 Rebekah L. I. Crake1†
Rebekah L. I. Crake1† Eleanor R. Burgess1†
Eleanor R. Burgess1† Janice A. Royds2
Janice A. Royds2 Elisabeth Phillips1
Elisabeth Phillips1 Margreet C. M. Vissers3
Margreet C. M. Vissers3 Gabi U. Dachs1*
Gabi U. Dachs1*- 1Mackenzie Cancer Research Group, Department of Pathology and Biomedical Science, University of Otago Christchurch, Christchurch, New Zealand
- 2Department of Pathology, University of Otago, Dunedin, New Zealand
- 3Centre for Free Radical Research, Department of Pathology and Biomedical Science, University of Otago Christchurch, Christchurch, New Zealand
Gliomas are a heterogeneous group of cancers that predominantly arise from glial cells in the brain, but may also arise from neural stem cells, encompassing low-grade glioma and high-grade glioblastoma. Whereas better diagnosis and new treatments have improved patient survival for many cancers, glioblastomas remain challenging with a highly unfavorable prognosis. This review discusses a super-family of enzymes, the 2-oxoglutarate dependent dioxygenase enzymes (2-OGDD) that control numerous processes including epigenetic modifications and oxygen sensing, and considers their many roles in the pathology of gliomas. We specifically describe in more detail the DNA and histone demethylases, and the hypoxia-inducible factor hydroxylases in the context of glioma, and discuss the substrate and cofactor requirements of the 2-OGDD enzymes. Better understanding of how these enzymes contribute to gliomas could lead to the development of new treatment strategies.
Introduction
Gliomas and glioblastomas are brain cancers with significant morbidity and mortality, and limited treatment options. Our review will briefly describe these neoplasms, then concentrate on a super-family of enzymes, the 2-oxoglutarate dependent dioxygenase enzymes (2-OGDD), with dozens of members currently known (Figure 1). 2-OGDDs participate in numerous processes including collagen and hormone synthesis, fatty acid metabolism, stress signaling, epigenetic modifications and oxygen sensing (2–7). We will discuss specific members of the 2-OGDD family that have attracted recent interest, including the DNA demethylases [ten-eleven translocases (TET)], the histone demethylases [Jumonji-C domain-containing demethylases (JmjC)], and the hypoxia-inducible factor (HIF) hydroxylases. These enzymes require molecular oxygen and 2-oxoglutarate [2-OG, produced by isocitrate dehydrogenase (IDH)] as substrates, and non-ferrous iron (Fe2+) and vitamin C (ascorbate) as cofactors. Decreased availability of either substrate or co-factors reduces 2-OGDD enzyme activity, resulting in these enzymes acting as cellular sensors of energy metabolism, oxygen availability, and iron homeostasis (2–4, 8–14). Finally, we consider the potential means of modifying the activity of the 2-OGDDs, specifically by modulating ascorbate availability.
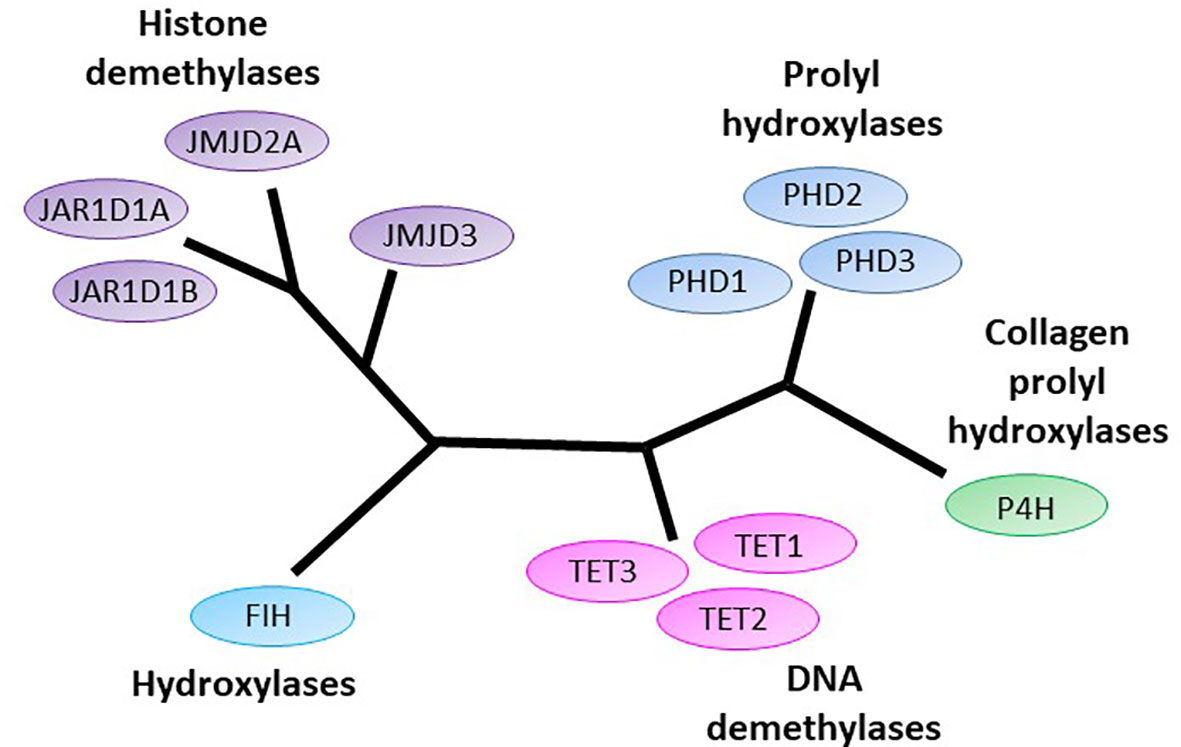
Figure 1 The human 2-oxoglutarate dependent dioxygenases implicated in initiation and progression of gliomas. 2-oxoglutarate dependent dioxygenases (2-OGDDs) include the DNA demethylases Ten-eleven translocases (TETs), the Jumonji-C domain containing histone demethylases (JMJD and JAR1Ds), the prolyl hydroxylases (PHDs) and hydroxylases that control hypoxic response (factor inhibiting HIF, FIH), and the collagen prolyl hydroxylases (P4H). Colors are used to indicate close phylogenetic relationships [adapted from Johansson (1)].
Glioma and Glioblastoma and Treatment Options
Gliomas are a heterogeneous group of neoplasms that arise from glial cells in the brain (15, 16), or neural stem cells (17). Low-grade gliomas, grades II and III, include astrocytomas and oligodendrogliomas which predominantly develop from astrocytes or oligodendrocytes, respectively. Glioblastomas (GBM) are grade IV and can develop either as a high-grade lesion (primary GBM), or from astrocytoma progression (secondary GBM) (15, 16).
Gliomas accounted for 1.6% of cancer diagnoses and 2.5% of cancer related deaths worldwide in 2018 (18). Astrocytomas, oligodendrogliomas and secondary GBM tend to develop in younger individuals (median age 35, 45, and 38 years, respectively), compared to primary GBM (median age of 55 years) (17, 19). Gliomas are identified by magnetic resonance imaging (MRI) or computed tomography (CT/CAT). Although treatment options are available as detailed below, high grade gliomas are incurable (20–23). Some low grade gliomas can be cured, although these are rare (17). Following diagnosis, the median survival for patients with oligodendroglioma can be up to 16 years, for astrocytoma 5–8 years, and for GBM only 15–31 months (17, 24).
The current standard treatment for gliomas focuses on extending patient survival and includes maximal safe debulking surgery followed by radiotherapy and concomitant or adjuvant chemotherapy (20), usually with the alkylating agent temozolomide (20, 21, 25, 26). Debulking surgery is performed to reduce intracranial pressure and neurological symptoms, while resected tissue is utilized for tumor classification (17, 22, 23). Post-treatment recurrence is common with approximately 80% of gliomas recurring in close proximity to the primary site (27). Gliomas treated with temozolomide often hypermutate at recurrence leading to treatment resistance, as evidenced by the mere 20% of recurring gliomas showing response to the same agent (17, 28). Dissemination beyond the brain is uncommon, but some high grade gliomas may spread into the meninges or opposing brain hemisphere (17).
Besides traditional oncogenic drivers, gliomas are characterized by deregulated epigenetics and high levels of hypoxia (low oxygen), processes which are largely regulated by 2-OGDDs.
2-OGDD’s and Epigenetic Reprogramming
A significant proportion of 2-OGDDs in mammals are demethylases involved in epigenetic reprogramming (6, 29). Methylation and demethylation of DNA and histones are the fundamental processes guiding epigenetic inheritance and regulating transcriptional activation and repression (30, 31). Consequently, aberrant modifications to these epigenetic processes, causing hyper- and hypo-methylating events, have emerged as hallmarks of cancer progression (32, 33). Demethylases regulate the epigenome through either the conversion of methylated cytosine (5-mC) to hydroxymethylcytosine (5-hmC) on DNA (Figure 2), or the removal of methyl groups from lysine residues on histones (Figure 3).
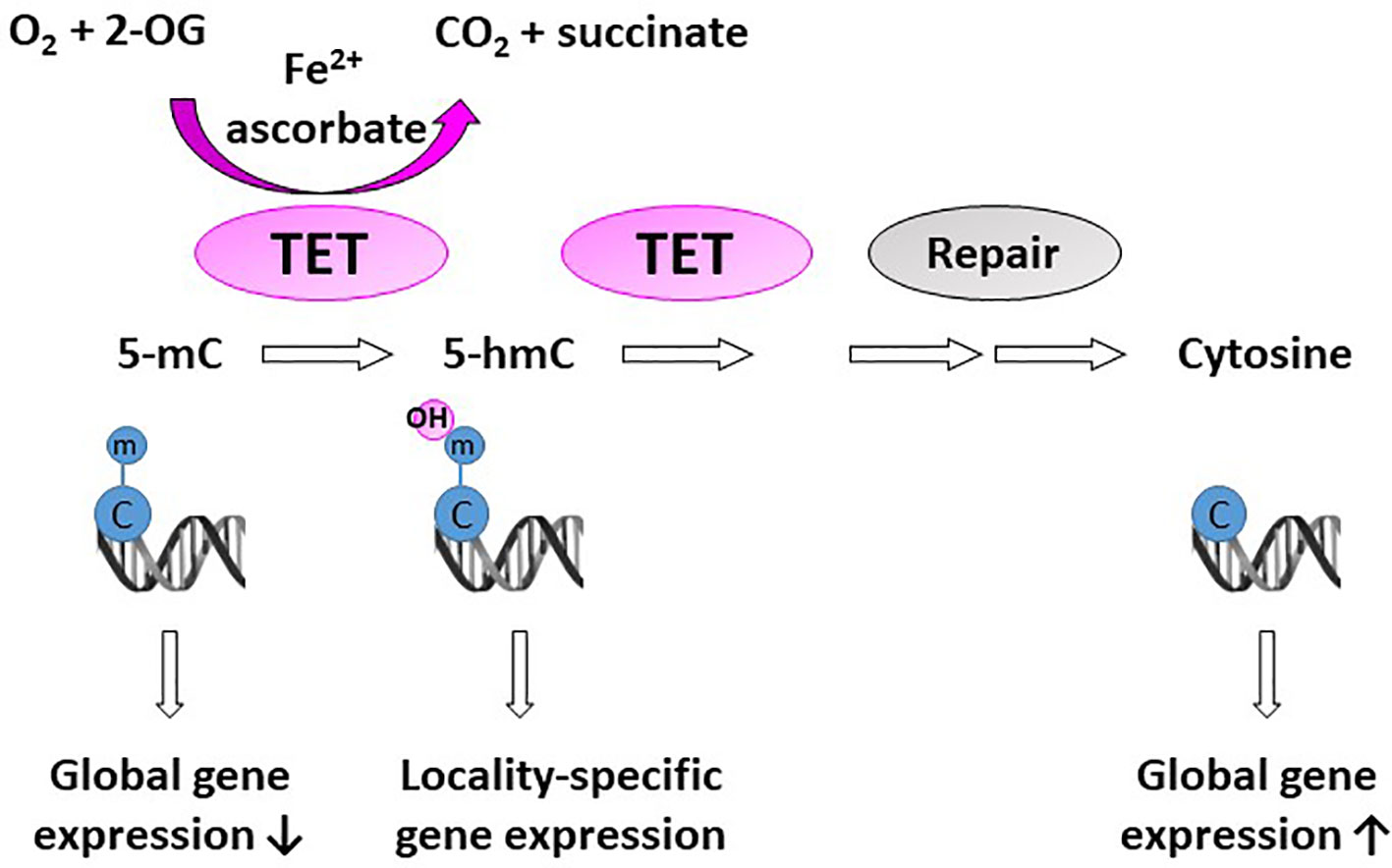
Figure 2 Activity of DNA demethylases. The ten-eleven translocases (TET1-3) hydroxylate methyl cytosine (5-mC). Hydroxy-methyl cytosine (5-hmC) are further converted to several intermediates, which are excised and repaired by thymine DNA glycosylase and base-excision repair (Repair) to generate an unmethylated cytosine. Methylation at CpG islands tends to repress gene expression, but some evidence suggests that gene expression is increased when 5-mC is converted to 5-hmC in gene promoters and enhancers (locality-specific); however, the exact biological role of 5-hmC is not yet known.
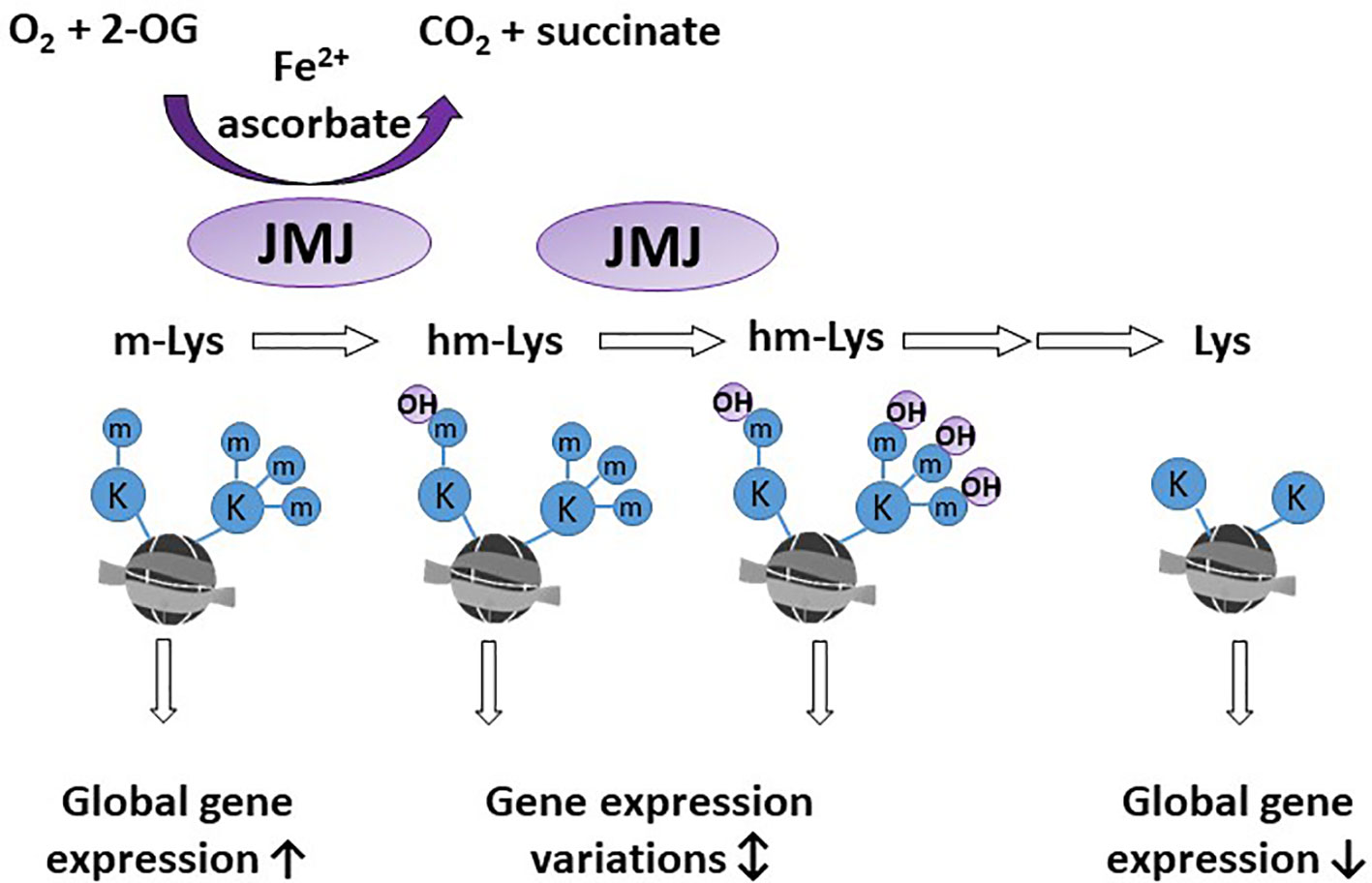
Figure 3 Activity of histone demethylases. Jumonji-C domain-containing demethylases (JMJ) act on lysine residues of the histone tails. Methylated lysine (m-Lys, mK) are hydroxylated (hm-Lys, hmK), which via several intermediates, leads to demethylated lysine residues. The effect of m-Lys and hm-Lys on histone structure and subsequent gene expression is very complex. Global gene expression tends to be increased in the more open methylated histone structure, compared to compact, demethylated histones.
Global Methylcytosine Status in Glioma
Epigenetic modifications are considered key mechanisms regulating occurrence and prognosis of glioma (34), and are used as classifiers of glioma subtypes (15, 16). Early genome-wide 5-mC analyses revealed that many low-grade gliomas and secondary GBMs contained large numbers of hypermethylated loci referred to as the glioma CpG island methylator phenotype (G-CIMP), and this was closely associated with the presence of somatic IDH1 mutations, and improved prognosis (35–37). IDH1 mutations are considered to occur early during the genesis of glioma, persisting during progression to secondary GBM, but they rarely appear in primary GBM (24, 38). Although initial reports suggested that G-CIMP remains stable during disease progression (35, 37), more recent analyses have shown their loss upon recurrence of IDH1-mutant gliomas (39–42). Loss of G-CIMP at recurrence resembled genome-wide 5-mC patterns seen in IDH1 wild-type primary GBM, and was associated with poorer outcomes (41). A novel 7-CpG signature has been identified in non-G-CIMP primary GBMs, where high-risk signatures correlated with poorer overall survival in patients treated with temozolomide and radiation (43), suggesting that even in the absence of G-CIMP and IDH1 mutations, 5-mC marks may be prognostic.
Interestingly however, earlier approaches for quantifying methylation, such as those used in the identification of G-CIMP (35, 36), relied on bisulfite conversion techniques, which cannot differentiate between 5-mC and 5-hmC. It was the introduction of oxidative bisulfide chemistry that made the distinction between 5-mC and 5-hmC possible (44), and consequently, led to the discovery of 5-hmC-specific binding proteins that are not only involved in DNA repair, but also transcriptional regulation (45, 46). These findings suggest that 5-hmC, in addition to being an intermediate in DNA demethylation, may have its own unique epigenetic role.
Many studies report a loss of global 5-hmC content in glioma compared to healthy brain tissues (47–52). Clinically, lower levels of 5-hmC have been associated with high tumor grade and poorer prognosis in glioma (48, 50, 53). More recent investigations moved beyond measuring global 5-hmC levels, to delineate, with base resolution, specific genomic locations to show 5-hmC patterns; all reporting higher than expected levels of 5-hmC at intronic CpG dinucleotides of high-grade gliomas and GBMs (50, 51, 54). Higher intronic 5-hmC levels correlated with elevated expression of the corresponding gene (54), with 5-hmC levels enriched within enhancer elements (50, 54), and 5-hmC levels associated with histone marks for open chromatin (50, 51). Thus, despite global loss of 5-hmC, genomic locations associated with transcriptional regulation and expression were commonly enriched for 5-hmC in glioma.
Ten-Eleven Translocases (TETs)
The TET family consists of three members (TET1/2/3). TET enzymes are involved in both passive and active DNA demethylation (55). During active DNA demethylation, oxidized cytosine intermediates (5-hmC) are excised by thymine DNA glycosylase and repaired with base-excision repair (BER) to generate an unmethylated cytosine (56, 57) (Figure 2). Passive DNA demethylation on the other hand occurs during cell replication, when 5-mC at CpG sites are not recognized and replaced with unmethylated cytosine (58). In humans, TET1 is located on chromosome 10q21.3, TET2 on chromosome 4q24, and TET3 on chromosome 2p13.1. It is noteworthy that 10q21.3 is commonly deleted in gliomas (59–61).
TET2 is the most studied isoform in glioma. In comparison to normal human brain tissue, TET2 gene and protein expression is reduced in GBM and other gliomas (62, 63). TET2 expression is significantly decreased with increased grade, and lower TET2 was associated with poorer overall survival (62). Similar observations have been reported for the other two isoforms (53). In glioma, reduction in TET3 expression was associated with a genome-wide reduction in 5-hmC levels compared to normal brain, and decreased TET3 expression correlated with poorer prognosis (64). Together, these findings imply a tumor suppressive role for TET enzymes in glioma.
Numerous studies highlight the potential mechanisms by which TET expression and activity are dysregulated in gliomas. A likely mechanism for reduced TET activity is the indirect inhibition via mutated IDH1/2 (65). Mutant IDH1 enzymes often exhibit neomorphic activity, converting isocitrate into 2-hydroxyglutarate (2-HG), instead of 2-OG, which is required for TET activity (66, 67). In IDH1-mutant gliomas it has been suggested that 2-HG generation is responsible for the presence of G-CIMP, likely due to reduced TET demethylase activity (35, 36). Independent of IDH status, a complete absence of 5-hmC immunoreactivity was associated with nuclear exclusion of TET1 in 61% of gliomas (52). Transcription of TET2 may be repressed by zinc finger E-box-binding homeobox 1 (ZEB1) in gliomas. ZEB1 levels were inversely correlated with TET2 levels in tumors, and physical binding of ZEB1 to the TET2 promoter in glioma cells was observed in vitro (62). However, reductions in TET2 expression and activity are unlikely to be due to TET2 mutations, as direct sequencing of TET2 revealed very few mutations and minimal association of mutations with levels of 5-hmC in gliomas (68, 69). In humans, intragenic CpG sites within TET2 showed higher levels of 5-mC and lower levels of 5-hmC in GBM compared to normal brain, although within the TET2 promoter low levels of both 5-mC and 5-hmC were detected at CpG sites of normal brain and GBM (63). Therefore, it is unlikely that lower TET2 expression in glioma is transpiring as a result of promoter methylation-mediated transcriptional suppression, but may be an effect of transcription prevention due to intragenic DNA methylation. Interestingly though, in a cohort of low-grade gliomas that had promoter hypermethylation, all were wild-type IDH1/2 (68). From this we hypothesize that in the absence of IDH1/2 mutations, a small portion of IDH wildtype tumors may instead remodel TET2 promoter methylation in an attempt to disrupt TET2 activity. Taken together, these studies demonstrate the tumor suppressive role of TET enzymes in glioma, and highlight the potential mechanisms by which TET expression and activity may be dysregulated. Restoration of TET function to resemble healthy brain tissue may aid in regulating epigenetic processors that can counteract glioma progression and improve treatment outcomes.
Jumonji-C Domain Containing Histone Demethylases
Many 2-OGDDs are from the large family of evolutionary conserved jumonji-C (JmjC) domain-containing proteins that are responsible for catalysing the removal of methyl groups from lysine residues on histones (29, 70) (Figure 3). There are several subfamilies of JmjC domain-containing histone lysine demethylases with substrate specificity to different methylated lysine residues, and this specificity is mediated by the functional domains unique to each subfamily (2, 29). The dynamic interplay between histone methylation and demethylation can result in either gene activation [via histone 3 lysine 4 (H3K4), H3K36 and H3K79] or inactivation (H3K9 and H3K27) (71, 72).
JmjC-domain containing demethylase isoforms are heterogeneously expressed in different brain locations and adult brain cell types (73, 74). Under hypoxic conditions, JMJD3 expression increased in neurons (75), but the modulation of specific histone methylation marks in response to varying oxygen tensions, and ranges of iron and 2-OG levels in brain cells, has yet to be determined.
In gliomas, changes in JmjC-dependent demethylase expression have been associated with differences in genome-wide histone methylation patterns (76, 77), and lower global histone methylation was associated with poorer patient survival (78). Conflicting reports on expression patterns of JmjC-dependent demethylases in glioma and GBM tissues have been published (77, 79–83). Overall, whether JmjC-dependent demethylases promote or suppress gliomagenesis is likely enzyme-dependent, although the majority of studies show these enzymes to promote tumor progression.
Introduction of the IDH1R132H mutation in astrocytoma cells has been associated with both global histone hypermethylation (84), and enrichment of specific histone methylation marks (85). Mutant IDH1 enzymes generate the oncometabolite 2-HG instead of 2-OG, and 2-HG has been shown to competitively inhibit histone demethylase activity (65, 86). Despite the fact that reduced histone demethylase activity has been linked with higher histone methylation in astrocytoma cells, most clinical gliomas show increased levels of JmjC-dependent demethylases and lower histone methylation (76–81). Thus, further investigations of the biological mechanisms causing increased expression of JmjC-dependent demethylases in glioma is needed.
2-OGDDs and the Hypoxic Pathway
Gliomas are highly hypoxic tumors and this is associated with poor patient prognosis (23, 87–89). Hypoxia induces adverse tumor characteristics including genomic instability, decreased apoptotic potential, increased expression of oncogenes and increased angiogenesis, which have all been described in gliomas (90). These characteristics are driven by the hypoxia inducible factors (HIFs) which are regulated by microenvironmental oxygen levels and the 2-OGDD enzymes, prolyl hydroxylases (PHD) and factor inhibiting HIF (FIH) (Figure 4) (91). As these HIF hydroxylases are dependent on oxygen for optimal function their activity is likely impaired in gliomas (92–95).
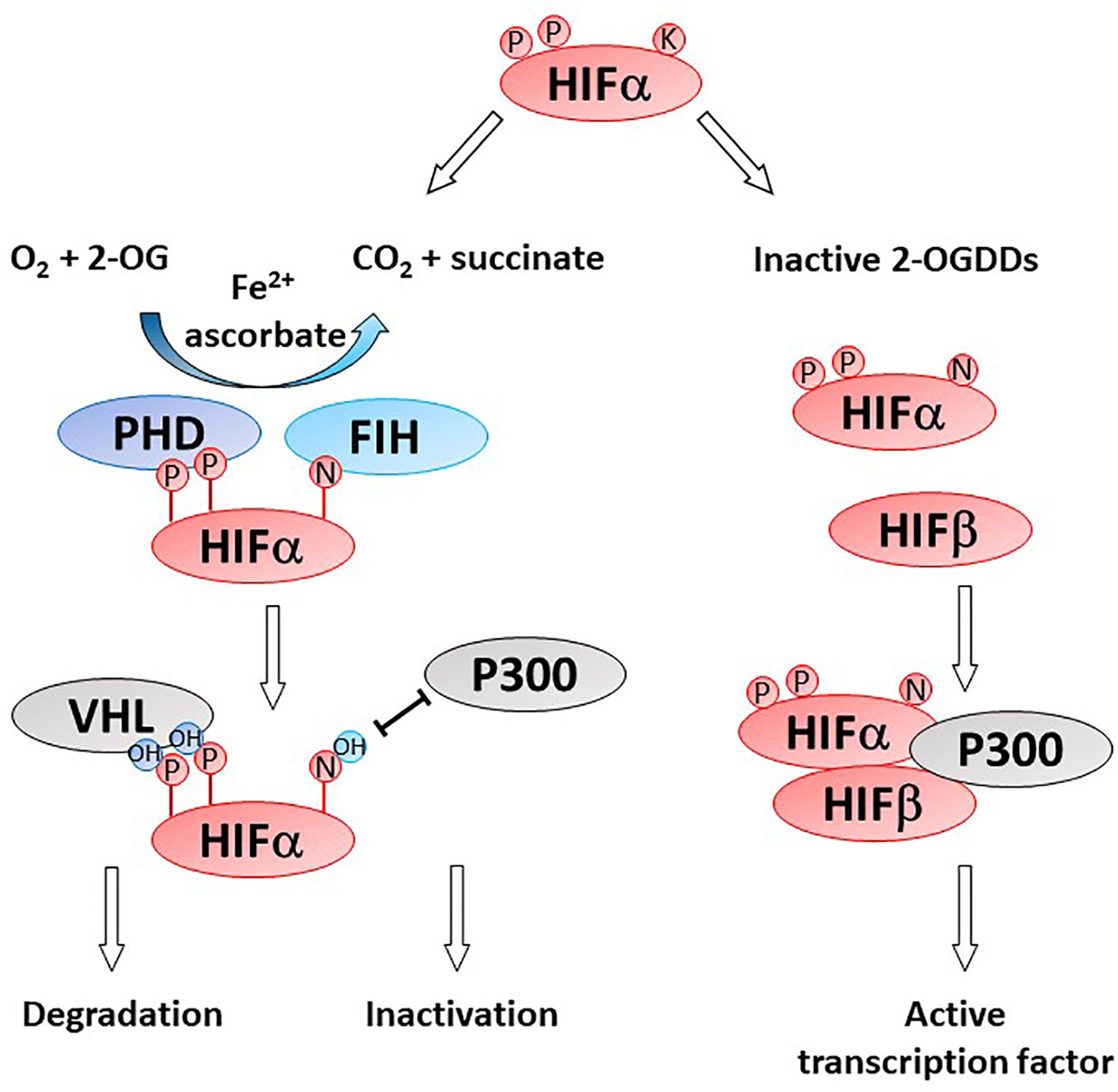
Figure 4 Activity of HIF-hydroxylases. Response to low oxygen (hypoxia) is regulated via the prolyl (PHD) and asparaginyl hydroxylases (factor inhibiting HIF, FIH). Specific proline (P) and asparagine (N) residues on the alpha subunit of the hypoxia inducible factor (HIFα) are hydroxylated by PHD and FIH, respectively. Proline hydroxylation enables recognition by von Hippel Lindau (VHL) as part of the proteasome and HIFα degradation. Asparagine hydroxylation prevent binding of the cofactor P300, thereby inactivating the HIF transcription factor. In the absence of one or more of the OGDD substrates or cofactors, PHD and FIH enzymes become inactive, enabling the accumulation of HIFα and formation of an active HIF transcription factor via binding to HIFβ. Genes under HIF control regulate cancer pathways such as angiogenesis, metastasis, glycolysis, etc.
Hypoxia-Inducible Factors
Hypoxic conditions typical of most solid tumors result in accumulation of the HIF transcription factors (11, 96). The HIFs (HIF-1,2,3) are heterodimeric transcription factors, consisting of one of three oxygen-sensitive α-subunits and a constitutive β-subunit (also known as aryl hydrocarbon receptor nuclear translocator (ARNT)) (97–99). HIF-1α is located on chromosome 14q23 (100); HIF-2α, also known as endothelial PAS domain protein 1 (EPAS1), is on 2p21, and HIF-3α is on 19q13 (101). Active HIF complexes accumulate in the nucleus and bind to specific hypoxic response elements (HREs) in promoter regions of HIF target genes, inducing their expression (102, 103). HREs contain the consensus sequence 5’CGTG3’, targets of CpG methylation, and CpG methylation blocks HIF-1 binding and transactivation (104). Even though HIF-1 and HIF-2 have identical HREs, their response to hypoxia, tissue distribution, target genes and their pro- or anti-tumor effects are distinct (105). Binding of HIF-1 and HIF-2 to their canonical HREs vary according to histone modifications, with HIF-1 preferentially associating with H3K4me3 modifications and HIF-2 with H3K4me1 (106). These binding patterns were interpreted as HIF-1 binding predominantly at regulatory regions within promoters, and HIF-2 binding to enhancer regions (106).
HIF’s modulate expression of hundreds of genes, and as a result, promote tumor growth and spread, adaptation to the tumor microenvironment, and resistance to chemo- and radio-therapy (107, 108). HIF activity also affects DNA methylation, histone acetylation and regulates noncoding RNAs (109), demonstrating the complexities of the hypoxic pathway.
HIF-1α has been proposed to drive glioma progression from low-grade astrocytoma to high-grade GBM (23, 110). Higher HIF-1α expression in human astrocytoma and GBM has been correlated with worse prognosis (111–114). Moreover, expression of HIF-1α, and its downstream target genes vascular endothelial growth factor (VEGF), glucose transporter (GLUT1), and carbonic anhydrase (CA9), show increased expression in higher grade gliomas compared to lower grade (115). In addition, increases in VEGF expression were shown to localize to hypercellular and necrotic regions that form as result of decreased oxygen and nutrient delivery (113, 116).
In comparison to HIF-1α, HIF-2α may be a specifically attractive target in GBMs, as it is expressed in glioma stem cells but not normal neuronal progenitors and it is activated by long-term hypoxia, in addition to its association with poor patient survival (117).
HIF Hydroxylases
HIF activity is regulated at the post-translational level by two families of HIF hydroxylases, the prolyl hydroxylase domain (PHD) and factor inhibiting HIF (FIH) 2-OGDDs (Figure 4). PHDs hydroxylate specific proline residues on the HIF-α subunit targeting the subunit for proteasomal degradation (9, 92, 118). FIH hydroxylates an asparagine residue and prevents binding of the co-activators CBP/p300 and nuclear translocation (93, 119). The instability of HIF-α and the inhibition of transcriptional activators binding results in a reduction in both HIF-α protein and expression of target genes. Protein hydroxylation was considered irreversible, but a recent study showed evidence that FIH-mediated asparagine hydroxylation may be reversible by as yet unknown cellular enzymes (120).
The PHDs and the FIH enzymes have specific roles in oxygen and metabolic sensing due to their exquisite requirements for molecular oxygen and 2-OG, respectively (11). PHDs have higher affinity for oxygen (9, 13), bind unusually tightly to Fe2+ (121), and have a lower affinity for ascorbate compared to FIH (3), possibly due to the narrower opening to the enzyme active site of PHDs (122). PHD2 and FIH bind 2-OG with distinct residues, with FIH sharing more homology with JmjC-domain containing protein family of enzymes (94), suggesting evolutionary divergence (122). The HIF hydroxylases therefore have different sensitivities to loss of enzyme substrates and/or co-factors.
PHD2 (encoded by EGLN1) is generally acknowledged as the primary mediator of HIF-1α protein degradation, with PHD1 and 3 more likely to be involved in fine-tuning of the hypoxic response (123). This may be reflected in the subcellular localisation of each isoform, with PHD1 detected exclusively in the nucleus, PHD2 (and FIH) in the cytoplasm, and PHD3 in both compartments (124). Interestingly, PHD2 is activated, rather than inhibited, by the R-enantiomer of 2-HG as it mimics 2-OG, suggesting that IDH mutations may lead to HIF-α degradation (125–127). It has been shown, using immunohistochemistry, that HIF-1α and IDHR132H expression are not related, supporting the ability of 2-HG to activate PHD2 (128).
FIH (also known as HIF1AN) is mapped to chromosome 10q24 (93). Full or partial deletions of chromosome 10q often occur in gliomas, and the frequency of these deletions increases with tumor grade (59–61). Thus, FIH mRNA expression often decreases with increasing tumor grade (129, 130). Investigation of FIH in GBMs has shown that FIH reduces the interaction between p300 and HIF-1α which is essential for transcriptional activity (131), and correspondingly higher FIH expression was associated with reduced GLUT1 and VEGF expression in GBM cells (131). Loss of FIH through deletion of chromosome 10q24 may increase the hypoxic response, and thus contribute to an aggressive and treatment resistant glioma phenotype associated with hypoxia (23, 87, 88). While PHD2 is activated by the mutant IDH metabolite, 2-HG, FIH is inhibited, resulting in p300 interacting with HIF-α and inducing target gene expression (125). However, FIH has been shown to interact with a glioma tumor suppressor gene (ANKDD1A), which increased FIH activity, thereby reducing HIF-α activation and preventing HIF target gene expression (132).
Substrate and Cofactor Requirements of 2-OGDDs
2-OGDDs contain a non-heme iron (Fe2+) in the active site, and utilize 2-OG (also known as α-ketoglutarate) and molecular oxygen as substrates (Figure 5). Ascorbate is an essential cofactor, and is thought to be required to maintain the active site Fe2+ in the reduced state (11–14, 133, 134).
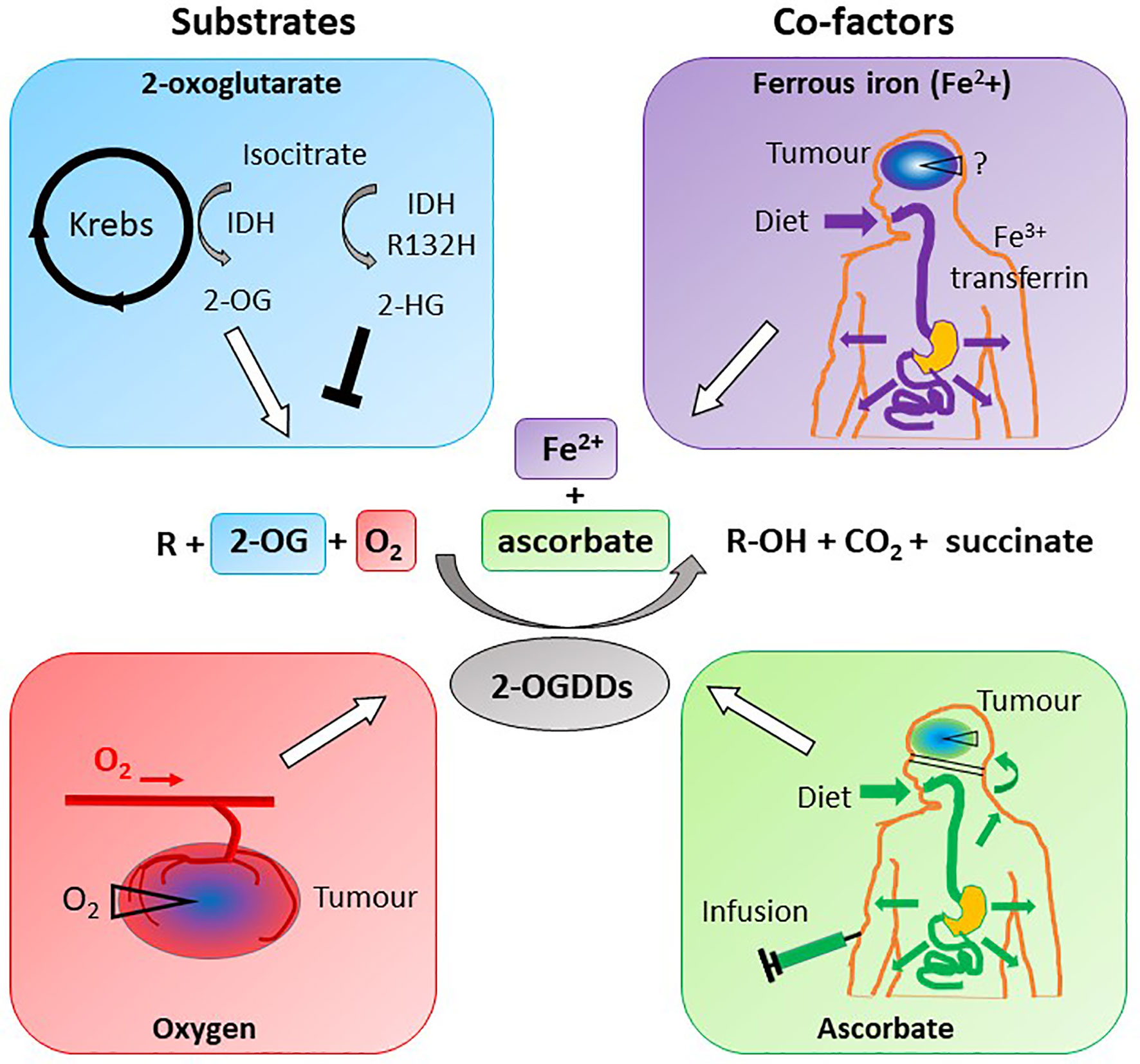
Figure 5 Substrate and cofactor requirements of the 2-oxoglutarate dependent dioxygenases. These enzymes hydroxylate nucleotides or amino acids (R), forming a hydroxylated product (R-OH). 2-OGDDs require 2-oxoglutarate (2-OG) and molecular oxygen (O2) as substrates, and ferrous iron (Fe2+) and ascorbate as cofactors. Isocitrate from the Krebs cycle is converted to 2-OG by isocitrate dehydrogenase (IDH), but mutant IDH (IDHR132H) produces the competitive inhibitor 2-hydroxyglutarate (2-HG). Low oxygen (hypoxia) is a characteristic of most solid tumors, thus reducing activity of 2-OGDDs. Iron obtained from the diet is transported via transferrin and taken up across the BBB via transferrin receptors; the content in tumors is unknown. Ascorbate is obtained via a healthy diet or infusions, and cannot cross the BBB but will travel via the choroid plexus. Cellular ascorbate uptake is via sodium vitamin C transporters; tumors may be low in the vitamin.
Molecular Oxygen as 2-OGDD Substrate and its Availability in Gliomas
Oxygen is an absolute requirement for all 2-OGDDs, but not all 2-OGDDs function as oxygen sensors. The PHDs have a Km for oxygen of 230-250 μM, slightly above dissolved oxygen concentrations in air (20.9% O2 in gas phase ~ 200 μM dissolved O2) (135), and this property makes these proteins the predominant cellular oxygen sensors. These Km values are significantly greater than for FIH at 90 μM, and the collagen prolyl 4-hydroxylases (C-P4H) at 40 μM (13, 135, 136).
Tissue partial pressure of oxygen (PO2) in the human body range from 104–108 mmHg in the alveola of the lung, to arterial PO2 of 90 mmHg and venous PO2 of 40 mmHg, to most tissues that have between tissue PO2 of 20–70 mmHg (137). Oxygen levels in the brain are heterogeneous and difficult to measure, with levels of 30–48 mmHg in normal brain recorded. Hypoxia is defined as <5 mmHg, and levels below 2.5 mmHg are known to induce clinical radioresistance (138–145). Theoretical modelling suggests the minimum oxygen level requirement for brain tissue is 17 mmHg and that a critical range for hypoxic injury is 4–11 mmHg (140).
Hypoxia is a prominent feature of gliomas. Oxygen levels in tumors have been measured using a range of techniques, including injection of hypoxia markers (146–149), direct measurements using polarographic O2 microelectrodes (145, 150) and MRI/CT scans (151). Using these techniques, oxygen levels were observed to be lower in gliomas and peritumoral regions compared to normal brain tissues, and readings below 2.5 mmHg were more common in tumors of increasing grade (145, 150, 152), with the most severe hypoxia detected in GBMs (23, 88, 89). A review of six studies reported a median PO2 of 13 mmHg in gliomas (144). This suggests that in high grade gliomas, the activity of 2-OGDD enzymes is likely to be reduced, but hypoxia is not routinely measured.
As ionizing radiation remains the mainstay of glioma treatment, and since hypoxia governs radioresistance, considerable clinical effort has been focused on reducing tumor hypoxia. With regards to 2-OGDD activity, these strategies may also improve enzyme activity, and are thus briefly discussed here. In an attempt to improve tumor oxygenation, patients have been given pure oxygen in a pressurized environment (hyperbaric oxygen) to breath (153). Alternatively, patients breathed 95% O2 with 5% CO2 (carbogen), which, together with hypercapnic-induced vasodilation, increases the amount of dissolved plasma O2 at the capillary level (154). Carbogen was tested in combination with nicotinamide, which is believed to prevent transient cessations in blood flow, thus inhibiting the development of acute hypoxia (154). Drugs have been developed to improve oxygen delivery (eg trans sodium crocetinate, TSC) (155), to normalize the tumor vasculature (e.g., anti-VEGF bevacizumab) (156), or to reduce oxygen consumption rate via mitochondrial poisons (e.g., anti-parasitic drugs atovaquone, ivermectin, proguanil, mefloquine, and quinacrine) and tested in patients with GBM (157). Overall, the results of these clinical trials have been disappointing and none of the approaches have been adopted into clinical practice, and actual oxygen measurements were largely lacking.
2-Oxoglutarate and Oncometabolites as 2-OGDD Substrates in Gliomas
The substrate 2-oxoglutarate (2-OG) is a product of the reaction in which isocitrate dehydrogenase enzymes (IDH) convert isocitrate to 2-OG (Figure 5). This reaction occurs in the TCA cycle via IDH3 and in the cytosol via IDH1 and IDH2. A number of IDH1 and IDH2 mutations have been reported in glioma, with the most common a base substitution in codon 132 of IDH1 resulting in an arginine to histidine replacement (IDH1R132H) (24, 67). IDH1R132H has been identified in more than 70% of grade II/III astrocytic and oligodendroglial diffuse gliomas, and in more than 80% of secondary GBM, but rarely in primary GBM (24, 158, 159). IDH1R132H enzymes generate 2-HG, instead of 2-OG (66, 67), which binds competitively to 2-OGDD enzymes and inhibits their function (Figure 5). Studies have, however, also reported that accumulation of 2-HG does not universally occur in all IDH1R132H and, conversely, that some wild type IDH cells accumulate 2-HG (160, 161). Further research is required to understand the impact of IDH mutations on patients with glioma, and how 2-HG accumulation interacts with other 2-OGDD substrates and co-factors to influence their activity in these tumors.
In gliomas with IDH1R132H mutations, drugs that inhibit the mutant IDH1 enzymes may improve 2-OGDD activity by reducing 2-HG production. A number of mutant IDH1 inhibitors are currently being evaluated in clinical trials with glioma patients, including AG-120 (Ivosidenib), AG-881 (Vorasidenib), BAY1436032 (Bayer), and DS-1001b (NCT02746081) (162–166). Pre-clinical investigations have reported anti-proliferative effects, reductions in tumor growth rates, and lower levels of 2-HG in both glioma cells and tumors from animal models (167–170). To date, 2-HG levels have only been measured in the plasma of human glioma patients following intervention with Ivosidenib, reporting no difference compared to those without treatment (163). Despite this, interest remains high and data from a Phase I study of Ivosidenib and Vorasidenib in patients with recurrent, non-enhancing, IDH1-mutant, low-grade glioma is currently pending (162). In addition to inhibitors, vaccines against mutant IDH1 have been tested in mice (171, 172). Mice with mutant IDH1R132H gliomas treated with the vaccine showed longer survival than non-immunized mice, and had higher levels of peripheral anti- IDH1R132H antibodies, IFN-γ, and CD8+ T cells (172). However, 2-HG levels in tumors were not measured, and thus the vaccine effect on 2-OGDDs remains to be tested.
Iron as Cofactor for 2-OGDDs and Availability in the Brain
Iron is the most abundant transition metal in the brain (173) and is more concentrated in some regions than in others (range; 13.5–1.75 µmol/g dry weight) (174), including the iron-rich substantia nigra, caudate nucleus and globus pallidus (175). Most gliomas arise in the frontal/temporal lobe, areas rich in glial cells and, potentially, relatively lower in iron. Astrocytes secrete hepcidin, which modulates the expression of ferroportin and other iron regulatory proteins, and thereby function as iron sensors to regulate and communicate the iron requirement of the brain through paracrine signaling (176–178).
Iron uptake into the brain is tightly regulated through the endothelial cells and neighboring astrocytes in the blood brain barrier (BBB) (176, 179, 180), primarily through the transferrin bound iron (TBI) pathway (174, 181, 182) however, when this pathway becomes saturated, non-TBI pathways are used (183). Through the canonical TBI uptake pathway, ferric iron (Fe3+) forms halotransferrin (174), which is able to pass through the BBB by binding to transferrin receptors on the apical surface of brain microvascular endothelial cells (BMVECs). Within the BMVEC, excess iron is stored in the cytosolic labile iron pool, which is the principle source of metals for metabolism (184). Efflux of iron through the abluminal membrane into the brain interstitium occurs through ferroportin (185, 186), expressed on the basolateral side of the BMVEC cells (186, 187). Once iron has crossed the BBB barrier, it is taken up by neurons, astrocytes, oligodendrocytes and microglia through TBI and nonTBI uptake pathways (183).
Iron plays a role in carcinogenesis, with proteins that modulate and regulate iron metabolism often dysregulated in gliomas (188–191). GBM cancer-stem-like cells were shown to upregulate transferrin expression, and to extract iron more effectively from the tumor microenviroment than non-stem-like tumor cells in an ex vivo explant model (192). The Fe2+ content in gliomas has not been reported but reduced levels may impede 2-OGDD function in tumors and cancer stem cells.
Ascorbate as Cofactor for 2-OGDDs and its Availability in the Brain
As a cofactor for 2-OGDDs, ascorbate acts to reduce Fe3+ back to active Fe2+ (Figure 5). This activity appears to be specific to ascorbate as alternative reducing agents such as glutathione or N-Acetylcysteine are unable to substitute for ascorbate in 2-OGDD activity (12, 193, 194). Ascorbate is also involved in stabilizing cysteine residues in PHD enzymes, preventing intramolecular oxidation and supporting catalytic activity (126).
Normal brain tissue has one of the highest ascorbate levels of all tissues in the body, reaching intracellular concentrations of 2–10 mM depending on the cell type (195–197). In times of ascorbate insufficiency, the brain is one of the last tissues to lose ascorbate, supporting its importance to brain function (195, 198). The specific vitamin C transporters (sodium-dependent vitamin C transporters) are not expressed on the endothelial cells lining the BBB (198), and ascorbate enters the central nervous system through the choroid plexus where it can diffuse through the cerebrospinal fluid to the brain (195, 198). Cells within the brain express ascorbate transporters allowing intracellular ascorbate accumulation (198).
Data on ascorbate content in gliomas is limited to a single study that reported ascorbate levels in astrocytomas from eleven patients (199). While ascorbate levels were not different between astrocytoma tissue and non-neoplastic tissue, DNA content was significantly higher in astrocytoma (tumor) tissues indicating increased cell density (or cellularity) of the tissues. This suggests that intracellular ascorbate per cell may be reduced in astrocytoma tissue compared to normal, non-necrotic tissue (199). These intriguing findings need to be confirmed.
Ascorbate and Epigenetic Reprogramming
Evidence for the effects of ascorbate on epigenetic reprogramming is largely from embryonic cells (200, 201), but data in gliomas is missing. In patients with myeloid malignancies, oral ascorbate supplementation resulted in an increase in the ratio of 5-hmC compared to 5-mC in mononuclear myeloid cells (202). Here, DNA demethylation was not associated with changes in TET expression (202, 203), but instead appear to result from ascorbate-mediated restoration of endogenous TET activity, which was supported by studies with Tet2-deficient mice (204). Gliomas show lower TET expression independent of TET mutations (63, 68, 69), and we hypothesize that ascorbate may compensate for lower TET expression by upregulating residual TET2 function as was observed in a case study of acute myeloid leukemia (205).
Low-grade primary gliomas commonly harbor IDH mutations that persist during progression to secondary GBM (24). In vitro, ascorbate was able to circumventing competitive inhibition by 2-HG in colon cancer and HOXA9-immortalized mouse bone marrow cells with IDH1R132H mutations (206, 207). One in vitro study reported the effects of ascorbate on epigenetic marks in LN229 glioma cells, showing increased TET3 mRNA expression, as well as increased 5-hmC, but these cells did not harbor an IDH mutation (64). Yet, these findings, together with reported associations between higher 5-hmC levels and better prognosis in patients with GBM (50), support the notion that sufficient ascorbate levels in glioma tumors may induce demethylation activity and promote more favorable outcomes, although whether sufficient ascorbate can overcome high levels of 2-HG remains to be determined.
In addition to TET-mediated effects, ascorbate induced H3K9me2/3 and H3K36me2/3 demethylation via JmjC-dependent demethylases in embryonic stem, embryonic fibroblast and Th17 cells from mice (208–210). Ascorbate caused reductions in H3K9 methylation and increased expression of the JmjC-dependent demethylases, JHDM2A-C and JHDM3B, as well as widespread DNA demethylation at CpG island boundaries in human embryonic stem cells (211). However, investigations in gliomas are lacking, despite the well-established link between glioma formation and global 5-hmC deficiency.
Ascorbate and the Hypoxic Pathway
The relationship between intracellular ascorbate levels and HIF pathway activity has been investigated in numerous cancers, but not yet in gliomas. In vitro investigations of intracellular ascorbate levels and HIF pathway activity have been performed in varying cancer types, findings from which have guided further in vivo studies (212–215). In relevant mouse models (using ascorbate-dependent Gulo-/- mice), increased ascorbate intake or administration was associated with increased tumor ascorbate levels, reduced HIF pathway activity and reduced tumor growth (216). In clinical samples of endometrial (217), colorectal (218), thyroid (214), papillary cell renal cell carcinomas (215, 219), and breast cancer (220), high tumor ascorbate levels were associated with low HIF-1α protein levels and low HIF target gene expression. This relationship was not evident in clear cell renal cell carcinoma (215, 219), that have a mutated von Hippel-Lindau factor which prevents proteasomal degradation of hydroxylated HIF-α (219). Higher levels of tumor ascorbate were associated with improved disease-free survival in patients with colorectal cancer (218) and improved disease-specific survival in patients with breast cancer (220).
High Dose Ascorbate as Cancer Treatment
Infusion with high dose ascorbate as an alternative or complementary therapy for cancer is widespread (221), but lacks evidence of efficacy (222, 223), despite early promising data (224, 225). Since then, pharmacokinetic data have demonstrated that infusion results in supra-physiological plasma ascorbate levels that are not achievable by oral administration (226–229). Case studies and small clinical trials continue to surface that suggest there may be circumstances under which high dose ascorbate infusion can provide a clinical benefit (230–239).
Preclinical Models of High Dose Ascorbate Treatment in Gliomas
In a mouse xenograft glioma model, analysis of ascorbate levels in plasma, tumor, and cerebrospinal fluid samples showed that ascorbate increased 1 h post intraperitoneal injection with 4 g/kg of ascorbate (240). One study, using an intracranial GL261 glioma mouse model, reported that radiation treatment slowed tumor growth, whereas ascorbate treatment made no difference, and the combination of ascorbate and radiotherapy induced faster progression (241), in conflict to in vitro findings of radio-sensitisation by ascorbate in numerous glioma cell lines, including GL261 cells (242–244). However, ascorbate levels in the intracranial model were not measured, and thus the impact of ascorbate on glioma response to radiation remain uncertain.
Clinical Trials in Patients With Gliomas and GBMs
Preclinical data led to phase I clinical trials administering intravenous ascorbate to glioma patients, with and without standard radiotherapy and temozolomide (245). High dose vitamin C (HDVC) was found to be safe and well tolerated, reaching target 20 mM plasma levels (240, 245), but tumor ascorbate levels were not measured. A trend of improved overall survival was reported, but participant numbers were too small to determine statistical significance (240). Two case reports for the use of HDVC infusions in patients with glioma have also been reporting (237, 246).
Previous research has shown an association between a lower proportion of methylation at the O-6-methylguanine-DNA methyltransferase (MGMT) promoter in glioma tumors and poorer patient prognosis (247). MGMT is a DNA repair enzyme responsible for resistance to temozolomide, and hypermethylation of the MGMT promoter is evident in 40–45% of gliomas (248, 249), with higher methylation levels in low-grade gliomas compared to GBMs (53). Interestingly, in glioma patients with low methylation levels at the MGMT promoters, HDVC infusions resulted in improved overall survival (240), but unfortunately, ascorbate levels in the glioma tissue were not measured. Plasma ascorbate levels do not necessarily reflect tumor ascorbate levels due reduced functioning vasculature and BBB in gliomas. Overall, the clinical worth of HDVC in cancer remains unproven.
Conclusion
The superfamily of 2-OGDD enzymes play a vital role in glioma progression and patient prognosis, being involved in epigenetic modifications and oxygen sensing. Limiting supplies of one or more of their substrates or cofactors in gliomas is likely although reported measurements are rare. Restoration of epigenetic modifications offers a promising target in the treatment of cancer, as these alterations are reversible, as opposed to genetic mutations. Attempts at increasing tumor oxygenation to improve effectiveness of radiation and chemotherapy in glioma are not (yet) in clinical practice (250), and new strategies are sought. Ascorbate infusion is a safe and cheap option that may be able to normalize 2-OGDD function in a subset of glioma tumor subtypes. However, this will likely depend on mutation status and on the ability to increase intracellular ascorbate levels in these tumors. Future research will need to confirm ascorbate status of clinical glioma tumors, on measuring 5-hmC levels and HIF activity in clinical samples, and on determining an optimal ascorbate dose for patients, before embarking on phase III trials to determine clinical efficiency.
Author Contributions
Conceptualization, RC, EB, JR, and GD. Funding acquisition, EP and GD. Supervision, EP, MV, and GD. Writing—original draft, RC and EB. Writing—review and editing, JR, MV, and GD. All authors contributed to the article and approved the submitted version.
Funding
We received funding from the Mackenzie Charitable Foundation (EP and GD), the Canterbury Medical Research Foundation (RC, EP, and GD) and the University of Otago (RC and GD).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to acknowledge receipt of the doctoral scholarship from the University of Otago (EB).
References
1. Johansson C, Tumber A, Che K, Cain P, Nowak R, Gileadi C, et al. The roles of Jumonji-type oxygenases in human disease. Epigenomics (2014) 6(1):89–120. doi: 10.2217/epi.13.79
2. McDonough MA, Loenarz C, Chowdhury R, Clifton IJ, Schofield CJ. Structural studies on human 2-oxoglutarate dependent oxygenases. Curr Opin Struct Biol (2010) 20:659–72. doi: 10.1016/j.sbi.2010.08.006
3. Ozer A, Bruick RK. Non-heme dioxygenases: Cellular sensors and regulators jelly rolled into one? Nat Chem Biol (2007) 3:144–53. doi: 10.1038/nchembio863
4. Schofield CJ, Ratcliffe PJ. Oxygen sensing by HIF hydroxylases. Nat Rev Mol Cell Biol (2004) 5:343–54. doi: 10.1038/nrm1366
5. Wu Y-C, Ling Z-Q. The role of TET family proteins and 5-hydroxymethylcytosine in human tumors. Histol Histopathol (2014) 29:991–7. doi: 10.14670/HH-29.991
6. Islam MS, Leissing TM, Chowdhury R, Hopkinson RJ, Schofield CJ. 2-Oxoglutarate-Dependent Oxygenases. Annu Rev Biochem (2018) 87:585–620. doi: 10.1146/annurev-biochem-061516-044724
7. Vissers MC, Das AB. Ascorbate as an Enzyme Cofactor. In: Vitamin C. CRC Press Taylor and Francis Group, Florida, USA. (2020) pp . CRC Press Taylor and Francis Group (2020):71–98
8. Grosso G, Bei R, Mistretta A, Marventano S, Calabrese G, Masuelli L, et al. Effects of vitamin C on health: A review of evidence. Front Biosci (2013) 18:1017–29. doi: 10.2741/4160
9. Epstein ACR, Gleadle JM, McNeill LA, Hewitson KS, O’Rourke J, Mole DR, et al. C. elegans EGL-9 and mammalian homologs define a family of dioxygenases that regulate HIF by prolyl hydroxylation. Cell (2001) 107:43–54. doi: 10.1016/S0092-8674(01)00507-4
10. Mandl J, Szarka A, Bánhegyi G. Vitamin C: update on physiology and pharmacology. Br J Pharmacol (2009) 157:1097–110. doi: 10.1111/j.1476-5381.2009.00282.x
11. Vissers MCM, Kuiper C, Dachs GU. Regulation of the 2-oxoglutarate-dependent dioxygenases and implications for cancer. Biochem Soc Trans (2014) 42:945–51. doi: 10.1042/BST20140118
12. Dickson KM, Gustafson CB, Young J II, Züchner S, Wang G. Ascorbate-induced generation of 5-hydroxymethylcytosine is unaffected by varying levels of iron and 2-oxoglutarate. Biochem Biophys Res Commun (2013) 439:522–7. doi: 10.1016/j.bbrc.2013.09.010
13. Koivunen P, Hirsilä M, Günzler V, Kivirikko K II, Myllyharju J. Catalytic Properties of the Asparaginyl Hydroxylase (FIH) in the Oxygen Sensing Pathway Are Distinct from Those of Its Prolyl 4-Hydroxylases. J Biol Chem (2004) 279:9899–904. doi: 10.1074/jbc.M312254200
14. Nytko KJ, Spielmann P, Camenisch G, Wenger RH, Stiehl DP. Regulated function of the prolyl-4-hydroxylase domain (PHD) oxygen sensor proteins. Antioxid Redox Signal (2007) 9:1329–38. doi: 10.1089/ars.2007.1683
15. Louis DN, Perry A, Reifenberger G, von Deimling A, Figarella-Branger D, Cavenee WK, et al. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: a summary. Acta Neuropathol (2016) 131:803–20. doi: 10.1007/s00401-016-1545-1
16. Wesseling P, Capper D. WHO 2016 Classification of gliomas. Neuropathol Appl Neurobiol (2018) 44:139–50. doi: 10.1111/nan.12432
17. Behin A, Hoang-Xuan K, Carpentier AF, Delattre JY. Primary brain tumours in adults. Lancet (2003) 361:323–31. doi: 10.1016/S0140-6736(03)12328-8
18. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A, et al. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin (2018) 68:394–424. doi: 10.3322/caac.21492
19. Watanabe K, Tachibana O, Sato K, Yonekawa Y, Kleihues P, Ohgaki H. Overexpression of the EGF Receptor and p53 Mutations are Mutually Exclusive in the Evolution of Primary and Secondary Glioblastomas. Brain Pathol (1996) 6:217–23. doi: 10.1111/j.1750-3639.1996.tb00848.x
20. Stupp R, Mason WP, Van Den Bent MJ, Weller M, Fisher B, Taphoorn MJB, et al. Radiotherapy plus Concomitant and Adjuvant Temozolomide for Glioblastoma. N Engl J Med (2005) 352:987–96. doi: 10.1056/NEJMoa043330
21. Stupp R, Hegi ME, Mason WP, van den Bent MJ, Taphoorn MJ, Janzer RC, et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol (2009) 10:459–66. doi: 10.1016/S1470-2045(09)70025-7
22. Annovazzi L, Mellai M, Schiffer D. Chemotherapeutic drugs: DNA damage and repair in glioblastoma. Cancers (2017) 9:57–73 doi: 10.3390/cancers9060057
23. Huang WJ, Chen WW, Zhang X. Glioblastoma multiforme: Effect of hypoxia and hypoxia inducible factors on therapeutic approaches (review). Oncol Lett (2016) 12:2283–8. doi: 10.3892/ol.2016.4952
24. Yan H, Parsons DW, Jin G, McLendon R, Rasheed BA, Yuan W, et al. IDH1 and IDH2 Mutations in Gliomas. N Engl J Med (2009) 360:765–73. doi: 10.1016/S0513-5117(09)79085-4
25. Van Den Bent MJ, Hegi ME, Stupp R. Recent developments in the use of chemotherapy in brain tumours. Eur J Cancer (2006) 42:582–8. doi: 10.1016/j.ejca.2005.06.031
26. Hirst TC, Vesterinen HM, Sena ES, Egan KJ, Macleod MR, Whittle IR. Systematic review and meta-analysis of temozolomide in animal models of glioma: Was clinical efficacy predicted. Br J Cancer (2013) 108:64–71. doi: 10.1038/bjc.2012.504
27. Hou LC, Veeravagu A, Hsu AR, Tse VCK. Recurrent glioblastoma multiforme: a review of natural history and management options. Neurosurg Focus (2006) 20:E5. doi: 10.3171/foc.2006.20.4.2
28. Touat M, Li YY, Boynton AN, Spurr LF, Iorgulescu JB, Bohrson CL, et al. Mechanisms and therapeutic implications of hypermutation in gliomas. Nature (2020) 580:517–23. doi: 10.1038/s41586-020-2209-9
29. Dong C, Zhang H, Xu C, Arrowsmith CH, Min J. Structure and function of dioxygenases in histone demethylation and DNA/RNA demethylation. IUCrJ (2014) 1:540–9. doi: 10.1107/S2052252514020922
30. Shi Y. Histone lysine demethylases: Emerging roles in development, physiology and disease. Nat Rev Genet (2007) 8:829–33. doi: 10.1038/nrg2218
31. Fu Y, He C. Nucleic acid modifications with epigenetic significance. Curr Opin Chem Biol (2012) 16:516–24. doi: 10.1016/j.cbpa.2012.10.002
32. Berdasco M, Esteller M. Developmental Cell Review Aberrant Epigenetic Landscape in Cancer: How Cellular Identity Goes Awry. (2010) 19:698–711. doi: 10.1016/j.devcel.2010.10.005
33. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell (2011) 144:646–74. doi: 10.1016/j.cell.2011.02.013
34. Gusyatiner O, Hegi ME. Glioma epigenetics: From subclassification to novel treatment options. Semin Cancer Biol (2018) 51:50–8. doi: 10.1016/j.semcancer.2017.11.010
35. Noushmehr H, Weisenberger DJ, Diefes K, Phillips HS, Pujara K, Berman BP, et al. Identification of a CpG Island Methylator Phenotype that Defines a Distinct Subgroup of Glioma. Cancer Cell (2010) 17:510–22. doi: 10.1016/j.ccr.2010.03.017
36. Turcan S, Rohle D, Goenka A, Walsh LA, Fang F, Yilmaz E, et al. IDH1 mutation is sufficient to establish the glioma hypermethylator phenotype. Nature (2012) 483:479–83. doi: 10.1038/nature10866
37. Laffaire J, Everhard S, Idbaih A, Crinière E, Marie Y, de Reyniès A, et al. Methylation profiling identifies 2 groups of gliomas according to their tumorigenesis. Neuro Oncol (2011) 13:84–98. doi: 10.1093/neuonc/noq110
38. Parsons DW, Jones S, Zhang X, Lin JC, Leary RJ, Angenendt P, et al. An integrated genomic analysis of human glioblastoma multiforme. Science (2008) 321:1807–12. doi: 10.1126/science.1164382
39. Bai H, Harmanci AS, Erson-Omay EZ, Li J, Coşkun S, Simon M, et al. Integrated genomic characterization of IDH1-mutant glioma malignant progression. Nat Genet (2015) 48:59–66. doi: 10.1038/ng.3457
40. Mazor T, Pankov A, Johnson BE, Hong C, Hamilton EG, Bell RJA, et al. DNA Methylation and Somatic Mutations Converge on the Cell Cycle and Define Similar Evolutionary Histories in Brain Tumors. Cancer Cell (2015) 28:307–17. doi: 10.1016/j.ccell.2015.07.012
41. de Souza CF, Sabedot TS, Malta TM, Stetson L, Morozova O, Sokolov A, et al. A Distinct DNA Methylation Shift in a Subset of Glioma CpG Island Methylator Phenotypes during Tumor Recurrence. Cell Rep (2018) 23:637–51. doi: 10.1016/j.celrep.2018.03.107
42. Nomura M, Saito K, Aihara K, Nagae G, Yamamoto S, Tatsuno K, et al. DNA demethylation is associated with malignant progression of lower-grade gliomas. Sci Rep (2019) 9:1–12. doi: 10.1038/s41598-019-43790-7
43. Yin AA, He Y-L, Etcheverry A, Liu Y-H, Aubry M, Barnholtz-Sloan J, et al. Novel predictive epigenetic signature for temozolomide in non-G-CIMP glioblastomas. Clin Epigenet (2019) 11:76. doi: 10.1186/s13148-019-0670-9
44. Booth MJ, Ost TWB, Beraldi D, Bell NM, Branco MR, Reik W, et al. Oxidative bisulfite sequencing of 5-methylcytosine and 5- hydroxymethylcytosine. Nat Protoc (2013) 8:1841–51. doi: 10.1038/nprot.2013.115
45. Spruijt CG, Gnerlich F, Smits AH, Pfaffeneder T, Jansen PWTC, Bauer C, et al. Dynamic readers for 5-(Hydroxy)methylcytosine and its oxidized derivatives. Cell (2013) 152:1146–59. doi: 10.1016/j.cell.2013.02.004
46. Iurlaro M, Ficz G, Oxley D, Raiber EA, Bachman M, Booth MJ, et al. A screen for hydroxymethylcytosine and formylcytosine binding proteins suggests functions in transcription and chromatin regulation. Genome Biol (2013) 14. doi: 10.1186/gb-2013-14-10-r119
47. Jin S-G, Jiang Y, Qiu R, Rauch TA, Wang Y, Schackert G, et al. 5-Hydroxymethylcytosine Is Strongly Depleted in Human Cancers but Its Levels Do Not Correlate with IDH1 Mutations. (2011) 71:7360–5. doi: 10.1158/0008-5472.CAN-11-2023
48. Kraus T, Globisch D, Wagner M, Eigenbrod S, Widmann D, Münzel M, et al. Low values of 5-hydroxymethylcytosine (5hmC), the ‘sixth base,’ are associated with anaplasia in human brain tumors. Int J Cancer (2012) 131:1577–90. doi: 10.1002/ijc.27429
49. Kraus T, Kolck G, Greiner A, Schierl K, Guibourt V, Kretzschmar HA, et al. Loss of 5-hydroxymethylcytosine and intratumoral heterogeneity as an epigenomic hallmark of glioblastoma. Tumor Biol (2015) 36:8439–46. doi: 10.1007/s13277-015-3606-9
50. Johnson KC, Houseman EA, King JE, Von Herrmann KM, Fadul CE, Christensen BC. 5-Hydroxymethylcytosine localizes to enhancer elements and is associated with survival in glioblastoma patients. Nat Commun (2016) 7:1–11. doi: 10.1038/ncomms13177
51. Fernandez AF, Bayó GF, Sierra MI, Urdinguio RG, Tora EG, García MG, et al. Loss of 5hmC identifies a new type of aberrant DNA hypermethylation in glioma. Orig Artic Hum Mol Genet (2018) 27:3046–59. doi: 10.1093/hmg/ddy214
52. Waha A, Müller T, Gessi M, Waha A, Isselstein LJ, Luxen D, et al. Nuclear exclusion of TET1 is associated with loss of 5- hydroxymethylcytosine in IDH1 wild-type gliomas. Am J Pathol (2012) 181:675–83. doi: 10.1016/j.ajpath.2012.04.017
53. Orr BA, Haffner MC, Nelson WG, Yegnasubramanian S, Eberhart CG. Decreased 5-Hydroxymethylcytosine is associated with neural progenitor phenotype in normal brain and shorter survival in malignant glioma. PloS One (2012) 7:e41036. doi: 10.1371/journal.pone.0041036
54. Glowacka WK, Jain H, Okura M, Maimaitiming A, Mamatjan Y, Nejad R, et al. 5-Hydroxymethylcytosine preferentially targets genes upregulated in isocitrate dehydrogenase 1 mutant high-grade glioma. Acta Neuropathol (2018) 135:617–34. doi: 10.1007/s00401-018-1821-3
55. Kohli RM, Zhang Y. TET enzymes, TDG and the dynamics of DNA demethylation. Nature (2013) 502:472–9. doi: 10.1038/nature12750
56. He YF, Li BZ, Li Z, Liu P, Wang Y, Tang Q, et al. Tet-mediated formation of 5-carboxylcytosine and its excision by TDG in mammalian DNA. Science (2011) 333:1303–7. doi: 10.1126/science.1169786
57. Dalton SR, Bellacosa A. DNA demethylation by TDG. Epigenomics (2012) 4:459–67. doi: 10.2217/epi.12.36
58. Otani J, Kimura H, Sharif J, Endo TA, Mishima Y. Cell Cycle-Dependent Turnover of 5-Hydroxymethyl Cytosine in Mouse Embryonic Stem Cells. PloS One (2013) 8:82961. doi: 10.1371/journal.pone.0082961
59. Fults D, Pedone CA, Thompson GE, Uchiyama CM, Gumpper KL, Iliev D, et al. Microsatellite deletion mapping on chromosome 10q and mutation analysis of MMAC1, FAS, and MXI1 in human glioblastoma multiforme. Int J Oncol (1998) 12:905–10. doi: 10.3892/ijo.12.4.905
60. Ichimura K, Ohgaki H, Kleihues P, Collins VP. Molecular pathogenesis of astrocytic tumours. J Neurooncol (2004) 70:137–60. doi: 10.1007/s11060-004-2747-2
61. Sonoda Y, Murakami Y, Tominaga T, Kayama T, Yoshimoto T, Sekiya T. Deletion Mapping of Chromosome 10 in Human Glioma. Japan J Cancer Res (1996) 87:363–7. doi: 10.1111/j.1349-7006.1996.tb00231.x
62. Chen B, Lei Y, Wang H, Dang Y, Fang P, Wang J, et al. Repression of the expression of TET2 by ZEB1 contributes to invasion and growth in glioma cells. Mol Med Rep (2017) 15:2625–32. doi: 10.3892/mmr.2017.6288
63. García MG, Carella A, Urdinguio RG, Bayón GF, Lopez V, Tejedor JR, et al. Epigenetic dysregulation of TET2 in human glioblastoma. Oncotarget (2018) 9:25922–34. doi: 10.18632/oncotarget.25406
64. Carella A, Tejedor JR, García MG, Urdinguio RG, Bayón GF, Sierra M, et al. Epigenetic downregulation of TET3 reduces genome-wide 5hmC levels and promotes glioblastoma tumorigenesis. Int J Cancer (2020) 146:373–87. doi: 10.1002/ijc.32520
65. Xu W, Yang H, Liu Y, Yang Y, Wang P, Kim SH, et al. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of α-ketoglutarate-dependent dioxygenases. Cancer Cell (2011) 19:17–30. doi: 10.1016/j.ccr.2010.12.014
66. Dang L, White DW, Gross S, Bennett BD, Bittinger MA, Driggers EM, et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature (2009) 462:739–44. doi: 10.1038/nature08617
67. Cohen AL, Holmen SL, Colman H. IDH1 and IDH2 mutations in gliomas. Curr Neurol Neurosci Rep (2013) 13:345. doi: 10.1007/s11910-013-0345-4
68. Kim YH, Pierscianek D, Mittelbronn M, Vital A, Mariani L, Hasselblatt M, et al. TET2 promoter methylation in low-grade diffuse gliomas lacking IDH1/2 mutations. J Clin Pathol (2011) 64:850–2. doi: 10.1136/jclinpath-2011-200133
69. Kraus T, et al. Genetic characterization of ten-eleven-translocation methylcytosine dioxygenase alterations in human glioma. J Cancer (2015) 6:832–42. doi: 10.7150/jca.12010
70. Klose RJ, Kallin EM, Zhang Y. JmjC-domain-containing proteins and histone demethylation. Nat Rev Genet (2006) 7:715–27. doi: 10.1038/nrg1945
71. Martin C, Zhang Y. The diverse functions of histone lysine methylation. Nat Rev Mol Cell Biol (2005) 6:838–49. doi: 10.1038/nrm1761
72. Mosammaparast N, Shi Y. Reversal of Histone Methylation: Biochemical and Molecular Mechanisms of Histone Demethylases. Annu Rev Biochem (2010) 79:155–79. doi: 10.1146/annurev.biochem.78.070907.103946
73. Smith SMC, Kimyon RS, Watters JJ. Cell-type-specific Jumonji histone demethylase gene expression in the healthy rat CNS: Detection by a novel flow cytometry method. ASN Neuro (2014) 6:193–207. doi: 10.1042/AN20130050
74. Karuppagounder SS, Kumar A, Shao DS, Zille M, Bourassa MW, Caulfield JT, et al. Metabolism and epigenetics in the nervous system: Creating cellular fitness and resistance to neuronal death in neurological conditions via modulation of oxygen-, iron-, and 2-oxoglutarate-dependent dioxygenases. Brain Res (2015) 1628:273–87. doi: 10.1016/j.brainres.2015.07.030
75. Lee HY, Choi K, Oh H, Park YK, Park H. HIF-1-dependent induction of jumonji domain-containing protein (JMJD) 3 under hypoxic conditions. Mol Cells (2014) 37:43–50. doi: 10.14348/molcells.2014.2250
76. Liau BB, Sievers C, Donohue LK, Gillespie SM, Flavahan WA, Miller TE, et al. Adaptive Chromatin Remodeling Drives Glioblastoma Stem Cell Plasticity and Drug Tolerance. Cell Stem Cell (2017) 20:233–246.e7. doi: 10.1016/j.stem.2016.11.003
77. Li M, Cheng J, Ma Y, Guo H, Shu H, Huang H, et al. The histone demethylase JMJD2A promotes glioma cell growth via targeting Akt-mTOR signaling. Cancer Cell Int (2020) 20:101. doi: 10.1186/s12935-020-01177-z
78. Liu BL, Cheng JX, Zhang X, Wang R, Zhang W, Lin H, et al. Global histone modification patterns as prognostic markers to classify glioma patients. Cancer Epidemiol Biomarkers Prev (2010) 19:2888–96. doi: 10.1158/1055-9965.EPI-10-0454
79. Dai B, Hu Z, Huang H, Zhu G, Xiao Z, Wan W, et al. Overexpressed KDM5B is associated with the progression of glioma and promotes glioma cell growth via downregulating p21. Biochem Biophys Res Commun (2014) 454:221–7. doi: 10.1016/j.bbrc.2014.10.078
80. Banelli B, Carra E, Barbieri F, Würth R, Parodi F, Pattarozzi A, et al. The histone demethylase KDM5A is a key factor for the resistance to temozolomide in glioblastoma. Cell Cycle (2015) 14:3418–29. doi: 10.1080/15384101.2015.1090063
81. Staberg M, Rasmussen RD, Michaelsen SR, Pedersen H, Jensen KE, Villingshøj M, et al. Targeting glioma stem-like cell survival and chemoresistance through inhibition of lysine-specific histone demethylase KDM2B. Mol Oncol (2018) 12:406–20. doi: 10.1002/1878-0261.12174
82. Frescas D, Guardavaccaro D, Bassermann F, Koyama-Nasu R, Pagano M. JHDM1B/FBXL10 is a nucleolar protein that represses transcription of ribosomal RNA genes. Nature (2007) 450:309–13. doi: 10.1038/nature06255
83. Ene C, Edwards L, Riddick G, Baysan M, Woolard K, Kotliarova S, et al. Histone Demethylase Jumonji D3 (JMJD3) as a Tumor Suppressor by Regulating p53 Protein Nuclear Stabilization. PloS One 7 (2012)7:e51407. doi: 10.1371/journal.pone.0051407
84. Lu C, Ward PS, Kapoor GS, Rohle D, Turcan S, Abdel-Wahab O, et al. IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature (2012) 483:474–8. doi: 10.1038/nature10860
85. Turcan S, Makarov V, Taranda J, Wang Y, Fabius AWM, Wu W, et al. Mutant-IDH1-dependent chromatin state reprogramming, reversibility, and persistence. Nat Genet (2018) 50:62–72. doi: 10.1038/s41588-017-0001-z
86. Chowdhury R, Yeoh KK, Tian YM, Hillringhaus L, Bagg EA, Rose NR, et al. The oncometabolite 2-hydroxyglutarate inhibits histone lysine demethylases. Nat Publ Gr (2011) 12:463–9. doi: 10.1038/embor.2011.43
87. Monteiro AR, Hill R, Pilkington GJ, Madureira PA. The role of hypoxia in glioblastoma invasion. Cells (2017) 6:45–64. doi: 10.3390/cells6040045
88. Spence AM, Muzi M, Swanson KR, O’Sullivan F, Rockhill JK, Rajendran JG, et al. Regional hypoxia in glioblastoma multiforme quantified with [ 18F] fluoromisonidazole positron emission tomography before radiotherapy: Correlation with time to progression and survival. Clin Cancer Res (2008) 14:2623–30. doi: 10.1158/1078-0432.CCR-07-4995
89. Kaynar MY, Sanus GZ, Hnimoglu H, Kacira T, Kemerdere R, Atukeren P, et al. Expression of hypoxia inducible factor-1α in tumors of patients with glioblastoma multiforme and transitional meningioma. J Clin Neurosci (2008) 15:1036–42. doi: 10.1016/j.jocn.2007.07.080
90. Jensen RL. Brain tumor hypoxia: tumorigenesis, angiogenesis, imaging, pseudoprogression, and as a therapeutic target. J Neuro-oncol Neurooncol (2009) 92:317–35. doi: 10.1007/s11060-009-9827-2
91. Semenza GL. Defining the role of hypoxia-inducible factor 1 in cancer biology and therapeutics. Oncogene (2010) 29:625–34. doi: 10.1038/onc.2009.441
92. Ivan M, Kondo K, Yang H, Kim W, Valiando J, Ohh M, et al. HIFα targeted for VHL-mediated destruction by proline hydroxylation: Implications for O2 sensing. Science (2001) 292:464–8. doi: 10.1126/science.1059817
93. Mahon PC, Hirota K, Semenza GL. FIH-1: A novel protein that interacts with HIF-1α and VHL to mediate repression of HIF-1 transcriptional activity. Genes Dev (2001) 15:2675–86. doi: 10.1101/gad.924501
94. Hewitson KS, McNeill LA, Riordan MV, Tian Y-M, Bullock AN, Welford RW, et al. Hypoxia-inducible Factor (HIF) Asparagine Hydroxylase Is Identical to Factor Inhibiting HIF (FIH) and Is Related to the Cupin Structural Family*. (2002)277:26351–5. doi: 10.1074/jbc.C200273200
95. Jaakola P, Mole DR, Tian Y, Wilson MI, Gielbery J, Gaskell, et al. Targeting of HIF-alpha to to the von Hippel–Lindau Ubiquitylation Complex by O2-Regulated Prolyl Hydroxylation. Science (2001) 292:468–72. doi: 10.1126/science.1059817
96. Kaelin WG, Ratcliffe PJ. Oxygen Sensing by Metazoans: The Central Role of the HIF Hydroxylase Pathway. Mol Cell (2008) 30:393–402. doi: 10.1016/j.molcel.2008.04.009
97. Schödel J, Oikonomopoulos S, Ragoussis J, Pugh CW, Ratcliffe PJ, Mole DR. High-resolution genome-wide mapping of HIF-binding sites by ChIP-seq. Blood (2011) 117. doi: 10.1182/blood-2010-10-314427
98. Semenza GL. Hypoxia-inducible factors: Mediators of cancer progression and targets for cancer therapy. Trends Pharmacol Sci (2012) 33:207–14. doi: 10.1016/j.tips.2012.01.005
99. Mole DR, Blancher C, Copley RR, Pollard PJ, Gleadle JM, Ragousis J, et al. Genome-wide association of hypoxia-inducible factor (HIF)-1α and HIF-2α DNA binding with expression profiling of hypoxia-inducible transcripts. J Biol Chem (2009) 284:16767–75. doi: 10.1074/jbc.M901790200
100. Semenza GL, Rue EA, Iyer NV, Pang MG, Kearns WG. Assignment of the hypoxia-inducible factor 1α gene to a region of conserved synteny on mouse chromosome 12 and human chromosome 14q. Genomics (1996) 34:437–9. doi: 10.1006/geno.1996.0311
101. https://www.ensembl.org/index.html?redirect=no. (Accessed on 26/02/2021).
102. Maxwell PH, Dachs GU, Gleadle JM, Nicholls LG, Harris AL, Stratford IJ, et al. Hypoxia-inducible factor-1 modulates gene expression in solid tumors and influences both angiogenesis and tumor growth. Proc Natl Acad Sci U S A (1997) 94:8104–9. doi: 10.1073/pnas.94.15.8104
103. Dachs GU, Patterson AV, Firth JD, Ratcliffe PJ, Stuart Townsend KM, Stratford IJ, et al. Targeting gene expression to hypoxic tumor cells. Nat Med (1997) 3:515–20. doi: 10.1038/nm0597-515
104. Wenger RH, Grassmann M. Molecular Biology of Hypoxia-Inducible Factor-1. Mol Biol Hematopoiesis (1999) pp:269–77. doi: 10.1007/978-1-4615-4797-6_34
105. Keith B, Johnson RS, Simon MC. HIF1 α and HIF2 α: sibling rivalry in hypoxic tumour growth and progression. Nat Rev Cancer (2012) 12:9–22. doi: 10.1038/nrc3183
106. Smythies JA, Sun M, Masson N, Salama R, Simpson PD, Murray E, et al. Inherent DNA binding specificities of the HIF-1α and HIF-2α transcription factors in chromatin. EMBO Rep (2019) 20. doi: 10.15252/embr.201846401
107. Majmundar AJ, Wong WJ, Simon MC. Hypoxia-inducible factors and the response to hypoxic stress. Mol Cell (2010) 2:294–309. doi: 10.1016/j.molcel.2010.09.022
108. Chowdhury R, Hardy A, Schofield CJ. The human oxygen sensing machinery and its manipulation. Chem Soc Rev (2008) 37:1308–19. doi: 10.1039/b701676j
109. Choudhry H, Harris AL. Advances in Hypoxia-Inducible Factor Biology. Cell Metab (2018) 27:281–98. doi: 10.1016/j.cmet.2017.10.005
110. Jensen RL. Hypoxia in the tumorigenesis of gliomas and as a potential target for therapeutic measures. Neurosurg Focus (2006) 20:E24. doi: 10.3171/foc.2006.20.4.16
111. Korkolopoulou P, Patsouris E, Konstantinidou AE, Pavlopoulos PM, Kavantzas N, Boviatsis E, et al. Hypoxia-inducible factor 1a/vascular endothelial growth factor axis in astrocytomas. Associations with microvessel morphometry, proliferation and prognosis. Neuropathol Appl Neurobiol (2004) 30:267–78. doi: 10.1111/j.1365-2990.2003.00535.x
112. Chen W, Cheng X, Wang X, Wang J, Wen X, Xie C, et al. Clinical implications of hypoxia-inducible factor-1alpha and caveolin-1 overexpression in isocitrate dehydrogenase-wild type glioblastoma multiforme. Oncol Lett (2019) 17:2867–73. doi: 10.3892/ol.2019.9929
113. Zagzag D, Zhong H, Scalzitti JM, Laughner E, Simons JW, Semenza GL. Expression of hypoxia-inducible factor 1α in brain tumors: Association with angiogenesis, invasion, and progression. Cancer (2000) 88:2606–18. doi: 10.1002/1097-0142(20000601)88:11<2606::AID-CNCR25>3.0.CO;2-W
114. Lo Dico A, Martelli C, Diceglie C, Lucignani G, Ottobrini L. Hypoxia-Inducible Factor-1alpha Activity as a Switch for Glioblastoma Responsiveness to Temozolomide. Front Oncol (2018) 8:249. doi: 10.3389/fonc.2018.00249
115. Jensen RL, Ragel BT, Whang K, Gillespie D. Inhibition of hypoxia inducible factor-1a (HIF-1a) decreases vascular endothelial growth factor (VEGF) secretion and tumor growth in malignant gliomas. J Neurooncol (2006) 78:233–47. doi: 10.1007/s11060-005-9103-z
116. Brat DJ, Castellano-Sanchez AA, Hunter SB, Pecot M, Cohen C, Hammond EH, et al. Pseudopalisades in Glioblastoma Are Hypoxic, Express Extracellular Matrix Proteases, and Are Formed by an Actively Migrating Cell Population. Cancer Res (2004) 64:920–7. doi: 10.1158/0008-5472.CAN-03-2073
117. Renfrow JJ, Soike MH, Debinski W, Ramkissoon SH, Mott RT, Frenkel MB, et al. Hypoxia-inducible factor 2α: A novel target in gliomas. Future Med Chem (2018) 10:2227–36. doi: 10.4155/fmc-2018-0163
118. Yu F, White SB, Zhao Q, Lee FS. HIF-1α binding to VHL is regulated by stimulus-sensitive proline hydroxylation. Proc Natl Acad Sci U S A (2001) 98:9630–5. doi: 10.1073/pnas.181341498
119. Lando D, Peet DJ, Gorman JJ, Whelan DA, Whitelaw ML, Bruick RK. FIH-1 is an asparaginyl hydroxylase enzyme that regulates the transcriptional activity of hypoxia-inducible factor. Genes Dev (2002) 16:1466–71. doi: 10.1101/gad.991402
120. Rodriguez J, Haydinger CHD, Peet DJ, Nguyen L, von Kriegsheim A. Asparagine hydroxylation is a reversible post-translational modification. Mol Cell Proteomics (2020) 13:1777–89. doi: 10.1101/2020.03.22.002436
121. McNeill LA, Flashman E, Buck MRG, Hewitson KS, Clifton IJ, Jeschke G, et al. Hypoxia-inducible factor prolyl hydroxylase 2 has a high affinity for ferrous iron and 2-oxoglutarate. Mol Biosyst (2005) 1:321–4. doi: 10.1039/b511249b
122. McDonough MA, Li V, Flashman E, Chowdhury R, Mohr C, R Lié nard BM, et al. Cellular oxygen sensing: Crystal structure of hypoxia-inducible factor prolyl hydroxylase (PHD2). Proc Natl Acad Sci USA (2006) 103:9814–9. doi: 10.1073/pnas.0601283103
123. Berra E, Benizri E, Ginouvès A, Volmat V, Roux D, Pouysségur J. HIF prolyl-hydroxylase 2 is the key oxygen sensor setting low steady-state levels of HIF-1α in normoxia. EMBO J (2003) 22:4082–90. doi: 10.1093/emboj/cdg392
124. Metzen E, Berchner-Pfannschmidt U, Stengel P, Marxsen JH, Stolze I, Klinger M, et al. Intracellular localisation of human HIF-1α hydroxylases: Implications for oxygen sensing. J Cell Sci (2003) 116:1319–26. doi: 10.1242/jcs.00318
125. Ježek P. 2-hydroxyglutarate in cancer cells. Antioxid Redox Signaling (2020) 33:903–26. doi: 10.1089/ars.2019.7902
126. Losman J-A, Koivunen P, Kaelin WG. 2-Oxoglutarate-dependent dioxygenases in cancer. Nat Rev Cancer (2020) 20:710–26. doi: 10.1038/s41568-020-00303-3
127. Koivunen P, Lee S, Duncan CG, Lopez G, Lu G, Ramkissoon S, et al. Transformation by the (R)-enantiomer of 2-hydroxyglutarate linked to EGLN activation. Nature (2012) 483:484–8. doi: 10.1038/nature10898
128. Williams SC, Karajannis MA, Chiriboga L, Golfinos JG, Von Deimling A, Zagzag D, et al. R132H-mutation of isocitrate dehydrogenase-1 is not sufficient for HIF-1α upregulation in adult glioma. Acta Neuropathol (2011). doi: 10.1007/s00401-010-0790-y
129. Womeldorff M, Gillespie D, Jensen RL. Hypoxia-inducible factor-1 and associated upstream and downstream proteins in the pathophysiology and management of glioblastoma. Neurosurg Focus (2014) 37:1–17. doi: 10.3171/2014.9.FOCUS14496
130. Rhodes DR, Yu J, Shanker K, Deshpande N, Varambally R, Ghosh D, et al. ONCOMINE: A Cancer Microarray Database and Integrated Data-Mining Platform. Neoplasia (2004) 6:1–6. doi: 10.1016/S1476-5586(04)80047-2
131. Wang E, Zhang C, Polavaram N, Liu F, Wu G, Schroeder MA, et al. The Role of Factor Inhibiting HIF (FIH-1) in Inhibiting HIF-1 Transcriptional Activity in Glioblastoma Multiforme. PloS One (2014) 9:e86102. doi: 10.1371/journal.pone.0086102
132. Feng J, Zhang Y, She X, Sun Y, Fan L, Ren X, et al. Hypermethylated gene ANKDD1A is a candidate tumor suppressor that interacts with FIH1 and decreases HIF1α stability to inhibit cell autophagy in the glioblastoma multiforme hypoxia microenvironment. Oncogene (2019) 38:103–19. doi: 10.1038/s41388-018-0423-9
133. Kuiper C, Vissers MCM. Ascorbate as a cofactor for Fe-and 2-oxoglutarate dependent dioxygenases: Physiological activity in tumour growth and progression. Front Oncol (2014) 4:359. doi: 10.3389/fonc.2014.00359
134. Rebouche, Charles J. Ascorbic acid and carnitine biosynthesis. Am J Clin Nutr (1991) 54:1147S–52S. doi: 10.1093/ajcn/54.6.1147s
135. Hirsilä M, Koivunen P, Günzler V, Kivirikko K II, Myllyharju J. Characterization of the human prolyl 4-hydroxylases that modify the hypoxia-inducible factor. J Biol Chem (2003) 278:30772–80. doi: 10.1074/jbc.M304982200
136. Myllyharju J, Kivirikko K II. Characterization of the iron- and 2-oxoglutavate binding sites of human prolyl 4-hydroxylase. EMBO J (1997) 16:1173–80. doi: 10.1093/emboj/16.6.1173
137. Ortiz-Prado E, Dunn JF, Vasconez J, Castillo D, Viscor G. Partial pressure of oxygen in the human body: a general review. Am J Blood Res (2019) 9:1–14.
138. Charbel FT, Hoffman WE, Misra M, Hannigan K, Ausman J II. Cerebral interstitial tissue oxygen tension, pH, HCO3, CO2. Surg Neurol (1997) 48:414–7. doi: 10.1016/S0090-3019(96)00473-9
139. Baker NA. A galvanic cell suitable for monitoring cortical oxygen in man. Med Biol Eng (1975) 13:443–9. doi: 10.1007/BF02477117
140. Hoffman WE, Charbel FT, Edelman G. Brain tissue oxygen, carbon dioxide, and pH in neurosurgical patients at risk for ischemia. Anesth Analg (1996) 82:582–6. doi: 10.1213/00000539-199603000-00027
141. Assad F, Schultheiss R, Leniger-Follert E, Wüllenweber R. Measurement of Local Oxygen Partial Pressure (PO2) of the Brain Cortex in Cases of Brain Tumors. Berlin, Heidelberg: Springer (1984), 263–70. doi: 10.1007/978-3-642-69360-1_45
142. Meixensberger J, Dings J, Kuhnigk H, Roosen K. Studies of tissue PO2 in normal and pathological human brain cortex. Acta Neurochir Suppl (Wien) (1993) 59:58–63. doi: 10.1007/978-3-7091-9302-0_10
143. Dings J, Meixensberger J, Jäger A, Roosen K. Clinical Experience with 118 Brain Tissue Oxygen Partial Pressure Catheter Probes. Neurosurgery (1998) 43:1082–94. doi: 10.1097/00006123-199811000-00045
144. Vaupel P, Höckel M, Mayer A. Detection and characterization of tumor hypoxia using pO2 histography. Antioxid Redox Signaling (2007) 9:1221–35. doi: 10.1089/ars.2007.1628
145. Rampling R, Cruickshank G, Lewis AD, Fitzsimmons SA, Workman P. Direct measurement of pO2 distribution and bioreductive enzymes in human malignant brain tumors. Radiat Oncol Biol Phys (1994) 29:427–31. doi: 10.1016/0360-3016(94)90432-4
146. Qian Y, Von Eyben R, Liu Y, Chin FT, Miao Z, Apte S, et al. 18F-EF5 PET-based Imageable Hypoxia Predicts Local Recurrence in Tumors Treated With Highly Conformal Radiation Therapy. Int J Radiat Oncol Biol Phys (2018) 102:1183–92. doi: 10.1016/j.ijrobp.2018.06.287
147. Valk PE, Mathis CA, Prados M, Gilbert JC, Budinger TF. Hypoxia in human gliomas: Demonstration by PET with fluorine-18-fluoromisonidazole. Artic J Nucl Med (1993) 33:2133–7.
148. Evans SM, Jenkins KW, Chen HI, Jenkins WT, Judy KD, Hwang WT, et al. The relationship among hypoxia, proliferation, and outcome in patients with de nouo glioblastoma: A pilot study. Transl Oncol (2010) 3:160–9. doi: 10.1593/tlo.09265
149. Koch CJ. Measurement of absolute oxygen levels in cells and tissues using oxygen sensors and 2-nitroimidazole EF5. Methods Enzymol (2002) 352:3–31. doi: 10.1016/S0076-6879(02)52003-6
150. Collingridge DR, Piepmeier JM, Rockwell S, Knisely JPS. Polarographic measurements of oxygen tension in human glioma and surrounding peritumoural brain tissue. Radiother Oncol (1999) 53:127–31. doi: 10.1016/S0167-8140(99)00121-8
151. Kaur B, Khwaja FW, Severson EA, Matheny SL, Brat DJ, Van Meir EG, et al. Hypoxia and the hypoxia-inducible-factor pathway in glioma growth and angiogenesis. Neuro Oncol (2005) 7:134–53. doi: 10.1215/S1152851704001115
152. Cruickshank GS, Rampling R. Peri-tumoural hypoxia in human brain: peroperative measurement of the tissue oxygen tension around malignant brain tumours. Acta Neurochir Suppl (Wien) (1994) 60:375–7. doi: 10.1007/978-3-7091-9334-1_101
153. Stępień K, Ostrowski RP, Matyja E. Hyperbaric oxygen as an adjunctive therapy in treatment of malignancies, including brain tumours. Med Oncol (2016) 33:1–9. doi: 10.1007/s12032-016-0814-0
154. Miralbell R, Mornex F, Greiner R, Bolla M, Storme G, Hulshof M, et al. Accelerated radiotherapy, carbogen, and nicotinamide in glioblastoma multiforme: Report of European Organization for Research and Treatment of Cancer Trial 22933. J Clin Oncol (1999) 17:3143–9. doi: 10.1200/JCO.1999.17.10.3143
155. Gainer JL, Sheehan JP, Larner JM, Jones DR. Trans sodium crocetinate with temozolomide and radiation therapy for glioblastoma multiforme. J Neurosurg (2017) 126:460–6. doi: 10.3171/2016.3.JNS152693
156. Bindra RS, Chalmers AJ, Evans S, Dewhirst M. GBM radiosensitizers: dead in the water…or just the beginning? J Neuro-Oncol (2017) 134:513–21. doi: 10.1007/s11060-017-2427-7
157. Mudassar F, Shen H, O’Neill G, Hau E. Targeting tumor hypoxia and mitochondrial metabolism with anti-parasitic drugs to improve radiation response in high-grade gliomas. J Exp Clin Cancer Res (2020) 39:1–17. doi: 10.1186/s13046-020-01724-6
158. Balss J, Meyer J, Mueller W, Korshunov A, Hartmann C, Von Deimling A. Analysis of the IDH1 codon 132 mutation in brain tumors. Acta Neuropathol (2008) 116:597–602. doi: 10.1007/s00401-008-0455-2
159. Bleeker FE, Lamba S, Leenstra S, Troost D, Hulsebos T, Vandertop WP, et al. IDH1 mutations at residue p.R132 (IDH1R132) occur frequently in high-grade gliomas but not in other solid tumors. Hum Mutat (2009) 30:7–11. doi: 10.1002/humu.20937
160. Fan J, Teng X, Liu L, Mattaini KR, Looper RE, Vander Heiden MG, et al. Human phosphoglycerate dehydrogenase produces the oncometabolite D-2-hydroxyglutarate. ACS Chem Biol (2015) 10:510–6. doi: 10.1021/cb500683c
161. Terunuma A, Putluri N, Mishra P, Mathé EA, Dorsey TH, Yi M, et al. MYC-driven accumulation of 2-hydroxyglutarate is associated with breast cancer prognosis. J Clin Invest (2014) 124:398–412. doi: 10.1172/JCI71180
162. Mellinghoff I, Maher E, Wen P, Cloughesy T, Peters K, Choi C, et al. A phase 1, multicenter, randomized, openlabel, perioperative study of ag-120 (ivosidenib) and AG-881 in patients with recurrent, nonenhancing, IDH1-mutant, low-grade glioma. Neuro Oncol (2018). doi: 10.1093/neuonc/noy148.973
163. Fan B, Mellinghoff IK, Wen PY, Lowery MA, Goyal L, Tap WD, et al. Clinical pharmacokinetics and pharmacodynamics of ivosidenib, an oral, targeted inhibitor of mutant IDH1, in patients with advanced solid tumors. Invest New Drugs (2020) 38:433–44. doi: 10.1007/s10637-019-00771-x
164. Natsume A, Wakabayashi T, Miyakita Y, Narita Y, Mineharu Y, Arakawa Y, et al. Phase I study of a brain penetrant mutant IDH1 inhibitor DS-1001b in patients with recurrent or progressive IDH1 mutant gliomas. J Clin Oncol (2019) 15:2004. doi: 10.1200/jco.2019.37.15_suppl.2004
165. Karpel-Massler G, Nguyen TTT, Shang E, Siegelin MD. Novel IDH1-Targeted Glioma Therapies. CNS Drugs (2019) 33:1155–66. doi: 10.1007/s40263-019-00684-6
166. Han S, Liu Y, Cai SJ, Qian M, Ding J, Larion M, et al. IDH mutation in glioma: molecular mechanisms and potential therapeutic targets. Br J Cancer (2020) 122:1580–9. doi: 10.1038/s41416-020-0814-x
167. Urban DJ, Martinez NJ, Davis MI, Brimacombe KR, Cheff DM, Lee TD, et al. Assessing inhibitors of mutant isocitrate dehydrogenase using a suite of pre-clinical discovery assays. Sci Rep (2017) 7:12758. doi: 10.1038/s41598-017-12630-x
168. Nakagawa M, Nakatani F, Matsunaga H, Seki T, Endo M, Ogawara Y, et al. Selective inhibition of mutant IDH1 by DS-1001b ameliorates aberrant histone modifications and impairs tumor activity in chondrosarcoma. Oncogene (2019) 38:6835–49. doi: 10.1038/s41388-019-0929-9
169. Nicolay B, Narayanaswamy R, Amatangelo MD, Aguado E, Nagaraja R, Murtie J, et al. EXTH-34. Combined use of the pan IDH mutant inhibitor AG-881 with radiation therapy shoes added benefit in an orthotopic IDH1 mutant glioma model in vivo. Neuro Oncol (2017) 19: vi79. doi: 10.1093/neuonc/nox168.326
170. Pusch S, Krausert S, Fischer V, Balss J, Ott M, Schrimpf D, et al. Pan-mutant IDH1 inhibitor BAY 1436032 for effective treatment of IDH1 mutant astrocytoma in vivo. Acta Neuropathol (2017) 133:629–44. doi: 10.1007/s00401-017-1677-y
171. Schumacher T, Bunse L, Pusch S, Sahm F, Wiestler B, Quandt J, et al. A vaccine targeting mutant IDH1 induces antitumour immunity. Nature (2014) 512:324–7. doi: 10.1038/nature13387
172. Pellegatta S, Valletta L, Corbetta C, Patanè M, Zucca I, Riccardi Sirtori F, et al. Effective immuno-targeting of the IDH1 mutation R132H in a murine model of intracranial glioma. Acta Neuropathol Commun (2015) 3:4. doi: 10.1186/s40478-014-0180-0
173. Ward RJ, Zucca FA, Duyn JH, Crichton RR, Zecca L. The role of iron in brain ageing and neurodegenerative disorders. Lancet Neurol (2014) 13:1045–60. doi: 10.1016/S1474-4422(14)70117-6
174. Bradbury MW. Transport of iron in the blood-brain-cerebrospinal fluid system. J Neurochem (1997) 69:443–54. doi: 10.1046/j.1471-4159.1997.69020443.x
175. Berg D, Hochstrasser H. Iron metabolism in parkinsonian syndromes. Mov Disord (2006) 21:1299–310. doi: 10.1002/mds.21020
176. Beydoun R, Hamood MA, Gomez Zubieta DM, Kondapalli KC. Na+/H+ Exchanger 9 Regulates Iron Mobilization at the Blood-Brain Barrier in Response to Iron Starvation *. (2017) 292:4293–301. doi: 10.1074/jbc.M116.769240
177. McCarthy RC, Kosman DJ. Glial Cell Ceruloplasmin and Hepcidin Differentially Regulate Iron Efflux from Brain Microvascular Endothelial Cells. PloS One (2014) 9:89003. doi: 10.1371/journal.pone.0089003
178. Du F, Qian C, Ming Qian Z, Wu X-M, Xie H, Yung W-H, et al. Hepcidin directly inhibits transferrin receptor 1 expression in astrocytes via a cyclic AMP-protein kinase a pathway. Glia (2011)59:936–45. doi: 10.1002/glia.21166
179. Duck KA, Simpson IA, Connor JR. Regulatory mechanisms for iron transport across the blood-brain barrier. Biochem Biophys Res Commun (2017) 494:70–5. doi: 10.1016/j.bbrc.2017.10.083
180. Torti SV, Torti FM. Cellular iron metabolism in prognosis and therapy of breast cancer. Crit Rev Oncog (2013) 18:435–48. doi: 10.1615/CritRevOncog.2013007784
181. Khan A II, Liu J, Dutta P. Iron transport kinetics through blood-brain barrier endothelial cells. Biochim Biophys Acta Gen Subj (2018) 1862:1168–79. doi: 10.1016/j.bbagen.2018.02.010
182. Malecki EA, Devenyi AG, Beard JL, Connor JR. Existing and emerging mechanisms for transport of iron and manganese to the brain. J Neurosci Res (1999) 56:113–22. doi: 10.1002/(SICI)1097-4547(19990415)56:2<113::AID-JNR1>3.0.CO;2-K
184. Cabantchik Z II. Labile iron in cells and body fluids: physiology, pathology, and pharmacology. Front Pharmacol (2014) 5:45. doi: 10.3389/fphar.2014.00045
185. McCarthy RC, Kosman DJ. Iron transport across the blood-brain barrier: Development, neurovascular regulation and cerebral amyloid angiopathy. Cell Mol Life Sci (2015) 72:709–27. doi: 10.1007/s00018-014-1771-4
186. McCarthy RC, Kosman DJ. Ferroportin and exocytoplasmic ferroxidase activity are required for brain microvascular endothelial cell iron efflux. J Biol Chem (2013) 288:17932–40. doi: 10.1074/jbc.M113.455428
187. Wu LJC, Leenders AGM, Cooperman S, Meyron-Holtz E, Smith S, Land W, et al. Expression of the iron transporter ferroportin in synaptic vesicles and the blood-brain barrier. Brain Res (2004) 1001:108–17. doi: 10.1016/j.brainres.2003.10.066
188. Weston C, Klobusicky J, Weston J, Connor J, Toms SA, Marko NF. Aberrations in the Iron Regulatory Gene Signature Are Associated with Decreased Survival in Diffuse Infiltrating Gliomas. PloS One (2016) 11:e0166593. doi: 10.1371/journal.pone.0166593
189. Legendre C, Garcion E. Iron metabolism: a double-edged sword in the resistance of glioblastoma to therapies. Trends Endocrinol Metab (2015) 26:322–31. doi: 10.1016/j.tem.2015.03.008
190. Magri S, Pinton L, Masetto E, Cassandro S, Pozzuoli A, Belluzzi E, et al. Role of iron metabolism in the immunosuppression mediated by myeloid cells in glioblastoma patients. Ann Oncol (2019) 30:xi56. doi: 10.1093/annonc/mdz452.029
191. Vela D. Hepcidin, an emerging and important player in brain iron homeostasis. J Trans Med (2018) 16:25. doi: 10.1186/s12967-018-1399-5
192. Schonberg DL, Miller TE, Wu Q, Flavahan WA, Das NK, Hale JS, et al. Preferential Iron Trafficking Characterizes Glioblastoma Stem-like Cells. Cancer Cell (2015) 28:441–55. doi: 10.1016/j.ccell.2015.09.002
193. Flashman E, Davies SL, Yeoh KK, Schofield CJ. Investigating the dependence of the hypoxia-inducible factor hydroxylases (factor inhibiting HIF and prolyl hydroxylase domain 2) on ascorbate and other reducing agents. Biochem J (2010) 427:135–42. doi: 10.1042/BJ20091609
194. Minor EA, Court BL, Young J II, Wang G. Ascorbate induces ten-eleven translocation (Tet) methylcytosine dioxygenase-mediated generation of 5-hydroxymethylcytosine. J Biol Chem (2013) 288:13669–74. doi: 10.1074/jbc.C113.464800
195. Harrison FE, May JM. Vitamin C function in the brain: vital role of the ascorbate transporter SVCT2. Free Radic Biol Med (2009) 46:719–30. doi: 10.1016/j.freeradbiomed.2008.12.018
196. Figueroa-Méndez R, Rivas-Arancibia S. Vitamin C in health and disease: Its role in the metabolism of cells and redox state in the brain. Front Physiol (2015) 6. doi: 10.3389/fphys.2015.00397
197. Hornig D. Distribution of ascorbic acid, metabolites and analogues in man and animals. (1975) 258:103–18. doi: 10.1111/j.1749-6632.1975.tb29271.x
198. Nualart F. Vitamin C Transporters, Recycling and the Bystander Effect in the Nervous System: SVCT2 versus Gluts. J Stem Cell Res Ther (2014) 04:209. doi: 10.4172/2157-7633.1000209
199. Landolt H, Langemann H, Probst A, Gratzl O. Levels of water-soluble antioxidants in astrocytoma and in adjacent tumor-free tissue. J Neurooncol (1994) 21:127–33. doi: 10.1007/BF01052896
200. Blaschke K, Ebata KT, Karimi MM, Zepeda-Martínez JA, Goyal P, Mahapatra S, et al. Vitamin C induces Tet-dependent DNA demethylation and a blastocyst-like state in ES cells. Nature (2013) 500:222–6. doi: 10.1038/nature12362
201. Hore TA, Von Meyenn F, Ravichandran M, Bachman M, Ficz G, Oxley D, et al. Retinol and ascorbate drive erasure of Epigenetic memory and enhance reprogramming to naïve pluripotency by complementary mechanisms. Proc Natl Acad Sci USA (2016) 113:12202–7. doi: 10.1073/pnas.1608679113
202. Gillberg L, Ørskov AD, Nasif A, Ohtani H, Madaj Z, Hansen JW, et al. Oral vitamin C supplementation to patients with myeloid cancer on azacitidine treatment: Normalization of plasma vitamin C induces epigenetic changes. Clin Epigenet (2019) 11:143. doi: 10.1186/s13148-019-0739-5
203. Peng D, Ge G, Gong Y, Zhan Y, He S, Guan B, et al. Vitamin C increases 5-hydroxymethylcytosine level and inhibits the growth of bladder cancer. Clin Epigenet (2018) 10. doi: 10.1186/s13148-018-0527-7
204. Cimmino L, Dolgalev I, Wang Y, Yoshimi A, Martin GH, Wang J, et al. Restoration of TET2 Function Blocks Aberrant Self-Renewal and Leukemia Progression. Cell (2017) 170:1079–1095.e20 doi: 10.1016/j.cell.2017.07.032.
205. Das AB, Kakadia PM, Wojcik D, Pemberton L, Browett PJ, Bohlander SK, et al. Clinical remission following ascorbate treatment in a case of acute myeloid leukemia with mutations in TET2 and WT1. Blood Cancer J (2019) 9:82. doi: 10.1038/s41408-019-0242-4
206. Gerecke C, Schumacher F, Berndzen A, Homann T, Kleuser B. Vitamin C in combination with inhibition of mutant IDH1 synergistically activates TET enzymes and epigenetically modulates gene silencing in colon cancer cells. Epigenetics (2020) 15:307–22. doi: 10.1080/15592294.2019.1666652
207. Mingay M, Chaturvedi A, Bilenky M, Cao Q, Jackson L, Hui T, et al. Vitamin C-induced epigenomic remodelling in IDH1 mutant acute myeloid leukaemia. Leukemia (2018) 32:11–20. doi: 10.1038/leu.2017.171
208. Wang T, Chen K, Zeng X, Yang J, Wu Y, Shi X, et al. The histone demethylases Jhdm1a/1b enhance somatic cell reprogramming in a vitamin-C-dependent manner. Cell Stem Cell (2011) 9:575–87. doi: 10.1016/j.stem.2011.10.005
209. Ebata KT, Mesh K, Liu S, Bilenky M, Fekete A, Acker MG, et al. Vitamin C induces specific demethylation of H3K9me2 in mouse embryonic stem cells via Kdm3a/b. Epigenet Chromatin (2017) 10:36. doi: 10.1186/s13072-017-0143-3
210. Song MH, Nair VS, Oh K II. Vitamin C enhances the expression of IL17 in a Jmjd2-dependent manner. BMB Rep (2017) 50:49–54. doi: 10.5483/BMBRep.2017.50.1.193
211. Chung TL, Brena RM, Kolle G, Grimmond SM, Berman BP, Laird PW, et al. Vitamin C promotes widespread yet specific DNA demethylation of the epigenome in human embryonic stem cells. Stem Cells (2010) 28:1848–55. doi: 10.1002/stem.493
212. Kuiper C, Dachs GU, Currie MJ, Vissers MCM. Intracellular ascorbate enhances hypoxia-inducible factor (HIF)-hydroxylase activity and preferentially suppresses the HIF-1 transcriptional response. Free Radic Biol Med (2014) 69:308–17. doi: 10.1016/j.freeradbiomed.2014.01.033
213. Dachs G, Campbell EJ, Vissers MC. Ascorbate availability affects tumor implantation-take rate and increases tumor rejection in gulo-/- mice. Hypoxia (2016) 4:41. doi: 10.2147/HP.S103088
214. Jóźwiak P, Ciesielski P, Zaczek A, Lipińska A, Pomorski L, Wieczorek M, et al. Expression of hypoxia inducible factor 1α and 2α and its association with Vitamin C level in thyroid lesions. J Biomed Sci (2017) 24:83. doi: 10.1186/s12929-017-0388-y
215. Wohlrab C, Kuiper C, Vissers MCM, Phillips E, Robinson BA, Dachs GU. Ascorbate modulates the hypoxic pathway by increasing intracellular activity of the HIF hydroxylases in renal cell carcinoma cells. Hypoxia (2019) 7:17–31. doi: 10.2147/HP.S201643
216. Campbell EJ, Vissers MCM, Bozonet S, Dyer A, Robinson BA, Dachs GU. Restoring physiological levels of ascorbate slows tumor growth and moderates HIF-1 pathway activity in Gulo –/– mice. Cancer Med (2015) 4:303–14. doi: 10.1002/cam4.349
217. Kuiper C, Molenaar IG, Dachs GU, Currie MJ, Sykes PH, Vissers MCM. Low ascorbate levels are associated with increased hypoxia-inducible factor-1 activity and an aggressive tumor phenotype in endometrial cancer. Cancer Res (2010) 70:5749–58. doi: 10.1158/0008-5472.CAN-10-0263
218. Kuiper C, Dachs GU, Munn D, Currie MJ, Robinson BA, Pearson JF, et al. Increased tumor ascorbate is associated with extended disease-free survival and decreased hypoxia-inducible factor-1 activation in human colorectal cancer. Front Oncol (2014) 4:10. doi: 10.3389/fonc.2014.00010
219. Wohlrab C, Vissers MCM, Phillips E, Morrin HR, Robinson BA, Dachs GU. The Association Between Ascorbate and the Hypoxia-Inducible Factors in Human Renal Cell Carcinoma Requires a Functional Von Hippel-Lindau Protein. Front Oncol (2018) 8:574. doi: 10.3389/fonc.2018.00574
220. Campbell EJ, Dachs GU, Morrin HR, Davey VC, Robinson BA, Vissers MCM. Activation of the hypoxia pathway in breast cancer tissue and patient survival are inversely associated with tumor ascorbate levels. BMC Cancer (2019) 19:307. doi: 10.1186/s12885-019-5503-x
221. Padayatty SJ, Sun AY, Chen Q, Espey MG, Drisko J, Levine M. Vitamin C: Intravenous Use by Complementary and Alternative Medicine Practitioners and Adverse Effects. PloS One (2010) 5:e11414. doi: 10.1371/journal.pone.0011414
222. Creagan ET, Moertel CG, O’Fallon JR, Schutt AJ, O’Connell MJ, Rubin J, et al. Failure of high-dose vitamin C therapy to benefit patients with advanced cancer. N Engl J Med (1979) 301. doi: 10.1056/NEJM197909273011303
223. Moertel CG, Fleming TR, Creagan ET, Rubin J, O’Connell MJ, Ames MM. High-dose vitamin C versus placebo in the treatment of patients with advanced cancer who have had no prior chemotherapy. N Engl J Med (1985) 312. doi: 10.1056/NEJM198501173120301
224. Cameron E, Pauling L. Supplemental ascorbate in the supportive treatment of cancer: Reevaluation of prolongation of survival times in terminal human cancer. Proc Natl Acad Sci (1978) 75:4538–42. doi: 10.1073/pnas.75.9.4538
225. Cameron E, Campbell A. The orthomolecular treatment of cancer II. Clinical trial of high-dose ascorbic acid supplements in advanced human cancer. Chem Interact (1974) 9:285–315. doi: 10.1016/0009-2797(74)90019-2
226. Padayatty S, Sun H, Wang Y, Riordan, Hugh D, Hewitt SM, et al. Vitamin C Pharmacokinetics : Implications for Oral and IV use. Ann Intern Med (2004) 140:533–8. doi: 10.7326/0003-4819-140-7-200404060-00010
227. Stephenson CM, Levin RD, Spector T, Lis CG. Phase I clinical trial to evaluate the safety, tolerability, and pharmacokinetics of high-dose intravenous ascorbic acid in patients with advanced cancer. Cancer Chemother Pharmacol (2013) 72:139–46. doi: 10.1007/s00280-013-2179-9
228. Nielsen TK, Højgaard M, Andersen JT, Poulsen HE, Lykkesfeldt J, Mikines KJ, et al. Elimination of Ascorbic Acid following High-Dose Infusion in Prostate Cancer Patients: A Pharmacokinetic Evaluation. Basic Clin Pharmacol Toxicol (2014) 116:(15AD). doi: 10.1111/bcpt.12323
229. Hoffer LJ, Levine M, Assouline S, Melnychuk D, Padayatty SJ, Rosadiuk K, et al. Phase I clinical trial of i.v. ascorbic acid in advanced malignancy. Ann Oncol (1969) 19. doi: 10.1093/annonc/mdn377
230. Drisko JA, Chapman J, Hunter VJ. The Use of Antioxidants with First-Line Chemotherapy in Two Cases of Ovarian Cancer. J Am Coll Nutr (2003) 22:118–23. doi: 10.1080/07315724.2003.10719284
231. Padayatty SJ, Riordan HD, Hewitt SM, Katz A, Hoffer LJ, Levine M, et al. Intravenously administered vitamin C as cancer therapy: Three cases. CMAJ (2006) 174:937–42. doi: 10.1503/cmaj.050346
232. Mikirova N, Casciari J, Rogers A, Taylor P. Effect of high-dose intravenous vitamin C on inflammation in cancer patients. J Transl Med (2012) 10. doi: 10.1186/1479-5876-10-189
233. Welsh J, Wagner BA, van’t Erve TJ, Zehr PS, Berg DJ, Halfdanarson TR, et al. Pharmacological ascorbate with gemcitabine for the control of metastatic and node-positive pancreatic cancer (PACMAN): results from a phase I clinical trial. Cancer Chemother Pharmacol (2013) 71:765–75. doi: 10.1007/s00280-013-2070-8
234. Ma Y, Chapman J, Levine M, Polireddy K, Drisko J, Chen Q. High-dose parenteral ascorbate enhanced chemosensitivity of ovarian cancer and reduced toxicity of chemotherapy. Sci Transl Med (2014) 6:222ra18–222ra18. doi: 10.1126/scitranslmed.3007154
235. Seo M-S, Kim J-K, Shim J-Y. High-Dose Vitamin C Promotes Regression of Multiple Pulmonary Metastases Originating from Hepatocellular Carcinoma. Yonsei Med J (2015) 56:1449–52. doi: 10.3349/ymj.2015.56.5.1449
236. Raymond YCF, Glenda CSL, Meng LK. Effects of High Doses of Vitamin C on Cancer Patients in Singapore: Nine Cases. Integr Cancer Ther (2016) 15:197–204. doi: 10.1177/1534735415622010
237. Mikirova N, Hunnunghake R, Scimeca RC, Chinshaw C, Ali F, Brannon C, et al. High-dose intravenous vitamin C treatment of a child with neurofibromatosis type 1 and optic pathway glioma: A case report. Am J Case Rep (2016) 17:774–81. doi: 10.12659/AJCR.899754
238. Polireddy K, Dong R, Reed G, Yu J, Chen P, Williamson S, et al. High dose parenteral ascorbate inhibited pancreatic cancer growth and metastasis: Mechanisms and a phase I/IIa study. Sci Rep (2017) 7:1–15. doi: 10.1038/s41598-017-17568-8
239. Wang F, He M-M, Wang Z-X, Li S, Jin Y, Ren C, et al. Phase I study of high-dose ascorbic acid with mFOLFOX6 or FOLFIRI in patients with metastatic colorectal cancer or gastric cancer. BMC Cancer (2019) 19. doi: 10.1186/s12885-019-5696-z
240. Schoenfeld JD, Sibenaller ZA, Mapuskar KA, Wagner BA, Cramer-Morales KL, Furqan M, et al. O 2 ·– and H 2 O 2 -Mediated Disruption of Fe Metabolism Causes the Differential Susceptibility of NSCLC and GBM Cancer Cells to Pharmacological Ascorbate. Cancer Cell (2017) 31:487–500.e8. doi: 10.1016/j.ccell.2017.02.018
241. Grasso C, Fabre M-S, Collis SV, Castro ML, Field CS, Schleich N, et al. Pharmacological doses of daily ascorbate protect tumors from radiation damage after a single dose of radiation in an intracranial mouse glioma model. Front Oncol (2014) 4:356. doi: 10.3389/fonc.2014.00356
242. Herst PM, Broadley KW, Harper JL, McConnell MJ. Pharmacological concentrations of ascorbate radiosensitize glioblastoma multiforme primary cells by increasing oxidative DNA damage and inhibiting G2/M arrest. Free Radic Biol Med Biol Med (2012) 52:1486–93. doi: 10.1016/j.freeradbiomed.2012.01.021
243. Ma E, Chen P, Wilkins HM, Wang T, Swerdlow RH, Chen Q. Pharmacologic ascorbate induces neuroblastoma cell death by hydrogen peroxide mediated DNA damage and reduction in cancer cell glycolysis. Free Radic Biol Med (2017) 113:36–47. doi: 10.1016/j.freeradbiomed.2017.09.008
244. Castro ML, McConnell MJ, Herst PM. Radiosensitisation by pharmacological ascorbate in glioblastoma multiforme cells, human glial cells, and HUVECs depends on their antioxidant and DNA repair capabilities and is not cancer specific. Free Radic Biol Med (2014) 74:200–9. doi: 10.1016/j.freeradbiomed.2014.06.022
245. Bodeker KL, Allen BG, Smith MC, Monga V, Sandhu S, Hohl RJ, et al. First-in-human phase 1 clinical trial of pharmacological ascorbate combined with radiation and temozolomide for newly diagnosed glioblastoma. Clin Cancer Res (2019) 25:6590–7. doi: 10.1158/1078-0432.CCR-19-0594
246. Baillie N, Carr AC, Peng S. The use of intravenous vitamin C as a supportive therapy for a patient with glioblastoma multiforme. Antioxidants (2018) 7. doi: 10.3390/antiox7090115
247. Hegi ME, Diserens A-C, Gorlia T, Hamou M-F, de Tribolet N, Weller M, et al. MGMT Gene Silencing and Benefit from Temozolomide in Glioblastoma. N Engl J Med (2005) 352:997–1003. doi: 10.1056/NEJMoa043331
248. Pegg AE, Dolan ME, Moschel RC. Structure, Function, and Inhibition of O6-Alkylguanine-DNA Alkyltransferase. Prog Nucleic Acid Res Mol Biol (1995) 51:167–223. doi: 10.1016/S0079-6603(08)60879-X
249. Esteller M, Garcia-Foncillas J, Andion E, Goodman SN, Hidalgo OF, Vanaclocha V, et al. Inactivation of the DNA-repair gene MGMT and the clinical response of gliomas to alkylating agents. N Engl J Med (2000) 343:1350–4. doi: 10.1056/NEJM200011093431901
Keywords: hypoxia, brain cancer, PHD, HIF-1, TET, IDH, ascorbate
Citation: Crake RLI, Burgess ER, Royds JA, Phillips E, Vissers MCM and Dachs GU (2021) The Role of 2-Oxoglutarate Dependent Dioxygenases in Gliomas and Glioblastomas: A Review of Epigenetic Reprogramming and Hypoxic Response. Front. Oncol. 11:619300. doi: 10.3389/fonc.2021.619300
Received: 20 October 2020; Accepted: 25 January 2021;
Published: 25 March 2021.
Edited by:
Yaohua Liu, Shanghai First People’s Hospital, ChinaReviewed by:
Chuanlu Jiang, Second Affiliated Hospital of Harbin Medical University, ChinaMaite Verreault, INSERM U1127 Institut du Cerveau et de la Moelle Épinière (ICM), France
Copyright © 2021 Crake, Burgess, Royds, Phillips, Vissers and Dachs. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Gabi U. Dachs, Z2FiaS5kYWNoc0BvdGFnby5hYy5ueg==
†These authors have contributed equally to this work