by in utero electroporation also induced Group 3 medulloblastoma in mice (82). The first mouse model of Group 4 medulloblastoma was recently developed by overexpression of an activated SRC combined with p53 inactivation in the developing cerebellum (83). It is worthy of note that in some animal models induced tumors simultaneously possess multiple molecular characteristics. For example, the GTML (Glt1-tTA/TRE-MYCN-Luc) model in which MYCN aberration is driven by the glutamate transporter 1 (Glt1) promoter expressed in hindbrain progenitors develops tumors that closely resemble Group 3, but also shows the features of WNT, SHH, and Group 4 medulloblastoma (24).
Various medulloblastoma mouse models have been used to dissect mechanisms, including those influenced by the tumor microenvironment, that regulate the progression from precancerous lesions to medulloblastoma tumors (110). In general, tumors not only consist of the heterogenous tumor cell population but also of the extracellular matrix (ECM) surrounding the cells, resident and infiltrating cells such as tumor-associated fibroblasts, endothelial cells, pericytes, adipocytes, and immune cells including lymphocytes and macrophages as well as soluble factors, including cytokines, chemokines, growth factors, matrix remodeling enzymes and inflammatory enzymes (111, 112). The tumor microenvironment is known to contribute to tumor progression, metastasis formation and therapeutic response (113–122). In brain tumors, macrophages are the most abundant type of immune cells and are particularly high in Shh-driven medulloblastoma. In humans, decreased macrophage numbers are correlated with significant poorer outcome and indeed, a recent study in NeuroD2:SmoA1 mice and derivative mouse lines was able to demonstrate that tumor-associated macrophages have properties that kill tumor cells (123). The NeuroD2:SmoA1 medulloblastoma model was also used to show that blocking TGF-β signaling promoted memory T cell development thereby conferring antitumor immunity (124) and in Atoh1-Cre;Ptch1fl/fl mice, tumor astrocyte-derived Shh induced the proliferation of medulloblastoma tumor cells (125). Cancer stem cells reside in specialized, anatomically distinct niches within the tumor microenvironment (126) and medulloblastoma stem cells (Nestin+, Prominin+) are closely associated with capillaries in the perivascular niche. Using mice infected with RCAS-Shh RCAS-SHH in combination with RCAS-N-myc-T50A or RCAS-AKT-Myr Δ11–60 of Ntv-a wild-type p53 and Ntv-a p53-null background, Hambardzumyan et al. showed that similar to human medulloblastomas, nestin-expressing perivascular stem cells survive radiation, activate PI3K/Akt signaling, undergo PTEN/p53-dependent cell cycle arrest and shortly thereafter re-enter the cell cycle (127). Medulloblastoma mouse models have also been used to elucidate pathways involved in tumor angiogenesis, in medulloblastoma metastasis, and in cell senescence and reprogramming (110).
Most mouse models of glioma are generated by altering key signaling pathways disrupted in human gliomas, including Ras, EGFR, Akt, Rb, Pten, Nf1 and platelet-derived growth factor (PDGF) (Table 1). GFAP-V12Ha-ras mice were generated by overexpressing oncogenic V12Ha-ras in astrocytes, and 95% of these mice died from low- and high-grade astrocytoma within 2-6 months (88). Further expressing a mutant EGFRVIII or inactivating PTEN in GFAP-V12Ha-ras mice demonstrated earlier tumor onset, higher tumor grade and a dramatic reduction in survival (89, 90). Introduction of activated Ras (KRas) into neural progenitors with the RCAS/TVA system, combined with activated Akt or PTEN loss induced high-grade gliomas in mice that resembled human GBMs (92, 93). Further deleting ink4a/arf increased tumor incidence and grades in these mice (128). Silencing of Bcl6 in neuronal precursor cells suppressed, but did not abolish, the formation of tumors in a somatic KrasG12V-driven glioma mouse model (99, 129). Transgenic S100β-v-erbB mice in which a transforming allele of EGFR, v-erbB, is expressed under the control of murine S100β promotor developed low-grade oligodendroglioma, and further deleting ink4a/arf or p53 increased tumor grade and penetrance (91). An Nf1+/-; p53+/- mouse model shows a range of astrocytoma stages, from low-grade astrocytoma to glioblastoma multiforme (84). Conditionally deleting NF1 in glial progenitors and astrocytes of p53 null mice dramatically increased the penetrance of induced astrocytoma and the incidence of non-CNS neoplasms (85). Further loss of Pten in glial progenitors and astrocytes of this mouse model significantly accelerated tumor growth and animal mortality (86). TgGFAPT121 mice generated by a truncated SV40 T antigen (T121) to inactivate the Rb pathway in astrocytes develop high grade astrocytoma and die perinatally (87). PDGF B-chain (PDGF-B) is another common target used to generate glioma models. PDGF-B delivered to nestin-positive neural progenitors or GFAP positive astrocytes induced low grade glioma in mice. Loss of Ink4a–Arf dramatically shortened tumor latency and enhanced malignancy of gliomas. p53 loss can also enhance PDGF-B driven glioma in mouse models (94–96). A more recent DIPG model was developed by overexpressing PDGF-B and H3.3K27M together with p53 loss in nestin-positive neural progenitors. The induced tumors in the brainstem of these mice demonstrated DIPG-like features that recapitulate the histopathological and molecular characteristic of human DIPG (96, 130). The MADR method was used to generate pediatric glioma mice modeling simultaneous H3f3a, Pdgfra, and Trp53 mutations with two missense mutation variants G34R or K27M that recapitulated human tumor heterogeneity and developmental hierarchy (98). Other recent pediatric brain tumor models have also been successful in capturing tumor heterogeneity and spatiotemporal characteristics of pediatric gliomas. PiggyBac transposon systems not only can circumvent the loss or inactivation of episomal plasmids delivered to glial cells via in utero electroporation but also allow for expression of multiple oncogenes in selected cell populations at different times in brain development (131). Using in utero electroporation of piggyBac transposons, Chen and colleagues generated rat tumor models by directing HRasV12 and AKT to different cell populations. Using the same transgene under the control of different promoters resulted in tumors ranging from glioblastoma multiforme to anaplastic oligoastrocytomas and atypical teratoid/rhabdoid-like tumors that could be distinguished at the cellular and the molecular level (100, 131). Moreover, targeting different genes, PTEN or NF1, in the same lineage resulted in distinct neuropathologies and when PTEN, NF1 and P53 were targeted simultaneously caused the formation of GBM (101).
The generation of ependymoma GEMMS began recently. RELAFUS1 fusion gene expressed in nestin, GFAP, or BLBP positive cells in the mouse brain induced tumors which recapitulate the histology and transcriptome panel of human ST-EPN-RELA ependymomas (102). The YAP1-MAMLD1 fusion gene delivered to mice by in utero electroporation drove tumor formation and tumors share histological and molecular characteristics of human ST-EPN-YAP1 (103). Recently, Eder and colleagues reported that ectopic expression of active nuclear YAP1 (nlsYAP5SA) or conditional deletion of YAP1’s negative regulators LATS1 and LATS2 kinases in neural progenitor cells in ventricular zone also induced tumors which display molecular and ultrastructural characteristics of human ependymoma (104, 105).
Zebrafish Brain Tumor Models
Zebrafish are an alternative model to study human cancer as they can develop tumors that are histologically and genetically similar to those in humans (132). Zebrafish models are also amenable to high-throughput screening for drug discovery as well as transplantation of primary patient tumors. This makes zebrafish a cost- and time-effective alternative to other in vivo tumor models such as rodents. In recent years, several pediatric brain tumor models have been developed in zebrafish and have been used to identify molecular mechanisms driving tumor formation. This includes the analysis of unique and shared molecular pathways driving pediatric HGG within and outside the brainstem (133) and to identify three molecular subgroups of DIPG (134). Ependymoma, glioma and choroid plexus carcinoma cells from mouse models of pediatric brain tumors were conditioned to grow at 34°C and used for orthotopic xenografts in zebrafish. These cells not only readily formed tumors but also spinal metastasis. The tumors retained the histological characteristics of the corresponding mouse tumor and formed tumor vasculature by recruiting fish endothelial cells (135). Lin et al. used zebrafish to experimentally validate subgroup-specific enhancers in medulloblastoma (13), Modzelewska et al. used tumors grown in zebrafish to demonstrate that MEK inhibitors can reverse the growth of embryonal brain tumors derived from oligoneural precursor cells (14) and Idilli et al. used them to study telomere maintenance mechanisms in pediatric brain tumors (136). With protocols for developing zebrafish tumor models evolving, long-term orthotopic transplantation of tumor cells is now possible (137). This allows for the long-term in vivo studies of tumor cell behaviors including tumor invasion and dissemination as well as testing for more durable response of tumors to novel anticancer therapeutics and the development of cancer drug resistance. Overall, zebrafish may provide an opportunity to develop pediatric brain tumor models in a timely and affordable manner for preclinical drug discovery in a model system with intact blood-brain barrier.
In Vitro Brain Tumor Models
Cancer Cell Lines
Cancer cell lines play an important role in brain tumor research. The cells lines can be established directly from patients’ samples or from animal models. These cells often retain original tumor features, are easy to grow and propagate, and can be stored for a long time. They are well-suited models to explore a tumors’ molecular features in vitro and predict the tumors’ response to therapeutic regimens. Cancer cell lines are particularly useful in high-throughput drug screening to identify and evaluate potential targets for chemotherapies. Most established pediatric brain tumor cell lines are medulloblastoma cell lines. Less than half of these cell lines have been molecularly defined, among which the majority represent the SHH or Group 3 subtypes; only a few are for WNT or Group 4 tumors (Table 2). In addition, around half SHH medulloblastoma cell lines have mutations in TP53, and almost all Group 3 cell lines bear MYC amplification, while only a small part of SHH and Group 3 medulloblastoma patients typically have these mutations (24, 55, 145, 150). This discrepancy might be because the medulloblastomas with TP53 mutation and MYC amplification are more aggressive with poorer prognosis, and more aggressive cells are easier to grow in vitro. Similarly, although gliomas are the most common brain tumors in children, most gliomas are low grade gliomas with less malignancy and more favorable prognosis. Moreover, some high grade gliomas, such as DIPG, have a limited tissue availability due to tumor locations and established glioma cell lines are fewer than medulloblastoma (150). In recent years, however, with the refinement of surgical skills and advances in DIPG biology, biopsy becomes more feasible in DIPG patients and new patient derived DIPG cell lines are becoming available (Table 2).
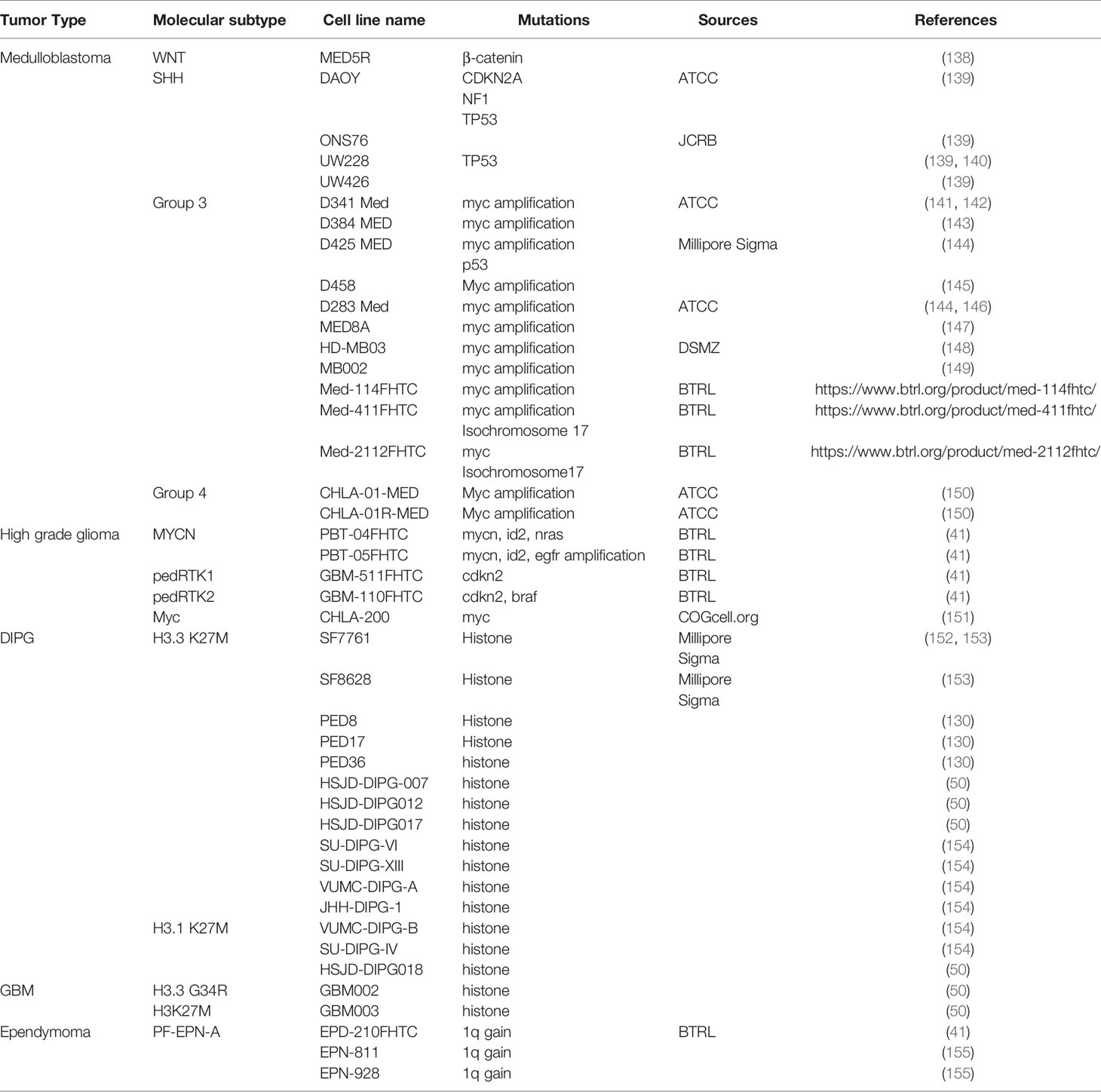
Table 2 Established pediatric brain tumor cell lines with defined molecular characteristics.
Cell culture in vitro has intrinsic drawbacks. Most cell lines are maintained as a monolayer culture in serum containing media, and a genetic and phenotypic drift from original tumors will gradually occur with passage (156). The cell lines are homogenous populations that cannot fully recapitulate the heterogeneity of tumors, and are not suitable to study tumor host interactions during tumor development. In monolayer culture, all cells receive the same level of nutrition and oxygen, which is different from tumor growth in vivo. Moreover, tumor cell lines are typically grown on borosilicate glass or clear plastics in vitro, which are much more rigid compared to extracellular matrix on which cells are naturally grown in vivo (157).
The advent of neurosphere cultures addressed some limitations of traditional cell cultures. Neurospheres are typically cultured in serum-free medium and can maintain tumor heterogeneity and preserve the phenotype and genotype of primary tumors (158). Some pediatric brain tumors, including DIPG, have been successfully cultured in neurospheres and used to generate xenografts that recapitulate the histological features and infiltrative growth of original patient tumors (24, 130, 152, 159). Neurosphere formation also can independently predict clinical outcome in malignant glioma (160). Therefore, neurospheres are a more representative and reliable cell model compared to traditional cell lines (36). However, neurosphere culture has some limitations, too. For example, the lack of a tumor microenvironment highly enriches glioma stem cell (GSC)-like cells, which only represent a relatively small subpopulation in native tumors (158, 161).
Three-Dimensional Culture
3D culture such as spheroids and scaffold-based cultures are other techniques that have been developed to overcome the limitations with traditional monolayer culture. In scaffold-based 3D cultures, extracellular matrix can be synthesized to simulate a tumor’s natural microenvironment and a gradient of oxygen and nutrient level can be constituted to mimic a tumor’s hypoxic core in vivo. Moreover, gene expression panels in 3D culture more closely resemble human tumors in vivo (157, 162, 163). To date, there are two major types of 3D culture: anchorage-dependent and anchorage-independent 3D models.
Anchorage-Independent 3D Models
Anchorage-independent 3D models are achieved mainly by self-assembly of cells grown in special tissue culture plates, such as hanging drop microplates and low attachment plates; they do not need any scaffold to facilitate the culture. The hanging drop culture is a well-known 3D culture technology. Typically, there is a micro-hole at the top of wells, which allows the medium to pass through and form a small droplet. Since there is no surface available for the cells in the droplet to attach, these cells tend to form spheroids. Spheroids can also be generated when they are grown in ultra-low attachment plates. Ependymoma cell lines cultured in ultra-low attachment plates better recapitulated the histological and transcriptional features of the primary tumors when compared to a monolayer (164). In addition, magnetic levitation is a newly developed method for spheroid formation. In this method, cells coated with magnetic nanoparticles are cultured in a magnetic field and the cells are floated toward the air/liquid interface within a low adhesion plate to form spheroids (162, 163, 165). Larger tumor spheroids formed by anchorage-independent models may consist of a peripheral layer with proliferating cells, an intermediate layer with quiescent cells and an inner necrotic core, which may closer reproduce human tumor architecture in vivo (165, 166). In recent years, with the refinement of technologies on spheroid culture, anchorage-independent 3D models have become common methods for cancer drug discovery, even applicable for high-throughput drug screening. Spheroids can be established with a few different types of cells and are especially suitable for studies about cell-cell interactions during brain tumor development (167). However, there is typically no extracellular matrix (ECM) in spheroids, and thus they are unsuitable for studying cell-host interactions which is a key game player in tumor pathologies.
Anchorage-Dependent 3D Models
The cells inside the body are usually surrounded by ECM, a network of extracellular molecules, which not only provides the structural scaffold for the surrounding cells, but also plays an important role in cell proliferation, differentiation, migration, survival and adhesion (168). The composition of ECM is highly heterogeneous and tissue-specific. The brain ECM ingredients include proteoglycans, hyaluronic acids, tenascins, collagen, fibronectin, vitronectin and laminin (162, 169). In anchorage-dependent 3D models, cells are encapsulated into scaffold materials, which can mimic the composition and key physical properties of ECM. Hydrogels are the most commonly used scaffolds for anchorage-dependent 3D models. Hydrogels are water-swollen networks of polymers and can mimic salient components of ECM. The highly hydrated and porous nature of hydrogel make them ideal to encapsulate cells and render ECM-like functions, such as supporting cell survival, growth, differentiation and modulating the response to chemotherapy, immunotherapy and radiation therapy (161).
Hydrogels may come from natural sources or can be synthetic. The widely used natural hydrogels for neural cell culture are collagen I and Matrigel. Matrigel is extracted from the Engelbreth-Holm-Swarm (EHS) mouse sarcoma, a tumor rich in ECM components, such as laminin, collagen, heparan sulfate proteoglycans, entactin/nidogen, and several growth factors. Matrigel is minimally processed and it can better mimic in vivo ECM (170). However, two major components of Matrigel are laminin and collagen, which are in low concentration in the brain ECM (158). Thus, collagen and Matrigel are not ideal choices as in vivo-like 3D scaffolds for brain tumor cells. In addition, collagen and Matrigel are derived from natural sources, they are heterogenous and not well defined, and exhibit considerable batch-to-batch variability. Moreover, collagen and Matrigel are available in liquid form and require handling at cold temperatures (below 10°C) to avoid premature gelation. The need for handling these hydrogels at low temperatures makes them poorly suited for common liquid handling equipment used for high-throughput screens in drug discovery (158, 162). Some of these limitations might be overcome by synthetic hydrogels. Synthetic hydrogels are derived from polymeric materials, such as polyethylene glycol (PEG), polylactic acid (PA) and polyglycolic acid (PGA). These hydrogels can simulate the composition and function of ECM; they often have engineered tunable properties to achieve desired stiffness and porosity, to enhance cell proliferation and differentiation by encapsulating bioactive molecules, such as growth factors or hormones. However, these polymers are biological inert, so they must be modified by addition of cell adhesion ligands or mixing with other natural ECM components to acquire the properties of cell adhesion (158, 162, 163). To date, PEG is a widely used synthetic hydrogel in neural cell 3D culture. A PEG-based hydrogel has been successfully used to grow GBM cell lines. In this system, the PEG-based hydrogel was modified with CRGDS and a MMP-cleavable peptide to facilitate cell proliferation, migration; hyaluronic acid (HA) was also added to mimic brain extracellular matrix (171). A synthetic MAX8 β-hairpin hydrogel was successfully used to culture pediatric medulloblastoma cell lines in a high-throughput screening setting (172, 173). Hydrogel-based models are not suitable for long-term culture since they degrade fast. Solid porous scaffolds can be adapted to bypass this issue. Solid porous scaffolds are prepared from natural or synthetic polymers with mechanical stability and pore interconnectivity. The cells can be directly added to these solid porous scaffolds and maintain their 3D properties with continuous supply of nutrients. A recently developed tunable 3D brain tissue model integrated the porous scaffold with hydrogels. In this model, the donut shaped silk fibroin protein scaffold was infused with ECM hydrogels and brain tumor cells can grow into spheroids within the stiff silk scaffold, or migrate toward the central hydrogel. Thus, the outer-ring scaffold can be used to anchor neuronal cells, and the central soft hydrogel allows axonal penetration and connectivity (174, 175). A pediatric anaplastic ependymoma has been successfully cultured by this model (176). In addition, culturing glioblastoma tumor-initiating cells (TICs) in microscale alginate hydrogel tubes (AlgTubes) has been reported. This culture system allows for long-term and scalable production of glioblastoma cells for drug discovery (177). Self-assembling peptide (SAP) hydrogels are an evolving field for neural cell culture. These synthetic peptides can self-assemble under physiological conditions and support neural cell attachment, differentiation and synapse formation. SAP hydrogels are highly versatile, their material properties can be modulated by substituting amino acids, extending or shortening the peptide sequence, or by the addition of functional epitopes. A widely used peptide hydrogel is RADA16. However, peptide-based hydrogels may have poor mechanical properties, and some exhibit impaired cell viability caused by low pH, making it difficult to culture sensitive brain tumor cells (163).
Brain Organoids
Organoids are an emerging technology to study pediatric brain tumors. Organoids are typically generated with embryonic stem cells (ESC) or induced pluripotent stem cells (iPSC) and have the potential to grow into a 3D architecture in a way similar to in vivo tissue development by virtue of their capacity to self-renew and differentiate. Early organoid models were typically heterogeneous and lacked reproducibility since it was difficult to control the differentiation pattern of stem cells. However, with technical advances on directed differentiation, stems cells can now be differentiated into virtually any specific lineages. This has significantly moved forward the application of organoid models including in biomarker and drug discovery (163). Significant effort is being made in developing neural-based spheroids with cerebral organoids being one of the early ones (178). Cerebral organoids can be used as platforms for human brain tumor cells, or tumors can be initiated in cerebral organoids by introducing oncogenes and/or disrupting tumor repressor genes using gene editing technologies. Human cerebellar organoids derived from iPS cells electroporated with Otx2/c-MYC induced Group 3 medulloblastoma (179). Injecting cancer stem cells derived from GBM patients into cerebral organoids or genetic engineering of cerebral organoids by introducing HRasG12V and disrupting p53 initiated tumorigenesis that closely recapitulated patient GBMs (180, 181). Organoids can also be established from patient brain tumors. Hubert et al. generated GBM organoids directly from patient samples that present hypoxic gradients and regional tumor heterogeneity (182). Organoids can theoretically resemble any in vivo brain niche with preserved cell distribution, can retain genetic and phenotypic stabilities, and are capable of long term culture; this makes them a valuable model to discover tumor initiation and progression, and a more accurate tool to predict the responses to tumor treatments. However, organoid cultures typically lack blood vessels and immune cells, which makes them unsuitable for testing tumor treatments targeting angiogenesis, or studying the contribution of immune system on tumorigenesis and relevant therapies. In addition, although organoids have proper cell composition and functions, they typically lack correct anatomical organization (162, 183). Another drawback of organoids and also found in spheroid models is that they often have a necrotic core, which sets a limit on the culture size and longevity. To overcome this limitation, microfluidic devices can be incorporated into 3D models. Microfluidic devices are designed for cell cultures under perfusion and allow for steady supplies of oxygen and nutrients while at the same time removing waste (163).
Conclusions
A precise in vivo pediatric brain tumor model is the one, which can faithfully recapitulate tumor’s histopathological and molecular features; exhibit tumor’s spatiotemporal characterization; demonstrate a tumor’s microenvironment; predict patients’ response to treatments; show high rate of incidence and short latency; and is reproducible, timesaving and cost-effective (184). Such accuracy in tumor models can best be achieved when genetic insults match the cell of origin and are introduced at developmental stages that are critical to tumor development. For effective in vitro drug discovery of novel cancer therapeutics, in vitro brain tumor models should not only recapitulate tumor biology but culture methods should also be suitable for high-throughput screening (HTS). New technologies and with it the possibilities of more complex screening platforms may be integrated to optimize the model systems for pediatric brain tumors. For example, the recently developed brain cancer-on-a-chip models incorporate multiple tissue types in 3D cultures into microphysiological system (MPS) and provide precise control of a cellular microenvironment and real-time monitoring on cell behavior and response. Nevertheless, while brain cancer-on-a-chip models can better mimic the physiological function of brain, challenges remain. Brain tumors demonstrate profound inter- and intra-tumoral heterogeneity and cellular plasticity to adapt their phenotypes to the surrounding. With more accurate in vitro and in vivo tumor models, however, it is possible to improve the current low approval rate of anticancer drugs, to offer more treatment options for pediatric brain tumor patients.
Although pediatric brain tumor models have been expanded immensely in the past decades, there is no single model that meets all criteria and thus, experimental design and purpose will need to guide the choice of the brain tumor model (22, 24). The rapid advancement of genomic characterization of pediatric brain tumors and with it new genomic signatures of tumor subgroups add to the complexity of developing precise pediatric brain tumor models. Moreover, in recent years the genome landscape of pediatric brain tumors, both somatic and epigenetic, has been complemented by the analysis of tumor transcriptomes. Despite the plethora of data generated through such approaches, the finding that impaired differentiation of specific neural progenitors is a common mechanism underlying pediatric cancers (185) provides hope that a rational approach towards developing in vitro and in vivo pediatric brain tumor models can achieve a manageable library of research platforms for the development of impactful therapeutic interventions for pediatric brain cancers.
Author Contributions
ZL and SL perceived and wrote the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the Nemours Foundation and the DoBelieve Foundation.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Karen Sperle for suggestions and critical reading of the manuscript.
References
1. Ostrom QT, Gittleman H, Truitt G, Boscia A, Kruchko C, Barnholtz-Sloan JS. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2011-2015. Neuro Oncol (2018) 20(suppl_4):iv1–iv86. doi: 10.1093/neuonc/noy131
2. Louis DN, Perry A, Reifenberger G, von Deimling A, Figarella-Branger D, Cavenee WK, et al. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: a summary. Acta Neuropathol (2016) 131(6):803–20. doi: 10.1007/s00401-016-1545-1
3. Wells EM, Packer RJ. Pediatric brain tumors. Continuum (Minneap Minn) 21(2 Neuro-oncol) (2015) 21(2 Neuro-oncology):373–96. doi: 10.1212/01.CON.0000464176.96311.d1
4. Dang M, Phillips PC. Pediatric Brain Tumors. Continuum (Minneap Minn) (2017) 23(6, Neuro-oncology):1727–57. doi: 10.1212/CON.0000000000000545
5. Udaka YT, Packer RJ. Pediatric Brain Tumors. Neurol Clin (2018) 36(3):533–56. doi: 10.1016/j.ncl.2018.04.009
6. Pollack IF, Agnihotri S, Broniscer A. Childhood brain tumors: current management, biological insights, and future directions. J Neurosurg Pediatr (2019) 23(3):261–73. doi: 10.3171/2018.10.PEDS183772018.10.PEDS18377
7. Wong CH, Siah KW, Lo AW. Corrigendum: Estimation of clinical trial success rates and related parameters. Biostatistics (2019) 20(2):366. doi: 10.1093/biostatistics/kxy072
8. Wong CH, Siah KW, Lo AW. Estimation of clinical trial success rates and related parameters. Biostatistics (2019) 20(2):273–86. doi: 10.1093/biostatistics/kxx069
9. Filbin MG, Sturm D. Gliomas in Children. Semin Neurol (2018) 38(1):121–30. doi: 10.1055/s-0038-1635106
10. Sturm D, Pfister SM, Jones DTW. Pediatric Gliomas: Current Concepts on Diagnosis, Biology, and Clinical Management. J Clin Oncol (2017) 35(21):2370–7. doi: 10.1200/JCO.2017.73.0242
11. Ferris SP, Hofmann JW, Solomon DA, Perry A. Characterization of gliomas: from morphology to molecules. Virchows Arch (2017) 471(2):257–69. doi: 10.1007/s00428-017-2181-4
12. Penman CL, Faulkner C, Lowis SP, Kurian KM. Current Understanding of BRAF Alterations in Diagnosis, Prognosis, and Therapeutic Targeting in Pediatric Low-Grade Gliomas. Front Oncol (2015) 5:54. doi: 10.3389/fonc.2015.00054
13. Lin CY, Erkek S, Tong Y, Yin L, Federation AJ, Zapatka M, et al. Active medulloblastoma enhancers reveal subgroup-specific cellular origins. Nature (2016) 530(7588):57–62. doi: 10.1038/nature16546
14. Modzelewska K, Boer EF, Mosbruger TL, Picard D, Anderson D, Miles RR, et al. MEK Inhibitors Reverse Growth of Embryonal Brain Tumors Derived from Oligoneural Precursor Cells. Cell Rep (2016) 17(5):1255–64. doi: 10.1016/j.celrep.2016.09.081
15. Diaz AK, Baker SJ. The genetic signatures of pediatric high-grade glioma: no longer a one-act play. Semin Radiat Oncol (2014) 24(4):240–7. doi: 10.1016/j.semradonc.2014.06.003
16. Pajtler KW, Witt H, Sill M, Jones DT, Hovestadt V, Kratochwil F, et al. Molecular Classification of Ependymal Tumors across All CNS Compartments, Histopathological Grades, and Age Groups. Cancer Cell (2015) 27(5):728–43. doi: 10.1016/j.ccell.2015.04.002
17. Khatua S, Ramaswamy V, Bouffet E. Current therapy and the evolving molecular landscape of paediatric ependymoma. Eur J Cancer (2017) 70:34–41. doi: 10.1016/j.ejca.2016.10.013
18. Vitanza NA, Partap S. Pediatric Ependymoma. J Child Neurol (2016) 31(12):1354–66. doi: 10.1177/0883073815610428
19. Northcott PA, Robinson GW, Kratz CP, Mabbott DJ, Pomeroy SL, Clifford SC, et al. Medulloblastoma. Nat Rev Dis Primers (2019) 5(1):11. doi: 10.1038/s41572-019-0063-610.1038/s41572-019-0063-6
20. Huang SY, Yang JY. Targeting the Hedgehog Pathway in Pediatric Medulloblastoma. Cancers (Basel) (2015) 7(4):2110–23. doi: 10.3390/cancers7040880
21. Cavalli FMG, Remke M, Rampasek L, Peacock J, Shih DJH, Luu B, et al. Intertumoral Heterogeneity within Medulloblastoma Subgroups. Cancer Cell (2017) 31(6):737–54 e6. doi: 10.1016/j.ccell.2017.05.005
22. Neumann JE, Swartling FJ, Schuller U. Medulloblastoma: experimental models and reality. Acta Neuropathol (2017) 134(5):679–89. doi: 10.1007/s00401-017-1753-3
23. Huszthy PC, Daphu I, Niclou SP, Stieber D, Nigro JM, Sakariassen PO, et al. In vivo models of primary brain tumors: pitfalls and perspectives. Neuro Oncol (2012) 14(8):979–93. doi: 10.1093/neuonc/nos135
24. Dobson THW, Gopalakrishnan V. Preclinical Models of Pediatric Brain Tumors-Forging Ahead. Bioengineer (Basel) (2018) 5(4):81. doi: 10.3390/bioengineering5040081
25. Day CP, Merlino G, Van Dyke T. Preclinical mouse cancer models: a maze of opportunities and challenges. Cell (2015) 163(1):39–53. doi: 10.1016/j.cell.2015.08.068
26. Simeonova I, Huillard E. In vivo models of brain tumors: roles of genetically engineered mouse models in understanding tumor biology and use in preclinical studies. Cell Mol Life Sci (2014) 71(20):4007–26. doi: 10.1007/s00018-014-1675-3
27. Koestner A, Swenberg JA, Wechsler W. Transplacental production with ethylnitrosourea of neoplasms of the nervous system in Sprague-Dawley rats. Am J Pathol (1971) 63(1):37–56.
28. Barth RF, Kaur B. Rat brain tumor models in experimental neuro-oncology: the C6, 9L, T9, RG2, F98, BT4C, RT-2 and CNS-1 gliomas. J Neurooncol (2009) 94(3):299–312. doi: 10.1007/s11060-009-9875-7
29. Kruse CA, Molleston MC, Parks EP, Schiltz PM, Kleinschmidt-DeMasters BK, Hickey WF. A rat glioma model, CNS-1, with invasive characteristics similar to those of human gliomas: a comparison to 9L gliosarcoma. J Neurooncol (1994) 22(3):191–200. doi: 10.1007/BF01052919
30. Ko L, Koestner A, Wechsler W. Morphological characterization of nitrosourea-induced glioma cell lines and clones. Acta Neuropathol (1980) 51(1):23–31. doi: 10.1007/BF00688846
31. Benda P, Lightbody J, Sato G, Levine L, Sweet W. Differentiated rat glial cell strain in tissue culture. Science (1968) 161(3839):370–1. doi: 10.1126/science.161.3839.370
32. Cuatico W, Cho JR, Spiegelman S. Molecular evidence for a viral etiology of human CNS tumors. Acta Neurochir (Wien) (1976) 35(1-3):149–60. doi: 10.1007/BF01405943
33. Rabotti GF, Raine WA. Brain Tumours Induced in Hamsters Inoculated Intracerebrally at Birth with Rous Sarcoma Virus. Nature (1964) 204:898–9. doi: 10.1038/204898a0
34. Ogawa K, Hamaya K, Fujii Y, Matsuura K, Endo T. Tumor induction by adenovirus type 12 and its target cells in the central nervous system. Gan (1969) 60(4):383–92.
35. Robertson FL, Marques-Torrejon MA, Morrison GM, Pollard SM. Experimental models and tools to tackle glioblastoma. Dis Model Mech (2019) 12(9):dmm040386. doi: 10.1242/dmm.040386
36. Patrizii M, Bartucci M, Pine SR, Sabaawy HE. Utility of Glioblastoma Patient-Derived Orthotopic Xenografts in Drug Discovery and Personalized Therapy. Front Oncol (2018) 8:23. doi: 10.3389/fonc.2018.00023
37. Okada S, Vaeteewoottacharn K, Kariya R. Application of Highly Immunocompromised Mice for the Establishment of Patient-Derived Xenograft (PDX) Models. Cells (2019) 8(8):889. doi: 10.3390/cells8080889
38. Zarzosa P, Navarro N, Giralt I, Molist C, Almazan-Moga A, Vidal I, et al. Patient-derived xenografts for childhood solid tumors: a valuable tool to test new drugs and personalize treatments. Clin Transl Oncol (2017) 19(1):44–50. doi: 10.1007/s12094-016-1557-2
39. Hermans E, Hulleman E. Patient-Derived Orthotopic Xenograft Models of Pediatric Brain Tumors: In a Mature Phase or Still in Its Infancy? Front Oncol (2019) 9:1418. doi: 10.3389/fonc.2019.01418
40. Giannini C, Sarkaria JN, Saito A, Uhm JH, Galanis E, Carlson BL, et al. and PDGFRA gene amplifications retained in an invasive intracranial xenograft model of glioblastoma multiforme. Neuro Oncol (2005) 7(2):164–76. doi: 10.1215/S1152851704000821
41. Brabetz S, Leary SES, Grobner SN, Nakamoto MW, Seker-Cin H, Girard EJ, et al. A biobank of patient-derived pediatric brain tumor models. Nat Med (2018) 24(11):1752–61. doi: 10.1038/s41591-018-0207-3
42. Smith KS, Xu K, Mercer KS, Boop F, Klimo P, DeCupyere M, et al. Patient-derived orthotopic xenografts of pediatric brain tumors: a St. Jude resource. Acta Neuropathol (2020) 140(2):209–25. doi: 10.1007/s00401-020-02171-5
43. Tsoli M, Shen H, Mayoh C, Franshaw L, Ehteda A, Upton D, et al. Correction to: International experience in the development of patient-derived xenograft models of diffuse intrinsic pontine glioma. J Neurooncol (2019) 141(2):265. doi: 10.1007/s11060-018-03060-4
44. Tsoli M, Shen H, Mayoh C, Franshaw L, Ehteda A, Upton D, et al. International experience in the development of patient-derived xenograft models of diffuse intrinsic pontine glioma. J Neurooncol (2019) 141(2):253–63. doi: 10.1007/s11060-018-03038-2
45. Vo BT, Kwon JA, Li C, Finkelstein D, Xu B, Orr BA, et al. Mouse medulloblastoma driven by CRISPR activation of cellular Myc. Sci Rep (2018) 8(1):8733. doi: 10.1038/s41598-018-24956-1
46. Pei Y, Moore CE, Wang J, Tewari AK, Eroshkin A, Cho YJ, et al. An animal model of MYC-driven medulloblastoma. Cancer Cell (2012) 21(2):155–67. doi: 10.1016/j.ccr.2011.12.021
47. Northcott PA, Lee C, Zichner T, Stutz AM, Erkek S, Kawauchi D, et al. Enhancer hijacking activates GFI1 family oncogenes in medulloblastoma. Nature (2014) 511(7510):428–34. doi: 10.1038/nature13379
48. Kawauchi D, Robinson G, Uziel T, Gibson P, Rehg J, Gao C, et al. A mouse model of the most aggressive subgroup of human medulloblastoma. Cancer Cell (2012) 21(2):168–80. doi: 10.1016/j.ccr.2011.12.023
49. Funato K, Major T, Lewis PW, Allis CD, Tabar V. Use of human embryonic stem cells to model pediatric gliomas with H3.3K27M histone mutation. Science (2014) 346(6216):1529–33. doi: 10.1126/science.1253799
50. Mohammad F, Weissmann S, Leblanc B, Pandey DP, Hojfeldt JW, Comet I, et al. EZH2 is a potential therapeutic target for H3K27M-mutant pediatric gliomas. Nat Med (2017) 23(4):483–92. doi: 10.1038/nm.4293
51. Johnson RA, Wright KD, Poppleton H, Mohankumar KM, Finkelstein D, Pounds SB, et al. Cross-species genomics matches driver mutations and cell compartments to model ependymoma. Nature (2010) 466(7306):632–6. doi: 10.1038/nature09173
52. Huang M, Tailor J, Zhen Q, Gillmor AH, Miller ML, Weishaupt H, et al. Engineering Genetic Predisposition in Human Neuroepithelial Stem Cells Recapitulates Medulloblastoma Tumorigenesis. Cell Stem Cell (2019) 25(3):433–46 e7. doi: 10.1016/j.stem.2019.05.013
53. Susanto E, Marin Navarro A, Zhou L, Sundstrom A, van Bree N, Stantic M, et al. Modeling SHH-driven medulloblastoma with patient iPS cell-derived neural stem cells. Proc Natl Acad Sci USA (2020) 117(33):20127–38. doi: 10.1073/pnas.1920521117
54. Ledford H. US cancer institute to overhaul tumour cell lines. Nature (2016) 530(7591):391. doi: 10.1038/nature.2016.19364
55. Roussel MF, Stripay JL. Modeling pediatric medulloblastoma. Brain Pathol (2020) 30(3):703–12. doi: 10.1111/bpa.12803
56. Huse JT, Holland EC. Genetically engineered mouse models of brain cancer and the promise of preclinical testing. Brain Pathol (2009) 19(1):132–43. doi: 10.1111/j.1750-3639.2008.00234.x
57. Niclou SP, Danzeisen C, Eikesdal HP, Wiig H, Brons NH, Poli AM, et al. A novel eGFP-expressing immunodeficient mouse model to study tumor-host interactions. FASEB J (2008) 22(9):3120–8. doi: 10.1096/fj.08-109611
58. Scheer N, Snaith M, Wolf CR, Seibler J. Generation and utility of genetically humanized mouse models. Drug Discovery Today (2013) 18(23-24):1200–11. doi: 10.1016/j.drudis.2013.07.007
59. Gibson P, Tong Y, Robinson G, Thompson MC, Currle DS, Eden C, et al. Subtypes of medulloblastoma have distinct developmental origins. Nature (2010) 468(7327):1095–9. doi: 10.1038/nature09587
60. Robinson G, Parker M, Kranenburg TA, Lu C, Chen X, Ding L, et al. Novel mutations target distinct subgroups of medulloblastoma. Nature (2012) 488(7409):43–8. doi: 10.1038/nature11213nature11213
61. Goodrich LV, Milenkovic L, Higgins KM, Scott MP. Altered neural cell fates and medulloblastoma in mouse patched mutants. Science (1997) 277(5329):1109–13. doi: 10.1126/science.277.5329.1109
62. Yang ZJ, Ellis T, Markant SL, Read TA, Kessler JD, Bourboulas M, et al. Medulloblastoma can be initiated by deletion of Patched in lineage-restricted progenitors or stem cells. Cancer Cell (2008) 14(2):135–45. doi: 10.1016/j.ccr.2008.07.003
63. Li P, Du F, Yuelling LW, Lin T, Muradimova RE, Tricarico R, et al. A population of Nestin-expressing progenitors in the cerebellum exhibits increased tumorigenicity. Nat Neurosci (2013) 16(12):1737–44. doi: 10.1038/nn.3553
64. Wetmore C, Eberhart DE, Curran T. Loss of p53 but not ARF accelerates medulloblastoma in mice heterozygous for patched. Cancer Res (2001) 61(2):513–6.
65. Uziel T, Zindy F, Xie S, Lee Y, Forget A, Magdaleno S, et al. The tumor suppressors Ink4c and p53 collaborate independently with Patched to suppress medulloblastoma formation. Genes Dev (2005) 19(22):2656–67. doi: 10.1101/gad.1368605
66. Ayrault O, Zindy F, Rehg J, Sherr CJ, Roussel MF. Two tumor suppressors, p27Kip1 and patched-1, collaborate to prevent medulloblastoma. Mol Cancer Res (2009) 7(1):33–40. doi: 10.1158/1541-7786.MCR-08-0369
67. Briggs KJ, Corcoran-Schwartz IM, Zhang W, Harcke T, Devereux WL, Baylin SB, et al. Cooperation between the Hic1 and Ptch1 tumor suppressors in medulloblastoma. Genes Dev (2008) 22(6):770–85. doi: 10.1101/gad.1640908
68. Lee Y, Miller HL, Russell HR, Boyd K, Curran T, McKinnon PJ. Patched2 modulates tumorigenesis in patched1 heterozygous mice. Cancer Res (2006) 66(14):6964–71. doi: 10.1158/0008-5472.CAN-06-0505
69. Hallahan AR, Pritchard JI, Hansen S, Benson M, Stoeck J, Hatton BA, et al. The SmoA1 mouse model reveals that notch signaling is critical for the growth and survival of sonic hedgehog-induced medulloblastomas. Cancer Res (2004) 64(21):7794–800. doi: 10.1158/0008-5472.CAN-04-1813
70. Hatton BA, Villavicencio EH, Tsuchiya KD, Pritchard JI, Ditzler S, Pullar B, et al. The Smo/Smo model: hedgehog-induced medulloblastoma with 90% incidence and leptomeningeal spread. Cancer Res (2008) 68(6):1768–76. doi: 10.1158/0008-5472.CAN-07-5092
71. Dey J, Ditzler S, Knoblaugh SE, Hatton BA, Schelter JM, Cleary MA, et al. A distinct Smoothened mutation causes severe cerebellar developmental defects and medulloblastoma in a novel transgenic mouse model. Mol Cell Biol (2012) 32(20):4104–15. doi: 10.1128/MCB.00862-12
72. Mao J, Ligon KL, Rakhlin EY, Thayer SP, Bronson RT, Rowitch D, et al. A novel somatic mouse model to survey tumorigenic potential applied to the Hedgehog pathway. Cancer Res (2006) 66(20):10171–8. doi: 10.1158/0008-5472.CAN-06-0657
73. Lee Y, Kawagoe R, Sasai K, Li Y, Russell HR, Curran T, et al. Loss of suppressor-of-fused function promotes tumorigenesis. Oncogene (2007) 26(44):6442–7. doi: 10.1038/sj.onc.1210467
74. Zhu G, Rankin SL, Larson JD, Zhu X, Chow LM, Qu C, et al. PTEN Signaling in the Postnatal Perivascular Progenitor Niche Drives Medulloblastoma Formation. Cancer Res (2017) 77(1):123–33. doi: 10.1158/0008-5472.CAN-16-1991
75. Tong WM, Ohgaki H, Huang H, Granier C, Kleihues P, Wang ZQ. Null mutation of DNA strand break-binding molecule poly(ADP-ribose) polymerase causes medulloblastomas in p53(-/-) mice. Am J Pathol (2003) 162(1):343–52. doi: 10.1016/S0002-9440(10)63825-4
76. Browd SR, Kenney AM, Gottfried ON, Yoon JW, Walterhouse D, Pedone CA, et al. N-myc can substitute for insulin-like growth factor signaling in a mouse model of sonic hedgehog-induced medulloblastoma. Cancer Res (2006) 66(5):2666–72. doi: 10.1158/0008-5472.CAN-05-2198
77. McCall TD, Pedone CA, Fults DW. Apoptosis suppression by somatic cell transfer of Bcl-2 promotes Sonic hedgehog-dependent medulloblastoma formation in mice. Cancer Res (2007) 67(11):5179–85. doi: 10.1158/0008-5472.CAN-06-4177
78. Swartling FJ, Grimmer MR, Hackett CS, Northcott PA, Fan QW, Goldenberg DD, et al. Pleiotropic role for MYCN in medulloblastoma. Genes Dev (2010) 24(10):1059–72. doi: 10.1101/gad.1907510
79. Hill RM, Kuijper S, Lindsey JC, Petrie K, Schwalbe EC, Barker K, et al. and P53 defects emerge at medulloblastoma relapse and define rapidly progressive, therapeutically targetable disease. Cancer Cell (2015) 27(1):72–84. doi: 10.1016/j.ccell.2014.11.002
80. Dhar SS, Zhao D, Lin T, Gu B, Pal K, Wu SJ, et al. MLL4 Is Required to Maintain Broad H3K4me3 Peaks and Super-Enhancers at Tumor Suppressor Genes. Mol Cell (2018) 70(5):825–41 e6. doi: 10.1016/j.molcel.2018.04.028
81. Jenkins NC, Rao G, Eberhart CG, Pedone CA, Dubuc AM, Fults DW. Somatic cell transfer of c-Myc and Bcl-2 induces large-cell anaplastic medulloblastomas in mice. J Neurooncol (2016) 126(3):415–24. doi: 10.1007/s11060-015-1985-9
82. Kawauchi D, Ogg RJ, Liu L, Shih DJH, Finkelstein D, Murphy BL, et al. Novel MYC-driven medulloblastoma models from multiple embryonic cerebellar cells. Oncogene (2017) 36(37):5231–42. doi: 10.1038/onc.2017.110
83. Forget A, Martignetti L, Puget S, Calzone L, Brabetz S, Picard D, et al. Aberrant ERBB4-SRC Signaling as a Hallmark of Group 4 Medulloblastoma Revealed by Integrative Phosphoproteomic Profiling. Cancer Cell (2018) 34(3):379–95 e7. doi: 10.1016/j.ccell.2018.08.002
84. Reilly KM, Loisel DA, Bronson RT, McLaughlin ME, Jacks T. Nf1;Trp53 mutant mice develop glioblastoma with evidence of strain-specific effects. Nat Genet (2000) 26(1):109–13. doi: 10.1038/79075
85. Zhu Y, Guignard F, Zhao D, Liu L, Burns DK, Mason RP, et al. Early inactivation of p53 tumor suppressor gene cooperating with NF1 loss induces malignant astrocytoma. Cancer Cell (2005) 8(2):119–30. doi: 10.1016/j.ccr.2005.07.004
86. Kwon CH, Zhao D, Chen J, Alcantara S, Li Y, Burns DK, et al. Pten haploinsufficiency accelerates formation of high-grade astrocytomas. Cancer Res (2008) 68(9):3286–94. doi: 10.1158/0008-5472.CAN-07-6867
87. Xiao A, Wu H, Pandolfi PP, Louis DN, Van Dyke T. Astrocyte inactivation of the pRb pathway predisposes mice to malignant astrocytoma development that is accelerated by PTEN mutation. Cancer Cell (2002) 1(2):157–68. doi: 10.1016/s1535-6108(02)00029-6
88. Ding H, Roncari L, Shannon P, Wu X, Lau N, Karaskova J, et al. Astrocyte-specific expression of activated p21-ras results in malignant astrocytoma formation in a transgenic mouse model of human gliomas. Cancer Res (2001) 61(9):3826–36.
89. Ding H, Shannon P, Lau N, Wu X, Roncari L, Baldwin RL, et al. Oligodendrogliomas result from the expression of an activated mutant epidermal growth factor receptor in a RAS transgenic mouse astrocytoma model. Cancer Res (2003) 63(5):1106–13.
90. Wei Q, Clarke L, Scheidenhelm DK, Qian B, Tong A, Sabha N, et al. High-grade glioma formation results from postnatal pten loss or mutant epidermal growth factor receptor expression in a transgenic mouse glioma model. Cancer Res (2006) 66(15):7429–37. doi: 10.1158/0008-5472.CAN-06-0712
91. Weiss WA, Burns MJ, Hackett C, Aldape K, Hill JR, Kuriyama H, et al. Genetic determinants of malignancy in a mouse model for oligodendroglioma. Cancer Res (2003) 63(7):1589–95.
92. Holland EC, Celestino J, Dai C, Schaefer L, Sawaya RE, Fuller GN. Combined activation of Ras and Akt in neural progenitors induces glioblastoma formation in mice. Nat Genet (2000) 25(1):55–7. doi: 10.1038/75596
93. Hu X, Pandolfi PP, Li Y, Koutcher JA, Rosenblum M, Holland EC. mTOR promotes survival and astrocytic characteristics induced by Pten/AKT signaling in glioblastoma. Neoplasia (2005) 7(4):356–68. doi: 10.1593/neo.04595
94. Dai C, Celestino JC, Okada Y, Louis DN, Fuller GN, Holland EC. PDGF autocrine stimulation dedifferentiates cultured astrocytes and induces oligodendrogliomas and oligoastrocytomas from neural progenitors and astrocytes in vivo. Genes Dev (2001) 15(15):1913–25. doi: 10.1101/gad.903001
95. Becher OJ, Hambardzumyan D, Walker TR, Helmy K, Nazarian J, Albrecht S, et al. Preclinical evaluation of radiation and perifosine in a genetically and histologically accurate model of brainstem glioma. Cancer Res (2010) 70(6):2548–57. doi: 10.1158/0008-5472.CAN-09-2503
96. Barton KL, Misuraca K, Cordero F, Dobrikova E, Min HD, Gromeier M, et al. PD-0332991, a CDK4/6 inhibitor, significantly prolongs survival in a genetically engineered mouse model of brainstem glioma. PloS One (2013) 8(10):e77639. doi: 10.1371/journal.pone.0077639
97. Halvorson KG, Barton KL, Schroeder K, Misuraca KL, Hoeman C, Chung A, et al. A high-throughput in vitro drug screen in a genetically engineered mouse model of diffuse intrinsic pontine glioma identifies BMS-754807 as a promising therapeutic agent. PloS One (2015) 10(3):e0118926. doi: 10.1371/journal.pone.0118926
98. Kim GB, Rincon Fernandez Pacheco D, Saxon D, Yang A, Sabet S, Dutra-Clarke M, et al. Rapid Generation of Somatic Mouse Mosaics with Locus-Specific, Stably Integrated Transgenic Elements. Cell (2019) 179(1):251–67 e24. doi: 10.1016/j.cell.2019.08.013
99. Breunig JJ, Levy R, Antonuk CD, Molina J, Dutra-Clarke M, Park H, et al. Ets Factors Regulate Neural Stem Cell Depletion and Gliogenesis in Ras Pathway Glioma. Cell Rep (2015) 12(2):258–71. doi: 10.1016/j.celrep.2015.06.012
100. Chen F, Becker AJ, LoTurco JJ. Contribution of tumor heterogeneity in a new animal model of CNS tumors. Mol Cancer Res (2014) 12(5):742–53. doi: 10.1158/1541-7786.MCR-13-0531
101. Chen F, Rosiene J, Che A, Becker A, LoTurco J. Tracking and transforming neocortical progenitors by CRISPR/Cas9 gene targeting and piggyBac transposase lineage labeling. Development (2015) 142(20):3601–11. doi: 10.1242/dev.118836
102. Ozawa T, Arora S, Szulzewsky F, Juric-Sekhar G, Miyajima Y, Bolouri H, et al. A De Novo Mouse Model of C11orf95-RELA Fusion-Driven Ependymoma Identifies Driver Functions in Addition to NF-kappaB. Cell Rep (2018) 23(13):3787–97. doi: 10.1016/j.celrep.2018.04.099
103. Pajtler KW, Wei Y, Okonechnikov K, Silva PBG, Vouri M, Zhang L, et al. YAP1 subgroup supratentorial ependymoma requires TEAD and nuclear factor I-mediated transcriptional programmes for tumorigenesis. Nat Commun (2019) 10(1):3914. doi: 10.1038/s41467-019-11884-5
104. Eder N, Roncaroli F, Domart MC, Horswell S, Andreiuolo F, Flynn HR, et al. Author Correction: YAP1/TAZ drives ependymoma-like tumour formation in mice. Nat Commun (2020) 11(1):4934. doi: 10.1038/s41467-020-18851-5
105. Eder N, Roncaroli F, Domart MC, Horswell S, Andreiuolo F, Flynn HR, et al. YAP1/TAZ drives ependymoma-like tumour formation in mice. Nat Commun (2020) 11(1):2380. doi: 10.1038/s41467-020-16167-y
106. Kim H, Kim M, Im SK, Fang S. Mouse Cre-LoxP system: general principles to determine tissue-specific roles of target genes. Lab Anim Res (2018) 34(4):147–59. doi: 10.5625/lar.2018.34.4.147
107. von Werder A, Seidler B, Schmid RM, Schneider G, Saur D. Production of avian retroviruses and tissue-specific somatic retroviral gene transfer in vivo using the RCAS/TVA system. Nat Protoc (2012) 7(6):1167–83. doi: 10.1038/nprot.2012.060
108. Weber J, Rad R. Engineering CRISPR mouse models of cancer. Curr Opin Genet Dev (2019) 54:88–96. doi: 10.1016/j.gde.2019.04.001
109. Wu X, Northcott PA, Croul S, Taylor MD. Mouse models of medulloblastoma. Chin J Cancer (2011) 30(7):442–9. doi: 10.5732/cjc.011.10040
110. Tamayo-Orrego L, Charron F. Recent advances in SHH medulloblastoma progression: tumor suppressor mechanisms and the tumor microenvironment. F1000Res (2019) 8(F1000 Faculty Rev):1823. doi: 10.12688/f1000research.20013.1
111. Balkwill FR, Capasso M, Hagemann T. The tumor microenvironment at a glance. J Cell Sci (2012) 125(23):5591–6. doi: 10.1242/jcs.116392
112. Egeblad M, Nakasone ES, Werb Z. Tumors as organs: complex tissues that interface with the entire organism. Dev Cell (2010) 18(6):884–901. doi: 10.1016/j.devcel.2010.05.012S1534-5807(10)00248-0
113. Dickreuter E, Cordes N. The cancer cell adhesion resistome: mechanisms, targeting and translational approaches. Biol Chem (2017) 398(7):721–35. doi: 10.1515/hsz-2016-0326
114. Holle AW, Young JL, Spatz JP. In vitro cancer cell-ECM interactions inform in vivo cancer treatment. Adv Drug Delivery Rev (2016) 97:270–9. doi: 10.1016/j.addr.2015.10.007S0169-409X(15)00232-X
115. Henke E, Nandigama R, Ergun S. Extracellular Matrix in the Tumor Microenvironment and Its Impact on Cancer Therapy. Front Mol Biosci (2019) 6:160. doi: 10.3389/fmolb.2019.00160
116. Holohan C, Van Schaeybroeck S, Longley DB, Johnston PG. Cancer drug resistance: an evolving paradigm. Nat Rev Cancer (2013) 13(10):714–26. doi: 10.1038/nrc3599nrc3599
117. Paolillo M, Schinelli S. Extracellular Matrix Alterations in Metastatic Processes. Int J Mol Sci (2019) 20(19):4947. doi: 10.3390/ijms20194947
118. Junttila MR, de Sauvage FJ. Influence of tumour micro-environment heterogeneity on therapeutic response. Nature (2013) 501(7467):346–54. doi: 10.1038/nature12626nature12626
119. McMillin DW, Negri JM, Mitsiades CS. The role of tumour-stromal interactions in modifying drug response: challenges and opportunities. Nat Rev Drug Discovery (2013) 12(3):217–28. doi: 10.1038/nrd3870nrd3870
120. Li F, Simon MC. Cancer Cells Don’t Live Alone: Metabolic Communication within Tumor Microenvironments. Dev Cell (2020) 54(2):183–95. doi: 10.1016/j.devcel.2020.06.018
121. Turley SJ, Cremasco V, Astarita JL. Immunological hallmarks of stromal cells in the tumour microenvironment. Nat Rev Immunol (2015) 15(11):669–82. doi: 10.1038/nri3902nri3902
122. Hanahan D, Coussens LM. Accessories to the crime: functions of cells recruited to the tumor microenvironment. Cancer Cell (2012) 21(3):309–22. doi: 10.1016/j.ccr.2012.02.022
123. Maximov V, Chen Z, Wei Y, Robinson MH, Herting CJ, Shanmugam NS, et al. Tumour-associated macrophages exhibit anti-tumoural properties in Sonic Hedgehog medulloblastoma. Nat Commun (2019) 10(1):2410. doi: 10.1038/s41467-019-10458-9
124. Gate D, Danielpour M, Rodriguez J Jr., Kim G-B, Levy R, Bannykh S, et al. T-cell TGF-β signaling abrogation restricts medulloblastoma progression. Proc Natl Acad Sci USA (2014) 111(33):E3458–E66. doi: 10.1073/pnas.1412489111
125. Liu Y, Yuelling LW, Wang Y, Du F, Gordon RE, O’Brien JA, et al. Astrocytes Promote Medulloblastoma Progression through Hedgehog Secretion. Cancer Res (2017) 77(23):6692–703. doi: 10.1158/0008-5472.Can-17-1463
126. Plaks V, Kong N, Werb Z. The cancer stem cell niche: how essential is the niche in regulating stemness of tumor cells? Cell Stem Cell (2015) 16(3):225–38. doi: 10.1016/j.stem.2015.02.015
127. Hambardzumyan D, Becher OJ, Rosenblum MK, Pandolfi PP, Manova-Todorova K, Holland EC. PI3K pathway regulates survival of cancer stem cells residing in the perivascular niche following radiation in medulloblastoma. Vivo Genes Dev (2008) 22(4):436–48. doi: 10.1101/gad.1627008
128. Uhrbom L, Dai C, Celestino JC, Rosenblum MK, Fuller GN, Holland EC. Ink4a-Arf loss cooperates with KRas activation in astrocytes and neural progenitors to generate glioblastomas of various morphologies depending on activated Akt. Cancer Res (2002) 62(19):5551–8.
129. Xu L, Chen Y, Dutra-Clarke M, Mayakonda A, Hazawa M, Savinoff SE, et al. BCL6 promotes glioma and serves as a therapeutic target. Proc Natl Acad Sci USA (2017) 114(15):3981–6. doi: 10.1073/pnas.1609758114
130. Welby JP, Kaptzan T, Wohl A, Peterson TE, Raghunathan A, Brown DA, et al. Current Murine Models and New Developments in H3K27M Diffuse Midline Gliomas. Front Oncol (2019) 9:92. doi: 10.3389/fonc.2019.00092
131. Chen F, Becker A, LoTurco J. Overview of Transgenic Glioblastoma and Oligoastrocytoma CNS Models and Their Utility in Drug Discovery. Curr Protoc Pharmacol (2016) 72:14 37 1–14 37 12. doi: 10.1002/0471141755.ph1437s72
132. Casey MJ, Stewart RA. Pediatric Cancer Models in Zebrafish. Trends Cancer (2020) 6(5):407–18. doi: 10.1016/j.trecan.2020.02.006
133. Wu G, Diaz AK, Paugh BS, Rankin SL, Ju B, Li Y, et al. The genomic landscape of diffuse intrinsic pontine glioma and pediatric non-brainstem high-grade glioma. Nat Genet (2014) 46(5):444–50. doi: 10.1038/ng.2938
134. Buczkowicz P, Hoeman C, Rakopoulos P, Pajovic S, Letourneau L, Dzamba M, et al. Genomic analysis of diffuse intrinsic pontine gliomas identifies three molecular subgroups and recurrent activating ACVR1 mutations. Nat Genet (2014) 46(5):451–6. doi: 10.1038/ng.2936
135. Eden CJ, Ju B, Murugesan M, Phoenix TN, Nimmervoll B, Tong Y, et al. Orthotopic models of pediatric brain tumors in zebrafish. Oncogene (2015) 34(13):1736–42. doi: 10.1038/onc.2014.107
136. Idilli AI, Pagani F, Kerschbamer E, Berardinelli F, Bernabe M, Cayuela ML, et al. Changes in the Expression of Pre-Replicative Complex Genes in hTERT and ALT Pediatric Brain Tumors. Cancers (Basel) (2020) 12(4):1028. doi: 10.3390/cancers12041028
137. Casey MJ, Modzelewska K, Anderson D, Goodman J, Boer EF, Jimenez L, et al. Transplantation of Zebrafish Pediatric Brain Tumors into Immune-competent Hosts for Long-term Study of Tumor Cell Behavior and Drug Response. J Vis Exp (2017) 123:e55712. doi: 10.3791/55712
138. Othman RT, Kimishi I, Bradshaw TD, Storer LC, Korshunov A, Pfister SM, et al. Overcoming multiple drug resistance mechanisms in medulloblastoma. Acta Neuropathol Commun (2014) 2:57. doi: 10.1186/2051-5960-2-57
139. Triscott J, Lee C, Foster C, Manoranjan B, Pambid MR, Berns R, et al. Personalizing the treatment of pediatric medulloblastoma: Polo-like kinase 1 as a molecular target in high-risk children. Cancer Res (2013) 73(22):6734–44. doi: 10.1158/0008-5472.CAN-12-43310008-5472.CAN-12-4331
140. Keles GE, Berger MS, Srinivasan J, Kolstoe DD, Bobola MS, Silber JR. Establishment and characterization of four human medulloblastoma-derived cell lines. Oncol Res (1995) 7(10-11):493–503.
141. Snuderl M, Batista A, Kirkpatrick ND, Ruiz de Almodovar C, Riedemann L, Walsh EC, et al. Targeting placental growth factor/neuropilin 1 pathway inhibits growth and spread of medulloblastoma. Cell (2013) 152(5):1065–76. doi: 10.1016/j.cell.2013.01.036
142. Friedman HS, Burger PC, Bigner SH, Trojanowski JQ, Brodeur GM, He XM, et al. Phenotypic and genotypic analysis of a human medulloblastoma cell line and transplantable xenograft (D341 Med) demonstrating amplification of c-myc. Am J Pathol (1988) 130(3):472–84.
143. Friedman GK, Moore BP, Nan L, Kelly VM, Etminan T, Langford CP, et al. Pediatric medulloblastoma xenografts including molecular subgroup 3 and CD133+ and CD15+ cells are sensitive to killing by oncolytic herpes simplex viruses. Neuro Oncol (2016) 18(2):227–35. doi: 10.1093/neuonc/nov123
144. Thompson EM, Keir ST, Venkatraman T, Lascola C, Yeom KW, Nixon AB, et al. The role of angiogenesis in Group 3 medulloblastoma pathogenesis and survival. Neuro Oncol (2017) 19(9):1217–27. doi: 10.1093/neuonc/nox033
145. Ivanov DP, Coyle B, Walker DA, Grabowska AM. In vitro models of medulloblastoma: Choosing the right tool for the job. J Biotechnol (2016) 236:10–25. doi: 10.1016/j.jbiotec.2016.07.028S0168-1656(16)31438-9
146. Friedman HS, Burger PC, Bigner SH, Trojanowski JQ, Wikstrand CJ, Halperin EC, et al. Establishment and characterization of the human medulloblastoma cell line and transplantable xenograft D283 Med. J Neuropathol Exp Neurol (1985) 44(6):592–605. doi: 10.1097/00005072-198511000-00005
147. Northcott PA, Shih DJ, Peacock J, Garzia L, Morrissy AS, Zichner T, et al. Subgroup-specific structural variation across 1,000 medulloblastoma genomes. Nature (2012) 488(7409):49–56. doi: 10.1038/nature11327
148. Milde T, Lodrini M, Savelyeva L, Korshunov A, Kool M, Brueckner LM, et al. HD-MB03 is a novel Group 3 medulloblastoma model demonstrating sensitivity to histone deacetylase inhibitor treatment. J Neurooncol (2012) 110(3):335–48. doi: 10.1007/s11060-012-0978-1
149. Bandopadhayay P, Bergthold G, Nguyen B, Schubert S, Gholamin S, Tang Y, et al. BET bromodomain inhibition of MYC-amplified medulloblastoma. Clin Cancer Res (2014) 20(4):912–25. doi: 10.1158/1078-0432.CCR-13-2281
150. Xu J, Margol A, Asgharzadeh S, Erdreich-Epstein A. Pediatric brain tumor cell lines. J Cell Biochem (2015) 116(2):218–24. doi: 10.1002/jcb.24976
151. Xu J, Erdreich-Epstein A, Gonzalez-Gomez I, Melendez EY, Smbatyan G, Moats RA, et al. Novel cell lines established from pediatric brain tumors. J Neurooncol (2012) 107(2):269–80. doi: 10.1007/s11060-011-0756-5
152. Hashizume R, Smirnov I, Liu S, Phillips JJ, Hyer J, McKnight TR, et al. Characterization of a diffuse intrinsic pontine glioma cell line: implications for future investigations and treatment. J Neurooncol (2012) 110(3):305–13. doi: 10.1007/s11060-012-0973-6
153. Chan KM, Fang D, Gan H, Hashizume R, Yu C, Schroeder M, et al. The histone H3.3K27M mutation in pediatric glioma reprograms H3K27 methylation and gene expression. Genes Dev (2013) 27(9):985–90. doi: 10.1101/gad.217778.113
154. Grasso CS, Tang Y, Truffaux N, Berlow NE, Liu L, Debily MA, et al. Functionally defined therapeutic targets in diffuse intrinsic pontine glioma. Nat Med (2015) 21(6):555–9. doi: 10.1158/1535-7163.TARG-15-LB-B06
155. Donson AM, Amani V, Warner EA, Griesinger AM, Witt DA, Levy JMM, et al. Identification of FDA-Approved Oncology Drugs with Selective Potency in High-Risk Childhood Ependymoma. Mol Cancer Ther (2018) 17(9):1984–94. doi: 10.1158/1535-7163.MCT-17-1185
156. Lee J, Kotliarova S, Kotliarov Y, Li A, Su Q, Donin NM, et al. Tumor stem cells derived from glioblastomas cultured in bFGF and EGF more closely mirror the phenotype and genotype of primary tumors than do serum-cultured cell lines. Cancer Cell (2006) 9(5):391–403. doi: 10.1016/j.ccr.2006.03.030
157. Kapalczynska M, Kolenda T, Przybyla W, Zajaczkowska M, Teresiak A, Filas V, et al. 2D and 3D cell cultures - a comparison of different types of cancer cell cultures. Arch Med Sci (2018) 14(4):910–9. doi: 10.5114/aoms.2016.63743
158. Caragher S, Chalmers AJ, Gomez-Roman N. Glioblastoma’s Next Top Model: Novel Culture Systems for Brain Cancer Radiotherapy Research. Cancers (Basel) (2019) 11(1):44. doi: 10.3390/cancers11010044
159. Monje M, Mitra SS, Freret ME, Raveh TB, Kim J, Masek M, et al. Hedgehog-responsive candidate cell of origin for diffuse intrinsic pontine glioma. Proc Natl Acad Sci USA (2011) 108(11):4453–8. doi: 10.1073/pnas.1101657108
160. Laks DR, Masterman-Smith M, Visnyei K, Angenieux B, Orozco NM, Foran I, et al. Neurosphere formation is an independent predictor of clinical outcome in malignant glioma. Stem Cells (2009) 27(4):980–7. doi: 10.1002/stem.15
161. Xiao W, Sohrabi A, Seidlits SK. Integrating the glioblastoma microenvironment into engineered experimental models. Future Sci OA (2017) 3(3):FSO189. doi: 10.4155/fsoa-2016-0094
162. Langhans SA. Three-Dimensional in Vitro Cell Culture Models in Drug Discovery and Drug Repositioning. Front Pharmacol (2018) 9:6. doi: 10.3389/fphar.2018.00006
163. Lovett ML, Nieland TJF, Dingle YTL, Kaplan DL. Innovations in 3D Tissue Models of Human Brain Physiology and Diseases. Adv Funct Mater (2020) n/a(n/a):1909146. doi: 10.1002/adfm.201909146
164. Amani V, Donson AM, Lummus SC, Prince EW, Griesinger AM, Witt DA, et al. Characterization of 2 Novel Ependymoma Cell Lines With Chromosome 1q Gain Derived From Posterior Fossa Tumors of Childhood. J Neuropathol Exp Neurol (2017) 76(7):595–604. doi: 10.1093/jnen/nlx040
165. Nath S, Devi GR. Three-dimensional culture systems in cancer research: Focus on tumor spheroid model. Pharmacol Ther (2016) 163:94–108. doi: 10.1016/j.pharmthera.2016.03.013S0163-7258(16)30021-3
166. Sant S, Johnston PA. The production of 3D tumor spheroids for cancer drug discovery. Drug Discovery Today Technol (2017) 23:27–36. doi: 10.1016/j.ddtec.2017.03.002
167. Costa EC, Moreira AF, de Melo-Diogo D, Gaspar VM, Carvalho MP, Correia IJ. 3D tumor spheroids: an overview on the tools and techniques used for their analysis. Biotechnol Adv (2016) 34(8):1427–41. doi: 10.1016/j.biotechadv.2016.11.002
168. Frantz C, Stewart KM, Weaver VM. The extracellular matrix at a glance. J Cell Sci (2010) 123(Pt 24):4195–200. doi: 10.1242/jcs.023820
169. Mouw JK, Ou G, Weaver VM. Extracellular matrix assembly: a multiscale deconstruction. Nat Rev Mol Cell Biol (2014) 15(12):771–85. doi: 10.1038/nrm3902nrm3902
170. Hughes CS, Postovit LM, Lajoie GA. Matrigel: a complex protein mixture required for optimal growth of cell culture. Proteomics (2010) 10(9):1886–90. doi: 10.1002/pmic.200900758
171. Wang C, Tong X, Yang F. Bioengineered 3D brain tumor model to elucidate the effects of matrix stiffness on glioblastoma cell behavior using PEG-based hydrogels. Mol Pharm (2014) 11(7):2115–25. doi: 10.1021/mp5000828
172. Worthington P, Drake KM, Li Z, Napper AD, Pochan DJ, Langhans SA. Beta-hairpin hydrogels as scaffolds for high-throughput drug discovery in three-dimensional cell culture. Anal Biochem (2017) 535:25–34. doi: 10.1016/j.ab.2017.07.024
173. Worthington P, Drake KM, Li Z, Napper AD, Pochan DJ, Langhans SA. Implementation of a High-Throughput Pilot Screen in Peptide Hydrogel-Based Three-Dimensional Cell Cultures. SLAS Discovery (2019) 24(7):714–23. doi: 10.1177/2472555219844570
174. Tang-Schomer MD, White JD, Tien LW, Schmitt LI, Valentin TM, Graziano DJ, et al. Bioengineered functional brain-like cortical tissue. Proc Natl Acad Sci USA (2014) 111(38):13811–6. doi: 10.1073/pnas.1324214111
175. Chwalek K, Sood D, Cantley WL, White JD, Tang-Schomer M, Kaplan DL. Engineered 3D Silk-collagen-based Model of Polarized Neural Tissue. J Vis Exp (2015) 105):e52970. doi: 10.3791/52970
176. Sood D, Tang-Schomer M, Pouli D, Mizzoni C, Raia N, Tai A, et al. 3D extracellular matrix microenvironment in bioengineered tissue models of primary pediatric and adult brain tumors. Nat Commun (2019) 10(1):4529. doi: 10.1038/s41467-019-12420-1
177. Li Q, Lin H, Rauch J, Deleyrolle LP, Reynolds BA, Viljoen HJ, et al. Scalable Culturing of Primary Human Glioblastoma Tumor-Initiating Cells with a Cell-Friendly Culture System. Sci Rep (2018) 8(1):3531. doi: 10.1038/s41598-018-21927-4
178. Lancaster MA, Knoblich JA. Generation of cerebral organoids from human pluripotent stem cells. Nat Protoc (2014) 9(10):2329–40. doi: 10.1038/nprot.2014.158
179. Ballabio C, Anderle M, Gianesello M, Lago C, Miele E, Cardano M, et al. Modeling medulloblastoma in vivo and with human cerebellar organoids. Nat Commun (2020) 11(1):583. doi: 10.1038/s41467-019-13989-3
180. Ogawa J, Pao GM, Shokhirev MN, Verma IM. Glioblastoma Model Using Human Cerebral Organoids. Cell Rep (2018) 23(4):1220–9. doi: 10.1016/j.celrep.2018.03.105
181. Linkous A, Balamatsias D, Snuderl M, Edwards L, Miyaguchi K, Milner T, et al. Modeling Patient-Derived Glioblastoma with Cerebral Organoids. Cell Rep (2019) 26(12):3203–11 e5. doi: 10.1016/j.celrep.2019.02.063
182. Hubert CG, Rivera M, Spangler LC, Wu Q, Mack SC, Prager BC, et al. A Three-Dimensional Organoid Culture System Derived from Human Glioblastomas Recapitulates the Hypoxic Gradients and Cancer Stem Cell Heterogeneity of Tumors Found In Vivo. Cancer Res (2016) 76(8):2465–77. doi: 10.1158/0008-5472.CAN-15-2402
183. Drost J, Clevers H. Organoids in cancer research. Nat Rev Cancer (2018) 18(7):407–18. doi: 10.1038/s41568-018-0007-6
184. Perrin S. Preclinical research: Make mouse studies work. Nature (2014) 507(7493):423–5. doi: 10.1038/507423a
Keywords: medulloblastoma, glioma, pediatrics, preclinical models, in vivo models, in vitro models, cancer
Citation: Li Z and Langhans SA (2021) In Vivo and Ex Vivo Pediatric Brain Tumor Models: An Overview. Front. Oncol. 11:620831. doi: 10.3389/fonc.2021.620831
Received: 23 October 2020; Accepted: 15 March 2021;
Published: 01 April 2021.
Edited by:
Jing He, Guangzhou Medical University, ChinaReviewed by:
Moise Danielpour, Cedars Sinai Medical Center, United StatesXinhui Du, Zhengzhou University, China
Copyright © 2021 Li and Langhans. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sigrid A. Langhans, c2lncmlkLmxhbmdoYW5zQG5lbW91cnMub3Jn