 Hiroyuki Takeda
Hiroyuki Takeda
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oncol. , 25 March 2021
Sec. Gastrointestinal Cancers
Volume 11 - 2021 | https://doi.org/10.3389/fonc.2021.602194
This article is part of the Research Topic The Treatment of RAS or BRAF Mutated Metastatic Colorectal Cancer: Challenges and Perspectives View all 8 articles
BRAF mutations constitute an important poor prognostic factor in metastatic colorectal cancer (mCRC) and the development of treatments in this context is of great necessity to prolong patient survival. Although the association between BRAF mutations and microsatellite instability (MSI) has been known for several years, previous clinical trials have revealed that the former has a limited prognostic impact and that immune checkpoint inhibitors offer a significant survival benefit to mCRC patients with both characteristics. Furthermore, the genomic classification of BRAF mutations according to their molecular functions enables greater understanding of the characteristics of mCRC patients with BRAF mutations, with therapeutic strategies based on this classification made more ideal to improve poor prognosis through the delivery of targeted therapies. Recently, a phase III trial was conducted in previously treated mCRC patients with BRAF V600E–mutated tumors and revealed that the combination therapy approach of BRAF inhibition and anti–epidermal growth factor receptor antibody therapy with or without MEK inhibition was more efficacious than standard chemotherapy alone. This review discusses current treatment strategies and future perspectives in BRAF-mutated mCRC.
Colorectal cancer (CRC) is one of the most common cancers, with an associated mortality rate of 9.2% that makes it the second leading cause of cancer-associated deaths (1). Early-stage CRC can be curable under surgery, whereas metastatic or recurrent CRC is usually unresectable and carries a poor prognosis. In recent years, treatments targeting vascular endothelial growth factor (VEGF) receptors and epidermal growth factor receptors (EGFRs) have been reported to significantly extend the survival of metastatic CRC (mCRC). Furthermore, various biomarker studies, including concerning the RAS gene, have been also conducted and some findings have been deemed clinically useful in practice. In particular, BRAF mutations have been shown collectively to be a remarkably poor prognostic factor (2, 3). BRAF is part of the RAS–RAF–MEK intracellular signaling pathway and its mutation is considered a genetic aberration that activates a signal that promotes tumor growth. BRAF is a kinase protein located downstream of EGFR, suggesting that patients with BRAF-mutated mCRC are less likely to benefit from anti-EGFR agents. However, a few clinical trials to date have suggested additional benefits of anti-EGFR therapy, so controversy therefore remains (4, 5). The use of fluorouracil/folinic acid, oxaliplatin, and irinotecan (FOLFOXIRI) plus bevacizumab is considered to be a promising treatment regimen to prolong survival for BRAF V600E–mutated mCRC. However, recent reports indicate that further investigation is required (6). This article summarizes the data of previous clinical trials in BRAF-mutated mCRC, discusses current treatment strategies, and offers future perspectives.
BRAF is a serine/threonine-specific protein kinase involved in the signaling cascade of the mitogen-activated protein kinase (MAPK) pathway, which promotes cell growth and differentiation (Figure 1). Activated RAF proteins trigger the activation of MEK1/2 and further activate ERK. Subsequently, ERK phosphorylates transcription factors and regulates significant cellular activity (7, 8). Approximately 15% to 30% of all CRCs are thought to contribute to cancer in the serrated pathway, a multistage carcinogenic mechanism that is an alternative to the traditional adenomatous carcinoma model. Morphologically, serrated lesions can be classified as hyperplastic polyps, serrated adenomas/polyps, and classic serrated adenomas, respectively. These lesions exhibit a high incidence of BRAF mutations and also present the CpG island methylator phenotype (CIMP), which causes methylation of mismatch repair genes and can progress to microsatellite instability (MSI) (9).
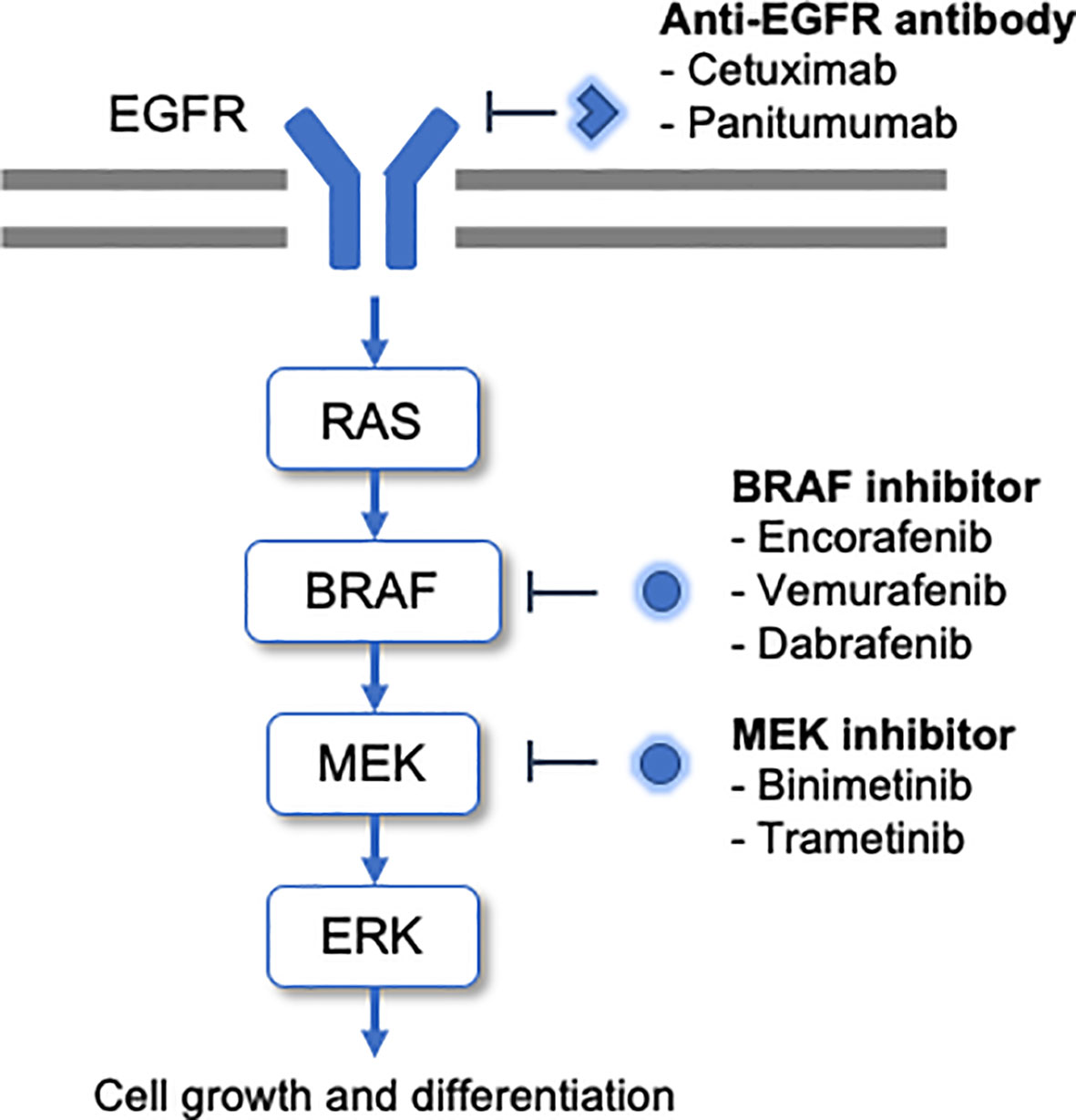
Figure 1 BRAF mutations promote the activation of this pathway and tumorigenesis. The molecular-targeted drugs developed to date for BRAF-mutated CRC are shown alongside the cascade.
BRAF mutations are found in 10% to 15% of CRCs, including early-stage cancers (10). In addition, the frequency of BRAF mutations in mCRC has been reported to be 8% to 10% according to prior retrospective analyses of several clinical trials (3, 11, 12). BRAF mutations are a key poor prognostic factor in this population, affecting the progression-free survival (PFS) and overall survival (OS) alike (13–15). The prognostic impact of BRAF mutations is generally similar in between CRCs and melanomas, whereas in lung cancers, there are conflicting results among studies, possibly due to their low frequency (16, 17). Patients with BRAF-mutated CRC are more likely to be female, have tumors on the right side, exhibit more peritoneal metastases, and show more mucinous histology in comparison with patients with wild-type CRC (18–20). Most BRAF variants present with mutations in BRAF V600E. Most recently, it has become recognized that clinical features differ depending upon the site of the mutation.
Most BRAF-mutated mCRC have mutations in V600E, but 2% to 3% show other mutations (19, 21). Non-V600 BRAF-mutant mCRC was reported to involve different clinical characteristics relative to V600 BRAF-mutant mCRC; typically, patients with non-V600 BRAF-mutant mCRC are slightly younger and are less female, with low-grade tumors and with primary tumors located on the left side more often (19). In terms of prognosis, the median OS was longer in patients with non-V600 BRAF-mutant mCRC as compared with among both V600 BRAF-mutant mCRC and BRAF wild-type mCRC patients. In patients with NSCLC, the prognostic impact of non-V600 mutations is not consistent across reports (22, 23), while in patients with melanoma, there was no significant survival difference between non-V600 mutations, V600 BRAF-mutation, and BRAF wild-type (24).
Based on recent analysis, BRAF mutations can be classified into three groups according to their function (25, 26). Class 1, encompassing V600 mutations, is marked by high kinase activity and exhibit MEK/ERK signaling activation as RAS-independent monomers. Class 2 mutations involve intermediate kinase activity and exhibit RAS-independent activation of MEK/ERK signaling in a dimer with BRAF. Meanwhile, class 3 mutations exhibit reduced kinase activity and are dependent on RAS to activate signaling (27). Notably, class 3 mutations are more likely to be associated with long-term survival relative to both class 1 and class 2 mutations (28). Moreover, it has been reported that patients with BRAF mutations may respond differently to treatments depending on the mutation class. BRAF mutations of classes 2 and 3 are not responsive to RAF inhibitors, whereas class 3 mutants are RAS-dependent and potentially effective for EGFR inhibition (27). Confirming this, a large series analysis of non-V600 BRAF-mutant CRC patients receiving anti-EGFR therapy revealed that patients with class 3 BRAF mutations responded to anti-EGFR therapy, while those with class 2 BRAF mutations did not responded (29). On the contrary, two studies have reported that non-V600 BRAF mutations are unable to be mitigated with anti-EGFR therapy (30, 31). Owing to the rarity of non-V600 BRAF mutations in this population, the efficacy of anti-EGFR therapy and the optimal regimen for non-V600 BRAF-mutant CRC remains unclear. A multicenter phase II trial of the combination of the MEK inhibitor binimetinib, the BRAF inhibitor encorafenib, and the anti-EGFR antibody drug cetuximab is currently ongoing, assessing patients with non-V600E BRAF-mutant mCRC who have not received any previous anti-EGFR antibody drugs. This study will also ultimately test the same regimen in class 3 patients with prior anti-EGFR antibody exposure (32).
Acquired BRAF alterations also may occur. BRAF amplification has been reported in patients with BRAF-mutated mCRC who have developed resistance to combination RAF/MEK inhibition. This alteration results in the onset of resistance to the RAF/EGFR or RAF/MEK combination through sustained MAPK pathway activity (33). Thus, therapeutic strategies to address the BRAF mutation class and resistance through BRAF amplification are also attracting attention regarding non-BRAF V600E.
Clinical trials assessing the impact of conventional chemotherapy, including both as first-line and second-line treatment, are shown in Table 1. The efficacy of anti-EGFR therapy for BRAF-mutated mCRC is controversial. In the pooled analysis of CRYSTAL and OPUS, two randomized clinical trials examining the effects of adding cetuximab to doublet chemotherapy, anti-EGFR treatment displayed numerical benefits in the objective response rate (ORR), PFS, and OS for BRAF-mutated mCRC, although no significant difference was recorded. Anti-EGFR plus chemotherapy presented an ORR of 21.9%, median PFS of 7.1 months, and median OS of 14.1 months; in contrast, chemotherapy alone demonstrated an ORR of 13.2%, median PFS of 3.7 months, and median OS of 9.9 months (3).
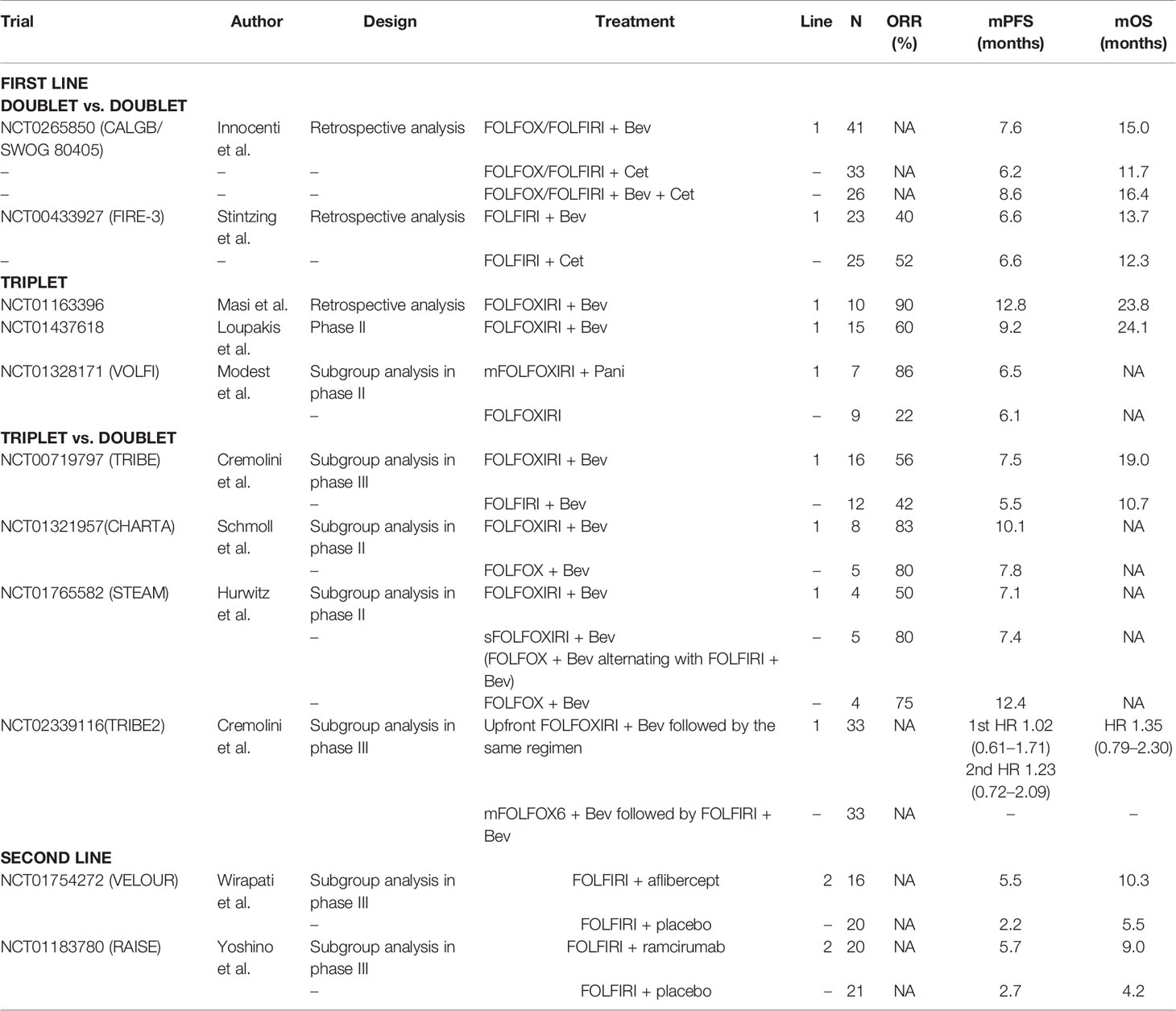
Table 1 Efficacy of conventional chemotherapies for BRAF-mutated metastatic colorectal cancer.
Two meta-analyses to date have evaluated the influence of anti-EGFR therapy in BRAF-mutated mCRC. Although these reports suggested a trend existed toward better hazard ratios (HRs) concerning PFS and OS in the context of EGFR therapy, it did not show a statistically significant difference (4, 5). Recently, the VOLFI trial (AIO KRK0109), a randomized phase II trial investigating the addition of panitumumab to triplet chemotherapy with FOLFOXIRI, reported results for the subgroup of patients with BRAF-mutated mCRC. The ORR of the FOLFOXIRI plus panitumumab arm was 86% and was higher as compared with that of 22% in the FOLFOXIRI-alone arm. In contrast with the effect of ORR, the prolongation of PFS was modest, as PFS was 6.5 and 6.1 months for the FOLFOXIRI plus panitumumab and FOLFOXIRI-alone arms, respectively (34). These results support that anti-EGFR therapy may show some efficacy for the treatment of BRAF-mutated mCRC, although the magnitude of efficacy is less than that of RAS/BRAF wild-type mCRC. Further accumulation of evidence and molecular analyses may be necessary to determine whether anti-EGFR therapy truly offers a degree of efficacy that is beneficial enough.
The efficacy of anti-VEGF therapy for BRAF-mutated mCRC has been also discussed. A biomarker analysis of a phase III study—the first report of the efficacy of bevacizumab in previously untreated patients with mCRC—included 10 patients with BRAF mutations. The median survival was 16 months among seven patients in the bevacizumab group and eight months for three patients in the placebo group, with an HR of 0.11 [95% confidence interval (CI): 0.01–1.06] (35). However, as the number of patients included was very small, it is not clear whether bevacizumab had the same effect in BRAF-mutated mCRC that it does in other types of mCRC. As second-line treatment, the efficacy of anti-VEGF targets against BRAF-mutated tumors has been reported in a relatively large number of patients. Among 41 patients with BRAF mutation, the effect of ramucirumab, a monoclonal antibody drug targeting VEGFR2, relative to that of placebo was demonstrated in a subgroup analysis of the RAISE trial, with HRs of 0.54 (95% CI: 025–1.13) and 0.55 (95% CI: 0.28–1.08) for PFS and OS, respectively (36). A subgroup analysis of BRAF mutants in the VELOUR trial, a phase III study that proved the efficacy of aflibercept, also suggested that the efficacy of anti-VEGF therapy was preserved, with an HR of 0.59 (95% CI: 0.22–1.58) for PFS and that of 0.42 (95% CI: 0.16–1.09) for OS (37).
Meanwhile, in the CALGB/SWOG80405 and FIRE-3 trials, where bevacizumab was compared with cetuximab as first-line treatment, retrospective analyses were conducted for BRAF-mutated mCRC (14, 38). For BRAF-mutated mCRC, the median PFS in the bevacizumab group and the cetuximab group were similar in these studies; the median PFS outcomes in the bevacizumab group were 7.6 months and 6.6 months, and those in the cetuximab group were 6.2 months and 6.6 months, respectively. Further, the median OS tended to be slightly longer for bevacizumab, garnering median OS outcomes of 15.0 and 13.7 months in the bevacizumab group and 11.7 and 12.3 months in the cetuximab group. Ultimately, however, it remains uncertain which therapeutic option (bevacizumab or anti-EGFR antibody medication) as first-line treatment is more favorable for addressing BRAF-mutated mCRC.
FOLFOXIRI plus bevacizumab is considered to be one of the leading regimens against BRAF-mutated mCRC based on evidence from both retrospective and prospective trials involving small numbers of patients as well as the results of a subgroup analysis of the TRIBE trial. A retrospective analysis of a phase II study of FOLFOXIRI plus bevacizumab indicated that the response rate was 90%, the PFS was 12.8 months, and the OS was 23.8 months among 10 patients with BRAF-mutated mCRC (39). Elsewhere, a prospective study was conducted involving 15 patients with BRAF-mutated mCRC and FOLFOXIRI plus bevacizumab ensured a response rate of 60%, a PFS of 9.2 months, and an OS of 24.1 months, with these results being comparable to those of the previous report (40). In the TRIBE trial, FOLFOXIRI plus bevacizumab (triplet arm) versus FOLIRI plus bevacizumab (doublet arm) in the context of mCRC were compared. A subgroup analysis of BRAF-mutated tumors was also completed to confirm the previously described efficacy of the triplet regimen for BRAF-mutated mCRC. The results showed that the response rate was 56% in the triplet group (n = 16 patients) and 42% in the doublet group (n = 12 patients), PFS outcomes of 7.5 months versus 5.5 months and OS outcomes of 19.0 months versus 10.7 months, indicating that the triplet group was more favorable for BRAF-mutated tumors (6). In the subgroup analysis of the STEAM and CHART trials, the HRs for the median PFS were 0.8 (95% CI: 0.2–3.0) and 0.72 (95% CI: 0.25–2.07), respectively, which were not significantly different but suggested that the triplet regimen was a slightly better option (41, 42). A pooled analysis including these studies reported an HR of 0.65 (95% CI: 0.33–1.26) for the triplet group, indicating no significant difference, but suggesting that the triplet regimen was again preferable (41).
However, recently, a subgroup analysis of the TRIBE2 trial presented a different trend as compared with in previous studies (43). A total of 33 patients with BRAF-mutated mCRC were present in both the experimental and control groups, with an HR for the PFS of 1.02 (95% CI: 0.61–1.71), while the HR for PFS2, defined as the time from randomization to disease progression on any treatment given after the first instance of disease progression, was 1.23 (95% CI: 0.72–2.09). Furthermore, the OS presented an HR of 1.35 (95% CI: 0.79–2.30), which indicates considerable uncertainty regarding whether the triplet therapy approach is beneficial for BRAF-mutated mCRC. A meta-analysis of these five studies using individual patient data reported an HR of 1.11 (95% CI: 0.75–1.73) for the OS of the triplet regimen versus the doublet regimen, with an uncertain level of benefit noted in patients with BRAF-mutated mCRC (44). In addition, using real-world data from the United States, a retrospective evaluation of triplet and doublet regimens for patients with BRAF-mutated mCRC was conducted and showed that 16 patients receiving the triplet regimen and 423 patients receiving the doublet regimen showed median OS outcomes of 13.8 months and 15.5 months (p = 0.38), respectively (45). In summary, the benefit of the triplet plus bevacizumab regimen in BRAF-mutated tumors is controversial and adequate prospective validation in BRAF-mutated tumors is becoming more crucial. Also, there is insufficient evidence of the additive effect of anti-VEGF–targeted drugs in BRAF-mutated mCRC, but no negative evidence is currently available; thus, anti-VEGF drugs are recommended for patients for whom anti-VEGF therapy is accessible. There is no specific regimen recommended for first-line therapy of BRAF-mutated tumors, as the National Comprehensive Cancer Network guidelines recommend using the same regimen as that for the RAS wild type tumors.
Therapeutic strategies encompassing BRAF and MEK inhibitors for melanoma and thyroid cancer with BRAF mutation have been successful and several BRAF and MEK inhibitors have already been approved to date by the United States Food and Drug Administration for these diseases (46). In this section, the current development of BRAF and MEK inhibitors for the management of BRAF-mutated mCRC will be described (Table 2). In contrast with in melanoma, monotherapy with BRAF inhibitors has not been quite as successful in mCRC. For example, a phase I dose-escalation study of encorafenib was conducted in 18 patients with BRAF V600E–mutated mCRC and none of the patients showed a confirmed response. In 12 patients, however, the best response was achieved with stable disease, with a median PFS of 4.0 months (47). Elsewhere, the effects of vemurafenib were assessed in 21 patients with BRAF-mutated mCRC, with only one partial response. The best response with stable disease was seen in seven patients and the median PFS was 2.1 months (48). In a basket study of vemurafenib in BRAF V600E–mutated cancers, 10 patients with mCRC who received vemurafenib monotherapy experienced no response. Both in vitro and in vivo studies have reported that the cause of the failure to respond to BRAF inhibition alone was the rapid reactivation of ERK via the feedback activation of EGFR. Given this, the combination of BRAF inhibitor and anti-EGFR antibody medications was expected to increase efficacy in mCRC and the combination of vemurafenib and cetuximab was therefore attempted in another cohort. Ultimately, however, one of the 27 patients experienced a partial response (49). Moreover, in a phase I/II trial of another anti-EGFR antibody combination, 13 patients received the combination of vemurafenib and panitumumab, and two patients responded to treatment (50).
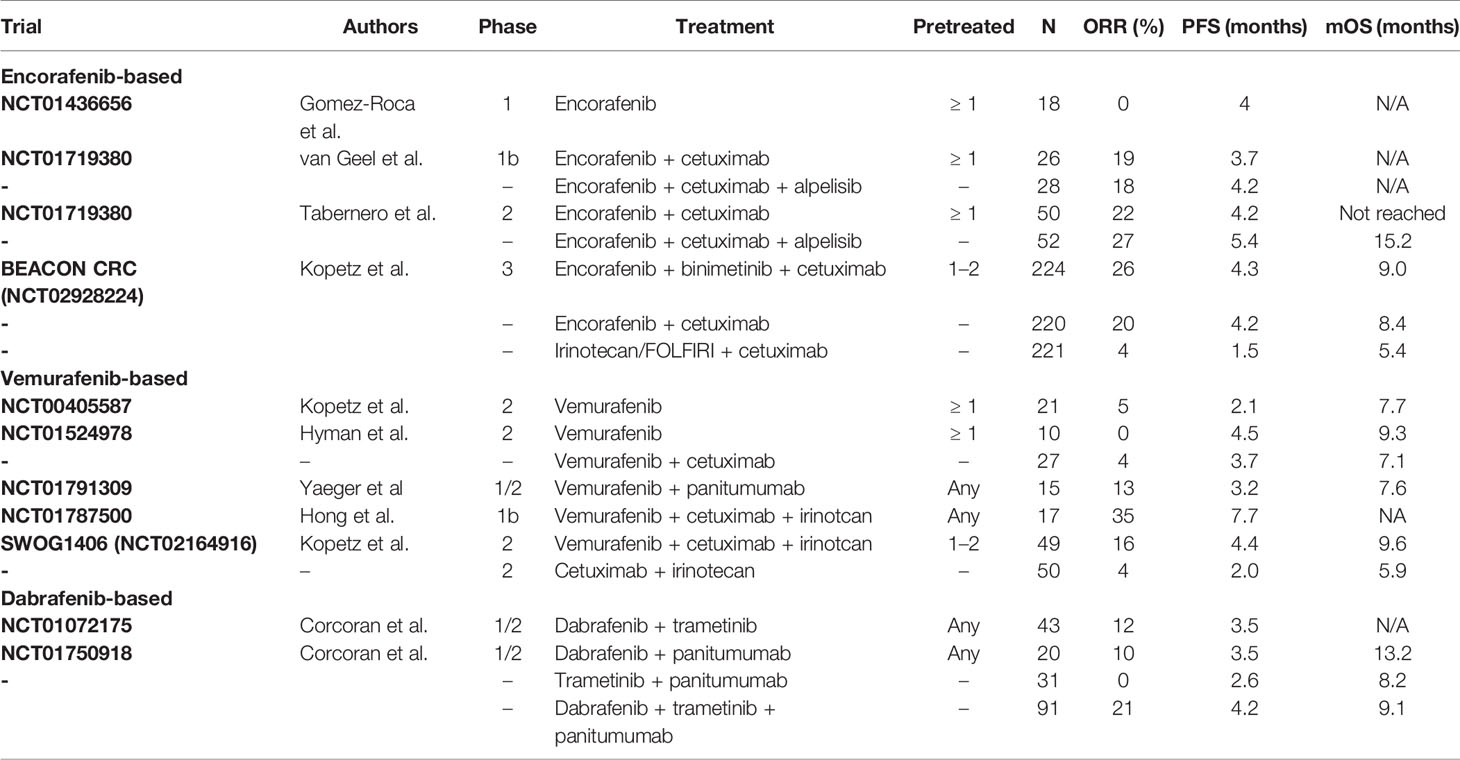
Table 2 Efficacy of targeted therapies for BRAF V600E–mutated mCRC.
Other BRAF inhibitors were also tested in combination with anti-EGFR antibody drugs. In a phase Ib study of the combination of encorafenib and cetuximab in patients with BRAF-mutated mCRC, five of 26 (18%) patients showed a response. The trial also included the PI3K inhibitor alpelisib in combination with encorafenib and cetuximab, with responses observed in five of 28 (18%) patients; for these two study populations, the median PFS outcomes were 3.7 months and 4.2 months, respectively (51). Subsequently, a phase II part of the same study enrolled 50 patients in the encorafenib plus cetuximab arm and 52 patients in the encorafenib plus cetuximab plus alpelisib arm, respectively, and reported response rates of 22% and 27% and median PFS outcomes of 4.2 months and 5.4 months, respectively (52). Vemurafenib has also been assessed in combination with irinotecan, a cytotoxic anticancer agent: a phase I trial of vemurafenib plus cetuximab with or without irinotecan in BRAF-mutated mCRC showed promising outcomes, with a response rate of 35% and a median PFS of 7.7 months (53). In the subsequent phase II study, however, the results were not as expected, with a response rate of just 16% and a median PFS of 4.4 months (54).
As the inhibition of BRAF leads to the activation of MEK-dependent signaling, simultaneously inhibiting MEK while administering a BRAF inhibitor is expected to result in a better therapeutic response. Henceforth, further studies that have attempted to inhibit both BRAF and MEK will be described. In a phase I/II study of the MEK inhibitor trametinib in combination with dabrafenib, five of 43 patients (12%) with BRAF-mutated mCRC achieved a partial response (55). MEK inhibitors were further studied in combination with anti-EGFR therapy: a phase I/II study of dabrafenib and trametinib plus panitumumab in comparison with dabrafenib plus panitumumab or trametinib plus panitumumab, achieved a response rate of 21% and a median PFS of 4.2 months with the triplet regimen (56). Recently, a large phase III (BEACON CRC) trial demonstrated that encorafenib plus cetuximab with or without binimetinib was superior in terms of the response rate, PFS, and OS compared to FOLFIRI/irinotecan plus cetuximab. As a result of this study, the Food and Drug Administration approved encorafenib plus cetuximab for the treatment of BRAF V600E–mutated mCRC (57). The updated National Comprehensive Cancer Network guidelines also recommend combining encorafenib and cetuximab for patients with BRAF V600E–mutated mCRC who previously progressed on first-line therapy.
In previous research, BRAF-mutated mCRC showed a higher percentage of MSI than BRAF wild-type mCRC (12.6% vs. 3%) (12). A pooled analysis of four phase III studies in mCRC reported significantly worse PFS and OS outcomes for BRAF-mutant mCRC in comparison with BRAF wild-type mCRC with microsatellite-stable tumors, but no significant difference was apparent between BRAF-mutant and BRAF wild-type mCRC with MSI-high tumors (13). Therefore, MSI may have a stronger impact on prognosis as compared with BRAF mutation. Several clinical trials testing the effects of anti–programmed cell death protein 1 (PD-1) antibody in MSI-high/mismatch repair-deficient mCRC included patients with BRAF mutations. These results showed that BRAF mutation was not a predictor for the efficacy of anti–PD-1 antibody in MSI-high mCRC. In the KEYNOTE-164 phase II trial of pembrolizumab in previously treated MSI-high mCRC, there were nine patients included with BRAF V600E mutations in cohort A (who received two or more lines of treatment) and five patients with BRAF V600E mutations in cohort B (who received one or more lines of treatment) and the response rates were 55% and 20%, respectively. The response rates for BRAF-mutant and BRAF wild-type mCRC were 42% and 38%, respectively, and seemed to be independent of the BRAF mutation status (58). In the recently reported phase III KEYNOTE-177 trial of pembrolizumab versus chemotherapy in MSI-high mCRC, a subgroup analysis of patients with BRAF V600E mutations showed an HR of 0.48 (95% CI: 0.27–0.86) for PFS, similar to that for BRAF wild-type mCRC (HR: 0.50, 95% CI 0.31–0.80) (59).
The phase II trial of nivolumab or the combination of nivolumab plus ipilimumab in MSI-high mCRC, CheckMate 142, also included patients with BRAF-mutated mCRC and reported a relatively high response rate. In this trial, patients with BRAF-mutated mCRC were grouped into three arms; patients who received two or more lines of treatment were assigned to the nivolumab group (n = 25 patients) and patients who received one or more lines of treatment (n = 29 patients) or no prior treatment (n = 17 patients) were assigned to the nivolumab plus ipilimumab group. The response rates reported for these patients were 25%, 55%, and 76%, respectively, and were comparable to those for BRAF wild-type patients (41.4%, 55%, and 62%, respectively) (60–62). Therefore, BRAF-mutated mCRC with concomitant MSI-high should be preferred for treatment strategies according to the MSI-high status.
Several clinical trials in BRAF V600E–mutated mCRC are currently ongoing (Table 3). Of these, there are two prospective trials in the first-line treatment. The randomized phase II trial AIO-KRK-0116 (NCT04034459) compares the efficacy of FOLFOXIRI plus cetuximab to FOLFOXIRI plus bevacizumab in patients with untreated BRAF-mutated mCRC (63). Another one is the phase II trial, ANCHOR-CRC (NCT03693170), which will evaluate the efficacy of the combination of encorafenib, binimetinib, and cetuximab, the regimen in the BEACON CRC trial, in the first-line treatment of patients with BRAF V600E–mutated mCRC (64). Results of the first stage of this trial were recently presented at the virtual 22nd ESMO World Congress on Gastrointestinal Cancer; 40 patients were ultimately evaluated for efficacy, with the response rate of 50% (95% CI: 33.8–66.2), tumor shrinkage in 85%, and the median PFS of 4.9 months (95% CI: 4.4–8.1) was observed, and the study is now in the second stage (65). Although not specific to first-line therapy, a phase II trial, the IMPROVEMENT study (NCT03727763) is also ongoing to evaluate the efficacy and the safety of the combination of FOLFIRI with cetuximab and vemurafenib (66).
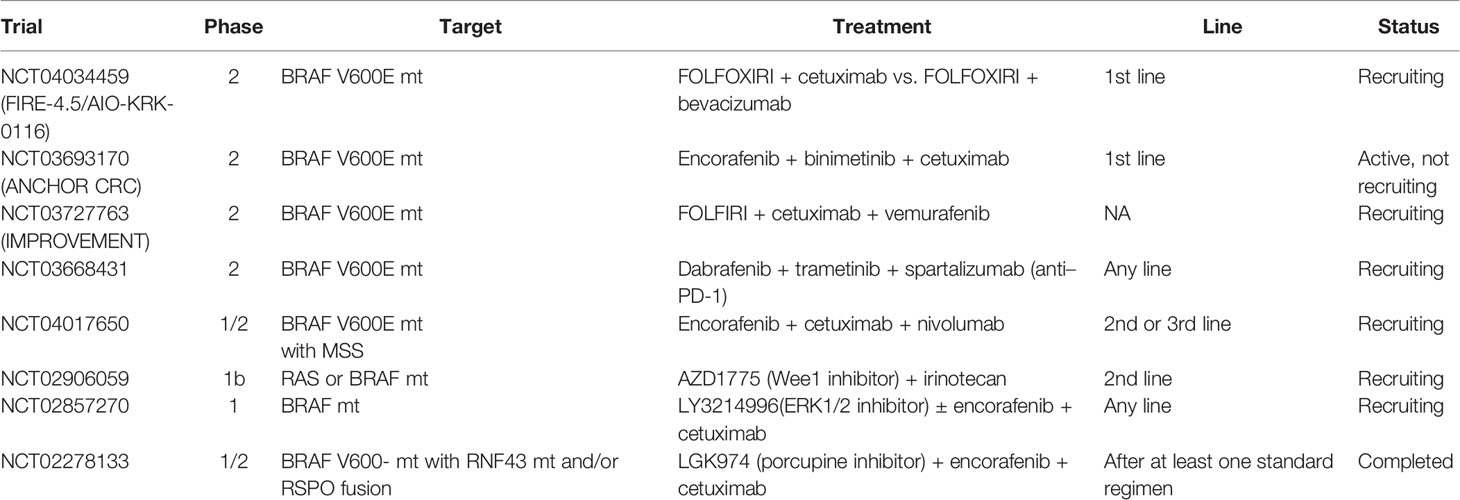
Table 3 Ongoing trials for BRAF-mutated mCRC.
Combination therapy with immune checkpoint inhibitors and targeted therapies is being investigated in two trials. NCT03668431 is a phase II trial investigating the combination of dabrafenib and trametinib with the PD-1 inhibitor, PDR001 (spartalizumab) in previously treated BRAF-mutated mCRC patients (67). A similar trial of anti–PD-1 antibody medication in combination with BRAF inhibition (NCT04017650) is ongoing in patients receiving second- and third-line treatments with encorafenib in combination with cetuximab and nivolumab (68).
Preclinical data indicate that Wee1 and ERK1/2 are located downstream of BRAF in the MAP kinase signaling cascade and are potentially important therapeutic targets, and based on this, AZD1775 (Wee1 inhibitor) and LY3214996 (ERK1/2 inhibitor) have been tested in phase I trials (69–73). In addition, activation of the Wnt pathway by RNF43 mutation may contribute to the resistance of BRAF-mutated mCRC to BRAF inhibitors. Therefore, a phase I/II trial of combination therapy of Wnt pathway inhibition with WNT974 (porcupine inhibitor) and BRAF inhibition has been conducted for BRAF-mutant CRC with RNF43 mutation (74–76).
Although clinical trials have not progressed since then, preclinical studies have reported that CDK1 and MCL-1 are involved in apoptosis resistance in BRAF-mutant CRC (77, 78). Combination therapy with these inhibitors and BRAF inhibitors is expected to be developed.
The future development of treatments for BRAF-mutated mCRC is likely to be based on combination of BRAF inhibitors and anti-EGFR agents for untreated BRAF-mutated mCRC, while further investigation of combination therapy with novel agents to overcome resistance to BRAF inhibitors is expected to promoted.
Advances in genetic analysis techniques and the development of therapies via clinical trials have significantly improved the treatment of BRAF-mutated mCRC to date. In BRAF-mutant mCRC with MSI-high, the administration of immune checkpoint inhibitors as first-line treatment led to a significant survival benefit. In contrast, standard of care in first-line treatment for microsatellite-stable BRAF-mutated mCRC remains chemotherapy, as the optimal regimen is still uncertain and controversial. As second-line therapy, the combination of encorafenib plus cetuximab was demonstrated to provide a survival benefit, which is one of the most promising regimens for BRAF V600E–mutated mCRC. However, even with this new treatment, the OS with second-line therapy is only about 10 months, which is not sufficiently longer than that in RAS/BRAF wild-type mCRC. Therefore, additional therapeutic strategies must be elucidated to control the disease in the longer term; such may require the adoption of molecularly targeted therapies in first-line treatment or the further investigation of the molecular mechanisms of resistance to BRAF treatment.
HT drafted the manuscript and YS revised it precisely. All authors contributed to the article and approved the submitted version.
YS has received consulting fees from Takeda Pharmaceutical and Daiichi Sankyo and honoraria from Takeda Pharmaceutical, Taiho Pharmaceutical, Chugai Pharma, Yakult Honsha, Sanofi, Bayer Yakuhin, Bristol-Myers Squibb Japan, Merk Biopharma, Lilly Japan, Nippon Kayaku, Kyowa Kirin.
The remaining author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors would like to thank enago™ for proofreading the manuscript.
1. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin (2018) 68:394–424. doi: 10.3322/caac.21492
2. Richman SD, Seymour MT, Chambers P, Elliott F, Daly CL, Meade AM, et al. KRAS and BRAF Mutations in Advanced Colorectal Cancer Are Associated With Poor Prognosis but Do Not Preclude Benefit From Oxaliplatin or Irinotecan: Results From the MRC FOCUS Trial. J Clin Oncol (2009) 27:5931–7. doi: 10.1200/JCO.2009.22.4295
3. Bokemeyer C, Cutsem EV, Rougier P, Ciardiello F, Heeger S, Schlichting M, et al. Addition of cetuximab to chemotherapy as first-line treatment for KRAS wild-type metastatic colorectal cancer: Pooled analysis of the CRYSTAL and OPUS randomised clinical trials. Eur J Cancer (2012) 48:1466–75. doi: 10.1016/j.ejca.2012.02.057
4. Rowland A, Dias MM, Wiese MD, Kichenadasse G, McKinnon RA, Karapetis CS, et al. Meta-analysis of BRAF mutation as a predictive biomarker of benefit from anti-EGFR monoclonal antibody therapy for RAS wild-type metastatic colorectal cancer. Br J Cancer (2015) 112:1888–94. doi: 10.1038/bjc.2015.173
5. Pietrantonio F, Petrelli F, Coinu A, Di Bartolomeo M, Borgonovo K, Maggi C, et al. Predictive role of BRAF mutations in patients with advanced colorectal cancer receiving cetuximab and panitumumab: A meta-analysis. Eur J Cancer (2015) 51:587–94. doi: 10.1016/j.ejca.2015.01.054
6. Cremolini C, Loupakis F, Antoniotti C, Lupi C, Sensi E, Lonardi S, et al. FOLFOXIRI plus bevacizumab versus FOLFIRI plus bevacizumab as first-line treatment of patients with metastatic colorectal cancer: updated overall survival and molecular subgroup analyses of the open-label, phase 3 TRIBE study. Lancet Oncol (2015) 16:1306–15. doi: 10.1016/S1470-2045(15)00122-9
7. Dhillon AS, Hagan S, Rath O, Kolch W. MAP kinase signalling pathways in cancer. Oncogene (2007) 26:3279–90. doi: 10.1038/sj.onc.1210421
8. Montagut C, Settleman J. Targeting the RAF–MEK–ERK pathway in cancer therapy. Cancer Lett (2009) 283:125–34. doi: 10.1016/j.canlet.2009.01.022
9. De Palma F, D’Argenio V, Pol J, Kroemer G, Maiuri M, Salvatore F. The Molecular Hallmarks of the Serrated Pathway in Colorectal Cancer. Cancers (2019) 11:1017. doi: 10.3390/cancers11071017
10. Seppälä TT, Böhm JP, Friman M, Lahtinen L, Väyrynen VMJ, Liipo TKE, et al. Combination of microsatellite instability and BRAF mutation status for subtyping colorectal cancer. Br J Cancer (2015) 112:1966–75. doi: 10.1038/bjc.2015.160
11. Tol J, Nagtegaal ID, Punt CJA. BRAF Mutation in Metastatic Colorectal Cancer. N Engl J Med (2009) 361:98–9. doi: 10.1056/NEJMc0904160
12. Seligmann JF, Fisher D, Smith CG, Richman SD, Elliott F, Brown S, et al. Investigating the poor outcomes of BRAF-mutant advanced colorectal cancer: analysis from 2530 patients in randomised clinical trials. Ann Oncol (2017) 28:562–8. doi: 10.1093/annonc/mdw645
13. Venderbosch S, Nagtegaal ID, Maughan TS, Smith CG, Cheadle JP, Fisher D, et al. Mismatch Repair Status and BRAF Mutation Status in Metastatic Colorectal Cancer Patients: A Pooled Analysis of the CAIRO, CAIRO2, COIN, and FOCUS Studies. Clin Cancer Res (2014) 20:5322–30. doi: 10.1158/1078-0432.CCR-14-0332
14. Innocenti F, Ou F-S, Qu X, Zemla TJ, Niedzwiecki D, Tam R, et al. Mutational Analysis of Patients With Colorectal Cancer in CALGB/SWOG 80405 Identifies New Roles of Microsatellite Instability and Tumor Mutational Burden for Patient Outcome. J Clin Oncol (2019) 37:1217–27. doi: 10.1200/JCO.18.01798
15. Modest DP, Ricard I, Heinemann V, Hegewisch-Becker S, Schmiegel W, Porschen R, et al. Outcome according to KRAS-, NRAS- and BRAF-mutation as well as KRAS mutation variants: pooled analysis of five randomized trials in metastatic colorectal cancer by the AIO colorectal cancer study group. Ann Oncol (2016) 27:1746–53. doi: 10.1093/annonc/mdw261
16. Baik CS, Myall NJ, Wakelee HA. Targeting BRAF -Mutant Non-Small Cell Lung Cancer: From Molecular Profiling to Rationally Designed Therapy. Oncologist (2017) 22:786–96. doi: 10.1634/theoncologist.2016-0458
17. Safaee Ardekani G, Jafarnejad SM, Tan L, Saeedi A, Li G. The Prognostic Value of BRAF Mutation in Colorectal Cancer and Melanoma: A Systematic Review and Meta-Analysis. PLoS One (2012) 7:e47054. doi: 10.1371/journal.pone.0047054
18. Tran B, Kopetz S, Tie J, Gibbs P, Jiang Z-Q, Lieu CH, et al. Impact of BRAF mutation and microsatellite instability on the pattern of metastatic spread and prognosis in metastatic colorectal cancer: Metastatic Pattern in BRAF Mutant CRC. Cancer (2011) 117:4623–32. doi: 10.1002/cncr.26086
19. Jones JC, Renfro LA, Al-Shamsi HO, Schrock AB, Rankin A, Zhang BY, et al. Non-V600BRAF Mutations Define a Clinically Distinct Molecular Subtype of Metastatic Colorectal Cancer. J Clin Oncol (2017) 35:2624–30. doi: 10.1200/JCO.2016.71.4394
20. Cremolini C, Di Bartolomeo M, Amatu A, Antoniotti C, Moretto R, Berenato R, et al. BRAF codons 594 and 596 mutations identify a new molecular subtype of metastatic colorectal cancer at favorable prognosis. Ann Oncol (2015) 26:2092–7. doi: 10.1093/annonc/mdv290
21. Ciardiello F, Normanno N, Maiello E, Martinelli E, Troiani T, Pisconti S, et al. Clinical activity of FOLFIRI plus cetuximab according to extended gene mutation status by next-generation sequencing: findings from the CAPRI-GOIM trial. Ann Oncol (2014) 25:1756–61. doi: 10.1093/annonc/mdu230
22. Marchetti A, Felicioni L, Malatesta S, Grazia Sciarrotta M, Guetti L, Chella A, et al. Clinical Features and Outcome of Patients With Non–Small-Cell Lung Cancer Harboring BRAF Mutations. J Clin Oncol (2011) 29:3574–9. doi: 10.1200/JCO.2011.35.9638
23. Litvak AM, Paik PK, Woo KM, Sima CS, Hellmann MD, Arcila ME, et al. Clinical Characteristics and Course of 63 Patients with BRAF Mutant Lung Cancers. J Thorac Oncol (2014) 9:1669–74. doi: 10.1097/JTO.0000000000000344
24. Kim DW, Haydu LE, Joon AY, Bassett RL, Siroy AE, Tetzlaff MT, et al. Clinicopathological features and clinical outcomes associated with TP53 and BRAF N on- V 600 mutations in cutaneous melanoma patients: TP53 and BRAF Non-V600 Mutations. Cancer (2017) 123:1372–81. doi: 10.1002/cncr.30463
25. Zheng G, Tseng L-H, Chen G, Haley L, Illei P, Gocke CD, et al. Clinical detection and categorization of uncommon and concomitant mutations involving BRAF. BMC Cancer (2015) 15:779. doi: 10.1186/s12885-015-1811-y
26. Yao Z, Yaeger R, Rodrik-Outmezguine VS, Tao A, Torres NM, Chang MT, et al. Tumours with class 3 BRAF mutants are sensitive to the inhibition of activated RAS. Nature (2017) 548:234–8. doi: 10.1038/nature23291
27. Dankner M, Rose AAN, Rajkumar S, Siegel PM, Watson IR. Classifying BRAF alterations in cancer: new rational therapeutic strategies for actionable mutations. Oncogene (2018) 37:3183–99. doi: 10.1038/s41388-018-0171-x
28. Schirripa M, Biason P, Lonardi S, Pella N, Pino MS, Urbano F, et al. Class 1, 2, and 3 BRAF -Mutated Metastatic Colorectal Cancer: A Detailed Clinical, Pathologic, and Molecular Characterization. Clin Cancer Res (2019) 25:3954–61. doi: 10.1158/1078-0432.CCR-19-0311
29. Yaeger R, Kotani D, Mondaca S, Parikh AR, Bando H, Van Seventer EE, et al. Response to Anti-EGFR Therapy in Patients with BRAF non-V600–Mutant Metastatic Colorectal Cancer. Clin Cancer Res (2019) 25:7089–97. doi: 10.1158/1078-0432.CCR-19-2004
30. Johnson B, Loree JM, Jacome AA, Mendis S, Syed M, Morris VK II, et al. Atypical, Non-V600 BRAF Mutations as a Potential Mechanism of Resistance to EGFR Inhibition in Metastatic Colorectal Cancer. JCO Precis Oncol (2019) 3:PO.19.00102. doi: 10.1200/PO.19.00102
31. Shinozaki E, Yoshino T, Yamazaki K, Muro K, Yamaguchi K, Nishina T, et al. Clinical significance of BRAF non-V600E mutations on the therapeutic effects of anti-EGFR monoclonal antibody treatment in patients with pretreated metastatic colorectal cancer: the Biomarker Research for anti-EGFR monoclonal Antibodies by Comprehensive Cancer genomics (BREAC) study. Br J Cancer (2017) 117:1450–8. doi: 10.1038/bjc.2017.308
32. Kotani D, Bando H, Taniguchi H, Masuishi T, Komatsu Y, Yamaguchi K, et al. BIG BANG study (EPOC1703): multicentre, proof-of-concept, phase II study evaluating the efficacy and safety of combination therapy with binimetinib, encorafenib and cetuximab in patients with BRAF non-V600E mutated metastatic colorectal cancer. ESMO Open (2020) 5:e000624. doi: 10.1136/esmoopen-2019-000624
33. Ahronian LG, Sennott EM, Van Allen EM, Wagle N, Kwak EL, Faris JE, et al. Clinical Acquired Resistance to RAF Inhibitor Combinations in BRAF-Mutant Colorectal Cancer through MAPK Pathway Alterations. Cancer Discov (2015) 5:358–67. doi: 10.1158/2159-8290.CD-14-1518
34. Modest DP, Martens UM, Riera-Knorrenschild J, Greeve J, Florschütz A, Wessendorf S, et al. FOLFOXIRI Plus Panitumumab As First-Line Treatment of RAS Wild-Type Metastatic Colorectal Cancer: The Randomized, Open-Label, Phase II VOLFI Study (AIO KRK0109). J Clin Oncol (2019) 37:3401–11. doi: 10.1200/JCO.19.01340
35. Ince WL, Jubb AM, Holden SN, Holmgren EB, Tobin P, Sridhar M, et al. Association of k-ras, b-raf, and p53 Status With the Treatment Effect of Bevacizumab. JNCI J Natl Cancer Inst (2005) 97:981–9. doi: 10.1093/jnci/dji174
36. Yoshino T, Portnoy DC, Obermannová R, Bodoky G, Prausová J, Garcia-Carbonero R, et al. Biomarker analysis beyond angiogenesis: RAS/RAF mutation status, tumour sidedness, and second-line ramucirumab efficacy in patients with metastatic colorectal carcinoma from RAISE—a global phase III study. Ann Oncol (2019) 30:124–31. doi: 10.1093/annonc/mdy461
37. Wirapati P, Pomella V, Vandenbosch B, Kerr P, Maiello E, Jeffery GM, et al. Velour trial biomarkers update: Impact of RAS, BRAF, and sidedness on aflibercept activity. J Clin Oncol (2017) 35:3538–8. doi: 10.1200/JCO.2017.35.15_suppl.3538
38. Stintzing S, Miller-Phillips L, Modest DP, Fischer von Weikersthal L, Decker T, Kiani A, et al. Impact of BRAF and RAS mutations on first-line efficacy of FOLFIRI plus cetuximab versus FOLFIRI plus bevacizumab: analysis of the FIRE-3 (AIO KRK-0306) study. Eur J Cancer (2017) 79:50–60. doi: 10.1016/j.ejca.2017.03.023
39. Masi G, Loupakis F, Salvatore L, Fornaro L, Cremolini C, Cupini S, et al. (irinotecan, oxaliplatin, fluorouracil, and folinate) as first-line treatment for metastatic colorectal cancer: a phase 2 trial. Lancet Oncol (2010) 11:845–52. doi: 10.1016/S1470-2045(10)70175-3
40. Loupakis F, Cremolini C, Salvatore L, Masi G, Sensi E, Schirripa M, et al. FOLFOXIRI plus bevacizumab as first-line treatment in BRAF mutant metastatic colorectal cancer. Eur J Cancer (2014) 50:57–63. doi: 10.1016/j.ejca.2013.08.024
41. Shui L, Wu Y-S, Lin H, Shui P, Sun Q, Chen X, et al. Triplet Chemotherapy (FOLFOXIRI) Plus Bevacizumab Versus Doublet Chemotherapy (FOLFOX/FOLFIRI) Plus Bevacizumab in Conversion Therapy for Metastatic Colorectal Cancer: a Meta-Analysis. Cell Physiol Biochem (2018) 48:1870–81. doi: 10.1159/000492508
42. Hurwitz HI, Tan BR, Reeves JA, Xiong H, Somer B, Lenz H, et al. Phase II Randomized Trial of Sequential or Concurrent FOLFOXIRI-Bevacizumab Versus FOLFOX-Bevacizumab for Metastatic Colorectal Cancer (STEAM). Oncologist (2019) 24:921–32. doi: 10.1634/theoncologist.2018-0344
43. Cremolini C, Antoniotti C, Rossini D, Lonardi S, Loupakis F, Pietrantonio F, et al. Upfront FOLFOXIRI plus bevacizumab and reintroduction after progression versus mFOLFOX6 plus bevacizumab followed by FOLFIRI plus bevacizumab in the treatment of patients with metastatic colorectal cancer (TRIBE2): a multicentre, open-label, phase 3, randomised, controlled trial. Lancet Oncol (2020) 21:497–507. doi: 10.1016/S1470-2045(19)30862-9
44. Cremolini C, Antoniotti C, Stein A, Bendell J, Gruenberger T, Rossini D, et al. Individual Patient Data Meta-Analysis of FOLFOXIRI Plus Bevacizumab Versus Doublets Plus Bevacizumab as Initial Therapy of Unresectable Metastatic Colorectal Cancer. J Clin Oncol (2020) 38:3314–24. doi: 10.1200/JCO.20.01225
45. Braithwaite M, Nevala-Plagemann CD, Baron K, Haaland B, Pappas LM, Garrido-Laguna I. Real-world outcomes of patients with BRAF-mutated mCRC treated in the United States. J Clin Oncol (2020) 38:4030–0. doi: 10.1200/JCO.2020.38.15_suppl.4030
46. Subbiah V, Baik C, Kirkwood JM. Clinical Development of BRAF plus MEK Inhibitor Combinations. Trends Cancer (2020) 6:797–810. doi: 10.1016/j.trecan.2020.05.009
47. Gomez-Roca CA, Delord J, Robert C, Hidalgo M, von Moos R, Arance A, et al. Encorafenib (Lgx818), an Oral Braf Inhibitor, in Patients (Pts) with Braf V600E Metastatic Colorectal Cancer (Mcrc): Results of Dose Expansion in an Open-Label, Phase 1 Study. Ann Oncol (2014) 25:iv182. doi: 10.1093/annonc/mdu333.38
48. Kopetz S, Desai J, Chan E, Hecht JR, O’Dwyer PJ, Maru D, et al. Phase II Pilot Study of Vemurafenib in Patients With Metastatic BRAF -Mutated Colorectal Cancer. J Clin Oncol (2015) 33:4032–8. doi: 10.1200/JCO.2015.63.2497
49. Hyman DM, Puzanov I, Subbiah V, Faris JE, Chau I, Blay J-Y, et al. Vemurafenib in Multiple Nonmelanoma Cancers with BRAF V600 Mutations. N Engl J Med (2015) 373:726–36. doi: 10.1056/NEJMoa1502309
50. Yaeger R, Cercek A, O’Reilly EM, Reidy DL, Kemeny N, Wolinsky T, et al. Pilot Trial of Combined BRAF and EGFR Inhibition in BRAF-Mutant Metastatic Colorectal Cancer Patients. Clin Cancer Res (2015) 21:1313–20. doi: 10.1158/1078-0432.CCR-14-2779
51. van Geel RMJM, Tabernero J, Elez E, Bendell JC, Spreafico A, Schuler M, et al. A Phase Ib Dose-Escalation Study of Encorafenib and Cetuximab with or without Alpelisib in Metastatic BRAF -Mutant Colorectal Cancer. Cancer Discovery (2017) 7:610–9. doi: 10.1158/2159-8290.CD-16-0795
52. Tabernero J, Geel RV, Guren TK, Yaeger RD, Spreafico A, Faris JE, et al. Phase 2 results: Encorafenib (ENCO) and cetuximab (CETUX) with or without alpelisib (ALP) in patients with advanced BRAF- mutant colorectal cancer (BRAFm CRC). J Clin Oncol (2016) 34:3544–4. doi: 10.1200/JCO.2016.34.15_suppl.3544
53. Hong DS, Morris VK, El Osta B, Sorokin AV, Janku F, Fu S, et al. Phase IB Study of Vemurafenib in Combination with Irinotecan and Cetuximab in Patients with Metastatic Colorectal Cancer with BRAFV600E Mutation. Cancer Discov (2016) 6:1352–65. doi: 10.1158/2159-8290.CD-16-0050
54. Kopetz S, McDonough SL, Lenz H-J, Magliocco AM, Atreya CE, Diaz LA, et al. Randomized trial of irinotecan and cetuximab with or without vemurafenib in BRAF-mutant metastatic colorectal cancer (SWOG S1406). J Clin Oncol (2017) 35:3505–5. doi: 10.1200/JCO.2017.35.15_suppl.3505
55. Corcoran RB, Atreya CE, Falchook GS, Kwak EL, Ryan DP, Bendell JC, et al. Combined BRAF and MEK Inhibition With Dabrafenib and Trametinib in BRAF V600–Mutant Colorectal Cancer. J Clin Oncol (2015) 33:4023–31. doi: 10.1200/JCO.2015.63.2471
56. Corcoran RB, André T, Atreya CE, Schellens JHM, Yoshino T, Bendell JC, et al. Combined BRAF, EGFR, and MEK Inhibition in Patients with BRAFV600E -Mutant Colorectal Cancer. Cancer Discov (2018) 8:428–43. doi: 10.1158/2159-8290.CD-17-1226
57. Kopetz S, Grothey A, Yaeger R, Van Cutsem E, Desai J, Yoshino T, et al. Encorafenib, Binimetinib, and Cetuximab in BRAF V600E-Mutated Colorectal Cancer. N Engl J Med (2019) 381:1632–43. doi: 10.1056/NEJMoa1908075
58. Le DT, Kim TW, Van Cutsem E, Geva R, Jäger D, Hara H, et al. Phase II Open-Label Study of Pembrolizumab in Treatment-Refractory, Microsatellite Instability-High/Mismatch Repair-Deficient Metastatic Colorectal Cancer: KEYNOTE-164. J Clin Oncol (2020) 38:11–9. doi: 10.1200/JCO.19.02107
59. Andre T, Shiu K-K, Kim TW, Jensen BV, Jensen LH, Punt CJA, et al. Pembrolizumab versus chemotherapy for microsatellite instability-high/mismatch repair deficient metastatic colorectal cancer: The phase 3 KEYNOTE-177 Study. J Clin Oncol (2020) 38:LBA4–4. doi: 10.1200/JCO.2020.38.18_suppl.LBA4
60. Lenz H-J, Lonardi S, Zagonel V, Van Cutsem E, Limon ML, Wong KYM, et al. Nivolumab plus low-dose ipilimumab as first-line therapy in microsatellite instability-high/DNA mismatch repair deficient metastatic colorectal cancer: Clinical update. J Clin Oncol (2020) 38:11–1. doi: 10.1200/JCO.2020.38.4_suppl.11
61. Overman MJ, McDermott R, Leach JL, Lonardi S, Lenz H-J, Morse MA, et al. Nivolumab in patients with metastatic DNA mismatch repair-deficient or microsatellite instability-high colorectal cancer (CheckMate 142): an open-label, multicentre, phase 2 study. Lancet Oncol (2017) 18:1182–91. doi: 10.1016/S1470-2045(17)30422-9
62. Overman MJ, Lonardi S, Wong KYM, Lenz H-J, Gelsomino F, Aglietta M, et al. Durable Clinical Benefit With Nivolumab Plus Ipilimumab in DNA Mismatch Repair–Deficient/Microsatellite Instability–High Metastatic Colorectal Cancer. J Clin Oncol (2018) 36:773–9. doi: 10.1200/JCO.2017.76.9901
63. ClinicalTrials.gov. NCT04034459_FOLFOXIRI Plus Cetuximab vs. FOLFOXIRI Plus Bevacizumab 1st-line in BRAF-mutated mCRC (AIO-KRK-0116). Available at: https://clinicaltrials.gov/ct2/show/NCT04034459.
64. ClinicalTrials.gov. NCT03693170_Encorafenib, Binimetinib and Cetuximab in Subjects With Previously Untreated BRAF-mutant ColoRectal Cancer (ANCHOR-CRC). Available at: https://clinicaltrials.gov/ct2/show/NCT03693170.
65. Grothey A, Tabernero J, Taieb J, Yaeger R, Yoshino T, Maiello E, et al. LBA-5 ANCHOR CRC: a single-arm, phase 2 study of encorafenib, binimetinib plus cetuximab in previously untreated BRAF V600E-mutant metastatic colorectal cancer. Ann Oncol (2020) 31:S242–3. doi: 10.1016/j.annonc.2020.04.080
66. ClinicalTrials.gov. NCT03727763_Cetuximab and Vemurafenib Plus FOLFIRI for BRAF V600E Mutated Advanced Colorectal Cancer (IMPROVEMENT). Available at: https://clinicaltrials.gov/ct2/show/NCT03727763.
67. ClinicalTrials.gov. NCT03668431_Dabrafenib + Trametinib + PDR001 In Colorectal Cancer. Available at: https://clinicaltrials.gov/ct2/show/NCT03668431.
68. ClinicalTrials.gov. NCT04017650_Encorafenib, Cetuximab, and Nivolumab in Treating Patients With Microsatellite Stable, BRAFV600E Mutated Unresectable or Metastatic Colorectal Cancer. Available at: https://clinicaltrials.gov/ct2/show/NCT04017650.
69. Sharma A, Madhunapantula SV, Gowda R, Berg A, Neves RI, Robertson GP. Identification of Aurora Kinase B and Wee1-Like Protein Kinase as Downstream Targets of V600EB-RAF in Melanoma. Am J Pathol (2013) 182:1151–62. doi: 10.1016/j.ajpath.2012.12.019
70. Bhagwat SV, McMillen WT, Cai S, Zhao B, Whitesell M, Shen W, et al. ERK Inhibitor LY3214996 Targets ERK Pathway–Driven Cancers: A Therapeutic Approach Toward Precision Medicine. Mol Cancer Ther (2020) 19:325–36. doi: 10.1158/1535-7163.MCT-19-0183
71. Morris EJ, Jha S, Restaino CR, Dayananth P, Zhu H, Cooper A, et al. Discovery of a novel ERK inhibitor with activity in models of acquired resistance to BRAF and MEK inhibitors. Cancer Discov (2013) 3:742–50. doi: 10.1158/2159-8290.CD-13-0070
72. ClinicalTrials.gov. NCT02906059_Study of Irinotecan and AZD1775, a Selective Wee 1 Inhibitor, in RAS or BRAF Mutated, Second-line Metastatic Colorectal Cancer. Available at: https://clinicaltrials.gov/ct2/show/NCT02906059.
73. ClinicalTrials.gov. NCT02857270_A Study of LY3214996 Administered Alone or in Combination With Other Agents in Participants With Advanced/Metastatic Cancer. Available at: https://clinicaltrials.gov/ct2/show/NCT02857270.
74. Wang Y, Palmer M, Jaeger S, Bagdasarian L, Qiu S, Woolfenden S, et al. Abstract 2140: Dual Wnt and EGFR-MAPK dependency of BRAF V600E -mutant colorectal cancer. In: Molecular and Cellular Biology. Philadelphia, PA: American Association for Cancer Research (2015). p. 2140–0. doi: 10.1158/1538-7445.AM2015-2140
75. Chen G, Gao C, Gao X, Zhang DH, Kuan S-F, Burns TF, et al. Wnt/β-Catenin Pathway Activation Mediates Adaptive Resistance to BRAF Inhibition in Colorectal Cancer. Mol Cancer Ther (2018) 17:806–13. doi: 10.1158/1535-7163.MCT-17-0561
76. ClinicalTrials.gov. NCT02278133_Study of WNT974 in Combination With LGX818 and Cetuximab in Patients With BRAF-mutant Metastatic Colorectal Cancer (mCRC) and Wnt Pathway Mutations. Available at: https://clinicaltrials.gov/ct2/show/NCT02278133.
77. Zhang P, Kawakami H, Liu W, Zeng X, Strebhardt K, Tao K, et al. Targeting CDK1 and MEK/ERK Overcomes Apoptotic Resistance in BRAF-Mutant Human Colorectal Cancer. Mol Cancer Res (2018) 16:378–89. doi: 10.1158/1541-7786.MCR-17-0404
Keywords: colorectal cancer, BRAF mutation, BRAF V600E, BRAF non-V600E, microsatellite instability (MSI)
Citation: Takeda H and Sunakawa Y (2021) Management of BRAF Gene Alterations in Metastatic Colorectal Cancer: From Current Therapeutic Strategies to Future Perspectives. Front. Oncol. 11:602194. doi: 10.3389/fonc.2021.602194
Received: 02 September 2020; Accepted: 09 March 2021;
Published: 25 March 2021.
Edited by:
Claudia Von Arx, Imperial College London, United KingdomReviewed by:
Antonio Ieni, University of Messina, ItalyCopyright © 2021 Takeda and Sunakawa. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yu Sunakawa, y.suna0825@gmail.com
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.